

Abstract
Pseudoxanthoma elasticum (PXE), a prototype of heritable ectopic mineralization disorders, is caused by mutations in the ABCC6 gene encoding a putative efflux transporter ABCC6. It was recently demonstrated that the absence of ABCC6-mediated ATP release from the liver, and consequently reduced PPi levels, underlie the pathogenesis of PXE. Given that tissue-nonspecific alkaline phosphatase (TNAP), encoded by ALPL, is the enzyme responsible for degrading PPi, we hypothesized that reducing TNAP levels either by genetic or pharmacological means would lead to amelioration of the ectopic mineralization phenotype in the Abcc6−/− mouse model of PXE. Thus, we bred Abcc6−/− mice to heterozygous Alpl+/− mice that display approximately 50% plasma TNAP activity. The Abcc6−/−Alpl+/− double mutant mice showed 52% reduction of mineralization in the muzzle skin compared to the Abcc6−/− Alpl+/+ mice. Subsequently, oral administration of SBI-425, a small molecule inhibitor of TNAP, resulted in 61% reduction of plasma TNAP activity and 58% reduction of mineralization in the muzzle skin of Abcc6−/− mice. By contrast, SBI-425 treatment of Enpp1 mutant mice, another model of ectopic mineralization associated with reduced PPi, failed to reduce muzzle skin mineralization. These results suggest that inhibition of TNAP might provide a promising treatment strategy for PXE, a currently intractable disease.
Keywords: Pseudoxanthoma elasticum, ectopic mineralization, mouse model, inorganic pyrophosphate, tissue-nonspecific alkaline phosphatase
INTRODUCTION
Ectopic mineralization - inappropriate biomineralization in soft connective tissues - is a widespread pathological problem encountered frequently during physiological aging and found in several common disorders (obesity, diabetes, chronic kidney disease, etc.) causing significant morbidity and mortality (Giachelli, 1999). In addition, ectopic mineralization is seen in several rare genetic disorders with defined gene defects. Pseudoxanthoma elasticum (PXE), a multisystem disorder with a protean phenotypic spectrum, is one example of a heritable ectopic mineralization disorder. The clinical manifestations in PXE are primarily evident in the skin, eyes, and cardiovascular system (Neldner, 1988; Uitto et al., 2017). PXE is a late-onset and slowly progressive disease with systemic manifestations for which there is no effective treatment.
PXE is caused in most cases by biallelic loss-of-function mutations in the ABCC6 gene (Bergen et al., 2000; Le Saux et al., 2000; Ringpfeil et al., 2000). The ABCC6 gene encodes ABCC6, a putative transmembrane efflux transporter protein primarily expressed in the liver (Belinsky and Kruh, 1999; Scheffer et al., 2002). The metabolic features of PXE have been determined to be a consequence of defective ABCC6 transporter activity in hepatocytes (Jiang et al., 2009; Li et al., 2017). It was recently shown that under physiologic conditions ABCC6 mediates adenosine triphosphate (ATP) release from the liver to the extracellular space. Outside the liver, but still within the confines of the liver vasculature, pericellular ATP is rapidly hydrolyzed into adenosine monophosphate and inorganic pyrophosphate (PPi) by ENPP1, an ectonucleotidase (Jansen et al., 2014; Jansen et al., 2013). As a result, plasma levels of PPi are reduced to approximately 30-40% of normal in patients with PXE and in Abcc6−/− murine models (Jansen et al., 2014; Jansen et al., 2013; Li et al., 2017). ENPP1 mutations are associated with type 1 generalized arterial calcification of infancy (GACI) while ABCC6 mutations are also associated with type 2 GACI, heritable ectopic mineralization disorders in which low or absent levels of PPi are associated with early onset of extensive vascular mineralization that causes significant mortality in infancy (Li et al., 2014a; Nitschke et al., 2012; Rutsch et al., 2003). Collectively, ABCC6-mediated ATP release and subsequent conversion to plasma PPi by ENPP1 links the shared pathogenetic pathway from mutations in the ABCC6 and ENPP1 genes to plasma PPi deficiency and eventually ectopic mineralization in PXE and GACI (Uitto et al., 2017).
PPi has been recognized as a potent mineralization inhibitor for four decades (Orriss et al., 2016; Meyer, 1984), and deficiency of PPi levels was recently established as the underlying cause for PXE as a result of ABCC6 mutations. These results suggest that therapeutics capable of counteracting ectopic mineralization by targeting PPi deficiency could be of use for treating PXE. One approach towards increasing PPi levels involves inhibition of tissue-nonspecific alkaline phosphatase (TNAP, encoded by the ALPL gene), a membrane-bound enzyme that degrades PPi. The early development of Abcc6−/− mice has provided a model system to explore potential treatment modalities for PXE (Klement et al., 2005). In this study, two experiments were performed to attempt to inhibit TNAP activity. As a proof of concept, a genetic model, Abcc6−/−Alpl+/− mice, was used to determine the effect of reduced TNAP activity on ectopic mineralization. Subsequently, Abcc6−/− mice were treated with SBI-425 (Pinkerton et al., 2018), a small molecule inhibitor of TNAP, to test whether SBI-425 provides sustained and titratable inhibition of TNAP activity and consequently might counteract ectopic mineralization. The results demonstrated that inhibition of TNAP activity attenuated ectopic mineralization in the Abcc6−/− mouse model. However, the therapeutic effect of TNAP inhibition was not caused by increased plasma PPi levels, suggesting there are other mechanisms by which TNAP inhibition attenuates ectopic mineralization. Interestingly, inhibition of TNAP activity by SBI-425 had no effect on ectopic mineralization in the Enpp1asj and Enpp1asj-2J mutant mouse models of GACI which also display extremely low levels of plasma PPi (Li et al., 2013; Li et al., 2014b).
