

Abstract
Here we report a transcription factor decoy strategy for targeted activation of eight large silent polyketide synthase and non-ribosomal peptide synthetase gene clusters, ranging from 50 to 134 kilobases in multiple streptomycetes, and characterization of a novel oxazole family compound produced by a 98-kb biosynthetic gene cluster. Due to its simplicity and ease, this strategy can be readily scaled up for discovery of natural products in streptomycetes.
Graphical abstract
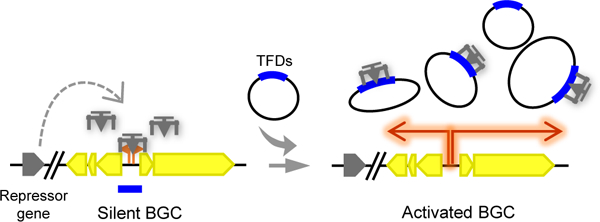
Discovery of novel antibiotics remains an important task for modern biotechnology, especially with the emergence and prevalence of antibiotic resistance1. A rich source of pharmaceutical agents, microbial natural products are synthesized by the enzymes encoded in biosynthetic gene clusters (BGCs) within genomes. Recent advances in large-scale genome sequencing and bioinformatics has revealed the existence of a vast number of uncharacterized BGCs that outnumbers those already known by approximately ten-fold2,3. However, these BGCs are mostly transcriptionally silent under conventional culture conditions due to tight regulation in their native hosts. Many methods have been developed to fully exploit this largely untapped reservoir of natural products, either by cloning and expressing the entire BGCs in heterologous hosts or manipulating regulators directly in native hosts4–6. However, it remains an overwhelming challenge to activate large BGCs (>50 kilobases, kb) such as modular polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) BGCs in Streptomyces species, which often span up to hundreds of kb of DNA.
To overcome this key limitation, we developed a transcription factor decoy (TFD) strategy for the targeted and high-throughput activation of silent PKS and NRPS BGCs in streptomycetes. TFDs are DNA molecules designed to interfere with gene regulation by mimicking regulatory DNAs that are bound to regulators and thus prevent the latter from binding to their cognate DNA targets. This could result in the de-repression of a target silent BGC as well as de-activation of a target naturally active BGC (Fig. 1a). A related strategy was reported in mammalian cells using short double-stranded oligodeoxynucleotides that contain consensus binding sequences to specific TFs to achieve transcriptional de-activation or de-repression of certain genes. A large number of TFDs have been successfully tested in vitro or in preclinical animal studies, among which are the NF-κB and STAT3 targeting TFDs that have entered into clinical trials7,8. Unlike the TFDs used in mammalian cells, it is difficult to precisely predict the regulatory DNAs in a target BGC. In addition, it is desirable to keep the regulatory elements as intact as possible so that the reporter can faithfully report the expression levels. Therefore, in our strategy, the entire intergenic regions as well as an extension of ~100 bp to both the up- and downstream ORFs in a target BGC are cloned as the TFD DNAs and introduced into the native hosts by a genetically stable multicopy plasmid. Of note, “regulatory DNA” refers to the native copy while the “TFD DNA” refers to the plasmid-borne copies of that regulatory DNA. For a low-throughput protocol, each TFD is transformed into the native host separately, while for high-throughput, a TFD plasmid library is constructed, with the aid of a built-in reporter gene to screen for highly expressed colonies (Fig. 1b).
Fig. 1. Workflow of the TFD strategy.
