

Abstract
AS160 and Tbc1d1 are key Rab GTPase–activating proteins (RabGAPs) that mediate release of static GLUT4 in response to insulin or exercise-mimetic stimuli, respectively, but their cooperative regulation and its underlying mechanisms remain unclear. By employing GLUT4 nanometry with cell-based reconstitution models, we herein analyzed the functional cooperative activities of the RabGAPs. When both RabGAPs are present, Tbc1d1 functionally dominates AS160, and stimuli-inducible GLUT4 release relies on Tbc1d1-evoking proximal stimuli, such as AICAR and intracellular Ca2+. Detailed functional assessments with varying expression ratios revealed that AS160 modulates sensitivity to external stimuli in Tbc1d1-mediated GLUT4 release. For example, Tbc1d1-governed GLUT4 release triggered by Ca2+ plus insulin occurred more efficiently than that in cells with little or no AS160. Series of mutational analyses revealed that these synergizing actions rely on the phosphotyrosine-binding 1 (PTB1) and calmodulin-binding domains of Tbc1d1 as well as key phosphorylation sites of both AS160 (Thr642) and Tbc1d1 (Ser237 and Thr596). Thus, the emerging cooperative governance relying on the multiple regulatory nodes of both Tbc1d1 and AS160, functioning together, plays a key role in properly deciphering biochemical signals into a physical GLUT4 release process in response to insulin, exercise, and the two in combination.
Keywords: glucose transporter type 4 (GLUT4), GTPase activating protein (GAP), membrane trafficking, microscopic imaging, signal transduction, insulin, AMP-activated kinase (AMPK), Akt PKB
Introduction
Two Tbc1d-family Rab GTPase-activating proteins (RabGAPs),2 the Akt substrate of 160 kDa (AS160)/Tbc1d4 (hereafter referred to as AS160) and Tbc1d1, have been suggested to be key regulators of insulin- and exercise-mediated redistribution of the glucose transporter GLUT4 from its intracellular storage compartment(s) to the cell surface (1, 2). These two proteins share ∼50% identical primary sequences with similar domain structures, including two phosphotyrosine-binding (PTB) domains, a putative calmodulin-binding domain (CBD), and a GAP domain (3–5). They also share multiple Akt phosphorylation sites, but Tbc1d1 has a key AMPK phosphorylation site (Ser237 in humans), which apparently contributes to the unique regulatory aspects of Tbc1d1 (2, 6). Another notable difference between these two proteins is tissue distribution (i.e. AS160 is expressed in multiple differentiated tissues, including both adipocytes and skeletal muscles, whereas Tbc1d1 is highly expressed in skeletal muscles) (7, 8). Thus, skeletal muscle cells express both AS160 and Tbc1d1, and GLUT4 regulation in skeletal muscle cells might therefore be more complex than that in adipocytes. Although previous biochemical observations suggested that Tbc1d1 actions dominate AS160 actions because ectopic expression of Tbc1d1 strongly inhibits insulin-responsive GLUT4 translocation in 3T3-L1 adipocytes, the functional cooperative activities of these two RabGAPs are unclear.
We previously determined the respective functional roles of the two RabGAPs in intracellular GLUT4 behavior, employing a novel GLUT4 nanometry approach, which is based on single molecule imaging with Quantum dot (QD) fluorescent nanocrystals, combined with cell-based reconstitution models of GLUT4 behavior (9, 10). By ectopically and independently expressing either AS160 or Tbc1d1, we confirmed that each of these proteins has the capability to independently trigger stimuli-responsive GLUT4 liberation processes from its static states, which is the first step necessary for initiating the entire GLUT4-trafficking itinerary. However, the proximal upstream signals for triggering the liberation process differed markedly between these two RabGAPs. AS160 governs insulin-responsive liberation in an Akt-mediated phosphorylation-dependent manner (10). In contrast, Tbc1d1 has at least two distinct regulatory modes, AMPK-responsive and insulin-responsive, and this protein temporarily acquires the insulin-responsive ability for GLUT4 liberation after exercise-mimetic stimuli, such as AICAR pretreatment or increases in cytosolic Ca2+ concentrations (“regulatory mode shift” of Tbc1d1) (9). Importantly, mutants of the PTB1 domain of Tbc1d1, including an obesity-related R125W (11, 12), failed to undergo the regulatory mode shift for insulin responsiveness acquisition, indicating the necessity of this domain in the process. These clear determinations of the respective functional roles of AS160 and Tbc1d1 in GLUT4 liberation allowed us to explore the functional cooperative activities between these two RabGAPs. Therefore, in the present study, we performed detailed analyses of GLUT4 behavior, focusing especially on GLUT4 liberation processes, in the co-presence of AS160 and Tbc1d1 to reveal functional cooperation between these two proteins.
Results
GLUT4 behavior in the co-presence of AS160 and Tbc1d1
To analyze GLUT4 behavior in the co-presence of AS160 and Tbc1d1 in detail, we herein took advantage of the cell-based reconstitution model employing 3T3-L1 fibroblasts exogenously expressing Myc-GLUT4, HA-sortilin, and AS160 and Tbc1d1, the first two alone and the other two in combination (Fig. 1A). We labeled Myc-GLUT4 with QD-conjugated anti-Myc antibodies, and after internalization we tracked individual QD fluorescence. We evaluated intracellular GLUT4 behavior based on the movement speed of GLUT4 molecules at 5, 15, and 30 min after each stimulus. Because our QD-based single molecule analysis allows us to quantitatively describe the “states” of the GLUT4 behavior (e.g. static or released) with an overall average of many GLUT4 molecules in certain cells under a specific treatment condition (e.g. before or after insulin stimulation), as described previously (13), continuous tracking of the same GLUT4 molecules throughout the period of delivery of stimuli is not required. We first confirmed our previous observations in cells expressing either AS160 or Tbc1d1. Insulin can liberate static GLUT4, leading to faster movement in cells expressing AS160 but not Tbc1d1 within 5 min (Fig. 1B (left) and Fig. S1 (top)), whereas the AMPK activator AICAR can liberate static GLUT4 in cells expressing Tbc1d1 only at a later time point (15 min) (Fig. 1B (middle) and Fig. S1 (middle)). When these two RabGAPs coexist, only AICAR (i.e. not insulin) exerts GLUT4-releasing activity (Fig. 1B (right) and Fig. S1 (bottom)). We also analyzed GLUT4 behavior in differentiated 3T3-L1 adipocytes, which endogenously express abundant AS160 but less Tbc1d1, and found similar tendencies in the GLUT4 behaviors in response to insulin and AICAR (Fig. S2) (i.e. insulin liberates static GLUT4 in control adipocytes as described previously (Fig. S2, top), but in adipocytes expressing only HaloTag-Tbc1d1 (by electroporating Tbc1d1 plasmid and AS160 siRNA), AICAR but not insulin liberates static GLUT4 (Fig. S2, middle)). In addition, in adipocytes expressing both AS160 and Tbc1d1 (by electroporating the HaloTag-Tbc1d1 plasmid), AICAR, but not insulin, tends to exert GLUT4-releasing activity in adipocytes exogenously expressing Tbc1d1 (Fig. S2, bottom). These results suggest that Tbc1d1 plays a dominant role in the liberation of static GLUT4 when both RabGAPs are present. Diffusion coefficient maps further illustrate the released status of GLUT4 molecules under each experimental condition (Fig. 1C).
