

Summary
Palmitoylation is a post-translational modification (PTM) involving the thioesterification of cysteine residues with a 16 carbon saturated fatty acid. Little is known about rates of depalmitoylation or the parameters that dictate these rates. Here we report a modular strategy to synthesize quenched fluorogenic substrates for the specific detection of depalmitoylase activity and for mapping the substrate specificity of individual depalmitoylases. We demonstrate that human depalmitoylases APT1 and APT2, and TgPPT1 from the parasite Toxoplasma gondii have distinct specificities that depend on amino acid residues distal to the palmitoyl cysteine. This information informs the design of optimal and non-optimal substrates as well as isoform selective substrates to detect the activity of a specific depalmitoylase in complex proteomes. In addition to providing tools for studying depalmitoylases, our findings identify a previously unrecognized mechanism for regulating steady state levels of distinct palmitoylation sites by sequence-dependent control of depalmitoylation rates.
Keywords: Chemical probes, Depalmitoylases, Combinatorial libraries, Dynamic palmitoylation, Thioesterases, fluorogenic peptides, substrate specificity.
Graphical Abstract
eTOC
Amara et al. describe a method for preparing positional scanning libraries of fluorogenic palmitoylated peptide substrates. This allowed identification of residues that are distal to the palmitoylation site that impact turnover. This information allowed the design of substrates that are selective for a specific depalmitoylatin enzyme.
Protein palmitoylation is the post-translational addition of a palmitate (S-palmitoylation) onto the sulfhydryl group of a cysteine residue to form a thioester linkage(Linder and Deschenes, 2007). Protein palmitoylation can be constitutive or dynamic, with transition rates of palmitoylation cycles that vary between minutes to hours(Ahearn et al., 2011; Wedegaertner and Bourne, 1994). Palmitoylation affects fundamental cellular processes such as cell signaling, neuronal development, cellular growth and differentiation(el-Husseini Ael and Bredt, 2002; Smotrys and Linder, 2004), and is therefore subject to multiple layers of regulation. Enzymatic addition of palmitate to proteins is mediated by palmitoyl acyl transferases (PATs). The modification increases their hydrophobicity and facilitates protein-protein interactions, membrane association and segregation into lipid rafts, protein trafficking, protein function and protein stability(Linder and Deschenes, 2007; Mitchell et al., 2006). Additionally altered rates of protein palmitoylation can give rise to neuronal ceroid lipofuscinosis and is implicated in other human disease such as cancer, Alzheimer’s, Huntington’s disease, schizophrenia and mental retardation(Resh, 2012).
Depalmitoylation plays an integral part in maintaining turnover rates of dynamically palmitoylated proteins. Changes in substrate turnover rates modulate protein activity. In response to receptor stimulation for example, Gα proteins such as Gsα and Giα undergo increased turnover(Smotrys and Linder, 2004). Depalmitoylated Gα redistributes to the membrane to facilitate signal transduction(Goddard and Watts, 2012). Similarly, depalmitoylation of eNOS and PSD-95, triggered by receptor agonists, results in their activation(el-Husseini Ael and Bredt, 2002). Ras family small GTPases are also dynamically palmitoylated. Maintaining a rapid turnover rate is crucial for N- and H-Ras localization to the golgi and plasma membrane. Oncogenic H-Ras shows an increased turnover rate resulting from increased depalmitoylation(Baker et al., 2003) and blocking depalmitoylation results in prolonged membrane association and the loss of activation of downstream signaling pathways(Dekker et al., 2010). The identity of the depalmitoyase responsible for the depalmitoylation of Ras and other dynamically palmitoylated proteins in vivo remain unclear.
Depalmitoylases, such as acyl protein thioestrases (APTs) or palmitoyl protein thioesterases (PPTs), remove palmitate from proteins. Members from this family belong to the metabolic serine hydrolase (SH) superfamily, utilizing a serine residue for the catalytic hydrolysis of ester, thioester or amide bonds(Long and Cravatt, 2011). To date, four mammalian depalmitoylases have been characterized. These are PPT1 and PPT2(Sugimoto et al., 1996; Verkruyse and Hofmann, 1996), which are lysosomal enzymes involved in protein degradation, and APT1(Duncan and Gilman, 1998) and APT2(Tomatis et al., 2010), which are enzymes that are thought to be responsible for depalmitoylation of cytosolic proteins. Recently, three additional metabolic SH members, ABHD17A-C, were shown to depalmitoylate the postsynaptic density protein PSD-95 and N-Ras and thus are potentially additional depalmitoylases(Lin and Conibear, 2015; Yokoi et al., 2016). The metabolic SH family consists of over 100 members in humans, many of which are yet to be characterized and potentially include additional depalmitoylases. Advances in metabolic labeling and enrichment methods have accelerated the annotation of palmitoylated substrates(Foe et al., 2015; Hang et al., 2007; Hannoush and Arenas-Ramirez, 2009; Martin et al., 2011). Pulse chase experiments that use the palmitoyl analogue 17-octadecynoic acid (17-ODYA) can be used to distinguish stably palmitoylated proteins from those that have short turnover times(Jones et al., 2012; Martin et al., 2011). These studies revealed subsets of proteins, mostly involved in intercellular signaling, cell growth and cancer that show inherently dynamic palmitoylation.
Tools to measure depalmitoylation activity and map substrate preferences of these enzymes will be necessary to understand how these enzymes recognize their substrates and facilitate the hydrolysis of the thioester linkage. Recently, Kathayat at al. described a fluorescence probe capable of measuring depalmitoylation activity in living cells. Hydrolysis of the thioester linkage of the acylated cysteine in the probe results in an intermolecular rearrangement that leads to an increase in fluorescence. The application of this probe in live cells allowed measurement of changes in depalmitoylation activity as a result of hormone stimulation, further supporting the role of dynamic palmitoylation in the regulation of cellular signaling. However, to address solubility issues, the acyl chain in this probe was reduced to 7 carbons, compromising the specificity of the probe to long-chain thioesterases. Moreover, the intermolecular reaction needed for the fluorescence turn on hampers the application of the probe for the kinetic measurements of hydrolysis rates. The requirement for the secondary reaction triggered by the hydrolyzed cysteine also limits this scaffold to modifications to residues on the C-terminal side of the palmitoylation site only.
To address the current limitations of existing chemical probes for depalmitoylases, we developed a design that places a fluorophore on the N-terminus of a peptide that has a palmitoylated cysteine containing a quencher group attached to the lipid chain through a click chemistry handle. The fluorescence is quenched on the intact, palmitoylated peptide but is de-quenched upon cleavage of the thioester bond by a depalmitoylase. Because the chemistry is modular and amenable to solid phase peptide synthesis (SPPS), it is possible to rapidly make substrates with diverse peptide sequences flanking the palmitoylation site. We demonstrate the applicability of this approach by measuring the kinetic properties of APTs in vitro and in situ using complex cell lysates or whole organ homogenates. We further show that the strategy enables synthesis of positional scanning libraries of substrates that allow mapping of specificity determinants of individual depalmitoylases. Our data provide evidence that depalmitoylases possess distinct substrate specificities based on the residues flanking the palmitoylated cysteine residue. We show that the sequence surrounding the palmitoylated cysteine modulates the affinity for the depalmitoylase and, to a larger extent, the rate of depalmitoylation. This relationship between substrate sequence and rate of depalmitoylation highlights a mechanism by which depalmitoylases may maintain specific steady state turnover rates across diverse, but overlapping protein substrates.
Results:
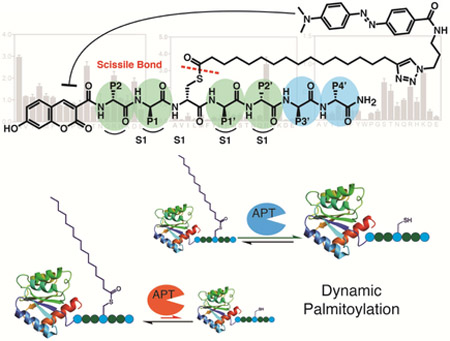
The lack of efficient methods to selectively measure depalmitoylase activity motivated us to design a modular reporter assay that was amenable to SPPS and that could be used to make diverse palmitoylated peptide substrates. We envisioned an optimal assay that would make use of a cysteine-based substrate containing a palmitoylated thioester whose hydrolyzed product could be detected using a standard fluorescence detector (Fig. 1a). The hydrophobicity of the palmitoyl chain, or similar long-chain mimics, poses a significant challenge for achieving sufficient aqueous solubility of a small reporter molecule. Substitution of the long palmitoyl chain with a shorter octanoyl lipid, such as is used in the common esterase substrate 4-nitrophenyl octanoate (4-NPO, Fig. 1b), slightly increases solubility in aqueous or detergent containing buffers but potentially compromises the selectivity towards long-chain-fatty-acyl hydrolases. However, we should note that we have not systematically tested various lipid chain lengths and it is possible that depalmitoylase enzymes have distinct lipid chain length preferences. Regardless, we used the 18-carbon chain lipid 17-ODYA because this has been used extensively as a mimetic for palmitate in proteomic studies. To increase the solubility we used morpholine as the C-terminal amide to generate a general Quenched Substrate for thiolEsters, (QStE; Fig. 1b). The fluorophore 7-hydroxy-3-carboxycoumarin was positioned at the N-terminus of the peptide while the 4-(dimethylaminoazo)benzene-4-carboxylic acid (DABCYL) quencher was attached to the lipid thioester, using the alkyne functionality. Our QStE substrate, as well as the oxyester analogue, Quenched Substrate for Esterases (QSE; Fig. 1b) were soluble in aqueous buffer at concentrations up to 100 μM, and in detergent supplemented buffer (CHAPS, 10 mM) at concentrations up to 500 μM. We measured the excitation spectra of the QStE and QSE substrates and found that the excitation of the 7-hydroxy-3-carboxycoumarin dye shifted from 384 to 410 nM as a result of the amide (Fig 1c). DABCYL has a wide absorbance range between 350-550 nm (Fig. 1c) allowing effective quenching of the coumarin dye. We observed a 100-fold increase in fluorescence upon hydrolysis of the thioester or ester bond in the substrates (Fig. 1d). This large increase of fluorescence results in a substrate with a broad dynamic range that enables sensitive detection of hydrolysis.
Figure 1: Design of a quenched fluorogenic substrate for depalmitoylases.

a) Overall design of the fluorescently quenched substrate to detect depalmitoylase activity. The substrate contains a S-palmitoyled cysteine carrying a quencher molecule that is released upon thioester hydrolysis by the depalmitoylase, producing a fluorescent peptide product. b) Chemical structures of the thioester (QStE) and ester (QSE) fluorogenic substrates, Palmostatin B, the T. gondii PPT1 specific inhibitor JCP-174 and the general colorimetric esterase substrate 4-nitropehyl octanoate (4-NPO) c) Absorbance spectra of QStE, QSE (at 1 mM) and the precursor fluorophore (Coumarin amide) and quencher (DABCYL-N3). d) Fluorescence spectra of QStE and QSE (at 10 μM, Ex=410 nm, Em=450 nm), before and after chemical hydrolysis with 5% hydroxylamine (HA) or 0.1M sodium hydroxide (NaOH) for 30 minutes. e) Measurement of QStE hydrolysis (at 10 μM) by the indicated recombinant depalmiylases (HsAPT1 at 50 nM, HsAPT2 at 150 nM, and TgPPT1 at 100 nM), esterases (MGLL and PLA2 at 500 nM) and proteases (Trypsin and Papain at 100 nM and Collagenase IV at 1 mg/ml). The catalytically dead TgPPT1 is included as a negative control (TgPPT1 S128A at 100 nM). f) Plot of the hydrolysis of QStE and 4-NPO in mouse liver homogenates in the presence of the general depalmitoylase inhibitor Palmostatin B. Lysates (20 μg) were incubated with DMSO or Palmostatin B (10 μM) for 30 minutes on ice before the addition of substrates (5 μM). g) Specific activity of depalmitoylases measured with QStE in different organ homogenates and cell line lysates. QStE was added to lysates (20 μg) at varying concentrations and the initial rates of hydrolysis were used to calculate the specific activity. h) Relative activity of depalmitoylases measured with QStE in PC3 cell lysate, treated with the isotype-selective inhibitors ML348 and ML349, the general serine hydrolase inhibitor phenylmethylsulfonyl fluoride (PMSF), the serine hydrolase inhibitor hexadecylsulfonyl fluoride (HDSF) and Palmostatin B (PalmB). Lower panel shows the residual activity of APT1 and APT2 (as indicated) after treatment with each inhibitor by labeling lysates with the ABP fluorophosphonate-rhodamine (FP-Rho). Lysates (at 20 μg) were incubated with DMSO or inhibitors (10 μM) for 30 minutes on ice before the addition of QStE (5 μM). Error bars represent S.D. of three replicates. Statistical significance is calculated using ordinary oneway ANOVA compared with DMSO (**** P=0.001, n.s. P=0.9999). See also figure S3.
