

Abstract

Sialic acid sugars on mammalian cells regulate numerous biological processes, while aberrant expression of sialic acid is associated with diseases such as cancer and pathogenic infection. Inhibition of the sialic acid biosynthesis may therefore hold considerable therapeutic potential. To effectively decrease the sialic acid expression, we synthesized C-5-modified 3-fluoro sialic acid sialyltransferase inhibitors. We found that C-5 carbamates significantly enhanced and prolonged the inhibitory activity in multiple mouse and human cell lines. As an underlying mechanism, we have identified that carbamate-modified 3-fluoro sialic acid inhibitors are more efficiently metabolized to their active cytidine monophosphate analogues, reaching higher effective inhibitor concentrations inside cells.
Sialic acids are a family of complex nine-carbon sugars abundantly expressed at the termini of mammalian glycans on cell-bound and secreted glycoproteins and glycolipids.1 The negative charge of sialic acids can facilitate the binding and transport of ions, enhance the viscosity of mucins, and stabilize proteins and membranes. Additionally, they mask underlying galactose residues, thereby regulating protein half-life2 and recycling.1 Sialoglycans are recognized by sialic acid-binding immunoglobulin-like lectins (Siglecs), a family of immunoregulatory receptors,3,4 and selectins that mediate trafficking of immune cells.5 Although sialic acids play an important role in numerous physiological processes, they are also associated with several pathologies. For example, certain viruses (e.g., influenza) target sialoglycans to infect host cells6 and some pathogenic bacteria (e.g., Pseudomonas) decorate their surface with sialic acids to escape the host immune system.7 Additionally, overexpression of sialoglycans on the surface of cancer cells has been indicated to mediate resistance to apoptosis,8,9 support immune evasion,10 and facilitate metastatic spread.11,12 Hence, in these cases, reducing the (over)expression of sialic acid holds considerable therapeutic potential. Lowering of sialic acid expression can be achieved by the use of bacterial sialidases, enzymes that cleave sialic acids from sialoglycans. However, bacterial sialidases are hard to obtain with high purity, can be immunogenic, and their effect is short-lived as intracellular sialoglycan biosynthesis is unaffected.13,14 Small-molecule inhibitors of sialic acid biosynthesis or glycan sialylation might overcome these issues.15,16 As a target, sialyltransferases (STs) are especially promising as they catalyze a late stage reaction in sialoglycan formation. Inhibiting ST therefore affects both the de novo synthesis of sialic acid and its salvage pathway. Furthermore, cancer cells are frequently hypersialylated as a result of oncogene-induced expression of STs.8 A family of 20 STs catalyze sialic acid transfer to glycans inside the Golgi with substrate and linkage specificity resulting in a variety of α2,3-, α2,6-, and α2,8-linked sialic acid-containing glycans.1 This sialylation reaction can be inhibited using a fluorinated sialic acid substrate [cytidine monophosphate (CMP)–SiaFAc].17 Although closely resembling the natural CMP–sialic acid substrate, a fluoride is thought to destabilize the oxocarbenium transition state needed for sialic acid transfer to penultimate carbohydrates to occur.18,19 Additionally, accumulation of CMP–Sia induces feedback inhibition of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE), which is involved in N-acetylmannosamine (ManNAc) biosynthesis, the metabolic precursor of sialic acid.20,21 CMP–SiaFAc is, however, not cell-permeable and hence not active in cell culture or suitable for use in vivo. This hurdle was overcome by Paulson and co-workers by feeding a peracetylated metabolic precursor (SiaFAc, 1) which enables passive diffusion over the cell membrane (Figure 1a).22 Inside the cell, SiaFAc is deacetylated by esterases and CMP-activated by the CMP N-acetylneuraminic acid synthetase (CMAS) to prepare the active CMP–SiaFAc. We and others have shown that 1 inhibits sialylation with high specificity in cancer cells in vitro and in vivo, resulting in reduced tumor growth and metastasis.23−26 During these studies, we found that the sensitivity of different cell lines for SiaFAc varied from low micromolar to high micromolar concentrations. These findings prompted us to improve the inhibitory potency of the lead compound SiaFAc by introducing modifications to the sialic acid backbone (Figure 1b). It is well documented that C-5-modified sialic acids are utilized as substrates by cells for the sialylation reaction, and hence, this position was selected for modification.27,28 We have designed and synthesized 17 different fluorinated sialic acid analogues and tested their effects in multiple cell lines. We found that replacement of the natural N-acetamide substituent by a carbamate drastically improved the inhibitory activity of the lead compound. Several of the carbamate analogues showed potent and long-lasting inhibition of sialic acid expression in the low micromolar range in both human and mouse cancer cell lines. Remarkably, the carbamate-modified sialic acids also showed potent effects in cancer cell lines with very poor sensitivity for the lead compound SiaFAc (1). Investigation of the mechanism of action in silico and in vitro showed that the carbamate-based inhibitors are more efficiently metabolized toward their active CMP analogues, reaching higher effective intracellular inhibitor concentrations.
