

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in genes encoding the polycystin (PC) 1 and 2 proteins. The goal of this study was to determine the role of calcium in regulating cyst growth. Stromal interaction molecule 1 (STIM1) protein expression was 15-fold higher in PC1-null proximal tubule cells (PN) than in heterozygote (PH) controls and 2-fold higher in an inducible, PC1 knockout, mouse model of ADPKD compared to a non-cystic match control. IP3 receptor protein expression was also higher in the cystic mice. Knocking down STIM1 with siRNA reduced cyst growth and lowered cAMP levels in PN cells. Fura2 measurements of intracellular Ca2+ showed a dramatic reduction in thapsigargin-stimulated release of ER Ca2+ following STIM1 silencing or application of 2-APB, consistent with altered ER Ca2+ movement; the protein expression of the Ca2+-dependent adenylyl cyclases (AC) AC3 and AC6 was up- and down-regulated, respectively. Like STIM1 knockdown, application of the calmodulin inhibitor W7 lowered cAMP levels, further indicating that STIM1 regulates AC3 via Ca2+ We conclude that the high levels of STIM1 in ADPKD cells play a role in supporting cyst growth and promoting high cAMP levels and an increased release of Ca2+ from the ER. Thus, our results provide novel therapeutic targets for treating ADPKD.
Keywords: calcium, autosomal dominant polycystic kidney disease, STIM, cysts, cAMP
1. INTRODUCTION:
Polycystins and ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common dominant genetic disorder in humans [1]. A hallmark of the disease is the progressive enlargement of multiple renal cysts that leads to a decline in renal function and culminates in renal failure in 50% of all patients [2]. Mutations in the pkd1 and pdk2 genes are associated with ADPKD. These genes encode the polycystins, PC1 & 2, the latter, referred to as TRPP2 (transient receptor potential polycystic), functions as a non-selective cation channel[3] [4]. Malfunction of either PC1 or PC2 leads to cyst formation [5]
cAMP and ADPKD.
A key component of cyst formation is cAMP. cAMP-dependent signal transduction cascades are involved in regulating normal renal function [6]. For example, it is well known that during water conservation, arginine vasopressin (AVP) binds to the AVP receptor 2 (V2R) in the basolateral membrane and activates adenylate cyclase, increasing intracellular cAMP and Ca2+ levels [7] [8]. We and many others have detected elevated cAMP levels in animal- and cell-based models of ADPKD [9]. Whereas increased cAMP does not cause cells to proliferate in normal kidneys, it does so in ADPKD [10]. Thus, one scenario for how cysts grow is that dysregulation of Ca2+ and cAMP signaling, caused by tonic activation by AVP in ADPKD, stimulates cyst growth [9]. Indeed, clinical trials have tested the efficacy of V2R inhibition as a treatment for ADPKD, and remaining hydrated is recommended for patients to avoid activating AVP signaling [11]. Although there is now a drug approved for ADPKD, Tolvaptan [12], because of its potential serious side-effects, there is still a need for new therapies..
Calcium and ADPKD.
Misregulation of Ca2+ is associated with cyst formation in ADPKD [13], with some investigators reporting that disruption of Ca2+ signaling is the primary event that supports increased cyst growth [14]. Indeed, our groups’ early work was among the first to show that PC2 is involved in the movement of Ca2+ [15]. PC1 and 2 operate in concert at three locations within the cell: in the ER to regulate inositol triphosphate receptor (IP3R)-induced Ca2+ release, at the plasma membrane to regulate store-operated calcium entry (SOCE) via store-operated Ca2+ channels (SOC), and at the primary cilium [5] to perhaps sense fluid flow. Several studies have shown that a reduction in the function of either PC1 or 2 leads to dysregulation of Ca2+ signaling (see [9] for a review).
However, the precise details of how this misregulation of Ca2+ leads to aberrant cAMP signaling occurs are still very controversial. One scenario is that Ca2+ restriction in ADPKD cells causes cAMP-dependent activation of the B-Raf/mito gen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway, which results in increased cell growth [16]. Likewise, increased Ca2+ influx into ADPKD cells has been shown to restore more normal cAMP signaling, reducing cell growth [17]. On the other hand, we have evidence that elevated intracellular Ca2+ fuels cyst growth using a model ADPKD cell line (PN18= pkd1 knockout; PH= pkd1 heterozygote) clonally isolated from single parental clones obtained from a pkdfl/− mouse that had been manufactured in the ImmortoMouse containing the H-2Kb-tsA58 gene. The null cells (PN) stably express the Cre recombinase, and the control cells (PH) are from the original clone, which is heterozygous for the expression of PC1 [18, 19]. Others have shown that expression of the C-terminal fragment of PC1 can cause an increase in basal levels of intracellular Ca2+ and induce abnormal Ca2+ oscillations, which also result in increases in cell signaling [14]. Thus, there appears to be a dichotomy of thought on how Ca2+ plays a role in ADPKD with some thinking that Ca2+ restriction causes cyst growth and others that enhanced release of Ca2+ from the ER is the major factor which fuels cyst growth. It is important to mention it is known that excessive influx of Ca2+ contributes to the growth of certain cancers indicating that Ca2+ is a factor in fueling cell growth[20–22].
Calcium and cAMP.
The link between Ca2+ and increases in cAMP occurs through Ca2+-dependent adenylyl cyclases (ACs)[23]. Basically, there are two types of cyclases that respond to Ca2+: One is activated by Ca2+ via calmodulin (e.g., AC3); the other type is inhibited by Ca2+ (e.g., AC5/6) [24]. It is already known that AC6 is expressed at higher levels in ADPKD cells that lack PC1 [23] than in normal cells in which PC1 is functioning appropriately. Thus, as suggested in the literature, Ca2+ restriction would be expected to increase AC6-mediated production of cAMP [23]. In contrast, we have suggested that enhanced Ca2+ signaling via excessive release of Ca2+ from the ER occurs in ADPKD [25]. The higher levels of Ca2+ would activate AC3. Thus, we proposed an alternate hypothesis: that enhanced release of Ca2+ from the ER stimulates AC3 to elevate cAMP. AC3 is particularly relevant because it is associated with the primary cilium, particularly in the sensory system [26]. Furthermore, AC3 is normally expressed in renal epithelium [27]. Thus, our published data have prompted a shift in our understanding of the role of AC3 in ADPKD.
Role of STIM1:
The ER is a major storage area for Ca2+ and plays a key role in signal transduction [28]. ER Ca2+ is tightly regulated and one of the key factors in sensing ER calcium is STIM1. When STIM1 senses a reduction in ER Ca2+, it is translocated to the plasma membrane, where it activates store-operated Ca2+ entry (SOCE), increasing the movement of Ca2+ into the cell via Ca2+ release-activated Ca2+ channel modulator protein 1 (Orail), and perhaps other Ca2+ channels[28]. The Ca2+ that enters the cytosol is then returned to the ER via the sarco/ER Ca2+-ATPase (SERCA) Ca2+ pump [29]. It has been suggested that enhanced levels of STIM1 and excessive influx of Ca2+ contribute to the growth of certain cancers [20, 22, 30]
We have shown previously that PC1 binds to the IP3R and sequesters STIM1 in the ER [25, 31] reducing Ca2+-dependent cell signaling. In the absence of PC1 STIM1 is located primarily at the plasma membrane where it tonically increases SOCE [25]. We have created a renal-specific STIM1-knockout mouse that is normal in all respects except that it cannot concentrate its urine, pointing to a key role for STIM1 in the ability of the mice to regulated water balance [32]
Having uncovered a role for STIM1 in renal physiology and in view of the need to understand more about the role of Ca2+ in ADPKD, we conducted the present study in mice and cellular models of ADPKD. It is important to note that strategies are needed to restore normal Ca2+ metabolism in ADPKD patients in specific organs where ADPKD malfunctions, without affecting Ca2+ metabolism throughout the entire body.
