

SUMMARY
WNT7A and WNT7B control CNS angiogenesis and blood-brain barrier formation by activating endothelial Wnt/β-catenin signaling. The GPI-anchored protein RECK and adhesion G protein-coupled receptor GPR124 critically regulate WNT7-specific signaling in concert with FZD and LRP co-receptors. Here, we demonstrate that primarily the GPR124 ectodomain, but not its transmembrane and intracellular domains, mediates RECK/WNT7-induced canonical Wnt signaling. Moreover, RECK is the predominant binding partner of GPR124 in rat brain blood vessels in situ. WNT7A and WNT7B, but not WNT3A, directly bind to purified recombinant soluble RECK, full-length cell surface RECK, and the GPR124:RECK complex. Chemical cross-linking indicates that RECK and WNT7A associate with 1:1 stoichiometry, which stabilizes short-lived, active, monomeric, hydrophobic WNT7A. In contrast, free WNT7A rapidly converts into inactive, hydrophilic aggregates. Overall, RECK is a selective WNT7 receptor that mediates GPR124/FZD/LRP-dependent canonical Wnt/β-catenin signaling by stabilizing active cell surface WNT7, suggesting isoform-specific regulation of Wnt bioavailability.
Graphical Abstract
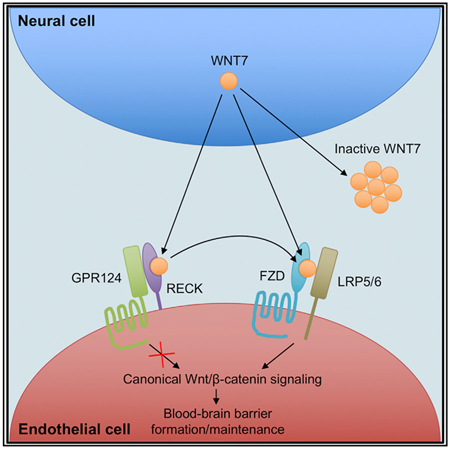
In Brief
Canonical Wnt/β-catenin signaling in brain endothelium is highly specialized and requires the WNT7-specific co-activators RECK and GPR124 in addition to the Wnt co-receptors FZD and LRP. Here, Vallon et al. demonstrate direct binding of WNT7 to RECK resulting in stabilization of active WNT7 and increased FZD:WNT7 complex formation.
INTRODUCTION
Canonical Wnt/β-catenin signaling is activated by Wnt-mediated heterodimerization of Frizzled (FZD1-10) and low-density lipoprotein receptor-related proteins 5 or 6 (LRP5/6) co-receptors, which stabilizes the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) activator β-catenin to transcribe Wnt target genes (Clevers and Nusse, 2012; Janda et al., 2017). In the CNS, canonical Wnt signaling critically regulates embryonic CNS angiogenesis and blood-brain barrier (BBB) formation (Daneman et al., 2009; Stenman et al., 2008; Zhou et al., 2014), postnatal BBB function (Wang et al., 2012), and adult pathologic BBB integrity (Chang et al., 2017). Wnt7a and Wnt7b expressed in embryonic neuroepithelium activate endothelial canonical Wnt signaling (Daneman et al., 2009; Stenman et al., 2008). Wnt activity correlates with its hydrophobicity (Kakugawa et al., 2015; Zhang et al., 2012), a result of its palmitoylation and essential for FZD binding (Janda et al., 2012).
Fzd4, the major canonical FZD in CNS endothelium (Daneman et al., 2009; Zhang et al., 2014), regulates postnatal retinal angiogenesis, blood-retinal barrier formation, and cerebellar BBB integrity (Wang et al., 2012; Zhou et al., 2014). CNS endothelial cells also express Fzd1 and Fzd8 (Daneman et al., 2009; Zhang et al., 2014), which may act redundantly with Fzd4, as no single Fzd knockout (KO) affects embryonic CNS vasculature (Wang et al., 2016). CNS endothelial canonical Wnt signaling is highly specialized and requires WNT7-specific co-activators GPR124 and RECK or alternatively the FZD4-specific ligand Norrin and co-receptor TSPAN12 (Choet al., 2017; Junge et al., 2009; Ulrich et al., 2016; Wang et al., 2012; Zhou and Nathans, 2014; Zhou etal., 2014).
GPR124 (TEM5/ADGRA2) is an orphan adhesion family G protein-coupled receptor (GPCR) with characteristic large ectodomain (ECD) (Hamann et al., 2015). Murine Gpr124 KO profoundly impairs embryonic CNS angiogenesis and BBB maturation in forebrain and ventral neural tube (Kuhnert et al., 2010), similar to Wnt7a−/−; Wnt7b−/− mice (Daneman et al., 2009; Stenman et al., 2008). The Gpr124 KO is rescued by endothelial-specific constitutive Wnt/β-catenin signaling (Zhou and Nathans, 2014). Conditional adult Gpr124 KO is well tolerated under homeostasis, but exacerbates BBB breakdown and CNS hemorrhage during ischemic stroke and glioblastoma, which is also rescued by constitutive β-catenin activation (Chang et al., 2017).
The glycosylphosphatidylinositol (GPI)-anchored protein reversion-inducing cysteine-rich protein with kazal motifs (RECK) is a metalloproteinase inhibitor (Oh et al., 2001) and regulates canonical Wnt/β-catenin signaling (Vanhollebeke et al., 2015; Cho et al., 2017; Ulrich et al., 2016). While global Reck KO mice die at midgestation prior to CNS angiogenesis (Oh et al., 2001), endothelial-specific Reck KO mice die later (de Almeida et al., 2015) with CNS angiogenesis defects and hemorrhage similar to Gpr124−/− or Wnt7a−/−; Wnt7b−/− mice (Cho et al., 2017; Daneman et al., 2009; Kuhnert et al., 2010; Stenman et al., 2008). GPR124, RECK, FZDs, and LRP5/6 synergistically mediate WNT7-specific canonical Wnt reporter activation in vitro (Vanhollebeke et al., 2015). Zebrafish CNS endothelial tip cell formation requires both gpr124 and reck cell-autonomously (Vanhollebeke et al., 2015), endothelial Gpr124 and Reck cooperate during murine CNS angiogenesis and BBB formation, recombinant GPR124 and RECK fragments interact physically in vitro, and mouse Reck KO is rescued by endothelial constitutive Wnt signaling activation (Cho et al., 2017).
Here, we demonstrate that canonical WNT7 signaling is primarily regulated by the GPR124 ECD, but not by its transmembrane domain (TMD) and intracellular domain (ICD), suggesting extracellular GPR124 action rather than intrinsic GPR124 signal transduction. Importantly, WNT7 directly binds RECK and the GPR124:RECK complex, which stabilizes WNT7 in its active, monomeric, hydrophobic form and enhances WNT7 binding to FZD.
RESULTS
GPR124-Mediated Canonical WNT7 Signaling Does Not Require GPR124 Signal Transduction
We confirmed RECK as a predominant binding partner of GPR124 in rat brain blood vessels in situ by combined chemical cross-linking/affinity chromatography (Figures 1A and S1A). To characterize the GPR124:RECK binding interface, recombinant soluble GPR124 ECD (sGPR124) or sGPR124 mutants lacking individual subdomains were tested for binding to recombinant soluble (ΔGPI) Fc-tagged RECK (sRECK-Fc) by Protein A pulldown. While sGPR124 Δhormone receptor domain (ΔHRM) still bound sRECK-Fc, albeit less efficiently than wild-type sGPR124, mutants lacking the leucine-rich repeat domain (ΔLRR) or the GPCR autoproteolysis-inducing domain (ΔGAIN) did not bind sRECK-Fc (Figures 1B and S1B). Cross-linking mass spectrometry of the sGPR124:sRECK-Fc complex revealed extensive interactions between GPR124 LRR/GAIN and RECK cystine knot motif (CK)/EGF2 domains, confirming the pull-down studies and further suggesting interaction between GPR124 GAIN and RECK EGF2 (Figures 1C and S1C; Table S1).
Figure 1. Canonical RECK/GPR124/WNT7 Signaling Does Not Require Intrinsic GPR124 Signal Transduction.

(A) Co-purification of RECK with GPR124 by αGPR124 affinity chromatography from rat brain blood vessels. Where indicated, proteins in isolated brain blood vessels were cross-linked in situ using DSP. Arrows depict proteins excised and identified by mass spectrometry.
(B) Binding of purified sGPR124 proteins (5 nM) to purified sRECK-Fc (50 nM) as assessed by Protein A pull-down.
(C) Cross-linking mass spectrometric analysis of the purified sGPR124:sRECK-Fc complex. Red lines indicate mass spectrometry-inferred cross-links between adjacent lysine residues within the protein complex.
(D) Analysis of GPR124 mutants in Super TOP-Flash (STF) canonical Wnt/β-catenin reporter assay. Cells were co-transfected with the indicated expression constructs and STF/RLuc reporters for 48 hr. STF activity was normalized with RLuc activity and cell surface expression levels (OD410) of the corresponding GPR124 mutant. mbECD is a fusion of the GPR124 ECD and the VEGFR2 TMD. FL, full-length; mb, membrane-bound. Mean (n = 3) ± SD. *p < 0.05 versus RECK/WNT7B only.
(E and F) Activity of sGPR124-Fc (E) or sGPR124 deletion mutant proteins (F) in STF assays. Cells were co-transfected with indicated expression constructs and STF/RLuc reporters. 24 hr after transfection indicated proteins were added at 0, 10, 100, or 1,000 nM (wedges). 24 hr later STF activity was determined and normalized to RLuc activity. Mean (n = 3) ± SD. WT, wild-type. *p < 0.05 versus no protein.
All western blots (WB) were performed under reducing conditions. All data are representative of at least two independent experiments with similar results. LRR, leucine-rich repeat; CT, C-terminal; IG, immunoglobulin; HRM, hormone receptor; GAIN, GPCR autoproteolysis-inducing; CK, cystine knot motif; CRD, cysteine-rich domain; EGF, epidermal growth factor; s, soluble ECD; Fc, IgG Fc fragment.
GPR124/WNT7 co-transfection of HEK293 cells activates the canonical Wnt/β-catenin luciferase reporter Super TOP-Flash (STF), which is further enhanced by additional RECK co-transfection (Cho et al., 2017; Vanhollebeke et al., 2015) (Figure S1D). HEK293 cells endogenously express RECK, but not GPR124 protein (Figure S1E). GPR124/WNT7-mediated STF activation was completely abolished in HEK293 RECK−/− cells (generated by CRISPR/Cas9 KO), and restored upon RECK transfection (Figures S1E and S1F), indicating that GPR124 absolutely requires RECK to induce canonical WNT7 signaling.
Whether the GPR124:RECK complex intrinsically transduces a WNT7-dependent signal into the cell that synergizes with FZD/WNT7/LRP signaling is unknown. We thus tested GPR124 mutants for RECK/WNT7-dependent STF activation, which was normalized to GPR124 mutant cell surface expression (Figures 1D, S2A, and S2B). GPR124 ECD deletion completely abrogated reporter activation, but surprisingly ICD deletion only partially impaired signaling by 22% (Figure 1D). Further, the GPR124 ECD fused to the single-pass TMD of VEGFR2 without the tyrosine kinase domain (membrane-bound ECD [mbECD]) mediated RECK/WNT7-dependent STF activation, albeit less than full-length GPR124 (Figure 1D). Addition of purified recombinant soluble GPR124 ECD (sGPR124-Fc, Figure S1B) to the culture medium was fully sufficient to activate RECK/WNT7-dependent STF to the same extent as full-length GPR124 (Figure 1E). Purified recombinant sGPR124 subdomain deletion mutants (Figures 1F and S1B) variably activated RECK/WNT7 signaling: ΔLRR and ΔGAIN mutants were inactive and ΔHRM partially active (Figure 1F), correlating with RECK binding (Figures 1B and 1C). Although variably expressed on the cell surface, all RECK single domain deletion mutants were inactive or only minimally active in STF assays (Figures S2C and S2D).
