

Abstract
Voltage-gated sodium channels are protein complexes comprised of one pore forming α subunit and two, non-pore forming, β subunits. The voltage-gated sodium channel β subunits were originally identified to function as auxiliary subunits, which modulate the gating, kinetics, and localization of the ion channel pore. Since that time, the five β subunits have been shown to play crucial roles as multifunctional signaling molecules involved in cell adhesion, cell migration, neuronal pathfinding, fasciculation, and neurite outgrowth. Here, we provide an overview of the evidence implicating the β subunits in their conducting and non-conducting roles. Mutations in the β subunit genes (SCN1B-SCN4B) have been linked to a variety of diseases. These include cancer, epilepsy, cardiac arrhythmias, sudden infant death syndrome/sudden unexpected death in epilepsy, neuropathic pain, and multiple neurodegenerative disorders. β subunits thus provide novel therapeutic targets for future drug discovery.
Keywords: Voltage-gated sodium channel, α subunit, β subunit, cell adhesion, neuronal pathfinding, channelopathy, sudden unexpected death in epilepsy, Dravet Syndrome
14.1. The basics of the voltage-gated sodium channel β subunits
There are five voltage-gated sodium channel (VGSC) β subunits, which are encoded by four genes, SCN1B-SCN4B (O’Malley and Isom 2015).SCN1B encodes the β1 subunit and the developmentally regulated splice variant, β1B, while the β2, β3, and β4 subunits are encoded by SCN2B-SCN4B, respectively (Table 1) (Isom et al. 1992; Isom et al. 1995a; Kazen-Gillespie et al. 2000; Morgan et al. 2000; Patino et al. 2011; Qin et al. 2003; Yu et al. 2003). β subunits each contain a large, extracellular V-set immunoglobulin (Ig) domain, making them part of the Ig superfamily of cell adhesion molecules (CAMs) (Brackenbury and Isom 2011; O’Malley and Isom 2015). β1B differs from the other subunits in that it is the only one that is not a type I transmembrane protein, but rather, a soluble, secreted CAM expressed during embryonic development in brain and throughout development, into adulthood, in heart. The C-terminal domain of β1B is encoded by a retained intron, resulting in a unique polypeptide sequence that does not contain a transmembrane segment (Patino et al. 2011). β subunit Ig domains are stabilized by two completely conserved cysteine residues in the extracellular portion, maintaining the β-sheet structure, as established by the X-ray crystal structures of β3 and β4 (Gilchrist et al. 2013; Namadurai et al. 2014).
Table 1:
VGSC genes and their encoded proteins.
| VGSC α subunits |
VGSC β subunits |
||
|---|---|---|---|
| Gene | Protein | Gene | Protein |
| SCN1A | Nav1.1 | SCN1B | β1 |
| SCN2A | Nav1.2 | SCN1B | β1B |
| SCN3A | Nav1.3 | SCN2B | β2 |
| SCN4A | Nav1.4 | SCN3B | β3 |
| SCN5A | Nav1.5 | SCN4B | β4 |
| SCN8A | Nav1.6 | ||
| SCN9A | Nav1.7 | ||
| SCN10A | Nav1.8 | ||
| SCN11A | Nav1.9 | ||
VGSCs are comprised of one pore-forming α subunit and two different β subunits, either a β1 or β3 and a β2 or β4 (Fig. 1) (O’Malley and Isom 2015). β1 and β3 non-covalently associate with α, while β2 and β4 associate with α by cysteine disulfide bonds, Cys-26 and Cys-58, respectively, both of which are located in the extracellular Ig domain (Chen et al. 2012; Gilchrist et al. 2013; McCormick et al. 1998; Meadows et al. 2001; Spampanato et al. 2004). While there is no biochemical evidence to show that β1B associates with α subunits by co-immunoprecipitation, co-expression of β1B and Nav1.5 in heterologous systems results in plasma membrane retention of β1B and increased sodium current density, implicating association with α (Patino et al. 2011; Watanabe et al. 2008). Heterologous co-expression of β1B with Nav1.2 results in changes in current activation and inactivation, while co-expression with Nav1.3 results in subtle alternation of current properties, again suggesting a functional association (Kazen-Gillespie et al. 2000; Patino et al. 2011).
Fig. 1:
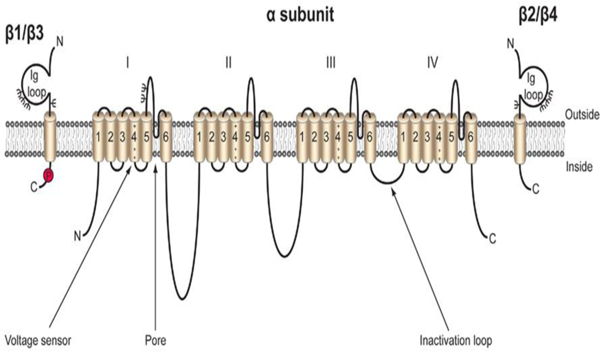
Cartoon diagram of the VGSC. VGSCs are comprised of one pore-forming, or α subunit, and one or two non-pore forming β subunits. The α subunit is made up four domains each of which contain six transmembrane segments. The voltage sensor is located in transmembrane segment four of each domain (Catterall). There are five β subunits, β1-β4, and the developmentally regulated β1B. β1-β4 all contain an intracellular C-terminal domain, a single transmembrane domain, and a large extracellular immunoglobulin (Ig) domain (Isom et al. 1994). β1B also possesses an Ig domain, but does not contain an intracellular or transmembrane domain, resulting in a soluble, secreted protein (Patino et al. 2011). β1 and β3 are non-covalently linked to the α subunit, while β2 and β4 are linked by disulfide bonds. Each β subunit is heavily glycosylated, denoted by Ψ, and β1 also contains an intracellular phosphorylation site at tyrosine 181 (Isom and Catterall 1996; Malhotra et al. 2004). Figure reproduced from (Brackenbury and Isom 2011).
VGSC β subunits are expressed in many tissues and cell types, not all of which are excitable (Brackenbury and Isom 2011). The expression of each specific β subunit is also developmentally regulated. In rodent brain, β1B and β3 are most highly expressed during embryonic development and early life. This differs from heart, in which β1B and β3 expression continues into adult life (Kazen-Gillespie et al. 2000; Patino et al. 2011; Shah et al. 2001). β1 and β2 display peak expression in brain during adulthood (Isom et al. 1992; Isom et al. 1995a). The developmental regulation of β4 subunit expression is yet to be determined. β subunits are localized to a variety of specific sub-cellular compartments. In the brain and peripheral nervous system, β subunits are highly enriched the axon initial segment and nodes of Ranvier (Buffington and Rasband 2013; Chen et al. 2002; Chen et al. 2004; Dhar Malhotra et al. 2001; O’Malley et al. 2009). These sites are important in the initiation and propagation of action potentials in neurons and have a high density of VGSC α subunit expression (Brackenbury et al. 2010). In heterologous cells, the β1 C-terminal domain interacts with the scaffolding protein, ankryin-G, in a tyrosine phosphorylation-dependent manner and a similar mechanism is proposed at the axon initial segment and nodes of Ranvier (Malhotra et al. 2000). In cardiomyocytes, the phosphorylation of β1 may regulate its sub-cellular localization. Tyrosine phosphorylated β1 subunits are localized to the intercalated disks, while non-phosphorylated β1 subunits are localized to t-tubules (Malhotra et al. 2004).
In addition to phosphorylation, the β subunits are post-translationally modified by glycosylation and proteolytic cleavage. All 5 β subunits are highly N-linked glycosylated (Isom et al. 1992). This heavy glycosylation of mature β subunits accounts for about 12 kilodaltons (kDa) of the ~36 kDa total molecular weight. β subunit glycosylation impacts their surface expression and channel modulatory properties (Johnson et al. 2004). Lastly, the transmembrane β subunits are also substrates for sequential cleavage by the β-site amyloid precursor protein cleaving enzyme-1 (BACE1) and γ-secretase (Wong et al. 2005). Initially, BACE1 cleaves β subunits on the extracellular side of the membrane, shedding the Ig domain, which may function as a soluble CAM, similar to β1B. Subsequently, γ-secretase cleaves the β subunits in the lumen of the membrane, generating an intracellular domain (ICD) (Fig. 2) (Haapasalo and Kovacs 2011; Wong et al. 2005). Evidence shows that the β2 subunit ICD translocates to the nucleus where it increases expression of the Nav1.1 α subunit (Kim et al. 2007). A similar mechanism has been proposed, but not shown, for the other β subunits. Sequential cleavage of β subunits may play important roles in mediating neurite outgrowth, migration, and cell adhesion (Brackenbury and Isom 2011; Kim et al. 2005).
Fig. 2:
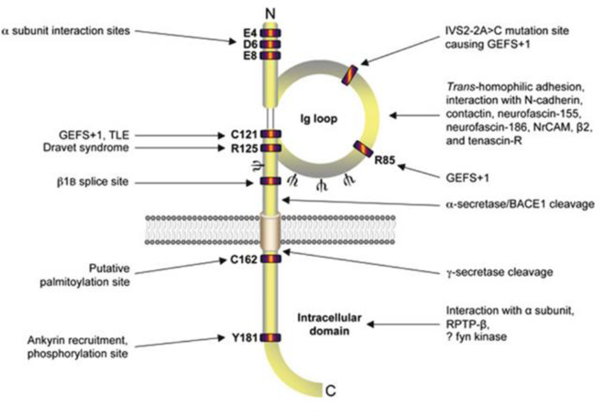
Cartoon diagram of β1/β1B topology. Both intracellular and extracellular residues on β1 are important for interacting with the α subunit (McCormick et al. 1998; Spampanato et al. 2004). Epilepsy-linked mutation sites are clustered in the Ig domain (Audenaert et al. 2003; Meadows et al. 2002; Patino et al. 2009; Scheffer et al. 2007; Wallace et al. 2002; Wallace et al. 1998). The alternative splice site for β1B, ankyrin interaction site (Kazen-Gillespie et al. 2000; Patino et al. 2011; Qin et al. 2003), α-secretase/BACE1/γ-secretase cleavage sites (Wong et al. 2005), N-glycosylation sites (Ψ) (McCormick et al. 1998), tyrosine phosphorylation site (Malhotra et al. 2004), and the putative palmitoylation and fyn kinase interaction site are designated (Brackenbury et al. 2008; McEwen and Isom 2004). Figure reproduced from (Brackenbury and Isom 2011).
