

Abstract
The tumor suppressor p16Ink4a, encoded by the INK4a gene, is an inhibitor of cyclin D-dependent kinases 4 and 6, CDK4 and CDK6. This inhibition prevents the phosphorylation of the retinoblastoma protein (pRb), resulting in cellular senescence through inhibition of E2F-mediated transcription of S phase genes required for cell proliferation. The p16Ink4a plays an important role in tumor suppression, whereby its deletion, mutation, or epigenetic silencing is a frequently observed genetic alteration in prostate cancer. To assess its roles and related molecular mechanisms in prostate cancer initiation and progression, we generated a mouse model with conditional deletion of p16Ink4a in prostatic luminal epithelium. The mice underwent oncogenic transformation and developed prostatic intraepithelial neoplasia (PIN) from eight months of age, but failed to develop prostatic tumors. Given the prevalence of aberrant androgen signaling pathways in prostate cancer initiation and progression, we then generated R26hARL/wt:p16L/L: PB-Cre4 compound mice, in which conditional expression of the human AR transgene and deletion of p16Ink4a co-occur in prostatic luminal epithelial cells. While R26hARL/wt:PB-Cre4 mice showed no visible pathological changes, R26hARL/wt:p16L/L: PB-Cre4 compound mice displayed an early onset of high-grade PIN (HGPIN), prostatic carcinoma, and metastatic lesions. Strikingly, we observed tumors resembling human sarcomatoid carcinoma with intermixed focal regions of signet ring cell carcinoma (SRCC) in the prostates of the compound mice. Further characterization of these tumors showed they were of luminal epithelial cell origin, and featured characteristics of epithelial to mesenchymal transition (EMT) with enhanced proliferative and invasive capabilities. Our results not only implicate a biological role for AR expression and p16Ink4a deletion in the pathogenesis of prostatic SRCC, but also provide a new and unique genetically engineered mouse (GEM) model for investigating the molecular mechanisms for SRCC development.
Introduction
Mounting evidence has shown aging to be one of the most important risk factors for human prostate cancer (reviewed in [1]). Aging leads to decreased regenerative capability and an increased risk of malignant transformation (reviewed in [2]). The tumor suppressor p16INK4a has been shown to play a critical role in cellular aging and proliferation [3, 4]. An inhibitor of cyclin D-dependent kinases, 4 and 6, p16INK4a prevents phosphorylation of the retinoblastoma protein (pRb), which inhibits the transcription of E2F-regulated genes required for cell cycle entry at the G1/S checkpoint [3, 5]. The suppressive role of p16Ink4a on cell cycle progression is frequently disrupted in tumor cells, either by deletions or inactivating mutations of p16Ink4a [6] or pRb [7]. The expression of p16Ink4a is generally low in normal tissue [8]. The tumor suppressor role of p16Ink4a can be initiated by a number of distinct stimuli, including oncogenic stress, DNA-damage, and aging [9]. It has been shown that deletion of p16Ink4a occurs in human prostate cancer [10, 11]. Conversely, a suppressive growth effect of the induced expression of p16Ink4a has been observed in a prostate cancer xenograft model [12]. These lines of evidence suggest a pivotal role of p16Ink4a in prostate tumorigenesis.
Androgen signaling, mediated through the AR and its ligands, testosterone and 5α-dihydrotestosterone (DHT), plays a critical role in prostate tumorigenesis [13–17]. The AR is a member of the nuclear hormone receptor superfamily [18, 19]. The unbound AR forms a complex with heat-shock proteins (HSPs) [20, 21]. Upon binding to a ligand, the AR dissociates from the HSPs and translocates into the nucleus, where it binds to the androgen responsive element (ARE), recruits cofactors [22], and activates its targeted gene expression, which in turn controls prostate development and tumorigenesis. Aberrant activation of the AR directly promotes prostate cancer cell growth and survival, and androgen deprivation therapy (ADT) has been a standard treatment for prostate cancer for many decades [23]. However, many patients eventually relapse after therapy and develop castration resistant prostate cancer (CRPC) [13, 15, 24]. An important re-occurring element in prostate cancer is hormone refractoriness, the reemergence of androgen signaling through a variety of molecular mechanisms [13, 15]. Global gene expression profiling has shown the AR to be the only gene to be consistently up-regulated in CRPC [24]. Alterations of AR activity through mutation [25], amplification [26], overexpression [24], or cross-talk between AR and other growth factor pathways [27] directly contribute to tumor progression and CRPC development [28].
While abnormal expression and deletion of p16Ink4a have been observed in human prostate cancers [10, 11], its roles and related molecular mechanisms in prostate cancer initiation and progression are poorly understood. In addition, there is no mouse model that can directly assess the role of p16Ink4a and the collaborative effect of p16Ink4a suppression and AR activation in prostate tumorigenesis. In this study, we first developed a conditional p16Ink4a deletion mouse strain, in which the mouse p16Ink4a gene was selectively deleted in prostatic luminal epithelium. Intriguingly, deletion of p16Ink4a alone was sufficient to induce oncogenic transformation and mouse prostatic intraepithelial neoplasia (PIN) from eight months of age. However, the PIN lesions failed to progress to prostatic tumors in the mice, suggesting that additional hits are required for tumor formation. Given the prevalent role of androgen signaling in prostate cancer initiation and progression, we generated R26hARL/wt:p16L/L: PB-Cre4 compound mice, where conditional expression of the human AR transgene and deletion of p16Ink4a co-occur in prostatic luminal epithelial cells. The R26hARL/wt:p16L/L: PB-Cre4 compound mice revealed an earlier onset of PIN as well as prostatic carcinomas. Moreover, we identified a rare form of highly malignant tumor, sarcomatoid carcinoma with signet ring cell carcinoma (SRCC) in R26hARL/wt:p16L/L: PB-Cre4 compound mice [29–31]. These results not only implicate a biological role for transgenic AR expression and p16Ink4a deletion in the pathogenesis of prostatic SRCC but also provide a new and unique GEM model for investigating the molecular mechanisms for SRCC development.
Results
Conditional deletion of p16Ink4a in mouse prostate epithelium induces oncogenic transformation and prostatic intraepithelial neoplasia formation
Aberrant expression of p16Ink4a has been detected in human prostate cancer samples [10, 11, 32]. To directly investigate the biological role of p16Ink4a in prostate tumorigenesis, we generated a mouse line, p16L/L:PB-Cre4, in which p16Ink4a was conditionally deleted in prostatic luminal epithelium using a modified probasin promoter, ARR2PB, (Fig 1A) [33]. Using genomic PCR approaches, we confirmed the Cre/LoxP-mediated recombination of p16Ink4a in the mouse prostate (Fig 1B). The p16L/L:PB-Cre4 offspring were born at the expected Mendelian ratios and appeared normal with no obvious differences from their wild-type littermates at birth. Per the recommendations of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee [34], we assessed p16L/L:PB-Cre4 mice from birth and identified pathological lesions resembling low-grade prostatic intraepithelial neoplasia (LGPIN) in eight month old p16L/L:PB-Cre4 mice (Fig 1C). These lesions originated predominantly in the dorsal/lateral prostate (D/LP) and ventral prostate (VP) lobes (Fig 1C1 and Fig 1C2, respectively), consisting of atypical proliferative patterns with occasional stratification of cells and papillary structures. These atypical lesions contained epithelial cells that appeared larger than adjacent normal cells and lacked normal polarity (Fig 1C1’-2’). Development of these lesions in p16L/L:PB-Cre4 mice suggests a tumor suppressive role of p16Ink4a in prostatic epithelium. However, the animals with these lesions failed to develop prostate tumors after twenty-four months of age (Fig 2E). This implies that other genetic and/or epigenetic alterations are required in combination with p16Ink4a deletion for the formation of a prostate cancer.
Fig 1. Generation of p16Ink4a conditional deletion and AR conditional transgenic mice.

