

SUMMARY
Inflammasome-activated caspase-1 cleaves gasdermin D to unmask its pore forming activity, the predominant consequence of which is pyroptosis. Here, we report an additional biological role for gasdermin D in limiting cytosolic DNA surveillance. Cytosolic DNA is sensed by Aim2 and cyclic GMP-AMP synthase (cGAS) leading to inflammasome and type I interferon responses, respectively. We found that gasdermin D activated by the Aim2 inflammasome suppressed cGAS-driven type I interferon response to cytosolic DNA and Francisella novicida in macrophages. Similarly, interferon-β (IFN-β) response to F. novicida infection was elevated in gasdermin D-deficient mice. Gasdermin D-mediated negative regulation of IFN-β occurred in a pyroptosis-, interleukin-1 (IL-1)-, and IL-18-independent manner. Mechanistically, gasdermin D depleted intracellular potassium (K+) via membrane pores, and this K+ efflux was necessary and sufficient to inhibit cGAS-dependent IFN-β response. Thus, our findings have uncovered an additional interferon regulatory module involving gasdermin D and K+ efflux.
Graphical Abstract
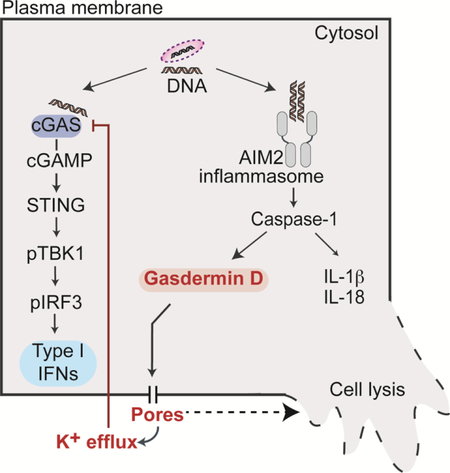
eTOC blurb
Gasdermin D is a pore forming protein, which upon activation by inflammasome complexes mediates pyroptotic cell death and IL-1 release. Banerjee et al., demonstrate a previously unknown regulatory role for gasdermin D-driven K+ efflux in reining in cGAS-dependent type I interferon response to cytosolic DNA.
INTRODUCTION
Inflammasome complexes trigger the enzymatic activity of caspase-1, which activates interleukin-1β (IL-1β) and IL-18 (Martinon et al., 2002). Caspase-1 also targets gasdermin D, a pore forming protein (Kayagaki et al., 2015; Shi et al., 2015). The pore-forming activity of gasdermin D resides in its N-terminal domain and is inhibited by its C-terminal domain. Caspase-1 cleaves gasdermin D at the linker region between these two domains liberating the N-terminal domain, which migrates to the plasma membrane forming pores with an inner diameter of 10 – 15 nm (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). An important consequence of gasdermin D activation is a lytic form of cell death called pyroptosis (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). However, new evidence points out that gasdermin D executes additional functions independent of cell death; gasdermin D pores mediate the release of IL-1β and IL-18 without inducing cell death in response to certain ligands (Evavold et al., 2018). Additionally, following cytosolic LPS sensing by caspase- 11, gasdermin D activates the NLRP3 inflammasome by inducing potassium (K+) efflux (Kayagaki et al., 2015; Rühl and Broz, 2015; Schmid-Burgk et al., 2015). Central to these distinct functions of gasdermin D is its membrane pore forming activity. However, if gasdermin D executes any additional immune functions is largely unknown.
A key surveillance mechanism in the cytosol, in addition to inflammasomes, is the cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) synthase (cGAS) pathway. cGAS is a sensor for cytosolic DNA, and the binding of DNA by cGAS triggers its nucleotidyl transferase activity leading to the synthesis of cGAMP from ATP and GTP. cGAMP stimulates the transcription of type I interferon genes via the STING adaptor-TBK1 kinase-IRF3 transcription factor axis (Burdette et al., 2011; Wu et al., 2013). The type I interferon response elicited by cGAS plays important roles in host defense (Schneider et al., 2014). However, it is increasingly appreciated that the sustained production of type I interferons at high amounts is detrimental to the host, particularly during infections with intracellular bacteria such as Francisella novicida, Mycobacterium tuberculosis, and Listeria monocytogenes (Auerbuch et al., 2004; Henry et al., 2010; Mayer-Barber et al., 2014; McNab et al., 2015; Storek et al., 2015). Therefore, the magnitude and duration of cGAS-driven type I interferon responses should be kept in check. However, how the host restrains cGAS signaling during infections is poorly defined.
Here, we demonstrate that gasdermin D activated by the Aim2 inflammasome complex suppresses cytosolic DNA-induced production of type I interferons in macrophages. Consistent with this finding, mice lacking gasdermin D displayed enhanced IFN-β response to F. novicida infection. Pyroptosis and the extracellular release of IL-1 cytokines are dispensable for the inhibition of IFN-β by gasdermin D. Mechanistically, gasdermin D-induced membrane pores leaked intracellular potassium (K+) ions, and this K+ efflux in turn impaired type I interferon responses to cytosolic DNA and F. novicida. Gasdermin D-K+ efflux axis targeted cGAS to reduce cGAMP synthesis and thus, IFN-β production. In summary, this study uncovers a previously unrecognized key role for gasdermin D in restraining cytosolic DNA-elicited interferon responses. Collectively, an emerging theme from the findings of this work and the recent studies (Evavold et al., 2018; Kayagaki et al., 2015) is that the fundamental pore forming ability of gasdermin D and the consequent ionic fluxes confer gasdermin D additional biological functions independent of the terminal cell lytic event.
RESULTS
Aim2 inflammasome inhibits IFN-β response to cytosolic DNA in vitro and in vivo
DNA in the cytosol is sensed by cGAS and Aim2 (absent in melanoma 2). In contrast to cGAS that triggers transcriptional induction of the type I interferons, Aim2, upon directly binding DNA, forms an inflammasome complex by oligomerizing ASC and caspase-1 (Hornung and Latz, 2010). Infection with F. novicida, a cytosolic bacterium, or transfection of the synthetic double stranded DNA, poly(dA:dT), into the cytosol, induced IL-1β secretion in wild-type bone marrow derived macrophages (BMDMs), and this response was abolished in Aim2−/− BMDMs (Fig. 1A). In stark contrast, Aim2−/− BMDMs secreted higher amounts of IFN-β than wild-type BMDMs in response to F. novicida (Fig. 1B). Similarly, poly(dA:dT)-induced IFN-β secretion was elevated in Aim2−/− BMDMs compared to wild-type BMDMs, which is consistent with the earlier observations (Fig. 1B) (Corrales et al., 2016; Fernandes-Alnemri et al., 2010; Gray et al., 2016; Hornung et al., 2009; Jones et al., 2010; Rathinam et al., 2010; Wang et al., 2017). In line with their enhanced production of IFN-β, F. novicida- and poly(dA:dT)-stimulated Aim2−/− BMDMs, compared to wild-type BMDMs, expressed higher amounts of interferon stimulated gene (ISG) products such as viperin and ISG15 (Fig. 1C). Aim2 has been shown to inhibit colon tumorigenesis independently of the inflammasome complex (Man et al., 2015; Wilson et al., 2015). To test if the interferon-suppressive role of Aim2 is inflammasome-dependent or - independent, we used Asc−/− and Casp1−/−Casp11−/− macrophages. Asc−/− and Casp1−/−Casp11−/− BMDMs phenocopied Aim2-deficient BMDMs and produced more IFN-β after poly(dA:dT) transfection or F. novicida infection (Fig. 1D and 1E). Moreover, unlike poly(dA:dT) and F. novicida, TLR ligands (LPS and poly(I:C)) and RNA virus (Sendai) that do not engage the Aim2 inflammasome induced normal amounts of IFN-β production in Aim2−/−, Asc-/s-, or Casp1−/−Casp11−/−BMDMs (Fig. 1D-F). Taken together, these data clearly demonstrate that Aim2-mediated inhibition of IFN-β is dependent on its inflammasome-activating ability (Corrales et al., 2016; Wang et al., 2017).