QStE and QSE are probes of thioesterase and esterase activity of deplamitoylases
To confirm the specificity of our reporter substrate, we incubated QStE with several serine-, cysteine- and metallo-proteases, as well as representatives of the serine hydrolase lipases and the established human depalmitoylases, HsAPT1 and HsAPT2, and TgPPT1 from T. gondii (Fig. 1e). Importantly, the thioester substrate QStE was not hydrolyzed by fatty acid hydrolases such as phospholipase A2 (PLA2) or monoacylglycerol lipase (MGLL) but was readily hydrolyzed by APT1 and APT2 as well as TgPPT1 but not the catalytically inactive mutant TgPPT1S128A, thus confirming its high degree of selectivity for acyl thioesterases that process long chain lipid modifications. To further test the selectivity of QStE we incubated the substrate in mouse liver lysates that contain diverse esterase activities. For comparison we used the general substrate 4-NPO, which has often been used to measure in vitro activity of depalmitoylases(Child et al., 2013; Dekker et al., 2010) (Fig. 1f). These results demonstrated that the substrate 4-NPO was more effectively processed by the lysates but this activity was only slightly reduced by treatment with the broad-spectrum depalmitoylase inhibitor Palmostatin B(Rusch et al., 2011) (PalmB, Fig. 1b). The substrate QStE, on the other hand, showed robust processing which was completely blocked by PalmB suggesting that it is mainly processed by depalmitoylases while the substrate 4-NPO is a general substrate of diverse esterases.
Although thioesterases are characterized by their preference for thioester-containing substrates, many thioesterases exhibit esterase activity as well. APT1 for example, was first classified as a lysophospholipase(Lu et al., 1996) but later shown to prefer thioacylated substrates(Duncan and Gilman, 1998, 2002). We compared the catalytic efficiencies of the recombinant APTs, HsAPT1, HsAPT2 and TgPPT1 for hydrolysis of the thioester substrate QStE and the ester substrate QSE (Table 1). Indeed the catalytic efficiencies of all three enzymes were 10-30 times greater for QStE compared to QSE, validating their intrinsic preference for thioester hydrolysis. Interestingly, TgPPT1 exhibits similar kinetic rates for both substrates however a large increase in Km for QSE reduces the catalytic efficiency by roughly 10-fold compared to QStE. Km values were generally higher for QSE than for QstE across all three enzymes, suggesting a molecular landscape within the binding pocket that is best suited for the larger thioester compared to the oxyester linkage. The ability of TgPPT1 to hydrolyze both the ester and thioester linkages with comparable rates suggests it may have a dual role as an esterase and a thioesterase.
Table 1:
Catalytic rate constants and efficiencies of the QStE and QSE substrates for the indicated depalmitoylating enzymes.
| Enzyme | Substrate | km (μM) | kcat (s−1) | kcat/km (s−1M−1) |
|---|---|---|---|---|
| TgPPT1 | QStE QSE |
5.7±1.5 42.6±5.6 |
(1.7±0.1) × 10−2
(1.6±0.1) × 10−2 |
3,000 380 |
| HsAPT1 | QStE QSE |
14.5±2.8 19.1 ±5.7 |
(4.6±0.4) × 10−2
(0.3±0.1) × 10−2 |
3,200 160 |
| HsAPT2 | QStE QSE |
19.3±3.2 37.5±15.2 |
(4.4±0.3) × 10−2
(0.26±0.06) × 10−2 |
2,300 69 |
Depalmitoylation activities differ across different organs and cell types according to the expression levels of depalmitoylases(Breuza et al., 2016; Gao et al., 2013; Garland et al., 2018). We therefore compared the processing of the QStE substrate in various organs and common cell lines that express varying levels of depalmitoylases. We measured the highest activity in the spleen (Fig. 1g), where previous studies identified high mRNA expression levels for the lysosomal enzymes PPT1/2(Soyombo and Hofmann, 1997). Heart tissue extracts had the lowest activity, consistent with the reported low mRNA levels of depalmitoylases in these tissues. Specific activity also varied across multiple cancer cell lines. Both prostate cancer cell lines LNCaP and PC3 had the highest levels of substrate processing activity, which was substantially elevated compared to HEK293 cells. Specific inhibition of APT1, APT2 or PPT1 with the isoform-selective inhibitors ML348, ML349(Adibekian et al., 2010) and HDSF(Das et al., 2000) respectively in PC3 cell lysate resulted in only minor reductions in QStE processing suggesting these enzymes only make up a small part of the total activity measured by this substrate (Fig. 1h). Selective inhibition of APT1 or APT2 was confirmed by competitive labeling with the fluorescent probe fluorophosphonate-rhodmine (FP-Rho). Again, we found that the broad-spectrum inhibitor PalmB was able to completely block processing of the substrate. Although PPT1, APT1 and APT2 are the most well characterized depalmitoylases, evidence suggests that other thioesterases such as ABHD17 proteins or yet still uncharacterized depalmitoylases may contribute to the total activity measured by QStE. Notably, inhibition by PMSF, a general serine hydrolase inhibitor that does not inhibit APTs and PPTs, did not reduce the specific activity measured by QStE ruling out the possibility of overlapping esterase activity from other serine hydrolases present in the lysate. A similar moderate decrease in activity upon knockdown of APT1was reported by Kathayat et al.(Kathayat et al., 2016).
Generation of combinatorial libraries of quenched fluorogenic palmitoylated peptide substrates
The hypothesis that depalmitoylases possess substrate specificity is supported by the fact that ABHD17, APT2 and APT1 display specificity for processing of palmitoylation sites on distinct substrates(Hernandez et al., 2017; Kong et al., 2013; Yokoi et al., 2016). More recent studies(Kathayat et al., 2016) show that the addition of lysine at the C-terminal side of an acylated cysteine confers substrate preference for APT1 over APT2 and accelerates the hydrolysis rate. Furthermore, sequence variance in Ras isoforms surrounding known palmitoylation sites influence palmitoylation turnover rates and possibly the preference for processing by specific depalmitoylases(Lin et al., 2017). Using the modular chemistry of the QStE substrate, we reasoned that it should be possible to make extended peptides containing defined sequences flanking the palmitoylated cysteine. While it is possible to attach the palmitate to a free cysteine after synthesis of the peptides, we found that direct synthesis of the DABCYL quencher-modified palmitoylated cysteine as an Fmoc-protected building block allowed more uniform and flexible incorporation into peptides using SPPS. The 7-hydroxy-3-carboxycoumarin fluorophore can be added as the final step of the synthesis by acylation of the amino terminus. As with synthesis of QStE, we used 17-ODYA as the acyl thioester and installed the DABCYL via click chemistry to produce the building block 20 (Fig. 2a and Scheme S6). To facilitate an efficient Fmoc SPPS we used Rink amide resin with HCTU as a coupling agent with 5 equivalents of each amino acid in a 20-minute coupling cycle (Fig. 2a). To prevent thioester hydrolysis, we replaced the standard piperidine deprotection method with 1% DBU, using four 1-minute treatments for each coupling cycle following the addition of the cysteine building block. The reaction time was carefully adjusted to allow sufficient deprotection while reducing the extent of S→N acyl transfer and side product accumulation. The coupling of the coumarin fluorophore on the N-terminus was achieved using NHS-activated 7-hydroxycoumarin (compound 6, Scheme S2) in DMF containing 5% sodium bicarbonate buffer at pH 8 for 30 minutes.
Figure 2: Positional scanning libraries highlight substrate specificities of APTs at positions P1 and P2.

a) Synthesis scheme for preparation of quenched fluorogenic substrates using solid phase peptide synthesis (SPPS). Peptide libraries were synthesized on a rink amide resin, the palmitoyl mimic containing DABCYL was synthesized as an Fmoc protected cysteine analogue (compound 20 or C20) and coupled instead of cysteine in the sequence. The N-terminus was capped with the flourophore 7-hydroxy-3-carboxycoumarin. Peptide positions spanning the thioester-containing cysteine analogue from the N-terminus side are referred to as P1, P2.., and from the C-terminus side as P1’, P2’... Libraries contain combinatorial mixtures (marked X) of all natural amino acids (excluding cysteine) at position P2 or position P1 and an isokinetic mixture of the same amino acids at the adjacent position (marked IK). b) Plots of fold-change in hydrolysis rates for each P2 scanning sublibrary relative to the reference peptide NAC20KKNT. Error bars represent S.D. of three replicates c). Same as in b) for the P1 positional scanning sublibraries. d) Effective concentration of each library was evaluated by chemical cleavage of the thioester bond by incubation with 5% HA for 30 minutes. Concentrations were estimated using a standard curve generated from non-quenched substrates. DBU = 1,8-Diazabicyclo(5.4.0)undec-7-ene. HA = hydroxylamine. See also figure S4.
Our initial library was based on the sequence of the confirmed target of TgPPT1, Gliding-associated protein 45 (GAP45)(Child et al., 2013). Palmitoylation of Gap45 occurs at Cys5 and plays an important role in trafficking and membrane localization together with a myristoylation that occurs at Gly-2(Frenal et al., 2010). We wanted our library to contain variable amino acid residues on each side of the cysteine (containing the scissile bond), to account for the specificity that might be conferred by the immediate chemical environment surrounding the cysteine. In analogy to the protease convention proposed by Schecter and Berger(Schechter and Berger, 1967), the substrate amino acid positions from the N-terminal side of the cysteine are denoted P1 and P2, and correspond to the protein sub-sites S1 and S2. The substrate amino acid positions from the C-terminal side of the cysteine are labeled P1’ and P2’ and correspond to the protein sub-sites S1’ and S2’ (Fig 2a). The sequence used for the library contained residues 3-9 (NACKKNT) of the native GAP45 palmitoylation site, excluding the terminal methionine and myristoylated Gly-2. TgPPT1 as well as APT1 and APT2 had comparable, moderate activity for depalmitoylation of the parent GAP45 peptide substrate NAC20KKNT with catalytic efficiencies of 3.5 × 104, 6.3 × 104 and 2.1 × 104 s−1M−1 respectively (Table S1), providing a convenient starting point for the analysis of sequence specificity of all three enzymes.
Mapping substrate specificity using positional scanning substrate libraries
We synthesized positional scanning combinatorial libraries in which either the P1 or the P2 position was scanned through all natural amino acids (excluding cysteine) while the other position contained an isokinetic mixture of the same 19 amino acids (Fig. 2b,c). After synthesis, the libraries were analyzed by LCMS for integrity and effective concentrations measured by hydrolysis with hydroxylamine (HA, Fig. 2d). For enzymatic analysis, all libraries were used at high molar ratio of substrate over enzyme. All three enzymes were allowed to process the libraries and the rates of depalmitoylation were measured. Changes in the amino acid composition in either position P1 or P2 resulted in up to 3-fold changes to the rate of hydrolysis. All three enzymes showed similar substrate preferences with a moderate preference for small aliphatic residues at position P2 and elevated rates with polar residues at position P1. A sharp decrease in hydrolysis was observed with negatively charged residues at P1 and bulky aromatic residues at P1 and P2. The similarities in amino acid preference were more pronounced between APT1 and APT2, in accordance with the 68% sequence identity and active site similarities reported in crystal structure analysis of these related enzymes(Won et al., 2016). To better characterize and compare the distinct preferences between the orthologous depalmitoylases, we plotted the rate of hydrolysis on a common hydrophobicity scale (Kyte and Doolittle hydropathy scale(Kyte and Doolittle, 1982) (Fig. 3a). A preference for small residues is seen at P2 (red circles), with glycine showing the highest rate for APT1 while the slightly bigger alanine is preferred by TgPPT1. Moderate rates were measured for aliphatic hydrophobic residues, as well as positively charged residues. A sharp decrease in hydrolysis was observed for the aromatic residues tryptophan and tyrosine, as well as negatively charged residues such as glutamate and aspartate. At position P1, TgPPT1 preferred serine and alanine while most aliphatic, hydrophobic residues were better tolerated by APT1. P1 Arginine significantly increased hydrolysis by APT1 while both arginine and aspartate had elevated hydrolysis by TgPPT1. While substrate preferences between APT1 and APT2 overlap, subtle differences do exist. Specifically, APT2 has an increased tolerance for larger aliphatic and aromatic residues and a decreased tolerance for alanine at the P2 position. At P1, APT2 prefers alanine and serine over the rest of the aliphatic hydrophobic residues.
Figure 3: Validation of substrate specificity using fluorogenic peptides designed from the library screening data.

a) Rates of hydrolysis for each amino acid substitution, measured with P1 or P2 libraries plotted relative to a hydrophobicity scale (adopted from Kyte and Doolittle(Kyte and Doolittle, 1982)). Small aliphatic residues (red circles) are favored at positions P1 and P2 and result in fast hydrolysis rates, positively charged residues (green circles) as well as hydrophobic aliphatic residues (yellow circles) result in moderate hydrolysis rates for all three depalmitoylases. b) Michaelis Menten plots showing rates of hydrolysis of the indicated fluorogenic peptides synthesized based on data from the positional scanning libraries. Each point represents mean and standard deviation of three replicates c) Catalytic rates and efficiencies of the fluorogenic peptides derived from the Michaelis Menten plots in (b). See also table S1 and figure S1 and S2.
