

Abstract
The role of beta and α-cells to glucose control are established, but the physiological role of δ-cells is poorly understood. Delta-cells are ideally positioned within pancreatic islets to modulate insulin and glucagon secretion at their source. We review the evidence for a negative feedback loop between delta and β-cells that determines the blood glucose set point and suggest that local δ-cell-mediated feedback stabilizes glycemic control.
Introduction
Over half of U.S. adults are now estimated to have diabetes or pre-diabetes (44). Type 1 diabetes (T1D) is caused by the autoimmune destruction of β-cells, whereas Type 2 diabetes (T2D) results from peripheral insulin resistance precipitated by factors associated with lifestyle and genetic predisposition. Both diseases are characterized by absolute (T1D) or relative (T2D) insulin deficiency. Consequently, pancreatic β-cells have been studied intently for decades. Less appreciated is that excess glucagon secretion from pancreatic α-cells is responsible for as much as half of the hyperglycemia in diabetes (77), which is the immediate cause for most diabetes-related complications. Successful diabetes management therefore requires effective strategies not only to restore insulin or improve insulin action but to prevent glucagon-induced hepatic glucose production from aggravating hyperglycemia. Here, we make the case that pancreatic δ-cells provide crucial feedback control of α- and β-cells to coordinate insulin and glucagon secretion in healthy islets that breaks down in diabetes.
The Pancreatic Islet is Home to More Than β-Cells
The principal endocrine output of the pancreatic islets are insulin and glucagon. During and shortly after feeding, nutrients absorbed across the intestinal epithelia stimulate insulin secretion. Conversely, under catabolic conditions that occur between meals or during a fast, β-cells are silent as α-cell activity increases to safeguard against hypoglycemia. Healthy islets are capable of balancing insulin and glucagon output with tremendous precision. This is illustrated by continuous glucose monitoring (CGM) experiments in mice (78) that reveal the narrow range within blood glucose is maintained over multiple diurnal cycles despite ad libitum food access. Similarly, a healthy human pancreas maintains euglycemia over 87,000 meals consumed in a lifetime.1 Although α- and β-cells each possess the ability to sense glucose in a cell-autonomous fashion, it is no coincidence that they are organized in close proximity within the islets of Langerhans. This arrangement enables careful coordination between insulin and glucagon at their source by a potent combination of paracrine, neural, and endocrine inputs (FIGURE 1A). Among the most prominent of these signals is somatostatin released by pancreatic δ-cells (62), which make up ~5–10% of the endocrine cells within the islet.
FIGURE 1.

Pancreatic δ-cells
A: pancreatic δ-cells receive input from numerous paracrine, endocrine, and neural inputs, and translate this into appropriate inhibition of glucagon and insulin release by α- and β-cells. Select stimulatory and inhibitory inputs are given for each of the islet cell types. B: schematic representation of the profiles of insulin, glucagon, and somatostatin secretion as a function of blood glucose.
Discovery of δ-Cells
Pancreatic δ-cells were first recognized as a distinct cell type from α- and β-cells based on alcohol- and aqueous-based histological staining methods, and were originally referred to as A1 cells, C cells, gamma cells, or D cells (3). These cells had no known function and were considered to represent a distinct functional stage of α- or β-cells. This changed with the discovery that somatostatin, a novel hypothalamic peptide named for its ability to suppress somatic growth by inhibiting growth hormone (11), is found in all pancreatic δ-cells (21, 32). Synthetic somatostatin peptide was subsequently confirmed to potently inhibit insulin and glucagon secretion from isolated islets (13). However, the pancreatic δ-cell has long been understudied, in part because it has been challenging to quantify the impact of local δ-cell-dependent feedback on α- and β-cells. The physiological importance of δ-cell-mediated feedback on insulin and glucagon release is only now coming into focus.
Paracrine Cross Talk Within Pancreatic Islets
Although β- and α-cell activity is inextricably tied to glucose levels, many signals from inside and outside the islet ultimately contribute to shape the final insulin and glucagon output. Insulin release from β-cells is triggered when glucose exceeds a threshold of ~7 mM glucose (16, 80) (FIGURE 1B). Glucose-stimulated insulin secretion (GSIS) can be further amplified via the actions of incretin hormones such as glucagon-like peptide-1 (GLP-1). GLP-1 activates its cognate GLP-1 receptor (GLP1R), a Gαs-coupled class B GPCR abundantly expressed on β-cells. This amplifying pathway is largely ineffective below the glucose threshold of β-cells, preventing incretins from stimulating insulin release under low glucose. This greatly reduces the risk of insulin-induced hypoglycemia, which underlies the success and safety of incretin-based therapies in treating T2D.
Control of glucagon release is complex and follows a biphasic response to glucose (FIGURE 1B). Maximal glucagon secretion occurs under low glucose and decreases toward a nadir around 5 mM glucose (24, 43, 80). Alpha-cells are directly suppressed by glucose via an α-cell-intrinsic mechanism that involves KATP channel inhibition (80). On top of the direct stimulatory effects of low glucose, α-cells respond robustly to adrenergic stimulation as part of the counterregulatory response to hypoglycemia (75). A rise in glucose beyond 5 mM activates an ER Ca2+ store-operated mechanism that, paradoxically, facilitates glucagon release under hyperglycemic conditions (41, 80). It has long been known that glucagon stimulates insulin secretion (66, 67), even though the systemic actions of glucagon functionally oppose those of insulin. This effect is mediated by the glucagon receptor (GCGR), a class B GPCR related to the incretin receptors that, like GLP1R, is Gαs-coupled and expressed by β-cells (FIGURE 1). Glucagon essentially acts in an incretin-like fashion to amplify GSIS within the islet. In fact, the most recent evidence suggests that glucagon from α-cells may be required for full GSIS in response to hyperglycemia (59). Glucagon secretion is suppressed by β-cell-dependent inhibitory control (28, 29), which is often attributed to the direct inhibitory actions of β-cell-derived factors such as insulin, GABA, and Zn2+ on α-cells (22, 43, 53, 60, 80, 83). However, none of these β-cell-derived factors has consistently emerged as the predominant inhibitor of α-cell activity. Therefore, the possibility of β-cell-dependent activation of δ-cells that inhibits α-cells via somatostatin during hyperglycemia is likely. Collectively, this illustrates how hormone release from the islets is controlled by a complex—at times paradoxical—web of paracrine interactions.