Figure 1.

(a) Working model of metabolic ST inhibitors. The acetylated sialic acid derivative is taken up via passive diffusion and deacetylated by intracellular esterases. CMP activation in the nucleus by CMAS produces the active inhibitor CMP–SiaFAc, which competitively blocks STs and induces feedback inhibition of de novo sialic acid biosynthesis. (b) Structure of C-3-fluorinated ST inhibitors 1–18 with C-5 amide or carbamate modifications.
Results and Discussion
Synthesis of C-5-Modified Fluorinated Sialic Acid Analogues
The SiaFAc (1) derivatives 2–18 (Figure 1b) were prepared from a common C-5 Boc-protected precursor 12 (Scheme 1) to allow divergent derivatization of the C-5 amine after Boc deprotection. Thioglycoside 19(29) was reacted with bromine to afford a mixture of the corresponding glycosyl bromide and glycal 20. The reaction of the mixture under basic conditions then afforded glycal 20 in a good overall yield (78%). Electrophilic fluorination was performed using Selectfluor in a mixture of H2O/dimethylformamide (DMF) affording axial-fluoride 21.30 Finally, 21 was acetylated to afford 12 in a high yield (95%). The Boc inhibitor 12 was modified in a two-step sequence of Boc deprotection followed by acylation of the amine. Previously, we reported an analogous method to prepare nonfluorinated C-5-modified sialic acid derivatives in a good yield.31 In the case of fluoride analogue 12, however, the yields of the two-step sequence were considerably lower (6–90%, Scheme 1). Presumably, the presence of the electron-withdrawing fluoride rendered the C-5 amine less reactive, giving rise to various side reactions. Nevertheless, including precursor 12, a total of 17 derivatives (2–18) of parent acetamide 1 were prepared. Included are derivatives 5, 6, and 7 containing an alkyne or azide group which is amendable for modification using the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. To explore whether modifications introduced in this manner are tolerated in the sialic acid metabolism, derivative 14 was prepared from 7, using benzyl azide and CuI/tris((1-benzyl-4-triazolyl)methyl)amine (TBTA).
Scheme 1. Synthesis of C-5-Modified ST Inhibitors.

(i) Br2, DCM, r.t., 2.5 h; (ii) TEA, DCM, r.t., 16 h, 78% (over two steps); (iii) Selectfluor, 1:3 H2O/DMF, 60 °C, 3 h, 72% (based on recovery); (iv) Ac2O, py, r.t., 48 h, 95%; (v) TFA, DCM, H2O, r.t., 2 h; (vi) activated acyl substituents (A), TEA, DCM, r.t.; 2: Side product in a reaction of 12 → 11 90%; 3: A = chloroacetyl chloride, 16 h, 60%; 4: A = acetoxyacetyl chloride, 16 h, 43%; 5: A = azidoacetic acid N-hydroxysuccinimide (NHS) ester, 23 h, 16%; 6: A = 4-pentynoic acid NHS ester, 16 h, 7%; 7: A = N-propargyloxycarbonyl-succinimide, 15 h, 40%; 8: A = allyl chloroformate, 21.5 h, 16%; 9: A = methyl chloroformate, 16 h, 33%; 10: A = ethyl chloroformate, 16 h, 16%; 11: A = isobutyl chloroformate, 16 h, 33%; 13: A = benzyl chloroformate, 21.5 h, 6%; 15: A = n-butyl chloroformate, 16 h, 32%; 16: A = 2-methoxymethyl chloroformate, 16 h, 90%; 17: A = 2,2,2-trichloroethoxycarbonyl chloroformate, 16 h, 17%; 18: A = 2-fluoroethyl chloroformate, 16 h, 60%; and (vii) 14: benzyl azide, TBTA, CuI, Cu, DMF, H2O, tBuOH, r.t., 16 h, 46%.
