

Abstract
Purpose:
To characterize the ocular phenotype of DICER1 syndrome
Design:
Prospective, single-center, case-control study
Subjects, Participants, and/or Controls:
One hundred and three patients with an identified germline, pathogenic DICER1 variant (DICER1 -carriers) and 69 family control subjects underwent clinical and ophthalmic examination at the National Institutes of Health between 2011 and 2016.
Methods:
All participants were evaluated with a comprehensive ophthalmic exam including best corrected visual acuity, slit-lamp biomicroscopy and a dilated fundus examination. A subset of patients returned for a more detailed evaluation including spectral-domain optical coherence tomography, color fundus photography, fundus autofluorescence imaging, visual field testing, full field electroretinogram and genetic testing for inherited retinal degenerative diseases.
Main Outcome Measures:
Visual acuity and examination findings
Results:
Most DICERl-carriers (97%) maintained a visual acuity of 20/40 or better in both eyes. Twenty three DICERl-carriers (22%) had ocular abnormalities compared with four (6%) family controls (P=0.005). These abnormalities included retinal pigment abnormalities (N=6, 5.8%), increased cup-to-disc ratio (N=5, 4.9%), optic nerve abnormalities (N=2, 1.9%), epiretinal membrane (N=2, 1.9%) and drusen (N=2, 1.9%). Overall, we observed a significant difference (p= 0.03) in the rate of retinal abnormalities in DICERl-carriers (N=11, 11%) vs. controls (N=1;1.5%). One patient had an unexpected diagnosis of retinitis pigmentosa with a novel variant of unknown significance in PRPF31, and one had optic nerve elevation in the setting of increased intracranial pressure of unclear etiology. Three patients (3%) had DICERl-related ciliary body medulloepithelioma (CBME), two of which were identified during routine examination, a significantly higher rate than that previously reported.
Conclusions:
Ophthalmologists should be aware of the ophthalmic manifestations of the DICER1 syndrome and individuals and families should be counseled on the potential signs and symptoms. We recommend that children with a germline pathogenic variant in DICER1, especially those under the age of 10 years, undergo annual dilated ophthalmic examination, looking for evidence of CBME, signs of increased intracranial pressure and perhaps changes in the retinal pigment epithelium.
Précis
Ciliary body medulloepitheliomas have previously been described in patients with DICER1 syndrome. In addition to these findings, DICER1-carriers should be monitored for changes in the optic nerve and retina.
Introduction
The DICER1 syndrome is a recently-recognized tumor predisposition disorder that arises from pathogenic germline variants in DICER1. DICER1 is a widely expressed gene essential to the processing of mature microRNA (miRNA), single-standed RNA molecules critical to regulating gene expression and protein synthesis.1,2 The disorder is associated with a variety of rare malignancies including pleuropulmonary blastoma (PPB), Sertoli-Leydig cell tumor, embryonal rhabdomyosarcoma of the cervix, pineoblastoma, pituitary blastoma and thyroid cancer. The non-malignant DICER1 phenotype includes macrocephaly, multi-nodular goiter, nasal chondromesenchymal hamartoma and cystic nephroma.3–8
Ciliary body medulloepithelioma (CBME) is a primitive neuroepithelial neoplasm typically arising from the nonpigmented ciliary epithelium, with 75–90% of cases presenting in the first decade of life,9 typically with visual symptoms or abnormalities on ophthalmologic examination. These tumors are divided into benign and malignant by their histopathology, however, the most important feature for prognosis is extraocular extension, although distant metastasis and mortality are rare. CBME presents as a fleshy mass with clear cysts, leading to secondary glaucoma in approximately 50% of patients, as well as cataract and retinal detachment10. Although it is associated with the DICER1 syndrome, fewer than 1% of patients with PPB manifest CBME and conversely, 5% of patients with CBME have a history of ppb6,9,11,12. Current consensus guidelines for the surveillance of individuals with a DICER1 pathogenic variant suggest visual acuity screening and routine eye examination from age 3 years to at least age 10 years13.
In recent years, the role of Dicer enzymes in the retinal pigment epithelium (RPE) has been an area of interest for ophthalmic researchers. Donor human eyes with geographic atrophy in the setting of age-related macular degeneration (ARMD) have been found to have reduced DICER1 messenger RNA (mRNA) in macular RPE versus controls14. Multiple animal models have been created to further study the retinal effects of downregulating this miRNA processing enzyme.
The full spectrum of the human DICER1 -associated ocular phenotype is currently unknown. To better define this, we investigated the prevalence of ocular anomalies detected on routine clinical evaluation in a cohort of prospectively-ascertained individuals who harbor a pathogenic variant in DICER1 and in family controls. Select individuals were more thoroughly evaluated with additional ophthalmic examinations and testing at the National Eye Institute (NEI).
