

Abstract
Multidrug resistance is highly conserved in mammalian, fungal, and bacterial cells, is characterized by resistance to several unrelated xenobiotics, and poses significant challenges to managing infections and many cancers. Eukaryotes use a highly conserved set of drug efflux transporters that confer pleiotropic drug resistance (PDR). To interrogate the regulation of this critical process, here we developed a small molecule–responsive biosensor that couples transcriptional induction of PDR genes to growth rate in the yeast Saccharomyces cerevisiae. Using diverse PDR inducers and the homozygous diploid deletion collection, we applied this biosensor system to genome-wide screens for potential PDR regulators. In addition to recapitulating the activity of previously known factors, these screens identified a series of genes involved in a variety of cellular processes with significant but previously uncharacterized roles in the modulation of yeast PDR. Genes identified as down-regulators of the PDR included those encoding the MAD family of proteins involved in the mitotic spindle assembly checkpoint (SAC) complex. Of note, we demonstrated that genetic disruptions of the mitotic spindle assembly checkpoint elevate expression of PDR-mediating efflux pumps in response to exposure to a variety of compounds that themselves have no known influence on the cell cycle. These results not only establish our biosensor system as a viable tool for investigating PDR in a high-throughput fashion, but also uncover critical control mechanisms governing the PDR response and a previously uncharacterized link between PDR and cell cycle regulation in yeast.
Keywords: biosensor, drug resistance, gene regulation, multidrug transporter, microarray
Introduction
Multidrug resistance (MDR)3 is a highly conserved process in mammalian, fungal, and bacterial cells that is characterized by resistance to a variety of unrelated xenobiotics (1–3). Such resistance is primarily conferred through complex interactions between a network of transcriptional regulators and genes encoding downstream transmembrane ATP-binding cassette (ABC) efflux pumps (4). More than a dozen proteins in this network, also termed pleiotropic drug resistance (PDR), have been identified in Saccharomyces cerevisiae. Among the most extensively studied of these are plasma membrane-bound pumps Pdr5, Snq2, and Yor1 (5). Pdr5, a transporter with hundreds of verified substrates, is a functional homolog of Candida albicans Cdr1 and mammalian P-glycoprotein (MDR1); two transporters implicated in clinical resistance to a variety of drugs (3, 6). Snq2 shares structural similarity to Pdr5 and was the first ABC transporter in yeast that was implicated in drug resistance (7), whereas Yor1 belongs to the cystic fibrosis transmembrane conductance regulator family of transporters. All are major drivers of drug resistance in yeast (8).
Hyperactivation of the PDR is the leading cause of resistance to newly developed antifungal azole and echinocandin drugs in many fungal pathogens (3), whereas overexpression of MDR1, the human homolog of Pdr5, creates substantial challenges in chemotherapy as drugs are pumped out of cancer cells and cannot reach therapeutic concentrations (9). As a result, molecules that modulate the activity of these pumps are among the leading candidates to overcome widespread drug resistance (10, 11).
The genetic, structural, and functional similarity of S. cerevisiae PDR regulators and pumps to their homologs in humans and pathogenic fungi have made yeast a valuable model to study PDR regulation, as well as mutations in the pumps themselves that lead to multidrug resistance and ABC protein diseases such as cystic fibrosis (12).
Activation of the PDR transcriptional cascade is a rapid and complex process. Extensive study has revealed Pdr1 and Pdr3, two zinc finger transcription factors with partially overlapping roles, regulate more than half of all known pumps: Pdr5, Snq2, and Yor1 included. These regulators act through binding to conserved motifs in the promoters called PDR elements (4). Yrr1, another transcription factor responsive to a different set of stimuli controls the expression of Snq2 and Yor1 through binding of Yrr1-response element (YRRE) (13). Additional transcription regulators, such as Yap1 (yes-associated protein 1), Yrm1 (yeast reveromycin resistance modulator), and Msn2 (multicopy suppressor of SNF1 mutation), have each been demonstrated to play a role in cellular response to chemical, oxidative, and hypoxic stress (4). Although these transcriptional regulators are critical in determining the level of PDR expression, a detailed understanding of the entire regulatory cascade that causes xenobiotic stress to induce increased pump expression has yet to be developed. In particular, a specific compound or stress signal usually activates only a certain set of transporter genes, and for transporters with identical or highly similar promoter response elements, transcriptional response can still be quite different in response to the same stimulus (5, 14). The current set of known PDR regulators does not fully account for the specificity and diversity of PDR. Many factors that act upstream of and in conjunction with known PDR transcription factors are yet to be identified and characterized.
To search, on a genome-wide level, for additional proteins that participate in the transcriptional cascade responsible for drug sensing and transporter activation, we developed a novel biosensor system that couples the growth rate of yeast cells to the expression of a specific PDR transporter. We then applied this system to screens of the yeast homozygous-diploid deletion collection under drug treatment and identified deletion mutants that showed either diminished or enhanced PDR activation. Using this approach, we have discovered a series of genes with significant and previously uncharacterized roles in the modulation of the yeast PDR. Enriched among these hits are genes known to be involved in multiple areas of yeast biology, including regulation of cellular process and response to external stimuli, as well as cellular signaling and phospholipid metabolism.
Among the previously uncharacterized regulators of the PDR identified here are the Mad family of proteins involved in the mitotic SAC complex, the disruption of which has been explored as an anticancer strategy (15). We demonstrate that such disruption of mitotic spindles leads to elevated PDR response for certain compounds due to hyperactivation of transporters, causing cells in which Mad proteins are deleted to be significantly more resistant to several drugs.
This work establishes a novel chemical genomic means of interrogating transcriptional factors involved in PDR and applies this system to uncover multiple novel contributors to PDR regulation and a novel link between cell cycle regulation and PDR. These results not only increase our understanding of yeast biology but also provide novel targets for possible therapeutic intervention.
Results
Development of a biosensor system that couples PDR transporter expression to strain growth
One key challenge in dissecting the regulation of pleiotropic drug resistance in a high-throughput manner is linking the transcriptional response of PDR to a selectable phenotype, thereby allowing the system to be perturbed by genetic and chemical methods to determine the factors involved in PDR regulation. Toward this end, a plasmid-based biosensor that conferred a growth advantage to yeast cells upon exogenous chemical treatment and subsequent induction of transporter transcription was constructed (Fig. 1A). This system consists of a yeast CEN/ARS plasmid on which the promoter of the PDR transporter being investigated (PPDR) was cloned upstream of the gene for imidazole glycerol-phosphate dehydratase (his3), a protein essential for histidine biosynthesis, and the cyc1 terminator (TCYC1). The promoters tested include those of pdr5, snq2, and yor1, three of the best-characterized ABC transporters in yeast. Promoters were selected as the 1 kb immediately upstream of the start codon as, for each of these promoters, this region has been demonstrated sufficient for PDR-dependent expression (Table S1) (16–18). Strains transformed with biosensor constructs were grown in minimal defined media lacking histidine and containing varying concentrations of 3-amino-1,2,4-triazole (3-AT, 4), a potent and specific competitive inhibitor of His-3 (19). The inclusion of an inhibitory but sublethal concentration of 3-AT in the growth media allows the system to be tuned for the different expression levels of each promoter being examined.