C-5 Carbamate-Fluorinated Sialic Acids Inhibit Sialylation with Increased Potency Compared to C-5 Amide Analogues
The inhibitor potency of 1–18 at different concentrations was first assessed in murine B16-F10 tumor cells using lectins specific for α2,3-linked (MALII, Table 1) or α2,6-linked sialic acids (SNA-I, Table S1). The EC50 values were determined for both linkage types, defined as the concentration where a 50% decrease in lectin binding compared to control was observed (Figure 2a,b). Strikingly, carbamates 7–12 were all more potent inhibitors than acetamide 1. Carbamate 7 was one of the most active inhibitors with an average 37-fold increase in potency compared to the amide analogues. A direct comparison of carbamate 7 with amide 6 shows a 56-fold increase in inhibitory potency for the former. Amide 6 only differs from 7 in the substitution of the carbamate oxygen for a CH2. On the basis of this structural difference, we hypothesize that the carbamate group enables an additional binding interaction of STs resulting in improved inhibition. In line with these findings, we previously showed that the nonfluorinated analogue of 7, a carbamate, was better incorporated into the glycocalyx than the nonfluorinated homologue of 5, an amide.31 This finding is also supported by a later study on the metabolic precursor of sialic acid, where an increased metabolic incorporation was observed for mannosamine carbamates over amides with the same side chain length.32 In addition to linkage type, linear 2–3 carbon substituents (7, 8, 10, and 18) on the carbamate seem to afford more potent inhibitors than 1 (9) or 4 (15) carbons, or the introduction of a more bulky substituent (11–14). In an attempt to modulate carbamate hydrogen bonding properties, 16–18 were synthesized, but these did not further improve the inhibitory effect observed with 7. Chloroacetamide 3 was toxic, potentially because of its reactive nature. For these reasons, compounds 3 and 13–18 were not used for subsequent experiments.
Table 1. EC50 Values in Micromolar for Inhibition of α2,3-Linked Sialic Acida.

Cell lines were cultured for 3 days with 0–204.8 μM amide or carbamate-fluorinated sialic acids or DMSO vehicle control. The cells were stained with biotinylated MALII lectin that recognizes α2,3-linked sialic acids and streptavidin-PE. Lectin binding was determined by flow cytometry and is presented as mean percentage lectin binding ±SEM normalized to the control (n = 3). The relative inhibitory potency was calculated for the B16-F10 cell line by dividing the EC50 of SiaFAc (1) by the EC50 of the compound of interest.
Figure 2.

(a,b) Amide and carbamate-modified fluorinated sialic acids inhibit sialylation of B16-F10 cells. B16-F10 cells were treated with 0.1–204.8 μM amide (red) or carbamate (blue)-fluorinated sialic acids or dimethyl sulfoxide (DMSO) vehicle control. After 3 days, the cells were stained with biotinylated MALII (a) or SNA-I (b) that recognize α2,3-linked or α2,6-linked sialic acids, respectively, followed by streptavidin-PE staining. Binding of lectins was determined by flow cytometry and data are presented as mean percentage lectin binding ± standard error of the mean (SEM) normalized to the control (n = 3). The EC50 values were extrapolated for all compounds (Tables 1 and S1). (c–f) Recovery of sialylation after acetamide and carbamate-fluorinated sialic acid treatment. B16-F10 cells were incubated for 3 days with 51.2 μM acetamide or carbamate-fluorinated sialic acids or DMSO control. Fluorinated sialic acids were removed from the culture and the cells were re-seeded. During a period of 6 days, sialylation was assessed daily by flow cytometry using MALII or SNA-I lectins. Graphs show recovery of α2,3-sialylation (c,d) or α2,6-sialylation (e,f) in time presented as mean percentage lectin binding ±SEM normalized to control (n = 3).
To assess if the improved inhibition of carbamate derivatives is not restricted to the B16-F10 cell line, the experiments were extended to human THP-1, HEK293, and HeLa cell lines, as well as murine 9464D and EL4 cells (Tables 1 and S1). In line with the findings for B16-F10 cells, the carbamates inhibited sialylation with significantly higher efficacy compared with the amide analogues in all of the tested cell lines. Remarkably, the carbamates also showed good potency in 9464D and EL4 cancer cells that showed very poor sensitivity to the lead compound 1. Overall, no significant preference was observed for the inhibition of α2,3-linked (Table 1) over α2,6-linked sialic acid (Table S1). Next, a toxicity profile of 1–2 and 4–12 was established by monitoring the metabolic activity of cells after 3 days of treatment. Importantly, none of the compounds were toxic at concentrations <51.2 μM and most inhibitors were not even toxic at concentrations as high as 204.8 μM (Figure S1). Finally, we showed that all sialic acid mimetics were highly specific as only inhibition of sialylation, but not overall glycosylation was observed (Figure S2). Altogether, these data indicate that carbamate-modified ST inhibitors can selectively and potently block sialylation in a dose-dependent manner without causing cellular toxicity in vitro.