RESULTS
Reduced TNAP activity attenuated connective tissue mineralization in Abcc6−/− mice
To explore the potential efficacy of inhibition of TNAP activity for counteracting ectopic mineralization in PXE, Abcc6−/− mice were crossed with Alpl+/− mice to generate Abcc6−/−Alpl+/− mice. If reduced TNAP activity in the Abcc6−/−Alpl+/− mice resulted in elevated plasma levels of PPi, a natural substrate of TNAP, it was expected to ameliorate the degree of ectopic mineralization in the Abcc6−/− mice. The Alpl−/− mice and patients with genetic deficiency of TNAP develop hypophosphatasia, caused by accumulation in the extracellular matrix of PPi, a potent mineralization inhibitor (Fedde et al., 1999; Whyte et al., 2012; Russell, 1965; Russell et al., 1971). Consequently, these mice die during the early postnatal period. As compared to wild type mice, the Alpl+/− mice have ~50% plasma TNAP activity but exhibit normal skeletal development. Thus, the Abcc6−/− mice were crossed with Alpl+/− mice and the corresponding Abcc6−/−Alpl+/+ and Abcc6−/−Alpl+/− mice were used to assess the effects of a controlled 50% reduction of TNAP activity on plasma PPi levels and ectopic mineralization. Wild type C57BL/6J mice served as baseline of normal plasma PPi levels and negative controls of ectopic tissue mineralization. All mice were kept on standard control diet and euthanized at 12 weeks of age for analysis.
Mineralization of the dermal sheath of vibrissae in the muzzle skin, an early and reliable biomarker in the overall mineralization process in the Abcc6−/− mice, was examined by two independent assays. One piece of muzzle skin was processed for semi-quantitative histopathologic examinations. A mineralization-specific stain, von Kossa, revealed robust mineralization in the dermal sheath of vibrissae in the Abcc6−/−Alpl+/+ mice. The Abcc6−/−Alpl+/− mice showed significantly reduced mineralization in the muzzle skin (Fig. 1a). Another piece of muzzle skin was solubilized and the amount of calcium was quantitated using a chemical assay of calcium. The calcium assay showed a significant reduction of 52% in the amount of calcium in the muzzle skin in the Abcc6−/−Alpl+/− mice (Fig. 1b).
Figure 1. Genetic proof of concept study demonstrated that reduced TNAP activity in the Alpl+/− mice inhibited ectopic connective tissue mineralization in the Abcc6−/− mice.
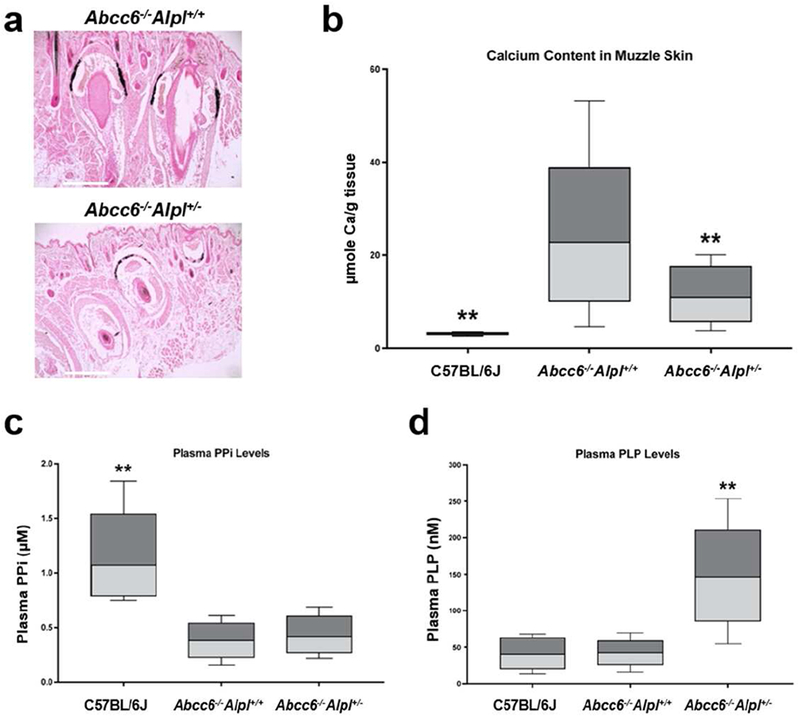
(a) Muzzle skin biopsies were processed for histopathology followed by von Kossa stains. The Abcc6−/−Alpl+/− mice (lower panel) developed less ectopic mineralization of the dermal sheath of vibrissae in the muzzle skin compared to Abcc6−/−Alpl+/+ mice (upper panel); (b) The amount of calcium in the muzzle skin of Abcc6−/−Alpl+/− mice was significantly decreased as compared to Abcc6−/−Alpl+/+ mice; (c) Plasma PPi levels in the Abcc6−/−Alpl+/+ mice were 30% of that in wild type mice; however, plasma PPi levels in the Abcc6−/−Alpl+/− mice were not statistically different from that in the Abcc6−/− Alpl+/− mice; (d) Plasma levels of PLP were significantly increased in the Abcc6−/−Alpl+/− mice. Values were expressed as mean ± SD; n = 12 - 15 mice per group. **p < 0.01, compared with Abcc6−/−Alpl+/+ mice. Scale bar, 0.4 mm.
Plasma PLP levels, but not PPi levels, were increased in Abcc6−/−Alpf+/− mice
To further examine the consequences of reduced TNAP activity on ectopic mineralization, PPi levels were determined in plasma of wild type C57BL/6J, Abcc6−/−Alpl+/+ and Abcc6−/−Alpl+/− mice. Consistent with previous results (Jansen et al., 2014), the Abcc6−/−Alpl+/− mice had approximately 30% plasma PPi levels of that in wild type mice (Fig. 1c). Reduced TNAP activity in Abcc6−/−Alpl+/− mice did not result in a significance increase in plasma PPi levels (Fig. 1c). The plasma levels of another natural substrate of TNAP, pyridoxal 5’-phosphate (PLP), were also determined. The results demonstrated a significant, 2.4-fold, increase of plasma PLP levels in the Abcc6−/−Alpl+/− mice as compared to Abcc6−/−Alpl+/+ mice (Fig. 1d). Therefore, plasma PLP but not PPi levels served as a reliable biomarker for inhibition of TNAP activity.