Figure 1. Aim2 inflammasome activation suppresses cytosolic DNA-induced type I interferon production.

(A and B) IL-1β (A) and IFN-β (B) in the supernatants of Pam3CSK4-primed wild-type (WT) and Aim2−/− BMDMs stimulated with F. novicida (MOI=50) or poly(dA:dT) for 6 h.
(C) Immunoblots of viperin, ISG15 and β-actin in the lysates of WT and Aim2−/− iBMDMs stimulated with F. novicida (MOI=100), poly(dA:dT), or Sendai virus for 8 h.
(D to F) IFN-β in the supernatants of BMDMs from WT, Asc−/− (D), Casp1−/−Casp11−/− (E), and Aim2−/− (F) mice stimulated with indicated treatments.
(G-J) Cytokine quantities assessed 24 h post-infection (p.i) in the plasma of WT or indicated mutant mice infected s.c. with 5 × 105 CFU of F. novicida.
(K) Plasma IFN-β in WT and Aim2−/− mice 6 h after intraperitoneal infection with 1 × 109 CFU of E. coli BL21.
Combined data from three independent experiments are shown as mean ± SEM (A, B, D, E, and F). *, P <0.05; ns, not significant; two-way ANOVA followed by the Sidak’s post-test (A, B, D, and E) or unpaired two-tailed t test (G-K). In G-K, each circle represents a mouse and the horizontal lines represent mean and data are from one experiment representative of three. IFN-β data are presented as units/ml or pg/ml depending upon the recombinant IFN-β standard used in the ELISA.
We examined if Aim2 exhibits a similar suppressive effect on cGAS-induced IFN-β response to cytosolic DNA during bacterial infections in vivo. In line with the in vitro results in macrophages, plasma IFN-β amounts at 24 h p.i were significantly higher in F. novicida-infected mice lacking Aim2 than in wild-type mice (Fig. 1G). On the other hand, wild-type mice had increased amounts of IL-1β in plasma compared to Aim2−/− mice (Fig. 1H). We further tested the involvement of the inflammasome complex in Aim2 suppression of cGAS signaling by monitoring F. novicida-induced IFN-β production in Casp1−/−Casp11−/− mice. F. novicida-infected Casp1−/−Casp11−/− mice had significantly elevated amounts of IFN-β but reduced amounts of IL- 18 in plasma relative to wild-type mice (Fig. 1I and 1J). Furthermore, E. coli, which engages the caspase-11-noncanonical inflammasome but not the Aim2 inflammasome (Kailasan Vanaja et al., 2014; Rathinam et al., 2012), induced comparable amounts of IFN-β production in wild-type and Aim2−/− mice (Fig. 1K). Thus, it is evident from all these data that the Aim2 inflammasome functions to limit cytosolic DNA-induced type I interferon responses in vivo.
Aim2 restricts cytosolic DNA-driven type I interferon responses via gasdermin D
Gasdermin D is a recently identified substrate for inflammatory caspases including caspase-1 (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). This raises two possibilities by which the Aim2 inflammasome may exert its regulatory effect on IFN-β production; first, caspase-1 activated by Aim2 may directly regulate cGAS-induced IFN-β production without involving gasdermin D. Consistent with this possibility, a recent study shows that, during viral infections, caspase-1 depletes cellular concentrations of functional cGAS by cleaving it and thus inhibiting type I interferon production (Wang et al., 2017). However, in our study, strong caspase-1 activation by Aim2 inflammasome ligands, F. novicida and poly(dA:dT), and NLRP3 stimuli, nigericin and ATP, in wild-type BMDMs did not result in a reduction in cGAS protein or its cleavage (Fig. 2A). An alternative possibility is that gasdermin D activated by the Aim2 inflammasome (Fig. S1A) might mediate the suppression of IFN-β. To test this, we examined poly(dA:dT)- and F. novicida-induced IFN-β secretion in wild-type and Gsdmd−/− immortalized BMDMs (iBMDMs). We found that, while IL-1β secretion and cell death were reduced, IFN-β production was markedly higher in Gsdmd−/− cells, phenocopying Aim2−/− cells (Fig. 2B-D). The augmented production of IFN-β in response to cytosolic DNA was also observed in primary BMDMs from Gsdmd−/− mice (Fig. S1B). Furthermore, the hyper-production of IFN-β by F. novicida-infected Gsdmd−/− BMDMs was sustained over a period of time (Fig. 2E). In this kinetic analysis, Gsdmd−/− BMDMs displayed, as expected, reduced LDH release upon F. novicida infection. However, there was no difference in the intracellular ATP content of F. novicida- infected wild-type and Gsdmd−/− BMDMs (Fig. 2F). In other words, an increase in cytosolic DNA- induced IFN-β production was evident in Gsdmd−/− BMDMs without a concomitant increase in intracellular ATP. E. coli induces IFN-β via TLR4 (Kailasan Vanaja et al., 2014; Rathinam et al., 2012) and elicits gasdermin D activation and cell death via caspase-11 (Fig. 2G) (Kayagaki et al., 2015). E. coli triggered comparable IFN-β production in wild-type, Gsdmd−/− and Casp11−/− macrophages, indicating that the TLR4-driven IFN-β production was not sensitive to gasdermin D-mediated inhibition (Fig. 2G). Sendai virus-induced IFN-β was also comparable between wild- type and Gsdmd−/− cells (Fig. 2H). Like IFN-β, cGAS-dependent production of IL-6 was also higher in F. novicida-infected Gsdmd−/− macrophages (Fig. 2I-K). IFN-β production per cell was measured by intracellular staining followed by flow cytometry for viperin, as a surrogate for IFN-β. This showed that viperin expression on a per-cell basis as indicated by the mean fluorescence intensity was higher in F. novicida- or poly(dA:dT)-, but not IFN-β-, stimulated gasdermin D-deficient cells compared to wild-type cells (Fig. S1C). Unlike Gsdmd−/− BMDMs, Nlrp3−/− BMDMs produced wild-type amounts of IFN-β upon F. novicida infection (Fig. S1D). These results suggest that the Aim2 inflammasome down-regulates type I interferon responses via gasdermin D.
Figure 2. Gasdermin D is required for inflammasome-mediated suppression of type I interferon production.

(A) Immunoblotting for cGAS and β-actin in the lysates and the cleaved caspase-1 (p20) in the methanol chloroform-precipitated supernatants of Pam3CSK4-primed WT BMDMs stimulated with F. novicida, poly(dA:dT), nigericin, or ATP.
(B-D) IFN-β secretion (B), LDH release (C), and IL-1β secretion (D) by Pam3CSK4-primed WT and Gsdmd−/− iBMDMs stimulated with F. novicida or poly(dA:dT) for 6 h.
(E-F) IFN-β secretion (E), LDH release, and cellular ATP (F) in WT and Gsdmd−/− primary BMDMs infected with F. novicida (MOI=100) at 4, 6, 8 and 12 h p.i.
(G) IFN-β secretion and LDH release by WT, casp11−/− and Gsdmd−/− primary BMDMs infected with EHEC (MOI=50) at 16 h p.i.
(H) IFN-β in the supernatants of WT and Gsdmd−/− BMDMs stimulated with Sendai virus or LPS for 6 h.
(I-K) IFN-β (I) or IL-6 (J and K) in the supernatants of BMDMs of indicated genotypes infected with F. novicida (MOI of 25 and 50) or Sendai virus for 6 h.
