

Abstract
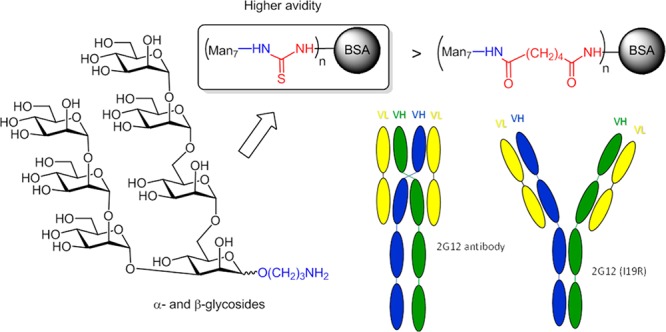
Novel neoglycoproteins containing oligomannosidic penta- and heptasaccharides as structural variants of oligomannose-type N-glycans found on human immunodeficiency virus type 1 gp120 have been prepared using different conjugation methods. Two series of synthetic ligands equipped with 3-aminopropyl spacer moieties and differing in the anomeric configuration of the reducing mannose residue were activated either as isothiocyanates or as adipic acid succinimidoyl esters and coupled to bovine serum albumin. Coupling efficiency for adipic acid connected neoglycoconjugates was better than for the thiourea-linked derivatives; the latter constructs, however, exhibited higher reactivity toward antibody 2G12, an HIV-neutralizing antibody with exquisite specificity for oligomannose-type glycans. 2G12 binding avidities for the conjugates, as determined by Bio-Layer Interferometry, were mostly higher for the β-linked ligands and, as expected, increased with the numbers of covalently linked glycans, leading to approximate KD values of 10 to 34 nM for optimized ligand-to-BSA ratios. A similar correlation was observed by enzyme-linked immunosorbent assays. In addition, dendrimer-type ligands presenting trimeric oligomannose epitopes were generated by conversion of the amino-spacer group into a terminal azide, followed by triazole formation using “click chemistry”. The severe steric bulk of the ligands, however, led to poor efficiency in the coupling step and no increased antibody binding by the resulting neoglycoconjugates, indicating that the low degree of substitution and the spatial orientation of the oligomannose epitopes within these trimeric ligands are not conducive to multivalent 2G12 binding.
Introduction
Glycosidic epitopes present on the gp120 component of the human immunodeficiency virus (HIV) type 1 envelope protein (Env) constitute major targets for broadly neutralizing antibodies (bnAbs).1−4 Part of this glycan “canopy” is composed mainly of high-mannose glycans, ranging from Man5GlcNAc2 to Man9GlcNAc2, and is referred to also as the “mannose patch”.5−7 Significant effort has been expended in the production of synthetic glycans and glycopeptides to mimic the high glycan density on the viral surface.8,9 Examples include Man9GlcNAc2 ligands conjugated to the Neisseria meningitidis outer membrane protein complex via a cyclic peptide scaffold,10 multivalent attachment of Man9 to the virus-like particle bacteriophage Qβ11 and dendrimeric oligomannoside ligands linked to bovine serum albumin (BSA),12 keyhole limpet hemocyanin (KLH),13,14 or diphtheria toxoid (CRM197).15 In many cases, the copper-assisted cycloaddition reaction between suitable azide and alkyne reactants (“click chemistry”) has been used for covalent attachment of the ligands.11,12,14,16−19
In those cases where immunization studies have been performed with these synthetic antigens, elicited serum antibodies have shown high affinity for their cognate glycans but weak or no binding to natively glycosylated gp120.20,21 The reason(s) for these outcomes are not clear; one possible factor is B-cell tolerance, due to immune recognition of the presented oligomannosides as “self”,22 leading to antibody responses capable of binding the synthetic molecules but not natural oligomannose chains. Furthermore, the clustering of oligomannosides on the synthetic antigens may create immunogenic neo-epitopes that reduce the likelihood of eliciting antibodies with the proper specificity to HIV.
Not long ago, a surrogate of oligomannose glycans on HIV was identified in a bacterial lipooligosaccharide (LOS) fragment. The chemical structure of this lipooligosaccharide, isolated from the phytopathogenic Rhizobium radiobacter Rv3 strain, was elucidated and revealed the presence of an α-Man-(1 → 2)-α-Man-(1 → 2)-α-Man-(1 → 3)-α-Man oligomannose fragment that resembles the D1 arm of HIV oligomannosides (Figure 1A).23 The antigenic similarity to oligomannose was shown by binding to 2G12, the first antiglycan HIV-1 neutralizing antibody described and the only one described so far to exclusively bind oligomannose, specifically the D1 arm.24−27 Notably, immunization of mice with heat-killed bacteria from the Rv3 strain elicited antibodies that were cross-reactive with HIV-1 gp120. Subsequently, a crystal structure of the bacterial carbohydrate backbone complexed to 2G12 was determined, providing additional evidence for the structural homology between the bacterial glycan and mammalian oligomannose on the HIV-1 surface.28 The obtained crystal structure of the bacterial ligand was then used to model and construct ligands that more closely resemble oligomannose, by including a D3-arm surrogate to position 6 of the central mannose unit (Figure 1B).
Figure 1.

(A) Structure of Man9GlcNAc2 N-glycan. (B) Structure of the bacterial lipooligosaccharide isolated from R. radiobacter Rv3. Synthetic extensions are added to the D3-arm (marked in blue). Dashed lines indicate substoichiometric substitution.
Recently, we have communicated the chemical synthesis of the bacterial pentasaccharide LOS core comprising the central α-Man-(1 → 5)-linked Kdo2GlcNAc2 unit29 followed by the synthesis of the oligomannosidic mimetics in both anomeric configurations. (Note: the reducing end mannose is α-linked in the Rhizobium radiobacter Rv3 lipooligosaccharide in contrast to the β-linkage in N-glycoproteins.30) A small library of 2 pentamannosides and 4 heptamannosides has been prepared as spacer-equipped ligands as well as their respective BSA conjugates. The envisaged increased antigenicity of the modified D3-arm was verified in the crystal structure of a heptamannoside ligand bound to PGT128, one of several glycan-specific antibodies described more recently with broad HIV-neutralizing activity.31 Moreover, a BSA neoglycoconjugate containing the heptasaccharide α-Man-(1 → 2)-α-Man-(1 → 2)-α-Man-(1 → 3)-[α-Man-(1 → 2)-α-Man-(1 → 6)-α-Man-(1 → 6)]-β-Man as ligand induced modest neutralizing antibody responses in human-antibody transgenic rats.30
Parallel to our studies to design glycoconjugates that can elicit antibodies of similar specificity and neutralizing activity to PGT128 and related antibodies, we have also set out to assess the impact of these modified bacterial oligomannoside mimetics toward recognition by 2G12. Herein we present the synthesis and 2G12 binding properties of additional neoglycoconjugates, including a clustered multivalent presentation of these oligomannoside epitopes. Specifically, we evaluate the influence of two different spacer groups and conjugation methods with respect to coupling efficiency and antibody binding properties.
Results and Discussion
Synthesis of Thiourea and Adipic Amide Linked Neoglycoconjugates
The previously described30 anomeric oligomannosides 1, 3, 5, 7, 9, and 11, equipped with a 3-aminopropyl spacer group, were directly activated for coupling to BSA. Alternatively they were converted in good to excellent yields into the corresponding 3-azidopropyl derivatives 2, 4, 6, 8, 10, and 12 (Schemes 1 and 2)32 to enable “click chemistry” via 1,3-dipolar cycloaddition reactions.33,34
Scheme 1. Synthesis of Anomeric Mannopentaoside Azidopropyl Spacer Derivatives 2 and 4.

Reagents and conditions: K2CO3, imidazole-1-sulfonyl azide·HCl, 0.1 M CuSO4·5H2O, 2:1 MeOH–H2O, rt 24 h.
Scheme 2. Synthesis of Anomeric Mannoheptaoside Azidopropyl Spacer Derivatives 6, 8, 10, and 12.

Reagents and conditions: K2CO3, imidazole-1-sulfonyl azide·HCl, 0.1 M CuSO4·5H2O, 2:1 MeOH–H2O, rt, 24 h.
The azidopropyl spacer derivatives were purified using gel chromatography on LH-20 resin and fully characterized by NMR and HRMS. Notably, the NMR data of the 3-amino- and 3-azidopropyl group, respectively, in compounds 1–12 indicated a restricted motional freedom for the α-anomeric ligands, as seen from signal splitting of the geminal N-linked methylene protons at ∼3.40 ppm and C-linked CH2 signals in the aliphatic region. The β-anomers, in contrast, consistently showed magnetic equivalence of these protons, indicative of greater motional freedom.
In previous reports, we reacted the terminal amino group-containing oligomannose ligands with thiophosgene and then coupled the resulting isothiocyanates to BSA to yield thiourea-linked neoglycoconjugates (Scheme 3).21,30 To achieve a sufficient ligand density, a high molar excess of the spacer glycoside in the coupling reaction was needed, resulting in ligand to BSA ratios of up to 6.4:1, as measured by MALDI-TOF mass spectrometry.30 The low coupling efficiency of the isothiocyanate conjugation experiments can be explained by a lower concentration of the protein used and the competing, reversible reaction of the activated ligand with sulfhydryl groups leading to unstable dithiocarbamate derivatives.35
Scheme 3. Synthesis of Thiourea-Linked Neoglycoconjugates 13–1830.

As an alternative conjugation method, the 3-aminopropyl spacer derivatives were extended by an adipate linker followed by conversion into BSA conjugates. First, the lead heptasaccharides 5 and 7 were subjected to a two-step procedure starting with disuccinimido adipate (DSAP) reagent 19 to give the activated intermediate monoester derivative followed by reaction with BSA to furnish the adipic bisamide linked neoglycoconjugates 20 and 21, respectively (Scheme 4).36 Conjugation using the adipic amide protocol led to a significantly improved coupling efficiency (Table 1). The coupling efficiency decreased slightly when we attempted to prepare conjugates containing ≥8 oligomannoside moieties, most likely because the most reactive amino groups of the protein had already been substituted (entries 10 and 13). In a previous study, three lysine residues (Lys 235, Lys 437, and Lys 455) had been identified as the most reactive and sterically accessible sites in squaric-acid based synthesis of BSA neoglycoconjugates.37,38 In general, reactivity of the β-linked heptamannoside 7 was slightly better than for its α-anomeric counterpart 5 (entries 1 and 6; 9 and 12), which is consistent with the NMR finding that the α-spacer group had a reduced conformational flexibility (vide supra).
