

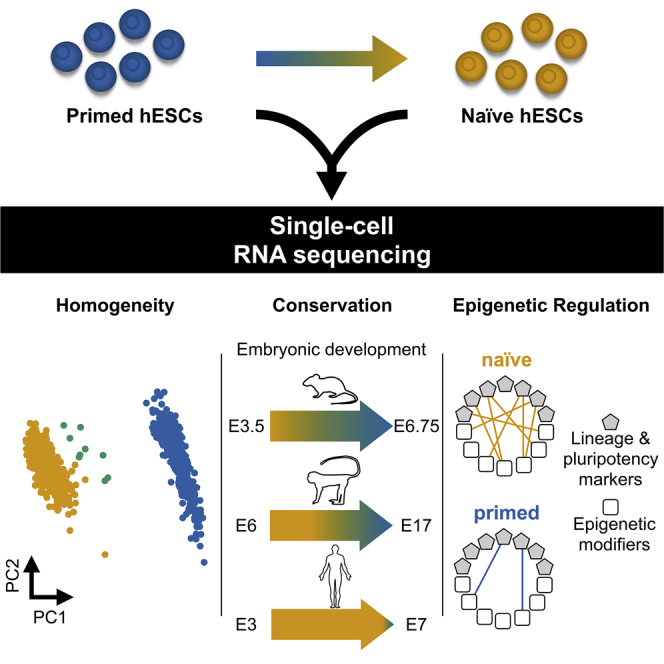
Summary
Conventional human embryonic stem cells are considered to be primed pluripotent but can be induced to enter a naive state. However, the transcriptional features associated with naive and primed pluripotency are still not fully understood. Here we used single-cell RNA sequencing to characterize the differences between these conditions. We observed that both naive and primed populations were mostly homogeneous with no clear lineage-related structure and identified an intermediate subpopulation of naive cells with primed-like expression. We found that the naive-primed pluripotency axis is preserved across species, although the timing of the transition to a primed state is species specific. We also identified markers for distinguishing human naive and primed pluripotency as well as strong co-regulatory relationships between lineage markers and epigenetic regulators that were exclusive to naive cells. Our data provide valuable insights into the transcriptional landscape of human pluripotency at a cellular and genome-wide resolution.
Keywords: human embryonic stem cells, pluripotency, single-cell RNA-seq, naive, primed, heterogeneity, early embryonic development, evolution
Graphical Abstract
Highlights
-
•
A single-cell RNA-seq resource of naive and primed human embryonic stem cells (hESCs)
-
•
Naive and primed hESCs are homogeneous except for a naive intermediate subpopulation
-
•
Naive and primed pluripotency signatures are conserved between species
-
•
Pluripotency and lineage markers correlate with epigenetic machinery in naive hESCs
Messmer et al. demonstrate that the single-cell transcriptomes of naive and primed human embryonic stem cells (hESCs) are mostly homogeneous. The study defines an expression signature that is conserved across species and shows differential epigenetic regulation between naive and primed pluripotency.
Introduction
Human and mouse embryonic stem cells (ESCs) are both derived from the inner cell mass (ICM) of the pre-implantation epiblast but differ in transcriptomic, epigenetic, and morphological features that correspond to consecutive stages of ontogeny. Mouse ESCs (mESCs) are marked by early developmental characteristics such as expression of the core pluripotency network, including Oct4, Klf4, or Dppa3; the activity of both X chromosomes in females; global DNA hypomethylation; and apolar morphology of the dome-shaped mESC colonies and, therefore, show the characteristics of naive pluripotency (Boroviak and Nichols, 2017). In contrast, primed or conventional human ESCs (hESCs) are developmentally more advanced and resemble murine post-implantation epiblast or mouse epiblast stem cells, thus they are considered to be primed pluripotent (Brons et al., 2007, Tesar et al., 2007).
Several groups have aimed to capture naive pluripotency in humans and to establish culture conditions closely recapitulating the signature of human ICM cells. These studies attempted to induce a naive state in hESCs by reprogramming primed hESCs with cytokines or small molecules (Gafni et al., 2013, Hanna et al., 2010, Takashima et al., 2014, Theunissen et al., 2014, Ware et al., 2014) or by directly culturing hESCs isolated from pre-implantation ICM cells under conditions that favor naive stemness (Guo et al., 2016). Among these, the stimulation of NANOG and KLF2 expression in 2 inhibitors (PD0325901 and CHIR99021) + Leukemia inhibitory factor (2i+Lif) conditions (inhibition of mitogen-activated protein extracellular signal-regulated kinase [ERK] and glycogen synthase kinase-3 beta) and subsequent restriction of protein kinase C (PKC) activity yielded hESCs with a close resemblance to the human blastocyst (Guo et al., 2017, Huang et al., 2014, Takashima et al., 2014). These reprogrammed naive hESCs express naive pluripotency markers, including OCT4, SOX2, and KLF2 and KLF4 (Boroviak and Nichols, 2017), and their metabolic and epigenetic profiles resemble the phenotype of mESCs rather than the primed state of conventional hESCs (Takashima et al., 2014).
There is still incomplete understanding of the transcriptional features that drive naive and primed pluripotency in ESCs (Ware, 2017, Weinberger et al., 2016). Studies exploring transcriptional identity and heterogeneity in mESCs have found significant variability associated with different states of pluripotency (Klein et al., 2015, Kolodziejczyk et al., 2015, Kumar et al., 2014). In a recent in vivo study of early mouse development (Mohammed et al., 2017), transcriptional noise was suggested to contribute to cell fate decision-making. However, although certain key pluripotency genes are much less variably expressed in the naive state (e.g., NANOG), single-cell RNA sequencing (scRNA-seq) suggests that overall heterogeneity in gene expression in mESC lines is independent of the respective culture condition and pluripotency state (Kolodziejczyk et al., 2015).
Our understanding of in vivo lineage commitment in humans is much more limited. By studying transcriptional profiles of developmental stages embryonic day 3 (E3) to E7 of human preimplantation embryos, the first lineage decisions between trophectoderm, primitive endoderm, and epiblast have been described (Petropoulos et al., 2016, Stirparo et al., 2018). Furthermore, a recent study has investigated the primed-to-naive cellular state transition process and found that genes related to hemogenic endothelium development were overrepresented in naive hESCs, resulting in higher differentiation potency into hematopoietic lineages (Han et al., 2018). Nonetheless, the extent and details of hESC heterogeneity have not been systematically characterized, and it is unclear whether the variability in gene expression is important for differentiation. To address these questions, we performed scRNA-seq of primed hESCs and reprogrammed naive hESCs to investigate the heterogeneity within each subpopulation and to compare their molecular phenotypes with in vivo transcriptome studies of embryogenesis.