The δ-Cell Provides Paracrine Feedback Within the Islet
Delta-cells are organized together with α-cells around the mouse islet periphery, where they envelop a core of β-cells. In humans, they are intermingled with the other endocrine cell types. δ-Cells release a 14-amino acid form of somatostatin (Sst-14) that is proteolytically cleaved from a larger precursor (50). Somatostatin is also released from other peripheral sites, notably from enteroendocrine D cells in the gastrointestinal (GI) tract, which primarily secrete a larger, 28-amino acid form (Sst-28). Circulating somatostatin concentrations are largely unaffected by pancreatectomy and therefore do not reflect pancreatic δ-cell activity (25, 74). Instead, the GI tract is the major source of circulating somatostatin, which may influence islet β- and α-cells (14, 42). Nevertheless, circulating somatostatin (~5–25 pM) is an order of magnitude below the IC50 and EC50 values of somatostatin receptors (55, 56), suggesting that δ-cells are the predominant source of somatostatin within the islet. Therefore, the main function of the pancreatic δ-cell is to provide feedback control of neighboring β- and α-cells via local circulation and the interstitial compartment (13, 65). The inhibitory actions of somatostatin are mediated via five somatostatin receptors, SSTR1 to SSTR5. These are class A GPCRs that generally inhibit their target cells by activating the inhibitory Gαi-protein or G-protein-coupled inwardly rectifying K+ (GIRK) channels (50). Islets or β-cells have been suggested to express each of the somatostatin receptors, mostly by antibody-dependent methods that critically depend on the quality and careful validation of the reagents (reviewed in Refs. 9, 38, 50, 51). However, comprehensive α-, β-, and δ-cell transcriptomes do not support much of this prior work. Although it is true that somatostatin receptors are expressed by each of the islet cell types (FIGURE 1A), abundant expression of Sstr2 by α-cells is the only aspect that unequivocally holds up from these earlier reports (reviewed in Refs. 1, 20). Mouse β-cells abundantly (but not exclusively) express Sstr3, which is rarely mentioned in reviews of islet somatostatin receptors. Somatostatin secretion is stimulated dose-dependently by glucose in a linear fashion (FIGURE 1B) (65, 82). But even below the glucose threshold for β-cells, α-cells are activated on the addition of somatostatin antagonists, suggesting that significant basal somatostatin tone restrains α-cell activity across the glucose spectrum. In addition to glucose, sulfonylureas, amino acids, and cAMP are all capable of stimulating somatostatin secretion (2, 10, 23, 34, 69). However, until recently, the physiological cues that govern δ-cell activity during normal glucose metabolism were not understood.
Urocortin3 is Required for Normal Glucose-Stimulated Somatostatin Secretion
We discovered that normal glucose-stimulated somatostatin secretion (GSSS) requires the peptide hormone Urocortin 3 (UCN3), which is related to corticotropin-releasing hormone (CRH) and activates the type 2 CRH receptor (CRHR2). UCN3 is the third most abundant β-cell hormone after insulin and amylin, and is co-released with insulin from β-cells (40, 78). UCN3 selectively activates δ-cells, which express the α isoform of CRHR2 (20, 33), to release somatostatin. This finding closed a novel intra-islet negative feedback loop that is initiated by β-cell release of UCN3 to promote δ-cell somatostatin secretion, which inhibits β-cells. Indeed, Ucn3-null mice show impaired somatostatin secretion, which can be fully rescued by addition of synthetic UCN3 peptide (78). Predictably, the impaired somatostatin secretion enables exaggerated first- and second-phase GSIS, which is also immediately normalized upon perfusion with synthetic UCN3. This Ucn3-null phenotype closely resembles the phenotype of somatostatin-null mice, which also demonstrate an exaggerated first- and second-phase GSIS that is acutely normalized by the supplementation of synthetic somatostatin peptide (26). The similar phenotype of the null mice offers strong support for the participation of UCN3 and somatostatin in the same feedback loop. Blocking endogenous UCN3 with the selective CRHR2 peptide antagonist Astressin2b (Ast2b) (56) prevents GSSS from mouse and human islets (78). This demonstrates that, during hyperglycemia, endogenous Ucn3 released from β-cells is necessary and sufficient for somatostatin secretion, which proceeds to inhibit β-cells. Taken together, these observations have established that UCN3 activates a β-cell-dependent, δ-cell-mediated negative feedback loop to attenuate insulin secretion.
Bidirectional Exchange of Paracrine Signals Within the Islet
The paracrine role of δ-cell and the viability of the UCN3-mediated negative feedback loop rests on the ability of δ-cells to efficiently receive and relay signals within the islet. Cross talk is often considered to occur via intra-islet capillary circulation, but there is no consensus whether circulation in the rodent islets favors mantle-to-core communication (46, 49) or vice versa (8, 48, 68). Several factors favor the model of a feedback loop mediated by UCN3 and somatostatin that relies on a bidirectional exchange of signals between in the mantle and β-cells in the core. First, there is emerging evidence that islet blood flow is dynamically regulated (19). Second, somatostatin and/or UCN3 may reach their target cells by diffusion through the interstitial space. Third, β-cells within an islet are electrically coupled via connexin-36 gap junctions (5, 27), implying that not every individual β-cell needs to be directly suppressed by somatostatin to ensure effective inhibition of all β-cells within an islet. Fourth, mouse δ-cells have axon-like projections that enable the release and receipt of signals at some distance from the cell body and make them readily distinguishable from β- and α-cells (FIGURE 2A). Finally, adult human islets feature a more random intermingling of α-, β-, and δ-cells (FIGURE 2B) that would only be more conducive to the bidirectional feedback between β- and δ-cells first identified in mouse islet. Interestingly, human δ-cells lack the characteristic axon-like projections of mouse δ-cells and are significantly more compact than mouse δ-cells (FIGURE 2C), which may represent a morphological correlate of the distribution of δ-cells throughout the human islet.
