

Abstract
Significance: Proline catabolism refers to the 4-electron oxidation of proline to glutamate catalyzed by the enzymes proline dehydrogenase (PRODH) and l-glutamate γ-semialdehyde dehydrogenase (GSALDH, or ALDH4A1). These enzymes and the intermediate metabolites of the pathway have been implicated in tumor growth and suppression, metastasis, hyperprolinemia metabolic disorders, schizophrenia susceptibility, life span extension, and pathogen virulence and survival. In some bacteria, PRODH and GSALDH are combined into a bifunctional enzyme known as proline utilization A (PutA). PutAs are not only virulence factors in some pathogenic bacteria but also fascinating systems for studying the coordination of metabolic enzymes via substrate channeling.
Recent Advances: The past decade has seen an explosion of structural data for proline catabolic enzymes. This review surveys these structures, emphasizing protein folds, substrate recognition, oligomerization, kinetic mechanisms, and substrate channeling in PutA.
Critical Issues: Major unsolved structural targets include eukaryotic PRODH, the complex between monofunctional PRODH and monofunctional GSALDH, and the largest of all PutAs, trifunctional PutA. The structural basis of PutA-membrane association is poorly understood. Fundamental aspects of substrate channeling in PutA remain unknown, such as the identity of the channeled intermediate, how the tunnel system is activated, and the roles of ancillary tunnels.
Future Directions: New approaches are needed to study the molecular and in vivo mechanisms of substrate channeling. With the discovery of the proline cycle driving tumor growth and metastasis, the development of inhibitors of proline metabolic enzymes has emerged as an exciting new direction. Structural biology will be important in these endeavors.
Keywords: proline dehydrogenase, aldehyde dehydrogenase 4A1, proline utilization A, substrate channeling, protein oligomerization
Introduction
Proline metabolism has multifaceted roles in cancer, stress protection, protein chaperoning, cell signaling, programmed cell death, nutrient adaptation and survival, and pathogen virulence. This review focuses on the structural biology of proline catabolism. Many other aspects of proline metabolism have been reviewed elsewhere. For example, Phang's group has discussed the complex connections between proline metabolism and cancer (66, 96, 100–105). Several articles discuss proline as a stress substrate and the role of proline metabolism in reactive oxygen species (ROS) generation (11, 61, 96, 103, 104). Proline metabolism has myriad roles in plants (41, 60, 76, 107, 126, 148). Other proline-related subjects include neurochemistry and neurological disorders (29, 140, 145), inborn errors of proline metabolism (42, 99, 140), nutrition (143, 144), the proline biosynthetic enzyme Δ1-pyrroline-5-carboxylate (P5C) synthase (32, 93, 108), and archaeal proline dehydrogenase (PRODH) (47).
Proline catabolism is an important part of overall metabolism. The oxidation of proline by the catabolic enzymes PRODH and l-glutamate γ-semialdehyde dehydrogenase (GSALDH) (Fig. 1A) is directly coupled to the electron transport chain via reduction of membrane-bound ubiquinone (85, 135). Proline catabolism thus harvests energy for the cell, particularly under nutrient depletion conditions, and in this way, l-proline supports the growth of bacteria (25, 48, 87, 133, 141).
FIG. 1.

The reactions and enzymes of proline metabolism. (A) Proline catabolism. (B) Proline biosynthesis from glutamate. (C) Diagram depicting the subcellular locations of proline metabolic enzymes in humans and the proline cycle. The gray oval represents the mitochondria. γ-GPR, γ-glutamate phosphate reductase; G5K, glutamate 5-kinase; GSAL, l-glutamate-γ-semialdehyde; GSALDH, l-glutamate γ-semialdehyde dehydrogenase; OAT, ornithine δ-aminotransferase; ORN, ornithine; P5C, Δ1-pyrroline-5-carboxylate; P5CS, P5C synthase; PRODH, proline dehydrogenase.
Proline catabolism in eukaryotes has been implicated in ROS generation, which has profound effects on oxidation–reduction homeostasis and cell survival. In humans, increased expression of PRODH causes significant ROS formation, which activates intrinsic and extrinsic apoptotic pathways (28, 68). PRODH is a p53-inducible tumor suppressor protein (67, 106). Paradoxically, PRODH also produces transient ROS, inducing survival pathways (147). For example, in Caenorhabditis elegans, PRODH generates ROS signals and induces the homologues of p38 MAP kinase and Nrf2, leading to increased expression of antioxidant enzymes and extended life span (147). Thus, PRODH plays a pivotal role in cell signaling processes that impact cell survival and death.
Hyperprolinemia disorders are caused by inborn errors in proline catabolism. Genetic mutations that disable PRODH activity result in type I hyperprolinemia, which is a risk factor for schizophrenia (24, 46). Certain mutations in the gene encoding GSALDH (ALDH4A1) cause type II hyperprolinemia, an inherited autosomal recessive disorder characterized by a deficiency in GSALDH activity (8, 30, 36, 113, 122, 130). Type II hyperprolinemia is causally linked to neurologic manifestations, including intellectual developmental disorders (99, 122).
Proline catabolism is also emerging as an important factor in survival and virulence mechanisms of pathogens. Helicobacter pylori (54, 87, 88) and procyclic trypanosomatids (56) have evolved to occupy proline-rich environmental niches and use proline as a major source of energy. Mycobacterium tuberculosis responds to hypoxia by increasing PRODH activity to produce P5C, which detoxifies methylglyoxal to 2-acetyl-1-pyrroline (13). Furthermore, both catabolic enzymes, as well as two proline biosynthetic enzymes, have been found to be essential for optimal growth of M. tuberculosis and are potential drug design targets (112). In the fungal pathogen, Cryptococcus neoformans, GSALDH is required for optimal production of the major virulence factors (58). Bacterial GSALDHs are being considered components of vaccines against Staphylococcus aureus (62). Finally, a recent study has implicated proline catabolism in the virulence of Ehrlichia chaffeensis, the causative agent of human monocytic ehrlichiosis (23).
Structural biology contributes to our understanding of protein function. Currently, the Protein Data Bank (PDB) contains over seventy structures of proline catabolic enzymes and domains. These structures include many biologically relevant ligands, including substrates, cofactors, substrate analogs, inhibitors, covalent inactivators, and DNA. The following pages describe these structures and their implications for understanding the functions of proline catabolic enzymes.
The Enzymes of Proline Metabolism
Proline catabolism
Proline metabolism refers to the five enzyme-catalyzed reactions that catabolize and synthesize l-proline (1, 95) (Fig. 1). The catabolic arm consists of the two enzymes PRODH and GSALDH (Fig. 1A). PRODH (EC 1.5.5.2, formerly EC 1.5.99.8) catalyzes the flavin-dependent oxidation of proline to P5C. The two-electron reduction of the enzyme-bound flavin occurs concomitant with the oxidation of proline. The electrons stored in the reduced flavin cofactor are subsequently transferred to the membrane electron transport chain, thus regenerating the oxidized enzyme for another round of catalysis. As with most other flavoenzyme dehydrogenases, the flavin remains associated with the enzyme during the entire catalytic cycle, although the cofactor is noncovalently bound. Flavin adenine dinucleotide (FAD) is the flavin cofactor commonly found in PRODHs, although at least one bacterial PRODH utilizes FMN (43). The hydrolysis of P5C generates GSAL, which is the substrate for GSALDH (EC 1.2.1.88). GSALDH is a member of the aldehyde dehydrogenase (ALDH) superfamily (ALDHSF) and is also known as ALDH4A1.
The nomenclature for the second enzyme of proline catabolism is somewhat confusing. Although GSAL is the substrate being oxidized, the enzyme is often called Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH, EC 1.5.1.12). In 2013, the Nomenclature Committee of The International Union of Biochemistry and Molecular Biology (IUBMB) deleted EC 1.5.1.12 in favor of EC 1.2.1.88 and recommended the name GSALDH. Nevertheless, readers will see P5CDH used in the literature, including recent articles from this author's laboratory. Also, some PDB entries deposited before 2014 list EC 1.5.1.12 in the header of the coordinate file (e.g., PDB code 4LGZ, deposited June 30, 2013). More recent depositions list EC 1.2.1.88. In summary, the second enzyme of proline catabolism has three names: GSALDH, P5CDH, and ALDH4A1. Herein, the IUBMB-recommended GSALDH will be used.
Proline biosynthesis
The main proline biosynthetic pathway starts with glutamate (Fig. 1B). Although P5C and GSAL are intermediates in both proline biosynthesis and catabolism, biosynthesis is not the reverse of catabolism. Rather, three different enzymes catalyze the transformation of glutamate to proline. Glutamate 5-kinase (G5K, EC 2.7.2.11) catalyzes the transfer of a phosphoryl group from ATP to the γ-carboxylate of l-glutamate, generating l-glutamate 5-phosphate (or γ-glutamate phosphate). The enzyme γ-glutamate phosphate reductase (γ-GPR, EC 1.2.1.41) catalyzes the reversible NADPH-dependent reduction of l-glutamate-5-phosphate to GSAL and inorganic phosphate. The IUBMB-accepted name for this enzyme is glutamate-5-semialdehyde dehydrogenase, which refers to the reverse reaction. This name reminds us that the enzyme is member of the ALDHSF (ALDH18A1 in humans, ALDH19 in bacteria and plants). Cyclization of GSAL with loss of water generates P5C, which is the substrate for P5C reductase (EC 1.5.1.2, P5CR, PYCR in humans). P5CR catalyzes the NADPH-dependent reduction of P5C to l-proline.
Bifunctional proline metabolic enzymes
In certain organisms, two proline metabolic enzyme activities are combined into a bifunctional enzyme, allowing coordination of sequential metabolic reactions. In some bacteria, the catabolic enzymes PRODH and GSALDH are combined into a single polypeptide chain known as proline utilization A (PutA) (64, 129). In plants and animals, the biosynthetic enzymes G5K and γ-GPR are combined into the bifunctional enzyme P5C synthase (P5CS) (32, 93, 108). Bifunctional enzymes often exhibit substrate channeling, which can improve kinetic efficiency, protect reactive intermediates, and prevent cross talk between competing pathways, such as proline catabolism and biosynthesis. See a recent review from Becker's group on substrate channeling in proline metabolism (5).
Subcellular localization and larger metabolic context
The enzymes described above belong to a larger picture of metabolism (Fig. 1C). Subcellular localization of proline metabolic enzymes is an important aspect of this larger context. These concepts are summarized for the human enzymes here. Good reviews addressing this topic more thoroughly are available (103). The additional isoforms found in plants and their unique localization have been reviewed recently (32, 148). Also, the roles of proline in the energy metabolism of trypanosomatids have been reviewed (19, 20).
Proline catabolism occurs in mitochondria (Fig. 1C). PRODH is an inner mitochondrial membrane enzyme. This location facilitates the transfer of electrons from the reduced FAD in PRODH to the mitochondrial electron transport chain for ATP production. The mitochondrial localization of PRODH is also essential for its role as an ROS generator that activates apoptosis (28, 68). GSALDH is located in the mitochondrial matrix. The end product of proline catabolism, glutamate, can be converted to the citric acid cycle intermediate α-ketoglutarate (αKG) by the mitochondrial enzyme glutamate dehydrogenase. Thus, proline catabolism is linked to one of the major hubs of metabolism.
Proline biosynthesis is split between the mitochondrion and the cytosol (Fig. 1C). The two-step conversion of Glu to GSAL by the bifunctional enzyme P5CS occurs inside mitochondria. Proline can also be synthesized from ornithine (ORN). In this route, ornithine δ-aminotransferase (OAT) catalyzes the interconversion of ORN and GSAL. This reaction connects proline metabolism with arginine metabolism and the urea cycle (82). Although P5CR is considered to be cytosolic, at least one isoform (PYCR1) colocalizes with mitochondrial markers (109). Phang et al. have suggested the possibility that P5CR associates with mitochondrial outer membranes or the cytosolic surface of plasma membranes (103).
The segregation of PRODH from P5CR is an important element of the “proline cycle” paradigm proposed by Phang's group in the 1980s (38, 39, 95). P5C is the central metabolite of the cycle, so Phang has advocated classifying proline metabolic enzymes as either P5C producing (PRODH, P5CS, OAT) or proline utilizing (P5CR, GSALDH) (103). The basic idea is that these enzymes form a metabolic shuttle that transfers reducing equivalents between mitochondria and the cytosol (Fig. 1C). Inside mitochondria, PRODH extracts electrons from proline for the electron transport chain or to produce ROS. The P5C generated by PRODH can be transported outside the mitochondria, where it is reduced back to proline by P5CR. Proline then enters the mitochondria to complete the cycle. In addition, Phang's group has shown that P5C in the cytosol stimulates the pentose phosphate pathway (PPP) to produce NADPH in response to consumption by P5CR (97, 98). Thus, a major redox function of proline metabolism is to transfer reducing potential into mitochondria and oxidizing potential out of mitochondria. This function underlies the multifaceted roles of proline metabolism in cancer and other diseases (66, 90, 96, 100–105). In particular, the proline cycle has recently been shown to play a key role in tumor cell growth (65) and metastasis (31).
Proline Dehydrogenase
The PRODH fold
The fold of PRODH has been determined from crystal structures of monofunctional PRODHs (69, 138) from Thermus thermophilus and Deinococcus radiodurans (DrPRODH), as well as structures of the Escherichia coli PutA (EcPutA) PRODH domain (59) and full-length PutAs (50, 51, 71, 117, 121). PRODH adopts a variant of the (βα)8 barrel fold (Fig. 2). This basic fold was first discovered in a crystal structure of triosephosphate isomerase (TIM), so it is often referred to as the TIM barrel (7). The TIM barrel is ubiquitous in nature and supports many diverse enzymatic functions and cofactors. Wierenga provides an introduction to TIM barrels (139), and Nagano et al. have reviewed the functional diversity of TIM barrels (86).