Figure 1.
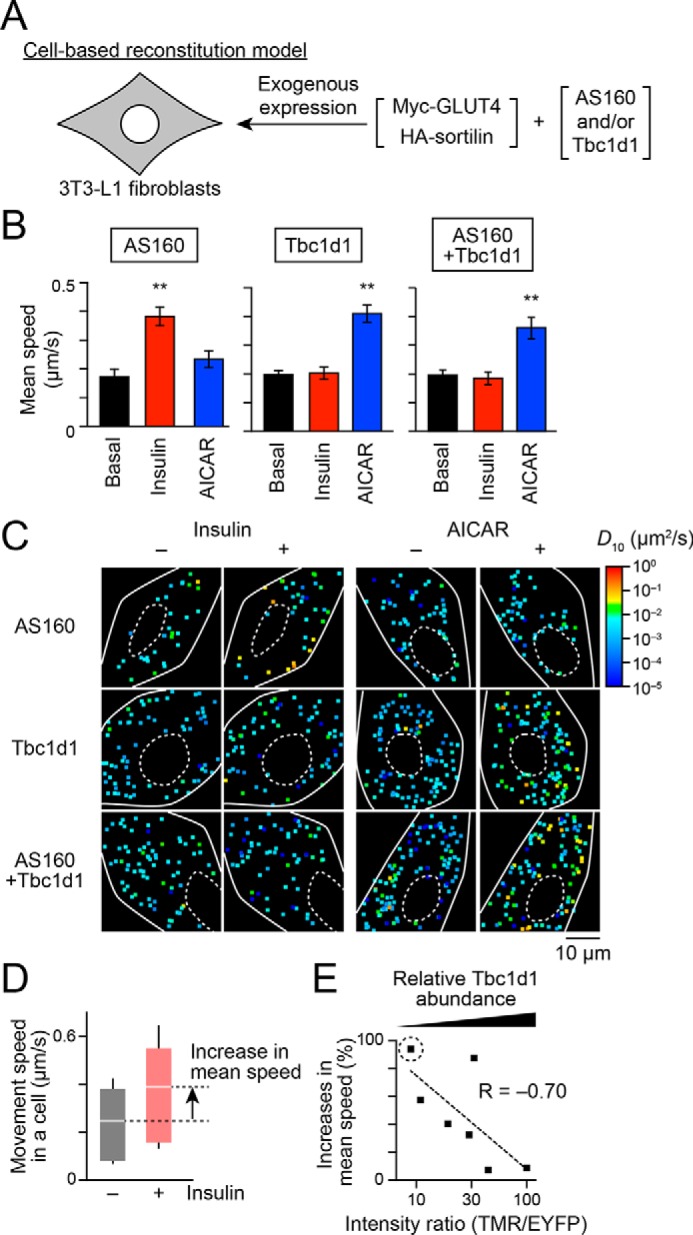
Dominant actions of Tbc1d1 in insulin-responsive GLUT4 liberation in cells co-expressing AS160 and Tbc1d1. A, cell-based reconstitution model. In this study, 3T3-L1 fibroblasts exogenously expressing Myc-GLUT4, HA-sortilin, and both Tbc1d1 and AS160, or either one alone, were used. B, mean speed of intracellular movement of GLUT4 under the indicated conditions in cells expressing Tbc1d1 (left), AS160 (middle), or both (right). The cells were simulated without (black) or with insulin (red; 100 nm) or AICAR (blue; 1 mm) for 30 min. **, p < 0.01 by Dunnett's multiple comparison versus basal states (n = 6–8). C, representative diffusion coefficient maps of GLUT4 movement in cells under the indicated conditions. D, box plots of GLUT4 movement speeds in a cell expressing EYFP-AS160 and HaloTag-Tbc1d1 before (black) and after (red) insulin stimulation (100 nm, 30 min). HaloTag-Tbc1d1 was stained with HaloTag TMR ligand, and epifluorescence of TMR and EYFP was acquired just before analyzing single-molecule GLUT4 behavior. The ratio of HaloTag-Tbc1d1 to EYFP-AS160 was calculated on the basis of their fluorescence intensities. We also calculated the percentage increase in the mean movement speed of GLUT4 in response to insulin stimulation (arrow). Intensity ratio (TMR/EYFP) and percentage increase in the movement speed in this cell were 9.4 and 94.1%, respectively. White lines and error bars represent mean and S.D., respectively. E, relationship between insulin-responsive GLUT4 liberation and relative Tbc1d1 abundance to that of AS160. Each dot represents a cell. A dot surrounded by a dotted circle represents the data shown in D.
To further explore this observation, we next analyzed the relationships between the intensity ratio of the two exogenously expressed RabGAPs and actual executive functions (insulin-responsive liberation of static GLUT4). We herein estimated the relative ratio of exogenously expressed Tbc1d1 to AS160 in cells co-expressing EYFP-AS160 and HaloTag-Tbc1d1 at various ratios (Fig. S3). The cells were labeled with QD-conjugated anti-Myc antibodies, followed by staining with HaloTag TMR ligand. Epifluorescence of EYFP and TMR was acquired, and single-molecule analyses of GLUT4 behavior within the same cell were then performed. We also prepared cells expressing either HaloTag-Tbc1d1 or EYFP-AS160 to correct the bleed-through of the fluorescence (see “Experimental procedures” for details). With this approach, we cannot estimate the abundance ratio at the specific region in the cells due to time-to-time changes in the ratio at that point. However, in the present study, such apparently rough estimation of the ratio would be more suitable for analyzing the overall behavior of many GLUT4 molecules within the cells. With this approach, we found that cells with an increased intensity ratio of Tbc1d1 relative to AS160 showed dampening of insulin-responsive increases in the speed of GLUT4 movement; in other words, by plotting insulin-induced percentage increases in the mean speed of GLUT4 movement, which resulted from GLUT4 liberation, versus the relative ratio of Tbc1d1 to AS160 obtained from each cell, we identified a negative correlation between these two values (Fig. 1, D and E). These calculations revealed an inhibitory impact on Tbc1d1 favoring insulin-induced AS160-mediated liberation in a relative ratio–dependent manner.
Stimulus-inducible phosphorylation status in the co-presence of AS160 and Tbc1d1
To examine the potential impact of Tbc1d1 on AS160 phosphorylation (and/or vice versa) in response to insulin stimulation, we next analyzed whether the co-presence of AS160 and Tbc1d1 induces changes in the phosphorylation profiles of the proteins. For this analysis, we established multiplex assays of the phosphorylation signals to quantitatively compare phosphorylation statuses among many treatment conditions in a single run of the assay. These assays utilize 1) capture antibody–conjugated fluorescent magnetic beads, 2) biotinylated antibodies for detection of the protein/modification of interest, and 3) R-phycoerythrin–labeled streptavidin and detection of the types of beads and intensities of R-phycoerythrin on the basis of flow cytometry–based analysis (Fig. 2A). With this approach, we can analyze phosphorylation in cells expressing EYFP-AS160 and HaloTag-Tbc1d1 by using 1) a mixture of two different fluorescent beads conjugated to anti-EGFP and anti-HaloTag antibodies, respectively, and 2) biotinylated antibodies for phospho-specific or total antibodies for AS160 and Tbc1d1 in a 96-well plate format. For validation of this assay, we compared the phosphorylation status of the same samples by Western blotting (Fig. 2B) and multiplex assays (Fig. 2C, filled circles). Results obtained from these two independent assays showed a similar tendency (Fig. 2C), and the two values showed positive correlations for all of the phosphorylation sites analyzed (Fig. 2D), indicating that our multiplex assay can appropriately detect the phosphorylation status of AS160 and Tbc1d1. We represented the phosphorylation status with the normalized phosphorylation obtained by the ratio of phosphorylation/total (Fig. 2E), and we found no significant changes in the phosphorylation of Tbc1d1 and AS160 between the co-presence of these two proteins and the presence of either one alone, except for AS160 phosphorylation at Ser704, in the cell-based reconstitution model employing 3T3-L1 fibroblasts (Fig. 2F).
