

Abstract
The naturally occurring channelrhodopsin variant anion channelrhodopsin-1 (ACR1), discovered in the cryptophyte algae Guillardia theta, exhibits large light-gated anion conductance and high anion selectivity when expressed in heterologous settings, properties that support its use as an optogenetic tool to inhibit neuronal firing with light. However, molecular insight into ACR1 is lacking owing to the absence of structural information underlying light-gated anion conductance. Here we present the crystal structure of G. theta ACR1 at 2.9 Å resolution. The structure reveals unusual architectural features that span the extracellular domain, retinal-binding pocket, Schiff-base region, and anion-conduction pathway. Together with electrophysiological and spectroscopic analyses, these findings reveal the fundamental molecular basis of naturally occurring light-gated anion conductance, and provide a framework for designing the next generation of optogenetic tools.
Subject terms: X-ray crystallography, Biophysical chemistry
The crystal structure of anion channelrhodopsin-1 (ACR1) from the algae Guillardia theta provides insights into the basis of anion conductance.
Main
Most organisms depend on light for energy and information. Motile organisms typically capture light using rhodopsin proteins, largely classified into two groups: microbial (type I) and animal (type II)1,2, both exhibiting seven-transmembrane helices and a retinal-based chromophore, but with different effector mechanisms. Animal rhodopsins primarily work as G-protein-coupled receptors that recruit secondary messengers to control effectors such as ion channels that modulate cellular activity, whereas channel and pump microbial rhodopsins can directly provide effector functionality as transmembrane current1,2. Heterologous expression of single-component microbial opsin genes targeted to specific cells of animals defines an experimental approach (optogenetics3) for biology, enabling control of specific cells in behaving organisms with light.
Both channel and pump-encoding opsins are established in optogenetics. Variants of the channel subtype (cation-conducting channelrhodopsins, CCRs) elicit light-triggered cation currents (usually excitatory in neurons). Indeed, light-triggered cation currents are excitatory in the natural host as well; plant behaviours initially observed by botanists more than 150 years ago (movement of single-celled algae excited by light)4 were later found to be due to CCRs, with the initially known member of this subclass (Chlamydomonas reinhardtii ChR1) identified as a cation channel in 20025. Many CCRs have been discovered or designed5–14, and currently available CCRs offer a palette of diversity in absorption spectrum, photocurrent magnitude, light sensitivity and on/off-kinetics12,15.
The development of inhibitory optogenetics initially lagged, but has made strides in recent years3,4,16. Light-induced neuronal inhibition with microbial opsins was first achieved with inward Cl− pumps and outward H+ pumps such as Natronomonas pharaonis halorhodopsin (NpHR) and archaerhodopsin-3 (AR3)17,18. Although widely used, these pumps move only one ion per photon (versus hundreds for channels), thereby exhibiting reduced efficacy15,16. In 2014, anion-conducting channelrhodopsins (ACRs) were created19,20 on the CCR backbone, guided by structural modelling; subsequently, in 2015, naturally occurring ACRs were isolated from chlorophyte algae21 (GtACR1 and GtACR2). The designed ACRs have been developed further22–24, and additional natural ACRs have been found by genome mining25–27. ACRs can translocate 104–105 ions per second21 and can exhibit 102–104-fold higher light sensitivity than inhibitory pumps19–21. After the first demonstration in 2015 of ACRs as inhibitory optogenetic tools that could successfully modulate animal behaviour (with a designed ACR called iC++22), both ACR classes have been widely applied in mice, flies and fish22,23,28–30.
Despite progress in ACR-based inhibitory optogenetics, little is known about the structural basis of radically different ion-selectivity involved in anion conduction. Homology models of GtACR1 were built27,31–33 using the structure of the C1C2 CCR34, but precise structural information on ACRs remained completely lacking. A high-resolution crystal structure would be beneficial, not only to enhance fundamental understanding, but also to provide a foundation for expanding the toolbox of optogenetics (as rapidly resulted from the first CCR crystal structure34 in 2012).
Here we obtain and characterize the crystal structure for GtACR1 at 2.9 Å resolution. This information, together with electrophysiological and spectroscopic analyses, revealed unique natural ACR structure–function relationships that span the extracellular domain, retinal-binding pocket, Schiff base region, and anion-conduction pathway. These features advance our understanding of natural channelrhodopsin biology, and reveal a path for the design and creation of new tools for optogenetics.
Structure determination
To understand the structural basis of light-activated anion conduction, we purified (Extended Data Fig. 1a) and crystallized the best-characterized natural ACR, GtACR1. To improve crystallizability, we truncated 13 C-terminal residues; the resulting construct (residues 1–282) showed similar photocurrents to full-length GtACR1 in human HEK293 cells (Extended Data Fig. 1b) and robust expression in neurons (Extended Data Fig. 1c). Crystals were obtained by lipidic cubic phase analysis (Extended Data Fig. 1d); the structure was determined by molecular replacement, using coordinates of C1C2 (Protein Data Bank accession 3UG9)34, and refined to 2.9 Å resolution (Extended Data Fig. 2).
Extended Data Fig. 1. Crystallography.

a, Size exclusion chromatogram of the purified GtACR1 protein used for crystallography. Similar results were seen in more than 20 independent experiments. b, Electrophysiology of full-length GtACR1 (left) and the final crystallization construct (right); whole-cell voltage-clamp recordings in five cells held at −70 mV, with 513 nm light at 1.0 mW mm−2 irradiance delivered with timing as shown with green-coloured bars, while cells were held at resting potentials from −95 mV (lowest trace) to +5 mV (uppermost trace) in steps of 10 mV. Similar results were seen in 3–5 cells from each group, and no significant difference was seen in resting potential, input resistance, reversal potential or photocurrent magnitude. c, Confocal images of cultured hippocampal neurons expressing full-length GtACR1 (left) and the final crystallization construct (right). Similar results were seen in more than five cells from 3–5 coverslips. Note the markedly reduced aggregation of the truncated construct. d, Crystals of GtACR1. Similar crystals were generated in more than 200 experiments. e, Lattice packing of GtACR1 crystals, viewed parallel to the x axis (left) and the y axis (right). f, Different amino acid configurations at different chains within the asymmetric unit of GtACR1. g, C-terminal interactions among different chains within the asymmetric unit of GtACR1.
Extended Data Fig. 2. Structural analysis of GtACR1.

a, 2Fo − Fc maps (blue mesh, contoured at 1σ) for the retinal-binding pockets of chains A–D. b, 2Fo − Fc maps (blue mesh, contoured at 1σ) for the lipid molecules. c, 2Fo − Fc maps (blue mesh, contoured at 1σ) and Fo − Fc maps (green and red meshes, contoured at 3.0σ and −3.0σ, respectively) for the Schiff base region of chains A–D. Water molecules are shown as red spheres. d, Table describing data collection and refinement statistics of GtACR1. Dataset was collected from 80 crystals. Values in parentheses are for the highest-resolution shell.
Crystals belonged to the P21 space group, containing four GtACR1 protomers (chains A–D) in the asymmetric unit (Extended Data Fig. 1e). Chains A/B and chains C/D were each associated as dimers, with the two dimer molecules arranged anti-parallel. Each protomer showed almost identical conformation except for orientation of certain residues facing the membrane (for example, Trp150, Phe168, Tyr201 and Leu232; Extended Data Fig. 1f), with a notable C-terminal difference. Although the C termini of chains B/C were ordered until Pro273 with similar conformations, those of chains A/D were ordered until Asp278 and Glu280, respectively, and the last 6–8 residues exhibited completely different conformations (Extended Data Fig. 1g). Except for the disordered 3 N-terminal and 2–9 C-terminal residues, GtACR1 itself (residues 4–278 in chain A, 4–273 in chain B, 4–273 in chain C, and 4–280 in chain D), all-trans retinal (ATR), 5 lipids and 4 water molecules were all clearly resolved in the electron density map (Extended Data Fig. 2).
