

Abstract
The entry of foreign DNA into many mammalian cell types triggers the innate immune system, a complex set of responses directed at preventing infection by pathogens. One aspect of the response is the potent epigenetic silencing of incoming viral DNAs1, including the extrachromosomal DNAs formed immediately after infection by retroviruses. These unintegrated viral DNAs are very poorly transcribed in all cells, even in permissive cells, in contrast to the robust expression observed after integration2–5. The factors responsible for this poor expression have not yet been identified. To explore the mechanisms responsible for repression of unintegrated viral DNAs, we performed a genome-wide CRISPR-Cas9 screen for genes required for silencing an integrase-deficient MLV-GFP reporter virus. Our screen identified a DNA-binding protein, NP220; the three proteins of the HUSH complex (MPP8, TASOR, PPHLN1), which silences proviruses in heterochromatin6 and retrotransposons7,8; histone methyltransferase SETDB1; and other host factors that are required for silencing. Further tests by chromatin immunoprecipitation (ChIP) showed that NP220 is the key protein that recruits HUSH, SETBD1, and histone deacetylases HDAC1 and HDAC4 to silence the unintegrated retroviral DNA. Knockout of NP220 accelerates the replication of retroviruses. These experiments have revealed the molecular machinery utilized for the silencing of extrachromosomal retroviral DNA.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Retrovirus infection begins with the reverse transcription of the RNA genome in the cytoplasm to form a linear double-stranded DNA that is soon delivered into the nucleus9. A portion of this linear DNA gives rise to two circular DNA forms containing one or two tandem copies of the Long Terminal Repeat (LTR) sequences at the ends of the linear DNA10,11. The linear DNA is inserted into the host genome to form the integrated provirus, which is actively transcribed and produces progeny virus. In contrast, the unintegrated DNAs are transcriptionally silent, do not replicate, and disappear over time. To probe the mechanism of silencing of unintegrated retroviral DNAs, we infected HeLa cells with integrase-deficient (IND184A) or integrase-proficient (INWT) MLV-Luc viruses and monitored expression of the luciferase reporter. Comparable levels of retroviral DNA were produced, but expression of the reporter was ~30-fold lower from IND184A virus than from INWT virus (Extended Data Fig. 1a and 1b). Treatment of the cells with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) dramatically increased luciferase expression of unintegrated IND184A MLV-Luc DNA without significant effect on integrated WT control (Extended Data Fig. 1c and 1d). Chromatin immunoprecipitation (ChIP) experiments showed that H3 histones on unintegrated viral DNA were largely deacetylated, and carried repressive H3K9me3 marks but not H3K27me3 marks. In contrast, H3 histones on integrated retroviral DNA were heavily acetylated and barely methylated at H3K9 or H3K27 (Extended Data Fig. 1e-j).
To identify host factors responsible for silencing of unintegrated retroviral DNA, we performed an unbiased genome-wide CRISPR-Cas9 screen, selecting for knock-out of host factors that relieve the silencing (Fig. 1a). HeLa cells transduced with a CRISPR sgRNA library were infected with integrase-deficient MLV-GFP reporter virus, and GFP-positive cells were isolated by sorting (Fig. 1b). After a second round of selection, targeted genes whose knockout was enriched in the selected cells were identified by sequencing of sgRNAs. Five genes stood out: NP220 (also known as ZNF638); all three subunits of the HUSH complex (MPP8, TASOR, and PPHLN1); and SETDB1, a histone methyl transferase (HMT) (Fig. 1c and Supplementary Table 1).
Fig. 1: CRISPR-Cas9 screen to identify host factors responsible for silencing of unintegrated retroviral DNA.
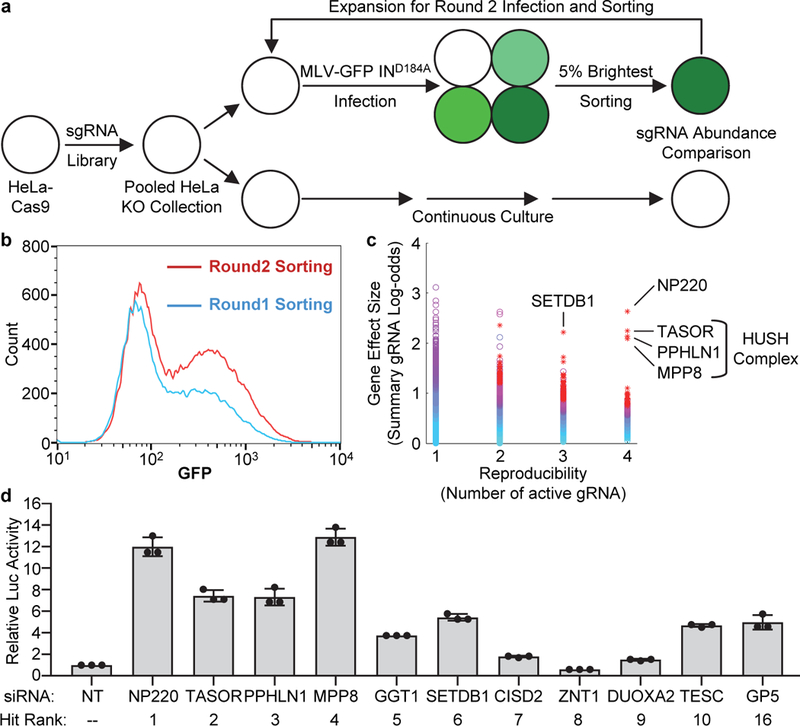
a, Flowchart of the genome-wide CRISPR-Cas9 screen strategy. A pooled collection of HeLa KO cells was infected with integrase-deficient MLV-GFP virus, and the 5% brightest GFP-positive cells were isolated by sorting. These cells were subjected to a second round of infection and selection, and DNAs were recovered for analysis of the resident sequences encoding the CRISPR guide RNAs. b, GFP signals detected by FACS during Round 1 and Round 2 sorting.c, Candidate genes essential for the silencing were identified by analyzing the fold-change in abundance over control and number of enriched sgRNAs per gene using software HiTSelect. CRISPR screen (a), GFP monitoring (b), and sgRNA analysis (c) were performed only once.d, Validation of candidate genes from the screen. Hela cells were transfected with indicated siRNA and then infected with integrase-deficient (IND184A) MLV-Luc virus. Luciferase activities were measured and activity in cells transfected with non-targeting (NT) siRNA was set as 1. Data presented are mean ± SD from three independent experiments (n = 3).
NP220 is a nuclear double-stranded DNA (dsDNA)-binding protein with preference for cytidine clusters12, and contains a DNA-binding domain (DB) and a single C-terminal C2H2-type zinc finger (ZnF) motif (Fig. 2a). Knockdown or knockout of NP220 resulted in a dramatic increase in expression of luciferase from unintegrated MLV DNA (Fig. 1d and 2b, Extended Data Fig. 2a and 2b) without affecting viral DNA levels (Extended data Fig. 1k). Re-expression of wild-type NP220, but not deletion mutants lacking either the DNA binding domain or the zinc finger motif, restored silencing in the NP220 knockout cells (Fig. 2c). Thus, both the DNA binding domain and zinc finger of NP220 were required for silencing. The HUSH complex was identified for its role in silencing proviruses integrated into heterochromatin6 and also young retrotransposons7. Knockout of MPP8, TASOR, or PPHLN1 subunits similarly increased reporter expression from unintegrated DNA (Fig. 2d-g). The MPP8 subunit is known to exhibit methyl H3K9-binding activity, and tryptophan 80 (W80) is critical for this binding6,13,14. Re-expression of wild-type MPP8, but not substitution mutant W80A, restored the repression of unintegrated DNA in the KO line (Fig. 2e). SETDB1 is an HMT responsible for generating H3K9me3 marks. Knockout or knockdown of SETDB1, but not other HMTs (SETDB2, SUV39H1, SUV39H2, EHMT1, EHMT2, or EZH2) again relieved the silencing of unintegrated retroviral DNA (Fig. 2h and Extended Data Fig. 2c-d).
Fig. 2: NP220, HUSH complex, and SETDB1 are required for the silencing of unintegrated MLV DNA.
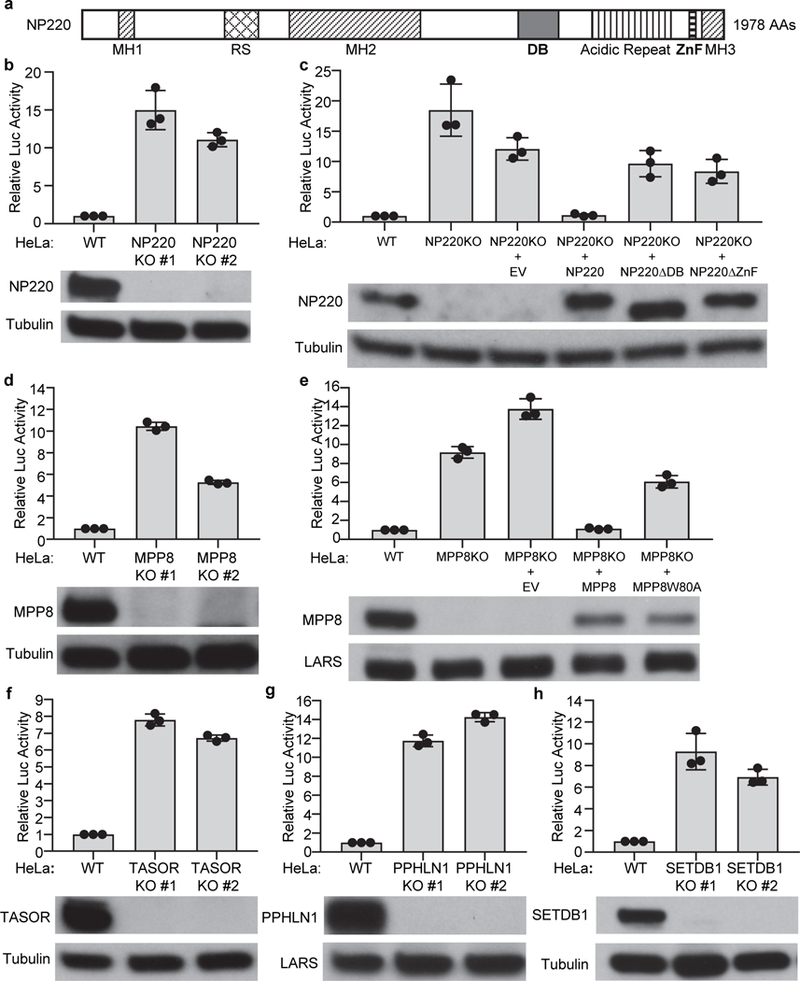
a, Schematic domain organization of NP220. MH1, MH2, and MH3: domains homologous to matrin 3; RS: arginine- and serine-rich domain; DB: DNA-binding domain; ZnF: Zinc finger motif. b-h NP220 (b and c), MPP8 (d and e), TASOR (f), PPHLN1 (g), and SETDB1(h) are required for the silencing of unintegrated MLV DNA. Indicated cells were infected with integrase-deficient (IND184A) MLV-Luc virus. Luciferase activities were measured and activity in parental (WT) HeLa cells was set as 1 (top panels). The expression of indicated proteins was determined by Western blot (bottom panels). EV - empty vector; NP220ΔDB - NP220 with DNA binding domain deletion; NP220ΔZnF - NP220 with zinc finger deletion; MPP8W80A – MPP8 with W80A mutation deficient in H3K9me3 binding activity. Data are represented as mean ± SD from three independent experiments (n = 3).