Methods
Study recruitment, phenotyping and DICER1 genetic testing.
Individuals with a history of a DICERl-associated tumor or who harbored a known pathogenic DICER1 variant were recruited to an on-going natural history study of the DICER1 syndrome at the National Cancer Institute (NCI) (NCT-01247597). After genetic counseling, probands and their family members underwent germline DICER1 genetic testing as previously reported.15 All participants or their guardians completed detailed questionnaires about past medical and surgical history, including eye-associated problems. Pathogenic DICER1-mutation carriers (hereafter, “DICER1 -carriers”) and family controls (no detected pathogenic germline DICER1 mutations by sequencing, or inferred negative by pedigree analysis) were invited to undergo a three-day evaluation at the National Institutes of Health (NIH) Clinical Center (CC), including comprehensive ophthalmic evaluation, between 2011 and 2016.
Study participants were assessed with a review of any previous examination records, medical and ocular histories, slit-lamp biomicroscopy and a dilated funduscopic examination. Best-corrected visual acuity was measured using the Early Treatment of Diabetic Retinopathy Study (ETDRS) chart recorded as Snellen Acuity or age-appropriate pediatric vision testing methods, including Allen figures, HOTV charts, fix and follow (FF) or central, steady and maintained (CSM). Spectral domain optical coherence tomography (OCT) (SD-OCT: Cirrus HD-OCT; Carl Zeiss Meditec, Dublin, CA) as well as color fundus photography (Topcon and Optos ultrawide-field retinal imaging device; Dunfermline, Scotland) were obtained per the examining physician’s discretion. All participants provided written, informed consent, and the study was approved by the Institutional Review Boards of the National Cancer Institute and the National Eye Institute. All study protocols adhered to the tenets of the Declaration of Helsinki and complied with the Health Insurance Portability and Accountability Act.
Focused ophthalmic evaluation and genetic testing.
After reviewing data from the comprehensive CC evaluation, DCER1-carriers with any significant ophthalmic abnormality (N=7) were invited back for a more detailed evaluation at the NEI. Seven patients were consented to the Genetics of Inherited Eye Disease Protocol (NCT-02471287) and underwent a comprehensive ocular examination, including best-corrected visual acuity testing, slit-lamp biomicroscopy, and dilated fundus examination. In addition, patients also completed visual field testing, electroretinography (ERG) and retinal imaging, including SD-OCT, fundus autofluorescence imaging, and color fundus photography. International Society for Clinical Electrophysiology of Vision (ISCEV) standard full-field flash ERGs were recorded from corneal bipolar Burian-Allen electrodes (Hansen Ophthalmic Laboratories, Iowa City, IA) using a commercial electrophysiology system (LKC, Gaithersburg, MD).16,17 Following genetic counseling, four patients with findings suggesting retinal degeneration underwent Clinical Laboratory Improvement Amendments (CLIA) clinical molecular genetic testing with a 280- gene inherited retinal dystrophy panel (Molecular Vision Laboratory; Hilsboro, Oregon; CLIA Lab ID: 38D2059762). Variants were interpreted using (1) publicly available population and disease databases, including the Exome Aggregation Consortium (ExAC),18 1,000 Genomes (1,000G)19 Exome Sequencing Project (ESP),20 Human Gene Mutation Database (HGMD)21 and ClinVar,22 as well as (2) in silico prediction tools, including Polymorphism Phenotyping v2 (PolyPhen-2)23 and Sorting Intolerant from Tolerant (SIFT).24 The Fisher exact test, chi-square and t-tests were used for determining statistical significance.
Results
Cohort Demographics
One hundred and three DICERl-carriers and 69 family controls were evaluated at the outpatient eye clinic at the NIH CC between 2011 and 2016. There was a significant difference in gender distribution (p = 0.04; chi-square). The mean age at exam was 27.0 years for DICERl-carriers and 37.9 years for controls (Table 1), a statistically significant difference (p = 0.001; two-tailed t- test).
Table 1.