Results
We assayed the transcriptomes of single primed and naive hESCs (WiCell WA09-NK2) to investigate gene expression heterogeneity and to identify potential subpopulations within different human pluripotency states. In total, we collected 480 hESCs grown under naïve titrated 2 inhibitors (PD0325901 and CHIR99021) + Leukemia inhibitory factor + inhibitor Gö6983 (t2iL+Gö) conditions (Takashima et al., 2014) and 480 hESCs grown under primed (E8) culture conditions (Chen et al., 2011). Single cells were separated and collected using fluorescence-activated cell sorting (FACS), and full-length cDNAs were prepared using the switch mechanism at the 5′ end of RNA templates (Smart-seq2) protocol (Picelli et al., 2014), followed by Nextera XT library preparation (Figure 1A). We removed low-quality cells and normalized for cell-specific bias prior to further analyses (STAR Methods; Figure S1A).
Figure 1.

Naive and Primed Human ESCs Exhibit Strong Differences in Gene Expression
(A) Naive and primed human ESCs were cultured in N2B27 supplemented with t2iL+Gö or in E8 medium, dissociated into single cells, and sorted into 96-well plates loaded with RLT lysis buffer and External RNA Controls Consortium (ERCC) spike-ins. RNA-seq libraries were prepared using the SmartSeq2 protocol and submitted for sequencing.
(B) PCA plot of hESC expression profiles, constructed from batch-corrected and normalized log expression values of highly variable genes detected across the entire dataset. Cells are colored by their condition, and the percentage of variance explained by the first two principal components is shown.
(C) Smear plot of log2-fold changes in expression between the naive and primed conditions, where differential expression (DE) genes were detected using edgeR at a false discovery rate (FDR) of 5%.
Naive and Primed hESCs Form Distinct Phenotypic Clusters
To confirm that scRNA-seq can recapitulate known differences between naive and primed conditions, we performed dimensionality reduction on all cells in the dataset using principal-component analysis (PCA) on highly variable genes (STAR Methods). We observed strong separation between naive and primed cells on the first principal component (Figure 1B), indicating that the difference between conditions is the dominant factor of variation. Differential expression analysis between naive and primed conditions identified a number of genes that were strongly upregulated under each condition (Figure 1C). This included the previously reported naive pluripotency and ground state marker genes KLF17, DPPA5, DNMT3L, GATA6, TBX3, IL6ST, DPPA3, and KLF5 (Blakeley et al., 2015, Dunn et al., 2014, Guo et al., 2017, Shahbazi et al., 2016, Theunissen et al., 2016, Yan et al., 2013). Although KLF4 has been described as a marker for both naive and primed cells (Ware, 2017), we only observed its expression in naive hESCs, consistent with other studies (Weinberger et al., 2016). In primed hESCs, we observed upregulation of established marker genes of primed pluripotency, such as CD24, ZIC2, and SFRP2, but not OTX2 or TFT (Buecker et al., 2014, Guo et al., 2016, Shakiba et al., 2015). Shared pluripotency markers, including SOX2, OCT4, and NANOG, did not significantly differ between the naive and primed population.
We also identified additional (and only recently suggested; Collier et al., 2017) markers of naive and primed hESCs (Table 1; Figure S1B; Table S1). The naive markers included genes that have been implicated in germ cell function (e.g., HORMAD1 for meiotic progression; Chen et al., 2005); KHDC3L as a regulator of imprinting (Parry et al., 2011); the alkaline phosphatases ALPP and ALPPL2, which are generally used as markers of pluripotent cells (Martí et al., 2013); as well as putative regulatory genes such as ZNF729. Some of these are also expressed in the early embryo; e.g., TRIM60 (Choo et al., 2002) and HORMAD1 (Chen et al., 2005). Primed markers included a number of genes related to later developmental stages; e.g., SOX11 for neuronal development (Bergsland et al., 2006), CYTL1 for chondrogenesis and expressed at implantation (Ai et al., 2016), HMX2 (an NK-like [NKL] homeobox gene) for organogenesis (Wang et al., 2001), and THY1 for hematopoietic stem cells (Majeti et al., 2007). We also found regulators of key signaling pathways, such as DUSP6 (a negative regulator of mitogen-activated protein kinase [MAPK] signaling) (Muda et al., 1996) and the receptor tyrosine phosphatase PTPRZ1 (Levy et al., 1993). We validated a number of these genes at the protein level using proteomics (Figure S1C) and in bulk RNA-seq data of the hESC lines UCLA1, WIBR3, and SHEF6 under naive and primed conditions (Table 1; Figure S1D; Pastor et al., 2016, Theunissen et al., 2016, Guo et al., 2017).
Table 1.
Markers of Naive and Primed Pluripotency in hESCs
| Naive | logFC (WA09-NK2) | logFC∗ (UCLA1)a | logFC∗ (WIBR3)b | logFC∗ (SHEF6)c | Primed | logFC∗ (WA09-NK2) | logFC∗ (UCLA1)a | logFC∗ (WIBR3)b | logFC∗ (SHEF6)c |
|---|---|---|---|---|---|---|---|---|---|
| KHDC1L | 14.49 | 8.82 | 13.23 | 9.36 | DUSP6 | −13.88 | −6.29 | −9.69 | −7.09 |
| FAM151A | 14.13 | 7.57 | 10.49 | 9.20 | FAT3 | −9.80 | −8.44 | −10.76 | −7.65 |
| HORMAD1 | 15.03 | 7.09 | 10.94 | 10.25 | THY1 | −13.60 | −8.78 | −8.47 | −6.26 |
| ALPPL2 | 20.17 | 7.32 | 11.26 | 8.82 | STC1 | −11.82 | −8.79 | −12.14 | −7.57 |
| ZNF729 | 14.07 | 4.25 | 11.15 | 4.83 | KLHL4 | −12.58 | −7.10 | −13.17 | −5.09 |
| KHDC3L | 19.58 | 6.88 | 12.78 | 9.54 | ZDHHC22 | −15.53 | −9.02 | −7.86 | −9.44 |
| TRIM60 | 18.53 | 7.90 | 12.19 | 8.33 | NEFM | −13.20 | −4.64 | −7.33 | −5.26 |
| MEG8 | 17.57 | 8.23 | 8.89 | not DE | HMX2 | −10.58 | not DE | not DE | not DE |
| OLAH | 10.80 | 7.58 | 12.87 | 7.97 | PLA2G3 | −15.29 | −6.31 | −5.74 | −8.07 |
| LYZ | 17.31 | 5.47 | 7.94 | 4.70 | PTPRZ1 | −10.55 | −8.33 | −12.72 | −9.63 |
| HYAL4 | 17.01 | 5.92 | 9.13 | 5.66 | CYTL1 | −14.54 | −7.60 | −9.43 | −9.09 |
| ALPP | 16.58 | 4.55 | 10.03 | 9.29 | SOX11 | −10.20 | −6.33 | −8.60 | −4.97 |
Log fold change between the primed and naive population; adjusted p < 0.005 for all shown DE genes.
