

Abstract
Tissue fibrosis is a major cause of mortality that results from the deposition of matrix proteins by an activated mesenchyme. Macrophages accumulate in fibrosis, but the role of specific subgroups in supporting fibrogenesis has not been investigated in vivo. Here we used single-cell RNA sequencing (scRNA-seq) to characterize the heterogeneity of macrophages in bleomycin-induced lung fibrosis in mice. A novel computational framework for the annotation of scRNA-seq by reference to bulk transcriptomes (SingleR) enabled the subclustering of macrophages and revealed a disease-associated subgroup with a transitional gene expression profile intermediate between monocyte-derived and alveolar macrophages. These CX3CR1+SiglecF+ transitional macrophages localized to the fibrotic niche and had a profibrotic effect in vivo. Human orthologues of genes expressed by the transitional macrophages were upregulated in samples from patients with idiopathic pulmonary fibrosis. Thus, we have identified a pathological subgroup of transitional macrophages that are required for the fibrotic response to injury.
Fibrosing diseases are a major cause of mortality. Whether toxin-related, genetic, malignant, infectious, autoimmune or idiopathic, they can affect any organ1. Lung fibrosis is a particularly vexing clinical problem because of the lack of effective therapies and a poor understanding of its etiology. The most prevalent form, idiopathic pulmonary fibrosis, has a median survival of only 3 years, and approved treatments are limited2. With respect to the pathogenesis of fibrosis in multiple organs, attention has focused on fibroblast activation, given the central role of fibroblasts in the deposition of matrix proteins such as collagen3–6. However, the mechanisms of fibroblast activation and proliferation are not fully understood.
The healthy adult lung contains two types of resident macrophages: a self-renewing population of embryonically-derived macrophages known as alveolar macrophages and a separate population of interstitial macrophages present near the larger airways and in the lung interstitium7,8. In injury models of fibrosis, monocyte-derived cells are recruited to tissue sites and have a profibrotic effect9–11. However, the diversity of these cells and the mechanisms by which they mediate the profibrotic effect are incompletely understood, in part because the study of specific transcriptomic subsets has been limited by bulk sequencing approaches. In particular, the kinetically evolving heterogeneity inherent to disease cannot be parsed with the limited range of flow cytometric markers used to study populations in bulk. Because scRNA-seq can resolve diversity within cellular populations, we reasoned that it should enhance understanding of macrophage heterogeneity and specify subsets for functional analysis in fibrosis.
A current limitation in single cell analysis is that there are no prevalent methods for identification of cell types other than manual annotation, which suffers from subjectivity. Moreover, referencing conventional markers is challenged by incomplete mRNA detection. We developed a computational tool for unbiased annotation as well as clustering of scRNA-seq, called SingleR (single cell recognition). SingleR assigns cellular identity for single cell transcriptomes by comparison to reference datasets of pure cell types sequenced by microarray or RNA-seq. Here, we used SingleR to identify macrophages from a mixed sample of lung cells at baseline and after bleomycin-induced lung injury in mice. In addition, we applied a novel hierarchical clustering approach based on SingleR correlations to subcluster macrophages in fibrosis, which enabled the identification of cells with transitional gene expression between monocyte-derived and alveolar macrophages. We used markers identified by the subclustering analysis to investigate the functional properties of these transitional macrophages.
Our studies revealed that monocyte-derived, disease-associated macrophages transitioning to alveolar identity localized to the fibrotic niche and exerted a profibrotic effect. Specific ablation of these macrophages prevented lung fibrosis. Mechanistically, the scar-associated macrophages were the predominant source of the mitogen Pdgf-aa, which was necessary for proliferation of fibroblasts. These studies uncover a role for macrophages in the paracrine regulation of fibroblasts and provide a new perspective on myeloid-mesenchymal crosstalk in response to injury.
Results
Reference datasets enhance annotation of scRNA-seq
In order to improve annotation of cell types in scRNA-seq, we developed a computational method called SingleR, which correlated single cell transcriptomes with reference transcriptomic datasets and improved its inferences iteratively (Fig. 1a, Supplementary Information 1). The SingleR pipeline is based on correlating reference bulk RNA-seq datasets of pure cell types with single-cell gene expression. First, we validated SingleR using the Immunological Genome Project (ImmGen) database12 to annotate mouse scRNA-seq and Encode13 and Blueprint Epigenomics14 transcriptomes to annotate human data. In the first step of the SingleR pipeline, a Spearman coefficient was calculated for single-cell gene expression with each of the samples in the reference dataset, using only the variable genes in the reference dataset. Using only variable genes among the cell types increased the ability to distinguish closely related cell types. This process was performed iteratively using only the top cell types from the previous step and the variable genes among them until only one cell type remained (for details see Methods, Supplementary Information 1 and the SingleR Github repository at https://github.com/dviraran/SingleR ).
Figure 1. Reference-based annotation of scRNA-seq.
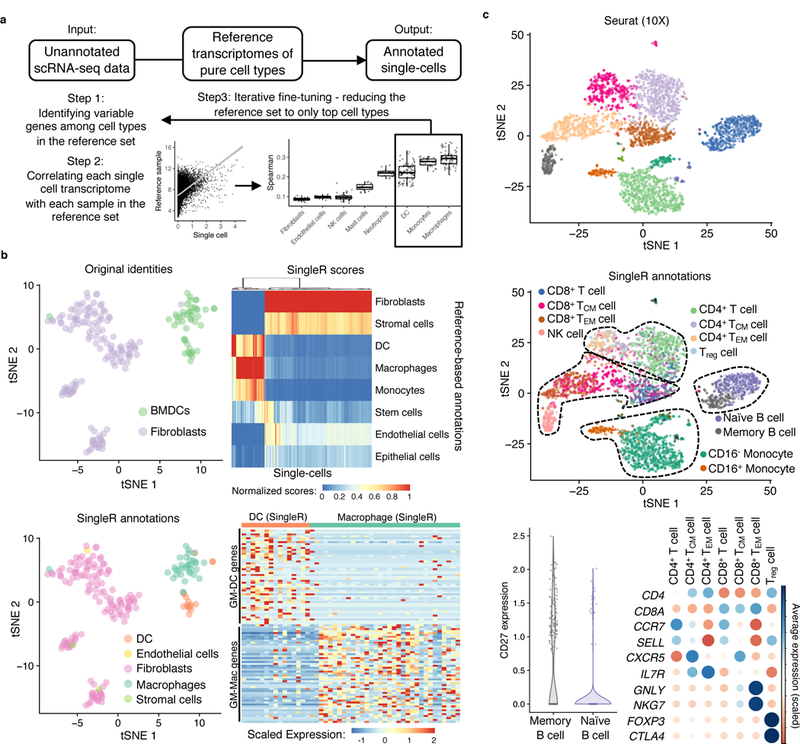
a, Schematic of SingleR, a protocol for cell-type annotation by reference to transcriptomes of pure cell types. b, tSNE plot of a published scRNA-seq dataset of fibroblast and bone marrow-derived dendritic cells15 (top left). Heatmap of SingleR scores for top correlated cell types (top right). SingleR annotation of cell identity indicated on t-SNE plot (bottom left). Expression in single cells of genes derived by differential expression analysis of published microarray data from bone marrow-derived, GM-CSF-cultured macrophages (GM-Macs) and DCs (GM-DCs)18 (bottom right). c, t-SNE plots of a published scRNA-seq dataset of PBMC20 analyzed by Seurat without (top) and with (middle) SingleR annotations indicated for individual cells. Cell type markers expressed by SingleR annotation (bottom). TCM = central memory T cell; TEM=effector memory T cell.
To validate SingleR in mouse, we analyzed a published study of scRNA-seq of mouse bone marrow-derived dendritic cells (BMDC) and fibroblasts15. When applied to the ImmGen database12 as a reference library, SingleR identified 33 of the 48 cells thought in the study to be BMDCs as macrophages (Fig. 1b and Supplementary Information 2). Of note, cells in the study15 were grown in culture in the presence of the growth factor GM-CSF and then suspended for flow cytometric sorting based on CD11c expression, a protocol16,17 that has been shown not to result in a pure population of DCs but also results in a proportion of cells bearing macrophage markers18,19. As such, SingleR could make the distinction between macrophages and dendritic cells in an unbiased fashion based on the granularity and breadth of the transcriptomic comparison. In a separate validation, we applied SingleR using the Blueprint and Encode databases as reference comparators for a dataset generated by scRNA-seq analysis of human peripheral blood mononuclear cells20. In comparison to marker-based and reference-based approaches20–22, which are limited in their ability to distinguish cell states, SingleR differentiated cellular subtypes such as memory and naïve B and T cells (Fig. 1c and Supplementary Fig. 1). SingleR performed similarly when applied to >50 publicly available scRNA-seq datasets (Supplementary Information 2 and http://comphealth.ucsf.edu/SingleR/). These analyses show that reference-based comparison enhances cell type annotation of scRNA-seq.