FIGURE 2.

Projection of a 3D reconstruction of a pancreatic islet from a transgenic reporter strain
A: projection of a 3D reconstruction of a pancreatic islet from a transgenic reporter strain, captured by confocal microscope. β-Cells are visualized by the nuclear expression of an mCherry under control of the Ins1 promoter. δ-Cells are visualized by the expression of Cre recombinase under control of the somatostatin (Sst) promotor, which leads to their irreversible expression of yellow fluorescent protein (YFP; green). B: projection of a 3D reconstruction of a human pancreatic islet, captured by confocal microscope and stained for somatostatin (green), insulin (red), and glucagon (white). Nuclei (dapi) are counterstained in blue. Note how the human δ-cells are notably more compact compared with the axon-like mouse δ-cells. C: the difference in morphology of mouse and human δ-cells and β-cells from the same islet quantified their circularity, defined as the normalized ratio of the area over the perimeter of the cell outline. Each cell outline was determined in Nikon Elements, and circularity was calculated as . A value of 1 indicates a perfect circle. Mouse δ-cells stand out for their elongated morphology, which manifests as a significant reduction in circularity, compared with β-cells, whereas human δ-cells are similarly compact to human β-cells. Numbers in-between parentheses reflect the number of cells quantified from 3D confocal reconstructions of intact islets from two individual subjects for each species.
The β-Cell as a Blueprint for the δ-Cell
The discovery that UCN3 is required for normal GSSS raised the question whether δ-cells respond directly to glucose at all, or whether GSSS can be fully accounted for by the paracrine actions of UCN3. To develop a working model of the relative contributions of glucose and paracrine signals such as UCN3 on δ-cell activity, it is helpful to turn to the β-cells. β-Cells share an immediate precursor with δ-cells in pancreas development (70), and the mechanistic basis of their activation has been extensively studied. β-Cell activation is triggered by the uptake of glucose by passive diffusion through glucose transporters, followed by its stepwise catabolism via the TCA cycle and oxidative phosphorylation yielding ATP. The increased ATP-to-ADP ratio closes ATP-sensitive K+ leak (KATP) channels, which causes accumulation of K+ and depolarization of the β-cell. This triggers opening of voltage-gated Na+ and L-type Ca2+ channels. The resulting Ca2+ influx stimulates exocytosis of secretory granules (reviewed in Ref. 61).
A number of studies have suggested that β- and δ-cells share common mechanisms of activation. Diazoxide, which keeps the KATP channel complex in the open conformation to prevent depolarization, and the L-type calcium channel blocker isradipine block both insulin and somatostatin secretion when applied to intact islets (10, 78). Importantly, exogenous UCN3 fails to rescue both diazoxide- and isradipine-mediated inhibition of somatostatin secretion (78). This demonstrates that the closure of δ-cell KATP channels and the influx of Ca2+ via L-type channels in response to glucose is necessary for normal somatostatin secretion. However, somatostatin release from islets null for Ucn3 is only modestly (but significantly) stimulated by glucose but can be fully rescued by synthetic UCN3 (78). This proves that the bulk of “glucose-stimulated” somatostatin release actually depends on local UCN3. Overall, this favors a model where δ- and β-cells use similar mechanisms to trigger hormone release in response to glucose and to further amplify it by Gαs-mediated signaling. Where δ-cells differ from β-cells is in the identity of the signals that amplify glucose-stimulated hormone secretion, with locally released UCN3 the principal paracrine signal to stimulate δ-cells, whereas β-cells respond instead to incretins and glucagon (FIGURE 1A).
The δ-Cell as a Modulating Hub That Shapes Islet Cell Activity
Although UCN3 is the principal paracrine signal to stimulate somatostatin secretion, δ-cells respond to a multitude of paracrine, endocrine, and neural signals. For example, the potent insulinostatic actions of the hunger hormone ghrelin (17, 18, 54, 76, 84) are mediated indirectly via the stimulation of somatostatin release from δ-cells (1, 20). And long-chain free fatty acids, such as palmitate, stimulate insulin secretion not just directly via the stimulation of GPR40 and enhanced β-cell intracellular metabolic rate (35, 37), but also indirectly by suppressing somatostatin secretion via the inhibitory receptor GPR120 expressed by δ-cells (72). Adrenosympathetic inputs (i.e., catecholamines) stimulate α-cells via β1 adrenergic receptors as part of the counterregulatory response to hypoglycemia. Simultaneously, β- and δ-cells are inhibited via α2 adrenergic receptors, which suppress insulin secretion and facilitate de-repression of α-cells from somatostatin-mediated inhibition, respectively (20, 57). δ-Cells are also suppressed by cholinergic inputs from autonomic innervation in mouse islets or from acetylcholine release by human α-cells (58). Recent transcriptomes from mouse (1, 4) and human (39) δ-cells have validated the δ-cell-selective expression of these receptors, and suggest furthermore that receptors for leptin (LEPR) and dopamine (DRD2) are expressed by human, but not mouse, δ-cells. Collectively, these observations cast the δ-cell as a central hub within the islet that translates inputs from paracrine and endocrine signals, nutrients, and neurotransmitters into appropriate intra-islet feedback inhibition via somatostatin (FIGURE 1) (39).
Local Feedback Inhibition by δ-Cells Determines the Set Point for Plasma Glucose
The physiological significance of δ-cell paracrine signaling is highlighted by the role of the UCN3-induced, δ-cell-mediated negative feedback loop in postnatal development. Full expression of endogenous UCN3 does not occur until 2 wk postpartum (P14) and coincides with a notable attenuation of plasma insulin and rise in glucose levels at this young age in mice (6, 79). To establish causality, we generated a doxycycline-inducible β-cell-specific bitransgenic mouse model to induce endogenous levels of UCN3 specifically within insulin-expressing cells with an onset and duration of our choosing (78). We induced UCN3 prematurely by administering doxycycline to pregnant dams incapable of UCN3 induction from E10.5 onward. This resulted in a premature increase in plasma glucose in bitransgenic offspring, which reflects the premature onset of UCN3-driven, somatostatin-mediated inhibition of insulin (FIGURE 3, A AND B). In control littermates, this feedback does not set in until full expression of endogenous UCN3 after P14. This experiment established that the onset of local inhibitory feedback by pancreatic δ-cells on insulin release determines the homeostatic set point for plasma glucose. Since the induction of UCN3 is restricted to pancreatic β-cells and remains undetectable in blood, we successfully isolated the effect of pancreatic δ-cells from the potentially confounding contributions of somatostatin by other sources, such as the enteroendocrine D cells responsible for most of the circulating somatostatin (25, 74).