Identification of a Subcluster in the Naive hESCs Population
We observed a small group of naive cells between the main naive and primed clusters (Figure 1B). We identified these cells by hierarchical clustering within the naive population, yielding a separate cluster of 9 cells. Despite being labeled as naive, this cluster was distinguishable from the other cells in the naive population as well as from the primed population (Figure 2A). These cells expressed some naive markers (DPPA3 and TFCP2L1; Figure 2B) but also exhibited primed-like characteristics (downregulation of KLF4 and KLF7) (Figure S2A); thus we labeled them “intermediate.” This subpopulation does not consist of doublets from the single-cell sorting procedure because they uniquely express genes that are absent in the primed population and other naive cells.
Figure 2.

The Naive Subpopulation Is Transcriptionally Distinct from the Other Naive and Primed Cells
(A) Heatmap of the top 50 genes with the strongest differential expression between the naive and intermediate cells (top) or between the intermediate and primed cells (bottom). The box for each cell (column) and gene (row) is colored according to the log2-fold change from the average expression for each gene.
(B) Log2 expression profiles of selected marker genes across cells in the naive, intermediate, and primed populations. Each point represents a cell in the corresponding population.
(C) Normalized protein expression of DPPA5 against ABCG2 in naive and primed hESCs. Protein expression was determined using immunofluorescence staining of cytospin-fixed cells.
(D) Representative immunofluorescence images of naive and primed hESCs using DPPA5 and ABCG2 antibodies. The scale bar represents 20 μm.
One question is whether this intermediate population arises from primed cells that were not fully transformed into the naive state or from naive cells that have acquired a more primed state. To investigate this, we specifically examined the expression of imprinted genes such as MEG3, PEG3, and SNRPN. Loss of imprinting has been reported under all current naive hESCs culture conditions, whereas conventional hESCs rarely show imprinting defects (Guo et al., 2017, Guo et al., 2016, Pastor et al., 2016). When lost, imprinting cannot be restored in non-germline cells, which can directly affect the expression level of the imprinted genes. We found similar expression of imprinted genes in the intermediate cluster compared with naive hESCs (Figure S2B), indicating that the subpopulation cells originate from the naive cells rather than being reprogramming-refractory remnants of the primed population that would not yet have undergone global DNA demethylation and loss of imprinting.
A number of genes were also uniquely upregulated in the intermediate population compared with both the naive and primed population. This includes ABCG2, CLDN4, VGLL1, GATA2, GATA3, and ERP27 (Figure S2C; see Table S2 for the full list), with significant over-representation of genes involved in morphological structure formation, development, and signaling (see Figure S2D for the Gene Ontology [GO] analysis). This suggests that the intermediate population is a separate state from the naive and primed conditions. Indeed, the transcription of NANOG was strongly downregulated in the intermediate population compared with both naive and primed cells (Figure 2B). In this respect, the subpopulation state shares some transcriptional features with the recently proposed state of formative pluripotency (Smith, 2017). Immunofluorescence staining based on high expression of ABCG2 and low expression of DPPA5 supported the existence of the intermediate population within the naive condition (Figures 2C and 2D).
Subclusters with Lineage-Specific Gene Expression Profiles Are Not Present in Naive or Primed hESCs
To study transcriptional heterogeneity within the naive and primed conditions, we applied t-distributed stochastic neighbor embedding (t-SNE) (van der Maaten and Hinton, 2008) to the cells under each condition after removing all intermediate population cells from the naive condition. We did not observe any distinct clustering within each condition; instead, the major driver of heterogeneity in each condition was the cell cycle (Figure S3A).
To focus only on heterogeneity related to embryonic development, we constructed the t-SNE plots using only a set of 184 endoderm-, ectoderm-, and mesoderm-specific markers (Table S3). The aim was to enrich for any weak population structure related to early fate commitment. However, we still did not observe any clusters corresponding to the different germ layers in either the naive or primed populations (Figures 3A, 3B, and S3B). This suggests that the primed cells remain in a mostly homogeneous undifferentiated state and have yet to begin the process of committing to differentiate into the germ layers.
Figure 3.

Naive and Primed Populations Do Not Exhibit Lineage-Associated Structure, but Correlations between Lineage Markers and Epigenetic Regulators Are Stronger under the Naive Condition
(A and B) Gene expression of germ layer-specific marker genes in the (A) naive and (B) primed population, visualized using tSNE on the batch-corrected normalized log expression values. Each point in the scatterplot represents a cell, which is colored by the expression of respective mesoderm (SNAI1), ectoderm (ITGA6), or endoderm (PAF1) markers.
(C and D) Heatmaps of the strongest correlation values between selected pluripotency and lineage markers (rows) and epigenetic markers (columns) for the naive (C) and the primed (D) population. The correlation values were bound at [−0.5, 0.5].
The homogeneity of both the naive and primed conditions suggests that it is possible to explore co-regulatory relationships via gene-gene correlations within each population. In particular, we focused on epigenetic modulators because of their importance in controlling cellular memory and their relevance for early embryonic development. Within each condition, we computed pairwise correlations between the expression profiles of 704 epigenetic modulators with a set of 94 developmental markers (Table S4; Figure S3C). We observed strong correlations in the naive population (Figure 3C) that were much weaker in the primed population (Figure 3D). This indicates that the expression of the epigenetic machinery is more distinctly linked to the naive gene expression network and particularly to regulators related to de novo DNA and histone methylation (e.g., DNMT3A/B and EHMT1).
A Naive-to-Primed Axis Can Identify Pluripotency Transitions in Other Species
To integrate our data with previous in vivo studies, we defined a naive-to-primed axis based on empirically defined marker genes that were strongly differentially expressed between the two conditions (STAR Methods). Cells from other scRNA-seq datasets were mapped onto this axis based on the proportion of naive-primed markers (or homologous equivalents in non-human data) they expressed. As a proof of concept, we mapped the previously described intermediate population onto the naive-to-primed signature map (Figure S4A). The subpopulation hESCs were located close to the naive axis but expressed a lower proportion of signature markers than the residual naive population. This is consistent with our hypothesis that the intermediate population originates from naive cells but has lost some features of naive pluripotency.