To confirm the integrity of the library screening results, we synthesized and purified individual peptides with sequences that were predicted to have enhanced or reduced rates of hydrolysis for each of the depalmitoylases. As expected, substitutions at P1 and P2 with optimal amino acids resulted in increased rates of hydrolysis, as seen for the peptides ASC20KKNT and GSC20KKNT (Fig. 3b). We observed up to 2-fold enhancement in Kcat values and up to 10-fold increase in catalytic efficiencies (Kcat/Km) for hydrolysis by TgPPT1 or APT1 compared with the original peptide NAC20KKNT (Fig. 3c). Substitutions at P2 reflected distinct specificity trends seen in the P2 library, with ASC20KKNT preferred by TgPPT1 while GSC20KKNT was preferred by APT1. A sharp decrease in the Km values for both of these substrates for APT1, despite less favorable substitution of serine over alanine at P1, underscores the importance of the P2 residue for binding kinetics. Further demonstrating the importance of the P2 residue, we found that a single amino acid substitution of tryptophan at this position dramatically reduced the hydrolysis rate of the parent substrate by both TgPPT1 and APT1 (Fig. 3b,c) and the catalytic efficiency dropped by nearly 70-fold for hydrolysis of WAC20KKNT by TgPPT1.
Encouraged by the substantial effect of amino acid sequences on substrate recognition and depalmitoylation rates for the P1 and P2 libraries, we explored the importance of P1’ and P2’ using positional scanning libraries starting from the optimal sequence ASC20KKNT. After synthesis, we analyzed the libraries by LCMS for integrity and effective concentrations were measured by hydrolysis with HA (Fig. 4c). Analysis of the initial rates of hydrolysis revealed a striking enhancement for arginine at positions P2’ for all three enzymes (Fig. 4b). Interestingly, most amino acid substitutions resulted in rates that were lower then that of the reference peptide ASC20KKNT (Fig. 4a,b). Positively charged residues, in particular lysine and arginine, are preferred at position P1’ as well (Fig. 4d, green circles). Subtle differences between the enzymes include a preference for the polar aliphatic residues serine and threonine by TgPPT1 (red circles) and to a lesser extent by APT2. Moderate rates were also observed for APT2 with isoleucine at P1’ and alanine at P2’.
Figure 4: Residues in the P1’ and P2’ positions of fluorogenic substrates dramatically impact rate of hydrolysis.

Plots of fold-change from the rate measured for the optimal substrate ASC20KKNT in which the C-terminal position P1’ (a) or P2’ (b) are scanned through the indicated amino acids (one letter codes). Hydrolysis was measured and initial rates calculated from the time curves. Error bars represent standard deviation of three replicates. c) Effective concentration of each library was evaluated by chemical cleavage of the thioester bond by incubation with 5% HA for 30 minutes. Concentrations were estimated using a standard curve generated from non-quenched substrates. d) Plots of the rates of hydrolysis for each amino acid substitution, measured for the P1’ or P2’ positional scanning libraries relative to hydrophobicity scale. Polar interactions with positively charged residues (green circles) are favored at both positions, polar aliphatic residues (red circles) are favored at position P1’ only by TgPPT1. Moderate preference for alanine at P2’, isoleucine and threonine at P1’ (yellow circles) is seen only for APT2. See also figure S4
Exploring the importance of arginine at P2’ and the effect of positive charge at both P1’ and P2’ positions, we synthesized individual peptide sequences in which the lysine at P1’ or P2’ was replaced with either arginine or alanine. Replacing lysine at P2’ with arginine in the optimized peptides ASC20KKNT or GSC20KKNT further increased their rate of hydrolysis by all three enzymes. This increased rate was due to a 2-fold enhancement in the Kcat values for the substrates for TgPPT1 and APT1 (Fig. 5a,b). Eliminating the positive charge from both P1’ and P2’ positions (GAC20AANT) resulted in a sharp, 40-fold decrease in hydrolysis rate and a 100-fold decrease in catalytic efficiency compared with the most optimal sequence (GAC20KRNT; Fig. 5c). Furthermore, we found that hydrolysis of suboptimal sequences containing bulky aromatic P1 and P2 residues could be substantially improved by substitution of arginine at P2’ (compare WYC20KRNT to WYC20KKNT; Fig. 5d).
Figure 5: Engineering of fluorogenic peptide substrates based on substrate specificity profiles.

Michaelis Menten plots comparing the kinetic curves of optimized peptides, measured with (a) TgPPT1 or (b) HsAPT1. Each point represents the mean and standard deviation of three replicates. c) Catalytic efficiencies of the fluorogenic peptides derived from the Michaelis Menten plots in (a) and (b). d) Michaelis Menten plot comparing kinetic curves of suboptimized and least optimized peptides measured for HsAPT1. e) Activity of the selective substrate AHC20DRNT for HsAPT1 and HsAPT2. f) Catalytic efficiencies of the fluorogenic peptides derived from the Michaelis Menten plots in (d) and (e). g) Plot of Kcat values for the indicated fluorogenic substrates for TgPPT1 HsAPT1 and HsAPT2. Error bars represent S.D. of three replicates. See also table S1 and figures S1, S2 and S6.
The substitution of negatively charged residues at position P1’ (GVC20DHNT) reduced the Kcat value (0.03 ± 0.001 s−1, APT1) and increased the Km compared to the parent substrate (Fig. 5f) while substitution of negatively charged residues at both positions P1 and P2 along with bulky aromatic residues at positions P1’ and P2’ (DEC20WYNT) resulted in a substrate with extremely low Kcat values for all the depalmitoylases (0.003 ± 0.001 s−1, APT1) and over 1,000-fold decreased catalytic efficiency compared with the reference sequence NAC20KKNT (19 vs. 61,000 s−1M−1, respectively for APT1). Taken together these results demonstrate that we were able to construct and characterize peptide substrates with hydrolysis rates that span over three orders of magnitude for each enzyme (Fig. 5g), confirming the importance of sequence recognition in the control of rates of depalmitoylation.
Sequence selectivity and redundancy
Our libraries suggest that while there is substantial overlap in sequence preferences between APT1 and APT2, there are some differences that could explain how these two enzymes could have divergent sets of substrates. We therefore sought to explore these subtle differences to determine if this information could be used to selectively monitor APT1 and APT2 activities. Substrates that we synthesized with the most preferred amino acids favored fast hydrolysis but have overall low selectivity. To achieve an APT1-selective peptide we focused on amino acids that were moderately favorable for APT1 but poorly accepted by ATP2, such as in the peptide AHC20DRNT (Fig. 5e). Incubation of this substrate with APT1 resulted in a moderate rate of hydrolysis but, importantly, the substrate had a rate of hydrolysis by APT2 that was two orders of magnitude lower (Fig. 5f). Applying the same approach to find APT2-selective sequences resulted in substrates that were only poorly processed by APT2 such as FRC20KANT, PAC20EANT and SYC20IANT (Fig. 5g, Table S1). APT2 exhibited lower efficiencies and lower Kcat values with all of the short peptides, a result of its lower thermal stability compared with APT1(Won et al., 2016). Importantly, consistent with the substrate screening data, the peptide WRC20NRHV, derived from one of the palmitoylation sites of the transcriptional activator Scribble, was was most effectively processed by APT2 (Fig. S6).
Application of selective substrates in complex proteomes
To validate the specificity trends generated by our library screens, and to probe depalmitoylation activity in situ, we measured substrate hydrolysis in T. gondii lysates as well as in tumor tissue homogenates. While we believe that the general QStE probe has the potential to be cell permeable and thus could be used in intact cells, the current version contains a low wavelength coumarin dye that is not suitable for use due to high background fluorescence in cells. In T, gondii lysates, the optimal peptides ASC20KKNT and ASC20KRNT showed increased rate of hydrolysis compared with the GAP45 peptide NAC20KKNT, in line with our findings for purified TgPPT1 (Fig. 6a). Importantly, treatment with the TgPPT1-selective inhibitor JCP174(Child et al., 2013) (Fig. 1b) reduced the rate of hydrolysis by approximately 80% and a genetic knockout of TgPPT1 (TgΔppt1) reduced hydrolysis rates to a similar extent (Fig. 6b). Pretreatment with PalmB completely abolished activity in the lysate, confirming that the majority of the signal from the optimized substrates is the result of TgPPT1 activity but that some low level processing of the substrate by other depalmitoylases is likely occuring.
Figure 6: Specificity of fluorogenic peptides in complex proteomes.

a) Activity of endogenous depalmitoylases measured in T. gondii lysates (at 5 μg) using the optimized fluorgenic substrates (at 2.5 μM). Error bars represent the S.D. of three replicates. b) Normalized rate of hydrolysis for the ASC20KRNT substrate (at 2.5 μM) measured in Ku80 T. gondii lysates (at 5 μg) untreated (WT) or treated with JCP174 (JCP174, 10 μM) or Palmostatin B (PalmB, 10 μM) and untreated PPT1 knockout (ΔPPT1) T. gondii lysates (at 5 μg). Error bars represent S.D. of three biological replicates and 3 technical replicates. Statistical significance is calculated using one-way ANOVA (**** p < 0.0001), (*** p = 0.0003), (* p = 0.0252). c) Activity of endogenous depalmitoylases measured in mammary breast tumor homogenate (at 5 μg) using optimized and non-optimal fluorogenic substrates (at 2.5 μM). Error bars represent the S.D. of three replicates. d) and e) Normalized rate of hydrolysis for ASC20KKNT and AHC20DRNT (at 2.5 μM) measured in tumor homogenates treated with vehicle (DMSO) or the inhibitors ML348, ML349 and Palmostatin B (at 10 μM). Error bars represent S.D. of three biological replicates and 3 technical replicates. Statistical significance is calculated using one-way ANOVA (**** p = 0.0001), (ns p = 0.6711). See also figure S5.
We also tested the substrates in homogenates derived from mammary tumor tissues to determine if the profiles of specificity were retained in complex proteomes that likely contain many depalmitoylases. We found that the optimal substrate GAC20KRNT had increased hydrolysis in the lysate compared with NAC20KRNT (Fig. 6c). Furthermore, the non-optimal GAC20AANT that lacks positive charge at the P1’ and P2’ positions show dramatically reduced hydrolysis in the extract. We then wanted to determine if the substrate that we designed to be selective for APT1 could be used to specifically monitor its activity in situ. As expected, hydrolysis of the non-selective substrate ASC20KKNT could be reduced using both the APT1-specific inhibitor ML348 as well as the APT2-specific inhibitor ML349 (Fig. 6d). In contrast, only the APT1-specific inhibitor ML348 could reduce the hydrolysis activity of the APT1-selective peptide AHC20DRNT confirming the high degree of selectivity of the substrate for APT1 over APT2 (Fig. 6e). Because ML348 does not result in complete inhibition of AHC20DRNT hydrolysis it is likely that this substrate remains at least weakly active against additional depalmitoylases.
Discussion:
Dynamic palmitoylation regulates protein activity during normal cellular processes as well as in disease. Depalmitoylases are fundamental for maintaining palmitoylation steady states of these dynamic processes. Changes in turnover rates by increased exposure to depalmitoylases, for example through receptor stimulation or activating mutations, facilitates signal transduction of heterotrimeric G proteins or the intercellular trafficking of neuronal proteins. It is unclear however, which types of regulation govern the dynamics of depalmitoylation and repalmitoylation cycles. While there is yet no evidence of mechanisms that directly alter the activity of either PAT or APTs, It has been suggested that expression levels and sub-cellular localization may serve to set overall palmitoylation steady state levels(Won et al., 2018). Here, we demonstrate a mechanism for controlling rates of substrate hydrolysis of depalmitoylating enzymes that relies on sequence recognition of residues that surround the palmitoylation site. These residues not only affect binding affinity for the enzyme but also dictate overall catalytic rates of depalmitoylation. We find distinct and overlapping specificities among the homologous human depalmitoylases APT1, APT2 as well as the parasite enzyme TgPPT1. The ability to discriminate between different palmitoylated substrates and vary depalmitoylation rates may serve as a means by which depalmitoylases maintain diverse steady state turnover rates across large pools of substrates.
Similarities between APT1 and APT2, both in sequence identity and in active site structure, suggest these enzymes likely overlap for many substrates in vivo. Several studies in cellular assays however, have shown specific substrates that undergo selective processing by APT1(Lin et al., 2017; Tian et al., 2012), APT2(Hernandez et al., 2017; Tomatis et al., 2010) or neither(Lin and Conibear, 2015; Vallejo et al., 2017). Co-crystal structures of APT1 and APT2 bound with their isoform-selective inhibitors, ML348 and ML349, shed light on binding topology, highlighting the importance of a flexible β5-α2 loop and small G3 helix for the formation of the hydrophobic channel that accommodates the acyl chain(Won et al., 2016). While the flexibility in loop conformation most likely accounts for the selectivity towards long chain fatty acids, the overall surface polarity and binding landscape is nearly identical with only a few mutations across the active site. It seems that any selectivity between these two enzymes would not arise solely from the difference in binding affinities but from additional unknown factors. Different rates of hydrolysis may be one such factor that differentiates two or more potential substrates with comparable binding affinities. As is the case for the short peptide AHC20DRNT, which is hydrolyzed by APT1 with a rate that is over 60 times higher then APT2 while both enzymes bind the substrates with comparable Km vales. Differences in hydrolysis rates by different APTs towards the same substrate, even when not achieving complete selectivity may still account for the change in phenotype seen upon knockout or inhibition of a single APT enzyme.