(L and M) IFN-β in the plasma (L) and the bacterial load in the spleen and liver (M) of WT and Gsdmd−/− mice infected s.c. with 1.5 × 105 CFU of F. novicida at 24 h p.i.
(N) Survival of WT and Gsdmd−/− mice (n=5) infected s.c. with 2.5 × 102 CFU of F. novicida.
(O)Survival of WT and Aim2−/− mice (n=5) infected s.c. with 2.5 × 102 CFU of F. novicida and injected i.p. with 250 μg of an isotype control or anti-IFNAR antibody 12 h p.i.
Combined data from three independent experiments are shown as mean ± SEM (B-K). *, P <0.05; ns, not significant; two-way ANOVA followed by the Sidak’s post-test (B, C, D, G, and K), unpaired two-tailed t test (L and M), or Mantel-Cox test (N and O). In L and M, each circle represents a mouse and the horizontal lines represent mean. Data in L-O are from one experiment representative of three (L and M) or two (N and O). IFN-β data are presented as units/ml or pg/ml depending upon the recombinant IFN-β standard used in the ELISA. See also Figure S1.
To test the role of gasdermin D in type I interferon response to F. novicida infection in vivo, wild-type and Gsdmd−/− mice were infected with F. novicida, and plasma IFN-β was measured at 24 h p.i. Similar to our observations in BMDMs, gasdermin D-deficient mice had significantly elevated IFN-β in plasma relative to wild-type mice (Fig. 2L). However, the bacterial loads in the spleens and livers of wild-type and Gsdmd−/− mice were similar suggesting that the increased IFN-β in Gsdmd−/− mice was not due to higher bacterial burden (Fig. 2M). Wild-type and Gsdmd−/− mice were infected with F. novicida and monitored for survival: gasdermin D deficiency compromised the survival of the host as gasdermin D-deficient mice succumbed to F. novicida infection earlier than WT mice (Fig. 2N). Next, we sought to understand if the elevated IFN-β response in inflammasome-deficient mice impact host resistance to F. novicida. To this end, type I IFN signaling in F. novicida-infected Aim2−/− mice was partially blocked with the administration of very low dose of anti-type-1 interferon receptor (IFNAR) antibody (250 μg/mouse) just once (12 h p.i.), and the survival of mice was monitored. Anti-IFNAR antibody- treated Aim2−/− mice survived significantly longer than Aim2−/− mice treated with an isotype control antibody (Fig. 2O). This data suggest that the elevated IFN-β in the absence of Aim2 and gasdermin D increases the susceptibility of the host to F. novicida infection. Consistent with our result with anti-IFNAR antibody, a latest report showed that the genetic ablation of IFNAR1 in Aim2-deficient mice protected Aim2−/− mice from F. novicida infection (Zhu et al., 2018). Overall, gasdermin D activated by the Aim2 inflammasome reins in the host-detrimental type I interferon expression, promoting anti-bacterial defense.
Gasdermin D targets cGAS to suppress cytosolic DNA-induced IFN-β production
Upon binding DNA, cGAS synthesizes the second messenger cGAMP, which binds to STING. STING triggers the activation of TBK1, which in turn phosphorylates IRF3 to initiate IFN-β transcription (Sun et al., 2013). To define which step of this pathway is affected by gasdermin D, we examined the above-described events in cGAS signaling in wild-type and Gsdmd−/− macrophages. F. novicida and poly(dA:dT), but not LPS and Sendai virus, induced phosphorylation of TBK1 and IRF3 was substantially higher in Gsdmd−/− (Fig. 3A) and Aim2−/− (Fig. 3B) macrophages compared to wild-type cells. These results indicate that the upstream cGAS or STING activation itself is likely to be affected by gasdermin D. To explore this, cGAS binding of DNA was assessed by immunoprecipitation of cGAS from F. novicida-infected BMDMs followed by PCR analysis of bacterial DNA bound to cGAS. This approach revealed that cGAS in Gsdmd−/− BMDMs bound to significantly higher quantity of F. novicida DNA than cGAS in wild-type BMDMs suggesting that gasdermin D affects cGAS binding of DNA (Fig. 3C- D). Upon binding DNA, cGAS undergoes oligomerization forming aggregates, which can be visualized as punctate staining (Cui et al., 2017; Sun et al., 2013; Wassermann et al., 2015; Xia et al., 2016). We compared cGAS puncta formation in WT and Gsdmd−/− BMDMs by immunofluorescence microscopy with a cGAS antibody that was validated in cGAS-deficient BMDMs (Fig. S2). F. novicida and poly(dA:dT) induced cGAS puncta staining (Fig. 3E), and this puncta formation was observed at a higher frequency in Gsdmd−/− BMDMs (Fig. 3F-I). Consistent with the increased DNA binding and oligomerization of cGAS in Gsdmd−/− BMDMs, the enzymatic activity of cGAS was also enhanced in Gsdmd−/− BMDMs; the cellular quantity of cGAMP in F. novicida- and poly(dA:dT)-stimulated Gsdmd−/− macrophages were higher than that of wild-type macrophages (Fig. 3J). Furthermore, the nonreducing polyacrylamide gel electrophoresis (PAGE) analysis showed elevated amounts of STING dimers in F. novicida- and poly(dA:dT)-, but not cGAMP-stimulated Gsdmd−/− cells (Fig. 3K), which is in concordance with the higher cGAS activation in the absence of gasdermin D. Taken together, these data suggest that gasdermin D targets cGAS to suppress cytosolic DNA-induced IFN-β production.
Figure 3. Gasdermin D suppresses cytosolic DNA-induced IFN-β by targeting cGAS.

(A) Immunoblots of pIRF3, pTBK1, IRF3, TBK1, and β-actin in the lysates of WT and Gsdmd−/− BMDMs stimulated with indicated treatments for indicated times. Med, medium; Fn, F. novicida.
(B) Immunoblots of pIRF3, pTBK1, IRF3, and TBK1 in the lysates of WT and Aim2−/− BMDMs stimulated with indicated treatments for 8 h.
(C-D) PCR analysis of F. novicida DNA recovered from cGAS immunoprecipitates from WT and Gsdmd−/− BMDMs stimulated with F. novicida for 6 h. Agarose gel image of PCR products (C). Fold increase in cGAS-associated F. novicida DNA in Gsdmd−/− BMDMs over that of wild-type BMDMs as revealed by quantitative PCR (D). Fn, F. novicida.
(E-I) Confocal images of WT and Gsdmd−/− BMDMs stimulated with F. novicida or poly(dA:dT) for 5 h. Cells were stained with an antibody for cGAS (green), cholera toxin B for plasma membrane (magenta), and DAPI for nucleus and DNA (blue) (E-H). Arrowheads indicate cGAS puncta. Three different fields per genotype for F. novicida and poly(dA:dT) stimulations are shown in F and H, respectively (merged images). Enlarged images of the boxed areas in F are shown in G. Quantification was done by counting the cells containing cytosolic cGAS puncta in 30 fields with approximately 20 cells each (I). Scale bar, 5 μM (E) or 10 μM (F and H).
(J) cGAMP amounts in WT and Gsdmd−/− BMDMs stimulated with F. novicida or poly(dA:dT) for 5 h as measured by LC-MS.
(K) STING monomers and dimers in the lysates of WT and Gsdmd−/− BMDMs stimulated with F. novicida (MOIs of 50 and 100), poly(dA:dT), or cGAMP for 6 h as assessed by nonreducing PAGE and immunoblotting. Med, medium.
Combined data from three independent experiments are shown as mean ± SEM. *, P <0.05; unpaired two-tailed t test (D) or two-way ANOVA followed by the Sidak’s post-test (I and J). Scale represents 5 μM (E) or 10 μM (F and H). See also Figure S2.