2. METHODS:
Cell culture and reagents.
pkd1-null (PN) and control heterozygous (PH) cells were cultured as previously described[18, 19]. PN and PH cells were grown in 10-cm culture dishes under permissive conditions (33°C), with γ-interferon in the culture medium. Cells were then transferred to non-permissive conditions at 37°C in γ-interferon-free culture medium and evaluated at full confluency. Forskolin (#11018), IBMX (15879), 2APB (D9754) were purchased from Sigma; W7 was purchased from Tocris (#0369); adenylate cyclase 3 (SC588) and Ezrin (SC58758), PC2 (Sc28331), HSP27 (SC 13132), HSP70 (SC66048), STIM1 (SC6889), IP3R-I/II/III (SC3777518) and β-actin (SC47778) were purchased from Santa Cruz Biotech, TX,USA. HSP90 (ADI-SPA-830F) was purchased from Enzo Life Sciences, NY, USA. AC6 (GTX47798) was purchased from GeneTex, Irvine, CA, USA.
Mouse strain and treatment:
All animal use complied with the guiding principles of the Johns Hopkins University Institutional Animal Care and Use Committee, and the protocols for this work were approved by this Committee. Pkd1fl/fl;Pax8rtTA;TetO-cre mice on a C57BL/6 background[33] were provided by the Baltimore PKD Center and used to test the steady-state protein levels of STIM1. Mice of both sexes were used in this study. Pkd1fl/fl;Pax8rtTA;TetO-cre mice were injected IP with doxycycline resuspended in sterile water (4 μg of doxycycline/g body weight) on postnatal day (PND) 11, PND12, and PND13. This treatment produces very rapid and aggressive cyst growth[34, 35]. On PND21, the mice were euthanized.
Western blotting.
Cells cultured, harvested and processed as previously described [36]. In brief, the cells were solubilized in lysis buffer (150 mM Tris-HCl [pH 7.4] with 50 mM NaCl, 1% NP40, and Halt protease inhibitor) (Thermo Scientific, #78438). The cell lysates were centrifuged at 10,000×g for 10 min at 4°C to pellet insoluble material, and the supernatants were collected. The protein concentrations were measured with a Bio-Rad Protein Assay (Biorad, #500-0006), and the supernatants were then denatured in 2× Laemmli buffer at 37°C for 20 min and run on 3-8% SDS-PAGE gels (Thermo Scientific, #EA03785) before transfer to a polyvinylidene fluoride membrane (Bio-Rad). The membranes were incubated with primary antibodies overnight and then washed with TBS-Tween 20 buffer. An HRP-conjugated secondary antibody from GE Healthcare (NA934V; 1:10,000) was incubated with the membranes for 1 h, and then ECL Prime (GE Healthcare) was used for detection on film from Denville Scientific (E3018). The membranes were then stripped using Restore Western blot buffer (Pierce, VWR) and reprobed for protein loading controls.
cAMP assay:
PN and PH cells were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (DMEMZF12) medium supplemented with 3% FBS and γ-interferon (5 U/ml; Sigma-Aldrich) at 33°C and 5% CO2 and plated in a 6-well plate for 24 h. The cells were then changed into γ-interferon-free medium and maintained at 37°C for 4 days before being used in the experiment. cAMP levels were measured with a direct cAMP Enzyme Immunoassay Kit (Sigma-Aldrich, #CA200) according to the manufacturer’s protocol. The results are expressed as pmole/ml. Statistical analysis was performed using a two-tailed Student’s t-test.
Fura-2 Ca2+ imaging assay:
PN cells were maintained in DMEM/F12 medium supplemented with 3% FBS and y-interferon (5 U/ml; Sigma-Aldrich) at 33°C and 5% CO2 and plated in 35×10-mm cell culture dishes for 24 h. The cells were then changed into y-interferon-free medium and maintained at 37°C for 4 days. Confluent cells were treated with 2-ABP (10 μM) or DMSO for 16 h before being used in the experiment. On Day 5, the cells were washed three times in imaging buffer (20 mM HEPES with 126 mM NaCl, 4.5 mM KCl, 2 mM MgCl2, and 10 mM glucose at pH 7.4), then loaded with the cell permeant acetoxymethyl (AM) ester of the calcium indicator Fura-2 (Fura-2/AM) at 37°C for 60 min. Fura-2/AM was first dissolved in 1 mg/ml pluronic/DMSO and then diluted to 5 μM in imaging buffer containing 2 mM CaCl2 or calcium free imaging buffer in case of no calcium experiments. They were placed on the stage of a Zeiss inverted microscope equipped with a Sutter Lambda 10-2 controller and filter wheel assembly. For ATP stimulation experiments, the cells were exposed to 100 μM ATP diluted in the imaging buffer. A Zeiss FluorArc mercury lamp was used to excite the cells at 340 and 380 nm, and the emission response was measured at 510 nm. Cell fluorescence was measured in response to excitation for 1000 ms at 340 nm and 200 ms at 380 nm once every 4 s. Image acquisition, image analysis, and filter wheel control were performed using IPLab software.
siRNA knockdown of STIM1:
PN cells were cultured as described above. They were seeded onto six-well culture plates and grown to 50-60% confluence in complete growth medium at 33°C. They were then transferred to non-permissive conditions at 37°C, in γ-interferon- and antibiotic-free culture medium. Mouse STIM1 siRNA or scrambled siRNA (Origene#SR419122) was transfected into the cells using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer’s instructions, for various periods of time and using various siRNA concentrations. The 72-h time incubation time and 1-nM concentration provided the best knockdown of STIM1 protein expression and were used in subsequent experiments.
3. RESULTS:
PC1 regulates STIM and the IP3R.
To understand the role of STIM1 in ADPKD, we utilized a model ADPKD cell line as described above [18, 19]. We chose this cell line because the PH cells, containing PC1, and the PN, the PC1 null cells, originated from the same clone. Thus, they are from the same genetic background. All the cells are of proximal tubule origin [19]. To understand its role in ADPKD, we first measured the steady-state levels of STIM1 (Fig. 1A, B) in PN vs PH cells. Interestingly, there was a nearly 15-fold higher levels of resting levels of STIM1 in PN cells as compared to PH cells. To verify that this phenomenon was indeed of relevance to ADPKD, we harvested kidneys from mice representing a mouse model of ADPKD, the pkd1fl/fl;Pax8rtTA;TetO-cre mouse. These mice have an inducible TetO-cre with a floxed pkd1 gene. When they are treated with doxycycline, the functional product of the pkd1 gene is knocked out, resulting in a very aggressive model of PKD, with numerous cysts appearing in both the kidney cortex and medulla at 3 weeks of age[35]. Pkd1fl/fl;Pax8rtTA;TetO-cre mice were injected daily from PND11 to PND13 with doxycycline or DMSO (as a control). Kidneys were harvested on PND21, and STIM1 protein levels were assessed. It is noteworthy that we saw an approximately 2-fold higher level of STIM1 in the kidneys injected with doxycycline than in the untreated animals (Fig. 1C, D). Aggressive cyst formation was noted in the kidneys from the mice that received doxycycline.