Co-IP experiments were used to probe the interaction of NP220 with the HUSH complex. Immunoprecipitation (IP) of NP220 resulted in efficient co-IP of MPP8 and TASOR (Fig. 3a, lanes 7 and Extended Data Fig. 3a), independent of DNA or RNA (Extended Data Fig. 4b). The N-terminal 471 amino acids of NP220 were sufficient to mediate the co-IP with MPP8 (Extended Data Fig. 3b). In MPP8 KO cells, NP220 did not co-IP either MPP8 or TASOR (Fig. 3a, lane 9), consistent with MPP8 serving as the direct partner of NP220. In TASOR KO cells, levels of both TASOR and MPP8 were very low (Fig. 3a, lane 4), and neither was detected as interacting with NP220 (Fig. 3a, lane 10). In PPHLN1 KO cells, the levels of MPP8 and TASOR were increased (Fig. 3a, lane 5), and IP of NP220 resulted in correspondingly increased co-IP of both (Fig. 3a, lane 11).
Fig. 3: NP220 recruits HUSH complex and SETDB1 to silence unintegrated MLV DNA.
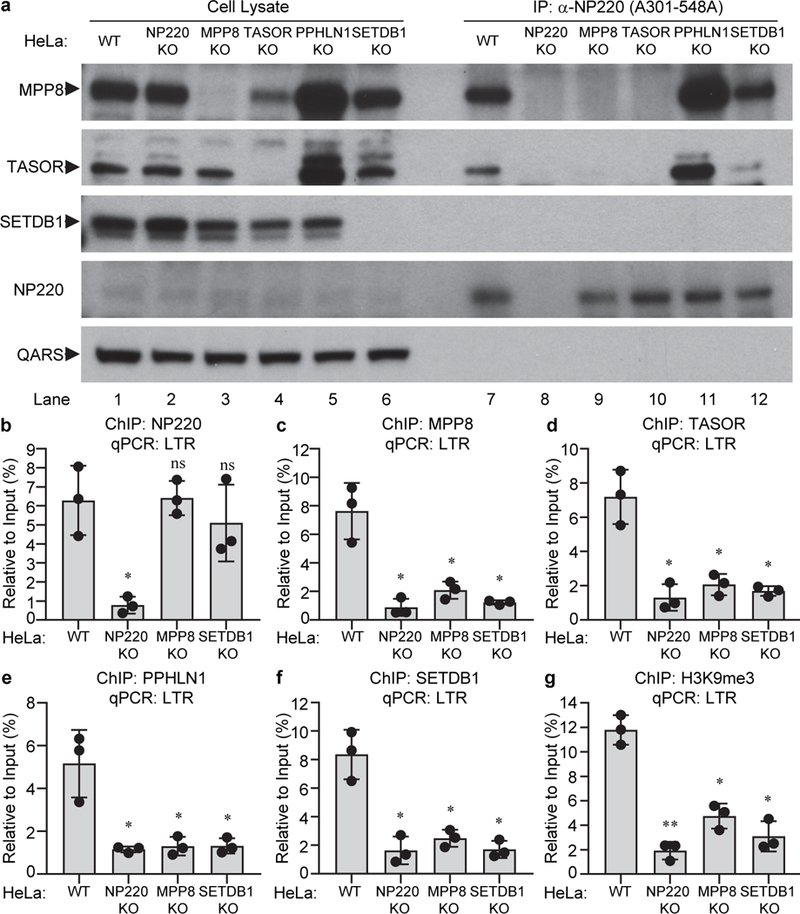
a, NP220 interacts with HUSH complex. Endogenous NP220 was immunoprecipitated (IP) from the indicated HeLa cell lines, and coimmunoprecipitating proteins were probed by Western blot. Images are representative of two independent experiments with similar results. b-g, NP220 recruits HUSH complex and SETDB1 to deposit H3K9me3 on unintegrated MLV DNA. Indicated HeLa cell lines were infected with integrase-deficient (IND184A) MLV-Luc virus. ChIP was performed using antibodies to NP220 (b), MPP8 (c), TASOR (d), PPHLN1 (e), SETDB1 (f), H3K9me3 (g), followed by qPCR targeting LTR. qPCR data from each ChIP were calculated as percent of input DNA. Data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05; **: p < 0.01. P values are from paired two-sided t-tests. Exact p values are presented in the accompanying source data.
ChIP assays were performed to monitor binding of these proteins to unintegrated viral DNAs. NP220 bound to both linear and circular unintegrated DNA (Fig. 3b and Extended Data Fig. 3e). NP220 KO eliminated the ChIP signal (Fig. 3b), and re-expression of WT NP220 or the ΔZnF mutant restored binding, while expression of the ΔDB mutant did not (Extended Data Fig. 3c). Importantly, NP220 bound to DNA even when expression of any of the HUSH subunits, or SETDB1 was eliminated, suggesting that NP220 is the primary DNA-binding component (Fig. 3b). We also detected robust binding of each subunit of the HUSH complex (MPP8, TASOR, and PPHLN1) to unintegrated viral DNA in wild-type cells, but not in MPP8 KO cells nor in SETDB1 KO cells (Fig. 3c-e). HUSH binding required histone methylation, because re-expression of MPP8 mutant W80A, lacking the methylation binding activity, in the MPP8 KO line showed only poor binding to the viral DNA (Extended Data Fig. 3d). None of the HUSH subunits or SETDB1 bound to viral DNA in NP220 KO cells (Fig. 3c-f), suggesting NP220 plays a key role in bringing the HUSH complex and SETDB1 to DNA.
Knockout of NP220, MPP8, or SETDB1 significantly decreased H3K9me3 modification on unintegrated retroviral DNA (Fig. 3g), indicating that NP220, HUSH, and SETDB1 are all required to mediate H3K9 trimethylation on unintegrated retroviral DNA. Knockdown of HDAC1 or HDAC4 also increased the expression of unintegrated retroviral DNA (Fig. 4a and 4b), and increased the levels of acetylated histone H3 (Extended Data Fig. 4c). Co-IP assays showed that endogenous NP220 bound HDAC4, but not other HDACs (Fig. 4d and Fig. 4a). The NP220-HDAC4 interaction was independent of DNA or RNA (Extended Data Fig. 4b). NP220 with the zinc finger deletion (ΔZnF) did not co-IP with HDAC4 (Fig. 4e), indicating that the C-terminal zinc finger motif is critical for this interaction. ChIP assays showed that both HDAC1 and HDAC4 were bound to unintegrated retroviral DNA, and that the binding was reduced in NP220 KO cells (Fig. 4f and 4g). Re-expression of wild-type NP220 in NP220 KO cells decreased the levels of acetylated H3 on unintegrated DNA, but mutant NP220 ΔDB without DNA-binding activity or mutant NP220 ΔZnF without HDAC4-binding activity failed to do so (Fig. 4h). The results indicate that NP220 utilizes its C-terminal zinc finger to recruit HDAC4, and likely HDAC1, to deacetylate histone H3 and thereby silence the expression of unintegrated retroviral DNA. Apparently deacetylation is mechanistically upstream of H3 methylation: HDAC inhibition and H3 acetylation led to decreased H3K9me3 (Extended Data Fig. 4c), while SETDB1 knockout and H3K9 demethylation did not lead to H3 acetylation (Extended Data Fig. 4d).
Fig. 4: NP220 recruits HDAC1 and HDAC4 to deacetylate histone H3 on unintegrated retroviral DNA.
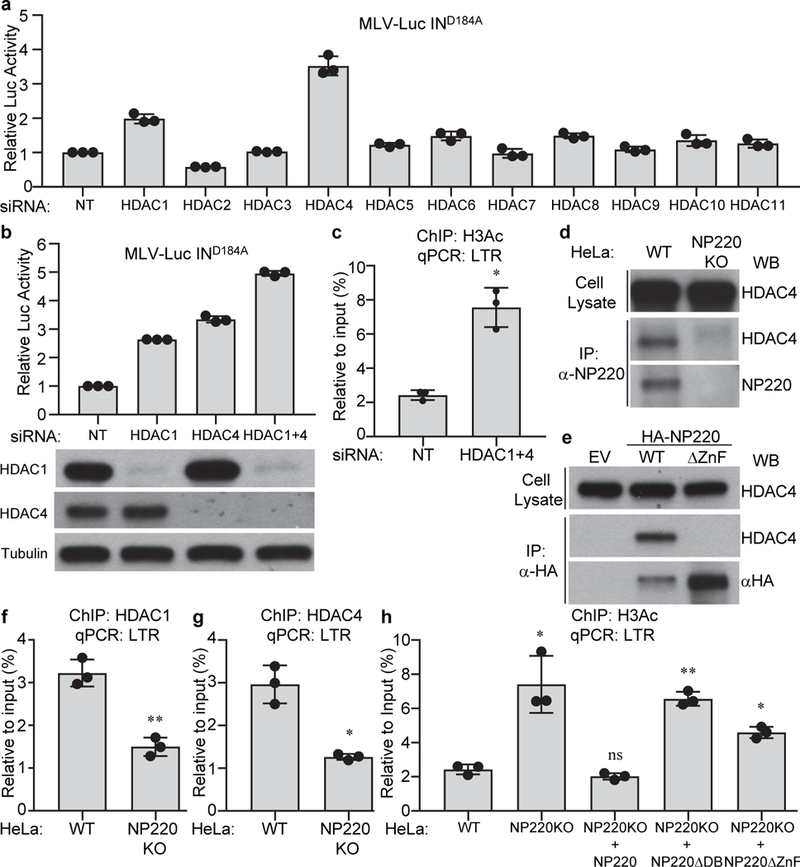
a-c, HDAC1/4 are required for the silencing of unintegrated retroviral DNA. Hela cells were transfected with indicated siRNAs and then infected with integrase-deficient (IND184A) MLV-Luc virus. (a, b) Luciferase activities were measured and luciferase activity in non-targeting (NT) siRNA transfected cells was set as 1. The expression of HDAC1 and HDAC4 was determined by Western blot (b, bottom panels). (c) ChIP was performed using antibodies to pan-acetyl H3 followed by qPCR targeting LTR. d, e, NP220 interacts with HDAC4. (d) Endogenous NP220 was immunoprecipitated (IP) from indicated HeLa cell lines. (e) HA-tagged NP220 or NP220 with Zinc finger deletion (ΔZnF) were introduced into NP220 KO HeLa cells, and then immunoprecipitated (IP) by HA antibody. The coimmunoprecipitating HDAC4 was probed by Western blot. Images are representative of two independent experiments with similar results. f-h, NP220 recruits HDAC1 and HDAC4 to deacetylate histone H3 on unintegrated retroviral DNA. Indicated cells were infected with integrase-deficient (IND184A) MLV-Luc virus. ChIP was performed using antibodies to HDAC1 (f), HDAC4 (g), pan-acetyl H3 (h), followed by qPCR targeting LTR. In (a-c and f-h), data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05; **: p < 0.01. P values are from paired two-sided t-tests. Exact p values are presented in the accompanying source data.