C-5 Carbamate-Fluorinated Sialic Acids Show Long-Lasting Inhibition of Sialylation
Previously, we found that the recovery time of sialylation after treatment with 64 μM SiaFAc (1) was about 2–3 days, whereas the recovery time after enzymatic sialidase treatment was less than a day.23 To determine the recovery times of the new amide and carbamate inhibitors, B16-F10 cells were incubated for 3 days with 51.2 μM fluorinated sialic acids and recovery of sialylation was monitored over time by lectin staining. Recovery to normal sialylation levels took approximately 2–3 days for all amide derivatives (Figure 2c,e) and about 5–6 days for the carbamates (Figure 3d,f). A prolonged inhibition for carbamates was also observed at a lower concentration of 25.6 μM (Figure S3). Our previous data showed that pretreatment of B16-F10 cells with 1 impaired metastasis formation in vitro and tumor outgrowth in vivo.23,24 Hence, it will be interesting to further explore the therapeutic potential of the carbamate-modified ST inhibitors. Altogether, the improved inhibitory potency combined with the ability to block sialylation for a prolonged period of time could make C-5 carbamate-modified ST inhibitors valuable tools in glycobiology research and potentially in cancer treatment.
Figure 3.
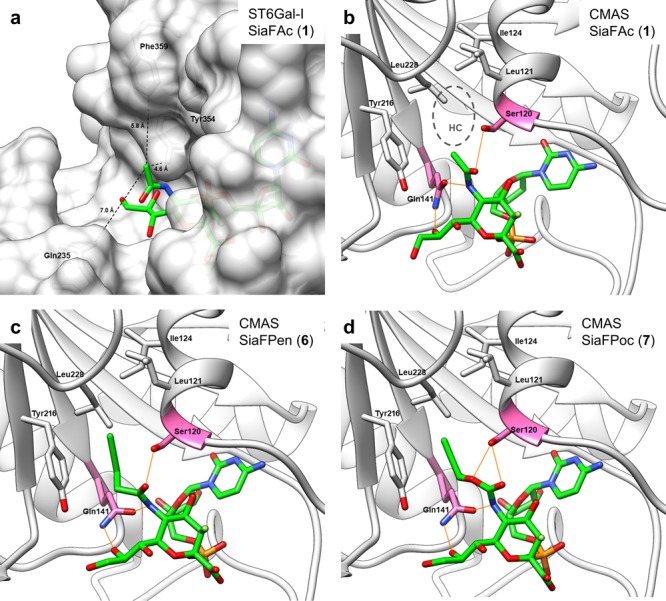
(a) CMP–SiaFAc (from 5BO9, green carbons) was superposed onto cocrystalized CMP in the ST6Gal-I binding pocket (4JS2, white carbons and surface). Distances (Å) to nearest residues Gln235, Tyr354, and Phe359 are indicated with black dashes. (b) Cocrystalized CMP–Sia (green carbons) in the CMAS binding pocket (1QWJ, domain D, white carbons). Between the HC (Leu121, Ile124, Tyr216, and Leu228) and the sialic acid pyranose ring, there is a hydrophilic gate (Ser120, Gln141, pink carbons). Hydrogen bonds are depicted with orange lines. (c,d) Energy-minimized poses of SiaFPen (6) [green carbons, (c)] and SiaFPoc (7) [green carbons, (d)] in the CMAS binding pocket (1QWJ, domain D, white carbons). Hydrogen bonds are depicted with orange lines. The hydrophilic gate accommodates the carbamate [SiaFPoc, (d)] more favorably than the amide [SiaFPen, (c)] because of an additional polar interaction with Ser120.
C-5 Carbamate-Fluorinated Sialic Acids are More Efficiently Metabolized to Their CMP Analogue than the Corresponding Amides
To better understand the improved potency of the carbamate analogues compared to their amide counterparts, we performed in silico experiments. It is well established that fluorinated sialic acids are processed by CMAS to their corresponding CMP derivatives, which then act as competitive inhibitors for the donor substrate (CMP–sialic acid) of all STs. Hence, we first investigated if the alterations in the C-5 substituents of fluorinated CMP–sialic acids would be expected to alter binding to STs. Using the crystal structure of ST6Gal-I as a model, we found that the C-5 substituent is directed into the solvent (Figure 3a). The nearest residues were between 4.6 and 7.0 Å away from the acyl carbon atom (oxygen for the carbamates), implying that differences in ST binding were unlikely to cause the difference in potency. An alternative explanation holds that the C-5 carbamate derivatives show enhanced CMP activation by CMAS relative to the C-5 amide analogues. To this end, the CMP analogues of compounds 1, 6, and 7 were docked into the crystal structure of murine CMAS, which is almost completely homologue to human CMAS (Figure 3b–d). Within the CMAS binding pocket, the C-5 acetyl moiety was directed toward a hydrophobic cavity (HC) formed by Leu121, Ile124, Tyr216, and Leu22833 (Figure 3b), which in the crystal structure held the natural methyl group, but is big enough to fit larger moieties. Additionally, Ser120 and Gln141 formed a hydrophilic cavity. Gln141 made polar interactions with N5 (N-acetyl) and O8 and Ser120 with the N-acetyl carbonyl oxygen (Figure 3b). Modeling amide and carbamate modifications into the CMAS crystal structure indicated that additional interactions of the carbamates with this hydrophilic gate could arise. As shown for CMP–SiaFPen 6 (Figure 3c) and CMP–SiaFPoc 7 (Figure 3d), the alkoxy oxygen of the carbamate could make an additional polar interaction with Ser120. The C-5 carbamate derivatives may therefore have a lower KM leading to a higher Kcat/KM and increased intracellular concentrations of the fluorinated CMP sialic acids. The increased concentration is expected to improve ST inhibition as well as feedback inhibition of the de novo sialic acid biosynthesis.
