

Abstract
Daily oral antiretroviral (ARV) drugs for pre-exposure prophylaxis (PrEP) has proven efficacy for diverse groups of high-risk individuals. However, daily dosing regimen has augmented non-adherence. These experiments comparatively investigated the long-acting (LA) PrEP potency of subcutaneous (SubQ) administrated tenofovir alafenamide (TAF) and emtricitabine (FTC) loaded nanoparticles (NPs) to solution in humanized (hu) mice. TAF+FTC NPs and TAF+FTC solution (each drug at 200 mg/kg) were administered to hu-CD34-NSG mice (n=3/time point) for plasma and tissue pharmacokinetic parameter estimation using LC-MS/MS. NP enhanced tissue ARV assimilation compared to plasma. The same dose was administered for PrEP efficacy in HIV-1 challenged hu-BLT mice (n=5/group). The hu-BLT mice were vaginally challenged with a transmission-founder (T/F) virus at 5 × 105 TCID50 inoculation, on day 4, 7 and 14 post-SubQ treatments (PT) and were compared to infected-untreated-control hu-BLT mice. By 21 days PT, 100% TAF+FTC solution-treated and control-untreated mice were infected. However, TAF+FTC NPs resulted in significant (p=0.0002) protection from HIV-1 (day 4: 80%, day 7 and 14: 60%, respectively) compared to control mice. This proof-of-concept study demonstrated detectable TAF/FTC vaginal levels among TAF+FTC NP-treated hu-BLT mice correlating with prolonged PrEP efficacy, thus establishing long-acting TAF+FTC NPs as a potential PrEP modality.
Keywords: Nanoparticles, tenofovir alafenamide, emtricitabine, HIV-1 prevention, Long-acting
Graphical Abstract
Introduction
Presently, approximately 36.9 million people worldwide are living with human immunodeficiency virus-1 (HIV-1) [1]. Orally administered antiretroviral (ARV) drugs for pre-exposure prophylaxis (PrEP) have demonstrated efficacy in diverse groups of high-risk individuals [2-6]. Indeed, HIV protection in men who have unprotected sex with men (MSM), injection drug use, and serodiscordant couples all have shown significant benefit from oral ARVs therapy (ART). However, several clinical trials with oral coformulation of two nucleoside/nucleotide reverse transcriptase inhibitors (NRTI), i.e. tenofovir disoproxil fumarate+emtricitabine (TDF+FTC) have been terminated early for futility predominantly due to adherence issues [7, 8]. The use of “on demand” TDF+FTC (Truvada™) has also demonstrated significant efficacy for MSM [9]. In the Intervention Préventive de I'Exposition aux Risques avec et pour les Gays (iPERGAY) study of ARV prophylaxis, TDF+FTC prevented contraction of HIV-1 in MSM taken before and after sexual activity. An international survey among high-risk individuals has documented positive responses for a long-acting (LA) preventative option [10]. Thus, an injectable LA delivery system would be advantageous as a preventative strategy and may lead to significantly improved adherence.
Reportedly compared to TDF, tenofovir alafenamide (TAF), a hydrophilic tenofovir (TFV) prodrug, is metabolized to TFV intracellularly [11]. TAF demonstrates high intracellular TFV concentrations as well as its active metabolite i.e. TFV-diphosphate (TFV-dp), compared to plasma and therefore, TAF dose is significantly lower (one-tenth of TDF dosage) [11, 12]. Even though TAF is useful for treatment in a low dosage, use of TAF+FTC coformulation for PrEP is still investigational. It has been hypothesized that limited efficacy of TAF+FTC coformulation for women could be due to low levels of active TFV in cervical, and vaginal tissue[13-15]. Therefore, designing a TAF+FTC LA delivery system would significantly boost the PrEP efficacy of TAF+FTC coformulation and at the same time could further mitigate other side-effects associated with the use of these NRTI combinations, such as mitochondrial toxicity, renal function and bone mineral density [16].
The current study was designed to evaluate LA efficacy of a novel poly(lactide co-glycolide) (PLGA) based TAF+FTC nanoformulation, compared to TAF+FTC coformulated in solution. The polymer i.e. PLGA and the combination ARV i.e. TAF+FTC, used for the nanoformulation were all Food and Drug Administration (FDA) approved compounds. The 200 mg/kg dosage of TAF+FTC nanoparticles (NPs) and as solution was compared for PrEP efficacy and simultaneously TFV and FTC PK from plasma, PBMC active metabolites, and tissue drug levels. Results from these experiments in humanized (hu)-BLT mice demonstrate TAF+FTC NPs provides LA NP efficacy, by prolonging enhanced vaginal tissue drug accumulation (i.e. at highest risk) and therefore, could significantly improve PrEP efficacy and adherence for women.
Materials and Methods:
Materials
PLGA (75:25 lactide:glycolide ratio; Mw 4,000-15,000), poly (vinyl alcohol) (PVA) (M.W. 13,000-23,000), dichloromethane (DCM), acetonitrile (ACN), N-(2-Hydroxyethyl)piperazine-N’ – (2-ethanesulfonic acid) (HEPES) buffer, potassium dihydrogen phosphate (KH2PO4) and Phosphate Buffered Saline (PBS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO), and water optima® LC-MS were purchased from Fisher BioReagents (Fair Lawn, NJ, USA). Pluronic F127 (PF-127) was purchased from D-BASF (Edinburgh, UK). TAF and FTC (~100% purity) were generously provided by Gilead Sciences Inc. (Foster City, CA, USA). High glucose dulbecco's modified eagle medium with L-glutamine (HiDMEM), fetal bovine serum (FBS) and antibiotic-antimycotic (100X) were purchased from Thermo Scientific (Miami, OK, USA). All reagents were used as received without further purification.
TAF+FTC NP preparation and characterization
The TAF+FTC NPs were prepared according to our previously reported method, with some modifications [17, 18]. Briefly, TAF+FTC NPs were formulated under sterile conditions by using a water-in-oil-in-water (w-o-w) emulsion methodology. The primary or inner aqueous phase containing 50 mg FTC in 1 mM HEPES buffer (pH 9) was first emulsified with organic phase containing PLGA and PF-127 at 1:1 ratio along with TAF at 50 mg/100 mg PLGA in DCM and sonicated to get water-in-oil (w-o) emulsion. The above w-o emulsion was further emulsified with outer aqueous phase containing 1 % PVA (as a stabilizer), followed by sonication, producing the final water-in-oil-in-water (w-o-w emulsion). The w-o-w emulsion was stirred overnight in a hood to evaporate the organic phase. Further, the free unbound components were removed by dialysis in sterile water-methanol (10:0.5 ratio) solvent, using a dialysis membrane (Slide-A-lyzer Cassette 2G, 20K MWCO Thermo Scientific, Rockford, IL USA). The TAF+FTC NPs underwent lyophilization and stored at 4°C until used.
For physical characterization, an appropriate amount of freeze-dried TAF+FTC NP was dispersed in ultrapure water at room temperature (RT). The size, polydispersity index (PDI) and surface charge were analyzed using a ZetaPlus Zeta Potential Analyzer instrument (Brookhaven Instruments Corporation, Holtsville, NY, USA). The above studies were performed in triplicate on five different batches of NPs.
The percentage encapsulation efficiency (%EE) and drug loading (%DL) was evaluated by high performance liquid chromatography (HPLC) analysis (Shimadzu, Kyoto, Japan). Briefly, TAF+FTC NP (1 mg) was dissolved in DMSO (40% DMSO final concentration in mobile phase) and was centrifuged (14000×g for 5 mins at 4°C) and filtered through Amicon®Ultra Centrifugal filters (MWCO 30KDa; Merck KGaA, Darmstadt, Germany). A similar procedure was followed with TAF+FTC solution (concentration range from 500 to 0.48 μg/mL of each drug in the mixture) to generate the standard curve (r2=0.99). The TAF and FTC loading concentrations in 1 mg of TAF+FTC NPs, was estimated based on its respective standard curve determined by HPLC analysis (method: Isocratic; Mobile phase: 25mM KH2PO4 45%: ACN 55%; TAF and FTC: absorbance maximum at 260 and 280 nm respectively, retention time 4 min and 3.3 min respectively). Intra-day and inter-day variability were < 15%. The %EE (Equation 1) and % DL (Equation 2) were estimated by the following formulas:
| Equation 1 |
| Equation 2 |
The topography of the TAF+FTC NP was evaluated using scanning electron microscopic (SEM) imaging on a Hitachi S-4700 Field-emission SEM (New York, NY, USA) following methods described previously [17, 18].