SingleR identifies a transitional macrophage in fibrosis
We next applied SingleR to enhance annotation of a lung scRNA-seq dataset obtained from whole lungs of adult, male mice (10–12 weeks age) dissociated into single cell suspension and flow-sorted on live cells, using 6 steady-state mice and 3 mice 14 days after a single tracheal instillation of bleomycin, the fibrotic phase of the injury model (Fig. 2a, Supplementary Table 1 and Supplementary Fig. 2). Using the ImmGen database as the reference compendium of datasets for SingleR, we annotated the lung cells and superimposed these cellular identities on the clustering analysis of bleomycin and steady-state cells, pooled together (Fig. 2b and Supplementary Fig. 3). The number of lung macrophages was higher in the bleomycin-treated condition than at steady state (Fig. 2a,b). Because macrophages and dendritic cells have similar transcriptomic profiles23–25, we repeated SingleR analysis using published RNA-seq datasets of mouse lung macrophages and dendritic cells26,27 to isolate the macrophages in the pooled bleomycin-injured and steady-state samples (Fig. 2c and Supplementary Fig. 4). SingleR identified two subtypes of macrophages: alveolar and interstitial macrophages (Fig. 2c). The annotation scores for alveolar and interstitial macrophages were strongest at the poles of the macrophage cluster, suggesting a continuum of gene expression between alveolar and interstitial macrophage (Fig. 2d). To quantify this continuum, we performed a similarity analysis28 by comparison of each cell to mouse RNA-seq datasets of alveolar and interstitial macrophages27. This analysis indicated a gradient of cellular identity in the macrophage cluster between alveolar and interstitial macrophage (Fig. 2d).
Figure 2. Reference datasets reveal heterogeneity of macrophage subtypes in lung fibrosis.
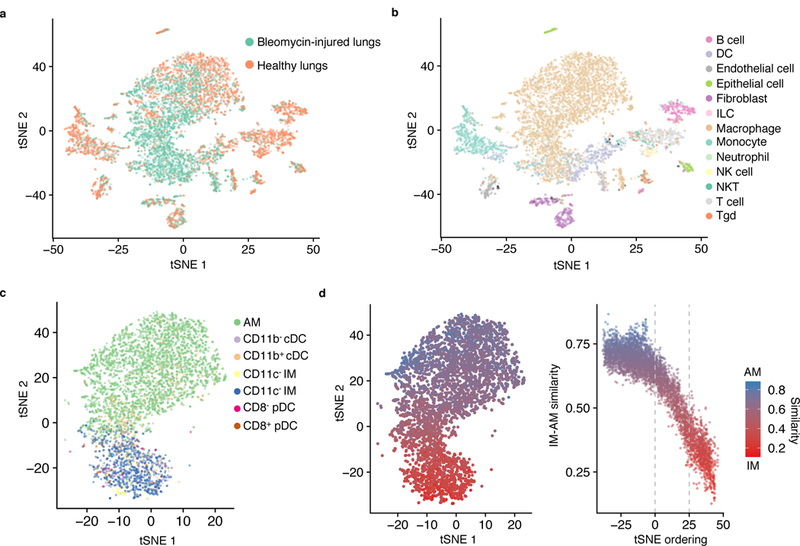
a-c, t-SNE plots of single-cell suspensions from mouse whole lung sequenced by Drop-seq color-coded by condition (a); annotated by SingleR using the ImmGen database as reference (b); and annotated by SingleR with lung-specific myeloid datasets26,27 (c). Data shown pool replicates (n=3 biologically independent mice for bleomycin-injured and n=6 biologically independent mice for healthy control). d, Quantification of similarity in gene expression of individual cells to bulk RNA-seq profiles of alveolar macrophages (AM) and CD11c+ interstitial macrophages (IM)27.
We next used SingleR annotation scores for each single cell compared with each sample in the reference dataset in order to hierarchically cluster the macrophages. That is, the full range of SingleR correlation scores for each cell with reference to ImmGen were used for unsupervised hierarchical clustering of the macrophages from the lung scRNA-seq dataset. This analysis identified three distinct subclusters (C1, C2 and C3), all three detected at varying levels in both steady-state and bleomycin-treated mice (Fig. 3a): cluster C1 was equally present in steady-state lung and bleomycin injury and corresponded to the cells that were annotated as alveolar macrophages by the lung-specific datasets (Fig. 2c); on the other hand, Clusters C2 and C3 were enriched in cells isolated from mice with bleomycin-induced fibrosis and expressed Cx3cr1, Ccr2, Mafb and MHCII genes, consistent with monocytic origin (Fig. 3b and Supplementary Table 2). Notably, 45% of cluster C2 cells (but only 7.3% of cluster C1 cells and 4.7% of cluster C3 cells) expressed at least 33% of cluster C1 genes and at least 33% of cluster C3 genes (Fig. 3c), suggesting that cluster C2 was intermediate in gene expression between C1 and C3. Consistent with this finding, comparison to 2 separate published datasets of lung macrophages27,29, enabling differential expression analysis between alveolar and interstitial macrophage genes, indicated that C1 had higher expression of alveolar macrophage genes than cluster C2, which in turn had higher alveolar macrophage genes than cluster C3 (Supplementary Fig. 5).
Figure 3. Hierarchical clustering by reference annotation reveals a transitional macrophage in fibrosis.
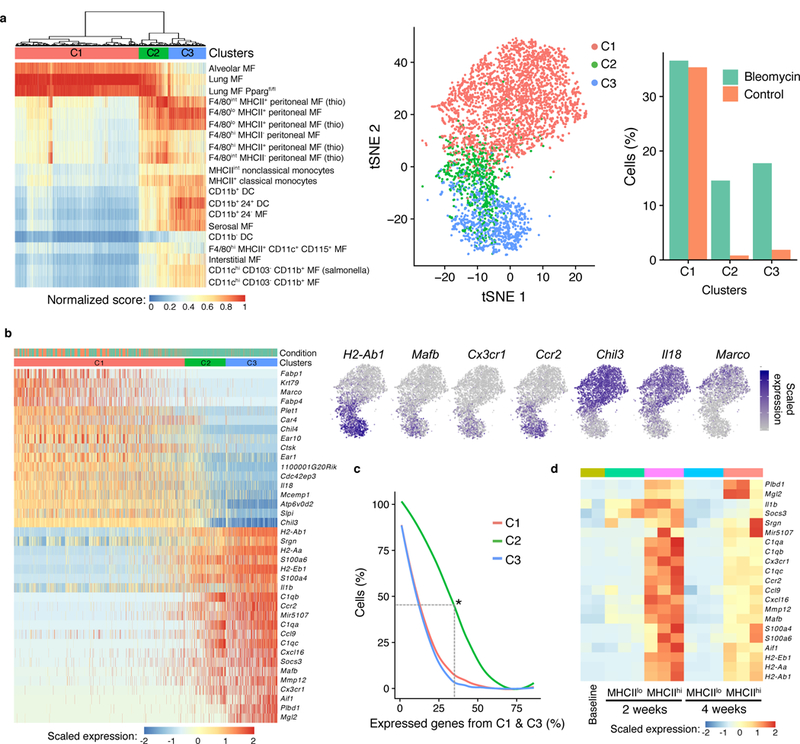
a, Heatmap of SingleR annotation scores derived by reference to the ImmGen database with clusters superimposed on the t-SNE plot. MF = macrophages. b, Heatmap of genes differentially expressed between C1 and C3 with examples superimposed on the t-SNE plot. Cells are ordered by the number of non-zero C3 genes. c, Percentage of cells in each cluster that express genes in b (differentially expressed between C1 and C3). Asterisk indicates that 45% of C2 cells express at least 33% of C1 genes and at least 33% of C3 genes. d, Heatmap of the expression of C3 genes in bulk RNA-seq of alveolar macrophages (SiglecF+CD11c+) sorted on MHCIIlo at baseline and both MHCIIlo and MHCIIhi at two time points after bleomycin lung injury (n=2 mice biologically independent mice for baseline, n=3 biologically independent mice at 2 weeks, and n=3 biologically independent mice at 4 weeks).