GtACR1 structure and comparison with C1C2 and CrChR2
GtACR1 exhibits a unique N-terminal extracellular domain (residues 4–29), a 7-transmembrane domain (residues 30–249), and a C-terminal region (residues 250–280) (Fig. 1). In comparing GtACR1 with the CCRs C1C2 (PDB accessions 3UG9 and 4YZI)13,34 and CrChR235, we observed both similarities (despite relatively low sequence identities of 28% and 27%, respectively; Extended Data Fig. 3) and notable distinctions. Although there were aspects of architectural commonality between GtACR1 and C1C2 dimers and between GtACR1 and CrChR2 dimers (root mean square deviation (r.m.s.d.) values of 2.10 Å and 1.87 Å respectively over all Cα atoms), and between corresponding monomers (r.m.s.d. values of 1.62 Å and 1.39 Å), many crucial differences with GtACR1 were apparent (Fig. 2a, b).
Fig. 1. Overall structure of GtACR1.
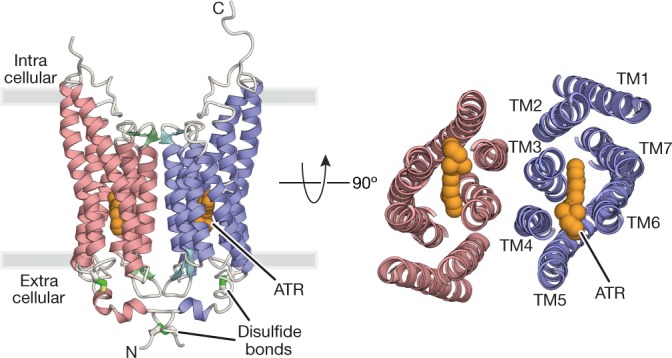
Crystal structure of the GtACR1 dimer, viewed parallel to the membrane (left) and from the extracellular side (right). Disulfide bonds are shown using a stick model (green), and ATR (orange) is depicted by a sphere model.
Extended Data Fig. 3. Structure-based sequence alignment of microbial opsin genes.

The sequences are GtACR1 (GenBank accession AKN63094.1), GtACR2 (GenBank AKN63095.1), ZipACR (GenBank APZ76709.1), PsuACR1 (GenBank ID: KF992074.1), the chimaeric channelrhodopsin between CrChR1 and CrChR2 (C1C2, PDB code 3UG9)34, CrChR1 (GenBank 15811379), CrChR2 (GenBank 158280944), ChR1 from Volvox carteri (VcChR1, UniProtKB B4Y103), ChR1 from V. carteri (VcChR2, UniProtKB ID: B4Y105), Chrimson (GenBank ID: AHH02126.1), ChR from Tetraselmis striata (TsChR, GenBank ID: KF992089.1), HsBR (PDB code 1C3W)59, HsHR (PDB code 1E12)48, and Krokinobacter eikastus rhodopsin 2 (KR2, PDB code 3X3B)60. The sequence alignment was created using PROMALS3D61 and ESPript 362 servers. Secondary structure elements for GtACR1 are shown as coils and arrows. ‘TT’ represents turns. Cysteine residues forming intermolecular and intramolecular disulfide bridges are highlighted in green and yellow, respectively. The residues of retinal-binding pockets are coloured pink. The residues in the Schiff base region are coloured cyan. The residues forming the ECS2 and CCS are coloured orange and blue, respectively.
Fig. 2. Structural comparison of GtACR1 with C1C2.
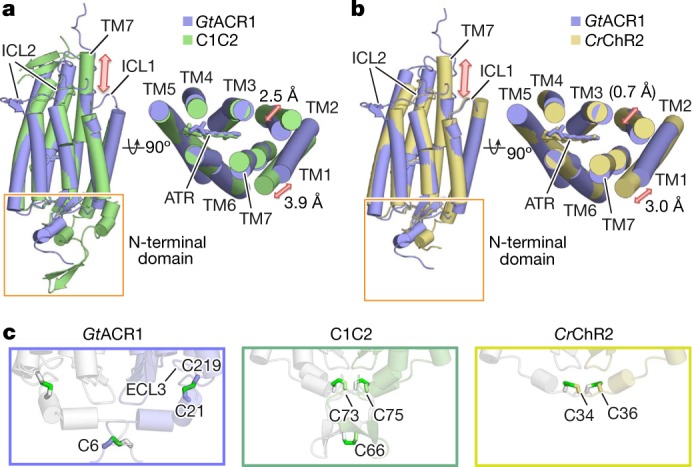
a, b, Side (left) and extracellular (right) view of GtACR1 (blue) superimposed onto C1C2 (green) (a), and CrChR2 (yellow) (b). Red arrows mark the differences between the structures. c, Magnified view of N termini of GtACR1, C1C2 and CrChR2 as delimited by orange boxes in a and b. Green sticks denote disulfide bonds; note intramolecular disulfides bonds in GtACR1 (C219-to-C21) compared to the exclusively intermolecular disulfide bonds in C1C2 (at C73, C75, and C66) and CrChR2 (at C34 and C36).
First, although transmembrane helix 7 (TM7) of C1C2–CrChR2 protrudes approximately 18 Å from the membrane and its following C-terminal region exhibits a β-sheet (Fig. 2a, b), TM7 of GtACR1 does not protrude (resembling more pump-type rhodopsins such as bacteriorhodopsin and halorhodopsin) (Extended Data Fig. 4) and its C-terminal region displays a random coil (Fig. 2a, b; Extended Data Fig. 1g); although lacking secondary structure, this region has several hydrogen-bonding interactions with TM5, TM6, intracellular loop 2 (ICL2) and ICL3, and thus could be important in assembly/structural integrity (Extended Data Fig. 5a). To test this, we truncated the corresponding 29 residues from GtACR1 as crystallized; this almost abolished expression, consistent with the prediction that the C terminus is important for folding and/or stability (Extended Data Fig. 5b).
Extended Data Fig. 4. Structural comparison among GtACR1, HsBR, HsHR, C1C2 and CrChR2.

a, b, Side view and extracellular view of the superimposed transmembrane regions of GtACR1 (blue) and HsBR (cyan) (a), GtACR1 (blue) and HsHR (beige) (b), C1C2 (green) and CrChR2 (yellow) (c). The ATRs are shown as stick models, and are coloured orange (GtACR1), salmon (HsBR), light-yellow (HsHR), green (C1C2) and yellow (CrChR2).
Extended Data Fig. 5. Interactions between N- and C-terminal regions and the 7-TM domain.

a, Interactions between the C-terminal region and the 7-TM domain. Hydrogen bonds are shown by dashed lines. b, Fluorescent size-exclusion chromatography traces of the full-length GtACR1 (1–295), the crystallized construct (1–282), and the C-terminal truncated construct (∆C: 1–253), showing possible importance of the C terminus in proper folding and/or stability. Similar results were observed in three independent experiments. c, Interactions between the N-terminal region and the ECL1. Hydrogen bonds are shown by dashed lines. d, Fluorescent size-exclusion chromatography traces of wild-type and C-to-S mutants of GtACR1. Labels indicate estimated elution positions of the aggregate, GtACR1–eGFP, and free eGFP; C-to-S mutants show decreased (<1/3) expression compared to the wild type. Similar results were observed in three independent experiments. e, Stained SDS–PAGE gel image of wild-type and N-terminal 6-amino-acid-truncated GtACR1 in the presence and absence of reducing reagent (β-mercaptoethanol); the wild type runs as a mixer of monomer and dimer in β-mercaptoethanol,whereas N-terminal-truncated GtACR1 stays monomeric even in the absence of β-mercaptoethanol. This experiment was performed once, but similar experiments with different concentrations of β-mercaptoethanol were performed three times, all with similar results. f–h, Dimer interfaces of GtACR1 (f), C1C2 (g) and CrChR2 (h) viewed at two angles from the side; note reduced interface area (outlined) for GtACR1. For gel source data, see Supplementary Fig. 1.