Scheme 4. Synthesis of Adipic Amide-Linked Neoglycoconjugates 20 and 21.

Table 1. Coupling Efficiency of Anomeric Heptamannoside Spacer Derivatives 5 and 7 to BSAc.

Thiourea-based glycoconjugates are marked in blue; adipic acid connected conjugates in red. aMolar ratio of aminopropyl glycoside to BSA used in the conjugation reaction. bEfficiency defined as the percentage of reacted glycoside.
2G12 Binding to Neoglycoconjugates
Apparent binding affinities of the anti-HIV antibody 2G12 for the constructed BSA conjugates, ranging in ligand density from 1.5 to 8.3 ligands per BSA molecule, were determined by Biolayer Interferometry (BLI). In a first series of experiments performed with select thiourea-linked glycoconjugates, 14, 16c, and 17 were identified as being bound most strongly by 2G12, with approximate KD values ranging from 19 to 34 nM (Figure 2). Substantially lower binding was observed for the α-anomeric pentasaccharide conjugate 13 (183 nM) and in particular for the β-anomeric heptasaccharide antigen 18 (394 nM), harboring the artificial α-(1 → 4)-linkage for the D3 arm-like extension. These differences are not explained by differences in coupling density as conjugates 14 and 17, both of which are bound well by 2G12, carry an average number of 3.3 and 3.4 glycans per BSA, respectively, that matches that of the poorly bound conjugates 13 and 18, which contain on average 3.5 and 3.1 glycans per BSA, respectively. Of the β-linked thiourea glycoconjugates assayed in this first set of experiments, compound 16c was bound most strongly, with an apparent KD of 23 nM, which, however, is about an order of magnitude higher than the reported39KD (2.5 nM) for the interaction between 2G12 and HIV-1 gp140. With the exception of conjugate 13, the dissociation rates determined in the BLI experiments displayed a similar trend as the KD values.
Figure 2.
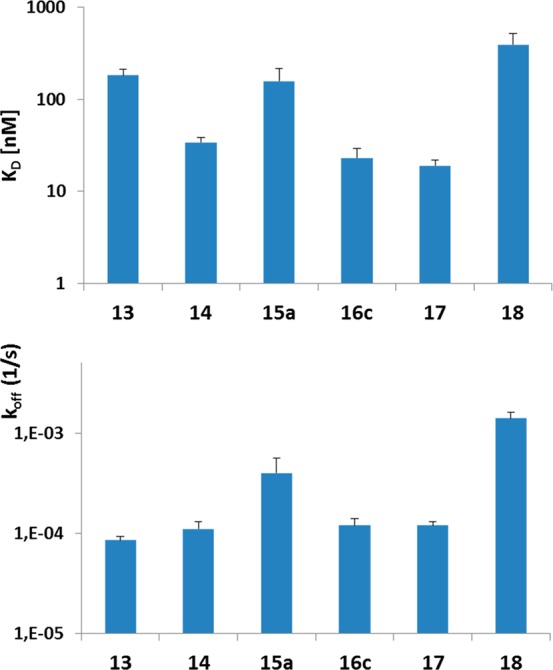
Apparent binding avidities determined by BLI for select thiourea-linked glycoconjugates to 2G12. KD, koff, and SEM (error bars) are based on 3–4 independent experiments, performed at glycoconjugate concentrations ranging from 16 μg/mL to 1 mg/mL (corresponding to a concentration range of 0.2 to 14 μM). Only measurements with a good curve fit (R2 > 0.85) were used for KD and koff determination.
To assess the optimum ligand density for 2G12 binding, thiourea- and adipic acid-linked neoglycoconjugates of select substitution degrees containing the D3-arm extended heptasaccharide unit in both anomeric forms were then evaluated. No binding was observed for conjugates containing fewer than two ligands per BSA molecule (16a, 20a; data not shown), which is consistent with the need for 2G12 to bind bivalently to achieve avid interaction with antigen.25,27 Conjugates 16b and 20b bound to 2G12 with low affinity (KD > 1 μM), precluding accurate determination of KD and koff (data not shown). At comparable ligand densities, the thiourea constructs were superior binders relative to the adipic acid conjugates. The better binding of the thiourea derivatives is also discernible when comparing the apparent KD values (Table 2). To achieve a similar apparent avidity, adipic acid conjugates required a higher ligand/BSA ratio (compare conjugate 15c and 20c, as well as 16c and 21b, respectively). Furthermore, and similar to the coupling efficiency (Table 1), conjugates with the branching mannose in the β-anomeric configuration displayed a tendency to be bound more strongly by 2G12 than their α-anomeric counterparts.
Table 2. BLI Data for Binding of Thiourea and Adipic Acid Conjugates to 2G12.
| Conjugate | Concentration range (μM) | Measurementsa | Experiments | kon ± SEMb (M–1 s–1) | koff ± SEMb (s–1) | KD ± SEMb (nM) |
|---|---|---|---|---|---|---|
| 13 | 0.23–14.4 | 18 | 4 | 5.1 ± 0.3 × 102 | 8.5 ± 0.8 × 10–5 | 183 ± 30 |
| 14 | 0.45–7.2 | 15 | 3 | 3.8 ± 0.5 × 103 | 1.1 ± 0.2 × 10–4 | 34 ± 4 |
| 15a | 0.23–14.4 | 23 | 4 | 4.5 ± 1.0 × 103 | 4.0 ± 1.6 × 10–4 | 157 ± 61 |
| 15c | 0.84–6.7 | 12 | 3 | 3.2 ± 0.1 × 104 | 5.8 ± 0.8 × 10–4 | 19 ± 3 |
| 16c | 0.22–13.9 | 26 | 4 | 1.1 ± 0.1 × 104 | 1.2 ± 0.2 × 10–4 | 23 ± 6 |
| 17 | 0.22–14.2 | 26 | 4 | 1.0 ± 0.1 × 104 | 1.2 ± 0.1 × 10–4 | 19 ± 3 |
| 18 | 0.45–7.1 | 16 | 4 | 4.5 ± 0.7 × 103 | 1.4 ± 0.2 × 10–3 | 394 ± 125 |
| 20c | 0.84–6.8 | 12 | 3 | 2.6 ± 0.2 × 104 | 1.4 ± 0.1 × 10–3 | 71 ± 2 |
| 20d | 0.81–6.5 | 12 | 3 | 1.5 ± 0.1 × 104 | 2.1 ± 0.4 × 10–3 | 319 ± 112 |
| 21b | 0.85–6.8 | 9 | 3 | 1.3 ± 0.1 × 104 | 7.4 ± 1.1 × 10–4 | 76 ± 17 |
| 21c | 0.81–6.5 | 5 | 2 | 3.2 ± 0.1 × 104 | 2.9 ± 0.1 × 10–4 | 10 ± 1 |
| 32 | 0.45–7.2 | 10 | 3 | 1.5 ± 0.1 × 104 | 6.1 ± 0.3 × 10–4 | 46 ± 4 |
| 33 | 0.46–7.3 | 2 | 2 | 7.3 ± 6.8 × 103 | 2.0 ± 1.1 × 10–3 | 274c |
| 34 | 0.43–6.8 | 3 | 2 | 7.6 ± 4.3 × 104 | 1.6 ± 0.6 × 10–3 | 23 ± 4 |
| 35 | 0.45–7.2 | 2 | 2 | 1.9 ± 0.4 × 104 | 2.3 ± 1.3 × 10–3 | 121c |
Only measurements with R2 ≥ 0.85 were included.
SEM, standard error of the mean.
Calculated as koff/kon.
In summary, the optimum ligand density was found to be in a range of 4–6 ligands per BSA molecule for the thiourea adducts, whereas the adipic acid linked conjugates required a higher copy number to attain comparable binding avidities for 2G12.
Comparison of the Antigenic Properties of Synthetic Oligomannose Glycosides to Natural Oligomannose Using 2G12 and the 2G12 I19R Mutant
A mutant version of 2G12 was used to compare the antigenic properties of selected synthetic oligomannose glycosides with those of HIV glycans. 2G12 exists naturally as a domain-exchanged antibody, resulting in two conventional antibody-combining sites and a homodimeric VH/VH′ interface formed by crossover of the antibody heavy chains. The multivalent binding surface thus created accounts for the high selectivity of 2G12 for clustered oligomannose chains such as presented on the HIV envelope glycoprotein surface.25 Others have shown that mutating Ile19 (Kabat numbering) in the 2G12 heavy chain to Arg yields a nondomain exchanged version with an archetypical Y-shaped architecture.40 The I19R mutant (2G12-I19R) binds oligomannose chains in the same manner as wild-type 2G12, but does so substantially less avidly and is unable to neutralize HIV. To evaluate the antigenic presentation of oligomannosides on select neoglycoconjugates described above, wild-type 2G12 and mutant 2G12-I19R were assessed by ELISA for binding to the adipic acid linked glycoconjugates compared to recombinant HIV gp140. Substantial binding of 2G12-I19R to gp140 and the glycoconjugates was only observed when the avidity of the antibody was increased by first precomplexing it with an anti-Fc antibody, as observed also by others.40 Both antibodies bound most strongly to the β-linked neoglycoconjugate 21c; slightly lower binding was observed for the equivalent α-linked anomer neoglycoconjugate 20d at comparable ligand density (Figure 3). However, and noticeably, similar to the results obtained in BLI experiments, binding of wild-type 2G12 to the neoglycoconjugates was significantly lower than to gp140, illustrating that even at higher ligand density, the neoglycoconjugates do not entirely mimic the organization of natural oligomannose on the HIV envelope glycoprotein. In contrast, binding of 2G12-I19R to neoglycoconjugates 20b and 21b was stronger than to gp140 (Figure 3), indicating that these glycoconjugates could represent unique vaccine candidates for priming the elicitation of 2G12-like antibody responses.
Figure 3.