Co-culture STF assays revealed that GPR124:RECK cis (same cell), but not trans (opposing cells) interactions promoted canonical WNT7 signaling (Figure S2E). Although the metalloproteinase inhibitor function of RECK (Oh etal., 2001) could potentially mediate GPR124/WNT7-dependent STF activation, the pan-metalloproteinase inhibitor GM6001 (Ilomastat) did not mimic RECK-mediated GPR124/WNT7 signaling (Figure S2F). We also formally confirmed that GPR124/RECK/WNT7A-induced STF activation correlates with increased cytosolic β-catenin levels (Figure S2G).
RECK Is a WNT7 Receptor
Despite remarkable convergence of Wnt7, Gpr124, and Reck KO phenotypes, it is not known if WNT7 functionally couples to GPR124/RECK via direct binding or through more indirect mechanisms (Noda et al., 2016). RECK contains a FZD-like cysteine-rich domain (CRD) (Figure S3A) (Pei and Grishin, 2012) that could directly bind WNT7, analogous to Wnt binding to the CRD of FZDs (Janda et al., 2012). Potential WNT7 binding to sRECK-Fc or sGPR124-Fc was tested using a co-pull-down assay. HEK293 RECK−/− cells stably transfected with WNT7A (HEK293 RECK−/− WNT7A cells) were cultured with sRECK-Fc or sGPR124-Fc protein added to the culture medium to potentially capture newly secreted WNT7A. Protein A pull-down of sRECK-Fc from conditioned medium (CM) resulted in copurification of WNT7A (Figure 2A). In contrast, WNT7A did not appreciably bind sGPR124-Fc. WNT7B released from HEK293 RECK−/− WNT7B cells also bound to sRECK-Fc, but not to VEGFR2 ECD-Fc used as negative control (Figure S3B). Analogous studies using HEK293 RECK−/− WNT3A cells showed expected binding of WNT3A to sFZD4-Fc and sFZD8-Fc positive controls but not to sRECK-Fc or sGPR124-Fc (Figure 2B), further asserting the specificity of the RECK:WNT7 interaction. Furthermore, sRECK-Fc protein dose-dependently inhibited RECK/GPR124/WNT7B- (Figure 2C) but not WNT3A-mediated STF activation (Figure S3C). We also used soluble RECK deletion mutants to map domains binding WNT7A. Only RECK ΔCK and ΔCRD mutants were secreted from HEK293 cells (Figure S1B and data not shown) and thus usable for binding studies; both sRECK ΔCK and ΔCRD were defective in WNT7A binding (Figure 2D).
Figure 2. RECK Is a WNT7 Receptor.

(A) WNT7A binds to RECK and FZD8. HEK293 RECK−/− WNT7A cells were cultured in medium supplemented with indicated purified proteins (25 nM) for 72 hr and CM subjected to Protein A pull-down.
(B) WNT3A binds to FZD4 and FZD8, but not to GPR124 and RECK. HEK293 RECK−/− WNT3A cells were cultured in medium supplemented with indicated purified proteins (25 nM) for 72 hr and CM subjected to Protein A pull-down.
(C) Super TOP-Flash (STF) canonical Wnt/β-catenin reporter gene assay. HEK293 RECK+/+ or RECK−/− cells were co-transfected with the indicated expression constructs and STF/RLuc reporters. 24 hr after transfection sRECK-Fc protein was added at 0, 10, 100, or 1,000 nM (wedges). 24 hr later STF activity was normalized with RLuc activity. Mean (n = 3) ± SD. *p < 0.05 versus no protein.
(D) WNT7A binding to RECK is mediated by RECK cystine knot motif (CK) and cysteine-rich domains (CRD). HEK293 RECK−/− WNT7A cell culture medium was supplemented with the indicated proteins (3 nM) and conditioned for 96 hr. CM were subjected to Protein A pull-down.
(E and F) Cell surface RECK binds to WNT7A, but not to WNT3A. HEK293 RECK−/− WNT7A (E) or WNT3A (F) cells were transfected with the indicated expression constructs. 48 hr after transfection, SNAP tags were specifically biotinylated (bSNAP) and cell surface proteins cross-linked in situ using DTSSP (100 μM). Cells were lysed, subjected to streptavidin pull-down, and cross-links reversed. WCL, whole cell lysate.
All western blots (WB) were performed under reducing conditions. All data are representative of at least two independent experiments with similar results. CM, conditioned medium; s, soluble ECD; Fc, IgG Fc fragment.
Because sRECK and sGPR124 might not completely recapitulate WNT7 binding of the cognate full-length proteins, we also assessed WNT7A binding to full-length cell surface GPR124 and RECK. HEK293 RECK−/− WNT7A cells were transfected with SNAP-GPR124 or SNAP-RECK fusion constructs, which were active in STF assays and similarly expressed on the cell surface (Figures S3D and S3E). The SNAP tag was specifically biotinylated (bSNAP) and cell surface protein complexes stabilized in situ by the non-cell-permeable, thiol-cleavable chemical cross-linker DTSSP. Streptavidin pull-down from cell lysates under denaturing conditions followed by cross-link reversal showed co-pull-down of WNT7A with bSNAP-RECK, but not bSNAP-GPR124 (Figure 2E). Without prior cell surface cross-linking, WNT7A was not pulled down by bSNAP-RECK (Figure 2E). On HEK293 RECK−/− WNT3A cells bSNAP-FZD4, but not bSNAP-RECK and bSNAP-GPR124 bound WNT3A (Figure 2F), further confirming specificity of the interaction between cell surface RECK and WNT7A.
WNT7A Binds to FZDs and LRP5/6
We also assessed WNT7A binding to the archetypal Wnt receptors FZD4 and FZD8, both expressed in CNS endothelium, with Fzd4 being predominantly expressed (Daneman et al., 2009; Zhang et al., 2014). Interestingly, neither full-length SNAP-Fzd4 expressed on the cell surface (Figures 2E and S3E), nor recombinant sFZD4-Fc detectably bound transfected WNT7A, and only trace WNT7A binding was seen with sFZD4-His (Figures 2A and S3F; Table S2). However, STF reporter activation in Lrp5/WNT7A-co-transfected HEK293 RECK−/− cells was modestly increased by Fzd4 (Figure S3G), suggesting low affinity WNT7A binding to FZD4. In contrast, sFZD8 bound WNT7A even more efficiently than sRECK (Figure 2A) and both sFZD4 and sFZD8 bound WNT3A (Figure 2B). GPR124/RECK/WNT7 over-expression activated the STF reporter in parental, but not in FZD1/2/4/5/7/8 KO HEK293T cells (Voloshanenko et al., 2017), whereas both cell lines mediated similar STF activation upon Fzd8/WNT7 overexpression (Figure S3H), indicating that endogenous FZDs mediate GPR124/RECK/WNT7 signaling. Both recombinant LRP5 and LRP6 ECDs bound WNT7A in HEK293 RECK−/− WNT7A CM (Figures S3F and S4A; Table S2). Further, recombinant Dickkopf-1 (DKK1), an LRP5/6 antagonist (Mao et al., 2001), dose-dependently inhibited RECK/GPR124/WNT7-mediated STF activation (Figure S4B).
RECK Binding Stabilizes Short-Lived, Active, Monomeric, Hydrophobic WNT7A
Interestingly, WNT7A binding to RECK or FZD8 was only observed when sRECK or sFZD8 proteins had been added to the medium of WNT7A-expressing cells to capture newly secreted WNT7A (Figure 3A). In contrast, WNT7A in harvested, cell-free WNT7A CM did not bind recombinant sRECK or sFZD8 (Figure 3A), indicating that active WNT7A is very short-lived. Moreover, neither commercially available recombinant WNT7A (PeproTech) nor WNT7A/7B CM mediated GPR124/RECK-dependent STF activation in HEK293 (Figures S4C and S4D). In contrast, recombinant WNT3A protein (PeproTech) and WNT3A CM activated STF (Figures S4C and S4D).
Figure 3. RECK Binding Stabilizes Short-Lived, Active, Monomeric, Hydrophobic WNT7A.

(A) RECK and FZD8 only bind newly secreted WNT7A. Culture media of HEK293 RECK−/− WNT7A cells were supplemented with the indicated proteins (100 nM) and conditioned for 72 hr (“during conditioning”). Alternatively, the indicated proteins were added to the CM after harvesting after 72h (“after conditioning”). CM were subjected to Protein A pull-down.
(B and C) CM from HEK293 RECK−/− WNT7A (B) or WNT3A (C) cells were analyzed by reducing (+DTT) or non-reducing (−DTT) SDS-PAGE/WB. Where indicated CM were subjected to Triton X-114 phase separation.
(D) sRECK-Fc protein stabilizes newly secreted, monomeric, hydrophobic WNT7A. Culture medium of HEK293 RECK−/− WNT7A cells was supplemented with indicated proteins (100 nM) and conditioned for 72 hr (“during conditioning”). Alternatively, indicated proteins were added to CM after harvesting after 72 hr (“after conditioning”). CM aliquots were subjected to Triton X-114 phase separation and remaining CM to Protein A pull-down followed by Triton X-114 phase separation of pull-downs and unbound supernatants. Irrelevant lanes (between lanes 4 and 5) have been removed from the WB.
(E) RECK and WNT7A form a 1:1 complex. Expi293F cells were co-transfected with sRECK-His and WNT7A. 96 hr after transfection CM was harvested and sRECK-His:WNT7A complex purified by combined chemical cross-linking (BS3)/tandem affinity purification. Purified complex was analyzed by reducing SDS-PAGE/WB.
(F) Free, hydrophobic WNT7A is highly unstable. sRECK-Fc:WNT7A, sFZD8-Fc-Biotin:WNT7A, and sFZD8-Fc-Biotin:WNT3A complexes were isolated from the corresponding HEK293 RECK−/− WNT7A/WNT3A CM (96 hr, 100 nM recombinant protein) using Protein A or streptavidin agarose. Bound Wnt proteins were eluted by low pH, neutralized, diluted into PBS/10% FBS and incubated at 37°C. At indicated time points Wnt eluate was subjected to Triton X-114 phase separation.
All data are representative of at least two independent experiments with similar results. CM, conditioned medium; WB, western blot; T, total; De, detergent phase; Aq, aqueous phase; s, soluble ECD; Fc, IgG Fc fragment.
Wnt activity correlates with its hydrophobicity, a result of its palmitoylation and essential for FZD binding (Janda et al., 2012). Loss of Wnt hydrophobicity by removal of the lipid group and/or aggregation is inactivating (Kakugawa et al., 2015; Zhang et al., 2012). To determine hydrophobicity/activity of CM-derived WNT7A, we used the detergent Triton X-114, which dissolves in aqueous buffers at 4°C, but separates into a detergent phase above 20°C, drawing hydrophobic molecules from the aqueous buffer into the detergent phase (Bordier, 1981).