Figure 1.
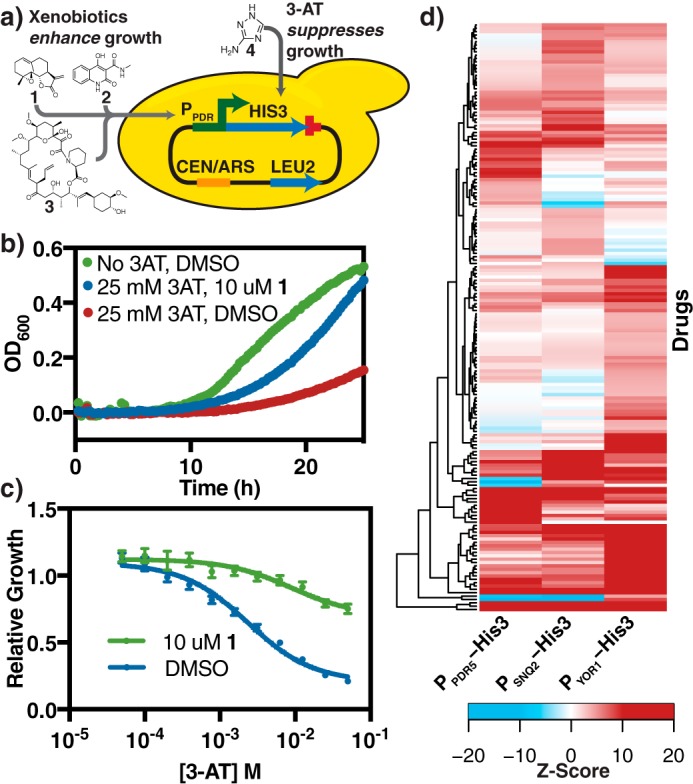
Construction of a biosensor system that couples PDR transporter expression and strain growth. a, schematic of the biosensor system and structures of inducing compounds used. The biosensor plasmids are outlined in Table S6. PPDR refers to promoters of PDR transporters pdr5, snq2, or yor1. Yeast strains transformed with these plasmids are grown in YNB media, without leucine and histidine. In addition, chemicals that induce the PDR response, such as parthenolide (1), cbf_5236571 (2), and FK506 (3) were added to the media. Media also contained 3-AT (4), a potent competitive inhibitor of His3 to further control the growth and dynamic range of the system. b, growth curve of JLY31, a PDR5 biosensor strain showing growth suppression by 4 (25 mm) and rescue by induction with 1 (10 μm). Curves represent the average of 4 biological replicates. c, dose-response of JLY31 under increasing concentrations of 4. Relative growth = AUC4/AUCno 4. Curves represent the best fit dose-response curve, and error bars represent S.D. (n = 4). These data are identical to Fig. S1a, whereas analogous plots for compounds 1-3 with all biosensor strains are shown in Fig. S1, b–i. d, heat map of drugs that significantly induces PDR transporters (Z ≥ 3 for one or more biosensor construct). A survey of 800 natural products on induction of the JLY31–33 biosensor strain was conducted. A Z-score was calculated to reflect the strain growth, normalized to quality control-adjusted growth of the same strain treated with DMSO.
The utility of this system was demonstrated by examining growth induction by a series of chemicals (Fig. 1B, Table 1, Fig. S1). In these experiments, a strain possessing the construct in which his3 expression was driven by PPDR5 (JLY31) shows significantly increased growth in the presence of 3-AT upon treatment with 10 μm parthenolide (1), a terpenoid natural product, as compared with DMSO control. This improved growth phenotype persisted in a dose-dependent manner across a range of 3-AT concentrations (Fig. 1C). We observed similar growth induction upon treatment with 1 in strains carrying PSNQ2 (JLY32) and PYOR1 (JLY33) biosensor constructs (Table 1, Fig. S1).
Table 1.
IC50 values for 4 in strains containing biosensor constructs under inducing (drug-treated) and non-inducing (DMSO vehicle) conditions
All inducers were added at 10 μm. Dose-response curves are displayed in Fig. S1.
| Drug | Promoter | IC50 3-AT (m) |
|
|---|---|---|---|
| Drug treated | DMSO vehicle | ||
| 1 | PPDR5 | 8.8 × 10−3 | 2.3 × 10−3 |
| 1 | PSNQ2 | 1.4 × 10−3 | 2.3 × 10−3 |
| 1 | PYOR1 | 5.7 × 10−3 | 2.2 × 10−4 |
| 2 | PPDR5 | 8.9 × 10−7 | 1.3 × 10−6 |
| 2 | PSNQ2 | 1.3 × 10−6 | 6.0 × 10−7 |
| 2 | PYOR1 | 5.4 × 10−4 | 8.8 × 10−5 |
| 3 | PPDR5 | 5.3 × 10−3 | 2.0 × 10−3 |
| 3 | PSNQ2 | 2.4 × 10−3 | 1.1 × 10−3 |
| 3 | PYOR1 | 5.0 × 10−4 | 1.8 × 10−4 |
Previous studies demonstrated that the transcriptional induction of the PDR can vary significantly from compound to compound. Notably, FK506 (3), an immunosuppressant, induced pdr5 and snq2 expression, whereas cbf_5236571 (2) specifically induced snq2 with little effect on pdr5 (20). This biosensor system recapitulated these results, with 3 inducing growth in all three systems tested, whereas 2 strongly induces only the PSNQ2 and PYOR1 systems with growth being slightly suppressed in the PPDR5 case (Table 1, Fig. S1).
To examine the generality of these results, we treated biosensor strains JLY31-33 with a library of 800 natural products (Fig. 1D, supporting Data). For each strain–drug combination, a Z-score was calculated to reflect the change in strain growth upon treatment with drug, normalized to quality control-adjusted growth (see “Experimental procedures”) of the same strain treated with DMSO. A high positive Z-score reflects a strong growth advantage under drug treatment and suggests the compound is a strong inducer of the specific PDR promoter. In contrast, a negative Z-score suggests a growth disadvantage, which can result from a combination of compound toxicity and little or no induction. We observed broad induction across varieties of compounds with 175 (22%) compounds leading to significant induction (Z>3) of at least one promoter. There was no apparent correlation between chemical structure and transporter induction for any of the three PDR promoters tested (Fig. S2). The varied induction profiles observed for each pump in response to structurally diverse natural products further underscores the complexity of PDR regulation.