Second, the N-terminal domain of GtACR1 has a short helix–loop–helix forming hydrogen-bonding interactions with extracellular loop 1 (ECL1) (Extended Data Fig. 5c), whereas that of C1C2 has three helices and two β-strands, tethered to ECL1 via both hydrogen bonding and a Zn2+ ion13. Notably, C1C2, CrChR2 and GtACR1 all have several (2–3) N-terminal cysteine residues, but with different positions and functions. Cys66, Cys73 and Cys75 of C1C2, and Cys34 and Cys36 of CrChR2 form three and two intermolecular disulfide bridges respectively (Fig. 2c). Previous studies had predicted that the residue corresponding to Cys73 in C1C2 (Cys34 in CrChR2) would be Cys21 in GtACR126 and would form an intermolecular disulfide bridge32. However, the GtACR1 structure revealed that Cys21 forms not an intermolecular, but instead a new intramolecular, disulfide bridge with Cys219 on ECL3, whereas Cys6 forms an intermolecular disulfide bridge (Figs. 1, 2c). Gel-filtration chromatography and SDS–PAGE (Extended Data Fig. 5d, e) further support the conclusion that Cys21 and Cys219 are more important for folding and expression, and Cys6 for dimerization.
Third, ICL2 of GtACR1 has a β-sheet that is unique among microbial rhodopsins, extending from the protein core (Fig. 2a, b), in contrast to ICL2 of the C1C2–CrChR2 dimer, which is a random coil close to the protein core involved in dimerization34. Notably, because of these differences in the N terminus and ICL2, the interface area of the GtACR1 dimer (1,315 Å2) is smaller than that for the C1C2 (2,027 Å2) or CrChR2 (1,688 Å2) dimers (Extended Data Fig. 5f–h). This property is concordant with our finding that loss of the intermolecular disulfide bridge markedly affects GtACR1 dimerization in SDS–PAGE analysis (Extended Data Fig. 5e), whereas the loss of the disulfide in C1C2–CrChR2 has minimal effect on dimerization36–38.
Finally, we note a feature of overall GtACR1 structure; the extracellular ends of TM1/TM2 are notably tilted compared to those of CCRs (Fig. 2a, b). These tilts remodel the extracellular vestibule, forming a novel ion-conducting pathway. This unanticipated structural feature appears of substantial importance for understanding the unique ion-conduction properties of GtACR1 (below).
Retinal-binding pocket
In all rhodopsins, retinal is covalently bound to a TM7 lysine residue, forming the Schiff base. GtACR1 and C1C2 contain similar configurations of all-trans-retinal and 15-anti-retinal34,39,40 (Fig. 2a). Here we focus on comparison with C1C2, because the 2017 CrChR2 CCR structure was almost identical to the well-studied 2012 C1C2 CCR structure (Extended Data Fig. 4c; r.m.s.d. value of 0.82 Å over all Cα atoms), and was also reported as a mixture of two states D480 and D470 (absorbing light at 480 and 470 nm, respectively)35,36,41 making it difficult to compare to GtACR135,36. The GtACR1 structure reveals that most residues forming the retinal-binding pocket (RBP) are not conserved between GtACR1 and CCRs (Fig. 3a, b; Extended Data Fig. 3); in C1C2, ATR is enclosed by 16 residues (Fig. 3b), but 11 are not conserved in GtACR1 (Fig. 3a). To analyse the function of these residues, we measured absorption spectra and photocurrents in 10 mutants.
Fig. 3. RBP of GtACR1.

a, b, RBP of GtACR1 (a) and C1C2 (b). c, Effects of mutations (on residues comprising the GtACR1 RBP) on off-kinetics (top, fast closing; bottom, slow closing). Colour codes summarize the role of each residue in setting kinetics, wavelength or both. Data are mean and s.e.m; n = 10 for wild type (WT), 7 for C102A, 4 for M105A, M105I and C133R, and 5 for the rest. *P < 0.05, **P = 0.0021, ****P < 0.0001, Kruskal–Wallis with Dunn’s test. d, Absorption spectra of wild-type GtACR1 and the C237A mutant. Spectra were measured in one experiment. e, Traces of the wild-type GtACR1 and four kinetics-shifted mutants. Scale bar denoted by corresponding colour.
Previous studies reported that GtACR1 has five spectroscopically distinguishable intermediate states: K, L, M, N and O (with L and M as conducting states), and with opening and closing regulated by two different mechanisms (coupled fast-opening–slow-closing and slow-opening–fast-closing)31,33. Confirming previous measurements, we observed that wild-type GtACR1 photocurrent peaks at λmax = 514 nm with biphasic decay (τoff1:54 ± 4.5 ms; τoff2:280 ± 25ms), and that mutant GtACR1(C102A) shows decelerated τoff2 (32 ± 12 s)31,33 (Fig. 3c, e). Notably like C102A, C102S also exhibits decelerated τoff2 (17 ± 2.5 s). M105A, M105I and E163Q show markedly slowed τoff1 (120 ± 30 ms, 90 ± 3.9 ms and 110 ± 22 ms, respectively), suggesting that Met105 and Glu163 are involved in the slow-opening–fast-closing mechanism (Fig. 3c, e).
Notably, studies of Halobacterium salinarum bacteriorhodopsin (HsBR) predict that mutation of certain residues would affect the energy barrier for the transition from K to L intermediates42 (closed to open in GtACR131). Thr198, which interacts with the β-ionone ring of ATR in C1C2, corresponds to Cys133 in GtACR1 (Fig. 3a, b). In HsBR and CrChR2, mutations in residues surrounding the β-ionone affect biophysical properties; for example, M118A in HsBR changes the absorption spectrum (λmax shifting from 551 to 474 nm)43, and T159C in CrChR2 affects conductance and kinetics44. However, the GtACR1(C133A) and GtACR1(C133R) mutants exhibited only slightly blue-shifted spectra, with kinetics and photocurrents comparable to wild-type levels (Fig. 3c, e; Extended Data Figs. 6–8). Thus, the RBP of the GtACR1 β-ionone may be unusually robust (which could also depend on additional non-conserved residues around Cys133, such as Thr134 and Phe160; Fig. 3a, b; Extended Data Fig. 3). Another interesting RBP residue is Cys237, which affects key properties including absorption, kinetics and selectivity when mutated to alanine (Fig. 3c–e; Extended Data Figs. 6–8); notably, the mutant exhibits only a single fast component of current decay (τoff1:87 ± 4.1 ms), suggesting involvement of this residue in the slow-closing mechanism (likely along with the Cys102 residue31; Fig. 3c–e).
Extended Data Fig. 6. Conductances, reversal potentials, absorption spectra and kinetics of wild-type GtACR1 and mutants.

a–c, Photocurrents (a), reversal potentials (b) and absorption spectra (c) of wild-type GtACR1 and ten mutants of the retinal-binding pocket. λmax values are listed in the table (c, bottom). Photocurrents are measured in whole-cell voltage-clamp recordings held at −70 mV, with 513 nm light at 1.0 mW mm−2 irradiance. Data are mean and s.e.m.; n = 9 for WT, 6 for E163Q, 5 for C102A, M105A, C133A, C133R, C153A, E163A and C237A, and 4 for the rest. *P < 0.05, **P < 0.01, one-way ANOVA followed by Dunnett’s test. Reversal potentials are measured with identical light stimulation while cells were held at resting potentials from −95 mV to +15 mV in steps of 10 mV. Data are mean and s.e.m. n = 10 for WT and C237A, 6 for E163A and E163Q, 5 for C102A, M105A, C133A and C153A, and 4 for the rest. **P = 0.0022, one-way ANOVA followed by Dunnett’s test. Spectra measurement was performed in two independent trials, with wild type as a positive control. d, Comparison of fast closing (left) and slow closing (right) coefficients of wild-type and Y72F mutant GtACR1. Data are mean and s.e.m. n = 10 for WT and 5 for Y72F. P = 0.7 for both graphs, two-tailed t-test.