14.1.1. Modulation of the ion channel pore by β subunits
VGSC β subunits are traditionally known for their functions in modulating the gating and kinetics of the VGSC pore (Calhoun and Isom 2014). In Xenopus oocytes, expression of Nav1.2 mRNA alone results in sodium currents that are activated and inactivated much slower than those recorded in neurons. Co-injection of β1 or β2 mRNA altered the sodium current parameters (Isom et al. 1992). Co-expression β1 with Nav1.2 increased peak sodium current density, shifted the voltage-dependence of inactivation negatively, and accelerated activation and inactivation in comparison to Nav1.2 is alone (Isom et al. 1992). Co-expression of β2 with Nav1.2 also resulted in increased peak sodium current density and accelerated current inactivation. Expression of the three subunits together, β1, β2, and Nav1.2, yielded the largest peak sodium currents and the most rapidly inactivating channels, most closely mimicking that observed in neurons (Isom et al. 1995a).
In addition to modulating VGSC activity, β1 can also act on Kv1.1, Kv1.2, Kv1.3, Kv1.6, Kv4.2, Kv4.3, and Kv7.2 in Xenopus oocytes or heterologous cells (Deschenes and Tomaselli 2002; Nguyen et al. 2012). Interestingly, β1 and Kv4.2 co-immunoprecipitate from mouse brain. Kv4.2 is a major contributor to A-type potassium current. Knockdown of β1 decreases A-type potassium current and prolongs action potential waveforms in cultured cortical neurons. β1 expression also increases the stability of Kv4.2 in HEK293 cells, leading to increased total and cell surface expression of Kv4.2 (Marionneau et al. 2012).
In addition to oocytes, heterologous mammalian cell lines have been utilized to study VGSC modulation by the β subunits. Although these models are more physiologically relevant, they cannot fully replicate VGSC activity in native excitable cells, such as neurons and cardiomyocytes. In Chinese Hamster Lung (CHL) cells, co-expression of Nav1.2 with β1 increases peak current density and causes a negative shift in the voltage-dependence of activation and inactivation, although to a lesser extent than observed in Xenopus oocytes (Isom et al. 1992; Isom et al. 1995b; Patino et al. 2009). Co-expression of Nav1.2 and β2 in CHL cells does not recapitulate the results observed in Xenopus oocytes, instead resulting in sodium currents that are unchanged or reduced in comparison to the expression of Nav1.2 alone (Kazarinova-Noyes et al. 2001; McEwen et al. 2004). In addition to altering kinetics of the ion channel pore, co-expression of β1 and/or β2 with Nav1.2 affects α subunit surface expression. Co-expression of β1 with Nav1.2 increases α subunit cell surface expression (Isom et al. 1995a). When β2 is added to the experiment, the cell surface expression of α subunits is even further increased, even though β2 cannot generate this effect in the absence of β1 (Kazarinova-Noyes et al. 2001). The modulatory and localization effects of β subunits on α subunits are impacted by the presence of other Ig-superfamily CAMs. In the case of the CAM, contactin, co-expression with Nav1.2 and β1 increases α subunit cell surface expression and sodium current density approximately four-fold over that observed with Nav1.2 plus β1. This is also displayed with NF186, although to a lesser extent than the effects observed with contactin. β1B co-expression with Nav1.2 in CHL cells also increases α subunit surface expression and peak sodium current density, although, this combination only has a modest effect on channel activation and inactivation (Kazen-Gillespie et al. 2000; Patino et al. 2011).
The effects of the β subunits on a variety of α subunits have also been studied in Chinese Hamster Ovary (CHO) cells and Human Embryonic Kidney (HEK) cells. β1 or β3 co-expression with Nav1.3 in CHO cells results in a negative shift in the voltage dependence of inactivation, but does not influence the rate of inactivation. In this same system, co-expression of β2 with Nav1.3 had no effect on the gating or kinetics of the ion channel pore (Meadows et al. 2002). Co-expression of β3 with Nav1.5 in CHO cells results in a negative shift in the voltage dependence of inactivation, but decreases the rate of inactivation (Ko et al. 2005). Co-expression of β1B with Nav1.3 in CHO cells has no effect on Nav1.3 cell surface expression or sodium current density, different from the large effect of β1B observed in CHL cells (Kazen-Gillespie et al. 2000; Patino et al. 2011). In HEK cells, co-expression of Nav1.5 and β4 results in a negative shift in the voltage dependence of inactivation in comparison to expression of Nav1.5 alone (Medeiros-Domingo et al. 2007). The β4 subunit, when expressed with Nav1.2 or Nav1.4, induces a negative shift in the voltage dependence of activation (Yu et al. 2003). This is also the case for co-expression of β4 with Nav1.1, although this results in increased levels of non-inactivating current (Aman et al. 2009). Also in HEK cells, β1 and β3 subunits each modulate activity, cell surface expression, and glycosylation state of Nav1.7. β1 or β3 co-expression with Nav1.7 resulted in shifted activation and inactivation and increased sodium current density. Co-expression of β1 also resulted in alternative glycosylation of Nav1.7, while co-expression with β3 led to increased expression of fully-glycosylated Nav1.7 (Laedermann Cé et al. 2013). Overall, studies on β subunit modulation of VGSCs in heterologous systems have revealed cell type, β subunit, and α subunit specific effects.
The most physiologically relevant method to study VGSC modulation by the β subunits is to utilize primary cells, e.g. neurons or cardiomyocytes. In these native cells, β subunit effects are, in general, more modest than observed in heterologous over-expression systems. Scn1b-null mice, lacking both β1 and β1B, model the epileptic encephalopathy Dravet syndrome, and exhibit spontaneous seizures, ataxia, and premature death around post-natal day (P) 19 (Chen et al. 2004). Acutely isolated P10-P18 Scn1b-null pyramidal and bipolar hippocampal neurons show no differences in VGSC activity compared to age-matched wild-type animals (Chen et al. 2004; Patino et al. 2009). However, slice recordings from this age range revealed hyperexcitability in the Scn1b-null CA3 hippocampal region as well as epileptiform activity in the hippocampus and cortex, suggesting altered VGSC activity in axons or dendrites (Patino et al. 2009). There are altered sodium currents and decreased excitability in cultured Scn1b-null cerebellar granule neurons (CGNs) (Brackenbury et al. 2010). In contrast, acutely isolated Scn1b-null dorsal root ganglion (DRG) neurons are hyperexcitable (Brackenbury et al. 2010; Lopez-Santiago et al. 2011). These results suggest that the effects of β1 and β1B in brain are neuronal cell-type specific, consistent with that observed in heterologous cells. Similar to that observed in heterologous systems, β1/ β1B expression in vivo affects the expression of α subunits, especially Nav1.1 and Nav1.3. In the Scn1b-null hippocampal CA3 region, Nav1.1 expression is decreased, while Nav1.3 expression is increased (Chen et al. 2004). β2 also modulates VGSC gating and kinetics in vivo. Acutely isolated Scn2b-null hippocampal neurons display a negative shift in the voltage dependence of inactivation in comparison to neurons from age-matched, wild-type mice (Chen et al. 2002). Acutely isolated Scn2b-null small-fast DRG neurons have decreased sodium current density and decreased rates of TTX-sensitive sodium current activation and inactivation (Lopez-Santiago et al. 2006). Importantly, the β4 intracellular domain is postulated to play a role in resurgent sodium current, or the influx of sodium ions through the ion channel pore during repolarization. β4 knockdown in mouse CGNs showed reduced resurgent sodium current and repetitive firing (Bant and Raman 2010). Furthermore, expression of a β4 intracellular domain peptide in CA3 neurons, which do not endogenously express β4 subunits, generates resurgent sodium current (Grieco et al. 2005). This activity is particularly important in high-frequency firing neurons. Scn4b-null mice have defects in sodium current modulation. Scn4b-null mice have reduced resurgent sodium current and repetitive firing in medium spiny neurons of the striatum, as well as increased failure rates of inhibitory postsynaptic currents with repetitive stimulation (Miyazaki et al. 2014). β1 and β1B are also implicated in regulating resurgent sodium current in the cerebellum, as Scn1b-null CGNs have normal transient sodium current, but decreased resurgent sodium current, even though the overall protein expression of β4 is unchanged (Brackenbury et al. 2010). Together, these data indicate that modulation of sodium current by the β subunits in vivo is cell-type-, subcellular domain-, β subunit-, and α subunit-specific.
VGSC β subunits are also important regulators of excitability in the heart. In ventricular cardiomyocytes isolated from Scn1b-null mice, transient and persistent sodium currents are increased due to increased Scn5a and Nav1.5 expression, resulting in prolongation of action potential repolarization and the QT interval (Lin et al. 2014; Lopez-Santiago et al. 2007). Furthermore, Scn1b-null mice display increased susceptibility to polymorphic ventricular arrhythmias. Scn1b-null ventricular cardiomyocytes also have increased tetrodotoxin (TTX)-sensitive sodium current, increased Nav1.3 mRNA levels, increased incidence of delayed after-depolarizations, delayed Ca2+ transients, and frequent spontaneous Ca2+ release. Addition of TTX prevented the majority of changes in Ca2+ handling, indicating mutations in Scn1b may result in disrupted intracellular Ca2+ homeostasis in ventricular myocytes (Lin et al. 2014). Scn2b deletion in mice leads to atrial and ventricular arrhythmias and increased levels of atrial fibrosis. These animals exhibit region-specific effects in heart. Scn2b-null ventricular myocytes show reduced sodium and potassium currents, with conduction slowing in the right ventricle compared to wild-type. Scn2b-null atria had normal levels of sodium current compared to wild-type (Bao et al. 2016). Scn3b-null mice also show abnormal cardiac excitability, with ventricular tachycardia from electrical stimulation that is not observed in wild-type mice. Scn3b-null hearts also demonstrate atrial tachycardia during atrial burst pacing (Hakim et al. 2008).