FIGURE 3.

The homeostatic set point for glucose
A: in the absence of UCN3 in young neonatal mice, the homeostatic set point for glucose is determined by the balance between insulin and glucagon action. B: after the onset of UCN3 expression in mouse β-cells, β-cell activation leads to the co-secretion of UCN3 with insulin. This activates feedback inhibition that curbs insulin secretion and effectively reduces insulin action.
The Benefit of Feedback Inhibition by δ-Cells
As a field, the focus on restoring β-cell mass and function to increase insulin output and better manage diabetes makes it easy to forget that negative feedback regulation is a fundamental principle in biology to which β-cells are no exception. What then is the benefit of a δ-cell-mediated feedback mechanism that inhibits insulin secretion? Unlike incretin hormones, which can only amplify insulin secretion during hyperglycemia and are therefore relatively safe from stimulating insulin during hypoglycemia, insulin itself has a very real potential to cause dangerous episodes of hypoglycemia. Indeed, insulin-induced hypoglycemia is a major risk factor that contributes to the death of too many patients who manage their diabetes with insulin (15). Somatostatin-mediated feedback control on β-cells is the mechanism by which healthy islets prevent excess insulin release. This feedback control must be robust because even a single hypoglycemic episode can be fatal. Our working model is that the benefit of δ-cell-mediated feedback 1) prevents hyperinsulinemia-induced hypoglycemia and 2) ensures stable euglycemia with minimal deviations from the glucose set point.
Insulin secretion under hyperglycemia is pulsatile and driven by β-cell autonomous mechanisms (31, 45). Somatostatin secretion is also pulsatile, synchronized with β-cells, but trails insulin release by 30 s to a few minutes (28, 29, 65). Although somatostatin secretion is triggered by glucose alone below the glucose threshold of β-cells (FIGURE 1B), the majority of somatostatin release during hyperglycemia depends on UCN3 from β-cells (78), which may account for the delay in somatostatin secretion. A model where somatostatin secretion directly depends on the paracrine actions of β-cell-derived UCN3 ensures 1) synchronicity of δ- and β-cell activity, 2) proportionality between the degree by which plasma glucose and insulin have deviated from their set point and the strength of the ensuing negative feedback, 3) δ-cell-mediated feedback control of insulin secretion is activated with an intrinsic delay. The purpose of negative feedback is not to prevent the initiation of insulin secretion in the face of hyperglycemia; that would lead to diabetes. A delay in somatostatin secretion ensures that the initial insulin secretion in response to hyperglycemia proceeds uninhibited. But thereafter, what we consider to be GSIS is in fact the net result of the simultaneous stimulation of β-cells with glucose and inhibition with somatostatin (FIGURE 4A). We propose that such feedback inhibition on β-cells is instrumental in precisely attenuating insulin secretion in anticipation of the return of plasma glucose to its homeostatic set point (FIGURE 4A). This prevents overshooting of insulin, which would cause a hypoglycemic excursion.
FIGURE 4.
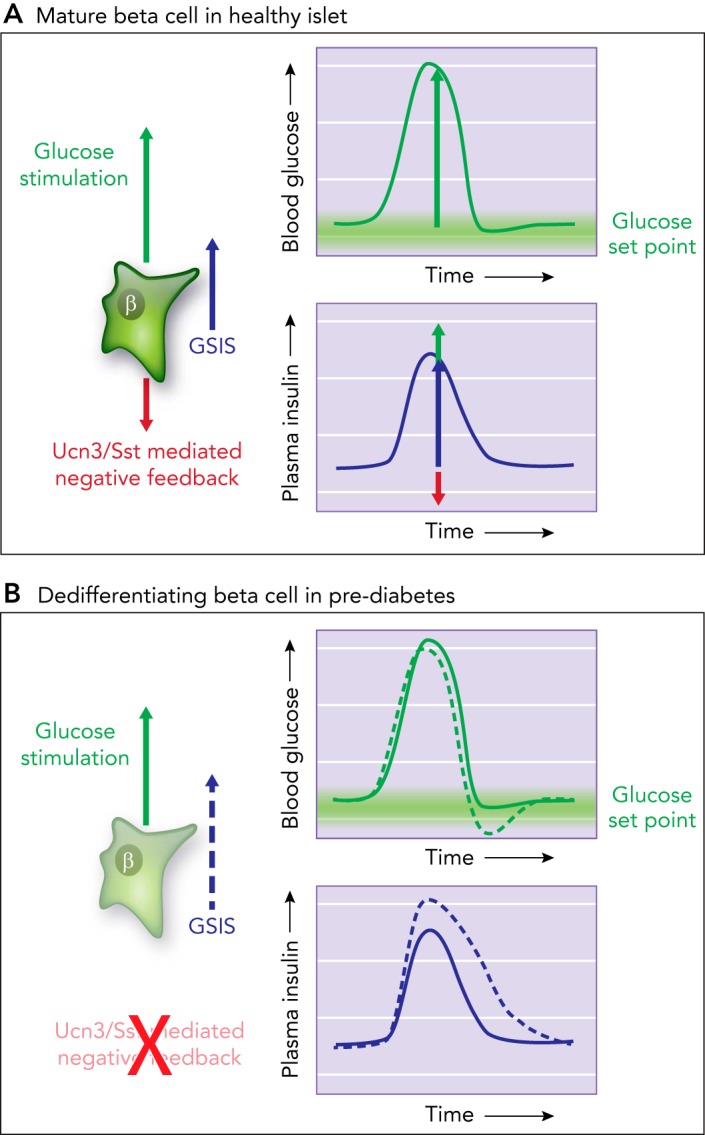
Model of tonic feedback inhibition on β- and α-cells
A: model how tonic feedback inhibition on β- and α-cells ensures the timely attenuation of insulin (or glucagon) secretion as glucose is restored to its homeostatic equilibrium. B: when this local feedback breaks down, insulin secretion is des-inhibited. This prolongs insulin action, which causes glucose values to overshoot their glucose set point and contributes to hyperglycemia in diabetes.