We next examined silencing of retroviruses other than MLV. Knockout or knockdown of NP220 increased the expression of unintegrated DNA of human immunodeficiency virus type 1 (HIV-1) and Mason-Pfizer monkey virus (MPMV), but not Rous sarcoma virus (RSV) (Extended Data Fig. 5a-e). Knockout of the components of the HUSH complex had no or minimal effect on silencing of unintegrated DNAs of HIV-1, MPMV, or RSV (Extended Data Fig. 5c-e). Knockdown of HDAC1/4 relieved the silencing of unintegrated HIV-1 and MPMV DNA (Extended Data Fig. 5f-g). We conclude that the silencing of unintegrated DNAs of the various retrovirus genera is mediated by a distinctive variety of host factors.
NP220 preferentially binds to cytidine clusters in dsDNA at the consensus sequence of CCCCC(G/C)12. The MLV U3 promoter region contains 5 close matches to consensus NP220 binding sites (Fig. 5a and Extended Data Fig. 6a). By electrophoresis mobility shift assays (EMSA), incubation of the NP220 DNA-binding domain (NP220DB) with a 84-bp biotin-labeled DNA fragment of the MLV LTR containing two potential NP220 binding sites (Bio-DNA84) produced a mobility shifted band, and the resulting shift was sensitive to competition by cold probe (DNA84), but not by a mutant probe with deletions of the NP220 binding sites (DNA72) (Fig. 5b). These results indicate that NP220 specifically binds the cytidine clusters in MLV DNA. To test the importance of these DNA sequences in NP220-mediated silencing of unintegrated DNA, we generated a panel of variant reporters (Fig. 5a). Mutations M1 and M2 had no significant effect on the LTR promoter activity when integration was allowed, but allowed higher expression of unintegrated DNAs (Extended Data Fig. 6b). Expression of unintegrated MLV DNA with replacement of the U3 region with the RSV U3, which lacks cytidine clusters, or with deletion or mutation of the NP220 binding sites, was less sensitive to NP220 KO (Fig. 5c and Extended Data Fig. 6c). ChIP assays showed that the association of NP220 with viral DNA, like the silencing of expression, decreased upon the replacement, deletion, or mutation of the NP220-binding DNA sequences (Fig. 5d). In the case of HIV-1, we identified 5 consensus NP220 binding sequences in the U3 promoter region (Extended Data Fig. 7a and b). Deletion of these putative binding sequences made HIV-1 unintegrated DNA less responsive to NP220 KO (Extended Data Fig. 7c and d). It should be noted that the last three binding sequences overlap with SP1 binding sites, and deletion of these sequences severely diminished HIV-1 LTR basal promoter activity (Extended Data Fig. 7e).
Fig. 5: The binding specificity of NP220 to unintegrated retroviral DNA.
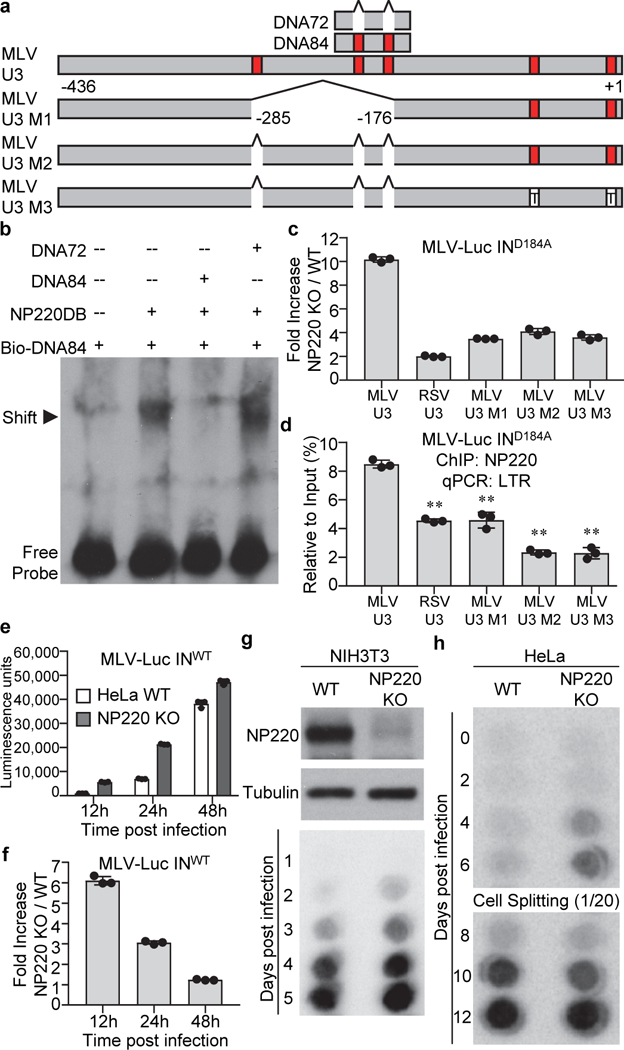
a, Locations of putative NP220 binding sites in MLV U3 region. Putative NP220 binding sites are indicated in red. T: C nucleotides mutated to T. b, NP220 binds a DNA fragment from MLV U3 region. Indicated DNA fragments were incubated with NP220 DNA binding domain (NP220DB) and shifted bands were probed with an antibody recognizing biotin. c, d, Specific DNA sequences are responsible for NP220 silencing and binding unintegrated retroviral DNA. Indicated cells were infected with integrase-deficient (IND184A) MLV-Luc virus bearing indicated deletions or mutations in the U3 region. (c) Luciferase activities were measured and fold increase (NP220 KO / WT) was calculated as the ratio of luciferase activity in KO cells to that in WT cells. (d) ChIP was performed to assess the association of NP220 with unintegrated MLV-Luc DNA. RSV U3: replacement of MLV U3 with RSV U3. **: p < 0.01. P values are from paired two-sided t-tests. Exact p values are presented in the accompanying source data. e, f, Indicated HeLa cells were infected with integrase-proficient (INWT) MLV-Luc virus. (e) At indicated times post infection, luciferase activities were measured. (f) Fold increase (NP220 KO / WT) was calculated as the ratio of luciferase activity in KO cells to that in WT cells. g, h, Knockout of NP220 increases the rate of MLV replication in a spreading infection. (Panel g, upper) The expression of NP220 was determined by Western blot. Indicated cells were infected with replication-competent ecotropic (g) or amphotropic (h) MLV. Viral spreading was monitored by assay for reverse transcriptase in the culture medium. In (c-f), data presented are mean ± SD from three independent experiments (n = 3). In (b, g, and h), images are representative of two independent experiments with similar results.
NP220 not only silenced nonintegrating viral DNA vectors, but also impacted infection by integration-competent vectors and even replicating viruses. MLV DNA was silenced, marked with histone deacetylation, and bound by NP220, and NP220 KO markedly enhanced the expression of viral DNA at 12 h post-infection, when most of the viral DNA is unintegrated, but not at 48 h post-infection, when most of the viral DNA is integrated into the host genome (Fig. 5e-f and Extended Data Fig. 6d-g). The rate of MLV and HIV-1 spread in NP220 KO cells was faster than in control cells (Fig. 6g-h and Extended Data Fig. 7f).
We have here defined the mechanisms and the machinery by which silencing is imposed on unintegrated retroviral DNAs (Extended Data Fig. 8). These findings define new functions for NP220 and the HUSH complex. The restriction is sufficiently strong that many viruses have evolved means to evade or inactivate this machinery: the Vpr and Vpx proteins from primate immunodeficiency viruses mediate degradation of the HUSH complex to relieve silencing of proviruses15,16, and the ICP0 gene of HSV-1 relieves HDAC-mediated silencing of viral DNA17,18. The silencing mechanisms and machinery uncovered here may have wide-ranging impacts on the design of non-integrating retroviral vectors for gene therapy and the gene expression following plasmid DNA transfection.
Methods
Cell Lines
Mammalian cell lines HeLa (CCL-2), NIH3T3 (CRL-1658), HEK293T (CRL-11268), COS-7 (CRL-1651), and chicken cell line DF-1 (CRL-12203) were purchased from ATCC. All these lines and their derivatives were maintained at 37°C and 5% CO2 in DMEM supplemented with 10% inactivated fetal bovine serum, 2 mM glutamine, penicillin, and streptomycin. MT-4 (#120) cells were obtained through the NIH AIDS Reagent Program and cultured at 37°C and 5% CO2 in RPMI 1640 supplemented with 10% inactivated fetal bovine serum.
