

Mycobacterium tuberculosis, one of the world’s leading causes of death, must acquire nutrients, such as iron, from the host to multiply and cause disease. Iron is an essential metal and M. tuberculosis possesses two different systems to acquire iron from its environment: siderophore-mediated iron acquisition (SMIA) and heme-iron acquisition (HIA), involving uptake and degradation of heme to release ferrous iron.
KEYWORDS: BCG, Mycobacterium bovis, Mycobacterium tuberculosis, Tween 80, heme, hemin, iron, mycobactin, siderophore, tyloxapol
ABSTRACT
Mycobacterium tuberculosis, one of the world’s leading causes of death, must acquire nutrients, such as iron, from the host to multiply and cause disease. Iron is an essential metal and M. tuberculosis possesses two different systems to acquire iron from its environment: siderophore-mediated iron acquisition (SMIA) and heme-iron acquisition (HIA), involving uptake and degradation of heme to release ferrous iron. We have discovered that Mycobacterium bovis BCG, the tuberculosis vaccine strain, is severely deficient in HIA, and we exploited this phenotypic difference between BCG and M. tuberculosis to identify genes involved in HIA by complementing BCG’s defect with a fosmid library. We identified ppe37, an iron-regulated PPE family gene, as being essential for HIA. BCG complemented with M. tuberculosis ppe37 exhibits HIA as efficient as that of M. tuberculosis, achieving robust growth with <0.2 µM hemin. Conversely, deletion of ppe37 from M. tuberculosis results in a strain severely attenuated in HIA, with a phenotype nearly identical to that of BCG, requiring a 200-fold higher concentration of hemin to achieve growth equivalent to that of its parental strain. A nine-amino-acid deletion near the N terminus of BCG PPE37 (amino acids 31 to 39 of the M. tuberculosis PPE37 protein) underlies BCG’s profound defect in HIA. Significant genetic variability exists in ppe37 genes across different M. tuberculosis strains, with more than 60% of sequences from completely sequenced M. tuberculosis genomes having mutations that result in altered PPE37 proteins; furthermore, these altered PPE37 proteins are nonfunctional in HIA. Our findings should allow delineation of the relative roles of HIA and SMIA in M. tuberculosis pathogenesis.
INTRODUCTION
Mycobacterium tuberculosis, the primary etiologic agent of tuberculosis (TB), is one of the world’s leading causes of death, killing 1.7 million persons annually worldwide (1). M. tuberculosis, like other pathogens, must acquire nutrients from the host in order to multiply and cause disease, with iron being one such nutrient. Iron is an essential metal for most forms of life, and pathogens have developed elaborate systems for obtaining iron from their host; counteracting this, the host has developed systems for limiting iron availability during infection, a process termed nutritional immunity (2–4). Pathogens have evolved many ways to acquire iron from the host: (i) siderophore-mediated iron acquisition (SMIA) from transferrin and lactoferrin; (ii) heme-iron acquisition (HIA) involving surface receptors that bind heme or hemoproteins, such as hemoglobin or a bacterial secreted hemophore, followed by intracellular degradation of heme by heme oxygenases to release Fe2+; (iii) direct binding of transferrin via a receptor followed by extraction and transport of iron; (iv) transport of free Fe2+; and (v) iron acquisition from ferritin (5–9).
While some pathogens have multiple iron acquisition systems (e.g., Staphylococcus aureus has five [10]), until recently a well-characterized SMIA system (11–14) was believed to be the sole means by which M. tuberculosis acquires iron from its environment. However, we (15, 16) along with others (17) have demonstrated the existence of a second M. tuberculosis iron acquisition system that allows growth in submicromolar concentrations of hemin and hemoglobin, and we have identified several genes involved in HIA (Rv0203, mmpL11, and mhuD), that when deleted result in partial attenuation in the ability of M. tuberculosis to utilize iron from hemin (15, 18, 19). However, as attenuation is only partial, additional genes are almost certainly critical to HIA in M. tuberculosis.
Interestingly, we have discovered that Mycobacterium bovis BCG, the TB vaccine strain which has a genome that is nearly identical to that of M. tuberculosis (yet severely attenuated in comparison to M. tuberculosis, largely due to specific deletions) (20–22), is highly attenuated in HIA. In a series of studies (unpublished data), we found that none of the HIA genes we have identified thus far can be implicated as responsible, suggesting that the phenotypic difference between BCG and M. tuberculosis in HIA arises from differences outside the previously identified genomic regions. Although BCG’s defect in HIA is severe, it is not complete; growth with hemin is possible using high concentrations of hemin.
In this study, we have utilized the phenotypic difference in HIA between BCG and M. tuberculosis to complement BCG’s defect using a fosmid library of M. tuberculosis Erdman genomic DNA. By this approach, we demonstrate that ppe37 is an essential gene for efficient HIA by M. tuberculosis and that a defective BCG ppe37, encoding a protein with a nine-amino-acid (aa) deletion near the N terminus, is the cause of BCG’s inability to efficiently utilize hemin as an iron source. This nine-amino-acid deletion is also present in all virulent M. bovis strains. We have also found that more than 60% of M. tuberculosis strains out of a set of 146 with completely sequenced genomes have frameshift mutations in ppe37, compared with the sequence of the H37Rv reference strain, that encode altered PPE37 proteins. Furthermore, we have shown that the ppe37 genes with frameshift mutations are incapable of complementing BCG’s HIA defect, indicating that they are nonfunctional, and that two M. tuberculosis strains (HN878 and CDC1551) with ppe37 variants are defective in HIA. In contrast to a recent report, we did not find a role for ppe36 in HIA (23). Finally, as a practical matter, we demonstrate that the commonly used detergent, Tween 80, at relatively low concentrations significantly interferes with efficient HIA, whereas the detergent tyloxapol does not interfere with HIA except at very high concentrations.
RESULTS
BCG is defective in HIA.
We have previously shown that BCG mbtB and M. tuberculosis mbtB mutants with defective SMIA due to disrupted siderophore biosynthesis do not show sustained growth in standard 7H9 medium containing ∼130 μM Fe3+ (as ferric ammonium citrate) unless it is supplemented with exogenous siderophore; however, the mutants are capable of several generations of growth in unsupplemented 7H9 medium until their stored iron is depleted, and then growth is halted (15, 16). We also discovered a heme-iron acquisition (HIA) system in M. tuberculosis that allows for robust growth using submicromolar concentrations of hemin or hemoglobin (15, 16). Unlike M. tuberculosis, BCG grows very poorly with hemin as an iron source (Fig. 1). BCG’s defect in HIA is severe, with almost no growth apparent at less than 1 µM hemin in contrast to excellent growth of M. tuberculosis even at concentrations as low as 0.078 µM. Interestingly, even though M. tuberculosis grows very efficiently with hemin, it is apparently highly resistant to the toxic effects of heme, as no inhibition is observed with up to even 40 µM hemin; in contrast, the pathogen Staphylococcus aureus is strongly inhibited by 10 µM hemin (24). While BCG’s defect is severe, it is not complete; growth to ∼50% of the level in the presence of exogenous siderophore (mycobactin J) was obtained at the high concentration of 40 µM hemin (Fig. 1A). As can be seen from the poor growth in the absence of added supplement (Fig. 1A and B) other than the ferric ammonium citrate present in 7H9 medium, any other potential source of iron present in the medium (e.g., from the oleic acid-albumin-dextrose-catalase [OADC] supplement) that BCG and M. tuberculosis might be capable of using is of little consequence.
FIG 1.
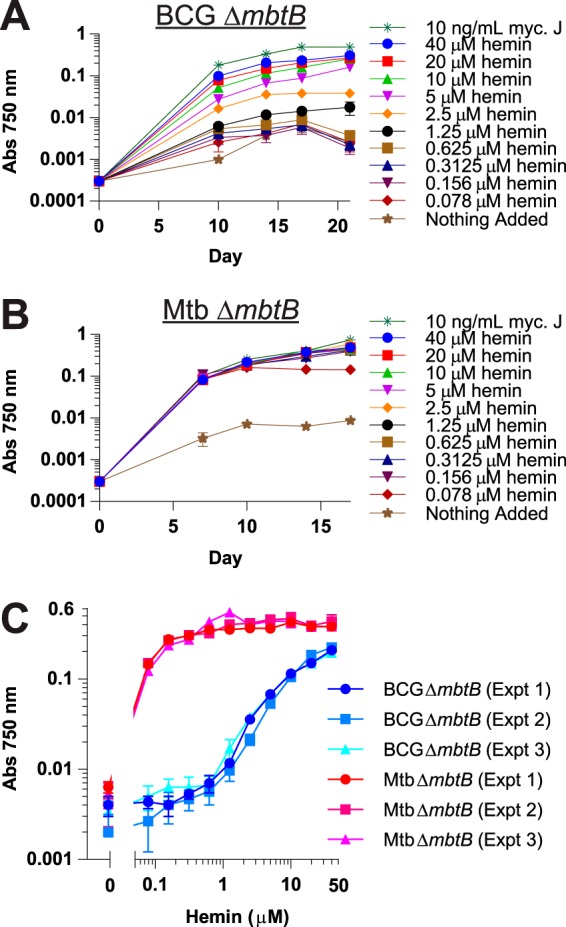
BCG is defective in HIA. BCG ΔmbtB (A) and M. tuberculosis ΔmbtB (B) were grown in 7H9–OADC–0.01% TLX medium with various concentrations of hemin (0.078 to 40 µM), 10 ng/ml mycobactin J (myc. J), or no additional supplement in 96-well plates. Cultures were inoculated to an initial calculated A750 of 0.00025, and growth was determined by measuring absorbance at 750 nm at various time points up to 21 days. Bacterial density measurements with the 96-well plate reader are 4-fold less than measurements made with a cuvette-based spectrophotometer. (C) BCG ΔmbtB and M. tuberculosis ΔmbtB were grown at various hemin concentrations, as indicated in panels A and B, and growth was measured at 14 days. Shown are three independent experiments (Expt) for each strain. Data are the means ± standard errors from triplicate wells for each condition. In most instances, the error bars are smaller than the symbols.
Complementation of the BCG HIA defect with M. tuberculosis fosmid libraries.
To identify genes involved in HIA, we exploited the phenotypic difference in HIA between BCG and M. tuberculosis using fosmid libraries of M. tuberculosis Erdman genomic DNA to complement BCG’s defect. To generate fosmid libraries, we first constructed an Escherichia coli-mycobacterium integrating shuttle fosmid vector, pMycInt(apr)-sacB-FOS3 (Fig. 2A), derived from the commercially available E. coli fosmid vector pSMART FOS (Lucigen). In addition to the features of pSMART-FOS (i.e., efficient cloning of large genomic DNA inserts using phage packaging extract, the presence of transcription terminators to improve insert stability in E. coli, and the ability to be maintained at single copy or to be amplified to medium copy number in E. coli), pMycInt(apr)-sacB-FOS3 includes the mycobacterial attP-int region from pMC1s (25) to allow stable site-specific integration into the mycobacterial genome, an apramycin resistance gene (replacing the chloramphenicol resistance gene of pSMART-FOS) that is suitable for use in both E. coli and mycobacteria, and a sacB stuffer fragment in the multicloning site to facilitate cloning of genomic DNA (gDNA) inserts.
FIG 2.

Complementation of the BCG HIA defect with M. tuberculosis fosmid libraries identifies ppe37 as an essential gene for efficient HIA. (A) E. coli-mycobacterium integrating shuttle fosmid vector, pMycInt(apr)-sacB-FOS3, used for the construction of fosmid libraries. Features are as follows: sacB, sucrase gene for sucrose counterselection; int, mycobacterial attP-int region for stable site-specific integration into the mycobacterial genome; cos, lambda cos site to allow packaging into phage lambda particles; sopABC (parABSF), partition system of the bacterial F plasmid; incC, incompatibility region of the bacterial F plasmid; repE, replication initiation protein for the bacterial F plasmid; ori2 (oriS), single-copy origin of replication of bacterial F plasmid; oriV, inducible (with TrfA) origin of replication. (B) Genomic map of fosmid clones with enhanced HIA displaying a 65.2-kb region of the M. tuberculosis Erdman chromosome (nucleotides 2336070 to 2401279; genes, ERDMAN_2301 to ERDMAN_2368) with the location and orientation of the genomic DNA inserts from 10 unique fosmid clones with enhanced HIA (arrows labeled fosmid A through J). The 14.9-kb region that all 10 fosmids have in common is boxed. (C) Map of the 14.9-kb common region showing the five subregions (PCR A through E) tested for enhanced HIA. Only subregion E conferred enhanced HIA on BCG. (D) PPE37 protein alignment. BCG PPE37 (BCGT_1943; GenBank accession number AHM07863.1) is missing 9 amino acids (ARAWHALAA, amino acids 31 to 39; shown in red) and has one additional difference (S188I; in red) from the sequence of the M. tuberculosis PPE37 protein (ERDMAN_2339, GenBank accession number WP_003411053.1).
To avoid selecting clones complemented with an intact mbtB gene, we isolated high-molecular-weight gDNA from M. tuberculosis ΔmbtB::Km or the double mutant, M. tuberculosis ΔmbtB::Km ΔmhuD::apr. The original purpose for using the double mutant was to have a selectable marker (the apramycin resistance gene present at the site of the mhuD deletion) in the fosmid library as a control to measure the frequency of marker transfer. However, as the final fosmid vector pMycInt(apr)-sacB-FOS3 has an apramycin resistance gene, rather than a hygromycin resistance gene as originally designed (see Materials and Methods), this was not possible. Genomic DNA fragments of ∼35 to 45 kb were ligated into PmeI-digested pMycInt(apr)-sacB-FOS3, and ligations were packaged into phage particles to generate two fosmid libraries in E. coli (M. tuberculosis ΔmbtB::Km, 1,637 clones; M. tuberculosis ΔmbtB::Km ΔmhuD::apr, 9,346 clones). Based on an estimated average insert size of ∼40 kb, 15-fold coverage of the M. tuberculosis Erdman genome was obtained with the M. tuberculosis ΔmbtB::Km library, and 85-fold coverage was obtained with the M. tuberculosis ΔmbtB::Km ΔmhuD::apr library (M. tuberculosis Erdman genome size, 4.39 Mb [26]).