The long-term stability of TAF+FTC NPs was evaluated on three batches of freeze dried TAF+FTC NP (same batches) kept at 4°C for 1 year. The 1-year old freeze-dried TAF+FTC NPs (5 mg) were dissolved in 1 mL 40% DMSO in mobile phase, following the same method used for fresh freeze-dried NPs. The size, PDI, surface charge, TAF:FTC ratio, %DL and %EE were evaluated. Mean ± standard error of mean (SE) obtained for three batches of TAF+FTC NPs 1-year old batch was compared to the fresh batch data (Supplementary Table 1).
Endosomal release study
To evaluate endosomal drug release profile of TAF+FTC NP, each drug at 100 μg/mL entrapped in TAF+FTC NP was dissociated in simulated endosomal solution (i.e. 20 mM citrate buffer, pH 5.5 at 37°C). After brief vortexing, a 100μL supernatant aliquot was collected at respective times (0.03, 0.1, 0.25, 0.5, 1, 4, 8, 16, 24 and 96 h). To maintain sink conditions, at each time-point the volume collected was replaced with fresh medium. The aliquot collected was subsequently centrifuged (20,817 ×g for 5 mins at 4°C) for supernatant collection. All the supernatants and standards were filtered through spin filter units (30k MWCO, Amicon® Ultra-0.5mL, MilliporeSigma, Burlington, MA, USA), prior to HPLC analysis as described above in HPLC chromatography section. Mean % cumulative release of TAF and FTC at respective time-points was calculated as described in earlier publication [18], on 3 batches of TAF+FTC NP.
Cytotoxicity assay
To evaluate in-vitro cytotoxicity, TZM-bl cell line and CellTiter-Glo® luminescent assay method was used, as described previously [18]. Briefly, TZM-bl cells at 104 cells/well, were treated for 96 h in triplicate respectively with TAF+FTC NP or TAF+FTC solution, at different concentrations (20, 10, 1, 0.1, 0.01 μg/mL). Positive and negative controls included 5% DMSO and 1×PBS treated cells (in triplicate), respectively. To determine cytotoxicity, CellTiter-Glo® luminescent cell viability assay kit (Promega, Madison, WI, USA) was used and manufacturer protocol was followed. The luminescence of the samples was read using a Synergy II multimode reader with Gen5™ software (BioTek, Winooski, VT, USA). The percentage cytotoxicity was based on % normalized viability against the untreated negative control group. The experiment was carried out using three independent NPs batches and the results are presented as mean ± SE.
Intracellular kinetics studies
The in-vitro time-dependent drug uptake/retention studies were performed on TZM-bl cells. The 6-well plates were seeded with TZM-bl cells (104 cells/well). The treatment groups were treated with 10 μg/mL of TAF+FTC NPs or TAF+FTC solution, for 24 h. For time-dependent uptake studies, at the respective times 1, 4, 8, and 16 h (before treatment wash-off) respective plates were washed with warm 1×PBS and processed as explained below. However, for retention studies, post 24 h treatment, the TAF+FTC NP or solution was washed-off by warm 1×PBS (thrice) and were maintained in fresh complete HiDMEM i.e. hiDMEM supplemented with 10% FBS and 1× antibiotic-antimycotic, until respective harvesting time (1, 4, 8, 16, 24, and 96 h after wash, corresponding to 25, 28, 32, 40, 48, and 120 h, respectively). At respective time points, cells were washed again with 1×PBS. After the washing, plates were air dried in biosafety cabinet and stored at −80°C until analysis. The samples were further processed and were analyzed by methodology as described in LC-MS/MS section below.
Pharmacokinetic (PK) assessment in humanized mice
CD34+ humanized NOD.Cg-PrkdcscidIL2rgtm1Wjl/Szj (NOD/SCID/IL2rgnull, NSG) mice (hu-CD34-NSG) were purchased from Jackson Laboratory (Bar Harbor, ME, USA) to be used for PK studies. It closely resembles the hu-BLT mice, which were used for PrEP studies. The mice were acclimatized for a week before the start of the experiments. Further, all mice in ARV NPs and ARV solution treatment group, received subcutaneously (SubQ) TAF+FTC NPs and TAF+FTC solution (at 200 mg/kg each of TAF and FTC), respectively. TAF+FTC NPs were suspended in 0.6 mL of 5% dextrose. At 1, 4, and 12 h followed by 1, 2, 4, 7, 10, and 14 days after TAF+FTC NP post-treatment (PT), mice (n=3/time point) were sacrificed by carbon dioxide inhalation and cervical dislocation. Similarly, mice (n=3/time point) receiving 200 mg/kg TAF+FTC solution SubQ were sacrificed at 4, 8, 24, 48 and 72 h. From all sacrificed mice, blood and organs included vagina and colon were harvested. The blood was collected into a dipotassium ethylenediaminetetraacetic acid (K2EDTA) containing collection tubes (BD Microtainer® MAP; Franklin Lakes, NJ, USA), centrifuged at 2100 RPM for 20 minutes, plasma harvested and frozen until analysis by LC-MS/MS [19]. The peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-Paque (GE Healthcare Life Sciences, Uppsala, Sweden) density gradient separation method per manufacturer's protocol and were kept frozen until analysis by LC-MS/MS. TFV and FTC were analyzed from plasma and tissues, whereas active metabolites were analyzed from PBMCs. Phoenix WinNonlin (Certara Inc., Princeton, NJ, USA) was used to determine PK parameters of the plasma and tissue concentration-time data using non-compartmental analysis. These tissue concentration-time data were used to determine the amounts of TFV and FTC at specific times at the site of infection and related tissues.
LC-MS/MS analysis of drugs
The plasma and tissue TFV and FTC concentration were analyzed by following our previously reported method [19]. Briefly, 100 μL plasma or tissue homogenate (homogenized in deionized [1:1 w/v] using titanium beads) was mixed with 25 μL of internal standard spiking solution followed by 100 μL of 1% trifluoroacetic acid. Samples were vortexed and placed into solid-phase extraction (SPE) cartridges. The eluent was evaporated to dryness under a stream of nitrogen, reconstituted with 100 μL of 50% ACN in water and 5 μL was injected into the LC-MS/MS instrument. Chromatographic separation was carried out using a Restek Pinnacle DB Biph (2.1mm × 50mm, 5μm) column with isocratic mobile phase consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) (48:52 v/v) at a flow rate of 0.250 mL/min. The dynamic range of the validated assay was 1 to 2000 ng/mL for TFV and FTC.
For TFV-dp and FTC-triphosphate (FTC-tp) analysis, 500 μL 70% methanol in water was added to PBMC pellet (approximately 106 cells), vortexed, sonicated and centrifuged. Two hundred microliters supernatant was mixed with 50 μL internal standard spiking solution (1μg/mL 2-chloroadenosine triphosphate and 250 ng/mL TFV adenine-13C (U)), evaporated to dryness at 45°C and reconstituted with 100 μL 2mM ammonium formate containing 0.05% v/v triethylamine, 5 μL was injected on to LC-MS/MS operated in negative mode. Kinetex C18 (75×4.6 mm, 2.6μ) column with isocratic mobile phase containing 2mM ammonium formate+0.05% triethylamine: ACN (95:5) at a flow rate of 0.25 mL/min was used to produce the chromatographs. The dynamic calibration range for both the analytes was 1 to 1000 ng.