Figure 2.


Analysis of phosphorylation signals of AS160 and Tbc1d1 by multiplex assay. A, schematic drawing for multiplex assays of phosphorylation signals. B, representative Western blotting of HaloTag-Tbc1d1 or EYFP-AS160 (phosphorylation or total) of cell lysates treated under the indicated conditions. Note that total EYFP-AS160 was obtained by its EYFP fluorescence. Some cells were pretreated with LY294002 (LY; 50 μm) for 30 min before stimulation with insulin or AICAR for 10 min. C, quantification of HaloTag-Tbc1d1 or EYFP-AS160 (phosphorylation or total) analyzed by Western blotting (open squares) and multiplex assay (filled circles). These values were obtained from the same samples as those shown in B. D, correlation between results obtained with the two assay systems. The values were obtained from the data shown in C and are presented as ratios (phosphorylation/total). E, -fold changes in phosphorylation of HaloTag-Tbc1d1 and EYFP-AS160 analyzed by multiplex assays. **, p < 0.01; ***, p < 0.001 by Dunnett's multiple comparison versus basal states (n = 3). F, phosphorylation signals in the co-presence of AS160 and Tbc1d1. The values are shown as ratios (phosphorylation/total) and as -fold increases, as compared with untreated cells expressing either AS160 or Tbc1d1. *, p < 0.05 by two-way repeated measures analysis of variance for the expressed proteins (n = 3). n.d., not detected. Error bars, S.E.
Rapid insulin response to sequential/combined exercise-mimetic stimulation in the co-presence of AS160 and Tbc1d1
We have previously observed that Tbc1d1 has the unique capability of temporarily acquiring insulin responsiveness by sequential or combined application of exercise-mimetic stimuli (such as AMPK activation or Ca2+) and the insulin-designated “regulatory mode shift” of Tbc1d1 (9). Therefore, we next analyzed whether such exercise-mimetic stimuli can equivalently induce the regulatory mode shift in cells co-expressing AS160 and Tbc1d1. First, we applied sequential stimulation with AICAR and insulin to the cells (Fig. 3A). As observed previously, insulin resulted in the liberation of static GLUT4 in the Tbc1d1-expressing cells acquiring regulatory mode shift status by prior exposure to AICAR, and statistically significant enhancement of the speed of movement was observed at 30 min, but not at 5 min, of insulin stimulation (Fig. 3B, left). Such a regulatory mode shift phenomenon was similarly observed in cells co-expressing Tbc1d1 and AS160, although interestingly, significant liberation was observed even after just 5 min of insulin stimulation (Fig. 3B, right). Detailed evaluation of the cellular contents of the two RabGAPs revealed that the accelerating potency of AS160 to insulin-responsive GLUT4 liberation depended on the amount of AS160 relative to that of Tbc1d1 (Fig. 3C).
Figure 3.
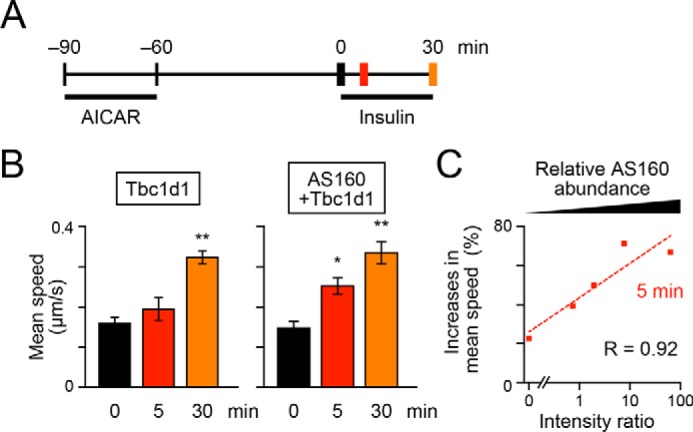
Accelerating potency of AS160 on insulin-responsive GLUT4 liberation in response to sequential treatments with AICAR and insulin. A, treatment and acquisition protocols. Cells were first stimulated with AICAR (1 mm) for 30 min and then washed for 1 h, followed by stimulation with insulin (100 nm). Imaging was performed before (black) and after (red, 5 min; orange, 30 min) insulin stimulation. B, mean speeds of intracellular movements of GLUT4 in cells expressing Tbc1d1 (left) or both AS160 and Tbc1d1 (right). *, p < 0.05; **, p < 0.01 by Dunnett's multiple comparison versus before insulin stimulation (n = 5–7). C, relationship between insulin-responsive GLUT4 liberation at 5 min of insulin stimulation and relative AS160 abundance. Error bars, S.E.
Similarly, in differentiated adipocytes expressing exogenous Tbc1d1 (by transfection) with marginal AS160 (by siRNA-mediated knockdown) due to prior AICAR treatment, insulin liberated static GLUT4 at 30 min (Fig. S4A, left), indicating that the regulatory mode shift can be recapitulated in 3T3-L1 adipocytes under this experimental condition. Furthermore, in adipocytes co-expressing Tbc1d1 and AS160, significant liberation was observed at an earlier time point (15 min) of insulin stimulation (Fig. S4A, right).
Given that prior treatment with AICAR enables the liberated GLUT4 to complete the entire itinerary, it is possible that after withdrawal of AICAR, the traveling GLUT4 finally reaches a distinct storage compartment different from that in which GLUT4 was originally located and that the rapid AS160-mediated actions affect the repackaged GLUT4. To address this issue, we also analyzed the effects of combined stimuli consisting of experimental rises in intracellular Ca2+ concentrations ([Ca2+]i), which were locally induced by photolysis of the caged Ca2+ compound nitrophenyl-EGTA (NPE) in the continuous presence of insulin for the indicated periods (Fig. 4A). As observed previously, we detected significant liberation of static GLUT4 after 10 min of exposure to elevated [Ca2+]i in cells expressing Tbc1d1 accompanied by insulin (Fig. 4B, left), implying that [Ca2+]i changes induced the regulatory mode shift (9). Interestingly, however, significant liberation of static GLUT4 was observed immediately after just 0.5 min of [Ca2+]i elevation in cells co-expressing AS160 and Tbc1d1 (Fig. 4B, right). Such accelerating potency was again dependent on the relative ratio of Tbc1d1 to AS160 (Fig. 4C). Similar accelerating potency was also observed in differentiated adipocytes under the aforementioned experimental conditions (Fig. S4B). These observations strongly suggested that AS160 can modulate the insulin sensitivity of the Tbc1d1-dependent [Ca2+]i-involved GLUT4 liberation in cells co-expressing AS160 and Tbc1d1, and dynamic GLUT4 repackaging is not always necessary for rendering the regulatory mode shift phenomenon.