The fosmid libraries were electroporated into BCG ΔmbtB ΔpanCD (a pantothenate auxotroph in addition to being defective in siderophore biosynthesis), and clones with enhanced HIA were selected on agar plates containing 10 or 50 µM hemin. As a control, we tested the libraries for their ability to complement the panCD mutation by plating a portion of the electroporations on plates containing mycobactin but lacking pantothenate, obtaining 9 panCD complemented clones from a calculated total of 2,095 transformants plated (1 panCD complemented clone per 233 transformants). Clones that grew on plates with hemin were obtained at a similar frequency (127 HIA clones from a calculated total of 12,920 transformants plated; 1 HIA clone per 102 transformants). We analyzed the growth of 25 clones from hemin plates in broth culture, and 24 displayed an efficient HIA phenotype similar to that of M. tuberculosis ΔmbtB (data not shown).
Mapping of fosmid genomic DNA sequences that complement the BCG HIA defect.
To identify the M. tuberculosis genes in the BCG fosmid clones that were responsible for enhanced HIA, we amplified both the 5′ and 3′ ends of the fosmid gDNA inserts separately in hemi-nested PCRs and sequenced the PCR products. To amplify the fosmid gDNA inserts with unknown sequence, we designed PCR primers with 7-mer sequences on the 3′ end that occur with high frequency and relatively even distribution in the M. tuberculosis genome (see Table S1 in the supplemental material). For 17 out of the 24 clones with enhanced HIA, both the 5′ and 3′ ends of the fosmid gDNA inserts were successfully identified by this approach, with 10 of the 17 fosmids having unique gDNA inserts. The unique gDNA inserts ranged in size from 27.6 to 36.9 kb (mean, 31.4 kb; median, 31.0 kb) with the chromosomal orientation evenly split (5 fosmid clones contain gDNA inserts with the plus strand 5′ to 3′, and the other five clones have genomic DNA inserts with the minus strand 5′ to 3′). Most significantly, all 10 unique gDNA inserts share a region of 14.9 kb (Fig. 2B).
A functional PPE37 is required for efficient HIA by BCG.
The 14.9-kb common region contains many genes of unknown function, so to identify the gene or genes responsible for enhanced HIA, we amplified five subregions by PCR, cloned them into pMycInt(apr)-sacB-FOS3, transformed the plasmids into BCG ΔmbtB ΔpanCD, and selected clones with enhanced HIA on agar plates containing hemin as for the original fosmid libraries. Only one of the subregions (Fig. 2C, PCR E) conferred enhanced HIA on BCG (data not shown). Two of the three genes in this subregion, hisG and hisE, are involved in histidine biosynthesis and appeared unlikely to be involved in HIA. However, the third gene, ppe37 (alternate gene names, irg2 and Rv2123), was an excellent candidate for HIA involvement as it is part of the IdeR regulon and is induced by low iron (27, 28). It thus appeared that ppe37 might be solely responsible for the enhanced HIA phenotype.
A comparison of the genomic sequences of M. tuberculosis and BCG revealed that although ppe37 is present in the BCG genome, the protein encoded by the BCG gene lacks 9 amino acids (ARAWHALAA, amino acids 31 to 39 of the M. tuberculosis PPE37 protein) near the N terminus of the protein, suggesting a possible reason for the defective HIA phenotype in BCG (Fig. 2D). BCG PPE37 differs in one additional way from the M. tuberculosis PPE37 protein (Ser → Ile at position 188 of M. tuberculosis PPE37). A blastp search of protein databases revealed that 12 of 12 BCG PPE37 protein sequences and 62 of 62 PPE37 protein sequences from virulent M. bovis (from which the BCG vaccine was derived) contain the identical 9-amino-acid deletion and the S188I variation. A strain originally identified as M. bovis MAL010093 has a PPE37 sequence 100% identical to the M. tuberculosis PPE37 sequence, but this strain has recently been reported to be Mycobacterium africanum and not M. bovis (29) although the databases have not been updated to reflect this recharacterization. M. africanum, Mycobacterium microti, and Mycobacterium canettii possess PPE37 proteins that are identical, or nearly so, to the M. tuberculosis PPE37 protein, and none of the sequences have the 9-amino-acid deletion or the S188I variation. PPE37 appears to be unique to M. tuberculosis complex mycobacteria. The best matches outside the M. tuberculosis complex have much less similarity (Mycobacterium marinum, 56% identity; Mycobacterium haemophilum, 55% identity). Mycobacterium leprae has a pseudogene (ML1308c) for ppe37 while Mycobacterium smegmatis does not possess a ppe37 orthologue; indeed, it has only two PE/PPE pairs in its genome which correspond to orthologues in the M. tuberculosis esx-1 and esx-3 regions (30).
It was recently reported that PPE37 is exported, with the protein found primarily in the cell membrane fraction, and that it possesses a eukaryotic-type signal sequence from residues 1 to 40, although the details for how the signal sequence was determined were not reported (31). We analyzed the M. tuberculosis PPE37 sequence using SignalP, TatP, LipoP, and TargetP to predict potential secretion signals. SignalP does not predict a signal peptide for PPE37 using any of the organism parameters (Gram positive, Gram negative, or eukaryotic); however, using the Gram-positive bacteria parameters, the D-score which is used to discriminate signal peptides, for residues 1 to 38 was higher than what is considered ideal for nonsecretory proteins (0.270 versus ∼0.1; D-score of >0.450 to discriminate signal peptides). TatP does not predict a Tat signal peptide for PPE37 but, like SignalP. identifies a potential cleavage site between amino acids 38 and 39. Finally, PPE37 was not identified as a lipoprotein using LipoP, but a potential cleavage site for a classical signal peptide cleaved by signal peptidase I was predicted between amino acids 29 and 30. Thus, PPE37 does not have a conventional signal peptide that can be detected by the SignalP, TatP, or LipoP algorithm, but the higher score than nonsecretory proteins and the cleavage site predictions suggest that it may possess a nonclassical signal peptide. As PPE37 has two transmembrane domains and as the protein was detected in the cell membrane fraction (31), it is likely to be an integral membrane protein. Interestingly, the cleavage site predictions occur either in the amino acids deleted from the BCG PPE37 protein or immediately adjacent to the deletion, suggesting that the BCG PPE37 protein might be processed differently. TargetP, which is designed for predicting the subcellular location of eukaryotic proteins, in contrast to the other algorithms, does predict a signal peptide for PPE37; however, the signal peptide length is predicted to be 15 aa (not 40 aa, as reported in reference 31), and the reliability class score for the prediction was very poor. Moreover, as TargetP is intended for eukaryotic proteins, its relevance to PPE37 is questionable.
To confirm that ppe37 alone is responsible for the efficient HIA phenotype, we cloned M. tuberculosis ppe37 along with its promoter region (131 bp of sequence upstream of the start codon) into pMycInt(apr)-sacB-FOS3 and transformed the plasmid into BCG ΔmbtB ΔpanCD (the same strain used for fosmid complementation [see above]) and BCG ΔmbtB; we included BCG ΔmbtB after we observed that the panCD mutation had a significant (and unexpected) impact on sensitivity to manganese protoporphyrin IX (MnPPIX) (see below). The resulting strains had an HIA phenotype identical to that of the original fosmid clones and the subregion E clone (Fig. 3A). Complementation of BCG with M. tuberculosis ppe37 dramatically increased the efficiency of HIA in broth resulting in a greater than a 100-fold decrease in the concentration of hemin allowing 50% of maximal growth (50% effective concentration [EC50]), with values of 14.1 versus 0.09 µM for BCG ΔmbtB and 31.4 versus 0.13 µM for BCG ΔmbtB ΔpanCD (Fig. 3B). Transformation of BCG ΔmbtB with an empty vector as a control caused a small, but statistically significant, increase in efficiency of HIA (EC50 of 14.1 versus 3.0 µM). In contrast, the two noncomplemented BCG ΔmbtB ΔpanCD strains grew somewhat more poorly than BCG ΔmbtB with hemin (Fig. 3A and B). Supplementation of the medium of these two strains with pantothenate is the likely cause for the increased EC50s as we found in a head-to-head comparison that supplementation of BCG ΔmbtB with pantothenate increases the hemin EC50 (data not shown).
FIG 3.
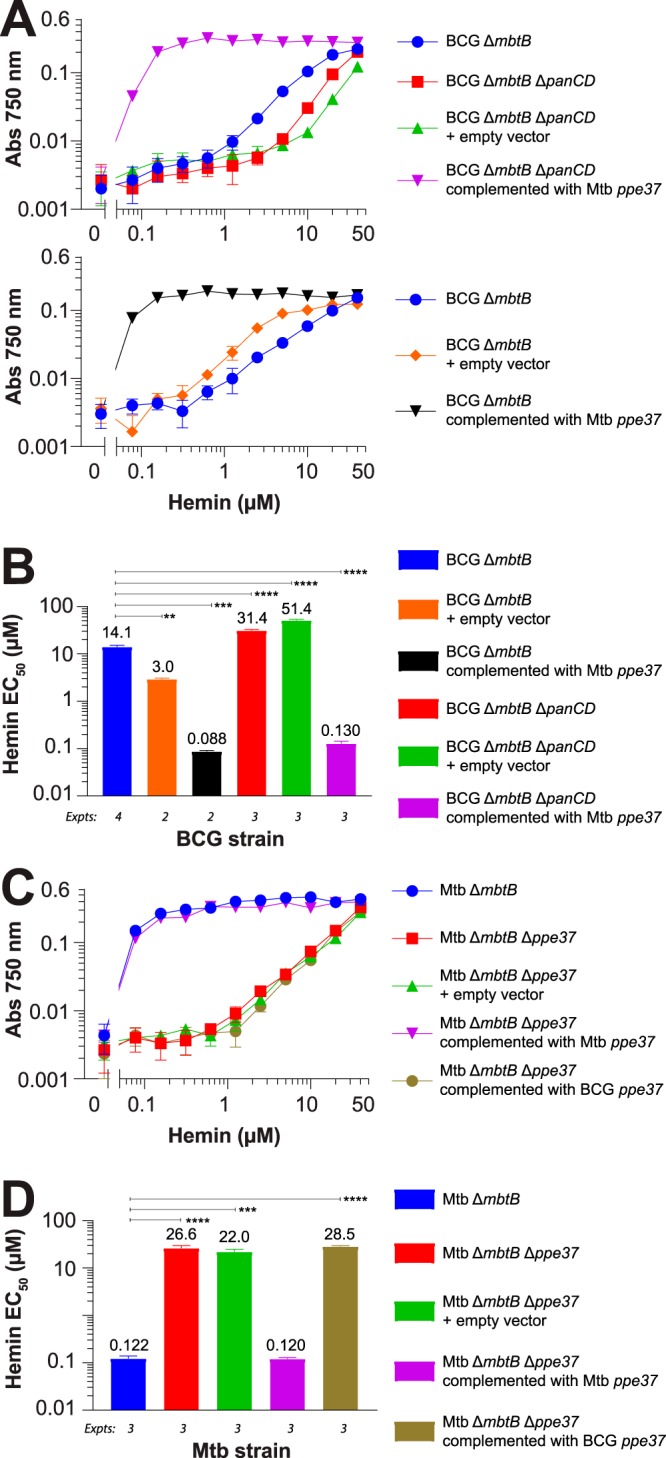
PPE37 is essential for efficient HIA in broth. Bacteria were grown in 7H9–OADC–0.01% TLX medium with various concentrations of hemin (0.078 to 40 µM), 10 ng/ml mycobactin J, or no additional supplement in 96-well plates. Cultures were inoculated to an initial calculated A750 of 0.00025, and growth was determined by measuring absorbance at 750 nm at 14 days. Bacterial density measurements with the 96-well plate reader are 4-fold less than measurements made with a cuvette-based spectrophotometer. (A and C) One representative experiment (out of 2 to 4) is shown for BCG ΔmbtB and BCG ΔmbtB ΔpanCD strains and M. tuberculosis ΔmbtB strains, as indicated. Data are the means ± standard errors from triplicate wells for each condition. In many instances, the error bars are smaller than the symbols. (B and D) The EC50 for hemin (concentration allowing 50% of maximal growth) was calculated by fitting the data from panels A and C to a four-parameter concentration-versus-response curve using nonlinear regression (Prism, version 7) and is shown for BCG ΔmbtB and BCG ΔmbtB ΔpanCD strains and M. tuberculosis ΔmbtB strains, as indicated. Data are the means ± standard errors from 2 to 4 independent experiments for each strain (number of experiments indicated below the bars). Adjusted P values are as follows: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (by one-way ANOVA with Tukey’s multiple-comparison test).
PPE37 is essential for efficient HIA by M. tuberculosis.
To determine the role of ppe37 in M. tuberculosis, we constructed M. tuberculosis ΔmbtB::Km Δppe37::hyg, replacing 1.2 kb of the ppe37 gene (encoding amino acids 46 through 445) with a hygromycin resistance cassette via allelic exchange (Fig. S1). Remarkably, we found that M. tuberculosis ΔmbtB Δppe37 has an HIA phenotype nearly identical to that of BCG ΔmbtB (Fig. 3). As was the case for BCG ΔmbtB, complementation of M. tuberculosis ΔmbtB Δppe37 with M. tuberculosis ppe37 completely restored an efficient HIA phenotype, while complementation with BCG ppe37 had no effect (Fig. 3C and D). These experiments measured growth at 14 days, a time at which growth has plateaued at the lowest concentration of hemin tested (0.078 µM), presumably due to consumption of hemin (Fig. 1B), and therefore the EC50s calculated on day 14 may be underestimated. Thus, for three strains that grow efficiently with hemin, we measured growth with hemin at 7 days (so that less hemin would be consumed) and expanded the studies also to measure the EC50 for mycobactin J under the same conditions (Fig. S2). The EC50s for hemin at 7 days (61 to 90 nM) compared with the levels at 14 days were unchanged for BCG ΔmbtB complemented with M. tuberculosis ppe37 and were up to 2-fold less for the M. tuberculosis strains. Differences in EC50s among the three strains at 7 days were not statistically significant. Even though growth of M. tuberculosis with hemin is very efficient, we found that growth with mycobactin J in iron-rich 7H9 medium (∼130 μM Fe3+) is two orders of magnitude more efficient (mycobactin J EC50 of 0.3 to 0.6 nM [0.25 to 0.55 ng/ml] versus 61 to 90 nM for the hemin EC50). These results are very similar to those obtained by Jones et al. comparing growth with exomycobactin (also called exochelin or carboxymycobactin [12, 32]) instead of mycobactin J to growth with hemin (33). In contrast to hemin, which is degraded inside the bacterium to release iron, exomycobactin can shuttle iron from the medium to the bacterium and then be recycled (33), which may account in part for the greater efficiency of exomycobactin. Detergent-solubilized mycobactin J, as used here, seems to function similarly to exomycobactin, but it is not known whether mycobactin J is recycled.