Genome-wide, multiplexed interrogation of PDR reveals multiple candidate regulators of the PDR process
With a phenotypic screen for transporter induction established, we performed a genome-wide screen using the yeast deletion library to identify mutants that affect the transcriptional induction of PDR transporters. We transformed a barcoded yeast homozygous deletion collection with each of the PPDR5, PSNQ2, and PYOR1 biosensor constructs (pCH81–83). The pooled transformants were grown in media containing an inhibitory but sublethal concentration of 3-AT with either an inducing compound or DMSO as a control. The optimal concentrations of chemicals applied in each screen were determined through titrations that sampled biosensor response across a broad range of drug and 3-AT concentrations. After 6 generations of growth, the relative abundance of each deletion mutant in the treatment and control pools was quantified by microarray hybridization and analysis (Fig. 2A). A strain overrepresented in the treatment group has a positive-fold-change (fc) and the gene deleted in this mutant is, therefore, a putative down-regulator of the induction of the promoter being examined. Conversely, negative fc values identified putative up-regulators of pump induction. A total of 9 screens were conducted: PPDR5, PSNQ2, and PYOR1 biosensors each with compounds 1, 2, and 3 (Fig. 2B, supporting Data). These screens identified 314 hits, or deletions that were significantly different (log2(|fc|)>0.75 and a p < 0.01) in at least one of the conditions tested.
Figure 2.

Multiplexed interrogation of PDR regulators reveals multiple candidate regulators of the PDR process. a, schematic of the homozygous deletion collection screening with biosensor system. Cultures were harvested after 6 generations of growth and relative abundance of each mutant in the treatment and control conditions were quantified by microarray analysis. b, Venn diagrams representing the overlapping hits across the 3 biosensor systems for each of the three inducers (1–3). Numbers represent ORFs that were significantly up-regulated (red), down-regulated (blue), or contra-regulated (purple). c, heat map of deletion mutants for which a log2(|fc|)>0.75 and p < 0.01 for at least one condition. All data were calculated from 4 biological replicates.
Although many deletion mutants only met the significance cutoff in a single condition, a number of hits appear general, demonstrating a modulation of multiple PDR proteins with multiple inducers (Fig. 2C). We observed a relatively smaller number of hits in screening of PPDR5 with 2, and these hits have no overlap with PSNQ2 and PYOR1. This is to be expected as 2 does not induce a growth phenotype in the PPDR5 system (Table 1, Fig. S1d). This suggests that this system has a low false-positive rate and the false-positives observed do not overlap with hits in other experiments.
Notably, the screens identified many known PDR regulators and factors that mediate cellular response to chemicals and stress. Yrr1, a known transcriptional regulator of Snq2 and Yor1, was identified as a hit in the screen with 2 on these two transporters (21). Analysis of gene ontology enrichment of hits identified in these screens shows enrichment for terms such as regulation of response to stimulus, regulation of response to stress, and regulation of signaling, further demonstrating that the biosensor technology identified known elements of the PDR process (Table S2).
In addition to the known PDR-related pathways, several cellular processes were enriched in observed hits that were not previously associated with PDR, including mitotic SAC and cell cycle control, negative regulation of chromatin silencing, regulation of transcription, and regulation of primary metabolic process. To validate that the hits identified were truly modulating PDR induction and not artifacts of the biosensor system, we assayed the change in transcript levels upon xenobiotic treatment directly using quantitative PCR (qPCR). We focused the validation efforts on hits identified in two or more screens. Individual deletion mutants were treated for 1 h with 50 μm of the compound to be screened during exponential growth. qPCR assays were performed to determine the fold-induction of each PDR transporter (Fig. 3, Table S3). Deletion of an up-regulator is expected to lead to less PDR induction, and thus a relative induction value lower than 1, whereas down-regulator deletion mutants are expected to have values higher than 1. The results of these individual experiments correlate well with those found in the growth assays, suggesting that the growth assays are a viable proxy for transcription (Fig. 3, Table S3).
Figure 3.
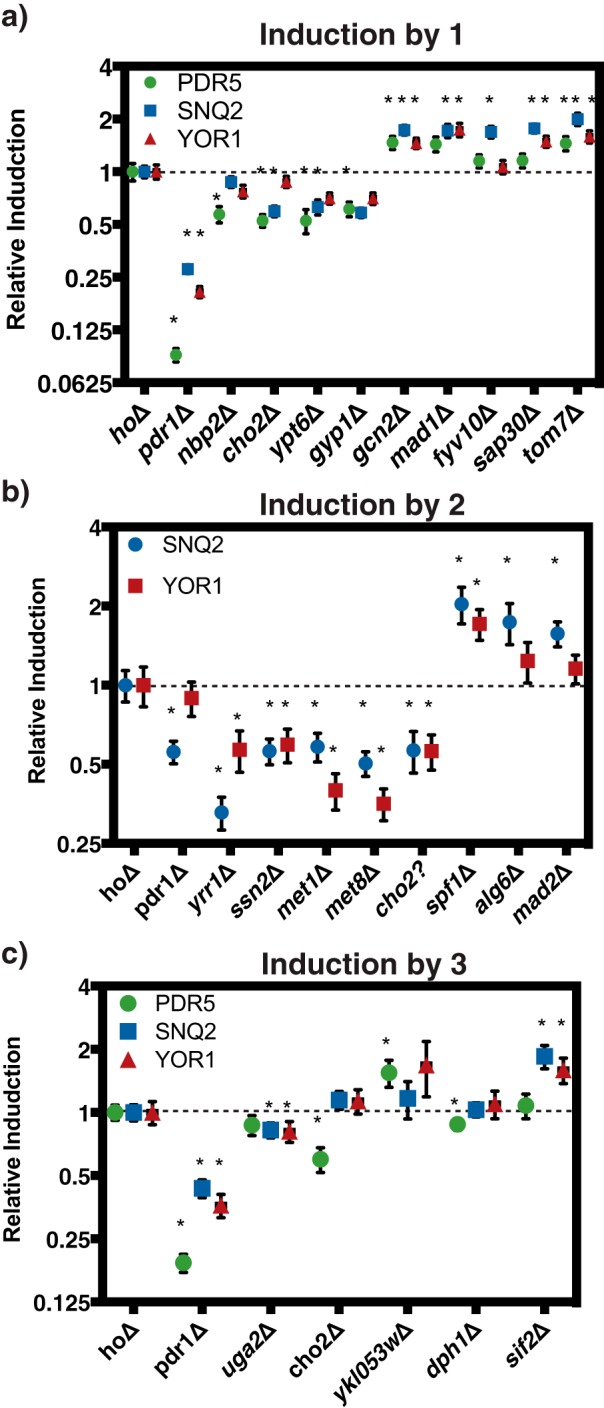
qPCR validation of PDR transcriptional responses confirms screen hits. Strains were grown to mid-exponential phase and treated with the indicated compounds at 50 μm for 1 h. Relative induction was calculated by comparing the fold-induction of the transporter gene between hoΔ and the deletion strain. Error bars represent S.D. (n = 3). *, indicates p < 0.05, based on Student's t test on the fold-induction of the transporter gene between hoΔ and the deletion strain. Treatment with (a) 1, (b) 2, and (c) 3.