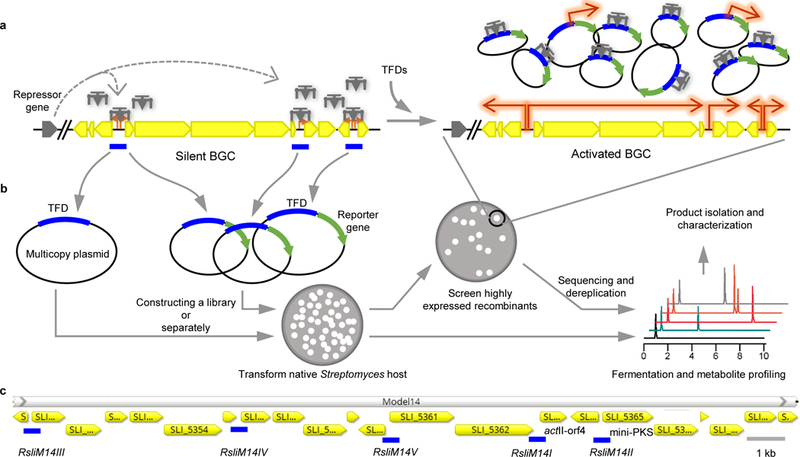
a, Mechanism for activation of a silent BGC by TFD sequestration of repressors. When transformed into native hosts, the TFDs encoded in the multicopy plasmid sequester the cognate repressors, rendering the BGC de-repressed (i.e. activated). b, Workflow for low-throughput and high-throughput protocols, respectively. c, The actinorhodin BGC in Streptomyces lividans 66. The blue bars indicate the regulatory DNAs. In the label RsliM14III, R=regulatory DNA, sli=Streptomyces lividans, M14=cluster 14, III=third; italicized font indicates TFD DNA while normal font indicates the recombinant strain harboring this TFD DNA. The same labelling scheme is used for other intergenic DNA regions of any target BGCs in this work.
As a proof of concept, we first applied the low-throughput TFD strategy to two known pigment-producing BGCs, actinorhodin (act) (Fig. 1c) and undecylprodigiosin (red) that are normally silent in Streptomyces lividans 66 (Supplementary Fig. 1a,b). We cloned all potential regulatory DNAs as TFDs and transformed each TFD into S. lividans separately, followed by cell cultivation and product detection. The regulatory DNAs from the operons containing either positive regulator genes or core structural genes were demonstrated to be most effective. By introducing tens of copies of such TFDs, negative regulators were sequestered away from their cognate, genomic copies of regulatory DNAs, resulting in de-repression of the activator genes, such as those well-characterized pathway-specific activators, ActII-ORF4 (RsliM14I), RedD (RsliM18I), and RedZ (RsliM18III) (Supplementary Fig. 1c,d), whose overexpression was proven to be able to activate corresponding gene clusters9. Unexpectedly, introduction of the act mini-PKS TFD (RsliM14II) also led to high actinorhodin production (Supplementary Fig. 1), in spite of the fact that no negative regulation on this regulatory DNA was reported, and the only binding protein ActII-ORF4 was also absent when the act BGC was silent (http://dbscr.hgc.jp/prom/actI-orf1.html). This observation suggests other unknown regulation pathways may exist, which nevertheless could be bypassed by the TFD strategy.