There is very little free heme/hemin in nature; it is almost always bound to hemoglobin, hemopexin, albumin, or other proteins or lipids. Our growth medium contains 5 mg/ml bovine serum albumin (75 µM) (from the 10% OADC supplement), which should complex all of the added hemin. However, as hemopexin has a much stronger affinity for hemin than albumin, M. tuberculosis in vivo may encounter hemin bound to hemopexin. Therefore, we examined whether M. tuberculosis could use hemin bound to hemopexin, and we found that it could use it just as efficiently as hemin bound to albumin (Fig. S3).
To define better the cause of the apparently nonfunctional BCG ppe37, we constructed derivatives of M. tuberculosis ppe37 encoding proteins with deletions of 3, 6, or 9 amino acids at positions 31 to 39 and transformed these into BCG ΔmbtB ΔpanCD and M. tuberculosis ΔmbtB Δppe37. None of the plasmids encoding deletions in this region were capable of complementing either strain (Table 1, Experiment 1).
TABLE 1.
Complementation of BCG ΔmbtB ΔpanCD and M. tuberculosis ΔmbtB Δppe37 with plasmids encoding PPE37 deletion mutants and variants
| Expt no. and plasmid | Variation or deletiona | Growth with the indicated supplement(s) (CFU)b
|
|||
|---|---|---|---|---|---|
| BCG ΔmbtB ΔpanCD |
M. tuberculosis ΔmbtB Δppe37 |
||||
| Myc + Pan | Hm + Pan | Myc | Hm | ||
| Expt 1 | |||||
| pMycInt(apr)-FOS3-Mtb PPE37 | None | 40 | 37 | 22 | 21 |
| pMycInt(apr)-Mtb PPE37 | None | >200c | >300 | 42 | 66 |
| pMycInt(apr)-Mtb PPE37Δ31–39 | Deletion of nt encoding aa 31 to 39 | >200 | 0 | 104 | 0 |
| pMycInt(apr)-Mtb PPE37Δ31–33 | Deletion of nt encoding aa 31 to 33 | >300 | 0 | 32 | 0 |
| pMycInt(apr)-Mtb PPE37Δ31–36 | Deletion of nt encoding aa 31 to 36 | >300 | 0 | 116 | 0 |
| pMycInt(apr)-Mtb PPE37Δ37–39 | Deletion of nt encoding aa 37 to 39 | >300 | 0 | 121 | 0 |
| pMycInt(apr)-Mtb PPE37Δ34–39 | Deletion of nt encoding aa 34 to 39 | >300 | 0 | 96 | 0 |
| pMycInt(apr)-Mtb PPE37Δ34–36 | Deletion of nt encoding aa 34 to 36 | >300 | 0 | 66 | 0 |
| Expt 2 | |||||
| pMycInt(apr)-Mtb PPE37 | None | 300 | 208 | ND | ND |
| pMycInt(apr)-Mtb HN878 PPE37 | Frameshift mutation C481del (same as M. tuberculosis 49-02) | 342 | 0 | ND | ND |
| pMycInt(apr)-Mtb EAI 91_0079 PPE37 | C1218T plus frameshift mutation G1219del (same as M. tuberculosis 26105) | 271 | 0 | ND | ND |
| pMycInt(apr)-Mtb IO T17X PPE37 | None | 310 | 272 | ND | ND |
| pMycInt(apr)-Mtb CDC1551 PPE37 | Large deletion of ppe37 gene | 420 | 0 | ND | ND |
| Expt 3 | |||||
| pMycInt(apr)-Mtb PPE37 | None | 390 | 244 | ND | ND |
| pMycInt(apr)-Mtb IO T17X PPE37 | None | 474 | 328 | ND | ND |
| pMycInt(apr)-Mtb IO T17X PPE37 (GT1016-7del) | GT1016-7del (same as M. tuberculosis F11) | 370 | 0 | ND | ND |
Compared with the M. tuberculosis Erdman ppe37 sequence (see Fig. S6 and Table S2 in the supplemental material).
Supplements added to 7H10 plates are as follows: Myc, 50 ng/ml mycobactin J; Hm, 50 µM hemin; Pan, 50 µg/ml pantothenate. ND, not done.
For the BCG plates in experiment 1 with a large number of colonies, the colonies had begun to coalesce, and so only an estimate was possible.
To determine if the mbtB deletion had any effect on the HIA phenotype, we constructed M. tuberculosis Δppe37 by restoring the mbtB locus of M. tuberculosis ΔmbtB Δppe37. Iron-depleted medium is required for studies examining HIA in strains producing siderophores to avoid growth due to SMIA. However, it is technically very challenging to produce mycobacterial growth medium that is sufficiently depleted of iron to restrict growth of siderophore-producing strains completely as even concentrations significantly less than 1 µM iron support robust growth. For example, in this study, we found that as little as 0.078 µM hemin provides sufficient iron for significant growth of M. tuberculosis (Fig. 1 and 3). Thus, we first determined the appropriate concentration of iron chelator to add to our iron-depleted medium to prevent growth using various doses of either deferoxamine, as used by Kurthkoti et al. (34), or bipyridyl (also called dipyridyl), as used by Mitra et al. (23). Consistent with its ability to chelate ferric iron (the same as exomycobactin/mycobactin) and its low cell permeability, deferoxamine at ≥50 µM strongly inhibited wild-type M. tuberculosis Erdman growth due to SMIA from trace iron in the medium. However, even at a deferoxamine concentration of 400 µM, M. tuberculosis growth via HIA with 0.2 µM hemin occurred (data not shown). In contrast to results with deferoxamine, a significantly higher concentration of bipyridyl (200 µM) was required to completely inhibit M. tuberculosis growth, and growth inhibition was not strictly due to inhibiting SMIA as no growth was obtained with 0.2 µM hemin in the presence of 200 µM bipyridyl. That bipyridyl is a ferrous iron chelator (exomycobactin/mycobactin have low affinity for ferrous iron) and also membrane permeant likely explain its lack of specificity for SMIA.
Consequently, to bind trace iron and inhibit SMIA, we included 200 µM deferoxamine in our iron-depleted medium for studies of HIA in mycobacterial strains producing siderophores. Doing so, we found that M. tuberculosis Δppe37 is highly attenuated in HIA, with a phenotype similar to that of M. tuberculosis ΔmbtB Δppe37 (Fig. S4); thus, the loss of siderophores due to the deletion of mbtB has no apparent effect on the HIA phenotype.
PPE37 has minimal effect on inhibition by MnPPIX.
The heme mimetics, GaPPIX and MnPPIX, are non-iron metalloporphyrins in which Ga and Mn, respectively, replace Fe in the protoporphyrin ring and have been demonstrated to have similar MICs against both Mycobacterium smegmatis and BCG (GaPPIX is 2-fold more potent than MnPPIX) (35). GaPPIX and MnPPIX are inhibitory to many different bacteria and are believed to be taken up by heme uptake systems although it has also been proposed that the targets of these molecules may be localized to the bacterial surface, and thus a heme uptake system may not be required for them to exhibit inhibitory activity (35). The inhibitory activities of both GaPPIX and MnPPIX are likely due to their acting as metabolic poisons of bacterial respiration by inserting into the heme binding site of cytochromes and generating reactive oxygen species. As expression of a functional PPE37 has such a dramatic effect on HIA, we sought to determine if PPE37 is involved in MnPPIX inhibition (Fig. 4). In the BCG ΔmbtB strain, complementation with M. tuberculosis ppe37 resulted in a reduced MnPPIX 95% inhibitory concentration (IC95), with a value of 6.2 versus 18.9 µM (P < 0.05). However, this result was not significantly different from that with BCG ΔmbtB transformed with an empty vector (IC95 of 8.9 µM). Interestingly, all three BCG ΔmbtB ΔpanCD strains (with no plasmid, with an empty vector, or complemented with M. tuberculosis ppe37) exhibited much greater sensitivity to MnPPIX than BCG ΔmbtB (IC95 of 2.1 to 2.9 µM versus 18.9 µM) and a steeper dose response; however, there was no significant difference among the BCG ΔmbtB ΔpanCD strains complemented or not complemented with M. tuberculosis PPE37. The cause for the greater MnPPIX sensitivity in BCG ΔmbtB ΔpanCD is unknown, but it is not simply due to the presence of pantothenate in the medium as supplementation of BCG ΔmbtB with pantothenate did not decrease the IC95 (data not shown). All five of the M. tuberculosis strains tested were more resistant to inhibition by MnPPIX than BCG ΔmbtB (IC95 of 31 to 54 µM versus 18.9 µM) although the differences did not reach statistical significance. Deletion of ppe37 from M. tuberculosis ΔmbtB had no significant impact on MnPPIX sensitivity. Thus, ppe37 appears to have little role in MnPPIX inhibition in both M. tuberculosis and BCG.
FIG 4.

PPE37 has little role in MnPPIX inhibition. Bacteria were grown in 7H9–OADC–0.01% TLX medium with 10 ng/ml mycobactin J and various concentrations of MnPPIX inhibitor (0.078 to 40 µM) or no inhibitor in 96-well plates. Cultures were inoculated to an initial calculated A750 of 0.00025, and growth was determined by measuring absorbance at 750 nm at 14 days. Bacterial density measurements with the 96-well plate reader are 4-fold less than measurements made with a cuvette-based spectrophotometer. (A and C) One representative experiment (out of 2 to 4) is shown for BCG ΔmbtB and BCG ΔmbtB ΔpanCD strains (A) and M. tuberculosis ΔmbtB strains, as indicated. Data were normalized to the amount of growth with no inhibitor and are the means ± standard errors from triplicate wells for each condition. In many instances, the error bars are smaller than the symbols. Since the logarithim of 0 is not defined, growth with no inhibitor was plotted at a concentration of 1 × 10−4 µM. (B and D) The IC95 for MnPPIX (concentration inhibiting growth by 95%) is shown for BCG ΔmbtB and BCG ΔmbtB ΔpanCD strains and M. tuberculosis ΔmbtB strains, as indicated. To calculate the IC95, data from panels A and C were fit to a log [inhibitor]-versus-response curve using nonlinear regression (Prism, version 7). Data are the means ± standard errors from 2 to 4 independent experiments for each strain (number of experiments indicated below the bars). Adjusted P values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by one-way ANOVA with Tukey’s multiple-comparison test).
As other investigators have utilized GaPPIX rather than MnPPIX in HIA studies (23), we sought to compare the inhibitory activities of the two non-iron metalloporphyrins. Dose-response curves for GaPPIX and MnPPIX indicate that M. tuberculosis is only modestly more sensitive to GaPPIX (Fig. S5), as previously observed for M. smegmatis and BCG by Stojiljkovic et al. (35). We tested a total of seven different M. tuberculosis strains and mutants for sensitivity to GaPPIX and MnPPIX, as shown in Fig. S5, and found that the GaPPIX IC95 ranged from 1.4-fold to 3.6-fold less (median, 2.0-fold) than the MnPPIX IC95 (data not shown).
Genetic variation of ppe37 in M. tuberculosis.
Recently, Ahmad et al. reported significant variability in PPE37 from an analysis of 28 M. tuberculosis strains with completed genomes and presented a protein alignment of PPE37 proteins from three representative strains (H37Rv, F11, and 49-02) that demonstrated that the F11 sequence is identical to that of H37Rv PPE37 on the N terminus but has an altered C terminus and that the 49-02 sequence is identical to that of H37Rv PPE37 on the C terminus but has an altered N terminus (31). However, further explanation of the variability was not discussed. Therefore, to further investigate the genetic variability of ppe37 in M. tuberculosis, we did a blastn search using the nucleotide sequence of the 5.4-kb hisE-ppe37-metH region from the M. tuberculosis H37Rv reference genome (identical to the sequence from M. tuberculosis Erdman, the strain used in our study) and analyzed 146 sequences from M. tuberculosis strains with completely sequenced genomes for sequence differences. Our analysis revealed that the majority of ppe37 sequences differ from the reference H37Rv ppe37 sequence by frameshift mutations which result in altered PPE37 proteins of various lengths (Fig. S6 and Table S2). Fifty-six out of 146 (38%) ppe37 genes encode full-length PPE37 proteins (473 amino acids) identical to the H37Rv reference sequence or with one single nucleotide polymorphism (SNP). The largest set of ppe37 variants (52 of 146, or 36%), which includes the gene from strain F11, have a 2-nucleotide (nt) deletion at position 1016 to 1017, which results in a 496-amino-acid protein identical to the first 339 amino acids of the H37Rv PPE37 protein (∼72% of the full-length protein) with an altered C terminus. The second largest set of ppe37 variants (36 of 146, or 25%), which includes the gene from strain 49-02 (Beijing lineage [36]), has a deletion of a single nucleotide at position 481, which results in a protein identical to the first 169 amino acids of the H37Rv PPE37 protein (∼36% of the full-length protein), and the frameshift results in an early stop after amino acid 176. Alternatively, there is a GTG triplet located 11 nucleotides downstream of the normal ppe37 GTG initiation codon, which, if used as an alternative start codon, will result in a 468-amino-acid protein with the 305 amino acids of the C terminus identical to the sequence of the H37Rv PPE37 protein but with an N-terminal sequence with no meaningful homology to the H37Rv N-terminal sequence. A single strain, M. tuberculosis 26105, has a frameshift different from that of the other variants which results in a 410-amino-acid protein (identical to the first 406 amino acids of H37Rv PPE37) and one strain, M. tuberculosis CDC1551, has a deletion of the majority of the ppe37 gene, as previously noted (30).