(A) A scheme was shown of the conditional p16Ink4a deletion target construct. (B) Genomic PCR was used to confirm the p16Ink4a targeting allele (blue solid arrow) and deleted allele (blue empty arrow) of different mouse tissues. (C) Representative H&E of the prostate tissue was shown for dorsal/lateral prostate (DLP), C1, and ventral prostate (VP), C2, for p16L/L:PB-Cre4 mice. (D) A scheme was shown of the conditional AR transgene target construct and the p16Ink4a deletion target construct. For the AR transgene construct, a PGK-neomycin cassette was flanked with loxP sites (LSL), red triangles, inserted between the CAG promoter and a FLAG-tagged AR coding sequence. For the p16nkK4a deletion, two LoxP sites, red triangles, were inserted to flank exon 1 alpha. Targeting constructs were shown, upper and Cre-driven recombined alleles for both constructs are shown below. (E) Genomic PCR was used to confirm the AR (black solid arrow) or p16Ink4a (blue solid arrow) targeting allele and AR recombined (black empty arrow) or p16Ink4a deleted (blue empty arrow) allele from different tissues of R26hARL/wt:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice. (F-H) Immunofluorescence (IF) assays. Prostate tissues of R26hARL/wt:PB-Cre4, p16L/L:PB-Cre4, and R26hARL/wt:p16L/L:PB-Cre4 mice were used for IF analyses and tissues sections were probed for AR (red, F1, G1, H1) or p16INK4a (green, F2, G2, H2) and DAPI (blue, F3, G3, H3), respectively.
Fig 2. Histological and immunohistochemistry analyses of the prostate from R26hARL/wt:PB-Cre4, p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4.

(A-B) Representative H&E images of the prostate tissue was shown for AP, DLP, and VP for R26hARL/wt:PB-Cre4 (A1-A3, magnification A1’-A3’) and R26hARL/wt:p16L/L:PB-Cre4 mice (B1-B3, magnification B1’-B3’). Scale bar used was 100 μm or 25 μm. (C). Representative H&E and IHC images of adjacent prostate tissue sections of AP from the p16L/L:PB-Cre4 mice were shown for staining with different antibodies as labeled in right bottom corner. (D). Representative H&E and IHC images of adjacent prostate tissue sections from the p16L/L: R26hARL/wt:PB-Cre4 mice were shown for staining with different antibodies (please see S1B and S1B’ Fig for positive staining of synaptophysin). Scale bar used was at 25 μm for all panels (C1-D9). (E) Pathological abnormalities of R26hARL/wt:PB-Cre4, p16L/L:PB-Cre4, and R26hARL/wt:p16L/L: PB-Cre4 mice reported for different time points.
Simultaneous expression of AR and deletion of p16Ink4a in the prostate accelerates oncogenic transformation in the mouse prostate
Genetic alterations of AR frequently occur during the course of prostate cancer initiation and progression in the presence of other genetic and epigenetic changes [24, 35, 36]. Given the prevalence of alterations in androgen signaling in human prostate cancers, we generated R26hARL/wt:p16L/L:PB-Cre4 compound mice, in which conditional AR expression and p16Ink4a deletion co-occur in prostate epithelium (Fig 1D). While the AR transgene and/or p16Ink4a target alleles were detected in various tissues isolated from R26hARL/wt:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice (black or blue solid arrows, Fig 1E), the activated AR transgene or p16Ink4a deleted alleles were detected exclusively in prostate tissues.(Black or blue empty arrows, Fig 1E). Using co-immunofluorescence (co-IF), we assessed the expression of the AR transgene and p16Ink4a deletion in the prostate tissues isolated from mice of each genotype. The expression of transgenic human AR protein was only detected in prostatic epithelial cells of R26hARL/wt:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice (Fig 1F1 and Fig 1H1). Similarly, p16Ink4a expression presented in prostatic luminal epithelium of R26hARL/wt:PB-Cre4 mice (Fig 1F2) but was absent in the samples of p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice (Fig 1G2 and Fig 1H2). These data confirmed conditional expression of the AR transgene and deletion of p16Ink4a co-occur in the prostate of the R26hARL/wt:p16L/L:PB-Cre4 compound mice.
The R26hARL/wt:p16L/L: PB-Cre4 mice and their littermates, R26hARL/wt:PB-Cre4, were born at the expected Mendelian ratios. They appeared normal with no obvious physical differences from their wild-type littermates at birth. Gross and pathological analyses showed no obvious abnormalities in the prostate of R26hARL/+:PB-Cre4 mice (Fig 2E), which is consistent with previous reports [37, 38]. All prostatic lobes displayed normal prostatic epithelium consisting of a single uniform layer of both luminal and basal cells (Fig 2A1-3’). Intriguingly, we observed that R26hARL/wt:p16L/L:PB-Cre4 compound mice developed PIN in all prostatic lobes (Fig 2B1-3 versus Fig 1C1-2), suggesting that the atypical lesions observed in the p16 knockouts were indeed precursor lesions. Specifically, pathological changes resembling high grade PIN (HGPIN) occurred within the anterior prostate (AP), containing atypical cells filling the lumen of the ducts, large pleomorphic nuclei with prominent nucleoli, and nuclear hyperchromasia (Fig 2B1-3’). Using immunohistochemical (IHC) approaches, the cellular properties of atypical cells within PIN lesions of p16L/L:PB-Cre4 mice and R26hARL/wt:p16L/L: PB-Cre4 compound mice were assessed. Positive nuclear staining of human AR protein only appeared in atypical cells of the prostate tissues of R26hARL/wt:p16L/L: PB-Cre4 compound mice (Fig 2C2 and Fig 2D2). However, positive staining of mouse AR revealed in the nucleus of luminal cells of both p16L/L:PB-Cre4 (Fig 2C3) and R26hARL/wt:p16L/L: PB-Cre4 compound mice (Fig 2D3). Loss of p16Ink4a staining was observed in prostate tissues of both p16L/L:PB-Cre4 (Fig 2C4) and R26hARL/wt:p16L/L:PB-Cre4 compound mice (Fig 2D4), whereas the p16Ink4a expression was detected in R26hARL/wt:PB-Cre4 mice (S1A and S1A’ Fig). Most of the atypical cells within PIN lesions from both p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice showed positive staining for E-cadherin (Fig 2C5 and Fig 2D5) and cytokeratin-8, CK8, (Fig 2C6 and Fig 2D6), which was consistent with their luminal epithelial origin. A scattered subset of cells were immunoreactive to the basal cell markers cytokeratin-5, CK5, (Fig 2C7 and Fig 2D7) and p63 (Fig 2C8 and Fig 2D8), but no staining for the neuroendocrine cell marker synaptophysin (Fig 2C9 and Fig 2D9) was seen in the prostate tissues from mice of either genotype. Taken together, the above results demonstrate that simultaneous expression of AR and deletion of p16Ink4a accelerates, and is necessary for oncogenic transformation in the mouse prostate, and that the atypical cells within the PIN lesions of the both p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice are prostatic luminal cell in origin.
Development of prostate tumors possessing simultaneous expression of AR transgene and loss of p16Ink4a expression in prostate epithelium
Based on the reported observations that HGPIN lesions can develop into prostate adenocarcinoma [39, 40], we continued examining p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice at advanced age. There was no observable progression of the focal atypical lesions in p16L/L:PB-Cre4 mice after twenty four months (Fig 2E and Fig 3A), suggesting the deletion of p16Ink4a alone in the prostate is not sufficient for formation of malignancy in mice. In contrast, prostate carcinoma developed in R26hARL/wt:p16L/L:PB-Cre4 compound mice (Fig 3A). The invasive prostatic carcinomas were observed at eight months of age and increased incidence was observed in R26hARL/wt:p16L/L:PB-Cre4 mice over twelve months of age (Fig 2E).
Fig 3. Synergistic effect of conditional AR expression and p16Ink4a deletion accelerates formation of adenocarcinoma.

(A) Pathological abnormalities of the prostate were listed for both p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice. (B-C) Representative image of prostatic tumors from R26hARL/wt:p16L/L:PB-Cre4 mice. Scale bar used was at 200 μm. B1-C4. Pathological dissemination of typical morphological characteristics of HGPIN (B1, C1) adenocarcinoma (B2, C2 and C3) and regions of necrosis (B3) and sarcomatoid carcinoma (B4, C4). D1-D8. Representative H&E and IHC images of adjacent prostate tissue sections of adenocarcinoma lesions from the R26hARL/wt:p16L/L:PB-Cre4 mice were shown for staining with different antibodies (right bottom corner). Scale bar used was at 25 μm for all panels (B1-D8).