Figure 4.
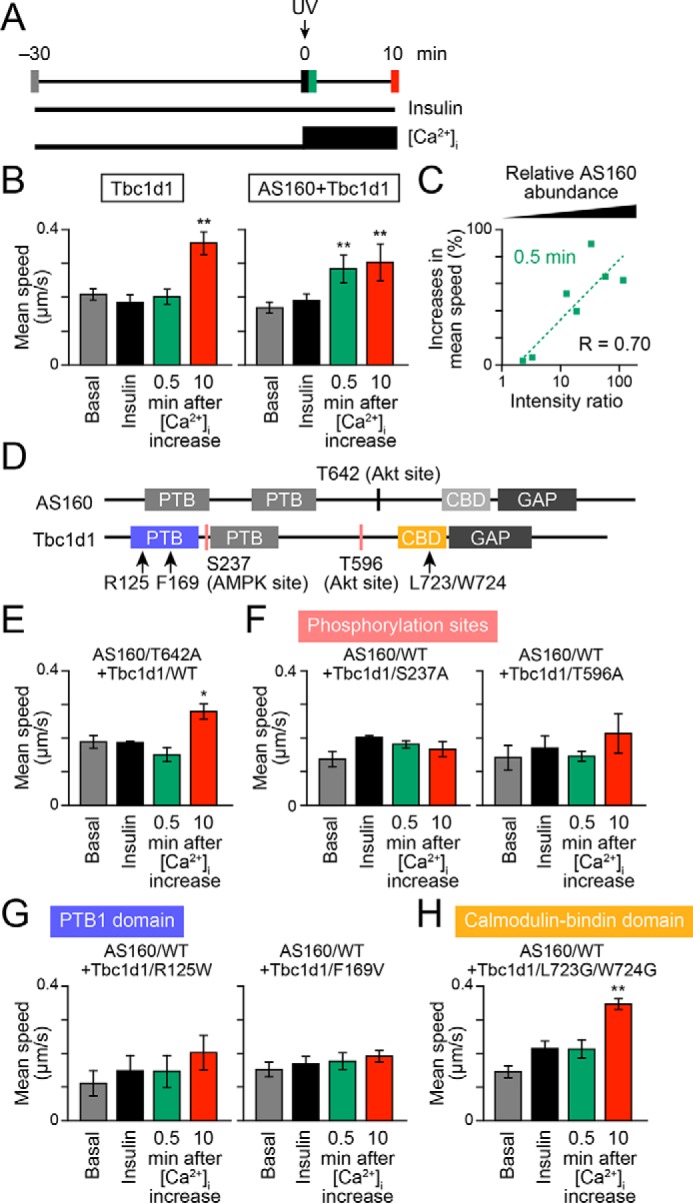
Accelerating potency of AS160 to insulin-responsive GLUT4 liberation in response to treatment with insulin and Ca2+ combined. A, treatment and acquisition protocols. Cells were first stimulated with insulin (100 nm) for 30 min, and then, at time 0, photolysis of the caged Ca2+ compound NPE was induced (UV; arrow). Imaging was performed in the basal (gray), insulin-stimulated (black), and after (green, 0.5 min; red, 10 min) photolysis states. B, mean speeds of intracellular movements of GLUT4 in cells expressing Tbc1d1 (left) or both AS160 and Tbc1d1 (right). **, p < 0.01 by Dunnett's multiple-comparison versus insulin-stimulated states (just before photolysis) (n = 6–8). C, relationship between insulin-responsive GLUT4 liberation at 0.5 min of photolysis and relative AS160 abundance. D, schematic domain structures of human AS160 and Tbc1d1. Mutated amino acids are shown as text. E–H, effects of AS160 (E) or Tbc1d1 (F–H) mutants on GLUT4 liberation in response to combined treatment with insulin and Ca2+. *, p < 0.05; **, p < 0.01 by Dunnett's multiple comparison versus insulin-stimulated states (n = 5–7). Error bars, S.E.
Functional domains critical to the cooperative actions
AS160 and Tbc1d1 both have several domains and key phosphorylation sites that are potentially critical for their functions (Fig. 4D). Therefore, we next analyzed the impacts of several pivotal mutations in AS160 and/or Tbc1d1 on their GLUT4 liberation properties. In this set of experiments, we utilized combined stimuli comprising insulin and Ca2+ to explore the molecular mechanisms underlying their cooperative operations. First, a nonphosphorylation AS160 mutant of a critical Akt phosphorylation site (T642A) failed to achieve the potency needed to liberate GLUT4 within 0.5 min after increases in [Ca2+]i (Fig. 4E), suggesting that Thr642 phosphorylation is necessary for the AS160-triggered impact. Second, the nonphosphorylation Tbc1d1 mutants of either the AMPK site (S237A) or the Akt site (T596A) completely failed to stimulate GLUT4 liberation triggered by [Ca2+]i transients (Fig. 4F), implying that Tbc1d1 phosphorylation, serving as a pivotal GLUT4-releasing regulator in cells co-expressing AS160 and Tbc1d1, is an absolute requirement.
Consistent with the notion that Tbc1d1 serves as the major regulator, Tbc1d1 mutation of the PTB1 domain, including an obesity-related (R125W) and a loss-of-function mutation (F169V), resulted in complete loss of the insulin responsiveness acquisition, and, furthermore, no increases in GLUT4 velocity (due to the liberation deficiency) were observed even in the co-presence of AS160 (Fig. 4, F and G). These results are consistent with our previous study investigating the regulatory mode of Tbc1d1 actions using cells expressing Tbc1d1 alone (9).
Finally, we found that mutations in Leu-Trp, which are conserved in the middle of CBD in human Tbc1d1 (Leu723-Trp724) and AS160 (Leu842-Trp843) (4), to Gly-Gly resulted in a total loss of the rapid response (i.e. at 0.5 min), whereas the releasing capability at a later time point was preserved (Fig. 4H).
To clarify the potential alterations in insulin-dependent phosphorylation profiles of AS160 and Tbc1d1 by expressing the aforementioned mutants, we employed our homemade multiplex assay and found no obvious differences in phosphorylation statuses among the cells expressing each of the Tbc1d1 mutants with AS160 except for AS160 phosphorylation at Ser704 induced by ionomycin treatment (Fig. 5). Because AS160 is reported to form a homodimer via its PTB domains (14) and AS160 and Tbc1d1 are highly homologous proteins (5), it is possible that AS160 and Tbc1d1 can form heterodimers in a manner dependent on their PTB domains. To assess this possibility, we took advantage of our multiplex assays to detect heterodimer formation (Fig. S5A). In this assay, total EYFP-AS160 in cell lysates was captured by anti-EGFP antibody–conjugated fluorescent magnetic beads, and associated HaloTag-Tbc1d1 was detected by biotinylated anti-Tbc1d1 antibody. Obvious signals can be detected when EYFP-AS160 and HaloTag-Tbc1d1 were both expressed in cells (Fig. S5B), suggesting that AS160 and Tbc1d1 are associated. However, no significant changes were observed in responses to insulin, AICAR, and ionomycin or among Tbc1d1 mutants (R125W and F169V) (Fig. S5C).
Figure 5.