Demographics and ocular characteristics of DICERl-carriers and family control
| DICER1-carriers | Controls | |
|---|---|---|
| Total | 103 | 69 |
| Male | 47 (46%) | 43 (62%) |
| Female | 56 (54%) | 26 (38%) |
| Age (mean, years) | 27.0 | 37.9 |
| Age (range, years) | 0.8 – 73.9 | 0.9 – 74.3 |
| BCVA | 20/12.5 – 20/80 | 20/12.5 – 20/25 |
| Ophthalmic Findings | ||
| Optic Nerve Abnormality | 7 | 2 |
| Retinal Pigmentary Abnormality | 6 | 0 |
| Cilliary Body Tumor | 3 | 0 |
| Macular Drusen | 2 | 1 |
| Epiretinal Membrane | 2 | 0 |
| Color Blindness | 1 | 0 |
| Choroidal Nevus | 1 | 0 |
| Retinal Degeneration | 1 | 0 |
| Iris Nevus | 0 | 1 |
BCVA= best corrected visual acuity
Ocular characteristics of controls
Of the 69 family controls, only four (6%) had ophthalmic abnormalities greater than refractive error or age appropriate cataract formation (Table 1): increased cup-to-disc ratio (N=2), iris nevus (N=1) and drusen (N=1). Best corrected visual acuity for this group ranged from 20/12.5 to 20/25.
Ocular characteristics of DICER1-carriers
Ninety-six of the 103 (93%) DICER1-carriers who could participate in ETDRS testing maintained acuity of 20/40 or better in both eyes; four pediatric patients were noted to have age- appropriate acuity of FF and CSM, without a Snellen equivalent. Only two patients had best- corrected visual acuity measured to be worse than 20/60 in one eye (excluding the one patient with a history of enucleation): an 18-year-old boy with anisometropia of five diopters leading to amblyopia with a visual acuity of 20/60, and a 57-year-old with long-standing history of amblyopia, leading to 20/100 acuity.
Twenty-three DICER1-carriers (22%) had an ocular abnormality that exceeded simple refractive error or age-related cataracts, compared with 6% of controls (p=0.005; Table 1). Of these, eleven DICER1-carriers (11%) had retinal findings (drusen, RPE, retinal degeneration) compared with one control (1.5%), a significant difference (p = 0.03). One participant with red-green color blindness had a strong family history of color blindness and an otherwise normal examination. One DICER1-carrier (with DICER1 c.1507G>T: p.E503X) developed CBME and underwent enucleation at an outside institution at age four, 47 years prior to NIH evaluation; his presenting symptoms were unknown. He has not been diagnosed with other malignancies. Additionally, two NIH patients developed a CBME after their initial visit.
One 7-year-old boy (with DICER1 c.3658C>T: p.Q1220X) with a history of type I PPB as an infant reported vision loss in one eye during his yearly pediatric well-child visit, five years after evaluation at NIH and one year after a normal well child check with the pediatrician. Ophthalmologic examination found hand motion vision, a sluggish right pupil, sensory exotropia, cataract and a right ciliary body mass. He underwent enucleation, and subsequent pathology identified total cataract with focal capsular rupture and phacoantigenic uveitis, focal temporal tractional retinal detachment attached to a cyclitic membrane covering a ciliary body tumor. This tumor was a medulloepithelioma, confined to the eye with no malignant features. (Figure 1) A second patient, a 7-year-old girl with DICER1 c.1408G>T:p.E470X and no other history of malignacny, expressed no visual complaints, but was noted to have new-onset strabismus by her mother, and when directly queried, the child reported blurry vision. Ophthalmic examination revealed light perception vision, sensory exotropia, a shallow anterior chamber with anteriorly displaced lens and a large mass filling the posterior chamber, 4.5 years after her initial NIH examination. Ten months prior to this at her pediatrician well-child check, her mother reported a significant increase in blinking and the patient’s acuity was slightly asymmetric at 20/20 and 20/25 at that time. She underwent enucleation, and pathology confirmed abnormal proliferation of primitive neuroepithelial cells surrounded by myxoid stroma centered at the ciliary body, consistent with medulloepithelioma.
Figure 1.
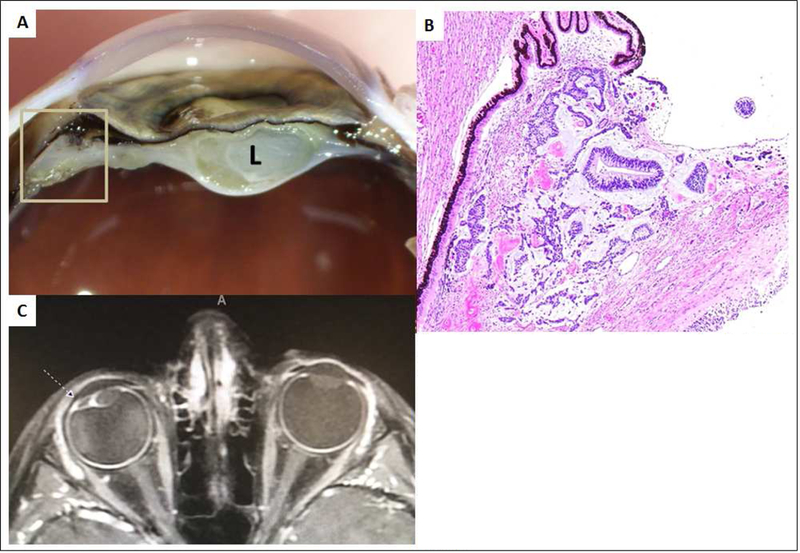
7-year-old boy, DICER1 carrier with medulloepithelioma of the ciliary body. A: Macroscopic photograph of the anterior portion of the eye after sectioning demonstrating a tan-white lesion (inside square) adjacent to the ciliary body with a membrane extending around the cataractous lens (L). B: Microscopic photograph of the tumor (same site as in the previous photo inside the square) adjacent to the pigmented epithelium of ciliary body. The tumor is composed by tubular structures of neoplastic neuroepithelium seen on a basophilic loose stroma. Hematoxylin and Eosin stain. Original magnification 4X. C: T1 weighted axial image with contrast demonstrating the ciliary body tumor (dashed arrow). Note the displacement of the lens laterally.