Intriguingly, intracystic adenocarcinomas, invasive adenocarcinomas, and sarcomatoid carcinomas were observed (Fig 3B and Fig 3C). The co-existence of these tumor types in the R26hARL/wt:p16L/L:PB-Cre4 mice could be evidence of disease progression. These neoplastic lesions included characteristic regions of HGPIN (Fig 3B1 and Fig 3C1), multi-focal, poorly differentiated adenocarcinomas (Fig 3B2 and Fig 3C2), areas of increased proliferative activity and necrosis (Fig 3B3), as well as moderately differentiated adenocarcinoma (Fig 3C3). Adjacent to lesions of intracystic adenocarcinomas were regions classified as invasive sarcomatoid carcinoma, which was the dominant pathologic feature in all cases (Fig 3B4 and Fig 3C4).
The cellular properties of prostatic adenocarcinoma were assessed using IHC in tumor tissues of R26hARL/wt:p16L/L:PB-Cre4 mice. A typical nuclear staining for the transgenic AR was observed in most tumor cells, providing a direct link between the expression of the human AR transgene and tumor development (Fig 3D2). The tumor cells also showed positive staining for endogenous mouse AR, E-cadherin, and CK8 (Fig 3D3-5), the hallmarks of prostatic luminal epithelium. These tumor cells were negative for the basal epithelial cell markers p63 and CK5 (Fig 3D6-7), as well as for the neuroendocrine cell marker synaptophysin (Fig 3D8). These results demonstrate that the prostatic adenocarcinomas possess the same luminal prostatic cell properties as observed in PIN lesions of R26hARL/wt:p16L/L:PB-Cre4 mice, indicating a luminal epithelial origin.
In this study, diffuse, poorly differentiated lesions with biphasic epithelial and mesenchymal properties were observed in the prostatic stroma of R26hARL/wt:p16L/L:PB-Cre4 mice over twelve months of age (Fig 2E). These are classified as sarcomatoid carcinomas and are biphasic malignant neoplasms with features of epithelial-mesenchymal transition (EMT) (reviewed in [34] [41]). As shown in Fig 4A and Fig 4B, these lesions have tumor cells with a spindle-cell morphology (Fig 4A1 and Fig 4B1), focal regions of necrosis (Fig 4A2), and increased mitotic figures (Fig 4A3-3’ and 4B3-3’), indicative of a highly proliferative cell population in these sarcomatoid carcinomas. Invasion of the spindle-like tumor cells into surrounding muscle layers also appeared within the tumor samples (Fig 4B2). Development of this type of invasive prostatic carcinoma in this model suggests a collaborative role of AR activation and p16Ink4a deletion in prostate cancer initiation and progression.
Fig 4. Simultaneous conditional expression of AR transgene and loss of p16Ink4a expression induce sarcomatoid carcinoma development.
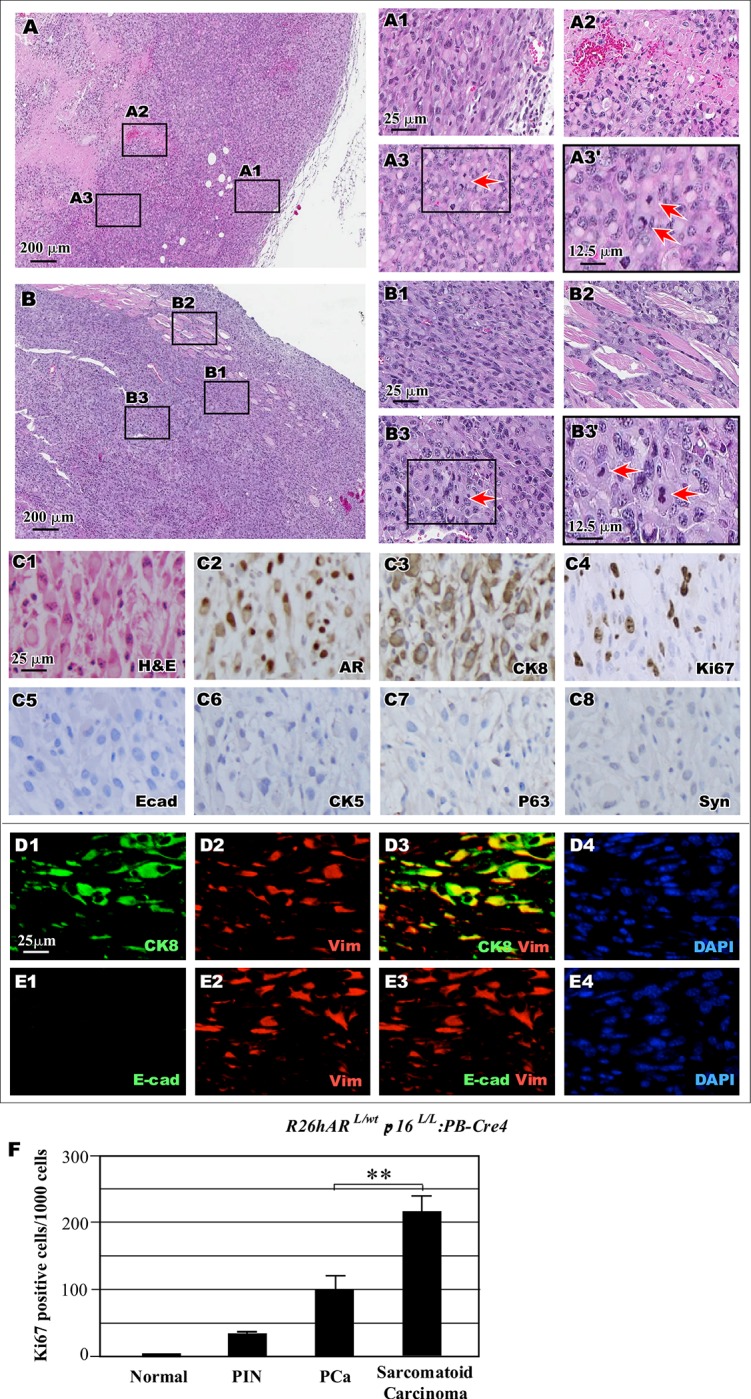
(A-B) Representative images of two tumor samples display the region of sarcomatoid carcinoma. Scale bar = 200 μm. Representative images of different lesions are shown, including sarcomatoid carcinoma (A1, B1), area of necrosis (A2), invasion into muscle (B2), and images of mitotic figures (A3, B3, see the arrows). (C) Representative H&E and IHC images from the R26hARL/wt:p16L/L:PB-Cre4 mice were shown for staining with different antibodies as labeled in the right bottom corner. (D-E) Co-IF from the R26hARL/wt:p16L/L:PB-Cre4 mice of secretory epithelial cell marker CK8 (green), mesenchymal cell marker vimentin (red), and DAPI (blue), upper panels; Co-IF of secretory epithelial cell marker E-cadherin (green) and mesenchymal cell marker vimentin (red) and DAPI (blue), lower panels. F. R26hARL/wt:p16L/L: PB-Cre4 mouse tissues were stained and quantified for Ki67 for the above tissues were measured in total 1,000 epithelial cells. Representative data shows mean and standard deviation. ** p < 0.005 Scale bar = 25 μm and magnifications were at 12.5 μm (A1-E4).
Our observation of sarcomatoid carcinomas adjacent to areas of PIN and invasive adenocarcinoma in the prostate tissues of R26hARL/wt:p16L/L:PB-Cre4 mice suggests that the sarcomatoid carcinomas may have developed from PIN and prostatic adenocarcinomas. To test this hypothesis, we assessed the properties of sarcomatoid carcinoma (Fig 4C1). IHC revealed positive nuclear staining for mouse AR in spindle-like tumor cells, suggesting an origin of the AR-positive luminal epithelial cells (Fig 4C2). The tumor cells also showed positive staining for CK8 (Fig 4C3) but not E-cadherin, CK5, p63, or synaptophysin (Fig 4C5-8). Using co-IF, co-expression of CK8 and the mesenchymal cell marker vimentin in tumor cells was observed (Fig 4D1-4). Loss of E-cadherin, an epithelial marker, expression was confirmed in these CK8- and vimentin-positive cells (Fig 4E1-4). The expression of AR and CK8 but loss of E-cadherin suggests that the sarcomatoid carcinomas retain luminal epithelial cell properties and undergo EMT, providing a clear evidence of disease progression.