Effects of Tbc1d1 mutants on phosphorylation signals of AS160 and Tbc1d1 analyzed by multiplex assays. Cells expressing Myc-GLUT4-mCherry, HA-sortilin, EYFP-AS160, and WT or the indicated mutants of HaloTag-Tbc1d1 were stimulated as follows. In A, cells were first stimulated without (black) or with AICAR (1 mm) for 30 min (blue) and then washed for 1 h (green), followed by stimulation with insulin (100 nm) for 5 min (red). In B, cells were stimulated without (black) or with ionomycin (1 μg/ml) (cyan) or ionomycin + insulin (100 nm) for 5 min (orange). The values are shown as ratios (phosphorylation/total) and as -fold increases, as compared with untreated cells. **, p < 0.01 by two-way repeated measures analysis of variance (n = 3). n.d., not detected. Error bars, S.E.
Discussion
Our present study provides compelling evidence that AS160 and Tbc1d1 cooperatively regulate stimuli-responsive GLUT4-releasing activities, based on single-molecule analysis of GLUT4 behavior in cell-based reconstitution models (Fig. 6) and in 3T3-L1 adipocytes (Figs. S2 and S4). This approach is highly suitable for experiments of this type because our single-molecule strategy allows complex GLUT4-trafficking itineraries to be dissected in each experimentally traceable step, and our experimental reconstitution model can definitively describe the actions of specific regulatory factors by manipulating ectopic expression levels or combinations of the key factors, including AS160 and Tbc1d1. We herein focused on the stimulus-responsive GLUT4 release from the static compartment, given the known functions of AS160 and Tbc1d1 (9, 10). Although the possibility that AS160 and Tbc1d1 are involved in other GLUT4-trafficking processes, such as final fusion steps at the plasma membrane (15, 16), cannot be ruled out, the initial stimulus-dependent liberation process is obviously critical for the entire GLUT4-trafficking itinerary because the trigger for liberation from the static compartment is essential for GLUT4 to ultimately reach the plasma membrane.
Figure 6.
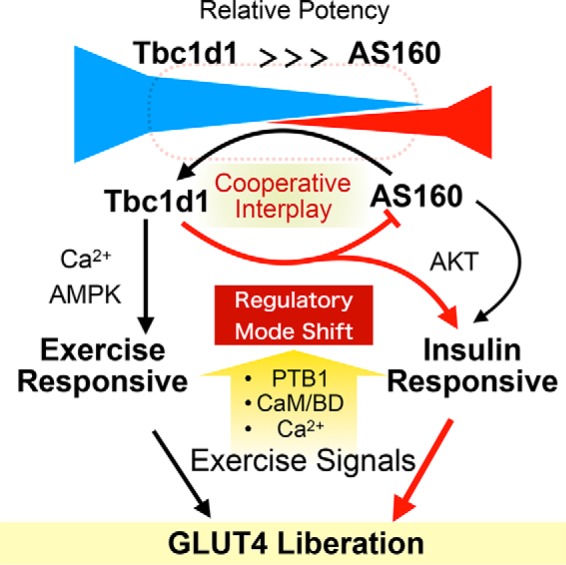
Schematic depiction of cooperative governance for stimulus-dependent GLUT4 liberation process via Tbc1d1 and AS160. Under coexistence of Tbc1d1 and AS160 conditions, Tbc1d1 functionally dominates AS160, given that stimuli-dependent GLUT4 liberation predominantly relies on Tbc1d1-evoking proximal stimuli, such as AMPK activation (AICAR) and an increase in intracellular Ca2+. However, AS160 modulates sensitivity to external stimuli in the Tbc1d1-dominated GLUT4 release process via promoting the regulatory mode shift of Tbc1d1, resulting in efficient acquisition of insulin responsiveness, which requires an intact PTB1 domain and Ser237 phosphorylation of Tbc1d1. This potentiating action of AS160 also relies on the CBD of both Tbc1d1 and Thr642 (Akt site) of AS160. This cooperative governance, which relies on combined regulatory modes involving both Tbc1d1 (e.g. PTB1, CBD, Ser237 phosphorylation) and AS160 (e.g. Thr642 phosphorylation), plays a key role in deciphering biochemical signals (e.g. phosphorylation and [Ca2+]i) into physical GLUT4 release processes in response to insulin and exercise-related stimuli.
We first confirmed that Tbc1d1 functionally dominates AS160 in terms of triggering insulin-responsive GLUT4 liberation (Fig. 1), which is consistent with the results of previous studies employing 3T3-L1 adipocytes ectopically expressing Tbc1d1 (5, 17). Regarding skeletal muscle, previous studies revealed the soleus muscle to demonstrate more insulin-sensitive responsiveness and to have a higher ratio of AS160 to Tbc1d1 than extensor digitorum longus muscle (2, 18–20), and our present observations may, at least partially, explain the mechanistic rationale for determining insulin sensitivity. Although molecular details of the dominant functions of Tbc1d1 regarding insulin-responsive GLUT4 liberation remain unclear, some Tbc1d1 mutants, such as S237A and L724G/W725G, showed a slight tendency to liberate static GLUT4 in response to insulin (Fig. 4, F and H), suggesting involvement of composite actions via multiple sites of Tbc1d1 in the observed inhibitory effects.
Dominant actions of Tbc1d1 in cells co-expressing AS160 and Tbc1d1 also contribute to the temporal acquisition of insulin responsiveness after exercise-mimetic stimuli, a phenomenon termed the regulatory mode shift, which is a characteristic intrinsic property of Tbc1d1 (9). One of the striking observations made in the present study is that AS160 can positively modulate Tbc1d1-mediated GLUT4-releasing activities after exercise mimetic stimuli, which suggests a potentiating action of AS160 on insulin sensitivity even in the presence of Tbc1d1. This action would presumably be mediated by as yet unknown functional interactions between AS160 and Tbc1d1, because mutations in either protein resulted in loss of the insulin sensitization actions. One critical domain is the PTB domain of Tbc1d1 as well as its phosphorylation sites. Although functional aspects of the PTB domain of Tbc1d1 have not as yet been adequately studied, PTB domains of AS160 reportedly regulate homodimerization, 14-3-3 binding, and GLUT4-vesicle fusion with the plasma membrane (14, 15, 21). Because AS160 and Tbc1d1 are highly homologous proteins (5), it is possible that Tbc1d1 can also form homodimers and, more intriguingly, that AS160 and Tbc1d1 can form heterodimers in a manner dependent on their PTB domains. In the present study, we detected an association between AS160 and Tbc1d1 by using our multiplex assays, but we found no significant changes in this association among various stimulation conditions or Tbc1d1 mutants. However, changes in the association may occur transiently at specific subcellular locations along with other associating proteins (20), and it would presumably be difficult to detect such localized changes with the current approach, which uses whole cell lysates. Indeed, Tbc1d1 reportedly interacts through PTB domains with the cytoplasmic domain of the insulin-responsive aminopeptidase, and the resulting association is influenced by phosphorylation of Tbc1d1 by Akt or AMPK (22). Furthermore, recent biochemical studies have revealed that AMPK containing the α1, but not the α2, isoform of the catalytic subunit form a complex with Tbc1d1 via PTB domains (23). Although dimerization of AS160 via its C-terminal GAP domain is reported to be unrelated to GAP catalytic activity (24), it is still possible that PTB domain-mediated heterodimerization and associations with other proteins, such as IRAP and AMPK, can modulate the functional balances between the two RabGAPs. Further optimization of our multiplex assay for evaluating complex formation comprising the two RabGAPs and other associating protein(s) would be necessary to clarify this point. Another important domain identified in the present study is CBD of Tbc1d1. Although our previous analysis in cells expressing only Tbc1d1 revealed that the CBD mutant exhibited no obvious defect in GLUT4 liberation in response to transient increases in [Ca2+]i in the presence of insulin stimulation (9), this domain appears to have some involvement in the functional interaction with AS160. Although functional roles of the CBD of Tbc1d1 have not been identified, the CBD of AS160 can bind with calmodulin in a Ca2+-dependent manner and thereby regulate contraction- but not insulin-stimulated glucose uptake in skeletal muscle (4, 25).