Figure 1:
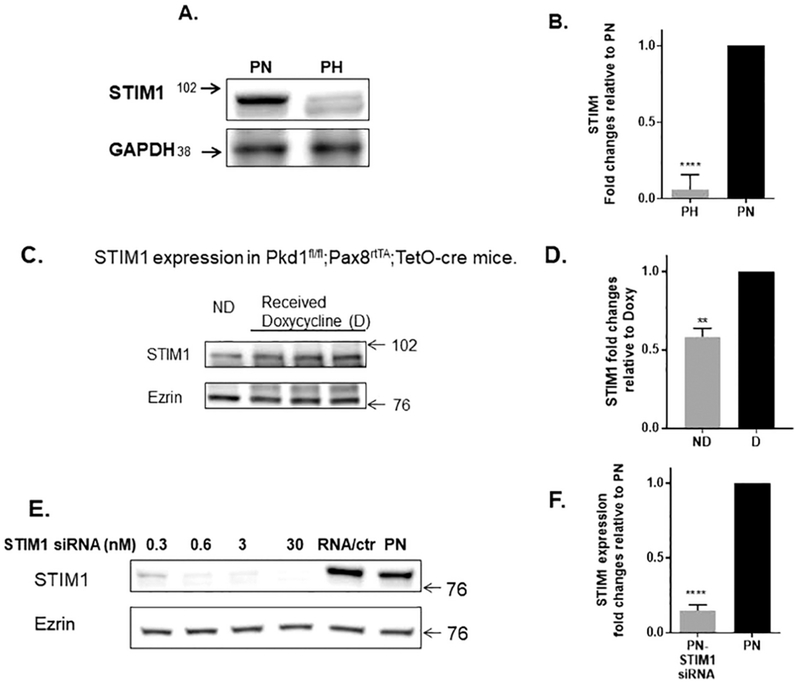
A-B). Steady State Levels of STIM1: Western blot showing expression of STIM1 in PN and PH cells. Note that STIM1 protein expression is 15 fold higher in PN vs. PH cells B) Columns represent averages ± standard errors of the STIM1expression in PN and PH cells. Experiment was repeated 6 times. C-D) STIM1 expression in pkd1fl/fl ;Pax8rta;TetO-cre mice. C) Representative image is of PN21 kidney tissue isolated from mice with no cysts (ND=pkd1 Δneo mouse not induced with doxycycline) and following treatment with doxycycline injected intraperitoneally (4 μg of doxycycline/g body weight) on postnatal days 11, 12, and 13. D) Columns represent averages ± standard errors (N=3). Note that STIM1 levels increase when the animals are treated with doxycycline to induce the PC 1 null phenotype. E-F). STIM1 Silencing. Mouse STIM1siRNA or scrambled siRNA (Orgene#SR419122) was transfected into cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions for various periods of time and concentrations. Note that the 3nM concentration and 72h time point showed better knockdown of STIM1. Columns represent averages ± standard errors of the STIM1 expression. Experiment was repeated 4 times. All data were analyzed by non-parametric t test. *P<0.05, **P<0.01, ****P< 0.0001.
Given that STIM1 was upregulated in PN cells when compared to PH cells, we silenced STIM1 using siRNA in order to begin to address the role of STIM1 in ADPKD. STIMI levels could be reduced effectively by using this approach (Fig 1E, F); they were reduced by approximately 85%.
Next, we determined the steady state levels of IP3R in the same mice kidneys depicted in Fig. 1C-D. Interestingly, inducing cyst formation by applying doxycycline to knockout PC1, causes a 5-fold increase in the steady state levels of the IP3R (Fig. 2A) . This indicates that key proteins involved in Ca2+ signaling in cells such as STIM1 and IP3R [28]are upregulated in ADPKD kidneys. On the other hand, there is no difference in expression in PN vs PH cells (Fig. 2B). Fig. 2C shows that silencing STIM1 in PN cells has no effect on IP3R protein expression. In contrast, 2-aminoethyl diphenyl borate (2-APB) which is known to be a non-specific modulator of SOCE and ER Ca2+ release [37] did cause an approximately 20% reduction in IP3R protein levels.
Figure 2: Steady State Levels of IP3R:
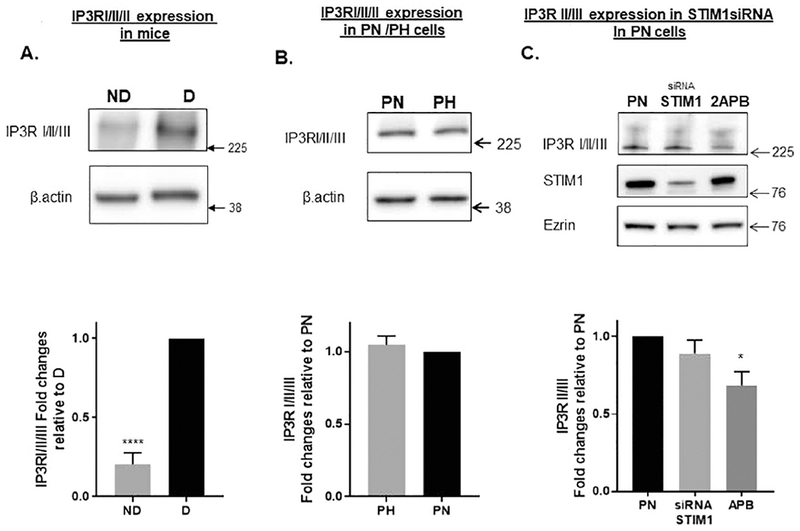
(A) IP3R expression in pkd1fl/fl ;Pax8rta;TetO-cre mice. Representative image is of PN21 kidney tissue isolated from mice with no cysts (pkd1 Δneo mouse not induced with doxycycline) and following treatment with doxycycline injected intraperitoneally (4 μg of doxycycline/g body weight) on postnatal days 11, 12, and 13. Columns represent averages ± standard errors (N=3). Note that IP3R levels increase when the animals are treated with doxycycline to induce the PC1 null phenotype. (B) Western blot showing expression of IP3R in PN and PH cells. Note that there is no difference in IP3R protein expression in PH vs. PN cells Columns represent averages ± standard errors of the IP3R expression in PN and PH cells. Experiment was repeated 6 times. (C). STIM1 Silencing and 2-APB treatment. Cells were treated STIM1siRNA or scrambled siRNA. Columns represent averages ± standard errors of the STIM1 expression. Experiment was repeated 4 times. All data were analyzed by non-parametric t test. *P<0.05. ****P< 0.0001. Note that 2-APB treatment reduced IP3R expression to a small extent.
Knockdown of STIM1 reduces cyst size.
One of the key questions that we needed to address was whether STIM1 plays a role in cyst growth. To answer this question, we grew PH and PN cells in 3D culture and measured the size of the cysts that formed. Fig. 3 shows that in PH cells no cysts developed. In contrast, large cysts developed in PN cells, as observed after 15-18 days in culture. PN cells were used in all subsequent experiments. As expected, the cysts grew larger when the mice were treated with the adenylyl cyclase activator forskolin, indicating that the cyst growth is indeed cAMP-dependent, as shown previously [38]. Knocking down STIM1 with siRNA inhibited cyst growth by ~58% when the cysts were viewed at 15 days. This is an important finding, in that it shows that when the cysts are treated at 10 days of culture, when cysts are already growing, knocking down STIM1 arrests their development. The inhibition of cyst growth was partially overcome when the cells with STIM1 knockdown were also treated with forskolin. Next, we applied 2-APB. Fig 3 shows that 2-APB application inhibited cyst growth to a greater magnitude then STIM1 silencing. Forskolin treatment caused a further dramatic inhibition of cyst growth when applied with 2-APB. These data suggest that maneuvers that affect Ca2+ metabolism such as STIM1 silencing and 2-APB reduce cyst growth.