In order to determine the directionality of the transitional state of C2 cells across time in the model, we performed bulk RNA-seq of SiglecF+CD11c+MHCIIlo and SiglecF+CD11c+MHCIIhi cells, paralleling clusters C1 and C2, respectively. These cells were sorted from lungs of bleomycin-injured, 10–12-week-old, wild-type male mice. SiglecF+CD11c+MHCIIhi macrophages had higher expression of genes identified by scRNA-seq in cells from clusters C2 and C3 compared to SiglecF+CD11c+MHCIIlo cells at 2 weeks after injury (Fig. 3d and Supplementary Fig. 6a). However, the expression of these genes in SiglecF+CD11c+MHCIIhi cells at 4 weeks after injury was reduced to levels more similar to those detected in SiglecF+CD11c+ alveolar macrophages at steady-state (Fig. 3d). These data suggest that between the 2- and 4-week time points, SiglecF+CD11c+MHCIIhi cells, which correspond to cluster C2 identified by SingleR, transition toward a steady-state alveolar macrophage profile (corresponding to cluster C1).
Cluster C2 and C3 markers are found in alternate lung fibrosis models
In order to test the relevance of the identified gene expression pattern to other models of fibrosis, we used a mouse model of lung fibrosis induced by epithelial telomere dysfunction. In this model, SPC-CreERT2 mice are crossed with a Trf1fl/fl allele for tamoxifen-inducible deletion of Trf1, a shelterin complex protein that protects telomeres. Trf1 deletion after tamoxifen induction leads to age-dependent telomere shortening and is associated with cellular senescence and progressive fibrosis through the lifespan30. Whole lung lysates from SPC-CreERT2 / Trf1fl/fl mice harvested at 3 and 9 months after initiation of tamoxifen treatment and Trf1fl/fl control mice harvested at 9 months after tamoxifen were submitted for gene expression profiling by microarray. We detected a progressive increase in the expression of genes characteristic of clusters C2 and C3 in the lung lysates from tamoxifen-treated SPC-CreERT2 / Trf1fl/fl mice compared to control, with peak expression at 9 months (Fig. 4a), coincident with peak fibrosis in this model. Although these data represent whole lung rather than isolated macrophages, together with the known increase in macrophages during the fibrotic period in this model30, the results are consistent with a common genetic signature and a generalizable pattern of macrophage response during fibrogenesis.
Figure 4. Markers of macrophages identified by scRNA-seq are found in multiple fibrosis models.
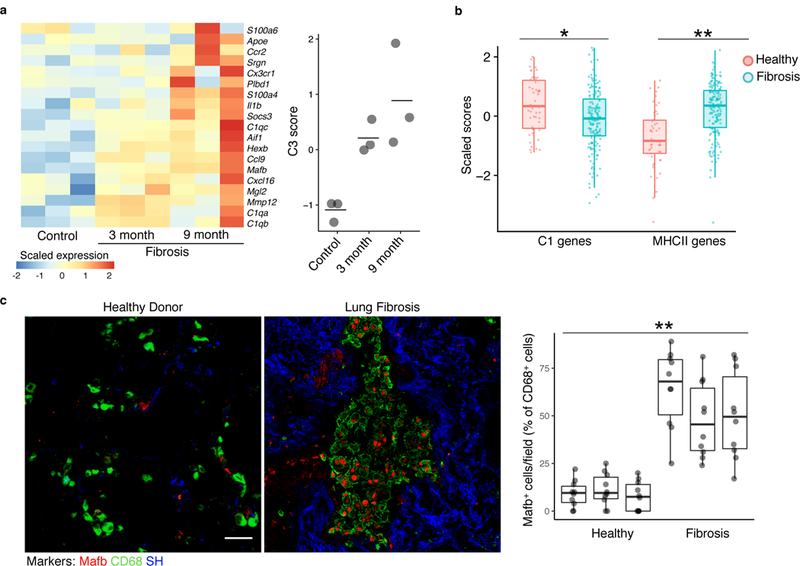
a, Heatmap of C3 genes from bleomycin-induced fibrosis measured by microarray in an epithelial telomere dysfunction model of progressive lung fibrosis (n=3 biologically independent mice in each condition, center value is mean). b, Gene set enrichment analysis scores of human orthologues of cluster C1 genes and of MHCII genes in bulk RNA-seq samples from patient lung biopsy specimens32. Points indicate individual microarray samples from n=167 and n=50 patients and controls, respectively. c, Immunofluorescence of human fibrotic and healthy control lung (representative images are shown; quantitation is for n=3 independent fibrotic patient lung and n=3 independent healthy lung samples, 10 images per sample). Scale bar, 50 μm. SH=second harmonic imaging of collagen. Box plot center line is median, box limits are upper and lower quartiles, and whiskers denote the largest and smallest values no more than 1.5 times the interquartile range from the limits. Wilcoxon test 2-tailed p-values are presented. * p<0.01, ** p<0.0001.
We then asked if the gene expression profiles identified in mouse were relevant for human lung fibrosis. To address this question, we performed single-sample gene set enrichment analysis (ssGSEA)31 to quantify the expression of human orthologues of genes of interest in a published microarray dataset of 167 lung samples from patients with lung fibrosis and 50 healthy controls32. This analysis showed a significant decrease in the expression of genes associated with alveolar macrophages (cluster C1) and a significant increase in the expression of MHC II genes (cluster C2 and C3) in the human fibrosis samples compared to the healthy controls (Fig. 4b). Moreover, immunofluorescence of lung tissue indicated the expression of MAFB, a marker detected specifically in clusters C2 and C3, in CD68+ macrophages from patients with idiopathic lung fibrosis compared to healthy controls (Fig. 4c). Taken together, these studies support the relevance of the C2 and C3 markers identified in the bleomycin model to multiple contexts of fibrosis, mouse and human.
Cluster C2 macrophages localize to the fibrotic niche
Next, we tried to identify the anatomic niche of macrophage clusters C1, C2 and C3. Because Cx3cr1 was expressed in clusters C2 and C3 in the bleomycin-treated mice, we used mice expressing the tamoxifen-inducible allele Cx3cr1-CreERT233, which were crossed with Rosa26-loxp-STOP-loxp-TdTomato reporter mice to mark Cx3cr1-expressing cells with the fluorophore TdTomato. Tamoxifen was given every other day starting 1 day before bleomycin treatment until lung analysis on days 7 or 14, while control mice received the same tamoxifen schedule through day 14 without bleomycin to determine the baseline levels of TdTomato+ cells. At baseline, TdTomato+ cells represented 6% of the live lung cells by flow cytometry, likely representing the interstitial macrophages present in steady-state lung (Fig. 5a and Supplementary Fig. 6b). In bleomycin-treated mice we observed an increase in the percentage of TdTomato+ cells among total lung live cells by flow cytometry at both day 7 (23%) and day 14 (38%) compared to baseline (Fig. 5a). Of note, the expression of the alveolar macrophage marker SiglecF on TdTomato+ cells increased (as assessed by mean fluorescence intensity) across the time series (Fig. 5a), indicating the acquisition of an alveolar macrophage marker by monocyte-derived macrophages across time.
Figure 5. Transitional macrophages expressing CX3CR1, MHCII, and SiglecF localize to sites of fibroblast accumulation after lung injury.
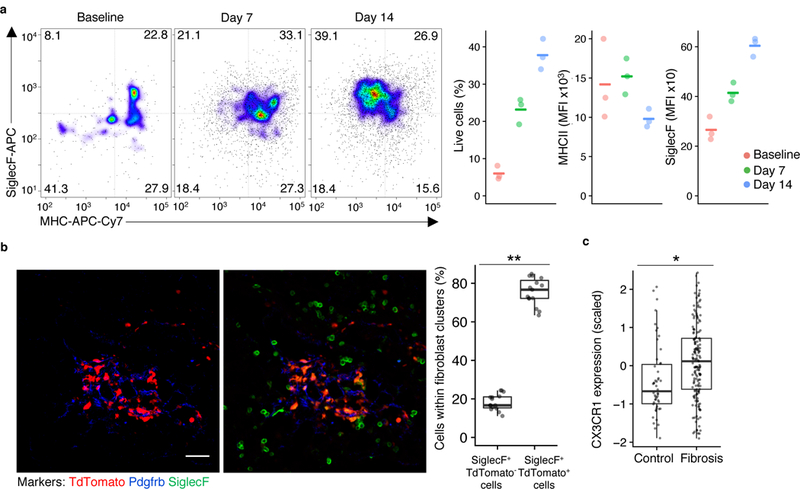
a, Flow cytometry of TdTomato+ lung cells in Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice with tamoxifen administration before and after injury followed by flow cytometry, with representative pseudocolor plots and values plotted for TdTomato+ cells at three different time points (n=3 biologically independent mice per group, mean is marked). b, Lung immunofluorescence at 14 days after injury in Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice treated as in a (representative images are shown; quantitation is for n=3 biologically independent mice, 5 images per mouse). Scale bar, 50 μm. c, Expression of CX3CR1 in bulk RNA-seq samples from patient lung biopsy specimens32. Box plot center lines are median, box limits are upper and lower quartiles, and whiskers denote the largest and smallest values no more than 1.5 times the interquartile range from the limits. Wilcoxon test 2-sided p-values are presented. * p <0.001; ** p < 0.0001.