FIG. 2.

The PRODH fold. The structure of DrPRODH complexed with THFA is shown (PDB code 4H6Q). FAD is colored yellow; THFA is colored gray. The strands and helices of the (βα)8 barrel are labeled. Upper inset: Close-up view of the FAD isoalloxazine and THFA. The N5 atom of the FAD is the hydride acceptor. The C5 atom of THFA represents the hydride donor of proline. The distance between these two atoms in the structure is 3.2 Å. Lower inset: Sequence alignment of α8 residues from diverse PRODHs. At, Arabidopsis thaliana; Dr, Deinococcus radiodurans; Ec, Escherichia coli; FAD, flavin adenine dinucleotide; Gs, Geobacter sulfurreducens; Hs, Homo sapiens; PDB, Protein Data Bank; PRODH, proline dehydrogenase; Sc, Saccharomyces cerevisiae; Tc, Trypanosoma cruzi; THFA, l-tetrahydrofuroic acid. This figure and others were created with PyMOL (26). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
PRODH exhibits a notable deviation from the classic TIM barrel fold. In the classic TIM barrel, α8 sits alongside β8 and packs between helices 1 and 7. In contrast, α8 in PRODH lies above the carboxyl end of the barrel with its long axis perpendicular to β-strands (Fig. 2). This deviation is significant because α8 contains universally conserved residues that bind the substrate proline (Fig. 2, lower inset).
The active site in TIM barrels is usually located at the carboxyl termini of the strands of the barrel; PRODH adheres to this paradigm (Fig. 2). The isoalloxazine of the FAD consists of three fused six-membered rings and is the redox center of the cofactor. The re face of the isoalloxazine packs tightly against strands 4–6 (Fig. 2, upper inset). The dimethylbenzene edge of the isoalloxazine contacts the β7-α7 loop. The pyrimidine edge forms hydrogen bonds with the β3-α3, β4-α4, and β5-α5 loops. The si face of the isoalloxazine forms one wall of the proline-binding site.
The proline site of PRODH
The proline-binding site of PRODH has been characterized by high-resolution X-ray crystallography of PRODH and PutAs complexed with the proline analog l-tetrahydrofuroic acid (THFA) (69, 71, 89, 117, 149). The binding pose is identical in all the structures. THFA binds between the si face of the isoalloxazine and α8 (Fig. 2). The five-membered ring of THFA is approximately parallel to the isoalloxazine and centered over the middle ring of the cofactor. In this position, the C5 atom of THFA is close to the N5 of the flavin. The C5-N5 distance in the three structures ranges from 3.2 to 3.6 Å (Fig. 2, upper inset). This geometry is consistent with a hydride transfer mechanism, since C5 of THFA represents the C atom of proline that is oxidized, and N5 is the electron acceptor of the FAD. Biochemical studies have shown that PRODH uses hydride transfer rather than a radical mechanism (83).
The PRODH/PutA-THFA structures imply a conserved set of interactions that maintain proline in the optimal pose for hydride transfer. Eleven highly conserved residues contact the inhibitor (and presumably proline), as shown for Geobacter sulfurreducens PutA (GsPutA) (Fig. 3A, B). Helix α8 plays an important role in substrate recognition by providing four of these residues: two Arg residues that bind the inhibitor carboxylate, a Tyr residue that packs against the THFA ring, and a Glu that ion pairs to the second Arg. This quartet is part of a conserved sequence motif (Fig. 2, lower inset). The other interacting residues belong to either the strands of the barrel or to β-α loops of the barrel. Most of the residues are identically conserved in PutAs and PRODH. One exception is the Tyr that forms a water-mediated hydrogen bond to the heteroatom of THFA (represents the amino group of Pro); some PutAs have an Asn at this position.
FIG. 3.

The proline binding site of PRODH deduced from structures of enzyme-THFA complexes. (A) PRODH active site of GsPutA complexed with THFA (PDB code 4NMA). (B) Schematic diagram of interactions between PRODH and proline in the Michaelis complex. Dotted lines denote hydrogen bonds and ion pairs. Thick solid lines denote nonpolar contacts. (C) Comparison of the open (cyan) and THFA-bound closed (gray) PRODH active sites in GsPutA (PDB codes 4NM9 and 4NMA). The arrows show the directions of conformational changes that accompany THFA binding. Figure adapted from Singh et al. (117) and Tanner (128). GsPutA, Geobactr sulfurreducens proline utilization A. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Two ion pairs are notable in the THFA complex (Fig. 3A, B). The two Arg residues that bind the inhibitor carboxylate each ion pair to conserved Glu residues. The first Arg in the sequence ion pairs with a Glu residue in the β1-α1 loop of the PRODH barrel (Arg421-Glu149 in GsPutA). The second Arg ion pairs with the Glu of the α8 sequence motif (Arg422-Glu425 in GsPutA). These ion pairs facilitate substrate binding by stabilizing the Arg residues for interaction with the substrate carboxylate group. It may be that these ion pairs help ameliorate the electrostatic repulsion that results from bringing the two Arg guanidinium groups close together (4.7 Å).
Molecular motions that accompany proline binding and P5C release
It is notable that so many residues cluster around such a small substrate in PRODH (Fig. 3A, B). Thus, the active site of the Michaelis complex is very crowded. Furthermore, the structures show that the inhibitor, and presumably proline, is completely buried, which implies that protein motion is required for substrate binding and product release.
The nature of the molecular motions involved in substrate binding and product release has been deduced from crystal structures. Conformational changes are inferred by comparing crystal structures determined in the presence and absence of THFA. This comparison has been made for GsPutA and DrPRODH. The two systems reveal a common theme of active site dynamics.
The THFA-free structures are profoundly different from the THFA complexes. In the absence of THFA, the active site adopts an open state in which α8 is shifted away from the isoalloxazine and the ion pair between the first Arg of the α8 motif and Glu of the β1-α1 loop is broken (Fig. 3C). The shift of α8 is 3.5 Å in GsPutA and 1.7 Å in DrPRODH. Rupture of the ion pair is associated with a remodeling of the β1-α1 loop. The structures imply a movement of the β1-α1 Glu residue by more than 5 Å between the two states (Fig. 3C).
The structures imply the following scenario for substrate binding and product release. In the resting oxidized enzyme, the active site adopts an open conformation in which α8 and the β1-α1 loop are retracted, which leaves the si face of the isoalloxazine exposed. On proline binding, α8 and the β1-α1 loop converge on the proline-binding site resulting in ion pair formation between the β1-α1 Glu and the first Arg of the α8 motif.
Why does the active site open to release P5C? Here one needs to consider the conformation of the reduced flavin. Reduction induces a severe butterfly bending of the isoalloxazine, which appears to initiate a cascade of events that leads to product release (69). Bending of the ring system pushes the N5–N10 axis of the flavin toward the newly formed P5C, creating steric clash in the highly crowded active site. Retraction of α8 and rupture of the Arg-Glu ion pair alleviate this clash, creating an opening that allows product dissociation.
The Arg-Glu ion pair that links α8 to the β1-α1 loop is a central player in the conformational changes associated with proline binding and P5C release. Because it is formed in the closed E-S complex and broken in the ligand-free, open state, the ion pair appears to function as the active site gate. Furthermore, because the ion pairing residues are identically conserved in monofunctional PRODHs and PutAs, the gate is probably a universal aspect of PRODH catalysis.
Recognition of the quinone electron acceptor by PRODH
Whereas substrate recognition of proline has been studied extensively, the structural basis for recognition of electron acceptors is not as well understood. The only evidence for how quinones bind to PRODH comes from a structure of covalently inactivated GsPutA complexed with menadione bisulfite (MB) (117). Inactivation by the mechanism-based inactivator N-propargylglycine (Fig. 4A) locks PutA into a conformation that resembles the proline-reduced state (124, 137). The inactivation results in a 3-carbon covalent link between an active site Lys and the FAD N5 (Fig. 4A). Since this conformation is stable under laboratory (oxidizing) conditions, it was used to grow crystals of the PutA-quinone complex. MB is a small quinone that has high solubility because of the -SO3- functional group. The combination of using the covalently inactivated enzyme and MB facilitated the first structure determination of a PRODH-quinone complex (117).
FIG. 4.

The quinone binding site of PRODH as seen in a structure of inactivated GsPutA complexed with MB. (A) Structures of (1) N-propargylglycine and (2) the covalently modified FAD resulting from inactivation by N-propargylglycine. In N-propargylglycine-inactivated GsPutA, Lys203 makes a covalent link with the FAD. (B) Interactions for MB (green) bound to inactivated GsPutA (PDB code 4NMF). The distances between MB and the N5 and N10 atoms of the FAD are indicated. (C) Comparison of the PRODH active sites of GsPutA-THFA (yellow protein, pink THFA) and inactivated GsPutA-MB (gray protein, green MB), highlighting the proximity of the proline and quinone sites and the structural differences involving α8 and Glu149. MB, menadione bisulfite. Figure adapted from Singh et al. (117). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
MB binds next to the middle ring of the isoalloxazine si face, with the benzoquinone and isoalloxazine forming a ∼40° angle (Fig. 4B). A carbonyl O atom of MB is 3.2 and 4.0 Å from the flavin N10 and N5 atoms, respectively (Fig. 4B). The close approach between MB and the isoalloxazine is consistent with direct electron transfer from the flavin to the quinone without an intermediary electron carrier (83). The 1,4-naphthoquinone is surrounded by several aromatic and nonpolar residues, which is expected for a nonpolar ligand.
Although the proline and quinone sites overlap, the protein conformations of the two complexes are rather different (Fig. 4C). THFA (i.e., proline) binds in the closed state, whereas MB binds to an open state. In the MB complex, helix α8 is shifted to the open position and the ion pair gate is broken. The open state is required to accommodate the large ring system of MB.
Kinetic studies of PRODHs
Three assays are used to measure the catalytic activity of PRODH. The proline:2,6-dichlorophenolindophenol (DCPIP) oxidoreductase assay leverages a dye-coupled reaction that measures the reduction of DCPIP. Phenazine methosulfate is used as a secondary electron acceptor that mediates electron transfer from the flavoenzyme to the terminal electron acceptor DCPIP. DCPIP is a general-purpose dye used in many biochemical assays. It is blue in the oxidized form and colorless in the reduced form. Reduction of DCPIP is monitored by a decrease in absorbance near 600 nm. The extinction coefficient used varies from laboratory to laboratory, but is typically in the range of ɛ600 = 16,100–19,100 M−1 cm−1 (14, 115, 136). A more direct assay is to measure the reduction of the terminal electron acceptor without a mediator. Quinones such as coenzyme Q1 (CoQ1) (ɛ278 = 14,500 M−1 cm−1) (4, 85, 121) and menadione (ɛ262 = 14,000 M−1 cm−1) (85, 117) are convenient. This assay is perhaps more relevant than the DCPIP assay because the electron acceptor better resembles a biological quinone. Note that the kinetic parameters for proline depend on the quinone used (85). The third assay follows the reaction of P5C/GSAL with o-aminobenzaldehyde (oAB) (80, 85, 121, 155). The oAB assay monitors the increase in absorbance at 443 nm (ɛ443 = 2590 M−1 cm−1). This assay is useful for studying substrate channeling, since it allows one to monitor the release of the intermediate of proline catabolism.
These methods have been used to determine the kinetic parameters of a few monofunctional PRODHs (Table 1). The parameters depend on the type of assay used, so caution is advised when comparing results for different enzymes. Nevertheless, the Km for proline tends to be in the millimolar range, while that of the quinone is usually lower, in the micromolar range. The competitive inhibition constant of THFA (Ki) tends to be in the low millimolar range for monofunctional PRODHs and PutAs (85, 138). DrPRODH has atypically high Km and Ki (69).
Table 1.
Steady-State Kinetic Parameters of Monofunctional Proline Dehydrogenases
l-Glutamate γ-Semialdehyde Dehydrogenase
Catalytic mechanism and fold
GSDAH (or P5CDH) belongs to the ALDHSF. ALDHs catalyze the NAD(P)+-dependent oxidation of aldehydes to carboxylic acids. The classification of ALDHs into families and subfamilies based on amino acid and gene sequences has been described (120, 131, 146). The current analysis reveals 24 ALDH gene families in eukaryotes (132). Humans have 19 functional genes and 3 pseudo genes. ALDH families 1–9, 16, and 18 are found in humans. GSALDH is known as ALDH4A1.
ALDHs have a conserved catalytic mechanism (49). The mechanism begins with nucleophilic attack by the catalytic cysteine on the aldehyde (Fig. 5A) producing a hemithioacetal intermediate (Fig. 5B). Hydride transfer to NAD+ generates NADH and the acyl-enzyme intermediate (Fig. 5C). Hydrolysis of the acyl-enzyme intermediate yields the carboxylic acid product (Fig. 5D) and regenerates the resting enzyme.
FIG. 5.

The catalytic mechanism of ALDH superfamily enzymes. The residue numbers refer to human GSALDH. (A) The catalytic Cys attacks the carbonyl of the aldehyde substrate to form the hemithioacetal intermediate. (B) Hydride transfer from the hemithioacetal intermediate to NAD+ generates NADH and the acyl-enzyme intermediate. (C) A water molecule attacks the acyl-enzyme intermediate to generate the carboxylic acid product. (D) The products dissociate from the enzyme, and NAD+ binds to prepare the active site for another round of catalysis. Figure adapted from Luo et al. (72). ALDH, aldehyde dehydrogenase.