Next we mapped published scRNA-seq datasets of pre-implantation embryos from mice (Mohammed et al., 2017), cynomolgus monkeys (Nakamura et al., 2016), and humans (Petropoulos et al., 2016) onto our naive-to-primed axis. For mouse and monkey embryos, we observed a gradual loss of naive marker expression and an increase in primed marker expression (Figures 4A and 4B). This is consistent with the transition from naive to primed pluripotency and suggests that the relevant genes are conserved across species. Equal proportions of naive-primed markers were expressed at approximately E5 (mice) and E9–E13 (monkeys). In contrast, we did not observe any clear shift to primed pluripotency in humans before E7 (Figure 4C), consistent with the similarity of in vivo naive pluripotency with in vitro reprogrammed naive pluripotency under the applied culture conditions (Takashima et al., 2014).
Figure 4.

Cells Shift from a Naive-like to a Primed-like Expression Pattern during Early Embryonic Development
Naive and primed markers were identified from the DE analysis of the hESC data. New cells are mapped onto the naive-primed axis based on the proportions of naive and primed markers that they express. This was performed for cells derived from mouse embryos (A), cynomolgus monkey embryos (B), and human pre-implantation embryos (C). For each plot, the density of cells is represented by the color of the pixels. Cells on the red line have equal proportions of expressed primed and naive markers.
See also Figure S4.
We also defined a naive-to-intermediate axis using the identified unique markers of our intermediate population instead of the primed markers. We observed a shift from the naive expression pattern to that of the intermediate population after E5 in the human data (Figure S4B). This suggests that the intermediate population may also be present in vivo and relevant to human embryonic development.
Discussion
By sequencing the transcriptomes of single naive and primed hESCs, we identified discrete expression signatures of the two pluripotency states. In addition to recovering existing markers (Ware, 2017, Weinberger et al., 2016), we defined genes that are highly specific to each population. These expression markers are well conserved across species, as we were able to show by mapping mouse, monkey, and human sequencing data onto our naive-to-primed signature axis.
Another aim of this study was to clarify the heterogeneity and developmental progression of each pluripotency state in hESCs. We found that both naive and primed states of cultured hESCs were comparably homogeneous, except for a small subpopulation of cells in the naive state with transcriptional features of primed pluripotency. This was surprising because the primed state was expected to be more differentiated and possibly showing signatures of early lineage commitments, as suggested by in vivo work in mice (Mohammed et al., 2017). However, the comparably low levels of heterogeneity of naive and primed pluripotency in vitro have also been observed in mESC lines (Kolodziejczyk et al., 2015) and could be a reflection of the medium favoring one particular cellular phenotype. Therefore, these artificial states may be a misleading representation of primed pluripotency, which is more heterogeneous in vivo.
We observed that cell cycle-related effects were the most prominent source of variability within both the naive and the primed population. It is possible that specific cell cycle states may play a major role in contributing to cell fate decisions by introducing transcriptional noise. We also found that naive hESCs showed stronger correlations of pluripotency and lineage markers to epigenetic regulators than primed hESCs. Given the major epigenetic resetting observed during early embryonic development (i.e., from fertilization to the formation of the naive ICM cells; Iurlaro et al., 2017), the naive transcriptional state may have a unique need to be tightly coupled to the expression of the epigenetic machinery. In contrast, primed hESCs represent a later developmental stage in which the epigenetic machinery may be less strictly controlled as the epigenome is re-established in a more heterogeneous and cell type-specific manner. Future work exploring the epigenetic dynamics in early mouse and human embryonic development by single-cell epigenomics will help to dissect these mechanisms in more detail.
We also identified a subpopulation of naive hESCs that showed both features of naive and primed states. Indeed, we assume that the naive state in hESCs is temporally limited and that cells are prone to exit it. The existence of “formative” pluripotency has recently been suggested (Smith, 2017). This state may represent a cellular phase where cells acquire differentiation competency and are marked by the expression of early post-implantation factors such as OTX2, SOX3, and POU3F1 and the transient loss of NANOG expression. Interestingly, the intermediate population is characterized by significantly decreased NANOG transcription, although we did not detect significant upregulation of OTX2, SOX3, and POU3F1. It remains to be seen whether this subpopulation corresponds to cells exiting naive pluripotency toward formative pluripotency and whether this represents a real in vivo state or arises because of culture-specific conditions.
Our study provides important insights into the transcriptomic heterogeneity of naive and primed hESCs. The identification of specific markers may contribute to studying the reprogramming dynamics during the primed-to-naive transitions and delineate key transcriptional events leading to human naive pluripotency. Finally, we catalog and compare pluripotency identity across species to characterize transitions between different pluripotency states that mark specific temporal windows of embryonic development.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human DPPA5/ESG1 Antibody | R&D Systems | AF3125-SP; RRID: AB_2094168 |
| Human ABCG2 Antibody | R&D Systems | MAB995-SP; RRID: AB_2220316 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human LIF | Stem Cell Institute, Cambridge | N/A |
| CHIR99021 | Stem Cell Institute, Cambridge | N/A |
| PD0325901 | Stem Cell Institute, Cambridge | N/A |
| Gö6983 | TOCRIS | 2285 |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 |
| MEM non-essential amino acids | Thermo Fisher Scientific | 11140050 |
| TeSR-E8 | STEMCELL Technologies | 05990 |
| B27 | Thermo Fisher Scientific | 17504044 |
| N2 | Stem Cell Institute, Cambridge | N/A |
| DMEM-F12 | Thermo Fisher Scientific | 11320-033 |
| Neurobasal | Thermo Fisher Scientific | 21103049 |
| Accutase | STEMCELL Technologies | 07920 |
| Triton X-100 | Sigma-Aldrich | T9284 |
| Tween20 | Sigma-Aldrich | P9416 |
| DAPI | Thermo Fisher Scientific | 62248 |
| RNase Inhibitor | Clontech | 2313A |
| ERCC RNA Spike-In Mix | Ambion | 4456740 |
| dNTPs | Thermo Fisher Scientific | 10319879 |
| Critical Commercial Assays | ||
| AMPure XP beads | Beckman Coulter | A63880 |
| Nextera XT DNA Sample Preparation Kit | Illumina | FC-131-1096 |
| Nextera XT Index Kit | Illumina | FC-131-1001 |
| Superscript II reverse transcriptase | Invitrogen | 18064-014 |
| Deposited Data | ||
| Raw sequence data and count tables | This study | E-MTAB-6819 |
| Experimental Models: Cell Lines | ||
| Human WA09 Embryonic stem cells | Takashima et al., 2014 | N/A |
| Oligonucleotides | ||
| Oligo-dT30VN | Picelli et al., 2014 | N/A |
| AAGCAGTGGTATCAACGCAGAGTACT30VN | ||
| Template Switching Oligo | Picelli et al., 2014 | N/A |
| AAGCAGTGGTATCAACGCAGAGTACATrGrG+G | ||
| ISPCR oligo |
Picelli et al., 2014 |
N/A |
| AAGCAGTGGTATCAACGCAGAGT | ||
| Software and Algorithms | ||
| R version 3.5.1 (Feather Spray) | The R Project | https://www.r-project.org |
| scran | Lun et al., 2016b | https://bioconductor.org/packages/release/bioc/html/scran.html |
| scater | McCarthy et al., 2017 | https://bioconductor.org/packages/release/bioc/html/scater.html |
| Rtsne | van der Maaten and Hinton, 2008 | https://cran.r-project.org/web/packages/Rtsne/index.html |
| edgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| subread | Liao et al., 2013 | http://subread.sourceforge.net |
| Rsubread | Liao et al., 2014 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wolf Reik (wolf.reik@babraham.ac.uk).