SBI-425, a prototypic small molecule inhibitor of TNAP, attenuated connective tissue mineralization in Abcc6−/− mice
Based on the demonstration in the genetic proof of concept study that reduced TNAP activity attenuated ectopic mineralization in Abcc6−/− mice, a treatment trial with SBI-425, a selective and orally bioavailable TNAP inhibitor, was initiated. SBI-425 does not inhibit placental, intestinal, or germ-cell alkaline phosphatase, while demonstrating significantly improved pharmacokinetic profiles as compared to the previously identified chemical inhibitors of TNAP (Pinkerton et al., 2018; Sheen et al., 2015). The ability of SBI-425 to target and inhibit TNAP was previously confirmed in mouse models that overexpress human TNAP in vascular smooth muscle cells and endothelial cells and demonstrated significantly reduced aortic calcification and cardiac hypertrophy, without deleterious effects on skeletal mineralization (Sheen et al., 2015; Romanelli et al., 2017). To investigate the effects of SBI-425 for PXE, the Abcc6−/− mice were fed a diet supplemented with SBI-425 at two doses, 7.5 and 75 mg/kg body weight/day, starting at 4 weeks of age followed by an additional 8 weeks. As a result of treatment, plasma TNAP activity was reduced by 61% in mice treated with 75 mg/kg/day SBI-425 with concomitant increase of plasma PLP levels (Fig. 2a).
Figure 2. Treatment of mice with SBI-425 resulted in decreased TNAP activity and increased PLP levels in plasma.
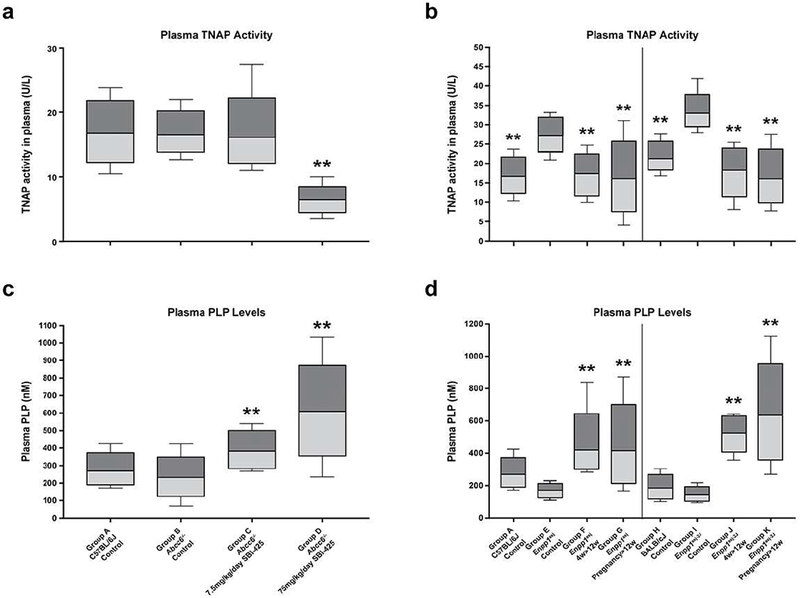
Plasma TNAP activities and PLP levels in the Abcc6−/− mice (a) and Enpp1 mutant mice (b; left panel, Enpp1asj; right panel, Enpp1asj-2J mice). Treatment with 75 mg/kg/day SBI-425 resulted in reduction of plasma TNAP activity with concomitant increase of PLP levels in the Abcc6−/− mice (Group D) as compared to the Abcc6−/− control mice (Group B); Note significant increase of plasma TNAP activity in the Enpp1asj and Enpp1asj-2J mice (Group E and I) as compared to their respective wild type controls (Group A and H). Treatment with 75 mg/kg/day SBI-425 starting at 4 weeks of age (Group F and J) or in pregnant mothers (Group G and K) resulted in significant reduction of TNAP activities and increase of PLP levels in the progeny when examined at 12 weeks of age. Values were expressed as mean ± SD; n = 7 - 20 mice per group. **p < 0.01, compared with Abcc6−/− (Group B), Enpp1asj (Group E), or Enpp1asj-2J (Group I) mice on control diet.
Histologic examination revealed significant reduction in the muzzle skin calcification in the Abcc6−/− mice treated with 75 mg/kg/day SBI-425 (Fig. 3a). The same mice treated with 7.5 mg/kg/day SBI-425 had similar levels of ectopic mineralization to the Abcc6−/− mice without treatment. The therapeutic effect of SBI-425 was substantiated by measuring the calcium content in muzzle skin biopsies using a chemical assay. The results confirmed a significant, 58%, reduction in the amount of calcium in the Abcc6−/− mice treated with 75 mg/kg/day SBI-425, as compared to the untreated Abcc6−/− mice (Fig. 3b).
Figure 3. Treatment with SBI-425 attenuated ectopic connective tissue mineralization in Abcc6−/− mice but not in Enpp1asj and Enpp1asj-2j mice.