Extended Data Fig. 8. Representative traces of the I–V measurement of wild-type GtACR1 and mutants.

Voltage clamp traces corresponding to the I–V relationships in Extended Data Fig. 7 between −95 mV and +15 mV.
The Schiff-base region
In C1C2, two carboxylates (TM3 Glu162, TM7 Asp292) are within 4 Å of the Schiff-base nitrogen, which forms a direct hydrogen bond with Asp292 (Fig. 4a). However, in GtACR1, the TM3 residue is Ser97, and the TM7 Asp234 has a conformation quite different from Asp292 of C1C2, possibly owing to local interactions with Tyr72 and Tyr207. Notably, the overall architecture of the Schiff-base region in GtACR1 is more similar to halorhodopsins (Fig. 4a). However, in GtACR1, there is no clear electron density, suggesting water or Cl− within hydrogen-bonding distance of the Schiff-base (Supplementary Discussion), and presumably the protonated Schiff base forms at least a weak hydrogen bond with Asp234 (Fig. 4a). Therefore, we undertook structure-guided functional characterization of Tyr72, Tyr207 and Asp234.
Fig. 4. The protonated Schiff base region of GtACR1 and its counterions.
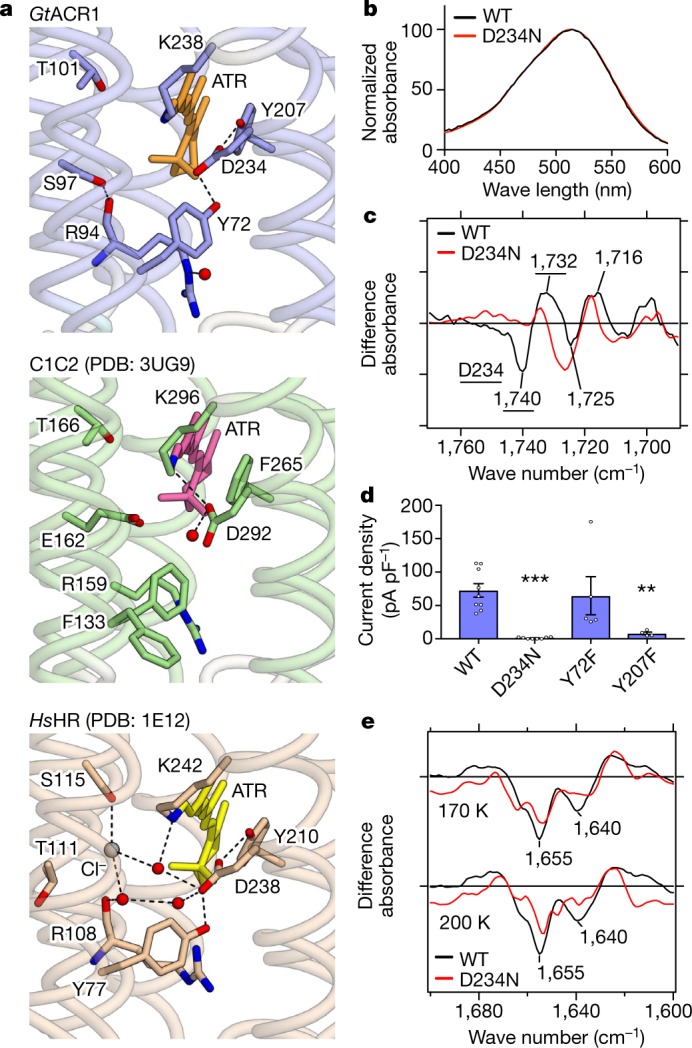
a, Structures of the Schiff base in GtACR1 (top), C1C2 (middle) and HsHR (bottom). Red spheres and dashed lines represent water molecules and hydrogen bonds, respectively; in GtACR1, D234 forms hydrogen bonds with the protonated Schiff base, Y72 and Y207, more similarly to HsHR than C1C2. b, Similar absorption spectra of wild-type GtACR1 and the D234N mutant, suggesting D234 protonation in the dark (see also Extended Data Fig. 9a). c, Light-induced difference FTIR spectra at 77 K. Note disappearance of the 1,740(−)/1,732(+) cm−1 peak pair (assigned to C = O vibration of a protonated carboxylate39,45) in D234N. Findings in b and c hold from at least pH 5–9 (Extended Data Fig. 9c). d, Current densities of wild-type GtACR1and three mutants. Note D234N abolishes the photocurrent (surprising if protonated in the dark), and Y207 (but not Y72) is essential (consistent with the importance of the local hydrogen-bonded network). Data are mean and s.e.m.; n = 9 for WT, 8 for D234N, 5 for Y72F and 4 for Y207F. **P = 0.01, ***P = 0.0006, one-way ANOVA followed by Dunnett’s test. e, Light-induced difference FTIR spectra of wild type and D234N at 170 K and 200 K. Decreased intensity of negative bands at 1,640 and 1,655 cm−1 reveals smaller conformational change of transmembrane helices in D234N. All spectroscopy experiments were performed once.
First, we analysed protonation of Asp-234 using ultraviolet-visible (UV-vis) and low-temperature Fourier-transform infrared (FTIR) spectroscopy. Both assays strongly suggested the protonation of Asp234 in the dark, for the following reasons: first, wild-type and D234N mutants showed almost identical UV-vis absorption spectra (Fig. 4b; Extended Data Fig. 9a); and second, the light-induced difference-FTIR spectra at 77 K showed that a peak pair at 1,740(−)/1,732(+) cm−1 in the wild type, assigned to C = O vibration of a protonated carboxylate39,45, disappears in the D234N mutant (Fig. 4c; Extended Data Fig. 9b). Because the wild-type λmax of the UV-vis spectra and intensity of the FTIR peak-pair remain unchanged from pH 5–9 (Extended Data Fig. 9c), Asp234 is therefore presumed to be protonated over a wide pH range, concordant with previous Raman spectroscopy39.
Extended Data Fig. 9. Spectroscopic characterization of wild-type GtACR1 and the D234N mutant.

a, Absorption spectra of wild-type GtACR1 (top left) and the D234N mutant (top right) measured from pH 3.0 to 10.0. The λmax value at each pH is listed in the table (bottom). b, Difference FTIR spectra of wild-type GtACR1 and the D234N mutant measured at 77 K, 170 K and 200 K. c, Difference FTIR spectra of wild-type GtACR1 in the 1,690–1,770 cm−1 region measured at pH 5.0, 7.0 and 9.0. Forty identical recordings at 77 K and seven identical recordings at 170 K and 200 K were averaged.
However, surprisingly, electrophysiology revealed that D234N nearly abolishes the photocurrent (Fig. 4d). Generally, the effects of aspartate-to-asparagine mutation are small when aspartate is protonated, but in the uniquely configured GtACR1 Schiff-base environment involving close apposition of Asp234, the small difference between aspartate-hydroxyl and asparagine-amino could rearrange the hydrogen-bond network around the Schiff base and thus disturb light-induced conformational changes. This concept is supported by difference FTIR spectra in the amide-I region at 170 K and 200 K (Fig. 4e): the intensity of negative bands at 1,640 and 1,655 cm−1 decreases in D234N, revealing that the conformational change of transmembrane helices in D234N is significantly smaller than in the wild type. Just as with D234N, the nearby Y207F mutation also causes loss-of-function (Fig. 4d). Considering that Phe207 naturally occurs in fully functional C1C2 and even in other natural ACRs including GtACR2 and the ZipACR variant with divergent sequences27 (Extended Data Fig. 3), the precisely arranged hydrogen-bond network of the Schiff-base region thus appears essential for channel activity.