Gasdermin D suppresses cGAS-dependent type I interferon response by triggering potassium (K+) efflux
We next sought to define the mechanism involved in the cGAS-suppressive function of gasdermin D. Whereas the formation of plasma membrane pores and the cessation of metabolic activity during pyroptosis are not sensitive to glycine, the swelling and terminal rupture of cells are delayed by glycine (Conos et al., 2017; Evavold et al., 2018; Russo et al., 2016). Glycine greatly reduced F. novicida-induced pyroptotic lysis as assessed via LDH release (Fig. 4A) or CytoTox-Fluor assay (Fig. 4B). More relevantly, glycine-treated cells, which were resistant to gasdermin D-induced lysis, did not produce excessive amounts of IFN-β in response to F. novicida (Fig. 4C). The Aim2 inflammasome catalyzes the maturation of IL-1β and IL-18 via caspase-1 and mediates their release in a gasdermin D-dependent manner. Therefore, it is possible that active IL-1β and/or IL-18 can act in an autocrine or paracrine fashion to control the type I interferon responses as shown in the case of M. tuberculosis infection (Mayer-Barber et al., 2014). To test this idea, we compared F. novicida-induced IFN-β production in IL-1R- and IL-18R-deficient BMDMs with that of wild-type BMDMs. In contrast to the phenotype in Aim2- and gasdermin D-deficient cells, IFN-β response in IL-1R- and IL-18R- deficient BMDMs was comparable to that of wild-type BMDMs (Fig. 4D). Furthermore, to address if there is any redundancy between IL-1β and IL-18 in regulating IFN-β, IL-1β signaling in IL-18R-deficient BMDMs was blocked with IL-1R antagonist, and IFN-β response to cytosolic DNA was tested. As shown in Fig. 4E, the inhibition of IL-1R signaling with IL-1R antagonist did not affect F. novicida-induced IFN-β secretion in IL-18R-deficient and WT BMDMs, thus ruling out a role for IL-1β and IL-18 in gasdermin D control of IFN-β.
Figure 4. Gasdermin D suppresses type I interferon production by inducing K+ efflux.

(A) Cell death in WT BMDMs infected with F. novicida for 6, 12, 24 and 36 h in the presence or absence of glycine as assessed by LDH release.
(B) Cell death in WT BMDMs infected with F. novicida for 6 h in the presence or absence of glycine as assessed by CytoTox-Fluor assay.
(C)LDH release and secretion of IFN-β by WT BMDMs infected with F. novicida for 6 h in the presence or absence of glycine.
(D)IFN-β in the supernatants of WT, Il1r1−/− or Il18r1−/− BMDMs following infection with F. novicida for 6 h.
(E)IFN-β in the supernatants of F. novicida-infected WT and Il18r1−/− BMDMs treated with DMSO or IL-1R antagonist at the indicated concentrations.
(F-H) Intracellular K+ as assessed by APG4 staining in WT, Aim2−/− (F), Casp1−/−Casp11−/− (G), and Gsdmd−/− (H) in uninfected and F. novicida-infected BMDMs at 6 h p.i. (in the presence of glycine).
(I to L) IFN-β secretion, cell death (assessed by LDH release assay), and cell viability (assessed by CellTiter-Glo assay) in WT BMDMs stimulated with poly(dA:dT) or F. novicida as indicated for 6 h in DMEM (I and K) or isosmotic minimal medium (J and L) supplemented with indicated amounts of KCl.
(M) IFN-β in the supernatants of Gsdmd−/− iBMDMs reconstituted with the empty vector (EV), WT gasdermin D, or I105N mutant gasdermin D in response to poly(dA:dT) transfection and LPS stimulation.
Combined data from three (B-M) or two (A) independent experiments are shown as mean ± SEM. *, P <0.05; ns, not significant; two-way ANOVA followed by the Sidak’s post-test. IFN-β data are presented as units/ml or pg/ml depending upon the recombinant IFN-β standard used in the ELISA. See also Figure S3.
The N terminal fragment of gasdermin D binds to plasma membrane phospholipids forming pores with an inner diameter of 10 – 15 nm (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). These pores permit ionic fluxes across the plasma membrane dissipating intracellular ionic gradients. It is well documented that the formation of gasdermin D- pores triggering ionic fluxes is an early event in the cell lytic cascade and is succeeded by water influx, rise in osmotic pressure, cell swelling, the extensive loss of membrane integrity, and eventual lysis (Bergsbaken et al., 2009; de Vasconcelos et al., 2018; Evavold et al., 2018; Fink and Cookson, 2005). Thus, gasdermin D-mediated ion fluxes and the pyroptotic cell death are kinetically separable events (de Vasconcelos et al., 2018; Evavold et al., 2018; Russo et al., 2016). Recent reports show that ionic perturbations induced by pore forming proteins govern cellular functions. For instance, ionic fluxes driven by mixed lineage kinase-like (MLKL), the pore forming effector of the necroptotic pathway, has been shown to facilitate plasma membrane repair as well as activate NLRP3 inflammasome prior to cell death (Conos et al., 2017; Gong et al., 2017; Gutierrez et al., 2017). Taking all this into consideration, we reasoned that gasdermin D-mediated ionic fluxes—particularly the efflux of potassium (K+), which is the most abundant ion in the intracellular milieu with a steep gradient across the plasma membrane—may play a role in the negative regulation of cGAS-dependent type I interferon responses. First, we assessed if inflammasome-mediated gasdermin D activation during F. novicida infection triggers potassium efflux by utilizing a highly specific potassium stain, APG4 (Eil et al., 2016; Prindle et al., 2015). We observed that the intracellular K+ dropped significantly following infection with F. novicida in wild-type macrophages but not in Aim2−/−, Casp1−/−Casp11−/−, and Gsdmd−/− macrophages (Fig. 4F-H). The reduction in intracellular K+ in wild-type cells was not due to cell lysis as these experiments were performed in the presence of glycine, which prevented cell lysis but not K+ efflux (Fig. S3A).
Next to assess if potassium efflux is required for the gasdermin D-mediated suppression of interferon responses, K+ efflux was diminished by increasing the extracellular concentration of K+ with KCl (Muñoz-Planillo et al., 2013) and its effect on IFN-β secretion was tested. The incubation of wild-type macrophages in DMEM or isosmotic minimal medium supplemented with increasing concentrations of KCl to block K+ efflux increased poly(dA:dT)-induced IFN-β secretion without affecting pyroptosis or cell viability (Fig. 4I and 4J). The blockade of K+ efflux similarly enhanced IFN-β secretion following F. novicida infection without affecting pyroptosis or cell viability (Fig. 4K and 4L). These results clearly indicate that gasdermin D attenuates cGAS- induced type I interferons by inducing K+ efflux. An isoleucine-to-asparagine mutation at position 105 (I105N) in the N terminus of gasdermin D compromises its capacity to efficiently oligomerize, form pores and presumably induce ionic flux (Aglietti et al., 2016). We stably reconstituted Gsdmd−/− iBMDMs with the wild-type or I105N mutant gasdermin D and tested the effect of this mutation on type I interferon production (Fig. S3B). Wild-type gasdermin D strongly reduced poly(dA:dT)-elicited IFN-β response in Gsdmd−/− iBMDMs whereas I105N gasdermin D failed to suppress IFN-β response (Fig. 4M) further supporting a role for gasdermin D-driven pores and ionic flux in the inhibition of cGAS signaling.