Optic nerve abnormalities were noted in a total of seven DICER1-carriers. Five of these had an increased cup to disc ratio without any signs of glaucoma or retinal nerve fiber layer (RNFL) thinning on OCT. Two patients were found to have elevation of the optic nerve head and were asked to return for more comprehensive testing. One patient with optic nerve findings was a 7- year-old boy who complained of headaches, transient blurring of vision and occasional ringing in his ears. His acuity at the time of his assessment was 20/20 OD and 20/25 OS with no color vision deficits as measured by Ishihara color plates. His anterior segment exam was normal, however his dilated exam demonstrated retinal vascular tortuosity and disc elevation without evidence of optic nerve head drusen (Figure 2A). His symptoms and clinical exam suggested increased intracranial pressure. His subsequent magnetic resonance imaging/magnetic resonance venography (MRI/MRV) was normal and lumbar puncture opening pressure was elevated at 37 mm H20; cerebral spinal fluid (CSF) laboratory testing was otherwise within normal limits. He had improvement of both his headache and disc elevation with acetozolamide treatment and continues to be monitored closely. The other participant with optic nerve abnormalities was a 65- year-old woman with optic nerve head drusen who maintained good visual acuity in the range of 20/25–20/30 but developed paracentral scotomas in the setting of RNFL thinning on OCT (Figure 2B).
Figure 2.
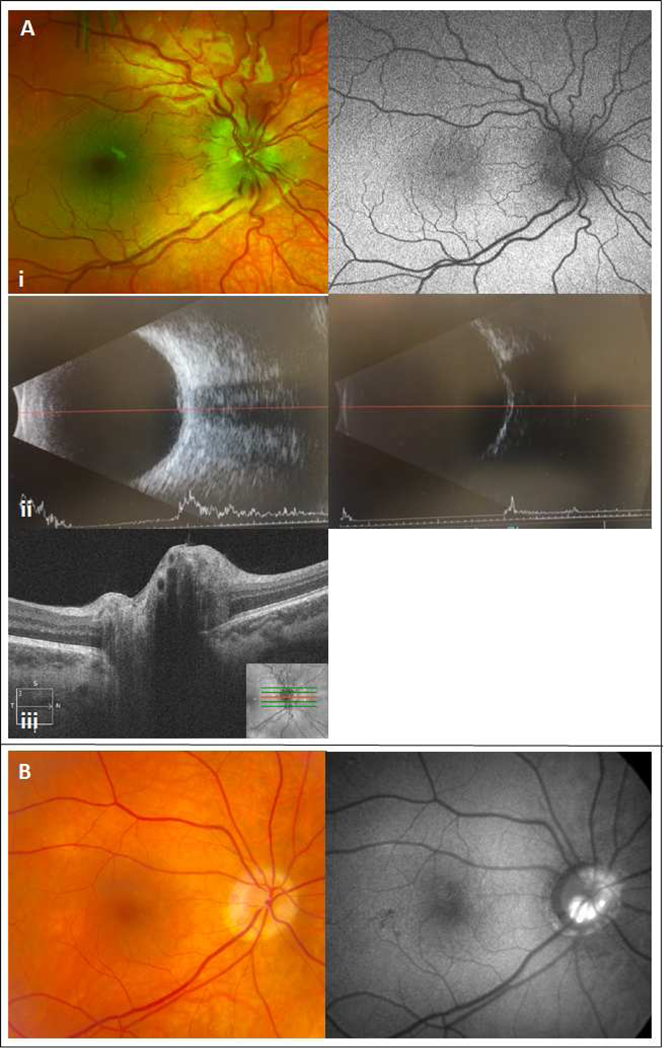
Panel A: 7-year-old boy with headaches and transient blurred vision. Examination of the fundus revealed disc elevation with retinal vascular tortuosity and no hyperautofluorecence at the nerve head (i). There was no obvious hyper- or hypo-reflective lesion in the peripapillary area on b-scan imaging in high (ii left) or low gain (ii right). OCT does not demonstrate any classic optic nerve head drusen features (iii). Panel B: 65-year-oid female with optic nerve head drusen noted on direct visualization and hyperautofluorescent lesions on imaging.