We examined the proliferative activity of the cells in tumor lesions from R26hARL/wt:p16L/L: PB-Cre4 mouse prostates. Using IHC, we measured the expression of the proliferation marker Ki67 in normal and aberrant pathological lesions in each sample. An increase in Ki67 staining in areas of PIN, prostatic adenocarcinoma, and sarcomatoid carcinoma was observed when compared to normal prostatic tissues (Fig 4F). Notably, there were approximately 200 Ki67- positive cells per 1000 cells within the sarcomatoid carcinoma lesions (Fig 4F), which was significantly higher than those in PIN or prostatic adenocarcinoma lesions. These observations are consistent with the frequent appearance of mitotic figures presented in Fig 4A3’ and Fig 4B3’, and demonstrate the highly proliferative nature of the sarcomatoid carcinoma cells.
Development of signet ring cell carcinoma in the prostate and metastatic sites of R26hARL/wt:p16L/L:PB-Cre4 mice
Intermixed with regions of both the prostate adenocarcinoma and sarcomatoid carcinoma lesions of R26hARL/wt:p16L/L:PB-Cre4 compound mice was a tumor subtype with a particularly unique phenotype. Upon pathological examination of the tumor samples a distinct population of cells resembling signet ring cells (SRC) was observed, featuring large eosinophilic vacuoles displacing the nucleus to the cell periphery [42]. These prostatic SRCs appeared in various proportions in sarcomatoid carcinoma including up to 100% of some regions (Fig 5A1-2 and Fig 5B1-2). High magnification visualization of these tumor lesions showed typical SRC morphology (Arrows, Fig 5A1’-2’ and Fig 5B1’-2’). It has been shown that intracytoplasmic mucins within the large eosinophilic vacuoles were frequently detected in SRCs [43]. To evaluate the status of neutral mucinous components within the SRCs in the R26hARL/wt: p16L/L:PB-Cre4 mouse tumors, we performed Periodic Acid Schiff (PAS) and PAS-Diastase (PAS-D) staining. As shown in S1C to S1D’ Fig, positive staining of both PAS and PAS-Diastase were revealed in liver tissues of wild type mice. However, there was no positive staining with PAS and PAS-D in prostate tissues with SRC lesions (Fig 5C’ and Fig 5D’). Staining with Alcian Blue was also negative in the above prostate tissues containing SRCC (Fig 5E and Fig 5E’), in the contrast to the positive control samples (S1E and S1E’ Fig). These results suggest the absence of mucinous components and proteoglycans in SRCs of R26hARL/wt:p16L/L: PB-Cre4 mice. Identification of prostatic SRCC in the R26hARL/wt:p16L/L:PB-Cre4 mouse model is novel and provides a useful tool for investigating signet ring cell carcinomas in the future. Using IHC approaches, we further assessed the cellular properties of the prostatic SRCs. Notably, the SRCs were immunoreactive with antibodies against vimentin (Fig 5F and Fig 5F’), AR (Fig 5G1), and CK8 (Fig 5G2), but not for E-cadherin (Fig 5G3), CK5 (Fig 5G4), p63 (Fig 5G5), or synapotphysin (Fig 5G6) respectively. These results demonstrate a prostatic SRC expression pattern similar to the spindle-like sarcomatoid carcinoma cells, suggesting that these tumor lesions may be derived from the same cell origin.
Fig 5. Histological and Immunochemical identification of signet ring cell carcinoma.

(A-B) Representative image of the prostate signet ring cell carcinomas, SRCCs, in R26hARL/wt:p16L/L:PB-Cre4 mice. Scale bar = 200 μm. A1-B2. Pathological characterization of typical morphological characteristics of SRCC was highlighted in two different tumor samples with magnified views (A1’- B2’). (C-F) Representative view is shown for PAS/PAS (C-D, magnified view C’-D’) staining, Alcian Blue staining (E, magnified view E’), and vimentin immunoreactivity (F, magnified view F’). Scale bar used was at 50 μm and magnified views scale bar used was at 12.5 μm. (G). Representative view of IHC images of adjacent prostate tissue sections of signet ring cell carcinoma lesion from the R26hARL/wt:p16L/L:PB-Cre4 mice are shown for staining with different antibodies (right bottom corner). Scale bar used was at 12.5 μm. (H-J) Representative images of H&E of metastatic lesions in the lung (H), liver (I), and Lymph node (J) with magnified views (H’-J’). (K) Pathological abnormalities of the different aged mice were listed for R26hARL/wt:p16L/L: PB-Cre4 mice. Scale = 50 μm, or 12.5 μm (C-F, H-J).
During the course of our investigation, we closely monitored the mice for metastases in distant organs. Interestingly, we identified metastatic lesions in three R26hARL/wt:p16L/L:PB-Cre4 mice over twelve months of age (Fig 5K). Histological analyses showed metastatic tumor lesions, in the lung (Fig 5H and Fig 5H’), liver (Fig 5I and Fig 5I’), and lymph nodes (Fig 5J and Fig 5J’). Each of these metastatic lesions was composed primarily of spindle-like cells with focal regions of SRCs. IHC analyses of the SRCs in metastatic lesions revealed similar cellular properties to those in the primary tumors of R26hARL/wt:p16L/L:PB-Cre4 mice. SRCs in metastatic tissues from the lung were positive for AR and CK8, but negative for E-cadherin and CK5 (S2 Fig). These data confirm the prostatic cell origin of the metastatic disease and provide an additional line of evidence demonstrating the highly aggressive features of prostatic SRCC in R26hARL/wt:p16L/L: PB-Cre4 compound mice.
Identification of downstream targets that promote prostate tumor initiation and progression
In search for the molecular basis by which transgenic AR expression and p16INK4a deletion regulate prostate tumorigenesis, we performed RNA-sequencing analysis to examine the global transcriptome profiles in the mouse prostate tumor samples. Given that HGPIN and prostate tumor development respectively occurred in p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice, we focused specifically on those mice. RNA transcriptome profiling yielded differentially expressed genes (DEGs) of 960 genes that were upregulated with a fold change ≥ 5 and 537 genes that were downregulated at a fold change ≥ 5 from samples of the p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice in comparison to those from age and sex-matched wild type littermates (S1 Table). The change in expression of these genes reflects the activity of either p16Ink4a deletion alone or in combination with transgenic AR expression in the mouse prostate. According to gene set enrichment analysis (GSEA), the hallmark EMT signature was the most significantly enriched in the R26hARL/wt:p16L/L:PB-Cre4 compound mice (S3B Fig) [44]. Forty-nine DEGs within our data set overlapped with the hallmark EMT signature gene set (200 genes) (S3A Fig). Using real-time quantitative RT-PCR, qRT-PCR, alteration of a subset of these genes was validated (Fig 6A). We confirmed the statically significant upregulation of Spp1, Timp1, Twist, Sfrp4, Foxg1, Adam12, Cd44, and Itgb1 in the R26hARL/wt:p16L/L:PB-Cre4 mice in comparison to p16L/L: PB-Cre4 and wild type mice (Fig 6B). IHC analyses showed increased expression of Spp1 and CD44 in the prostates of the R26hARL/wt:p16L/L:PB-Cre4 mice relative to the p16L/L: PB-Cre4 mice (Fig 6C1-2’ versus Fig 6D1-2’). The above data provide a molecular basis for the effect of transgenic AR expression and p16Ink4a deletion in prostate cancer progression.
Fig 6. Co-expression of AR transgene and loss of p16Ink4a expression induces an EMT signature.

(A) Heatmap showing a subset of differentially expressed genes (DEGs) that corresponded to an overlap of hallmark EMT genes comparing p16L/L: PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice. (B) qRT-PCR confirmation of the gene alterations in prostate tissues of p16L/L: PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4. The data is presented as the mean ± (SD) (n = 3) ** p < 0.005 and * p < 0.01 by student’s T test. (C-D) Representative view of IHC images of adjacent prostate tissue sections of sarcomatoid carcinoma lesion from the R26hARL/wt:p16L/L: PB-Cre4 mice (C1-C2) and p16L/L: PB-Cre4 mice (D1-D2) and their magnified views (C1’-D2’). Scale bar = 50 μm or 12.5 μm.