14.1.2. The β subunits as cell adhesion molecules
All five β subunits have an extracellular Ig domain and belong to the Ig superfamily of CAMs (Isom and Catterall 1996). Importantly, β subunits have also been shown experimentally to function as CAMs (Isom 2002). An especially large body of work in this area has been completed on the β1 subunit. In Drosophila S2 cells expressing either β1 or β2, large aggregates form, suggesting these molecules can participate in trans homophilic cell adhesion in vitro (Malhotra et al. 2000). Upon β1-β1 trans homophilic cell adhesion in Drosophila S2 cells, ankyrin is recruited to the point of cell-cell contact (Meadows et al. 2001). Ankyrin binds to the β1 subunit via the intracellular C-terminal domain in a tyrosine phosphorylation-dependent manner. When residue Y181 of β1 is phosphorylated, ankyrin is unbound, while when Y181 is not phosphorylated, ankyrin binds to β1, indicating that downstream signaling events occur in response to cell-cell adhesion (Malhotra et al. 2002). In addition, β1 subunits can form heterophilic interactions with other CAMs, including contactin, N-cadherin, NrCAM, neurofascin-155, neurofascin-186, and the VGSC β2 subunit as well as the extracellular matrix protein, tenascin-R (Fig. 3) (McEwen and Isom 2004; Xiao et al. 1999). β2 subunits can also participate in heterophilic adhesion in vitro with both tenascin-R and tenascin-C (Srinivasan et al. 1998; Xiao et al. 1999). Drosophila S2 cells expressing β3 subunits do not aggregate, suggesting that β3 does not participate in trans homophilic adhesion (McEwen et al. 2009). In contrast, β3 subunits expressed in HEK cells participate in trans heterophilic adhesion with other CAMs, although this does not result in β3-ankyrin binding (McEwen et al. 2009; McEwen and Isom 2004; Ratcliffe et al. 2001). The function of β4 in cell adhesion remains more poorly understood (Brackenbury and Isom 2011). Insights from crystallographic, mutagenic, and photo-crosslinking studies have revealed the structural importance of an antiparallel interface between β4 subunits in trans homophilic adhesion (Shimizu et al. 2016). Recent evidence shows that β4 Ig domains interact in a parallel manner involving a disulfide bond between cysteine 58 and hydrophobic and hydrogen bonding interactions between residues 30 through 35. Deletion of the β4 N-terminal domain led to decreased cell adhesion and increased association with the α subunit, revealing the importance of β4 cis dimerization (Shimizu et al. 2017).
Fig. 3:
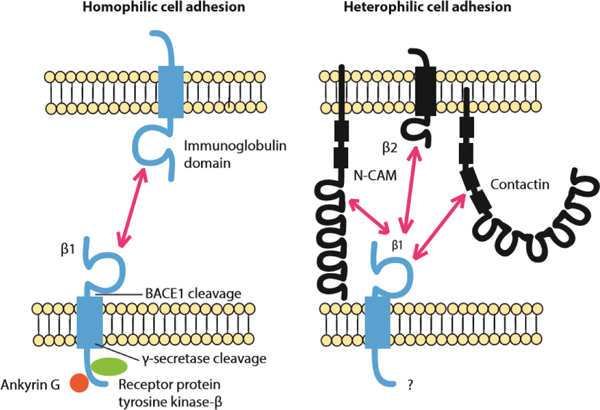
β1 participates in homophilic and heterophilic cell adhesion (Malhotra et al. 2000). Left: Schematic of β1-β1 homophilic cell adhesion and its downstream signaling. At points of cell-cell contact β1 binds to ankyrin in a phosphorylation-dependent manner. When β1 is not phosphorylated it is bound to ankyrin, while when tyrosine 181 is phosphorylated it is not bound to ankyrin (Malhotra et al. 2000; Malhotra et al. 2002). In rat brain β1 interacts with receptor protein tyrosine kinase-β which may contribute to regulating the β1 phosphorylation state (Ratcliffe et al. 2000). Right: β1 participates in heterophilic cell adhesion with N-CAM, VGSC β2 subunits, and contactin.
Consistent with the role of β1 and β2 in cell adhesion, these molecules have been identified to mediate neurite outgrowth in CGNs (Davis et al. 2004). In this series of experiments, CGNs were grown on a monolayer of CHL cells that either did, or did not, express β subunit proteins. When β1-β1 trans homophilic cell adhesion occurred between the CGN and the monolayer expressing β1, neurite length was longer than when it did not. In contrast, β2-mediated homophilic adhesion resulted in decreased neurite length while β4 had no effect on this biological output. These data suggest that β1-β1 trans homophilic cell adhesion initiates a signal transduction cascade to drive neurite outgrowth in vitro while β2-mediated signaling may be inhibitory. Cell adhesion-mediated neurite outgrowth has been shown to occur through two downstream pathways: either via an epidermal growth factor receptor (EGFR) or fibroblast growth factor receptor (FGFR) mediated signal transduction cascade, or through the fyn kinase pathway (Brackenbury et al. 2008). Inhibitors of FGFR and EGFR had no effect on β1-mediated neurite outgrowth in CGNs. In contrast, CGNs isolated from fyn-null mice grown on a CHL monolayer expressing β1 did not show extended neurite length, suggesting that β1-mediated neurite outgrowth signals through a pathway that involves fyn kinase. This hypothesis is further supported by results showing that β1 subunit peptides associate with fyn in detergent-resistant membrane fractions solubilized from mouse brain (Brackenbury et al. 2008). The proteolytic processing of β1 by BACE1 and γ-secretase is also important for β1-mediated neurite outgrowth, as inhibitors of γ-secretase block β1-mediated neurite outgrowth (Fig. 4) (Brackenbury and Isom 2011). The secreted Scn1b splice variant, β1B, increases neurite outgrowth to a similar extent as full-length β1 (Patino et al. 2011). Outside of the central nervous system, the β1 subunit can induce the growth of neurite-like features from cultured breast cancer cells, suggesting a possible developmental role for β1 in other cell-types (Nelson et al. 2014).
Figure 4:
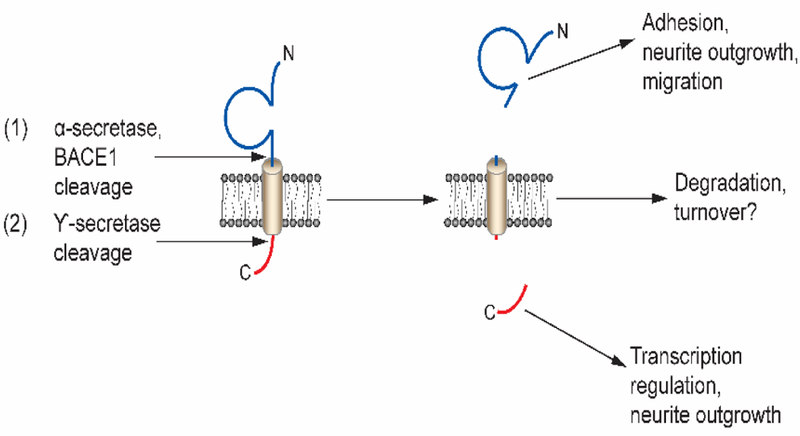
β subunits are sequentially cleaved by α-secretase and/or the β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) and subsequently by γ-secretase in the lumen of the membrane. Sequential cleavage generates a soluble extracellular N-terminal domain and intracellular C-terminal domain (Kim et al. 2005; Wong et al. 2005). The soluble N-terminal domain participates in cell adhesion and migration, while the intracellular domain modulates neurite outgrowth and, in the case of β2, regulates VGSC gene transcription in vitro (Davis et al. 2004; Kim et al. 2007; Kim et al. 2005; Miyazaki et al. 2007). Figure reproduced from (Brackenbury and Isom 2011).
β1/β1B-mediated cell adhesive activity has been implicated in neuronal development in vivo. In the Scn1b null mouse, there are fewer optic nerve nodes of Ranvier. At the ultrastructural level, optic nerve, spinal cord, and sciatic nerve nodes have abnormal architecture (Chen et al. 2004). Scn1b-null mice also have defects in neuronal pathfinding and fasciculation in multiple brain regions. In normal cerebellum, CGN axons project from the granule layer to the molecular layer, where they form parallel fibers. In the Scn1b null mouse, CGN axons are defasiculated, forming a disrupted molecular layer. Abnormal pathfinding and defasiculation are also observed in the Scn1b-null corticospinal tract and hippocampus. In a related model, dendritic arborization of pyramidal neurons in subiculum is reduced in Scn1b-C121W mutant animals (Reid et al. 2014). The Scn2b- and Scn3b-null mouse models do not have an apparent neurological phenotype, although Scn2b-null mice have increased seizure susceptibility and altered pain sensation (Chen et al. 2002; Hakim et al. 2010; Hakim et al. 2008; Lopez-Santiago et al. 2006; O’Malley and Isom 2015). CNS abnormalities in the Scn4b-null mouse model were recently described. Scn4b-null mice display deficits in balance and motor coordination and resurgent sodium current in null Purkinje neurons was reduced by approximately 50 percent. This was further validated using in vivo short hairpin RNA knockdown of β4 in adult Purkinje neurons (Ransdell et al. 2017). Overexpression of the β4 subunit in Neuro2a cells results in increased neurite outgrowth, dendrite formation, and filopodia-like protrusions, suggesting a role for β4 in neuronal pathfinding and migration (Oyama et al. 2006).
14.2. The role of β subunits in pathophysiology
14.2.1. Cancer
VGSC β subunits are expressed in prostate, breast, lung, and cervical cancers. This expression is subunit specific and varies by cancer type (Brackenbury 2012). β1 is found in breast, prostate, and cervical cancers, while β2 has been detected in breast and prostate cancers, and β3 in prostate and lung cancers (Chioni et al. 2009; Diss et al. 2008; Hernandez-Plata et al. 2012; Jansson et al. 2012; Roger et al. 2007).
β1 and β2 expression levels correspond with metastatic potential in prostate cancer cells (Chioni et al. 2009; Jansson et al. 2012). Experiments performed in vitro with breast cancer cells have shown that β1 expression enhances cell-cell and cell-substrate adhesion and decreases cell migration (Chioni et al. 2009). On the other hand, data suggest that β1 contributes to cell invasion during metastasis in breast cancer cells (Chioni et al. 2009). Overexpression of β1 increases vascular endothelial growth factor secretion and angiogenesis, and decreases apoptosis in endothelial cells (Andrikopoulos et al. 2011). β1 overexpression in an orthotopic mouse model of breast cancer increases tumor growth and metastasis (Nelson et al. 2014). In the well-defined prostate cancer cell line, LNCaP, β2 overexpression increases cell length, but reduces cell volume, which may result in increased cellular motility and invasion. In a wound healing assay, cells overexpressing β2 migrate farther than controls. To the contrary, over-expression of β2 decreases tumor formation and growth after tumor implantation into nude mice. Furthermore, β2 over-expression enhances invasion and growth on laminin (Jansson et al. 2012).
Unlike β1 and β2, β3 is postulated to function as a tumor suppressor because its amino acid sequence contains two p53 response elements. In p53-null mouse embryo fibroblasts, Scn3b is increased after andriamycin treatment and β3 expression induces p53-dependent apoptosis (Adachi et al. 2004). Less is known about the expression of β4 in cancer, although β4 expression levels are lower in cervical and prostate cancer cells in comparison to noncancerous cells (Diss et al. 2008; Hernandez-Plata et al. 2012). β4 co-expression with Nav1.5 has also been shown to play a role in CD4+ T cell development (Lo et al. 2012). These data suggesting roles for VGSC β subunits in cancer indicate that these molecules are important to the functioning of non-excitable, in addition to excitable, cells.