K+ efflux is sufficient to inhibit type I interferon response to cytosolic DNA
If gasdermin D activation dampens IFN-β production in wild-type BMDMs via K+ efflux as we hypothesize, then pharmacologically restoring K+ efflux in inflammasome- and gasdermin D- deficient cells would reinstate this inhibition and reduce cytosolic DNA-induced IFN-β. We tested this idea by utilizing two well-characterized K+ ionophores, nigericin and valinomycin (Page and Di Cera, 2006). RAW cells were used in these experiments as they lack Asc and therefore, are resistant to inflammasome-mediated pyroptosis following nigericin and valinomycin stimulation (Fig. S4A). Nigericin and valinomycin induced K+ efflux in RAW macrophages as assessed by APG4 staining (Fig. 5A and 5B). Consistent with its induction of K+ efflux, nigericin suppressed IFN-β production by RAW cells following F. novicida infection and poly(dA:dT) transfection (Fig. 5A and 5C). But nigericin did not inhibit TLR-mediated IFN-β response to LPS and poly(I:C) (Fig. 5C). This result suggest that the suppressive effect of nigericin on F. novicida- and poly(dA:dT)-induced IFN-β production is neither due to global shut down of IFN-β expression nor cytotoxicity. Similar to nigericin, valinomycin impaired RAW cell production of IFN-β following F. novicida and poly(dA:dT) stimulations (Fig. 5B). Unlike these K+ ionophores, the calcium ionophore, ionomycin, did not affect F. novicida-induced IFN-β secretion (Fig. S4B). Similarly, the calcium chelator, BAPTA-AM, had no effect on IFN-β response to cytosolic DNA (Fig. S4C). These results, thus, rule out a role for calcium influx in type I interferon inhibition by gasdermin D. Nigericin treatment reduced cGAMP quantity in F. novicida- and poly(dA:dT)- stimulated RAW cells, supporting the notion that gasdermin D targets cGAS via K+ efflux (Fig. 5D). In agreement with the impairment of cGAS activation by K+ efflux, nigericin reduced TBK1 and IRF3 activation in RAW macrophages in response to poly(dA:dT) but not LPS (Fig. 5E). These results indicate that K+ efflux targets the cytosolic cGAS-STING-mediated but not TLR- initiated type I interferon responses.
Figure 5. Induction of K+ efflux in inflammasome-deficient macrophages is sufficient to reduce cytosolic DNA-induced type I interferon response.

(A and B) IFN-β in the supernatants of and intracellular K+ (at 6 h post-stimulation) in RAW macrophages stimulated with F. novicida (MOI of 50) or poly(dA:dT) and treated at 1.5–2 h after stimulations with vehicle (ethanol), 2.5 or 5 μM nigericin (A) or 5 or 10 μM valinomycin (VM; B).
(C) IFN-β in the supernatants of RAW macrophages stimulated with poly(dA:dT), LPS (1 μg/ml) or poly(I:C) (40 μg/ml) and treated at 1.5–2 h after stimulations with 2.5 μM nigericin.
(D) cGAMP amounts in RAW macrophages 5 h after stimulated with F. novicida or poly(dA:dT) and treated at 1.5–2 h after stimulations with 2.5 μM nigericin as measured by LC-MS.
(E) Immunoblots of pIRF3, pTBK1, IRF3, TBK1, and β-actin in lysates of RAW macrophages stimulated with poly(dA:dT) or LPS and treated with vehicle (Veh) or 2.5 μM nigericin (Nig).
(F and G) IFN-β in the supernatants of and intracellular K+ in WT and Casp1−/−Casp11−/− BMDMs (measured at 6 h post-infection) infected with F. novicida and treated with vehicle (ethanol), 2.5 μM nigericin (F), or 5 μM valinomycin (VM; G) 1.5–2 h post-infection.
(H) IFN-β amounts in the supernatants of Casp1−/−Casp11−/− BMDMs (measured at 6 h post- stimulation) stimulated with poly(dA:dT), LPS, or poly(I:C) and treated with vehicle, 2.5 μM nigericin or 5 μM valinomycin (VM).
(I)IFN-β in the supernatants of F. novicida-, poly(dA:dT)-, or poly(I:C)-stimulated WT BMDMs incubated in isosmotic minimal medium containing 5 mM KCl and Casp1−/−Casp11−/− BMDMs incubated in isosmotic minimal medium containing 5 or 0 mM KCl.
(J and K) IFN-β in the supernatants of WT and Gsdmd−/− BMDMs (measured at 6 h post- infection) infected with F. novicida and treated with vehicle, nigericin (J), or valinomycin (VM; K) at the indicated concentrations 1.5–2 h post-infection.
(L) IFN-β in the supernatants of Gsdmd−/− BMDMs (measured at 6 h post-stimulation) stimulated with F. novicida, poly(dA:dT), LPS, or poly(I:C) and treated with vehicle, 2.5 μM nigericin, or 5 μM valinomycin (VM).
(M-O) IFN-β in the supernatants of indicated macrophages (at 6 h post-stimulation) stimulated with F. novicida (MOI of 50) or poly(dA:dT) and treated at 1.5–2 h after stimulations with vehicle (ethanol) or increasing concentrations of nigericin (0.0125–2.5 μM).
(P) IFN-β in the supernatants of and intracellular ATP in Gsdmd−/− BMDMs (at 6 h post- stimulation) stimulated with F. novicida or LPS. After 1 h of stimulation cells were incubated in isosmotic minimal medium containing 5 mM or 0 mM KCl and 1 h later, KCl was added back to cells incubated in isosmotic minimal medium without KCl to restore K+ concentration in the medium to 5 mM.
(Q) cGAMP synthesis by recombinant cGAS incubated with DNA (10 and 2 ng), radioactive ATP, and GTP with or without 10 mM KCl as analyzed by thin-layer chromatography (TLC) and autoradiography. Arrowhead indicates cGAMP. cGAMP in TLC were quantified and expressed, within each DNA concentration, as percent relative to ‘with KCl’ condition, which is considered as 100%.
Combined data from three (A-D and F-P) or two (Q) independent experiments are shown as mean ± SEM. *, P <0.05; two-way ANOVA followed by the Sidak’s post-test. IFN-β data are presented as units/ml or pg/ml depending upon the recombinant IFN-β standard used in the ELISA. See also Figures S4 and S5.
Next, we used Casp1−/−Casp11−/− macrophages to validate these findings. Compared to F. novicida-infected wild-type cells, Casp1−/−Casp11−/− BMDMs had higher intracellular K+ and secreted increased amounts of IFN-β (Fig. 5F and 5G). Addition of nigericin or valinomycin to Casp1−/−Casp11−/− BMDMs lowered intracellular K+ content and correspondingly reduced IFN-β production (Fig. 5F and 5G). Importantly, this IFN-β reduction upon nigericin and valinomycin treatments was not due to cell death as the caspase-1 deficiency and the use of very low amounts of nigericin and valinomycin circumvented nigericin- and valinomycin-induced pyroptosis (Fig. S4D). It should also be noted that while cytosolic DNA-induced IFN-β secretion by Casp1−/−Casp11−/− BMDMs was diminished by nigericin or valinomycin, LPS- and poly(I:C)-induced IFN-β secretion remained unaffected (Fig. 5H). Similarly, the incubation of Casp1−/− Casp11−/− macrophages in K+ free medium, which is known to trigger K+ efflux (Rühl and Broz, 2015), diminished their higher IFN-β production, in response to F. novicida and poly(dA:dT), to quantities comparable to that of wild-type cells (Fig. 5I). On the other hand, poly(I:C)-induced IFN-β production was not affected by the K+ content of the medium (Fig. 5I). The increased IFN-β secretion by F. novicida-infected gasdermin D-deficient macrophages was also reduced to wild-type amounts by nigericin and valinomycin without affecting cell death (Fig. 5J, 5K, and S4D). In contrast, IFN-β production by gasdermin D-deficient macrophages in response to TLR4 and TLR3 activation was not suppressed by nigericin and valinomycin (Fig. 5L).