Experimental Model and Subject Details
Human WA09-NK2 ESCs (Takashima et al., 2014) were kindly provided by Austin Smith and grown under naive or primed conditions.
Method Details
Cell culture and collection
Naive hESCs were grown in 6-well dishes on mouse embryonic fibroblasts in N2B27 supplemented with human LIF, 1μM Chiron, 1μM PD03 and 2μM Gö6983. 1 passage before sorting, cells were plated on 6-well plates coated with Matrigel (growth-factor reduced). Primed hESCs were grown in 6-well dishes coated with Vitronectin in E8 media. For collection, hESCs were dissociated with Accutase and sorted in 96 well plates containing lysis buffer on a BD Aria Cell sorter, gating for cell size and granularity. In each plate, 4 wells were left empty as negative controls. Plates were immediately spun down and frozen at −80°C until subsequent processing. This was performed in two batches – the first batch contained 96 cells from each condition, while the second batch contained 384 cells from each condition (480 cells in total per condition).
Library preparation and sequencing
Single-cells were sorted in 2uL of Lysis Buffer (0.2% v/v Triton X-100 (Sigma-Aldrich, cat. no. T9284) with 2U/ul RNase Inhibitor (Clontech, cat. no. 2313A)) in 96 well plates, spun down and immediately frozen at −80°C. cDNA from sorted single cells was prepared following the SmartSeq2 protocol (Picelli et al., 2014). Briefly, oligo-dT primer, dNTPs (ThermoFisher, cat. no. 10319879) and ERCC RNA Spike-In Mix (1:25,000,000 final dilution, Ambion, cat. no. 4456740) were added to the single-cell lysates, and Reverse Transcription and PCR were performed. The cDNA libraries for sequencing were prepared using Nextera XT DNA Sample Preparation Kit (Illumina, cat. no. FC-131-1096), according to the protocol supplied by Fluidigm (PN 100-5950 B1). Libraries from 96 single cells were pooled and purified using AMPure XP beads (Beckman Coulter). Pooled samples were sequenced on an Illumina HiSeq 2500 instrument, using paired-end 100-bp reads. On average, we obtained 2.1 × 106 reads per cell in batch 1 and 0.5 × 106 reads per cell in batch 2.
Immunofluorescence Analysis
Antibody staining was performed as previously described (Santos et al., 2003). Briefly, hESCs were cytospun, after fixation with 2% PFA for 30 minutes at room temperature. Cells were permeabilised with 0.5% Triton X-100 in PBS for 1h; blocked with 1% BSA in 0.05%Tween20 in PBS (BS) for 1h; incubation of the appropriate primary antibody diluted in BS; followed by wash in BS and secondary antibody. Secondary antibody was Alexa Fluor conjugated (Molecular Probes) diluted 1:1000 in BS and incubated for 30 minutes. Incubations were performed at room temperature unless otherwise stated. DNA was counterstained with 5 μg/mL DAPI in PBS. Single optical sections were captured with a Zeiss LSM780 microscope (63x oil-immersion objective). Fluorescence semi-quantification analysis was performed with Volocity 6.3 (Improvision).
Quantification and Statistical Analysis
Alignment and read counting
Read pairs were aligned to a reference consisting of the hg38 build of the human genome as well sequences for the ERCC spike-in transcripts. This was performed using the subread aligner v1.6.0 (Liao et al., 2013) in paired-end mode with unique alignment. Each read pair was then assigned to a gene in the Ensembl GRCh38 v91 annotation or to the spike-in transcripts. This was done using the featureCountsfunction in the Rsubread package v1.28.1 (Liao et al., 2014). Only reads with mapping quality scores above 10 were used for counting. Read counts from technical (sequencing) replicates of the same cell were added together prior to further analysis. On average, over 71% of reads mapped to the genome with over 59% mapped to exons.
Quality control on cells and genes
A range of quality metrics were computed for each cell (Figure S1A) using the calculateQCMetrics function in the scater package v1.6.3 (McCarthy et al., 2017). For each metric, outlier values were identified as those that were more than three median absolute deviations from the median. Low quality cells were identified in each batch, as those with small outlier values for the log-transformed total count; small outliers for the log-transformed number of expressed genes; large outliers for the proportion of read pairs assigned to mitochondrial genes; or large outliers for the proportion of read pairs assigned to spike-in transcripts. These cells were removed from the dataset prior to further analysis, leaving 414 naive and 423 primed cells remained for downstream analysis.
The cell cycle phase for each cell was identified using the cyclone classifier (Scialdone et al., 2015) implemented in the scran package v1.6.9. This was performed with a set of human marker genes, identified by training the classifier on a pre-existing hESC dataset (Leng et al., 2015).
Normalization of cell-specific biases
For the endogenous genes, cell-specific size factors were computed using the deconvolution method (Lun et al., 2016a) with pre-clustering. For each gene, the count for each cell was divided by the appropriate size factor. A pseudo-count of 1 was added, and the value was log2-transformed to obtain log-normalized expression values. This was repeated using the spike-in transcripts, where the size factor for each cell was proportional to the sum of counts for all spike-in transcripts (Lun et al., 2017).