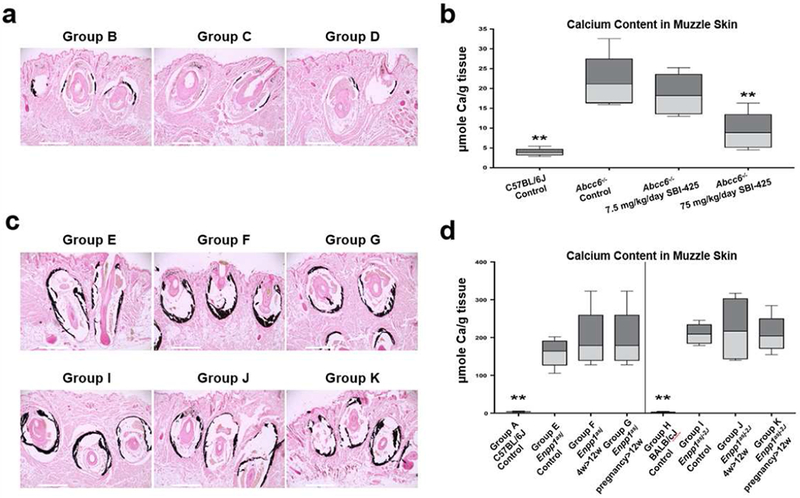
(a) Tissues were collected from Abcc6−/− mice at 12 weeks of age and analyzed by histopathology with von Kossa stains. The Abcc6−/− mice treated with 7.5 mg/kg/day SBI-425 (Group C) had similar levels of mineralization in the muzzle skin to that of Abcc6−/− control mice (Group B). In contrast, treatment with 75 mg/kg/day SBI-425 revealed marked decrease of mineralization (Group D); (b) The chemical assay of calcium demonstrated significant reduction in the amount of calcium in the muzzle skin in the Abcc6−/− mice treated with 75 mg/kg/day SBI-425 (Group D); (c) The Enpp1asj mice (upper panel) and Enpp1asj-2J mice (lower panel) treated with 75 mg/kg/day SBI-425 postnatally (Group F and J) and in pregnant mothers (Group G and K) had similar levels of mineralization in the muzzle skin to their respective mutant mice on the control diet (Group E and I); (d) The chemical assay of calcium did not reveal differences in the amount of calcium in the muzzle skin in the treated mice. Values were expressed as mean ± SD; n = 7 - 20 mice per group. **p < 0.01, compared with Abcc6−/− (Group B), Enpp1asj (Group E), or Enpp1asj-2J (Group I) mice on control diet. Scale bar, 0.4 mm.
TNAP inhibition by SBI-425 has no effect on bone microarchitecture in Abcc6−/− mice
Because reduced TNAP activity, especially complete loss of TNAP activity, is associated with hypophosphatasia, a heritable disease characterized by defective bone mineralization (Millan and Whyte, 2016; Whyte, 2016), the femurs were dissected from wild type C57BL/6J and Abcc6−/− mice for evaluation of their bone microarchitecture as a result of SBI-425 treatment. The femurs were examined by μCT analysis at the end of the treatment trial (Fig. 4). Marked differences in the bone density and trabecular microarchitecture were noted between the male and female mice, consistent with previous reports (Bouleftour et al., 2014; Pennypacker et al., 2009; Li et al., 2015; Li et al., 2016). Consequently, sex-matched comparisons of the μCT results were performed between wild type, untreated Abcc6−/− mice and the same mice fed with 75 mg/kg/day SBI-425. No differences were found between wild type and Abcc6−/− mice. SBI-425 treatment in Abcc6−/− mice did not result in any appreciable changes to bone microarchitecture (Fig. 4). Quantification of the bone microarchitecture did not show significant differences between control and treated mice across all trabecular bone parameters (Table 2). These results attest to the safety of SBI-425 for TNAP inhibition, at oral doses up to 75 mg/kg/day, without deleterious effects on bone morphometry.
Figure 4. MicroCT analysis does not reveal changes in trabecular bone microarchitecture in the Abcc6−/− mice treated with 75 mg/kg/day SBI-425.
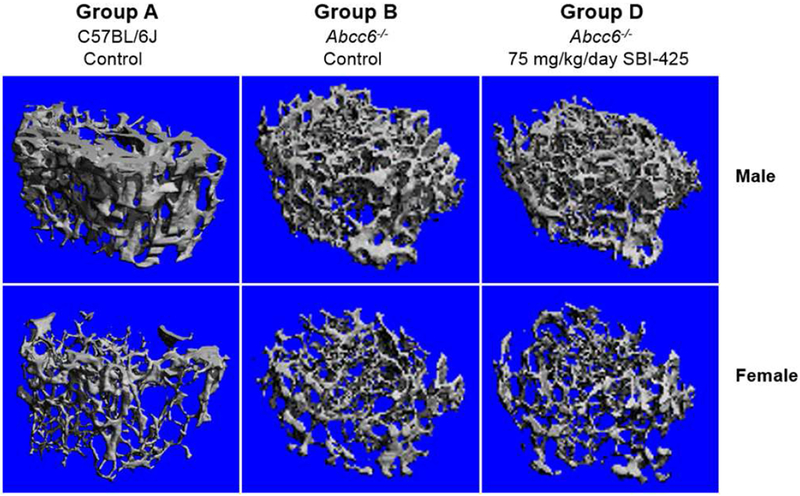
Note the distinct differences between male and female mice in each group. Changes were not found in Abcc6−/− mice (Group B) compared to wild type C57BL/6J mice (Group A). Treatment with 75 mg/kg/day SBI-425 in the Abcc6−/− mice (Group D) did not alter the femoral microarchitecture as compared to Abcc6−/− control mice (Group B). n = 3-5 per sex in each group. Their respective quantitative data were detailed in Table 2.
Table 2.
Trabecular bone phenotypes of the femurs by microCT analysis
| Group1) | Sex | BMD (mg/cm3) | BV/TV (%) | Tb.Th (μm) | Tb.N (1/mm) | Tb.Sp (μm) | SMI | Conn.D (TV/mm3) |
|---|---|---|---|---|---|---|---|---|
| A | M | 198.9 ± 70.5 | 14.6 ± 2.9 | 37.7 ± 20.2 | 4.4 ± 0.1 | 222.7 ± 6.0 | 2.1 ± 0.8 | 207.5 ± 106.1 |
| A | F | 100.2 ± 13.4 | 5.8 ± 2.5** | 30.6 ± 4.6 | 3.6 ± 0.2** | 277.5 ± 16.0** | 2.7 ± 0.5 | 124.2 ± 54.5 |
| B | M | 246.1 ± 12.2 | 17.8 ± 1.3 | 44.8 ± 7.7 | 5.7 ± 0.5 | 168.4 ± 13.6 | 1.5 ± 0.1 | 378.3 ± 118.0 |
| B | F | 125.0 ± 24.8** | 7.4 ± 2.0** | 33.2 ± 3.1* | 4.0 ± 0.3** | 247.4 ± 22.0** | 2.2 ± 0.3** | 185.1 ± 52.2* |
| D | M | 210.8 ± 62.2 | 14.4 ± 5.7 | 37.9 ± 6.4 | 5.5 ± 0.8 | 178.1 ± 23.5 | 1.6 ± 0.5 | 363.3 ± 132.5 |
| D | F | 150.0 ± 23.6 | 9.0 ± 2.0 | 34.5 ± 1.3 | 4.5 ± 0.4 | 218.6 ± 21.4* | 2.2 ± 0.2 | 241.4 ± 52.6 |
For a description of different groups, see Table 1.