Recently, two reports suggested that δ-cells are directly coupled to β-cells via gap junctions to explain the synchronicity of pulsatile insulin and somatostatin responses (12, 81), a possibility that had previously been ruled out (47). Indeed, this would lead to instant activation of δ-cells on β-cell activation and could not account for the delay in pulsatile somatostatin release compared with insulin. We have looked at the responses of several thousand δ-cells within intact islets and have yet to observe clear evidence of direct gap-junction connections between δ- and β-cells (Huising laboratory, unpublished observations).
Loss of UCN3 from β-Cells Early in Diabetes Increases Glycemic Volatility
UCN3 is one of the first β-cell markers to disappear in pre-diabetes (78). The mechanistic basis for this rapid downregulation is not well understood beyond the observation that treatment with pro-inflammatory cytokines in vitro (7) or exposure to a pro-inflammatory environment in the context of the non-obese diabetic (NOD) mouse model of T1D (63) causes a loss of β-cell UCN3 expression. Treatment with the β-cell toxin streptozotocin similarly causes the downregulation of UCN3, suggesting that the STZ-induced nitric oxide (NO) and reactive oxygen species (ROS) cause oxidative stress (73) that inhibits UCN3 expression (78). Regardless of the mechanism(s) responsible for the downregulation of UCN3 in diabetes, the loss of UCN3 deprives δ-cells of the principal signal they need to secrete somatostatin in response to hyperglycemia (78), even though δ-cell numbers are relatively unaffected in diabetes (30, 52, 64, 71). Indeed, restoration of endogenous levels of UCN3 in diabetic β-cells using a doxycycline-inducible mouse model secondary to the loss of UCN3 in T2D aggravated diabetes (78), likely by increasing somatostatin-mediated suppression of insulin. Loss of UCN3 during diabetes is therefore a partially adaptive response that maximizes insulin output in the face of increasing peripheral insulin resistance in T2D (78). However, this comes at the expense of local feedback inhibition of β-cells. Under circumstances where normal UCN3 and somatostatin-mediated feedback control of β-cells breaks down, GSIS truly becomes dependent on glucose alone. The absence of negative feedback initially allows for excess insulin secretion (FIGURE 4B), with extended insulin action causing plasma glucose to overshoot its set point, activating counterregulation and contributing to glycemic volatility. Indeed, by CGM of ob/ob mice, we observed markedly increased glycemic volatility in addition to severe hyperglycemia (78). More recently, it was shown that the onset of T1D in NOD mice and T2D in ZDF rats is characterized by a marked increase in the amplitude of glucose excursions (36, 85). This observation is consistent with the progressive loss of UCN3—and the feedback control it triggers—before the autoimmune-mediated demise of β-cells that causes full-blown hyperglycemia (63).
Summary and Conclusions
Pancreatic δ-cells are emerging as important contributors that are well-positioned to modulate insulin and glucagon secretion directly at their source. The intra-islet feedback inhibition that δ-cells provide to β- and α-cells is essential for precise control and coordination of insulin and glucagon secretion. These interactions are necessary for stable glycemic control and determine the homeostatic set point for glucose. In addition to their paracrine activation by UCN3, δ-cells receive selective inputs from multiple hormones, neurotransmitters, and nutrients, and integrate these into appropriate feedback modulation of insulin and glucagon secretion. Finally, loss of normal δ-cell-mediated feedback inhibition occurs early in diabetes and likely contributes significantly to glycemic volatility and other aspects of the pathophysiology of diabetes, including excess glucagon secretion during hyperglycemia. As we better appreciate the physiological contribution of δ-cells to glucose homeostasis, we would be remiss if we did not consider δ-cells and somatostatin as therapeutic targets to realign insulin and glucagon release in diabetes. Although sustained restoration of UCN3 in T2D aggravated hyperglycemia (78), this observation does not disqualify δ-cell-dependent feedback as a target in T2D. It merely indicates that continuous activation of δ-cell-dependent feedback in diabetes is no more advisable than the continuous administration of insulin in diabetes. Analogous to insulin, there is a need to align δ-cell release of somatostatin with the time its actions are most protective, whether by preventing episodes of hyperinsulinemic hypoglycemia or by curbing excess glucagon secretion in T2D.
Acknowledgments
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (NID DK-110276) and the Juvenile Diabetes Research Foundation (CDA-2-2013-54) (to M.O.H.). J.L.H. and G.M.N. were supported by a National Institute of General Medical Sciences-funded Pharmacology Training Program (T32 GM-099608).
M.O.H. receives grant support from Crinetics, Inc. to evaluate proprietary somatostatin-related compounds. None of this work is discussed in this paper.
M.O.H., T.v.d.M., and G.M.N. conceived and designed research; M.O.H. and M.S.P. analyzed data; M.O.H., T.v.d.M., J.L.H., M.S.P., and G.M.N. interpreted results of experiments; M.O.H. prepared figures; M.O.H. drafted manuscript; M.O.H., T.v.d.M., J.L.H., M.S.P., and G.M.N. edited and revised manuscript; M.O.H., T.v.d.M., J.L.H., M.S.P., and G.M.N. approved final version of manuscript; M.S.P. performed experiments.
Footnotes
Assuming three meals a day, average U.S. life expectancy of 79.56 yr (source: https://www.cia.gov/library/publications/the-world-factbook/fields/2102.html).