14.2.2. Cardiac arrhythmia
The VGSC β subunits are expressed in the human heart and conduction system. Here, SCN1B is expressed at the highest levels in atria and endocardium, while SCN2B and SCN3B are expressed throughout the human heart (Gaborit et al. 2007). In mouse ventricular cardiomyocytes, β2, β4, and tyrosine-phosphorylated β1 subunits are expressed at the intercalated disc along with Nav1.5, the predominant heart α subunit, (Maier et al. 2004; Malhotra et al. 2004). At t-tubules of ventricular cardiomyocytes, β2, β3, and non-phosphorylated β1 are co-expressed with Nav1.1, Nav1.3, and Nav1.6 α subunits (Dhar Malhotra et al. 2001; Maier et al. 2004; Malhotra et al. 2004). Cardiac VGSC β subunits are critical for action potential upstroke, conduction velocity, and excitation-contraction coupling, suggesting that abnormal expression of β subunits may contribute to cardiac disease states (Remme and Bezzina 2010).
Mutations in genes encoding VGSC β subunits are linked to multiple types of cardiac disease (Bao and Isom), including long QT syndrome (LQTS) (Medeiros-Domingo et al. 2007; Riuro et al. 2014), a ventricular arrhythmia in which there is delayed action potential repolarization, resulting in prolongation of the QT interval on the electrocardiogram. LQTS causes an increased risk of ventricular fibrillation (VF) and sudden cardiac death (Alders and Christiaans 1993). There is now an extensive list of LQTS mutations, including mutations in ion channel genes (Nakano and Shimizu 2016; Tester and Ackerman 2014). Two mutations, resulting in gain-of-function activity, have been identified in SCN1B and SCN4B, respectively (Medeiros-Domingo et al. 2007; Nakano and Shimizu 2016; Riuro et al. 2014), including β1B p.P213T, which results in increased late sodium current and action potential duration, shifted window current, and decreased rate of slow inactivation, and β4 p.L179F, which results in a positive shift in sodium current inactivation causing abnormal action potential repolarization (Medeiros-Domingo et al. 2007; Riuro et al. 2014).
Multiple mutations in SCN1B have also been linked to Brugada syndrome (BrS) (Holst et al. 2012; Hu et al. 2012; Watanabe et al. 2008; Yuan et al. 2014). BrS patients have an increased risk of sudden cardiac death due to VF (Watanabe et al. 2008). SCN1B mutations are associated with reductions in Nav1.5-generated sodium current density, hyperpolarized voltage-dependence of sodium current inactivation, and/or alterations in the rate of recovery from inactivation (Watanabe et al. 2008). A missense mutation in SCN2B, p.D211G, has been linked to BrS and results in reduced sodium current density by decreasing Nav1.5 cell surface expression (Riuro et al. 2013). Mutations in all four of the VGSC β subunit genes are linked to atrial fibrillation (AF) (Li et al. 2013; Olesen et al. 2011; Wang et al. 2010; Watanabe et al. 2009).
Mouse models lacking individual β subunits show the important roles of these subunits in cardiac function. Cardiac function in Scn1b-null mice is altered, even after blocking autonomic input. These animals exhibit action potential depolarization and prolonged QT intervals, suggesting a LQTS phenotype. Scn1b-null ventricular myocytes have increased transient and persistent sodium current in comparison to wild-type animals, and an increase in Nav1.5 transcript and protein levels (Lopez-Santiago et al. 2007). Scn1b-null mice also show increased TTX-sensitive sodium current in the ventricular myocyte midsection, concurrent with increased Scn3a mRNA levels. Cardiac-specific Scn1b-null mice also display increased Scn3a mRNA, lengthened action potential repolarization, delayed after repolarizations and Ca2+ transients, and frequent spontaneous release of Ca2+. Alterations in Ca2+ levels were blocked by TTX (Lin et al. 2014). Scn2b-null mice exhibit a mixed, Brugada-atrial fibrillation like phenotype. Scn2b-null ventricular myocytes have alterations in sodium and potassium current densities, particularly in the right ventricular outflow tract. Similar to Scn2b-null brain, total levels of Nav1.5 protein were found to be similar to those from wild-type animals, supporting the hypothesis that a main function of β2 in the ventricle is to chaperone VGSC α subunits to the cell surface without changing overall channel expression. In contract, Scn2b null atria had normal levels of sodium and potassium currents but increased levels of fibrosis. Lastly, Scn2b-null hearts display increased susceptibility to atrial fibrillation and repolarization dispersion compared to wild-type animals (Bao et al. 2016). Scn3b-null mice also show cardiac dysfunction. In both atria and ventricles, Scn3b-null mice display an increased susceptibility to arrhythmia, reduced peak sodium current, conduction abnormalities that are similar to Brugada syndrome models, bradycardia, AV block, and deficits in sinoatrial node recovery (Hakim et al. 2008). The role of β4 in cardiac function has yet to be reported using the null mouse model.
14.2.3. Epilepsy
Many mutations in VGSC genes have been linked to epilepsies, including SCN1B (Kaplan et al. 2016). There has as yet been no explicit neurological phenotype associated with SCN3B and no epilepsy phenotype linked to SCN4B. The mutation SCN1B p.C121W, identified in a patient with Generalized Epilepsy with Febrile Seizures plus (GEFS+), was one of the first epilepsy mutations ever identified (Wallace et al. 1998). GEFS+ patients initially experience febrile seizures, which then progress to persistent afebrile seizures (Wallace et al. 1998). The heterozygous p.C121W knock-in mouse has been shown to model the GEFS+ phenotype (Wimmer et al. 2010). The p.C121W mutation disrupts a key disulfide bond in the Ig loop (McCormick et al. 1998; Wallace et al. 1998). Although p.C121W traffics appropriately to the plasma membrane and its co-expression increases VGSC α subunit cell surface levels in culture, it is unable to participate in trans-homophilic cell adhesion or modulate sodium current in vitro (Meadows et al. 2002). Studies of p.C121W subcellular localization in cultured neurons showed that, unlike wildtype β1, mutant subunits do not traffic to specialized axonal subdomains including the AIS and nodes of Ranvier. Phenotypically, p.C121W homozygous mice model Dravet syndrome, displaying brain-region specific hyperexcitability, reduced dendritic arborization of pyramidal neurons in the subiculum, and an increased susceptibility to febrile and spontaneous seizures (Wimmer et al. 2010). Animals that are heterozygous for this mutation are more susceptible to hyperthermia-induced seizures than Scn1b(+/−) or Scn1b (+/+) animals. Even though β1-C121W is localized to the cell surface of neurons in vivo, they are incompletely glycosylated and do not interact with α subunits (Kruger et al. 2016). Additional GEFS+ mutations in SCN1B, p.R85C and p.R85H, have also been studied in heterologous cells in vitro (Xu et al. 2007). Both mutants have decreased expression compared to wild-type and are unable to modulate α subunits. Although of the two, only p.R85H has been shown to reach the plasma membrane (Patino et al. 2009; Xu et al. 2007).
The Scn1b-null mouse line is a model of Dravet Syndrome (DS), and mutations in Scn1b are linked to DS (Chen et al. 2004; Patino et al. 2009), a severe and intractable pediatric epileptic encephalopathy that typically presents within the first year of life with myoclonic seizures that can change etiology over time. DS patients also suffer from a variety of comorbidities including ataxia, behavioral and developmental delay, and a high risk of sudden unexpected death in epilepsy, or SUDEP (Gataullina and Dulac 2017). DS mutations in SCN1B are homozygous recessive. The first DS mutation identified in SCN1B was p.R125C. This mutation has abnormal trafficking and does not reach the cell surface in vitro, resulting in a functional null phenotype (Patino et al. 2009). An additional SCN1B DS mutations, p.I106F, was later identified, although the mechanism underlying this mutation remains unknown (Ogiwara et al. 2012). Scn1b-null mice further validate the role of β1 in DS. These animals have frequent spontaneous seizures and abnormal neuronal excitability and development, consistent with that observed in DS patients (Chen et al. 2004). In addition, Scn1b null mice die at ~P 21, and are thus a SUDEP model.
Heterozygous mutations in SCN1B have been linked to a variety of other epilepsies. These include p.R85C, p.R85H, p.R125L, and an in-frame deletion mutation (Fendri-Kriaa et al. 2011; Scheffer et al. 2007; Wallace et al. 1998). One mutation that is specific to the developmentally regulated splice variant, β1B, has also been identified, p.G257R, and is linked to idiopathic epilepsy in multiple pedigrees. In vitro, this mutation also has defects in membrane trafficking (Patino et al. 2011). Except for this mutation specific to β1B, all epilepsy-linked mutations in SCN1B code for amino acids in the Ig loop domain, suggesting the clinical relevance of cell adhesion in the pathogenesis of epilepsy.
Scn2b-null mice express approximately half of normal levels of cell surface TTX-sensitive VGSCs in brain. These animals are also more prone to pharmacologically induced seizures compared to wild-type animals (Chen et al. 2002). Additionally, a polymorphism in SCN2B (rs2298771) has been associated with idiopathic epilepsy (Baum et al. 2014). In conclusion, the β1 and β2 subunits play critical roles in epilepsy.
14.2.4. Neurodegenerative disorders
β subunits have been implicated in neurodegenerative disorders including amyotrophic lateral sclerosis (ALS), Alzheimer’s Disease (AD), Huntington’s disease (HD), Multiple Sclerosis (MS), and Parkinson’s disease (PD) (Calhoun and Isom 2014; O’Malley and Isom 2015). ALS is characterized by the degeneration of motor neurons in the spinal cord, motor cortex, and brainstem (Al-Chalabi et al. 2017). Differential gene expression of Scn1b and Scn3b have been observed in the Sod1 mouse model of ALS. Scn1b mRNA and protein are decreased, while there is increased Scn3b mRNA and protein in ventral dorsal horn. Neuronal hyperexcitability is found in ALS, thus, alterations in the expression of Scn1b and Scn3b, as well as the changes reported in the expression of Nav1.6, may explain hyperexcitability in ALS patients (Nutini et al. 2011).