Discussion
Loss of the CDK4/6 inhibitor, p16INK4a, due to its deletion or reduced expression through promoter hyper-methylation, has been observed in human prostate cancer samples [32, 45, 46]. Although the oncogenic role of p16Ink4a loss has been investigated in several tumor types [47–49], the loss of the tumor suppressor in prostate tumorigenesis has not been evaluated in a relevant mouse model. In this study, we developed a novel mouse model, p16L/L:PB-Cre4 mice, where the p16Ink4a gene was selectively deleted in prostatic luminal epithelial cells through Cre-LoxP-mediated recombination. We observed the development of mouse prostatic intraepithelial neoplasia in p16L/L:PB-Cre4 mice at eight months of age, providing the first line of evidence demonstrating that p16Ink4a deletion can initiate oncogenic transformation and PIN formation in the mouse prostate. In this study, we did not observe further severe pathologic changes in p16L/L:PB-Cre4 mice, suggesting that deletion of p16Ink4a alone is not sufficient to induce progression to prostate cancer. The precise mechanisms for this observation are currently unclear, potentially requiring additional genetic alterations for p16Ink4a-mediated tumor development in the prostate. The current mouse model, p16L/L:PB-Cre4 mice, is a biologically relevant experimental tool for identification of such alterations in the initiation of malignant transformation.
Aberrations of androgen signaling have been frequently identified in prostate cancer. Specifically, AR gene amplification or upregulation of the AR gene expression appears along with other genetic and epigenetic alterations in human prostate tumor tissues [35, 36]. Given the significance of both AR and p16Ink4a alterations in human prostate cancers, we developed R26hARL/wt:p16L/L:PB-Cre4 compound mice in which the deletion of the p16Ink4a gene and conditional expression of the human AR transgene co-occur in the prostatic luminal epithelium. We observed accelerated PIN development in R26hARL/wt:p16L/L:PB-Cre4 compound mice. We did not find any significant abnormality in the R26hARL/wt:PB-Cre4 mice. In contrast to 20% of the p16L/L:PB-Cre4 mice developing PIN lesions, the R26hARL/wt:p16L/L:PB-Cre4 compound mice showed PIN penetrance of 70%. This evidence demonstrates that the combined upregulation of AR and deletion of p16INK4a enhances oncogenic transformation in mouse prostate epithelium. Intriguingly, about 20% of the compound mice developed prostatic invasive carcinomas, sarcomatoid carcinomas, SRCCs, and metastatic tumors. Despite the aggressiveness of the above tumor phenotypes, the level of penetrance suggests that other genetic and epigenetic factors are required for tumor formation, progression, and metastasis.
In this study, we also observed aged mice showing increased tumor incidence and more aggressive tumor phenotypes, indicating a clear inverse correlation between aging and prognosis in the compound mice. All mice over 12 months of age bearing adenocarcinomas also developed invasive sarcomatoid carcinoma with SRCC, and metastatic disease. Not only do these data demonstrate a collaborative effect of AR expression and deletion of p16INK4a in promoting prostate cancer initiation and progression, but also provide a new and biological relevant mouse model that mimics the age-dependent human prostate cancer phenotypes.
As described in this study, we observed a variety of pathologic changes in the prostate of R26hARL/wt:p16L/L:PB-Cre4 compound mice. In addition to HGPIN and prostatic adenocarcinomas, we also identified sarcomatoid carcinoma that was the dominant pathologic feature and signet ring cell carcinoma lesions in the compound mice. Prostatic sarcomatoid carcinomas included spindle-like cells with highly proliferative and aggressive growth features, and invasion into the neighboring stroma. IHC analyses showed positive staining for AR in tumor cells within sarcomatoid carcinoma lesions, indicating that they originated from prostatic epithelium and, perhaps, PIN and/or prostatic adenocarcinomas. In agreement with these observations, earlier clinical studies have shown that prostate cancer patients with sarcomatoid carcinomas also had prior history and diagnosis of prostatic adenocarcinoma [50, 51]. A study of 42 patients showed approximately 66% of prostatic sarcomatoid cancer patients had prior pathologic diagnoses of prostate adenocarcinomas [52]. Deletion of prostate-specific erythroblast transformation-specific-related gene (ERG) has been detected in both the sarcomatoid and adjacent prostate adenocarcinoma, indicating that these two tumor types may be derived from the same cell origin [53]. We also detected positive staining for vimentin, a mesenchymal marker, in prostatic sarcomatoid tumor cells in the compound mice, suggesting the that these sarcomatoid carcinomas are undergoing the EMT process.
Most notably from this study, we identified an tumor type pathologically resembling signet ring cell carcinoma (SRCC), found as a part of the sarcomatoid carcinoma lesions in the R26hARL/wt:p16L/L:PB-Cre4 compound mice. SRCC is a rare type of highly malignant adenocarcinoma in humans but has not been described in mice. It is of epithelial origin and is characterized by cells featuring a crescent-shaped nucleus compressed against the cell membrane due to a large cytoplasmic vacuole [54]. Primary SRCC tumors are most often observed in the glandular cells of the stomach but also can be found in other tumor types, including prostate cancer [43, 55]. SRCC possesses invasive characteristics and is capable of colonizing at distant sites [29, 31, 56]. Previous studies have shown prostatic SRCCs to be highly aggressive with poor survival rates [29–31, 55].
As shown in this study, we observed tumor lesions containing the morphology of SRC in the prostate of R26hARL/wt:p16L/L:PB-Cre4 compound mice. IHC analyses further showed the prostatic SRCs to be AR and CK8-positive, suggesting they are derived from prostatic luminal cells. The positive staining with vimentin in the SRCs further indicates the EMT features of SRCs. We also identified SRCs in metastatic tumor sites, including the lung, liver, and lymph nodes. These multiple lines of experimental evidence indicate that prostatic SRCC in R26hARL/wt:p16L/L:PB-Cre4 compound mice share many pathological and biological features with human SRCCs. In this study, we also assessed the alterations of AR and p16Ink4a in human prostatic SRCC samples. Interestingly, from a human tissue microarray, we found 7 of 7 samples displayed positive staining for AR, and 5 out of 7 showed absence or reduced staining for p16Ink4a (S2 Table). These data imply a possible role of AR and p16Ink4a in SRCC pathogenesis, and warrant further investigation of patient samples.
Observation of sarcomatoid carcinoma and SRCC lesions with invasive and metastatic features in R26hARL/wt:p16L/L:PB-Cre4 compound mice supports the pattern of tumor progression and EMT development. This suggests a collaborative role of transgenic AR and p16Ink4a in enhancing tumor cell transdifferentiation and EMT promotion. We assessed this collaborative program by RNA sequencing and quantified real-time PCR approaches to analyze the changes of the transcriptional profiles from the prostate tumor samples isolated from p16L/L:PB-Cre4 and R26hARL/wt:p16L/L:PB-Cre4 mice. We observed an increase in the expression of a cluster of genes related to EMT and tumor progression in tumor samples isolated from R26hARL/wt:p16L/L:PB-Cre4 compound mice in comparison to those from p16L/L:PB-Cre4 mice. All together these data provide the molecular basis for future investigations to identify the specific regulators and signaling pathways that directly contribute to prostate cancer initiation and progression in R26hARL/wt:p16L/L:PB-Cre4 compound mice.
Materials and methods
Ethics statement
All animal experiments performed in this study were approved by the ethics committee of the Administrative Panel on Institutional Animal Care and Use Committee at Beckman Research Institute/City of Hope.
Mouse mating and genotyping
All mice that were used in this study were from a C57BL/6 background. Mice containing the conditional p16Ink4a allele (p16LoxP/LoxP), also named p16L/L, were kindly provided by Dr. Sharpless at University of North Carolina [57]. The AR transgenic mouse strain, R26hARLoxP/wt (R26hARL/wt) was generated previously [38]. Prostate specific conditional knockout mice were generated using PB-Cre4 mouse strain [33]. We generated p16L/wt:R26hARL/wt mice by intercrossing p16L/L with R26hARL/wt mice. The experimental mice were produced from intercrossing of p16L/wt: R26hARL/wt and p16L/wt: PB-Cre4 mice to generate three goal mice with genotypes: R26hARL/wt: PB-Cre4, p16L/L: PB-Cre4, and p16L/L:R26hARL/wt: PB-Cre4. Mice were genotyped by PCR as described previously [38, 57]. Detection of the AR targeting allele used the forward primer, 5’-TTCGGCTTCTGGCGTGTGAC-3’ with the reverse primer, 5’-CTCTGGAACAGATTCTGGAAA-3’ and 5’-AGCGCATCGCCTTCTATCGCCTTC-3’. The p16Ink4a conditional allele and deleted allele were detected with the forward, 5’-TACCACAGTTTGAACAGCGTGA-3’ with the reverse primer, 5’-AACCAACTTCCTCCTTCCCC-3’, and 5’-CCAAAACCACAACAGCTAGGA-3’. Genomic PCR conditions for AR recombined allele were 95°C for 3 min, then 95°C for 30 sec, 64°C for 30 sec, and 72°C for 60 sec for 35 cycles, then 72°C for 5 min. Genomic PCR conditions for p16Ink4a amplified at 95°C for 3 min, then 95°C for 45 sec, 58°C for 40 sec, and 72°C for 60 sec for 35 cycles, then 72°C for 5 min.