DNA Construction
Replication-defective retroviral vectors pNCA-Luc (MLV vector expressing firefly luciferase), pNCA-GFP (MLV vector expressing GFP), and pSARM-Luc (MPMV vector expressing firefly luciferase) have been described before19. pRCAS-Luc (RSV vector expressing luciferase) was constructed by replacing GFP in pRCAS-GFP with firefly luciferase. pNL4.3-Luc (HIV-1 vector expressing firefly luciferase) was obtained from NIH AIDS Reagent Program. Plasmid pCMV-intron expresses WT Gag and Pol from NB-tropic MLV. pMD2.G expresses the vesicular stomatitis virus (VSV) envelope glycoprotein. All the integrase-deficient constructs (pRCAS-Luc IND121A, pSARM-Luc IND127A, pCMV-intron IND184A, pNL4.3-Luc IND64A) were created by site directed mutagenesis.
pLvx-EF1-IRES-Neo was constructed by replacing the CMV promoter in pLvx-IRES-Neo (Clontech, #632181) with the EF1 promoter. Coding sequence for MPP8 (WT or with W80A mutation) were cloned into pLvx-EF1-IRES-Neo to produce pLvx-EF1-IRES-Neo-MPP8 and pLvx-EF1-IRES-Neo-MPP8W80A. NP220 CDS with silent mutation in the sgRNA targeting region (WT, or with DNA binding domain deletion ΔDB, or with zinc finger motif deletion ΔZnF) were also cloned into pLvx-EF1-IRES-Neo to produce pLvx-EF1-IRES-Neo-NP220, pLvx-EF1-IRES-Neo-NP220 ΔDB, pLvx-EF1-IRES-Neo-NP220 ΔZnF).
MLV-Luc reporter vectors bearing deletions or mutations of putative NP220 binding sites were constructed as follows: the MLV U3 region in the 3’LTR of pNCA-Luc was replaced with RSV U3 to generate MLV-Luc RSV U3; MLV-Luc U3 M1 was constructed by deleting −176 to −285 (relative to the first nucleotide of R region) in the 3’LTR U3 region; MLV-Luc U3 M2 was constructed by deleting the first 3 putative NP220 binding sites (shown in Extended Data Fig. 6a); MLV-Luc U3 M3 was constructed based on MLV-Luc U3 M2 by further mutating the fourth and fifth putative NP220 binding sites (shown in Extended Data Fig. 6a) to TCTTCG and ACTTCT respectively. We mutated rather than deleted the fourth and fifth binding sites because they are located near the TATA box. All the mutations or deletions were introduced into MLV-Luc reporter vectors by overlapping PCR.
pNL4.3-Luc U3 M1, M2, and M3 were constructed by deleting the first 3, 5, and all 6 putative NP220 binding sites (shown in Extended Data Fig. 7a), respectively. All the mutations or deletions were introduced into MLV-Luc reporter vectors by overlapping PCR.
Ecotropic MLV Infectious Molecular Clone (pNCS) has been deposited to Addgene (Plasmid #17362). Amphotropic MLV Infectious Molecular Clone (pNCA-Ampho) has been described before20. MLV HIV-1 NL4–3 Infectious Molecular Clone (pNL4–3) was obtained from NIH AIDS Reagent Program.
DNA Transfection, Virus package and Infection
All the DNA transfections were performed using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
To package HIV-1 or MPMV based VSV-G pseudotyped retroviruses, viral vectors (pNL4.3-Luc or pSARM-Luc, or their derivative mutants) together with pMD2.G, were transfected into HEK293T cells. To package MLV vector based VSV-G pseudotyped viruses, viral vectors (pNCA-GFP or pNCA-Luc) together with pCMV-intron (expressing MLV Gag and Gag-Pol or GagPol with mutant integrase) and pMD2.G were transfected into HEK293T cells. To package RSV based VSV-G pseudotyped retroviruses, viral vectors (pRCAS-Luc and its derivative mutants) together with pMD2.G, were transfected into DF-1 cells. To package lentiviral vector based VSV-G pseudotyped viruses, viral vectors together with pCMVdeltaR8.2 (expressing HIV-1 Gag and Gag-Pol) and pMD2.G were transfected into HEK293T cells. 40 h after transfection, supernatants were collected and filtered through a 45 μm membrane to produce virus preparations.
Unless otherwise indicated, viruses were 3-fold diluted with cell culture medium containing 20 mM HEPES (pH 7.5) and 4 μg/ml polybrene. 3 h after infection, cell culture medium was changed.
Luciferase Assay
Luciferase activity was assayed 40 h after infection, using the Luciferase Assay System (Promega).
Reverse Transcription
Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific).
Quantitative PCR (qPCR)
Quantitative PCR was performed in ABI 7500 Fast Real-Time PCR System using FastStart Universal SYBR Green Master (Rox). The PCR cycle program was: 1) 50°C 2 min, 1 cycle; 2) 95°C 10 min, 1 cycle; 3) 95 °C 15 s -> 60 °C 30 s -> 72 °C 30 s, 40 cycles; 4) 72°C 10 min, 1 cycle. The primers used for real time PCR are listed in Supplementary Table 2.
Chromatin immunoprecipitation (ChIP)
2*106 cells were seeded in 10-cm dishes and infected with VSV-G pseudotyped, integrase-deficient or integrase-proficient, MLV-Luc virus for two days. The virus used for infection was pretreated with 5 U/ml DNase (Promega, M6101) supplemented with 10 mM MgCl2 at 37 OC for 1 hour to remove any residual plasmid DNA. Cells were crosslinked in 1% formaldehyde for 10 min, quenched in 0.125 M glycine for 5 min, and lysed in 1 ml of ChIP lysis buffer (50 mM Tris-HCl pH 8.0, 1% SDS, 10 mM EDTA, complemented with Protease inhibitor cocktail). Cell lysates were sonicated using Branson 450 Digital Sonifier (12% power amplitude, 30” X 8 times, on ice for 60” between each sonication) to produce an average chromatin fragment size of 200–800 base pairs and centrifuged at 13000 rpm at 4 °C for 20 min. The supernatant of ~50 μg sonicated chromatin was then immunoprecipitated overnight using 5 μg specific antibodies in ChIP dilution buffer (10 mM Tris-HCl pH 8.0, 1% Triton X-100,0.1% SDS, 150 mM NaCl, 2 mM EDTA). Next day, 25 μl of Dynabeads (12.5 μl protein A + 12.5 μl protein G) were added and incubated for an additional 2 h. The beads were washed twice each in ChIP low salt buffer (20 mM Tris-HCl pH 8.0, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA), ChIP high salt buffer (20 mM Tris-HCl pH 8.0, 1% Triton X-100, 0.1% SDS, 500 mM NaCl, 2 mM EDTA), ChIP LiCl buffer (10 mM Tris-HCl pH 8.0, 1% NP-40, 250 mM LiCl, 1 mM EDTA), and TE buffer (10 mM Tris-HCl pH8.0, 1 mM EDTA). Protein-DNA complex was eluted from beads in 200 μl elution buffer (TE buffer containing 1% SDS, 100 mM NaCl, 5 mM DTT), treated with RNase A (1 μg/elution, 37°C, 1 h) and Proteinase K (15 μg/elution, 37°C, 2 h), reverse crosslinked (65°C overnight), and purified using QIAquick PCR purification kit according to the manufacturer’s instructions (Qiagen). Quantitative PCRs were performed with indicated primers. For all ChIP assays, ChIP with histone H3 and control rabbit IgG antibodies were included as positive control and negative control, respectively. qPCR data from each ChIP with specific antibodies were calculated as percent to input DNA.
CRISPR-mediated gene knockout
Four sgRNAs per gene were selected from the Human CRISPR Knockout Pooled Library (Brunello, Addgene#73178) and cloned into LentiCRISPRv2GFP (for MPP8 KO) or lentiCRISPR v2 (for all the other genes). Hela cells were transfected with pool of four plasmids using lipofectamine 2000 for two days. For MPP8 knockout, the brightest 1% GFP positive cells were sorted out by FACS; For the knockout of other genes, the transfected cells were selected in 1 μg/ml puromycin for two weeks. Single cells from the resulting pool cells were seeded in 96-well plate and specific gene knockout clones were screened by Western blotting using specific antibodies.
To rescue the expression of MPP8 in MPP8 KO cells, KO cells were transduced with pLvx-EF1-IRES-Neo-MPP8 (wild type or with indicated mutation) and selected in 800 μg/ml G418 for two weeks. To rescue the expression of NP220 in NP220 KO cells, KO cells were transduced with pLvx-EF1-IRES-Neo-NP220 (wild type or with indicated deletion) and selected in 800 μg/ml G418 for two weeks.
To knockout NP220 in MT-4 cells, four target sequences were selected from the Human CRISPR Knockout Pooled Library (Brunello, Addgene#73178). To knockout NP220 in NIH3T3 cells, four targets were selected from the Mouse CRISPR Knockout Pooled Library (Brie, Addgene#73633). For each knockout, a mix of four crRNAs was synthesized by Integrated DNA Technologies (IDT). The crRNAs were first annealed with ATTO550 tagged Alt-R CRISPR-Cas9 tracrRNA (IDT# 1075927) and then mixed with S.p. Cas9 Nuclease (IDT# 1081058) at room temperature for 20 min to form RNP. The final concentrations of crRNA:tracrRNA duplex and Cas9 nuclease were 24 μM and 20.8 μM. For NIH3T3 cells, 5 μL of the RNP were mixed with 3.5 * 105 NIH3T3 cells and RNP transfection was performed using 4D-Nucleofector System (Lonza) with program EN-158. For MT-4 cells, 5 μL of the RNP were mixed with 106 MT-4 cells and RNP transfection was performed using 4D-Nucleofector System (Lonza) with program CA-137. 24 hours after transfection, 1% brightest ATTO 550 cells were sorted out by FACS and expanded. The knockout efficiency was confirmed by Western blotting using NP220 specific antibody.
siRNA transfection
All siRNAs were obtained from Dharmacon (SMARTpool: ON-TARGETplus siRNA). Targeted gene and catalog numbers are as follows: HDAC1(L-003493–00), HDAC2(L-003495–02), HDAC3(L-003496–00), HDAC4(L-003497–00), HDAC5(L-003498–00), HDAC6(L-003499–00), HDAC7(L-009330–00), HDAC8(L-003500–00), HDAC9(L-005241–00), HDAC10(L-004072–00), HDAC11(L-004258–00), NP220(human, L-013715–02), NP220(mouse, L-047870–01), MPP8(L-021680–02), TASOR(L-020306–02), PPHLN1(L-021306–02), GGT1(L-005884–01), CISD2(L-032593–02), ZNT1(L-007522–01), DUOXA2(L-032210–02), TESC(L-020896–01), GP5(L-012671–00), SETDB1(L-020070–00), SETDB2(L-014751–00), SUV39H1(L-009604–00), SUV39H2(L-008512–00), EHMT1(L-007065–02), EHMT2(L-006937–00), EZH2(L-004218–00), non-targeting control siRNA(NT, D-001810–10).