Attributing depalmitoylation of individual substrates to a specific depalmitoylase in vivo remains a difficult task. A clearer understanding of substrate specificities could explain or event predict the selectivity or redundancy of distinct depalmitoylases towards specific palmitoylated substrates. Our in vitro analysis of short acylated peptides provides compelling evidence for the ability of APTs to recognize sequence elements and consequently vary their rate of depalmitoylation of specific protein substrates. The general trends that we observed for purified enzymes in vitro were similar to what we observed in situ, in complex proteomes. Sequence elements of biologically relevant protein substrates contain valuable information that determines its dynamic nature. For example, two palmitoylation sites are predicted for the tumor suppressor Scribble (Scrib), responsible for its localization to the basolateral membrane in polarized cells (Chen et al., 2016). Depalmitoylation of Scrib is selectively performed by APT2 and upregulated expression of this depamitoylase in epithelial cancers results in mislocalization of Scrib to the cytosol (Dow et al., 2003; Hernandez et al., 2017). The observed preference for APT2 over APT1 can potentially be explained by the amino acid at the P1’ position of the predicted palmitoylation sites Cys4 (LKCIP) and Cys10 (WRCNR) with both isoleucine and asparagine at P1’ being preferred by APT2 over APT1. Indeed, while both APT2 and APT1 were able to cleave the Scrib-derived peptide containing Cys4 (Fig. S6), APT2 showed enhanced activity compared to APT1, consistent with the sequence selectivity determined using the library screening approach. While the overall cleavage rates differed by a relatively small amount, this difference, combined with substrate localization and expression levels of the APTs could all be key determinants in overall selectivity. As we have suggested, there are likely multiple variables that mediate specific substrate repertoires for each depalmitoylase, with primary sequence of the substrate being one of those parameters.
In many cases, overlapping activity is observed among the established depalmitoylases. N-Ras and H-Ras undergo rapid turnover with membrane occupancy halflives of under 5 minutes for the mono-palmitoylated N-Ras and roughly 20 minutes for H-Ras, which is palmitoylated on two cysteines(Rocks et al., 2005). Turnover of the palmitate is effectively blocked with PalmB, leading to decreased downstream signaling, but not by selective inhibition or knockdown of APT1, APT2 or both(Vujic et al., 2016). The amino acid sequence surrounding the confirmed palmitoylation sites(Collins et al., 2017) of N-Ras, Cys80 (QGCMG) and H-Ras, Cys184 and Cys181 (PGCMSCK), include amino acids that are only moderately preferred by APT1 and APT2, with no apparent differences between the enzymes. Indeed in a recent study ABHD17A-C proteins were identified as the major depalmitoylases that process N-Ras, although the triple knockdown of the ABHDs did not completely block depalmitoylation(Lin and Conibear, 2015).
The difference in hydrolysis rates exhibited by different depalmitoylases for the same substrate suggests that these enzymes exhibit some level of substrate selectivity. In this study, we provide evidence that recognition of residues flanking the palmitoylated cysteine is one way these enzymes may acheive substrate specificity. Our data suggests that while binding affinities may not differ significantly between different substrates, selectivity can be achieved through differences in overall rate of substrate hydrolysis. This suggests a degree of plasticity in how the enzyme binds its substrates. How this plasticity is achieved is not clear but one hypothesis may be through subtle differences in substrate binding conformations, which determine how the thioester bond is presented to the catalytic serine. There is some evidence from the analysis of APT1 and APT2 co-crystalized with different inhibitors to suggest differences in substrate/inhibitor conformation can exist(Won et al., 2016). Further studies will be required to better understand the exact molecular determinants of substrate specificity for these enzymes.
In conclusion, our combinatorial fluorescent peptide libraries provide a flexible, highly modulate platform suitable to generate tools that can be used to map substrate specificities of established and predicted depalmitoylases as well as to monitor their activity in complex proteomes. We are optimistic that such an understanding of substrate specificities and how dynamic palmitoylation is regulated will provide new tools for rational design of selective inhibitors and potentially new therapeutic strategies for biological processes that involve palmitoylation as a primary means of regulation. A comprehensive understanding of substrate specificities, whether selective of overlapping, would provide a means to potentially alter protein function through modulation of palmitoylation steady states. The ability to induce the increase or decrease in levels of a specific palmitoylation site on a defined protein target (through genetic engineering) would allow a clear analysis of the roles of those PTMs in diverse biological processes. Such efforts could enable not only a map of where palmitoylation occurs on proteins but also a clear understanding of the functional relevance of each of those sites.
Significance
Protein palmitoylation is a widespread post translational modification involving a palmitoyl lipid attached to a cysteine through a thioester bond. The reversibility of this modification enables dynamic regulation of protein function (activity, localization, etc.) through cycles of depalmitoylation and repalmitoylation. In this study we have developed libraries of synthetic palmitoylated peptide substrates that can be used to identify elements distal to the palmitoylated cysteine residues that regulate hydrolysis rates by diverse depalmitoylating enzymes. We find that the human depalmitoylating enzymes APT1 and APT2 and the Toxoplasma gondii TgPPT1 display overlapping but distinct patterns of substrate specificity. This finding highlights a novel mechanism of regulation for the depalmitoylation of substrates and provides evidence that selectivity can be achieved through the peptide sequences in the vicinity of the palmitoylated cysteine. Furthermore, knowledge of the substrate specificity of a depalmitoylase can be used to generate selective substrates and potentially to modify known palmitoylation sites on native substrates. This would allow direct studies of the function of specific palmitoylation events on a given substrate by altering the rate at which that palmitate is removed. This will ultimately help to identify specific palmitoylation events that are critical for regulation of important biological processes and potentially therapeutic agents that can be used to alter these events.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Bogyo (mbogyo@stanford.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILES
Parasite strains and cell lines
T. gondii strains ΔKu80 and ΔKu80Δppt1(Child et al., 2013), were maintained by passage through monolayers of human foreskin fibroblasts (HFFs), cultured in Dulbecco’s modified medium (DMEM) supplemented with 10% FetalPlex™ animal serum (Gemini Bio Products), 2 mM L-Glutamine and 10 U/mL penicillin G, and 100 μg/mL streptomycin (Pen/Strep) and incubated at 37 °C with 5% CO2. Cell lines were cultured in 10 cm. tissue culture-treated dishes and incubated at 37 °C with 5% CO2. SKOV3 (ATCC HTB-77) cells were cultured in McCoy’s media supplemented with 10% fetal calf serum and Pen/Strep. PC-3 (ATCC CRL-1435) and LnCaP (ATCC CRL-1740) cells were cultured in RPMI supplemented with 10% fetal calf serum and Pen/Strep. OVCAR-3 (ATCC HTB-161) cells were cultured in RPMI supplemented with 20% fetal calf serum and Pen/Strep. MDA-MB-231 (ATCC HTB-26) and MCF7 (ATCC HTB-22) cells were cultured in DMEM supplemented with 10% fetal calf serum, Pen/Strep and 2 mM L-glutamine.
Mice strains
All animal care and experimentation was conducted under protocols agreed upon by the Administrative Panel on Laboratory Animal Care at Stanford University. 14-week old virgin C57BL/6 females and 21-week old virgin C57BL/6 MMTV-PyMT (Jackson Laboratories, strain B6.FVB-Tg(MMTV-PyVT)634Mul/LellJ) were euthanized under CO2 and organs were harvested for depalmitoylase activity assays. MMTV-PyMT females develop palpable mammary tumors, which metastasize to the lungs.
METHOD DETAILS
Compound synthesis
All reagents were purchased from commercial sources and were used without further purification. All solvents were purchased from Fisher Scientific (HPLC grade). Reaction products were purified by normal phase silica-gel flash column chromatography (60 Å, 230-400 mesh) or by high performance liquid chromatography (Agilent 1260 Infinity System, Agilent Technologies) using a ZORBAX 300SB-C18 reverse phase column. Structure characterization was based on NMR spectroscopy, recorded on a 500 MHz Bruker AVANCE system or a Varian 400 MHz (400/100) or a Varian 500 MHz (500/125), as well as mass spectrometry, measured using Surveyor Plus liquid chromatography system coupled to a Finnigan LTQ mass detector (Thermo-Fisher Scientific) or an Agilent 1100 series LCMS using an API 150EX single-quadrupole mass spectrometer. Spectroscopic data was analyzed using MestReNova 11.0.1 (Mestrelab Research). Chemical shifts are given in ppm (δ) relative to tetramethylsilane as an internal standard, coupling constantans are given in Hz. Recombinant protein purification was done on a ÄKTAexplorer system, Amersham Pharmacia Biotech.
Synthesis Procedures and Product Characterization
3-azidopropane-1-amine (1):
To a stirring aqueous solution of 3-bromopropyl-1-amine (1 eq.), lithium azide (LiN3, 3 eq.) was added in one portion. The solution was stirred at RT for 10 minutes then refluxed for 12 hours. After completion of the reaction, the stirring solution was allowed to cool and ⅔ of the water were evaporated under vacuum. The resulting solution was gently stirred on ice and KOH pellets were added until a phase separation was observed. Phases were separated and the aqueous layer was further extracted with EA. Organic extracts were pulled together and dried with MgSO4, EA was evaporated carefully under vacuum yielding pure product 1 as colorless oil. 1H NMR (500 MHz, Chloroform-d) δ 3.36 (t, J = 6.7 Hz, 2H), 2.79 (t, J = 6.8 Hz, 2H), 1.71 (p, J = 6.8 Hz, 2H). 13C NMR (126 MHz, Chloroform-d) δ 49.14, 39.31, 32.43.
2,5-dioxocyclopentyl (E)-4-((4-(dimethylamino)phenyl)diazenyl)benzoate (2):
DABCYL acid (1 eq.) was dissolved in DMF. NHS (1.2 eq.) and EDC (1.2 eq.) were added and the reaction was stirred at RT for 16 hours. DMF was then evaporated under vacuum and the residue was dissolved in EA. The solution was washed with water and brine, dried with MgSO4 and evaporated under vacuum. The crude material was used in the next reaction without further purification. 1H NMR (400 MHz, Chloroform-d) δ 8.23 (d, J = 8.3 Hz, 2H), 8.01 (s, 1H), 7.91 (d, J = 8.2 Hz, 4H), 6.76 (d, J = 8.8 Hz, 2H), 3.12 (d, J = 1.2 Hz, 6H), 2.95 (s, 3H), 2.88 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 169.27, 131.65, 125.85, 124.59, 122.37, 111.45, 40.27, 25.69. ESI [M+H]+ calcd. 367.13; found 367.3.
(E)-N-(3-azidopropyl)-4-((4-(dimethylamino)phenyl)diazenyl)benzamide (3):
Compound 2 (1 eq.) was dissolved in DMF, then compound 1 (3 eq.) was added together with DIPEA (3 eq.). The reaction was stirred at RT and monitored by TLC until completion (~4 hours). DMF was evaporated under vacuum and the resulting residue was purified by column chromatography (2-5% MeOH:DCM). 1H NMR (500 MHz, Chloroform-d) δ 7.84 – 7.81 (m, 2H), 7.80 (s, 4H), 6.72 – 6.67 (m, 2H), 3.49 (t, J = 6.7 Hz, 2H), 3.40 (t, J = 6.5 Hz, 2H), 3.04 (s, 6H), 1.87 (p, J = 6.6 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 203.75, 192.21, 152.86, 143.58, 134.27, 127.78, 125.42, 122.23, 111.48, 49.86, 49.52, 40.28, 37.72, 28.72. ESI [M+H]+ calcd. 352.18; found 352.4.
Ethyl 7-hydroxy-2-oxo-2H-chromene-3-carboxylate (4):
Compound 4 was synthesized according to reported procedure(el-Husseini Ael and Bredt, 2002). 2,4-dihydroxy benzaldehyde (1 eq.) and diethyl malonate (1 eq.) were dissolved in ethanol. Piperidine (0.2 eq.) was then added and the solution was heated under reflux for 12 hours. After cooling, a white precipitate appeared and was collected by filtration. The crude product was recrystallized in ethanol to afford 4 as a white solid. 1H NMR (500 MHz, Methanol-d4) δ 8.68 (s, 1H), 7.65 (d, J = 8.7 Hz, 1H), 6.87 (dd, J = 8.6, 2.3 Hz, 1H), 6.74 (d, J = 2.2 Hz, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, Methanol-d4) δ 164.91, 159.59, 159.05, 151.17, 133.02, 115.64, 113.09, 112.15, 106.41, 103.14, 62.46, 14.54. ESI [M+H]+ calcd. 235.05; found 235.0.