Analysis of CM from HEK293 RECK−/− WNT7A/WNT3A cells by non-reducing (−DTT) SDS-PAGE/western blotting revealed that WNT7A/WNT3A exist as both (1) monomeric and/or non-covalently aggregated species (~45 kDa), as well as (2) covalent aggregates (>120 kDa) (Figures 3B and 3C, lane 2). Both WNT7A and WNT3A covalent aggregates collapsed to monomers upon DTT reduction (+DTT), indicating intermolecular disulfide bonds (Figures 3B and 3C, lane 1 versus lane 2). Interestingly, both low and high MW WNT7A species partitioned into the aqueous, but not the detergent phase upon Triton X-114 phase separation (Figure 3B, lanes 5–7), correlating with lack of STF activation by WNT7A CM (Figure S4C). In contrast, low MW WNT3A exclusively partitioned into the detergent phase (Figure 3C, lanes 5–7), correlating with STF activation by WNT3A CM (Figure S4C). To determine, if CM-derived low MW WNT7A in non-reducing SDS-PAGE is a monomer and/or oligomer under non-denaturing conditions, we performed size exclusion chromatography (Figure S4E). The ~45 kDa WNT7A species was present in elution fractions corresponding to 100–500 kDa indicating that WNT7A predominantly forms large aggregates in CM in vitro. Similar aggregation might occur in vivo as most WNT7A in E12.5 mouse forebrain extracts partitioned into the Triton X-114 aqueous phase as covalent aggregates (Figure S4F). In contrast, E12.5 mouse forebrain WNT3A equally partitioned into the Triton X-114 detergent and aqueous phases as low MW species (Figure S4G).
Next, HEK293 RECK−/− WNT7A cells were allowed to secrete WNT7A into culture medium supplemented with sGPR124-Fc or sRECK-Fc protein (i.e., “during conditioning”) followed by Triton X-114 phase separation of the CM. Non-reducing SDS-PAGE/western blotting of detergent and aqueous phases revealed that sRECK-Fc, but not sGPR124-Fc, stabilized hydrophobic/detergent phase WNT7A (Figure 3D, “Input” lanes 5–8). However, when WNT7A CM was harvested first and recombinant sRECK-Fc added under cell-free conditions (i.e., “after conditioning”), hydrophilic WNT7A did not convert to the hydrophobic form (Figure 3D, “Input” lanes 3 and 4).
Protein A pull-down from the CM (without Triton X-114) followed by Triton X-114 phase separation was performed to determine which WNT7A species bound sRECK-Fc. Triton X-114 phase separation selectively eluted WNT7A from the Protein A/sRECK-Fc/WNT7A beads (Figure S4H). When recombinant sRECK-Fc had been added to the culture medium of live cells (i.e., “during conditioning”), low MW, but not high MW WNT7A, was found in the Protein A pull-down eluate and exclusively partitioned into the detergent phase (Figure 3D, “Protein A-bound” lanes 7 and 8). Conversely, Triton X-114 phase separation of the unbound supernatants from the Protein A/sRECK-Fc pull-down showed depletion of WNT7A from the detergent, but not the aqueous phase (Figure 3D, “Protein A-unbound” lanes 7 and 8). No WNT7A was detectable in the Protein A pull-down when sRECK-Fc had been added to the CM after conditioning (Figure 3C, “Protein A-bound” lanes 3 and 4).
Hydrophobic WNT7A bound to RECK could be monomeric or oligomeric under non-denaturing conditions. To test if RECK binds monomeric or oligomeric WNT7A, the purified sRECK:WNT7A complex was covalently cross-linked with non-cleavable BS3. Western blotting revealed a cross-linker dose-dependent shift of non-cross-linked sRECK (~140 kDa) and WNT7A (~50 kDa) to a single, cross-linked band at ~190 kDa representing a 1:1 complex of sRECK and WNT7A, indicating that RECK directly binds monomeric WNT7A without participation of accessory proteins (Figure 3E).
Similar to WNT7A eluted from sRECK, WNT7A eluted from sFZD8 mainly partitioned into the Triton X-114 detergent phase (Figure S5A). sRECK stabilization of WNT7A in CM was, however, more efficient than equimolar sFZD8 or sLRP6 (Figure S5B). Combinations of different recombinant receptor ECDs in CM elicited modest additive, but not synergistic WNT7A stabilization. Further, GPR124 did not regulate WNT7A stabilization by RECK (Figure S5B).
The aggregation inactivation of WNT7A might be intrinsic to this protein or mediated by Wnt-inactivating enzymes (Kakugawa et al., 2015; Zhang et al., 2012). If Wnt-inactivating enzymes in HEK293 inactivated WNT7A, purified hydrophobic WNT7A should be stable under cell-free conditions similar to purified WNT3A (Willert et al., 2003). Thus, we purified hydrophobic WNT7A in complex with sRECK or sFZD8 and specifically eluted WNT7A by low pH followed by neutralization, incubation at 37°C, and Triton X-114 phase separation (Figure 3F). Notably, by 60 min post-elution, hydrophobic WNT7A had completely converted to the hydrophilic form, suggesting that aggregation inactivation is intrinsic to WNT7A and not mediated by Wnt-inactivating enzymes. In contrast, WNT3A eluted from sFZD8 was stable and hydrophobic at 37°C for at least 120 min (Figure 3F).
GPR124, RECK, and WNT7A Form a Ternary Complex
To test if RECK can assemble a ternary GPR124/RECK/WNT7A complex, sGPR124 and sRECK-Fc proteins were added to medium of HEK293 RECK−/− WNT7A cells during culture and allowed to form complexes. Upon Protein A pull-down from the CM, both sGPR124 and WNT7A co-purified with sRECK-Fc (Figure 4A), indicating ternary GPR124/RECK/WNT7A complex formation. However, WNT7A binding to sRECK-Fc was not regulated by sGPR124 (Figure 4A, lanes 2 and 3) and sRECK-Fc:sGPR124 complex formation was not regulated by WNT7A (Figure 4A, lanes 1 and 2). Neither sGPR124 nor WNT7A co-purified with negative control sEPHB4-Fc (Figure 4A, lanes 4 and 5). Because sRECK-Fc pull-down might show independent sRECK-Fc:WNT7A and sRECK-Fc:sGPR124 complexes without ternary complex formation, we performed a control sGPR124 pull-down. sRECK and WNT7A both co-purified with sGPR124 (Figure S5C), confirming ternary GPR124/RECK/WNT7A complex formation, as GPR124 does not directly bind WNT7A (Figure 2A).
Figure 4. RECK Promotes FZD8:WNT7A Complex Formation.

(A) GPR124, RECK, and WNT7A form a ternary complex. HEK293 RECK−/− (control) or HEK293 RECK−/− WNT7A cells were cultured in medium supplemented with the indicated proteins (50 nM) for 72 hr. CM were harvested and subjected to Protein A pull-down.
(B) GPR124 does not regulate cell surface RECK:WNT7A complex formation. Cells were transfected with indicated expression constructs for 48 hr. SNAP tags were specifically biotinylated (bSNAP) and cell surface proteins cross-linked in situ by DTSSP (100 μM). Cells were lysed, subjected to streptavidin pull-down, and cross-links reversed. WCL, whole cell lysate. β2AR, β2-adrenergic receptor.
(C) FZD/GPR124/RECK/WNT7A quaternary complex formation is not detectable in vitro. sRECK-His:WNT7A complex was purified from HEK293 RECK−/− WNT7A CM (72 hr, 100 nM sRECK-His) using Ni-NTA agarose. Indicated purified proteins/sRECK:WNT7A complex (1 μM each) were allowed to form complexes and subjected to Protein A pull-down. Faint non-specific sRECK binding in all lanes.
(D) FZD8 elutes WNT7A from RECK. sRECK-Fc:WNT7A complex was isolated from HEK293 RECK−/− WNT7A CM (72 hr, 100 nM sRECK-Fc) by Protein A agarose. Beads were incubated with 0.1, 1, or 10 μM sFZD4/sFZD8 protein (wedges) or glycine, pH 2.9 for 1 hr (eluate 1) and then with 1% SDS (eluate 2).
(E) WNT7A binding to RECK and FZD8 is mutually exclusive. HEK293 RECK−/− WNT7A cells were cultured in medium supplemented with indicated proteins (25 nM) for 72 hr. CM were subjected to sequential Ni-NTA agarose (1st) and Protein A agarose (2nd) pull-downs.
(F) RECK dose-dependently enhances FZD8:WNT7A complex formation. HEK293 RECK−/− WNT7A cells were cultured in medium supplemented with sFZD8-His and sRECK-Fc proteins as indicated for 72 hr. CM were subjected to Ni-NTA pull-down.
(G) Model of RECK/GPR124-regulated canonical WNT7 signaling. GPR124 ECD binding to RECK strongly enhances RECK/WNT7-induced canonical Wnt signaling by an extracellular mechanism that does not involve intrinsic GPR124 signal transduction or regulation of RECK:WNT7 complex formation. The RECK cystine knot motifs (blue) and cysteine-rich domain (purple) mediate binding of RECK:GPR124 to WNT7, stabilizing WNT7 in its active, monomeric, hydrophobic form. Free WNT7 rapidly converts into an inactive, aggregated, hydrophilic state. The GPR124:RECK complex acts as a stabilizing receptor for WNT7 increasing bioavailability of active, monomeric cell surface WNT7. GPR124:RECK-bound WNT7 is eventually transferred to FZD and LRP5/6 co-receptors by transient direct or indirect interactions. Alternatively, GPR124/RECK/WNT7 could form a stable multi-protein receptor complex with FZD/LRP5/6.
All western blots (WB) were performed under reducing conditions. All data are representative of at least two independent experiments with similar results. CM, conditioned medium; s, soluble ECD; Fc, IgG Fc fragment.
See also Figure S5.
Potential regulation of RECK:WNT7A complex formation by GPR124 on the cell surface was investigated by co-transfection of HEK293 RECK−/− WNT7A cells with SNAP-RECK and GPR124. SNAP-RECK was specifically biotinylated (bSNAP-RECK) and cell surface protein complexes stabilized in situ using the thiol-cleavable chemical cross-linker DTSSP. Streptavidin pull-down of bSNAP-RECK from cell lysates followed by cross-link reversal revealed similar WNT7A co-pull-down with and without GPR124 co-transfection (Figure 4B). WNT7A did not co-purify with negative control bSNAP-β2AR (Figure 4B).
Because GPR124 regulation of RECK/WNT7 signaling was not mediated by modulating RECK:WNT7A complex formation, we tested if GPR124 regulates RECK cell surface expression. HEK293 RECK−/− cells were transfected with GPR124 and/or RECK and cell surface proteins isolated by cell surface bio-tinylation/streptavidin pull-down. Interestingly, RECK cell surface, but not total expression levels were drastically increased by GPR124 co-expression (Figure S5D). In contrast, cell surface GPR124 was slightly decreased by RECK. However, cell surface RECK expression was not decreased by Gpr124 conditional KO (Gpr124Δ/−) in primary brain endothelial cells (Figure S5E), indicating potential functional differences between overexpressed and endogenous GPR124.
RECK Promotes FZD8:WNT7A Complex Formation
To address potential formation of a multi-protein receptor complex consisting of GPR124, RECK, FZD4/FZD8, and WNT7A, we mixed the corresponding purified ECDs with purified sRECK:WNT7A complex. Protein A pull-down of sFZD4-Fc or sFZD8-Fc from the protein mixture did not detectably co-precipitate sGPR124 or sRECK (Figure 4C). However, WNT7A co-purified with sFZD8-Fc possibly by relay of WNT7A from sRECK to sFZD8-Fc, because the resultant sFZD8-Fc:WNT7A complex lacked sRECK (Figure 4C, lane 8). Relay of WNT7A from sRECK to sFZD8-Fc was GPR124-independent (Figure 4C, lanes 7 and 8). Consistently, sFZD8, but not sFZD4, dose-dependently eluted WNT7A from sRECK-Fc:WNT7A immobilized on Protein A agarose, suggesting mutually exclusive, sequential binding of WNT7 to RECK and then FZD (Figure 4D).