Retinal changes in DICERl-carriers included epiretinal membranes (N=2), drusen (N=2), RPE abnormalities (N=6) and retinitis pigmentosa (N=1). Patients with RPE abnormalities ranged from small focal areas of RPE dropout to congenital hypertrophy of the RPE (CHRPE), without any diffuse changes suggestive of a retinal degeneration. The one patient with a small, flat choroidal nevus was 16 years old at the time of evaluation and had no characteristics worrisome for melanoma: no overlying orange pigment, surrounding halo or associated subretinal fluid. The patient with CHRPE was a 33-year-old woman with no personal or family history of familial adenomatous polyposis. One 37-year-old female DICERl-carrier reported a diagnosis of retinitis pigmentosa prior to her study enrollment (Figure 3). Her fundus exam demonstrated classic waxy pallor of the optic nerve, retinal atrophy with bony spicules and vascular attenuation. Her electroretinogram responses were severely reduced and delayed, confirming a diagnosis of retinitis pigmentosa. Genetic testing revealed a novel missense variant in PRPF31 (NM_015629.3:c.829A>C; NP_056444:p.Ser277Arg), altering a deeply conserved Serine residue in the Nop domain. This variant was absent from population and disease databases (ExAC, gnomAD, ESP, 1000G, HGMD, ClinVar) and was predicted to be damaging by in silico prediction programs (SIFT and Polyphen-2). Her mother, who is also a DICERl-carrier, underwent comprehensive ophthalmic testing. She had no retinal degeneration, and did carry the PRPF31 missense variant, therefore, the classification of this PRPF31 allele is a variant of uncertain significance. However, retinal degenerations associated with small nuclear riboproteins such as PRPF31 are known to exhibit incomplete penetrance.
Figure 3.
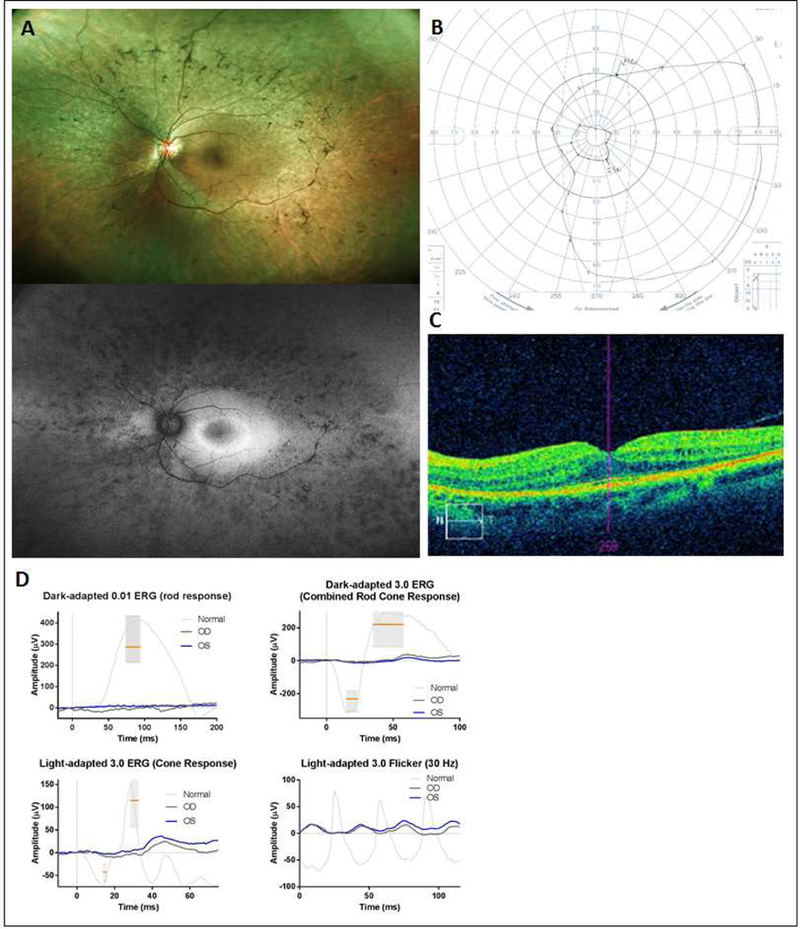
37-year-old DICER1-carrier woman presenting with retinitis pigmentosa. Fundus examination revealed classic features of retinal atrophy, bony spicules and vessel attenuation (top panel A) with peripheral hypoautofluorescence with a central hyperautoflu orescent ring (lower panel A). Visual acuity was preserved at 20/20 in each eye with a constricted visual field (B) and macular cystic changes and loss of photoreceptor IS/OS band demonstrated on OCT (C). Full-field ERG showed nearly unrecordable scotopic responses with severely reduced photopic responses, consistent with a rod-cone dystrophy (D).