For siRNA transfection, 105 HeLa cells were seeded in 6-well plates. 24 h later, siRNA were transfected into cells by Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s protocol. After another 24 h, the same siRNA transfection was performed for the second time. 6 hours after second transfection, the siRNA transfected cells were infected with virus for further experiments.
Co-immunoprecipitation and immunoblotting
For co-immunoprecipitation, ~ 5*106 HeLa cells were lysed in 1ml Pierce™ IP Lysis Buffer for 10 min and the lysate was clarified by centrifugation at 4 °C for 15 min at 12000 rpm. For nuclease treatment, cell lysates were treated with Benzonase (250 U/ml supplemented with 10 mM MgCl2), or DNase (5 U/ml supplemented with 10 mM MgCl2), or RNase A (5 μg/ml). 40 μl Dynabeads (20 μl protein A + 20 μl protein G beads) were first mixed with 1 μg specific antibody for 10 min at room temperature and then washed twice with TBST. Antibody coated Dynabeads were incubated with 800 μl cell lysate at 4 °C for 4 h. The beads were then washed 3 times with TBST. The bound proteins on the beads were eluted with 40 μl 1X SDS sample buffer and subjected to SDS-PAGE and Western blot analysis.
For immunoblotting, cells were lysed in RIPA Lysis and Extraction Buffer for 10 min. The lysate was clarified by centrifugation at 4°C for 15 min at 12000 rpm. The samples were heated at 95 °C in SDS sample buffer and resolved by SDS-PAGE electrophoresis, transferred to a PVDF membrane and probed with specific antibodies by Western blot.
Genome-wide CRISPR-Cas9 screen
The Human CRISPR Knockout Pooled Library (Brunello, 2 vector system) was obtained from Addgene and the screen was performed broadly as described before21. The CRISPR sgRNA library virus was packaged by transfecting HEK293T cells with library DNA, HIV-1 Gag-Pol expressing plasmid pCMVΔR8.2, and pMD2.G. HeLa cells were transduced with lentiCas9-Blast virus and two days after transduction, cells were selected in 5 μg/ml blasticidin for two weeks to get pooled HeLa-Cas9 cells. 108 Hela-Cas9 cells were transduced with CRISPR sgRNA library virus at M.O.I. ~ 0.3. Two days after transduction, cells were selected in medium containing 1 μg/ml puromycin for two weeks to get the collection of pooled HeLa knockout cells, and cells were cultured in medium containing 5 μg/ml blasticidin and 1 μg/ml puromycin during the whole process of screening. 2*107 pooled KO HeLa cells were infected with integrase deficient (IND184A) MLV-GFP virus (3-fold dilution) and 5% brightest GFP cells were sorted out by FACS. The sorted out cells were expanded for 2 weeks and then the above infection/sorting procedure was performed for the second time. Genomic DNA was extracted from the resulting selected cells and the control cells (transduced with the sgRNA library, cultured in parallel but without infection/sorting). The abundance of sgRNAs in the control cells and the sorted cells were analyzed by PCR followed by next generation sequencing (NGS). The PCR to amplify the sgRNA was performed as described21 (step 32 −33), except that the reverse primer for control sample is: 5’- CAAGCAGAAGACGGCATACGAGATACTGTATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTatTCTACTATTCTTTCCCCTGCACTGT-3’ and the reverse primer for sorted sample is: 5’-CAAGCAGAAGACGGCATACGAGATAGGTCGCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgatTCTACTATTCTTTCCCCTGCACTGT-3’. The PCR products were purified by Zymo-Spin V with Reservoir and the samples were deep-sequenced on the Illumina Miseq. The sgRNA counts and abundance were analyzed as described21. The degree of sgRNA enrichment and gene hit rank was analyzed by software HiTSelect22.
Bacterial protein purification
The gene fragment encoding the NP220 DNA binding domain (NP220DB, corresponding to amino acid residues 1240 – 1478 of NP220) was cloned into the pGEX-5X-3 vector. The protein was overexpressed in Escherichia coli BL21 (DE3) Star strain (NEB). The cells were induced with 0.1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 37 °C. The harvested cells were resuspended in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% NP-40 and lysed with 0.25 mg/ml lysozeme for1 h followed by sonication. Cell lysates were centrifuged for 30 min at 4 °C before incubating with Glutathione Sepharose beads at 4 °C for 2 h. The beads were washed 5 times with PBS and protein was eluted by elution buffer containing 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2 mM CaCl2, and 10 μg/ml Factor Xa Protease (NEB).
Electrophoresis mobility shift assays (EMSA)
DNA probe Bio-DNA84 was 5’ end-labeled with biotin. EMSA was performed using LightShift™ Chemiluminescent EMSA Kit (Thermo Fisher). 20 fmol Bio-DNA84 was incubated with 800 ng bacterially expressed NP220DB protein at room temperature in a total volume of 20 μl. For DNA competitions, unlabeled DNA (DNA84 or DNA72) was added at same time as probe. Binding reactions were analyzed by electrophoresis on 5% native polyacrylamide gels. The sequence of DNA84 is: AGGATATCTGTGGTAAGCAGTTCCTGCCCCGGCTCAGGGCCAAGAACAGATGGTCCCCAGATGCGGTCCAGCCCTCAGCAGTTT (NP220 binding sites are bold, underlined), and the sequence of DNA72 is: AGGATATCTGTGGTAAGCAGTTCCTGCTCAGGGCCAAGAACAGATGGTATGCGGTCCAGCCCTCAGCAGTTT. All the DNA duplexes were ordered from IDT.
MLV Replication and Reverse Transcriptase Assay
Ecotropic MLV and amphotropic MLV viruses were produced by transfecting HEK293T cells with pNCS and pNCA-Ampho DNAs, respectively, using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
For ecotropic MLV replication in NIH3T3 cells, NIH3T3 WT or NP220 KO cells (104 per well) were seeded in 6-well plates and infected with ecotropic MLV at low multiplicity of infection (MOI). Culture medium was changed at 3 h post infection. Culture supernatants (50 μl) were taken every day for 5 days post infection and assayed for reverse transcriptase activity to monitor the production of progeny virus.
For amphotropic MLV replication in HeLa cells, HeLa WT or NP220 KO cells (3×104 per well) were seeded in 6-well plates and infected with amphotropic MLV at low multiplicity of infection (MOI). Culture medium was changed at 3 h post infection. Culture supernatants (50 μl) were taken every two days post infection. Cells were split 1:20 at 6 d post infection. The culture medium was assayed for reverse transcriptase activity to monitor the production of progeny virus.
For the reverse transcriptase (RT) assay, 5 ul of culture supernatant was incubated with 20 μl Hot/Cold Mix at room temperature for 40 min and then 5 μl of the reaction mix was spotted on DEAE paper. The DEAE paper was washed with 2X SSC buffer for 20 min twice, dried under heat lamp, exposed and visualized by phosphor imaging (GE). The formula for the buffers are as following: Hot/Cold Mix [1 ml Cold Mix, 72 μl Hot Mix, 2 μl MnCl2 (0.5 M), 1 μl P32 dTTP, add all the reagents in order]; Cold Mix [60 mM Tris-HCl pH 8.3, 75 mM NaCl, 0.06% NP40]; Hot Mix [7.6 mg/ml Oligo dT, 16.6 μM dTTP, 166 μg/ul poly(A), 0.5 M DTT]; Oligo dT (GE healthcare, Cat. # 27–7858-2); poly(A) (GE healthcare, Cat. # 27–4110).
HIV-1 Replication
HIV-1 viruses were produced by transfecting HEK293T cells with pNL4.3 using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. For HIV-1 replication in MT-4 cells, MT-4 WT or MT-4 NP220 KO cells (106) were transduced with HIV-1 virus (1 ng p24) by spin infection (3500 rpm at room temperature for 2 h) and then cultured at 37OC. 3 hours post infection, cells were washed twice with PBS and then cultured in 1 ml medium in 24-well plate. For every two days, culture supernatants (50 μl) were taken out for p24 measurement,and half of cells and medium (500 μl) were transferred to a new well containing 500 μl fresh medium. p24 levels were determined using HIV1 p24 ELISA Kit (abcam, ab218268), to monitor the production of progeny virus.
Statistical analysis
Sample size is provided in the figure legends. Statistical significance was determined by a two-tailed Student’s t-test. The experiments were not randomized. The investigators were not blinded to allocation during experiments or in outcome assessment.
Extended Data
Extended Data Fig. 1: Silencing of unintegrated, but not integrated, retroviral DNA.