Feature selection and dimensionality reduction
Feature selection was performed by computing the variance of the normalized log-expression values across cells for each endogenous gene or spike-in transcript. To represent technical noise, a mean-dependent trend was fitted to the variances of the spike-in transcripts using the trendVar function in the scran package (Lun et al., 2016b). This was done separately for each batch of cells to ensure that large variances were not driven by uninteresting batch effects. The decomposeVar function was used to obtain the biological component of the variance by subtracting the fitted value of the trend (i.e., the technical component) from the total variance of each gene. The combineVar function was then used to consolidate statistics across batches.
Batch effects were removed from the log-expression matrix using the removeBatchEffects function from the limma package v3.34.9 (Ritchie et al., 2015). This involved performing a linear regression on the log-expression profile of each gene and setting the blocking term for the batch to zero, which was possible for this data due to the balanced naive-primed composition of each batch. Principal component analysis was applied to the corrected expression matrix, only using the genes with positive biological components. This was performed with the denoisePCA function from scran to determine the number of principal components to retain. t-SNE was performed on the retained PCs using the Rtsne package v0.13.
Testing for differential expression between conditions
Counts for the naive and primed cells within each batch were pooled to obtain four sets of pseudo-bulk counts (Lun and Marioni, 2017). Low-abundance genes with average counts below 5 were removed and normalization was performed on the remainders with the trimmed mean of M-values method (Robinson and Oshlack, 2010). Genes were tested for differential expression (DE) between naive and primed conditions using the quasi-likelihood framework in the edgeR package v3.20.9 (Y. Chen et al., 2016). The experimental design was parameterized using an additive design containing a condition term and the batch blocking factor. DE genes were defined as those with significant differences between conditions at a FDR of 5%.
To validate the identified marker genes, DE genes in three different bulk RNA-seq datasets (Pastor et al., 2016, Theunissen et al., 2016, Guo et al., 2017) were identified using the quasi-likelihood framework. This used the procedure described above with the only difference being that DE genes were defined as having absolute log-fold changes significantly greater than 0.5 at a FDR of 5% (McCarthy and Smyth, 2009). This ensured that the DE analysis focused on genes with strong differences in expression. The results of the analysis for each dataset were visualized using volcano plots. For comparison, we highlighted the top 200 naive markers and the top 200 primed markers from our single-cell data (ranked by p value) on each plot.
Detecting the intermediate population
Dimensionality reduction was performed as previously described using only the cells in the naive condition. The retained principal components were used for hierarchical clustering of the cells with the hclust function in R, using Ward linkage on the Euclidean distances. Clusters of cells were identified using a simple tree cut, where the optimal number of clusters was determined by maximizing the average silhouette width. The cluster of cells located between the bulk of cells from the naive and primed conditions in the PCA plot was denoted as the intermediate population.
The intermediate population was characterized by testing for differential expression relative to the other naive cells or to the primed cells. This was done using t tests on the log-expression values (Soneson and Robinson, 2018) after blocking on the batch. For each contrast, several candidates were chosen from the top set of DE genes for further validation by immunofluorescence staining.
Exploring lineage-related heterogeneity
Dimensionality reduction was performed for each condition as previously described. For the naive condition, cells in the intermediate population were removed. PCA plots were colored according to the expression of CDK1 to represent cell cycle activity (Figure S3A). Dimensionality reduction in each condition was also repeated using only genes that were specific for the germ layers (Table S3) to detect potential early lineage commitment. Again, cells in the intermediate population were excluded. The resulting t-SNE plots were colored by expression of the mesoderm marker SNAI1 (Evseenko et al., 2010), the ectoderm marker ITGA6 (Brafman et al., 2013) or the endoderm marker PAF1 (Ponnusamy et al., 2009) to visualize any lineage-related substructure and by the expression of cell-cycle marker CDK1 (Figure S3C).
Correlations with epigenetic modulators
Pairwise correlations of selected lineage and pluripotency markers to epigenetic modulators (Table S4) were calculated using the correlatePairs function from the scran package. This involved computing Spearman’s rank correlation coefficients between the log-expression profiles of lineage marker genes (endoderm, ectoderm, mesoderm, trophectoderm, core pluripotency, naive pluripotency, formative pluripotency, primed pluripotency and germline) and genes comprising the epigenetic machinery. P-values for all pairs were combined and corrected for multiple testing.
To visualize the correlations, we computed the average absolute correlation across naive and primed conditions for each gene pair. We then selected the top 25 lineage/pluripotency markers and the top 25 epigenetic modifiers with the largest average absolute correlations. For each condition, a heatmap of the correlation values between all pairs of the selected genes was constructed using the pheatmap function from the pheatmap package v1.0.8.
Mapping temporal trajectories in early embryos
Naive marker genes were defined from our data as those that were DE relative to primed cells (using the pseudo-bulk statistics, above) at a FDR of 5% and with a log2-fold change of 10; were present in at least 25% of naive cells; and were present in no more than 5% of primed cells. Similarly, primed marker genes were defined as those that were DE relative to naive cells at a FDR of 5% and with a log2-fold change of −10; were present in at least 25% of primed cells; and were present in no more than 5% of naive cells.
A marker gene was considered to be expressed in a cell from a different dataset if its (normalized) count was greater than 10. For each cell, we calculated the proportion of naive markers that were expressed. This was repeated for the primed markers. Cells were mapped onto the “naïve-primed axis” based on these proportions. Large naive proportions and small primed proportions indicate that the cell is naive, and vice versa for primed cells.
Mapping onto the naive-primed axis was performed for cells collected from human pre-implantation embryos (Petropoulos et al., 2016), mouse embryos (Mohammed et al., 2017), and cynomolgus monkey embryos (Nakamura et al., 2016). Mouse homologs for the marker genes were identified using the getLDS function from the biomaRt package (Durinck et al., 2005), using the homology relationships predicted by Ensembl. Monkey homologs for marker genes were identified as those with the same gene symbol. As a control, we also performed remapping using the naive, primed and intermediate population cells in our own dataset.
To assess the expression of intermediate population genes in the human pre-implantation embryos (Petropoulos et al., 2016), an intermediate-naive axis was constructed similarly to the naive-primed axis. Intermediate population marker genes were considered uniquely expressed in the subpopulation by a log2-fold change of 5 against both the naive and the primed population at a FDR of 5%, and by their presence in less than 25% of the naive and the primed cells.
Data and Software Availability
Code availability
All analysis code is available at https://github.com/MarioniLab/NaiveHESC2016.
Deposition of sequencing data
The accession number for the scRNA-seq data reported in this paper is ArrayExpress: E-MTAB-6819.