Figure 3: Cysts growth in pkd1−/− mouse derived proximal tubule cells.
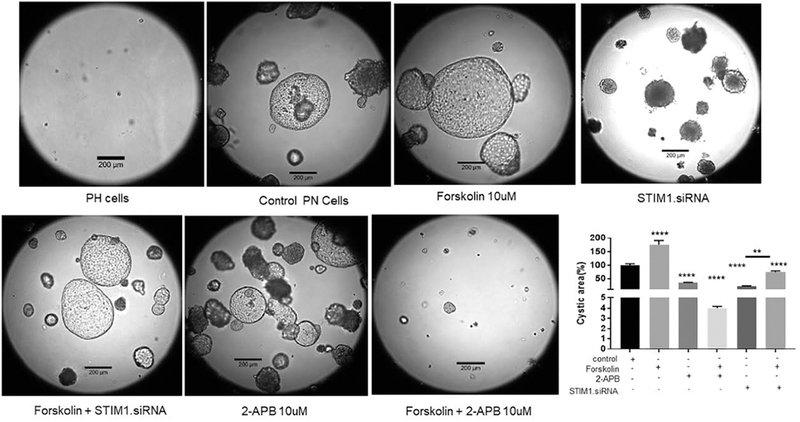
Cells were treated with Control (opti-MEM), Lipo –control (transfection reagent control), RNA control, STIM1 siRNA 3nM. SiRNA treated once on Day 11. Pics were taken on Day 18th. Columns represent mean ± SEM (n=6-10). Average cyst from control group was considered 100%, and the rest of the cysts were compared with this cyst.**P<0.01, ****P<0.0001.
SOCE is elevated in PC1 null cells.
As mentioned above, STIM1 plays a major role in the regulation of Ca2+ [39]. In order to explore the role of STIM1 in regulating Ca2+ in renal cells, we measured intracellular Ca2+ (Fig. 4A) using Fura2 as we had done previously [31]. As a first step, we evaluated the impact of PC1 on SOCE by removing Ca2+ from the extracellular solution. As shown in Fig. 4A&B there is as expected a small decrease in intracellular Ca2+. We treated the cells with thapsigargin, a specific inhibitor of the SERCA pump that, when applied, allows Ca2+ to leak out of the ER through independent Ca2+-permeable pathways [40]. Note that the increase in intracellular Ca2+ induced by thapsigargin Fig. 4 A&C was larger in PN vs PH cells as we showed previously [41]. Adding thapsigargin in the absence of extracellular Ca2+ is well-known to activate SOCE [42]. Fig 4 A&D shows a large transient increase in intracellular Ca2+ following an abrupt increase in extracellular Ca2+ to 5 mM which indicates Ca2+ entry across the plasma membrane. Note again that SOCE is much high in PN vs PH cells suggesting that a function PC1 inhibits SOCE [25].
Figure 4: Intracellular Ca2+ in PN vs. PH cells.
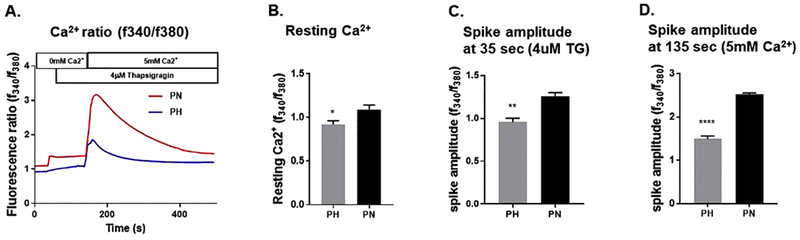
A) Representative traces of intracellular Ca2+ release in response to Thapsigargin (TG) in PN or PH cells. Cells were kept in zero Ca2+ for approximately one hour prior to the imaging experiment to induce SOCE and subsequently treated with 5 mM Ca2+ for the time indicated. Intracellular Ca2+ (F340/F380) levels obtained by ratiometric Fura-2 AM analysis of PN/PH cells. B) Graph summarizes resting calcium levels. C) Summarizes the average spike amplitude of Ca2+ release in response to TG in absence of extracellular calcium. D) Summarizes the average spike amplitude of Ca2+ release in response to 5 mM extracellular Ca2+ in presence of TG. Amplitude was measured as standard deviation of signal base to peak fluorescence ratio. Asterisk indicates significance between the two groups. *P < 0.05 **P < 0.01, ****P < 0.0001 (n=4-5).
Knockdown of STIM1 reduces Ca2+ release.
We first treated the cells with ATP, which causes an increase in intracellular Ca2+. As we had observed previously in MDCK cells [31], exposing the cells to ATP causes a transient increase in intracellular Ca2+ over basal levels (Fig. 5A-C). Silencing STIM1 had no effect on intracellular Ca2+, nor did it affect the Ca2+ transient induced by ATP. Given that purinergic receptors are themselves Ca2+ channels in the plasma membrane [43], the observation that the magnitude of the ATP-induced increase Ca2+ was unchanged can most likely be explained by the similar basal Ca2+ levels before and after STIM1 silencing. We next applied 2-APB, which produced a response, identical to that of knocking down STIM1. There was no change in either resting Ca2+ or in the ATP-induced increase in intracellular Ca2+ (Fig. 5D-F).
Figure 5: ATP-induced changes in intracellular Ca2+ in PN cells treated with STIM1siRNA or with 2-APB.
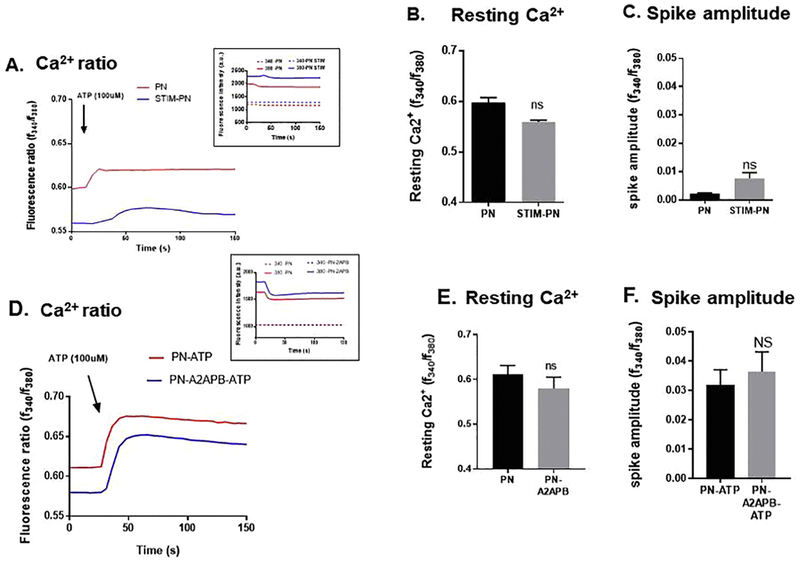
A) Representative traces of intracellular Ca+2 in response to 100 μM ATP in STIM1siRNA-PN/PH cells. Inset shows the individual tracings of 340 and 380 nm. B) Graph summarizes the average resting levels of intracellular Ca+2. C) Amplitude of Ca+2 release in response to ATP. (D) Representative traces of intracellular Ca+2 release in response to ATP (100μM) in PN cells untreated and treated with APB, 10 μM. E) Graph summarizes resting Ca2+ levels and F) graph summarizes the average spike amplitude of Ca2+ release in response to ATP. Note that neither STIM1 silencing or 2-APB had any effect on the resting Ca+2 or ATP induced increased in intracellular Ca+2. Amplitude was measured as standard deviation of signal base to peak Δf/f. Amplitude was measured as standard deviation of signal base to peak Δf/f. Asterisk indicates significance between the two groups (student’s t-test,, n=4-5). Measurements were made in standard Ca2+ solutions.