We next performed lung immunofluorescence analysis of Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice at day 14 after bleomycin injury to determine the spatial organization of the macrophages in the fibrotic lung. TdTomato+SiglecF+ macrophages were detected in direct contact with clusters of fibroblasts as defined by areas delimited by multiple adjacent Pdgfrb+ cells (Fig. 5b). On the other hand, TdTomato-SiglecF+ alveolar macrophages (cluster C1) were detectable throughout the lung except in regions where Pdgfrb+ fibroblasts accumulated at sites of injury (Fig. 5b). The fact that TdTomato+ cells within the scar were also SiglecF+ suggests that the macrophages localizing to fibrotic zones were in a transitional state representative of cluster C2. Human CX3CR1 mRNA was also more highly expressed in lung microarray samples from 167 patients with lung fibrosis than in samples from 50 healthy controls (Fig. 5c)32, underscoring the relevance of the C2 cluster for human disease. Taken together, these results show that macrophage cluster C2 localizes to the fibrotic niche.
CX3CR1-expressing macrophages drive lung fibrosis
Because of the proximity of transitional macrophages to fibroblasts at fibrotic sites, we investigated whether the CX3CR1+ macrophages provided trophic or mitogenic support to fibroblasts. Macrophage-derived Pdgf has been shown to support fibroblast growth in co-culture34. In the lung scRNA-seq dataset, Pdgfa was uniquely expressed in the fibrosis-associated clusters C2 and C3 (Fig. 6a). Furthermore, Pdgfa mRNA was more highly expressed by bulk RNA-seq in SiglecF+CD11c+MHCIIhi (cluster C2) macrophages than SiglecF+CD11c+MHCIIlo macrophages (cluster C1) at two weeks (Fig. 6a). Lung immunofluorescence analysis of Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice at day 14 after bleomycin injury indicated that the homodimeric form of Pdgfa, Pdgf-aa, was highly and uniquely expressed in TdTomato+ cells adjacent to fibroblasts that were positive for expression of Pdgfra, the receptor for Pdgf-aa (Fig. 6b).
Figure 6. Pdgf-aa is a macrophage-derived trophic factor for fibroblasts.
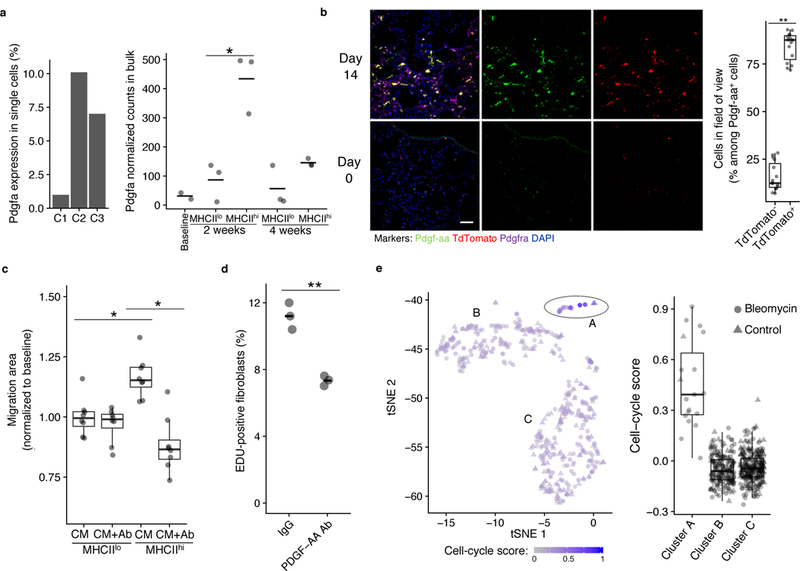
a, Pdgfa expression in lung scRNA-seq macrophage clusters and by bulk RNA-seq of SiglecF+CD11c+ lung macrophages after bleomycin injury. Mean is marked. Wald test 2-sided p-value is presented (for the lung scRNA-seq data, n=3 biologically independent mice for bleomycin-injured and n=6 biologically independent mice for healthy control; for the bulk RNA-seq data, n=2 biologically independent mice for baseline, n=3 biologically independent mice for all other conditions). b, Lung immunofluorescence at baseline (bottom) and 14 days after injury (top) in tamoxifen-induced Cx3cr1-CreERT2 / Rosa26loxpSTOPloxp-TdTomato mice (representative images are shown; quantitation is for the 14-day time point in n=3 biologically independent mice, 5 images per mouse). Scale bar, 50 μm. c, 3T3 fibroblast gap closure in response to conditioned media (CM) from lung macrophages sorted by MHCII expression, with and without Pdgf-aa blocking antibody (Ab; n=8 biologically independent mice total, each point a separate assay). d, EDU quantitation in 3T3 fibroblasts co-cultured with TdTomato+ cells sorted from tamoxifen-induced Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice 14 days after injury. Mean is marked (n=3 biologically independent co-cultures). e, t-SNE of SingleR-annotated fibroblasts from the lung scRNA-seq dataset showing intensity of cell cycle signature (n=3 biologically independent mice for bleomycin, n=6 biologically independent mice for control). Box plot center lines are median, box limits are upper and lower quartiles, and whiskers denote the largest and smallest values no more than 1.5 times the interquartile range from the limits. Wilcoxon test 2-sided p-values are presented. * p < 0.01; ** p < 0.0001.
To test whether cluster C2 cells supported fibroblast migration or proliferation through production of Pdgf-aa, we performed in vitro assays with 3T3 fibroblasts, an embryonic mouse fibroblast cell line. Conditioned media from SiglecF+CD11c+MHCIIhi macrophages (corresponding to cluster C2) sorted from wild-type mice 14 days after bleomycin injury and cultured for 2 days enhanced gap closure by 3T3 fibroblasts compared to conditioned media from SiglecF+CD11c+MHCIIlo macrophages (corresponding to cluster C1) isolated from the same mice (Fig. 6c). Antibody blockade of Pdgf-aa inhibited the enhanced gap closure seen in SiglecF+CD11c+MHCIIhi compared to SiglecF+CD11c+MHCIIlo conditioned media (Fig. 6c), suggesting the fibroblast response was dependent on Pdgf-aa secreted by SiglecF+CD11c+MHChi macrophages. In a second assay, when TdTomato+ cells sorted 14 days after bleomycin injury in tamoxifen-induced Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-TdTomato mice were co-cultured with 3T3 fibroblasts, Pdgf-aa blocking antibody decreased the proliferation of fibroblasts as measured by EDU uptake (Figure 6d).
Analysis of the lung scRNA-seq dataset including cells from both bleomycin-treated and steady-state lung pooled together and annotated by SingleR showed three clusters of fibroblasts, cluster A, B, and C (Fig. 6e). Pdgfra was not detected in the lung scRNA-seq dataset, which may reflect the limitation of the depth of sequencing with the single-cell method; however, a significant expansion of Pdgfra+ fibroblasts after bleomycin treatment has been reported in a study analyzing the fibroblast compartment in this model of fibrosis 35. Next, we used gene set analysis36 to quantify cell cycle genes in the lungs of bleomycin-treated mice compared to steady state 14 days after bleomycin treatment. Cluster A showed the highest expression of cell cycle genes and was predominantly composed of cells from bleomycin-treated mice (Figure 6e).
To investigate the role of CX3CR1+ cells in the proliferation of fibroblasts in vivo, Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-Diphteria Toxin A mice were treated with tamoxifen between day 8 and day 21 after bleomycin injury to induce diphtheria toxin A-dependent deletion of CX3CR1+ cells during the fibrotic phase of bleomycin lung injury. At 21 days after bleomycin injury, bleomycin-injured, tamoxifen-induced Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-Diphteria Toxin A mice had significantly fewer macrophages quantified by immunofluorescent labeling of either Mertk+ or SiglecF+ cells in the lung (Supplementary Fig. 7), significantly decreased numbers of cells expressing the fibroblast markers Pdgfra+ and Pdgfrb+ (Fig. 7a), and significantly decreased lung collagen as measured by hydroxyproline assay compared to bleomycin-injured wild-type mice (Fig. 7b). This result indicates that CX3CR1+ macrophages were required for the expansion of the fibroblast compartment and the deposition of fibrotic scar.