Acknowledgments
We are grateful to members of the Marioni and Reik lab for helpful discussions. We would like to thank Amanda J. Collier for maintaining hESC cultures. We thank the WT Sanger sequencing facility for assistance with high-throughput sequencing. The work was supported by core funding from Cancer Research UK (award 17197 to J.C.M.), EMBL (to J.C.M.), an UKRI Rutherford Fund Fellowship (to F.v.M.), BBSRC (BB/K010867/1 to W.R.), and the Wellcome Trust (095645/Z/11/Z to W.R.).
Author Contributions
The experiments were designed by F.v.M. and W.R. and executed by F.v.M., A.S., F.S., and H.M. Computational analysis was performed by T.M., A.T.L.L., and J.C.M. The manuscript was written by T.M., F.v.M., A.T.L.L., J.C.M., and W.R. with input from all other authors.
Declaration of Interests
The authors declare no competing interests.
Published: January 22, 2019
Footnotes
Supplemental Information includes three figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.099.
Contributor Information
Aaron Tin Long Lun, Email: aaron.lun@cruk.cam.ac.uk.
John C. Marioni, Email: marioni@ebi.ac.uk.
Wolf Reik, Email: wolf.reik@babraham.ac.uk.
Supplemental Information
References
- Ai Z., Jing W., Fang L. Cytokine-Like Protein 1(Cytl1): A Potential Molecular Mediator in Embryo Implantation. PLoS ONE. 2016;11:e0147424. doi: 10.1371/journal.pone.0147424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsland M., Werme M., Malewicz M., Perlmann T., Muhr J. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 2006;20:3475–3486. doi: 10.1101/gad.403406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley P., Fogarty N.M.E., Del Valle I., Wamaitha S.E., Hu T.X., Elder K., Snell P., Christie L., Robson P., Niakan K.K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development. 2015;142:3613. doi: 10.1242/dev.131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T., Nichols J. Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development. 2017;144:175–186. doi: 10.1242/dev.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brafman D.A., Phung C., Kumar N., Willert K. Regulation of endodermal differentiation of human embryonic stem cells through integrin-ECM interactions. Cell Death Differ. 2013;3:369–381. doi: 10.1038/cdd.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons I.G.M., Smithers L.E., Trotter M.W.B., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S.M., Howlett S.K., Clarkson A., Ährlund-Richter L., Pedersen R.A., Vallier L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Buecker C., Srinivasan R., Wu Z., Calo E., Acampora D., Faial T., Simeone A., Tan M., Swigut T., Wysocka J. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:838–853. doi: 10.1016/j.stem.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-T., Venditti C.A., Theiler G., Stevenson B.J., Iseli C., Gure A.O., Jongeneel C.V., Old L.J., Simpson A.J.G. Identification of CT46/HORMAD1, an immunogenic cancer/testis antigen encoding a putative meiosis-related protein. Cancer Immun. 2005;5:9. [PubMed] [Google Scholar]
- Chen G., Gulbranson D.R., Hou Z., Bolin J.M., Ruotti V., Probasco M.D., Smuga-Otto K., Howden S.E., Diol N.R., Propson N.E. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lun A.T.L., Smyth G.K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438. doi: 10.12688/f1000research.8987.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo K.-B., Chen H.-H., Liu T.Y.-C., Chang C.-P. Different modes of regulation of transcription and pre-mRNA processing of the structurally juxtaposed homologs, Rnf33 and Rnf35, in eggs and in pre-implantation embryos. Nucleic Acids Res. 2002;30:4836–4844. doi: 10.1093/nar/gkf623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier A.J., Panula S.P., Schell J.P., Chovanec P., Plaza Reyes A., Petropoulos S., Corcoran A.E., Walker R., Douagi I., Lanner F., Rugg-Gunn P.J. Comprehensive Cell Surface Protein Profiling Identifies Specific Markers of Human Naive and Primed Pluripotent States. Cell Stem Cell. 2017;20:874–890.e7. doi: 10.1016/j.stem.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S.J., Martello G., Yordanov B., Emmott S., Smith A.G. Defining an essential transcription factor program for naïve pluripotency. Science. 2014;344:1156–1160. doi: 10.1126/science.1248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- Evseenko D., Zhu Y., Schenke-Layland K., Kuo J., Latour B., Ge S., Scholes J., Dravid G., Li X., MacLellan W.R., Crooks G.M. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2010;107:13742–13747. doi: 10.1073/pnas.1002077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni O., Weinberger L., Mansour A.A., Manor Y.S., Chomsky E., Ben-Yosef D., Kalma Y., Viukov S., Maza I., Zviran A. Derivation of novel human ground state naive pluripotent stem cells. Nature. 2013;504:282–286. doi: 10.1038/nature12745. [DOI] [PubMed] [Google Scholar]
- Guo G., von Meyenn F., Santos F., Chen Y., Reik W., Bertone P., Smith A., Nichols J. Naive Pluripotent Stem Cells Derived Directly from Isolated Cells of the Human Inner Cell Mass. Stem Cell Reports. 2016;6:437–446. doi: 10.1016/j.stemcr.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., von Meyenn F., Rostovskaya M., Clarke J., Dietmann S., Baker D., Sahakyan A., Myers S., Bertone P., Reik W. Epigenetic resetting of human pluripotency. Development. 2017;144:2748–2763. doi: 10.1242/dev.146811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Chen H., Huang D., Chen H., Fei L., Cheng C., Huang H., Yuan G.-C., Guo G. Mapping human pluripotent stem cell differentiation pathways using high throughput single-cell RNA-sequencing. Genome Biol. 2018;19:47. doi: 10.1186/s13059-018-1426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J., Cheng A.W., Saha K., Kim J., Lengner C.J., Soldner F., Cassady J.P., Muffat J., Carey B.W., Jaenisch R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K., Maruyama T., Fan G. The naive state of human pluripotent stem cells: a synthesis of stem cell and preimplantation embryo transcriptome analyses. Cell Stem Cell. 2014;15:410–415. doi: 10.1016/j.stem.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iurlaro M., von Meyenn F., Reik W. DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 2017;43:101–109. doi: 10.1016/j.gde.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk A.A., Kim J.K., Tsang J.C.H., Ilicic T., Henriksson J., Natarajan K.N., Tuck A.C., Gao X., Bühler M., Liu P. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 2015;17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.M., Cahan P., Shalek A.K., Satija R., DaleyKeyser A., Li H., Zhang J., Pardee K., Gennert D., Trombetta J.J. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516:56–61. doi: 10.1038/nature13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N., Chu L.