Next, we addressed the effect of STIM1 on ER Ca2+ release. Fig. 6A-B shows again that intracellular Ca2+ is higher in PN vs PH cells and that there is no effect of STIM1 silencing on resting Ca2+. Fig. 6 A&C shows that thapsigargin induce ER Ca2+ release is higher in PN vs. PH cells as we showed previously [41]. Importantly knockdown of STIM1 reduced the thapsigargin-induced release of Ca2+ from the ER in the PN cells to levels near to those observed in PH cells. Likewise, 2-APB had a similar dramatic effect. 2APB considerably slowed the thapsigargin-induced release of Ca2+ from the ER. Our data suggest that both STIM1 knockdown and 2-APB operate via a similar mechanism that alters the transport of Ca2+ across the ER membrane.
Figure 6: Thapsigargin-induced changes in intracellular Ca2+in PH/PN cells and in PN cells treated with STIM1siRNA or with 2-APB.
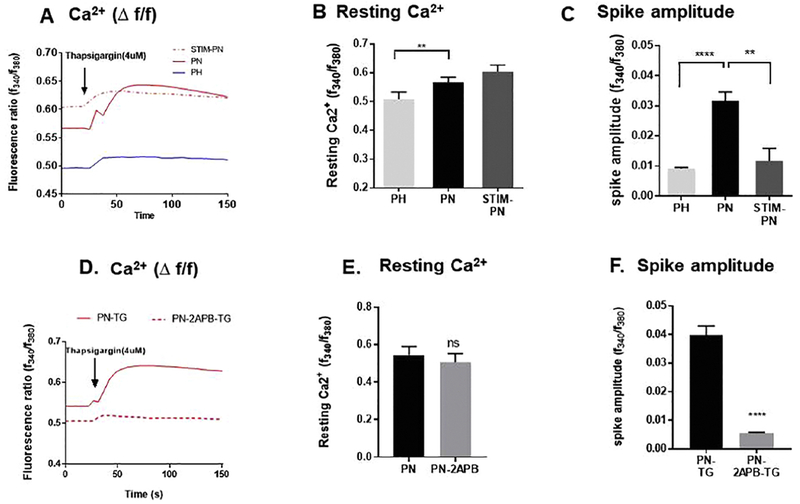
A) Representative traces of intracellular Ca+2 release in response to 4 μM thapsigargin in PN/PH cells and in STIM1siRNA treated PN cells. B) Graph summarizes the average resting levels of Ca+2. C) Amplitude of Ca+2 release in response to Thapsigargin. (D) Representative traces of intracellular Ca+2 release in response to thapsigargin in PN cells and cells treated with APB, 10 μM. E) Graph summarizes resting Ca2+ levels and F) graph summarizes the average spike amplitude of Ca2+ release in response to thapsigargin. Note that STIM1 silencing or 2-APB treatment in particular significantly reduced the thapsigargin-induced release of Ca+2 from the ER. Asterisk indicates significance between the two groups (n=4-5). (student’s t-test, **P < 0.01, ****P <0.0001, n=4). Measurements were made in standard Ca2+ solutions
STIM1 silencing affects the protein levels of AC6, AC3 and PC2.
To further explore the role of STIM1 in cyst formation, we next determined the steady-state protein levels of two Ca2+-dependent adenylyl cyclases, AC3 and AC6. AC3 is activated by increasing intracellular Ca2+ via Ca2+-calmodulin[44], whereas AC6 is inhibited at higher Ca2+ [23]. Our western blots showed that both AC3 and AC6 were expressed in these cells (Fig. 7A, B). Knocking down STIM1 increased the steady-state levels of AC3 and reduced those of AC6. The data in total suggest that knocking down STIM1 can alter the protein expression of these two adenylyl cyclases but to a small extent.
Figure 7: Protein levels of adenylyl cyclase 3, 6:

A) Western blot showing expression of adenylate cyclase 3 (AC3) in PN and cells where STIM1 was knocked down (PN/S cells). Columns represent averages ± standard errors of the AC3 expression. B) Western blot showing expression of AC6 in PN and cells where STIM1 was knocked down (PN/S cells). Columns represent averages ± standard errors of the AC6 expression. C) Western blot showing expression of PC2 in PN/PH cells. Columns represent averages ± standard errors of the PC2 expression. D) Western blot showing expression of PC2 (left panel) and STIM1 (right panel) in PN where STIM1 was knocked down (PN/S cells). Experiment was repeated for four times. Columns represent averages ± standard errors of the AC3 expression. *P< 0.05, ** P< 0.01, ***P<0.001, ****P<0.0001. Data were analyzed by non parametric t test. Note that STIM1 silencing increases the steady state protein expression of AC3 but decreases that of AC6.
We also determined whether changes occurred in PC2 protein expression. The first thing to notice is that PC2 expression is approximately 2-fold higher in PN (PC1-null) vs PH cells (Fig. 7C). This is not surprising in light of our previous work which showed that functional PC1 downregulates the expression of PC2 via aggresomal degradative pathways [36]. What is surprising is the downregulation of PC2 levels when either 2-APB is applied or STIM1 silenced (Fig. 7D Left panel). This could suggest that these experimental maneuvers may mimic the role of PC1 by promoting the degradation of PC2 but more experiments will be needed to show this conclusively. Fig. 7D, right panel also shows that 2-APB treatment does not affect the protein expression of STIM1.
Knockdown of STIM1 reduces cAMP levels.
We observed (Fig. 1, above) a nearly 15 fold increase in STIM1 in PN cells over the level in PH cells. To determine whether the large amount of STIM1 present in the PN cells plays a role in increasing cAMP levels, we examined the role of STIM1 in regulating cAMP levels. Our data show (Fig. 8A) that cAMP levels were higher in PN cells containing STIM1 than in PH cells in which STIM1 was silenced.
Figure 8. Steady State Levels of cAMP:
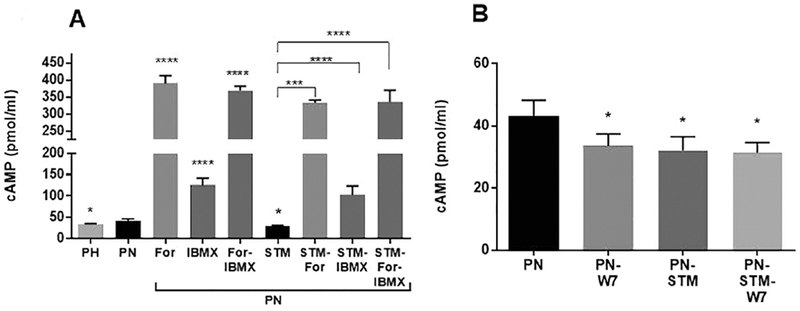
A) At day five confluent cells were treated with Forskolin (100μ M) or IBMX (100μ M) for 30min before harvesting the cells for assay. Cyclic AMP levels were measured with a direct cAMP Enzyme immunoassay Kit based on the manufacturer’s protocol. Results are expressed as pmole/ml. Columns represent averages ± SEs. *P < 0.05, ***P<0.001, ****P<0 0001. Statistical analysis was performed using a 2-tailed Student t-test. n=3-6: Note that forskolin causes a large increase in cAMP. A smaller increase occurs with IBMX. However, the increase induced by forskolin alone is similar to that of IBMX plus forskolin. STIM1 silencing reduces the levels of cAMP compared to untreated PN cells. Forskolin and IBMX increase cAMP to a lesser extent when STIM1 is knocked down. B): At day four confluent cells were treated with W7 (50uM) for 16h. Note that both STIM1 knockdown and W7 treatment reduced cAMP levels about the same amount. Treating cells with W7 when STIM1 is silenced abolishes the W7 effect indicating that both are working via a common pathway.