For in vitro intracellular TFV, FTC, TFV-dp and FTC-tp determination, 60 μL 0.5% ethylenediaminetetraacetic acid (EDTA) in water was added and vortexed to detach the cells from well plate. To the detached cells, 140 μL methanol was added to lyse the cells. Well plates were vortexed and the contents were centrifuged at 14000 rpm for 5 mins, 4°C. The 100 μL supernatant was mixed with internal standard spiking standard solution (1μg/mL 2-chloroadenosine triphosphate, 250 ng/mL FTC-13C15N2 and 250 ng/mL TFV adenine-13C (U)), dried at 45°C and reconstituted with 100 μL 2mM ammonium formate. The same sample was used for drug and metabolite quantification using different LC-MS/MS assays. The respective LC-MS methods described above for TFV and FTC, TFV-dp and FTC-tp were used for quantification.
An Exion HPLC system (Applied Biosystems, Foster City, CA, USA) coupled with AB Sciex API 5500 Q Trap with an electrospray ionization (ESI) source (Applied Biosystems, Foster City, CA, USA) was used. The LC-MS/MS system was controlled by Analyst 1.6.1 software. Average inter-day and intra-day variability of both the assays was < 15% according to the FDA analysis guidelines [20].
Generation of hu-BLT mice model
To generate humanized bone marrow, liver, thymus mice (hu-BLT) for PrEP studies, the 6 to 8-week-old NOD.Cg-PrkdcscidIL2rgtm1Wjl/Szj (NOD/SCID/IL2rgnull, NSG) mice were purchased from the Jackson Laboratory and kept in pathogen-free conditions at University of Nebraska-Lincoln Life Sciences Annex. The above-mentioned hu-BLT mice were obtained by following previously published protocols [21, 22]. Those hu-BLT mice that had > 50% the human leukocytes to total leukocytes ratio in the peripheral blood, were considered for challenge with HIV-1.
Ethics Statement
All the hu-BLT mice PrEP experiments performed, adhered to the NIH Guide for the Care and Use of Laboratory Animals and the protocol used was approved by Institutional Animal Care and Use Committee (IACUC, Protocol #1322, University of Nebraska-Lincoln; NE, USA)[23]. All PK in vivo experiments were performed in accordance with Creighton University IACUC approved protocol (Protocol #0989).
HIV-1 vaginal challenge in hu-BLT mouse
The treated mice received TAF+FTC NPs (equivalent to 200 mg/kg each TAF and FTC) SubQ in 0.6 ml 5% dextrose (D5W) on day 0. Groups of treated mice (n=5/time point) were challenged intravaginal with one HIV transmission/founder (T/F) virus (SUMA.c/2821) at 5 × 105 TCID50 in 20 μL as separate groups on days 4, 7, 14 days, respectively post-treatment (PT). Similarly, TAF+FTC solution at 200 mg/kg, was administered to a separate group (n=5) of hu-BLT mice and were challenged with HIV virus as above on day 4 PT. The untreated-infected control mice (n=5) received only D5W on day 0 and were challenged on day 4.
Another group of hu-BLT mice received TAF+FTC NPs (equivalent to 100 mg/kg each TAF and FTC) in 0.6 ml D5W SubQ on day 0. The hu-BLT mice groups were challenged similarly to the treatment mice described above on day 4, 7, and 14 PT and compared to a group of control, untreated hu-BLT mice.
Plasma viral load (pVL)
To evaluate pVL, plasma was collected from hu-BLT mice starting two weeks post-viral challenge followed by weekly collection. Plasma viral RNA (vRNA) was extracted from plasma using QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA) following manufacturers protocol. Similarly, vRNA was extracted from AcroMetrix™ HIV-1 Panel copies/mL standards (Thermo Fisher Scientific, Waltham, MA, USA). The pVL was estimated by the one-step Reverse transcription polymerase chain reaction (RT-PCR) analysis, performed using TaqMan® Fast Virus 1-Step Master Mix (ThermoScientific, Waltham, MA, USA) along with primers: forward 5’-GCCTCAATAAAGCTTGCCTTGA-3’, reverse 5’-GGGCGCCACTGCTAGAGA-3’ and a probe 5’-/FAM/CCAGAGTCACACAACAGACGGGCACA/BHQ_1/-3’ targeting the gag region of HIV-1. The RT-PCR cycle (45 cycles) was performed on an Applied Biosystems® 7500 Real-Time PCR Systems (ThermoScientific, Waltham, MA, USA). Interpretation of fluorescence data was estimated by the RT-PCR instrument’s software. The viral copy numbers of unknown samples were calculated based on the regression analysis of the Ct values of the standard known viral copies/mL. For the regression analysis, the standard curves of known pVL with r2 ≥ 0.98 were considered for evaluation of unknown pVL [24]. The detection limit of the assay was 800 copies/mL of plasma.
HIV-1 vRNA detection in tissues using in situ hybridization (ISH)
The previously published ISH method was used to detect vRNA in tissues [24, 25]. In brief, harvested tissue i.e. spleen, female reproductive track (FRT), cervical, axillary, human thymic organoids, and mesenteric lymph nodes, were fixed in 4% paraformaldehyde and embedded in O.C.T. (Baxter Healthcare corp.; Waukegan, IL, USA). Approximately 6-μm tissue sections of the tissues were cut and adhered to a SuperFrost Plus slide (Fisher Scientific; Lenexa, KS, USA) and air-dried. The sections were then rehydrated, permeabilized, and acetylated prior to hybridization to 35S-labeled HIV anti-sense riboprobes that covered >90% of HIV-1 genome [22] and sense riboprobes were used as a negative control. The tissue sections were washed, RNase digested, and coated with nuclear track emulsion. After 7 days of exposure, they were developed and counterstained with H&E stain (Thermo Fisher Scientific; Waltham, MA, USA).
Statistical Analysis
All experimental data were represented as mean ± SE, if not specified. Two-way ANOVA based statistical significance testing method was used for TAF+FTC NP stability study and the result depicted adjusted P-value >0.9 for all the studied parameters. To evaluate HIV-1 viral infectivity, Kaplan-Meier curve fitting (a nonparametric statistic) and all other statistical analysis were performed using GraphPad Prism 7 software (La Jolla). A P-value of ≤ 0.05 was considered statistically significant.
Results:
TAF+FTC NP Characterization
The interfacial polymer deposition by the w-o-w emulsion solvent evaporation method resulted in well-defined TAF+FTC NPs. The dynamic light scattering analysis demonstrated TAF+FTC NPs averaging 233.2 ± 12.8 nm in size, with polydispersity index (PDI) of 0.11 ± 0.05, and −19.1 ± 4.1 as surface charge (n=5; Supplementary Table 1). The %EE for TAF and FTC into the polymeric nanoparticles averaged 69.2 ± 14.5% and 65.9 ± 18.2%, respectively (n=5). Morphological analysis by SEM image (Supplementary figure 1) demonstrated the obtained NPs were well-defined spherical particles with very uniform size distribution as indicated by low PDI value (< 0.2), which in turn is in good agreement with the SEM image. Further, 1year stability studies (Supplementary Table 1) shows no significant differences observed in physicochemical properties of TAF+FTC NPs upon 1 year storage at 4°C.
Endosomal release kinetics
To assess endosomal release kinetics of TFV+FTC from TAF+FTC NP, release over time in simulated endosomal pH (pH=5.5) was evaluated by HPLC (Supplementary Figure 2). The above study demonstrated that in the endosomal condition, both TFV and FTC drug follows similar release kinetics (one-phase decay kinetics) i.e. after initial fast drug release the drugs tends to release relatively constantly. Evidently FTC shows delayed release (rate constant, K= 4 h−1) compared to TFV (rate constant, K= 2.65 h−1). This could be due to the fact that FTC (highly aqueous soluble drug) gets in the inner core of the NP, whereas TAF has aqueous and organic phase solubility persisting throughout the encapsulation. However, the effective time constant (teff) evaluated to be 22.6 mins and 15 mins respectively for TAF and FTC until plateau phase reached (~73%). All together approximately 73% of total TAF+FTC content were released during this study. Also, the half-life calculated to be 15.7 and 10.2 mins for TFV and FTC release from NP in the simulated endosomal condition. These results are consistent with PLGA nano-formulations characteristic biphasic drug-release [26]. Therefore, once in endosome ~73% of entrapped drug will be released within ~30 mins and the remaining amount will be released over-time to maintain the equilibrium.