Discussion
DICER1 is an endonuclease critical for the production of miRNA, which helps regulate gene expression in embryologic and early developmental stages.1 The DICER1 syndrome is an autosomal dominant tumor-predisposition disorder that features a mixture of rare and common tumors primarily affecting children and young adults, including pleuropulmonary blastoma, cystic nephroma, Sertoli-Leydig cell tumors, thyroid cancer and multinodular goiter. Medulloepithelioma of the ciliary body is known to be associated with the DICER1 syndrome, and was the primary reason for ophthalmic evaluation. A 2011 review of 299 patients in the International PPB Registry identified three patients who had both PPB and CBME, and one patient whose father had medulloepithelioma12. Presenting signs and symptoms include decreased visual acuity or leukocoria, leading to identification of a mass on the iris or ciliary body. In our two incident pediatric CBME patients, the presenting symptom was vision loss of unknown duration, noted upon direct questioning, and strabismus noted by a parent. Both patients had documented normal undilated ocular examinations by their pediatricians within the one year prior to ciliary body tumor diagnosis; one with only mildly asymmetric acuity. MRI imaging is important to identify extent of invasion, while ocular imaging with ultrasound biomicroscopy (UBM) may help identify the typical heterogeneous, cystic features of CBME. Definitive diagnosis, however, is based on pathology in most cases. We did not observe any genotype-phenotype or clinical correlates in the three patients in this study with CBME, who all harbored different germline truncating DICER1 variants. Our ability to draw firm conclusions on CBME correlations is limited by the small number of affected individuals and their young age. It has been reported that fewer than 1% of patients with PPB manifest CBME, which is lower than our experience (3%) with this cohort.
There are other DICER1 -associated tumors that may present with ocular signs and symptoms. DICERl-carriers are at increased risk of developing pituitary blastoma. de Kock et al. described twelve pituitary blastoma cases with germline and/or somatic DICER1 mutations25. Patients most often presented with ophthalmoplegia and Cushing disease; only a minority (17%, N=2) demonstrated proptosis and visual disturbance. None of the participants in our study were found to have proptosis or ophthalmoplegia, and none harbored a known pituitary blastoma. Similarly, Sabbaghian et al described a pathogenic DICER1 germline variant in a patient with a pineoblastoma.26 Although none of the patients in the current study had a known pineoblastoma, we did identify a 7-year-old boy DICER1 -carrier who presented with disc elevation (Figure 2) in the setting of idiopathic increased intracranial pressure (ICP). It is unclear if this boy’s ICP is associated with his DICER1 status; data from additional patients is needed. Our findings show that the ocular phenotype of the DICER1 syndrome is not fully established. This highlights the importance of a full dilated examination in DICER1-carriers, specifically to evaluate the optic nerve for disc edema or other signs of increased ICP.
The 65-year-old female DICERl-carrier with identified optic nerve head drusen and RNFL thinning (Figure 2B) likely falls into the 1% of the general population that may present with this finding. Optic nerve head drusen is bilateral in 75–85% of patients and presents most often in whites.27,28 As optic nerve drusen enlarge, they can compress nerve fibers leading to visual field defects, as seen in our patient. We do not believe this finding is related to the DICERl syndrome, however further observations are necessary.
We report, for the first time, a significant difference in retinal abnormalities in DICERl-carriers versus controls. We identified 11 DICERl-carriers (11%; versus 1.5% in controls; p =0.03) with retinal findings ranging from drusen, RPE abnormalities to retinal degeneration. Although some of these retinal findings are certainly common in the general population (e.g. CHRPE, RPE drop-out), some may be DICERl-associated. Some authors speculate that the processing of miRNA is critical to the normal health and development of the retina.29 If true, then some of the retinal changes we observe may be DICERl-associated. Our observations merit long-term follow-up to determine the natural history of these changes. In addition, replication of our observations in other cohorts is needed. Awareness of these retinal differences, especially in younger people, may aid in the identification of previously unrecognized, at-risk individuals.