Enzymes in a superfamily share a common three-dimensional fold. The ALDH fold consists of three domains: NAD+ binding, catalytic, and oligomerization, as shown for human GSALDH in Figure 6A. The NAD+-binding domain adopts the Rossmann fold. The core of this domain is a parallel five-stranded β-sheet having strand order 32145. This topology differs slightly from the classic Rossmann fold sheet, which has six strands arranged as 321456. The catalytic domain is an α/β structure and derives its name from the fact that it provides the essential Cys residue. The oligomerization domain is a β-substructure that mediates domain-swapped dimerization as well as higher order assembly into hexamers for some GSALDHs. It is a bipartite structure that consists of a β hairpin that protrudes from the Rossmann domain and a β-strand that is part of the C-terminus of the polypeptide chain. The two elements of the oligomerization domain are far apart in sequence. For example, in human GSALDH, the hairpin consists of residues 183–196, while the C-terminal β-strand consists of residues 545–550. The active site is located in the crevice between the NAD+-binding and catalytic domains.
FIG. 6.

The ALDH fold as seen in GSALDH. (A) Ribbon drawing of the protomer of human GSALDH (PDB code 3V9G). Glu and NAD+ from PDB codes 3V9K and 2J5N, respectively, have been docked to the structure to indicate the locations of these binding sites. The NAD+-binding, catalytic, and oligomerization domains are colored red, blue, and green, respectively. The interdomain linkers are colored yellow. (B) Interactions for Glu bound to mouse GSALDH. (C) Schematic diagram of interactions between GSAL and GSALDH implied from the structure of mouse GSALDH complexed with Glu. (D) Interactions for NAD+ bound to TtGSALDH (PDB code 2J5N). Figure adapted from Srivastava et al. (122) and Pemberton and Tanner (92). TtGSALDH, Thermus thermophilus l-glutamate γ-semialdehyde dehydrogenase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The ALDH topology is complicated by the fact that the catalytic domain is inserted between the last strand of the Rossmann domain and the C-terminal strand of the oligomerization domain [see Fig. 3 of Lagautriere et al. (55) for a topology diagram]. This results in two linkers connecting the catalytic and NAD+-binding domains (yellow in Fig. 6A). Linker 1 consists of about five residues and connects strand 5 of the Rossmann sheet to the beginning of the catalytic domain. Linker 2 consists of ∼20 residues that connect the end of the catalytic domain to a short helix that precedes the β-strand of the oligomerization domain. The interdomain linkers participate in binding GSAL and NAD+.
Recognition of GSAL by GSALDH
The structural basis of aldehyde recognition has been determined from high-resolution crystal structures of monofunctional GSALDHs complexed with the product Glu (45, 122) and other carboxylic acids having various aliphatic chain lengths (92). Glu binds in an extended conformation and forms several interactions with the enzyme (Fig. 6B, C). The backbone of the product forms several hydrogen bonds with a Gly-Ser motif that is located at the N-terminal end of interdomain linker 2. This part of linker 2 is known as the aldehyde anchor loop because of its key role in stabilizing the base of the aldehyde substrate in the active site (92). In addition, the α carboxylate of the product (or GSAL) forms a direct hydrogen bond with the Ser residue that is immediately C-terminal to the catalytic Cys and a water-mediated hydrogen bond with an Lys residue that is immediately N-terminal to the catalytic Cys. The aliphatic chain of Glu packs into an “aromatic box” consisting of two conserved Phe side chains, the second of which belongs to interdomain linker 2. The aromatic box is present in all ALDHs, however, the number and type of aromatic residues vary among ALDHs (110). One of O atoms of the ɛ-carboxylate of Glu occupies the oxyanion hole, another conserved structural element of ALDH substrate recognition. This atom represents the carbonyl O of GSAL and forms hydrogen bonds with a conserved Asn and the backbone N-H of the catalytic Cys. The hydrogen bond donors of the oxyanion hole stabilize the negative charge of the hemithioacetal intermediate.
Recognition of NAD+ by GSALDH
Recognition of NAD(P)+ has been studied with crystallography using monofunctional GSALDHs (44, 45, 55, 91, 122) and PutAs (50, 71, 121). The entire cofactor has been resolved with strong electron density in M. tuberculosis GSALDH [PDB code 4IHI, (55)], T. thermophilus GSALDH (TtGSALDH, PDB code 2J5N), and one PutA [PDB codes 5KF6, 5KF7 (71)]. In the other structures, the electron density is weak for the nicotinamide half of the NAD+, implying conformational disorder. This is common for ALDHs (74, 94). A structure of TtGSALDH complexed with NADP+ was also determined as part of a study of cofactor specificity (PDB code 2EHQ) (44). Density for NADP+ is strong in that structure, including the density for the 2′-phosphoryl group.
NAD(P)+ bound to GSALDH exhibits the canonical pose expected for a Rossmann fold enzyme (16) (Fig. 6D). The adenine ring packs between the α-helices that follow the third and fourth strands of the Rossmann fold. The hydroxyls of the adenosine ribose hydrogen bond with a conserved Lys from β2 of the Rossmann fold. The pyrophosphate hydrogen bonds to a conserved Ser residue located at the N-terminus of α4. This residue is part of a conserved sequence motif of FTGS. The nicotinamide ribose hydrogen bonds with a conserved Glu residue from the catalytic domain. The nicotinamide ring is wedged between the catalytic Cys and the methyl group of the Thr in the FTGS motif. The distance between the S atom of the catalytic Cys and the C4 of the nicotinamide is 2.8–3.2 Å, which is consistent with the accepted mechanism, in which a hydride ion transfers from the hemithioacetal intermediate to the C4 of NAD+. The nicotinamide is also close to interdomain linker 1. This linker contains the conserved sequence EXGG. The Glu of this motif is the catalytic residue that activates the water molecule for hydrolysis of the acyl-enzyme, while the main-chain carbonyl of X is a hydrogen bond acceptor of the nicotinamide carboxamide. The latter interaction helps position the cofactor for stereospecific hydride transfer.
Oligomeric state and quaternary structure of GSALDH
The oligomeric states and quaternary structures of monofunctional GSALDHs have been extensively characterized using small-angle X-ray scattering (SAXS), analytical ultracentrifugation, and X-ray crystallography (73, 91, 127). These studies have shown that GSALDHs form dimers or hexamers. Examples of dimeric GSALDHs include the enzymes from human, mouse, Bacillus halodurans, and Bacillus licheniformis (73). Hexameric GSALDHs are from Saccharomyces cerevisiae (91), T. thermophilus (73), D. radiodurans (73), and M. tuberculosis (55). The hexamer appears to be unique to GSALDH, as it has not been seen in other ALDHs. Also, the dimer-of-dimers tetramer formed by some ALDHs (74, 127) has not been observed in GSALDHs.
Like all other ALDHs, GSALDHs form a domain-swapped dimer in which the oligomerization domain of one protomer engages the other protomer (Fig. 7A). The domain swapping results in the formation of an intermolecular β-sheet involving the C-terminal strand of the oligomerization domain of one protomer and the final strand of the catalytic domain of the other protomer (Fig. 7A, left). Another major part of the dimer interface is located on the face opposite to that of the intermolecular β-sheet and consists of extensive nonpolar and electrostatic interactions involving side chains of two α-helices (Fig. 7A, right).
FIG. 7.

GSALDH oligomers. (A) The domain-swapped dimer as exemplified by Bacillus halodurans GSALDH (PDB code 3QAN). (B) The GSALDH hexamer as observed for TtGSALDH (PDB code 2BHQ). Figure adapted from Tanner (127). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In the hexameric GSALDHs, three of the domain-swapped dimers assemble into a trimer-of-dimers hexamer (Fig. 7B). The major interface between dimers is formed by the oligomerization and NAD+-binding domains. Thus, the oligomerization domain stabilizes both the domain-swapped dimer and the hexamer. Three copies of this interface line the inside surface of the tunnel that surrounds the threefold axis of the hexamer (Fig. 8A). For reference, this interface buries 1100 Å2 of surface area in TtGSALDH, while the interfacial area of the domain-swapped dimer is 2900 Å2.
FIG. 8.
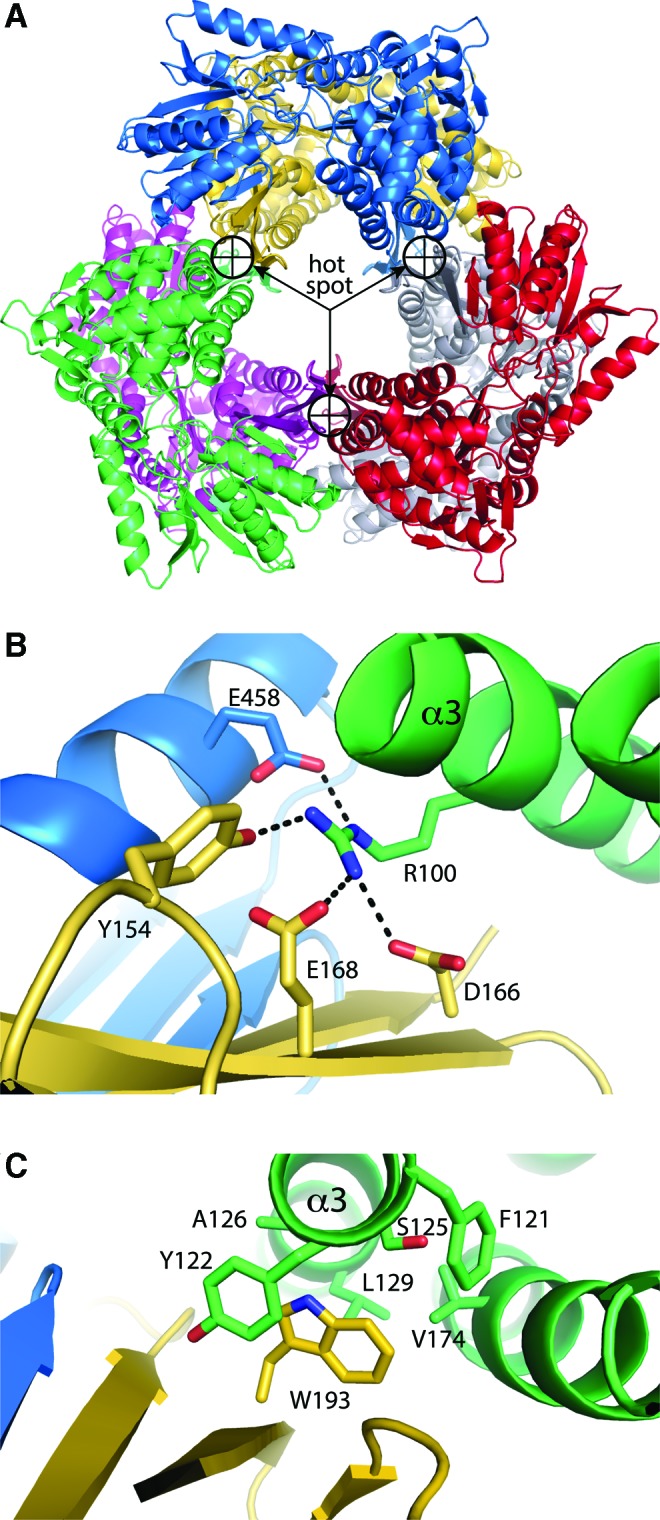
The hexamerization hot spot of GSALDH. (A) The hexamer of yeast GSALDH showing the location of the hexamerization hotspot (PDB code 4OE6). (B) Close-up view of the hot spot of TtGSALDH. Arg100 occupies the center of the hot spot and is essential for hexamer formation. (C) Close-up view of the hot spot of yeast GSALDH. Trp193 occupies the center of the hotspot and is essential for hexamer formation. Figure adapted from Luo et al. (73) and Pemberton et al. (91). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
SAXS and site-directed mutagenesis have been used to identify residues that are essential for hexamerization of GSALDH (73, 91). These studies identified a microenvironment within the major dimer–dimer interface that harbors interactions needed for hexamerization, that is, a hexamerization hot spot (Fig. 8A). Interestingly, the type of interactions within the hotspot is variable. In TtGSALDH and D. radiodurans GSALDH, the hot spot contains an Arg residue that forms electrostatic interactions with four residues from two other protomers (Fig. 8B). In yeast GSALDH, the hotspot contains a Trp that packs into a nonpolar hole formed by residues from another dimer in the hexamer (Fig. 8C). Mutation of either the Arg or Trp dramatically changes the oligomeric state from predominantly hexamer to predominantly dimer. Thus, although the hotspot occupies the same region of space in the two hexamers, the noncovalent interactions are fundamentally different.
The oligomerization of GSALDHs provides a counterexample to the sequence-structure paradigm of biochemistry. Simply stated, the idea is that sequence similarity implies structural similarity. The closer two proteins are in sequence, the more likely they will share a common three-dimensional structure. Paradoxically, global sequence identity and kingdom of life are poor predictors of the oligomeric state of GSALDH. For example, the Bacillus enzymes are 50% identical to TtGSALDH, and yet they form dimers in solution, whereas TtGSALDH is hexameric. The human and Bacillus enzymes are dimeric despite sharing only 30% identity. Furthermore, Put2p and human GSALDH are eukaryotic enzymes that have 42% identical amino acid sequences, yet Put2p is hexameric and human GSALDH is dimeric.
The failure of global sequence identity to predict the oligomeric state of GSALDH probably reflects the fact that the hexamer is stabilized by a hot spot, which by definition involves very few residues (15, 81). Prediction of oligomeric state thus requires structure-based analysis of local regions of the sequence within the hot spot rather than global alignment identities. This analysis is complicated by the fact that the chemical nature of the hot spot residue is variable, being Arg and Trp in the two cases that have been analyzed. More structures of GSALDHs will help to identify the full range of residues and interactions that stabilize the GSALDH hexamer, which will allow for better predictions of the oligomeric state and quaternary structure from sequence.