Intracellular uptake, conversion and release kinetics
To investigate the intracellular uptake, conversion and release of TAF+FTC from TAF+FTC NPs, TZM-bl cells were treated for 24 h to maximize uptake and thereafter cells were washed-off and were maintained in the fresh media to evaluate the TAF+FTC release/retention and conversion to its metabolites (Supplementary figure 3). This study verifies that TAF+FTC NPs were internalized and release its drug content instantaneously (as predicted by the endosomal release study, Supplementary Figure 2). At the cellular level, TFV shows sustained release and longer retention than FTC (Supplementary Figure 3A and D). The intracellular free TAF is converted to its active TFV-dp increases over time and attains equilibrium. Compared to solution, NP demonstrates enhanced accumulation and retention of TFV and TFV-dp. Similarly, intracellular FTC and FTC-tp release and retention increases and attains equilibrium over time (Supplementary Figure 3D and E). However, FTC is known to be eliminated quickly due to highly aqueous solubility [16]. This study also shows similar trends, compared to TAF, FTC has smaller accumulation and retention (Supplementary Figure 3A and D). Both active metabolites demonstrated comparable retention in both cases of treatment as NP or in solution (Supplementary Figure 3C and E). Therefore, nanoencapsulation improves and significantly maintained the high drug/active-drug metabolite for effective inhibition of reverse transcriptase activity.
In vitro cytotoxicity
The cytotoxicity of TAF+FTC NP in comparison to TAF+FTC solution was evaluated on TZM-bl cells to evaluate TAF+FTC NP toxic effect on endothelial cells upon SubQ administration. These endothelial cells would be the first cell type that TAF+FTC nanoformulation will encounter upon SubQ administration.
The cytotoxicity potency of TAF+FTC NP is comparable to that of TAF+FTC solution (Supplementary Figure 4). At highest concentration (TAF+FTC each at 20 μg/mL), TAF+FTC NP showed slight but non-significant reduction in % viability (~88%), whereas TAF+FTC solution showed significant cytotoxics the viability is reduced to ~60%. Therefore, cytotoxicity study on TZMbl cells suggests that nano-encapsulation improves the therapeutic-toxic window of TAF and FTC.
Tissue penetrability and in vivo PK study in hu-CD34-NSG mice
To evaluate the in vivo PK of TFV and FTC, and respective active metabolites in hu-CD4-NSG mice, plasma and tissue drug levels were subjected to non-compartmental analysis. Table 1 summarizes the PK results for 200 mg/kg TAF+FTC NP formulation and 200 mg/kg TAF+FTC in solution. TAF+FTC NPs administered SubQ resulted in significantly higher (p<0.05) TFV and FTC maximum plasma concentration (Cmax) compared to 200 mg/kg in solution (Figure 1C). Similarly, the lower dosage treatment i.e. 100 mg/kg TAF+FTC NP formulation PK study (supplementary table 2) reveals that at the site of infection (i.e. vaginal tissue) FTC drugs concentration after day 4 (i.e. day 7and 14) was under detection limit of LC-MS/MS.
Table 1.
PK parameters values obtained from non-compartmental modeling of LC-MS/MS data.
| Tissue type |
PK parameter | TAF+FTC NP (200 mg/kg) |
TAF+FTC solution (200 mg/kg) |
|||
|---|---|---|---|---|---|---|
| TFV | FTC | TFV | FTC | |||
| Plasma | Cmax (ng/mL) | 15954 ± 2,716# | 34026 ± 3,140# | 942 ± 137 | 850 ± 242 | |
| AUCall (Day×μg/mL) | 2.5 ± 0.4 | 3.9 ± 0.8 | 0.6 ± 0.08 | 0.2 ± 0.05 | ||
| CL (L/Day/kg) | 78.7 | 49.7 | 335.0 | 1031 | ||
| Vd (L/kg) | 145.6 | 204.3 | 285.3 | 149.3 | ||
| t1/2 (h) | 30.8 | 68.4 | 14.2 | 2.4 | ||
| MRTlast (h) | 13.5 | 13.5 | 13.4 | 5.9 | ||
| Liver | Cmax (μg/g) | 454.1 ± 15.9# | 50.2 ± 2.2# | 145.7 ± 13.2 | 1.8 ± 0.4 | |
| AUCall(Day×μg/g) | 138.7 ± 22.4 | 4.6 ± 0.2 | 92.3 ± 15.1 | 0.6 ± 0.1 | ||
| CL (g/Day/kg) | 1440 | 41709 | 2166 | 341035 | ||
| Vd (g/kg) | 5005 | 402295 | 698.4 | 287890 | ||
| MRTlast (h) | 10.6 | 10.7 | 11.1 | 12.7 | ||
| Spleen | Cmax (μg/g) | 49.1 ± 2.7# | 42.3 ± 0.8# | 3.5 ± 0.4 | 5.1 ± 1.9 | |
| AUCall(Day×μg/g) | 29.5 ± 3.0# | 6.7 ± 0.4 | 3.5 ± 0.8 | 3.4 ± 0.3 | ||
| CL (g/Day/kg) | 6709 | 28263 | 55231 | 48082 | ||
| Vd (g/kg) | 18242 | 247557 | 50110 | 71287 | ||
| MRTlast (h) | 41.1 | 27.6 | 20.7 | 18.8 | ||
| Colon | Cmax (μg/g) | 19.0 ± 1.9# | 28.2 ± 0.3# | 6.4 ± 0.9 | 2.2 ± 1.1 | |
| AUCall(Day×μg/g) | 28.7 ± 3.5# | 4.3 ± 0.6# | 7.3 ± 0.6 | 0.7 ± 0.2 | ||
| CL (g/Day/kg) | 6948 | 45284 | 25149 | 277926 | ||
| Vd (g/kg) | 50527 | 187930 | 26875 | 201348 | ||
| MRTlast (h) | 27.2 | 53.5 | 22.3 | 8.9 | ||
| Vagina | Cmax (μg/g) | 58.4 ± 23.4# | 54.1 ± 13.2# | 4.0 ± 1.1 | 4.5 ± 1.0 | |
| AUCall(Day×μg/g) | 10.9 ± 2.1 | 5.4 ± 1.1 | 2.2 ± 0.4 | 1.6 ± 0.4 | ||
| CL (g/Day/kg) | 18345 | 35791 | 92448 | 118762 | ||
| Vd (g/kg) | 46675 | 256605 | 39558 | 91640 | ||
| t1/2(h) | 42.3 | 119.3 | 6.5 | 12.8 | ||
| MRTlast (h) | 26.3 | 13.6 | 10.0 | 11.2 | ||
| Drug level (ng/G) | Day 3 | * | * | ND | 68 ± 17 | |
| Day 4 | 163.7 ± 44.9 | 19.4 ± 18.6 | * | * | ||
| Day 7 | 53.8 ± 14.0 | 90.2 ± 47.3 | * | * | ||
| Day | 11.1 ± 2.2 | ND | * | * | ||
| PBMC (active metabolites) | Cmax(pmol/106 cells) | 182 ± 60# | 182 ± 27# | 10.1 ± 9.5 | 6.2 ± 5.7 | |
| AUCall(Day×pmol) | 215.8 ± 46.7# | 88.6 ± 8.9# | 9.5 ± 2.6 | 8.7 ± 3.7 | ||
Data presented as mean ± SE; PK = Pharmacokinetic; ND=not detected; Cmax = maximum concentration; AUCall = area under the concentration-time curve; TAF= tenofovir alafenamide; TFV= tenofovir; FTC= emtricitabine; NP= nanoparticle; CL = Clearance; Vd = Volume of Distribution; MRT = Mean Residence Time;
indicates sample collection not scheduled for particular time point/treatment.
indicates p < 0.05.