The ocular phenotype of DICERl deficiency has been investigated in a variety of animal models. Heterozygous Dicer1-mutant zebrafish have normal RPE cells with no evidence of degeneration by age 20 months.30 In mice, loss of Dicer1 impairs the processing of long double-stranded Alu RNAs, which is hypothesized to contribute to the formation of geographic atrophy in age-related macular degeneration.14 In our study, two DICERl-carriers over the age of 65 years had retinal findings consistent with a diagnosis of age-related macular degeneration (ARMD). Since the number of patients over the age of 50 years in this group was small (N=25), it is difficult to ascertain whether or not there is an increased incidence of ARMD in DICERl-carriers.
It is unclear whether the retinitis pigmentosa in one DICER1 -carrier in our study is attributable to the novel missense variant in PRPF31 or to her germline loss-of-function DICER1 variant. While the identified PRPF31 variant is rare and predicted to be pathogenic by in silico prediction tools, there currently is insufficient evidence for pathogenicity. Pathogenic germline variation in PRPF31 accounts for 5–10% of autosomal-dominant retinitis pigmentosa. Family studies consistently show incomplete penetrance, with some mutation carriers having no vision problems.33 The incomplete penetrance in families with PRPF31-associated retinitis pigmentosa further complicates the interpretation of the PRPF31 variant of uncertain significance identified in the mother-daughter duo in this cohort. There is no additional family history of retinitis pigmentosa. If the retinal changes are not PRPF31-related, then perhaps they arise from discrete DICER1 somatic “second hit” mutations in the retina. Careful documentation and genetic testing of additional cases of retinitis pigmentosa in DICER1-carriers are needed to establish an association.
Our study is the largest evaluation of the ocular phenotype in DICER1-carriers, with family controls, to date. A strength of this study is the detailed phenotyping examination that all patients underwent at the NIH CC. However, we recognize that this may lead to an over-estimation of the ocular phenotype findings. Replication of our findings is needed in other cohorts. Recommendations on surveillance in DICER1-carriers have recently been published and suggest individuals and families should be counseled regarding visual acuity changes, strabismus or leukocoria with annual routine dilated ophthalmologic exam with visual acuity screening from 3 years of age through at least 10 years of age.13 Given that self-reported ocular symptoms are often limited in the pediatric population, we agree that all children under the age of ten years undergo an annual dilated eye examination. This can generally be done unsedated and should focus on evidence of ciliary body tumors (e.g., cataract, mass, secondary glaucoma, retinal detachment), retinal pigment abnormalities, as well as optic nerve changes that may give insight into intracranial pressure status. There are no routine ophthalmic imaging studies recommended in asymptomatic individuals, though providers should be aware of the ophthalmic DICER1- associated phenotypes and the need for thorough examination by an ophthalmologist. Providers should have a low threshold to add ancillary testing if there is a suspicion of ciliary body mass or intracranial mass causing increased ICP. A limitation of this study is the lack of ultrasound biomicroscopy (UBM), specifically looking at the ciliary body in this cohort. Future work evaluating DICER1 -carriers with UBM may help us gain a better understanding of the incidence of CBME as well as identify occult lesions before developing ophthalmic symptoms and therefore UBM should be considered when available.
Limitations of our study include marginally significant differences in male/female ratios in cases and controls. This is likely to have limited relevance given the lack of known predilection for retinal problems based on sex. The case cohort is significantly younger than controls, a difference that strengthens our findings.
In summary, we found that 22% of DICERl-carriers harbored an ocular abnormality, a significant difference when compared with 6% of family controls. Although the retinal changes we observed are similar to those found in Dicerl animal models, additional validation is needed to show that these changes are truly attributable to DICER1. Children with a pathogenic germline DICER1 variant warrant annual dilated eye exams, and patients and families must be educated about the ocular signs and symptoms that could prompt further ophthalmic evaluation.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Acknowledgments:
The authors thank the many patients, families, and treating physicians who participate in the National Cancer Institute DICER1-related Pleuropulmonary Blastoma Cancer Predisposition Syndrome study, the International Ovarian and Testicular Stromal Tumor Registry, and the International Pleuropulmonary/Blastoma/DICER1 Registry. The authors also wish to gratefully acknowledge the National Cancer Institute, National Eye Institute Intramural Research Programs, and the Pine Tree Apple Classic Fund.