We also determined potential competition between FZD and RECK for WNT7A binding. Equimolar amounts of sFZD4-Fc/sFZD8-Fc and sRECK-His proteins were added to the medium of HEK293 RECK−/− WNT7A cells and allowed to capture newly secreted WNT7A. In sequential pull-downs, WNT7A preferentially bound to sFZD8-Fc over sRECK-His, but preferred sRECK-His over sFZD4-Fc (Figure 4E). Although WNT7A preferentially bound sFZD8, sRECK-Fc markedly increased sFZD8:WNT7A complexes in CM of HEK293 RECK−/− WNT7A cells (Figure 4F), suggesting that RECK might present WNT7A to FZDs in a manner that favors FZD binding, ultimately leading to relay of WNT7A from RECK to FZD.
DISCUSSION
Despite remarkable convergence of Gpr124, Wnt7a/7b and Reck KO phenotypes (Cho et al., 2017; Daneman et al., 2009; de Almeida et al., 2015; Kuhnert et al., 2010; Stenman et al., 2008) and their synergistic activation of Wnt signaling (Cho et al., 2017; Vanhollebeke et al., 2015; Zhou and Nathans, 2014), molecular mechanisms of the GPR124/RECK/WNT7 intersection have been elusive. Here, we identified GPI-anchored RECK as a specific WNT7 receptor. During the revision of this manuscript, RECK was reported as a WNT7 receptor based on proximity ligation assays and micromolar binding of WNT7 peptides to the RECK CK domain (Eubelen et al., 2018). Our study also implicates the RECK CK domain in WNT7 binding. However, Wnt proteins bind to the CRD of FZD receptors (Janda et al., 2012) and RECK contains a FZD-like CRD (Pei and Grishin, 2012) that we identified as also essential for WNT7 binding. Competitive elution of WNT7A from RECK by the FZD8 ECD without stable RECK/WNT7/FZD8 ternary complex formation infers that the RECK binding site on WNT7 is similar to, or overlaps with the FZD binding site on Wnts (Janda et al., 2012). Potentially, both RECK CK and CRD domains cooperatively modulate WNT7 binding by direct or allosteric mechanisms.
The current results also provide mechanistic insight into GPR124 regulation of canonical RECK/WNT7 signaling. A signalosome model proposes that the GPR124 ICD associates with Dishevelled (DVL) that bridges to FZDs, while the GPR124 ECD recruits WNT7 via RECK (Eubelen et al., 2018). Our study supports an essential role of the GPR124 ECD, but reveals ICD function as largely dispensable for WNT7 signaling, because (1) a GPR124 ΔICD mutant retains strong activity in a canonical Wnt reporter assay, (2) membrane-tethered GPR124 ECD fused to the VEGFR2 TMD still mediates WNT7 signaling, and (3) recombinant soluble GPR124 ECD augments RECK/WNT7 signaling to the same extent as full-length GPR124. Thus, GPR124 does not appear to intrinsically transduce a canonical RECK/WNT7 signal into the cell, suggesting that GPR124/RECK act primarily in an extracellular fashion. GPR124 ICD deletion leaving 28 residual membrane-proximal amino acids has been reported to completely abrogate GPR124 function by preventing DVL recruitment (Eubelen et al., 2018); we deleted a larger portion of the GPR124 ICD, leaving only 10 membrane-proximal amino acids with the resultant protein being robustly active in canonical Wnt reporter assays. Cell surface proximity ligation studies suggest that extracellular GPR124 activity modulates RECK/WNT7 binding (Eubelen et al., 2018). Our biochemical studies do not indicate GPR124 regulation of soluble RECK:WNT7A complex formation in CM or cell surface RECK:WNT7A association. Overexpressed GPR124 upregulated RECK cell surface expression, which could indirectly increase cell surface RECK:WNT7 complexes, but Gpr124Δ/− primary brain endothelial cells do not exhibit a corresponding cell surface RECK decrease. Soluble GPR124 ECD structure-function analysis implicates the GPR124 LRR/GAIN and to a lesser extent HRM domains in RECK/WNT7 signaling, correlating with their requirement in GPR124:RECK complex formation, agreeing with full-length receptor mutants (Cho et al., 2017), but further indicating GAIN domain participation.
CNS endothelium expresses mRNAs for at least three canonical Fzds (Fzd4 and to a lesser degree Fzd1 and Fzd8) and non-canonical Fzd6 (Daneman et al., 2009; Zhang et al., 2014) that might redundantly mediate canonical Wnt signaling. Fzd4, the predominant Fzd in CNS endothelium, bound WNT7A with low affinity. In contrast, FZD8 robustly bound WNT7A. While GPR124 and/or RECK did not enhance FZD4:WNT7 association, RECK drastically enhanced FZD8:WNT7 binding, potentially reflecting mechanistic differences between FZD4 and FZD8 or technical limitations of our assays. On the other hand, FZD4 might preferentially bind Norrin (Wang et al., 2012), while other FZDs such as FZD1 and FZD8 could potentially possess tropism for WNT7. However, our results by no means exclude WNT7 binding to FZD4 by yet undescribed mechanisms.
Crucially, RECK binding stabilizes WNT7A in its active, monomeric, hydrophobic state. The obligate palmitoylation of Wnt proteins underlies their hydrophobicity, FZD binding, and activity (Janda et al., 2012; Kakugawa et al., 2015; Zhang et al., 2012). Unlike WNT3A, active, monomeric, hydrophobic WNT7A is very short-lived and rapidly converts into an inactive, aggregated, hydrophilic form after secretion, unless bound by RECK or other receptors. Wnt-inactivating enzymes convert active WNT3A to an inactive, aggregated, hydrophilic protein (Kakugawa et al., 2015; Zhang et al., 2012). However, the short half-life of active, hydrophobic WNT7A is intrinsic to this protein, since purified hydrophobic WNT7A eluted from RECK or FZD8 rapidly became hydrophilic, potentially underlying short-range action of WNT7A in vivo.
Immunofluorescence co-localization (Eubelen et al., 2018) or binding of RECK/GPR124 ECD fragments to GPR124/RECK/WNT7/FZD/LRP-transfected cells (Cho et al., 2017) have suggested WNT7-induced formation of FZD/LRP/GPR124/RECK multi-receptor complexes or signalosomes. The current biochemical studies with recombinant ECDs did not reveal multi-protein receptor complex formation, but might not have been sufficiently sensitive to detect transient complexes. Cell surface RECK:WNT7 interaction may be similarly short-lived, given the ultimate preference of WNT7 for FZD. The presentation of WNT7 to FZD by RECK in a manner favoring FZD:WNT7 complex formation could link GPR124/RECK/WNT7 and FZD/WNT7/LRP signaling, with GPR124:RECK stabilizing and presenting cell surface WNT7, thus directly or indirectly promoting FZD/WNT7/LRP association (Figure 4G).
Overall, our identification of RECK as a GPR124-associated WNT7 receptor that forms a 1:1 complex with active, monomeric, hydrophobic WNT7 extends the known structural and functional diversity of Wnt receptors. Conceivably, RECK could also modulate WNT7 action in non-vascular and non-CNS compartments. Stabilization of specific Wnts by accessory receptors, as exemplified by RECK and WNT7, may be generally utilized as a strategy broadly employed for multi-tiered control of Wnt signaling as well as other growth factor pathways.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Calvin J. Kuo (cjkuo@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and maintenance
HEK293/HEK293T cells and sub cell lines were maintained in DMEM supplemented with 10% FBS and 1x penicillin/streptomycin (ThermoFisher Scientific) at 37°C and 8% CO2. HEK293T FZD1/2/4/5/7/8 KO cells were provided by M. Boutros and O. Voloshanenko (Voloshanenko et al., 2017). Expi293F cells were obtained from ThermoFisher Scientific and maintained in Expi293 Expression Medium according to the manufacturer’s recommendations.
Rodent strains, maintenance, and breeding
Rodents were housed in 12-hour light and dark cycles in a pathogen-free animal facility. All animal experiments were performed in accordance with the National Institutes of Health guidelines for use and care of live animals and were approved by the Institutional Animal Care and Use Committee at Stanford University. Male Sprague Dawley rats were obtained from Charles River Laboratories. The mouse Gpr124 KO (Gpr124−) and floxed exon 1 (Gpr124fl) alleles were generated and genotyped as previously described (Kuhnert et al., 2010). Transgenic mice expressing the CreER(T2) fusion protein under control of a VE-cadherin promoter (Cdh5-CreER) were obtained from Ralf Adams and genotyped as previously described (Wang et al., 2010). Gpr124fl/−; Cdh5-CreER mice were obtained by crossing Gpr124fl/fl with Gpr124+/−; Cdh5-CreER mice. All mutant mice had been backcrossed to the C57BL/6 strain. E12.5 mouse embryos were obtained by setting up wild-type breeding pairs for timed pregnancies. The morning a female was found with a vaginal plug was considered E0.5. At E12.5 pregnant females were sacrificed and embryos dissected.
Primary brain endothelial cells
Primary brain endothelial cells were isolated from wild-type and Gpr124fl/−; Cdh5-CreER mice as previously described (Perrière et al., 2005) and cultured in 6-well plates until confluent (~7d). Cell culture medium was supplemented with 1 μM 4-hydroxytamoxifen for the first three days to induce conditional KO of Gpr124.
METHOD DETAILS
Generation of HEK293 RECK−/− cells
The RECK gene in HEK293 cells was disrupted using CRISPR/Cas9. The Cas9 coding sequence from pLentiCRISPR, a gift from Feng Zhang (Addgene plasmid #52961) (Sanjana et al., 2014) was fused to the T2A peptide and GFP coding sequences and sub cloned into the adenoviral shuttle vector pAdd2 (pAdd2-Cas9-T2A-GFP). The pLentiCRISPR vector was modified by replacing the Cas9 insert with mCherry yielding the construct pLenti-sgRNA-mCherry-P2A-Puro. Human RECK exon 1 without 5′ UTR was used as input sequence to identify RECK-specific single guide (sg) RNAs using the Zhang lab’s web tool (http://crispr.mit.edu). The two highest scored sgRNAs (sgRECK#1 and #2) were synthesized as double stranded DNAs with BsmBI-compatible 5′ overhangs by the Stanford Protein and Nucleic Acid (PAN) core facility. sgRECK#1 (sense: caccgGAGCAGCGCACCTCGCAGAG, antisense: aaacCTCTG CGAGGTGCGCTGCTCc) and sgRECK#2 (sense: caccgCACATACATCACGGCACATT, antisense: aaacAATGTGCCGTGATGTAT GTGc) were cloned into pLenti-sgRNA-Cherry-P2A-Puro. HEK293 cells were seeded into 6-well plates, incubated for 24h, and transfected with 3 μg Cas9 and/or 3 μg sgRECK#1 or sgRECK#2. 72h after transfection RECK expression levels were analyzed by western blotting. Cas9/sgRECK#1 and Cas9/sgRECK#2 co-transfected cells showed reduced RECK expression compared to Cas9, sgRECK#1, and #2 only transfected cells (data not shown). Since sgRECK#1 suppressed RECK expression more efficiently than sgRECK#2, single clones were isolated from Cas9/sgRECK#1 co-transfected HEK293 cells after 48h by limiting dilution cloning in a 96-well plate. Single clones were expanded in 6-well plates and screened for complete loss (homozygous deletion) of RECK expression by western blotting. Clone number 13 (HEK293 RECK−/− cells) was further expanded and used for subsequent experiments.