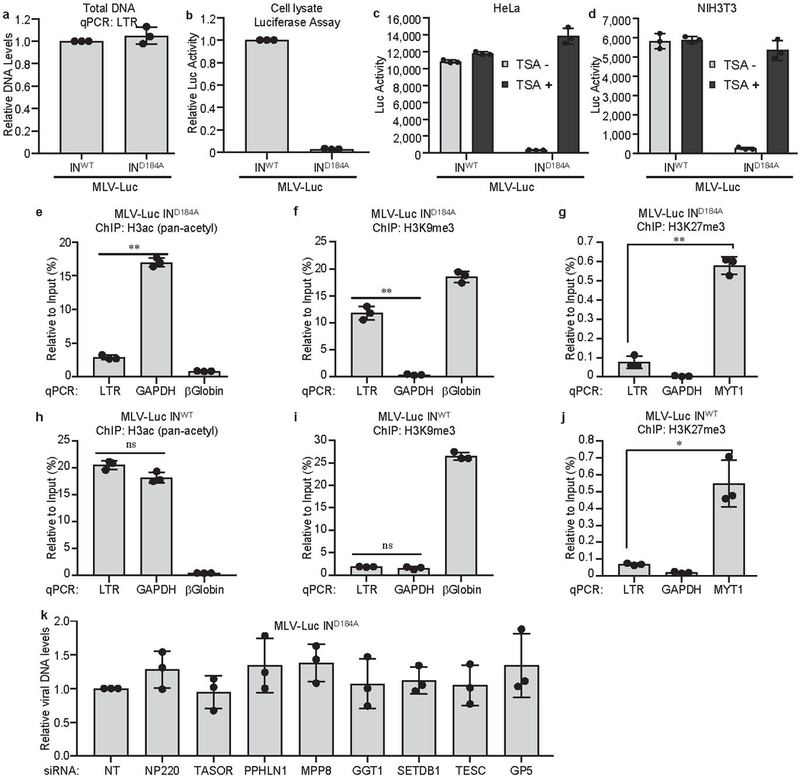
a, b, Silencing of unintegrated retroviral DNA. HeLa cells were infected with VSV-G pseudotyped, integrase-proficient (INWT) or integrase-deficient (IND184A) MLV-Luc virus. Total viral DNA levels (a) and luciferase activities (b) were measured 40 h post infection. The viral DNA level and luciferase activity from INWT MLV-Luc infection were set as 1. Data presented are mean ± SD from three independent experiments (n = 3).c, d, Silencing of unintegrated retroviral DNA is dependent on histone deacetylation. HeLa (c) and NIH3T3 (d) cells were infected with VSV-G pseudotyped, integrase-proficient (INWT) or integrase-deficient (IND184A) MLV-Luc virus and treated with DMSO (TSA-) or 1μM HDAC inhibitor Trichostatin A (TSA+). Luciferase activities were measured 40 h post infection. Data presented are mean ± SD from three independent experiments (n = 3). e-j, Repressive epigenetic marks are present on unintegrated retroviral DNA, while active epigenetic marks are present on integrated retroviral DNA. HeLa cells were infected with VSV-G pseudotyped, integrase-deficient (IND184A) (e-g) or integrase-proficient (INWT) (h-j) MLV-Luc virus. At 40 h post infection, ChIP, using antibodies to pan-acetyl H3, H3K9me3, or H3K27me3, followed by qPCR using indicated primers, was performed to assess the H3ac (e, h), H3K9me3 (f, i), and H3K27me3 (g, j) modifications across the LTR of unintegrated and integrated MLV-Luc DNA. qPCR data from each ChIP were calculated as percent of input DNA. Data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05; **: p < 0.01. P values are from paired two-sided t-tests. Exact p values can be found in the accompanying source data. k, Knockdown of indicated genes have no effect on viral DNA levels. Hela cells were first transfected with indicated siRNA and then infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. Viral DNA levels were measured 40 h post infection. DNA levels in HeLa cells transfected with non-targeting (NT) control siRNA was set as 1. Data presented are mean ± SD from three independent experiments (n = 3).
Extended Data Fig. 2: NP220 and SETDB1 are required for the silencing of unintegrated MLV DNA.
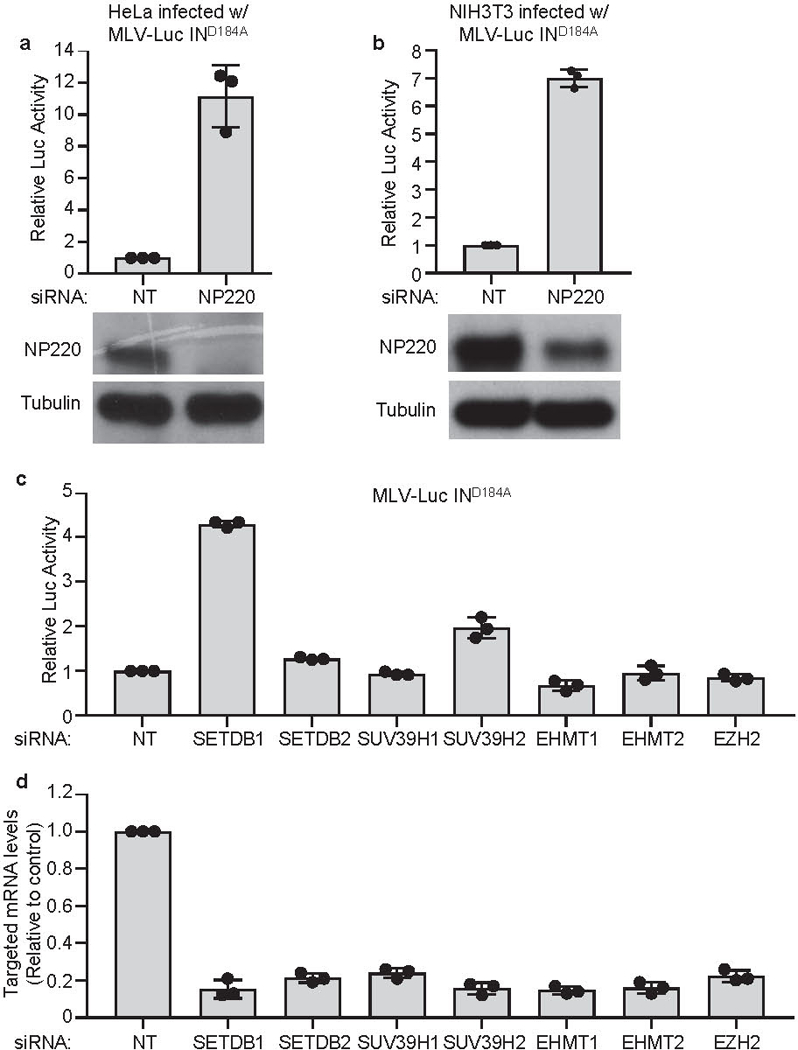
a, b, Hela (a) or NIH3T3 (b) cells were transfected with indicated siRNAs and then infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. Luciferase activities were measured 40 h post infection and luciferase activity in non-targeting (NT) control siRNA transfected cells was set as 1 (top panels). Data presented are mean ± SD from three independent experiments (n = 3). The expression of NP220 was determined by Western blot (bottom panels). c, Hela cells were first transfected with indicated siRNAs targeting histone methyltransferases and then infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. Luciferase activities were measured 40 h post infection and luciferase activity in non-targeting (NT) control siRNA transfected HeLa cells was set as 1. Data presented are mean ± SD from three independent experiments (n = 3). d, Knockdown efficiency of siRNAs used in (c). Hela cells were transfected with indicated siRNAs targeting histone methyltransferases and mRNA levels of siRNA targeted genes were measured by RT-qPCR. mRNA levels in non-targeting (NT) control siRNA transfected HeLa cells was set as 1. Data presented are mean ± SD from three independent experiments (n = 3).
Extended Data Fig. 3: NP220 recruits HUSH complex and SETDB1 to silence unintegrated MLV DNA.
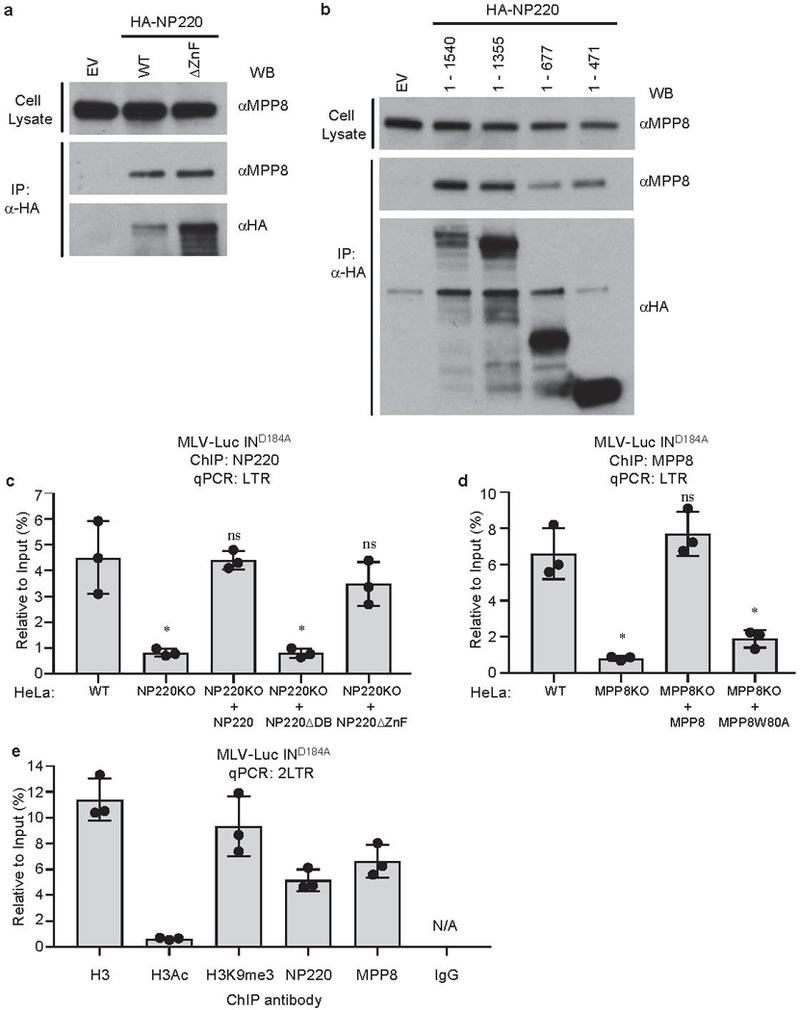
a, b, HA-tagged NP220, NP220 with zinc finger deletion (ΔZnF) (a) or indicated fragments of NP220 (b) were introduced into NP220 KO HeLa cells, and then immunoprecipitated (IP) by HA antibody. The coimmunoprecipitated MPP8 was probed by Western blot. Images are representative of two independent experiments with similar results. c, Parental HeLa cells (WT), NP220 KO HeLa cell line, and NP220 KO HeLa cells reconstituted with indicated variants of NP220 were infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. At 40 h post infection, ChIP was performed to assess the association of NP220 across the LTR of unintegrated MLV-Luc DNA. qPCR data from each ChIP were calculated as percent to input DNA. Data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05. P values are from paired two-sided t-tests. Exact p values can be found in the accompanying source data. d, Parental HeLa cells (WT), MPP8 KO HeLa cell line, and MPP8 KO HeLa cells reconstituted with indicated variants of MPP8 were infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. At 40 h post infection, ChIP was performed to assess the association of MPP8 with the LTR of unintegrated MLV-Luc DNA. qPCR data from each ChIP were calculated as percent to input DNA. Data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05. P values are from paired two-sided t-tests. Exact p values can be found in the accompanying source data. e, HeLa cells were infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. At 40 h post infection, ChIP was performed using indicated antibodies followed by qPCR using primers targeting 2LTR circles. qPCR data from each ChIP were calculated as percent to input DNA. Data presented are mean ± SD from three independent experiments (n = 3).