To determine if these M. tuberculosis PPE37 variants are functional in HIA, we transformed plasmids encoding four different PPE37 variants into BCG ΔmbtB ΔpanCD. Three of the four variants were obtained by amplifying the ppe37 genes from genomic DNA from M. tuberculosis HN878 (same mutation as M. tuberculosis 49-02), M. tuberculosis EAI 91_0079 (same mutation as M. tuberculosis 26105), and M. tuberculosis CDC1551. The fourth variant was obtained by introducing a two-nucleotide deletion at position 1016 to 1017 (M. tuberculosis F11 variant) into a ppe37 sequence identical to that of H37Rv (from M. tuberculosis IO T17X). None of the plasmids encoding PPE37 variants were capable of complementing the HIA defect of BCG (Table 1, Experiments 2 and 3). Although this demonstrates that the PPE37 variants are nonfunctional in BCG, it was possible that compensating mutations in other genes may still allow strains with these HIA-defective ppe37 genes to grow efficiently with hemin as an iron source. We therefore sought to determine whether M. tuberculosis HN878 and M. tuberculosis CDC1551 are capable of efficient HIA. As M. tuberculosis HN878 and M. tuberculosis CDC1551 produce siderophores, iron-depleted medium is required for studies examining HIA with these strains to avoid growth due to SMIA. Using iron-depleted medium containing 200 µM deferoxamine to bind trace iron, we were able to demonstrate that M. tuberculosis HN878 and M. tuberculosis CDC1551 are both highly deficient in HIA (Fig. S7A), similar to results with our M. tuberculosis ΔmbtB Δppe37 and M. tuberculosis Δppe37 mutants in the Erdman background (Fig. S4B), although M. tuberculosis CDC1551 appears to grow somewhat better than the others at high concentrations of hemin. Similarly, virulent M. bovis, which has the same PPE37 sequence as BCG (see above), is also highly attenuated in HIA in iron-depleted medium (Fig. S7B).
Role of pe22-ppe36 in HIA.
Recently, Mitra et al. identified ppe36, ppe62, and Rv0265c as playing a role in HIA by M. tuberculosis by screening a transposon mutant library of an avirulent M. tuberculosis H37Rv mutant (mc26206, a ΔleuCD ΔpanCD strain) for resistance to the toxic heme mimetic, GaPPIX (23). The authors found that ppe36 is essential for heme utilization by M. tuberculosis. Thus, we constructed an M. tuberculosis ΔmbtB::Km Δppe36::hyg mutant to compare the role of ppe36 and ppe37 in HIA (Fig. S8). We also constructed two additional mutants with a deletion of pe22 (adjacent to ppe36) or pe22-ppe36. In contrast to Mitra et al., we found that deletion of ppe36 had no effect on HIA by M. tuberculosis, and deletion of ppe36 did not affect MnPPIX inhibition either (Fig. S8F). To determine if the mbtB deletion had any effect on the HIA phenotype, we construted M. tuberculosis Δppe36 by restoring the mbtB locus of M. tuberculosis ΔmbtB Δppe36. We found that M. tuberculosis Δppe36 has no defect in HIA, similar to M. tuberculosis ΔmbtB Δppe36 (Fig. S7A).
Effect of detergent on HIA and MnPPIX inhibition.
Although the detergent tyloxapol (TLX) is often used in broth cultures of mycobacteria to disperse the bacteria, the use of Tween 80 is much more common. Thus, we compared the HIA phenotypes of our BCG and M. tuberculosis strains in medium containing the usual concentration of Tween 80 (0.05%) (Fig. S9) with the HIA phenotypes when the strains are grown in our standard growth medium, which contains 0.01% TLX (Fig. 3). We found that Tween 80 had a very strong effect on HIA. Strains lacking a functional PPE37 that grew poorly with hemin in the presence of 0.01% TLX (e.g., BCG ΔmbtB and M. tuberculosis ΔmbtB Δppe37) had enhanced growth in the presence of 0.05% Tween 80 (hemin EC50 decreased by 3- to 8-fold in most cases). In contrast, strains with a functional PPE37 that grew well with hemin in the presence of 0.01% TLX grew less well in the presence of 0.05% Tween 80 (hemin EC50 increased by 8- to 14-fold). These two opposing effects of Tween 80 detergent were such that complementation of BCG ΔmbtB and M. tuberculosis ΔmbtB Δppe37 with a functional M. tuberculosis PPE37 resulted in only a 4-fold improvement of the hemin EC50 in the presence of 0.05% Tween 80 whereas there was a >100-fold improvement of the hemin EC50 in the presence of 0.01% TLX. Because of the profound effect of Tween 80 on the HIA phenotype, we examined the phenotype of selected BCG and M. tuberculosis strains on agar plates in the absence of all detergent (with the exception of a small amount of TLX carried over from the inoculating cultures). On agar plates, strains with a functional PPE37 grew with hemin concentrations as low as 1 µM (the lowest concentration tested), while strains that lacked a functional PPE37 had barely visible growth even with 50 µM hemin (Fig. 5). Thus, the phenotype on plates is much more similar to the phenotype in broth cultures with 0.01% TLX than it is to that in broth cultures with 0.05% Tween 80, and the phenotypic differences between the strains with and without a functional PPE37 may be even greater on agar plates.
FIG 5.

PPE37 is essential for HIA on agar plates. Bacterial cultures were normalized to an A550 of 0.1 (∼2 × 106 CFU/ml), and 10-µl aliquots of undiluted bacteria (100) or 10−1 and 10−2 dilutions, as indicated to the left of each photograph, were spotted on 7H10 agar plates with no additional supplement, various concentrations of hemin (1, 5, 10, or 50 μM), or 50 ng/ml mycobactin J, as indicated to the right of each photograph. Plates were photographed at 14 days.
We also tested the effect of Tween 80 on MnPPIX inhibition. All BCG and M. tuberculosis strains were more sensitive to MnPPIX when grown in the presence of 0.05% Tween 80 than with 0.01% TLX; in many cases a >10-fold reduction in IC95 was observed (Fig. S9). The dose response for MnPPIX in Tween 80 was much steeper than that in TLX, and the IC95 spanned a narrow range for all strains tested (1.2 to 2.2 µM). As with the experiments done with TLX (Fig. 4), no role for ppe37 in MnPPIX inhibition was found.
As these differences between detergents were substantial and may have implications for mycobacterial physiology distinct from HIA, we sought to examine further the influence of detergent on HIA and MnPPIX inhibition by measuring the growth of BCG ΔmbtB, M. tuberculosis ΔmbtB, and their parental strains (BCG and M. tuberculosis) in medium containing either TLX or Tween 80 in concentrations ranging from 0.00078% to 0.2% detergent and supplemented with selected concentrations of mycobactin J, MnPPIX, or hemin (Fig. 6). For the BCG and M. tuberculosis parental strains, TLX up to the highest concentration of 0.2% had a relatively minor influence on growth under all conditions, and sensitivity to MnPPIX was not significantly increased by increasing the TLX concentration (Fig. 6A). In contrast to the minor influence of TLX, concentrations of Tween 80 of ≥0.1% had a noticeable growth-inhibitory effect on both BCG and M. tuberculosis (Fig. 6B). Approximately 4- to 8-fold less Tween 80 was needed to inhibit HIA by M. tuberculosis ΔmbtB (significant inhibition starting at 0.025%) compared with the amount of Tween 80 required for general growth inhibition. TLX was also capable of significantly inhibiting HIA but only at the highest concentration tested (0.2%, a concentration far greater than what is typically used to disperse mycobacteria). MnPPIX inhibition also displayed a Tween 80 dose response, with concentrations as low as 0.003% increasing inhibition; BCG ΔmbtB was the most sensitive of the four strains tested. Similar to MnPPIX inhibition, growth of BCG ΔmbtB with 5 µM hemin was enhanced with concentrations of Tween 80 as low as 0.003%. Thus, at low concentrations of detergent (less than or equal to what is typically used), Tween 80 has notable effects on both inhibition with MnPPIX and growth with hemin, whereas TLX has almost no effect over a very wide range of concentrations.
FIG 6.

Tween 80 has a significant impact on HIA and MnPPIX inhibition, whereas tyloxapol does not. Bacteria were grown in 7H9-OADC medium containing various concentrations of tyloxapol (A) or Tween 80 (B) ranging from 0.00078% to 0.2% in 96-well plates. The media were either not supplemented or supplemented with 10 ng/ml mycobactin J, 10 ng/ml mycobactin J plus 2.5 µM MnPPIX (Myc. + MnPPIX), 0.625 µM hemin, or 5 µM hemin, as indicated at the top of each graph. Cultures were inoculated to an initial calculated A750 of 0.00025, and growth was determined by measuring absorbance at 750 nm at 14 days. Data are the means ± standard errors from triplicate wells for each condition. In many instances, the error bars are smaller than the symbols. Interestingly, 2.5 µM MnPPIX in the presence of 0.1% Tween 80 was not inhibitory to any of the four strains. The reason for this is unknown, but the effect was reproducible across independent experiments.
DISCUSSION
We have shown that ppe37 is essential for efficient HIA in M. tuberculosis and that a defective ppe37 in BCG, encoding a protein with a nine-amino-acid deletion, prevents efficient HIA. The small deletion found in the BCG ppe37 gene occurs in all BCG strains as well as in virulent M. bovis. Deletion of ppe37 from M. tuberculosis ΔmbtB results in a strain with an HIA phenotype nearly identical to that of BCG ΔmbtB. Although the defect in HIA is severe for both BCG and the M. tuberculosis ppe37 mutant, far greater than the defects caused by deletion of Rv0203, mmpL11 (15), or mhuD (19), it is not complete. In broth culture utilizing a low concentration of tyloxapol detergent (0.01%), growth is possible using high concentrations of hemin (EC50 of ≥10 µM). However, on agar plates, in the absence of detergent, the HIA defect of BCG and the M. tuberculosis ppe37 mutant appears to be even more severe (barely perceptible growth even with 50 µM hemin). In contrast, strains with a functional ppe37 (M. tuberculosis and BCG complemented with M. tuberculosis ppe37) can utilize low concentrations of hemin very efficiently in broth (EC50 of < 0.1 µM) and on agar plates.
In the host, to prevent tissue damage arising from oxygen radical reactions catalyzed by free heme, extracellular hemoglobin and heme are tightly complexed with haptoglobin and hemopexin, respectively (37). Albumin, the most abundant protein in plasma, is also involved in scavenging free heme, but due to its lower affinity for heme, albumin-bound heme is quickly transferred to hemopexin under normal conditions. However, what is perhaps more important for an intracellular pathogen, such as M. tuberculosis, than the state of the extracellular heme pool is the availability of heme intracellularly and, in particular, in the phagosome. The intracellular labile heme pool is difficult to measure but has been calculated to be ∼600 nM in IMR90 human lung fibroblasts and ∼400 nM in HEK293 cells (38). M. tuberculosis utilizes hemin in broth (bound by albumin, as a large excess of albumin is present in the OADC supplement, or complexed with hemopexin) with very high efficiency (EC50 of <100 nM), so if M. tuberculosis can gain access to the aforementioned concentrations of labile heme in vivo, these concentrations would appear to be in the range that would support its growth. Considering the necrosis caused by intracellular growth of M. tuberculosis in host cells and the resulting tissue damage, it seems likely that M. tuberculosis would have access to the intracellular labile heme pool at least during some stages of infection.
The ppe37 gene is a member of the PE/PPE gene families which encompass 168 genes in M. tuberculosis H37Rv, or 7% of the total coding potential of the genome (39, 40). These mycobacterium-specific genes are largely found in slow-growing mycobacteria, with fast-growing mycobacteria possessing far fewer PE/PPE genes (e.g., M. smegmatis has 4 genes) (30). The genes are often found in pe-ppe pairs that appear to be coexpressed (40) although individual genes are also present in the genome, as is the case with ppe37. There are 69 ppe genes present in the M. tuberculosis H37Rv genome which encode proteins with a relatively conserved N-terminal sequence of ∼180 amino acids with a Pro-Pro-Glu (PPE) motif and a variable C-terminal sequence (39). PPE37 is further classified in the 10-member PPE-PPW subfamily (sublineage II) which possess highly conserved Gly-Phe-X-Gly-Thr and Pro-X-X-Pro-X-X-Trp sequence motifs in the C-terminal region (30, 39). The closest paralogs encoded in the M. tuberculosis genome are PPE2, PPE46, PPE1, and PPE11 (∼60% identity and ∼70% similarity for the N-terminal 180 amino acids and much less similarity over the rest of the protein sequences). While knowledge of the function of PE/PPE genes has continued to expand, many PE/PPE genes still have unknown functions (40). Many PE/PPE proteins have been localized to the mycobacterial membrane, cell wall, and/or culture filtrate; a few proteins have classical Sec pathway signal peptides, and others have been shown to be secreted by the type VII secretion system (39).
M. tuberculosis ppe37 (alternative gene name irg2) was shown many years ago to be part of the IdeR regulon and induced by low-iron conditions (28, 41), but otherwise minimal specific information regarding the function of ppe37 has been published. In addition to low iron, exposure to nitric oxide, hydrogen peroxide, and the antibiotic ethambutol also increase expression of ppe37 (42). Significant induction of ppe37 expression occurs during infection of murine bone marrow-derived macrophages as well as in the mouse lung (measured at 3 and 8 weeks postchallenge) (43), suggesting an important role for ppe37 in vivo. Two previous studies have attempted to determine the function of PPE37. In one study, expression of M. tuberculosis PPE37 by M. smegmatis resulted in a lower level of proinflammatory cytokines secreted by infected macrophages, but the mechanism by which expression of recombinant PPE37 interfered with the proinflammatory cytokine response was not determined (44). Recently, a second study examining the function of PPE37 demonstrated that PPE37 appears to be exported as it is predominantly located in the cell membrane fraction of M. tuberculosis grown under low-iron conditions (31). The authors also ascribed different host-modulating functions to the N-terminal and C-terminal segments of PPE37 when the segments are transfected into the human monocytic THP-1 cell line. It will be of interest to see how these host-modulating properties function in the context of an M. tuberculosis infection. In both of these prior studies, there is no obvious link to the role of PPE37 in HIA that we describe here. As the deletion of residues 31 to 39 in BCG PPE37 results in a nonfunctional protein for HIA, it is quite possible that PPE37 possesses an N-terminal signal sequence (albeit a nonclassical one that is below the SignalP cutoff) and that the loss of function is due to the BCG protein not being exported properly due to modification of the signal peptide. We have further shown that deletion of as few as three amino acids from M. tuberculosis PPE37 in the region spanning amino acids 31 to 39 completely prevents complementation of defective HIA in both BCG ΔmbtB ΔpanCD and M. tuberculosis ΔmbtB Δppe37, demonstrating that this region is critical to HIA.