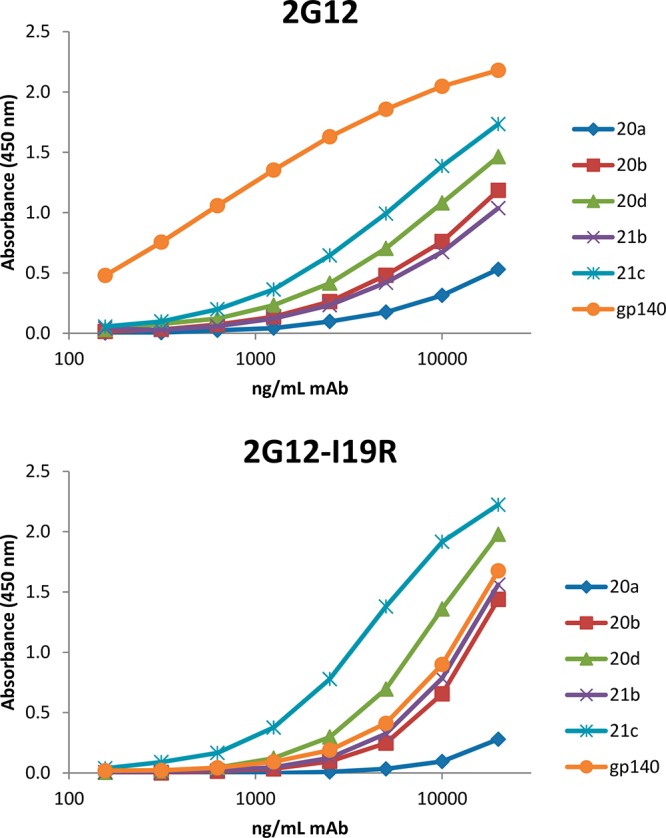
Binding of adipic acid linked neoglycoconjugates 20a, 20b, 20d, 21b, and 21c to wild-type 2G12 (top) and to the 2G12-I19R mutant (bottom) as determined by ELISA. Plates were coated with equal amounts of gp140 and neoglycoconjugates. This experiment has been performed twice with similar results.
Synthesis of Neoglycoconjugates Containing Clustered/Dendrimeric Oligomannose Epitopes
In the past, multivalent presentation of oligomannoside ligands for the interaction with the extended paratope of 2G12 has frequently been based on pentyl, 3-oxapentyl, or hexyl groups terminated by azido or amino groups.11,21,26,14,15 The inherent flexibility of such long-chain spacer groups, however, increases the entropic penalty in the binding process. In contrast, rigidification of ligands may contribute to free energy gains in the binding process to proteins.41−43 We thus set out to assess the impact of the short three-carbon aglycon directly tethered to a triazole scaffold, which should significantly confine the conformational space of the oligomannose fragments.
To this end, the known tris-propyne substituted amine 23(44,45) was used as reagent for the cycloaddition reaction.46 As alternate chain-elongated scaffold the di-5-aminopentanoic amide derivative 24 was also synthesized. Thus, the Boc-protecting group from 22 was cleaved by treatment with TFA to give 23, followed by HATU-promoted elongation of the liberated amino group with a second Boc-protected 5-amino pentanoic acid residue to furnish 24 in 67% yield (Scheme 5).
Scheme 5. Synthesis of Tris-Alkyne Substituted Linker Derivative 24.

Reagents and conditions: (a) TFA, DCM, 0 °C, 3 h; (b) DMF, Boc-5-amino-pentanoic acid, NMO, HATU, rt, 16 h, 67%.
The ensuing click reactions with the azide-terminated derivatives 2 and 4 were performed in moderate yields of 35–46% only, most likely due to the steric congestion present in the dense arrangement of the triazole-tethered oligomannosides (Scheme 6). The oligomannoside ligands were used in 3.3–3.6-fold excess to enforce the complete formation of three triazole rings. However, incompletely substituted reaction products remained in the reaction mixture. Separation of the 3-fold-substituted products from under-derivatized species and unreacted starting material could be achieved by size exclusion chromatography on Sephadex LH-20. The degree of substitution was determined from the 1H NMR signal intensity of the Boc group relative to the triazole protons, which agreed fully with the mass data obtained by MALDI-TOF measurements. The high steric hindrance of the ligands had a detrimental effect on the conjugation experiments, which were performed with the α-anomeric pentamannoside 2 as model compound. First, compound 2 was subjected to the click reaction with the short-chain linker 22 in a moderate 46% yield. The resulting triazole derivative 25 was then treated with TFA—to expose the terminal amino group—giving 26 in 97% yield. Activation of pentasaccharide ligand 26 with squaric acid47−49 under nonoptimized conditions mainly resulted in recovery of unreacted protein and a small fraction of substituted product with a very low ligand density (0.1 ligand/BSA, data not shown). Use of the chain extended version 29, derived from the Boc-protected precursor 27 upon acid treatment, led to somewhat better results and provided squaric acid conjugate 31 with a ligand copy number of 0.5 per BSA. Due to the improved incorporation of the ligand when using the extended aminopentanoic amide linker, thiourea conjugates were similarly prepared from the derivatives 29 and 30, which gave conjugates 32 with a copy number of 0.8, and 33 with 0.5 ligand/BSA ratio, respectively. Similar to the results obtained with the monovalent ligands (Table 1), the adipic amide conjugation method performed better, providing conjugates 34 and 35 with ligand/protein ratios of 3:1 and 1.3:1, respectively (Table 3).
Scheme 6. Synthesis of Pentamannoside-Dendron Containing Neoglycoconjugates.

Table 3. Coupling Efficiency for Anomeric Pentamannoside Dendron Derivatives 29 and 30 to BSA.
| Entry | Spacer glycoside | Protein concentration (nM) | Molar ratio | Conjugate | Ligand/BSA ratio | Efficiency [%] |
|---|---|---|---|---|---|---|
| 1 | 29 | 54 | 20 | 31 | 0.5 | 2.5 |
| 2 | 29 | 17 | 21 | 32 | 0.8 | 3.8 |
| 3 | 30 | 10 | 21 | 33 | 0.5 | 2.4 |
| 4 | 29 | 25 | 30 | 34 | 3.0 | 10.0 |
| 5 | 30 | 45 | 21 | 35 | 1.3 | 6.2 |
As measured by BLI, wild-type 2G12 bound the α-configured thiourea conjugate 32 with an apparent KD value of 46 nM, whereas the adipic amide conjugate 34, with a significantly better substitution degree, was bound only slightly better (23 nM). A similar trend was observed for the β-configured derivatives 33 and 35 with apparent KD values of 274 nM and 121 nM, respectively (Table 2). Hence, the avidities of the interactions between 2G12 and these multivalent constructs appear to be determined not only by the ligand densities of the conjugates but also, and perhaps not surprisingly, by the precise organization of the oligomannosides on the carrier molecule.
Conclusions
In this study we have undertaken a comparative evaluation of BSA conjugates prepared from a series of anomeric 3-aminopropyl oligomannose spacer glycosides based on the structure of a bacterial mimetic of HIV-1 glycans. Conjugation reactions using thiourea or adipic amide linkages revealed a substantially higher coupling efficiency for the latter approach. However, when compared based on matching ligand density, the thiourea constructs enabled higher avidity binding of the HIV-neutralizing antibody 2G12, with apparent KD values in the low nanomolar range and increased avidities for conjugates with higher ligand densities. The anomeric configuration of the spacer-extended central mannose unit had a minor influence, with slightly better binding observed for ligands harboring a reducing end mannose in the β-anomeric form.
The sterically crowded ligands clustered on tris-triazole substituted scaffolds showed unexpectedly low reactivity in the conjugation reactions and inferior binding to the antibody 2G12, illustrating the challenges in constructing antigens with appropriate spatial orientation for binding to this antibody. Further work will extend these studies to include additional anti-HIV glycan-specific neutralizing antibodies, to help inform the utility of oligomannose mimicry for the elicitation of HIV-neutralizing antibodies with oligomannose specificity.
Experimental Section
General Methods
Thin layer chromatography was performed on Merck precoated plates: generally, on 5 × 10 cm2, layer thickness 0.25 mm, Silica Gel 60F254; alternatively, on HPTLC plates with 2.5 cm concentration zone (Merck). Spots were detected by dipping reagent (anisaldehyde-H2SO4). For column chromatography silica gel (0.040–0.063 mm) was used. Optical rotations were measured with a PerkinElmer 243 B Polarimeter. [α]D20 values are given in units of 10–1 deg cm2 g–1. NMR spectra were recorded on a Bruker Avance III 600 instrument (600.22 MHz for 1H, 150.93 MHz for 13C) using standard Bruker NMR software. 1H spectra were referenced to δ = 0 using the TMS signal for solutions in CDCl3 and DSS for solutions in D2O (external calibration to 2,2-dimethyl-2-silapentane-5-sulfonic acid). 13C spectra were referenced to 77.00 (CDCl3) and 67.40 (D2O, external calibration to 1,4-dioxane) ppm. Assignments were based on COSY, HSQC, HMBC, and TOCSY spectra. ESI-MS data were obtained on a Waters Micromass Q-TOF Ultima Global instrument. MALDI-data were obtained on a Bruker Autoflex MALDI TOF/TOF instrument using 2,5-dihydroxyacetophenone as matrix.
The following
labeling of residues was used for NMR assignments:
General Procedure A for Diazotransfer Reactions
To a solution of the amine (1 equiv) in 2:1 MeOH–water was added K2CO3 (10 equiv), imidazole-1-sulfonyl azide hydrochloride (4 equiv) and a solution of CuSO4 × 5 H2O (0.1 M in water, 0.1 equiv). The reaction mixture was stirred at room temperature for 24 h. Subsequently the solution was filtered over Celite and the solvents were removed in vacuo. The crude product was purified using gel chromatography (Sephadex LH-20; 2:1 water–MeOH). Product containing fractions were lyophilized yielding the corresponding azides as colorless solids.
General Procedure B for Thiourea Based Conjugation
The amine ligand (0.002 mmol; 75 equiv) was dissolved in 0.1 M aq. NaHCO3 (2 mL) and a solution of thiophosgene (2 mL of a 0.006 M solution in CHCl3; 450 equiv) was added. The biphasic mixture was stirred vigorously for 2 h. The organic phase was removed with a pipet and the aqueous phase was extracted with CHCl3 three times. Traces of CHCl3 were removed by bubbling a stream of air through the remaining solution. The resulting aqueous phase was added to a solution of BSA (2 mg; 30 nmol; 1 equiv) in buffer A (2 mL of 0.3 M NaCl and 0.1 M NaHCO3) and was stirred for 62 h. Then the mixture was dialyzed against water three times and the residue was lyophilized yielding the product as a white solid. The amount of conjugated ligand per BSA was determined using MALDI-TOF mass spectroscopy.