Regarding phosphorylation signals of AS160 (Thr642 and Ser704) and Tbc1d1 (Ser237 and Thr596), in the present study, we found no significant differences in phosphorylation profiles between cells expressing either of these proteins alone and those expressing both proteins or among the Tbc1d1 mutants analyzed, except for Ser704 of AS160 under certain conditions. These results suggest that proximal phosphorylation status at least for the key sites examined in the present study cannot be the main factor responsible for functional synergism between AS160 and Tbc1d1. Although the difference was subtle, we observed statistically significant differences in the phosphorylation at Ser704 of AS160 in some cases. Phosphorylation of this site has been reported to be modulated by exercise and its related signals (26, 27). In fact, AS160 has several additional phosphorylation sites, and phosphorylation at some of these sites is reported to be sustained after several hours of exercise (28–32), including Ser341 as well as Ser704 (26). Moreover, exercise can augment insulin-stimulated phosphorylation of the specific sites of AS160. Thus, the possibility of differences in phosphorylation profiles among these sites, as well as contributions of these sites to the functional interactions, cannot be ruled out. We therefore need to further analyze the effects of the co-presence of AS160 and Tbc1d1 on phosphorylation of these sites and the exercise effects. In addition, we herein took advantage of a cell-based reconstitution model using 3T3L1 fibroblasts, but not skeletal muscle cell lines such as C2C12 cells (6), and basically utilized supraphysiological concentrations of insulin, indicating that analyses employing stimulation with physiological concentrations of insulin are necessary for evaluating exercise effects. Thus, caution must be exercised especially in interpreting data regarding phosphorylation profiles of these two RapGAPs. However, we can now effectively compare phosphorylation profiles among many treatment conditions by taking advantage of our multiplex assay. Detailed analyses of biochemical properties in the co-presence of the two RabGAPs with our multiplex assay will, in our view, be a powerful tool for elucidating the cooperative regulations governed by these proteins in skeletal muscle cells.
Meanwhile, it is clear, based on observations in skeletal muscle fibers, that there must be multiple other regulatory factors (8, 33–35) that might be inadequately expressed in the cell-based reconstitution model using undifferentiated 3T3L1 fibroblasts ectopically expressing three key components, GLUT4, sortilin, and AS160, and/or Tbc1d1. Nevertheless, this cell-based reconstitution model and our present findings can serve as a basis for further elucidating additional regulatory interventions regulating both GLUT4 liberation processes and the phosphorylation profiles of AS160 and Tbc1d1.
In summary, we herein clearly demonstrated functional cooperation between two key RabGAPs, AS160 and Tbc1d1, in insulin-responsive GLUT4 liberation. In this process, Tbc1d1 plays a dominant role, and AS160 may strictly regulate insulin sensitivity. Recently, we identified heterotypic endomembrane fusion as an initial step in the insulin-responsive GLUT4 trafficking in skeletal myofibers, which might be regulated by functional cooperation between AS160 and Tbc1d1 (20). Because such insulin-responsive heterotypic endomembrane fusion may occur in cells other than skeletal myofibers,3 further detailed mechanistic analyses of the heterotypic fusion regulated by AS160 and Tbc1d1 will provide knowledge promoting better understanding of GLUT4 behavior and exercise physiology.
Experimental procedures
Plasmids, siRNAs, and antibodies
All HaloTag expression vectors were purchased from Kazusa DNA Research Institute. Mutations were generated by PCR-based site-directed mutagenesis and confirmed by sequencing (PRISM 3130, Applied Biosystems). AS160 siRNAs were synthesized by Tomy Digital Biology. We used a mixture of three distinct siRNAs, with sequences of 5′-GCUCUGCGCCCGUAGACUA-3′, 5′-CCUAUGAGGUAGAAUAUCA-3′ (9), and 5′-GACUUAACUCAUCCAACGA-3′ (36). Anti-Myc mAb (9E10) was purified from hybridoma culture supernatants. Anti-phospho-Tbc1d1 (Ser237), anti-phospho-Tbc1d1 (Thr596), and anti-phospho-AS160 (Ser704) antibodies were generated by immunizing rabbits with keyhole limpet hemocyanin–conjugated peptides (H-CRPMRKSF-pS-QPGLRS-OH, H-CRRAN-pT-LSHFP-OH, and H-TSF-pS-APSFTA-OH, respectively), followed by immunoaffinity purification using SulfoLink Coupling Gel (Pierce). Anti-AS160 antibody was purchased from Sigma (SAB4200101). Anti-phospho-AS160 (Thr642) antibodies were purchased from Cell Signaling (for Western blotting) and Sigma (SAB4503896 for multiplex assay). Anti-Tbc1d1 antibody was purchased from Protein Tech (22124-1-AP). Anti-EGFP antibodies were purchased from MBL (587 and D153-3). HRP-conjugated secondary antibodies were commercially obtained from Pierce. Biotinylated antibodies were prepared with a biotin labeling kit-NH2 (DOJINDO).
Cell culture
3T3-L1 fibroblasts were plated onto glass-bottom dishes or standard culture dishes and differentiated into adipocytes (37). Fibroblasts were transfected with Lipofectamine 3000 (Thermo) with plasmid DNAs according to the manufacturer's instructions. Differentiated adipocytes were replated onto collagen-coated glass-bottom dishes. After a 24-h culture, adipocytes were electroporated with 10–25 μg of plasmid DNA and/or 250 pmol of siRNA with a CUY21EDITII electroporator (BEX Co.) and an electrode (LF513-5, BEX Co.) by applying a poration pulse at 400 V for 10 ms, followed by five pulses at −30 V for 10 ms at 50-ms intervals. Imaging experiments were performed after an additional 2 days of culture (13). All experiments were performed with cells exogenously expressing Myc-GLUT4 (fused to ECFP, EGFP, or mCherry), HA-sortilin, and both Tbc1d1 and AS160 or either one alone (fused to HaloTag or EYFP). Expression of HaloTag-fused proteins was confirmed by staining of cells with HaloTag TMR ligand (Promega). For the imaging experiments, the cells were immersed in a solution consisting of 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES-NaOH (pH 7.4), and 5.5 mm d-glucose.
QD labeling
QD-conjugated anti-Myc antibody was prepared as follows. First, the Fab′ fragment of anti-Myc antibody was prepared with a mouse IgG1 Fab and F(ab′)2 preparation kit (Pierce), followed by mild reduction with 2-mercaptoethanolamine. Maleimide-activated QD655 was also prepared by incubating Qdot 655 ITK Amino (PEG) Quantum dots (Thermo) with sulfo-SMCC (Pierce) at room temperature for 1 h. The resultant Fab′ fragment of anti-Myc antibodies and maleimide-activated QD655 were mixed, and the final concentrations of the QD655-conjugated antibodies were determined by optical density at 638 nm and then using the formula, A = ϵcL, where A is the absorbance, ϵ is the molar extinction coefficient (800,000 m−1 cm−1), c is the molar concentration, and L is the path length. For cellular labeling, the cells were serum-starved and incubated in the presence of 1.5–5 nm QD655-conjugated antibodies for 1 h in the presence of 1 nm insulin. The cells were extensively washed for at least 3 h to remove unbound QD-labeled antibodies.