7-hydroxy-2-oxo-2H-chromene-3-carboxylic acid (5):
Compound 4 (1 eq.) was dissolved in 2N NaOH and stirred for 1 hour. The solution was then acidified with 2N HCL until a precipitate appeared. The precipitate was collected by filtration, washed with water and dried, yielding pure compound 4 as a light yellow solid. 1H NMR (500 MHz, Methanol-d4) δ 8.79 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 6.92 (dd, J = 8.6, 2.2 Hz, 1H), 6.80 (d, J = 2.2 Hz, 1H). 13C NMR (126 MHz, Methanol-d4) δ 166.44, 162.63, 158.89, 151.94, 133.34, 115.87, 112.65, 112.40, 106.41, 103.27. ESI [M+H]+ calcd. 207.02; found 207.2.
2,5-dioxopyrrolidin-1-yl 7-hydroxy-2-oxo-2H-chromene-3-carboxylate (6):
Compound 5 (1 eq.) and NHS (1.2 eq.) were dissolved in DMF. EDC (1.2 eq.) was then added and the reaction was stirred at RT and monitored by TLC until completion (~3 hours). DMF was evaporated under vacuum and the resulting residue was diluted with EA and washed with 10% citric acid, water and brine. After evaporation the crude product was purified by column chromatography (2% MeOH:DCM). 1H NMR (500 MHz, Methanol-d4) δ 8.96 (s, 1H), 7.73 (d, J = 8.7 Hz, 1H), 6.92 (dd, J = 8.6, 2.2 Hz, 1H), 6.78 (d, J = 2.2 Hz, 1H), 2.93 (s, 4H). 13C NMR (126 MHz, MeOD) δ 171.82, 167.67, 160.07, 159.85, 153.98, 134.00, 116.05, 112.13, 108.14, 106.41, 103.37, 26.62. ESI [M+H]+ calcd. 304.04; found 304.1.
(9H-fluoren-9-yl)methyl (R)-(1-morpholino-1-oxo-3-(tritylthio)propan-2-yl)carbamate (7):
Fmoc-Cys(Trt)-OH (1 eq.) was dissolved in DMF. HBTU (2 eq.) and morpholine (2 eq.) were added and the reaction was stirred at RT for 30 minutes. DMF was then evaporated and the residue was dissolved in EA, washed with water and brine, then dried with MgSO4 and evaporated under vacuum. The crude product was purified by column chromatography (2% MeOH:DCM). 1H NMR (400 MHz, Chloroform-d) δ 7.79 – 7.72 (m, 2H), 7.60 (d, J = 7.5 Hz, 2H), 7.44 – 7.34 (m, 8H), 7.30 (q, J = 1.9, 1.4 Hz, 2H), 7.31 – 7.21 (m, 7H), 7.25 – 7.17 (m, 2H), 5.53 (d, J = 8.9 Hz, 1H), 4.49 (q, J = 7.2 Hz, 1H), 4.35 (qd, J = 10.5, 7.3 Hz, 2H), 4.21 (t, J = 7.1 Hz, 1H), 3.63 – 3.52 (m, 4H), 3.51 – 3.38 (m, 2H), 3.22 – 2.99 (m, 2H), 2.54 (d, J = 6.6 Hz, 2H). 13C NMR (101 MHz, Chloroform-d) δ 168.90, 155.62, 144.41, 143.80, 143.72, 141.26, 129.62, 128.02, 127.71, 127.09, 127.06, 126.87, 125.15, 119.96, 77.33, 77.01, 76.69, 67.15, 66.63, 66.52, 49.71, 47.09, 45.83, 42.49, 34.81. ESI [M+H]+ calcd. 655.26; found 655.4.
(R)-2-amino-1-morpholino-3-(tritylthio)propan-1-one (8):
Compound 7 was dissolved in a solution of 20% piperdine in DMF and stirred for 30 minutes. The solvent was evaporated under vacuum and the resulting residue was purified by column chromatography (1:1 EA:Hexane to EA to 5% MeOH:DCM). 1H NMR (400 MHz, Chloroform-d) δ 7.40 (dt, J = 8.2, 1.3 Hz, 6H), 7.29 – 7.24 (m, 1H), 7.28 – 7.18 (m, 6H), 7.22 – 7.13 (m, 2H), 3.61 – 3.41 (m, 5H), 3.38 – 3.23 (m, 1H), 3.17 (dd, J = 9.1, 4.9 Hz, 1H), 3.06 – 2.95 (m, 1H), 2.91 – 2.82 (m, 3H), 2.52 (ddd, J = 13.0, 8.9, 1.3 Hz, 1H), 2.41 (dd, J = 12.9, 4.8 Hz, 1H). 13C NMR (101 MHz, Chloroform-d) δ 171.77, 144.68, 129.64, 127.95, 127.00, 126.81, 77.50, 77.18, 76.86, 67.09, 66.66, 66.55, 50.83, 45.40, 42.27, 37.80. ESI [M+H]+ calcd. 433.19; found 433.2.
(R)-7-hydroxy-N-(1-morpholino-1-oxo-3-(tritylthio)propan-2-yl)-2-oxo-2H-chromene-3-carboxamide (9):
Compound 8 (1 eq.) was dissolved in DMF. Compound 6 (1.5 eq.) and DIPEA (2 eq.) were added. Reaction progress was monitored by TLC until completion (~4 hours), DMF was removed by evaporation and the crude mixture was purified by column chromatography (1-5% MeOH:DCM). 1H NMR (500 MHz, Methanol-d4) δ 9.38 (d, J = 7.9 Hz, 1H), 8.64 (s, 1H), 7.57 (d, J = 8.6 Hz, 1H), 7.29 (d, J = 7.8 Hz, 6H), 7.20 (t, J = 7.6 Hz, 6H), 7.13 (t, J = 7.2 Hz, 3H), 6.79 (dd, J = 8.6, 2.2 Hz, 1H), 6.69 (d, J = 2.2 Hz, 1H), 4.81 (t, J = 6.5 Hz, 1H), 3.56 – 3.30 (m, 6H), 3.28 – 3.22 (m, 0H), 3.15 – 3.07 (m, 1H), 2.59 – 2.48 (m, 2H). 13C NMR (126 MHz, Methanol-d4) δ 188.83, 170.18, 167.00, 159.07, 158.48, 155.19, 145.84, 133.51, 130.76, 129.11, 128.05, 121.56, 106.41, 103.16, 68.20, 67.65, 49.97, 47.37, 43.11, 42.04, 40.12, 35.20, 33.59. ESI [M+H]+ calcd. 621.2; found 621.2.
(R)-S-(2-(7-hydroxy-2-oxo-2H-chromene-3-carboxamido)-3-morpholino-3-oxopropyl) octadec-17-ynethioate (10):
Compound 9 (1 eq.) was dissolved in 5% TFA in DCM with triisopropylsilane (TIS, 2 eq.) and allowed to stir for 2 hours. The solvents were evaporated under vacuum and the crude was dissolved in neat TFA. 17-Octadecynoic acid (3 eq.) was dissolved in DCM with 2 drops of DMF and cooled in an ice bath. Oxalyl chloride (15 eq.) was then added drop wise, the ice bath was removed and the reaction mixture was stirred for 30 minutes. The solvents were evaporated under vacuum, the resulting acid chloride was the taken up in DCM and added to the stirring solution of compound 9. The mixture was stirred for 3 hours at RT and then the solvent was evaporated under vacuum. The resulting crude product was dissolved in 1:1 ACN:water and purified by HPLC (50-95% ACN:water over 15 minutes). 1H NMR (500 MHz, Methanol-d4) δ 9.54 (d, J = 8.1 Hz, 1H), 8.79 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 6.92 (dd, J = 8.6, 2.2 Hz, 1H), 6.81 (d, J = 2.3 Hz, 1H), 5.34 (td, J = 7.7, 4.5 Hz, 1H), 3.87 – 3.56 (m, 7H), 3.45 (dd, J = 14.3, 4.7 Hz, 1H), 3.26 (q, J = 7.4 Hz, 2H), 2.60 (t, J = 7.3 Hz, 2H), 2.21 – 2.14 (m, 3H), 1.64 (p, J = 7.3 Hz, 2H), 1.52 (p, J = 7.1 Hz, 2H), 1.47 – 1.21 (m, 22H). 13C NMR (126 MHz, Methanol-d4) δ 200.61, 189.43, 172.24, 169.91, 160.67, 159.29, 154.23, 141.19, 133.10, 130.38, 129.47, 128.54, 115.82, 106.41, 103.17, 79.04, 69.36, 67.70, 55.86, 44.66, 43.95, 43.81, 30.74, 30.52, 30.39, 30.24, 29.90, 29.75, 26.71, 19.03, 18.73, 17.29, 13.17. ESI [M+H]+ calcd. 641.32; found 641.4.
(R)-S-(2-(7-hydroxy-2-oxo-2H-chromene-3-carboxamido)-3-morpholino-3-oxopropyl) (E)-16-(1-(3-(4-((4-(dimethylamino)phenyl)diazenyl)benzamido)propyl)-1H-1,2,3-triazol-4-yl)hexadecanethioate (QStE):
Compound 10 (1 eq.) and compound 3 (1.5 eq.) were dissolved in DMF. Copper(II) sulfate (0.4 eq.) and ascorbic acid (0.4 eq.) were premixed in DMF and immediately added to the solution. The reaction mixture was heated to 40°C and stirred for 2 hours. All solvents were then evaporated under vacuum and the crude solid was dissolved in 1:1 ACN:water and purified by HPLC (50-95% ACN:water over 15 minutes). 1H NMR (500 MHz, DMSO-d6) δ 11.27 (s, 1H), 9.21 (d, J = 8.0 Hz, 1H), 8.81 (s, 1H), 8.65 (t, J = 5.6 Hz, 1H), 8.01 – 7.94 (m, 2H), 7.89 (s, 1H), 7.82 (dd, J = 8.7, 3.7 Hz, 4H), 6.92 – 6.80 (m, 3H), 5.23 (td, J = 7.0, 4.6 Hz, 1H), 4.39 (t, J = 6.9 Hz, 2H), 3.66 – 3.61 (m, 1H), 3.47 – 3.40 (m, 1H), 3.31 (dq, J = 16.1, 6.3, 5.5 Hz, 3H), 3.20 (dd, J = 14.0, 6.7 Hz, 1H), 3.08 (s, 5H), 2.61 – 2.51 (m, 3H), 2.55 (s, 3H), 2.09 (p, J = 6.8 Hz, 2H), 1.55 (s, 2H), 1.47 (q, J = 7.3 Hz, 2H), 1.26 (s, 2H), 1.24 – 1.20 (m, 11H), 1.17 (s, 7H), 1.14 – 1.10 (m, 6H). 13C NMR (126 MHz, DMSO) δ 199.22, 168.13, 166.48, 164.78, 161.74, 161.56, 158.86, 158.61, 157.16, 154.66, 153.53, 149.42, 147.51, 143.32, 135.33, 132.87, 129.05, 125.81, 122.59, 122.19, 115.18, 113.34, 112.26, 111.71, 102.53, 66.67, 48.69, 47.95, 46.25, 43.92, 42.82, 41.10, 40.69, 40.52, 40.35, 40.19, 40.02, 39.85, 39.69, 37.46, 31.13, 30.53, 29.77, 29.70, 29.67, 29.52, 29.39, 29.31, 28.81, 25.84, 25.73. ESI [M+H]+ calcd. 992.5; found 992.8.
(9H-fluoren-9-yl)methyl (S)-(3-(tert-butoxy)-1-morpholino-1-oxopropan-2-yl)carbamate (11):
Fmoc-Ser(tBu)-OH (1 eq.) was dissolved in DMF. HBTU (2 eq.) and morpholine (2 eq.) were added and the reaction was stirred at RT for 30 minutes. DMF was then evaporated and the residue was dissolved in EA, washed with water and brine then dried with MgSO4 and evaporated under vacuum. The crude product was purified by column chromatography (2% MeOH:DCM). 1H NMR (500 MHz, Chloroform-d) δ 7.79 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 5.77 (d, J = 8.2 Hz, 1H), 4.81 (td, J = 8.6, 4.9 Hz, 1H), 4.39 (d, J = 7.3 Hz, 2H), 4.23 (t, J = 7.2 Hz, 1H), 3.87 – 3.49 (m, 9H), 3.45 (t, J = 8.7 Hz, 1H), 1.19 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 169.71, 155.67, 143.89, 143.77, 141.29, 127.70, 127.05, 125.12, 119.98, 73.73, 67.07, 66.80, 63.70, 50.38, 47.14, 38.62, 27.40. ESI [M+H]+ calcd. 453.55; found [M+Na]+ 475.6.
(S)-2-amino-3-(tert-butoxy)-1-morpholinopropan-1-one (12):
Compound 11 was dissolved in a solution of 20% piperdine in DMF and stirred for 30 minutes. The solvent was evaporated under vacuum and the resulting residue was purified by column chromatography (1:1 EA:Hexane to EA to 5% MeOH:DCM). 1H NMR (400 MHz, Chloroform-d) δ 3.87 (t, J = 6.8 Hz, 1H), 3.71 – 3.57 (m, 6H), 3.52 (s, 2H), 3.37 (qd, J = 8.7, 6.8 Hz, 2H), 2.59 (s, 2H), 1.12 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 172.04, 73.41, 66.77, 65.54, 51.08, 45.99, 42.41, 27.40. ESI [M+H]+ calcd. 231.16; found [M+H]+ 231.3.