References
- 1.Adriaenssens AE, Svendsen B, Lam BY, Yeo GS, Holst JJ, Reimann F, Gribble FM. Transcriptomic profiling of pancreatic alpha, beta and δ-cell populations identifies δ-cells as a principal target for ghrelin in mouse islets. Diabetologia 59: 2156–2165, 2016. doi: 10.1007/s00125-016-4033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barden N, Alvarado-Urbina G, Côté JP, Dupont A. Cyclic AMP-dependent stimulation of somatostatin secretion by isolated rat islets of Langerhans. Biochem Biophys Res Commun 71: 840–844, 1976. doi: 10.1016/0006-291X(76)90907-4. [DOI] [PubMed] [Google Scholar]
- 3.Baskin DG. A historical perspective on the identification of cell types in pancreatic islets of Langerhans by staining and histochemical techniques. J Histochem Cytochem 63: 543–558, 2015. doi: 10.1369/0022155415589119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner C, van der Meulen T, Cacéres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse β-cells compared to human β-cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 15: 620, 2014. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benninger RK, Hutchens T, Head WS, McCaughey MJ, Zhang M, Le Marchand SJ, Satin LS, Piston DW. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca2+ wave dynamics. Biophys J 107: 2723–2733, 2014. doi: 10.1016/j.bpj.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 30: 261–264, 2012. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, Melton DA. Reversal of β cell de-differentiation by a small molecule inhibitor of the TGFβ pathway. eLife 3: e02809, 2014. doi: 10.7554/eLife.02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonner-Weir S, Orci L. New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes 31: 883–889, 1982. doi: 10.2337/diab.31.10.883. [DOI] [PubMed] [Google Scholar]
- 9.Braun M. The somatostatin receptor in human pancreatic β-cells. Vitam Horm 95: 165–193, 2014. doi: 10.1016/B978-0-12-800174-5.00007-7. [DOI] [PubMed] [Google Scholar]
- 10.Braun M, Ramracheya R, Amisten S, Bengtsson M, Moritoh Y, Zhang Q, Johnson PR, Rorsman P. Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic δ-cells. Diabetologia 52: 1566–1578, 2009. doi: 10.1007/s00125-009-1382-z. [DOI] [PubMed] [Google Scholar]
- 11.Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 179: 77–79, 1973. doi: 10.1126/science.179.4068.77. [DOI] [PubMed] [Google Scholar]
- 12.Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, Rorsman P. δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J Physiol 596: 197–215, 2018. doi: 10.1113/JP274581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown M, Rivier J, Vale W. Biological activity of somatostatin and somatostatin analogs on inhibtion of arginine-induced insulin and glucagon release in the rat. Endocrinology 98: 336–343, 1976. doi: 10.1210/endo-98-2-336. [DOI] [PubMed] [Google Scholar]
- 14.Colturi TJ, Unger RH, Feldman M. Role of circulating somatostatin in regulation of gastric acid secretion, gastrin release, and islet cell function. Studies in healthy subjects and duodenal ulcer patients. J Clin Invest 74: 417–423, 1984. doi: 10.1172/JCI111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cryer PE. Hypoglycemia-associated autonomic failure in diabetes: maladaptive, adaptive, or both? Diabetes 64: 2322–2323, 2015. doi: 10.2337/db15-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dean PM, Matthews EK. Glucose-induced electrical activity in pancreatic islet cells. J Physiol 210: 255–264, 1970. doi: 10.1113/jphysiol.1970.sp009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dezaki K, Damdindorj B, Sone H, Dyachok O, Tengholm A, Gylfe E, Kurashina T, Yoshida M, Kakei M, Yada T. Ghrelin attenuates cAMP-PKA signaling to evoke insulinostatic cascade in islet β-cells. Diabetes 60: 2315–2324, 2011. doi: 10.2337/db11-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dezaki K, Hosoda H, Kakei M, Hashiguchi S, Watanabe M, Kangawa K, Yada T. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes 53: 3142–3151, 2004. doi: 10.2337/diabetes.53.12.3142. [DOI] [PubMed] [Google Scholar]
- 19.Diez JA, Arrojo E Drigo R, Zheng X, Stelmashenko OV, Chua M, Rodriguez-Diaz R, Fukuda M, Köhler M, Leibiger I, Tun SBB, Ali Y, Augustine GJ, Barathi VA, Berggren PO. Pancreatic islet blood flow dynamics in primates. Cell Reports 20: 1490–1501, 2017. doi: 10.1016/j.celrep.2017.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, Huising MO. Comprehensive alpha, beta and δ-cell transcriptomes reveal that ghrelin selectively activates δ-cells and promotes somatostatin release from pancreatic islets. Mol Metab 5: 449–458, 2016. doi: 10.1016/j.molmet.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubois MP. Immunoreactive somatostatin is present in discrete cells of the endocrine pancreas. Proc Natl Acad Sci USA 72: 1340–1343, 1975. doi: 10.1073/pnas.72.4.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 54: 1808–1815, 2005. doi: 10.2337/diabetes.54.6.1808. [DOI] [PubMed] [Google Scholar]
- 23.Göpel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterisation of somatostatin-secreting δ-cells in intact mouse pancreatic islets. J Physiol 528: 497–507, 2000. doi: 10.1111/j.1469-7793.2000.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28: 84–116, 2007. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 25.Gutniak M, Grill V, Wiechel KL, Efendić S. Basal and meal-induced somatostatin-like immunoreactivity in healthy subjects and in IDDM and totally pancreatectomized patients. Effects of acute blood glucose normalization. Diabetes 36: 802–807, 1987. doi: 10.2337/diab.36.7.802. [DOI] [PubMed] [Google Scholar]
- 26.Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ, Christie MR, Persaud SJ, Jones PM. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58: 403–411, 2009. doi: 10.2337/db08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RK. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 61: 1700–1707, 2012. doi: 10.2337/db11-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellman B, Salehi A, Grapengiesser E, Gylfe E. Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem Biophys Res Commun 417: 1219–1223, 2012. doi: 10.1016/j.bbrc.2011.12.113. [DOI] [PubMed] [Google Scholar]