To confirm this hypothesis, the metabolic efficiency of the SiaFR derivatives toward their CMP-activated analogues by CMAS in time was established. To this end, intracellular CMP–SiaFR levels were analyzed in B16-F10 cells treated for different time points with SiaFR derivatives using reverse-phase ion pairing chromatography coupled to a triple quadrupole mass spectrometer operating in negative ion mode (Figure 4). These experiments revealed a more efficient conversion of the carbamate-based inhibitors SiaFPoc (7) and SiaFEt (10), over amides SiaFAc (1) and SiaFPen (6) (Figure 4a). The carbamate-based inhibitors also decreased the intracellular CMP–Sia pool faster than the amides (Figure 4b). These observations were consistent with the improved potency of the carbamate-based inhibitors (Table 1) as well as the in silico experiments (Figure 3). We therefore conclude that the improved potency of the C-5 carbamate-based inhibitors over the amides is due to a more efficient metabolism into the respective CMP analogues, yielding higher concentrations of the active inhibitor acting on STs and GNE.
Figure 4.

B16-F10 cells were incubated for indicated time points with 51.2 μM fluorinated sialic acid analogues or DMSO control. After sample preparation, the CMP–SiaFR (a) and CMP–Sia (b) levels were analyzed using reverse-phase ion pairing chromatography coupled to a triple quadrupole mass spectrometer operating in negative ion mode and presented as their abundance in the nucleotide sugar pool ±SEM (n = 2).
Conclusions
Sialic acids are involved in numerous biological processes ranging from neuronal development to immune cell trafficking. However, sialic acids are also the targets of several pathogens and aberrant sialylation of cancer cells mediates immune evasion, migration and metastasis. The highly effective inhibitors developed in this study are therefore interesting tools to study the broad biological functions of sialic acid sugars. Additionally, their specificity, high potency and prolonged inhibition qualifies them as good candidates for further therapeutic development. We showed that the carbamate based inhibitors are more efficiently metabolized towards their active CMP analogues, thereby reaching higher effective inhibitor concentrations inside cells. This mechanism could also be considered in the design of other sialylation inhibitors, as well as metabolic labeling reagents that utilize the same enzymatic machinery.
Experimental Section
1H and 13C NMR spectra were recorded on a Varian Inova 400 MHz or a Bruker AVANCE III 500 MHz spectrometer. The purity of the compounds (≥95%) was determined by nuclear magnetic resonance (NMR) spectroscopy. Mass spectra were recorded on a JEOL JMS-T100CS AccuTOF mass spectrometer. Automatic column chromatography was performed on Biotage Isolera Spektra One, using SNAP cartridges 10–50 g filled with normal silica (Biotage, 30–100 μm, 60 Å). TLC analysis was conducted on TLC silica gel, 60, F254, Merck, with detection by UV absorption (254 nm) where applicable and by spraying with 20% H2SO4 in MeOH followed by charring at ∼150 °C. Dichloromethane (DCM) was freshly distilled. All reactions were carried out under an argon atmosphere.
General Procedure for the Preparation of SiaFR (2–13, 15–18)
Boc inhibitor 12 (50 mg; 82 μmol) was dissolved in a 1:1:2 mixture of respectively DCM, H2O, and trifluoroacetic acid (TFA) (1.6 mL; 0.05 M). The mixture was stirred for 2 h at room temperature (r.t.) (TLC: (EtOAc/hept, 60:40 v/v) Rf = 0.09). The mixture was then diluted with H2O and concentrated in vacuo. The residue was dissolved in DCM (0.83 mL; 0.1 M) and successively activated acyl substituents (497 μmol; 6 equiv) and triethylamine (TEA) (35 μL; 248 μmol; 3 equiv) were added. After stirring at r.t. for 16 h, the mixture was diluted with DCM and washed successively with aq HCl (0.1 M) and sat. aq NaHCO3. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. Silica gel flash column chromatography (EtOAc in hept) afforded SiaFR 2–13, 15–18 (6–90%). Variations on this protocol can be found in the Supporting Information.