Histology and Immunohistochemistry, Immunofluorescence, PAS and PAS/D, Alcian Blue staining and histological analysis
The new guidelines recommended by The Mouse Models of Human Cancers Consortium Prostate Pathology Committee in 2013 were used for our pathological analyses [34]. Mouse tissues were fixed in 10% neutral-buffered formalin and processed into paraffin as previously described [38]. Mouse tissues were cut using 4 μm serial sections and rehydrated through a decreasing ethanol gradient. Histology was visualized by hematoxylin-eosin (H&E) staining performed as reported earlier [38]. Immunohistochemistry (IHC) included boiling the tissue in 0.01 M citrate buffer (pH 6.0) for antigen retrieval, and then incubated in 0.3% H2O2 for 15 min. Samples were blocked in 5% normal goat serum for 30 min and then incubated with primary antibodies diluted in 1% normal goat serum at 4°C overnight. Slides were incubated with biotinylated secondary antibodies for 1 hour, and then incubated with streptavidin ligated to horse radish peroxidase (HRP) (SA-5004, Vector Laboratories) for 30 min. Detection of HRP was with the 3,3'-diaminobenzidine (DAB) kit (SK-4100, Vector Laboratories). Nuclei were counterstained with 5% (w/v) Harris Hematoxylin and then mounted with Permount Medium (SP15-500, Fisher Scientific). Immunofluorescence (IF) of tissue samples were processed the same for IHC, except that the samples were incubated with fluorescent-conjugated secondary antibodies for 1 hour, and then mounted with Vectashield Mounting Medium with DAPI (H-1200, Vector Laboratories).
PAS staining was performed with the addition of 0.5% periodic acid (19840–0050, Acros) for 5 min at room temperature and then washed with distilled water. The tissue glycogen was digested with 0.5% Diastase (PAS/D) (AHDIA50, American MasterTech). Tissues were then incubated in Schiff ‘s reagent (26312-1A, Electron Microscopy Sciences) for 15 min at room temperature and then washed with tap water. Nuclei were counterstained with 5% (w/v) Harris Hematoxylin and mounted with Permount Medium (SP15-500, Fisher Scientific). Tissue samples for Alcian Blue staining were incubated in 1% Alcian Blue and 3% Acetic Acid at pH 2.5 for 30 min and then washed in distilled water. Counterstaining with nuclear-fast red solution was performed. After dehydration and clearing, the slides were mounted with Permount Medium (SP15-500, Fisher Scientific).
Antibodies
The primary antibodies used and their subsequent dilutions were: anti-P16 (rabbit, 1:1000, SC-1207, Santa Cruz), anti-human AR (mouse, 1:250, sc-7305, Santa Cruz Biotechnology), anti-CK5 (rabbit, 2400, PRB-160P, Covance), anti-CK8 (mouse, 1:2000, MMS-162P, Covance), anti-p63 (mouse, 1:2000, sc-8431, Santa Cruz), anti Ki67 (mouse, 1:1000, NCL-ki67, Novacastra), anti-E-cadherin (mouse, 1:200, Cat. No. c20820, BD Transduction Laboratoriesc, Sparks, MD, United States), anti-mouse/ human androgen receptor (rabbit, 1:250, sc-816, Santa Cruz), anti-synaptophysin (rabbit, 1:100, Cat. No. 18–0130, Invitrogen), anti-SPP1 (rabbit, 1:200, Cat. No. 91655, Abcam, Cambridge, MA, USA), CD44 (Rat, 1:50, sc-18849, Santa Cruz) and anti-Vimentin (Chicken, 1:2000, Cat. No. 919101, Biolegend). The biotinylated anti-rabbit or anti-mouse secondary antibody (BA-1000 or BA-9200, Vector Laboratories), or anti-rabbit or anti-mouse conjugated to AlexaFluor488 or to AlexaFluor594 (Molecular Probes) secondary antibody that were used for IHC or IF staining, respectively.
Microscope image acquisition
Images of H&E and IHC were acquired on an Axio Lab. A1 microscope using 10x and 40x Zeiss A-Plan objectives with a Canon EOS 1000D camera and using Axiovision software (Carl Zeiss). Images of immunofluorescence staining were acquired on a Nikon ECLIPSE E800 epi-fluorescence microscope using 20x and 40x Nikon Plan Fluor objectives with an QImaging RETIGA EXi camera and using QCapture software (QImaging).
RNA isolation and qRT-PCR assay
Mouse prostate tissues were homogenized in RNA-Bee (TEL-TEST, Inc., Friendswood, TX, USA), and total RNA was isolated as recommended by the manufacturer. Reverse transcription was carried out following our previous report [58]. For quantitative PCR, cDNA samples were mixed with SYBR greenER qPCR Super Mix Universal (11762, Invitrogen) and performed quantitative RT PCR according to the manufacture’s protocol. Relative mRNA levels were calculated by Delta Delta C(T) methods (KJ Livak, 2001, Method). Reactions were done in triplicate, and the values were normalized by PPIA (peptidylprolyl isomerase A) expression levels. Primers for SPP1 (5’- ATCTCACCATTCGGATGAGTCT- 3’; 5’- TGTAGGGACGATTGGAGTGAAA-3’), Timp1 (5’- TCTGGCATCCTCTTGTTGCT-3’; 5’- CATGAATTTAGCCCTTATGACCAG-3’), Twist (5’- GCTACGCCTTCTCCGTCTG -3’; 5’- GAAACAATGACATCTAGGTCTCCG-3’), Sfrp4 (5’- GTCACTGACCTTCCAGAAGATGT-3’; 5’- CCTTTTTGCACTTGCACCGA-3’), Adam12 (5’- TGGGGATGTGCCTCTTCAAC-3’; 5’- ATTCGTGCATTCCTCCGGTT-3’), Cd44 (5’- TCGATTTGAATGTAACCTGCCG-3’; 5’- CAGTCCGGGAGATACTGTAGC-3′), Itgb1 (5’- GGACGCTGCGAAAAGATGAA; 5’- CATTCTCCGCAAGATTTGGCA-3’), Foxc2 (5’- GCAACCCAACAGCAAACTTTC; 5’- GACGGCGTAGCTCGATAGG-3’) were synthesized and used in the qPCR reaction, respectively.
RNA sequencing
RNA sequencing was performed by the City of Hope Integrative Genomics core facility. cDNA synthesis and library preparation was performed using Kapa RNA mRNA HyperPrep kit (Kapa Biosystems, Cat KR1352) in accordance with the manufacturer supplied protocols. For gene set enrichment analysis (GSEA), genes were pre-ranked by the signed log 2 fold change. The preranked data were uploaded to GSEA and enrichment of hallmark gene sets were interrogated with 1,000 random permutations to obtain the P value, q value and normalized enrichment score (NES).
Statistical analyses
Cell numbers were counted manually using 40x photomicrographs as described in Fig 4F. Statistical analyses were performed using 2-tailed Student’s t test. The date was presented as the means ± S.D and cell numbers were made comparisons between groups, using a Student’s t test. p < 0.05 were considered significant.
Supporting information
(A) Representative P16Ink4a IHC images of prostate sample from the R26hARL/wt: PB-Cre4 mice. Scale bar = 100 μm or 25 μm. (B) IHC analyses of synaptophysin staining using mouse tissues as positive controls. Representative IHC images of positive synaptophysin staining showed in the cytoplasm of scattered neuron cells in the urethral urothelial and submucosal glands in mouse tissues samples as reported early (Toxicologic Pathology, 2005, 33:386–397). Scale bar = 100 μm or 25 μm. (C-D) Representative Periodic Acid Schiff (C-C’) and PAS-Diastase (D-D’) staining images from liver tissues of wild type mice were shown as positive controls in this study. Scale bar = 100 μm or 25 μm. (E) Representative Alcian Blue staining images from intestine tissues of wild type mice were shown as positive controls in this study. Scale bar = 100 μm or 25 μm.