- 29.Hellman B, Salehi A, Gylfe E, Dansk H, Grapengiesser E. Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology 150: 5334–5340, 2009. doi: 10.1210/en.2009-0600. [DOI] [PubMed] [Google Scholar]
- 30.Henquin JC, Ibrahim MM, Rahier J. Insulin, glucagon and somatostatin stores in the pancreas of subjects with Type-2 diabetes and their lean and obese non-diabetic controls. Sci Rep 7: 11015, 2017. doi: 10.1038/s41598-017-10296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoang DT, Hara M, Jo J. Design principles of pancreatic islets: glucose-dependent coordination of hormone pulses. PLoS One 11: e0152446, 2016. doi: 10.1371/journal.pone.0152446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hökfelt T, Efendić S, Hellerström C, Johansson O, Luft R, Arimura A. Cellular localization of somatostatin in endocrine-like cells and neurons of the rat with special references to the A1-cells of the pancreatic islets and to the hypothalamus. Acta Endocrinol Suppl (Copenh) 200: 5–41, 1975. [PubMed] [Google Scholar]
- 33.Huising MO, Pilbrow AP, Matsumoto M, van der Meulen T, Park H, Vaughan JM, Lee S, Vale WW. Glucocorticoids differentially regulate the expression of CRFR1 and CRFR2α in MIN6 insulinoma cells and rodent islets. Endocrinology 152: 138–150, 2011. doi: 10.1210/en.2010-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ipp E, Dobbs RE, Arimura A, Vale W, Harris V, Unger RH. Release of immunoreactive somatostatin from the pancreas in response to glucose, amino acids, pancreozymin-cholecystokinin, and tolbutamide. J Clin Invest 60: 760–765, 1977. doi: 10.1172/JCI108829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic β-cells through GPR40. Nature 422: 173–176, 2003. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 36.Korstanje R, Ryan JL, Savage HS, Lyons BL, Kane KG, Sukoff Rizzo SJ. Continuous glucose monitoring in female NOD mice reveals daily rhythms and a negative correlation with body temperature. Endocrinology 158: 2707–2712, 2017. doi: 10.1210/en.2017-00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kristinsson H, Bergsten P, Sargsyan E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim Biophys Acta 1853: 3248–3257, 2015. doi: 10.1016/j.bbamcr.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 38.Kumar U, Sasi R, Suresh S, Patel A, Thangaraju M, Metrakos P, Patel SC, Patel YC. Subtype-selective expression of the five somatostatin receptors (hSSTR1-5) in human pancreatic islet cells: a quantitative double-label immunohistochemical analysis. Diabetes 48: 77–85, 1999. doi: 10.2337/diabetes.48.1.77. [DOI] [PubMed] [Google Scholar]
- 39.Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, Kycia I, Robson P, Stitzel ML. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in Type 2 diabetes. Genome Res 27: 208–222, 2017. doi: 10.1101/gr.212720.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Chen P, Vaughan J, Lee KF, Vale W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc Natl Acad Sci USA 104: 4206–4211, 2007. doi: 10.1073/pnas.0611641104. . A correction for this article is available at doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu YJ, Vieira E, Gylfe E. A store-operated mechanism determines the activity of the electrically excitable glucagon-secreting pancreatic alpha-cell. Cell Calcium 35: 357–365, 2004. doi: 10.1016/j.ceca.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Loud FB, Holst JJ, Egense E, Petersen B, Christiansen J. Is somatostatin a humoral regulator of the endocrine pancreas and gastric acid secretion in man? Gut 26: 445–449, 1985. doi: 10.1136/gut.26.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PR, Cox R, Eliasson L, Rorsman P. A K ATP channel-dependent pathway within α-cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 5: e143, 2007. doi: 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menke A, Casagrande S, Geiss L, Cowie CC. Prevalence of and trends in diabetes among adults in the United States, 1988-2012. JAMA 314: 1021–1029, 2015. doi: 10.1001/jama.2015.10029. [DOI] [PubMed] [Google Scholar]
- 45.Merrins MJ, Poudel C, McKenna JP, Ha J, Sherman A, Bertram R, Satin LS. Phase analysis of metabolic oscillations and membrane potential in pancreatic islet β-cells. Biophys J 110: 691–699, 2016. doi: 10.1016/j.bpj.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami T, Fujita T, Miyake T, Ohtsuka A, Taguchi T, Kikuta A. The insulo-acinar portal and insulo-venous drainage systems in the pancreas of the mouse, dog, monkey and certain other animals: a scanning electron microscopic study of corrosion casts. Arch Histol Cytol 56: 127–147, 1993. doi: 10.1679/aohc.56.127. [DOI] [PubMed] [Google Scholar]
- 47.Nadal A, Quesada I, Soria B. Homologous and heterologous asynchronicity between identified alpha-, beta- and delta-cells within intact islets of Langerhans in the mouse. J Physiol 517: 85–93, 1999. doi: 10.1111/j.1469-7793.1999.0085z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nyman LR, Ford E, Powers AC, Piston DW. Glucose-dependent blood flow dynamics in murine pancreatic islets in vivo. Am J Physiol Endocrinol Metab 298: E807–E814, 2010. doi: 10.1152/ajpendo.00715.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohtani O, Ushiki T, Kanazawa H, Fujita T. Microcirculation of the pancreas in the rat and rabbit with special reference to the insulo-acinar portal system and emissary vein of the islet. Arch Histol Jpn 49: 45–60, 1986. doi: 10.1679/aohc.49.45. [DOI] [PubMed] [Google Scholar]
- 50.Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol 20: 157–198, 1999. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 51.Portela-Gomes GM, Stridsberg M, Grimelius L, Rorstad O, Janson ET. Differential expression of the five somatostatin receptor subtypes in human benign and malignant insulinomas - predominance of receptor subtype 4. Endocr Pathol 18: 79–85, 2007. doi: 10.1007/s12022-007-0014-8. [DOI] [PubMed] [Google Scholar]
- 52.Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia 24: 366–371, 1983. doi: 10.1007/BF00251826. [DOI] [PubMed] [Google Scholar]
- 53.Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 54: 1789–1797, 2005. doi: 10.2337/diabetes.54.6.1789. [DOI] [PubMed] [Google Scholar]
- 54.Reimer MK, Pacini G, Ahrén B. Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 144: 916–921, 2003. doi: 10.1210/en.2002-220819. [DOI] [PubMed] [Google Scholar]