These validated hits have diverse cellular functions, with many not previously linked to the PDR. The qPCR assays confirmed that known transcriptional up-regulators, such as Pdr1 for all three transporters and Yrr1 for Snq2 and Yor1, are important for PDR transcription induction, with their deletion leading to significant decreases in pump induction. Similarly, we observed similar reductions with the deletion of factors involved in phospholipid metabolism (Cho2), cell wall integrity (Nbp2), and RNA polymerase II mediator complex (Ssn2), validating their important role in up-regulating PDR transcription. Additionally, select genes involved in methionine metabolism also appear to behave as transcriptional up-regulators of the PDR process (Met-1, Met-8).
For several up-regulators, these data correlate well with large-scale drug screens that demonstrated that deletion of these genes lead to increased drug sensitivity. For example, an ssn2Δ mutant was shown to have decreased resistance to drugs such as benzopyrene, chitosan, and geldanamycin and a cho2Δ mutant was suggested to have decreased resistance to drugs such as tellurite, benomyl, and mycophenolic acid (22). Several membrane transporters appeared to act as down-regulators of PDR response, such as Tom7, a cellular transporter element involved in translocase of outer membrane complex, and Spf1, an endoplasmic reticulum membrane transporter important for intracellular membrane lipid composition. These qPCR results confirmed that the diverse groups of selected genes identified in the genome-wide screens are indeed regulators of the PDR process, and have established new connections between distinct cellular processes and PDR.
Disruption of spindle assembly checkpoint leads to elevated PDR activation
In both growth-based screens and subsequent qPCR validations, we observed previously uncharacterized involvement of cell cycle regulators in the PDR transcriptional response. In particular, the components of mitotic SAC, mad1–3, all appeared in growth screens and were validated by qPCR as down-regulators of the PDR process in response to exogenous drug treatment (Figs. 2 and 3). The mitotic SAC is a highly-conserved cell cycle surveillance mechanism that prevents abnormal chromosome segregation (23). Although the exact mechanism of MAD proteins remains unknown, these proteins are generally believed to form complexes that inhibit anaphase promoting complex (APC/C), an E3 ubiquitin ligase that leads to the degradation of multiple downstream cell-cycle proteins and enables sister-chromatid separation (24). Disruption of mitotic SAC leads to genome instability and has been implicated as the cause in many cancer cell lines (15) and inhibition of this complex has been proposed as a possible anticancer strategy. As deletion of genes for the MAD proteins leads to elevated PDR response, we set out to determine whether mad deletion strains were more resistant to chemical treatment and, if so, whether PDR plays a role in conferring the resistance.
To identify compounds suitable for this assay, we mined previous chemogenomic datasets measuring fitness changes of the same deletion collection screened here in response to treatment with 3,250 small molecules (25). We identified a collection of structurally diverse compounds that induced less of a fitness defect when MAD genes were deleted as compared with DMSO control. These compounds include: cbf_5328528 (5), paf C16 (6), k035-0031 (7), 0180-0423 (8), and N,N-dimethylsphingosine (9) (Fig. 4A). To avoid confounding results, we have verified that these compounds do not have any known connections to cell cycle. We monitored the growth of a reference hoΔ strain alongside the mad deletion strains, mad1Δ, mad2Δ, and mad3Δ treatment with 140 μm 5. The growth of all strains was significantly inhibited in this assay, but mad deletion strains grew better than hoΔ control (Fig. 4B). The dose-response curves of mad deletion strains when treated with 5 are shifted significantly to the right as compared with hoΔ, and mad deletion strains have a statistically significantly higher IC50 for 5, demonstrating that mad deletion strains are significantly more resistant to this compound (Fig. 4C, Table 2), consistent with the chemogenomic data. We observed a similar decrease in sensitivity of the MAD deletion mutants when treated with compounds 6-9. Next, we performed qPCR analysis on these samples to determine whether the transcription of PDR transporters was hyperactivated in the mad deletion strains. 5 induces all three transporters and is a strong inducer of pdr5 and yor1 (Fig. 4F). We observed that all three transporters are significantly more activated in mad deletion strains under 5 treatment (Fig. 4F), suggesting a potential PDR involvement in the observed resistance. Similarly, compounds 6-9 show increased induction of at least one pump in at least one of the mad deletion strains tested (Fig. 4F, Table S4).
Figure 4.

Disruption of spindle check point leads to elevated PDR activation. a, structures of compounds screened: cbf_5328528 (5), paf C-16 (6), k035-0031 (7), 0180-0423 (8), N,N-dimethylsphingosine (9). b, baseline corrected representative growth curve of hoΔ and mad deletion in BY4743 background under treatment with 140 μm 5. A600 were measured every 15 min over 30 h. c, dose-response of strains in b over increasing 5 concentrations. Relative growth = AUC5/AUCno drug. All wells contain the same DMSO concentration. AUC calculation was performed with a baseline corrected growth curve. The line represents best fit dose-response curve, and error bars represent S.D. (n = 3). d, baseline corrected representative growth curve of hoΔ and mad deletion in AD1–9 PDR transporter null background under 77.5 μm 5 treatment. e, dose-response of strains in d over increasing 5 concentrations. f, qPCR validation of transcription induction of all three transporters under the treatment of all five compounds in a. Methods are identical to those in Fig. 3. Error bars represent S.D. (n = 3). *, indicates p < 0.05.
Table 2.
IC50 in mad deletions strains in BY4743 and AD1–9 background
IC50 values were calculated based on best-fitting curve displayed. p values were calculated using a Student's t test with 3 biological replicates. Significant p values (<0.05) are italicized in bold.