Furthermore, detailed dose response experiments in WT, Gsdmd−/−, Casp1−/−Casp11−/−, and RAW macrophages showed that extremely low concentrations of nigericin (20–50 nM) were able to inhibit F. novicida- and poly(dA:dT)-induced IFN-β response without affecting cell viability and metabolic activity as indicated by CellTiter-Glo and PrestoBlue assays (Fig. 5M-O and S5A-C). Minimal concentrations of valinomycin (50–100 nM) also decreased IFN-β response to cytosolic DNA without reducing cell viability and metabolic activity (Fig. S5D). More importantly, K+ free media-induced reduction in IFN-β production was reversed when KCl was returned to culture medium (Fig. 5P) indicating that the cells remained viable during exposure to K+ free media. Likewise, nigericin-induced reduction in IFN-β production was reversed when the K+ ionophore was replaced from culture medium (Fig. S5E). Altogether, the effects of K+ efflux blockade and K+ efflux induction by pharmacological (K+ ionophores) and non-pharmacological (K+ free medium) means on IFN-β production in multiple cell types provide substantial evidence that K+ efflux is necessary and sufficient to inhibit cytosolic DNA-induced type I interferon response. Finally, we performed a cell-free cGAS enzymatic assay, in which recombinant cGAS was incubated with DNA, ATP, and GTP in the presence or absence of K+, and cGAMP production was monitored by thin-layer chromatography (TLC). In this cell-free assay, cGAS synthesis of cGAMP was substantially reduced in the absence of K+ (Fig. 5Q). This data reinforces that K+ is critical for cGAS catalytic activity and the depletion of intracellular K+ by gasdermin D pores impairs cGAS function independent of cell death.
DISCUSSION
The nucleotidyl transferase, cGAS, monitors the cytosol for foreign DNA during viral and bacterial infections and induces the production of type I interferons via the STING-TBK1-IRF3 pathway (Sun et al., 2013). Type I interferons are pleiotropic cytokines associated with immunopathology and autoimmunity when produced in excess, thus necessitating a critical need for the host to keep the cGAS pathway in check (Crow and Manel, 2015). Several mechanisms involving NLRX1, NLRC3, AKT, and autophagy as well as glutamylation and sumoylation of cGAS negatively regulate cGAS-STING signaling during viral infections (Chen et al., 2016). In this study, we demonstrated that gasdermin D activated by the inflammasome complex inhibited cGAS-mediated type I interferon response to cytosolic DNA by triggering K+ efflux. We propose the following model integrating the findings of this study with that of previous ones: cytosolic DNA activates cGAS-STING-IFN-β and Aim2 inflammasome axes. The Aim2 inflammasome activates gasdermin D, which forms plasma membrane pores leading to K+ efflux. The depletion of intracellular K+ halts cGAS sensing of cytosolic DNA and thus, type I interferon responses. These findings shed light on the crosstalk between type I interferon and inflammasome responses, two major outcomes of cytosolic immune surveillance.
The following independent lines of data in this study provide evidence that the interferon regulatory role of gasdermin D can be separated from pyroptosis: 1) Blockade of K+ efflux with KCl increased cytosolic DNA-induced IFN-β production in wild-type BMDMs without affecting cell death, 2) Restoring K+ efflux in inflammasome- and gasdermin D-deficient cells by pharmacological (K+ ionophores) and non-pharmacological (K+ free medium) approaches suppressed–without affecting cell death and metabolic activity –IFN-β response driven by cGAS but not TLR signaling, 3) K+ free media- and K+ ionophore-induced IFN-β suppression was reversible, 4) The effect of K+ on cGAMP synthesis in the cell-free cGAS enzymatic assay.
A growing body of evidence attributes new cellular roles to gasdermin D-like proteins that can be dissociated from their established cell death function. Gasdermin D is capable of mediating IL-1β and IL-18 secretion via its ring shaped plasma membrane pores without leading to cell death in specific cases (Evavold et al., 2018). During gasdermin D-initiated lytic cell death process, ionic flux via the plasma membrane pores well precedes cell lysis (Bergsbaken et al., 2009; de Vasconcelos et al., 2018; Fink and Cookson, 2005; Kovacs and Miao, 2017; Russo et al., 2016). This kinetics probably permits a sufficient time frame in which ionic fluxes can impact cellular functions before the cell enters an irreversible, terminal lytic phase. It is evident from this work that the collapse of intracellular K+ concentration by gasdermin D nanopores plays a cGAS-inhibitory role. In the noncanonical inflammasome pathway, K+ efflux induced by caspase- 11-cleaved gasdermin D triggers NLRP3 inflammasome activation prior to the demise of the cell (Kayagaki et al., 2015; Rühl and Broz, 2015; Schmid-Burgk et al., 2015). Similarly, ionic fluxes that occur during necroptosis also regulate cellular processes; K+ efflux occurring as a consequence of membrane pore formation by MLKL during necroptosis triggers NLRP3 activation (Conos et al., 2017; Gutierrez et al., 2017). Furthermore, the calcium influx elicited by MLKL has been shown to induce ESCRT-III machinery-mediated repair of the damaged plasma membrane (Gong et al., 2017). Thus, the ionic perturbations induced by pore forming effectors such as gasdermin D and MLKL during pyroptosis and necroptosis, respectively, regulate cellular processes independent of the terminal cell lytic event.
During microbial and sterile inflammation, the amount of N terminal fragment of gasdermin D that accumulates in a cell at any given time is a key determinant of whether that cell commits to pyroptosis or not. If the amount of N terminal fragment does not reach a particular threshold necessary to cause extensive membrane rupture–owing to submaximal inflammasome activation –the viability of the cell could be maintained for a longer duration. Speculatively, in this subpopulation of cells in which the amount of active gasdermin D is below a threshold necessary for cell lysis, the regulatory function of gasdermin D described in this study could become sustained and more prominent. It has been well documented that K+ plays important roles in cellular enzymatic reactions by enhancing the enzyme-substrate binding and catalysis (Gohara and Di Cera, 2016; Page and Di Cera, 2006). Taking these known K+ functions and our data from the in vitro enzymatic assay into account, we speculate that K+ facilitates cGAS-DNA interaction and thus, its enzymatic function, perhaps by directly influencing cGAS conformation. Supporting this idea that an optimal ionic milieu is key to cGAS function, latest studies discovered that the divalent cations, Mn2+ and Zn2+, increase the sensitivity of cGAS for DNA and its enzymatic activity, respectively (Du and Chen, 2018; Wang et al., 2018). Considering the profound causative link between type I interferons and autoimmune diseases such as lupus arising due to a dysregulated immune reaction to self nucleic acids, the identification of an additional interferon regulatory module involving gasdermin D and K+ efflux in this study may have therapeutic implications for these diseases.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Vijay Rathinam (Rathinam@uchc.edu)
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
C57BL/6J, Casp1−/−Casp11−/−, Aim2−/−, Il1r1−/−, and Il18r1−/− mice obtained from the Jackson Laboratory (Bar Harbor, ME) were bred and maintained in specific pathogen-free conditions in the animal facilities of UConn Health. Gsdmd−/− (Kayagaki et al., 2015), Aim2−/− (Jones et al., 2010), and Casp11−/− mice (Kayagaki et al., 2011) obtained from Genentech (South San Francisco, CA) were also bred and maintained in specific pathogen-free conditions in the animal facilities of UConn Health. Asc−/− (Rathinam et al., 2010), Nlrp3−/− (Rathinam et al., 2012), Mb21d1−/− (Suschak et al., 2016) mice were described previously. Both male and female mice of 8–24 weeks old were used. The animal protocols were carried out in accordance with the guidelines set forth by the UConn Health Institutional Animal Care and Use Committee.