Single-molecule imaging and particle tracking
Single-molecule imaging of QD was performed with an inverted microscope (IX81, Olympus) equipped with an electron-multiplying charge-coupled device camera (iXon Ultra, Andor Technology), a Nipkow disk confocal unit (CSU-X1, Yokogawa), a z-drift compensation module (Olympus), and an oil-immersion objective lens (UPLSAPO100xO, numerical aperture 1.4, Olympus) at 20 frames/s. Imaging was performed at ∼30 °C by using a stage heater and a lens heater (TOKAI HIT). QD655 fluorescence was excited at 488 or 532 nm with a solid-state laser and detected through a 655/12 bandpass filter (Semrock). Single-particle tracking was performed with G-Count (G-Angstrom) with a two-dimensional Gaussian fitting mode. We tracked each particle successfully fitted within a 13 × 13-pixel region of interest for at least 30 frames. When the signal in a frame was lost because of blinking, no fitting was performed until reappearance of the bright spot. When a bright spot did not reappear within 10 frames, tracking was aborted. We typically tracked 50–150 particles/cell and obtained the images in at least three independent experiments. We evaluated movements with mean velocities. The velocities for individual particle movements were calculated by linear fit of the displacement during four frames. Mean velocities were first calculated in a cell and then averaged among cells under the same treatment conditions. We found that QD in fixed cells showed nonnegligible velocities (∼0.19 μm/s), which were attributed to unavoidable instrumental noise. Thus, in figures representing mean velocities, we used corrected values instead of raw data by subtracting the noise. Because the velocities were calculated based on the movements within short periods of time and single-molecule fluorescence with a limited signal/noise ratio, such relatively large noise was inevitable. Diffusion coefficients of individual molecules were estimated as follows. First, mean square displacement (MSD) of individual particles for all accessible time lags τ was calculated with the equation,
| (Eq. 1) |
where N, Δt, and pi are the total number of positions measured, time interval of successive images, and position of the molecule in time frame i, respectively. The diffusion coefficient of the molecule was calculated by fitting the first 10 time points of the MSD values with the following,
| (Eq. 2) |
where D10 and C are the diffusion coefficient and instrumental noise, respectively. The MSD and D10 calculations and D10 map constructions were performed with a custom-written program based on LabVIEW and Vision (National Instruments).
Photolysis of a caged Ca2+ compound
The acetoxymethyl ester of the caged Ca2+ compound NPE (Thermo) was first dissolved in anhydrous DMSO at 10 mm and then diluted in serum-free Dulbecco's modified Eagle's medium. Cells were incubated for 30 min at 37 °C in serum-free Dulbecco's modified Eagle's medium containing 10 μm NPE-acetoxymethyl ester and 0.03% Cremophor EL (Sigma), and then washed with SolA. Photolysis of NPE was induced with a mercury lamp (U-ULS100HG, Olympus) through a 360-nm bandpass filter. The mercury lamp irradiation was gated with an electric shutter (SSH-R, Sigma Koki) with an opening duration of 0.1 s. Uncaging experiments were performed under yellow light illumination to prevent unintended photolysis of NPE.
Estimated ratio of the expression of exogenous AS160 to that of Tbc1d1
For estimating the ratio of exogenously expressed AS160 to that of Tbc1d1, we used 3T3-L1 fibroblasts expressing Myc-GLUT4-ECFP, HA-sortilin, and either HaloTag-Tbc1d1 + EYFP-AS160 or EYFP-Tbc1d1 + HaloTag-AS160. We used pBI bidirectional promotor vectors to express either EYFP-AS160 or EYFP-Tbc1d1 in conjunction with HA-sortilin and transfected a fixed amount of the pBI vector construct and variable amounts of the HaloTag vector construct. With these cells, we estimated the AS160/Tbc1d1 expression ratio based on their fluorescence, as described below. We first prepared cells expressing either EYFP or HaloTag proteins, and the cells expressing HaloTag proteins were then stained with 0.5 μm HaloTag TMR ligand for 15 min. Epifluorescent images of EYFP and TMR were taken through a U-MYFPHQ cube (Olympus; excitation, 490–500 nm; dichroic, 505 nm; emission, 515–560 nm; ch1) and a U-MWIGA3 cube (Olympus; excitation, 530–550 nm; dichroic, 570 nm; emission, 575–625 nm; ch2), respectively (Fig. S3). After acquisition, fluorescent intensities within cells and the area adjacent to the cell of interest (background fluorescence) were measured employing Fiji ImageJ, and background fluorescence was subtracted from observed fluorescence intensities within cells. We can represent the background-subtracted fluorescent intensities with a 2-by-2 matrix as follows,
where subscripts denote acquisition channels. Fractions of bleed-through at each channel can be calculated as follows.
In the experiments shown in Figs. 1E, 3C, and 4C, we observed EYFP and TMR epifluorescence with the same setting as that described above prior to single-molecule imaging of QD. Based on the fluorescence intensities, we calculated intensity ratios between EYFP and TMR as follows. Relationships between intensities acquired by each of the channels (Ich1 and Ich2) and true intensities of EYFP (IEYFP) and TMR (ITMR) are as follows.
| (Eq. 3) |
Therefore, we can calculate IEYFP and ITMR as follows.
| (Eq. 4) |
With our setting, we calculated EYFPch2/EYFPch1 and TMRch1/TMRch2 to be 0.108 and 0.009, respectively. Finally, the ratio of ITMR to IEYFP (RTMR/EYFP) was calculated by the following.
| (Eq. 5) |
We represented the relative ratio of the two RabGAPs as RTMR/EYFP. For example, Ich1 and Ich2 in the cells shown in Fig. S3 were 307 and 4253, respectively. Based on Equations 4 and 5, we calculated IEYFP, ITMR, and RTMR/EYFP to be 269, 4224, and 15.7, respectively. Note that these values do not represent absolute expression ratios. Because fluorescence intensities can vary among experimental days for various reasons, including expression efficiency, relationships between GLUT4 behaviors and expression ratios were compared with the data obtained only within each experimental day.