Figure 7. Cx3cr1-derived macrophages drive fibroblast accumulation and fibrosis after lung injury.
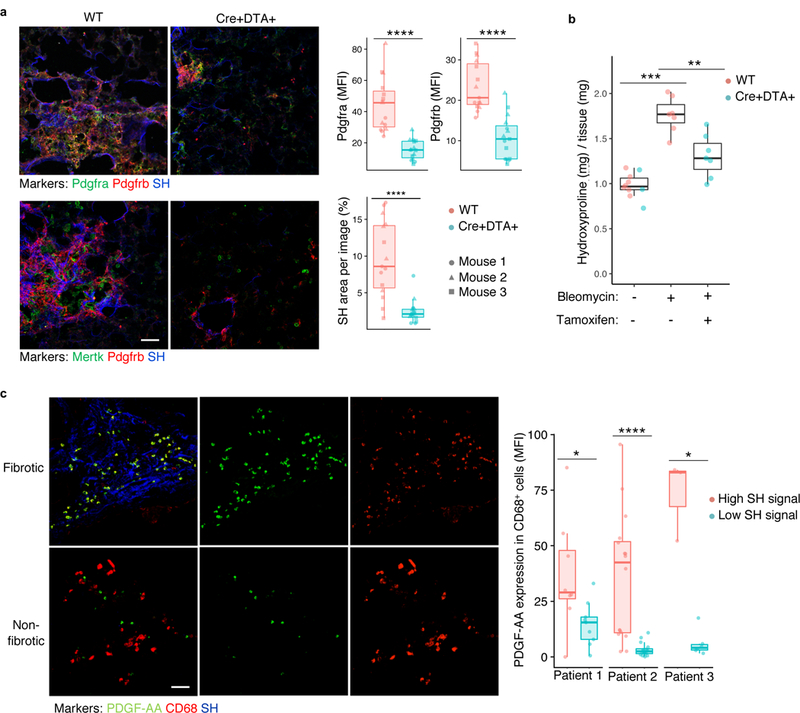
a, Lung immunofluorescence of fibroblast and macrophage markers 21 days after bleomycin injury in Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-Diphtheria Toxin A and wild-type mice (quantitation is for n=3 biologically independent mice per group, 5 images per mouse). b, Fibrosis measured by hydroxyproline assay for lung collagen content in mice treated as in a (blue points are Cx3cr1-CreERT2 / Rosa26loxpSTOPloxp-Diptheria Toxin A mice and red points are wild type; n=10, n=7, and n=7 biologically independent mice per group from left to right). Scale bar, 50 μm. c, Immunofluorescence of fibrotic (high SH) and non-fibrotic (low SH) regions from explanted human fibrotic lung, with quantification of individual images from 3 separate patients (8 high and 9 low regions in patient 1, 16 high and 16 low in patient 2, and 3 high and 6 low in patient 3). SH=second harmonic signal for collagen. Scale bar, 50 μm. Box plot center lines are median, box limits are upper and lower quartiles, and whiskers denote the largest and smallest values no more than 1.5 times the interquartile range from the limits. Wilcoxon test 2-sided p-values are presented. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
In human patients, one pathologic feature of idiopathic pulmonary fibrosis is the presence of normal tissue adjacent to regions of fibrotic foci, which are commonly found in the subpleural region of the lung37. In three lung explants displaying such regional heterogeneity, we found that PDGF-AA+ macrophages were associated with the fibrotic rather than non-fibrotic regions (Fig. 7c). Together, these results suggest the dependence of fibroblast proliferation, which is a key step in fibrogenesis38, on trophism exerted by PDGF-AA+ macrophages recruited to sites of injury.
Discussion
We show here that during fibrogenesis CX3CR1+SiglecF+ macrophages localize to sites of Pdgfra+ and Pdgfrb+ fibroblast accumulation at sites of bleomycin-induced injury in the lung. Deletion of these macrophages during the fibrotic period (days 8–21) eliminated Pdgfra+ or Pdgfrb+ fibroblasts and total lung collagen. Mechanistic studies indicated that fibroblast migration and proliferation were dependent on secretion of Pdgf-aa by SiglecF+CD11c+MHCIIhi and CX3CR1+ cells, respectively, suggesting that paracrine interactions between these macrophages and fibroblasts sustain fibroblast proliferation and tissue fibrosis.
Hierarchical clustering of correlation scores for macrophages with bulk datasets from ImmGen enabled the deconvolution of macrophage heterogeneity and identified three clusters of lung macrophages: alveolar macrophages (cluster C1), interstitial macrophages (cluster C3) and an intermediate cluster C2 bearing likeness to both alveolar and interstitial macrophages by gene expression. This analysis motivated the hypothesis that C2 is a transitional state arising after injury, which was supported by labeling and expression studies revealing kinetic transition toward increasing expression of the alveolar macrophage (C1) marker SiglecF by cells expressing the C2 and C3 marker Cx3cr1.
In cell culture systems, we found that antibody blockade of Pdgf-aa decreased the enhanced 3T3 fibroblast gap closure induced by conditioned media from MHCIIhi alveolar macrophages (corresponding to cluster C2) as well as the proliferation of 3T3 fibroblasts in co-culture with CX3CR1+ macrophages from bleomycin injured mice. Of note, one of the few FDA-approved therapies for lung fibrosis, the small molecule receptor tyrosine kinase inhibitor nintedanib, blocks PDGF receptor signaling39, and Pdgfra expressed by fibroblasts drives fibrosis in multiple organs in mice40–45, including lung fibrosis induced by bleomycin treatment46. These observations indicate that the Pdgf pathway is crucial to disease pathogenesis. Our results implicate the CX3CR1+SiglecF+ macrophage as the source of Pdgf-aa in the fibrotic niche.
A recent study reported an increase in the number of Ceacam1+Msr1+Ly6C−F4/80−Mac1+ monocytes, named segregated-nucleus containing atypical monocytes or ‘SatM’, in the bronchoalveolar lavage at day 13 after bleomycin treatment in mice11. SatM cells were found to be pro-fibrotic when transferred to the lung intravenously. However, macrophages were not tested, whereas our study characterizes these macrophage populations specifically. Furthermore, cells in that study were isolated by bronchoalveolar lavage, which does not efficiently recover cells from fibrotic regions. In the current study, we used dissociated whole lung for single cell analysis, which enabled an unbiased view of all cellular subtypes in the lung.
Our observation that ablation of macrophages expressing Cx3cr1 decreased fibroblasts and lung collagen content after bleomycin treatment has relevance for human disease. A similar macrophage gene expression profile was identified in an alternate mouse model of lung fibrosis mediated by telomere dysfunction, and analysis of human lung samples indicated increased expression of CX3CR1 and MHCII genes in patients with fibrosis as well as the expression of the C2 and C3 markers MAFB and PDGF-AA in regions of active fibrosis. These findings underscore the potential therapeutic significance of our functional studies in mice.
A limitation of using Cx3cr1-CreERT2 is that Cx3cr1 is expressed in both clusters C2 and C3, limiting the specificity of our studies. Our analysis suggests an evolution of cluster C3 towards cluster C1, with C2 representing a transitional state. Future studies should characterize the kinetics of this transition in greater depth and determine whether specific ablation of either cluster across time may be possible. Second, our annotation of cell states using SingleR relies on the particularities of the reference datasets. The ImmGen database is a large compendium of cell types from multiple organs, which greatly facilitated our clustering analysis by differential annotation. Nonetheless, we anticipate that the power of SingleR to identify new cell types or cell states will continue to increase as single cell and bulk RNA-seq datasets are amassed from a range of contexts.
In summary, this study identified a profibrotic macrophage subpopulation that localizes to sites of fibrotic scar with activating effects on the mesenchyme. Furthermore, the identification of markers specific for the profibrotic macrophages, including CX3CR1 and PDGF-AA, could be useful for therapeutic targeting in clinical fibrosis. Our results also demonstrate the value of using reference-based annotation of single cell datasets in order to map the heterogeneity of canonical cell types, such as macrophages, and to enhance the differentiation of transcriptomically unique subclusters within the data. Coupled with lineage tracing and functional studies, this approach should serve as a broadly applicable platform for study of specific cellular compartments in health and disease.