The ppe37 genes of M. tuberculosis, virulent M. bovis, and BCG are all transcribed at relatively low levels (when not induced by low iron), but PPE37 is often not detected by proteomics methods even with high coverage of the theoretical proteome, likely due to low expression and the general difficulty in detecting acidic PE/PPE proteins (45–49). However, one study which achieved 87% coverage of the BCG proteome by separately analyzing cell wall, plasma membrane, and cytoplasm fractions identified PPE37, indicating that the protein is expressed by BCG (50). Further studies will be required to determine the relative expression level and subcellular localization of BCG/M. bovis PPE37 and nonfunctional M. tuberculosis PPE37 variants compared with that of the functional M. tuberculosis PPE37.
Our study clearly demonstrates that ppe37 plays a major role in HIA, but its exact function in HIA is not yet known. A leading possibility, suggested by its likely location in the cell membrane, is that PPE37 plays a role in heme import from the periplasm to the cytosol. In this regard, the fact that ppe37 appears to play little role in inhibition by MnPPIX, a toxic heme mimetic, in both M. tuberculosis and BCG does not preclude PPE37 from functioning in heme import but may simply indicate that MnPPIX does not need to reach the cytosol to exert its inhibitory effect. M. tuberculosis exports heme via CcsAB to the periplasm for insertion into apocytochrome c to form mature cytochrome c as part of the electron transport chain (51). If MnPPIX that passes through the mycobacterial outer membrane (52) can compete with heme in the periplasm for insertion into apocytochrome c, thereby poisoning respiration, there would be no need for it first to be imported to the cytosol and then exported to the periplasm by CcsAB in order to inhibit bacterial growth. The alternative to PPE37 being involved in heme uptake would be that it plays a role in degradation of heme to release iron. We previously demonstrated that deletion of mhuD, encoding a cytosolic heme-degrading enzyme, from M. tuberculosis results in moderate attenuation of HIA but only at a low concentration of hemin (0.2 µM) (19) and with nothing like the severe HIA defect observed when ppe37 is deleted. Thus, M. tuberculosis clearly has additional means of releasing iron from heme that do not require MhuD. However, the localization of PPE37 in the cell membrane fraction (31) and the likely presence of a signal peptide in PPE37 argue against a role for PPE37 in cytosolic degradation of heme although this possibility cannot be ruled out at present. Finally, deletion of ppe37 (or expression of a nonfunctional PPE37 by BCG) might have a general effect on cell wall permeability which impacts HIA in a nonspecific manner; i.e., it might make the cell wall less permeable to diffusion of heme. However, this possibility seems unlikely as deletion of ppe37 would have to make the bacteria less permeable to heme without altering permeability to MnPPIX (i.e., the MnPPIX IC95 was not affected by deletion of ppe37). Furthermore, BCG is not known to be generally less permeable to other small molecules than M. tuberculosis, as far as we are aware, and if a defective PPE37 decreases cell wall permeability in a nonspecific manner, it would be expected to affect many different small molecules, not just heme.
There are three major classes of ppe37 variants present in M. tuberculosis strains with completely sequenced genomes (and two minor classes with one sequence each), with the majority of sequences (61%) possessing frameshift mutations compared with the reference M. tuberculosis H37Rv genome (and M. tuberculosis Erdman, the strain used in our study, which is identical to H37Rv in this region). At first glance, the PPE37 protein from strains such as F11 might have a reasonable chance of functioning in HIA as a large portion of the protein (∼72%) is identical to the H37Rv PPE37. However, the altered C terminus of the strain F11 PPE37 removes the highly conserved sequence motif that places the H37Rv PPE37 protein in the PPE-PPW subfamily. The PPE37 protein from strain 26105, which encodes a protein of 410 amino acids (identical to the first 406 amino acids of H37Rv PPE37, 82% of the total protein length), also lacks the C-terminal PPW sequence motif. The PPE37 protein from strains such as 49-02 would seem to have little chance in functioning in HIA. If translated from the normal start codon, the strain 49-02 PPE37 is severely truncated, not even encompassing the entire, relatively conserved PPE domain. If an alternate start codon is used for translation, a large portion of the C terminus would be identical to that of the H37Rv PPE37 (∼64% of the total protein length), but the N terminus would be altered, and, thus, the protein would no longer contain a PPE domain. Our BCG complementation studies have conclusively shown that all of the M. tuberculosis PPE37 variants which possess a frameshift mutation or a large deletion compared with the reference M. tuberculosis H37Rv genome are nonfunctional in HIA. In addition, we have demonstrated that M. tuberculosis with a ppe37 frameshift mutation (HN878) or large deletion (CDC1551), as well as virulent M. bovis (same PPE37 variation as BCG), is severely defective in HIA, indicating that these strains do not have other proteins that can functionally substitute for PPE37. Maintenance of HIA-defective PPE37 variants in all M. bovis strains and a majority of M. tuberculosis strains may suggest that PPE37 serves an additional function apart from its role in HIA, but what function, if any, remains unknown.
While we have found an essential role for ppe37 in efficient HIA by M. tuberculosis, we did not find a role for ppe36 in HIA, in stark contrast to Mitra et al. (23). The reason for these very different results is not entirely known. However, there are several differences between our studies that may explain the disparate conclusions. We have performed our studies using mbtB mutants of M. tuberculosis Erdman that are deficient in SMIA, thereby allowing us to analyze HIA phenotypes in standard (iron-rich) 7H9 medium (i.e., in the absence of added mycobactin J); hence, the mbtB mutants cannot utilize the ferric ammonium citrate present in the medium and must rely on hemin as their sole iron source. Mitra et al. used an avirulent M. tuberculosis H37Rv mutant (mc26206, ΔleuCD ΔpanCD strain) for their studies. As this strain has an intact SMIA system, analysis of the HIA phenotype required growth in a minimal medium (Hartmans de Bont [HdB]) supplemented with 20 µM iron chelator (2,2′-dipyridyl, also known as bipyridyl) to prevent growth from trace iron in the medium and 3 or 4 generations of growth in iron-free medium to deplete bacterial iron storage. Under these conditions, growth of the parental strain is relatively poor in the presence of a high concentration of hemin (10 µM), taking ∼27 days to reach stationary phase (optical density [OD] of ∼1), whereas growth in the presence of 10 µM ammonium ferric citrate is much more robust, reaching stationary phase by 11 days and achieving a significantly higher OD (∼2.3). In contrast, we found that M. tuberculosis ΔmbtB grows nearly as well with low concentrations of hemin (<1 µM) as it does when the medium is supplemented with mycobactin J. The poor growth with 10 µM hemin evident in the study of Mitra et al. may be due to their use of a membrane-permeant ferrous iron chelator to inhibit SMIA; in our hands this inhibitor was not specific for SMIA as it also prevented HIA. Interestingly, the original GaPPIX-resistant ppe36 transposon mutant from the study of Mitra et al. (Tn2) had only a slight growth defect when grown with hemin, in contrast to the isogenic ppe36 mutant which did not grow with hemin, so perhaps an unrecognized secondary mutation occurred during construction of the isogenic ppe36 mutant. Although the majority of our studies utilized mbtB mutants, we also constructed an M. tuberculosis Erdman ppe37 mutant with an intact SMIA system and confirmed that the HIA phenotype we observed was solely due to deletion of ppe37 and not to the combined deletions of mbtB and ppe37. Similarly, we constructed an M. tuberculosis Erdman ppe36 mutant with an intact SMIA system and found no difference in HIA phenotype compared to that of M. tuberculosis Erdman mbtB ppe36.
Whether the difference in HIA phenotype for the ppe36 mutants in the two studies is due to the strain of M. tuberculosis (Erdman versus H37Rv), the growth conditions used, the additional ΔleuCD ΔpanCD mutations, or some other factor will require further studies. We found that our BCG ΔmbtB ΔpanCD mutant, unexpectedly, grew somewhat less well with hemin than BCG ΔmbtB yet was significantly more sensitive to MnPPIX than BCG ΔmbtB. What influence, if any, the leuCD and panCD mutations present in the M. tuberculosis mc26206 strain used by Mitra et al. have on the HIA phenotype of their ppe36 mutant is unknown.
Broth cultures of mycobacteria usually contain detergent, most commonly Tween 80 at 0.05%, to disperse the bacteria and facilitate optical density measurements and other microbiological manipulations. Without detergent, mycobacteria form large clumps of cells that complicate analysis. In our lab, we have favored 0.01% TLX over 0.05% Tween 80 for dispersing mycobacterial cultures as it provides good dispersion, comparable to that of Tween 80 at least macroscopically, but is superior to Tween 80 for experiments involving analysis of proteins in culture filtrates. Surprisingly, we found that these two detergents had very different effects on the HIA phenotype. Tween 80 significantly inhibited HIA by M. tuberculosis ΔmbtB (a strain expressing functional PPE37) at concentrations of ≥0.025%. One explanation for this inhibition may be that the mycobacterial cell wall is significantly altered by the detergent such that PPE37 is extracted into the culture medium and/or its interactions with other cell wall components are detrimentally altered. We found that TLX can also significantly inhibit HIA by M. tuberculosis ΔmbtB but only at 0.2%, the highest concentration tested (20-fold higher than the normal amount of TLX used to disperse mycobacteria and 8-fold higher than the amount of Tween 80 required to inhibit HIA). Tween 80 had the oppossite effect on strains without a functional PPE37, enhancing growth with hemin compared with TLX. This effect of Tween 80 was observed at concentrations as low as 0.003% and increased MnPPIX inhibition was also noted at this concentration of Tween 80. This suggests that Tween 80 at very low concentrations increases the permeability of the cell wall, allowing greater diffusion of hemin and MnPPIX and possibly many other small molecules. TLX had little effect in this regard. Overall, the HIA phenotype in broth culture with 0.01% TLX closely resembles the HIA phenotype on agar plates (without detergent), while 0.05% Tween 80 minimizes the phenotypic differences between strains with strong and weak HIA.
Although our findings suggest that PPE37 is involved in the uptake of heme and not its degradation to release iron, the exact role of PPE37 in HIA, including what interactions it may have with other proteins involved in HIA, remains to be elucidated. The role of ppe37 and HIA in M. tuberculosis virulence also awaits further study as well as the relative roles of HIA and SMIA. We have shown that virulent M. bovis and M. tuberculosis strains with PPE37 variants are defective in HIA, indicating that it may be advantageous under some circumstances to not possess a highly efficient HIA system. But why some M. tuberculosis strains maintain an efficient HIA system and some do not remains a question. Although SMIA has been shown to play an important role in M. tuberculosis virulence in several studies (53–55), in one study, an M. tuberculosis mbtB mutant, defective in mycobactin biosynthesis, showed little attenuation in C57BL/6 mice challenged by aerosol (56), suggesting that sufficient iron was acquired in vivo through an alternate pathway (e.g., HIA). With the mutants constructed here, we are now positioned to perform side-by-side comparisons of mutants defective in SMIA (M. tuberculosis mbtB), HIA (M. tuberculosis ppe37), or both systems (M. tuberculosis mbtB ppe37) under various conditions and in different animal models.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains used in this study are listed in Table S3 in the supplemental material. M. tuberculosis strains were grown on Middlebrook 7H10 agar (BD, Sparks, MD) containing 10% (vol/vol) OADC (BD) and 0.5% (vol/vol) glycerol or as unshaken cultures in Middlebrook 7H9 broth (BD) supplemented with 10% (vol/vol) OADC, 0.2% (vol/vol) glycerol, and 0.01% (wt/vol) tyloxapol (7H9-OADC-0.01% TLX) at 37°C in an atmosphere of 5% CO2–95% air, except where indicated in the text and figure legends. Hygromycin (50 µg/ml), apramycin (50 µg/ml), calcium d-pantothenate (50 µg/ml), and mycobactin J (0.05 or 0.1 µg/ml in plates and 0.01 µg/ml in broth, except where indicated in the text and figure legends) were included as appropriate. Ferric mycobactin J was obtained from Allied Monitor, Inc. (Fayette, MO), and was solubilized as reported previously (16). Mn(III) protoporphyrin IX chloride (MnPPIX) and Ga(III) protoporphyrin IX chloride (GaPPIX) were obtained from Frontier Scientific, Inc. (Logan, UT). The ferrous iron chelator, 2,2′-bipyridyl (synonym, 2,2′-bipyridine, 2,2′-dipyridine, or 2,2′-dipyridyl), and the ferric iron chelator deferoxamine mesylate ([DFO] synonym, desferrioxamine) were obtained from Sigma-Aldrich. Fatty-acid-free bovine serum albumin (BSA) used in M. bovis cultures was obtained from bioWORLD (Dublin, OH). Escherichia coli strains were grown on Luria-Bertani or YT agar and Luria-Bertani broth at 37˚C, except for E. coli DY380, which was grown at 32˚C. Ampicillin (100 µg/ml), hygromycin (250 µg/ml), kanamycin (50 µg/ml), and apramycin (50 µg/ml) were included as appropriate.
Iron-depleted media.
For studies examining HIA in M. tuberculosis strains producing siderophores, an iron-depleted minimal medium (MM-OADC-0.01% TLX) similar to our standard 7H9-OADC-0.01% TLX medium except lacking ferric ammonium citrate, l-glutamic acid, pyridoxine, and biotin, was prepared. Trace iron was removed from all medium components, except for the divalent cations, by stirring with Chelex-100 resin at 4°C overnight. A sodium citrate-divalent cation solution was then added to the Chelex-100-treated medium, and the solution was sterilized by filtration through a 0.2-µm-pore-size filter. The final, iron-depleted medium contained 25 mM phosphate (2.5 g/liter disodium phosphate plus 1 g/liter monopotassium phosphate), 5 mM ammonium sulfate, 300 µM sodium citrate, 200 µM magnesium sulfate, 5 µM calcium chloride, 5 µM zinc acetate, 5 µM copper sulfate, 10% (vol/vol) OADC, 0.2% (vol/vol) glycerol, and 0.01% (wt/vol) tyloxapol (pH 6.8). Medium prepared in this manner contained very low iron levels, estimated as ∼0.3 µM by ferrozine iron assays (data not shown), and supported significantly less growth of M. tuberculosis strains than the same medium supplemented with FeCl3. However, over multiple batches of medium, iron levels could not be reduced sufficiently to prevent growth completely. Therefore, various concentrations of deferoxamine (ferric iron chelator) or 2,2′-bipyridyl (ferrous iron chelator) were added to the medium, as indicated in the text and figure legends, to inhibit SMIA due to residual trace iron in the medium.