Extended Data Fig. 4: Interaction between NP220 and HDACs.
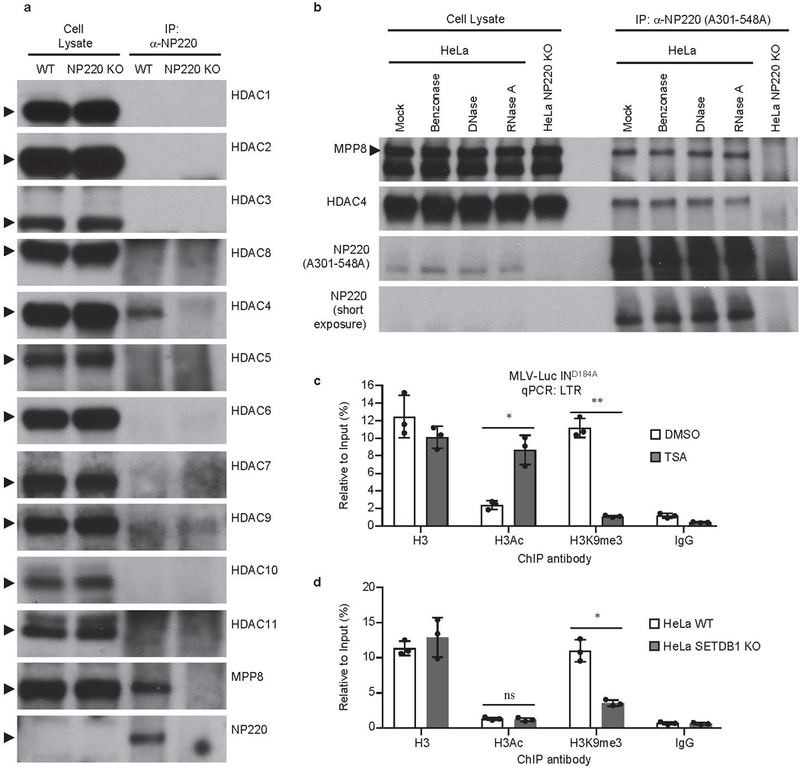
a, Screen for interaction between NP220 and HDACs. Endogenous NP220 were immunoprecipitated (IP) from indicated HeLa cell lines, and the indicated coimmunoprecipitated HDAC proteins and MPP8 were probed by Western blot using specific antibodies. MPP8 serves as a positive control. Images are representative of two independent experiments with similar results. b, NP220-MPP8 or NP220-HDAC4 interaction is independent of DNA or RNA. Cell lysates from indicated HeLa cell lines were treated with Benzonase, DNase, or RNase A, endogenous NP220 were immunoprecipitated (IP), and coimmunoprecipitating proteins were probed by Western blot using specific antibodies. Images are representative of two independent experiments with similar results.c, d, Relationship between deacetylation and H3K9me3 on unintegrated viral DNA. (c) HeLa cells were infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus and treated with DMSO (TSA-) or 1μM HDAC inhibitor Trichostatin A (TSA+). (d) Parental HeLa cells (WT) and SETDB1 KO HeLa cell line (SETDB1 KO) were infected with VSV-G pseudotyped, integrase-deficient (IND184A) MLV-Luc virus. At 40 h post infection, ChIP was performed using indicated antibodies followed by qPCR using primers targeting LTR. qPCR data from each ChIP were calculated as percent to input DNA. Data presented are mean ± SD from three independent experiments (n = 3). *: p < 0.05; **: p < 0.01. P values are from paired two-sided t-tests. Exact p values can be found in the accompanying source data.
Extended Data Fig. 5: NP220 mediates silencing of unintegrated retroviral DNA from HIV-1 and MPMV, but not RSV.
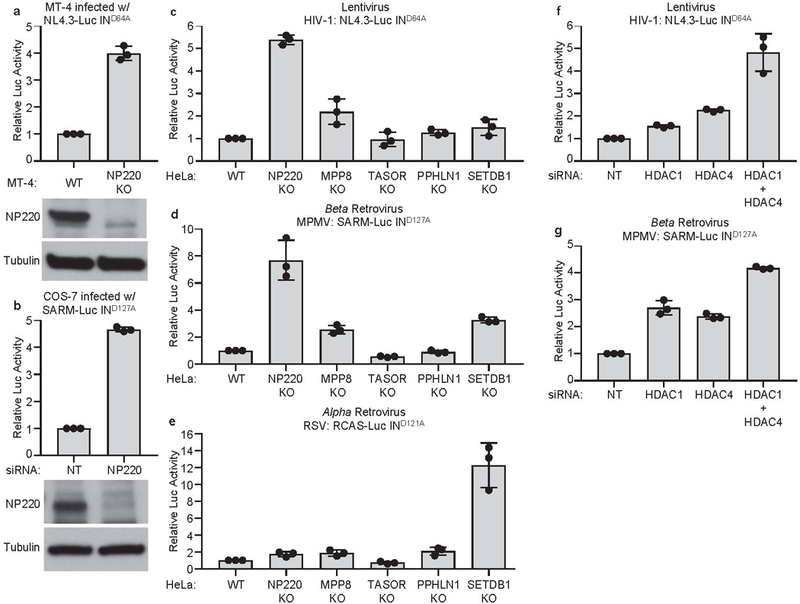
a, Parental MT-4 cells (WT) and NP220 KO MT-4 cell line (NP220 KO) were infected with VSV-G pseudotyped, integrase-deficient (IND164A) HIV-1 vector NL4.3-Luc. Luciferase activities were measured 40 h post infection and luciferase activity in parental (WT) MT-4 cells was set as 1 (top panel). Data are presented as mean ± SD from three independent experiments (n = 3). The expression of NP220 was determined by Western blot (bottom panel). b, COS-7 cells were first transfected with indicated siRNAs and then infected with VSV-G pseudotyped, integrase-deficient (IND127A) MPMV vector SARM-Luc. Luciferase activities were measured 40 h post infection and luciferase activity in non-targeting (NT) control siRNA transfected cells was set as 1 (top panel). Data presented are mean ± SD from three independent experiments (n = 3). The expression of NP220 was determined by Western blot (bottom panel). c, d, e, Indicated HeLa cell lines were infected with VSV-G pseudotyped, integrase-deficient HIV-1 vector NL4.3-Luc (c), MPMV vector SARM-Luc (d), or RSV vector RCAS-Luc (e). Luciferase activities were measured 40 h post infection. Luciferase activity in parental HeLa (WT) cells were set as 1. Data presented are mean ± SD from three independent experiments (n = 3). f, g, HDAC1/4 are involved in silencing of unintegrated HIV-1 and MPMV DNA. Hela cells were first transfected with indicated siRNAs, and then infected with VSV-G pseudotyped, integrase-deficient (IND64A) HIV-1 vector NL4.3-Luc (b) or VSV-G pseudotyped, integrase-deficient (IND127A) MPMV vector SARM-Luc (d). Luciferase activities were measured 40 h post infection and luciferase activities in HeLa cells transfected with control non-targeting (NT) siRNA were set as 1. Data are represented as mean ± SD from three independent experiments (n = 3).
Extended Data Fig. 6: The binding specificity of NP220 to unintegrated MLV DNA.
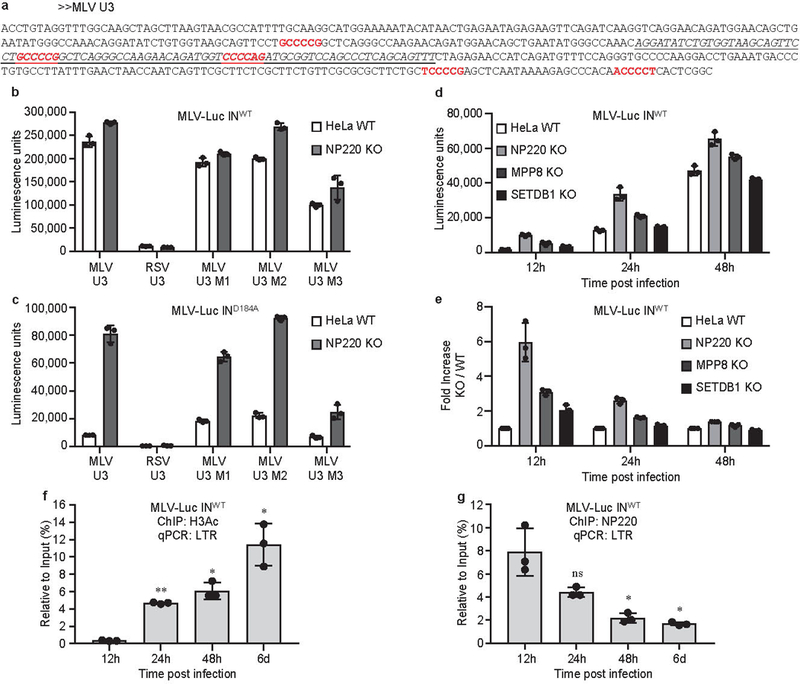
a, The sequence of MLV U3 region. Putative NP220 binding sites are indicated in red. The sequence of the 84-nt probe used for EMSA is italicized and underlined.b, c, Parental (WT) or NP220 knockout (NP220 KO) HeLa cells were infected with VSV-G pseudotyped, integrase-proficient (INWT) (b) or integrase-deficient (IND184A) (c) MLV-Luc virus bearing indicated deletions or mutations in the U3 region. Luciferase activities were measured 40 h post infection. Data presented are mean ± SD from three independent experiments (n = 3). d, e, Indicated HeLa cell lines were infected with VSV-G pseudotyped, integrase-proficient (INWT) MLV-Luc virus. (d) At indicated times post infection, luciferase activities were measured. (e) Fold increase (KO / WT) was calculated as the ratio of luciferase activities from KO cells to luciferase activities from WT cells. Data presented are mean ± SD from three independent experiments (n = 3). f, g, The dynamics of H3Ac deposition and NP220 association on viral DNA during the course of MLV infection. HeLa cells were infected with VSV-G pseudotyped, integrase-proficient (INWT) MLV-Luc virus. At indicated time points post infection, ChIP was performed using antibodies to H3Ac (f) and NP220 (g) followed by qPCR using primers targeting the LTR, to monitor the association of H3Ac (f) and NP220 (g) with viral DNA. qPCR data from each ChIP were calculated as percent of input DNA. Data presented are mean ± SD from three independent experiments (n = 3). ns: p > 0.05; *: p < 0.05; **: p < 0.01. P values are from paired two-sided t-tests. Exact p values can be found in the accompanying source data.