General Procedure C for Adipic Acid Based Conjugation
The amine ligand was dissolved in DMSO and N,N′-adipoylbis(succinimide) was added all at once followed by the addition of NEt3. The solution was stirred for 3 h at room temperature and then PBS-buffer (pH= 7.2) as well as EtOAc (1 mL) were added. The organic phase was removed with a pipet and the aqueous phase was washed two more times with EtOAc (1 mL each). Traces of EtOAc were removed by bubbling a stream of air through the remaining solution. The aqueous phase was subsequently added to BSA and the reaction mixture was stirred for 24 h at room temperature. In order to get rid of remaining free ligand and salts the crude reaction mixture was centrifuged using a spin filter (Amicon 0.5 M, 30 kDa) for 10 min (at 25 °C and 14 000 × g). The remaining solution was washed twice with PBS-buffer (0.45 mL each, 10 min at 25 °C and 14 000 × g) and was then removed from the filter by centrifugation. Lyophilization yielded the product as white solid. The amount of conjugated ligand/BSA was determined using MALDI-TOF mass spectroscopy.
General Procedure D for Click Reactions
The azide (3.3–3.6 equiv) was dissolved in 1:1 tertBuOH–water under argon and 24 (0.05 M in tertBuOH; 1 equiv) was added, followed by the addition of CuSO4 × 5 H2O (0.05 M in water; 1 equiv). The mixture was degassed by the freeze–pump–thaw method. Then Na-ascorbate (0.05 M in degassed water; 2 equiv) was added and the mixture was stirred at rt for 72 h. The mixture was filtered and the filtrate was concentrated. The crude product was purified by gel chromatography (Sephadex LH-20; 2:1 water–MeOH). The product containing fractions were lyophilized yielding the corresponding click products as solids. The structure of the products was verified by 1H NMR spectroscopy and MALDI-TOF mass spectroscopy.
General Procedure E for Boc Removal
The click-product was dissolved in 1:1 water–TFA (1 mL) and was stirred at 0 °C for 45 min. The solvent was removed in vacuo yielding the product.
General Procedure F for Conjugation of Dendritic Ligands
The multivalent amine ligand was dissolved in 0.1 M aq. NaHCO3, and a solution of thiophosgene (0.006 M in CHCl3) was added. The biphasic mixture was stirred vigorously for 2 h. The organic phase was removed with a pipet and the aqueous phase was extracted with CHCl3 three times. Traces of CHCl3 were removed by bubbling a stream of air through the remaining solution. The resulting aqueous phase was added to a solution of BSA in buffer A (0.3 M NaCl and 0.1 M NaHCO3) and was stirred for 62 h. In order to get rid of remaining free ligand and salts the reaction mixture was centrifuged using a spin filter (Amicon 0.5 M, 30 kDa) for 10 min (at 25 °C and 14 000 × g). The remaining solution was washed twice with water (0.45 mL each, 10 min at 25 °C and 14 000 × g) and was then removed from the filter by centrifugation. Lyophilization yielded the product as white solid. The amount of conjugated ligand/BSA was determined using MALDI-TOF mass spectroscopy.
Site-Directed Mutagenesis and Construction of mAb Expression Vectors
The sequences encoding the 2G12 heavy and light chains (omitting their authentic signal peptides) were amplified from the corresponding pDONR221 constructs50 by PCR using the primer combinations 2G12HC_BsaI_fw/2G12HC_BsaI_rv and 2G12LC_BsaI_fw/2G12LC_BsaI_rv (Sigma-Aldrich, USA), respectively (Supplementary Table S1). The fragments thus generated were then inserted into the BsaI sites of pICHα31160 (heavy chain) and pICHα26033 (light chain) as described.51,52 The domain-swapping point mutation I19R40 was introduced into the 2G12 heavy-chain sequence by QuikChange site-directed mutagenesis (Agilent Technologies, USA) with the complementary primer pair 2G12HC_I19R_fw and 2G12HC_I19R_rv (Supplementary Table S1) and Phusion DNA polymerase (Thermo Scientific, USA), using pDONR221–2G12HC50 as template. The mutated product was then cloned into pICHα31160 as outlined above. The fidelity of all wild-type and mutant 2G12 sequences was verified (Microsynth, Switzerland). For agroinfiltration of Nicotiana benthamiana, all constructs were transformed into the Agrobacterium tumefaciens strain GV3101::pMP90.
Expression of mAbs in N. benthamiana
N. benthamiana ΔXT/FT plants lacking plant-specific α1,3-fucosylation and β1,2-xylosylation were grown for 4–5 weeks at 24 °C with a 16 h light:8 h dark photoperiod. Infiltration with agrobacteria carrying the respective mAb expression vectors was then performed as reported previously.53 Briefly, overnight cultures were pelleted and then resuspended in infiltration buffer (25 mM Mes buffer (pH 5.6), 25 mM MgSO4, 0.1 mM acetosyringone) at an OD600 of 0.2 (1.0 OD600 corresponds to 5 × 108 cells/mL). Equal amounts of the strains carrying the respective heavy and light chain constructs were used. Infiltrated N. benthamiana leaves were harvested after 4–5 days.
Purification of mAbs Produced in N. benthamiana Leaves
Antibody extraction and purification was performed as described previously, with minor modifications.41,43 Briefly, infiltrated leaf material was homogenized under liquid nitrogen and extracted with 0.1 M Tris/HCl (pH 7.0) containing 0.5 M NaCl, 40 mM ascorbic acid and 1 mM EDTA (2 mL per gram leaf wet weight). The extract was clarified by a series of centrifugation and filtration steps. Antibodies were then purified by affinity chromatography on a column packed with 1 mL rProtein A Sepharose 4 Fast Flow (GE Healthcare, UK), using 0.1 M glycine/HCl (pH 3.0) for elution. Protein-containing eluate fractions were immediately neutralized by addition of 0.1 M Tris/HCl (pH 8.0), dialyzed against PBS containing 0.02% (v/v) NaN3 and then concentrated by ultrafiltration using Amicon YM30 centrifugal filter units (Merck, Germany).
ELISA
Enzyme-linked immunosorbent assay (ELISA) plates (96-well) were coated with 500 ng per well of HIV-1 92UG37 gp140 (Polymun Scientific, Austria), manufactured in CHO cells, or the respective conjugate in PBS for 16 h at 4 °C and then washed with PBS containing 0.05% Tween 20 (PBST). In the case of 2G12-I19R, antibody (20 μg/mL) was precomplexed with Fc-specific goat antihuman IgG F(ab′)2 fragments (10 μg/mL; Sigma-Aldrich) for 15 min at 4 °C. Samples were serially diluted (1:2) in PBST containing 1% BSA (dilution buffer) prior to addition to the wells. After incubation for 1 h and subsequent washing, bound antibodies were detected with 0.1 μg/mL goat antihuman kappa chain peroxidase conjugate (Sigma-Aldrich) in dilution buffer. After 1 h, plates were washed and then developed with 0.1 mg/mL 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) and 0.006% H2O2 in 35 mM citric acid/65 mM sodium phosphate (pH 5.0) for 15 min. Reactions were quenched by addition of 90 mM H2SO4 prior to analysis by spectrophotometry at 450 nm. All steps were performed at room temperature unless stated otherwise.
Bio-Layer Interferometry
All biolayer interferometry assays were performed on a ForteBio Octet QK system (Pall ForteBio) equipped with protein A biosensor tips (Pall ForteBio, Cat. No. 18–5010). The assay was performed at 30 °C in kinetics buffer (Pall ForteBio). HIV-1 antiglycan antibody 2G12 (Polymun Scientific, Austria) was immobilized onto protein A biosensors at 10 μg/mL for 60 s. Glycoconjugate samples were prepared in concentrations of 16–1000 μg/mL and kon and koff were measured, monitoring association (600 s) and dissociation (1200 s) in kinetics buffer. HIV-1 gp140 (250 μg/mL, corresponding to 3.5 μM) was used as positive control. BSA served as negative control. Data were processed and analyzed with the Octet data analysis software 6.4 (ForteBio) using a 1:1 binding model, which was chosen because the bivalent model gave very poor fits.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 6)]-α-d-mannopyranoside (2)