To tease this effect out further, we applied forskolin, which activates adenylyl cyclases directly;[24] as expected, we saw a large increase in cAMP levels. These data confirmed that the enhanced growth in forskolin-treated cysts depicted in Fig. 3 was indeed caused by increased cAMP levels. Silencing STIM1 reduced the forskolin-dependent increases in cAMP, suggesting that STIM1 could be having an effect on the maximum ability of adenylyl cyclases to generate cAMP in these cells.
Both the rate of production of cAMP via adenylyl cyclase and the rate of degradation by phosphodiesterase [45] determine the steady-state levels of cAMP in cells. To evaluate the role of phosphodiesterase, we applied the non-selective phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX). IBMX by itself increased cAMP levels, which were then further increased by the addition of forskolin plus IBMX. However, the stimulation of cAMP by forskolin was not significantly different in the presence of IBMX than it was in the absence of IBMX, indicating that phosphodiesterases do not contribute to the magnitude of the cAMP levels we observe under our experimental conditions.
To begin to address the role of Ca2+ in the STIM1-dependent decrease in cAMP, we applied the calmodulin inhibitor [7, 8] W7. W7 on its own reduced cAMP levels when applied at 50 μM (Fig. 8B). The level of reduction was the same when W7 was applied in combination with STIM1 knockdown, suggesting that STIM1 and calmodulin act via a similar pathway.
4. DISCUSSION:
STIM1 and IP3R.
Herein we show that STIM1 levels are elevated by almost 15-fold in a mouse-cell model of ADPKD derived from proximal tubules and 2-fold in kidneys from an inducible pkd−/− mouse model. We also show that the protein levels of the IP3R are upregulated by 5 fold in cystic vs. normal kidneys. To define the role of elevated STIM1 in supporting cyst growth, we knocked down STIM1 with siRNAi. Cysts from PN cells were grown in 3D culture and allowed to form cysts for 11 days. Importantly, in this experiment, the growth of the cysts was accelerated by forskolin, which increases cAMP levels, thus verifying the central role of cAMP in stimulating cyst growth in ADPKD [38]. siRNA was applied on Days 11-14. After knockdown under these conditions, we saw an approximately 50% reduction in cyst size. Because siRNA was applied to 3D culture medium and therefore may not have produced uniform knockdown of STIM1 in all cells, we chose to inhibit the STIM1 pathway with 2-APB. Application of 2-APB reduced the cyst size by more than 95%, demonstrating definitively that elevated STIM1 clearly supports cyst growth. These data are important because they suggest that ADPKD cells may share some characteristics with tumor cells in regard to cell proliferation and elevated STIM1.
STIM1 knockdown targets ER Ca2+ movement.
Misregulation of Ca2+ is associated with cyst formation in ADPKD[13], with some investigators reporting that disruption of Ca2+ signaling is the primary event that supports increased cyst growth[14]. Several studies have shown that a reduction in the function of either PC1 or PC2 leads to dysregulation of Ca2+ signaling (see [9] for a review). We show here that neither STIM1 knockdown nor the application of 2-APB has any effect on resting intracellular Ca2+ or on the increase of intracellular Ca2+ produced by ATP application in PN cells.
ATP stimulates both P2X and P2Y types of purinergic receptors which increase intracellular Ca2+[43, 46]. The former are non-selective Ca2+ channels, which cause an increase in intracellular Ca2+ directly. Whereas the latter are G-protein coupled and increase intracellular Ca2+ via IP3R[46]. Both P2X and P2Y receptor subtypes are present in proximal tubules and in cystic kidneys [47]. The question here is; which type in the PN cells does ATP stimulate? Schwiebert and collaborators [47] investigated the pattern of changes in intracellular Ca2+ induced by each of these receptors. They observed that when they stimulated P2Y receptors that the induced increase in intracellular Ca2+ was transient similar to what we observed previously when we applied ATP to MDCK cells [31]. On the other hand, when they stimulated P2X receptors a sustained increase in intracellular Ca2+ was observed similar to what we observed in Fig. 5 suggesting that our data is generated by a P2X response. The observation that the magnitude of the increase in intracellular Ca2+ induced by ATP also does not change following application of 2-APB or silencing STIM1 is also consistent with P2X receptor movement of Ca2+. As observed in Fig. 5, there is no change in the resting levels of intracellular Ca2+ when PN cells are treated with 2-APB or when STIM1 is silenced thus the gradient for Ca2+ to move into the cells is unchanged. Because the gradient is unchanged by these experimental maneuvers, when P2X receptors are activated, the increase in intracellular Ca2+ induced by ATP would also be the same, consistent with what we observed in Fig. 5. Interestingly, in a previous study, we exposed PN cells to histone deacetylase 6 (HDAC6) inhibitors, which resulted in a decrease in resting intracellular Ca2+[48]. Consistent with increased gradient for Ca2+ to enter the cells, the increase in intracellular Ca2+ induced by ATP was larger when HDAC6 was inhibited vs. untreated cells, again consistent with the movement of Ca2+ via P2X receptors.
In sharp contrast, we did see a large increase in SOCE in PN (PC1 null cells) vs PH (PC1 containing). Given the SOCE is a component of Ca2+ signaling [28], the data are consistent with enhanced Ca2+ signaling in cyst producing cells. Both STIM1 knockdown and the application of 2-APB had a profound effect on the release of Ca2+ from the ER that was induced by thapsigargin. Thapsigargin inhibits the SERCA pump[49]; therefore, the magnitude of the release of Ca2+ from the ER depends on the Ca2+ gradient between the lumen of the ER and the permeability of the ER membrane for Ca2+, the latter is facilitated by the IP3R [50]which is itself elevated in cystic mice. STIM1 is the sensor that monitors Ca2+ within the lumen of the ER and regulates SOCE [28]. Thus, STIM1 knockdown would be expected to limit the ability of the cell to replenish ER Ca2+[51].
2-APB has a complex effect on SOCE. For example, when STIM1 binds to the Ora1 channel, it forms puncta that represent concentrated formations at the plasma membrane at which STIM1 and Ora1 interact. 2-APB, under certain circumstances, can either inhibit or promote puncta formation[52]. Despite the uncertainty regarding this process, it is clear here that 2-APB dramatically reduced the release of Ca2+ from the ER that is induced by thapsigargin, consistent with 2-APB operating as an inhibitor.
STIM1 regulates the steady-state levels of PC2.
We have shown previously that that full-length PC1 that interacts with PC2 via a C-terminal coiled-coil domain regulates PC2 expression in vivo and in vitro by down-regulating PC2 expression via autophagy [36]. Thus, it is not suppressing, that PC2 protein levels are 2-fold higher in PN vs PH cells. What is novel is that knocking down STIM1 or application of 2-APB does indeed lower PC2 protein levels in PN cells by approximately 75%. The question is whether STIM1 plays a role in autophagy?
Interestingly, Jin and colleagues [53] studied the effect of STIM1 on autophagy and epithelial-mesenchymal transition (EMT) in podocytes in diabetic nephropathy. They found that, in podocytes cultured in the serum of diabetic nephrotic rats, autophagy decreased, whereas EMT increased and that both changes reverse after silencing STIM1. Interestingly, in ADPKD autophagy is defective [54] and EMT enhanced [55]in cystic kidneys. It will be interesting in future studies to see whether STIM1 silencing can also restore autophagy and EMT towards normal in ADPKD.