(S)-N-(3-(tert-butoxy)-1-morpholino-1-oxopropan-2-yl)-7-hydroxy-2-oxo-2H-chromene-3-carboxamide (13):
Compound 12 (1 eq.) was dissolved in DMF. Compound 6 (1.5 eq.) and DIPEA (2 eq.) were added. Reaction progress was monitored by TLC until completion (~4 hours), DMF was removed by evaporation under vacuum and the crude mixture was purified by column chromatography (5-15% MeOH:DCM). 1H NMR (400 MHz, Chloroform-d) δ 9.32 (d, J = 6.7 Hz, 1H), 8.51 (d, J = 0.7 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 6.80 – 6.71 (m, 2H), 5.15 – 5.05 (m, 1H), 3.76 (d, J = 9.0 Hz, 6H), 3.72 – 3.56 (m, 4H), 1.19 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 170.00, 165.05, 162.20, 161.46, 156.15, 153.20, 148.25, 133.36, 131.10, 102.95, 74.06, 66.77, 62.63, 59.81, 50.27, 49.86, 27.31. ESI [M+H]+ calcd. 419.17; found [M+H]+ 419.3.
(S)-2-(7-hydroxy-2-oxo-2H-chromene-3-carboxamido)-3-morpholino-3-oxopropyl hexadec-15-ynoate (14):
Compound 13 (1 eq.) was dissolved in 50% TFA in DCM and allowed to stir for 2 hours. The solvents were evaporated under vacuum and the crude was dissolved in neat TFA. 17-Octadecynoic acid (3 eq.) was dissolved in DCM with 2 drops of DMF and cooled in an ice bath. Oxalyl chloride (15 eq.) was added drop wise, the ice bath was removed and the reaction mixture was stirred for 30 minutes. The solvent was evaporated under vacuum, the resulting acid chloride was then taken up in DCM and added to the stirring solution of compound 13. The mixture was refluxed for 4 hours and then the solvent was evaporated under vacuum. The crude product was dissolved in 1:1 ACN:water and purified by HPLC (50-95% ACN:water over 15 minutes). 1H NMR (400 MHz, Chloroform-d) δ 9.54 (d, J = 7.9 Hz, 1H), 8.70 (s, 1H), 7.70 (d, J = 7.5 Hz, 1H), 6.82 (dd, J = 8.6, 2.3 Hz, 1H), 6.76 (d, J = 2.1 Hz, 1H), 3.85 – 3.73 (m, 1H), 3.68 – 3.59 (m, 8H), 3.34 (p, J = 1.6 Hz, 2H), 2.37 (t, J = 7.5 Hz, 2H), 2.24 (t, J = 7.5 Hz, 3H), 2.09 (dt, J = 1.6, 0.5 Hz, 2H), 1.53 (d, J = 10.5 Hz, 2H), 1.18 (d, J = 12.7 Hz, 22H). ESI [M+H]+ calcd. 625.34; found [M+H]+ 625.5.
(S)-2-(7-hydroxy-2-oxo-2H-chromene-3-carboxamido)-3-morpholino-3-oxopropyl (E)-14-(1-(3-(4-((4-(dimethylamino) phenyl)diazenyl)benzamido)propyl)-1H-1,2,3-triazol-4-yl)tetradecanoate (QSE):
Compound 14 (1 eq.) and compound 3 (1.5 eq.) were dissolved in DMF. Copper(II) sulfate (0.4 eq.) and ascorbic acid (0.4 eq.) were premixed in DMF and immediately added to the solution. The reaction mixture was heated to 40°C and stirred for 2 hours. All solvents were then evaporated under vacuum and crude solid was dissolved in 1:1 ACN:water and purified by HPLC (50-95% ACN:water over 15 minutes). 1H NMR (400 MHz, Chloroform-d) δ 9.66 (d, J = 7.7 Hz, 1H), 8.84 (s, 1H), 7.94 – 7.88 (m, 2H), 7.86 (d, J = 0.9 Hz, 4H), 7.67 (d, J = 8.5 Hz, 1H), 7.45 (s, 1H), 7.21 (d, J = 2.1 Hz, 1H), 7.14 (ddd, J = 8.5, 2.2, 0.9 Hz, 1H), 6.81 – 6.75 (m, 2H), 5.12 (t, J = 3.9 Hz, 1H), 4.84 (d, J = 2.8 Hz, 5H), 4.48 (t, J = 6.4 Hz, 2H), 3.99 – 3.82 (m, 2H), 3.72 – 3.63 (m, 9H), 3.50 (q, J = 4.5, 3.0 Hz, 2H), 3.12 (d, J = 0.9 Hz, 6H), 2.68 (t, J = 7.7 Hz, 2H), 2.60 (t, J = 7.4 Hz, 2H), 1.76 (p, J = 7.5 Hz, 2H), 1.65 – 1.57 (m, 2H), 1.48 – 1.36 (m, 1H), 1.27 – 1.22 (m, 16H). 13C NMR (101 MHz, Chloroform-d) δ 199.66, 167.74, 166.66, 165.92, 162.32, 159.99, 157.78, 154.13, 153.72, 149.10, 147.80, 144.73, 136.76, 132.31, 130.68, 127.83, 125.61, 122.22, 119.53, 118.91, 117.88, 113.66, 111.65, 110.21, 77.32, 77.00, 76.68, 58.39, 53.51, 49.52, 42.94, 42.59, 40.38, 34.34, 33.12, 29.94, 29.63, 29.49, 29.31, 29.29, 28.98, 28.98, 24.71. ESI [M+H]+ calcd. 976.52; found [M+H]+ 976.8.
Octadec-9-ynoic acid (15):
Oleic acid (2.825 gr, 10 mmol) was dissolved in diethyl ether (16 mL) and cooled to −10 °C. Bromine (1.92 gr, 12 mmol) was slowly added drop by drop, keeping the temperature below −5 °C throughout the addition. The solution was stirred an additional 15 minutes while allowing the temperature to warm to RT. Excess of the bromine was reacted with the scavenger 2-methyl-2-butene. n-propanol (40 mL) was then added together with pellets of potassium hydroxide (4 gr, 72 mmol) and the ether and pentacarbon unsaturated compounds from the scavenger were distilled off. When the distillate reached 60 °C, DMSO (3.6 mL) was added and the solution was refluxed at 100 °C for 1 hour. After cooling to RT the reaction mixture was poured onto an ice-cold solution of 2M HCl (50 mL). Precipitated stearolic acid was filtered, washed with cold water and dried, yielding 97% of product that was used for the next reaction without further purification. 1H NMR (400 MHz, Chloroform-d) δ 2.35 (t, J = 7.5 Hz, 2H), 2.17 – 2.10 (m, 3H), 1.69 – 1.59 (m, 2H), 1.58 – 1.42 (m, 4H), 1.41 – 1.19 (m, 18H), 0.91 – 0.86 (m, 3H). 13C NMR (101 MHz, Chloroform-d) δ 178.57, 80.35, 33.89, 31.83, 29.21, 29.15, 29.04, 28.94, 28.87, 28.75, 28.60, 24.61, 22.65, 18.74, 18.71, 14.09. ESI [M+H]+ calcd. 281.24; found [M+H]+ 281.1.
Octadec-9-yn-1-ol (16):
Stearolic acid (2.805 g, 10 mmol) was dissolved in anhydrous diethyl ether (15 mL) and was slowly added to a cooled suspension of LiAlH4 (760 mg, 20 mmol) in diethyl ether (15 mL). After 4 hours, water was added and the solution was acidified with sulfuric acid and extracted with diethyl ether. The ether was dried with MgSO4 and filtered though silica gel. Evaporation of the ether yielded product 16 at 99% yield. 1H NMR (400 MHz, Chloroform-d) δ 3.63 (td, J = 6.7, 0.8 Hz, 2H), 2.18 – 2.08 (m, 3H), 1.98 (dq, J = 18.9, 6.4 Hz, 1H), 1.60 – 1.23 (m, 25H), 0.94 – 0.84 (m, 3H). 13C NMR (101 MHz, Chloroform-d) δ 80.29, 80.15, 63.05, 32.77, 31.89, 31.84, 29.64, 29.31, 29.21, 29.16, 29.12, 29.11, 28.86, 28.77, 25.72, 25.69, 22.65, 18.75, 18.74, 14.09. ESI [M+H]+ calcd. 267.26; found [M+H]+ 267.3.
Octadec-17-yn-1-ol (17):
Potassium hydride (0.90 g, 22.5 mmol) was dissolved in 15 mL of anhydrous 1,3-diaminopropane (APA) under argon at room temperature to give a clear yellow solution of 1.5M Kapa. The solution was added to a well-stirred solution of 16 (1.33 g, 5 mmol) in APA (4.5 mL). The resulting orange suspension was stirred 16 hours, and then poured into 500 mL of 3N HCl cooled with ice chips. The product was extracted with diethyl ether, washed with 3N HCl, dried with MgSO4 and filtered through silica gel. The solvent was evaporated under vacuum to yield product 17 at 92%. 1H NMR (400 MHz, Chloroform-d) δ 3.64 (t, J = 6.6 Hz, 2H), 2.18 (td, J = 7.1, 2.7 Hz, 1H), 2.05 – 1.92 (m, 2H), 1.62 – 1.45 (m, 3H), 1.37 (q, J = 6.7, 5.9 Hz, 1H), 1.29 (s, 9H), 1.36 – 1.18 (m, 15H). 13C NMR (101 MHz, cdcl3) δ 68.00, 63.10, 32.81, 29.65, 29.59, 29.49, 29.42, 29.10, 28.76, 28.49, 25.73, 22.68, 18.39, 18.39. ESI [M+H]+ calcd. 267.26; found [M+H]+ 267.3.
Octadec-17-ynoic acid (18):
Compound 17 (1.33 g, 5 mmol) was dissolved in 10 mL of anhydrous DMF and a solution of PDC (6.58 g, 17.5 mmol) in 5 mL anhydrous DMF, was added slowly with vigorous stirring. After stirring for 12 hours, water (200 mL) was added and the solution was extracted with diethyl ether. The combined extracts were washed with water and dried with MgSO4. The crude was filtered through a plug of silica gel and after evaporation of the ether the final product was purified by column chromatography (20% EA:Hexane) to yield product 18 at 74%. 1H NMR (400 MHz, Chloroform-d) δ 2.34 (t, J = 7.5 Hz, 2H), 2.18 (td, J = 7.1, 2.7 Hz, 1H), 2.04 – 1.90 (m, 2H), 1.63 (t, J = 7.3 Hz, 2H), 1.60 – 1.45 (m, 1H), 1.35 (s, 1H), 1.35 – 1.23 (m, 23H). 13C NMR (101 MHz, Chloroform-d) δ 179.40, 84.82, 68.00, 33.92, 29.62, 29.61, 29.59, 29.57, 29.49, 29.42, 29.31, 29.23, 29.10, 29.05, 28.76, 28.49, 24.67, 22.68, 18.39, 14.10. ESI [M+H]+ calcd. 281.24; found [M+H]+ 281.1.
N-(((9H-fluoren-9-yl)methoxy)carbonyl)-S-(octadec-17-ynoyl)-L-cysteine (19):
TFA (2.5 mL, 5%) and TIS (1.5 mL, 3%) were added to a solution of Fmoc-Cys(Trt)-OH (292.6 mg, 0.5 mmol) in DCM (50 mL), and the mixture was stirred for 2 hours. The solvent was evaporated under vacuum, then toluene was added to the crude residue and the solution was evaporated again to dryness. The process was repeated three times until the crude appeared as dry white solid. The solid was then suspended in hexane, filtered though a sintered funnel and washed twice with hexane to remove triphenylmethane. The dry solid was dissolved in a solution of TFA (10 mL) and DCM (10 mL). 17-Octadecynoic acid 18 (420 gr, 1.5 mmol) was dissolved in chloroform (20 mL), thionyl chloride (0.65 mL, 9 mmol) was then added and the reaction was refluxed for 1 hour. The solvents were evaporated under vacuum and excess thionyl chloride was removed by adding toluene and evaporation to dryness three times. The resulting oily residue was dissolved in DCM (20 mL) and added to the stirring solution of Fmoc-Cys-OH. The reaction mixture was heated to 40 °C and stirred for 4 hours. The solvents were evaporated under vacuum and the crude was purified by dry loading flash chromatography in a two-step elution using 20% ethyl acetate in hexane to remove excess of compound 18 and then 4% methanol in DCM to elute product 19 in quantitative yields. 1H NMR (400 MHz, Chloroform-d) δ 7.75 (dt, J = 7.6, 1.0 Hz, 2H), 7.59 (t, J = 6.4 Hz, 2H), 7.39 (td, J = 7.5, 1.1 Hz, 2H), 7.30 (tt, J = 7.4, 1.2 Hz, 2H), 5.68 (d, J = 7.9 Hz, 1H), 4.60 (q, J = 7.1 Hz, 1H), 4.37 (dd, J = 7.5, 3.1 Hz, 2H), 4.23 (t, J = 7.2 Hz, 1H), 3.54 – 3.45 (m, 1H), 3.35 (dd, J = 14.2, 7.1 Hz, 1H), 2.56 (t, J = 7.5 Hz, 2H), 2.38 (dt, J = 26.6, 7.5 Hz, 1H), 2.20 – 2.11 (m, 1H), 1.63 (dq, J = 12.2, 7.5 Hz, 2H), 1.31 – 1.18 (m, 26H). 13C NMR (101 MHz, Chloroform-d) δ 199.29, 173.30, 163.48, 156.01, 143.77, 141.27, 131.90, 127.71, 127.07, 125.18, 119.96, 68.04, 67.40, 53.87, 47.07, 44.06, 43.84, 36.93, 31.83, 30.54, 29.82, 29.63, 29.60, 29.58, 29.50, 29.44, 29.41, 29.38, 29.22, 29.19, 29.16, 29.10, 28.92, 28.76, 28.49, 25.59, 23.87. ESI [M+H]+ calcd. 606.32; found [M+H]+ 606.7.