Figure 1.

The TFV (black) and FTC (red) concentration-time curves in vagina (A), respective active metabolites i.e. TFV-dp (black) and FTC-tp (red) in PBMCs (B), plasma (C), colon (D), obtained from hu-NSG mice receiving each drug at 200 mg/kg as TAF+FTC NP and the respective insert graphs presents the same studies on mice that received each drug at 200 mg/kg as TAF+FTC solution.
The non-compartmental analysis demonstrated prolonged plasma elimination half-life (t1/2) for both TFV and FTC from NP formulation, 31 and 68 h compared to 14.2 and 2.4 h for drugs in solution, respectively. Importantly, the NP formulation produced plasma AUCall that were 4.2 and 19.5 times higher for TFV and FTC than the drugs in solution at the same dosage, respectively (Table 1). Further, nano-encapsulation prolonged the vaginal tissue half-life averaging 6.5- and 9.3-fold higher for TFV and FTC, respectively compared to solution. Similarly, the NP formulation resulted in mean Cmax TFV-dp level was 18-fold higher and FTC-tp 29-fold higher compared to drugs in solution (Table 1 and Figure 1B).
At the site of infection, i.e. vaginal tissue the elimination half-lives (t1/2) of TFV and FTC were 42.3 and 119.3 h, respectively for NP formulation whereas TAF+FTC solution averaged 6.5 and 12.8 h. The vaginal tissue TFV levels averaged 164, 54, and 11 ng/G on days 4, 7, and 14 (Table 1 and Figure 1A). Mean FTC concentrations for the same days post-injection averaged 19, 90 ng/G, and not detected, respectively. In all of the tissues tested, the biodistribution of TFV and FTC upon TAF+FTC NP treatment, was enhanced compared to TAF+FTC in solution (Table 1 and Supplementary Table 3).
Further, at sites of HIV-1 infection (i.e. vaginal tissue and colon), simultaneous correlation analysis was performed between plasma vs vaginal or colon tissue for TAF+FTC NPs (Figure 2). Interestingly, correlation analysis reveals FTC has positive deviation towards vaginal tissue reflecting FTC's higher tendency to accumulate in the vaginal tissue compared to TFV. Tenofovir from TAF NPs tends to be retained in plasma (negative deviation towards vaginal tissue) compared to vaginal tissue (Figure 2A). However, colonic tissue FTC and TFV from TAF+FTC NPs assimilation correlation analysis demonstrated both drugs had positive deviation towards colon tissue (Figure 2B). Therefore, both drugs tend to accumulate more in the colon compared to plasma. This accumulation phenomenon confirms Kashuba et al. study report demonstrating plasma FTC obtains higher accumulation in vaginal tissues, whereas TFV accumulates in the colorectal tissues [27, 28].
Figure 2.

Correlation study between the concentration of TAF (black dots and line) and FTC (red dots and line) in plasma vs vaginal tissue (A) or colonic tissue (B) of TAF+FTC NP treated mice. Each dot represents respective drug concentrations simultaneously in plasma and tissue from each mouse under study.
PrEP experiments
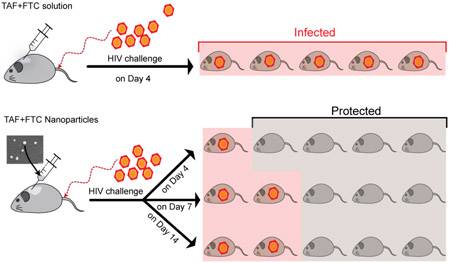
To evaluate PrEP efficacy, the hu-BLT mice were vaginal challenged with HIV-1 at different time-points PT with TAF+FTC NP (Figure 3A). The TAF+FTC NP treated mice demonstrated 80% protection when challenged at day 4; however, day 7 and 14 PT challenge attained 60% protection. Compared to control hu-BLT, mice treated with 200 mg/kg TAF+FTC NP were significantly (p=0.0002; Mantel-Cox log-rank test) protected (figure 3B). Of interest, the only day 4 challenged mouse that became infected, became infected after 28 days PT. These results demonstrate the long-acting potential of the NP formulation. This could have ramifications for monitoring PrEP patients with LA preparations. In contrast, all hu-BLT mice that received 100 mg/kg TAF+FTC NPs or 200 mg/kg TAF+FTC in solution became HIV-infected by Day 21 regardless of when the groups were infected (day 4, 7, or 14 PT), (Supplementary Figure 5).
Figure 3.

The protection study design schedule (A) and the Kaplan-Meier infectivity curve (B) of Hu-BLT mice receiving each drug at 200 mg/kg as TAF+FTC NPs or as TAF+FTC solution. Each mice group (n=5) were intravaginal challenged with T/F HIV-1 strain on 4, 7, or 14 days after PT of TAF+FTC NPs. The untreated-infected control mice group (n=5) and TAF+FTC solution group (n=5), were challenged on Day 4 post-SubQ treatment (PT).
To further verify the presence of vRNA at tissue level, the spleen, and lymph nodes were harvested post-experiment and analyzed for HIV-1 RNA using ISH (figure 4). The ISH results confirmed that those mice having > 800 copies/mL pVL, were also HIV-positive at the tissue level, whereas all the mice with non-detectable pVL were found to be HIV-negative.
Figure 4.

ISH detection of HIV-1 vRNA in lymph node and spleen of control and treated mice. The black silver grains clusters in the image represent the HIV-1 vRNA positive cells area. The tissue sections under consideration were H&E stained and vRNA hybridized was performed by 35S-labeled HIV anti-sense riboprobes that covers > 90% of HIV-1 genome. Day 4 tissues serve as representative for Day 14 HIV-protected animals. The representative untreated-infected control lymph node and spleen tissue section (row) were compared with respective tissues obtained from TAF+FTC NP treated and infected on day 4, 7 and 14 post-SubQ treatment (PT). The day 14 PT infected image represents tissue from infected mouse. The scale bar in the white (in first image) corresponds to 100 μm for all images.
Discussion:
A LA injectable delivery system could be an appropriate formulation for PrEP that could potentially maximize adherence and PrEP efficacy, in high-risk HIV patients. Previous studies with injectable, LA cabotegravir in macaques have documented SHIV protection [29, 30]. The vaginal tissue cabotegravir levels were above the protein-adjusted-inhibitory concentration for 90% of isolates (PA-IC90).
Maintenance of plasma ARV drug concentration is only possible when volunteers remain adherent to daily TDF+FTC regimen [27]. Due to non-adherence, several clinical trials in women have shown a lack of efficacy leading to early study termination [7, 8]. The aim of this research is to provide the LA platform to the FDA-approved ARVs. Evidence is accumulating that there is a correlation between the ARV drug plasma level and efficacy. However, significant differences in NRTI drug levels in vaginal tissue compared to rectal tissue were observed after oral administration of Truvada™[31]. Microbicide Trials Network (MTN-001) compared vaginal tissue TFV levels after oral and vaginal applications in HIV-negative women. The results demonstrated vaginal application of TFV gel achieved 130-times higher levels of TFV active metabolite (TFV-dp) in vaginal tissue compared to oral ingestion [32]. Yet, the 1% TFV gel demonstrated 39% protection possibly due to sporadic adherence [31]. Thus, even topical dosing indicates adherence is primarily important for TFV antiviral activity and could be responsible for the less than optimal results in clinical trials. The Partners-in-PrEP clinical trial randomized oral TDF or TDF/FTC PrEP among HIV-uninfected members of serodiscordant couples [2]. Plasma TFV levels ≥ 40 ng/mL were associated with an estimated 88% protective effect of PrEP against HIV-1 if tenofovir alone was ingested and 91% if TDF/FTC was ingested [33]. Therefore, to examine plasma and vaginal drug levels, a novel PrEP strategy was developed for LA PrEP.