It is common for a large BGC to contain several operons, but we selected only two regulatory DNAs in a target BGC as a basic criterion of the low-throughput protocol (Fig. 1b), one from a core structural gene operon and the other from a positive regulator gene operon. Next, we chose five uncharacterized PKS or NRPS BGCs from Streptomyces coelicolor A3(2) and Streptomyces albus J1074 (Supplementary Table 1) for activation, some of which possess uncommon traits: e.g., scoM1 encodes polyketide-type polyunsaturated fatty acid (PUFA) synthases (Supplementary Fig. 2). Based on HPLC analysis of fermentation extracts, we detected a new tripeptide peak from RsalbM14II of S. albus J1074 as a result of TFD activation (Supplementary Fig. 3a). In addition, we chose eight unknown BGCs from Streptomyces sp. F-5635 (designated as ssp8), which are either very large (42–134 kb) or contain interesting enzymes such as halogenases, CYP450s, or glycosyltransferases (Supplementary Table 1). Although this strain is not fully sequenced and hence annotation of some BGCs are incomplete, it does not affect the application of the TFD method. We cloned two regulatory DNAs from the sequenced regions of each BGC as TFDs and were able to detect new peaks from three of these eight target BGCs (Fig. 2a and Supplementary Fig. 3b,c). Compound 1, encoded by an incompletely sequenced BGC, was produced in large amount (~6 mg from 6 L fermentation) in Rssp8M28I. It was then purified and characterized by HRMS, 1-D and 2-D NMR (Supplementary Note 1), and was identified to be butyrolactol A, a broad antifungal compound comparable to the drug nystatin10. The polyol head group of 1 together with a tert-butyl tailed tetraene C16 carbon chain mimics glycolipid and is quite rare for a polyketide. Its biosynthesis has been proposed to involve sequential addition of hydroxymalonyl unit11. While this manuscript was in preparation, its full BGC was reported12. By dot-plotting analysis against this gene cluster and further pairwise alignments, we determined that ssp8M22, M28, and many small pieces in M29 from different contigs actually constitute this large PKS BGC (Supplementary Fig. 4), which greatly facilitated our study of this compound.
Fig. 2. Activation of BGCs in two streptomycetes.
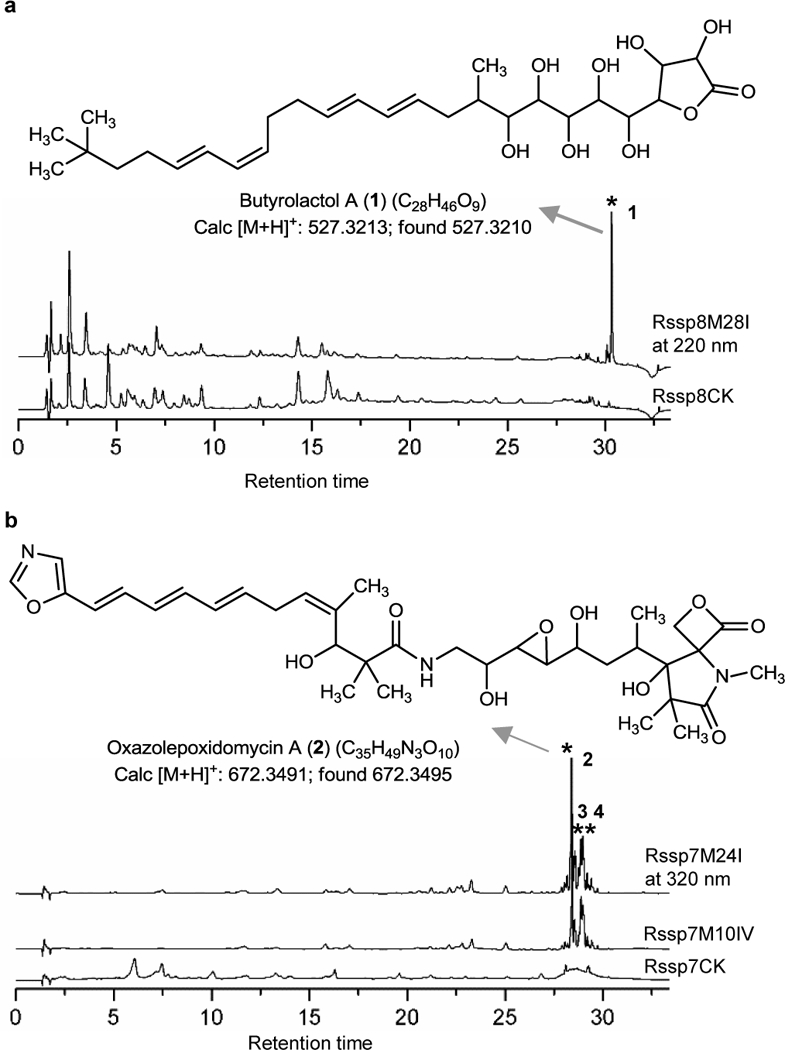
HPLC analysis of crude extracts and compounds isolated from successfully activated BGCs from Streptomyces sp. F-5635 (ssp8) (a) and Streptomyces sp. F-4335 (ssp7) (b). New peaks compared to the controls, including recombinants with empty plasmid or other TFDs, are labeled by *. Triplicate fermentation experiments were performed and results were reproducible. See Supplementary Notes 1 and 2 for structural characterization of compounds 1–4.