| Drug | Strain | BY4743 |
AD1–9 |
||
|---|---|---|---|---|---|
| IC50 | p | IC50 | p | ||
| μm | μm | ||||
| 5 | hoΔ | 111.6 | 78.3 | ||
| mad1Δ | 140.2 | 0.009 | 86.3 | 0.216 | |
| mad2Δ | 138.3 | 0.015 | 90 | 0.286 | |
| mad3Δ | 137.3 | 0.018 | 88.1 | 0.18 | |
| 6 | hoΔ | 8.6 | 7.84 | ||
| mad1Δ | 11.7 | 0.018 | 7.32 | 0.52 | |
| mad2Δ | 12.4 | 0.014 | 7.02 | 0.327 | |
| mad3Δ | 11.6 | 0.021 | 5.86 | 0.144 | |
| 7 | hoΔ | 53.5 | 43.76 | ||
| mad1Δ | 80.5 | 0.013 | 41.11 | 0.637 | |
| mad2Δ | 73.6 | 0.031 | 45.94 | 0.737 | |
| mad3Δ | 80 | 0.013 | 39.71 | 0.426 | |
| 8 | hoΔ | 252.8 | 0.95 | ||
| mad1Δ | 359.9 | 0.007 | 0.59 | 1 | |
| mad2Δ | 345.3 | 0.02 | 0.59 | 0.47 | |
| mad3Δ | 326.6 | 0.014 | 0.63 | 0.917 | |
| 9 | hoΔ | 12.3 | 0.6 | ||
| mad1Δ | 16.5 | 0.021 | 8.64 | 0.909 | |
| mad2Δ | 16.4 | 0.019 | 7.9 | 0.533 | |
| mad3Δ | 19.9 | 0.005 | 7.17 | 0.176 | |
To ascertain if the increased expression of the PDR transporters is responsible for the increased resistance, we constructed the same hoΔ, mad1Δ, mad2Δ, and mad3Δ deletions in the AD1–9 background (named hoΔ AD, mad1Δ, 2Δ, 3Δ AD, respectively). AD1–9 is a strain in which the genes for 9 plasma-membrane–bound PDR transporters, including pdr5, snq2, and yor1, are knocked out (26). If the observed increased resistance is due to increased expression of PDR transporters, we expected that the increased resistance phenotype will not persist in this background.
This hypothesis was confirmed in repeating the growth inhibition experiment in AD1–9 background. In these strains, both ho and mad deletions strains are again inhibited by 5, but the mad deletion strains with transporters knocked out no longer exhibit the increased resistance (Fig. 4D). The 5 dose-response curves of mad deletion strains are not significantly different from that of hoΔ AD, and the IC50 values of these strains are also not significantly different, suggesting these strains have similar sensitivities to 5 (Fig. 4E, Table 2). It is also notable that AD1–9 strains are more sensitive to 5, suggesting a PDR involvement in the resistance to this compound (Fig. 4F). To ensure this observation is not specific to a particular compound, we performed the same set of experiments with compounds 6–9. In all cases, deletion of MAD genes strains led to elevated resistance and increased PDR transporter induction. This increased resistance is no longer observed when PDR transporters are knocked out in the AD1–9 (Fig. 4F, Table 2, Fig. S4). These results demonstrate that disruption of the mitotic SAC complex leads to elevated PDR activation and increased drug resistance, revealing a novel link between cell-cycle regulation and pleiotropic drug resistance.
Discussion
In this study, we developed a biosensor technology that couples transcriptional induction of yeast pleiotropic drug response to growth rate, an approach that builds upon previous approaches with fluorescence-based reporters (27) by allowing screening of mutant pools in competitive growth assays. This system specifically captured dose-dependent PDR transcriptional response induced by a variety of compounds. Pooled screens of the homozygous diploid yeast deletion collection identified 314 putative PDR regulators spanning a broad range of functional areas, including, but not limited to, response to chemical stimulus, lipid metabolism, translation, RNA metabolism, and cell cycle. We subsequently confirmed 20 genes that had been identified in multiple screens as transcriptional regulators of the PDR process by demonstrating that their deletion directly affects transcript abundance upon exogenous chemical treatment. In particular, we discovered mitotic spindle checkpoint factors, Mad1–3, as down-regulators of the PDR process. Deletion of these genes leads to hyperactivation of PDR transporters upon exogenous compound treatment, leading to elevated resistance.
The involvement of cell-cycle regulators in PDR response has not been previously established, but is consistent with several previous observations. It was initially proposed more than a hundred years ago that defects in proper chromosomal segregation are tumorigenic (28). Subsequent work suggested that mutations in the SAC mechanism are important causes for aneuploidy and cancer (23). Many of these cells with impaired SAC function showed increased resistance to mitotic inhibitors in survival or growth assays, both in artificially induced conditions or naturally occurring cancer cell lines (29, 30). For instance, in a study with multidrug-resistant colon cancer cell lines where the expression of Mad2 was suppressed by more than 50%, a significantly enhanced PDR expression level was observed, leading the authors to suggest that elevated PDR in these SAC-impaired cells is the reason for increased drug resistance (31).
This study builds upon these observations, demonstrating that PDR activation due to SAC impairment could be another mechanism that cancer cells, many of which have mutations of SACs, become resistant to drug treatment. Mitotic proteins have been long pursued as cancer drug targets (32). More recent development efforts have focused on kinesins such as CENP-E, KSP, and kinases such as Plk1, Aurora A and B, and Mps1. Among these new targets, targeting Aurora B and Mps1 are directly targeting SAC inhibition (32). These results suggest additional considerations when thinking of the SAC members as drug targets. Specifically, by restoring the function of Mad1–3, cancers cells may not only have better regulated mitosis and proliferation, but may also become less resistant to traditional chemotherapies.
In addition to those involved in cell-cycle control, this work identified multiple other previously uncharacterized PDR regulators, many of which participate in processes that have a known connection to the PDR. For example, we have noticed spf1 and tom7, two genes related to transportation channels on mitochondrial outer membrane, as PDR regulators (33, 34). Defects in these factors lead to mitochondrial damage, which can activate retrograde signaling. In a Pdr3-dependent manner, retrograde signaling has been shown to then induce transcriptional activation of PDR genes to facilitate transporting damaging species outside the cells (35).
Another factor that lies in the intersection of PDR and other pathways is Cho2, encoding the phosphatidylethanolamine methyltransferase in phospholipids synthesis, which is involved in the cell wall integrity (CWI) pathway (36). In response to certain chemical stress, such as organic solvents, PDR and cell wall integrity were activated in a coordinated fashion to cope with the stress, suggesting potential co-regulation and cross-talk between these stress response pathways (37). Identification of Ssn2, a subunit of RNA polymerase II mediator complex, and Gcn2, a kinase for α-subunit of eIF2, as regulators of the PDR process is not unexpected as transcriptional regulation can have significant impact on gene expression (38). In both human cell lines and C. albicans, chromatin-remodeling complex, which Ssn2 and Gcn2 interact with, was observed to control expression of PDR transporters (39, 40).
In this study, we have presented the genome-wide dissection of the yeast PDR using a series of biosensors responsive to a variety of diverse small molecules. Through this work, we have confirmed the role of several proteins in PDR regulation and identified multiple additional factors with significant but previously uncharacterized regulatory roles. Furthermore, we have demonstrated that, in the presence of several toxic compounds, disruption of mitotic spindle checkpoint assembly leads to elevated PDR and increased resistance. These results not only demonstrate the biosensor system as a viable tool to investigate PDR, but also uncover novel control of the process and a connection to cell-cycle regulation.