(PDF)
(A-F) Representative H&E and IHC images of lung metastasis sample from the R26hARL/wt:p16L/L: PB-Cre4 mice were shown for staining with different antibodies (right bottom corner). Scale bar = 50 μm.
(PDF)
(A) A heatmap of 49 DEGs ≥ 5 fold in the R26hARL/wt:p16L/L: PB-Cre4 mice that overlapped with the list of hallmark EMT genes are listed with the accession numbers of each gene. This gene list was generated through GSEA pre-ranked analysis [44] of the DEGs that were altered comparing p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice. (B) Gene set Enrichment analysis (GSEA) plot of Hallmark EMT gene set, NES = 2.85 FDR (q-value) <0.000001.
(PDF)
Expression values were normalized to wild type mice and log2 transformed for the comparison between the RNA transcriptome alteration between p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice.
(PDF)
Seven clinical prostatectomy specimens with signet ring prostatic carcinoma component were mounted on one TMA (tissue microarray) and analyzed for AR, p16, CK8, CK5 and SPP1. “+” indicates pathologist determined classification of presence of staining, while “–”indicates pathologist determined absence of staining.
(PDF)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by NIHR01CA070297 for ZJS, NIHR01CA151623 for ZJS, NIHR01CA166894 for ZJS, NIHR21CA190021 for ZJS, NIHR01DK104941 for ZJS, and NIHP30CA033572 for JG. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Gronberg H. Prostate cancer epidemiology. Lancet. 2003;361(9360):859–64. Epub 2003/03/19. 10.1016/S0140-6736(03)12713-4 . [DOI] [PubMed] [Google Scholar]
- 2.Patel D, Scott CS. Inhibitor studies of purified haemopoietic (myeloid) cell esterases. Evidence for the existence of distinct enzyme species. Biochem Pharmacol. 1991;42(8):1577–85. Epub 1991/09/27. . [DOI] [PubMed] [Google Scholar]
- 3.Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375(6531):503–6. Epub 1995/06/08. 10.1038/375503a0 . [DOI] [PubMed] [Google Scholar]
- 4.D'Amico M, Wu K, Fu M, Rao M, Albanese C, Russell RG, et al. The inhibitor of cyclin-dependent kinase 4a/alternative reading frame (INK4a/ARF) locus encoded proteins p16INK4a and p19ARF repress cyclin D1 transcription through distinct cis elements. Cancer Res. 2004;64(12):4122–30. Epub 2004/06/19. 10.1158/0008-5472.CAN-03-2519 . [DOI] [PubMed] [Google Scholar]
- 5.Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci U S A. 1995;92(14):6289–93. Epub 1995/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarrard DF, Bova GS, Ewing CM, Pin SS, Nguyen SH, Baylin SB, et al. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer. 1997;19(2):90–6. Epub 1997/06/01. . [PubMed] [Google Scholar]
- 7.Kubota Y, Fujinami K, Uemura H, Dobashi Y, Miyamoto H, Iwasaki Y, et al. Retinoblastoma gene mutations in primary human prostate cancer. Prostate. 1995;27(6):314–20. Epub 1995/12/01. . [DOI] [PubMed] [Google Scholar]
- 8.Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10(15):2497–503. Epub 2011/07/22. 10.4161/cc.10.15.16776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15(2):203–11. Epub 1997/07/10. 10.1038/sj.onc.1201178 . [DOI] [PubMed] [Google Scholar]
- 10.Chakravarti A, DeSilvio M, Zhang M, Grignon D, Rosenthal S, Asbell SO, et al. Prognostic value of p16 in locally advanced prostate cancer: a study based on Radiation Therapy Oncology Group Protocol 9202. J Clin Oncol. 2007;25(21):3082–9. Epub 2007/07/20. 10.1200/JCO.2006.08.4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakravarti A, Heydon K, Wu CL, Hammond E, Pollack A, Roach M, et al. Loss of p16 expression is of prognostic significance in locally advanced prostate cancer: an analysis from the Radiation Therapy Oncology Group protocol 86–10. J Clin Oncol. 2003;21(17):3328–34. Epub 2003/08/30. 10.1200/JCO.2003.12.151 . [DOI] [PubMed] [Google Scholar]
- 12.Agus DB, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde DW, et al. Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999;91(21):1869–76. Epub 1999/11/05. . [DOI] [PubMed] [Google Scholar]
- 13.Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60(3 Suppl 1):132–8; discussion 8–9. . [DOI] [PubMed] [Google Scholar]
- 14.Culig Z, Klocker H, Bartsch G, Steiner H, Hobisch A. Androgen receptors in prostate cancer. J Urol. 2003;170(4 Pt 1):1363–9. 10.1097/01.ju.0000075099.20662.7f . [DOI] [PubMed] [Google Scholar]
- 15.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20(13):3001–15. 10.1200/JCO.2002.10.018 . [DOI] [PubMed] [Google Scholar]
- 16.Kyprianou N, Isaacs JT. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 1988;122(2):552–62. 10.1210/endo-122-2-552 [DOI] [PubMed] [Google Scholar]
- 17.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14(19):2410–34. [DOI] [PubMed] [Google Scholar]
- 18.Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240(4850):324–6. [DOI] [PubMed] [Google Scholar]
- 19.Lubahn DB, Joseph DR, Sar M, Tan J, Higgs HN, Larson RE, et al. The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol Endocrinol. 1988;2(12):1265–75. 10.1210/mend-2-12-1265 [DOI] [PubMed] [Google Scholar]
- 20.Sanchez ER, Faber LE, Henzel WJ, Pratt WB. The 56-59-kilodalton protein identified in untransformed steroid receptor complexes is a unique protein that exists in cytosol in a complex with both the 70- and 90-kilodalton heat shock proteins. Biochemistry. 1990;29(21):5145–52. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan WP, Vroman BT, Bauer VJ. Isolation of steroid receptor binding protein from chicken oviduct and production of monoclonal antibodies. J Steroid Biochem Mol Biol. 1992;43(1–3):37–41. [DOI] [PubMed] [Google Scholar]
- 22.Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5(10):1396–404. 10.1210/mend-5-10-1396 [DOI] [PubMed] [Google Scholar]
- 23.Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;168(1):9–12. . [DOI] [PubMed] [Google Scholar]
- 24.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–9. Epub 2004/01/02. 10.1038/nm972 . [DOI] [PubMed] [Google Scholar]
- 25.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–43. Epub 2012/06/23. 10.1038/nature11125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. Epub 2015/05/23. 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008;15(4):841–9. Epub 2008/08/01. 10.1677/ERC-08-0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Abronzo LS, Bose S, Crapuchettes ME, Beggs RE, Vinall RL, Tepper CG, et al. The androgen receptor is a negative regulator of eIF4E phosphorylation at S209: implications for the use of mTOR inhibitors in advanced prostate cancer. Oncogene. 2017;36(46):6359–73. Epub 2017/07/27. 10.1038/onc.2017.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chrysanthos NV, Nezi V, Zafiropoulou R, Papatheodoridis G, Manesis E. Metastatic Signet-Ring Cell Carcinoma of the Skin. Am J Gastroenterol. 2018. Epub 2018/06/13. 10.1038/s41395-018-0099-3 . [DOI] [PubMed] [Google Scholar]
- 30.Nichetti F, Morano F, Fabbri A, de Braud F, Di Bartolomeo M. Bilateral Parotid Gland Metastases from Gastric Signet-Ring Cell Carcinoma. Tumori. 2017:tj5000690 Epub 2017/12/02. 10.5301/tj.5000690 . [DOI] [PubMed] [Google Scholar]
- 31.Wei LY, Kong M, Zhang Z, Zhang XC. Breast metastasis of gastric signet-ring cell carcinoma. J Zhejiang Univ Sci B. 2017;18(11):1026–30. Epub 2017/11/10. 10.1631/jzus.B1700159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng W, Han Z, Zhu R, Liu P, Liu S. Association of p16 gene methylation with prostate cancer risk: a meta-analysis. J buon. 2015;20(4):1074–80. Epub 2015/09/30. . [PubMed] [Google Scholar]
- 33.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101(1–2):61–9. . [DOI] [PubMed] [Google Scholar]