Because many BGCs contain multiple regulatory DNAs, some of which are not easily identified, in order to increase the chance of finding one that works or works better, more regulatory DNAs should be evaluated. To this end, we designed a reporter-guided strategy for high-throughput screening of highly expressed TFD recombinant strains (Fig. 1b). Ten uncharacterized PKS or NRPS BGCs (30–112 kb) were chosen from two underexplored species, Streptomyces griseofuscus B-5429 and Streptomyces sp. F-4335 (designated as sgri and ssp7 respectively, see Supplementary Table 1), and all potential regulatory DNAs were cloned for library construction and screening, coupled with sequencing-based dereplication (Supplementary Figs. 5 and 6). Finally, fermentation and HPLC analyses showed that five of them were activated to produce new products (Figs. 2b and Supplementary Fig. 3d,e). Somewhat surprising is the case of Rssp7M10IV and Rssp7M24I, which showed relatively low sequence identity in their TFD DNAs but produced the same HPLC peaks (compounds 2–4 in Fig. 2b). By UV-Vis profile inspection (Supplementary Note 2), we deduced that 2 was a polyene polyketide produced by ssp7M24 instead of a polypeptide by ssp7M10. Subsequent RT-PCR experiments indicated that ssp7M24 was highly expressed in both recombinant strains Rssp7M10IV and Rssp7M24I, but expression of ssp7M10 was minimal in both strains (Supplementary Fig. 7), suggesting cross-regulation between ssp7M10 and ssp7M24. Such cases have also been documented in a few disparate BGCs, such as BGCs for synthesizing jadomycin and chloramphenicol in Streptomyces venezuelae ISP523013,14. In this aspect, it appears to be a drawback of this method for targeted activation, but it helps gain deeper insights into BGC regulation that may be elusive by other methods.
Compound 2 was purified (~3 mg from 8 L fermentation) and characterized by 1-D and 2-D NMR (Supplementary Note 2). Based on its structural similarity to the trans-AT PKS-synthesized oxazolomycin (the ozm cluster)15,16 which contains an unusual spiro system consisting of a β-lactone and a γ-lactam moiety, 2 was named oxazolepoxidomycin A (the oze cluster) and its biosynthetic mechanism was proposed accordingly (Supplementary Fig. 8). The striking structural difference between 2 and other oxazolomycins is the polyene system (Supplementary Fig. 9). In the oze pathway, the split bimodule encoded by OzeA and OzeB is putatively responsible for introduction of the first conjugated diene. In addition to its unprecedented bimodule organization, KS-DH-ACP-KS-KR, more surprising is the double bond position which indicates the occurrence of double bond migrations. It has been reported that double bond migration can proceed via either a canonical α,β-dehydration followed by double bond migration catalyzed by a DH-like domain (DH*) or enoyl-isomerase domain (EI) in a non-elongating module, or a direct β,γ-dehydration by single, dedicated modules which are organized as KS-DH-KR-ACP17–19. But neither situation applies to the oze pathway (Supplementary Fig. 10), suggesting that the combination of OzeA and OzeB may employ a novel mechanism for β,γ-dehydration, or other yet unidentified conserved amino acid residues could also influence double bond migration. At the end of the assembly line, an unusual terminal C domain was proposed to generate the characteristic spirocyclic terminus of this family, however, the mechanism remains to be elucidated. For installation of the epoxy group, there are two cytochrome P450 genes (ozeKL) located immediately after the trans-AT PKS genes in the oze cluster. Both of these are unlike the only P450 gene in the ozm cluster, which is far from the core region and thus not assigned any roles16. We presumed that two epoxidations occurred followed by a monensin-like epoxide hydrolysis20,21, based on the position of the epoxy group on 2 and its two adjacent hydroxyl groups, but the corresponding epoxide hydrolase needs to be identified. Further mechanistic studies are expected to address these questions.