Ion conducting pathway and constrictions
To identify the ion-conduction pathway, we calculated the full electrostatic surface potential of GtACR1 compared to C1C2. C1C2 has a cation-conducting pore pathway formed by TM1, TM2, TM3 and TM7, and GtACR1 has a pore pathway at approximately the same position (Fig. 5a, b) with three marked differences. First, in a pattern opposite to that of C1C2, the surface around the pore of GtACR1 is electropositive, suitable for cation exclusion and thus anion selectivity46 (Fig. 5a; Extended Data Fig. 10a); by contrast, C1C2 has 7 carboxylates along the ion-conducting pathway (Glu121, Glu122, Glu129, Glu136, Glu140, Glu162 and Asp292) and 14 carboxylates on intracellular/extracellular surfaces (Fig. 5b; Extended Data Fig. 10b, d), all contributing to electronegative surfaces in and around the pore suitable for anion exclusion/cation selectivity (Fig. 5b). In GtACR1, Glu122, Glu136 and Glu162 are replaced by Ala61, Ala75 and Ser97, respectively (Fig. 5a) and Glu140 is also not conserved (Extended Data Fig. 3). Also, as shown by previous and present FTIR, residues corresponding to Glu129/Asp292 (Glu68/Asp234) are neutralized45 (Fig. 4b, c). Finally, 12 protein-surface residues are replaced with arginine or lysine; the consistency of this pattern suggests these residues (and Arg94/Lys238) contribute to a suitable electrostatic environment for cation exclusion/anion conduction in GtACR1, confirming earlier predictions46 (Extended Data Figs. 3, 10a, c).
Fig. 5. Ion-conducting pathways of GtACR1 and C1C2.

a, b, Ion-conducting pathways of GtACR1 (a) and C1C2 (b). The surface is coloured by the electrostatic potential calculated using PDB accession 2PQR51 for both GtACR1 and C1C2. Green, purple and orange-dashed circles represent the extracellular constriction site (ECS), intracellular constriction site (ICS) and central constriction site (CCS), respectively. IV, intracellular vestibule.
Extended Data Fig. 10. Comparison of surface electrostatic potential of GtACR1 and C1C2.

a, b, Electrostatic potential surfaces of GtACR1 (a) and C1C2 (b) viewed from four angles. The surface is coloured on the basis of the electrostatic potential contoured from −15 kT (red) to +15 kT (blue). c, d, Representation of positively charged amino acids (lysine and arginine residues) in GtACR1 (c), and negatively charged amino acids (aspartate and glutamate residues) in C1C2 (d).
Second, extracellular vestibules of GtACR1 differ markedly from C1C2. C1C2 has two extracellular vestibules (EV1 and EV2) but only EV2 is connected to the ion-conducting pathway; EV1 is occluded by hydrogen bonding among Gln95, Glu136 and Glu140 (extracellular constriction site 1, ECS1) (Fig. 5b). However, Glu136 and Glu140 are not conserved in GtACR1, and the extracellular-side TM1 and TM2 are markedly tilted, as described above (Figs. 2c, 5a; Extended Data Fig. 3). Thus, pore size becomes much larger, and EV1 becomes connected to the GtACR1 pore-pathway. Furthermore, in contrast to EV1, EV2 of GtACR1 is disconnected because of interactions among Tyr81, Arg94 and Glu223 (extracellular constriction site 2, ECS2) (Figs. 5a, 6a), indicating that EV1 serves as the primary anion-entry pathway in GtACR1. Third, the anion-conducting pathway of GtACR1 is opened not only towards the extracellular side but also intracellularly. In C1C2, although the cation-conducting pathway is opened towards the extracellular side, the cytoplasmic side is occluded by intracellular (ICS) and central (CCS) constriction sites34. However, in GtACR1, residues forming the ICS in C1C2, including Tyr109, Glu122, His173 and Arg307, are replaced by Met 50, Ala61, Leu108 and Thr249, respectively, and the intracellular vestibule extends to the CCS (Figs. 5, 6b, c).
Fig. 6. Constriction sites of GtACR1.

a, The ECS separating EV1 and EV2. Hydrogen bonds are shown as dashed lines. b, Initial glimpse of a patent intracellular conduction pathway for a light-activated channel; architecture of the GtACR1 intracellular ion exit pore leading to the intracellular vestibule (IV). c, The CCS architecture: sole constriction site in the pore, which separates the extracellular and intracellular vestibules. d, Current densities of mutants in residues comprising the CCS. Note the importance of residues E68 and N239 for photocurrents. Data are mean and s.e.m. n = 9 for WT, 5 for Q46A, E68A, E68T and E239A, and 4 for the rest. *P < 0.05, **P < 0.01, one-way ANOVA followed by Dunnett’s test. e, Comparison of reversal potentials. Note the signature of increased cation flux (depolarized reversal potential), consistent with disrupted pore selectivity Data are mean and s.e.m. n = 10 for WT, 6 for Q46A and Q46C, 5 for E68A and 4 for the rest. *P = 0.014, one-way ANOVA followed by Dunnett’s test.
The channel is thus maintained in a closed state only by the CCS (Figs. 5, 6c). In C1C2, the CCS is formed by Ser102, Glu129 and Asn297. These three residues are conserved in GtACR1 (Gln46, Glu68 and Asn239) and its Gln46 on TM1 forms an additional hydrogen bond with Asn239, thereby further stabilizing the CCS. To test the function of these residues, we prepared 10 mutants of Gln46, Glu68 and Asn239, and measured activity by patch-clamp analysis. All Glu68 and Asn239 mutants exhibited smaller photocurrents, and Q46A showed comparable photocurrents but depolarized reversal-potential (Fig. 6d, e). Thus, all three CCS residues are important for anion-channel function, but with different roles: Glu68 and Asn239 for conductance, and Gln46 for selectivity.
Discussion
This high-resolution view into the inner workings of GtACR1 reveals that CCRs and natural ACRs share certain overall features, but also exhibit highly informative differences (especially in the architecture of the GtACR1 anion-conducting pathway, with exchange of one extracellular vestibule for another). The GtACR1 closed-state pore is also remarkable, almost entirely open with the exception of a single central constriction formed by Gln46, Asn239, Ser43 and Glu68; anions can be released intracellularly via the open conduction pore formed by Ala61, Leu108 and Thr249 (Figs. 5a, 6b). Thus, these data provide the first, to our knowledge, crystal structure of any channelrhodopsin revealing an open intracellular pore pathway.
Integration of structural, electrophysiological and spectroscopic analyses uncovered unique features of the Schiff base relevant to ChR (and halorhodopsin and bacteriorhodopsin) evolution. As in HsHR, a TM7 aspartate is coordinated by two tyrosine residues in GtACR1, and the TM3 glutamate in the CCR C1C2 is instead represented in both GtACR1 and HsHR by a neutral hydrophilic residue (Fig. 4a). Furthermore, a TM2 tyrosine (Tyr72, uniformly conserved among pump-type halorhodopsins and bacteriorhodopsins) is present in GtACR1 (and is almost 100% conserved among natural ACRs; Extended Data Fig. 3)27 but is dispensable for function; Y72F changes neither conductance (Fig. 4d) nor kinetics of the M-intermediate rise or decay (Extended Data Fig. 6d), characterized by fast or slow kinetics, respectively. This differs from bacteriorhodopsin, in which Y57F accelerates formation of the M-intermediate47. Because CCRs have replaced this residue (Extended Data Fig. 3), an evolutionary model is suggested in which natural ACRs such as GtACR1 evolved from light-driven Cl− pumps, and CCRs subsequently arose from natural ACRs via surface electrostatic remodelling4,46.