As M. bovis has an inactive pyruvate kinase (PykA) and cannot grow on glucose or glycerol as a carbon source (57–59), a modified iron-depleted medium was prepared for HIA studies of M. bovis. The iron-depleted minimal medium with albumin, MMA-pyruvate-0.01% TLX, was prepared as described above with the following changes: (i) OADC (which contains dextrose/glucose) was replaced with a fatty-acid-free BSA-NaCl solution (both components at the same concentration as in OADC), and (ii) glycerol was replaced with 40 mM sodium pyruvate.
Construction of BCG and M. tuberculosis mutants.
An unmarked BCG ΔmbtB mutant and an unmarked BCG ΔmbtB ΔpanCD double mutant were constructed in the BCG Tice parental strain via specialized transduction (60) as described below. The ΔmbtB and ΔpanCD mutations are essentially identical to previously described marked mutations (16) but without the antibiotic resistance markers. The ΔmbtB mutation is a 3.9-kb in-frame deletion that eliminates amino acids 51 to 1343 from the mbtB coding region. The ΔpanCD mutation is a 1,345-bp deletion that eliminates all of panC and panD. Allelic exchange substrates were constructed that included the final mutation (ΔmbtB or ΔpanCD), a hygromycin resistance (hyg) plus sacB cassette (hyg-sacB), and an internal (int) portion of the gene to be deleted (mbtBint or panCDint) and cloned into the allelic exchange vector phEX2 (15) (see Table S4 for a list of plasmids used in this study). The resulting plasmids (phEX2-ΔmbtB-hyg-sacB-mbtBint and phEX2-ΔpanCD-hyg-sacB-panCDint) were used to generate mycobacteriophage for performing allelic exchange. BCG clones that integrated the allelic exchange substrate into the chromosome were selected on 7H10 plates containing 50 µg/ml hygromycin and the appropriate supplements (mycobactin or mycobactin plus pantothenate) and confirmed to be sucrose sensitive (due to expression of sacB). In a second step, we briefly passaged hygromycin-resistant, sucrose-sensitive clones in the absence of hygromycin to allow recombination to occur between homologous regions flanking the hyg-sacB cassette (thus, eliminating the hyg-sacB cassette from the chromosome and leaving just the unmarked mutation) and then plated the clones on 7H10 plates containing 2% sucrose and appropriate supplements for counterselection. Correctly constructed mutants were identified by their phenotype (ΔmbtB strain, mycobactin auxotroph, Hygs sucrose sensitive; ΔpanCD strain, pantothenate auxotroph, Hygs sucrose resistant).
M. tuberculosis ΔmbtB (15), derived from M. tuberculosis Erdman, was used as the parental strain for construction of mutants with deletions of ppe37, ppe36, pe22, and pe22-ppe36 via specialized transduction (60), using a similar strategy for all, as described below. Upstream and downstream regions of 1.2 to 1.8 kb flanking the desired region marked for deletion were amplified by PCR and cloned by DNA assembly with two additional PCR products (a plasmid backbone and hyg) using commercially available DNA assembly reagents (Gibson Assembly Master Mix or NEBuilder HiFi DNA Assembly Master Mix, NEB) (61, 62) to generate intermediate allelic exchange substrate plasmids in which hyg is inserted between the upstream and downstream regions (i.e., plasmid backbone-upstream region-hyg-downstream region). The two additional PCR products used in the DNA assembly, a 2-kb plasmid backbone and a 1.1-kb hyg cassette, were amplified from pEX26, a derivative of pEX12A-glnA1 (15) in which the glnA1 fragment is replaced by hyg. After confirmation by restriction analysis and sequencing that the pEX26 derived plasmids were correct, they were digested with SwaI to obtain a linear allelic exchange substrate for recombination into the allelic exchange vector phEX3, a derivative of phEX2 in which the apramycin resistance gene has been removed, as previously described (15). The resulting plasmids were used to generate mycobacteriophage for performing allelic exchange. M. tuberculosis clones that integrated the allelic exchange substrate into the chromosome were selected on 7H10 plates containing 50 µg/ml hygromycin and mycobactin J. Correctly constructed mutants were identified by analysis of PCR products and their restriction digests (Fig. S1 and S8), as well as by DNA sequencing of the intact PCR products using primers annealing within hyg (hygFSP and hygRSP).
The mbtB locus of selected M. tuberculosis ΔmbtB strains was restored to wild type via specialized transduction using mycobacteriophage phEX3-mbtB, which contains a 7.1-kb region encompassing the entire mbtB gene along with 1.2 kb upstream and 1.7 kb downstream of mbtB. For the construction of phEX3-mbtB, a 7.1-kb mbtB PCR product was amplified from M. tuberculosis Erdman genomic DNA using primers mbtB-vec-FP and mbtB-vec-RP and cloned by DNA assembly with a 2-kb plasmid backbone from pEX26 (see above) to first generate the intermediate allelic exchange substrate plasmid, pEX-mbtB. After confirmation by restriction analysis and sequencing that pEX-mbtB was correct, phEX3-mbtB mycobacteriophage was prepared as described above for performing allelic exchange. Clones that integrated the allelic exchange substrate into the chromosome, thereby restoring the wild-type mbtB gene (and a functional SMIA system), were selected on 7H10 plates lacking mycobactin J.
Primers used in construction and confirmation of the mutants are listed in Table S1.
Construction of an E. coli-mycobacterium integrating shuttle fosmid vector, pMycInt(apr)-sacB-FOS3.
The commercially available E. coli fosmid vector pSMART FOS (Lucigen) allows the efficient cloning of large genomic DNA inserts (35 to 45 kb) and includes transcription terminators to improve insert stability. Also, while normally maintained as a single copy plasmid for stability, pSMART FOS has the advantage of being capable of amplification to medium copy number by inducing expression of TrfA in specialized E. coli strains to obtain high plasmid yields. To take advantage of these features for constructing mycobacterial genomic DNA fosmid libraries, we needed to make three modifications of pSMART FOS: (i) insertion of a stuffer fragment into the cloning site that can later be replaced with genomic DNA fragments to generate the library; (ii) replacement of the chloramphenicol resistance gene with an antibiotic resistance gene suitable for use in both E. coli and mycobacteria; and (iii) insertion of a mycobacterial genetic element that allows either replication of the fosmid as an extrachromosomal plasmid or allows integration into the chromosome. For the stuffer fragment, we inserted a sacB cassette flanked by PmeI restriction sites into the pSMART FOS cloning site to allow sucrose counterselection against clones taking up undigested vector. We originally chose to replace the chloramphenicol resistance gene of pSMART FOS with hyg as this is used successfully in many E. coli-mycobacterium shuttle plasmids. However, as this is a single-copy plasmid in E. coli (unless induced in specialized E. coli strains), the level of hygromycin resistance generated was barely above the natural hygromycin resistance of E. coli, rendering isolation of stable clones problematic. When we used an apramycin resistance cassette (apr) in place of hyg, high apramycin resistance was generated even when the plasmid was maintained as a single copy, thereby eliminating problems with plasmid stability. Our final plasmid also includes the attP-int region derived from pMC1s (25) which allows stable site-specific integration into the mycobacterial genome (63). In brief, the details of construction of the final E.coli-mycobacterium integrating shuttle fosmid vector, pMycInt(apr)-sacB-FOS3, are as follows: (i) the kanamycin resistance gene of pMC1s (a gift from Sabine Ehrt) was replaced with hyg by recombineering to generate pMC1s-hyg; (ii) a 3-kb apr-sacB cassette from pEX22 (a derivative of pEX12A-glnA1 in which the glnA1 fragment is replaced by a apr-sacB cassette) was isolated as an NsiI fragment, the ends blunted, and cloned into pSMART FOS (supplied by the company as a linearized, blunt fragment) generating pSMART FOS-apr-sacB; (iii) the chloramphenicol resistance gene of pSMART FOS-apr-sacB was replaced with attP-int-hyg from pMC1s-hyg by recombineering, generating pMycInt(hyg)-FOS; (iv) to replace hyg with apr (due to the difficulties with hygromycin resistance mentioned above), the apr-sacB stuffer fragment was first removed by digesting pMycInt(hyg)-FOS with PmeI and recircularizing the plasmid, generating pMycInt(hyg)-FOS2; (v) hyg of pMycInt(hyg)-FOS2 was then replaced with apr by recombineering to generate pMycInt(apr)-FOS2; and (vi) finally, a sacB cassette stuffer fragment was inserted into the PmeI site of pMycInt(apr)-FOS2, generating the final plasmid used for library construction, pMycInt(apr)-sacB-FOS3 (Fig. 2A). pSMART FOS-derived plasmids were maintained in E. coli Replicator FOS (which allows induction of the fosmid to medium copy number with arabinose) except for the steps requiring recombineering, which were done in E. coli DY380.
Preparation of high-molecular-weight M. tuberculosis genomic DNA.
High-molecular-weight genomic DNA was prepared from M. tuberculosis ΔmbtB and M. tuberculosis ΔmbtB ΔmhuD (the ΔmhuD deletion is marked with an apramycin resistance cassette) by two extractions of bacterial cell pellets (from 200 ml of culture) with 40 ml and 20 ml of CHCl3-MeOH (1:1) to remove lipids, followed by resuspension in 20 ml of phenol-CHCl3 (1:1) and extraction of nucleic acids into 20 ml of 10 mM Tris-1 mM EDTA ([TE] pH 8) buffer. Nucleic acids in the aqueous phase were precipitated with 0.1 volume of 3 M sodium acetate and 0.8 volume of isopropanol, washed with 70% ethanol, and redissolved in 0.5 ml of TE buffer with RNase to degrade RNA.
DNA suitable for construction of fosmid libraries in pMycInt(apr)-sacB-FOS3 (size-selected fragments of 35 to 45 kb, end repaired) was prepared as follows: (i) initial size selection on a 10% to 50% sucrose gradient (64); (ii) end repair of size-selected fragments with T4 polynucleotide kinase and T4 DNA polymerase (NEBNext End Repair kit) to obtain 5′-phosphorylated, blunt ends; and (iii) a second round of size selection on a 10% to 50% sucrose gradient. The first sucrose gradient was prepared by layering 5.6 ml of a 10% sucrose solution (20 mM Tris, 5 mM EDTA, 1 M NaCl, 10% [wt/vol] sucrose, pH 7.5) on top of 5.6 ml of a 50% sucrose solution (20 mM Tris, 5 mM EDTA, 1 M NaCl, 50% [wt/vol] sucrose, pH 7.5) in a 13.2-ml (14 by 89 mm) ultracentrifuge tube, and the gradient was formed by diffusion in the horizontal position (65). High-molecular-weight M. tuberculosis gDNA (0.2 ml) was mixed with 50 µl of 50% sucrose solution, incubated at 60°C for 15 min, and loaded on the top of the gradient. The gradient was centrifuged at 30,000 rpm (154,000 × g) in a Sorvall TH-641 rotor at 20°C for 18 h. Fractions of 500 µl were carefully removed from the top, diluted with two volumes of double-distilled H2O (ddH2O), and then precipitated with isopropanol prior to analysis on agarose gels. Fractions containing fragments of the appropriate size were combined, end repaired with T4 polynucleotide kinase and T4 DNA polymerase, and subjected to a second round of sucrose density size selection to improve the size specificity. The second sucrose gradient was similar to the first except that lower concentrations of NaCl (0.3 M) and EDTA (1 mM) were used to avoid needing to dilute the mixture with ddH2O prior to isopropanol precipitation.
Construction of fosmid libraries.
The fosmid vector, pMycInt(apr)-sacB-FOS3, was digested with PmeI and dephosphorylated with alkaline phosphatase (FastAP; Thermo Scientific), and the 10-kb vector backbone fragment ws purified (Zymoclean Gel DNA Recovery; Zymo Research) after separation from the sacB cassette on an agarose gel. The vector was ligated to end-repaired gDNA fragments of ∼35 to 45 kb (10:1 molar ratio of vector to insert gDNA) overnight at room temperature with T4 DNA ligase (NEB) and then heat inactivated at 70°C for 15 min. The fosmid ligation reaction mixtures were packaged using MaxPlax Lambda packaging extract (Epicentre) according to the manufacturer’s instructions. The packaged fosmid libraries were transfected into E. coli Replicator FOS (cultured in LB, 0.2% maltose, and 10 mM MgSO4 for phage uptake), and the transfected bacteria were plated on yeast extract-tryptone (YT) plates containing 50 µg/ml apramycin and 5% sucrose [to counterselect against undigested pMycInt(apr)-sacB-FOS3]. Fosmid clones were pooled separately for each library and grown overnight in 200 ml of LB containing 50 µg/ml apramycin without arabinose induction. A 10-ml aliquot of overnight culture was diluted 20-fold in LB containing 50 µg/ml apramycin and 0.01% arabinose and grown for 8 h to induce the fosmid copy number. Fosmid library DNA was prepared separately from both uninduced and arabinose-induced libraries using a NucleoBond MidiPrep kit (Clontech).
Complementation of BCG HIA defect with M. tuberculosis fosmid libraries.
Fosmid library DNA (from uninduced libraries; see above) or the parental fosmid vector was electroporated into BCG ΔmbtB ΔpanCD (a pantothenate auxotroph in addition to being defective in siderophore biosynthesis). To first test the efficiency of electroporation, control electroporations were plated on 7H10 agar containing 50 µg/ml apramycin, 50 µg/ml pantothenate, and 50 ng/ml mycobactin J, generating ∼5 × 104 transformants per µg of pMycInt(apr)-sacB-FOS3 vector DNA (∼12 kb in size), but only ∼200 to 250 transformants per µg of fosmid library DNA due to the larger size of the fosmids (∼40 to 50 kb in size). Due to the low efficiency of electroporation for the fosmid libraries, a total of 19 electroporations were performed using up to 4 µg of uninduced fosmid library DNA per electroporation (both M. tuberculosis ΔmbtB and M. tuberculosis ΔmbtB ΔmhuD fosmid gDNA libraries were used). To select clones with enhanced HIA, electroporations were plated on 7H10 agar containing 50 µg/ml apramycin, 50 µg/ml pantothenate, and 10 or 50 µM hemin. Immediately after electroporation (parameters: 1.25 kV, 25 µF, 1,000 Ω, 0.1-cm cuvette [Bio-Rad]), bacteria were resuspended in 7H9–OADC (BBL)–0.01% TLX containing 50 ng/ml mycobactin J and 50 µg/ml pantothenate, incubated at 37°C overnight, washed to remove mycobactin and pantothenate from the culture, and plated as described above. The enhanced HIA phenotype observed on plates was confirmed in broth culture.