In conclusion, we developed a TFD strategy to successfully activate eight silent PKS/NRPS BGCs in multiple streptomycetes (Supplementary Table 2) and characterized a novel compound oxazolepoxidomycin A that is produced by a 98 kb BGC, demonstrating its potential for natural product discovery. This strategy directly manipulates regulator-binding DNAs and is complementary to other existing methods that manipulate regulators such as overexpression or deletion of regulator genes. Compared to the latter, this strategy is simpler and easier to perform and more generally applicable because it remains a challenge to identify all the regulators in a target BGC. Furthermore, it is time-consuming to delete negative regulators, although overexpression of an activator is relatively simple. All of these strategies take advantage of the native host system, such as the intact biosynthetic machineries, precursor supply and product export; once the regulation is rewired, production should be highly efficient. However, a limitation of our strategy is also apparent; similar to the CRISPR-Cas9 strategy22, it requires introduction of recombinant DNAs into the native hosts. For Streptomyces hosts of very low conjugation efficiency, the low-throughput protocol can be used, while in other cases, the high-throughput protocol should be applied to reach the full potential of this strategy. In the current version of this method, we did not explore combining multiple TFD DNAs from a BGC on one plasmid. One reason for this is that multiplying any individual regulatory DNA should be able to saturate the cognate regulators and thus liberate the whole regulon according to the regulon theory; another reason is to avoid combining TFDs of opposite effects which might give rise to neutralized outcomes. However, once individual effective TFDs are identified, we may be able to combine them together, or multiply them separately, or even apply both, to further improve compound production.
Online methods
Methods
Bacterial strains, culture conditions, and general remarks.
All Escherichia coli strains were cultured in LB broth at 37°C supplemented with appropriate antibiotics when needed. E. coli NEB5α was used for cloning, and E. coli ET12567/pUZ8002 for conjugation. Streptomyces strains used in this study and their corresponding culture conditions were listed in Supplementary Tables 3 and 4.
Construction of single TFD plasmid and TFD plasmid library, and corresponding Streptomyces recombinant strains.
The TFD plasmid was built either based on pKC113923 or its derivative pKCW101 (inserted the promoterless neo as a reporter gene). For single TFD plasmid construction, corresponding TFD fragment was PCR-amplified using primers listed in Supplementary Table 5, and then assembled to predigested pKC1139/BamHI+EcoRI or pKCW101/BamHI+EcoRI by Gibson Assembly24 (New England Biolabs). For TFD plasmid library construction, we pooled all PCR-amplified TFD fragments, adjusted DNA concentrations of each TFD fragment to 1:1 molar ratio, and finally carried out Gibson Assembly at 50°C for 30 min. The resulting reaction mixture was transformed into high-efficiency NEB5α competent cells (New England Biolabs). For single TFD plasmid construction, a couple of overnight colonies were picked for further culturing, plasmid extraction, and sequencing. For TFD plasmid library, 4 ml of LB/apramycin were added to the transformation mixture after 60 min recovery at 37°C, and then allowed to grow overnight. Plasmid library was extracted from overnight cultures and then immediately transformed into conjugation donor strain ET12567/pUZ800223 (in the morning), and allowed to grow for 6 hrs (in the afternoon). If mycelia were used for conjugation, spores were heat-shock treated to allow germination for 6 hrs in corresponding liquid medium (see Supplementary Table 3); if spores were used, spores were heat-shock treated just before performing conjugation in the afternoon. For verification of the coverage of the decoy plasmid library, PCR was performed using ET12567/pUZ8002/TFD plasmid library as template and all related TFD primers. All expected bands were amplified in our experiment, indicating all TFD plasmids were covered in the library. Conjugation conditions were listed in Supplementary Table 3. After incubating at 30°C for ~18 hrs, 300–500 exconjugants were grown out, which should be sufficient for library coverage. All colonies were scraped out and pooled together, then streaked out on fresh corresponding solid medium plates for one generation. Then all spores were collected and added in 20% glycerol for long-term storage at −20°C or −80°C. For fermentation analysis, spores were inoculated with a ratio of 1:100 into 4 ml of corresponding liquid media without any antibiotics in the afternoon, and the seed culture was allowed to grow overnight. Next morning, seed culture was transferred with a ratio of 1:100 into 50 ml of same liquid media with 25 μg/ml apramycin, and then allowed to grow for 3 days at 28°C, 250 rpm. Triplicates were performed for fermentation in order to confirm reproducibility.