To date, there is no literature clearly reporting vaginal TFV concentration that is necessary for protection against HIV-1. A study by Patterson et al. reported vaginal tissue concentrations after a single oral dose of Truvada™ demonstrated TFV concentration to be 6.8 ng/G on day 1 PT in the vaginal tissue [34]. Our PK study demonstrated 11.1 ± 2.2 ng/G on day 14 PT in the vagina (Table 1), which is comparable to the day 1 single oral dose of Truvada™. Therefore, the nanoencapsulation of ARV drugs substantially prolongs the ARV levels in tissues and supports the LA potential of the ARV NP. Veselinovic et al. determined TFV levels at steady-state after oral gavage of TDF (61.5 mg/kg) in humanized and non-humanized mice [35]. Median peak vaginal TFV level was reported to be 729 ng/G after oral administration in Rag(2) knockout mice. The current study demonstrates 200 mg/kg SubQ administration of TAF+FTC NPs led to higher accumulation of TFV (mean Cmax, 58 μg/G) in vaginal tissues (Table 1). Therefore, higher and more consistent ARV drug levels within vaginal tissue due to nanoencapsulation could potentially offer better protection.
The in-vitro studies show that nanoencapsulation of TAF+FTC NP reduces cytotoxic effect of the drug at higher concentrations (Supplementary Figure 4). Therefore, nanoencapsulation introduces properties like wide therapeutic-toxic window and improved intracellular retention/biphasic endosomal release (Supplementary Figure 3, 4) that could further improve the intracellular concentrations of TFV and FTC to inhibit viruses [16]. Another rate limiting factor for NRTI’s, is the conversion to their active forms to maintain intracellular metabolite concentration [16]. This study demonstrates, nanoencapsulation also improves intracellular drug delivery and active-drug conversion rate to maintain the drug metabolism dominance (Supplementary Figure 3). All through the study-period, the ARV drugs and metabolites demonstrated improved TAF/TFV and FTC accumulation and retention compared to solution. Therefore, nanocapsulation potentially could improve the high drug/active-drug concentration that could contribute to constantly maintain the effective drug-metabolite concentration needed for effective inhibition of reverse transcriptase activity.
The goal of this study was to comparatively determine the sustained-release efficacy of our novel LA ARVs nanoformulation to prevent vaginal HIV-1 infection in hu-BLT mouse model. In parallel, we evaluated tissue PK at the site of infection in hu-CD34-NSG mice. To the best of our knowledge, this is the first report of combining TAF+FTC in a single nanoformulation as a potential LA PrEP delivery modality for protection from HIV-1 infection. Furthermore, this is the first report of conducting simultaneous tissue PK for TFV and FTC as well as the active metabolites of TFV and FTC in PBMCs. For nanoformulated LA ARV loaded NP, we chose TAF/FTC combination over TDF/FTC, as TAF has been reported to be as effective as TDF in HIV-1 treatment but has shown less toxicity to kidneys and bone [11, 16]. Systemically, TAF penetrates into tissue better than TDF [36]. However, TAF/FTC is currently not FDA-approved for PrEP mainly due to low vaginal drug levels.
The major drawback of TAF/FTC coformulation was the low drug concentrations at the site of infection (i.e. cervical tissue or rectal tissue) compared to plasma concentrations [37]. However, present report evidently shows that nanoencapsulation enhances TAF/FTC penetration both in vaginal tissue and rectal/colon tissue when compared with solution (Figure 1A and D). The correlation analysis further shows FTC shows higher assimilation in the vaginal tissue. Interestingly, in colon tissue both TFV and FTC tends to assimilation more compared to plasma (Figure 2). Therefore, ARV nanoencapsulation potentially could enhance vaginal and rectal ARV drug concentration, overcoming the drawback of TAF/FTC coformulation for PrEP application.
The PK study augmented the TAF+FTC NP protection studies against HIV-1 challenge. Mean TFV and FTC vaginal drug levels from 200 mg/kg NP dosage on day 4 were 163.7 ng/G and 19.4 ng/G, respectively and that resulted in 80% protection (Figure 1 and 2). Mean PBMC TFV-dp and FTC-tp levels on day 4 for 200 mg/kg TAF+FTC NP dosage averaged 32.2 pmol/106 cells and 1.4 pmol/106 cells, respectively. Mean active metabolite levels from 200 mg/kg TAF+FTC solution on the last day (day 3) were 1.3 pmole/106 cells and 0.5 pmol/106 cells, for the TFV-dp and FTC-tp respectively. Anderson et al. demonstrated 30.7 fmol/106 PBMCs of TFV-dp produced 97% PrEP protection in a macaque model of rectal simian HIV [38]. Additionally, a TFV-dp concentration in PBMCs of 16 fmol/106 cells was associated with a 90% reduction in HIV acquisition in the iPrEx trial [4]. Using a chimeric animal model may not entirely mimic the human condition but we strived to use hu-BLT mice with > 50% human CD45 cells. Certainly, investigating the nanoformulation in a larger animal model would lend to a more accurate assessment of HIV-1 protection when assessing the efficacy of these combination nanoparticles. Even though, nanoformulation prolongs PK parameters of conventional ARVs by protecting it from non-specific degradation and clearance, the main drawback of nanoformulation is low drug to excipient (in present case polymers) ratio. Therefore, injection of a drug amount leads to a 10 times higher amount of excipient injection. Although, PLGA used for present nanoformulation is biocompatible and biodegradable, the presence of high polymeric components limits the amount of drug encapsulated per injection volume. To overcome this limitation, we are introducing minute modifying to nanoformulation to achieve enhanced %DL (~30 %).
This preliminary proof-of-concept study demonstrated nanoencapsulation of TAF+FTC in FDA-approved polymer prolonged the drug retention in humanized mice i.e. up to 14 days of study after a single SubQ dose. Moreover, these experiments demonstrate prolonged detectable drug concentrations substantiate HIV-1 protection over time, i.e. day 4 PT HIV challenge attended 80% protection and 60% protection achieved upon day 7 or 14 PT challenge. Therefore, LA TAF/FTC NP could overcome the drawback of oral TAF/FTC for PrEP as well as its LA potency could improve adherence. In future studies it would be interesting to study the PrEP efficacy study of LA TAF/FTC NP in larger animals such as macaques to closely evaluate efficacy in human. Additionally, HIV protection efficacy research using other ARV combination nanoencapsulated would be of interest. Also, an interesting future study could be SubQ delivery of the combination ARV nanoformulations for rectal challenge with HIV-1.
Supplementary Material
Supplementary Figure 1. Scanning electron microscopy (SEM) image of TAF+FTC NPs.
Supplementary Figure 2. TAF (A) and FTC (B) release kinetics from TAF+FTC NP in simulated endosomal fluid (pH 5.5). For the graph fitting, one-phase decay non-linear regression equation was used that analyzed the half-life, effective release constant (teff.) and plateau release phase. The release rate constant, ‘K’ for TAF and FTC was estimated to be 2.65 h−1 and 4.07 h−1. Each data point represents the mean ± SE obtained from three independent batche study (n=3).
Supplementary Figure 3. Intracellular uptake, conversion and retention kinetics study. (A) TAF uptake and retention kinetics; (B) TAF to TFV conversion and retention kinetics; (C) TFV to TFV-DP conversion and retention kinetics; (D) FTC uptake and retention kinetics; (E) FTC to FTC-TP conversion and retention kinetics. In all graphs, the left inverted bracket depicts the 24 h treatment period monitoring uptake, release and conversion kinetics of respective drugs (TAF or FTC) to respective active drug (TFV) or respective metabolites (TFV-dp or FTC-tp). Whereas, the right inverted bracket illustrates respective pro-drug (TAF), active drugs (TFV, FTC) and metabolites (TFV-DP or FTC-TP) retention period for another 96 h (120 h in total). The data represented as mean ± SE of data obtained from three independent batches (n=3), studied in triplicate.