Footnotes
Conflict of Interest: No conflicting relationship exists for any author.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill DA, Ivanovich J, Priest JR, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325(5943):965–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nature Reviews Cancer. 2014;14(10):662. [DOI] [PubMed] [Google Scholar]
- 3.Schultz KAP, Rednam SP, Kamihara J, et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clinical Cancer Research. 2017;23(12):e76–e82. [DOI] [PubMed] [Google Scholar]
- 4.Dehner LP, Messinger YH, Schultz KAP, Williams GM, Wikenheiser-Brokamp K, Hill DA. Pleuropulmonary blastoma: evolution of an entity as an entry into a familial tumor predisposition syndrome. Pediatric and Developmental Pathology. 2015;18(6):504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan NE, Bauer AJ, Doros L, et al. Macrocephaly associated with the DICER1 syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2017;19(2):244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doros L, Schultz K, Stewart D, et al. DICER1-related disorders. 2014 Apr 24. GeneReviews®[Internet] Seattle (WA): University of Washington, Seattle; 2016. [Google Scholar]
- 7.Doros L, Schultz KA, Stewart DR, et al. DICER1-related disorders. 2014. [Google Scholar]
- 8.Messinger YH, Stewart DR, Priest JR, et al. Pleuropulmonary blastoma: A report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer. 2015;121(2):276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaliki S, Shields CL, Eagle RC, et al. Ciliary body medulloepithelioma: analysis of 41 cases. Ophthalmology. 2013;120(12):2552–2559. [DOI] [PubMed] [Google Scholar]
- 10.Shields JA, Eagle RC Jr, Ferguson K, Shields CL. Tumors of the nonpigmented epithelium of the ciliary body: the Lorenz E. Zimmerman Tribute Lecture. Retina. 2015;35(5):957–965. [DOI] [PubMed] [Google Scholar]
- 11.Durieux E, Descotes F, Nguyen A-M, Grange JD, Devouassoux-Shisheboran M. Somatic DICER1 gene mutation in sporadic intraocular medulloepithelioma without pleuropulmonary blastoma syndrome. Human pathology. 2015;46(5):783–787. [DOI] [PubMed] [Google Scholar]
- 12.Priest JR, Williams GM, Manera R, et al. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma—a report from the International Pleuropulmonary Blastoma Registry. British Journal of Ophthalmology. 2011;95(7):1001–1005. [DOI] [PubMed] [Google Scholar]
- 13.Schultz KAP, Williams GM, Kamihara J, et al. DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clinical Cancer Research. 2018:clincanres. 30892017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471(7338):325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan NE, Bauer AJ, Schultz KAP, et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. The Journal of Clinical Endocrinology & Metabolism. 2017;102(5):1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marmor MF, Fulton A, Holder G, et al. ISCEV Standard for full-field clinical electroretinography (2008 update). Documenta Ophthalmologica. 2009;118(1):69–77. [DOI] [PubMed] [Google Scholar]
- 17.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Documenta ophthalmologica. 2015;130(1):1–12. [DOI] [PubMed] [Google Scholar]
- 18.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium GP. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Exome Variant Server NGESPE. http://evs.gs.washington.edu/EVS/.
- 21. https://portal.biobase-international.com/hgmd/pro/start.php.
- 22.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Research. 2016;44(Database issue):D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng PC, Henikoff S. Predicting Deleterious Amino Acid Substitutions. Genome Research. 2001;11(5):863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Kock L, Sabbaghian N, Plourde F, et al. Pituitary blastoma: a pathognomonic feature of germ- line DICER1 mutations. Acta neuropathologica. 2014;128(1):111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD. Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. Journal of medical genetics. 2012;49(7):417–419. [DOI] [PubMed] [Google Scholar]
- 27.Auw-Haedrich C, Staubach F, Witschel H. Optic disk drusen. Survey of ophthalmology. 2002;47(6):515–532. [DOI] [PubMed] [Google Scholar]
- 28.Sowka J, Gurwood A, Kabat A. Optic nerve head drusen. Handbook of Ocular Disease Management. 2001. [Google Scholar]
- 29.Sundermeier TR, Palczewski K. The impact of microRNA gene regulation on the survival and function of mature cell types in the eye. The FASEB Journal. 2016;30(1):23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akhtar S, Patnaik SR, Kotapati Raghupathy R, Al-Mubrad TM, Craft JA, Shu X. Histological characterization of the Dicer1 mutant zebrafish retina. Journal of ophthalmology. 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundermeier TR, Zhang N, Vinberg F, et al. DICER1 is essential for survival of postmitotic rod photoreceptor cells in mice. The FASEB Journal. 2014;28(8):3780–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohana R, Weiman-Kelman B, Raviv S, et al. MicroRNAs are essential for differentiation of the retinal pigmented epithelium and maturation of adjacent photoreceptors. Development. 2015;142(14):2487–2498. [DOI] [PubMed] [Google Scholar]
- 33.Rose A, Bhattacharya S. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31- associated retinitis pigmentosa. Clinical genetics. 2016;90(2):118–126. [DOI] [PubMed] [Google Scholar]