Reduced ER Ca2+ movement is associated with reduced cAMP levels.
Although neither STIM1 knockdown nor 2-APB treatment produced a change in resting Ca2+, the dramatic reduction in ER Ca2+ release should affect Ca2+ signaling in the cells. In renal cells, there are two types of adenylyl cyclases that respond to Ca2+, those activated by Ca2+ and those that are inhibited by it [24]. Our western blots confirmed that the PN cells contain both AC3, which is activated by Ca2+ through Ca2+-calmodulin, and AC6, which is inhibited by Ca2+ [24]. Interestingly, the steady-state levels of both cyclases were affected by STIM1 knockdown: AC3 levels increased significantly, whereas those of AC6 decreased. Measurement of cAMP showed that STIM1 knockdown reduced cAMP levels. The reduction was identical to that which occurred following the inhibition of calmodulin with W7. The observation that W7 and STIM1 knockdown were not synergistic suggests that STIM1 is regulating AC3 via calmodulin. Thus, the reduction in cAMP levels resulting from STIM1 knockdown is most likely the result of its effect in reducing AC6 levels and its ability to reduce AC3 activity via a calmodulin-dependent mechanism.
Here we did not see global changes in the intracellular Ca2+ in response to either STIM1 knockdown or 2-APB treatment. However, release of Ca2+ from the ER often occurs in the form of “puffs” or “sparks” that form the basis for Ca2+ waves [56]. In the kidney, oscillations in intracellular Ca2+ are critical for the fusion of vesicles containing AQP2 [7]. Thus, it is possible that localized transient reductions in the release of Ca2+ from the ER spark the movement of PC2 from the ER to the plasma membrane.
We envision a vicious cycle in which PC2 located at the ER causes excessive release of Ca2+ from the ER, coupled with increased SOCE maintaining the ER Ca2+ stores. We have observed that STIM1 protein levels are highly elevated in ADPKD cells, which would exacerbate this cycle. We suggest further that the elevated STIM1 contributes to the increased resting levels of cAMP via the fueling of cAMP-dependent cyst growth by calmodulin-dependent AC3. Knocking down STIM1 or treating cells with 2-APB breaks this cycle, reducing cAMP and reducing cyst growth.
Interestingly, STIM1 silencing caused a decrease in forskolin- and IBMX-dependent cAMP production; however, cAMP levels after STIM1 silencing in the presence of forskolin were still many times higher than control levels. The fact that STIM1 still dramatically reduced cyst growth in the presence of forskolin strongly suggests that STIM1 silencing inhibits the ability of cAMP to stimulate cyst growth. In the normal human kidney, signaling pathways that increase cAMP levels do not stimulate cell growth (see [10]). In ADPKD, increases in cAMP activate B-Raf, which in turn leads to an activation of ERK, turning on cell proliferation in response to cAMP. Thus, one likely outcome of STIM1 silencing is that it overrides the proliferation induced by cAMP via ERK signal transduction.
Conclusion:
One prevailing model for cyst growth is that Ca2+ restriction causes cAMP-dependent activation of the B-Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway, which results in increased cell growth [16]. We show here that both STIM1 and IP3R are expressed at significantly higher protein levels particularly in cystic mice kidneys compared to normal controls. We also found that several key components of Ca2+ signaling are elevated in PN (PC1 null) vs PH (PC1 containing) cells and propose that elevated ER Ca2+ release is actually the key to fueling cyst growth. One limitation of the study was that the studies were performed in mouse and not in human cells. However, we have uncovered a novel therapeutic pathway to inhibit cyst growth in ADPKD by lowering STIM1 levels, with the goal of restoring Ca2+ homeostasis which we expect may help with the development of new treatment strategies for ADPKD.
Highlights:
Silencing high levels of STIM1 in ADPKD cells reduces cysts, cAMP & ER Ca2+ release.
Acknowledgement:
The authors appreciate Deborah McClellan, Ph.D for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability: All data generated or analyzed during this study will be made available by the corresponding author upon reasonable request. All data generated or analyzed during this study are included in this published article. No data sets were generated.
Conflict of Interest: The authors have no conflict of interest pertaining to this work.
REFERENCES:
- [1].Torres VE, Harris PC, Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities, J.Intern.Med 261(1) (2007) 17–31. [DOI] [PubMed] [Google Scholar]
- [2].Calvet JP, Grantham JJ, The genetics and physiology of polycystic kidney disease, Semin. Nephrol 21(2) (2001) 107–123. [DOI] [PubMed] [Google Scholar]
- [3].Liu X, Vien T, Duan J, Sheu S-H, DeCaen PG, Clapham DE, Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium, eLife 7 (2018) e33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sutters M, Germino GG, Autosomal dominant polycystic kidney disease: molecular genetics and pathophysiology, J.Lab Clin.Med 141(2) (2003) 91–101. [DOI] [PubMed] [Google Scholar]
- [5].Ong AC, Harris PC, A polycystin-centric view of cyst formation and disease: the polycystins revisited, Kidney Int (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Devuyst O, Torres VE, Osmoregulation, vasopressin, and cAMP signaling in autosomal dominant polycystic kidney disease, Curr.Opin.Nephrol.Hypertens 22(4) (2013) 459–470. [DOI] [PubMed] [Google Scholar]
- [7].Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, Knepper MA, Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin, J. Biol. Chem 275(47) (2000) 36839–46. [DOI] [PubMed] [Google Scholar]
- [8].Hoffert JD, Chou CL, Fenton RA, Knepper MA, Calmodulin is required for vasopressin-stimulated increase in cyclic AMP production in inner medullary collecting duct, J. Biol. Chem 280(14) (2005) 13624–13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Torres VE, Vasopressin receptor antagonists, heart failure, and polycystic kidney disease, Annu. Rev. Med 66 (2015) 195–210. [DOI] [PubMed] [Google Scholar]
- [10].Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ, Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys, Kidney Int 63(6) (2003) 1983–94. [DOI] [PubMed] [Google Scholar]
- [11].Torres VE, Bankir L, Grantham JJ, A case for water in the treatment of polycystic kidney disease, Clin.J.Am.Soc.Nephrol 4(6) (2009) 1140–1150. [DOI] [PubMed] [Google Scholar]
- [12].Shoaf SE, Chapman AB, Torres VE, Ouyang J, Czerwiec FS, Pharmacokinetics and Pharmacodynamics of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease: Phase 2 Trials for Dose Selection in the Pivotal Phase 3 Trial, J. Clin. Pharmacol 57(7) (2017) 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Torres VE, Rossetti S, Harris PC, Update on autosomal dominant polycystic kidney disease, Minerva Med 98(6) (2007) 669–691. [PubMed] [Google Scholar]
- [14].Mangolini A, de Stephanis L, Aguiari G, Role of calcium in polycystic kidney disease: From signaling to pathology, World J Nephrol 5(1) (2016) 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG, Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents, Nature 408(6815) (2000) 990–4. [DOI] [PubMed] [Google Scholar]
- [16].Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP, Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype, J. Biol. Chem 279(39) (2004) 40419–30. [DOI] [PubMed] [Google Scholar]
- [17].Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP, Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells, J. Am. Soc. Nephrol 17(1) (2006) 178–187. [DOI] [PubMed] [Google Scholar]
- [18].Joly D, Ishibe S, Nickel C, Yu Z, Somlo S, Cantley LG, The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells, J. Biol. Chem 281(36) (2006) 26329–39. [DOI] [PubMed] [Google Scholar]