Compound 2 was synthesized according to general procedure A using the following amounts: Compound 1 (38 mg; 0.043 mmol) in 2:1 MeOH–water (3 mL), K2CO3 (59 mg; 0.43 mmol), imidazole-1-sulfonyl azide hydrochloride (36 mg; 0.172 mmol) and a solution of CuSO4 × 5 H2O (0.1 M in water; 10 μL; 4 μmol). Compound 2 was obtained as colorless solid (38 mg; 97%); 1H NMR (600 MHz, D2O): δ = 5.31 (d, J = 1.3 Hz, 1 H, H-1B), 5.26 (d, J = 1.6 Hz, 1 H, H-1C), 5.01 (d, J = 1.6 Hz, 1 H, H-1D), 4.87 (d, J = 1.6 Hz, 1 H, H-1E), 4.79 (s, J = 1.6 Hz, 1 H, H-1A), 4.07–4.06 (m, 2 H, H-2C, H-2A), 4.04 (dd, J = 1.7, 3.2 Hz, 1 H, H-2B), 4.03 (J = 1.8, 3.2 Hz, 1 H, H-2D), 3.97–3.94 (m, 3 H, H-3B, H-2E, H-6aA), 3.92 (dd, J = 3.2, 9.4 Hz, 1 H, H-3C), 3.87–3.53 (m, 25 H), 3.45–3.38 (m, 2 H, CH2CH2N3), 1.91–1.82 (m, 2 H, CH2CH2N3); 13C NMR (125 MHz, D2O): δ = 103.0 (C-1D), 101.6 (C-1B), 101.5 (C-1C), 100.7 (C-1A), 100.2 (C-1E), 79.6 (C-3A), 79.5 (C-2B), 79.3 (C-2C), 75.0 (C-5A), 74.1, 74.0 (d.i.), 73.5 (C-5B, C-5C, C-5D, C-5E), 71.9, 71.4, 71.1, 70.9, 70.8 (3 C) and 70.5 (7 C, C-2A, C-2E, C-2D, C-3E, C-3B, C-3C, C-3D), 67.8, 67.7, 67.6, and 67.5 (C-4B, C-4C, C-4D, C-4E), 65.5 (C-4A), 66.2 (C-6A), 65.7 (OCH2CH2), 61.9 and 61.8 (4 C, C-6B, C-6C, C-6D, C-6E), 49.0 (CH2CH2N3), 28.6 (CH2CH2N3). ESI-TOF HRMS: m/z calcd for C33H57N3O26 [M+NH4+]+: 929.3569; found: 929.3585.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 6)]-β-d-mannopyranoside (4)
Compound 4 was synthesized according to general procedure A using the following amounts: Compound 3 (5.0 mg; 0.006 mmol) in 2:1 MeOH–water (1 mL), K2CO3 (6.0 mg; 0.043 mmol), imidazole-1-sulfonyl azide hydrochloride (5.0 mg; 0.024 mmol) and a solution of CuSO4 × 5 H2O (0.1 M in water; 4 μL; 0.4 μmol). Compound 4 was obtained as colorless solid (4.7 mg; 91%); 1H NMR (600 MHz, D2O): δ = 5.30 (d, J = 1.4 Hz, 1 H, H-1B), 5.27 (d, J = 1.7 Hz, 1 H, H-1C), 5.01 (d, J = 1.7 Hz, 1 H, H-1D), 4.88 (d, J = 1.6 Hz, 1 H, H-1E), 4.64 (s, 1 H, H-1A), 4.10 (br d, J = 3.2 Hz, 1 H, H-2A), 4.07 (dd, J = 1.9, 3.1 Hz, 1 H, H-2C), 4.04 (dd, J = 1.7, 3.2 Hz, 1 H, H-2B), 4.03 (dd, J = 1.8, 3.3 Hz, 1 H, H-2D), 3.97–3.58 (m, 29 H), 3.51 (ddd, J = 1.8, 5.0, 9.8 Hz, 1 H, H-5A), 3.40 (t, J = 6.7 Hz, 2 H, CH2CH2N3), 1.88–1.84 (m, 2 H, CH2CH2N3). 13C NMR (125 MHz, D2O, selected data from HSQC-experiments): δ = 102.9 (C-1D), 101.4 (C-1B), 101.3 (C-1C), 100.6 (C-1A), 100.1 (C-1E), 81.6 (C-3A), 79.3 (C-2B), 79.2 (C-2C), 74.8 (C-5A), 67.6 (OCH2CH2), 66.5 (C-4A), 66.3 (C-6A), 48.8 (CH2CH2N3). ESI-TOF HRMS: m/z calcd for C33H57N3O26 [M+NH4+]+: 929.3569; found: 929.3569.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 6)-α-d-mannopyranosyl-(1 → 6)-α-d-mannopyranoside (6)
Compound 6 was synthesized according to general procedure A using following amounts: Compound 5 (8.0 mg; 0.006 mmol) in 2:1 MeOH-water (1 mL), K2CO3 (9.0 mg; 0.062 mmol), imidazole-1-sulfonyl azide hydrochloride (5.0 mg; 0.025 mmol) and a solution of CuSO4 × 5 H2O (0.1 M in water; 6 μL; 0.6 μmol). Compound 6 was obtained as colorless solid (6.0 mg; 84%); 1H NMR (600 MHz, D2O): δ = 5.30 (d, J = 1.3 Hz, 1 H, H-1B), 5.27 (d, J = 1.5 Hz, 1 H, H-1C), 5.11 (d, J = 1.4 Hz, 1 H, H-1F), 5.01 (d, J = 1.8 Hz, 1 H, H-1D or H-1G) and 5.00 (d, J = 1.7 Hz, 1 H, H-1D or H-1G), 4.86 (d, J = 1.3 Hz, 1 H, H-1E), 4.79 (d, J = 1.5 Hz, 1 H, H-1A), 4.08–4.06 (m, 2 H, H-2A, H-2C), 4.05–4.02 (m, 3 H, H-2B, H-2D, H-2G), 3.98 (dd, J = 1.7, 3.2 Hz, 1 H, H-2F), 3.97–3.53 (m, 38 H), 3.45–3.38 (m, 2 H, CH2CH2N3), 1.91–1.82 (m, 2 H, CH2CH2N3); 13C NMR (150 MHz, D2O, selected signals form HSQC-experiments): δ = 102.9 (2 C, C-1D, C-1G), 101.5 (2 C, C-1B, C-1C), 100.6 (C-1A), 100.2 (C-1E), 98.8 (C-1F), 79.6 (C-3A), 66.6 (C-6A), 65.6 (OCH2), 48.8 (CH2CH2N3); ESI-TOF HRMS: m/z calcd for C45H77N3O36 [M+NH4+]+: 1253.4625; found: 1253.4647.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 6)-α-d-mannopyranosyl-(1 → 6)-β-d-mannopyranoside (8)
Compound 8 was synthesized according to General procedure A using following amounts: Compound 7 (5.0 mg; 0.004 mmol) in 2:1 MeOH-water (1 mL), K2CO3 (5.6 mg; 0.04 mmol), imidazole-1-sulfonyl azide hydrochloride (0.0034 g; 0.016 mmol; 4 equiv) and a solution of CuSO4 × 5 H2O (0.1 M in water; 4 μL; 0.4 μmol). Compound 8 was obtained as colorless solid (4.4 mg; 90%); 1H NMR (600 MHz, D2O): δ = 5.30 (d, J = 1.5 Hz, 1 H, H-1B), 5.27 (d, J = 1.4 Hz, 1 H, H-1C), 5.11 (d, J = 1.4 Hz, 1 H, H-1F), 5.01 and 5.00 (2d, J = 1.8 Hz, each 1 H, H-1D, H-1G), 4.87 (d, J = 1.4 Hz, 1 H, H-1E), 4.64 (br s, 1 H, H-1A), 4.11 (br d, J = 3.2 Hz, 1 H, H-2A), 4.07 (dd, J = 1.8, 3.2 Hz, 1 H, H-2C), 4.05 (dd, J = 1.8, 3.1 Hz, 1 H, H-2B), 4.04 and 4.03 (2 x dd, J = 1.8, 3.3 Hz, each 1 H, H-2D, H-2G), 3.99–3.58 (m, 38 H) 3.51 (ddd, J = 1.8, 4.9, 9.8 Hz, 1 H, H-5A), 3.40 (t, J = 6.7 Hz, 2 H, CH2CH2N3), 1.88–1.83 (m, 2 H, CH2CH2N3); 13C NMR (150 MHz, D2O, selected signals from HSQC-experiments): δ = 103.0 (C-1D, C-1G), 101.4 (C-1B), 101.3 (C-1C), 100.6 (C-1A), 100.3 (C-1E), 98.8 (C-1F), 81.7 (C-3A), 74.7 (C-5A), 48.8 (CH2CH2N3). ESI-TOF HRMS: m/z calcd for C45H77N3O36 [M+H+]+: 1236.4360; found: 1236.4410.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 4)-α-d-mannopyranosyl-(1 → 6)-α-d-mannopyranoside (10)
Compound 2 was synthesized according to general procedure A using the following amounts: 9 (7.0 mg; 0.006 mmol) in 2:1 MeOH-water (1 mL), K2CO3 (8.0 mg; 0.058 mmol), imidazole-1-sulfonyl azide hydrochloride (5.0 mg; 0.023 mmol) and a solution of CuSO4 × 5 H2O (0.1 M in water; 6 μL; 0.6 μmol). 10 was obtained as colorless solid (6.5 mg; 91%); 1H NMR (600 MHz, D2O): δ = 5.45 (d, J = 1.6 Hz, 1 H, H-1F), 5.31 (d, J = 1.6 Hz, 1 H, H-1B), 5.27 (d, J = 1.7 Hz, 1 H, H-1C), 5.01 and 5.00 (2d, J = 1.7 Hz, 2 H, H-1G, H-1D), 4.86 (d, J = 1.6 Hz, 1 H, H-1E), 4.79 (d, J = 1.6 Hz, 1 H, H-1A), 4.07–4.02 (m, 6 H, H-2A, H-2C, H-2B, H-2F, H-2D, H-2G), 3.97- 3.55 (m, 39 H), 3.46–3.39 (m, 2 H, CH2CH2N3), 1.92–1.84 (m, 2 H, CH2CH2N3); 13C NMR (150 MHz, D2O, selected data from HSQC-experiments): δ = 102.8 (2 C, C-1D, C-1G), 101.4 (C-1B), 101.3 (C-1C), 100.7 (C-1F), 100.5 (C-1A), 99.9 (C-1E), 66.3 (OCH2CH2), 65.5 (C-6A), 48.7 (CH2CH2N3). ESI-TOF HRMS: m/z calcd for C45H77N3O36 [M+H+]+: 1236.4360; found: 1236.4412.
3-Azido-1-propyl α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 3)–[α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranosyl-(1 → 4)-α-d-mannopyranosyl-(1 → 6)-β-d-mannopyranoside (12)
Compound 12 was synthesized according to general procedure A using following amounts: 11 (6.0 mg; 0.005 mmol) in MeOH/water (2:1; 1 mL), K2CO3 (7.0 mg; 0.05 mmol), imidazole-1-sulfonyl azide hydrochloride (4.2 mg; 0.020 mmol) and a solution of CuSO4 × 5 H2O (0.1 M in water; 5 μL; 4 μmol) which gave 12 as colorless solid (3.6 mg; 59%); 1H NMR (600 MHz, D2O): δ = 5.46 (d, J = 1.6 Hz, 1 H, H-1F), 5.31 (d, J = 1.4 Hz, 1 H, H-1B), 5.27 (d, J = 1.6 Hz, 1 H, H-1C), 5.01 and 5.00 (2d, J = 1.7 Hz, 2 H, H-1G, H-1D), 4.87 (d, J = 1.2 Hz, 1 H, H-1E), 4.64 (br s, 1 H, H-1A), 4.10 (br. d, J = 3.2 Hz, 1 H, H-2A), 4.07–4.02 (m, 5 H, H-2C, H-2B, H-2F, H-2D, H-2G), 3.98- 3.59 (m, 38 H), 3.50 (ddd, J = 1.9, 5.8, 9.7 Hz, 1 H, H-5A), 3.43–3.39 (m, 2 H, CH2CH2N3), 1.89–1.84 (m, 2 H, CH2CH2N3); 13C NMR (150 MHz, D2O, selected data from HSQC-experiments): δ = 102.9 (2 C, C-1D, C-1G), 101.4 (C-1B), 101.3 (C-1C), 100.7 (C-1F), 100.6 (C-1A), 100.0 (C-1E), 81.5 (C-3A), 74.8 (C-5A), 48.7 (CH2CH2N3), 28.9 (CH2CH2N3); ESI-TOF HRMS: m/z calcd for C45H77N3O36 [M+H+]+: 1236.4360; found: 1236.4353.
Synthesis of BSA conjugates 13, 14, 15a, 15b, and 16a–c has been reported.25
Synthesis of BSA Conjugate 15c
The preparation was carried out according to general method B using 5 (6.0 mg; 5 μmol; 333 equiv), 2 mL aqu. NaHCO3 and 2 mL thiophosgene solution (12 μmol in CHCl3). After completion of isothiocyanate formation and removal of organic solvent, incubation with BSA (1 mg; 15 nmol) was performed in buffer A (0.5 mL). Processing as described afforded 5.0 mg of 15c. MALDI-TOF MS analysis revealed a ligand/BSA ratio of 6.4:1.