Further insight into the mechanism and development of anion conduction arises from the consideration of another unusual feature of the Schiff-base region: charge distribution. In the dark, the Schiff-base nitrogen is protonated and therefore requires a mechanism to stabilize the positive charge. In GtACR1, Glu68 and Asp234 provide the only carboxylates within 6 Å of this Schiff-base nitrogen (approximately 5.4 Å and 3.5 Å, respectively), but FTIR analyses indicate that both are also protonated in the dark45 (Fig. 4d, e). Cl− does not have a charge-stabilization role either, as Cl− is not bound to the Schiff-base region in GtACR133 (Fig. 4a; unlike in HsHR48). One possible explanation is that strongly polarized water could bind to the Schiff base (behaving as a hydroxyl ion, as proposed in HsBR and mutants2,49,50; Supplementary Discussion), and another possibility is that the partial-negative charge of the nearby Asp234 carbonyl is sufficient to weakly stabilize the positively charged Schiff base. As a result, the net charge in the Schiff-base region may represent the achievement of perhaps the most challenging evolutionary step in the adaptation to facilitate anion conduction (alongside the acquisition of positive surface electrostatic potential throughout the pore and vestibules; Fig. 4): namely, partial local positivity despite the obligate negative nature of the Schiff base counterion.
To advance our understanding of the molecular mechanism of light-gated anion conduction, additional studies (including structural resolution of natural or designed ACRs in fully open or intermediate states) will be required. This initial high-resolution structural information provides a framework for the further development of ACR-based optogenetic tools—for example, the creation of kinetic, spectral and selectivity variants that maintain the advantages of the GtACR1 backbone including strong photocurrents, just as the initial CCR structure34 allowed the development of new classes of optogenetic functionality4. Further insights into the evolutionary and functional relationships among different channelrhodopsin family members will continue to arise from the solution of structures that correspond to kinetic, spectral and selectivity variants, advancing basic understanding of this remarkable class of natural protein.
Methods
Sample sizes were determined based on previous literature and best practices in the field; no statistical methods were used to predetermine sample size. No experiments in animals were conducted in this paper and hence experiments were not randomized or blinded.
Cloning, protein expression and purification
The crystallization construct of GtACR1 was generated with several features to enhance protein purification and crystallogenesis. The flexible 13 amino acids at the C terminus were truncated after Gly282. A Flag tag followed by the 3C protease cleavage site was added to the N terminus and an enhanced GFP (eGFP) with a His10 tag and the 3C site was attached to the truncated C terminus via the 3C cleavage site. The finalized GtACR1 crystallization construct was expressed in Sf9 cells using the BestBac (Expression Systems) baculovirus system. Cell cultures were grown to a density of 4 × 106 cells ml−1, infected with GtACR1 baculovirus, and shaken at 27 °C for 18 h. Then, 20 μM all-trans retinal (ATR) (Sigma) was supplemented to the culture and incubation continued for 42 more hours, and cell pellets were collected and stored at −80 °C. To purify GtACR1, the pellets were lysed with a hypotonic lysis buffer (20 mM HEPES pH 7.5, 1 mM EDTA and protease inhibitors). The cell debris was then homogenized with a glass douncer in a solubilization buffer (1% n-dodecyl-β-d-maltopyranoside (DDM), 0.06% cholesteryl hemisuccinate tris salt (CHS), 20 mM HEPES pH 7.5, 500 mM NaCl, 20% glycerol, 10 mM imidazole and protease inhibitors) and solubilized for 2 h in 4 °C. The insoluble cell debris was removed by centrifugation (38,000g, 25 min), and the supernatant was mixed with the Ni-NTA agarose resin (Qiagen) for 2 h in 4 °C. The Ni-NTA resin was collected into a glass chromatography column, washed with 20 column volumes of a wash buffer (0.05% DDM, 0.01% CHS, 20 mM HEPES pH 7.5, 500 mM NaCl, 20% glycerol and 20 mM imidazole) and was eluted in a wash buffer supplemented with 250 mM imidazole. The Ni-NTA eluent was then supplemented with 2 mM CaCl2 and was loaded over anti-Flag M1 resin over 1 h. The protein was then washed with a Flag wash buffer (0.05% DDM, 0.01% CHS, 20 mM HEPES pH 7.5, 300 mM NaCl, 5% glycerol and 2 mM CaCl2) and eluted with a Flag elution buffer (0.05% DDM, 0.01% CHS, 20 mM HEPES pH 7.5, 300 mM NaCl, 5% glycerol, 0.2 mg ml−1 Flag peptide and 3 mM EDTA). After the cleavage of the Flag tag and eGFP-His10 by His-tagged 3C protease, the sample was reloaded onto the Ni-NTA column to capture the cleaved eGFP-His10. The flow-through containing GtACR1 was collected, concentrated and purified through gel-filtration chromatography in a final buffer (100 mM NaCl, 20 mM HEPES pH 7.5, 0.05% DDM and 0.01% CHS). Peak fractions were pooled and concentrated to 30 mg ml−1 (Extended Data Fig. 1b).
Crystallization
Purified GtACR1 protein was crystallized using the lipidic cubic phase (LCP) method as described previously34. Protein was mixed with monopalmitolein (Nu-chek) at a weight ratio of 1:1 (protein:lipid) using a coupled syringe mixing device. Then, 20–25 nl protein–LCP mixture drops were accurately dispensed on a 96-well sandwich plate and overlaid by 500 nl of precipitant solution by the Gryphon LCP robot (Art Robbins Instruments). Initial crystals were obtained in 10% (w/v) polypropylene glycol P 400 (PPG P400), 100 mM MES pH 6.0 and 100 mM potassium formate; the best crystals were obtained in 10–12% (w/v) polypropylene glycol P 400 (PPG P400), 100 mM MES pH 6.0, 100 mM potassium formate and 1–3% 1-butanol. Crystals were harvested using micromeshes (MiTeGen), and were flash-cooled in liquid nitrogen without any additional cryoprotection.
Data collection and structure determination
X-ray diffraction data were collected at Advanced Photon Source GM/CA-CAT beamline 23ID-B and 23ID-D using a micro beam size of 10 × 10 μm2, at a wavelength of 1.033 Å. Small wedge data, each consisting of 5–20°, were collected from single crystals, and 131 collected datasets were processed automatically using KAMO52. Each dataset was indexed and integrated using XDS53, and classified using the correlation coefficients between data sets. Eighty datasets in the best cluster were scaled and merged using XSCALE. The structure was determined by molecular replacement with the program MoRDa (Vagin and Lebedev; http://www.biomexsolutions.co.uk/morda), using the cation channelrhodopsin C1C2 (PDB accession 3UG9) and G11A mutant of SARS-CoV 3C-like protease (PDB accession 2PWX) as the search models. However, the 2PWX model was not fitted to electron density at all and removed. The resultant structure was iteratively refined using Refmac554, Phenix55 and MR-rosetta56, and manually rebuilt in Coot57. The final model contained 95.7, 4.1 and 0.3% in the favoured, allowed and outlier regions of the Ramachandran plot, respectively. Final refinement statistics are summarized in Extended Data Fig. 1. All molecular graphics figures were prepared with Cuemol (Ishitani; http://www.cuemol.org).
Electrophysiology
HEK293 cells (Thermo Fisher, authenticated by the vendor, not tested for mycoplasma contamination) were plated on poly-d-lysine coated glass coverslips (Fisher) at 10% confluency, and were transfected with 0.5 μg of a plasmid and 1 μl lipofectamine 2000 (Thermofisher Scientific) per well. After 24–48 h of transfection, cells were placed in an extracellular tyrode medium (150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES pH 7.4 and 10 mM glucose). A borosilicate patch pipette (Harvard Apparatus) with resistance of 3–6 MΩ was filled with intracellular medium (140 mM potassium-gluconate, 10 mM EGTA, 2 mM MgCl2 and 10 mM HEPES pH 7.2). The photocurrent and kinetic measurements were performed in voltage-clamp mode at membrane potential of −70 mV and −10 mV, respectively. Light was delivered with the Spectra X Light engine (Lumencor) connected to the fluorescence port of a Leica DM LFSA microscope, and a 513/15 filter was used for green light generation. To determine channel kinetics and photocurrent amplitudes, traces were first smoothed using a lowpass Gaussian filter with a −3 dB cutoff for signal attenuation and noise reduction at 1,000 Hz and then analysed in Clampfit software (Axon Instruments). Liquid junction potentials were corrected using the Clampex built-in liquid junction potential calculator as previously described22. Current density was calculated by dividing peak photocurrent amplitude by cell’s membrane capacitance, which was calculated from the Clampex built-in membrane test. Statistical analysis was performed with t-test or one-way ANOVA, and the Kruskal–Wallis test for non-parametric data, using Prism 7 (GraphPad) software.