Experimental procedures
Media and growth conditions
Two S. cerevisiae strains were primarily used in this study: BY4743 (MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0) and AD1–9 (MATα, pdr1–3, ura3, his1, Δyor1::hisG, Δsnq2::hisG, pdr5-Δ2::hisG, Δpdr10::hisG, Δpdr11::hisG, Δycf1::hisG, pdr3-Δ2::hisG, Δpdr15::hisG, pdr1-Δ3::hisG) (26). Yeast was grown in YNB (yeast nitrogen base) media in all experiments. YNB media contains 1.7 g/liter of yeast nitrogen base (MP Biomedicals, catalog number 114027512), 5 g/liter of ammonium sulfate, with selected amino acids based on specific experiments in the following concentrations: 20 mg/liter of histidine, 60 mg/liter of leucine, and 20 mg/liter of uracil. This media was further supplemented with 3-AT at various concentrations as indicated in the experiments.
Chemical reagents
The Microsource Pure Natural Products library (MSNP) was purchased from Discovery Systems. Parthenolide, 3-AT, paf C-16, and N,N-dimethylsphingosine were purchased from Sigma (catalog numbers P0667, 61-82-5, P4904, and SML0311, respectively). FK506 was purchased from Enzo Life Sciences (catalog number ALX-380-008). Cbf_5236571 and cbf_5328528 were purchased from ChemBridge. K035-0031 was purchased from Enamine (catalog number 50-138-184). 0180-0423 was purchased from ChemDiv.
Growth rate analysis of isogenic cultures and generation of dose-response curves
All growth experiments were conducted with GENios microplate reader system (Tecan) in which optical density (A600) was measured every 15 min over the entire course of the experiment. For experiments with isogenic strains, all growth assays were conducted in Nunc MicroWel 96-Well Microplates (Thermo Scientific) with 100 μl in each well. Pooled screens were conducted in Corning Costar 48-well cell culture plate (Sigma). Strains were inoculated into YNB media overnight, diluted into fresh media to allow for additional growth for 4–6 h, when they are in exponential growth, and then diluted to A600 = 0.01 to start the growth assays. The indicated concentrations of various drugs were added to plates using a Tecan D300e Digital Dispenser.
Relative growth was determined as previously reported (20). Briefly, A600 was measured every 15 min during the experiment. The mean of the first 10 measurements was used to baseline correct all subsequent measurements. Unless otherwise stated, all AUC measurements reported here are the sum of the baseline corrected A600 values over a 30-h period.
For dose-response curves, relative growth values for each condition were computed to compare the rate of growth of the same strain under the drug treatment versus no drug/DMSO control: relative growth = AUCdrug/AUCDMSO. The volumes of DMSO vehicle and drug solution were equivalent in all treatment versus control sample pairs. Modeling was completed in GraphPad Prism version 7.0 for Windows (GraphPad Software, La Jolla, CA), following standard practice.
Screen of MSNP collection
BY4743 hoΔ, was transformed with each of pCH81, pCH82, and pCH82 (Table S7). Strains were grown to stationary phase overnight in minimal media overnight and diluted to A600 = 0.01 into minimal media containing an inhibitory concentration of 3-AT (100 mm for strains with pCH81, 50 mm for strains with pCH82, and 2 mm for strains containing pCH83). 100 μl of diluted culture was placed into each well of a 96-well plate. 1 μl of each compound from all 800 compounds from the MSNP collection was inoculated into the growth media, leading to 1:100 dilution from the stock concentration. A600 was monitored every 15 min for 30 h. Each 96-well plate included 16 DMSO controls. Compound induction was quantified by calculating the following Z-score: Z = [AUCdrug − AUCmean(DMSO)]/AUCS.D.(DMSO). Compounds with a Z-score >3 were deemed to be significant inducers.
Screening of yeast homozygous deletion collection
To construct a homozygous deletion collection that contains the biosensor constructs, pCH81-83 were transformed into the pooled S. cerevisiae deletion collection using a standard lithium acetate protocol (41, 42). Each transformation resulted in ∼105 transformants that were subsequently pooled together. Pooled transformants were grown overnight to saturation and diluted to A600 = 0.01. Diluted cultures were left to recover for 4 h prior to challenge with both inducer and 3-AT, which were then added in the following concentrations: 200 μm 3-AT with 10 μm 1 for all three promoters, 200 μm 3-AT and 10 μm 2 for all three promoters, 800 μm 3-AT and 10 μm 3 for PPDR5, 1.6 mm 3-AT and 10 μm 3 for PSNQ2, and 320 μm 3-AT and 10 μm 3 for PYOR1. 700 μl of culture was grown in each well of a 48-well plate at 30 °C with orbital shaking in Infinite plate readers (Tecan).
Growth of the pooled culture was monitored every 15 min. To maintain cultures in log phase throughout the growth experiment, after 3 generations of competitive growth, cultures were diluted to A600 = 0.075, and grown for a further 3 generations after which 600 μl of culture was harvested saved to a 4 °C cooling station (Torrey Pines). This amounted to ∼6 culture doublings, or 6 generations of growth, from the beginning of the experiment. Pipetting events were triggered automatically by Pegasus Software and performed by a Freedom EVO work station (Tecan).
Genomic DNA from the pools was prepared using a YeastStar genomic DNA prep (Zymo Research) and deletion collection barcodes were amplified as previously reported (19). Barcode quantitation was performed using the Genflex Tag 16K Array v2 chip (Affymetrix), following standard procedures (41, 42). Relative abundance of each barcode in induced versus control conditions treated with an equivalent amount of DMSO was used to determine induction of the promoter being examined, as previously described (20). All p values were determined by a Student's t test for 4 biological replicates.
Quantitative RT-qPCR experiments
In experiments examining the transcriptional response of PDR promoters, individual strains were grown to saturation in minimal media overnight. Cultures were then diluted to A600 = 0.2 in fresh minimal media and grown for a further 4 h. Cells were then treated with drugs at the inducing concentration (Figs. 3 and 4f) for 1 h. 2 OD-equivalents of cells were then harvested for whole cell RNA extraction and DNA removal using a RiboPure RNA Purification Kit (ThermoFisher, catalog number AM1926). Purified RNA samples were reverse transcribed to single-stranded cDNA using High Capacity RNA-to-cDNA Kit (ThermoFisher, catalog number 4387406). These cDNA samples were used as template for quantitative PCR experiments. These qPCR included SYBR Green PCR Master Mix (ThermoFisher, catalog number 4309155), and primers at 5 μm concentrations. Primers were designed with the primer3 software package and tested as described previously (20). They are expected to yield ∼120 bp amplicons for PDR promoters and ACT1 (control). ACT1 was chosen as the control transcript for all experiments as its expression level was extremely stable across all conditions. qPCR were performed on a 7900HT Fast Real-Time PCR System (ThermoFisher) in 384-well format. qPCR data were analyzed using software integrated to the 7900HT system using automatic threshold determination. ΔCt values for each transport gene were calculated for each set of samples by comparing the difference between the Ct value of the same sample treated with drug versus DMSO control: ΔCttransporter = Ctdrug − CtDMSO. This ΔCttransporter value is then normalized for cell numbers by comparing it with ΔCtACT1: ΔΔCttransporter = ΔCtACT1 − ΔCttransporter. Student's t tests were applied on the ΔΔCt level by comparing the magnitude of ΔΔCt in a deletion strain to that in hoΔ control. In Figs. 3 and 4, a ΔΔΔCttransporter was further calculated by computing the difference between ΔΔCttransporter in a specific deletion strain and that in the hoΔ strain: ΔΔΔCttransporter, deletion strain = ΔΔCttransporter, deletion strain − ΔΔCttransporter, hoΔ. All fold-change values were calculated as the log2 of the ratio of the drug treatment condition: DMSO vehicle and p values are determined based on a Student's t test with the specified number of biological replicates.