Methyl 5-(tert-Butoxycarbamado)-2,4,7,8,9-penta-O-acetyl-3,5-dideoxy-3-fluoro-d-glycero-β-galacto-non-2-ulopyranosonate (12)
To a solution of 21 (789 mg; 1.39 mmol) in py (12 mL; 148 mmol; 107 equiv), Ac2O (6 mL; 63.6 mmol; 45.7 equiv) was slowly added. After stirring at r.t. for 24 h, the mixture was concentrated in vacuo using Tol for coevaporation. The residue was dissolved in EtOAc and washed successively with aq HCl (0.1 M) and sat. aq NaHCO3. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. Silica gel flash column chromatography (0 → 50% EtOAc in hept) afforded 12 (717 mg; 1.176 mmol; 95%) as a white foam.
Methyl 5-[(1-Benzyl-1H-1,2,3-triazol-4-yl)methylcarbamado]-2,4,7,8,9-penta-O-acetyl-3,5-dideoxy-3-fluoro-d-glycero-β-galacto-non-2-ulopyranosonate (14)
To a mixture of 7 (10 mg; 17 μmol) in a 1:9 mixture of H2O and tBuOH (0.19 mL; 0.09 M), Bn-N3 (4.5 mg; 34 μmol; 2 equiv) was added. A premixture of TBTA (29 mg), DMF (750 μL), and CuI (5.1 mg) was agitated until a homogeneous solution was obtained. The TBTA mixture (95 μL) was added to the H2O/tBuOH mixture and 10 mg of copper flakes was added. The reaction was stirred at r.t. for 16 h, filtered, and concentrated in vacuo. Silica gel flash column chromatography (0 → 80% EtOAc in hept) afforded 14 (5.7 mg; 17 μmol; 46%) as a white solid.
Methyl 5-(tert-Butoxycarbamado)-4,7,8,9-tetra-O-acetyl-2,6-anhydro-3,5-dideoxy-d-glycero-d-galacto-non-2-enonate (20)
To a solution of 19(31) (2.757 g; 4.22 mmol) in DCM (42.2 mL; 0.1 M), Br2 (0.239 mL; 4.64 mmol; 1.1 equiv) was slowly added. After 2.5 h of stirring at r.t., the reaction was diluted with DCM and washed with 10% aq Na2S2O3. The milky organic layer was dried over MgSO4, filtered, and the clear filtrate was extracted once more with 10% aq Na2S2O3 before drying over MgSO4 and filtering. The filtrate was concentrated in vacuo, dissolved in DCM (42.0 mL; 0.1 M), and TEA (1.699 g; 16.79 mmol; 4 equiv) was added. The reaction was stirred for 16 h at r.t. and concentrated in vacuo. The residue was dissolved in EtOAc and washed successively with aq HCl (0.1 M) and sat. aq NaHCO3. The organic layer was dried over MgSO4, filtered, and again concentrated in vacuo. Silica gel flash column chromatography (0 → 45% EtOAc in hept) afforded 20 (1.734 g; 3.26 mmol; 78% two steps) as a white solid.
Methyl 5-(tert-Butoxycarbamado)-4,7,8,9-penta-O-acetyl-3,5-dideoxy-3-fluoro-d-glycero-β-galacto-non-2-ulopyranosonate (21)
To a solution of 20 (1.724 g; 3.24 mmol) in a 1:3 mixture of H2O and DMF (32 mL; 0.1 M), Selectfluor (3.45 g; 9.73 mmol; 3 equiv) was added. The reaction was stirred at 60 °C for 3 h, quenched with sat. aq NaHCO3, and concentrated in vacuo—even though conversion was incomplete. The residue was dissolved in EtOAc and washed successively with aq. HCl (0.1 M) and sat. aq NaHCO3. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. Silica gel flash column chromatography (0 → 50% EtOAc in hept) afforded 21 (789 mg; 1.39 mmol; 72% based on the recovery of the starting material) as a white foam.
Acknowledgments
This work was supported by a VENI and ERC-Stg awarded to T.J.B. and a grant from the Dutch Cancer Society (KWF) KUN 2015-7604 awarded to G.J.A., T.J.B., and C.B. We thank the Centre for Molecular and Biomolecular Informatics within the Institute for Molecular Life Sciences at the Radboudumc for their contribution to the modeling studies. We thank Monique Van Scherpenzeel, CEO, at GlycoMScan, for the cellular nucleotide sugar analysis by mass spectrometry.
Glossary
Abbreviations
- CMAS
cytidine monophosphate N-acetylneuraminic acid synthetase
- CMP
cytidine monophosphate
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- GNE
UDP-GlcNAc 2-epimerase/ManNAc kinase
- ManNAc
N-acetyl mannosamine
- heptane
hept
- Siglec
sialic acid-binding immunoglobulin-like lectin
- ST
sialyltransferase
- TBTA
tris((1-benzyl-4-triazolyl)methyl)amine
- TEA
triethylamine
- Tol
toluene
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01757.