Differentiation of primary Bone-marrow Derived Macrophages
Primary bone-marrow derived macrophages (BMDMs) were generated by culturing bone marrow cells from wild-type and various mutant mice in DMEM with 10% fetal bovine serum (FBS) and 20% L929 supernatant. Cell culture media was replaced on days 3 and 6 with fresh DMEM with 10% FBS and 20% L929 supernatant. Cells were harvested and used for experiments after 6–7 days of culture.
Bacterial Strains
Bacteria and virus used in this study include Francisella tularensis subsp. novicida strain Utah 112 (F. novicida; BEI resources, NIAID, NIH), Enterohemorrhagic E. coli (Vanaja et al., 2016), E. coli BL21 (Vanaja et al., 2016), and Sendai virus (Cantrell strain; Charles River Laboratories).
METHOD DETAILS
Cell stimulations and infections
Francisella novicida was grown over night at 37°C in Mueller Hinton broth. BMDMs and RAW macrophages were infected at an MOI of 50 or 100 (unless otherwise indicated) and after 1 – 2 h of infection, medium was replaced with gentamicin (100 μg/ml) containing medium. Cells were infected with Sendai virus (10–20 HA units/ml). Cells were transfected with lipofectamine 2000 (2 μl/ml)-complexed poly(dA:dT) (1 µg/106 cells) or 2’−3’-cGAMP (2.5 µg/106 cells) or stimulated with poly(I:C) (40 µg/ml) or LPS (1 µg/ml). In certain experiments, cells were primed with 400 ng/ml Pam3CSK4 for 2–3 h before stimulations to assess inflammasome responses. The supernatants were collected 6–8 h post-stimulation unless otherwise indicated. In some experiments, glycine (50 mM) was added to cells at the time of or 1 h after stimulations. In experiments where K+ efflux was inhibited, increasing concentrations of KCl was added to the medium at the time of stimulations. In experiments where K+ efflux was induced, cells were treated with nigericin or valinomycin at the indicated concentrations 1.5–2 h after the stimulations or were incubated in K+ free medium with glycine throughout the duration of the experiment. Further, in some media replacement experiments, nigericin-containing media was replaced with DMEM without nigericin 1 h post addition of nigericin.
Inhibition or induction of K+ Efflux using isosmotic media
Isosmotic medium without or with defined concentrations of KCl was prepared as described previously (Rühl and Broz, 2015). Cells were infected with F. novicida and after 1 h of infection medium was replaced with gentamicin (100 μg/ml) containing buffer A (4.2 mM Na2CO3, 0.8 mM Na2HPO4, 1.3 mM CaCl2, 0.5 mM MgCl2, 10 mM D-glucose monohydrate, pH 7.4). For K+ efflux inhibition, cells were incubated in buffer A supplemented with 137 mM NaCl and 5 mM KCl, 117 mM NaCl and 25 mM KCl, or 92 mM NaCl and 50 mM KCl. For K+ efflux induction, cells were incubated in buffer A supplemented with 142 mM NaCl and glycine but without KCl. Cells were then stimulated and supernatants were collected as described above. Additionally, in some experiments 5 mM (isosmotic concentration) KCl was added to K+ free media 1 h post exposure to K+ free media.
ELISA, Cell Death Assay, and Immunoblotting
IFN-β (Rathinam et al., 2012) and IL-18 (Vanaja et al., 2016) were assessed by ELISA as described. IFN-β data are presented as units/ml or pg/ml depending upon the recombinant IFN-β standard used in the ELISA. IL-1β and IL-6 were assessed by Ready-Set-Go!® ELISA kits (eBioscience) according to the manufacturer’s instructions. Cell death was assessed by LDH cytotoxicity detection kit (Clontech) unless otherwise indicated according to the manufacturer’s instructions. Intracellular ATP and metabolic activity were measured with CellTiter-Glo assay (Promega) and PrestoBlue assay (Invitrogen), respectively, according to the manufacturer’s instructions. For western blotting analysis, cells were lysed with 1% NP-40 lysis buffer. Samples were run on polyacrylamide gels and transferred onto nitrocelluose membranes by using Trans- Blot Turbo Transfer System (Bio-Rad). Blots were then blocked and probed with appropriate primary and secondary antibodies, and protein bands were visualized with Bio-Rad Clarity ECL HRP substrate on a Syngene gel documentation box.
Liquid Chromatography-Mass Spectrometric measurement of cGAMP
Cells were stimulated with F. novicida or poly(dA:dT) for 5 h and snap-frozen. Cell extract prepared with methanol was filtered with 30 kDa filter to remove debris, dried, resuspended in water. c-di-AMP was added to the samples as an internal control. Samples were transferred to vials, capped and placed into the autosampler (kept at 4 °C). Aliquots of 10 or 20 µl from each vial was injected, along with cGAMP standards, onto the LC-MS/MS system (Agilent 1200 Autosampler and Binary pump 20) coupled to an ABI4000Q bench top mass spectrometer (MDS SCIEX). The MRM m/z transitions monitored for cGAMP and c-di-AMP were 675.5>506.0, and 659.0>524.0, respectively. Data were collected with the Analyst software.
Intracellular Potassium Measurement
Wild-type, Gsdmd−/−, Casp1−/−Casp11−/−, and Aim2−/− cells plated in 96-well clear bottom black plates were stimulated with F. novicida in glycine-containing media. After 6 h of stimulation, medium was replaced with PBS containing asante potassium green (APG)4 for 1 h, which is extremely specific for K+ (Eil et al., 2016; Prindle et al., 2015), and pluronic acid F-127, which aids in efficient permeation of APG4 into the cells. After 1 h of incubation at 37°C, cells were washed with PBS and the fluorescence was read with ClarioStar multimode microplate reader (BMG LABTECH, Germany).
Reconstitution of GsdmD−/− iBMDMs with GsdmD
HEK293T cells were transfected with pMSCV-puro (empty vector; EV), pMSCV-puro encoding 2xFLAG-HA-gasdermin D, or pMSCV-puro encoding 2xFLAG-HA-I105N gasdermin D mutant and retro viral packaging plasmids. Viral supernatants collected after 48 and 72 h of transfection were concentrated by centrifugation at 20,000 x g for 2 h and used to transduce Gsdmd−/− iBMDMs. Transduced cells were selected using puromycin, and the expression of wild-type and mutant gasdermin D was verified by western blotting with FLAG and gasdermin D antibodies.
Mice Infections
F. novicida was grown over night at 37°C in Mueller Hinton broth. C57BL/6 (wild-type; WT), Aim2-deficient (Rathinam et al., 2010), Casp1−/−Casp11−/−, and Gsdmd−/− mice were subcutaneously infected with 1.5 or 5 × 105 CFU of F. novicida. Cytokines in the plasma were analyzed at 24 h post-infection. Bacterial loads in spleen and liver at 24 h p.i. were assessed by serial dilution of plating of spleen and liver homogenates on Mueller-Hinton agar plates supplemented with 3% FBS. C57BL/6 (wild-type; WT) and Aim2−/− mice were infected with 1 × 109 CFU of E. coli BL21 by i.p. injection. IFN-β in the plasma was analyzed at 6 h post-infection. Mice were infected with 2.5 × 102 CFU of F. novicida in the survival studies. In some survival experiments, WT and Aim2−/− mice were injected i.p. with 250 μg of an isotype control or anti- IFNAR antibody (clone: MAR1–5A3) 12 h p.i.