Synthesis of adipic amide conjugates 20a–d and 21a–c
Compounds 20a–d were synthesized according to general procedure C using the following amounts: 5 (0.2 mg; 0.17 μmol; 5 equiv) and N,N′-adipoylbis(succinimide) (0.6 mg; 1.65 μmol; 50 equiv) in DMSO (0.2 mL), NEt3 (0.16 μL; 1.1 μmol). BSA (2.2 mg; 33 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 20a revealed a ligand/BSA ratio of 1.1:1.
5 (0.6 mg; 0.50 μmol; 10 equiv) and N,N′-adipoylbis(succinimide) (1.7 mg; 4.96 μmol; 100 equiv) in DMSO (0.2 mL), NEt3 (0.23 μL; 1.6 μmol). BSA (3.3 mg; 50 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 20b revealed a ligand/BSA ratio of 3.9:1.
5 (1 mg; 0.83 μmol; 20 equiv) and N,N′-adipoylbis(succinimide) (2.8 mg; 8.26 μmol; 200 equiv) in DMSO (0.3 mL), NEt3 (0.38 μL; 2.7 μmol). BSA (2.7 mg; 41 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 20c revealed a ligand/BSA ratio of 4.8:1.
5 (1 mg; 0.83 μmol; 50 equiv) and N,N′-adipoylbis(succinimide) (2.8 mg; 8.26 μmol; 500 equiv) in DMSO (0.3 mL), NEt3 (0.38 μL; 2.7 μmol). BSA (1.1 mg; 16 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 20d revealed a ligand/BSA ratio of 8:1.
Compounds 21a–c were synthesized according to general procedure C with a minor modification using the following amounts: 7 (1 mg; 0.83 μmol; 20 equiv) and N,N′-adipoylbis(succinimide) (2.8 mg; 8.26 μmol; 200 equiv) in DMSO (0.3 mL), NEt3 (0.38 μL; 2.7 μmol). For the work up H2O instead of PBS was added followed by washing with EtOAc. The intermediate was lyophilized and then conjugated to BSA (2.7 mg; 41 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 21a revealed a ligand/BSA ratio of 3.7:1.
7 (0.5 mg; 0.41 μmol; 20 equiv) and N,N′-adipoylbis(succinimide) (1.4 mg; 4.13 μmol; 200 equiv) in DMSO (0.2 mL), NEt3 (0.19 μL; 1.4 μmol). BSA (1.4 mg; 21 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 21b revealed a ligand/BSA ratio of 5:1.
7 (1 mg; 0.83 μmol; 40 equiv) and N,N′-adipoylbis(succinimide) (2.8 mg; 8.26 μmol; 400 equiv) in DMSO (0.3 mL), NEt3 (0.38 μL; 2.7 μmol). BSA (1.4 mg; 21 nmol) in 0.5 mL PBS-buffer. MALDI-TOF mass spectroscopic analysis of 21c revealed a ligand/BSA ratio of 8.3:1.
N-{[5-N-(tert-Butyloxycarbonylamino)-pentanoyl]-(5-aminopentanoyl)}tris[(propargyloxy)methyl]-aminomethane (24)
Compound 22 (0.3 g; 0.68 mmol) was dissolved in dry DCM (5 mL) under argon and was cooled to 0 °C. Then TFA (0.53 mL; 6.9 mmol) was added and the solution was stirred at room temperature for 3 h. The solvent was then removed and the residue 23 was coevaporated with CHCl3 three times.
Next, the free amine 23 was dissolved in dry DMF (1 mL) under Argon followed by the addition of Boc-5-aminopentanoic acid (0.165 g; 0.76 mmol), N-methylmorpholine (0.167 mL; 1.52 mmol) and HATU (0.289 g; 0.76 mmol). The mixture was stirred at rt for 16 h. The reaction was quenched by the addition of aq satd NH4Cl. DCM was added, phases were separated and the organic phase was washed with water and dried over Na2SO4. Concentration of the organic phase gave a crude product which was purified by column chromatography (DCM → 20:1 DCM-MeOH) to give 24 as a yellow oil (0.247 g; 67%); 1H NMR (600 MHz, CDCl3): 6.15 (bs, 1 H, NH), 5.78 [s, 1 H, C(CH2OR)3-NH], 4.80 (bs, 1 H, NH-Boc), 4.09 [d, J = 2.4 Hz, 6 H, C(CH2OCH2CCH)3], 3.78 [s, 6 H, C(CH2OCH2CCH)3], 3.18 (dt, JNH,CH2 = JCH2,CH2 = 6.3 Hz, 2 H, H-5′), 3.07 (m, 2 H, H-5), 2.42 (t, J = 2.2 Hz, 3 H, HCCCH2), 2.17–2.11 (m, 4 H, H-2, H-2’), 1.63–1.55 (m, 4 H, H-3, H-3′), 1.50–1.42 (m, 4 H, H-4, H-4′), 1.38 [bs, 9 H, (CH3)3C]; 13C NMR (150 MHz, CDCl3): 173.2 (C-1’), 173.1 (C-1), 79.5 (3 C, CH2CCH), 79.1 (CCH3), 74.7 (3 C, CH2CCH), 68.5 [3 C, C(CH2OCH2CCH)3], 59.3 [NHC(CH2R)3], 58.7 [3 C, C(CH2OCH2CCH)3], 38.9 (2 C, C-5, C-5′), 36.4 (C-2), 35.9 (C-2′), 29.5 (2 C, C-4, C-4′), 28.6 (3 C, CH3), 22.8 (C-3), 22.4 (C-3′); ESI-TOF HRMS: m/z calcd for C29H44N3O9 [M+H+]+: 534.3174 ; found: 534.3202.
Linker Derivative 25
Compound 25 was synthesized according to general procedure D slightly modified using following amounts: 2 (4.1 mg; 0.005 mmol; 3.6 equiv) in 2 mL water-tertBuOH (1:1), 22 (24 μL; 0.05 M in tertBuOH), CuSO4 × 5 H2O (24 μL; 0.05 M in water), Na-ascorbate (24 μL; 0.025 M in water). Product 25 (1.8 mg; 46%) was obtained as colorless solid. 1H NMR (600 MHz, D2O): 7.92 (s, H-triazole), anomeric protons: 5.29 (d, J = 1.0 Hz, 1 H), 5.24 (d, J = 1.6 Hz, 1 H), 4.98 (d, J = 1.3 Hz), 4.80 (d, J = 1.6 Hz) and 4.52 (bs, 1 H), selected linker signals: 2.92 (t, J = 6.6 Hz, 2 H, CH2NH), 1.45–1.38 (m, 2 H, COCH2CH2), 1.33 (s, 9 H, (CH3)3C).
Linker Derivative 26
Compound 26 was synthesized according to general procedure E using the following amounts: 25 (2.0 mg) afforded 26 (1.7 mg; 88%). 1H NMR (600 MHz, D2O): 7.93 (s, H-triazole), anomeric protons: 5.29 (d, J = 1.7 Hz, 1 H), 5.24 (d, J = 1.9 Hz, 1 H), 4.98 (d, J = 1.9 Hz), 4.80 (d, J = 1.4 Hz), 4.51 (d, J = 1.7 Hz, 1 H), selected linker signals: 2.91 (t, J = 7.0 Hz, 2 H, CH2NH), 1.59–1.46 (m, 4 H, COCH2CH2, CH2CH2NH). MALDI-TOF: m/z calcd for C117H197N11O82: [M+H+]+: 3069.1656; found: 3068.833.
Linker Derivative 27
Compound 27 was synthesized according to general procedure D using following amounts: 2 (5 mg; 0.006 mmol; 3.6 equiv) in 0.5 mL water-tertBuOH (1:1), 24 (30 μL; 0.05 M in tertBuOH), CuSO4 × 5 H2O (30 μL; 0.05 M in water), Na-ascorbate (60 μL; 0.05 M in degassed water). Product 27 (1.9 mg; 38%) was obtained as colorless solid followed by recovered 2 (1.9 mg; 38%). 1H NMR (600 MHz, D2O): 7.92 (s, H-triazole), anomeric protons: 5.28 (d, J = 1.5 Hz, 1 H), 5.24 (d, J = 1.9 Hz, 1 H), 4.98 (d, J = 2.0 Hz), 4.79 (d, J = 1.7 Hz) and 4.52 (bs, 1 H); selected linker signals: 2.96 (t, J = 7.1 Hz, 2 H, CH2NH), 2.10 (t, J = 8.7 Hz, 2 H, NCOCH2), 1.33 (s, 9 H, (CH3)3C).
MALDI-TOF: m/z calcd for C127H214N12O85 [M+Na+]+: 3290.27; found: 3290.95.
Linker Derivative 28
Compound 28 was synthesized according to general procedure D using the following amounts: 4 (7.2 mg; 0.008 mmol; 3.3 equiv) in 1:1 water–tertBuOH (0.6 mL), 24 (48 μL; 0.05 M in tertBuOH), CuSO4 × 5 H2O (48 μL; 0.05 M in water), Na-ascorbate (96 μL; 0.05 M in degassed water). Product 28 (2.5 mg; 35%) was obtained as colorless solid followed by recovered 4 (2.7 mg; 38%). 1H NMR (600 MHz, D2O): 7.97 (s, H-triazole), anomeric protons: 5.33 (d, J = 1.3 Hz, 1 H), 5.29 (d, J = 1.7 Hz, 1 H), 5.03 (d, J = 1.6 Hz), 4.89 (d, J = 1.6 Hz), 4.60 (bs, 1 H); selected linker signals: 3.02 (t, J = 6.8 Hz, 2 H, CH2NH),1.59–1.39 (m, 6 H, OCH2CH2, COCH2CH2, CH2CH2NH), 1.38 (s, 9 H, (CH3)3C).
MALDI-TOF: m/z calcd for C127H214N12O85 [M+K+]+: 3306.24; found: 3306.02.