- 55.Reubi JC, Schaer JC, Wenger S, Hoeger C, Erchegyi J, Waser B, Rivier J. SST3-selective potent peptidic somatostatin receptor antagonists. Proc Natl Acad Sci USA 97: 13973–13978, 2000. doi: 10.1073/pnas.250483897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivier J, Gulyas J, Kirby D, Low W, Perrin MH, Kunitake K, DiGruccio M, Vaughan J, Reubi JC, Waser B, Koerber SC, Martinez V, Wang L, Taché Y, Vale W. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J Med Chem 45: 4737–4747, 2002. doi: 10.1021/jm0202122. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-Diaz R, Caicedo A. Novel approaches to studying the role of innervation in the biology of pancreatic islets. Endocrinol Metab Clin North Am 42: 39–56, 2013. doi: 10.1016/j.ecl.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO, Caicedo A. Α-cells secrete acetylcholine as a non-neuronal paracrine signal priming β-cell function in humans. Nat Med 17: 888–892, 2011. doi: 10.1038/nm.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, Leibiger IB, Kenyon NS, Ricordi C, Pileggi A, Caicedo A, Berggren PO. Paracrine interactions within the pancreatic islet determine the glycemic set point. Cell Metab 27: 549–558.e4, 2018. doi: 10.1016/j.cmet.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rorsman P, Berggren PO, Bokvist K, Ericson H, Möhler H, Ostenson CG, Smith PA. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 341: 233–236, 1989. doi: 10.1038/341233a0. [DOI] [PubMed] [Google Scholar]
- 61.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 75: 155–179, 2013. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 62.Rorsman P, Huising MO. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat Rev Endocrinol 14: 404–414, 2018. doi: 10.1038/s41574-018-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab 25: 727–738, 2017. doi: 10.1016/j.cmet.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saito K, Yaginuma N, Takahashi T. Differential volumetry of A, B and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J Exp Med 129: 273–283, 1979. doi: 10.1620/tjem.129.273. [DOI] [PubMed] [Google Scholar]
- 65.Salehi A, Qader SS, Grapengiesser E, Hellman B. Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul Pept 144: 43–49, 2007. doi: 10.1016/j.regpep.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 66.Samols E, Marri G, Marks V. Interrelationship of glucagon, insulin and glucose. The insulinogenic effect of glucagon. Diabetes 15: 855–866, 1966. doi: 10.2337/diab.15.12.855. [DOI] [PubMed] [Google Scholar]
- 67.Samols E, Marri G, Marks V. Promotion of insulin secretion by glucagon. Lancet 2: 415–416, 1965. doi: 10.1016/S0140-6736(65)90761-0. [DOI] [PubMed] [Google Scholar]
- 68.Samols E, Stagner JI. Islet somatostatin–microvascular, paracrine, and pulsatile regulation. Metabolism 39, Suppl 2: 55–60, 1990. doi: 10.1016/0026-0495(90)90212-U. [DOI] [PubMed] [Google Scholar]
- 69.Schauder P, McIntosh C, Arends J, Arnold R, Frerichs H, Creutzfeldt W. Somatostatin and insulin release from isolated rat pancreatic islets stimulated by glucose. FEBS Lett 68: 225–227, 1976. doi: 10.1016/0014-5793(76)80441-3. [DOI] [PubMed] [Google Scholar]
- 70.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing β-cells in the mammalian pancreas. Nature 386: 399–402, 1997. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- 71.Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y, Unger RH. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes 31: 694–700, 1982. doi: 10.2337/diab.31.8.694. [DOI] [PubMed] [Google Scholar]
- 72.Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Sörhede Winzell M, Hammar M, Xu X, Smith DM, Morgan NG. GPR120 (FFAR4) is preferentially expressed in pancreatic δ-cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia 57: 1182–1191, 2014. doi: 10.1007/s00125-014-3213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 50: 537–546, 2001. [PubMed] [Google Scholar]
- 74.Taborsky GJ Jr, Ensinck JW. Contribution of the pancreas to circulating somatostatin-like immunoreactivity in the normal dog. J Clin Invest 73: 216–223, 1984. doi: 10.1172/JCI111194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taborsky GJ Jr, Mundinger TO. Minireview: The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153: 1055–1062, 2012. doi: 10.1210/en.2011-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Kahn SE, Cummings DE, Tschöp MH, D’Alessio D. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 59: 2145–2151, 2010. doi: 10.2337/db10-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122: 4–12, 2012. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, Huising MO. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med 21: 769–776, 2015. doi: 10.1038/nm.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and β-cells. PLoS One 7: e52181, 2012. doi: 10.1371/journal.pone.0052181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vieira E, Salehi A, Gylfe E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic α-cells. Diabetologia 50: 370–379, 2007. doi: 10.1007/s00125-006-0511-1. [DOI] [PubMed] [Google Scholar]
- 81.Vierra NC, Dickerson MT, Jordan KL, Dadi PK, Katdare KA, Altman MK, Milian SC, Jacobson DA. TALK-1 reduces delta-cell endoplasmic reticulum and cytoplasmic calcium levels limiting somatostatin secretion. Mol Metab 9: 84–97, 2018. doi: 10.1016/j.molmet.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 13, Suppl 1: 95–105, 2011. doi: 10.1111/j.1463-1326.2011.01450.x. [DOI] [PubMed] [Google Scholar]
- 83.Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, Sewing S, Rorsman P, Braun M. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 53: 1038–1045, 2004. doi: 10.2337/diabetes.53.4.1038. [DOI] [PubMed] [Google Scholar]
- 84.Yada T, Damdindorj B, Rita RS, Kurashina T, Ando A, Taguchi M, Koizumi M, Sone H, Nakata M, Kakei M, Dezaki K. Ghrelin signalling in β-cells regulates insulin secretion and blood glucose. Diabetes Obes Metab 16, Suppl 1: 111–117, 2014. doi: 10.1111/dom.12344. [DOI] [PubMed] [Google Scholar]
- 85.Zhang XD, Pechter D, Yang L, Ping X, Yao Z, Zhang R, Shen X, Li NX, Connick J, Nawrocki AR, Chakravarthy M, Li C. Decreased complexity of glucose dynamics preceding the onset of diabetes in mice and rats. PLoS One 12: e0182810, 2017. doi: 10.1371/journal.pone.0182810. [DOI] [PMC free article] [PubMed] [Google Scholar]