Identification of fosmid gDNA sequences that complement the BCG HIA defect.
PCR was used to identify the gDNA present in the integrated fosmid from complemented clones. Nested primer pairs were designed using Primer-BLAST and Primer3 (66–68) to amplify the gDNA insert. As the gDNA inserts for the fosmid library were quite large (35 to 45 kb), amplification of the entire insert was not possible. Thus, both the left and right fosmid-gDNA insert junctions were amplified separately in hemi-nested PCRs. To amplify the unknown gDNA inserts, we designed primers that contain 7-mer sequences that occur at high frequency in M. tuberculosis genomic DNA at the 3′ end of the primer and a random 20-nt sequence at the 5′ end. High-frequency 7-mers in the M. tuberculosis genome were identified with KAnalyze (69). The original TubercuList website (http://genolist.pasteur.fr/TubercuList/) (70) and SeqBuilder software (DNASTAR, Inc.) were used to determine the distribution of high-frequency 7-mer sites in the M. tuberculosis chromosome. The 7-mers with high-GC content were excluded as these sequences were often clustered in high-GC-content genes and were not very evenly distributed throughout the genome. Six 7-mers with high frequency and good distribution throughout the M. tuberculosis genome were selected for the primer design (Table S1).
Construction of complemented strains.
M. tuberculosis ppe37 (from M. tuberculosis Erdman gDNA) and BCG ppe37 (from BCG Tice gDNA) along with their promoter region (131 bp of sequence upstream of the start codon encompassing the entire hisE-ppe37 intergenic region) were amplified by PCR using primers ppe37-FP and ppe37-RP and cloned into the PmeI sites of the integrating fosmid vector, pMycInt(apr)-sacB-FOS3, by DNA assembly (thus replacing the sacB gene). Mutagenesis of the M. tuberculosis ppe37 gene to remove base pairs encoding various amino acids at positions 31 through 39 was achieved using in vivo assembly (IVA) cloning (71). First, the large (11.5-kb) complementation plasmid pMycInt(apr)-FOS3-Mtb PPE37, was amplified by PCR using primers pMycInt(apr)-F and pMycInt(apr)-R, generating a 5.1-kb PCR product including M. tuberculosis ppe37-attP-int-apr and lacking replication origins. This was joined with the origin of replication from pUC19 (738-bp PCR product amplified using primers pUC-pMycInt-F and pUC-pMycInt-R) by IVA cloning. The smaller plasmid generated, pMycInt(apr)-Mtb PPE37 (5.8 kb), was then mutated using IVA cloning by transformation of a PCR product of the entire plasmid amplified using a constant primer [ppe37(mut)-R] for all derivatives and a variable primer [ppe37(mut)-F1, -F2, -F3, -F4, -F5, or -F6] designed to delete from 9 to 27 bp from the ppe37 coding region. Correctly constructed plasmids were identified by restriction analysis and sequencing of the entire insert region.
Potential M. tuberculosis ppe37 variant genes, along with their promoter regions, were amplified from the genomic DNA of four different M. tuberculosis strains (HN878, CDC1551, East African Indian 91_0079, and Indo-Oceanic T17X [genomic DNA was obtained from BEI Resources]) using primers ppe37(NotI)-FP and ppe37(NotI)-RP [or ppe37(NotI)-CDC-RP for M. tuberculosis CDC1551 genomic DNA]. The downstream primers were designed to include sufficient sequence beyond the normal ppe37 stop codon to ensure that the full transcript is present for ppe37 frameshift variants (such as M. tuberculosis F11) where transcription proceeds past the normal stop codon. PCR products were cloned into the NotI sites of the integrating vector, pMycInt(apr)-Mtb PPE37, by DNA assembly (thus, replacing the M. tuberculosis Erdman ppe37 gene). Correctly constructed plasmids were identified by restriction analysis and sequencing of the entire insert region. The ppe37 genes from HN878, CDC1551, and East African Indian 91_0079 were confirmed to be variants (compared with the M. tuberculosis H37Rv reference genome); the ppe37 gene from Indo-Oceanic T17X was not a variant (i.e., it is identical to the H37Rv and Erdman sequence). The ppe37 GT1016-7 deletion variant (M. tuberculosis F11 variant) (Fig. S6 and Table S2) was obtained by mutagenesis of the plasmid pMycInt(apr)-Mtb IO T17X PPE37, using primers ppe37[GT1016-7del]-F and ppe37[GT1016-7del]-R, by IVA cloning. We electroporated plasmids expressing ppe37 or the parental plasmid, pMycInt(apr)-sacB-FOS3 (used as an empty vector control), into BCG ΔmbtB, BCG ΔmbtB ΔpanCD, and M. tuberculosis ΔmbtB Δppe37 and selected transformants on 7H10 plates containing 50 µg/ml apramycin and either 50 ng/ml mycobactin J or 50 µM hemin; plates for BCG ΔmbtB ΔpanCD also included 50 µg/ml pantothenate. Colonies were counted at 3 to 3.5 weeks, and the numbers in Table 1 represent ∼2% (BCG ΔmbtB ΔpanCD) or 4% (M. tuberculosis ΔmbtB) of the entire electroporation, plated on one or two plates.
Characterization of HIA phenotypes in broth culture.
Bacteria cultured to log phase in 7H9-OADC (BBL)–0.01% TLX broth supplemented with 10 ng/ml mycobactin J were diluted into 7H9-OADC (BBL)–0.01% TLX or 7H9-OADC (BBL)–0.05% Tween 80 broth to an initial calculated A750 of 0.002. The diluted bacterial cultures were then added to 96-well plates (100 µl inoculum added per well) containing 100 µl of the same medium without or with additional supplements (mycobactin J, mycobactin J plus MnPPIX inhibitor, or hemin at various concentrations as indicated in the figure legends). The 2-fold dilution results in a bacterial density with initial calculated A750 of 0.001 (based on measurements using a 1-cm cuvette) and an A750 of 0.00025 when adjusted for the 96-well plate reader measurements (i.e., bacterial density measurements are 4-fold less with the 96-well plate reader than with the cuvette-based spectrophotometer due to the shorter path length). The inner 60 wells of the 96-well plates were used for bacterial cultures, and the outer wells were filled with 200 µl of phosphate-buffered saline (PBS) to minimize evaporation. Growth was measured at 7 to 21 days, but in most instances after 14 days of growth. Fifty microliters of freshly prepared 50% formalin in PBS was added to each well (10% formalin final concentration) to inactivate the bacteria for 1 h; the formalin-killed cultures were transferred to a new 96-well plate (200 µl per well), and absorbance at 750 nm was measured in a Bio-Rad iMark microplate reader. Bacterial growth measurements at 750 nm avoid much of the absorbance due to hemin and MnPPIX, which have strong absorbance at the lower wavelengths typically used for bacterial measurements (15). However, as there is still some absorbance at 750 nm due to hemin and MnPPIX at the higher concentrations used, uninoculated medium-only controls were included for each growth condition for blank measurements that were subtracted from measurements of bacterial cultures. Calculations of the EC50 for hemin (concentration allowing 50% of maximal growth) and the IC95 for MnPPIX (concentration inhibiting growth by 95%) were performed using Prism, version 7.04 (GraphPad Software). The EC50 for hemin was calculated by fitting the data to a four-parameter [agonist]-versus-response curve with automatic outlier elimination (ROUT [robust regression and outlier removal] coefficient, 1%) using nonlinear regression. For each experiment, the top plateau parameter for all strains in the experiment was constrained to the value calculated for the strain (or strains) expressing a functional PPE37 that grew efficiently with hemin. To calculate the IC95 for MnPPIX, the data were first normalized to the amount of growth with no inhibitor and then fit to a four-parameter log [inhibitor]-versus-response curve using nonlinear regression. Since the logarithim of 0 is not defined, growth with no inhibitor was plotted at a concentration of 1 × 10−4 µM. Statistical analysis of differences in hemin EC50 and MnPPIX IC95 between strains was performed by one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (Prism, version 7.04).
Characterization of HIA phenotypes on agar plates.
Bacteria grown in 7H9-OADC (BBL)–0.01% TLX broth with 10 ng/ml mycobactin J were diluted to an A550 of 0.1 (estimated to contain 2 × 106 CFU/ml) with PBS, and then further 10-fold serial dilutions were prepared in PBS. Aliquots (10 µl) of bacterial dilutions were spotted on individual 7H10 plates containing no additional supplements, 50 ng/ml mycobactin J, or various concentrations of hemin (1, 5, 10, or 50 µM) and allowed to soak into the agar. Plates were incubated for 14 days and then photographed.
Sequence analysis.
The hisE-ppe37-metH region from the H37Rv reference genome sequence (GenBank accession number NC_000962.3) was identified using the MycoBrowser website (72). This region (NC_000962.3, nucleotides 2380663 to2386067, 5,405 bp) was used in a Microbial Nucleotide BLAST (73) against completed genomes of M. tuberculosis (taxid:1773), identifying 157 sequences of very high homology. In the process of analyzing these sequences, several sequences were excluded for suspected poor sequencing quality. The Haarlem/NITR202 sequence (GenBank accession number CP004886.1) has many ambiguous nucleotides, 54 mismatches, and 12 gap opens in the 5.4-kb region. The CAS/NITR204 sequence (GenBank accession number CP005386.1) has 14 mismatches and 22 gap opens in the 5.4-kb region. All other sequences had ≤3 mismatches and ≤3 gap opens. Thus, these two outliers were excluded from further analysis. Two other NITR strains sequenced as part of this same project were also excluded (Beijing/NITR203 [GenBank accession number NC_021054.1] and EAI5/NITR206 [NC_021194.1]) (74). For M. tuberculosis strain 2279 (GenBank accession number CP010336.1), we identified 3 deletions in the 5.4-kb hisE-ppe37-metH region, all within the ppe37 gene, more than any of the other strains. Therefore, as a test for sequencing quality, we did a similar BLAST search using the sequence of an essential gene, glnA1, similar in size to ppe37. Almost all of the strains have glnA1 sequences that are 100% identical to the H37Rv reference sequence, with five strains having one SNP (at different positions). However, M. tuberculosis strain 2279 has two separate 1-bp gaps near the middle of the glnA1 gene, resulting in frameshifts. These frameshifts in glnA1 are most likely sequencing errors since glnA1 is an essential gene; thus, the three deletions in the M. tuberculosis 2279 ppe37 are also suspect. M. tuberculosis strain RGTB327 (GenBank accession number CP003233.1) has two separate gaps of 1 and 3 bp near the middle of the glnA1 gene, resulting in a frameshift. Thus, strains 2279 and RGTB327 were excluded, leaving 151 sequences from completed genomes for analysis. The hisE-ppe37-metH region in six strains is in the opposite orientation compared with orientation in the reference H37Rv genome, so these six sequences were converted to their reverse complement sequence. DNAcollapser, part of the FaBox online fasta sequence toolbox (75), was used to collapse DNA sequences to a nonredundant set of haplotypes. Multiple sequence alignment of the haplotypes was performed with blastn, and the alignment was viewed in the NCBI Multiple Sequence Alignment Viewer, version 1.7.7, to identify differences between the reference H37Rv haplotype and the other haplotypes. Several strains (H37Rv, H37Ra, CCDC5079, and CCDC5180) have duplicate complete genomes in the database. Once the sequences from duplicate genomes were confirmed to be identical, only a single sequence for each strain was retained, thus eliminating five duplicate genomes, leaving 146 sequences in the final analysis (Fig. S6 and Table S2). The SignalP, version 4.1, TatP, version 1.0, LipoP, version 1.0, and TargetP, version 1.1, servers were used to analyze PPE37 for a potential secretion signal (76–79).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI135631. The following reagents were obtained through BEI Resources, NIAID, NIH: genomic DNA from Mycobacterium tuberculosis, strain HN878, NR-14867; genomic DNA from Mycobacterium tuberculosis, strain CDC1551, NR-48981; genomic DNA from Mycobacterium tuberculosis, strain East African Indian 91_0079, NR-44095; genomic DNA from Mycobacterium tuberculosis, strain Indo-Oceanic T17X, NR-44096; Mycobacterium tuberculosis, strain HN878, NR-13647; and Mycobacterium tuberculosis, strain CDC1551, NR-13649.
M.V.T. conceived and performed experiments and wrote the manuscript. S.N. performed experiments. M.A.H. conceived experiments and provided expertise, wrote the manuscript, and secured funding.
We declare that we have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00540-18.
REFERENCES
- 1.World Health Organization. 2017. Global tuberculosis report 2017. World Health Organization, Geneva, Switzerland.