Methods for cell culture, lectin staining and flow cytometry, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, in silico modeling, CMP–sialic acid quantification, and synthetic procedures including 1H and 13C NMR spectra; data on the effect of 1, 2, 4–12 on cell metabolic activity/viability, total cell surface glycosylation, recovery of sialylation after treatment with 25.6 μM fluorinated sialic acid mimetics, and EC50 values for inhibition of α2,6-linked sialic acid; and molecular formula strings (PDF)
(CSV)
Author Contributions
T.H. and J.F.A.P. contributed equally. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Varki A.; Schnaar R.; Schauer R.. Sialic Acids and Other Nonulosonic Acids. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor: NY; 2017. [Google Scholar]
- Yang W. H.; Aziz P. V.; Heithoff D. M.; Mahan M. J.; Smith J. W.; Marth J. D. An intrinsic mechanism of secreted protein aging and turnover. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 13657–13662. 10.1073/pnas.1515464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büll C.; Heise T.; Adema G. J.; Boltje T. J. Sialic Acid Mimetics to Target the Sialic Acid-Siglec Axis. Trends Biochem. Sci. 2016, 41, 519–531. 10.1016/j.tibs.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Macauley M. S.; Crocker P. R.; Paulson J. C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. 10.1016/s1471-4914(03)00071-6. [DOI] [PubMed] [Google Scholar]
- Stencel-Baerenwald J. E.; Reiss K.; Reiter D. M.; Stehle T.; Dermody T. S. The sweet spot: defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. 10.1038/nrmicro3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angata T.; Varki A.. Siglec interactions with pathogens. In Glycoscience: Biology and Medicine; Taniguchi N., Endo T., Hart G. W., Seeberger P. H., Wong C.-H., Eds.; Springer Japan: Tokyo, 2015; pp 633–642. [Google Scholar]
- Bull C.; Stoel M. A.; den Brok M. H.; Adema G. J. Sialic acids sweeten a tumor’s life. Cancer Res. 2014, 74, 3199–3204. 10.1158/0008-5472.can-14-0728. [DOI] [PubMed] [Google Scholar]
- Swindall A. F.; Bellis S. L. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 2011, 286, 22982–22990. 10.1074/jbc.m110.211375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büll C.; den Brok M. H.; Adema G. J. Sweet escape: Sialic acids in tumor immune evasion. Biochim. Biophys. Acta 2014, 1846, 238–246. 10.1016/j.bbcan.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Nakamori S.; Kameyama M.; Imaoka S.; Furukawa H.; Ishikawa O.; Sasaki Y.; Kabuto T.; Iwanaga T.; Matsushita Y.; Irimura T. Increased expression of sialyl lewis x antigen correlates with poor survival in patients with colorectal carcinoma: clinicopathological and immunohistochemical study. Cancer Res. 1993, 53, 3632–3637. [PubMed] [Google Scholar]
- Schultz M. J.; Swindall A. F.; Bellis S. L. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 2012, 31, 501–518. 10.1007/s10555-012-9359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlacek H. H.; Seiler F. R. Immunotherapy of neoplastic diseases with neuraminidase: Contradictions, new aspects, and revised concepts. Cancer Immunol. Immunother. 1978, 5, 153–163. 10.1007/bf00199623. [DOI] [Google Scholar]
- Moustafa I.; Connaris H.; Taylor M.; Zaitsev V.; Wilson J. C.; Kiefel M. J.; von Itzstein M.; Taylor G. Sialic Acid Recognition by Vibrio cholera Neuraminidase. J. Biol. Chem. 2004, 279, 40819–40826. 10.1074/jbc.m404965200. [DOI] [PubMed] [Google Scholar]
- Szabo R.; Skropeta D. Advancement of sialyltransferase inhibitors: therapeutic challenges and opportunities. Med. Res. Rev. 2016, 37, 219–270. 10.1002/med.21407. [DOI] [PubMed] [Google Scholar]
- Wang L.; Liu Y.; Wu L.; Sun X.-L. Sialyltransferase inhibition and recent advances. Biochim. Biophys. Acta 2016, 1864, 143–153. 10.1016/j.bbapap.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Burkart M. D.; Vincent S. P.; Wong C.-H. An efficient synthesis of CMP-3-fluoroneuraminic acid. Chem. Commun. 1999, 16, 1525–1526. 10.1039/a903362i. [DOI] [Google Scholar]