Like that of the Amyloid Precursor Protein (APP), most famously known for its potential implications in AD, β subunits are substrates for sequential cleavage by β-site APP cleaving enzyme-1 (BACE1) and γ-secretase, potentially linking the β subunits to AD (Wong et al. 2005). In AD pathology, APP is initially cleaved on the extracellular portion of the membrane by BACE1 and then subsequently cleaved in the lumen of the membrane by γ-secretase, generating the amyloid β (Aβ) peptide. Aβ then accumulates and forms amyloid plaques. BACE1 is ubiquitously expressed throughout the body, but is expressed at highest levels in the pancreas and brain (Cole and Vassar 2007). The expression of BACE1 increases with age in the cortex of AD patients (Evin et al. 2010). Interestingly, AD patients are at increased risk of seizures, further supporting a potential role of VGSCs in AD (Pandis and Scarmeas 2012). BACE1 cleavage of β2 reverses normal β2 modulation of VGSC β subunits. In BACE1-null mice, decreased cleavage of β2 (or possibly other BACE1 substrates, including other VGSC β subunits) may contribute to the increased neuronal excitability observed in AD patients (Kim et al. 2011). In addition, SCN3B mRNA is lower in AD brains with neurofibrillary tangles (NFTs), another pathological issue displayed in some AD cases, suggesting β subunits may be implicated in the formation of NFTs and hyperexcitability in AD (Dunckley et al. 2006).
β2 and β4 have been linked to HD, a genetic, neurodegenerative disease that affects motor coordination and mental ability. Ultimately, many of these patients lose their ability to walk and/or talk. In HD patient postmortem brain samples, SCN4B is downregulated in the striatum. This is mimicked in mouse models, where it has been shown to occur prior to loss of motor coordination. In vitro, β4 overexpression is implicated in neuronal development, suggesting that in HD, β4 dysregulation may contribute to neural degeneration. A decrease in β2 expression is also observed in the same mouse model of HD, but later in the pathogenesis of disease than observed for Scn4b (Oyama et al. 2006).
Although Scn2b-null mice have normal myelination, at least in the optic nerve, deletion of Scn2b is neuroprotective in the Experimental Allergic Encephalomyelitis (EAE) model of MS. Interestingly, Scn2b deletion in the EAE mouse model leads to decreased axonal degeneration, fewer demyelinated and dysmyelinated axons, reduced phenotypic severity, and increased survival (O’Malley et al. 2009). β2 may also be implicated in MS through sequential cleavage by BACE1 and γ-secretase. In cerebrospinal fluid from MS patients, there is decreased BACE1 activity and this biomarker in MS is linked to a more severe and prolonged disease state. Throughout MS progression, BACE1 expression continues to decline (Mattsson et al. 2009).
VGSC β1 subunits are also implicated in maintaining normal myelination. Scn1b-null mice phenotypically display abnormal optic nerve myelination, spinal cord dysmyelination, increased axonal degeneration, fewer optic nerve nodes of Ranvier, and defects in nodal ultrastructure in both the central and peripheral nervous systems. Loss of β1 expression, and thus adhesion, at nodes of Ranvier leads to abnormalities in the formation of paranodal junctions, suggesting β1 contributes to myelination (Chen et al. 2004).
Increased expression and glycosylation of the VGSC β4 subunit compared to wild-type animals has been identified in a mouse model of PD. Studies of neurite outgrowth in response to expression of WT vs. mutant β4 that could not be glycosylated showed that neurite outgrowth was accelerated, with an increased level of filopdia-like protrusions. Thus, the glycosylation state of β4 may be critical for neuronal morphology and may be involved in PD pathogenesis (Zhou et al. 2012). Overall, β subunits contribute to myelination and neurodegenerative disease states through a variety of mechanisms.
14.2.5. Neuropathic pain
A variety of factors can cause neuropathic pain, including genetic mutations and nerve injury. This leads to defects in nociception, the neuronal pathways implicated in sensing noxious stimuli. The VGSC β subunits are expressed in dorsal root ganglion (DRG) neurons and peripheral nerves, suggesting potential roles for these proteins in neuropathic pain (Lopez-Santiago et al. 2006). Behavioral pain phenotypes are difficult, if not impossible, to study in Scn1b-null mice due to their severe seizures and early post-natal death (Chen et al. 2004). In spite of this, Scn1b-null DRG neurons are hyperexcitable, suggesting that these mice may have some form of allodynia (Lopez-Santiago et al. 2011). On the other hand, Scn1b mRNA levels are increased in a model of chronic constrictive nerve injury, complicating the interpretation of the role of β1 in neuropathic pain (Blackburn-Munro and Fleetwood-Walker 1999).
Studies examining the role of β2 in neuropathic pain have also led to conflicting results. While Scn2b-null mice are less sensitive than wild-type littermates in models of inflammatory and neuropathic pain, β2 protein levels are increased in injured and non-injured wild-type neurons in spared nerve injury and spinal nerve ligation models in rat (Lopez-Santiago et al. 2006; Pertin et al. 2005). The latter occurs without a corresponding increase in mRNA levels (Pertin et al. 2005). Lastly, Scn2b mRNA levels are downregulated in cervical sensory ganglia after avulsion injury, but increased in a model of chronic constrictive nerve injury (Blackburn-Munro and Fleetwood-Walker 1999; Coward et al. 2001).
Scn3b mRNA expression is increased in multiple pain models, including in small C-fibers, in a chronic constrictive injury model in rats, in Aδ fibers in the streptozotocin model of diabetic neuropathy in rat, in the small and medium fibers in the sciatic nerve transection model, and finally, in the spared nerve injury model of neuropathic pain suggesting Scn3b may play a role in modulating pain (Shah et al. 2000; Shah et al. 2001; Takahashi et al. 2003)
Although there are little data to directly implicate β4 in pain, the C-terminal portion of β4 plays a role in generating resurgent sodium current in DRG neurons (Grieco et al. 2005). Paroxysmal Extreme Pain Disorder (PEPD) is an inherited neuropathic pain syndrome linked to gain-of-function mutations in SCN9A, encoding Nav1.7. When PEPD-linked Nav1.7 mutants are co-expressed with the C-terminal β4 peptide, differential enhancement of resurgent current is observed, suggesting a potential role for β4 in pain (Theile et al. 2011). In all, the β subunits contribute to pain phenotypes in a cell-type and subunit-specific manner.
14.2.6. Sudden Infant Death Syndrome (SIDS) and Sudden unexpected death in epilepsy (SUDEP)
Sudden Infant Death Syndrome, or SIDS, is the unexpected death of a child up to one year of age where a clear cause of death cannot be identified via autopsy (Krous et al. 2004). The mechanism of SIDS remains to be elucidated, but one out of ten cases is associated with cardiac ion channel gene mutations, including in genes encoding the β subunits (Van Norstrand and Ackerman 2009). p.V36M and p.V54G mutations in SCN3B and p.S206L in SCN4B have been linked to SIDS (Tan et al. 2010). Importantly, p.V36M in SCN3B has also been linked to idiopathic ventricular fibrillation, a potential fatal cardiac arrhythmia, and p.S206L in SCN4B also leads to abnormal excitability in rat ventricular myocytes (Tan et al. 2010; Valdivia et al. 2010). There has been one instance of SIDS in a child with a p.R214Q in β1B, which has also been associated with Brugada Syndrome (Hu et al. 2012). β1B modulates Nav1.5 function, potentially providing an underlying mechanism for SCN1B linked cardiac dysfunction (Patino et al. 2011). To date, no mutations in SCN2B have been linked to SIDS.
Some ion channel genes that have been linked to SIDS have also been linked to Sudden Unexpected Death in EPilepsy (SUDEP). SUDEP is defined as the sudden and unexpected death of a person with epilepsy without any identifiable cause of death during autopsy (Nashef et al. 2012). SUDEP occurs in up to 17% of epileptic patients and those diagnosed with Dravet syndrome (DS) are at an especially high risk for SUDEP (Ficker et al. 1998). Seizures that are difficult to treat by pharmacological intervention are also associated with increased SUDEP risk (Hesdorffer et al. 2012). Currently, there are no reliable biomarkers for SUDEP, but it is likely that death is initiated by dysfunction in multiple organ systems, including autonomic dysfunction, cardiac arrhythmia, central or obstructive apnea, hypoventilation, and pulmonary edema (Surges and Sander 2012). Several types of cardiac events are known to occur during or after seizure activity in epilepsy patients. These include asystole, atrial fibrillation, bradycardia, tachycardia, and T-wave alterations (Jansen and Lagae 2010). Epileptic activity may affect the autonomic nervous system, which is known to be a critical regulator of cardiac function. Dysregulation of the autonomic nervous system and spreading depression to the brain stem centers during an epileptic event can result in fatal cardiac abnormalities (Jansen and Lagae 2010; Massey et al. 2014; Surges and Sander 2012).
Multiple DS and epilepsy animal models also serve as models for SUDEP, including the previously discussed Scn1b-null mouse line (Chen et al. 2004). Additional models include Scn1a+/− mice, which model the haploinsufficiency observed in most DS patients, and the Kcna1 null mouse line, which deletes the voltage-gated potassium channel Kv1.1 (Glasscock et al. 2010; Oakley et al. 2011). Intriguingly, each of these SUDEP models presents with different cardiac alterations that may mechanistically contribute to SUDEP. Scn1b-null mice display increased cardiac sodium current and prolonged QT and RR intervals. Scn1b-null mice treated with atropine or propranolol do not show differences in QT interval compared to vehicle treated animals, indicating the cardiac phenotype may not be a result of an abnormal autonomic activity (Lopez-Santiago et al. 2007). Scn1a+/− mice also display increased cardiac sodium current, but additionally have bradycardia, focal discharges, a variable RR interval, and bundle branch block (Auerbach et al. 2013; Kalume et al. 2013). Kcna1-null mice display a cardiac phenotype as well, including atrioventricular (AV) block, bradycardia, premature ventricular contractions and altered heart rate variability (Glasscock et al. 2010). Contrary to Scn1b-null mice, treatment of Scn1a+/− and Kcna1-null animals with atropine reverses AV block, suggesting parasympathetic hyperexcitability in these models (Glasscock et al. 2010; Kalume et al. 2013). In summary, studies with animal models and patient mutations provide evidence that β subunits are likely key regulators in the pathogenesis of SIDS and SUDEP, although additional work must be completed to further understand and ultimately prevent SIDS and SUDEP events.
14.3. Conclusion
In conclusion, VGSC β subunits play critical roles in modulating the gating, localization, and kinetics of the VGSC pore as well as modulate the activities of some potassium channels. In addition, these non-pore-forming proteins function as CAMs and signaling molecules in both excitable and non-excitable cell types. Their importance as CAMs is implicated in neurite outgrowth, axonal pathfinding and fasciculation, and migration in cancerous cells. Sequential β subunit cleavage by BACE and γ-secretase also affects the expression of other genes. Mutations in the genes encoding β subunits are linked to a variety of devastating diseases, including epilepsy, SIDS and SUDEP, cancer, neuropathic pain, and some of the major neurodegenerative disorders (Fig. 5). Additional research needs to be completed in order to further understand the biology of these critical proteins and their potential as novel therapeutic targets for a wide variety of disease states.