- 2.Cassat JE, Skaar EP. 2013. Iron in infection and immunity. Cell Host Microbe 13:509–519. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hood MI, Skaar EP. 2012. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nairz M, Haschka D, Demetz E, Weiss G. 2014. Iron at the interface of immunity and infection. Front Pharmacol 5:152. doi: 10.3389/fphar.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Runyen-Janecky LJ. 2013. Role and regulation of heme iron acquisition in gram-negative pathogens. Front Cell Infect Microbiol 3:55. doi: 10.3389/fcimb.2013.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheldon JR, Heinrichs DE. 2015. Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol Rev 39:592–630. doi: 10.1093/femsre/fuv009. [DOI] [PubMed] [Google Scholar]
- 7.Sheldon JR, Laakso HA, Heinrichs DE. 2016. Iron acquisition strategies of bacterial pathogens. Microbiol Spectr 4(2):VMBF-0010-2015. doi: 10.1128/microbiolspec.VMBF-0010-2015. [DOI] [PubMed] [Google Scholar]
- 8.Segond D, Abi Khalil E, Buisson C, Daou N, Kallassy M, Lereclus D, Arosio P, Bou-Abdallah F, Nielsen Le Roux C. 2014. Iron acquisition in Bacillus cereus: the roles of IlsA and bacillibactin in exogenous ferritin iron mobilization. PLoS Pathog 10:e1003935. doi: 10.1371/journal.ppat.1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandrini S, Masania R, Zia F, Haigh R, Freestone P. 2013. Role of porin proteins in acquisition of transferrin iron by enteropathogens. Microbiology 159:2639–2650. doi: 10.1099/mic.0.071928-0. [DOI] [PubMed] [Google Scholar]
- 10.DeDent A, Kim HK, Missiakas D, Schneewind O. 2012. Exploring Staphylococcus aureus pathways to disease for vaccine development. Semin Immunopathol 34:317–333. doi: 10.1007/s00281-011-0299-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gobin J, Horwitz MA. 1996. Exochelins of Mycobacterium tuberculosis remove iron from human iron-binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J Exp Med 183:1527–1532. doi: 10.1084/jem.183.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gobin J, Moore CH, Reeve JR Jr, Wong DK, Gibson BW, Horwitz MA. 1995. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc Natl Acad Sci U S A 92:5189–5193. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macham LP, Ratledge C. 1975. A new group of water-soluble iron-binding compounds from mycobacteria: the exochelins. J Gen Microbiol 89:379–382. doi: 10.1099/00221287-89-2-379. [DOI] [PubMed] [Google Scholar]
- 14.Macham LP, Ratledge C, Nocton JC. 1975. Extracellular iron acquisition by mycobacteria: role of the exochelins and evidence against the participation of mycobactin. Infect Immun 12:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, Horwitz MA, Goulding CW. 2011. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci U S A 108:5051–5056. doi: 10.1073/pnas.1009516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tullius MV, Harth G, Maslesa-Galic S, Dillon BJ, Horwitz MA. 2008. A replication-limited recombinant Mycobacterium bovis BCG vaccine against tuberculosis designed for human immunodeficiency virus-positive persons is safer and more efficacious than BCG. Infect Immun 76:5200–5214. doi: 10.1128/IAI.00434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones CM, Niederweis M. 2011. Mycobacterium tuberculosis can utilize heme as an iron source. J Bacteriol 193:1767–1770. doi: 10.1128/JB.01312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nambu S, Matsui T, Goulding CW, Takahashi S, Ikeda-Saito M. 2013. A new way to degrade heme: the Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J Biol Chem 288:10101–10109. doi: 10.1074/jbc.M112.448399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Contreras H, Joens MS, McMath LM, Le VP, Tullius MV, Kimmey JM, Bionghi N, Horwitz MA, Fitzpatrick JA, Goulding CW. 2014. Characterization of a Mycobacterium tuberculosis nanocompartment and its potential cargo proteins. J Biol Chem 289:18279–18289. doi: 10.1074/jbc.M114.570119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, Pryor M, Duthoy S, Grondin S, Lacroix C, Monsempe C, Simon S, Harris B, Atkin R, Doggett J, Mayes R, Keating L, Wheeler PR, Parkhill J, Barrell BG, Cole ST, Gordon SV, Hewinson RG. 2003. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci U S A 100:7877–7882. doi: 10.1073/pnas.1130426100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Zhang Y, Zheng H, Pan Y, Liu H, Du P, Wan L, Liu J, Zhu B, Zhao G, Chen C, Wan K. 2013. Genome sequencing and analysis of BCG vaccine strains. PLoS One 8:e71243. doi: 10.1371/journal.pone.0071243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brosch R, Gordon SV, Garnier T, Eiglmeier K, Frigui W, Valenti P, Dos Santos S, Duthoy S, Lacroix C, Garcia-Pelayo C, Inwald JK, Golby P, Garcia JN, Hewinson RG, Behr MA, Quail MA, Churcher C, Barrell BG, Parkhill J, Cole ST. 2007. Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci U S A 104:5596–5601. doi: 10.1073/pnas.0700869104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitra A, Speer A, Lin K, Ehrt S, Niederweis M. 2017. PPE surface proteins are required for heme utilization by Mycobacterium tuberculosis. mBio 8:e01720-16. doi: 10.1128/mBio.01720-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, Anderson KL, Dunman PM, Joyce S, Skaar EP. 2007. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe 1:109–119. doi: 10.1016/j.chom.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. 2005. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyoshi-Akiyama T, Matsumura K, Iwai H, Funatogawa K, Kirikae T. 2012. Complete annotated genome sequence of Mycobacterium tuberculosis Erdman. J Bacteriol 194:2770. doi: 10.1128/JB.00353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gold B, Rodriguez GM, Marras SA, Pentecost M, Smith I. 2001. The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol Microbiol 42:851–865. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. 2002. ideR, an essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun 70:3371–3381. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zimpel CK, Brandao PE, de Souza Filho AF, de Souza RF, Ikuta CY, Ferreira Neto JS, Camargo NCS, Heinemann MB, Guimaraes AMS. 2017. Complete genome sequencing of Mycobacterium bovis SP38 and comparative genomics of Mycobacterium bovis and M. tuberculosis strains. Front Microbiol 8:2389. doi: 10.3389/fmicb.2017.02389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6:95. doi: 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmad J, Farhana A, Pancsa R, Arora SK, Srinivasan A, Tyagi AK, Babu MM, Ehtesham NZ, Hasnain SE. 2018. Contrasting function of structured N-terminal and unstructured C-terminal segments of Mycobacterium tuberculosis PPE37 protein. mBio 9:e01712-17. doi: 10.1128/mBio.01712-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horwitz LD, Horwitz MA. 2014. The exochelins of pathogenic mycobacteria: unique, highly potent, lipid- and water-soluble hexadentate iron chelators with multiple potential therapeutic uses. Antioxid Redox Signal 21:2246–2261. doi: 10.1089/ars.2013.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones CM, Wells RM, Madduri AV, Renfrow MB, Ratledge C, Moody DB, Niederweis M. 2014. Self-poisoning of Mycobacterium tuberculosis by interrupting siderophore recycling. Proc Natl Acad Sci U S A 111:1945–1950. doi: 10.1073/pnas.1311402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurthkoti K, Amin H, Marakalala MJ, Ghanny S, Subbian S, Sakatos A, Livny J, Fortune SM, Berney M, Rodriguez GM. 2017. The capacity of Mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. mBio 8:e01092-17. doi: 10.1128/mBio.01092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stojiljkovic I, Kumar V, Srinivasan N. 1999. Non-iron metalloporphyrins: potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol Microbiol 31:429–442. doi: 10.1046/j.1365-2958.1999.01175.x. [DOI] [PubMed] [Google Scholar]
- 36.Reiling N, Homolka S, Walter K, Brandenburg J, Niwinski L, Ernst M, Herzmann C, Lange C, Diel R, Ehlers S, Niemann S. 2013. Clade-specific virulence patterns of Mycobacterium tuberculosis complex strains in human primary macrophages and aerogenically infected mice. mBio 4:e00250-13. doi: 10.1128/mBio.00250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith A, McCulloh RJ. 2015. Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol 6:187. doi: 10.3389/fphys.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan X, Rietzschel N, Kwon H, Walter Nuno AB, Hanna DA, Phillips JD, Raven EL, Reddi AR, Hamza I. 2016. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc Natl Acad Sci U S A 113:E5144–E5152. doi: 10.1073/pnas.1609865113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fishbein S, van Wyk N, Warren RM, Sampson SL. 2015. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol 96:901–916. doi: 10.1111/mmi.12981. [DOI] [PubMed] [Google Scholar]
- 40.Delogu G, Brennan MJ, Manganelli R. 2017. PE and PPE genes: a tale of conservation and diversity. Adv Exp Med Biol 1019:191–207. doi: 10.1007/978-3-319-64371-7_10. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez GM, Gold B, Gomez M, Dussurget O, Smith I. 1999. Identification and characterization of two divergently transcribed iron regulated genes in Mycobacterium tuberculosis. Tuber Lung Dis 79:287–298. doi: 10.1054/tuld.1999.0219. [DOI] [PubMed] [Google Scholar]
- 42.Voskuil MI, Schnappinger D, Rutherford R, Liu Y, Schoolnik GK. 2004. Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis (Edinb) 84:256–262. doi: 10.1016/j.tube.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 43.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daim S, Kawamura I, Tsuchiya K, Hara H, Kurenuma T, Shen Y, Dewamitta SR, Sakai S, Nomura T, Qu H, Mitsuyama M. 2011. Expression of the Mycobacterium tuberculosis PPE37 protein in Mycobacterium smegmatis induces low tumour necrosis factor alpha and interleukin 6 production in murine macrophages. J Med Microbiol 60:582–591. doi: 10.1099/jmm.0.026047-0. [DOI] [PubMed] [Google Scholar]
- 45.Rienksma RA, Suarez-Diez M, Mollenkopf HJ, Dolganov GM, Dorhoi A, Schoolnik GK, Martins Dos Santos VA, Kaufmann SH, Schaap PJ, Gengenbacher M. 2015. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics 16:34. doi: 10.1186/s12864-014-1197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malone KM, Rue-Albrecht K, Magee DA, Conlon K, Schubert OT, Nalpas NC, Browne JA, Smyth A, Gormley E, Aebersold R, MacHugh DE, Gordon SV. 20 March 2018. Comparative ‘omics analyses differentiate Mycobacterium tuberculosis and Mycobacterium bovis and reveal distinct macrophage responses to infection with the human and bovine tubercle bacilli. Microb Genom doi: 10.1099/mgen.0.000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters JS, Calder B, Gonnelli G, Degroeve S, Rajaonarifara E, Mulder N, Soares NC, Martens L, Blackburn JM. 2016. Identification of quantitative proteomic differences between Mycobacterium tuberculosis lineages with altered virulence. Front Microbiol 7:813. doi: 10.3389/fmicb.2016.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schubert OT, Ludwig C, Kogadeeva M, Zimmermann M, Rosenberger G, Gengenbacher M, Gillet LC, Collins BC, Rost HL, Kaufmann SH, Sauer U, Aebersold R. 2015. Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 18:96–108. doi: 10.1016/j.chom.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 49.Yimer SA, Birhanu AG, Kalayou S, Riaz T, Zegeye ED, Beyene GT, Holm-Hansen C, Norheim G, Abebe M, Aseffa A, Tønjum T. 2017. Comparative proteomic analysis of Mycobacterium tuberculosis lineage 7 and lineage 4 strains reveals differentially abundant proteins linked to slow growth and virulence. Front Microbiol 8:795. doi: 10.3389/fmicb.2017.00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng J, Liu L, Wei C, Leng W, Yang J, Li W, Wang J, Jin Q. 2012. A comprehensive proteomic analysis of Mycobacterium bovis bacillus Calmette-Guerin using high resolution Fourier transform mass spectrometry. J Proteomics 77:357–371. doi: 10.1016/j.jprot.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 51.Small JL, Park SW, Kana BD, Ioerger TR, Sacchettini JC, Ehrt S. 2013. Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in Mycobacterium tuberculosis. mBio 4:e00475-13. doi: 10.1128/mBio.00475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niederweis M, Danilchanka O, Huff J, Hoffmann C, Engelhardt H. 2010. Mycobacterial outer membranes: in search of proteins. Trends Microbiol 18:109–116. doi: 10.1016/j.tim.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wells RM, Jones CM, Xi Z, Speer A, Danilchanka O, Doornbos KS, Sun P, Wu F, Tian C, Niederweis M. 2013. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog 9:e1003120. doi: 10.1371/journal.ppat.1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez GM, Smith I. 2006. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol 188:424–430. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, Tyagi AK. 2013. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J Infect Dis 208:1255–1265. doi: 10.1093/infdis/jit250. [DOI] [PubMed] [Google Scholar]
- 56.Tufariello JM, Chapman JR, Kerantzas CA, Wong KW, Vilcheze C, Jones CM, Cole LE, Tinaztepe E, Thompson V, Fenyo D, Niederweis M, Ueberheide B, Philips JA, Jacobs WR Jr. 2016. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc Natl Acad Sci U S A 113:E348–E357. doi: 10.1073/pnas.1523321113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keating LA, Wheeler PR, Mansoor H, Inwald JK, Dale J, Hewinson RG, Gordon SV. 2005. The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth. Mol Microbiol 56:163–174. doi: 10.1111/j.1365-2958.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- 58.Chavadi S, Wooff E, Coldham NG, Sritharan M, Hewinson RG, Gordon SV, Wheeler PR. 2009. Global effects of inactivation of the pyruvate kinase gene in the Mycobacterium tuberculosis complex. J Bacteriol 191:7545–7553. doi: 10.1128/JB.00619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noy T, Vergnolle O, Hartman TE, Rhee KY, Jacobs WR Jr, Berney M, Blanchard JS. 2016. Central role of pyruvate kinase in carbon co-catabolism of Mycobacterium tuberculosis. J Biol Chem 291:7060–7069. doi: 10.1074/jbc.M115.707430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tufariello JAnn, Bardarov S, Hatfull G, Larsen M, Bardarov S, Chan J, Pavelka MS, Sambandamurthy V, Jacobs WR. Jr, 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 61.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 62.Gibson DG, Smith HO, Hutchison CA 3rd, Venter JC, Merryman C. 2010. Chemical synthesis of the mouse mitochondrial genome. Nat Methods 7:901–903. doi: 10.1038/nmeth.1515. [DOI] [PubMed] [Google Scholar]
- 63.Lee MH, Pascopella L, Jacobs WR Jr, Hatfull GF. 1991. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci U S A 88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weis JH, Quertermous T. 1987. Size fractionation using sucrose gradients. Curr Protoc Mol Biol 1:5.3.1–5.3.8. [DOI] [PubMed] [Google Scholar]
- 65.Stone AB. 1974. A simplified method for preparing sucrose gradients. Biochem J 137:117–118. doi: 10.1042/bj1370117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 67.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3–new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Audano P, Vannberg F. 2014. KAnalyze: a fast versatile pipelined k-mer toolkit. Bioinformatics 30:2070–2072. doi: 10.1093/bioinformatics/btu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cole ST. 1999. Learning from the genome sequence of Mycobacterium tuberculosis H37Rv. FEBS Lett 452:7–10. doi: 10.1016/S0014-5793(99)00536-0. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Nafria J, Watson JF, Greger IH. 2016. IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Sci Rep 6:27459. doi: 10.1038/srep27459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kapopoulou A, Lew JM, Cole ST. 2011. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb) 91:8–13. doi: 10.1016/j.tube.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 73.Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, Madden TL, Matten WT, McGinnis SD, Merezhuk Y, Raytselis Y, Sayers EW, Tao T, Ye J, Zaretskaya I. 2013. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narayanan S, Deshpande U. 2013. Whole-genome sequences of four clinical isolates of Mycobacterium tuberculosis from Tamil Nadu, South India. Genome Announc 1:e00186-13. doi: 10.1128/genomeA.00186-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Villesen P. 2007. FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7:965–968. doi: 10.1111/j.1471-8286.2007.01821.x. [DOI] [Google Scholar]
- 76.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 77.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 78.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. 2005. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.