Kinetic studies of GSALDHs
The standard activity assay for ALDHs monitors the formation of NADH (ɛ340 = 6220 M−1 cm−1) (70, 84). The GSALDH kinetic parameters have been reported for two monofunctional GSALDHs (Table 2). Furthermore, a comprehensive study of the kinetics of human GSALDH has been reported (122). Some of the kinetic constants derived from global fitting of data to a Theorell-Chance mechanism are listed in Table 2. In the assumed kinetic scheme, NAD+ binds first, followed by GSAL, resulting in a ternary complex. Glu dissociates first, followed by NADH. Also, the enzyme is subject to substrate inhibition by P5C/GSAL (Ki = 112 μM). Release of NADH likely is the rate-limiting step (33, 122). The order of product dissociation means that NADH is bound to the enzyme during hydrolysis of the acyl-enzyme, which implies that the reduced nicotinamide recoils from the active site to allow the catalytic Glu and water of hydrolysis to gain access to the acyl-enzyme. The structural basis for this conformation change remains to be determined.
Table 2.
Steady-State Kinetic Parameters of Monofunctional l-Glutamate γ-Semialdehyde Dehydrogenases
Proline Utilization A
Classification of PutAs
PutAs can be classified according to domain architecture (type A, B, or C) and phylogeny (branch 1, 2, or 3) (Fig. 9). Type A PutAs have a minimal domain architecture consisting of N-terminal PRODH and C-terminal GSALDH modules (Fig. 9A). Type B PutAs have an additional C-terminal domain, named for its resemblance to domains in ALDHSF enzymes. Type C PutAs contain both the C-terminal ALDHSF domain and an N-terminal ribbon-helix-helix (RHH) DNA-binding domain. All three architectures are represented in branch 1 of the PutA phylogenetic tree, while branch 2 contains only type A PutAs, and branch 3 contains only type B PutAs (Fig. 9B). Organizing PutAs thus results in five classes, which are designated by a two-digit code consisting of the branch number and the domain architecture type: 1A, 1B, 1C, 2A, and 3B (Table 3).
FIG. 9.

Classification of PutAs according to domain architecture and global sequence identity. (A) The three domain architectures of PutAs. (B) Phylogenetic tree based on global sequence alignments of PutAs. PutAs with architecture types A, B, and C are indicated by black, blue, and red genus names, respectively. Large bold font denotes PutAs mentioned in the text. The alignments were calculated with Clustal Omega (116). The tree was made with DrawTree (27). The sequences used to make this tree are provided in Supplementary Data (Supplementary Data are available online at www.liebertpub.com/ars). PutAs, proline utilization A. Figure taken from Korasick et al. (51). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Table 3.
Oligomeric States and Quaternary Structures of Proline Utilization A
| PutA class | PDB code | Rg (Å) | Oligomeric state | Quaternary structure | |
|---|---|---|---|---|---|
| BjPutA | 1A | 3HAZ | 52 | Tetramer | Ring-shaped dimer of domain-swapped dimers |
| LpPutA | 1A | — | 47 | Dimer | Classic ALDH domain swapping |
| SmPutA | 1B | 5KF6-7 | 33/40 | Monomer/dimer | C-terminal ALDSF domain packs against the α- and GSALDH catalytic domains |
| EcPutA | 1C | — | 63 | Dimer | β-sheet from RHH domains |
| GsPutA | 2A | 4NMA-E, 4NM9 | 44 | Dimer | Classic ALDH domain swapping |
| BbPutA | 2A | 5UR2 | 46 | Dimer | Classic ALDH domain swapping |
| DvPutA | 2A | — | 46 | Dimer | Classic ALDH domain swapping |
| CfPutA | 3B | 5UX5 | 32/– | Monomer/dimer | Unknown |
ALDH, aldehyde dehydrogenase; ALDSF, aldehyde dehydrogenase superfamily; BbPutA, Bdellovibrio bacteriovorus proline utilization A; BjPutA, Bradyrhizobium japonicum proline utilization A; CfPutA, Corynebacterium freiburgense proline utilization A; DvPutA, Desulfovibrio vulgaris proline utilization A; EcPutA, Escherichia coli proline utilization A; GSALDH, l-glutamate γ-semialdehyde dehydrogenase; GsPutA, Geobacter sulfurreducens proline utilization A; LpPutA, Legionella pneumophila proline utilization A; PDB, Protein Data Bank; PutA, proline utilization A; RHH, ribbon-helix-helix; SmPutA, Sinorhizobium meliloti proline utilization A.
Major advances have been made in the structural biology of PutA since the author's 2008 review (128). Most notably, crystal structures of full-length PutAs representing four of the five PutA classes have been determined (Table 3). Only the structure of class 1C PutA remains unsolved, however; a high-quality model of EcPutA has been built using SAXS (118).
The PutA fold revealed from structures of type A PutAs
Crystal structures of three type A PutAs have been determined (Table 3): Bradyrhizobium japonicum PutA (BjPutA), GsPutA, and Bdellovibrio bacteriovorus PutA (BbPutA). These structures revealed the conserved fold of the PutA catalytic core (Fig. 10A). As expected, PutA contains (βα)8 barrel and ALDH folds that resemble monofunctional PRODH and GSALDH, respectively. The two active sites are separated by a linear distance of 41–45 Å and connected by a tunnel, which is used for channeling the intermediate P5C/GSAL. A unique feature of the PRODH barrel in PutA is an additional α-helix inserted between β5 and α5 of the barrel, which is not present in monofunctional PRODHs. Known as α5a, this helix contacts the FAD adenine and forms part of the substrate-channeling tunnel (Fig. 10B).
FIG. 10.

Structure of the class 2A PutA, GsPutA. (A) Ribbon drawing of the protomer (PDB code 4NM9). The pink surface represents the substrate-channeling tunnel. The domains are colored according to the legend. The dashes denote a disordered section of the linker domain. (B) Ribbon drawing of the dimer, with the two protomers colored green and blue. The pink surface represents the substrate-channeling tunnel. The inset shows a close-up view of the dimerization domain covering the substrate-channeling tunnel. Figure adapted from Singh et al. (117). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The structures also reveal that PutA is not a simple fusion of a PRODH to a GSALDH. The PRODH barrel and ALDH fold account for only 80–85% of the PutA polypeptide chain, while three smaller domains account for the remaining 15–20% (arm, α-domain, and PRODH-GSALDH linker, Fig. 10A). These ancillary domains help establish a precise spatial relationship between the two catalytic domains that is essential for substrate channeling.
The arm is located at the N-terminus of the polypeptide chain and consists of one to three α-helices that wrap around the PRODH barrel (Fig. 10A). The arm appears to act as a support structure for the U-shaped helical part of the PRODH-GSALDH linker. Branches 2 and 3 PutAs have only one α-helix in the arm (Fig. 10A). Branch 1 PutAs have longer arms: two α-helices in class 1A, and three α-helices in class 1B and 1C.
The α-domain connects the arm to the PRODH barrel (Fig. 10A). Although the secondary structure of the α-domain is conserved, its tertiary structure is not; the number and arrangement of α-helices differ among PutAs. Also, all PutA structures show some disorder in the α-domain, implying flexibility. In fact, the α-domain is completely disordered in the class 3B PutA from Corynebacterium freiburgense (CfPutA). Since the α-domain contacts the PRODH barrel and the GSALDH catalytic domain, it may be important for establishing the correct spacing between the two active sites. There is also evidence that the α domain is involved in membrane association (154, 155).
The third ancillary domain is formed by the peptide connecting the PRODH barrel to the NAD+-binding domain (“linker” in Fig. 10A). The linker is not a flexible tether but rather has a well-defined, conserved conformation. It starts at the C-terminus of α8 of the PRODH barrel and forms three α-helices arranged in a U-shape, which redirects the polypeptide chain toward the GSALDH Rossmann domain (Fig. 10A).
Oligomerization of type A PutAs: the core functional dimer
All type A PutAs form the classic ALDH domain-swapped dimer, in which the oligomerization domain of one protomer contacts the GSALDH catalytic domain of the opposite protomer (Fig. 10B). In addition, the oligomerization domain in type A PutA contacts the PRODH barrel, the PRODH-GSALDH linker, and the α-domain of the opposite protomer of the dimer. Through these quaternary structural contacts, the oligomerization domain helps fill the open space between the PRODH and GSALDH active sites (Fig. 10B). Importantly, a section of the substrate-channeling tunnel runs beneath the oligomerization domain (Fig. 10B). Thus, the oligomerization domain functions as a lid that helps sequester the intermediate in the tunnel. This is an example of quaternary structure playing an important role in the catalytic mechanism of an enzyme.
BjPutA differs from other type A PutA in that two of the domain-swapped dimers assemble into a ring-shaped dimer-of-dimers tetramer (121). The functional significance of this higher degree of oligomerization has been investigated using hot spot mutagenesis. Korasick et al. engineered a hot spot disruption mutant that cleanly produces dimeric BjPutA (51). The dimeric variant exhibited kinetic parameters similar to the wild-type enzyme, implying that tetramerization is not necessary for catalytic function in vitro. Furthermore, they showed that three other type A PutAs are domain-swapped dimers in solution, with no evidence for tetramer formation [BbPutA, Desulfovibrio vulgaris PutA (DvPutA), Legionella pneumophila PutA (LpPutA)] (Table 3). The conclusion of that study is that the domain-swapped dimer is the core structural and functional unit of type A PutAs.
The main substrate-channeling tunnel of PutA
The PutA structures reveal the structural basis of substrate channeling. The observation of a tunnel connecting the two active sites (Fig. 10A) strongly implies a substrate channeling mechanism. Indeed, kinetic measurements, such as transient time analysis, are consistent with substrate channeling (117, 121). Sophisticated approaches based on global fitting of steady- and transient-state kinetic measurements not only indicate a channeling mechanism but also show that the channeling step exhibits hysteresis (84), which is a form of allosteric regulation.
The physical characteristics of the substrate-channeling tunnel systems can be determined using software that finds and analyzes voids in protein structures (18). The substrate-channeling tunnel of PutA follows a curved, 75 Å long path (Fig. 11A). The central section of the tunnel has a length of ∼30 Å and a radius of 3.5–4.5 Å. The tunnel constricts near the two active sites. The central section runs parallel to two α-helices: α5a of the PRODH barrel and an α-helix of the GSALDH fold (770s helix in GsPutA and BjPutA, Fig. 10B inset). These helices help set the diameter and orientation of the central section. The tunnel is lined with charged and polar side chains, especially near the PRODH site and in the central section (Fig. S3 of (117)). Thus, the tunnel interior is a highly hydrophilic, which is appropriate for a zwitterionic intermediate.
FIG. 11.

The tunnel system of GsPutA. (A) Plot of the radius of the main tunnel of the resting enzyme (PDB code 4NM9) as a function of the distance from the flavin calculated using Mole (12). The locations of the ancillary tunnels are indicated. (B) Surface rendering of the tunnel system of the resting enzyme. Figure adapted from Singh et al. (117). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Tunnel blocking mutagenesis is an effective way to explore the functions of tunnels in proteins. In this method, one uses site-directed mutagenesis to increase the steric bulk of a side chain near the tunnel of interest. This method has been applied to the main tunnel of BjPutA. Arentson et al. targeted residues on the two helices that border the main tunnel in BjPutA (4). Mutating Asp779 to Tyr or Trp significantly decreased the overall rate of the PRODH-GSALDH channeling reaction. The crystal structures of D779Y and D779W revealed that the large side chains constrict in the central section of the tunnel, which impedes the channeling path of P5C/GSAL. These results provided additional evidence that the tunnel observed in the crystal structure is used for substrate channeling.
Ancillary tunnels of PutA
PutA in the resting state with the oxidized FAD contains six smaller tunnels that connect the main one to the bulk medium (Fig. 11B). The functions of these ancillary tunnels are unknown, but the GsPutA structures provide some hypotheses to test (117). For example, tunnel 2a closes when the proline analog THFA binds. This tunnel becomes blocked as a consequence of the conformational changes that occur when the ion pair gate closes (Fig. 3C). These observations suggest that tunnel 2a is the entryway for proline. Modeling studies suggest that tunnel 1, which flows along the FAD, may be used by quinones to access the reduced flavin. The proximity of tunnel 4 to the GSALDH site suggests the idea that it could be the escape route for the product Glu (Fig. 11B). Finally, the two waters of hydrolysis required per round of catalysis could enter the main tunnel via the ancillary tunnels; tunnels 3a and 3b perhaps fulfill this function.
The structures of type B PutAs reveal the fold and functions of the C-terminal domain
Architecture types B and C PutAs contain a C-terminal domain not found in type A PutAs (Fig. 9A). The function of this 100–200 residue domain has been an intriguing mystery of PutA biochemistry. Sequence analysis shows that all the residues needed for catalytic activity are present in the PRODH and GSALDH modules, which argues against the domain playing a direct role in catalysis. Homology modeling predicted that the C-terminal domain adopts a Rossmann-like fold similar to that found in ALDHs (70, 119), which suggested a role in NAD+ binding. However, this function was unlikely, since homology modeling and sequence alignments suggested that the Rossmann fold domain in the GSALDH module of type B and C PutAs is fully functional, and only one equivalent of NAD+ is required for the PutA catalytic cycle. Thus, the function of the C-terminal domain remained enigmatic for many years, until crystal structures were determined for the class 1B PutA from Sinorhizobium meliloti [SmPutA (71)] and the class 3B PutA, CfPutA (50).