- Montgomery A.; Szabo R.; Skropeta D.; Yu H. Computational characterisation of the interactions between human ST6Gal I and transition-state analogue inhibitors: insights for inhibitor design. J. Mol. Recognit. 2015, 29, 210–222. 10.1002/jmr.2520. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Benz J.; Greif M.; Engel A. M.; Sobek H.; Rudolph M. G. The structure of human α-2,6-sialyltransferase reveals the binding mode of complex glycans. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1826–1838. 10.1107/s0907444913015412. [DOI] [PubMed] [Google Scholar]
- Hinderlich S.; Weidemann W.; Yardeni T.; Horstkorte R.; Huizing M.. UDP-GlcNAc 2-Epimerase/ManNAc kinase (GNE): a master regulator of sialic acid synthesis. Topics in Current Chemistry; Springer, 2015; Vol. 366, pp 97–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala R.; Lehto V.-P.; Gahl W. A. Mutations in the human UDP-N-Acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am. J. Hum. Genet. 1999, 64, 1563–1569. 10.1086/302411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan C. D.; Antonopoulos A.; Lefort C. T.; Sonon R.; Azadi P.; Ley K.; Dell A.; Haslam S. M.; Paulson J. C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. 10.1038/nchembio.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull C.; Boltje T. J.; Wassink M.; de Graaf A. M. A.; van Delft F. L.; den Brok M. H.; Adema G. J. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol. Cancer Ther. 2013, 12, 1935–1946. 10.1158/1535-7163.mct-13-0279. [DOI] [PubMed] [Google Scholar]
- Büll C.; Boltje T. J.; van Dinther E. A. W.; Peters T.; de Graaf A. M. A.; Leusen J. H. W.; Kreutz M.; Figdor C. G.; den Brok M. H.; Adema G. J. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano 2015, 9, 733–745. 10.1021/nn5061964. [DOI] [PubMed] [Google Scholar]
- Macauley M. S.; Arlian B. M.; Rillahan C. D.; Pang P.-C.; Bortell N.; Marcondes M. C. G.; Haslam S. M.; Dell A.; Paulson J. C. Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J. Biol. Chem. 2014, 289, 35149–35158. 10.1074/jbc.m114.606517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büll C.; Boltje T. J.; Balneger N.; Weischer S. M.; Wassink M.; van Gemst J. J.; Bloemendal V. R. L. J.; Boon L.; van der Vlag J.; Heise T.; den Brok M. H.; Adema G. J. Sialic acid blockade suppresses tumor growth by enhancing T-cell–mediated tumor immunity. Cancer Res. 2018, 78, 3574–3588. 10.1158/0008-5472.can-17-3376. [DOI] [PubMed] [Google Scholar]
- Wratil P. R.; Horstkorte R.; Reutter W. Metabolic Glycoengineering with N -Acyl Side Chain Modified Mannosamines. Angew. Chem., Int. Ed. 2016, 55, 9482–9512. 10.1002/anie.201601123. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Xie R.; Dong L.; Chen X. Metabolic remodeling of cell-surface sialic acids: principles, applications, and recent advances. ChemBioChem 2016, 17, 11–27. 10.1002/cbic.201500344. [DOI] [PubMed] [Google Scholar]
- Chao C.-S.; Chen M.-C.; Lin S.-C.; Mong K.-K. T. Versatile acetylation of carbohydrate substrates with bench-top sulfonic acids and application to one-pot syntheses of peracetylated thioglycosides. Carbohydr. Res. 2008, 343, 957–964. 10.1016/j.carres.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Burkart M. D.; Zhang Z.; Hung S.-C.; Wong C.-H. A new method for the synthesis of fluoro-carbohydrates and glycosides using selectfluor. J. Am. Chem. Soc. 1997, 119, 11743–11746. 10.1021/ja9723904. [DOI] [Google Scholar]
- Büll C.; Heise T.; Beurskens D. M. H.; Riemersma M.; Ashikov A.; Rutjes F. P. J. T.; van Kuppevelt T. H.; Lefeber D. J.; den Brok M. H.; Adema G. J.; Boltje T. J. Sialic acid glycoengineering using an unnatural sialic acid for the detection of sialoglycan biosynthesis defects and on-cell synthesis of siglec ligands. ACS Chem. Biol. 2015, 10, 2353–2363. 10.1021/acschembio.5b00501. [DOI] [PubMed] [Google Scholar]
- Dold J. E. G. A.; Pfotzer J.; Späte A.-K.; Wittmann V. Dienophile-modified mannosamine derivatives for metabolic labeling of sialic acids: a comparative study. ChemBioChem 2017, 18, 1242–1250. 10.1002/cbic.201700002. [DOI] [PubMed] [Google Scholar]
- Krapp S.; Münster-Kühnel A. K.; Kaiser J. T.; Huber R.; Tiralongo J.; Gerardy-Schahn R.; Jacob U. The crystal structure of murine CMP-5-N-acetylneuraminic acid synthetase. J. Mol. Biol. 2003, 334, 625–637. 10.1016/j.jmb.2003.09.080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.