Fig. 5:
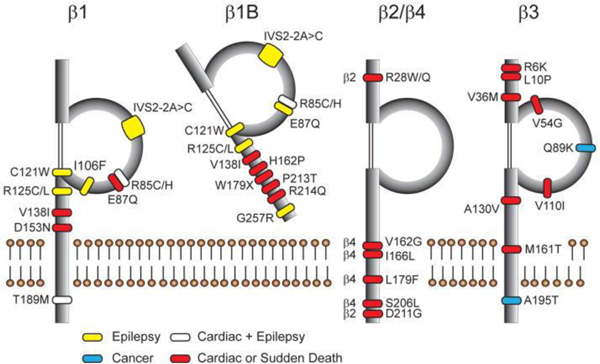
Disease-linked β subunit mutations. Figure reproduced from (O’Malley and Isom 2015).
References
- Adachi K, Toyota M, Sasaki Y, Yamashita T, Ishida S, Ohe-Toyota M, Maruyama R, Hinoda Y, Saito T, Imai K, Kudo R, Tokino T (2004) Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene 23: 7791–8. doi: 10.1038/sj.onc.1208067 [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A, van den Berg LH, Veldink J (2017) Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 13: 96–104. doi: 10.1038/nrneurol.2016.182 [DOI] [PubMed] [Google Scholar]
- Alders M, Christiaans I (1993) Long QT Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K (eds) GeneReviews(R). University of Washington, Seattle: [PubMed] [Google Scholar]
- University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved, Seattle (WA) [Google Scholar]
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM (2009) Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci 29: 2027–42. doi: 10.1523/jneurosci.4531-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrikopoulos P, Fraser SP, Patterson L, Ahmad Z, Burcu H, Ottaviani D, Diss JK, Box C, Eccles SA, Djamgoz MB (2011) Angiogenic functions of voltage-gated Na+ Channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling. J Biol Chem 286: 16846–60. doi: 10.1074/jbc.M110.187559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert D, Claes L, Ceulemans B, Lofgren A, Van Broeckhoven C, De Jonghe P (2003) A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61: 854–6. [DOI] [PubMed] [Google Scholar]
- Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, Yamakawa K, Meisler MH, Parent JM, Isom LL (2013) Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One 8: e77843. doi: 10.1371/journal.pone.0077843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS, Raman IM (2010) Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A 107: 12357–62. doi: 10.1073/pnas.1005633107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Isom LL NaV1.5 and Regulatory β Subunits in Cardiac Sodium Channelopathies. Cardiac Electrophysiology Clinics 6: 679–694. doi: 10.1016/j.ccep.2014.07.002 [DOI] [Google Scholar]
- Bao Y, Willis BC, Frasier CR, Lopez-Santiago LF, Lin X, Ramos-Mondragon R, Auerbach DS, Chen C, Wang Z, Anumonwo J, Valdivia HH, Delmar M, Jalife J, Isom LL (2016) Scn2b Deletion in Mice Results in Ventricular and Atrial Arrhythmias. Circ Arrhythm Electrophysiol 9. doi: 10.1161/circep.116.003923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L, Haerian BS, Ng HK, Wong VC, Ng PW, Lui CH, Sin NC, Zhang C, Tomlinson B, Wong GW, Tan HJ, Raymond AA, Mohamed Z, Kwan P (2014) Case-control association study of polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, SCN3A, SCN1B, and SCN2B and epilepsy. Hum Genet 133: 651–9. doi: 10.1007/s00439-013-1405-1 [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Fleetwood-Walker SM (1999) The sodium channel auxiliary subunits beta1 and beta2 are differentially expressed in the spinal cord of neuropathic rats. Neuroscience 90: 153–64. [DOI] [PubMed] [Google Scholar]
- Brackenbury WJ (2012) Voltage-gated sodium channels and metastatic disease. Channels (Austin) 6: 352–61. doi: 10.4161/chan.21910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL (2010) Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A 107: 2283–8. doi: 10.1073/pnas.0909434107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Davis TH, Chen C, Slat EA, Detrow MJ, Dickendesher TL, Ranscht B, Isom LL (2008) Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J Neurosci 28: 3246–56. doi: 10.1523/jneurosci.5446-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Isom LL (2011) Na Channel beta Subunits: Overachievers of the Ion Channel Family. Front Pharmacol 2: 53. doi: 10.3389/fphar.2011.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Rasband MN (2013) Na+ channel-dependent recruitment of Navbeta4 to axon initial segments and nodes of Ranvier. J Neurosci 33: 6191–202. doi: 10.1523/jneurosci.4051-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun JD, Isom LL (2014) The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb Exp Pharmacol 221: 51–89. doi: 10.1007/978-3-642-41588-3_4 [DOI] [PubMed] [Google Scholar]
- Catterall WA From Ionic Currents to Molecular Mechanisms. Neuron 26: 13–25. doi: 10.1016/S0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- Chen C, Bharucha V, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ 3rd, Kazen-Gillespie KA, Kazarinova-Noyes K, Shrager P, Saunders TL, Macdonald RL, Ransom BR, Scheuer T, Catterall WA, Isom LL (2002) Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A 99: 17072–7. doi: 10.1073/pnas.212638099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Calhoun JD, Zhang Y, Lopez-Santiago L, Zhou N, Davis TH, Salzer JL, Isom LL (2012) Identification of the cysteine residue responsible for disulfide linkage of Na+ channel alpha and beta2 subunits. J Biol Chem 287: 39061–9. doi: 10.1074/jbc.M112.397646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O’Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL (2004) Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 24: 4030–42. doi: 10.1523/jneurosci.4139-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioni AM, Brackenbury WJ, Calhoun JD, Isom LL, Djamgoz MB (2009) A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int J Biochem Cell Biol 41: 1216–27. doi: 10.1016/j.biocel.2008.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SL, Vassar R (2007) The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol Neurodegener 2: 22. doi: 10.1186/1750-1326-2-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward K, Jowett A, Plumpton C, Powell A, Birch R, Tate S, Bountra C, Anand P (2001) Sodium channel beta1 and beta2 subunits parallel SNS/PN3 alpha-subunit changes in injured human sensory neurons. Neuroreport 12: 483–8. [DOI] [PubMed] [Google Scholar]
- Davis TH, Chen C, Isom LL (2004) Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem 279: 51424–32. doi: 10.1074/jbc.M410830200 [DOI] [PubMed] [Google Scholar]
- Deschenes I, Tomaselli GF (2002) Modulation of Kv4.3 current by accessory subunits. FEBS Lett 528: 183–8. [DOI] [PubMed] [Google Scholar]
- Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL (2001) Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103: 1303–10. [DOI] [PubMed] [Google Scholar]
- Diss JK, Fraser SP, Walker MM, Patel A, Latchman DS, Djamgoz MB (2008) Beta-subunits of voltage-gated sodium channels in human prostate cancer: quantitative in vitro and in vivo analyses of mRNA expression. Prostate Cancer Prostatic Dis 11: 325–33. doi: 10.1038/sj.pcan.4501012 [DOI] [PubMed] [Google Scholar]
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, LaFleur BJ, Coon KD, Brown KM, Caselli R, Kukull W, Higdon R, McKeel D, Morris JC, Hulette C, Schmechel D, Reiman EM, Rogers J, Stephan DA (2006) Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging 27: 1359–71. doi: 10.1016/j.neurobiolaging.2005.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evin G, Barakat A, Masters CL (2010) BACE: Therapeutic target and potential biomarker for Alzheimer’s disease. Int J Biochem Cell Biol 42: 1923–6. doi: 10.1016/j.biocel.2010.08.017 [DOI] [PubMed] [Google Scholar]
- Fendri-Kriaa N, Kammoun F, Salem IH, Kifagi C, Mkaouar-Rebai E, Hsairi I, Rebai A, Triki C, Fakhfakh F (2011) New mutation c.374C>T and a putative disease-associated haplotype within SCN1B gene in Tunisian families with febrile seizures. Eur J Neurol 18: 695–702. doi: 10.1111/j.1468-1331.2010.03216.x [DOI] [PubMed] [Google Scholar]
- Ficker DM, So EL, Shen WK, Annegers JF, O’Brien PC, Cascino GD, Belau PG (1998) Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology 51: 1270–4. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S (2007) Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582: 675–93. doi: 10.1113/jphysiol.2006.126714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gataullina S, Dulac O (2017) From genotype to phenotype in Dravet disease. Seizure 44: 58–64. doi: 10.1016/j.seizure.2016.10.014 [DOI] [PubMed] [Google Scholar]
- Gilchrist J, Das S, Van Petegem F, Bosmans F (2013) Crystallographic insights into sodium-channel modulation by the beta4 subunit. Proc Natl Acad Sci U S A 110: E5016–24. doi: 10.1073/pnas.1314557110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL (2010) Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci 30: 5167–75. doi: 10.1523/jneurosci.5591-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM (2005) Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 45: 233–44. doi: 10.1016/j.neuron.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Kovacs DM (2011) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25: 3–28. doi: 10.3233/jad-2011-101065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim P, Brice N, Thresher R, Lawrence J, Zhang Y, Jackson AP, Grace AA, Huang CL (2010) Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol (Oxf) 198: 47–59. doi: 10.1111/j.1748-1716.2009.02048.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim P, Gurung IS, Pedersen TH, Thresher R, Brice N, Lawrence J, Grace AA, Huang CL (2008) Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog Biophys Mol Biol 98: 251–66. doi: 10.1016/j.pbiomolbio.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Plata E, Ortiz CS, Marquina-Castillo B, Medina-Martinez I, Alfaro A, Berumen J, Rivera M, Gomora JC (2012) Overexpression of NaV 1.6 channels is associated with the invasion capacity of human cervical cancer. Int J Cancer 130: 2013–23. doi: 10.1002/ijc.26210 [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Tomson T, Benn E, Sander JW, Nilsson L, Langan Y, Walczak TS, Beghi E, Brodie MJ, Hauser WA (2012) Do antiepileptic drugs or generalized tonic-clonic seizure frequency increase SUDEP risk? A combined analysis. Epilepsia 53: 249–52. doi: 10.1111/j.1528-1167.2011.03354.x [DOI] [PubMed] [Google Scholar]
- Holst AG, Saber S, Houshmand M, Zaklyazminskaya EV, Wang Y, Jensen HK, Refsgaard L, Haunso S, Svendsen JH, Olesen MS, Tfelt-Hansen J (2012) Sodium current and potassium transient outward current genes in Brugada syndrome: screening and bioinformatics. Can J Cardiol 28: 196–200. doi: 10.1016/j.cjca.2011.11.011 [DOI] [PubMed] [Google Scholar]
- Hu D, Barajas-Martinez H, Medeiros-Domingo A, Crotti L, Veltmann C, Schimpf R, Urrutia J, Alday A, Casis O, Pfeiffer R, Burashnikov E, Caceres G, Tester DJ, Wolpert C, Borggrefe M, Schwartz P, Ackerman MJ, Antzelevitch C (2012) A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm 9: 760–9. doi: 10.1016/j.hrthm.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL (2002) The role of sodium channels in cell adhesion. Front Biosci 7: 12–23. [DOI] [PubMed] [Google Scholar]