cGAS-DNA binding assay
Wild-type and Gsdmd−/− BMDMs were stimulated with F. novicida. After 6 h p.i., media was replaced with PBS containing 1% paraformaldehyde (PFA), and the cells were incubated for 10 min at room temperature. The crosslinking reaction was then quenched with 0.1 M Tris pH 7.4 in PBS. The cells were then collected and lysed in 0.5% Triton-X 100 lysis buffer. cGAS immunoprecipitation was performed by adding protein A beads and anti-cGAS antibody to the lysates. After overnight incubation at 4 °C, beads were washed twice with low salt, high salt, and lysis buffers. Then antibody-protein-DNA complexes were eluted with a freshly prepared elution buffer (1% SDS and 0.1 M NaHCO3 pH 8.0); Beads mixed with elution buffer was shaken on vortex for 15 min and then centrifuged at 13,000 rpm for 3 min. Eluted samples were then de- crosslinked by adding 0.3 M NaCl and incubating at 65 °C for 4 h. This was followed by protein digestion using proteinase K at 45 °C for 2 h. DNA was finally purified using phenol-chloroform- isoamyl alcohol precipitation. F. novicida DNA bound to cGAS was quantified by real time PCR with primers targeting the bacterial genome (mglA gene). The PCR products were also run on 1.5% agarose gels and imaged using a Syngene gel documentation box.
cGAS immunofluorescence staining
Wild-type and Gsdmd−/− BMDMs were infected with F. novicida or transfected poly(dA:dT). After 5 h, the cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X, blocked with 10% goat serum, incubated overnight at 4 oC with anti-cGAS antibody, washed, and stained for 1 h with fluorescently labeled anti-rabbit secondary antibody. Plasma membrane and nucleus were stained with cholera toxin B Alexa fluor 647 conjugate (CTB) (Life Technologies) and DAPI, respectively. The cells were visualized using a Zeiss LSM 780 microscope.
In vitro cGAS enzymatic assay
Recombinant mouse cGAS (catalytic domain) (28 ng) was mixed in a 10 μl reaction buffer (20 mM Tris-Cl [pH 7.4], 5 mM MgAc) with 10 or 2 ng of DNA, 12 μM each ATP and GTP, and 5.0 μCi of α−32P-ATP (800Ci/mmol). After 120 min of incubation at 37 °C, the reaction mixture was heated to 65 °C for 5 min, treated with calf-intest inal alkaline phosphatase, and analyzed by thin layer chromatography (TLC) as described before (Kranzusch et al., 2013).
Flow Cytometry
For flow cytometric quantification of viperin expression, cells were stimulated with F. novicida, poly(dA:dT), or IFN-β (500 IU/ml) for 16 h. Cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% saponin in PBS and stained with anti-viperin antibody in FACS buffer (PBS with 3% FBS and 0.1% sodium azide). Samples were run on LSR-II (BD Biosciences) and analyzed using FlowJo software.
Quantification and Statistical Analysis
Data were analyzed for statistical significance by two-way ANOVA followed by the Sidak’s post- test, unpaired two-tailed t test, or Mantel-Cox test as indicated with Prism Software. *, P <0.05; ns, not significant.
Supplementary Material
Highlights.
Inflammasome-activated Gasdermin D limits type I interferon responses to cytosolic DNA
Gasdermin D targets cGAS activation to inhibit IFN-β response to cytosolic DNA
Depletion of intracellular K+ by gasdermin D is responsible for limiting cGAS signaling
K+ efflux is sufficient to inhibit cGAS-dependent type I interferon responses
Acknowledgements:
We thank Drs. Vishva Dixit and Nobuhiko Kayagaki for Aim2−/−, Casp11−/−, and Gsdmd−/− mice; Dr. Feng Shao for wild-type and Gsdmd−/− iBMDM; and Drs. Thirumaladevi Kanneganti and Jonathan Kagan for reagents. Drs. Jan Beumer and Robert Parise for LC-MS for cGAMP. This work was supported by the NIH (AI119015 to V.R., AI083713 to K.A.F., and AI118896 to S.N.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing financial interests.
REFERENCES
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, and Portnoy DA (2004). Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, and Cookson BT (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol 7, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016). Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nature Immunology 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, Masters SL, Murphy JM, Schroder K, Vaux DL, et al. (2017). Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proceedings of the National Academy of Sciences 114, E961–E969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Woo S-R, Williams JB, McWhirter SM, Dubensky TW, and Gajewski TF (2016). Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. The Journal of Immunology 196, 3191–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nature Reviews Immunology 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou Y, Wang R, Wang J, Qu B, Shen N, et al. (2017). SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog 13, e1006156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, and Lamkanfi M (2018). Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 26, R568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature [DOI] [PubMed]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 8, eaat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, et al. (2016). Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature Immunology 11, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, and Cookson BT (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun 73, 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohara DW, and Di Cera E (2016). Molecular Mechanisms of Enzyme Activation by Monovalent Cations. J Biol Chem 291, 20840–20848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017). ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, and Stetson DB (2016). The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity 45, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SWG, Gale M, and Oberst A (2017). MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J Immunol 198, 2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer JD, Iwakura Y, Metzger DW, et al. (2010). Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. The Journal of Immunology 184, 3755–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, and Latz E (2010). Intracellular DNA recognition. Nature Reviews Immunology 10, 123–130. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, and Fitzgerald KA (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1- activating inflammasome with ASC. Nature 458, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, et al. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proceedings of the National Academy of Sciences 107, 9771–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailasan Vanaja S., Rathinam VAK, Atianand MK, Kalantari P, Skehan B, Fitzgerald KA, and Leong JM (2014). Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences 111, 7765–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L., Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase- 11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kovacs SB, and Miao EA (2017). Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, and Doudna JA (2013). Structure of Human cGAS Reveals a Conserved Family of Second-Messenger Enzymes in Innate Immunity. Cell Rep 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RKS, Gurung P, et al. (2015). Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417– 426. [DOI] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. (2014). Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, McNab F, Mayer-Barber K, Mayer-Barber K, Sher A, Sher A, Wack A, Wack A, and O’Garra A (2015). Type I interferons in infectious disease. Nature Reviews Immunology 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, and Núñez G (2013). K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page MJ, and Di Cera E (2006). Role of Na+ and K+ in Enzyme Function. Physiological Reviews 86, 1049–1092. [DOI] [PubMed] [Google Scholar]
- Prindle A, Liu J, Asally M, Ly S, Garcia-Ojalvo J, and Süel GM (2015). Ion channels enable electrical communication in bacterial communities. Nature 527, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature Immunology 11, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA (2012). TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell 150, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, and Dubyak GR (2016). Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. The Journal of Immunology 197, 1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühl S, and Broz P (2015). Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. European Journal of Immunology 45, 2927–2936. [DOI] [PubMed] [Google Scholar]
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J e201694696. [DOI] [PMC free article] [PubMed]
- Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, and Hornung V (2015). Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. European Journal of Immunology 45, 2911–2917. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Storek KM, Gertsvolf NA, Ohlson MB, and Monack DM (2015). cGAS and Ifi204 Cooperate To Produce Type I IFNs in Response to Francisella Infection. The Journal of Immunology 194, 3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suschak JJ, Wang S, Fitzgerald KA, and Lu S (2016). A cGAS-Independent STING/IRF7 Pathway Mediates the Immunogenicity of DNA Vaccines. J Immunol 196, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, and Rathinam VAK (2016). Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, Du X, Yang J, Li T, Wan Y, et al. (2018). Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity [DOI] [PubMed]
- Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G, et al. (2017). Inflammasome Activation Triggers Caspase-1-Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 46, 393–404. [DOI] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. (2015). Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host & Microbe 17, 799–810. [DOI] [PubMed] [Google Scholar]
- Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, Rogers AB, Brickey WJ, Wang Y, Schneider M, et al. (2015). Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, and Fan Z (2016). Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nature Immunology 17, 369–378. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Man SM, Karki R, Malireddi RKS, and Kanneganti T-D (2018). Detrimental Type I Interferon Signaling Dominates Protective AIM2 Inflammasome Responses during Francisella novicida Infection. Cell Rep 22, 3168–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.