Supplementary Figure 4. Cytotoxicity evaluation of TAF+FTC NP vs TAF+FTC solution on TZMbl cells at different concentrations (20, 10, 1, 0.1, 0.01 μg/mL) after 96 h of treatment. The normalized data against untreated control are represented as mean ± SE of 3 independent experiments (n=3, each experiment in triplicate).
Supplementary Figure 5. Protection efficiency evaluation based on Kaplan-Meier infectivity curve obtained from groups of Hu-BLT mice (n=5) SubQ received TAF+FTC NPs (each drug i.e. TAF or FTC @100 mg/kg). These treated mice were further challenged with T/F HIV-1 strain on 4, 7, or 14 days after SubQ respective treatment administration. Similarly, TAF+FTC solution (each drug i.e. TAF or FTC @100 mg/kg) treated mice (n=5) and untreated control mice (SubQ injection of D5W, n=5) groups were challenged on Day 4 after respective treatment.
Supplementary Table 1. Physicochemical properties of TAF+FTC NP and its stability after one year.
Supplementary Table 2. Pharmacokinetic parameters from non-compartmental modeling.
Supplementary Table 3. Mean ± SE of AUCall and Cmax of TFV and FTC from NP and solution (200 mg/kg treated concentration) in selected tissues.
Acknowledgements:
We thank Jim Rooney, M. D. and Gilead Sciences, Inc. for donation of the TAF and FTC drug powders. The work and publication have been funded by NIAID R01AI117740, 2015 (awarded to C.J.D.). No conflict of interest associated with any of the authors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].UNAIDS Global Statistics - 2015, in: UNAIDS Factsheet, 2016. [Google Scholar]
- [2].Baeten JM, Donnell D, Mugo NR, Ndase P, Thomas KK, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kidoguchi L, Coombs RW, Hendrix C, Marzinke MA, Frenkel L, Haberer JE, Bangsberg D, Celum C, Partners Pr EPST, Single-agent tenofovir versus combination emtricitabine plus tenofovir for pre-exposure prophylaxis for HIV-1 acquisition: an update of data from a randomised, double-blind, phase 3 trial, Lancet Infect Dis, 14 (2014) 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Baeten JM, Donnell D, Ndase P, Mugo NR, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kakia A, Odoyo J, Mucunguzi A, Nakku-Joloba E, Twesigye R, Ngure K, Apaka C, Tamooh H, Gabona F, Mujugira A, Panteleeff D, Thomas KK, Kidoguchi L, Krows M, Revall J, Morrison S, Haugen H, Emmanuel-Ogier M, Ondrejcek L, Coombs RW, Frenkel L, Hendrix C, Bumpus NN, Bangsberg D, Haberer JE, Stevens WS, Lingappa JR, Celum C, Partners Pr EPST, Antiretroviral prophylaxis for HIV prevention in heterosexual men and women, N Engl J Med, 367 (2012) 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernandez T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallas EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV, iPrEx Study T, Preexposure chemoprophylaxis for HIV prevention in men who have sex with men, N Engl J Med, 363 (2010) 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thigpen MC, Kebaabetswe PM, Paxton LA, Smith DK, Rose CE, Segolodi TM, Henderson FL, Pathak SR, Soud FA, Chillag KL, Mutanhaurwa R, Chirwa LI, Kasonde M, Abebe D, Buliva E, Gvetadze RJ, Johnson S, Sukalac T, Thomas VT, Hart C, Johnson JA, Malotte CK, Hendrix CW, Brooks JT, Group TDFS, Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana, N Engl J Med, 367 (2012) 423–434. [DOI] [PubMed] [Google Scholar]
- [6].Choopanya K, Martin M, Suntharasamai P, Sangkum U, Mock PA, Leethochawalit M, Chiamwongpaet S, Kitisin P, Natrujirote P, Kittimunkong S, Chuachoowong R, Gvetadze RJ, McNicholl JM, Paxton LA, Curlin ME, Hendrix CW, Vanichseni S, Bangkok Tenofovir Study G, Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, double-blind, placebo-controlled phase 3 trial, Lancet, 381 (2013) 2083–2090. [DOI] [PubMed] [Google Scholar]
- [7].Marrazzo JM, Ramjee G, Richardson BA, Gomez K, Mgodi N, Nair G, Palanee T, Nakabiito C, van der Straten A, Noguchi L, Hendrix CW, Dai JY, Ganesh S, Mkhize B, Taljaard M, Parikh UM, Piper J, Masse B, Grossman C, Rooney J, Schwartz JL, Watts H, Marzinke MA, Hillier SL, McGowan IM, Chirenje ZM, Team VS, Tenofovir-based preexposure prophylaxis for HIV infection among African women, N Engl J Med, 372 (2015) 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Van Damme L, Corneli A, Ahmed K, Agot K, Lombaard J, Kapiga S, Malahleha M, Owino F, Manongi R, Onyango J, Temu L, Monedi MC, Mak'Oketch P, Makanda M, Reblin I, Makatu SE, Saylor L, Kiernan H, Kirkendale S, Wong C, Grant R, Kashuba A, Nanda K, Mandala J, Fransen K, Deese J, Crucitti T, Mastro TD, Taylor D, Group FE-PS, Preexposure prophylaxis for HIV infection among African women, N Engl J Med, 367 (2012) 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Molina JM, Capitant C, Spire B, Pialoux G, Cotte L, Charreau I, Tremblay C, Le Gall JM, Cua E, Pasquet A, Raffi F, Pintado C, Chidiac C, Chas J, Charbonneau P, Delaugerre C, Suzan-Monti M, Loze B, Fonsart J, Peytavin G, Cheret A, Timsit J, Girard G, Lorente N, Preau M, Rooney JF, Wainberg MA, Thompson D, Rozenbaum W, Dore V, Marchand L, Simon MC, Etien N, Aboulker JP, Meyer L, Delfraissy JF, Group AIS, On-Demand Preexposure Prophylaxis in Men at High Risk for HIV-1 Infection, N Engl J Med, 373 (2015) 2237–2246. [DOI] [PubMed] [Google Scholar]
- [10].Eisingerich AB, Wheelock A, Gomez GB, Garnett GP, Dybul MR, Piot PK, Attitudes and acceptance of oral and parenteral HIV preexposure prophylaxis among potential user groups: a multinational study, PloS one, 7 (2012) e28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ray AS, Fordyce MW, Hitchcock MJ, Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus, Antiviral Res, 125 (2016) 63–70. [DOI] [PubMed] [Google Scholar]
- [12].Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T, Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases, Mol Pharmacol, 74 (2008) 92–100. [DOI] [PubMed] [Google Scholar]
- [13].Mascolini M, Oral TAF/FTC PrEP Prevents Vaginal SHIV Infection in Monkeys in: Gilead (Ed.) Conference on Retroviruses and Opportunistic Infections (CROI), NATAP, Boston, Massachusetts, 2018. [Google Scholar]
- [14].Garrett KL, Cottrell ML, Prince HM, Sykes C, Schauer A, Peery A, Rooney J, McCallister S, Gay C, Kashuba A, Concentrations of TFV and TFVdp in female mucosal tissues after a single dose of TAF., in: Gilead (Ed.) Conference on Retroviruses and Opportunistic Infections (CROI), CROI, Boston, MA, 2016. [Google Scholar]
- [15].Lerma GG, Tenofovir alafenamide PrEP protects monkeys from infection, but rectal and vaginal levels in humans may be too low, in: AIDS Map, NAM Publications, London, WC, 2016. [Google Scholar]
- [16].Holec AD, Mandal S, Prathipati PK, Destache CJ, Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics, Curr HIV Res, 15 (2017) 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mandal S, Prathipati PK, Kang G, Zhou Y, Yuan Z, Fan W, Li Q, Destache CJ, Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission, AIDS, 31 (2017) 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mandal S, Belshan M, Holec A, Zhou Y, Destache CJ, An Enhanced Emtricitabine-Loaded Long-Acting Nanoformulation for Prevention or Treatment of HIV Infection, Antimicrob Agents Chemother, 61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prathipati PK, Mandal S, Destache CJ, Simultaneous quantification of tenofovir, emtricitabine, rilpivirine, elvitegravir and dolutegravir in mouse biological matrices by LC-MS/MS and its application to a pharmacokinetic study, J Pharm Biomed Anal, 129 (2016) 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bioanalytical Method Validation in Guidance for Industry, in: F.a.D.A. Department of Health and Human Services (Ed.), CVM, CDER, 2013, pp. 2–9. [Google Scholar]