(E)-N-(((9H-fluoren-9-yl)methoxy)carbonyl)-S-(16-(1-(3-(4-((4-(dimethylamino)phenyl)diazenyl)benzamido)propyl)-1H-1,2,3-triazol-4-yl)hexadecanoyl)-L-cysteine (20):
compound 19 (302 mg, 0.5 mmol) was dissolved in degassed DMF (25 mL) together with compound 3 (210 mg, 0.6 mmol). Anhydrous copper(II) sulfate (20 mg, 0.125 mmol) and ascorbic acid (44 mg, 0.25 mmol) were added and the reaction mixture was stirred for 2 hours at 40 °C. The solvent was evaporated under vacuum and the crude residue was purified by flash chromatography using 3-5% methanol in DCM. Product 20 was obtained in 89% yield. 1H NMR (500 MHz, Chloroform-d) δ 7.94 – 7.87 (m, 3H), 7.84 (d, J = 8.4 Hz, 2H), 7.75 (dd, J = 7.7, 4.2 Hz, 3H), 7.62 – 7.55 (m, 4H), 7.45 – 7.35 (m, 4H), 7.31 (d, J = 7.6 Hz, 4H), 6.81 (d, J = 9.0 Hz, 2H), 5.79 (d, J = 7.9 Hz, 1H), 4.59 – 4.54 (m, 2H), 4.47 (t, J = 6.7 Hz, 2H), 4.36 (d, J = 7.9 Hz, 2H), 4.23 (d, J = 7.4 Hz, 1H), 3.48 (dd, J = 12.2, 6.4 Hz, 4H), 3.33 (dd, J = 14.2, 6.9 Hz, 1H), 3.15 (s, 6H), 2.69 (dd, J = 14.3, 6.7 Hz, 4H), 2.56 (s, 3H), 2.28 – 2.22 (m, 2H), 1.44 – 1.35 (m, 1H), 1.21 (s, 18H). 13C NMR (126 MHz, Chloroform-d) δ 196.28, 177.36, 172.25, 163.38, 163.38, 163.34, 156.21, 150.21, 146.56, 144.03, 141.47, 139.48, 129.98, 128.38, 127.93, 127.02, 127.00, 125.44, 124.97, 121.85, 120.19, 120.19, 112.69, 75.61, 74.32, 73.22, 55.55, 54.01, 50.18, 47.12, 44.27, 40.95, 37.12, 37.06, 37.06, 31.93, 31.93, 29.82, 29.74, 29.74, 29.33, 29.33, 29.04, 25.81. ESI [M+H]+ calcd. 957.5; found [M+H]+ 957.9.
Ethyl hexadecylfluorophosphonate (HDFP):
HDFP was synthesized according to published procedures(Martin et al., 2011). 1H NMR (400 MHz, Chloroform-d) δ 4.15 – 4.01 (m, 2H), 1.77 – 1.65 (m, 2H), 1.64 – 1.52 (m, 2H), 1.40 – 1.34 (m, 2H), 1.31 (t, J = 7.1 Hz, 3H), 1.25 (s, 24H), 0.91 – 0.83 (m, 3H). 13C NMR (101 MHz, Chloroform-d) δ 61.48, 31.91, 30.68, 30.51, 29.68, 29.67, 29.65, 29.64, 29.61, 29.57, 29.37, 29.08, 26.34, 24.94, 22.67, 22.38, 16.41, 14.10. ESI [M+H]+ calcd. 337.26; found [M+H]+ 337.7.
Recombinant enzymes
Recombinant enzymes, TgPPT1WT, TgPPT1S128A, HsAPT1 and HsAPT2 were overexpressed and purified according to published procedures(Child et al., 2013; Garland et al., 2018). Enzyme concentration was pre-determined by titration with the irreversible inhibitor ethyl hexadecylfluorophoshonate (HDFP). Phospholipase A2 (PLA2, EC 3.1.1.4), Trypsin (EC 3.4.21.4) and Papain (EC 3.4.22.2) were purchased from Sigma Aldrich. Collagenase IV (EC 3.4.24.3) was purchased from Worthington Biochemical Corporation. Recombinant Monoacylglycerol Lipase (MGLL, EC 3.1.1.23) was purchased from Creative Biomart.
Construction of combinatorial fluorogenic peptide libraries and individual fluorogenic substrates
Combinatorial peptide libraries were prepared on solid support using Syro II (Biotage®) automatic parallel peptide synthesizer. For each library, rink amide resin was weighted to match 7.5 μM scale. Fritted pipette tip was used as a vessel for synthesis, using 600 μL reaction volume and 700 μL wash volume for each step. Following Fmoc deprotection with 20% piperidine for 30 minutes, each amino acid (5 eq.) was dissolved in DMF and preactivated with HCTU (5 eq.) and 2,3,5-collidine (10 eq.) then incubated on resin for 20 minutes. The first amino acid in each library was double coupled and capped with acetic anhydride (5%) and pyridine (5%) in DMF for 20 minutes. The acylated building block C20 (synthesis procedure described in the supplementary section) was coupled using 3 equivalents with reaction time of 40 minutes. Each deprotection step following the addition of C20 was performed using (4×) 1-minute washes with a solution of 1% DBU in DMF. In each library one position was varied with all the natural L-amino acids (excluding cysteine) and an adjacent position was reacted with an isokinetic mixture (adopted from Craik et al.(Schneider and Craik, 2009)) of the same 19 amino acids. After deprotection of the last amino acid in the sequence, 7-hydroxycoumarin-3-carboxy acid N-succinimidyl ester was coupled in a solution of DMF with 5% sodium bicarbonate buffer (0.1M, pH 8.2) for 30 minutes. Peptides were dried and cleaved of the resin with cleavage cocktail (95% TFA, 2.5% TIS, 2.5% water) during 2.5 hours. Following cleavage, peptides were precipitated in cold diethyl ether and washed twice with cold ether, dried thoroughly and dissolved in DMSO at stock concentration of 100 mM. The integrity of each library was evaluated by LCMS analysis and effective concentrations were assessed by chemical hydrolysis by incubation with 5% hydrozylamine in reaction buffer (20 mM HEPES, 150 mM NaCl, 10 mM CHAPS, pH 7.4) for 30 minutes, after which fluorescence was measured (Ex = 410 nm, Em = 450 nm). Concentrations were interpolated using a concentration curve established by measuring the fluorescence of a free-hydroxyl intermediate of QSE (Compound 13, Scheme S4) at a range of concentrations. Individual fluorogenic substrates were synthesized using the same procedures and purified by RP-HPLC using a gradient of 20-95% acetonitrile in water containing 0.1% TFA. Detection of eluting peptides was done simultaneously on UV channels set to 230 nm and 500 nm. Characterization and purity was assessed by LCMS using a similar solvent system. Purified peptides were lyophilized and stock solutions were prepared in DMSO.
Library screening with purified proteins
For each library, DMSO stocks were diluted in reaction buffer (20 mM HEPES, 150 mM NaCl, 10 mM CHAPS, pH 7.4) to a final concentration of 300 μM, followed by the addition of purified recombinant depalmitoylase at a final concentration of 50 nM. Reactions were incubated at 37 °C and fluorescence was measured (E × = 410 nm, Em = 450 nm) over 30 minutes. Data points collected from three replicates were normalized with DMSO control and initial rates were calculated from a linear fit of the activity curves and expressed as relative fluorescence units per second (RFU/sec).
Steady-state kinetics
Parameters of steady state kinetics were determined by measuring the increase of fluorescence of hydrolyzed fluorogenic substrates. Stock solutions of fluorogenic substrates were diluted in reaction buffer (20 mM HEPES, 150 mM NaCl, 10 mM CHAPS, pH 7.4) and pipetted into 396-well black plates, at a series of concentrations ranging from 80 μM to 0.156 μM. Recombinant enzymes were then added to a final concentration between 20 nM to 1 nM. Reaction progress was recorded (Ex = 410 nm, Em = 450 nm) over a period of 30-60 minutes at 37 °C. Concentrations of hydrolyzed prod ucts were interpolated using a standard concentration curve. Reaction rates were determined by linear regression analysis of the initial linear phase of hydrolysis. Experiments were preformed in triplicates. The kinetic constants, Km and Kcat, were obtained by non-linear regression curve fit of Michaelis-Menten parameters; V0=[E]*Kcat*[S]/(Km+[S]), V0 (initial reaction rate), E (enzyme concentration), S (substrate concentration), Km (Michaelis constant) and Kcat (turnover number) to these data using GraphPad Prism. Catalytic efficiencies were calculated from the product of Kcat/Km.
Generation of lysates and tissue homogenates
T. gondii parasites were harvested by syringe lysis of infected HFF monolayers and lysed by sonication in phosphate buffered saline (PBS). Cell cultures were harvested from confluent plates and lysed by sonication in PBS. Mouse organs were harvested form 14-week old C57BL/6 virgin females, washed twice in cold PBS, minced with a scalpel then homogenized by sonication in cold PBS. Lysates were clarified, and the protein concentration was quantified with a BCA assay (Thermo Scientific). Tumors were harvested form thoracic or inguinal mammary fat pads of 21-week old virgin MMTV-Py MT females, washed twice in cold PBS, minced with a scalpel then incubated with Type-IV collagenase over 30 minutes at 37 °C. Cells were isolated by centrifugation and ly sed by sonication in cold PBS.
In situ depalmitoylase activity assays
Lysates were diluted in reaction buffer (20 mM HEPES, 150 mM NaCl, 10 mM CHAPS, pH 7.4), 5 μg was pipetted into each well in a 396-well black plate. DMSO or inhibitors (ML348, ML349, HDSF, PMSF or Palmostatin B) were added to a final concentration of 10 μM and incubated 30 minutes on ice. Fluorogenic substrates were then added at a final concentration of 2.5 μM. Reactions were incubated at 37 °C and fluorescence was measured (Ex = 410 nm, Em = 450 nm) over a period of 60 minutes. For determination of specific activity, 20 μg of lysate per reaction were incubated with QStE over a range of concentrations. Concentrations of hydrolyzed products were interpolated using a standard concentration curve. Reaction rates were determined by linear regression analysis of the initial linear phase of hydrolysis. Experiments were preformed in three technical replicates and three biological replicates. The kinetic constants, Km and Kcat, were obtained by nonlinear regression curve fit of Michaelis-Menten parameters; V0=[EL]*Kcat*[S]/(Km+[S]), V0 (initial reaction rate), EL (lysate concentration), S (substrate concentration), Km (Michaelis constant) and Kcat (turnover number) to these data using GraphPad Prism. Kcat represents the specific activity for each lysate.
FP-Rho competition assays
For competition assays, 20 μg of lysate was incubated with DMSO or inhibitors at 10 μM for 30 minutes on ice, then 1 μM of FP-Rho was added and samples were incubated for additional 20 min on ice. Reactions were quenched with reducing SDS sample buffer, and the entire sample was resolved by SDS-PAGE. A typhoon flat-bed scanner was used to scan the gel (532-nm laser, 610-nm filter, PMT800).