Linker Derivative 29
Compound 29 was synthesized according to general procedure E using the following amounts: 27 (3.5 mg) afforded 29 (3.4 mg; 100%). 1H NMR (600 MHz, D2O): 7.97 (s, H-triazole), anomeric protons: 5.34 (d, J = 0.9 Hz, 1 H), 5.29 (d, J = 0.9 Hz, 1 H), 5.03 (d, J = 1.3 Hz), 4.84 (d, J = 1.9 Hz), 4.57 (bs, 1 H), selected linker signals: 2.99 (t, J = 6.0 Hz, 2 H, CH2NH), 1.67–1.61 (m, 2 H, OCH2CH2), 1.53–1.37 (m, 4 H, COCH2CH2, CH2CH2NH).MALDI-TOF: m/z calcd for C122H206N12O83[M + Na]+: 3190.22; found: 3190.10.
Linker Derivative 30
Compound 30 was synthesized according to general procedure E using following amounts: 28 (2 mg) afforded 30 (1.9 mg; 100%); 1H NMR (600 MHz D,2O): 7.97 (s, H-triazole), anomeric protons: 5.33 (s, 1 H), 5.29 (s, 1 H), 5.04 (d, J = 1.2 Hz), 4.89 (d, J = 1.4 Hz), 4.60 (bs, 1 H); selected linker signals: 2.99 (t, J = 7.1 Hz, 2 H, CH2NH), 1.66–1.61 (m, 2 H, OCH2CH2), 1.50–1.38 (m, 4 H, COCH2CH2, CH2CH2NH). MALDI-TOF: m/z calcd for C122H206N12O83 [M + H]+: 3168.23; found: 3168.17.
BSA Conjugate 31
29 (1.7 mg; 0.5 μmol) was dissolved in a mixture of EtOH-water (2:1; 0.75 mL) followed by the addition of diethylsquarate (0.016 M in EtOH, 10 μL). Then aq. NaHCO3 (0.1 M, 0.2 mL) was added and the mixture was stirred for 16 h at room temperature. The intermediate was purified on LH-20 gel (2:1 water–MeOH) and product containing fractions were lyophilized. The activated ligand was dissolved in 0.5 mL of 0.5 M borate buffer (pH = 9) followed by the addition of BSA (1.8 mg; 27 nmol) and stirred for 66 h at room temperature.54 In order to get rid of remaining free ligand and salts the crude reaction mixture was centrifuged using a spin filter (Amicon 0.5 M, 30 kDa) for 10 min (at 25 °C and 14 000 × g). The remaining solution was washed twice with PBS buffer (0.45 mL each, 10 min at 25 °C and 14 000 × g) and was then removed from the filter by centrifugation. MALDI-TOF mass spectroscopy of 31 revealed an average ligand/BSA ratio of 0.53:1.
BSA Conjugate 32
Compound 32 was synthesized according to general procedure F using the following amounts: 29 (1.7 mg; 0.5 μmol) in 0.1 M NaHCO3 (1 mL), thiophosgene (1 mL, 0.006 M in CHCl3), BSA (1.7 mg; 26 nmol) in PBS buffer (0.5 mL). Product 32 was obtained as white solid (1.8 mg). MALDI-TOF mass spectroscopic analysis revealed a ligand/BSA ratio of 0.76:1.
BSA Conjugate 33
Compound 33 was synthesized according to general procedure F using the following amounts: 30 (1 mg; 0.3 μmol) in 0.1 M NaHCO3 (1 mL), thiophosgene (1 mL, 0.006 M in CHCl3), BSA (1 mg; 14 nmol) in buffer (0.5 mL). Product 33 was obtained as white solid in quantitative yield (1.1 mg). MALDI-TOF mass spectroscopic analysis revealed a ligand/BSA ratio of 0.46:1.
BSA Conjugate 34
Conjugate 34 was synthesized according to general procedure C using 29 (1 mg; 0.32 μmol), N,N′-adipoylbis(succinimide) (1.1 mg; 3.2 μmol) and NEt3 (0.01 μL; 0.69 mmol) in DMSO (0.2 mL), as well as 0.5 mg BSA in PBS-buffer (0.3 mL). MALDI-TOF mass spectrometry analysis of 34 revealed a ligand/BSA ratio of 3:1.
BSA Conjugate 35
Conjugate 35 was synthesized according to general procedure C using 30 (0.9 mg; 0.28 μmol), N,N′-adipoylbis(succinimide) (1.0 mg; 2.8 μmol) and NEt3 (0.01 μL; 0.94 mmol) in DMSO (0.2 mL), as well as 0.9 mg BSA in PBS-buffer (0.3 mL). MALDI-TOF mass spectrometry analysis of 35 revealed a ligand/BSA ratio of 1.3:1.
Acknowledgments
Financial support of this work by the Austrian Science Fund FWF (grant P26919–N28 to PK), the Canadian Institutes of Health Research (grant IBC-150408 to RP), and the National Institutes of Health (grant R01 AI134299 to RP) is gratefully acknowledged. Personal support by the Michael Smith Foundation for Health Research (Scholar Award to RP) is also acknowledged. We furthermore thank Jean-François Bruxelle for critical review of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00731.
NMR spectra of ligands, MALDI-TOF spectra of conjugates, BLI sensorgrams and oligonucleotide primers (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Doores K. J. (2015) The HIV glycan shield as a target for neutralizing antibodies. FEBS J. 282, 4679–4691. 10.1111/febs.13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiya S.; MacPherson I. S.; Krauss I. J. (2014) Recent strategies targeting HIV glycans in vaccine design. Nat. Chem. Biol. 10, 990–999. 10.1038/nchembio.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton D. R.; Hangartner L. (2016) Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu. Rev. Immunol. 34, 635–659. 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A.-J.; Vasiljevic S.; Pritchard L. K.; Harvey D. J.; Andev R. S.; Krumm S. A.; Struwe W. B.; Cupo A.; Kumar A.; Zitzmann N.; et al. (2016) Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glyoprotein. Cell Rep. 14, 2695–2706. 10.1016/j.celrep.2016.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doores K.; Bonomelli C.; Harvey D. J.; Vasiljevic S.; Dwek R. A.; Burton D. R.; Crispin M.; Scanlan C. N. (2010) Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. U. S. A. 107, 13800–13805. 10.1073/pnas.1006498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panico M.; Bouché L.; Binet D.; O’Connor M.-J.; Rahman D.; Pang P.-C.; Canis K.; North S. J.; Desrosiers R. C.; Chertova E.; et al. (2016) Mapping the complete glycoproteome of virion-derived HIV-1 gp120 provides insights into broadly neutralizing antibody binding. Sci. Rep. 6, 32956. 10.1038/srep32956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Pauthner M.; Andrabi R.; Rantalainen K.; Berndsen Z.; Diedrich J. K.; Menis S.; Sok D.; Bastidas R.; Park S.-K.; et al. (2018) Differential processing of HIV envelope glycans on the virus and soluble recombinant trimer. Nat. Commun. 9, 3693. 10.1038/s41467-018-06121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Tejada A.; Haynes B. F.; Danishefsky S. J. (2015) Designing synthetic vaccines for HIV. Expert Rev. Vaccines 14, 815–831. 10.1586/14760584.2015.1027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiya S.; MacPherson I. S.; Krauss I. J. (2014) Recent strategies targeting HIV glycans in vaccine design. Nat. Chem. Biol. 10, 990–999. 10.1038/nchembio.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss I. J.; Joyce J. G.; Finnefrock A. C.; Song H. C.; Dudkin V. Y.; Geng X.; Warren J. D.; Chastain M.; Shiver J. W.; Danishefsky S. J. (2007) Fully synthetic carbohydrate HIV antigens designed on the logic of the 2G12 antibody. J. Am. Chem. Soc. 129, 11042–11044. 10.1021/ja074804r. [DOI] [PubMed] [Google Scholar]
- Astronomo R. D.; Kaltgrad E.; Udit A. K.; Wang S. K.; Doores K.; Huang C. Y.; Pantophlet R.; Paulson J. C.; Wong C. H.; Finn M. G.; Burton D. R. (2010) Defining criteria for oligomannose immunogens for HIV using icosahedral virus capsid scaffolds. Chem. Biol. 17, 357–370. 10.1016/j.chembiol.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-K.; Liang P.-H.; Astronomo R. D.; Hsu T.-L.; Hsieh S.-L.; Burton D. R.; Wong C.-H. (2008) Targeting the carbohydrates on HIV-1: interaction pf oligomannose dendrons with human monoclonal antibody 2G12 and DC-SIGN. Proc. Natl. Acad. Sci. U. S. A. 105, 3690–3695. 10.1073/pnas.0712326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.; Song H.; Wang Y.; Stamatos N. M.; Wang L. X. (2006) Toward a carbohydrate-based HIV-1 vaccine: Synthesis and immunological studies of oligomannose-containing glycoconjugates. Bioconjugate Chem. 17, 493–500. 10.1021/bc0502816. [DOI] [PubMed] [Google Scholar]
- Li H.; Wang L.-X. (2004) Design and synthesis of a template-assembled oligomannose cluster as an epitope mimic for human HIV-neutralizing antibody 2G12. Org. Biomol. Chem. 2, 483–488. 10.1039/b314565d. [DOI] [PubMed] [Google Scholar]
- Kabanova A.; Adamo R.; Proietti D.; Berti F.; Tontini M.; Rappuoli R.; Costantino P. (2010) Preparation, characterization and immunogenicity of HIV-1 related high-mannose oligosaccharides-CRM 197 glycoconjugates. Glycoconjugate J. 27, 501–513. 10.1007/s10719-010-9295-0. [DOI] [PubMed] [Google Scholar]
- Liu C.-C.; Zhai C.; Zheng X.-J.; Ye X.-S. (2016) Altering the specificity of the antibody response to HIV gp 120 with a glycoconjugate antigen. ACS Chem. Biol. 11, 1702–1709. 10.1021/acschembio.6b00224. [DOI] [PubMed] [Google Scholar]
- Wang J.; Li H.; Zou G.; Wang L- X. (2007) Novel template-assembled oligosaccharide clusters as epitope mimics for HIV-neutralizing antibody 2G12. Design, synthesis, and antibody binding study. Org. Biomol. Chem. 5, 1529–1540. 10.1039/b702961f. [DOI] [PubMed] [Google Scholar]
- Bailey J. J.; Bundle D. R. (2014) Synthesis of high-mannose 1-thio glycans and their conjugation to protein. Org. Biomol. Chem. 12, 2193–2213. 10.1039/C3OB42194E. [DOI] [PubMed] [Google Scholar]