Library screening and dereplication.
In order to obtain single colonies, the collected spores of the TFD library were 100-fold serial diluted and spread onto corresponding freshly made solid medium with 100 μg/ml kanamycin (reporter gene). After incubation at 28°C for 4 days, 50 well-separated single colonies were picked for PCR using universal primers and sent for sequencing. Three individual colonies were chosen from each type for fermentation test after sequencing-based de-replicating.
Metabolite extraction, HPLC, and LC-MS analyses.
The supernatant of fermentation broth was adjusted to pH ~3 by adding HCl, and then mixed with equal volume of ethyl acetate. Organic phase was dried using rotovap, and then dissolved in 1 ml of methanol. After being filtered by 0.2 μm filter, the crude extract was injected into HPLC. All HPLC analyses were carried out on Agilent 1260, equipped with a diode array detector, using analytical column Kinetex SB-C18 (4.6×180 mm, 5 μm) with a flow rate of 1.0 ml/min. Detector wavelengths were set to 220, 260, 280, 300, and 320 nm. Two methods were used for analytical HPLC. Solvent A was water with 0.1% trifluoroacetic acid (TFA), and Solvent B was acetonitrile with 0.1% TFA. Flow rate was set to 1.0 ml/min. Method 1: 5–30% B in 20 min, 30–100% in 10 min, hold at 100% for 4 min, 100–5% B in 1 min, hold at 5% B for 5 min. Method 2: 5–50% B in 15 min, 50–100% B in 5 min, hold at 100% B for 4 min, 100–5% B in 1 min, hold 5% B for 5 min. For LC-MS analysis, ESI positive ion mode (Bruker, Amazon SL Ion Trap) was used, equipped with a Kinetex 2.6 μm XB-C18 100 Å (Phenomenex).
Isolation of Rssp8M28I product butyrolactol A (1).
Fermentation was performed using 6 L of MYM liquid medium. Crude extract was obtained by 1:1 ethyl acetate extraction and dissolved in methanol. After centrifugation to remove the insolubles, the sample was loaded onto a Sephadex LH-20 column pre-equilibrated by methanol. The first 30 min eluent was discarded while the subsequent fractions were collected in every 5 min. Each fraction was checked by HPLC and fractions containing the target peak (Frs. 4–6) were pooled together and finally subjected to semi-preparative HPLC for further purification. The freshly purified compound was injected onto HPLC to check purity, and re-injected after overnight to check stability. Partial acetonitrile was removed from the HPLC collections by rotovap, leaving mostly water, then sample was frozen at −80°C for more than 1 hr, and finally subjected to lyophilizer for overnight dry. About 6 mg of pure compound was obtained. Deuterated DMSO was used to dissolve the sample butyrolactol A for NMR experiment. NMR analysis was performed on an Agilent 600-MHz NMR spectrometer (Supplementary Note 1).