Abbreviations: M, male; F, female; BMD, bone mineral density; BV/TV, relative bone volume; Tb.Th, trabecular thickness; Tb.N, trabecular number; Tb.Sp, trabecular separation (marrow thickness); SMI, structure model index; Conn.D, connectivity density.
Values are expressed as mean ± SD. N = 3-5 males and females per group.
p< 0.05
p < 0.01, compared to the male mice in each group;
No statistical differences are noted in mice in Group B when compared to sex matched mice in Groups A and D.
SBI-425 has no effect on connective tissue mineralization in Enpp1 mutant mice
The Enpp1asj and Enpp1asj-2J mutant mice serve as models of GACI (Li et al., 2013; Li et al., 2014b). To test the potential efficacy of 75mg/kg/day dietary SBI-425 in counteracting ectopic mineralization these Enpp1 mutant mice were used. As compared to Abcc6−/− mice which have ~30% plasma PPi levels of wild type mice, the Enpp1asj-2J mice have essentially no plasma PPi due to a spontaneous large deletion and a small insertion in the Enpp1 gene resulting in complete loss of ENPP1 enzyme activity (Li et al., 2014b). By contrast, the Enpp1asj mice carry a missense p.V246D mutation with residual, ~15% of ENPP1 enzymatic activity and lowered plasma PPi levels (Li et al., 2013).
The Enpp1asj and Enpp1asj-2J mice were fed with SBI-425 starting at 4 weeks of age and followed for an additional 8 weeks. In addition, some pregnant mothers were treated with SBI-425 during pregnancy and nursing and the Enpp1 homozygous pups continuing the same treatment until 12 weeks of postnatal life. All mice were analyzed at 12 weeks of age. Increased plasma TNAP activity was observed in the Enpp1asj and Enpp1asj-2J mice, suggesting a compensatory mechanism to increase bone formation in the absence of ENPP1 (Mackenzie et al., 2012). As a result of SBI-425 treatment, plasma TNAP activities were significantly reduced accompanied by increased plasma PLP levels (Fig. 2b); however, analysis of calcium content and histology in muzzle skin from these mice showed that SBI-425 treatment failed to reduce the degree of ectopic mineralization (Fig. 3c,d).
DISCUSSION
PXE is the prototype of heritable disorders of ectopic mineralization, a group of autosomal recessive disorders, which also includes generalized arterial calcification of infancy (GACI) and arterial calcification due to CD73 deficiency (ACDC). These conditions have both overlapping and distinctive phenotypic features. GACI is characterized by severe, early-onset mineralization of the cardiovascular system and the majority of children with GACI die from cardiovascular collapse during the first six months after birth (Rutsch et al., 2008). By contrast, ACDC is a late-onset mineralization disorder that primarily affects the arteries of the lower extremities and periarticular ligaments (St Hilaire et al., 2011). Mutations in the ABCC6 gene cause the classic form of PXE and type 2 GACI, while ENPP1 mutations cause type 1 GACI. Significant recent progress has been made in understanding the pathomechanistic details of ectopic mineralization in these conditions, and this information has now provided a platform to develop treatments for PXE (Uitto et al., 2017). A particularly intriguing recent observation is that release of ATP from hepatocytes to the circulation is dependent on functional ABCC6, and in the absence of ABCC6 transporter activity, as in PXE, the extracellular pool of ATP is reduced and consequently less plasma PPi is generated by ENPP1 hydrolysis. Recent studies indicated that a unifying pathomechanistic feature in PXE, GACI and ACDC involves reduction in PPi levels allowing ectopic mineralization in the peripheral tissues to ensue (Uitto et al., 2017).
PPi levels are regulated by the opposing actions of ENPP1, which generates PPi, and TNAP, which degrades PPi. Since PPi is a powerful anti-mineralization factor, in this study we tested the hypothesis that inhibition of TNAP activity would increase plasma PPi levels and thereby counteract ectopic mineralization in a mouse model of PXE. A genetic model, Abcc6−/−Alpl+/− mice with ~50% reduction of plasma TNAP activity showed significant inhibition of mineralization. To titrate the level of TNAP activity required for inhibition of ectopic mineralization in Abcc6−/− mice, subsequent studies tested two different doses of a small molecule inhibitor of TNAP, SBI-425, which has been shown to reduce vascular mineralization while not adversely affecting bone mineralization in mouse models overexpressing human TNAP (Sheen et al., 2015). The Abcc6−/− mice treated with 75 mg/kg/day SBI-425 demonstrated 61% reduced plasma TNAP activity and ectopic mineralization in the muzzle skin while the same mice treated with 7.5 mg/kg/day SBI-425 did not. The therapeutic effect of SBI-425 did not cause deleterious consequences to the femoral bone in the treated mice. While significant reduction of ectopic mineralization was noted in Abcc6−/− mice either via genetics or pharmacological means for inhibition of TNAP, distinct mineralization was still present, indicating that reduction of TNAP activity up to 61% of the wild type level is not sufficient to completely arrest ectopic mineralization. The early postnatal lethality of the homozygous Alpl−/− mice prevented us to investigate whether a more drastic decrease of TNAP activity, <1% of wild type level, would have more pronounced therapeutic effects in Abcc6−/− mice (Fedde et al., 1999). In addition, though two different doses of SBI-425 was tested for titration, the efficacy for inhibition of TNAP activity did not achieve as high as 95% as we had anticipated for at the planning phase of this study. Further studies might require higher doses for more drastic reduction in TNAP activity and complete mineralization arrest. Please note that the investigational drug, the dose at 75 mg/kg/day and the Abcc6−/− mouse model for PXE in this study mirrors those in another study (Ziegler et al., 2017). Combined results of these independent and complementary studies suggested that inhibition of TNAP by SBI-425 could be turned into a viable approach for treatment of patients with PXE.