Extended Data Fig. 7: The binding specificity of NP220 to unintegrated HIV-1 DNA.
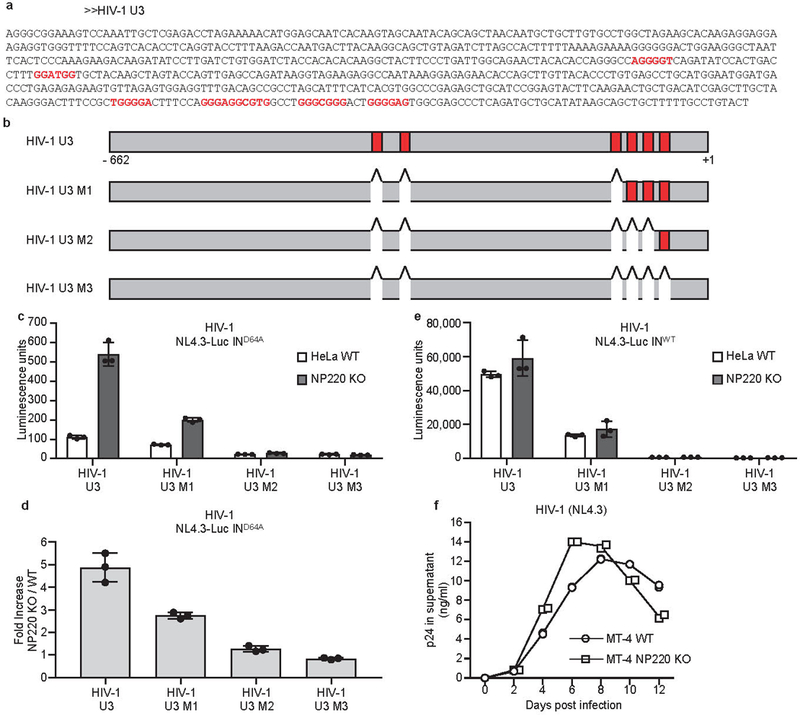
a, b, The sequences and locations of putative NP220 binding sites in HIV-1 U3 region of HIV-1 U3 region. Putative NP220 binding sites are indicated in red. c, d, Parental (WT) or NP220 knockout (NP220 KO) HeLa cells were infected with VSV-G pseudotyped, integrase-deficient (IND64A) MLV-Luc virus bearing indicated deletions or mutations in the U3 region. (c) Luciferase activities were measured 40 h post infection. (d) Fold increase (NP220 KO / WT) was calculated as the ratio of luciferase activities from KO cells to luciferase activities from WT cells. Data presented are mean ± SD from three independent experiments (n = 3).e, Parental (WT) or NP220 knockout (NP220 KO) HeLa cells were infected with VSV-G pseudotyped, integrase-proficient (INWT) HIV-Luc virus bearing indicated deletions or mutations in the U3 region. Luciferase activities were measured 40 h post infection. Data presented are mean ± SD from three independent experiments (n = 3). f, Knockout of NP220 increases the rate of HIV-1 spreading. (g) Parental (WT) and NP220 knockout (NP220 KO) MT-4 cells were infected with HIV-1(NL4.3). Viral spreading was monitored by assay for p24 concentration in the culture medium. Data presented are mean from two technical ELISA replicates and are representative of two independent experiments.
Extended Data Fig. 8: Schematic of the silencing of unintegrated retroviral DNAs.
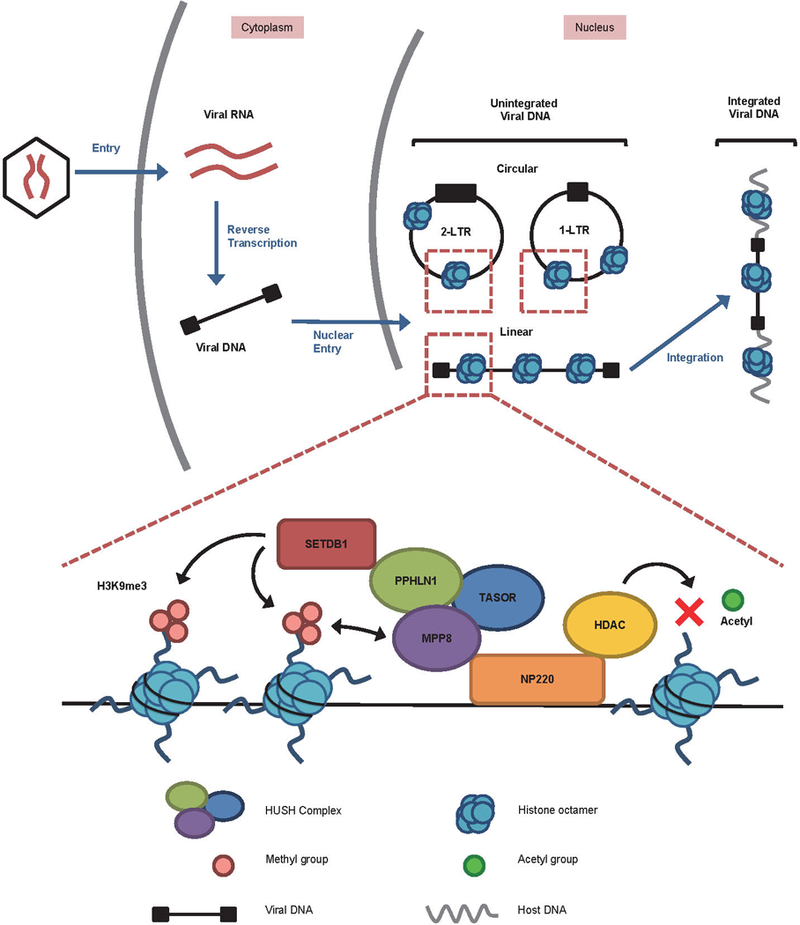
Retroviral infection results in the synthesis of a linear double stranded DNA in the cytoplasm, which is delivered into the nucleus to give rise to two circular forms and the integrated provirus (top). The unintegrated nuclear DNAs are rapidly loaded with nucleosomal histones (blue). In the case of MLV, NP220 binds to the unintegrated viral DNA and is responsible for attracting histone deacetylases (HDACs), the HUSH complex (consisting of MPP8, TASOR, and PPHLN1), and the histone methyltransferase SETDB1. HDACs remove the activation marks of histone acetylation, and SETDB1 introduces repressive H3K9me3 marks. MPP8 binds H3K9me3 to strengthen association with the viral chromatin.
Supplementary Material
Acknowledgments
Cell sorting was performed with the assistance of Amir Figueroa in the Core Facility of Department of Microbiology & Immunology at Columbia University. This work was supported by the Howard Hughes Medical Institute and by National Institutes of Health (NIH) grant R01 CA30488 (to S.P.G.).
Footnotes
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The following figures have associated raw data: Figures 3a-g, 4c-h, 5b, 5d; Extended Data Figures 1e-j, 3a-e, 4a-d, 6f-g. For gel source data, see Supplementary Fig. 1.
References
- 1.Orzalli MH & Knipe DM Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 68, 477–492, doi: 10.1146/annurev-micro-091313-103409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartzberg P, Colicelli J & Goff SP Construction and analysis of deletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for productive infection. Cell 37, 1043–1052 (1984). [DOI] [PubMed] [Google Scholar]
- 3.Schneider WM, Wu DT, Amin V, Aiyer S & Roth MJ MuLV IN mutants responsive to HDAC inhibitors enhance transcription from unintegrated retroviral DNA. Virology 426, 188–196, doi: 10.1016/j.virol.2012.01.034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakai H et al. Integration is essential for efficient gene expression of human immunodeficiency virus type 1. Journal of virology 67, 1169–1174 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakajima N, Lu R & Engelman A Human immunodeficiency virus type 1 replication in the absence of integrase-mediated dna recombination: definition of permissive and nonpermissive T-cell lines. Journal of virology 75, 7944–7955 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tchasovnikarova IA et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485, doi: 10.1126/science.aaa7227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu N et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232, doi: 10.1038/nature25179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbez-Masson L et al. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res 28, 836–845, doi: 10.1101/gr.228171.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varmus H Retroviruses. Science 240, 1427–1435 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Yoshimura FK & Weinberg RA Restriction endonuclease cleavage of linear and closed circular murine leukemia viral DNAs: discovery of a smaller circular form. Cell 16, 323–332 (1979). [DOI] [PubMed] [Google Scholar]
- 11.Kilzer JM et al. Roles of host cell factors in circularization of retroviral dna. Virology 314, 460–467 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Inagaki H et al. A large DNA-binding nuclear protein with RNA recognition motif and serine/arginine-rich domain. J Biol Chem 271, 12525–12531 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Chang Y, Horton JR, Bedford MT, Zhang X & Cheng X Structural insights for MPP8 chromodomain interaction with histone H3 lysine 9: potential effect of phosphorylation on methyl-lysine binding. Journal of molecular biology 408, 807–814, doi: 10.1016/j.jmb.2011.03.018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokura K, Sun L, Bedford MT & Fang J Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. The EMBO journal 29, 3673–3687, doi: 10.1038/emboj.2010.239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chougui G et al. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat Microbiol, doi: 10.1038/s41564-018-0179-6 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Yurkovetskiy L et al. Primate immunodeficiency virus Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. bioRxiv, doi: 10.1101/293001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu H & Roizman B The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. Journal of virology 83, 181–187, doi: 10.1128/JVI.01940-08 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poon AP, Gu H & Roizman B ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proceedings of the National Academy of Sciences of the United States of America 103, 9993–9998, doi: 10.1073/pnas.0604142103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 19.Wang GZ & Goff SP Postentry restriction of Mason-Pfizer monkey virus in mouse cells. Journal of virology 89, 2813–2819, doi: 10.1128/JVI.03051-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf D & Goff SP Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204, doi: 10.1038/nature07844 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863, doi: 10.1038/nprot.2017.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diaz AA, Qin H, Ramalho-Santos M & Song JS HiTSelect: a comprehensive tool for high-complexity-pooled screen analysis. Nucleic acids research 43, e16, doi: 10.1093/nar/gku1197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.