- Isom LL, Catterall WA (1996) Na+ channel subunits and Ig domains. Nature 383: 307–8. doi: 10.1038/383307b0 [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Catterall WA (1994) Auxiliary subunits of voltage-gated ion channels. Neuron 12: 1183–94. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839–42. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA (1995a) Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83: 433–42. [DOI] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA (1995b) Functional Co-expression of the 1 and Type IIA Subunits of Sodium Channels in a Mammalian Cell Line. Journal of Biological Chemistry 270: 3306–3312. doi: 10.1074/jbc.270.7.3306 [DOI] [PubMed] [Google Scholar]
- Jansen K, Lagae L (2010) Cardiac changes in epilepsy. Seizure 19: 455–60. doi: 10.1016/j.seizure.2010.07.008 [DOI] [PubMed] [Google Scholar]
- Jansson KH, Lynch JE, Lepori-Bui N, Czymmek KJ, Duncan RL, Sikes RA (2012) Overexpression of the VSSC-associated CAM, beta-2, enhances LNCaP cell metastasis associated behavior. Prostate 72: 1080–92. doi: 10.1002/pros.21512 [DOI] [PubMed] [Google Scholar]
- Johnson D, Montpetit ML, Stocker PJ, Bennett ES (2004) The sialic acid component of the beta1 subunit modulates voltage-gated sodium channel function. J Biol Chem 279: 44303–10. doi: 10.1074/jbc.M408900200 [DOI] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA (2013) Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 123: 1798–808. doi: 10.1172/JCI66220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DI, Isom LL, Petrou S (2016) Role of Sodium Channels in Epilepsy. Cold Spring Harb Perspect Med 6. doi: 10.1101/cshperspect.a022814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao ZC (2001) Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21: 7517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, Mattei LN, Rogers KE, Isom LL (2000) Cloning, localization, and functional expression of sodium channel beta1A subunits. J Biol Chem 275: 1079–88. [DOI] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM (2007) BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol 9: 755–64. doi: 10.1038/ncb1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM (2011) Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J Biol Chem 286: 8106–16. doi: 10.1074/jbc.M110.134692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Ingano LA, Carey BW, Pettingell WH, Kovacs DM (2005) Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem 280: 23251–61. doi: 10.1074/jbc.M412938200 [DOI] [PubMed] [Google Scholar]
- Ko SH, Lenkowski PW, Lee HC, Mounsey JP, Patel MK (2005) Modulation of Na(v)1.5 by beta1-- and beta3-subunit co-expression in mammalian cells. Pflugers Arch 449: 403–12. doi: 10.1007/s00424-004-1348-4 [DOI] [PubMed] [Google Scholar]
- Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, Cutz E, Hanzlick R, Keens TG, Mitchell EA (2004) Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 114: 234–8. [DOI] [PubMed] [Google Scholar]
- Kruger LC, O’Malley HA, Hull JM, Kleeman A, Patino GA, Isom LL (2016) beta1-C121W Is Down But Not Out: Epilepsy-Associated Scn1b-C121W Results in a Deleterious Gain-of-Function. J Neurosci 36: 6213–24. doi: 10.1523/jneurosci.0405-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann Cé J, Syam N, Pertin M, Decosterd I, Abriel H (2013) β1- and β3- voltage-gated sodium channel subunits modulate cell surface expression and glycosylation of Na(v)1.7 in HEK293 cells. Front Cell Neurosci 7. doi: 10.3389/fncel.2013.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, Fang WY, Yang YQ (2013) Mutations of the SCN4B-encoded sodium channel beta4 subunit in familial atrial fibrillation. Int J Mol Med 32: 144–50. doi: 10.3892/ijmm.2013.1355 [DOI] [PubMed] [Google Scholar]
- Lin X, O’Malley H, Chen C, Auerbach D, Foster M, Shekhar A, Zhang M, Coetzee W, Jalife J, Fishman GI, Isom L, Delmar M (2014) Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol. doi: 10.1113/jphysiol.2014.277699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WL, Donermeyer DL, Allen PM (2012) A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat Immunol 13: 880–7. doi: 10.1038/ni.2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Brackenbury WJ, Chen C, Isom LL (2011) Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J Biol Chem 286: 22913–23. doi: 10.1074/jbc.M111.242370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Meadows LS, Ernst SJ, Chen C, Malhotra JD, McEwen DP, Speelman A, Noebels JL, Maier SK, Lopatin AN, Isom LL (2007) Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol 43: 636–47. doi: 10.1016/j.yjmcc.2007.07.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Pertin M, Morisod X, Chen C, Hong S, Wiley J, Decosterd I, Isom LL (2006) Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J Neurosci 26: 7984–94. doi: 10.1523/jneurosci.2211-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA (2004) Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109: 1421–7. doi: 10.1161/01.cir.0000121421.61896.24 [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL (2000) Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem 275: 11383–8. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL (2002) Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem 277: 26681–8. doi: 10.1074/jbc.M202354200 [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Thyagarajan V, Chen C, Isom LL (2004) Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem 279: 40748–54. doi: 10.1074/jbc.M407243200 [DOI] [PubMed] [Google Scholar]
- Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL, Link AJ, Nerbonne JM (2012) The Sodium Channel Accessory Subunit Navβ1 Regulates Neuronal Excitability through Modulation of Repolarizing Voltage-Gated K(+) Channels. J Neurosci 32: 5716–27. doi: 10.1523/jneurosci.6450-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, Richerson GB (2014) SUDEP Mechanisms: The pathway to prevention. Nat Rev Neurol 10: 271–82. doi: 10.1038/nrneurol.2014.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Axelsson M, Haghighi S, Malmestrom C, Wu G, Anckarsater R, Sankaranarayanan S, Andreasson U, Fredrikson S, Gundersen A, Johnsen L, Fladby T, Tarkowski A, Trysberg E, Wallin A, Anckarsater H, Lycke J, Andersen O, Simon AJ, Blennow K, Zetterberg H (2009) Reduced cerebrospinal fluid BACE1 activity in multiple sclerosis. Mult Scler 15: 448–54. doi: 10.1177/1352458508100031 [DOI] [PubMed] [Google Scholar]
- McCormick KA, Isom LL, Ragsdale D, Smith D, Scheuer T, Catterall WA (1998) Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. J Biol Chem 273: 3954–62. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Chen C, Meadows LS, Lopez-Santiago L, Isom LL (2009) The voltage-gated Na+ channel beta3 subunit does not mediate trans homophilic cell adhesion or associate with the cell adhesion molecule contactin. Neurosci Lett 462: 272–5. doi: 10.1016/j.neulet.2009.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DP, Isom LL (2004) Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem 279: 52744–52. doi: 10.1074/jbc.M405990200 [DOI] [PubMed] [Google Scholar]
- McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL (2004) Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem 279: 16044–9. doi: 10.1074/jbc.M400856200 [DOI] [PubMed] [Google Scholar]
- Meadows L, Malhotra JD, Stetzer A, Isom LL, Ragsdale DS (2001) The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J Neurochem 76: 1871–8. [DOI] [PubMed] [Google Scholar]
- Meadows LS, Malhotra J, Loukas A, Thyagarajan V, Kazen-Gillespie KA, Koopman MC, Kriegler S, Isom LL, Ragsdale DS (2002) Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci 22: 10699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusie-Luna MT, Makielski JC, Ackerman MJ (2007) SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116: 134–42. doi: 10.1161/circulationaha.106.659086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H, Oyama F, Inoue R, Aosaki T, Abe T, Kiyonari H, Kino Y, Kurosawa M, Shimizu J, Ogiwara I, Yamakawa K, Koshimizu Y, Fujiyama F, Kaneko T, Shimizu H, Nagatomo K, Yamada K, Shimogori T, Hattori N, Miura M, Nukina N (2014) Singular localization of sodium channel beta4 subunit in unmyelinated fibres and its role in the striatum. Nat Commun 5: 5525. doi: 10.1038/ncomms6525 [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Oyama F, Wong HK, Kaneko K, Sakurai T, Tamaoka A, Nukina N (2007) BACE1 modulates filopodia-like protrusions induced by sodium channel beta4 subunit. Biochem Biophys Res Commun 361: 43–8. doi: 10.1016/j.bbrc.2007.06.170 [DOI] [PubMed] [Google Scholar]
- Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP (2000) beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci U S A 97: 2308–13. doi: 10.1073/pnas.030362197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Shimizu W (2016) Genetics of long-QT syndrome. J Hum Genet 61: 51–5. doi: 10.1038/jhg.2015.74 [DOI] [PubMed] [Google Scholar]
- Namadurai S, Balasuriya D, Rajappa R, Wiemhofer M, Stott K, Klingauf J, Edwardson JM, Chirgadze DY, Jackson AP (2014) Crystal structure and molecular imaging of the Nav channel beta3 subunit indicates a trimeric assembly. J Biol Chem 289: 10797–811. doi: 10.1074/jbc.M113.527994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, Tomson T (2012) Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 53: 227–33. doi: 10.1111/j.1528-1167.2011.03358.x [DOI] [PubMed] [Google Scholar]
- Nelson M, Millican-Slater R, Forrest LC, Brackenbury WJ (2014) The sodium channel beta1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int J Cancer 135: 2338–51. doi: 10.1002/ijc.28890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HM, Miyazaki H, Hoshi N, Smith BJ, Nukina N, Goldin AL, Chandy KG (2012) Modulation of voltage-gated K+ channels by the sodium channel beta1 subunit. Proc Natl Acad Sci U S A 109: 18577–82. doi: 10.1073/pnas.1209142109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutini M, Spalloni A, Florenzano F, Westenbroek RE, Marini C, Catterall WA, Bernardi G, Longone P (2011) Increased expression of the beta3 subunit of voltage-gated Na+ channels in the spinal cord of the SOD1G93A mouse. Mol Cell Neurosci 47: 108–18. doi: 10.1016/j.mcn.2011.03.005 [DOI] [PubMed] [Google Scholar]