Whereas Abcc6−/− mice had decreased plasma PPi concentrations compared to wild type mice, reduced TNAP activity did not significantly increase plasma PPi concentrations as we initially anticipated, potentially highlighting the contribution of other mechanisms, independent of PPi, to the therapeutic effect of TNAP inhibition on attenuation of ectopic mineralization. . Furthermore, in earlier studies where plasma TNAP levels were genetically increased by 10 to 15-fold, plasma PPi levels were not decreased (Narisawa et al., 2013; Sheen et al., 2015; Savinov et al., 2015). Therefore, plasma PPi levels are not a reliable marker for tracking phenotypic improvement. In this context, the levels of PLP, another physiological substrate for TNAP can be used reliably as a marker of therapeutic benefit (Whyte et al., 2012).
To investigate whether SBI-425 treatment has broader applications beyond PXE, the Enpp1asj and Enpp1asj-2J mice, models for GACI, were also treated with SBI-425. Previous studies suggested that three TNAP compounds, unrelated to SBI-425, inhibited TNAP activity and suppressed mineralization in cultured vascular smooth muscle cells derived from Enpp1−/− mice (Narisawa et al., 2007). Interestingly, SBI-425 administration at 75 mg/kg/day in both Enpp1asj and Enpp1asj-2J mice postnatally or prenatally did not affect the degree of ectopic mineralization. The lack of therapeutic effect of SBI-425 in the context of ENPP1 mutations could be at least partially explained by distinct pathophysiology of ABCC6 and ENPP1 in preventing ectopic mineralization under physiologic conditions. ENPP1 is the principal enzyme for generation of PPi by ATP hydyolysis. Upstream of ENPP1, ABCC6-mediated ATP release from hepatocytes is the main source of circulating PPi (Jansen et al., 2014). The work by us and others suggest that reduced PPi levels is the major factor leading to ectopic mineralization in PXE, but there might be an alternative, as yet unknown mechanism, independent of PPi, by which ABCC6 prevents ectopic mineralization under physiologic conditions (Zhao et al., 2017; Pomozi et al., 2017). Recent observations suggest that ABCC6 is also involved in extracellular nucleotide metabolism, suggesting a central role for ABCC6 in purinergic signaling in the development of ectopic mineralization (Kauffenstein et al., 2018).
Taken together, ectopic mineralization in PXE can be attenuated by inhibition of TNAP activity genetically or pharmacologically. The lack of restoration of plasma PPi levels does not explain why inhibition of TNAP activity results in attenuation of ectopic mineralization in PXE. Nevertheless, the results derived from the preclinical mouse studies suggest that inhibition of TNAP activity could reduce or slow down progression of the devastating consequences in patients with PXE. The mechanisms by which TNAP inhibition resulted in attenuation of ectopic mineralization in PXE but not in GACI remains unexplained. In this context, TNAP inhibition could be considered for other ectopic mineralization conditions only after careful testing in appropriate model systems.
MATERIALS AND METHODS
Mice and breeding
The Abcc6tm1JfK mouse was developed by targeted ablation of the Abcc6 gene (this mouse is referred to as Abcc6−/−) (Klement et al., 2005). Abcc6−/− mice were made congenic by backcrossing heterozygous mice for 10 generations with wild type C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME). The Alpl+/− mice (Alpltm1Jlm) were obtained from Sanford-Burnham-Prebys Medical Discovery Institute (Fedde et al., 1999) and subsequently backcrossed with C57BL/6J mice for four generations. Compound Abcc6 and Alpl mice were generated by intercrossing Abcc6−/− mice with Alpl+/− mice. The C57BL/6J-Enpp1asj/GrsrJ and BALB/cJ-Enpp1asj-2J/GrsrJ mice were obtained from The Jackson Laboratory. These Enpp1 mutant mice are referred in this publication as the Enpp1asj and Enpp1asj-2J mouse, respectively (Li et al., 2013; Li et al., 2014b). The wild type, heterozygous, and homozygous mice were generated from heterozygous matings. The C57BL/6J mice serve as wild type controls for Abcc6−/− and Enpp1asj mice while BALB/cJ mice serve as wild type controls for Enpp1asj-2J mice. All mice were maintained a standard rodent diet (Lab Diet 5010; PMI Nutrition, Brentwood, MO, USA). All protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Experimental design and treatments
In the proof of concept genetic study, the C57BL/6J, Abcc6−/−Alpl+/+, and Abcc6−/−Alpl+/− mice were fed a control diet throughout the experiments. At 12 weeks of age, these mice were euthanized for analysis. In the TNAP inhibitor SBI-425 treatment experiments, mice were divided into eleven groups (A to K) based on their Abcc6 or Enpp1 genotype and treatment regiments (Table 1). Four-week old wild type, Abcc6−/−, Enpp1asj, and Enpp1asj-2J mice were placed on either control diet or experimental diet supplemented with 0.003% or 0.03% SBI-425 and maintained on the same treatment regimen for an additional 8 weeks. The doses correspond to 7.5 or 75 mg/kg body weight/day SBI-425, assuming that a 20 g mouse consumes 5 g food per day. Groups G and K are Enpp1asj and Enpp1asj-2J mice, respectively, fed a SBI-425 containing diet (75 mg/kg/day) to the pregnant mothers and continuing the same treatment in pups until pups became 12 weeks of age. All mice were euthanized at 12 weeks of age for analysis.