Light-induced difference FTIR spectroscopy
Wild-type and D234N mutant GtACR1 were reconstituted into a mixture of POPE and POPG (molar ratio = 3:1) with a protein-to-lipid molar ratio of 1:30 by removing DDM with Bio-Beads (SM-2, Bio-Rad). The reconstituted samples were washed three times with buffers at pH 5.0 (2 mM citrate-NaOH), pH 7.0 (2 mM HEPES-NaOH) or pH 9.0 (2 mM borate-NaOH) with 1 mM NaCl. The pellet was re-suspended in the same buffer, with the concentration adjusted to 1.7 mg ml−1. A 60 µl aliquot was placed onto a BaF2 window and air-dried. FTIR spectroscopy was applied to the films hydrated with 1 µl H2O at 77 K, 170 K and 200 K as described previously58. In brief, the sample was placed in an Oxford DN-1704 cryostat mounted in the Bio-Rad FTS-40 spectrometer (instrumental resolution of FTIR is 2 cm−1). For the formation of photo-intermediates at 77 K, samples were illuminated at 500 nm (interference filter) from a 1-kW halogen-tungsten lamp for 2 min and photo-reversed with >600 nm light (R-62 cut-off filter, Toshiba) for 1 min. For formation of photo-intermediates at 170 K and 200 K, samples were illuminated with >500 nm light (Y-52 cut-off filter, Toshiba) for 1 min. For each measurement of FTIR spectroscopy, 256 interferograms were accumulated; 40 identical recordings at 77 K and 7 identical recordings at 170 K and 200 K were averaged.
Measurement of UV absorption spectra
Protein absorbance spectra were measured with an Infinite M1000 microplate reader (Tecan Systems Inc.) using 96 well plates (Thermofisher scientific). The GtACR1 samples were suspended in a buffer containing 100 mM NaCl, 0.05% DDM, 0.01% CHS, and 20 mM sodium citrate, sodium acetate, sodium cacodylate, HEPES, Tris, CAPSO or CAPS. pH was adjusted from 4 to 10 by the addition of NaOH or HCl. Recorded spectra value was averaged from 20 measurements from a single session.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at 10.1038/s41586-018-0511-6.
Supplementary information
This file contains a Supplementary Discussion.
Acknowledgements
We thank C. Lee, M. Lo, K. Geiselhart and M. Lima for technical support; K. K. Kumar, N. R. Latorraca, M. Inoue and K. Katayama for critical comments; and the APS beamline staff at 23ID-B and 23ID-D for assistance in data collection. We acknowledge support by the Stanford Bio-X and the Kwanjeong Foundation (Y.S.K.), JST PRESTO (JPMJPR1782 to H.E.K., JPMJPR15P2 to K.I.), the US Department of Energy, Scientific Discovery through Advanced Computing (SciDAC) program (R.O.D.), MEXT (17H03007 to K.I., 25104009/15H02391 for H.K.), J.S.T. CREST (JPMJCR1753, H.K.) and Mathers Charitable Foundation (B.K.K.). K.D. was supported by a grant for channelrhodopsin crystal structure determination from the NIMH (R01MH075957 to K.D.).
Reviewer information
Nature thanks P. Scheerer, L. Tian and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Fig. 7. Current–voltage (I–V) relationships of wild-type GtACR1 and mutants.

The I–V relationship between −95 mV and +15 mV was determined from the single current amplitude at the indicated potentials. Each measurement is normalized to the current amplitude measured at −25 mV. Data are mean and s.e.m. n = 10 for WT and C237A, 8 for E223A, 6 for Q46C, E163A and E163Q, 4 for E68S, E68T, C102S and M105I, and 5 for the rest.
Author contributions
Y.S.K. and H.E.K. contributed equally and either has the right to list himself first in bibliographic documents. Y.S.K. and H.E.K. expressed, purified and crystallized GtACR1, harvested crystals, and collected diffraction data. H.E.K. and K.Y. processed the diffraction data and solved the structure. Y.S.K. and L.E.F. performed electrophysiology. Y.S.K. measured UV-vis spectra. S.I. performed FTIR experiments under the guidance of K.I. and H.K. J.M.P. and R.O.D. provided input on structural considerations. C.R. and K.E.E. performed cell cultures and molecular cloning for electrophysiology. K.D. initiated and supervised this ChR structure/function project; Y.S.K., H.E.K., B.K.K. and K.D. planned and guided the work, and interpreted the data. Y.S.K., H.E.K. and K.D. prepared the manuscript and wrote the paper with input from all the authors.
Data availability
The protein coordinate and atomic structure factor have been deposited in the Protein Data Bank (PDB) under accession number 6CSM. The raw diffraction images have been deposited in the SBGrid Data Bank repository (ID: 569). All other data are available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yoon Seok Kim, Hideaki E. Kato
Contributor Information
Hideaki E. Kato, Email: hekato@stanford.edu
Karl Deisseroth, Email: deissero@stanford.edu.
Extended data
is available for this paper at 10.1038/s41586-018-0511-6.
Supplementary information
is available for this paper at 10.1038/s41586-018-0511-6.