Transient and stable transfection of cells
HEK293 and HEK293T cells were transfected using the calcium phosphate precipitation method as described (Graham and van der Eb, 1973). 24h before transfection 5 × 106 or 9 × 105 cells were seeded into 10 cm dishes or 6-well plate wells, respectively. Cells seeded into 10 cm dishes were transfected with 34 μg total DNA and cells seeded into 6-well plate wells with 6 μg total DNA. pBluescript was used as carrier DNA to keep DNA amounts constant. For stable transfections HEK293 cells were transfected with 6 μg expression plasmid (containing a neomycin resistance cassette) per well in 6-well plates. 48h after transfection cells were transferred to T75 flasks and selected for stable transfectants using 600 μg/ml geneticin (Chem-Impex International, Wood Dale, IL) for two weeks. Pools of stably transfected HEK293 cell clones were maintained in DMEM supplemented with 10% FBS and 600 μg/ml geneticin. Expi293F cells were transfected in 125 mL or 500 mL Erlenmeyer cell culture flasks using the ExpiFectamine 293 Transfection Kit (ThermoFisher Scientific) according to the manufacturer’s protocol.
Generation of GPR124 antibodies
Anti-mouse GPR124 ECD antibody
Anti-mouse GPR124 ECD antiserum was generated by immunizing rabbits (Rockland Immunochemicals, Pottstown, PA) with recombinant mouse GPR124 ECD, which had been cloned, expressed, and purified from stably transfected HEK293 cell conditioned medium as previously described (Vallon et al., 2010). Polyclonal antibody was purified from antiserum of best responder by antigen affinity chromatography.
Anti-human GPR124 ICD antibody
The human GPR124 ICD (aa 1067 − 1338, UniProtKB #Q96PE1) was cloned into the pFLAG-MAC bacterial expression vector (Sigma-Aldrich), expressed in E. coli BL21(DE3), and purified from E. coli lysates using αFLAG M2 agarose. Anti-human GPR124 ICD antiserum was generated by immunizing a rabbit (pAb productions, Hebertshausen, Germany) with the recombinant protein. Polyclonal antibody was purified from antiserum by antigen affinity chromatography.
Covalent coupling of antibodies to agarose beads
Rabbit anti-mouse GPR124 ECD antibody (500 μg antibody/100 μl beads), ChromPure rabbit IgG (Jackson ImmunoResearch #011-000-003, 500 μg antibody/100 μl beads), or carrier-free anti-WNT7A antibody (abcam #ab100792, 100 μg antibody/100 μl beads) were covalently coupled to agarose beads using AminoLink Plus Immobilization Kit (ThermoFisher Scientific) according to the manufacturer’s protocol.
Immunofluorescence staining
6 × 104 HEK293 cells per well were seeded into fibronectin-coated 8-chamber glass slides (ThermoFisher Scientific) and incubated for 24h. Cells were transfected with 4 ng indicated expression construct for 48h and fixed with 4% formaldehyde in PBS for 15 min. Fixed cells were washed three times with PBS (5 min each), blocked with 5% normal goat serum in PBS for 1h, and incubated with rabbit αFLAG antibody (Sigma-Aldrich, 10 μg/ml) in PBS/1% BSA for 1h. Cells were washed as before and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody (Jackson ImmunoResearch, 1:1000) in PBS/1% BSA for 1h. Cells were washed as before and coverslips mounted using VECTASHIELD HardSet Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). Stained cells were imaged using a fluorescence microscope with Apotome (Zeiss, Oberkochen, Germany).
Western blot analysis
Western blot analyses were performed using standard methods. Briefly, samples were supplemented (or eluted from agarose beads) with NuPAGE LDS Sample Buffer (ThermoFisher Scientific). Unless indicated otherwise samples were supplemented with 50 mM DTT (reducing conditions). NuPAGE® Novex® 4%−12% Bis-Tris Gels (ThermoFisher Scientific) were used for SDS-PAGE. PageRuler Plus Prestained Protein Ladder (ThermoFisher Scientific) was used as molecular weight marker. Separated proteins were transferred to PVDF membranes (EMD Millipore, Hayward, CA) and membranes were blocked with 5% non-fat dry milk in TBS/0.1 Tween 20 (TBST/milk). Membranes were incubated with primary antibodies diluted in TBST/milk or TBST/5% BSA (0.1 – 1 μg/ml). HRP-conjugated primary or secondary antibodies were diluted 1:10000 in TBST/milk. After antibody incubations (1h - overnight) blots were washed 3x with TBST for 10 min each. Bound antibodies were visualized using SuperSignal West Pico/Femto Chemoluminescent Substrates (ThermoFisher Scientific) and exposure of AccuRay Blue X-Ray Films (E&K Scientific).
GPR124 affinity chromatography
Brains from six 3-month-old Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) were dissected and meninges were removed using cotton swabs. Brains were washed once with ice-cold PBS and homogenized on ice in 10 mL PBS per brain using a 15 mL Dounce homogenizer. Homogenates were pooled, pelleted, and resuspended in 200 mL ice-cold PBS + 20% BSA. Suspension was centrifuged at 1000 x g, 4°C for 25 min and myelin top layer and supernatant were aspirated. Pellet was resuspended in 10 mL ice-cold PBS and passed through a 70 μm cell strainer. Strainer was washed three times with ice-cold PBS and inverted onto a 10 cm Petri dish. Blood vessels were rinsed off strainer using ice-cold PBS, transferred to a 50 mL conical tube, and pelleted. Blood vessels were split into two equal aliquots. One aliquot was resuspended in 10 mL ice-cold PBS/0.1 mM dithiobis(succinimidyl propionate) (DSP)/0.5% DMSO and one in 10 mL ice-cold PBS/0.5% DMSO only and incubated at 4°C under constant rotation for 2h. Blood vessels were pelleted, resuspended in 10 mL ice-cold TBS, and incubated at 4°C under constant rotation for 15 min. Blood vessels were washed once with ice-cold TBS, pelleted, weighed, and lysed in RIPA buffer (TBS, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, cOmplete Protease Inhibitor Cocktail [Roche, Indianapolis, IN]) at 1 mL lysis buffer per 100 mg tissue. Lysates were incubated on ice for 30 min vortexing occasionally. Lysates were cleared by centrifugation and incubated over night at 4°C under constant rotation with 10 μl anti-mouse GPR124 ECD or rabbit IgG agarose (5 μg antibody/μl beads). Agarose beads were transferred to mini spin columns (Thermo Fisher Scientific) and centrifuged at 3000 x g for 30 s. Flow throughs (unbound fractions) were saved. Beads were washed three times for 5 min with 0.5 mL RIPA buffer at 4°C under constant rotation. Washing was repeated as above with TBS/1% Triton X-100/1 M NaCl and then with 0.1 M sodium acetate, pH 5.3/1% Triton X-100. Bound proteins were eluted by incubating beads with 50 μl 0.1 M glycine/HCl, pH 2.0/1% Triton X-100 at 4°C under constant rotation for 5 min. Eluates were neutralized by adding 3.5 μl 1.5 M Tris/HCl, pH 8.8 and bound and unbound fractions were analyzed by SDS-PAGE followed by silver staining or western blotting. Silver-stained bands of proteins that co-purified with GPR124 upon in situ protein cross-linking were excised from gel and submitted to the Stanford University Mass Spectrometry (SUMS) core facility for identification.
Generation of conditioned media
5 × 105 HEK293 RECK−/− cells ± stable transfection with WNT3A, WNT7A, or WNT7B were seeded per well in 6-well plates. Where indicated, medium (DMEM + 10% FBS) was supplemented with recombinant protein and conditioned for 48h – 96h. Conditioned media were harvested and cleared by centrifugation.
Pull-down assays
Using conditioned media
Conditioned media (1 mL each) were supplemented with 100 μl 10xTBS (Protein A pull-down) or 10xPBS (Ni-NTA pull-down). 75 μl samples (inputs) were saved and remaining conditioned media were incubated with 10 μl Protein A or Ni-NTA agarose for 1h at room temperature under constant rotation. Agarose beads were washed six times with 2xTBS (Protein A) or 2xPBS/5 mM imidazole (Ni-NTA) and bound proteins were eluted by incubating beads with SDS-PAGE sample buffer for 10 min at room temperature under constant rotation. Eluates (pull-downs) and inputs were analyzed by western blotting.
Using recombinant protein mixtures
Recombinant proteins were added to TBS/0.1% Triton X-100 at indicated concentrations and incubated for 1h at room temperature or overnight at 4°C. Input samples were saved and remaining mixtures were incubated with 10 μl Protein A agarose for 1h at room temperature under constant rotation. Agarose beads were washed six times with TBS/0.1% Triton X-100 and bound proteins were eluted with reducing SDS-PAGE sample buffer for 10 min at room temperature. Inputs and eluates (pull-downs) were analyzed by western blotting.
Using whole cell lysates
HEK293 RECK−/− WNT7A/WNT3A cells seeded into 6-well plates were transfected with the indicated expression construct(s) and/or empty vector (600 ng expression vector(s) + 5.4 μg pBluescript). 48h after transfection SNAP tags were specifically auto-biotinylated (bSNAP) by incubating cells with 5 μM SNAP-Biotin substrate (New England Biolabs) for 30 min. Cells were washed twice with ice-cold HBS++ (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2) and incubated with 0.1 mM DTSSP cross-linker in HBS++ on ice for 2h. Unreacted cross-linker was quenched by adding 50 mM Tris/HCl, pH 7.4 for 15 min. Cells were scraped into 150 μl RIPA lysis buffer (TBS, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, cOmplete ULTRA Protease Inhibitor Cocktail [Roche], PhosSTOP Phosphatase Inhibitor Cocktail [Roche]) and incubated on ice for 30 min vortexing occasionally. Lysates were cleared by centrifugation and split 1:1 into input and pull-down aliquots. 10 μl streptavidin agarose was added to pull-down aliquots and incubated at 4°C under constant rotation for 1h. Beads were washed six times with TBS/2% SDS and bound proteins were eluted by incubating beads in reducing SDS-PAGE sample buffer supplemented with 3 mM biotin (Sigma) at 95°C for 10 min. Whole cell lysate inputs and streptavidin pull-downs were analyzed by western blotting.