Table 1.
Experimental groups of mice by genotype and treatment*
| Group | Genotype | No. of mice examined (M+F) | Treatment | Treatment started > treatment ended (weeks) |
|---|---|---|---|---|
| Set 1 (Abcc6−/− mice) | ||||
| A | C57BL/6J | 12 (6+6) | Control diet | 4 > 12 |
| B | Abcc6−/− | 12 (6+6) | Control diet | 4 > 12 |
| C | Abcc6−/− | 12 (6+6) | SBI-425 7.5 mg/kg/day | 4 > 12 |
| D | Abcc6−/− | 15 (9+6) | SBI-425 75 mg/kg day | 4 > 12 |
| Set 2 (Enpp1asj mice) | ||||
| E | Enpp1asj | 11 (6+5) | Control diet | 4 > 12 |
| F | Enpp1asj | 10 (4+6) | SBI-425 75 mg/kg/day | 4 > 12 |
| G | Enpp1asj | 20 (8+12) | SBI-425 75 mg/kg/day | pregnancy > 12 |
| Set 3 (Enpp1asj-2J mice) | ||||
| H | BALB/2J | 12 (6+6) | Control diet | 4 > 12 |
| I | Enpp1asj-2J | 8 (3+5) | Control diet | 4 > 12 |
| J | Enpp1asj-2J | 7 (3+4) | SBI-425 75 mg/kg/day | 4 > 12 |
| K | Enpp1asj-2J | 11 (4+7) | SBI-425 75 mg/kg/day | pregnancy > 12 |
Some mice with desired genotype were placed on either control diet or SBI-425 containing diets at 4 weeks of age and followed for another 8 weeks (4 > 12 weeks). Some pregnant mothers were placed on SBI-425 containing diets during pregnancy and nursing and continuing for the first 12 weeks of postnatal life of the offspring (pregnancy > 12 weeks). The mice were sacrificed at the age of 12 weeks for analysis. M, male; F, female.
Histopathological analysis
Muzzle skin biopsies (left side) from euthanized mice were collected and processed for histology. Tissue sections were stained with von Kossa using standard procedures.
Chemical quantitation of calcium
To quantify the mineral deposition in mouse tissues, muzzle skin biopsies (right side) were harvested and decalcified with 1.0 mol/L HCl for 48 hours at room temperature. Solubilized calcium was then determined by colorimetric analysis using the o’-cresolphthalein complexone method (calcium (CPC) LiquiColor; Stanbio Laboratory, Boerne, TX). The values were normalized to tissue weight.
Plasma inorganic pyrophosphate assay
Whole blood was collected by cardiac puncture into test tubes containing 100 μL CTAD and stored at room temperature after addition of 10 μL 15% trisodium EDTA (Sigma-Aldrich, St. Louis, MO). After centrifugation, plasma was depleted of platelets by filtration through a Centrisart I 300-kDa mass cutoff filter (Sartorius, New York, NY, USA), and stored at −80°C until further processing. PPi in plasma was measured by an enzymatic reaction using ATP sulfurylase to convert PPi into ATP in the presence of excess adenosine 5’-phosphosulfate (Sigma-Aldrich), a methodology adopted world-wide as described previously (Li et al., 2017; Jansen et al., 2014; Dedinszki et al., 2017; Pomozi et al., 2017; Bauer et al., 2018).
Plasma TNAP activity and PLP assays
In the proof of concept genetic study, the platelet-free plasma was used for determination of PLP levels by a stable isotope dilution LC-ESI-MS/MS method (van Zelst and de Jonge, 2012). In the TNAP inhibitor SBI-425 treatment experiments, heparin plasma was collected for determination of alkaline phosphatase activity using a colorimetric kit from Abcam (Cambridge, MA).
Microcomputed tomography
Microarchitecture of the distal trabecular bone and midshaft region of the right femur was analyzed. A 1.2 mm-thick region located proximal to the distal growth plate of femur was scanned at a 6 μm resolution using the micro-computed tomography (μCT) system (μCT35; Scanco Medical AG, Bassersdorf, Switzerland). The microstructural parameters were obtained through three-dimensional reconstruction and segmentation (using a Gaussian filter and a global threshold of 3685 Hounsfield units) using the manufacturer-provided software.
Statistical analysis
The data were analyzed using multivariable linear regression with the predictor of sex and treatment and their interactions for each group. Statistical significance was reached with p <0.05. All statistical computations were completed using R version 3.5.0 software.
ACKNOWLEDGMENTS
This study was supported by NIH/NIAMS grants R01AR055225 (JU), K01AR064766 (QL), and R01AR072695 (JU and QL). The authors acknowledge Penn Center for Musculoskeletal Disorders, supported by NIH/NIAMS P30AR050950, for assistance in analysis of bone morphometry. The authors thank Dian Wang, Ida Joely Jacobs, Koen van de Wetering, Diana Li, Wei-Ju Tseng, and Tingting Zhan for assistance. Carol Kelly helped in manuscript preparation.
Abbreviations:
- Asj
ages with stiffened joints
- GACI
generalized arterial calcification of infancy
- PLP
pyridoxal 5’-phosphate
- PPi
inorganic pyrophosphate
- PXE
pseudoxanthoma elasticum
- TNAP
tissue-nonspecific alkaline phosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state no conflict of interest. Anthony B. Pinkerton and Jose Luis Millan are inventors on a patent application covering SBI-425 (PCT WO 2013126608). John P. Sundberg has sponsored research with Takeda, Theravance, and Curadim and does consulting for Bioniz, none of which has any relevance to this project. Michael A. Levine is a member of a Clinical Advisory Board for Inozyme Pharma.