References
- 1.Zhang F, et al. The microbial opsin family of optogenetic tools. Cell. 2011;147:1446–1457. doi: 10.1016/j.cell.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ernst OP, et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 2014;114:126–163. doi: 10.1021/cr4003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015;18:1213–1225. doi: 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deisseroth K, Hegemann P. The form and function of channelrhodopsin. Science. 2017;357:eaan5544. doi: 10.1126/science.aan5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagel G, et al. Channelrhodopsin-1: a light-gated proton channel in green algae. Science. 2002;296:2395–2398. doi: 10.1126/science.1072068. [DOI] [PubMed] [Google Scholar]
- 6.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl Acad. Sci. USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F, et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat. Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat. Neurosci. 2009;12:229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 9.Gunaydin LA, et al. Ultrafast optogenetic control. Nat. Neurosci. 2010;13:387–392. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- 10.Yizhar O, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JY, Knutsen PM, Muller A, Kleinfeld D, Tsien RY. ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 2013;16:1499–1508. doi: 10.1038/nn.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klapoetke NC, et al. Independent optical excitation of distinct neural populations. Nat. Methods. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato HE, et al. Atomistic design of microbial opsin-based blue-shifted optogenetics tools. Nat. Commun. 2015;6:7177. doi: 10.1038/ncomms8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajasethupathy P, et al. Projections from neocortex mediate top-down control of memory retrieval. Nature. 2015;526:653–659. doi: 10.1038/nature15389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattis J, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat. Methods. 2011;9:159–172. doi: 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiegert JS, Mahn M, Prigge M, Printz Y, Yizhar O. Silencing neurons: tools, applications, and experimental constraints. Neuron. 2017;95:504–529. doi: 10.1016/j.neuron.2017.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 18.Chow BY, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344:420–424. doi: 10.1126/science.1252367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wietek J, et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344:409–412. doi: 10.1126/science.1249375. [DOI] [PubMed] [Google Scholar]
- 21.Govorunova EG, Sineshchekov OA, Janz R, Liu X, Spudich JL. Natural light-gated anion channels: a family of microbial rhodopsins for advanced optogenetics. Science. 2015;349:647–650. doi: 10.1126/science.aaa7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berndt A, et al. Structural foundations of optogenetics: determinants of channelrhodopsin ion selectivity. Proc. Natl Acad. Sci. USA. 2016;113:822–829. doi: 10.1073/pnas.1523341113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wietek J, et al. An improved chloride-conducting channelrhodopsin for light-induced inhibition of neuronal activity in vivo. Sci. Rep. 2015;5:14807. doi: 10.1038/srep14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wietek J, et al. Anion-conducting channelrhodopsins with tuned spectra and modified kinetics engineered for optogenetic manipulation of behavior. Sci. Rep. 2017;7:14957. doi: 10.1038/s41598-017-14330-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Govorunova EG, Sineshchekov OA, Spudich JL. Proteomonas sulcata ACR1: a fast anion channelrhodopsin. Photochem. Photobiol. 2016;92:257–263. doi: 10.1111/php.12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wietek J, Broser M, Krause BS, Hegemann P. Identification of a natural green light absorbing chloride conducting channelrhodopsin from Proteomonas sulcata. J. Biol. Chem. 2016;291:4121–4127. doi: 10.1074/jbc.M115.699637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govorunova EG, et al. The expanding family of natural anion channelrhodopsins reveals large variations in kinetics, conductance, and spectral sensitivity. Sci. Rep. 2017;7:43358. doi: 10.1038/srep43358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer SM, et al. Optogenetic and chemogenetic strategies for sustained inhibition of pain. Sci. Rep. 2016;6:30570. doi: 10.1038/srep30570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, et al. A central catecholaminergic circuit controls blood glucose levels during stress. Neuron. 2017;95:138–152. doi: 10.1016/j.neuron.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 30.Mohammad F, et al. Optogenetic inhibition of behavior with anion channelrhodopsins. Nat. Methods. 2017;14:271–274. doi: 10.1038/nmeth.4148. [DOI] [PubMed] [Google Scholar]
- 31.Sineshchekov OA, Govorunova EG, Li H, Spudich JL. Gating mechanisms of a natural anion channelrhodopsin. Proc. Natl Acad. Sci. USA. 2015;112:14236–14241. doi: 10.1073/pnas.1513602112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Sineshchekov OA, Wu G, Spudich JL. In vitro activity of a purified natural anion channelrhodopsin. J. Biol. Chem. 2016;291:25319–25325. doi: 10.1074/jbc.C116.760041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sineshchekov OA, Li H, Govorunova EG, Spudich JL. Photochemical reaction cycle transitions during anion channelrhodopsin gating. Proc. Natl Acad. Sci. USA. 2016;113:E1993–E2000. doi: 10.1073/pnas.1525269113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato HE, et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482:369–374. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volkov O, et al. Structural insights into ion conduction by channelrhodopsin 2. Science. 2017;358:eaan8862. doi: 10.1126/science.aan8862. [DOI] [PubMed] [Google Scholar]
- 36.Krause N, Engelhard C, Heberle J, Schlesinger R, Bittl R. Structural differences between the closed and open states of channelrhodopsin-2 as observed by EPR spectroscopy. FEBS Lett. 2013;587:3309–3313. doi: 10.1016/j.febslet.2013.08.043. [DOI] [PubMed] [Google Scholar]
- 37.Sattig T, Rickert C, Bamberg E, Steinhoff HJ, Bamann C. Light-induced movement of the transmembrane helix B in channelrhodopsin-2. Angew. Chem. Int. Edn Engl. 2013;52:9705–9708. doi: 10.1002/anie.201301698. [DOI] [PubMed] [Google Scholar]
- 38.Pescitelli G, et al. Exciton circular dichroism in channelrhodopsin. J. Phys. Chem. B. 2014;118:11873–11885. doi: 10.1021/jp505917p. [DOI] [PubMed] [Google Scholar]
- 39.Yi A, Mamaeva N, Li H, Spudich JL, Rothschild KJ. Resonance raman study of an anion channelrhodopsin: effects of mutations near the retinylidene Schiff base. Biochemistry. 2016;55:2371–2380. doi: 10.1021/acs.biochem.6b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hontani Y, et al. Reaction dynamics of the chimeric channelrhodopsin C1C2. Sci. Rep. 2017;7:7217. doi: 10.1038/s41598-017-07363-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruun S, et al. Light–dark adaptation of channelrhodopsin involves photoconversion between the all-trans and 13-cis retinal isomers. Biochemistry. 2015;54:5389–5400. doi: 10.1021/acs.biochem.5b00597. [DOI] [PubMed] [Google Scholar]
- 42.Maeda A, Tomson FL, Gennis RB, Balashov SP, Ebrey TG. Water molecule rearrangements around Leu93 and Trp182 in the formation of the L intermediate in bacteriorhodopsin’s photocycle. Biochemistry. 2003;42:2535–2541. doi: 10.1021/bi020532n. [DOI] [PubMed] [Google Scholar]
- 43.Greenhalgh DA, Farrens DL, Subramaniam S, Khorana HG. Hydrophobic amino acids in the retinal-binding pocket of bacteriorhodopsin. J. Biol. Chem. 1993;268:20305–20311. [PubMed] [Google Scholar]
- 44.Berndt A, et al. High-efficiency channelrhodopsins for fast neuronal stimulation at low light levels. Proc. Natl Acad. Sci. USA. 2011;108:7595–7600. doi: 10.1073/pnas.1017210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi A, et al. Structural changes in an anion channelrhodopsin: formation of the K and L intermediates at 80 K. Biochemistry. 2017;56:2197–2208. doi: 10.1021/acs.biochem.7b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berndt A, Deisseroth K. Expanding the optogenetics toolkit. Science. 2015;349:590–591. doi: 10.1126/science.aac7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Govindjee R, et al. Effects of substitution of tyrosine 57 with asparagine and phenylalanine on the properties of bacteriorhodopsin. Biochemistry. 1995;34:4828–4838. doi: 10.1021/bi00014a040. [DOI] [PubMed] [Google Scholar]
- 48.Kolbe M, Besir H, Essen LO, Oesterhelt D. Structure of the light-driven chloride pump halorhodopsin at 1.8 A resolution. Science. 2000;288:1390–1396. doi: 10.1126/science.288.5470.1390. [DOI] [PubMed] [Google Scholar]
- 49.Betancourt FM, Glaeser RM. Chemical and physical evidence for multiple functional steps comprising the M state of the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 2000;1460:106–118. doi: 10.1016/s0005-2728(00)00133-x. [DOI] [PubMed] [Google Scholar]
- 50.Facciotti MT, Rouhani S, Glaeser RM. Crystal structures of bR(D85S) favor a model of bacteriorhodopsin as a hydroxyl-ion pump. FEBS Lett. 2004;564:301–306. doi: 10.1016/S0014-5793(04)00208-X. [DOI] [PubMed] [Google Scholar]
- 51.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamashita K, Hirata K, Yamamoto M. KAMO: towards automated data processing for microcrystals. Acta Crystallogr. D. 2018;74:441–449. doi: 10.1107/S2059798318004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabsch W. Xds. Acta Crystallogr. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DiMaio F. Advances in Rosetta structure prediction for difficult molecular-replacement problems. Acta Crystallogr. D. 2013;69:2202–2208. doi: 10.1107/S0907444913023305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanimoto T, Furutani Y, Kandori H. Structural changes of water in the Schiff base region of bacteriorhodopsin: proposal of a hydration switch model. Biochemistry. 2008;42:2300–2306. doi: 10.1021/bi026990d. [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 60.Kato HE, et al. Structural basis for Na+ transport mechanism by a light-driven Na+ pump. Nature. 2015;521:48–53. doi: 10.1038/nature14322. [DOI] [PubMed] [Google Scholar]
- 61.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res.42, W320–W324 (2014). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains a Supplementary Discussion.
Data Availability Statement
The protein coordinate and atomic structure factor have been deposited in the Protein Data Bank (PDB) under accession number 6CSM. The raw diffraction images have been deposited in the SBGrid Data Bank repository (ID: 569). All other data are available from the corresponding authors upon reasonable request.