Online Methods
Mice
Ai14 (Rosa26-LSL-tdTomato), DTA (Gt(ROSA)26Sortm1(DTA)Lky), CX3CR1-CreERT2 (Cx3cr1tm2.1(cre/ERT2)Jung), and TRF1fl/fl, all on C57BL/6 background, wild type C57BL/6, and wild type 129S1 mice were obtained from the Jackson Laboratory. SPC-CreERT2rtTA mice, on C57BL/6 background, were obtained from Hal Chapman. Strain-appropriate controls were used in all experiments, and genotyping of the mice was performed by PCR. Cx3cr1-CreERT2 mice were administered 2 mg tamoxifen (Sigma) via IP injection every other day for Cre induction. Mice were maintained in specific-pathogen-free conditions in the Animal Barrier Facility of the University of California, San Francisco. All animal experiments were in accordance with protocols approved by the UCSF Institutional Animal Care and Use Committee.
Microfabrication of Devices
The PDMS Drop-seq droplet microfluidic device is fabricated with standard soft lithography techniques. Photoresist masters are created by spinning a layer of photoresist SU-8 (Microchem) onto a 3-inch silicon wafer (University Wafer), then baking at 95 °C for 20 minutes. Then, the photoresist is subjected to a 3-minute ultraviolet light exposure over Drop-seq (19) photolithography masks (CAD/Art Services) printed at 12,000 DPI. After ultraviolet exposure, the wafers are baked at 95 °C for 10 min, developed in fresh propylene glycol monomethyl ether acetate (Sigma Aldrich), rinsed with fresh propylene glycol monomethyl ether acetate, and baked at 95 °C for 1 minute to remove solvent. The microfluidic devices are fabricated by curing poly(dimethylsiloxane) (10:1 polymer-to-crosslinker ratio) over the photoresist master47. The devices are cured in a 65 °C oven for 2 hours and extracted with a scalpel, and inlet ports are added using a 0.75 mm biopsy core (World Precision Instruments). The device is bonded to a glass slide using O2 plasma treatment, and channels are treated with Aquapel (PPG Industries) to render them hydrophobic. Finally, the devices are baked at 65 °C for 20 min to dry the Aquapel before they are ready for use.
Drop-seq and data analysis
For scRNA-seq analysis of lung fibrosis, bleomycin (3 U/kg; Hospira) or water was instilled intratracheally to anesthetized male 129S1 mice, age 10–12 weeks. After 14 days, mice were sacrificed, and lungs were perfused with PBS and dissociated to a single-cell suspension in RPMI containing 0.13 U of liberase TM (Roche) and dissociated using gentleMACS (Miltenyi Biotec). Cells were passed through a 70 μm and 40 μm strainers followed by FACS sorting of DAPI- (live) cells. The single cell RNA-seq experiment is performed based on the Drop-seq protocol36. Briefly, the barcoded Drop-seq beads (ChemGenes corporation, MACOSKO-2011–10) and single cell suspensions from dissociated mouse lung are re-suspended to 100 beads/μl in PBS-BSA buffer and 120 cells/μl in Drop-seq lysis buffer (with additional 1 M NaCl added), respectively. Monodisperse droplets ∼1 nL in size were generated using the fabricated Drop-seq device. We used HFE7500 with 2% w/v ionic krytox as oil phase. The collected droplets were broken with perfluorooctanol (Sigma) in 30 ml of 6× SSC buffer. The beads were then washed and re-suspended in a reverse transcriptase mix for reverse transcription and the template switch reaction. Exonuclease I is used to remove unextended primers following the RT reaction. The beads were then washed, counted, aliquoted into PCR tubes as 6000 beads per PCR reaction, and PCR-amplified for 17 cycles: 95°C for 3 min; then 4 cycles of: 98°C for 20 sec, 65°C for 45 sec, 72°C for 3 min, and 13 cycles of: 98°C for 20 sec, 67°C for 20 sec, 72°C for 3 min; finally, 72°C for 5 min. The PCR reactions from one Drop-seq run were pooled and purified with AMPure XP beads; the amplified cDNA was quantified with Qubit dsDNA high sensitivity assay and checked on a BioAnalyzer High Sensitivity Chip (Agilent). The cDNA was fragmented and amplified for sequencing with the Nextera XT DNA sample prep kit (Illumina) using a primer that enabled the specific amplification of only the 3′ ends and Illumina index primers N70X. The libraries were purified with Ampure beads, quantified with Quabit dsDNA high sensitivity assay, checked on a BioAnalyzer High Sensitivity Chip, and then sequenced on Illumina Miseq or Hiseq2000. Paired-end sequence reads were processed mostly as previously described36. Briefly, cell barcode and UMI are extracted from Read 1. Read 2 was aligned to the mouse mm10 genome (UCSC) using Bowtie48. Reads mapping to exonic regions of genes of mouse mm10 genome were recorded. The number of transcripts for a given gene within a cell barcode was determined by counting unique UMIs and were compiled into a digital gene expression (DGE) matrix.
For initial clustering, we used Seurat v2.236 to cluster pooled data counts from 10 batches (42,000 cells, estimated based on the number of Drop-seq beads recovered, the concentration of cells, and droplet size). Cells with less than 500 non-zero genes were filtered out; genes expressed in only 1 cell were omitted. Altogether, the filtered data contained 8,366 cells and 13,861 genes (Supplementary Table 1). Expression data was log-normalized, and data were scaled to regress out differences in number of detected molecules. Variable genes were identified using the FindVariableGenes function with a x.low.cutoff = 0.0125, x.high.cutoff = 3 and y.cutoff = 0.5. t-SNE plot was computed using the top 10 principal components.
Cell Type Annotation of Single Cells (SingleR)
The SingleR pipeline is based on correlating gene expression of pure cell types with single-cell gene expression. SingleR can be used with any reference dataset. For mouse samples, we used the Immunological Genome Project (ImmGen) database, a collection of 830 microarray samples, which we classified to 20 main cell types and further annotated to 253 subtypes12; a dataset of 358 mouse RNA-seq samples annotated to 28 cell types collected, processed and shared, courtesy of Bérénice Benayoun, can also be used. For human samples we used the datasets from Blueprint Epigenomics, 144 RNA-seq pure immune samples annotated to 28 cell types14, and Encode, 115 RNA-seq pure stroma and immune samples annotated to 17 cell types13. More reference datasets for mouse and human are available with the SingleR R package. For specific applications, smaller datasets can be applicable (as demonstrated in figure 2c).
The annotation in SingleR is performed for each single cell independently. First, a Spearman coefficient is calculated for single-cell expression with each of the samples in the reference dataset. The correlation analysis is performed only on variable genes in the reference dataset. Next, multiple correlation coefficients per cell type according to the named annotations of the reference dataset are aggregated to provide a single value per cell type per single cell. SingleR uses the 80th percentile of correlation values, to prevent misclassification due to heterogeneity in the reference samples. Finally, in the fine-tuning step, SingleR reruns the correlation analysis, but only for the top cell types from the previous step. The analysis is performed only on variable genes between these cell types (see below). The lowest value cell type is removed (or values more than 0.05 below the top value), and then this step is repeated until only two cell types remain. The cell type corresponding to the top value after the last run is assigned to the single cell. Using only variable genes increases the ability to distinguish closely related cell types. Variable genes are defined as the top N genes that have a higher median expression in a cell type compared to each other cell type; we use a varying N, depending on the number of cell types used in the analysis. In the first round, N is 20 genes per cell type if there are 253 cell types (as in our ImmGen reference) or 71 genes per cell type if there are 28 cell types (as in our Blueprint+Encode reference). In the last fine-tuning step, N=333. More detail can be found in Supplementary Information 1 and at the SingleR Github repository https://github.com/dviraran/SingleR.
Clustering in Figure 3a was performed using Ward’s hierarchical agglomerative clustering method based on the SingleR scores across all cell types. Deconvolution analysis in Figure 2d was performed using the DeconRNA-seq package28 using the average expression of AM samples and average expression of IM3 samples from GSE9413527. Differential expression analysis (Figure 3b) was performed using the Seurat package v2.2 with default parameters.