QUANTIFICATION AND STATISTICAL ANALYSIS
See individual sections above for details on the statistics used for analysis.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GAPDH clone 6C5 | Millipore | MAB374 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant T. gondii TgPPT1W | (Child et al., 2013) | N/A |
| Recombinant T. gondii TgPPT1S128A | (Child et al., 2013) | N/A |
| Recombinant human HsAPT1WI | (Child et al., 2013; Garland et al., 2018) | N/A |
| Recombinant human HsAPT2WI | (Child et al., 2013; Garland et al., 2018) | N/A |
| Recombinant phospholipase A2 (PLA2) EC 3.1.1.4 | Sigma Aldrich | Cat#P6534 |
| Recombinant trypsin EC 3.4.21.4 | Sigma Aldrich | Cat#T 1426 |
| Recombinant papain EC 3.4.22.2 | Sigma Aldrich | Cat#P4762 |
| Recombinant monoacylglycerol lipase (MGLL) EC 3.1.1.23 | Creative Biomart | Cat#MGLL-2863H |
| Collagenase IV EC 3.4.24.3 | Worthington Biochemical Corp. | Cat#LS004186 |
| Hexadecylfluorophosphonate HDFP | This paper | N/A |
| JCP174 | (Child et al., 2013) | N/A |
| Palmostatin B | Sigma Aldrich | Cat#178501 |
| ML348 | (Adibekian et al.,2010) | N/A |
| ML349 | (Adibekian et al.,2010) | N/A |
| fluorophosphonate-rhodamine (FP-Rho) | (Lentz et al., 2016) | N/A |
| hexadecylsulfonyl fluoride (HDSF) | Santa Cruz Biotech. | Cat# sc-221708 CAS# 86855–26-7 |
| phenylmethylsulfonyl fluoride (PMSF) | Sigma Aldrich | Cat# P7626 CAS#329–98-6 |
| 4-nitropehyl octanoate (4-NPO) | Sigma Aldrich | Cat#21742–1G-F CAS#1956–10-1 |
| Quenched substrate for thioesterases QStE | This paper | N/A |
| Quenched substrate for esterases QSE | This paper | N/A |
| Critical Commercial Assays | ||
| BCA protein assay kit | Pierce | Cat# 23225 |
| Experimental Models: Cell Lines | ||
| Human: ovary adenocarcinoma cell line SKOV3 | (Child et al., 2013; Garland et al., 2018) | ATCC HTB-77 |
| Human: prostate adenocarcinoma cell line PC-3 | (Child et al., 2013; Garland et al., 2018) | ATCC CRL-1435 |
| Human: prostate carcinoma cell line LnCaP | (Child et al., 2013; Garland et al., 2018) | ATCC CRL-1740 |
| Human: ovary adenocarcinoma cell line OVCAR-3 | (Child et al., 2013; Garland et al., 2018) | ATCC HTB-161 |
| Human: breast adenocarcinoma cell line MDA-MB-231 | (Child et al., 2013; Garland et al., 2018) | ATCC HTB-26 |
| Human: breast adenocarcinoma cell line MCF7 | (Child et al., 2013; Garland et al., 2018) | ATCC HTB-22 |
| Experimental Models: Organisms/Strains | ||
| T. gondii strain ΔKu80 | (Child et al., 2013) | N/A |
| T. gondii strain ΔKu80Δppt1 | (Child et al., 2013) | N/A |
| Mouse: C57BL/6 MMTV-PyMT strain B6.FVB- Tg(MMTV-PyVT)634Mul/LellJ) | Jackson Laboratories | 022974 |
| Other | ||
| Syro II automated parallel peptide synthesizer | Biotage | N/A |
| HPLC 1260 Infinity System | Agilent Technologies | N/A |
| Finnigan LTQ mass detector | Thermo Fischer Sci. | N/A |
| HPLC 1100 series | Agilent Technologies | N/A |
| API 150EX single-quadrupole mass spectrometer | Applied Biosystems | N/A |
Highlights.
Synthesis of fluorescently quenched libraries of palmitoylated peptide substrates
Identification of peptide determinants of specificity for depalmitoylases
Synthesis of substrates with specificity for individual depalmitoylases
Application of substrates to measure depalmitoylase activity in complex proteomes
Acknowledgments
We would like to thank Brent Martin for providing the inhibitors ML348 and ML349; Chris Contag and Stephan Rogalla for providing the cancer cell lines used in this study; Vishwas Rao and Linhai Chen for experimental and conceptual assistance; and members of the Bogyo lab for helpful discussions. This work was funded by National Institutes of Health grant R01 GM111703 (to MB) and an EMBO Long-term Postdoctoral Fellowship (to NA).
Footnotes
Declaration of Interests
The authors declare no competing interests. - Here is the declaration of interests!!! Why are you asking for it again?
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adibekian A, Martin BR, Chang JW, Hsu KL, Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, et al. (2010). Characterization of a Selective, Reversible Inhibitor of Lysophospholipase 2 (LYPLA2) In Probe Reports from the NIH Molecular Libraries Program (Bethesda (MD)). [PubMed] [Google Scholar]
- Ahearn IM, Tsai FD, Court H, Zhou M, Jennings BC, Ahmed M, Fehrenbacher N, Linder ME, and Philips MR (2011). FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell 41, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Zheng H, Walker J, Coloff JL, and Buss JE (2003). Distinct rates of palmitate turnover on membrane-bound cellular and oncogenic H-ras. J Biol Chem 278, 19292–19300. [DOI] [PubMed] [Google Scholar]
- Breuza L, Poux S, Estreicher A, Famiglietti ML, Magrane M, Tognolli M, Bridge A, Baratin D, Redaschi N, and UniProt C (2016). The UniProtKB guide to the human proteome. Database (Oxford) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Zheng B, DeRan M, Jarugumilli GK, Fu J, Brooks YS, and Wu X (2016). ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat Chem Biol 12, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child MA, Hall CI, Beck JR, Ofori LO, Albrow VE, Garland M, Bowyer PW, Bradley PJ, Powers JC, Boothroyd JC, et al. (2013). Small-molecule inhibition of a depalmitoylase enhances Toxoplasma host-cell invasion. Nat Chem Biol 9, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MO, Woodley KT, and Choudhary JS (2017). Global, site-specific analysis of neuronal protein S-acylation. Sci Rep 7, 4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AK, Bellizzi JJ 3rd, Tandel S, Biehl E, Clardy J, and Hofmann SL (2000). Structural basis for the insensitivity of a serine enzyme (palmitoyl-protein thioesterase) to phenylmethylsulfonyl fluoride. J Biol Chem 275, 23847–23851. [DOI] [PubMed] [Google Scholar]
- Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Scholermann B, et al. (2010). Small-molecule inhibition of APT 1 affects Ras localization and signaling. Nat Chem Biol 6, 449–456. [DOI] [PubMed] [Google Scholar]
- Dow LE, Brumby AM, Muratore R, Coombe ML, Sedelies KA, Trapani JA, Russell SM, Richardson HE, and Humbert PO (2003). hScrib is a functional homologue of the Drosophila tumour suppressor Scribble. Oncogene 22, 9225–9230. [DOI] [PubMed] [Google Scholar]
- Duncan JA, and Gilman AG (1998). A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J Biol Chem 273, 15830–15837. [DOI] [PubMed] [Google Scholar]
- Duncan JA, and Gilman AG (2002). Characterization of Saccharomyces cerevisiae acyl- protein thioesterase 1, the enzyme responsible for G protein alpha subunit deacylation in vivo. J Biol Chem 277, 31740–31752. [DOI] [PubMed] [Google Scholar]
- el-Husseini Ael D, and Bredt DS (2002). Protein palmitoylation: a regulator of neuronal development and function. Nat Rev Neurosci 3, 791–802. [DOI] [PubMed] [Google Scholar]
- Foe IT, Child MA, Majmudar JD, Krishnamurthy S, van der Linden WA, Ward GE, Martin BR, and Bogyo M (2015). Global Analysis of Palmitoylated Proteins in Toxoplasma gondii. Cell Host Microbe 18, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, and Soldati-Favre D (2010). Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8, 343–357. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland M, Schulze CJ, Foe IT, van der Linden WA, Child MA, and Bogyo M (2018). Development of an activity-based probe for acyl-protein thioesterases. PLoS One 13, e0190255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard AD, and Watts A (2012). Regulation of G protein-coupled receptors by palmitoylation and cholesterol. BMC Biol 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, and Ploegh HL (2007). Chemical probes for the rapid detection of Fatty-acylated proteins in Mammalian cells. J Am Chem Soc 129, 2744–2745. [DOI] [PubMed] [Google Scholar]
- Hannoush RN, and Arenas-Ramirez N (2009). Imaging the lipidome: omega-alkynyl fatty acids for detection and cellular visualization of lipid-modified proteins. ACS Chem Biol 4, 581–587. [DOI] [PubMed] [Google Scholar]
- Hernandez JL, Davda D, Cheung See Kit M, Majmudar JD, Won SJ, Gang M, Pasupuleti SC, Choi AI, Bartkowiak CM, and Martin BR (2017). APT2 Inhibition Restores Scribble Localization and S-Palmitoylation in Snail-Transformed Cells. Cell Chem Biol 24, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ML, Collins MO, Goulding D, Choudhary JS, and Rayner JC (2012). Analysis of protein palmitoylation reveals a pervasive role in Plasmodium development and pathogenesis. Cell Host Microbe 12, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathayat RS, Elvira PD, and Dickinson BC (2016). A fluorescent probe for cysteine depalmitoylation reveals dynamic APT signaling. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, and Mukherjee AB (2013). Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J Biol Chem 288, 9112–9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, and Doolittle RF (1982). A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Lentz CS, Ordonez AA, Kasperkiewicz P, La Greca F, O’Donoghue AJ, Schulze CJ, Powers JC, Craik CS, Drag M, Jain SK, et al. (2016). Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infect Dis 2, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, and Conibear E (2015). ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DTS, Davis NG, and Conibear E (2017). Targeting the Ras palmitoylation/depalmitoylation cycle in cancer. Biochem Soc Trans 45, 913–921. [DOI] [PubMed] [Google Scholar]
- Linder ME, and Deschenes RJ (2007). Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol 8, 74–84. [DOI] [PubMed] [Google Scholar]
- Long JZ, and Cravatt BF (2011). The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev 111, 6022–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Verkruyse LA, and Hofmann SL (1996). Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc Natl Acad Sci U S A 93, 10046–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Wang C, Adibekian A, Tully SE, and Cravatt BF (2011). Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Vasudevan A, Linder ME, and Deschenes RJ (2006). Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res 47, 1118–1127. [DOI] [PubMed] [Google Scholar]
- Resh MD (2012). Targeting protein lipidation in disease. Trends Mol Med 18, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, and Bastiaens PI (2005). An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–1752. [DOI] [PubMed] [Google Scholar]
- Rusch M, Zimmermann TJ, Burger M, Dekker FJ, Gormer K, Triola G, Brockmeyer A, Janning P, Bottcher T, Sieber SA, et al. (2011). Identification of acyl protein thioesterases 1 and 2 as the cellular targets of the Ras-signaling modulators palmostatin B and M. Angew Chem Int Ed Engl 50, 9838–9842. [DOI] [PubMed] [Google Scholar]
- Schechter I, and Berger A (1967). On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27, 157–162. [DOI] [PubMed] [Google Scholar]
- Schneider EL, and Craik CS (2009). Positional scanning synthetic combinatorial libraries for substrate profiling. Methods Mol Biol 539, 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smotrys JE, and Linder ME (2004). Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev Biochem 73, 559–587. [DOI] [PubMed] [Google Scholar]
- Soyombo AA, and Hofmann SL (1997). Molecular cloning and expression of palmitoyl- protein thioesterase 2 (PPT2), a homolog of lysosomal palmitoyl-protein thioesterase with a distinct substrate specificity. J Biol Chem 272, 27456–27463. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Hayashi H, and Yamashita S (1996). Purification, cDNA cloning, and regulation of lysophospholipase from rat liver. J Biol Chem 271, 7705–7711. [DOI] [PubMed] [Google Scholar]
- Tian L, McClafferty H, Knaus HG, Ruth P, and Shipston MJ (2012). Distinct acyl protein transferases and thioesterases control surface expression of calcium-activated potassium channels. J Biol Chem 287, 14718–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomatis VM, Trenchi A, Gomez GA, and Daniotti JL (2010). Acyl-protein thioesterase 2 catalyzes the deacylation of peripheral membrane-associated GAP-43. PLoS One 5, e15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo D, Codocedo JF, and Inestrosa NC (2017). Posttranslational Modifications Regulate the Postsynaptic Localization of PSD-95. Mol Neurobiol 54, 1759–1776. [DOI] [PubMed] [Google Scholar]
- Verkruyse LA, and Hofmann SL (1996). Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem 271, 15831–15836. [DOI] [PubMed] [Google Scholar]
- Vujic I, Sanlorenzo M, Esteve-Puig R, Vujic M, Kwong A, Tsumura A, Murphy R, Moy A, Posch C, Monshi B, et al. (2016). Acyl protein thioesterase 1 and 2 (APT-1, APT- 2) inhibitors palmostatin B, ML348 and ML349 have different effects on NRAS mutant melanoma cells. Oncotarget 7, 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedegaertner PB, and Bourne HR (1994). Activation and depalmitoylation of Gs alpha. Cell 77, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Won SJ, Cheung See Kit M, and Martin BR (2018). Protein depalmitoylases. Crit Rev Biochem Mol Biol 53, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Davda D, Labby KJ, Hwang SY, Pricer R, Majmudar JD, Armacost KA, Rodriguez LA, Rodriguez CL, Chong FS, et al. (2016). Molecular Mechanism for Isoform-Selective Inhibition of Acyl Protein Thioesterases 1 and 2 (APT1 and APT2). ACS Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, and Fukata M (2016). Identification of PSD-95 Depalmitoylating Enzymes. J Neurosci 36, 6431–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.