Expression constructs and molecular cloning
All deletion mutants were generated using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Human GPR124 (UniProtKB #Q96PE1) lacking the endogenous signal peptide (aa 34 - 1338) was cloned into the mammalian expression vector p3xFLAG-CMV-9 (Sigma-Aldrich) as previously described (Vallon and Essler, 2006). ECD (Δaa 34 - 770) and ICD (Δaa 1078 - 1338) deletion mutants of GPR124 were cloned using p3xFLAG-CMV-9-GPR124 as template. C-terminally His-tagged human GPR124 ECD (sGPR124, UniProtKB #Q96PE1, aa 34 – 755 + DGGGSHHHHHH) was cloned into p3xFLAG-CMV-9 as previously described (Vallon and Essler, 2006). Subdomain deletion mutants of the GPR124 ECD, ΔLRR (Δaa 34 - 247), ΔHRM (Δaa 349 - 426), and ΔGAIN (Δaa 427 - 755) were cloned using p3xFLAG-CMV-9-sGPR124 as template. Human RECK (UniProtKB #095980) lacking the endogenous signal peptide (aa 23 – 971) was amplified by PCR from a cDNA clone (Incyte cDNA clone #LIFESEQ4097514, GE Healthcare Dharmacon, Lafayette, CO) and cloned into p3xFLAG-CMV-9. sGPR124-Fc and sRECK-Fc expression constructs were generated by fusing the mouse Igk signal peptide (aa 1 – 21) with human GPR124 ECD (aa 34 – 755) / human RECK ΔGPI (aa 23 – 942) and the Fc fragment of mouse IgG2a (aa 237 - 469). Fusion constructs were cloned into the E1 region of recombinant adenovirus type 5 (E1- and E3-deleted, replication deficient) and into pcDNA3.1(+) (Thermo Fisher Scientific). Adenoviral particles were generated by transfecting HEK293 cells with linearized adenoviral vectors as previously described (Yan et al., 2017). Adenoviruses encoding sFzd8-Fc, HA-Dkk1, and sVegfr2-Fc had been generated previously (Daneman et al., 2009; Kuhnert et al., 2004; Kuo et al., 2001). Untagged human Wnt expression constructs pcDNA-WNT3A, -WNT7A, and -WNT7B were gifts from Marian Waterman (Addgene plasmids #35908, #35914, and #35915) (Najdi et al., 2012). Domain deletion mutants of RECK, ΔCK (Δaa 37 - 338), ΔCRD (Δaa 343 - 476), ΔKazal1 (Δaa 632 - 677), ΔKazal2 (Δaa 708 - 750), ΔKazal3 (Δaa 753 - 787), and ΔEGF2 (Δaa 676 - 709) were cloned using pcDNA3.1-sRECK-Fcand p3xFLAG-CMV-9-RECKas templates. A C-terminally His-tagged soluble RECK expression construct (sRECK-His) was generated by inserting a His6 tag + stop codon into pcDNA3.1-sRECK-Fc immediately after the sRECK coding sequence using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent Technologies). GPR124 mbECD (membrane-bound ECD) and RECK GPI > TMD constructs were cloned by fusing the human VEGFR2 TMD (UniProtKB #P35968, aa 763 - 790) to human FKBP1A(UniProtKB#P62942,aa2 – 108, F36V mutation) and a stop codon. The VEGFR2 TMD-FKBP1A-stop fusion was inserted into p3xFLAG-CMV-9-sGPR124 between the spacer (DGGGS) and the His tag and into p3xFLAG-CMV-9-RECK between aa 942 and 943 of RECK by Gibson assembly cloning. The sGPR124-VEGFR2 TMD-FKBP1A fusion construct (GPR124 mbECD) mediated RECK/WNT7-induced STF activation without FKBP ligands as shown, but STF activation was increased two-fold upon addition of a dimerizing FKBP ligand (data not shown). Mouse Fzd4 expression construct pRK5-mFzd4 was a gift from Chris Garcia and Jeremy Nathans (Addgene plasmid #42256) (Yu et al., 2012). Mouse Lrp5 expression construct pRK5-Lrp5 was a gift from Aaron Hsueh (Yu et al., 2012). N-terminally SNAP-tagged expression constructs were cloned by inserting the SNAP tag (aa 1 – 182, pSNAPf, New England Biolabs) in frame into p3xFLAG-CMV-9-GPR124 and p3xFLAG-CMV-9-RECK (between the 3xFLAG tag and the GPR124/RECK insert) and into pRK5-mFzd4 (between the signal peptide and the mature protein) using Gibson assembly. N-terminally SNAP-tagged β2-adrenergic receptor (pSNAPf-ADRβ2) was from New England Biolabs. The mouse Fzd8 coding sequence (UniProtKB #Q61091) without signal peptide (aa 28 – 685) was synthesized by Integrated DNA Technologies and cloned into p3xFLAG-CMV-9.
Expression and purification of recombinant proteins
Recombinant sGPR124-Fc, sFZD8-Fc, HA-DKK1, and sVEGFR2-Fc were expressed in HEK293 cells transduced with the corresponding recombinant adenovirus at an MOI of ~1. Recombinant sGPR124 and sGPR124 subdomain deletion mutants were expressed in HEK293 cells stably transfected with p3xFLAG-CMV-9-sGPR124-His (wild-type or subdomain deletion). Cells were seeded into T225 flasks and cultured until confluent. Culture medium was replaced with serum-free DMEM (ThermoFisher Scientific) and, unless stably transfected, cells were transduced with adenovirus at the same time. Culture medium of adenovirally transduced HEK293 cells was conditioned until most cells showed adenoviral cytopathic effect (3 – 7 days). Culture medium of stably transfected cells was conditioned for 7 days. Medium conditioning of stably transfected HEK293 cells was repeated twice and conditioned media (CM) were pooled. Recombinant sRECK-Fc, sRECK-Fc subdomain deletion mutants, and sRECK-His proteins were expressed in Expi293F cells transiently transfected with the corresponding pcDNA3.1 expression construct. Serum-free culture medium of transfected Expi293F cells was conditioned for 5 days. CM were harvested and cleared by centrifugation. For purification of 3xFLAG-, HA-, and His-tagged proteins CM were concentrated 10 to 100 fold using Centricon Plus-70 centrifugal filters (MWCO 10 kDa, EMD Millipore). For purification of 3xFLAG-, HA-, and Fc-tagged proteins CM were supplemented with 1xTBS/0.1% Triton X-100. For purification of His-tagged proteins concentrated CM were diluted 1:10 in 2xPBS/10 mM imidazole. CM were incubated with 0.5 mL Protein A agarose (Fc-tagged proteins), 1 mL αFLAG M2 agarose (3xFLAG-tagged proteins), 1 mL αHA agarose (HA-tagged proteins), or 100 μl Ni-NTA agarose (His-tagged proteins) for2h at room temperature or overnight at 4°C under constant rotation or stirring. Protein A, αFLAG M2, and αHA agarose beads were recovered from CM using gravity flow columns and washed six times with 10 volumes 4xTBS/0.1% Triton X-100 and three times with 10 volumes 1xTBS. Bound proteins were eluted with 0.1 M glycine/HCl, pH 2.9 (Protein A agarose), pH 3.5 (αFLAG M2 agarose), or pH 2.5 (αHA agarose) and neutralized with 1 M Tris/HCl, pH 8.5. Ni-NTA agarose beads were recovered from CM, washed five times with 10 volumes 2xPBS/20 mM imidazole, and bound proteins were eluted with 2xPBS/250 mM imidazole. Eluates were concentrated using Amicon Ultra-4 centrifugal filters (MWCO 10 kDa, EMD Millipore) and dialyzed against PBS. Protein concentrations were determined using A280 measurements and the calculated extinction coefficients. Protein purity and integrity was assessed by SDS-PAGE followed by Coomassie Blue staining. Purified recombinant sEPHB4-Fc (#446-B4), sFZD4-Fc (#194-FZ), sLRP5-His (#7344-LR), and sLRP6-His (#2960-LR) were from R&D Systems (Minneapolis, MN). Recombinant His-tagged/biotinylated human FZD4 and mouse FZD8 cysteine-rich domains (sFZD4/sFZD8) were generated as previously described (Janda et al., 2012). Purified recombinant sFZD8-Fc was biotinylated using Sulfo-NHS-LC-Biotin (ThermoFisher Scientific) according to the manufacturer’s protocol.
Cross-linking mass spectrometry
Purified recombinant sGPR124 and sRECK-Fc proteins were mixed (1 μM each) in PBS. The homobifunctional, amine-reactive cross-linker DSS was dissolved in DMSO, added to the protein mixture (1 mM), and incubated at room temperature for 30 min. Unreacted DSS was quenched by adding 50 mM Tris/HCl, pH 7.4 for 15 min. Cross-linked and control samples were separated by SDS-PAGE followed by Coomassie Blue staining. The band corresponding to the sGPR124:sRECK-Fc complex was excised and submitted to the SUMS core facility for cross-linking mass spectrometric analysis. Intramolecular cross-links, cross-links within epitope tags, and cross-links with a negative log probability less than 2 are not shown. The cross-link map was generated using the xVis Crosslink Analysis Webserver (http://xvis.genzentrum.lmu.de/login.php) (Grimm et al., 2015).
Super TOP-Flash reporter gene assay
All Super TOPflash (STF) reporter gene assays were performed as co-culture assays of HEK293 cells stably expressing the indicated Wnt expression construct (“Wnt producing population”) and HEK293 cells transiently transfected with the STF reporter, Renilla luciferase, and indicated expression constructs (“Wnt reporting population”). Wnt producing population: HEK293 or HEK293 RECK−/− cells stably expressing the indicated Wnt expression construct were seeded in 96-well plates (1.5 × 104 cells/well) and incubated for 48h. Wnt reporting population: HEK293 or HEK293 RECK−/− cells were seeded in 6-well plates, incubated for 24h, and transfected with the indicated expression construct(s) and/or empty vector (60 ng total), 60 ng STF reporter (pGL4.49[luc2P/TCF-LEF/Hygro], Promega, Madison, WI), 60 ng Renilla luciferase (pRL-TK, Promega), and 5.82 μg pBluescript (carrier DNA). 24h after transfection “Wnt reporting population” was trypsinized, resuspended in fresh medium, and added to the “Wnt producing population” in the 96-well plate in a 1:1 ratio. “Wnt reporting population” was allowed to become adherent for 2h and medium was replaced with fresh medium supplemented or not with indicated recombinant proteins or chemical compounds. Cells were incubated for 24h and luciferase activities (firefly and Renilla) were measured using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase (STF) activity was normalized with Renilla luciferase activity and is shown relative to the activity in control cells (“non-Wnt producing population” + STF/Renilla/empty vector-transfected “Wnt reporting population”).
β-catenin stabilization assay
HEK293 RECK−/− cells stably transfected or not with WNT7A or WNT3A were seeded into 10 cm dishes and transfected with 340 ng STF reporter (pGL4.49[luc2P/TCF-LEF/Hygro], Promega), 340 ng Renilla luciferase (pRL-TK, Promega), 170 ng p3xFLAG-CMV-9-GPR124 + 170 ng p3xFLAG-CMV-9-RECK or 340 ng empty vector, and 33 μg pBluescript. 48h after transfection cells were scraped into 1 mL ice-cold 50 mM Tris/HCl, pH 7.4, 250 mM sucrose, 5 mM MgCl2, cOmplete protease inhibitor cocktail (Roche), and PhosSTOP phosphatase inhibitor cocktail (Roche). 18 μl of the cell suspensions were transferred to new tubes and used for Dual-Luciferase Reporter Assay (Promega). The remaining cells were lysed by passing the cell suspensions 20 times through a 25-gauge needle. Cell lysates were centrifuged at 100000xg, 4°C for 1h, pellets were discarded, and supernatants (cytosolic extracts) analyzed by western blotting.
Triton X-114 phase separation assay
Conditioned media (CM) were harvested, cleared by centrifugation, chilled on ice, supplemented with 2% Triton X-114 (from a 20% Triton X-114/TBS stock solution), and incubated on ice for 5 min. For combined Triton X-114 phase separation and WNT7A elution from Protein A/sRECK-Fc/WNT7A agarose, beads were resuspended in 1 mL ice-cold TBS/2% Triton X-114 and incubated on ice for 5 min. Forebrains from seven E12.5 mouse embryos were dissected on ice, weighed, and lysed in ice-cold TBS, 2% Triton X-114, cOmplete protease inhibitor cocktail (Roche), and PhosSTOP phosphatase inhibitor cocktail (Roche) (100 mg tissue/ml lysis buffer) by triturating with a 1 mL pipette until completely homogeneous. Lysate was incubated on ice for 30 min vortexing occasionally and cleared by centrifugation. CM/bead suspension/forebrain lysate was layered on a cushion of 100 μl TBS/6% sucrose/0.001% Phenol Red and incubated at 37°C for 5 min (phase separation and WNT7A elution). Phases and beads were separated by centrifugation at 2000 x g at room temperature for 5 min. Top aqueous phase was transferred to new tubes and sucrose interphase was aspirated leaving the bottom detergent phase and/or bead pellet. Detergent phase was separated from beads and Triton X-114 concentrations of aqueous (~0.1% Triton X-114) and detergent (~20% Triton X-114) phase were normalized by adding 2% Triton X-114 and 9 volumes TBS, respectively. Total CM/forebrain lysate before phase separation as well as aqueous and detergent phases were analyzed by non-reducing SDS-PAGE.