- [19].Wei F, Karihaloo A, Yu Z, Marlier A, Seth P, Shibazaki S, Wang T, Sukhatme VP, Somlo S, Cantley LG, Neutrophil gelatinase-associated lipocalin suppresses cyst growth by Pkd1 null cells in vitro and in vivo, Kidney Int 74(10) (2008) 1310–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Prevarskaya N, Skryma R, Shuba Y, Calcium in tumour metastasis: new roles for known actors, Nat.Rev.Cancer 11(8) (2011) 609–618. [DOI] [PubMed] [Google Scholar]
- [21].Wu Z, Qing J, Wang K, Xia Y, Zhang F, Suppression of stromal interaction molecule 1 inhibits SMMC7721 hepatocellular carcinoma cell proliferation by inducing cell cycle arrest, Biotechnol.Appl.Biochem (2014). [DOI] [PubMed] [Google Scholar]
- [22].Yang S, Zhang JJ, Huang XY, Orai1 and STIM1 are critical for breast tumor cell migration and metastasis, Cancer Cell 15(2) (2009) 124–134. [DOI] [PubMed] [Google Scholar]
- [23].Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE, Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease, J. Am. Soc. Nephrol 25(2) (2014) 232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hanoune J, Defer N, Regulation and role of adenylyl cyclase isoforms, Annu. Rev. Pharmacol. Toxicol 41(1) (2001) 145–174. [DOI] [PubMed] [Google Scholar]
- [25].Woodward OM, Li Y, Yu S, Greenwell P, Wodarczyk C, Boletta A, Guggino WB, Qian F, Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1, PLoS.One. 5(8) (2010) e12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bishop GA, Berbari NF, Lewis J, Mykytyn K, Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain, J. Comp. Neurol 505(5) (2007) 562–571. [DOI] [PubMed] [Google Scholar]
- [27].Rieg T, Kohan DE, Regulation of nephron water and electrolyte transport by adenylyl cyclases, Am. J. Physiol. Renal Physiol 306(7) (2014) F701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kurosaki T, Baba Y, Ca2+ signaling and STIM1, Prog.Biophys.Mol.Biol 103(1) (2010) 51–58. [DOI] [PubMed] [Google Scholar]
- [29].Jousset H, Frieden M, Demaurex N, STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum, J. Biol. Chem 282(15) (2007) 11456–11464. [DOI] [PubMed] [Google Scholar]
- [30].Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U, Iwatsubo M, Chen S, Goydos JS, Ishikawa Y, Iwatsubo K, Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration, PLoS.One. 9(2) (2014) e89292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Santoso NG, Cebotaru L, Guggino WB, Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway, Cell Physiol Biochem 27(6) (2011) 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cebotaru L, Cebotaru V, Wang H, Arend LJ, Guggino WB, STIM1fl/fl Ksp-Cre Mouse has Impaired Renal Water Balance, Cell. Physiol. Biochem 39(1) (2016) 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S, Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease, Nat.Genet 45(9) (2013) 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG, A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1, Nat. Med 13(12) (2007) 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Traykova-Brauch M, Schonig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice, Nat. Med 14(9) (2008) 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cebotaru V, Cebotaru L, Kim H, Chiaravalli M, Boletta A, Qian F, Guggino WB, Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway, J. Biol. Chem 289(10) (2014) 6404–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW Jr., Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry, J. Biol. Chem 283(28) (2008) 19265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hanaoka K, Guggino WB, cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells, J. Am. Soc. Nephrol 11(7) (2000) 1179–1187. [DOI] [PubMed] [Google Scholar]
- [39].Soboloff J, Spassova MA, Dziadek MA, Gill DL, Calcium signals mediated by STIM and Orai proteins--a new paradigm in inter-organelle communication, Biochim.Biophys.Acta 1763(11) (2006) 1161–1168. [DOI] [PubMed] [Google Scholar]
- [40].Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP, Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase, Proc. Natl. Acad. Sci. U. S. A 87(7) (1990) 2466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yanda MK, Liu Q, Cebotaru V, Guggino WB, Cebotaru L, Histone deacetylase 6 inhibition reduces cysts by decreasing cAMP and Ca(2+) in knockout mouse models of polycystic kidney disease, J. Biol. Chem 292(43) (2017) 17897–17908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJJC, Store-independent activation of Orai1 by SPCA2 in mammary tumors, 143(1) (2010) 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Menzies RI, Tam FW, Unwin RJ, Bailey MA, Purinergic signaling in kidney disease, Kidney Int (2016). [DOI] [PubMed] [Google Scholar]
- [44].Strait KA, Stricklett PK, Chapman M, Kohan DE, Characterization of vasopressin-responsive collecting duct adenylyl cyclases in the mouse, Am.J.Physiol Renal Physiol 298(4) (2010) F859–F867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chappe V, Mettey Y, Vierfond JM, Hanrahan JW, Gola M, Verrier B, Becq F, Structural basis for specificity and potency of xanthine derivatives as activators of the CFTR chloride channel, Br. J. Pharmacol 123(4) (1998) 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Unwin RJ, Bailey MA, Burnstock GJP, Purinergic signaling along the renal tubule: the current state of play, 18(6) (2003) 237–241. [DOI] [PubMed] [Google Scholar]
- [47].Schwiebert EM, Wallace DP, Braunstein GM, King SR, Peti-Peterdi J, Hanaoka K, Guggino WB, Guay-Woodford LM, Bell PD, Sullivan LP, Grantham JJ, Taylor AL, Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys, Am.J.Physiol Renal Physiol 282(4) (2002) F763–F775. [DOI] [PubMed] [Google Scholar]
- [48].Yanda MK, Liu Q, Cebotaru L, An inhibitor of histone deacetylase 6 activity, ACY-1215, reduces cAMP and cyst growth in polycystic kidney disease, Am. J. Physiol. Renal Physiol 313(4) (2017) F997–F1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Grubb BR, Gabriel SE, Mengos A, Gentzsch M, Randell SH, Van Heeckeren AM, Knowles MR, Drumm ML, Riordan JR, Boucher RC, SERCA pump inhibitors do not correct biosynthetic arrest of deltaF508 CFTR in cystic fibrosis, Am. J. Respir. Cell Mol. Biol 34(3) (2006) 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mak DO, McBride S, Foskett JK, Inositol 1,4,5-trisphosphate [correction of tris-phosphate] activation of inositol trisphosphate [correction of tris-phosphate] receptor Ca2+ channel by ligand tuning of Ca2+ inhibition, Proc.Natl. Acad. Sci.U.S.A 95(26) (1998) 15821–15825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL, Orail and STIM reconstitute store-operated calcium channel function, The Journal of Biological Chemistry 281(30) (2006) 20661–20665. [DOI] [PubMed] [Google Scholar]
- [52].Goto J, Suzuki AZ, Ozaki S, Matsumoto N, Nakamura T, Ebisui E, Fleig A, Penner R, Mikoshiba K, Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins, Cell Calcium 47(1) (2010) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jin J, Wu D, Zhao L, Zou W, Shen W, Tu Q, He QJIJCEP, Effect of autophagy and stromal interaction molecule 1 on podocyte epithelial-mesenchymal transition in diabetic nephropathy, 11(5) (2018) 2450–2459. [PMC free article] [PubMed] [Google Scholar]
- [54].Boletta A, Emerging evidence of a link between the polycystins and the mTOR pathways, Pathogenetics. 2(1) (2009) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y, Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks, Hum. Mol. Genet 18(13) (2009) 2328–43. [DOI] [PubMed] [Google Scholar]
- [56].Yip KP, Sham JS, Mechanisms of vasopressin-induced intracellular Ca2+ oscillations in rat inner medullary collecting duct, Am. J. Physiol. Renal Physiol 300(2) (2011) F540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]