- O’Malley HA, Isom LL (2015) Sodium channel beta subunits: emerging targets in channelopathies. Annu Rev Physiol 77: 481–504. doi: 10.1146/annurev-physiol-021014-071846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley HA, Shreiner AB, Chen GH, Huffnagle GB, Isom LL (2009) Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol Cell Neurosci 40: 143–55. doi: 10.1016/j.mcn.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley JC, Kalume F, Catterall WA (2011) Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia 52 Suppl 2: 59–61. doi: 10.1111/j.1528-1167.2011.03004.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Nakayama T, Yamagata T, Ohtani H, Mazaki E, Tsuchiya S, Inoue Y, Yamakawa K (2012) A homozygous mutation of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia 53: e200–3. doi: 10.1111/epi.12040 [DOI] [PubMed] [Google Scholar]
- Olesen MS, Jespersen T, Nielsen JB, Liang B, Moller DV, Hedley P, Christiansen M, Varro A, Olesen SP, Haunso S, Schmitt N, Svendsen JH (2011) Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res 89: 786–93. doi: 10.1093/cvr/cvq348 [DOI] [PubMed] [Google Scholar]
- Oyama F, Miyazaki H, Sakamoto N, Becquet C, Machida Y, Kaneko K, Uchikawa C, Suzuki T, Kurosawa M, Ikeda T, Tamaoka A, Sakurai T, Nukina N (2006) Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J Neurochem 98: 518–29. doi: 10.1111/j.1471-4159.2006.03893.x [DOI] [PubMed] [Google Scholar]
- Pandis D, Scarmeas N (2012) Seizures in Alzheimer Disease: Clinical and Epidemiological Data. Epilepsy Currents 12: 184–187. doi: 10.5698/1535-7511-12.5.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Brackenbury WJ, Bao Y, Lopez-Santiago LF, O’Malley HA, Chen C, Calhoun JD, Lafreniere RG, Cossette P, Rouleau GA, Isom LL (2011) Voltage-gated Na+ channel beta1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci 31: 14577–91. doi: 10.1523/jneurosci.0361-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O’Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL (2009) A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29: 10764–78. doi: 10.1523/jneurosci.2475-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertin M, Ji RR, Berta T, Powell AJ, Karchewski L, Tate SN, Isom LL, Woolf CJ, Gilliard N, Spahn DR, Decosterd I (2005) Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J Neurosci 25: 10970–80. doi: 10.1523/jneurosci.3066-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, D’Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM (2003) Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem 270: 4762–70. [DOI] [PubMed] [Google Scholar]
- Ransdell JL, Dranoff E, Lau B, Lo WL, Donermeyer DL, Allen PM, Nerbonne JM (2017) Loss of Navbeta4-Mediated Regulation of Sodium Currents in Adult Purkinje Neurons Disrupts Firing and Impairs Motor Coordination and Balance. Cell Rep 19: 532–544. doi: 10.1016/j.celrep.2017.03.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA (2000) A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci 3: 437–44. doi: 10.1038/74805 [DOI] [PubMed] [Google Scholar]
- Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA (2001) Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Leaw B, Richards KL, Richardson R, Wimmer V, Yu C, Hill-Yardin EL, Lerche H, Scheffer IE, Berkovic SF, Petrou S (2014) Reduced dendritic arborization and hyperexcitability of pyramidal neurons in a Scn1b-based model of Dravet syndrome. Brain 137: 1701–15. doi: 10.1093/brain/awu077 [DOI] [PubMed] [Google Scholar]
- Remme CA, Bezzina CR (2010) Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther 28: 287–94. doi: 10.1111/j.1755-5922.2010.00210.x [DOI] [PubMed] [Google Scholar]
- Riuro H, Beltran-Alvarez P, Tarradas A, Selga E, Campuzano O, Verges M, Pagans S, Iglesias A, Brugada J, Brugada P, Vazquez FM, Perez GJ, Scornik FS, Brugada R (2013) A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 34: 961–6. doi: 10.1002/humu.22328 [DOI] [PubMed] [Google Scholar]
- Riuro H, Campuzano O, Arbelo E, Iglesias A, Batlle M, Perez-Villa F, Brugada J, Perez GJ, Scornik FS, Brugada R (2014) A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 11: 1202–9. doi: 10.1016/j.hrthm.2014.03.044 [DOI] [PubMed] [Google Scholar]
- Roger S, Rollin J, Barascu A, Besson P, Raynal PI, Iochmann S, Lei M, Bougnoux P, Gruel Y, Le Guennec JY (2007) Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol 39: 774–86. doi: 10.1016/j.biocel.2006.12.007 [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, Xu R, Jackson G, Adams J, Connellan M, Petrou S, Wellard RM, Briellmann RS, Wallace RH, Mulley JC, Berkovic SF (2007) Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130: 100–9. doi: 10.1093/brain/awl272 [DOI] [PubMed] [Google Scholar]
- Shah BS, Stevens EB, Gonzalez MI, Bramwell S, Pinnock RD, Lee K, Dixon AK (2000) beta3, a novel auxiliary subunit for the voltage-gated sodium channel, is expressed preferentially in sensory neurons and is upregulated in the chronic constriction injury model of neuropathic pain. Eur J Neurosci 12: 3985–90. [DOI] [PubMed] [Google Scholar]
- Shah BS, Stevens EB, Pinnock RD, Dixon AK, Lee K (2001) Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta3, in rat CNS. J Physiol 534: 763–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Miyazaki H, Ohsawa N, Shoji S, Ishizuka-Katsura Y, Tosaki A, Oyama F, Terada T, Sakamoto K, Shirouzu M, Sekine S, Nukina N, Yokoyama S (2016) Structure-based site-directed photo-crosslinking analyses of multimeric cell-adhesive interactions of voltage-gated sodium channel beta subunits. Sci Rep 6: 26618. doi: 10.1038/srep26618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Tosaki A, Ohsawa N, Ishizuka-Katsura Y, Shoji S, Miyazaki H, Oyama F, Terada T, Shirouzu M, Sekine SI, Nukina N, Yokoyama S (2017) Parallel homodimer structures of the extracellular domains of the voltage-gated sodium channel beta4 subunit explain its role in cell-cell adhesion. J Biol Chem. doi: 10.1074/jbc.M117.786509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, MacDonald BT, Levin SI, Soltesz I, Benna P, Montalenti E, Isom LL, Goldin AL, Meisler MH (2004) A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci 24: 10022–34. doi: 10.1523/jneurosci.2034-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, Schachner M, Catterall WA (1998) Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proceedings of the National Academy of Sciences 95: 15753–15757. doi: 10.1073/pnas.95.26.15753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges R, Sander JW (2012) Sudden unexpected death in epilepsy: mechanisms, prevalence, and prevention. Curr Opin Neurol 25: 201–7. doi: 10.1097/WCO.0b013e3283506714 [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kikuchi S, Dai Y, Kobayashi K, Fukuoka T, Noguchi K (2003) Expression of auxiliary beta subunits of sodium channels in primary afferent neurons and the effect of nerve injury. Neuroscience 121: 441–50. [DOI] [PubMed] [Google Scholar]
- Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, Makielski JC, Ackerman MJ (2010) Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7: 771–8. doi: 10.1016/j.hrthm.2010.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester DJ, Ackerman MJ (2014) GENETICS OF LONG QT SYNDROME. Methodist Debakey Cardiovasc J 10: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Jarecki BW, Piekarz AD, Cummins TR (2011) Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J Physiol 589: 597–608. doi: 10.1113/jphysiol.2010.200915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia CR, Medeiros-Domingo A, Ye B, Shen WK, Algiers TJ, Ackerman MJ, Makielski JC (2010) Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res 86: 392–400. doi: 10.1093/cvr/cvp417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Norstrand DW, Ackerman MJ (2009) Sudden infant death syndrome: do ion channels play a role? Heart Rhythm 6: 272–8. doi: 10.1016/j.hrthm.2008.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Parasivam G, Barnett S, Wallace GB, Sutherland GR, Berkovic SF, Mulley JC (2002) Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 58: 1426–9. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC (1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel sz1 subunit gene SCN1B. Nat Genet 19: 366–370. [DOI] [PubMed] [Google Scholar]
- Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F, Liao Y, Fang F, Chen Q, Tu X, Wang QK (2010) Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophys Res Commun 398: 98–104. doi: 10.1016/j.bbrc.2010.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM (2009) Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol 2: 268–75. doi: 10.1161/circep.108.779181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kaab S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR (2008) Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118: 2260–8. doi: 10.1172/JCI33891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Reid CA, Mitchell S, Richards KL, Scaf BB, Leaw BT, Hill EL, Royeck M, Horstmann MT, Cromer BA, Davies PJ, Xu R, Lerche H, Berkovic SF, Beck H, Petrou S (2010) Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J Clin Invest 120: 2661–71. doi: 10.1172/jci42219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina N (2005) beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem 280: 23009–17. doi: 10.1074/jbc.M414648200 [DOI] [PubMed] [Google Scholar]
- Xiao ZC, Ragsdale DS, Malhotra JD, Mattei LN, Braun PE, Schachner M, Isom LL (1999) Tenascin-R is a functional modulator of sodium channel beta subunits. J Biol Chem 274: 26511–7. [DOI] [PubMed] [Google Scholar]
- Xu R, Thomas EA, Gazina EV, Richards KL, Quick M, Wallace RH, Harkin LA, Heron SE, Berkovic SF, Scheffer IE, Mulley JC, Petrou S (2007) Generalized epilepsy with febrile seizures plus-associated sodium channel beta1 subunit mutations severely reduce beta subunit-mediated modulation of sodium channel function. Neuroscience 148: 164–74. doi: 10.1016/j.neuroscience.2007.05.038 [DOI] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R (2003) Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci 23: 7577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Koivumaki JT, Liang B, Lorentzen LG, Tang C, Andersen MN, Svendsen JH, Tfelt-Hansen J, Maleckar M, Schmitt N, Olesen MS, Jespersen T (2014) Investigations of the Navbeta1b sodium channel subunit in human ventricle; functional characterization of the H162P Brugada syndrome mutant. Am J Physiol Heart Circ Physiol 306: H1204–12. doi: 10.1152/ajpheart.00405.2013 [DOI] [PubMed] [Google Scholar]
- Zhou TT, Zhang ZW, Liu J, Zhang JP, Jiao BH (2012) Glycosylation of the sodium channel beta4 subunit is developmentally regulated and involves in neuritic degeneration. Int J Biol Sci 8: 630–9. doi: 10.7150/ijbs.3684 [DOI] [PMC free article] [PubMed] [Google Scholar]