REFERENCES
- Bauer C, le Saux O, Pomozi V, Aherrahrou R, Kriesen R, Stolting S, et al. Etidronate prevents dystrophic cardiac calcification by inhibiting macrophage aggregation. Sci Rep 2018;8:5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky MG, Kruh GD. MOAT-E (ARA) is a full-length MRP/cMOAT subfamily transporter expressed in kidney and liver. Br J Cancer 1999;80:1342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet 2000;25:228–31. [DOI] [PubMed] [Google Scholar]
- Bouleftour W, Boudiffa M, Wade-Gueye NM, Bouet G, Cardelli M, Laroche N, et al. Skeletal development of mice lacking bone sialoprotein (BSP)--impairment of long bone growth and progressive establishment of high trabecular bone mass. PLoS One 2014;9:e95144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedinszki D, Szeri F, Kozak E, Pomozi V, Tokesi N, Mezei TR, et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med 2017;9:1463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res 1999;14:2015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachelli CM. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Pathol 1999;154:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RS, Kucukosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Nat Acad Sci USA 2013;110:20206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A, et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol 2014;34:1985–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Endo M, Dibra F, Wang K, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol 2009;129:348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffenstein G, Yegutkin GG, Khiati S, Pomozi V, Le Saux O, Leftheriotis G, et al. Alteration of extracellular nucleotide metabolism in pseudoxanthoma elasticum. J Invest Dermatol 2018; 10.1016/j.jid.2018.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement JF, Matsuzaki Y, Jiang QJ, Terlizzi J, Choi HY, Fujimoto N, et al. Targeted ablation of the Abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol 2005;25:8299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet 2000;25:223–7. [DOI] [PubMed] [Google Scholar]
- Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J. Mutant Enpp1asj mouse as a model for generalized arterial calcification of infancy. Dis Model Mech 2013;6:1227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brodsky JL, Conlin L, Pawel B, Glatz A, Gafni RI, et al. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: Genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol 2014a;134:658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Pratt CH, Dionne LA, Fairfield H, Karst SY, Sundberg JP, et al. Spontaneous asj-2J mutant mouse as a model for generalized arterial calcification of infancy: A large deletion/insertion mutation in the Enpp1 gene. PLOS One 2014b;9:e113542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sundberg JP, Levine MA, Terry SF, Uitto J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle 2015;14:1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Kingman J, Sundberg JP, Levine MA, Uitto J. Dual effects of bisphosphonates on ectopic skin and vascular soft tissue mineralization versus bone microarchitecture in a mouse model of generalized arterial calcification of infancy. J Invest Dermatol 2016;136:275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Kingman J, van de Wetering K, Tannouri S, Sundberg JP, Uitto J. Abcc6 knockout rat model highlights the role of liver in PPi homeostasis in pseudoxanthoma elasticum. J Invest Dermatol 2017;137:1025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie NC, Zhu D, Milne EM, van ‘t Hof R, Martin A, Darryl Quarles L, et al. Altered bone development and an increase in FGF-23 expression in Enpp1−/− mice. PloS One 2012;7:e32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys 1984;231:1–8. [DOI] [PubMed] [Google Scholar]
- Millan JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int 2016;98:398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa S, Harmey D, Yadav MC, O’Neill WC, Hoylaerts MF, Millan JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res 2007;22:1700–10. [DOI] [PubMed] [Google Scholar]
- Narisawa S, Yadav MC, Millan JL. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res 2013;28:1587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol 1988;6:1–159. [DOI] [PubMed] [Google Scholar]
- Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet 2012;90:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orriss IR, Arnett TR, Russell RG. Pyrophosphate: a key inhibitor of mineralisation. Curr Opin Pharmacol 2016;28:57–68. [DOI] [PubMed] [Google Scholar]
- Pennypacker B, Shea M, Liu Q, Masarachia P, Saftig P, Rodan S, et al. Bone density, strength, and formation in adult cathepsin K (−/−) mice. Bone 2009;44:199–207. [DOI] [PubMed] [Google Scholar]
- Pinkerton AB, Sergienko E, Bravo Y, Dahl R, Ma CT, Sun Q, et al. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg Med Chem Lett 2018;28:31–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomozi V, Brampton C, van de Wetering K, Zoll J, Calio B, Pham K, et al. Pyrophosphate supplementation prevents chronic and acute calcification in ABCC6-deficient mice. Am J Pathol 2017;187:1258–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A 2000;97:6001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli F, Corbo A, Salehi M, Yadav MC, Salman S, Petrosian D, et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS One 2017;12:e0186426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RG. Excretion of inorganic pyrophosphate in hypophosphatasia. Lancet 1965;2:461–4. [DOI] [PubMed] [Google Scholar]
- Russell RG, Bisaz S, Donath A, Morgan DB, Fleisch H. Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfecta, and other disorders of bone. J Clin Invest 1971;50:961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nature Genet 2003;34:379–81. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Boyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet 2008;1:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinov AY, Salehi M, Yadav MC, Radichev I, Millan JL, Savinova OV. Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer GL, Hu X, Pijnenborg AC, Wijnholds J, Bergen AA, Scheper RJ. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab Invest 2002;82:515–8. [DOI] [PubMed] [Google Scholar]
- Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res 2015;30:824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, et al. NT5E mutations and arterial calcifications. N Engl J Med 2011;364:432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Van de Wetering K, Varadi A, Terry SF. Novel insights into pathomechanisms and treatment development in heritable ectopic mineralization disorders. Summary of the PXE International Biennial Research Symposium. J Invest Dermatol 2017;137:790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zelst BD, de Jonge R. A stable isotope dilution LC-ESI-MS/MS method for the quantification of pyridoxal-5’-phosphate in whole blood. J Chromatogr B Analyt Technol Biomed Life Sci 2012;903:134–41. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 2012;366:904–13. [DOI] [PubMed] [Google Scholar]
- Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2016;12:233–46. [DOI] [PubMed] [Google Scholar]
- Zhao J, Kingman J, Sundberg JP, Uitto J, Li Q. Plasma PPi deficiency is the major, but not the exclusive, cause of ectopic mineralization in an Abcc6(−/−) mouse model of PXE. J Invest Dermatol 2017;137:2336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SG, Ferreira CR, MacFarlane EG, Riddle RC, Tomlinson RE, Chew EY, et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]