The structures of type B PutAs revealed the fold of the C-terminal domain. As shown for SmPutA, the C-terminal domain consists of an α/β domain with a protruding β-flap (Fig. 12A). The α/β domain exhibits the Rossmann dinucleotide-binding fold and thus resembles the NAD+-binding domain of the GSALDH module. The β-flap resembles the oligomerization domain found in all other ALDHSF enzyme structures reported to date, including type A PutAs. Because of these structural similarities, the PutA C-terminal domain is known as the C-terminal ALDHSF domain. Paradoxically, the Rossmann fold of the C-terminal domain does not bind NAD+, nor does the β-flap of the C-terminal domain mediate oligomerization.
FIG. 12.

The structure of the type B PutA SmPutA (PDB code 5KF6). (A) Structure of the C-terminal ALDHSF domain, along with a topology diagram of the Rossmann subdomain. (B) Structure of the protomer. The arm, α-domain, PRODH barrel, and linker are colored cyan (“PRODH module”). The GSALDH NAD+-binding and catalytic domains are colored red and blue, respectively. The C-terminal ALDHSF domain is colored gold. The pink surface represents the substrate-channeling tunnel. The asterisks denote the active sites. (C) Cartoon and surface representations of the dimer. On the left, the domains are colored as in (B). On the right, the two chains have different colors. (D) The separated protomers of the SmPutA dimer. The interaction surfaces are color coded according to modules/domains as in (B) PRODH module, cyan; NAD+ binding, red; GSALDH catalytic, blue; C-terminal ALDHSF, gold. Figure adapted from Luo et al. (71). SmPutA, Sinorhizobium meliloti proline utilization A. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The SmPutA and CfPutA structures show the tertiary structural interactions formed by the C-terminal ALDHSF domain, which provides insight into function (Fig. 12B). The catalytic core described above for type A PutA is conserved in type B PutA, as is the substrate-channeling tunnel. For ease of presentation, all the domains of the PRODH module—arm, α-domain, PRODH barrel, and linker—have the same color in Figure 12B. The Rossmann part of the C-terminal ALDHSF domain packs against the Rossmann NAD+-binding domain of the GSALDH module, while the β-flap of the C-terminal domain contacts the GSALDH catalytic domain (Fig. 12B). These tertiary structural relationships mimic the quaternary structure of type A PutAs. The interaction of the β-flap of the C-terminal domain with the GSALDH catalytic domain helps stabilize the aldehyde binding site. Thus, the C-terminal domain contributes indirectly to GSALDH activity, a conclusion that is supported by domain deletion analysis (50). Also, the β-flap of the C-terminal ALDHSF domain covers the substrate-channeling tunnel, again mimicking the quaternary structural interactions of the oligomerization domain of type A PutAs. Thus, the C-terminal domain plays an important role in coupling the two catalytic activities by helping to sequester the intermediate in the tunnel.
Type B PutAs form monomer–dimer equilibria in solution. For example, SAXS shows that SmPutA is primarily monomeric at a concentration of ∼1 mg/mL (8 μM), but a substantial population of dimers is observed at a higher enzyme concentrations (71). Dimerization of CfPutA was studied in more detail using analytical ultracentrifugation, revealing a more complex self-association behavior (50). Dimeric CfPutA was observed only when the active site ligands THFA and NAD+ were present, suggesting that ligand binding enhances self-association. This result is consistent with the morpheein model of enzyme hysteresis, in which substrate binding induces conformational changes that promote assembly of a high-activity oligomer (114). Whether other PutAs exhibit ligand-induced self-association is unknown.
Although type B PutAs dimerize, they do not form the classic domain-swapped dimer described above for type A PutAs. Recall that dimerization of type A PutA is mediated by a β-flap that protrudes from the GSALDH module (Fig. 10B). In type B PutA, this flap has been transplanted to the C-terminal ALDHSF domain, essentially converting quaternary structure into tertiary structure. Consequently, type B PutAs form a novel dimer, which has been characterized for SmPutA using a combination of SAXS and crystal structures (Fig. 12C).
SmPutA forms a symmetric and roughly spherical dimer with a radius of gyration of 40 Å and maximum dimension of 120 Å (Fig. 12C). The dimer interface buries 1800 Å2 of surface area from each chain. The twofold axis of the dimer runs next to Leu1147 of the C-terminal ALDHSF domain. A major part of the interface is formed by the packing of the first two α-helices of the C-terminal ALDHSF domain against the α- and GSALDH catalytic domains (Fig. 12D). The quaternary structure of the SmPutA dimer is different from all other ALDHSF members.
Class 1C (trifunctional) PutAs
Class 1C PutAs are unique in that they function as transcriptional repressors as well as bifunctional enzymes. Accordingly, the term “trifunctional” is sometimes used for 1C PutA. 1C PutAs are important in the history of PutA biochemistry. The 1C PutA from Salmonella typhimurium was the first PutA discovered (75, 77–79). Shortly thereafter, Wood's group characterized the 1C PutA, EcPutA (21, 22, 63, 142). Substrate channeling was first observed in trifunctional S. typhimurium PutA (125). These pioneering studies established three key features of trifunctional PutA: (i) the PRODH and GSALDH catalytic activities are combined into a single polypeptide chain, (ii) PutA is a membrane-associated enzyme, and (iii) PutA represses transcription of the put operon, which contains the putA gene and the gene encoding the proline transporter PutP. Today, EcPutA remains the best characterized PutA, mainly as a result of research from Becker's group (6, 10, 37, 83–85, 118, 124, 134, 150, 151, 153–157). Also, EcPutA and its domains have been used for structural studies, including the first structure of any PRODH (59, 124) and the first crystal structure of the PutA DNA-binding domain (57, 152).
1C PutAs are transcriptional repressors and therefore have a DNA-binding domain. The crystal structure of an EcPutA construct containing residues 1–669 suggested that the DNA-binding domain is located in the α-domain (residues 140–260) (59). Later studies disproved this hypothesis. In particular, sequence analysis predicted an RHH domain in EcPutA residues 1–50, and a domain construct consisting of EcPutA residues 1–47 was shown to bind the put intergenic DNA (37). A conserved Lys residue near the N-terminus is a key diagnostic sequence feature of the RHH in PutA (Lys9 in EcPutA). The RHH fold was subsequently confirmed by crystallography (57) and NMR (40) of 1C PutA DNA-binding domain constructs. Finally, the structure of the EcPutA RHH domain complexed with operator DNA was determined (152). These studies established that residues 1–50 of 1C PutAs contain an RHH DNA-binding domain.
Although a crystal structure of a full-length 1C PutA is not available, structural information has been obtained from SAXS, molecular dissection, and light scattering (118). EcPutA is dimeric in solution with a radius of gyration (Rg) of 63 Å. For comparison, dimeric and tetrameric type A PutAs have Rg values of 46 and 52 Å, respectively, and the SmPutA dimer has Rg of 40 Å (Table 3). SAXS shape reconstruction calculations reveal that EcPutA forms an elongated V-shaped particle having dimensions of 205 by 85 by 55 Å (Fig. 13). The particle consists of two large lobes connected by a 30-Å diameter cylinder. The maximum dimension of EcPutA far exceeds those of type A (140–150 Å) and type B (120 Å) PutAs. Thus, trifunctional PutA is the largest protein in the PutA family.
FIG. 13.

SAXS model of the 1C PutA EcPutA. The surface represents the SAXS shape reconstruction. The DNA-binding domain is colored orange (RHH dimer). The catalytic cores are colored as in Figure 12B: PRODH module, cyan; NAD+ binding, red; GSALDH catalytic, blue; C-terminal ALDHSF, gold. RHH, ribbon-helix-helix; SAXS, small-angle X-ray scattering. Figure adapted from (118). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
SAXS rigid body modeling has been used to generate models of the EcPutA dimer (118). The current working model shows the RHH domain mediating dimerization at the center of the particle, while the catalytic domains occupy the large outer lobes of the envelope (Fig. 13). The tertiary structure within the outer lobe likely resembles an SmPutA monomer. DNA is predicted to bind in a groove lined by the α-domains. Because there is evidence for the α-domain contacting the membrane (154, 155), the model implies that the DNA-binding and membrane-association interfaces are located on the same face of PutA. This led to the idea of a cloaking mechanism of gene regulation through which interaction of PutA with the membrane hides the DNA-binding surface from the put regulon, thereby activating transcription (118).
Kinetic studies of PutAs
The activity assays described above for PRODH and GSALDH can be used to measure the individual catalytic activities of PutA. Steady-state kinetic parameters for several PutAs have been determined (Table 4 and references therein).
Table 4.
Steady-State Kinetic Parameters for the Proline Dehydrogenase and l-Glutamate γ-Semialdehyde Dehydrogenase Activities of Proline Utilization A
| PRODH (variable substrate is Pro) | GSALDH (variable substrate is P5C) | |||||
|---|---|---|---|---|---|---|
| PutA | kcat (s−1) | Km (mM) | kcat/Km (M−1·s−1) | kcat (s−1) | Km (μM) | kcat/Km (M−1·s−1) |
| BjPutAa | 2 | 31 | 65 | 2.2 | 200 | 11,000 |
| BjPutAb | 5.6 | 150 | 37 | |||
| BjPutAc | 3.1 | 43 | 72 | 3.4 | 420 | 8100 |
| RcPutAd | 20.7 | 54.3 | 383 | 7.3 | 1530 | 4800 |
| RcPutAe | 1.0 | 5.6 | 179 | |||
| EcPutAf | 5.2 | 42 | 124 | 5.16 | 2000 | 2600 |
| EcPutAg | 12 | 100 | 120 | |||
| EcPutAg | 6.7 | 230 | 29 | |||
| EcPutAh | 0.43 | 1.5 | 287 | |||
| GsPutAi | 0.67 | 89 | 7 | 7.5 | 35 | 214,000 |
| HpPutAj | 8 | 146 | 56 | |||
| CfPutAk | 2.6 | 145 | 18 | 1.7 | 54 | 32,000 |
Proline parameters were determined at 23°C with CoQ1 fixed at 100 μM (121). The P5C parameters were determined at saturating NAD+ concentration.
DCPIP assay (52).
CoQ1 fixed at 250 μM as the electron acceptor in the PRODH assays (4).
Proline parameters were determined with [DCPIP] fixed at 75 μM (70). The P5C parameters were determined with [NAD+] fixed at 200 μM.
CoQ1 as the electron acceptor (3).
Proline parameters were determined by fitting to a two-site ping-pong mechanism with CoQ1 as the electron acceptor (85). The P5C parameters were obtained by fitting to an ordered ternary mechanism (84). kcat for the coupled PRODH-GSALDH reaction from Moxley et al. (84).
Parameters for proline were estimated from the oAB assay with membrane vesicles providing the electron acceptor for the FAD (85).
Proline parameters were determined with menadione fixed at 100 μM (117). The P5C parameters were determined with [NAD+] fixed at 200 μM.
DCPIP assay (53).
PRODH parameters were determined from the DCPIP assay (50).
oAB, o-aminobenzaldehyde; DCPIP, 2,6-dichlorophenolindophenol; FAD, flavin adenine dinucleotide; HpPutA, Helicobacter pylori proline utilization A; PRODH, proline dehydrogenase; RcPutA, Rhodobacter capsulatus proline utilization A.
Moxley and Becker used stopped-flow methods to study the proline:ubiquinone oxidoreductase reaction of EcPutA (83). The microscopic rates obtained with CoQ1 as the electron acceptor are consistent with a two-site ping-pong mechanism for the overall reaction. The spectroscopic signature of the FAD semiquinone was not observed, which is consistent with a hydride transfer mechanism in the oxidation of proline. The oxidative half-reaction is the rate-limiting step for kcat during catalytic turnover with CoQ1.
Kinetic measurements are also used to study substrate channeling in PutAs. Becker's group has reviewed this topic (5). The review by Anderson also has a section on the methods used to study substrate channeling (2). Here the approaches that have been used for PutAs are described.
Transient time analysis often is the first assay used to assess substrate channeling in PutAs. The time dependence of NADH production is monitored in an assay containing proline, an electron acceptor for the FAD, and NAD+. The experimental progress curve is compared to a theoretical one calculated from a free-diffusion (nonchanneling) model. This model assumes that P5C/GSAL is released to the bulk medium before binding to the GSALDH site. The transient time (τ) is defined as the lag time that precedes the attainment of a steady-state concentration of the intermediate (2). For the free-diffusion model, τ equals Km/Vmax of the second enzyme, that is, Km/Vmax of the GSALDH domain for P5C. Substrate channeling is indicated when the observed transient time is shorter than the one predicted from the model. This test has been used to obtain evidence for substrate channeling in BjPutA (121), GsPutA (117), Rhodobacter capsulatus proline utilization A (RcPutA) (70), SmPutA (71), and EcPutA (84).
It is also useful to perform transient time analysis on a nonchanneling control. Becker's group devised a clever control system for PutAs, known as the “mixed variants” (5). Two site-directed mutant PutAs are generated, one lacking PRODH activity and another lacking GSALDH activity. Mutating a conserved Arg residue of α8 eliminates PRODH activity (Fig. 2, lower inset). The GSALDH-deficient mutant has the catalytic Cys mutated to Ala. The transient time assay is then performed using an equimolar mixture of the two variants. Note that the mixed variants exhibit wild-type PRODH and GSALDH activities, however, functional PRODH and GSALDH active sites are not connected by a tunnel. Thus, P5C/GSAL generated in a PRODH active site must be released into the bulk medium before binding to a functional GSALDH site. Substrate channeling is indicated when the transient time for the wild-type PutA is shorter than that of the mixed variants.