- [21].Roncarolo MG, Carballido JM, Construction of human-SCID chimeric mice, Current protocols in immunology, Chapter 4 (2001) Unit 4 8. [DOI] [PubMed] [Google Scholar]
- [22].Wang LX, Kang G, Kumar P, Lu W, Li Y, Zhou Y, Li Q, Wood C, Humanized-BLT mouse model of Kaposi's sarcoma-associated herpesvirus infection, Proceedings of the National Academy of Sciences of the United States of America, 111 (2014) 3146–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guide for the Care and Use of Laboratory Animals (8th ed.), The National Academic Press, Washington, D.C., 2011. [Google Scholar]
- [24].Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT, Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells, Nature, 434 (2005) 1148–1152. [DOI] [PubMed] [Google Scholar]
- [25].Li Q, Gebhard K, Schacker T, Henry K, Haase AT, The relationship between tumor necrosis factor and human immunodeficiency virus gene expression in lymphoid tissue, J Virol, 71 (1997) 7080–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Makadia HK, Siegel SJ, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers, 3 (2011) 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cohen MS, Muessig KE, Smith MK, Powers KA, Kashuba AD, Antiviral agents and HIV prevention: controversies, conflicts, and consensus, AIDS (London, England: ), 26 (2012) 1585–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nicol MR, Kashuba AD, Pharmacologic opportunities for HIV prevention, Clin Pharmacol Ther, 88 (2010) 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Andrews CD, Yueh YL, Spreen WR, St Bernard L, Boente-Carrera M, Rodriguez K, Gettie A, Russell-Lodrigue K, Blanchard J, Ford S, Mohri H, Cheng-Mayer C, Hong Z, Ho DD, Markowitz M, A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge, Sci Transl Med, 7 (2015) 270ra274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Radzio J, Spreen W, Yueh YL, Mitchell J, Jenkins L, Garcia-Lerma JG, Heneine W, The long-acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge, Sci Transl Med, 7 (2015) 270ra275. [DOI] [PubMed] [Google Scholar]
- [31].Kashuba AD, Gengiah TN, Werner L, Yang KH, White NR, Karim QA, Abdool Karim SS, Genital Tenofovir Concentrations Correlate With Protection Against HIV Infection in the CAPRISA 004 Trial: Importance of Adherence for Microbicide Effectiveness, Journal of acquired immune deficiency syndromes (1999), 69 (2015) 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hendrix CW, Chen BA, Guddera V, Hoesley C, Justman J, Nakabiito C, Salata R, Soto-Torres L, Patterson K, Minnis AM, Gandham S, Gomez K, Richardson BA, Bumpus NN, MTN-001: randomized pharmacokinetic cross-over study comparing tenofovir vaginal gel and oral tablets in vaginal tissue and other compartments, PloS one, 8 (2013) e55013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Donnell D, Baeten JM, Bumpus NN, Brantley J, Bangsberg DR, Haberer JE, Mujugira A, Mugo N, Ndase P, Hendrix C, Celum C, HIV protective efficacy and correlates of tenofovir blood concentrations in a clinical trial of PrEP for HIV prevention, Journal of acquired immune deficiency syndromes (1999), 66 (2014) 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang H, Lu X, Yang X, Xu N, The efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate in antiretroviral regimens for HIV-1 therapy: Meta-analysis, Medicine, 95 (2016) e5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Patterson KB, Prince HA, Kraft E, Jenkins AJ, Shaheen NJ, Rooney JF, Cohen MS, Kashuba AD, Penetration of tenofovir and emtricitabine in mucosal tissues: implications for prevention of HIV-1 transmission, Science translational medicine, 3 (2011) 112re114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Veselinovic M, Yang KH, LeCureux J, Sykes C, Remling-Mulder L, Kashuba ADM, Akkina R, HIV pre-exposure prophylaxis: mucosal tissue drug distribution of RT inhibitor Tenofovir and entry inhibitor Maraviroc in a humanized mouse model, Virology, 464–465 (2014) 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cottrell ML, Garrett KL, Prince HMA, Sykes C, Schauer A, Emerson CW, Peery A, Rooney JF, McCallister S, Gay C, Kashuba ADM, Single-dose pharmacokinetics of tenofovir alafenamide and its active metabolite in the mucosal tissues, J Antimicrob Chemother, 72 (2017) 1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Anderson PL, Glidden DV, Bushman LR, Heneine W, Garcia-Lerma JG, Tenofovir diphosphate concentrations and prophylactic effect in a macaque model of rectal simian HIV transmission, J Antimicrob Chemother, 69 (2014) 2470–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Scanning electron microscopy (SEM) image of TAF+FTC NPs.
Supplementary Figure 2. TAF (A) and FTC (B) release kinetics from TAF+FTC NP in simulated endosomal fluid (pH 5.5). For the graph fitting, one-phase decay non-linear regression equation was used that analyzed the half-life, effective release constant (teff.) and plateau release phase. The release rate constant, ‘K’ for TAF and FTC was estimated to be 2.65 h−1 and 4.07 h−1. Each data point represents the mean ± SE obtained from three independent batche study (n=3).
Supplementary Figure 3. Intracellular uptake, conversion and retention kinetics study. (A) TAF uptake and retention kinetics; (B) TAF to TFV conversion and retention kinetics; (C) TFV to TFV-DP conversion and retention kinetics; (D) FTC uptake and retention kinetics; (E) FTC to FTC-TP conversion and retention kinetics. In all graphs, the left inverted bracket depicts the 24 h treatment period monitoring uptake, release and conversion kinetics of respective drugs (TAF or FTC) to respective active drug (TFV) or respective metabolites (TFV-dp or FTC-tp). Whereas, the right inverted bracket illustrates respective pro-drug (TAF), active drugs (TFV, FTC) and metabolites (TFV-DP or FTC-TP) retention period for another 96 h (120 h in total). The data represented as mean ± SE of data obtained from three independent batches (n=3), studied in triplicate.
Supplementary Figure 4. Cytotoxicity evaluation of TAF+FTC NP vs TAF+FTC solution on TZMbl cells at different concentrations (20, 10, 1, 0.1, 0.01 μg/mL) after 96 h of treatment. The normalized data against untreated control are represented as mean ± SE of 3 independent experiments (n=3, each experiment in triplicate).
Supplementary Figure 5. Protection efficiency evaluation based on Kaplan-Meier infectivity curve obtained from groups of Hu-BLT mice (n=5) SubQ received TAF+FTC NPs (each drug i.e. TAF or FTC @100 mg/kg). These treated mice were further challenged with T/F HIV-1 strain on 4, 7, or 14 days after SubQ respective treatment administration. Similarly, TAF+FTC solution (each drug i.e. TAF or FTC @100 mg/kg) treated mice (n=5) and untreated control mice (SubQ injection of D5W, n=5) groups were challenged on Day 4 after respective treatment.
Supplementary Table 1. Physicochemical properties of TAF+FTC NP and its stability after one year.
Supplementary Table 2. Pharmacokinetic parameters from non-compartmental modeling.
Supplementary Table 3. Mean ± SE of AUCall and Cmax of TFV and FTC from NP and solution (200 mg/kg treated concentration) in selected tissues.