Isolation of Rssp7M24I product oxazolepoxidomycin A (2).
Fermentation was performed using 8 L of R2 liquid medium. Same procedures were used, and finally about 3 mg of pure compound was obtained for NMR experiment after dissolving in deuterated DMSO (Supplementary Note 2).
Real time PCR (RT-PCR).
Total RNA were extracted from Rssp7CK, Rssp7M10IV and Rssp7M24I after culturing for 36 hrs (including the overnight seed culture) based on the protocol provided by PureLink™ RNA Mini Kit. Isolated RNA was reverse transcribed using SuperScript IV VILO Master Mix which includes first a gDNA removal. The internal reference gene is the hrdB homologous sigma factor in ssp7. Gene-specific primers are listed in Supplementary Table 5.
Supplementary Material
Acknowledgements
This work was supported by grant GM077596 (to H. Zhao) from the National Institutes of Health. Some of this data was collected in the Carl R. Woese Institute for Genomic Biology Core on a 600MHz NMR funded by NIH grant number S10-RR028833, LC-MS at MCB Metabolomics Center, and HRMS at SCS Mass Spectrometry Laboratory. B. Wang and F. Guo would dedicate this article to the memory of Keqian Yang, who made important contributions to their understanding of Streptomyces genetic regulation of secondary metabolism.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to H.Z.
Data availability. The DNA sequence encoding the oxazolepoxidomycin biosynthetic gene cluster from Streptomyces sp. NRRL F-4335 has been deposited to GenBank with accession code BK010686. All other data pertaining to this study are contained in the published article and its Supplementary Information files or are available from the corresponding author upon reasonable request.
References
- 1.Martens E & Demain AL J. Antibiot. 70, 520 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Ikeda H et al. Nat. Biotechnol. 21, 526 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Bentley SD et al. Nature 417, 141 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Ren H, Wang B & Zhao H Curr. Opin. Biotechnol. 48, 21–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nah H-J, Pyeon H-R, Kang S-H, Choi S-S & Kim E-S Front. Microbiol. 8(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rutledge PJ & Challis GL Nat. Rev. Microbiol. 13, 509–523 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Hecker M & Wagner AH Biochem. Pharmacol. 144, 29–34 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Gambari R Expert Opin. Ther. Pat. 21, 1755–1771 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Liu G, Chater KF, Chandra G, Niu G & Tan H Microbiol. Mol. Biol. Rev. 77, 112–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotake C et al. J Antibiot (Tokyo) 45, 1442–50 (1992). [DOI] [PubMed] [Google Scholar]
- 11.Chan YA et al. Proc. Natl. Acad. Sci. U S A 103, 14349–14354 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harunari E, Komaki H & Igarashi Y Beilstein J. Org. Chem. 13, 441–450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robertson AW et al. J. Am. Chem. Soc. 138, 2200–2208 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Xu G et al. J. Biol. Chem. 285, 27440–27448 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mori T et al. Tetrahedron Lett. 26, 1073–1076 (1985). [Google Scholar]
- 16.Zhao C et al. J. Biol. Chem. 285, 20097–20108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helfrich EJN & Piel J Nat. Prod. Rep. 33, 231–316 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Wagner DT et al. Structure 25, 1045–1055.e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gay DC, Spear PJ & Keatinge-Clay AT ACS Chem. Biol. 9, 2374–2381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shichijo Y et al. J. Am. Chem. Soc. 130, 12230–12231 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Gallimore AR et al. Chem. Biol. 13, 453–460 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Zhang MM et al. Nat. Chem. Biol. 13, 607–609 (2017). [Google Scholar]
References
- 23.Kieser T, Bibb MJ, Buttner MJ, Chater KF & Hopwood DA Practical Streptomyces Genetics, (John Innes Foundation Norwich, 2000). [Google Scholar]
- 24.Gibson DG et al. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.