Trapping of the intermediate is also used to assess substrate channeling. oAB is used to trap P5C/GSAL as described in the section on PRODH assays. Typically, the time dependence of P5C/GSAL generation is measured in the absence and presence of NAD+. Channeling is suggested when the amount of the intermediate detected decreases when NAD+ is included. This assay has been used to study BjPutA (121) and CfPutA (50).
The steady-state kinetic parameters for the coupled PRODH-GSALDH reaction are obtained by monitoring NADH using an assay containing proline, an electron acceptor for the FAD, and NAD+. The initial rates are recorded as a function of varying the concentration of proline or NAD+. The kinetic parameters for the coupled reaction have been measured for a few PutAs (Table 5).
Table 5.
Steady-State Kinetic Parameters for the Coupled Proline Dehydrogenase-l-Glutamate γ-Semialdehyde Dehydrogenase Reaction of Proline Utilization A
| PutA | kcat (s−1) | Km (mM) | kcat/Km (M−1·s−1) | Ki (mM) |
|---|---|---|---|---|
| BjPutA (121) | 0.5 | 24 | 21 | |
| BjPutA (4)a | 0.49 | 56 | 9 | 24 |
| SmPutA (71)a | 1.6 | 7 | 230 | 263 |
| EcPutA (84) | 0.73 | 20.8 | 35 | 83 |
Proline is the variable substrate.
Data were fit to a substrate inhibition model.
Finally, Becker's group has developed a sophisticated approach to determine how substrate channeling fits into the entire kinetic mechanism of PutA (84). This approach involves global analysis of steady- and transient-state kinetic data for the PRODH, GSALDH, and coupled PRODH-GSALDH reactions. The mechanism is quite complicated because each individual activity has two substrates, and proline is an inhibitor of GSALDH. A major result of this work is that the substrate-channeling step is rate limiting in the overall PRODH-GSALDH reaction. Furthermore, this step is slowest during the first turnover and speeds up 40-fold with subsequent turnovers. These results imply that substrate channeling in PutA is an activated process.
Future Challenges
Unsolved structural targets
Much progress has been made in the structural biology of proline catabolism, yet many challenges remain. The unsolved crystallography targets present opportunities for future research (Table 6). Among the monofunctional enzymes, there are no structures of eukaryotic PRODHs. Although the structures of bacterial PRODHs provide a good template for modeling the (βα)8 barrel, eukaryotic PRODHs have an N-terminal domain of unknown function. This domain has low sequence conservation among eukaryotic PRODHs and is without a homologue in the PDB. Furthermore, PRODH has emerged as a cancer drug target, and the structure of human PRODH would aid inhibitor discovery (31, 65).
Table 6.
Outstanding Proline Catabolism Structural Biology Targets
| Target | Significance |
|---|---|
| Eukaryotic PRODH | Cancer drug design target |
| PRODH complexed with ubiquinone | No structure of any PRODH or PutA complexed with a bona fide biological quinone; provides information about membrane association. |
| GSALDH complexed with P5C/GSAL | Addresses the question of whether GSALDH directly binds GSAL or converts P5C to GSAL in the active site |
| 1C PutA | The most complex of all PutAs; provides insight into the structural basis of transcriptional regulation and functional switching |
| 3B PutA dimer | Potentially unique dimer induced by ligand binding |
| PutA with P5C/GSAL in the tunnel | Determine the identity of the channeled species; reveal the roles of tunnel residues in facilitating channeling |
| PRODH-GSALDH protein–protein complex | Structural basis of PRODH-GSALDH intermolecular substrate channeling |
GSAL, l-glutamate-γ-semialdehyde.
The structure of a PRODH complexed with a bona fide quinone (e.g., ubiquinone) would be useful. Although the GsPutA-MB structure was a landmark result in that it provided the first evidence that the electron acceptor interacts directly with the FAD isoalloxazine, it is suboptimal because MB is a small water-soluble quinone that lacks an isoprenoid chain. A structure of PRODH or PutA complexed with ubiquinone would provide insight into how the enzyme interacts with the membrane.
Crystal structures have been determined for four of the five PutA classes (Table 3). Only trifunctional (class 1C) remains unsolved in its full-length form. The structure of trifunctional PutA complexed with DNA is needed to understand the structural basis of functional switching, that is, the mechanism by which 1C PutA switches between repressor and enzymatic functions (153).
Oligomeric states and quaternary structures of PutAs
PutAs exhibit an unexpectedly rich diversity of oligomeric state and quaternary structure. Three different dimers and a tetramer have been observed so far (Table 3). These oligomers represent different structural solutions to the problem of protecting the intermediates of proline catabolism. An outstanding question in this area concerns the quaternary structure of class 3B PutA. One class 3B PutA—CfPutA—exhibits ligand-induced dimerization, however, whether this dimer is the same one formed by the class 1B SmPutA is unknown. Also, it remains to be determined whether other PutAs also exhibit ligand-induced self-association and if this phenomenon regulates catalytic activity.
Substrate channeling
Substrate channeling is another area that is ripe for structural biology research. The functions of the ancillary tunnels of PutA are unknown. Tunnel blocking mutagenesis could be used to address this question.
The spatiotemporal details of P5C hydrolysis in PutA are unknown. Three limiting possibilities can be imagined: early, late, and middle. The “early” hypothesis refers to hydrolysis occurring in the PRODH site, which implies that GSAL is the channeled species. This hypothesis is supported by the observation of a conserved water molecule in the PRODH active site that appears to be poised to attack the newly formed P5C (Fig. 3A, B). “Late” hydrolysis means that P5C is the channeled intermediate and hydrolysis occurs in the GSALDH site. This hypothesis is consistent with the fact that the P5C/GSAL equilibrium favors P5C at physiological pH (9). The third option is that P5C is hydrolyzed in transit, possibly in the middle section of the main tunnel. Creative ways to trap intermediate states of PutA in crystallo are needed.
A related goal is to determine a structure of monofunctional GSALDH complexed with P5C/GSAL. This structure addresses the question of whether GSALDH directly binds GSAL or converts P5C to GSAL in the active site.
A novel aspect of PutA is that substrate channeling is an activated process (84). The substrate-channeling step of EcPutA is slowest during the first turnover and speeds up 40-fold with subsequent turnovers. This result implies that channeling is activated and implicates PutA as a new example of a hysteretic enzyme (34, 35). The structural basis for activation is unknown. Frieden proposed several causes of enzyme hysteresis, including isomerization, inhibitory ligand displacement, and oligomerization (35). The prevailing hypothesis for PutA is that isomerization associated with ligand binding (most likely proline) provokes conformational changes that assemble the PRODH active site and the tunnel system (84). Such isomerization may seal leaky parts of the tunnel system. This idea is based on structures of GsPutA showing that the binding of a proline analog is accompanied by remodeling of the tunnel system near the PRODH active site, and reduction of the FAD appears to close tunnel 4 near the GSALDH site (117). Capturing the activated enzyme in crystallo is a major goal. This could be achieved with kinetic crystallography approaches, in which combinations of substrates are diffused into the crystal followed by flash-cooling to trap intermediate states (17).
Finally, a recent study demonstrated for the first time that monofunctional PRODH and GSALDH physically interact and engage in substrate channeling (111). The structural basis of this interaction is a new challenge for structural biologists. While a crystal structure of the complex is the ultimate goal, lower resolution structural information may be within grasp. A model of the complex has been generated based on analogy to PutA (111). In this model, six copies of PRODH interact with a hexamer of GSALDH. Confirming the mass and stoichiometry of the complex would be useful. The interfaces in the model could be tested with site-directed mutagenesis. Finally, a low-resolution model of the complex may be achievable with SAXS.
Supplementary Material
Acknowledgment
Research reported in this publication was supported by the NIGMS of the National Institutes of Health under award numbers GM065546 and GM061068.
Abbreviations Used
- γ-GPR
γ-glutamate phosphate reductase
- ALDH
aldehyde dehydrogenase
- ALDHSF
aldehyde dehydrogenase superfamily
- BbPutA
Bdellovibrio bacteriovorus proline utilization A
- BjPutA
Bradyrhizobium japonicum proline utilization A
- CfPutA
Corynebacterium freiburgense proline utilization A
- CoQ1
coenzyme Q1
- DCPIP
2,6-dichlorophenolindophenol
- DrPRODH
Deinococcus radiodurans proline dehydrogenase
- DvPutA
Desulfovibrio vulgaris proline utilization A
- EcPutA
Escherichia coli proline utilization A
- FAD
flavin adenine dinucleotide
- G5K
glutamate 5-kinase
- GSAL
l-glutamate-γ-semialdehyde
- GSALDH
l-glutamate γ-semialdehyde dehydrogenase
- GsPutA
Geobacter sulfurreducens proline utilization A
- IUBMB
International Union of Biochemistry and Molecular Biology
- LpPutA
Legionella pneumophila proline utilization A
- MB
menadione bisulfite
- oAB
o-aminobenzaldehyde
- OAT
ornithine δ-aminotransferase
- ORN
ornithine
- P5C
Δ1-pyrroline-5-carboxylate
- P5CDH
Δ1-pyrroline-5-carboxylate dehydrogenase
- P5CS
P5C synthase
- PDB
Protein Data Bank
- PRODH
proline dehydrogenase
- PutA
proline utilization A
- RcPutA
Rhodobacter capsulatus proline utilization A
- RHH
ribbon-helix-helix
- ROS
reactive oxygen species
- SAXS
small-angle X-ray scattering
- SmPutA
Sinorhizobium meliloti proline utilization A
- THFA
l-tetrahydrofuroic acid
- TIM
triosephosphate isomerase
- TtGSALDH
Thermus thermophilus l-glutamate γ-semialdehyde dehydrogenase
References
- 1. Adams E. and Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem 49: 1005–1061, 1980 [DOI] [PubMed] [Google Scholar]
- 2. Anderson KS. Fundamental mechanisms of substrate channeling. Methods Enzymol 308: 111–145, 1999 [DOI] [PubMed] [Google Scholar]
- 3. Arentson BW, Hayes EL, Zhu W, Singh H, Tanner JJ, and Becker DF. Engineering a trifunctional proline utilization A chimaera by fusing a DNA-binding domain to a bifunctional PutA. Biosci Rep 36: e00413, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arentson BW, Luo M, Pemberton TA, Tanner JJ, and Becker DF. Kinetic and structural characterization of tunnel-perturbing mutants in Bradyrhizobium japonicum proline utilization A. Biochemistry 53: 5150–5161, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arentson BW, Sanyal N, and Becker DF. Substrate channeling in proline metabolism. Front Biosci 17: 375–388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baban BA, Vinod MP, Tanner JJ, and Becker DF. Probing a hydrogen bond pair and the FAD redox properties in the proline dehydrogenase domain of Escherichia coli PutA. Biochim Biophys Acta 1701: 49–59, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, Corran PH, Furth AJ, Milman JD, Offord RE, Priddle JD, and Waley SG. Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 angstrom resolution using amino acid sequence data. Nature 255: 609–614, 1975 [DOI] [PubMed] [Google Scholar]
- 8. Baumgartner MR, Rabier D, Nassogne MC, Dufier JL, Padovani JP, Kamoun P, Valle D, and Saudubray JM. Delta1-pyrroline-5-carboxylate synthase deficiency: neurodegeneration, cataracts and connective tissue manifestations combined with hyperammonaemia and reduced ornithine, citrulline, arginine and proline. Eur J Pediatr 164: 31–36, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Bearne SL. and Wolfenden R. Glutamate gamma-semialdehyde as a natural transition state analogue inhibitor of Escherichia coli glucosamine-6-phosphate synthase. Biochemistry 34: 11515–11520, 1995 [DOI] [PubMed] [Google Scholar]
- 10. Becker DF. and Thomas EA. Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry 40: 4714–4721, 2001 [DOI] [PubMed] [Google Scholar]
- 11. Ben Rejeb K, Abdelly C, and Savoure A. How reactive oxygen species and proline face stress together. Plant Physiol Biochem 80: 278–284, 2014 [DOI] [PubMed] [Google Scholar]
- 12. Berka K, Hanak O, Sehnal D, Banas P, Navratilova V, Jaiswal D, Ionescu CM, Svobodova Varekova R, Koca J, and Otyepka M. MOLEonline 2.0: interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res 40: W222–W227, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berney M, Weimar MR, Heikal A, and Cook GM. Regulation of proline metabolism in mycobacteria and its role in carbon metabolism under hypoxia. Mol Microbiol 84: 664–681, 2012 [DOI] [PubMed] [Google Scholar]
- 14. Bissbort SH, Vermaak WJ, Elias J, Bester MJ, Dhatt GS, and Pum JK. Novel test and its automation for the determination of erythrocyte acetylcholinesterase and its application to organophosphate exposure. Clin Chim Acta 303: 139–145, 2001 [DOI] [PubMed] [Google Scholar]
- 15. Bogan AA. and Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol 280: 1–9, 1998 [DOI] [PubMed] [Google Scholar]
- 16. Bottoms CA, Smith PE, and Tanner JJ. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci 11: 2125–2137, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bourgeois D. and Royant A. Advances in kinetic protein crystallography. Curr Opin Struct Biol 15: 538–547, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Brezovsky J, Chovancova E, Gora A, Pavelka A, Biedermannova L, and Damborsky J. Software tools for identification, visualization and analysis of protein tunnels and channels. Biotechnol Adv 31: 38–49, 2013 [DOI] [PubMed] [Google Scholar]
- 19. Bringaud F, Barrett MP, and Zilberstein D. Multiple roles of proline transport and metabolism in trypanosomatids. Front Biosci (Landmark Ed) 17: 349–374, 2012 [DOI] [PubMed] [Google Scholar]