- 34.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73(9):2718–36. Epub 2013/04/24. 10.1158/0008-5472.CAN-12-4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Network CGAR. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–25. 10.1016/j.cell.2015.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanbrough M, Leav I, Kwan PW, Bubley GJ, Balk SP. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc Natl Acad Sci U S A. 2001;98(19):10823–8. 10.1073/pnas.191235898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu C, Luong R, Zhuo M, Johnson DT, McKenney JK, Cunha GR, et al. Conditional expression of the androgen receptor induces oncogenic transformation of the mouse prostate. J Biol Chem. 2011;286(38):33478–88. Epub 2011/07/29. M111.269894 [pii] 10.1074/jbc.M111.269894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian J, Wollan P, Bostwick DG. The extent and multicentricity of high-grade prostatic intraepithelial neoplasia in clinically localized prostatic adenocarcinoma. Hum Pathol. 1997;28(2):143–8. Epub 1997/02/01. . [DOI] [PubMed] [Google Scholar]
- 40.Sakr WA, Haas GP, Cassin BF, Pontes JE, Crissman JD. The frequency of carcinoma and intraepithelial neoplasia of the prostate in young male patients. J Urol. 1993;150(2 Pt 1):379–85. Epub 1993/08/01. . [DOI] [PubMed] [Google Scholar]
- 41.Pang A, Carbini M, Moreira AL, Maki RG. Carcinosarcomas and Related Cancers: Tumors Caught in the Act of Epithelial-Mesenchymal Transition. J Clin Oncol. 2018;36(2):210–6. Epub 2017/12/09. 10.1200/JCO.2017.74.9523 . [DOI] [PubMed] [Google Scholar]
- 42.Guerin D, Hasan N, Keen CE. Signet ring cell differentiation in adenocarcinoma of the prostate: a study of five cases. Histopathology. 1993;22(4):367–71. Epub 1993/04/01. . [DOI] [PubMed] [Google Scholar]
- 43.Saito S, Iwaki H. Mucin-producing carcinoma of the prostate: review of 88 cases. Urology. 1999;54(1):141–4. Epub 1999/07/22. . [DOI] [PubMed] [Google Scholar]
- 44.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. Epub 2016/01/16. 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perinchery G, Bukurov N, Nakajima K, Chang J, Li LC, Dahiya R. High frequency of deletion on chromosome 9p21 may harbor several tumor-suppressor genes in human prostate cancer. Int J Cancer. 1999;83(5):610–4. Epub 1999/10/16. . [DOI] [PubMed] [Google Scholar]
- 46.Chi SG, deVere White RW, Muenzer JT, Gumerlock PH. Frequent alteration of CDKN2 (p16(INK4A)/MTS1) expression in human primary prostate carcinomas. Clin Cancer Res. 1997;3(10):1889–97. Epub 1998/11/17. . [PubMed] [Google Scholar]
- 47.Gadhikar MA, Zhang J, Shen L, Rao X, Wang J, Zhao M, et al. CDKN2A/p16 Deletion in Head and Neck Cancer Cells Is Associated with CDK2 Activation, Replication Stress, and Vulnerability to CHK1 Inhibition. Cancer Res. 2018;78(3):781–97. Epub 2017/12/13. 10.1158/0008-5472.CAN-17-2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Chen G, Cao R, Li J, He L, Guo X, et al. Allogeneic hematopoietic stem cell transplantation improves the prognosis of p16-deleted adult patients with acute lymphoblastic leukemia. Pharmacogenomics. 2017;18(1):77–84. Epub 2016/12/15. 10.2217/pgs-2016-0075 . [DOI] [PubMed] [Google Scholar]
- 49.Ortiz B, Fabius AW, Wu WH, Pedraza A, Brennan CW, Schultz N, et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc Natl Acad Sci U S A. 2014;111(22):8149–54. Epub 2014/05/21. 10.1073/pnas.1401952111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shannon RL, Ro JY, Grignon DJ, Ordonez NG, Johnson DE, Mackay B, et al. Sarcomatoid carcinoma of the prostate. A clinicopathologic study of 12 patients. Cancer. 1992;69(11):2676–82. Epub 1992/06/01. . [DOI] [PubMed] [Google Scholar]
- 51.Dundore PA, Cheville JC, Nascimento AG, Farrow GM, Bostwick DG. Carcinosarcoma of the prostate. Report of 21 cases. Cancer. 1995;76(6):1035–42. Epub 1995/09/15. . [DOI] [PubMed] [Google Scholar]
- 52.Hansel DE, Epstein JI. Sarcomatoid carcinoma of the prostate: a study of 42 cases. Am J Surg Pathol. 2006;30(10):1316–21. Epub 2006/09/27. 10.1097/01.pas.0000209838.92842.bf . [DOI] [PubMed] [Google Scholar]
- 53.Rodrigues DN, Hazell S, Miranda S, Crespo M, Fisher C, de Bono JS, et al. Sarcomatoid carcinoma of the prostate: ERG fluorescence in-situ hybridization confirms epithelial origin. Histopathology. 2015;66(6):898–901. Epub 2014/07/22. 10.1111/his.12493 . [DOI] [PubMed] [Google Scholar]
- 54.Warner JN, Nakamura LY, Pacelli A, Humphreys MR, Castle EP. Primary signet ring cell carcinoma of the prostate. Mayo Clin Proc. 2010;85(12):1130–6. Epub 2010/12/03. 10.4065/mcp.2010.0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujita K, Sugao H, Gotoh T, Yokomizo S, Itoh Y. Primary signet ring cell carcinoma of the prostate: report and review of 42 cases. Int J Urol. 2004;11(3):178–81. Epub 2004/03/11. . [DOI] [PubMed] [Google Scholar]
- 56.Paplomata E, Wilfong L. Signet ring cell carcinoma of the ampulla of Vater with leptomeningeal metastases: a case report. J Clin Oncol. 2011;29(21):e627–9. Epub 2011/05/25. 10.1200/JCO.2011.35.2385 . [DOI] [PubMed] [Google Scholar]
- 57.Monahan KB, Rozenberg GI, Krishnamurthy J, Johnson SM, Liu W, Bradford MK, et al. Somatic p16(INK4a) loss accelerates melanomagenesis. Oncogene. 2010;29(43):5809–17. Epub 2010/08/11. 10.1038/onc.2010.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hohaus S, Petrovick MS, Voso MT, Sun Z, Zhang DE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol. 1995;15(10):5830–45. Epub 1995/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative P16Ink4a IHC images of prostate sample from the R26hARL/wt: PB-Cre4 mice. Scale bar = 100 μm or 25 μm. (B) IHC analyses of synaptophysin staining using mouse tissues as positive controls. Representative IHC images of positive synaptophysin staining showed in the cytoplasm of scattered neuron cells in the urethral urothelial and submucosal glands in mouse tissues samples as reported early (Toxicologic Pathology, 2005, 33:386–397). Scale bar = 100 μm or 25 μm. (C-D) Representative Periodic Acid Schiff (C-C’) and PAS-Diastase (D-D’) staining images from liver tissues of wild type mice were shown as positive controls in this study. Scale bar = 100 μm or 25 μm. (E) Representative Alcian Blue staining images from intestine tissues of wild type mice were shown as positive controls in this study. Scale bar = 100 μm or 25 μm.
(PDF)
(A-F) Representative H&E and IHC images of lung metastasis sample from the R26hARL/wt:p16L/L: PB-Cre4 mice were shown for staining with different antibodies (right bottom corner). Scale bar = 50 μm.
(PDF)
(A) A heatmap of 49 DEGs ≥ 5 fold in the R26hARL/wt:p16L/L: PB-Cre4 mice that overlapped with the list of hallmark EMT genes are listed with the accession numbers of each gene. This gene list was generated through GSEA pre-ranked analysis [44] of the DEGs that were altered comparing p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice. (B) Gene set Enrichment analysis (GSEA) plot of Hallmark EMT gene set, NES = 2.85 FDR (q-value) <0.000001.
(PDF)
Expression values were normalized to wild type mice and log2 transformed for the comparison between the RNA transcriptome alteration between p16L/L:PB-Cre4 and R26hARL/wt:p16L/L: PB-Cre4 mice.
(PDF)
Seven clinical prostatectomy specimens with signet ring prostatic carcinoma component were mounted on one TMA (tissue microarray) and analyzed for AR, p16, CK8, CK5 and SPP1. “+” indicates pathologist determined classification of presence of staining, while “–”indicates pathologist determined absence of staining.
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.