Wnt stability assay
sRECK-Fc:WNT7A, sFZD8-Fc-Biotin:WNT7A, and sFZD8-Fc-Biotin:WNT3A complexes were generated by adding the corresponding Fc fusion protein (100 nM) to the culture medium of HEK293 RECK−/− WNT7A/WNT3A cells. After 96h conditioned media were harvested and protein complexes isolated using Protein A agarose (sRECK-Fc:WNT7A) or streptavidin agarose (sFZD8-Fc-Biotin:WNT7A/WNT3A). Bound proteins were eluted using ice-cold 0.1 M glycine/HCl, pH 2.9/0.5 M NaCl/2% Triton X-114 and immediately neutralized with Tris/HCl, pH 8.5. Low pH specifically eluted Wnt from the streptavidin/sFZD8-Fc-Biotin/Wnt beads. sRECK-Fc co-eluted with WNT7A from the Protein A/sRECK-Fc/WNT7A beads and was specifically eliminated by three consecutive Triton X-114 phase separations, replacing the aqueous phase with fresh TBS each time. Chilled eluates were diluted 1:100 into PBS/10% FBS and incubated at 37°C. At the indicated time points eluates were subjected to Triton X-114 phase separation and detergent (De) and aqueous (Aq) phases analyzed by non-reducing SDS-PAGE/Western blotting.
Combined chemical cross-linking/tandem affinity purification of the sRECK:WNT7A complex
Expi293F cells were co-transfected with sRECK-His and WNT7A expression constructs at a 9:1 ratio in 125 mL Erlenmeyer cell culture flasks (30 mL Expi293 Expression Medium). 96h after transfection conditioned media (CM) were harvested, cleared by centrifugation, and dialyzed against 2xPBS/10 mM imidazole. 1 mL CM aliquots were incubated with 10 μl Ni-NTA agarose for 1h at room temperature under constant rotation. Beads were washed three times with 2xPBS/20 mM imidazole and once with 1xPBS. Non-reversible, amine-reactive cross-linker BS3 (ThermoFisher Scientific) was added to the beads at indicated concentrations in 1 mL PBS and incubated for 30 min at room temperature under constant rotation. Bound protein complexes were eluted and residual BS3 cross-linker quenched by incubating beads with 100 μl 2xTBS/250 mM imidazole/0.1 Triton X-100 for 10 min. Eluates were incubated with 10 μl anti-WNT7A agarose for 1h at room temperature under constant rotation. Beads were washed four times with 2xTBS and bound protein complexes were eluted by incubating beads with non-reducing SDS-PAGE sample buffer for 10 min at room temperature. Eluates were supplemented with 50 mM DTT and analyzed by western blotting.
Cell surface ELISA
HEK293 RECK−/− cells seeded into 6-well plates were transfected with 300 ng indicated expression construct + 300 ng Renilla luciferase per well. 24h after transfection cells were seeded into two separate fibronectin-coated 96-well plates (6 × 104 cells/well) and incubated for 24h. One plate was used to determine Renilla luciferase activities using the Renilla Luciferase Assay System (Promega). Cells in the other plate were fixed with 4% formaldehyde in PBS for 15 min, washed three times with PBS (5 min each), and blocked with 5% normal donkey serum in PBS for 1h. Cells were incubated with polyclonal rabbit αFLAG antibody at 1 μg/ml in PBS/1% BSA for 1h, washed as before, and incubated with HRP-conjugated donkey anti-rabbit IgG antibody diluted 1:10000 in PBS/1% BSA for 1h. Cells were washed as before and 1-Step Ultra TMB Substrate (Thermo Fisher Scientific) was added. Reaction was stopped with 1M H2SO4 and OD410 or OD450 was measured. Background absorbance values from mock-transfected cells were subtracted from other values. OD410/OD450 values were normalized with mean values of Renilla luciferase activities (RLU).
Cell surface protein isolation
HEK293 cells were seeded in 6-well plates and transfected with 30 ng of each expression construct and/or empty vector for 48h. Primary brain endothelial cells were isolated from wild-type and Gpr124fl/−; Cdh5-CreER mice as previously described (Perrière et al., 2005) and cultured in 6-well plates until confluent (~7d). Brain endothelial cell culture medium was supplemented with 1 μM 4-Hydroxytamoxifen (Sigma) for the first three days to induce conditional KO of Gpr124. Cells were washed twice with ice-cold HBS++ (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2) and incubated with 0.4 mM Sulfo-NHS-LC-Biotin (ThermoFisher Scientific) in HBS++ on ice for 30 min. Unreacted biotin reagent was quenched by adding 50 mM Tris/HCl, pH 7.4 for 15 min. Cells were scraped into 150 μl RIPA lysis buffer (TBS, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, cOmplete ULTRA Protease Inhibitor Cocktail [Roche], PhosSTOP Phosphatase Inhibitor Cocktail [Roche]) and incubated on ice for 30 min vortexing occasionally. Lysates were cleared by centrifugation and split 1:1 into whole cell lysate and streptavidin pull-down aliquots. 10 μl streptavidin agarose were added to the pull-down aliquot and incubated at 4°C under constant rotation for 1h. Unbound supernatant (intracellular fraction) was saved, beads were washed six times with ice-cold RIPA lysis buffer, and bound proteins (cell surface fraction) were eluted by incubating beads in reducing SDS-PAGE sample buffer supplemented with 3 mM biotin (Sigma) at 95°C for 10 min. Whole cell lysate and cell surface/intracellular fractions were analyzed by western blotting.
Gel filtration
Gel filtration column (Superose 6 prep grade [GE Life Sciences, Pittsburgh, PA], 7.5 mL bed volume, 0.6 cm inner diameter) was calibrated using a mixture of α2-macroglubulin (720 kDa), IgG (150 kDa), and BSA (66 kDa). 100 μl WNT7A conditioned medium supplemented with 10% glycerol was loaded onto column and run at 0.12 ml/min (equilibration/elution buffer: 3xTBS). 1 min fractions were collected and proteins were precipitated with 10% trichloroacetic acid/0.02% sodium deoxycholate on ice for 1h. Precipitates were pelleted, washed once with ice-cold acetone, and dissolved in 30 μl non-reducing SDS-PAGE sample buffer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses comparing equal sample sizes and variances were performed using a two-tailed Student’s t test. All mean values represent biological replicates. n and p values are found in the figure legends. p values less than 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| αFLAG M2 agarose | Sigma-Aldrich | Cat# A2220; RRID: AB_10063035 |
| αFLAG M2-HRP | Sigma-Aldrich | Cat# A8592; RRID: AB_439702 |
| Rabbit αFLAG | Sigma-Aldrich | Cat# F7425; RRID: AB_439687 |
| αHA tag agarose | Sigma-Aldrich | Cat# A2095; RRID: AB_257974 |
| Rabbit αRECK | Cell Signaling Technology | Cat# 3433; RRID: AB_2238311 |
| Rabbit αWNT3A | Cell Signaling Technology | Cat# 2721; RRID: AB_2215411 |
| Rabbit αN-cadherin | ThermoFisher Scientific | Cat# PA1-20359; RRID: AB_2077427 |
| Mouse αβ-actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_626632 |
| Goat αVE-cadherin | Santa Cruz Biotechnology | Cat# sc-6458; RRID: AB_2077955 |
| Rabbit αWNT7A | abcam | Cat# ab100792; RRID: AB_10858110 |
| αWNT7A agarose | This paper | N/A |
| Rabbit αWNT7B | abcam | Cat# ab155313 |
| Mouse αβ-catenin | BD Biosciences | Cat# 610153; RRID: AB_397554 |
| Rabbit αGSK3β | Cell Signaling Technology | Cat# 9315; RRID: AB_490890 |
| Mouse αHis tag | ThermoFisher Scientific | Cat# MA1-21315; RRID: AB_557403 |
| Rabbit αSNAP tag | New England Biolabs | Cat# P9310; RRID: AB_10631145 |
| Goat αHuman Fc-HRP | Jackson ImmunoResearch | Cat# 109-035-098; RRID: AB_2337586 |
| Goat αMouse Fc-HRP | Jackson ImmunoResearch | Cat# 115-035-071; RRID: AB_2338506 |
| Donkey αRabbit IgG-HRP | Jackson ImmunoResearch | Cat# 711-035-152; RRID: AB_10015282 |
| Donkey αMouse IgG-HRP | Jackson ImmunoResearch | Cat# 715-035-150; RRID: AB_2340770 |
| Donkey αGoat IgG-HRP | Jackson ImmunoResearch | Cat# 705-035-003; RRID: AB_2340390 |
| Goat αRabbit IgG-Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 111-545-003; RRID: AB_2338046 |
| Rabbit αMouse GPR124 extracellular domain | This paper | N/A |
| Rabbit αGPR124 agarose | This paper | N/A |
| Rabbit IgG agarose | This paper | N/A |
| Rabbit αHuman GPR124 intracellular domain | This paper | N/A |
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | Agilent Technologies | Cat# 200131 |
| Ad sFzd8-Fc | Daneman et al., 2009 | N/A |
| Ad HA-Dkk1 | Kuhnert et al., 2004 | N/A |
| Ad sVegfr2-Fc (Ad Flk1-Fc) | Kuo et al., 2001 | N/A |
| Ad sGPR124-Fc | This paper | N/A |
| Ad sRECK-Fc | This paper | N/A |
| Chemicals, Peptides,and Recombinant Proteins | ||
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# T176 |
| SNAP-Biotin | New England Biolabs | Cat# S9110S |
| GM6001 | Enzo Life Sciences | Cat# BML-EI300 |
| DSS | ThermoFisher Scientific | Cat# 21655 |
| DSP | ThermoFisher Scientific | Cat# 22586 |
| DTSSP | ThermoFisher Scientific | Cat# 21578 |
| BS3 | ThermoFisher Scientific | Cat# 21580 |
| Human sGPR124 (sTEM5) | Vallon and Essler, 2006 | N/A |
| Human sGPR124-Fc | This paper | N/A |
| Human sRECK | This paper | N/A |
| Human sRECK-Fc | This paper | N/A |
| Human sFZD4 | Janda et al., 2012 | N/A |
| Mouse sFZD4-Fc | R&D Systems | Cat# 194-FZ |
| Mouse sFZD8 | Janda et al., 2012 | N/A |
| Mouse sFZD8-Fc | Daneman et al., 2009 | N/A |
| Mouse sLRP5 | R&D Systems | Cat# 7344-LR |
| Mouse sLRP6 | R&D Systems | Cat# 2960-LR |
| Mouse sVEGFR2-Fc (FLK1-Fc) | Kuo et al., 2001 | N/A |
| Mouse sEPHB4-Fc | R&D Systems | Cat# 446-B4 |
| Mouse HA-DKK1 | Kuhnert et al., 2004 | N/A |
| Mouse WNT3A | PeproTech | Cat# 315-20 |
| Human WNT7A | PeproTech | Cat# 120-31 |
| Critical Commercial Assays | ||
| Dual-luciferase reporter assay system | Promega | Cat# E1980 |
| Experimental Models:Cell Lines | ||
| Human: HEK293 cells | ATCC | Cat# CRL-1573 |
| Human: HEK293 RECK−/− cells | This paper | N/A |
| Human: HEK293 RECK−/− WNT3A cells | This paper | N/A |
| Human: HEK293 RECK−/− WNT7A cells | This paper | N/A |
| Human: HEK293 RECK−/− WNT7B cells | This paper | N/A |
| Human: HEK293T cells | ATCC | Cat# CRL-3216 |
| Human: HEK293T FZD1/2/4/5/7/8 KO cells | Voloshanenko et al., 2017 | N/A |
| Human: Expi293F cells | ThermoFisher Scientific | Cat# A14527 |
| Experimental Models:Organisms/Strains | ||
| Rat: Sprague Dawley | Charles River Laboratories | Cat# 001 |
| Mouse: Gpr124−: B6.129S-Gpr124tm1Cjku | Kuhnert et al., 2010 | N/A |
| Mouse: Gpr124fl: B6.129S-Gpr124flex1 | Kuhnert et al., 2010 | N/A |
| Mouse: Cdh5-CreER: C57BL/6-Tg(Cdh5-cre/ERT2)1Rha | Ralf Adams; Wang et al., 2010 | N/A |
| Oligonucleotides | ||
| hRECK sgRNA #1: GAGCAGCGCACCTCGCAGAG | This paper | N/A |
| hRECK sgRNA #2: CACATACATCACGGCACATT | This paper | N/A |
| Recombinant DNA | ||
| pFLAG-MAC-GPR124 ICD (aa 1067 – 1338, human) | This paper | N/A |
| p3xFLAG-CMV-9-GPR124 (aa 34 - 1338, human) | Vallon and Essler, 2006 | N/A |
| p3xFLAG-CMV-9-GPR124 ΔECD (Δaa 34 – 770) | This paper | N/A |
| p3xFLAG-CMV-9-GPR124 ΔICD (Δaa 1078 – 1338) | This paper | N/A |