- 20. Bringaud F, Riviere L, and Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol 149: 1–9, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Brown ED. and Wood JM. Conformational change and membrane association of the PutA protein are coincident with reduction of its FAD cofactor by proline. J Biol Chem 268: 8972–8979, 1993 [PubMed] [Google Scholar]
- 22. Brown ED. and Wood JM. Redesigned purification yields a fully functional PutA protein dimer from Escherichia coli. J Biol Chem 267: 13086–13092, 1992 [PubMed] [Google Scholar]
- 23. Cheng Z, Lin M, and Rikihisa Y. Ehrlichia chaffeensis proliferation begins with NtrY/NtrX and PutA/GlnA upregulation and CtrA degradation induced by proline and glutamine uptake. mBio 5: e02141, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clelland CL, Read LL, Baraldi AN, Bart CP, Pappas CA, Panek LJ, Nadrich RH, and Clelland JD. Evidence for association of hyperprolinemia with schizophrenia and a measure of clinical outcome. Schizophr Res 131: 139–145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Curtis J, Shearer G, and Kohl DH. Bacteriod proline catabolism affects N2 fixation rate of drought-stressed soybeans. Plant Physiol 136: 3313–3318, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeLano WL. The PyMOL User's Manual. Palo Alto, CA: DeLano Scientific, 2002 [Google Scholar]
- 27. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, and Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36: W465–W469, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, and Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res 61: 1810–1815, 2001 [PubMed] [Google Scholar]
- 29. Drew LJ, Crabtree GW, Markx S, Stark KL, Chaverneff F, Xu B, Mukai J, Fenelon K, Hsu PK, Gogos JA, and Karayiorgou M. The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int J Dev Neurosci 29: 259–281, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Efron ML. Familial hyperprolinemia. Report of a second case, associated with congenital renal malformations, hereditary hematuria and mild mental retardation, with demonstration of an enzyme defect. N Engl J Med 272: 1243–1254, 1965 [DOI] [PubMed] [Google Scholar]
- 31. Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grunewald TGP, and Fendt SM. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun 8: 15267, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fichman Y, Gerdes SY, Kovacs H, Szabados L, Zilberstein A, and Csonka LN. Evolution of proline biosynthesis: enzymology, bioinformatics, genetics, and transcriptional regulation. Biol Rev Camb Philos Soc 90: 1065–1099, 2015 [DOI] [PubMed] [Google Scholar]
- 33. Forte-McRobbie C. and Pietruszko R. Human glutamic-gamma-semialdehyde dehydrogenase. Kinetic mechanism. Biochem J 261: 935–943, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frieden C. Kinetic aspects of regulation of metabolic processes. The hysteretic enzyme concept. J Biol Chem 245: 5788–5799, 1970 [PubMed] [Google Scholar]
- 35. Frieden C. Slow transitions and hysteretic behavior in enzymes. Annu Rev Biochem 48: 471–489, 1979 [DOI] [PubMed] [Google Scholar]
- 36. Geraghty MT, Vaughn D, Nicholson AJ, Lin WW, Jimenez-Sanchez G, Obie C, Flynn MP, Valle D, and Hu CA. Mutations in the delta1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum Mol Genet 7: 1411–1415, 1998 [DOI] [PubMed] [Google Scholar]
- 37. Gu D, Zhou Y, Kallhoff V, Baban B, Tanner JJ, and Becker DF. Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J Biol Chem 279: 31171–31176, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hagedorn CH. and Phang JM. Catalytic Transfer of hydride ions from NADPH to oxygen by the interconversion of proline to delta-pyrroline-5-carboxylate. Arch Biochem Biophys 248: 166–174, 1986 [DOI] [PubMed] [Google Scholar]
- 39. Hagedorn CH. and Phang JM. Transfer of reducing equivalents into mitochondria by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch Biochem Biophys 225: 95–101, 1983 [DOI] [PubMed] [Google Scholar]
- 40. Halouska S, Zhou Y, Becker DF, and Powers R. Solution structure of the Pseudomonas putida protein PpPutA45 and its DNA complex. Proteins 75: 12–27, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hayat S, Hayat Q, Alyemeni MN, Wani AS, Pichtel J, and Ahmad A. Role of proline under changing environments: a review. Plant Signal Behav 7: 1456–1466, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu CA, Bart Williams D, Zhaorigetu S, Khalil S, Wan G, and Valle D. Functional genomics and SNP analysis of human genes encoding proline metabolic enzymes. Amino Acids 35: 655–664, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huijbers MM, Martinez-Julvez M, Westphal AH, Delgado-Arciniega E, Medina M, and van Berkel WJ. Proline dehydrogenase from Thermus thermophilus does not discriminate between FAD and FMN as cofactor. Sci Rep 7: 43880, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inagaki E, Ohshima N, Sakamoto K, Babayeva ND, Kato H, Yokoyama S, and Tahirov TH. New insights into the binding mode of coenzymes: structure of Thermus thermophilus [delta]1-pyrroline-5-carboxylate dehydrogenase complexed with NADP+. Acta Cryst F63: 462–465, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Inagaki E, Ohshima N, Takahashi H, Kuroishi C, Yokoyama S, and Tahirov TH. Crystal structure of Thermus thermophilus delta1-pyrroline-5-carboxylate dehydrogenase. J Mol Biol 362: 490–501, 2006 [DOI] [PubMed] [Google Scholar]
- 46. Jacquet H, Demily C, Houy E, Hecketsweiler B, Bou J, Raux G, Lerond J, Allio G, Haouzir S, Tillaux A, Bellegou C, Fouldrin G, Delamillieure P, Menard JF, Dollfus S, D'Amato T, Petit M, Thibaut F, Frebourg T, and Campion D. Hyperprolinemia is a risk factor for schizoaffective disorder. Mol Psychiatry 10: 479–485, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Kawakami R, Satomura T, Sakuraba H, and Ohshima T. l-Proline dehydrogenases in hyperthermophilic archaea: distribution, function, structure, and application. Appl Microbiol Biotechnol 93: 83–93, 2012 [DOI] [PubMed] [Google Scholar]
- 48. Kohl DH, Schubert KR, Carter MB, Hagedorn CH, and Shearer G. Proline metabolism in N2-fixing root nodules: energy transfer and regulation of purine synthesis. Proc Natl Acad Sci U S A 85: 2036–2040, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, and Vasiliou V. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev 64: 520–539, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Korasick DA, Gamage TT, Christgen S, Stiers KM, Beamer LJ, Henzl MT, Becker DF, and Tanner JJ. Structure and characterization of a class 3B proline utilization A: ligand-induced dimerization and importance of the C-terminal domain for catalysis. J Biol Chem 292: 9652–9665, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Korasick DA, Singh H, Pemberton TA, Luo M, Dhatwalia R, and Tanner JJ. Biophysical investigation of type A PutAs reveals a conserved core oligomeric structure. FEBS J 284: 3029–3049, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krishnan N. and Becker DF. Characterization of a bifunctional PutA homologue from Bradyrhizobium japonicum and identification of an active site residue that modulates proline reduction of the flavin adenine dinucleotide cofactor. Biochemistry 44: 9130–9139, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krishnan N. and Becker DF. Oxygen reactivity of PutA from Helicobacter species and proline-linked oxidative stress. J Bacteriol 188: 1227–1235, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krishnan N, Doster AR, Duhamel GE, and Becker DF. Characterization of a Helicobacter hepaticus putA mutant strain in host colonization and oxidative stress. Infect Immun 76: 3037–3044, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lagautriere T, Bashiri G, Paterson NG, Berney M, Cook GM, and Baker EN. Characterization of the proline-utilization pathway in Mycobacterium tuberculosis through structural and functional studies. Acta Crystallogr D Biol Crystallogr 70: 968–980, 2014 [DOI] [PubMed] [Google Scholar]
- 56. Lamour N, Riviere L, Coustou V, Coombs GH, Barrett MP, and Bringaud F. Proline metabolism in procyclic Trypanosoma brucei is down-regulated in the presence of glucose. J Biol Chem 280: 11902–11910, 2005 [DOI] [PubMed] [Google Scholar]
- 57. Larson JD, Jenkins JL, Schuermann JP, Zhou Y, Becker DF, and Tanner JJ. Crystal structures of the DNA-binding domain of Escherichia coli proline utilization A flavoprotein and analysis of the role of Lys9 in DNA recognition. Protein Sci 15: 1–12, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee IR, Lui EY, Chow EW, Arras SD, Morrow CA, and Fraser JA. Reactive oxygen species homeostasis and virulence of the fungal pathogen Cryptococcus neoformans requires an intact proline catabolism pathway. Genetics 194: 421–433, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee YH, Nadaraia S, Gu D, Becker DF, and Tanner JJ. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat Struct Biol 10: 109–114, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lehmann S, Funck D, Szabados L, and Rentsch D. Proline metabolism and transport in plant development. Amino Acids 39: 949–962, 2010 [DOI] [PubMed] [Google Scholar]
- 61. Liang X, Zhang L, Natarajan SK, and Becker DF. Proline mechanisms of stress survival. Antioxid Redox Signal 19: 998–1011, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lijek RS, Luque SL, Liu Q, Parker D, Bae T, and Weiser JN. Protection from the acquisition of Staphylococcus aureus nasal carriage by cross-reactive antibody to a pneumococcal dehydrogenase. Proc Natl Acad Sci U S A 109: 13823–13828, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ling M, Allen SW, and Wood JM. Sequence analysis identifies the proline dehydrogenase and delta 1-pyrroline-5-carboxylate dehydrogenase domains of the multifunctional Escherichia coli PutA protein. J Mol Biol 243: 950–956, 1994 [DOI] [PubMed] [Google Scholar]
- 64. Liu LK, Becker DF, and Tanner JJ. Structure, function, and mechanism of proline utilization A (PutA). Arch Biochem Biophys 632: 142–157, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu W, Hancock CN, Fischer JW, Harman M, and Phang JM. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides. Sci Rep 5: 17206, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu W. and Phang JM. Proline dehydrogenase (oxidase) in cancer. Biofactors 38: 398–406, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu Y, Borchert GL, Donald SP, Diwan BA, Anver M, and Phang JM. Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res 69: 6414–6422, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu Y, Borchert GL, Surazynski A, Hu CA, and Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 25: 5640–5647, 2006 [DOI] [PubMed] [Google Scholar]
- 69. Luo M, Arentson BW, Srivastava D, Becker DF, and Tanner JJ. Crystal structures and kinetics of monofunctional proline dehydrogenase provide insight into substrate recognition and conformational changes associated with flavin reduction and product release. Biochemistry 51: 10099–10108, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Luo M, Christgen S, Sanyal N, Arentson BW, Becker DF, and Tanner JJ. Evidence that the C-terminal domain of a type B PutA protein contributes to aldehyde dehydrogenase activity and substrate channeling. Biochemistry 53: 5661–5673, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Luo M, Gamage TT, Arentson BW, Schlasner KN, Becker DF, and Tanner JJ. Structures of proline utilization A (PutA) reveal the fold and functions of the aldehyde dehydrogenase superfamily domain of unknown function. J Biol Chem 291: 24065–24075, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Luo M, Gates KS, Henzl MT, and Tanner JJ. Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1. ACS Chem Biol 10: 693–697, 2015 [DOI] [PubMed] [Google Scholar]
- 73. Luo M, Singh RK, and Tanner JJ. Structural determinants of oligomerization of delta(1)-pyrroline-5-carboxylate dehydrogenase: identification of a hexamerization hot spot. J Mol Biol 425: 3106–3120, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Luo M. and Tanner JJ. Structural basis of substrate recognition by aldehyde dehydrogenase 7A1. Biochemistry 54: 5513–5522, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Maloy S. and Roth JR. Regulation of proline utilization in Salmonella typhimurium: characterization of put:Mu L(Ap, lac) operon fusions. J Bacteriol 154: 561–568, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mattioli R, Costantino P, and Trovato M. Proline accumulation in plants: not only stress. Plant Signal Behav 4: 1016–1018, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menzel R. and Roth J. Enzymatic properties of the purified putA protein from Salmonella typhimurium. J Biol Chem 256: 9762–9766, 1981 [PubMed] [Google Scholar]
- 78. Menzel R. and Roth J. Purification of the putA gene product. J Biol Chem 256: 9755–9761, 1981 [PubMed] [Google Scholar]
- 79. Menzel R. and Roth J. Regulation of genes for proline utilization in Salmonella typhimurium: autogenous repression by the putA gene product. J Mol Biol 148: 21–44, 1981 [DOI] [PubMed] [Google Scholar]
- 80. Mezl VA. and Knox WE. Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal Biochem 74: 430–440, 1976 [DOI] [PubMed] [Google Scholar]
- 81. Moreira IS, Fernandes PA, and Ramos MJ. Hot spots—a review of the protein-protein interface determinant amino-acid residues. Proteins 68: 803–812, 2007 [DOI] [PubMed] [Google Scholar]
- 82. Morris SM., Jr Arginine metabolism: boundaries of our knowledge. J Nutr 137: 1602S–1609S, 2007 [DOI] [PubMed] [Google Scholar]
- 83. Moxley MA. and Becker DF. Rapid reaction kinetics of proline dehydrogenase in the multifunctional proline utilization A protein. Biochemistry 51: 511–520, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moxley MA, Sanyal N, Krishnan N, Tanner JJ, and Becker DF. Evidence for hysteretic substrate channeling in the proline dehydrogenase and delta1-pyrroline-5-carboxylate dehydrogenase coupled reaction of proline utilization A (PutA). J Biol Chem 289: 3639–3651, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Moxley MA, Tanner JJ, and Becker DF. Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch Biochem Biophys 516: 113–120, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nagano N, Orengo CA, and Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol 321: 741–765, 2002 [DOI] [PubMed] [Google Scholar]