Bulk RNA-seq
For bulk RNA-seq analysis of lung fibrosis, bleomycin (3 U/kg; Hospira) or water was instilled intratracheally to anesthetized male 129S1 mice, age 10–12 weeks. Cells gated on SiglecF+CD11c+ and sorted on MHCIIhi or MHCIIlo (Supplementary Fig. 6a) were collected at 2 weeks and 4 weeks after injury, or at baseline for the MHCIIlo compartment. RNA was collected (RNeasy Micro, Qiagen) from these samples, and total RNA quality was assessed by spectrophotometer (NanoDrop, Thermo Fisher Scientific Inc., Waltham, MA) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Intact mRNA was isolated using the Dynabead mRNA Purification Kit for total RNA, according to manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA). Amplified cDNA was prepared using the NuGen Ovation RNA-Seq system V2 kit, according to the manufacturer’s protocol (NuGen Technologies, Inc., San Carlos, CA), and sequencing libraries were generated using the Nextera XT library preparation kit with multiplexing primers according to manufacturer’s protocol (Illumina, San Diego, CA). Library fragment size distributions were assessed using the Bioanalyzer 2100 and the DNA high-sensitivity chip (Agilent Technologies, Santa Clara, CA). Library sequence quality was assessed by sequencing single-end 50 base pair reads using the Illumina MiSeq platform, and libraries were pooled for high-throughput sequencing on the Illumina HiSeq 4000 by using equal numbers of uniquely mapped protein coding reads. RNA sequencing was performed on HiSeq 4000 machines (Illumina, San Diego, CA), followed by de-multiplexing of raw sequencing results, trimming of adapter sequences, and alignment to the reference genome using STAR software49. DESeq2 was used to normalize by size factor (reads per sample) as well as by library complexity, and we then applied Wald test to determine significance of differential expression.
Public gene expression data
Immunological Genome Project (ImmGen) raw expression data of phase 1 and 2 were downloaded as CEL files from GEO (GSE15907 and GSE37448), processed and normalized using the Robust Multi-array Average (RMA) procedure on probe-level data using Matlab functions. The analysis was performed using custom CDF file obtained from Brainarray50. Blueprint and Encode data were processed as we described previously51. Raw counts from the lung reference dataset were obtained from Gibbings et al.27 (GSE94135) and Altboum et al.26 (GSE49932). Counts were normalized and converted to TPM using R functions. Gene expression profile of GM-CSF derived bone marrow dendritic cell subsets (available at GSE6236118) was analyzed using the GEO2R tool for differentially expressed genes. Top 50 upregulated by fold-change of GM-DCs genes and top 50 GM-Macs were used in Figure 1b. Single-cell RNA-seq counts from Hashimshony et al.15 used in Figure 1b were downloaded from the ARCHS project52. 10X PBMC single-cell counts were downloaded directly from the 10X website (https://support.10xgenomics.com/single-cell-gene-expression/datasets ).
Analysis of published human lung microarray datasets
Yang et al.32 performed gene expression profiling for 167 lung tissues from idiopathic interstitial pneumonia subjects and 50 healthy controls. We downloaded raw CEL files GEO (Accession GSE32537) and processed them using custom CDFs from BrainArray. Normalization was performed using the Robust Multi-array Average (RMA) procedure on Affymetrix microarray data. Single-sample gene set enrichment analysis (ssGSEA31) was performed using human orthologues of C1 genes (Supplementary Table 3) and 15 human MHCII genes (HLA-DMA, HLA-DMB, HLA-DOA, HLA-DOB, HLA-DPA1, HLA-DPB1, HLA-DPB2, HLA-DQA1, HLA-DQA2, HLA-DQB1, HLA-DQB2, HLA-DRA, HLA-DRB1, HLA-DRB5, HLA-DRB6).
Microarray profiling in telomere dysfunction model of lung fibrosis
SPC-creERT2 / TRF1flox/flox mice and TRF1flox/flox litter-mate controls were subjected to tamoxifen administration beginning at 8 weeks age, as previously described30. Lungs perfused free of blood were harvested at 3 months and 9 months. Total RNA including small RNAs was extracted using miRNAeasy mini kit (Qiagen, Hilden, Germany). Total RNA was measured using the NanoDrop photospectrometer (NanoDrop, Wilmington, DE, USA). Agilent mouse microarray kit 8×60K v2 (G4852B) (Agilent technologies Inc., Santa Clara, CA, USA) was used. Gene expression profiling was performed by Onearray (Phalanx bio INC., San Diego, CA). All the samples passed the quality control parameters established by Agilent. Cy3-labeled aRNA was used for hybridization.
Single cell dissociation for FACS
Lungs were perfused with PBS and dissociated to a single cell solution in 10mM HEPES RPMI containing 0.2% collagenase (Wako Pure Chemical Industries), 0.1 mg/mL Dispase II (Roche), and 2000 U/mL DNase I (Merck). Cells were passed through a 70 μm strainer and stained at 4 °C for 30 minutes with following antibodies (1:100): SiglecF-APC (catalog number 50–1702-82, clone 1RNM44N, eBioscience), MHCII-APC-Cy7 (catalog number 47–5321-82, clone M5/114.15.2, eBioscience), and CD11c-PE (catalog number 557401, clone HL3, BD Biosciences). Cells were sorted either with BD FACSAria2 or SONY SH800 FACS. Data for analytical flow was analyzed using FlowJo software.
Immunostaining and Microscopy
Mouse lungs in the bleomycin model were perfused with PBS, inflated with 50% OCT, 10% sucrose in PBS and fixed overnight at 4 C in 4% PFA prior to freezing in OCT. Lungs from the telomere dysfunction model were perfused with PBS and frozen in OCT. For human samples OCT embedded tissues were cut to 15 μm sections, and sections were fixed with 4% PFA for 15 minutes at RT. OCT embedded tissues were cut to 15um sections, blocked with 3% donkey serum, 1% BSA, 0.3% TritonX and then stained with following antibodies (1:100 dilution): SiglecF (catalog number AF1706, R&D Systems), Mertk (catalog number AF591, R&D Systems), Pdgfrb (catalog number 14–1402-82, clone APB5, eBioscience), CD68 (catalog number ab955, clone KP1, Abcam), MafB (catalog number HPA005653, Sigma), PDGF-AA antibody (catalog number 07–1436, Millipore), and Pdgfra antibody (catalog number AF1062, R&D Systems). Secondary antibodies conjugated to Alexa fluorophores (Invitrogen and Abcam) were used at 1:200 dilution. Stained sections and 2-photon images of second harmonic signal were visualized at Zeiss LSM 780 NLO microscope. Image processing was done using ImageJ. For quantitation of Figure 5b, regions of fibroblast accumulation were delimited by Pdgf receptor staining. For quantitation of Figure 7a, mean fluorescent intensity of Pdgfra and Pdgfrb immunostaining was measured from cellular areas as defined by morphological tissue features. For quantitation of Figure 7c, % second harmonic signal was determined for each image, and the median across images (within patient) was defined as the threshold for categorization as fibrotic versus nonfibrotic. For hematoxylin and eosin (H&E) images, staining of frozen sections was performed using standard protocols, and images of whole sections were acquired on a VERSA automated slide scanner (Leica Biosystems) equipped with an Andor Zyla 5.5 sCMOS camera (Andor Technologies) acquired under NIH S10 grant OD021717; individual images were generated with the ImageScope software (Aperio Technologies, Vista, CA).
Fibroblast Migration Assay
3T3 mouse fibroblasts were seeded at 100% confluency into a 96-well plate with a central cell-free detection zone in the center of each well (Platypus technologies). 100 μl of conditioned media of macrophage cultures (48h culture in RPMI containing 10% FBS) were added per well. Pdgf-aa antibody (catalog number 07–1436, Millipore) was added at concentration 0.05 μg/ml. Cells were imaged via bright field microscopy at time points 0h and 24h. Image processing and migration quantification was done using ImageJ.
Fibroblast Proliferation Assay
Fibroblast proliferation by flow cytometric EDU assay was performed according to the manufacturer’s protocol (Click-iT Flow Cytometry, Invitrogen). 3T3 mouse fibroblasts, and TdTomato+ macrophages sorted from Cx3cr1-CreERT2 / Rosa26-loxp-STOP-loxp-Tdtomato mice 2 weeks after bleomycin injury, were seeded at a ratio of 1:3 in serum-free conditions in the presence of Pdgf-aa blocking antibody (catalog number 07–1436, Millipore) or IgG control (catalog number ab199376, lot GR3195151–2, Abcam). EDU was added at 48 hours, and cells were suspended two hours later for EDU detection in the TdTomato- (fibroblast) fraction on a SONY SH800 flow cytometer.
Measurement of hydroxyproline
Lung hydroxyproline content was measured as previously described6 three weeks after intratracheal instillation of bleomycin to sex-matched, male and female, 8–12-week-old C57BL/6J mice. Briefly, mouse lungs were incubated in 12 N HCl at 110°C for 18 h. Aliquots of the samples reconstituted in distilled water were added to 1.4% chloramine-T in 10% isopropanol and 0.5 M sodium acetate. Erlich’s solution was added, and the samples were incubated at 60°C for 10 min. Absorbance at 562 nm was measured and adjusted according to standard curves.