| p3xFLAG-CMV-9-sGPR124-His (aa 34 - 755, human) | Vallon and Essler, 2006 | N/A |
| p3xFLAG-CMV-9-sGPR124 ΔLRR-His (Δaa 34 - 247) | This paper | N/A |
| p3xFLAG-CMV-9-sGPR124 ΔHRM-His (Δaa 349 - 426) | This paper | N/A |
| p3xFLAG-CMV-9-sGPR124 ΔGAIN-His (Δaa 427 - 755) | This paper | N/A |
| p3xFLAG-CMV-9-sGPR124-VEGFR2 TMD-FKBP (mbECD) | This paper | N/A |
| pAd5ΔE1ΔE3-Igκ SP-sGPR124-Fc | This paper | N/A |
| Human RECK cDNA | GE Healthcare Dharmacon (Incyte) | Clone# LIFESEQ4097514 |
| p3xFLAG-CMV-9-RECK (aa 23 – 971, human) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔCK (Δaa 37 - 338) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔCRD (Δaa 343 - 476) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔKazal1 (Δaa 632 - 677) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔKazal2 (Δaa 708 - 750) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔKazal3 (Δaa 753 - 787) | This paper | N/A |
| p3xFLAG-CMV-9-RECK ΔEGF2 (Δaa 676 - 709) | This paper | N/A |
| pcDNA3.1-Igκ SP-sRECK-Fc (aa 23 - 942, human) | This paper | N/A |
| pcDNA3.1-Igκ SP-sRECK-Fc ΔCK (Δaa 37 - 338) | This paper | N/A |
| pcDNA3.1-Igκ SP-sRECK-Fc ΔCRD (Δaa 343 - 476) | This paper | N/A |
| pcDNA3.1-Igκ SP-sRECK-His | This paper | N/A |
| pAd5ΔE1ΔE3-Igκ SP-sRECK-Fc | This paper | N/A |
| p3xFLAG-CMV-9-sRECK-VEGFR2 TMD-FKBP (GPI > TMD) | This paper | N/A |
| pcDNA-WNT3A (human) | Najdi et al., 2012 | Addgene plasmid #35908 |
| pcDNA-WNT7A (human) | Najdi et al., 2012 | Addgene plasmid #35914 |
| pcDNA-WNT7B (human) | Najdi et al., 2012 | Addgene plasmid #35915 |
| pRK5-mFzd4 | Yu et al., 2012 | Addgene plasmid #42256 |
| pRK5-Lrp5 | Aaron Hsueh; Yu et al., 2012 | N/A |
| pSNAPf-ADRβ2 (human) | New England Biolabs | Cat# N9184 |
| pSNAPf | New England Biolabs | Cat# N9183S |
| p3xFLAG-CMV-9-SNAPf-GPR124 | This paper | N/A |
| p3xFLAG-CMV-9-SNAPf-RECK | This paper | N/A |
| pRK5-SNAPf-mFzd4 | This paper | N/A |
| p3xFLAG-CMV-9-Fzd8 (mouse) | This paper | N/A |
| pGL4.49[luc2P/TCF-LEF/Hygro] (STF reporter) | Promega | Cat# E4611 |
| pRL-TK (Renilla luciferase reporter) | Promega | Cat# E2241 |
| pLentiCRISPR | Sanjana et al., 2014 | Addgene plasmid #52961 |
| Software and Algorithms | ||
| xVis Crosslink Analysis Webserver | http://xvis.genzentrum.lmu.de/login.php; Grimm et al., 2015 | N/A |
| CRISPR design tool | http://crispr.mit.edu | N/A |
| Other | ||
| Bovine fibronectin | Sigma-Aldrich | Cat# F1141 |
| Normal donkey serum | Jackson ImmunoResearch | Cat# 017-000-121 |
| Normal goat serum | Jackson ImmunoResearch | Cat# 005-000-121 |
| Streptavidin agarose | ThermoFisher Scientific | Cat# 20347 |
| Protein A agarose | SeraCare | Cat# 5710-0005 |
| Ni-NTA agarose | QIAGEN | Cat# 30210 |
| Superose 6 PreD Grade | GE Life Sciences | Cat# 17048901 |
Highlights.
RECK is the predominant binding partner of GPR124 in rat brain blood vessels in situ
RECK/WNT7 signaling is primarily regulated by the GPR124 ectodomain
RECK is a WNT7 receptor that stabilizes short-lived, active, monomeric WNT7
RECK-bound WNT7 binds Frizzled receptors more readily than does free WNT7
ACKNOWLEDGMENTS
We are grateful to members of the Kuo laboratory and R. Nusse for helpful comments. We thank C. Janda for recombinant FZD4 ECD and M. Boutros and O. Voloshanenko for HEK293T FZD1/2/4/5/7/8 KO cells. We thank the Stanford University Mass Spectrometry (SUMS) core facility (https://mass-spec.stanford.edu) for protein ID and cross-linking mass spectrometric analyses. J.C. was supported by an American Heart Association Postdoctoral Fellowship (15POST23020039). K.Y. was supported by a Stanford University Dean’s Fellowship Award. K.C.G., D.S., and Y.M. were supported by HHMI. This work was also supported by an American Heart Association Innovative Science Award, Stanford Stroke Collaborative Action Network Pilot Grant, and NIH (NS064517, NS100904, and DK085527 to C.J.K.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.045.
REFERENCES
- Bordier C (1981). Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256, 1604–1607. [PubMed] [Google Scholar]
- Chang J, Mancuso MR, Maier C, Liang X, Yuki K, Yang L, Kwong JW, Wang J, Rao V, Vallon M, et al. (2017). Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 23, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C, Smallwood PM, and Nathans J (2017). Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron 95, 1056–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, and Nusse R (2012). Wnt/b-catenin signaling and disease. Cell 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, and Barres BA (2009). Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 106, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida GM, Yamamoto M, Morioka Y, Ogawa S, Matsuzaki T, and Noda M (2015). Critical roles for murine Reck in the regulation of vascular patterning and stabilization. Sci. Rep. 5, 17860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, Dumitru AC, Koehler M, Gut P, Alsteens D, Stainier DYR, et al. (2018). A molecular mechanism for Wnt ligand-specific signaling. Science 361, eaat1178. [DOI] [PubMed] [Google Scholar]
- Graham FL, and van der Eb AJ (1973). A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Grimm M, Zimniak T, Kahraman A, and Herzog F (2015). xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 43 (W1), W362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann J, Aust G, Araç D, Engel FB, Formstone C, Fredriksson R, Hall RA, Harty BL, Kirchhoff C, Knapp B, et al. (2015). International union of basic and clinical pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 67, 338–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Waghray D, Levin AM, Thomas C, and Garcia KC (2012). Structural basis of Wnt recognition by Frizzled. Science 337, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Dang LT, You C, Chang J, de Lau W, Zhong ZA, Yan KS, Marecic O, Siepe D, Li X, et al. (2017). Surrogate Wnt agonists that phenocopy canonical Wnt and β-catenin signalling. Nature 545, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, and Ye W (2009). TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 139, 299–311. [DOI] [PubMed] [Google Scholar]
- Kakugawa S, Langton PF, Zebisch M, Howell S, Chang TH, Liu Y, Feizi T, Bineva G, O’Reilly N, Snijders AP, et al. (2015). Notum deacylates Wnt proteins to suppress signalling activity. Nature 519, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, Nusse R, and Kuo CJ (2004). Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc. Natl. Acad. Sci. USA 101, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, Su H, Fruttiger M, Young WL, Heilshorn SC, and Kuo CJ (2010). Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CJ, Farnebo F, Yu EY, Christofferson R, Swearingen RA, Carter R, von Recum HA, Yuan J, Kamihara J, Flynn E, et al. (2001). Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc. Natl. Acad. Sci. USA 98, 4605–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, and Niehrs C (2001). LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411, 321–325. [DOI] [PubMed] [Google Scholar]
- Najdi R, Proffitt K, Sprowl S, Kaur S, Yu J, Covey TM, Virshup DM, and Waterman ML (2012). A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities. Differentiation 84, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Vallon M, and Kuo CJ (2016). The Wnt7’s tale: a story of an orphan who finds her tie to a famous family. Cancer Sci. 107, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, et al. (2001). The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 107, 789–800. [DOI] [PubMed] [Google Scholar]
- Pei J, and Grishin NV (2012). Cysteine-rich domains related to Frizzled receptors and Hedgehog-interacting proteins. Prot. Sci. 21, 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrière N, Demeuse P, Garcia E, Regina A, Debray M, Andreux JP, Couvreur P, Scherrmann JM, Temsamani J, Couraud PO, et al. (2005). Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J. Neurochem. 93, 279–289. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, and McMahon AP (2008). Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250. [DOI] [PubMed] [Google Scholar]
- Ulrich F, Carretero-Ortega J, Menéndez J, Narvaez C, Sun B, Lancaster E, Pershad V, Trzaska S, Véliz E, Kamei M, et al. (2016). Reck enables cerebrovascular development by promoting canonical Wnt signaling. Development 143, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon M, and Essler M (2006). Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin alpha(v)beta3 to glycosaminoglycans. J. Biol. Chem. 281, 34179–34188. [DOI] [PubMed] [Google Scholar]
- Vallon M, Rohde F, Janssen KP, and Essler M (2010). Tumor endothelial marker 5 expression in endothelial cells during capillary morphogenesis is induced by the small GTPase Rac and mediates contact inhibition of cell proliferation. Exp. Cell Res. 316, 412–421. [DOI] [PubMed] [Google Scholar]
- Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, Gauquier A, Cabochette P, Fukuhara S, Mochizuki N, et al. (2015). Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. eLife 4 10.7554/eLife.06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloshanenko O, Gmach P, Winter J, Kranz D, and Boutros M (2017). Mapping of Wnt-Frizzled interactions by multiplex CRISPR targeting of receptor gene families. FASEB J. 31, 4832–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rattner A, Zhou Y, Williams J, Smallwood PM, and Nathans J (2012). Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151, 1332–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chang H, Rattner A, and Nathans J (2016). Frizzled receptors in development and disease. Curr. Top. Dev. Biol. 117, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR 3rd, and Nusse R (2003). Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452. [DOI] [PubMed] [Google Scholar]
- Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. (2017). Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature 545, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Ye X, Guo N, and Nathans J (2012). Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development 139, 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Abreu JG, Yokota C, MacDonald BT, Singh S, Coburn KL, Cheong SM, Zhang MM, Ye QZ, Hang HC, et al. (2012). Tiki1 is required for head formation via Wnt cleavage-oxidation and inactivation. Cell 149, 1565–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, and Nathans J (2014). Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev. Cell 31, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, Taketo MM, and Nathans J (2014). Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 124, 3825–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.