- 87. Nagata K, Nagata Y, Sato T, Fujino MA, Nakajima K, and Tamura T. l-Serine, d- and l-proline and alanine as respiratory substrates of Helicobacter pylori: correlation between in vitro and in vivo amino acid levels. Microbiology 149: 2023–2030, 2003 [DOI] [PubMed] [Google Scholar]
- 88. Nakajima K, Inatsu S, Mizote T, Nagata Y, Aoyama K, Fukuda Y, and Nagata K. Possible involvement of put A gene in Helicobacter pylori colonization in the stomach and motility. Biomed Res 29: 9–18, 2008 [DOI] [PubMed] [Google Scholar]
- 89. Ostrander EL, Larson JD, Schuermann JP, and Tanner JJ. A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry 48: 951–959, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pandhare J, Cooper SK, and Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem 281: 2044–2052, 2006 [DOI] [PubMed] [Google Scholar]
- 91. Pemberton TA, Srivastava D, Sanyal N, Henzl MT, Becker DF, and Tanner JJ. Structural studies of yeast delta(1)-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): active site flexibility and oligomeric state. Biochemistry 53: 1350–1359, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pemberton TA. and Tanner JJ. Structural basis of substrate selectivity of delta(1)-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): semialdehyde chain length. Arch Biochem Biophys 538: 34–40, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Perez-Arellano I, Carmona-Alvarez F, Martinez AI, Rodriguez-Diaz J, and Cervera J. Pyrroline-5-carboxylate synthase and proline biosynthesis: from osmotolerance to rare metabolic disease. Protein Sci 19: 372–382, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Perez-Miller SJ. and Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 42: 7100–7109, 2003 [DOI] [PubMed] [Google Scholar]
- 95. Phang JM. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr Top Cell Reg 25: 92–132, 1985 [DOI] [PubMed] [Google Scholar]
- 96. Phang JM, Donald SP, Pandhare J, and Liu Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 35: 681–690, 2008 [DOI] [PubMed] [Google Scholar]
- 97. Phang JM, Downing SJ, and Yeh GC. Linkage of the HMP pathway to ATP generation by the proline cycle. Biochem Biophys Res Commun 93: 462–470, 1980 [DOI] [PubMed] [Google Scholar]
- 98. Phang JM, Downing SJ, Yeh GC, Smith RJ, Williams JA, and Hagedorn CH. Stimulation of hexose monophosphate pathway by pyrroline-5-carboxylate in cultured cells. J Cell Physiol 110: 255–261, 1982 [DOI] [PubMed] [Google Scholar]
- 99. Phang JM, Hu CA, and Valle D. Disorders of proline and hydroxyproline metabolism. In: Metabolic and Molecular Basis of Inherited Disease, edited by Scriver CR, Beaudet AL, Sly WS, and Valle D. New York: McGraw Hill, 2001, pp. 1821–1838 [Google Scholar]
- 100. Phang JM. and Liu W. Proline metabolism and cancer. Front Biosci (Landmark Ed) 17: 1835–1845, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Phang JM, Liu W, Hancock C, and Christian KJ. The proline regulatory axis and cancer. Front Oncol 2: 60, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Phang JM, Liu W, Hancock CN, and Fischer JW. Proline metabolism and cancer: emerging links to glutamine and collagen. Curr Opin Clin Nutr Metab Care 18: 71–77, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Phang JM, Liu W, and Zabirnyk O. Proline metabolism and microenvironmental stress. Annu Rev Nutr 30: 441–463, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Phang JM, Pandhare J, and Liu Y. The metabolism of proline as microenvironmental stress substrate. J Nutr 138: 2008S–2015S, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Phang JM, Pandhare J, Zabirnyk O, and Liu Y. PPARgamma and proline oxidase in cancer. PPAR Res 2008: 542694, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Polyak K, Xia Y, Zweier JL, Kinzler KW, and Vogelstein B. A model for p53-induced apoptosis. Nature 389: 300–305, 1997 [DOI] [PubMed] [Google Scholar]
- 107. Qamar A, Mysore KS, and Senthil-Kumar M. Role of proline and pyrroline-5-carboxylate metabolism in plant defense against invading pathogens. Front Plant Sci 6: 503, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rai AN. and Penna S. Molecular evolution of plant P5CS gene involved in proline biosynthesis. Mol Biol Rep 40: 6429–6435, 2013 [DOI] [PubMed] [Google Scholar]
- 109. Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, Shboul M, Tham PY, Kayserili H, Al-Gazali L, Shahwan M, Brancati F, Lee H, O'Connor BD, Schmidt-von Kegler M, Merriman B, Nelson SF, Masri A, Alkazaleh F, Guerra D, Ferrari P, Nanda A, Rajab A, Markie D, Gray M, Nelson J, Grix A, Sommer A, Savarirayan R, Janecke AR, Steichen E, Sillence D, Hausser I, Budde B, Nurnberg G, Nurnberg P, Seemann P, Kunkel D, Zambruno G, Dallapiccola B, Schuelke M, Robertson S, Hamamy H, Wollnik B, Van Maldergem L, Mundlos S, and Kornak U. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet 41: 1016–1021, 2009 [DOI] [PubMed] [Google Scholar]
- 110. Riveros-Rosas H, Gonzalez-Segura L, Julian-Sanchez A, Diaz-Sanchez AG, and Munoz-Clares RA. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem Biol Interact 202: 51–61, 2013 [DOI] [PubMed] [Google Scholar]
- 111. Sanyal N, Arentson BW, Luo M, Tanner JJ, and Becker DF. First evidence for substrate channeling between proline catabolic enzymes: a validation of domain fusion analysis for predicting protein-protein interactions. J Biol Chem 290: 2225–2234, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sassetti CM, Boyd DH, and Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48: 77–84, 2003 [DOI] [PubMed] [Google Scholar]
- 113. Scriver CR, Sly WS, Childs B, Beaudet AL, Valle D, Kinzler KW, and Vogelstein B. The Metabolic and Molecular Bases of Inherited Disease, 8th ed. New York: McGraw-Hill, 2001 [Google Scholar]
- 114. Selwood T. and Jaffe EK. Dynamic dissociating homo-oligomers and the control of protein function. Arch Biochem Biophys 519: 131–143, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Serrano H. and Blanchard JS. Kinetic and isotopic characterization of l-proline dehydrogenase from Mycobacterium tuberculosis. Biochemistry 52: 5009–5015, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, and Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Singh H, Arentson BW, Becker DF, and Tanner JJ. Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site. Proc Nat Acad Sci U S A 111: 3389–3394, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Singh RK, Larson JD, Zhu W, Rambo RP, Hura GL, Becker DF, and Tanner JJ. Small-angle X-ray scattering studies of the oligomeric state and quaternary structure of the trifunctional proline utilization A (PutA) flavoprotein from Escherichia coli. J Biol Chem 286: 43144–43153, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Singh RK. and Tanner JJ. Unique structural features and sequence motifs of proline utilization A (PutA). Front Biosci (Landmark Ed) 17: 556–568, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sophos NA. and Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact 143–144: 5–22, 2003 [DOI] [PubMed] [Google Scholar]
- 121. Srivastava D, Schuermann JP, White TA, Krishnan N, Sanyal N, Hura GL, Tan A, Henzl MT, Becker DF, and Tanner JJ. Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc Natl Acad Sci U S A 107: 2878–2883, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Srivastava D, Singh RK, Moxley MA, Henzl MT, Becker DF, and Tanner JJ. The three-dimensional structural basis of type ii hyperprolinemia. J Mol Biol 420: 176–189, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.This reference has been deleted
- 124. Srivastava D, Zhu W, Johnson WH, Jr, Whitman CP, Becker DF, and Tanner JJ. The structure of the proline utilization A proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction. Biochemistry 49: 560–569, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Surber MW. and Maloy S. The PutA protein of Salmonella typhimurium catalyzes the two steps of proline degradation via a leaky channel. Arch Biochem Biophys 354: 281–287, 1998 [DOI] [PubMed] [Google Scholar]
- 126. Szabados L. and Savoure A. Proline: a multifunctional amino acid. Trends Plant Sci 15: 89–97, 2010 [DOI] [PubMed] [Google Scholar]
- 127. Tanner JJ. SAXS fingerprints of aldehyde dehydrogenase oligomers. Data Brief 5: 745–751, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tanner JJ. Structural biology of proline catabolism. Amino Acids 35: 719–730, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tanner JJ. and Becker DF. PutA and proline metabolism. In: Handbook of Flavoproteins. Volume 1 Oxidases, Dehydrogenases and Related Systems, edited by Hille R, Miller SM, and Palfey BA. Berlin: De Gruyter, 2013, pp. 31–56 [Google Scholar]
- 130. Valle D, Goodman SI, Applegarth DA, Shih VE, and Phang JM. Type II hyperprolinemia. Delta1-pyrroline-5-carboxylic acid dehydrogenase deficiency in cultured skin fibroblasts and circulating lymphocytes. J Clin Invest 58: 598–603, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Vasiliou V. and Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics 2: 138–143, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Vasiliou V, Thompson DC, Smith C, Fujita M, and Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chem Biol Interact 202: 2–10, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Vílchez S, Manzanera M, and Ramos J. Control of expression of divergent Pseudomonas putida put promoters for proline catabolism. Appl Environ Microbiol 66: 5221–5225, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Vinod MP, Bellur P, and Becker DF. Electrochemical and functional characterization of the proline dehydrogenase domain of the PutA flavoprotein from Escherichia coli. Biochemistry 41: 6525–6532, 2002 [DOI] [PubMed] [Google Scholar]
- 135. Wanduragala S, Sanyal N, Liang X, and Becker DF. Purification and characterization of Put1p from Saccharomyces cerevisiae. Arch Biochem Biophys 498: 136–142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Watanabe S, Sueda R, Fukumori F, and Watanabe Y. Characterization of flavin-containing opine dehydrogenase from bacteria. PLoS One 10: e0138434, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. White TA, Johnson WH, Jr, Whitman CP, and Tanner JJ. Structural basis for the inactivation of Thermus thermophilus proline dehydrogenase by N-propargylglycine. Biochemistry 47: 5573–5580, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. White TA, Krishnan N, Becker DF, and Tanner JJ. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J Biol Chem 282: 14316–14327, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett 492: 193–198, 2001 [DOI] [PubMed] [Google Scholar]
- 140. Willis A, Bender HU, Steel G, and Valle D. PRODH variants and risk for schizophrenia. Amino Acids 35: 673–679, 2008 [DOI] [PubMed] [Google Scholar]
- 141. Wood JM. Genetics of l-proline utilization in Escherichia coli. J Bacteriol 146: 895–901, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wood JM. Membrane association of proline dehydrogenase in Escherichia coli is redox dependent. Proc Nat Acad Sci U S A 84: 373–377, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids 37: 1–17, 2009 [DOI] [PubMed] [Google Scholar]
- 144. Wu G, Bazer FW, Burghardt RC, Johnson GA, Kim SW, Knabe DA, Li P, Li X, McKnight JR, Satterfield MC, and Spencer TE. Proline and hydroxyproline metabolism: implications for animal and human nutrition. Amino Acids 40: 1053–1063, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wyse AT. and Netto CA. Behavioral and neurochemical effects of proline. Metab Brain Dis 26: 159–172, 2011 [DOI] [PubMed] [Google Scholar]
- 146. Yoshida A, Rzhetsky A, Hsu LC, and Chang C. Human aldehyde dehydrogenase gene family. Eur J Biochem 251: 549–557, 1998 [DOI] [PubMed] [Google Scholar]
- 147. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, and Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab 15: 451–465, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang L. and Becker DF. Connecting proline metabolism and signaling pathways in plant senescence. Front Plant Sci 6: 552, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang M, White TA, Schuermann JP, Baban BA, Becker DF, and Tanner JJ. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry 43: 12539–12548, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang W, Zhang M, Zhu W, Zhou Y, Wanduragala S, Rewinkel D, Tanner JJ, and Becker DF. Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2′-OH group in regulating PutA-membrane binding. Biochemistry 46: 483–491, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang W, Zhou Y, and Becker DF. Regulation of PutA-membrane associations by flavin adenine dinucleotide reduction. Biochemistry 43: 13165–13174, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zhou Y, Larson JD, Bottoms CA, Arturo EC, Henzl MT, Jenkins JL, Nix JC, Becker DF, and Tanner JJ. Structural basis of the transcriptional regulation of the proline utilization regulon by multifunctional PutA. J Mol Biol 381: 174–188, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhou Y, Zhu W, Bellur PS, Rewinkel D, and Becker DF. Direct linking of metabolism and gene expression in the proline utilization A protein from Escherichia coli. Amino Acids 35: 711–718, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zhu W. and Becker DF. Exploring the proline-dependent conformational change in the multifunctional PutA flavoprotein by tryptophan fluorescence spectroscopy. Biochemistry 44: 12297–12306, 2005 [DOI] [PubMed] [Google Scholar]
- 155. Zhu W. and Becker DF. Flavin redox state triggers conformational changes in the PutA protein from Escherichia coli. Biochemistry 42: 5469–5477, 2003 [DOI] [PubMed] [Google Scholar]
- 156. Zhu W, Gincherman Y, Docherty P, Spilling CD, and Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch Biochem Biophys 408: 131–136, 2002 [DOI] [PubMed] [Google Scholar]
- 157. Zhu W, Haile AM, Singh RK, Larson JD, Smithen D, Chan JY, Tanner JJ, and Becker DF. Involvement of the beta3-alpha3 loop of the proline dehydrogenase domain in allosteric regulation of membrane association of proline utilization A. Biochemistry 52: 4482–4491, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.