Construction of AD1–9 deletion strains
AD1–9 deletion background strain was a previous creation with all transporters individually knocked out (26). Furthermore, ho, Mad1–3 genes are knocked out in this background using genetic constructs and methods that were used to create the yeast deletion collection (43). Briefly, primer pairs that amplify regions that encode Δmad1–3::KanMX4 or Δho::KanMX4 respective BY4743 deletion strains were used to amplify knockout cassettes, with homologous regions on both end of the amplicon. This amplicon was then transformed into the AD1–9 background strain, and the transformants were selected for kanamycin resistance, followed by sequencing of the targeted region to confirm integration identity. The resulting stains contain Δmad1–3::KanMX4 or Δho::KanMX4 on AD1–9 background. Processed data are provided in the supporting Data.
Author contributions
J. L., K. K., U. S., R. W. D., M. E. H., and C. J. H. conceptualization; J. L., K. K., U. S., R. P. S. O., and C. J. H. data curation; J. L., K. K., U. S., R. P. S. O., and C. J. H. formal analysis; J. L. and C. J. H. supervision; J. L. and C. J. H. validation; J. L., K. K., U. S., R. P. S. O., A. M. A., J. H., R. W. D., and C. J. H. investigation; J. L. and K. K. visualization; J. L., K. K., R. P. S. O., A. M. A., J. H., R. W. D., and C. J. H. methodology; J. L., K. K., and C. J. H. writing-original draft; J. L., R. W. D., M. E. H., and C. J. H. project administration; J. L., K. K., R. P. S. O., A. M. A., M. E. H., and C. J. H. writing-review and editing; R. P. S. O. and A. M. A. resources; R. P. S. O., A. M. A., R. W. D., M. E. H., and C. J. H. funding acquisition.
Supplementary Material
Acknowledgment
We thank Angela Chu for thoughtful discussion and help in deletion collection.
This work was supported by National Institutes of Health Grant U01 GM110706. U. S., C. J. B. H., and M. E. H. own shares in Hexagon Bio. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–3, Tables S1–S7, and data.
All microarray data from genome-wide screens are available from EMBL-EBI ArrayExpress under accession number E-MTAB-6655.
- MDR
- multidrug resistance
- PDR
- pleiotropic drug resistance
- 3-AT
- 3-amino-1,2,4-triazole
- Fc
- fold-change
- qPCR
- quantitative PCR
- YNB
- yeast nitrogen base
- AUC
- area under curve.
References
- 1. Nikaido H. (2009) Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146 10.1146/annurev.biochem.78.082907.145923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kathawala R. J., Gupta P., Ashby C. R. Jr., and Chen Z.-S. (2015) The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist. Updat. 18, 1–17 10.1016/j.drup.2014.11.002 [DOI] [PubMed] [Google Scholar]
- 3. Prasad R., and Goffeau A. (2012) Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu. Rev. Microbiol. 66, 39–63 10.1146/annurev-micro-092611-150111 [DOI] [PubMed] [Google Scholar]
- 4. Paul S., and Moye-Rowley W. S. (2014) Multidrug resistance in fungi: regulation of transporter-encoding gene expression. Front. Physiol. 5, 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piecuch A., and Obłlk E. (2014) Yeast ABC proteins involved in multidrug resistance. Cell Mol. Biol. Lett. 19, 1–22 10.2478/s11658-013-0111-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gulshan K., and Moye-Rowley W. S. (2007) Multidrug resistance in fungi. Eukaryot. Cell 6, 1933–1942 10.1128/EC.00254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Decottignies A., Lambert L., Catty P., Degand H., Epping E. A., Moye-Rowley W. S., Balzi E., and Goffeau A. (1995) Identification and characterization of SNQ2, a new multidrug ATP binding cassette transporter of the yeast plasma membrane. J. Biol. Chem. 270, 18150–18157 10.1074/jbc.270.30.18150 [DOI] [PubMed] [Google Scholar]
- 8. Boujaoude L. C., Bradshaw-Wilder C., Mao C., Cohn J., Ogretmen B., Hannun Y. A., and Obeid L. M. (2001) Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J. Biol. Chem. 276, 35258–35264 10.1074/jbc.M105442200 [DOI] [PubMed] [Google Scholar]
- 9. Gillet J.-P., and Gottesman M. M. (2010) Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 596, 47–76 10.1007/978-1-60761-416-6_4 [DOI] [PubMed] [Google Scholar]
- 10. Denning D. W., and Bromley M. J. (2015) How to bolster the antifungal pipeline. Science 347, 1414–1416 10.1126/science.aaa6097 [DOI] [PubMed] [Google Scholar]
- 11. Shekhar-Guturja T., Gunaherath G. M., Wijeratne E. M., Lambert J.-P., Averette A. F., Lee S. C., Kim T., Bahn Y.-S., Tripodi F., Ammar R., Döhl K., Niewola-Staszkowska K., Schmitt L., Loewith R. J., Roth F. P., et al. (2016) Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat. Chem. Biol. 12, 867–875 10.1038/nchembio.2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown G. D., Denning D. W., Gow N. A., Levitz S. M., Netea M. G., and White T. C. (2012) Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv13 [DOI] [PubMed] [Google Scholar]
- 13. Le Crom S., Devaux F., Marc P., Zhang X., Moye-Rowley W. S., and Jacq C. (2002) New insights into the pleiotropic drug resistance network from genome-wide characterization of the YRR1 transcription factor regulation system. Mol. Cell Biol. 22, 2642–2649 10.1128/MCB.22.8.2642-2649.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghosh M., Shen J., and Rosen B. P. (1999) Pathways of As(III) detoxification in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 96, 5001–5006 10.1073/pnas.96.9.5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kops G. J., Weaver B. A., and Cleveland D. W. (2005) On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 5, 773–785 10.1038/nrc1714 [DOI] [PubMed] [Google Scholar]
- 16. Katzmann D. J., Hallstrom T. C., Mahé Y., and Moye-Rowley W. S. (1996) Multiple Pdr1p/Pdr3p binding sites are essential for normal expression of the ATP binding cassette transporter protein-encoding gene PDR5. J. Biol. Chem. 271, 23049–23054 10.1074/jbc.271.38.23049 [DOI] [PubMed] [Google Scholar]