-F., Barry C., Li Y., Choi J., Li X., Jiang P., Stewart R.M., Thomson J.A., Kendziorski C. Oscope identifies oscillatory genes in unsynchronized single-cell RNA-seq experiments. Nat. Methods. 2015;12:947–950. doi: 10.1038/nmeth.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J.B., Canoll P.D., Silvennoinen O., Barnea G., Morse B., Honegger A.M., Huang J.T., Cannizzaro L.A., Park S.H., Druck T. The cloning of a receptor-type protein tyrosine phosphatase expressed in the central nervous system. J. Biol. Chem. 1993;268:10573–10581. [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lun A.T.L., Marioni J.C. Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics. 2017;18:451–464. doi: 10.1093/biostatistics/kxw055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., Bach K., Marioni J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., Calero-Nieto F.J., Haim-Vilmovsky L., Göttgens B., Marioni J.C. Assessing the reliability of spike-in normalization for analyses of single-cell RNA sequencing data. Genome Res. 2017;27:1795–1806. doi: 10.1101/gr.222877.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R., Park C.Y., Weissman I.L. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí M., Mulero L., Pardo C., Morera C., Carrió M., Laricchia-Robbio L., Esteban C.R., Izpisua Belmonte J.C. Characterization of pluripotent stem cells. Nat. Protoc. 2013;8:223–253. doi: 10.1038/nprot.2012.154. [DOI] [PubMed] [Google Scholar]
- McCarthy D.J., Smyth G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–771. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Campbell K.R., Lun A.T.L., Wills Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H., Hernando-Herraez I., Savino A., Scialdone A., Macaulay I., Mulas C., Chandra T., Voet T., Dean W., Nichols J. Single-Cell Landscape of Transcriptional Heterogeneity and Cell Fate Decisions during Mouse Early Gastrulation. Cell Rep. 2017;20:1215–1228. doi: 10.1016/j.celrep.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muda M., Boschert U., Dickinson R., Martinou J.C., Martinou I., Camps M., Schlegel W., Arkinstall S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem. 1996;271:4319–4326. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- Nakamura T., Okamoto I., Sasaki K., Yabuta Y., Iwatani C., Tsuchiya H., Seita Y., Nakamura S., Yamamoto T., Saitou M. A developmental coordinate of pluripotency among mice, monkeys and humans. Nature. 2016;537:57–62. doi: 10.1038/nature19096. [DOI] [PubMed] [Google Scholar]
- Parry D.A., Logan C.V., Hayward B.E., Shires M., Landolsi H., Diggle C., Carr I., Rittore C., Touitou I., Philibert L. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am. J. Hum. Genet. 2011;89:451–458. doi: 10.1016/j.ajhg.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor W.A., Chen D., Liu W., Kim R., Sahakyan A., Lukianchikov A., Plath K., Jacobsen S.E., Clark A.T. Naive Human Pluripotent Cells Feature a Methylation Landscape Devoid of Blastocyst or Germline Memory. Cell Stem Cell. 2016;18:323–329. doi: 10.1016/j.stem.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S.P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell. 2016;165:1012–1026. doi: 10.1016/j.cell.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund Å.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Ponnusamy M.P., Deb S., Dey P., Chakraborty S., Rachagani S., Senapati S., Batra S.K. RNA polymerase II associated factor 1/PD2 maintains self-renewal by its interaction with Oct3/4 in mouse embryonic stem cells. Stem Cells. 2009;12:3001–3011. doi: 10.1002/stem.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F., Zakhartchenko V., Stojkovic M., Peters A., Jenuwein T., Wolf E., Reik W., Dean W. Epigenetic marking correlates with developmental potential in cloned bovine preimplantation embryos. Curr. Biol. 2003;13:1116–1121. doi: 10.1016/s0960-9822(03)00419-6. [DOI] [PubMed] [Google Scholar]
- Scialdone A., Natarajan K.N., Saraiva L.R., Proserpio V., Teichmann S.A., Stegle O., Marioni J.C., Buettner F. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015;85:54–61. doi: 10.1016/j.ymeth.2015.06.021. [DOI] [PubMed] [Google Scholar]
- Shahbazi M.N., Jedrusik A., Vuoristo S., Recher G., Hupalowska A., Bolton V., Fogarty N.N.M., Campbell A., Devito L., Ilic D. Self-organization of the human embryo in the absence of maternal tissues. Nat. Cell Biol. 2016;18:700–708. doi: 10.1038/ncb3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N., White C.A., Lipsitz Y.Y., Yachie-Kinoshita A., Tonge P.D., Hussein S.M.I., Puri M.C., Elbaz J., Morrissey-Scoot J., Li M. CD24 tracks divergent pluripotent states in mouse and human cells. Nat. Commun. 2015;6:7329. doi: 10.1038/ncomms8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Formative pluripotency: the executive phase in a developmental continuum. Development. 2017;144:365–373. doi: 10.1242/dev.142679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Robinson M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods. 2018;15:255–261. doi: 10.1038/nmeth.4612. [DOI] [PubMed] [Google Scholar]
- Stirparo G.G., Boroviak T., Guo G., Nichols J., Smith A., Bertone P. Integrated analysis of single-cell embryo data yields a unified transcriptome signature for the human pre-implantation epiblast. Development. 2018;145:dev158501–dev158514. doi: 10.1242/dev.158501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar P.J., Chenoweth J.G., Brook F.A., Davies T.J., Evans E.P., Mack D.L., Gardner R.L., McKay R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Friedli M., He Y., Planet E., O’Neil R.C., Markoulaki S., Pontis J., Wang H., Iouranova A., Imbeault M. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell. 2016;19:502–515. doi: 10.1016/j.stem.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L., Hinton G. Visualizing Data using t-SNE. JMLR. 2008;9:2579–2605. [Google Scholar]
- Wang W., Chan E.K., Baron S., Van de Water T., Lufkin T. Hmx2 homeobox gene control of murine vestibular morphogenesis. Development. 2001;128:5017–5029. doi: 10.1242/dev.128.24.5017. [DOI] [PubMed] [Google Scholar]
- Ware C.B. Concise Review: Lessons from Naïve Human Pluripotent Cells. Stem Cells. 2017;35:35–41. doi: 10.1002/stem.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware C.B., Nelson A.M., Mecham B., Hesson J., Zhou W., Jonlin E.C., Jimenez-Caliani A.J., Deng X., Cavanaugh C., Cook S. Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:4484–4489. doi: 10.1073/pnas.1319738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger L., Ayyash M., Novershtern N., Hanna J.H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016;17:155–169. doi: 10.1038/nrm.2015.28. [DOI] [PubMed] [Google Scholar]
- Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code availability
All analysis code is available at https://github.com/MarioniLab/NaiveHESC2016.
Deposition of sequencing data
The accession number for the scRNA-seq data reported in this paper is ArrayExpress: E-MTAB-6819.