Human Lung Tissues
Written informed consent was obtained from all subjects, and the study was approved by the UCSF institutional review board. Pulmonary fibrosis lung tissues were obtained at the time of lung transplantation from patients with a pathologic diagnosis of usual interstitial pneumonia and a consensus clinical diagnosis of idiopathic pulmonary fibrosis (IPF) according to available guidelines53. Age-similar, non-diseased, normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network54. Our studies indicate that these lungs are physiologically and pathologically normal. Lung fragments were inflated with 30% OCT suspended in PBS, embedded in OCT compound, mand frozen immediately after isolation in dry ice before storing at −80 °C until use.
Statistical analysis
All significance tests in this paper, unless otherwise stated, were assessed using the two-sided Wilcoxon rank-sum test.
Reporting Summary
Further information on research design is available in the Nature Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by a UCSF Marcus Award to M.B. and A.R.A., a National Institutes of Health grant (HL131560) to M.B, a Gruss Lipper Postdoctoral Fellowship to D.A., a UCSF Nina Ireland Program award to R.N. and P.J.W., a National Institutes of Health grant (HL139897) to P.J.W, and a National Institutes of Health award (National Institute of Allergy and Infectious Diseases Bioinformatics Support Contract HHSN272201200028C) to A.J.B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank D. Erle, A. Barczak, W. Eckalbar, and M. Adkisson of the UCSF Functional Genomics Core Facility, the UCSF Center for Advanced Technology, the Gladstone Institutes’ Histology & Light Microscopy Core, and D. Sheppard for his insightful comments on the manuscript. We thank the anonymous reviewers for their thoughtful comments.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. RNA-seq data have been deposited in the GEO depository under accession numbers GSE111664, GSE111690, and GSE114005 and at ArrayExpress under accession number E-MTAB-7142.
Code Availability
The SingleR open-source R package is maintained on GitHub R package and available from https://github.com/dviraran/SingleR. Code to reproduce the figures is also available in the GitHub repository.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
References
- 1.Rockey DC, Bell PD & Hill JA Fibrosis--A Common Pathway to Organ Injury and Failure. N Engl J Med 373, 96, doi: 10.1056/NEJMc1504848.(2015). [DOI] [PubMed] [Google Scholar]
- 2.Blackwell TS et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med 189, 214–222, doi: 10.1164/rccm.201306–1141WS.(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rock JR et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A 108, E1475–1483, doi: 10.1073/pnas.1117988108. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El Agha E et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 20, 261–273 e263, doi: 10.1016/j.stem.2016.10.004. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung C et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 188, 820–830, doi: 10.1164/rccm.201212–2297OC. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson NC et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19, 1617–1624, doi: 10.1038/nm.3282. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider C et al. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol 15, 1026–1037, doi: 10.1038/ni.3005. (2014). [DOI] [PubMed] [Google Scholar]
- 8.Tan SY & Krasnow MA Developmental origin of lung macrophage diversity. Development 143, 1318–1327, doi: 10.1242/dev.129122. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibbons MA et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 184, 569–581, doi: 10.1164/rccm.201010–1719OC. (2011). [DOI] [PubMed] [Google Scholar]
- 10.Misharin AV et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214, 2387–2404, doi: 10.1084/jem.20162152. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satoh T et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101, doi: 10.1038/nature20611. (2017). [DOI] [PubMed] [Google Scholar]
- 12.Heng TS, Painter MW & Immunological Genome Project C The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094, doi: 10.1038/ni1008–1091. (2008). [DOI] [PubMed] [Google Scholar]
- 13.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi: 10.1038/nature11247. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stunnenberg HG, International Human Epigenome C & Hirst M The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 167, 1145–1149, doi: 10.1016/j.cell.2016.11.007. (2016). [DOI] [PubMed] [Google Scholar]
- 15.Hashimshony T et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol 17, 77, doi: 10.1186/s13059-016-0938-8. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amit I et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263, doi: 10.1126/science.1179050. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shalek AK et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369, doi: 10.1038/nature13437. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helft J et al. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 42, 1197–1211, doi: 10.1016/j.immuni.2015.05.018. (2015). [DOI] [PubMed] [Google Scholar]
- 19.Guilliams M & Malissen B A Death Notice for In-Vitro-Generated GM-CSF Dendritic Cells? Immunity 42, 988–990, doi: 10.1016/j.immuni.2015.05.020. (2015). [DOI] [PubMed] [Google Scholar]
- 20.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049, doi: 10.1038/ncomms14049. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94, doi: 10.1038/nbt.4042. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 49, 708–718, doi: 10.1038/ng.3818. (2017). [DOI] [PubMed] [Google Scholar]
- 23.Gautier EL et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128, doi: 10.1038/ni.2419. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hume DA, Mabbott N, Raza S & Freeman TC Can DCs be distinguished from macrophages by molecular signatures? Nat Immunol 14, 187–189, doi: 10.1038/ni.2516. (2013). [DOI] [PubMed] [Google Scholar]
- 25.Randolph G & Merad M Reply to: “Can DCs be distinguished from macrophages by molecular signatures?”. Nat Immunol 14, 189–190, doi: 10.1038/ni.2517. (2013). [DOI] [PubMed] [Google Scholar]
- 26.Altboum Z et al. Digital cell quantification identifies global immune cell dynamics during influenza infection. Mol Syst Biol 10, 720, doi: 10.1002/msb.134947. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbings SL et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am J Respir Cell Mol Biol 57, 66–76, doi: 10.1165/rcmb.2016-0361OC. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong T & Szustakowski JD DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics 29, 1083–1085, doi: 10.1093/bioinformatics/btt090. (2013). [DOI] [PubMed] [Google Scholar]
- 29.Huang L, Nazarova EV, Tan S, Liu Y & Russell DG Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med 215, 1135–1152, doi: 10.1084/jem.20172020. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naikawadi RP et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 1, e86704, doi: 10.1172/jci.insight.86704. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verhaak RG et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest 123, 517–525, doi: 10.1172/JCI65833. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang IV et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 68, 1114–1121, doi: 10.1136/thoraxjnl-2012-202943. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yona S et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91, doi: 10.1016/j.immuni.2012.12.001. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X et al. Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757 e717, doi: 10.1016/j.cell.2018.01.015. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie T et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22, 3625–3640, doi: 10.1016/j.celrep.2018.03.010. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214, doi: 10.1016/j.cell.2015.05.002. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richeldi L, Collard HR & Jones MG Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952, doi: 10.1016/S0140-6736(17)30866-8. (2017). [DOI] [PubMed] [Google Scholar]
- 38.Noble PW, Barkauskas CE & Jiang D Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122, 2756–2762, doi: 10.1172/JCI60323. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wollin L et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 45, 1434–1445, doi: 10.1183/09031936.00174914. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson LE & Soriano P Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16, 303–313, doi: 10.1016/j.devcel.2008.12.003. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwayama T et al. PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 29, 1106–1119, doi: 10.1101/gad.260554.115. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horikawa S et al. PDGFRalpha plays a crucial role in connective tissue remodeling. Sci Rep 5, 17948, doi: 10.1038/srep17948. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes BJ et al. Activation of platelet-derived growth factor receptor alpha contributes to liver fibrosis. PLoS One 9, e92925, doi: 10.1371/journal.pone.0092925. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demaria M et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31, 722–733, doi: 10.1016/j.devcel.2014.11.012. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen YT et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80, 1170–1181, doi: 10.1038/ki.2011.208. (2011). [DOI] [PubMed] [Google Scholar]
- 46.Kitagawa M et al. Phf14, a novel regulator of mesenchyme growth via platelet-derived growth factor (PDGF) receptor-alpha. J Biol Chem 287, 27983–27996, doi: 10.1074/jbc.M112.350074. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin D, Xia Y & Whitesides GM Soft lithography for micro- and nanoscale patterning. Nat Protoc 5, 491–502, doi: 10.1038/nprot.2009.234. (2010). [DOI] [PubMed] [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25, doi: 10.1186/gb-2009-10-3-r25. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai M et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33, e175, doi: 10.1093/nar/gni179. (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aran D, Hu Z & Butte AJ xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220, doi: 10.1186/s13059-017-1349-1. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lachmann A et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 9, 1366, doi: 10.1038/s41467-018-03751-6. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raghu G et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183, 788–824, doi: 10.1164/rccm.2009-040GL. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ware LB et al. Assessment of lungs rejected for transplantation and implications for donor selection. Lancet 360, 619–620 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.