- 17. Cui Z., Hirata D., and Miyakawa T. (1999) Functional analysis of the promoter of the yeast SNQ2 gene encoding a multidrug resistance transporter that confers the resistance to 4-nitroquinoline N-oxide. Biosci. Biotechnol. Biochem. 63, 162–167 10.1271/bbb.63.162 [DOI] [PubMed] [Google Scholar]
- 18. Hallstrom T. C., and Moye-Rowley W. S. (1998) Divergent transcriptional control of multidrug resistance genes in Saccharomyces cerevisiae. J. Biol. Chem. 273, 2098–2104 10.1074/jbc.273.4.2098 [DOI] [PubMed] [Google Scholar]
- 19. James P., Halladay J., and Craig E. A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlecht U., Miranda M., Suresh S., Davis R. W., and St Onge R. P. (2012) Multiplex assay for condition-dependent changes in protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 9213–9218 10.1073/pnas.1204952109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X., Cui Z., Miyakawa T., and Moye-Rowley W. S. (2001) Cross-talk between transcriptional regulators of multidrug resistance in Saccharomyces cerevisiae. J. Biol. Chem. 276, 8812–8819 10.1074/jbc.M010686200 [DOI] [PubMed] [Google Scholar]
- 22. Cherry J. M., Hong E. L., Amundsen C., Balakrishnan R., Binkley G., Chan E. T., Christie K. R., Costanzo M. C., Dwight S. S., Engel S. R., Fisk D. G., Hirschman J. E., Hitz B. C., Karra K., Krieger C. J., et al. (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–D705 10.1093/nar/gkr1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bharadwaj R., and Yu H. (2004) The spindle checkpoint, aneuploidy and cancer. Oncogene 23, 2016–2027 10.1038/sj.onc.1207374 [DOI] [PubMed] [Google Scholar]
- 24. Musacchio A. (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 25, 3017 10.1016/j.cub.2015.10.050,10.1016/j.cub.2015.10.043 [DOI] [PubMed] [Google Scholar]
- 25. Lee A. Y., St. Onge R. P., Proctor M. J., Wallace I. M., Nile A. H., Spagnuolo P. A., Jitkova Y., Gronda M., Wu Y., Kim M. K., Cheung-Ong K., Torres N. P., Spear E. D., Han M. K., Schlecht U., et al. (2014) Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science 344, 208–211 10.1126/science.1250217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rogers B., Decottignies A., Kolaczkowski M., Carvajal E., Balzi E., and Goffeau A. (2001) The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 3, 207–214 [PubMed] [Google Scholar]
- 27. Yibmantasiri P., Bircham P. W., Maass D. R., Bellows D. S., and Atkinson P. H. (2014) Networks of genes modulating the pleiotropic drug response in Saccharomyces cerevisiae. Mol. Biosyst. 10, 128–137 10.1039/C3MB70351G [DOI] [PubMed] [Google Scholar]
- 28. Calkins G. N. (1914) Zur Frage der Entstehung maligner Tumoren. Science 40, 857–859 10.1126/science.40.1041.857 [DOI] [Google Scholar]
- 29. Yamada H. Y., and Gorbsky G. J. (2006) Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol. Cancer Ther. 5, 2963–2969 10.1158/1535-7163.MCT-06-0319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan K.-S., Koh C.-G., and Li H.-Y. (2012) Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 3, e411 10.1038/cddis.2012.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vasudevan S., Thomas S. A., Sivakumar K. C., Komalam R. J., Sreerekha K. V., Rajasekharan K. N., and Sengupta S. (2015) Diaminothiazoles evade multidrug resistance in cancer cells and xenograft tumour models and develop transient specific resistance: understanding the basis of broad-spectrum versus specific resistance. Carcinogenesis 36, 883–893 10.1093/carcin/bgv072 [DOI] [PubMed] [Google Scholar]
- 32. Salmela A.-L., and Kallio M. J. (2013) Mitosis as an anti-cancer drug target. Chromosoma 122, 431–449 10.1007/s00412-013-0419-8 [DOI] [PubMed] [Google Scholar]
- 33. Vashist S., Frank C. G., Jakob C. A., and Ng D. T. (2002) Two distinctly localized P-type ATPases collaborate to maintain organelle homeostasis required for glycoprotein processing and quality control. Mol. Biol. Cell 13, 3955–3966 10.1091/mbc.02-06-0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neupert W., and Herrmann J. M. (2007) Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749 10.1146/annurev.biochem.76.052705.163409 [DOI] [PubMed] [Google Scholar]
- 35. Gulshan K., Schmidt J. A., Shahi P., and Moye-Rowley W. S. (2008) Evidence for the bifunctional nature of mitochondrial phosphatidylserine decarboxylase: role in Pdr3-dependent retrograde regulation of PDR5 expression. Mol. Cell. Biol. 28, 5851–5864 10.1128/MCB.00405-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levin D. E. (2005) Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69, 262–291 10.1128/MMBR.69.2.262-291.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishida N., Jing D., Kuroda K., and Ueda M. (2014) Activation of signaling pathways related to cell wall integrity and multidrug resistance by organic solvent in Saccharomyces cerevisiae. Curr. Genet. 60, 149–162 10.1007/s00294-013-0419-5 [DOI] [PubMed] [Google Scholar]
- 38. Smith E., and Shilatifard A. (2010) The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 40, 689–701 10.1016/j.molcel.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dubey R., Lebensohn A. M., Bahrami-Nejad Z., Marceau C., Champion M., Gevaert O., Sikic B. I., Carette J. E., and Rohatgi R. (2016) Chromatin-remodeling complex SWI/SNF controls multidrug resistance by transcriptionally regulating the drug efflux pump ABCB1. Cancer Res. 76, 5810–5821 10.1158/0008-5472.CAN-16-0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lopes da Rosa J., and Kaufman P. D. (2012) Chromatin-mediated Candida albicans virulence. Biochim. Biophys. Acta 1819, 349–355 10.1016/j.bbagrm.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gietz R. D., and Schiestl R. H. (2007) Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 38–41 10.1038/nprot.2007.15 [DOI] [PubMed] [Google Scholar]
- 42. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 10.1038/nature00935 [DOI] [PubMed] [Google Scholar]
- 43. Deutschbauer A. M., Jaramillo D. F., Proctor M., Kumm J., Hillenmeyer M. E., Davis R. W., Nislow C., and Giaever G. (2005) Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 169, 1915–1925 10.1534/genetics.104.036871 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.