- Horiya S.; Bailey J. K.; Temme J. S.; Schlippe Y. V. G.; Krauss I. J. (2014) Directed evolution of multivalent glycopeptides tightly recognized by HIV antibody 2G12. J. Am. Chem. Soc. 136, 5407–5415. 10.1021/ja500678v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J. G.; Krauss I. J.; Song H. C.; Opalka D. W.; Grimm K. M.; Nahas D. D.; Esser M. T.; Hrin R.; Feng M.; Dudkin V. Y.; Chastain M.; Shiver J. W.; Danishefsky S. J. (2008) An oligosaccharide-based HIV-1 2G12 mimotope vaccine induces carbohydrate-specific antibodies that fail to neutralize HIV-1 virions. Proc. Natl. Acad. Sci. U. S. A. 105, 15684–15689. 10.1073/pnas.0807837105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astronomo R. D.; Lee H.-K.; Scanlan C. N.; Pantophlet R.; Huang C.-Y.; Wilson I. A.; Blixt O.; Dwek R. A.; Wong C.-H.; Burton D. R. (2008) A glycoconjugate antigen based on the recognition motif of a broadly neutralizing human immunodeficiency virus antibody, 2G12, is immunogenic but elicits antibodies unable to bind to self glycans of gp120. J. Virol. 82, 6359–6368. 10.1128/JVI.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Julien J.-P., Calarese D., Scanlan C., Lee H.-K., Rudd P., Wong C.-H., Dwek R. A., Burton D. A., and Wilson I. A. (2012). In Glycobiology and Drug Design, pp 187–215, American Chemical Society, Washington, DC. [Google Scholar]
- Clark B. E.; Auyeung K.; Fregolino E.; Parrilli M.; Lanzetta R.; De Castro C.; Pantophlet R. (2012) A bacterial lipooligosaccharide that naturally mimics the epitope of the HIV-neutralizing antibody 2G12 as a template for vaccine design. Chem. Biol. 19, 254–263. 10.1016/j.chembiol.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Trkola A.; Purtscher M.; Muster T.; Ballaun C.; Buchacher A.; Sullivan N.; Srinivasan K.; Sodroski J.; Moore J. P.; Katinger H. (1996) Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarese D. A.; Scanlan C. N.; Zwick M. B.; Deechongkit S.; Mimura Y.; Kunert R.; Zhu P.; Wormald M. R.; Stanfield R. L.; Roux K. H.; et al. (2003) Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300, 2065–2071. 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- Calarese D. A.; Lee H.-K.; Huang C.-Y.; Best M. D.; Astronomo R. D.; Stanfield R. L.; Katinger H.; Burton D. R.; Wong C.-H.; Wilson I. A. (2005) Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proc. Natl. Acad. Sci. U. S. A. 102, 13372–13377. 10.1073/pnas.0505763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan C. N. R.; Pantophlet R.; Wormald M. R.; Ollmann Saphire E.; Stanfield R.; Wilson I. A.; Katinger H.; Dwek R. A.; Rudd P. M.; Burton D. R. (2002) The broadly neutralizing anti-human immunodeficiency virus tpye 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J. Virol. 76, 7306–7321. 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield R. L.; De Castro C.; Marzaioli A. M.; Wilson I. A.; Pantophlet R. (2015) Crystal structure of the HIV neutralizing antibody 2G12 in complex with a bacterial oligosaccharide analog of mammalian oligomannose. Glycobiology 25, 412–419. 10.1093/glycob/cwu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trattnig N.; Farcet J.-B.; Gritsch P.; Christler A.; Pantophlet R.; Kosma P. (2017) Synthesis of a pentasaccharide fragment related to the inner core region of rhizobial and agrobacterial lipopolysaccharides. J. Org. Chem. 82, 12346–12358. 10.1021/acs.joc.7b02172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantophlet R.; Trattnig N.; Murrell S.; Lu N.; Chau D.; Rempel C.; Wilson I. A.; Kosma P. (2017) Bacterially derived synthetic mimetics of mammalian oligomannose prime antibody responses that neutralize HIV infectivity. Nat. Commun. 8, 1601. 10.1038/s41467-017-01640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejchal R.; Doores K. J.; Walker L. M.; Khayat R.; Huang P.-S.; Wang S.-K.; Stanfield R. L.; Julien J.-P.; Ramos A.; Crispin M.; et al. (2011) A potent and broadly neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103. 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard-Borger E. D.; Stick R. V. (2007) An efficient, inexpensive, and shelf-stable diazotransfer reagent: imidazole-1-sulfonyl azide hydrochloride. Org. Lett. 9, 3797–3800. 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Kawai Y.; Kitamoto N.; Osawa T.; Kato Y. (2009) Covalent modification of lysine residues by allyl isothiocyanate in physiological conditions: plausible transformation of isothiocyanate from thiol to amine. Chem. Res. Toxicol. 22, 536–542. 10.1021/tx8003906. [DOI] [PubMed] [Google Scholar]
- Mishra N. K.; Deepak Krishna R. N. V.; Sankararamakrishnan R.; Verma S. (2015) Controlling in vitro insulin amyloidosis with stable peptide conjugates: A combined experimental and computational study. J. Phys. Chem. B 119, 15395–15406. 10.1021/acs.jpcb.5b08215. [DOI] [PubMed] [Google Scholar]
- Jahouh F.; Saksena R.; Aiello D.; Napoli A.; Sindona G.; Kováč P.; Banoub J. H. (2010) Glycation sites in neoglycoconjugates from the terminal monosaccharide antigen of the O-PS of Vibrio cholerae O1, serotype Ogawa, and BSA revealed by matrix-assisted laser desorption-ionization tandem mass spectrometry. J. Mass Spectrom. 45, 1148–1159. 10.1002/jms.1796. [DOI] [PubMed] [Google Scholar]
- Huang B. X.; Kim H.-Y. (2004) Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1237–1247. 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Dotsey E. Y.; Gorlani A.; Ingale S.; Achenbach C. J.; Forthal D. N.; Feigner P. L.; Gach J. S. (2015) A high throughput protein microarray approach to classify HIV monoclonal antibodies and variants. PLoS One 10 (5), e0125581. 10.1371/journal.pone.0125581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doores K. J.; Fulton Z.; Huber M.; Wilson I. A.; Burton D. R. (2010) Antibody 2G12 recognizes di-mannose equivalently in domain- and nondomain-exchanged forms but only binds the HIV-1 glycan shield if domain exchanged. J. Virol. 84, 10690–10699. 10.1128/JVI.01110-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. (2009) Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure 17, 489–498. 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. A.; Chen W.; Gilson M. K. (2007) Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. U. S. A. 104, 1534–1539. 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl C.; Engström O.; Delaine T.; Håkansson M.; Genheden S.; Modig K.; Leffler H.; Ryde U.; Nilsson U. J.; Akke M. (2010) Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of galectin-3. J. Am. Chem. Soc. 132, 14577–14589. 10.1021/ja105852y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabre Y. M.; Contino-Pépin C.; Placide V.; Shiao T. C.; Roy R. (2008) Expeditive synthesis of glycodendrimer scaffolds based on versatile TRIS and mannoside derivatives. J. Org. Chem. 73, 5602–5605. 10.1021/jo8008935. [DOI] [PubMed] [Google Scholar]
- Marin M. J.; Rashid A.; Rejzek M.; Fairhurst S. A.; Wharton S. A.; Martin S. R.; McCauley J. W.; Wileman T.; Field R. A.; Russell D. A. (2013) Glyconanoparticles for the plasmonic detection and discrimination between human and avian influenza virus. Org. Biomol. Chem. 11, 7101–7107. 10.1039/c3ob41703d. [DOI] [PubMed] [Google Scholar]
- Tiwari V. K.; Mishra B. B.; Mishra K. B.; Mishra N.; Singh A. S.; Chen X. (2016) Cu-catalyzed click reaction in carbohydrate chemistry. Chem. Rev. 116, 3086–3240. 10.1021/acs.chemrev.5b00408. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Schroeter C.; Gabius S.; Brinck U.; Goerlach-Graw A.; Gabius H. J. (1991) Bioconjugate Chem. 2, 148–153. 10.1021/bc00009a003. [DOI] [PubMed] [Google Scholar]
- Wurm F. R.; Klok H.-A. (2013) Be squared: expanding the horizon of squaric acid-mediated conjugations. Chem. Soc. Rev. 42, 8220–8236. 10.1039/c3cs60153f. [DOI] [PubMed] [Google Scholar]
- Xu P.; Kelly M.; Vann W. F.; Qadri F.; Ryan E. T.; Kovác P. (2017) Conjugate vaccines from bacterial antigens by squaric acid chemistry: a closer look. ChemBioChem 18, 799–815. 10.1002/cbic.201600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos A.; Van Droogenbroeck B.; Hillmer S.; Grass J.; Kunert R.; Cao J.; Robinson D. G.; Depicker A.; Steinkellner H. (2011) Production of monoclonal antibodies with a controlled N-glycosylation pattern in seeds of Arabidopsis thaliana. Plant Biotechnol. J. 9, 179–192. 10.1111/j.1467-7652.2010.00540.x. [DOI] [PubMed] [Google Scholar]
- Loos A.; Gach J. S.; Hackl T.; Maresch D.; Henkel T.; Porodko A.; Bui-Minh D.; Sommeregger W.; Wozniak-Knopp G.; Forthal D. N.; et al. (2015) Glycan modulation and sulfoengineering of anti-HIV-1 monoclonal antibody PG9 in plants. Proc. Natl. Acad. Sci. U. S. A. 112, 12675–12680. 10.1073/pnas.1509090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemer M.; Mehofer U.; Torres Acosta J. A.; Verdianz M.; Henkel T.; Loos A.; Strasser R.; Maresch D.; Rademacher T.; Steinkellner H.; Mach L. (2014) The human anti-HIV antibodies 2F5, 2G12, and PG9 differ in their susceptibility to proteolytic degradation: down-regulation of endogenous serine and cysteine proteinase activities could improve antibody production in plant-based expression platforms. Biotechnol. J. 9, 493–500. 10.1002/biot.201300207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser R.; Stadlmann J.; Schahs M.; Stiegler G.; Quendler H.; Mach L.; Glossl J.; Weterings K.; Pabst M.; Steinkellner H. (2008) Generation of glyco-engineered Nicotiana benthamiana for the production of monoclonal antibodies with a homogeneous human-like N-glycan structure. Plant Biotechnol. J. 6, 392–402. 10.1111/j.1467-7652.2008.00330.x. [DOI] [PubMed] [Google Scholar]
- Hou S.-J.; Saksena R.; Kovac P. (2008) Preparation of glycoconjugates by dialkyl squarate chemistry revisited. Carbohydr. Res. 343, 196–210. 10.1016/j.carres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.