Western blotting and multiplex assays of phosphorylation signals
The cells were harvested in lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 20 mm sodium pyrophosphate, 10 mm NaF, 2 mm sodium orthovanadate, 1 mm EDTA, 1% Triton X-100, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, 6500 IU/ml aprotinin, phosphatase inhibitor mixture-1; Sigma), and after end-over-end rotation of the homogenates for 30 min, lysate supernatants were collected by centrifugation (15,000 rpm) for 20 min at 4 °C, and protein concentrations were measured using the bicinchoninic acid method with BSA as the standard (Pierce). After electrophoresis, EYFP fluorescence was detected with a Pharos FX Molecular Imager (Bio-Rad) equipped with a 488-nm laser. Western blotting was performed following standard procedures, and chemiluminescence was detected with an ImageQuant LAS4000 Mini system (GE Healthcare). Quantification was performed with ImageQuant TL software (GE Healthcare). For multiplex assays, total protein and phosphorylation levels of relevant proteins were determined with a Bio-Plex 200 Multiplex system (Bio-Rad). For this assay, total HaloTag-Tbc1d1 and EYFP-AS160 proteins in the lysate (2.5 μg of total protein) were captured by magnetic beads conjugated with anti-HaloTag or anti-EGFP antibodies, respectively, which had been prepared with a Bio-Plex amine-coupling kit (Bio-Rad) and Bio-Plex Pro magnetic COOH beads according to the manufacturer's instructions. After washing out the unbound molecules, the beads were incubated with 1 μg/ml biotinylated detection antibodies for 30 min at room temperature, followed by incubation with 2 μg/ml streptavidin-phycoerythrin (Bio-Rad or Vector Laboratories) for 10 min at room temperature.
Statistical analysis
Data with error bars are the mean ± S.E. except for the box plots. The statistical significance of differences between groups was assessed as shown in the figure legends. A p value less than 0.05 was considered to indicate a statistically significant difference.
Author contributions
H. H. and M. K. conceptualization; H. H., T. M., and T. I. data curation; H. H. and M. K. software; H. H., T. M., T. I., and M. K. formal analysis; H. H. and M. K. supervision; H. H. and M. K. funding acquisition; H. H. and M. K. validation; H. H. and M. K. investigation; H. H. and M. K. visualization; H. H. and M. K. methodology; H. H. and M. K. writing-original draft; H. H. and M. K. project administration; M. K. resources; M. K. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Natsumi Emoto for technical assistance.
This work was supported in part by Japan Society for the Promotion of Science Grant 17H02076 (to M. K.), Japan Agency for Medical Research and Development Grant 18058831 (to M. K.), a grant from the Japan Diabetes Society (to H. H.), and the Program for Fostering Researchers for the Next Generation in the Project for Establishing a Consortium for the Development of Human Resources in Science and Technology (to H. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
H. Hatakeyama and M. Kanzaki, unpublished observations.
- RabGAP
- Rab GTPase–activating protein
- AS160
- Akt substrate of 160 kDa
- CBD
- calmodulin-binding domain
- [Ca2+]i
- intracellular Ca2+ concentrations
- MSD
- mean square displacement
- PTB
- phosphotyrosine-binding
- QD
- quantum dot
- AMPK
- AMP-activated protein kinase
- EGFP
- enhanced green fluorescent protein
- ECFP
- enhanced cyan fluorescent protein
- NPE
- nitrophenyl-EGTA.
References
- 1. Bruss M. D., Arias E. B., Lienhard G. E., and Cartee G. D. (2005) Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 54, 41–50 10.2337/diabetes.54.1.41 [DOI] [PubMed] [Google Scholar]
- 2. Taylor E. B., An D., Kramer H. F., Yu H., Fujii N. L., Roeckl K. S., Bowles N., Hirshman M. F., Xie J., Feener E. P., and Goodyear L. J. (2008) Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J. Biol. Chem. 283, 9787–9796 10.1074/jbc.M708839200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kane S., Sano H., Liu S. C., Asara J. M., Lane W. S., Garner C. C., and Lienhard G. E. (2002) A method to identify serine kinase substrates: Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J. Biol. Chem. 277, 22115–22118 10.1074/jbc.C200198200 [DOI] [PubMed] [Google Scholar]
- 4. Kane S., and Lienhard G. E. (2005) Calmodulin binds to the Rab GTPase activating protein required for insulin-stimulated GLUT4 translocation. Biochem. Biophys. Res. Commun. 335, 175–180 10.1016/j.bbrc.2005.07.056 [DOI] [PubMed] [Google Scholar]
- 5. Roach W. G., Chavez J. A., Mîinea C. P., and Lienhard G. E. (2007) Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem. J. 403, 353–358 10.1042/BJ20061798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nedachi T., Fujita H., and Kanzaki M. (2008) Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 295, E1191–E1204 10.1152/ajpendo.90280.2008 [DOI] [PubMed] [Google Scholar]
- 7. Funai K., and Cartee G. D. (2008) Contraction-stimulated glucose transport in rat skeletal muscle is sustained despite reversal of increased PAS-phosphorylation of AS160 and TBC1D1. J. Appl. Physiol. 105, 1788–1795 10.1152/japplphysiol.90838.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castorena C. M., Mackrell J. G., Bogan J. S., Kanzaki M., and Cartee G. D. (2011) Clustering of GLUT4, TUG, and RUVBL2 protein levels correlate with myosin heavy chain isoform pattern in skeletal muscles, but AS160 and TBC1D1 levels do not. J. Appl. Physiol. 111, 1106–1117 10.1152/japplphysiol.00631.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hatakeyama H., and Kanzaki M. (2013) Regulatory mode shift of Tbc1d1 is required for acquisition of insulin-responsive GLUT4-trafficking activity. Mol. Biol. Cell 24, 809–817 10.1091/mbc.e12-10-0725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hatakeyama H., and Kanzaki M. (2011) Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic 12, 1805–1820 10.1111/j.1600-0854.2011.01279.x [DOI] [PubMed] [Google Scholar]
- 11. Meyre D., Farge M., Lecoeur C., Proenca C., Durand E., Allegaert F., Tichet J., Marre M., Balkau B., Weill J., Delplanque J., and Froguel P. (2008) R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum. Mol. Genet. 17, 1798–1802 10.1093/hmg/ddn070 [DOI] [PubMed] [Google Scholar]
- 12. Stone S., Abkevich V., Russell D. L., Riley R., Timms K., Tran T., Trem D., Frank D., Jammulapati S., Neff C. D., Iliev D., Gress R., He G., Frech G. C., Adams T. D., et al. (2006) TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum. Mol. Genet. 15, 2709–2720 10.1093/hmg/ddl204 [DOI] [PubMed] [Google Scholar]
- 13. Fujita H., Hatakeyama H., Watanabe T. M., Sato M., Higuchi H., and Kanzaki M. (2010) Identification of three distinct functional sites of insulin-mediated GLUT4 trafficking in adipocytes using quantitative single molecule imaging. Mol. Biol. Cell 21, 2721–2731 10.1091/mbc.e10-01-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dash S., Sano H., Rochford J. J., Semple R. K., Yeo G., Hyden C. S., Soos M. A., Clark J., Rodin A., Langenberg C., Druet C., Fawcett K. A., Tung Y. C., Wareham N. J., Barroso I., et al. (2009) A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc. Natl. Acad. Sci. U.S.A. 106, 9350–9355 10.1073/pnas.0900909106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan S. X., Ng Y., Burchfield J. G., Ramm G., Lambright D. G., Stöckli J., and James D. E. (2012) The Rab GTPase-activating protein TBC1D4/AS160 contains an atypical phosphotyrosine-binding domain that interacts with plasma membrane phospholipids to facilitate GLUT4 trafficking in adipocytes. Mol. Cell. Biol. 32, 4946–4959 10.1128/MCB.00761-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez E., and McGraw T. E. (2006) Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol. Biol. Cell 17, 4484–4493 10.1091/mbc.e06-07-0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chavez J. A., Roach W. G., Keller S. R., Lane W. S., and Lienhard G. E. (2008) Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 283, 9187–9195 10.1074/jbc.M708934200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. James D. E., Kraegen E. W., and Chisholm D. J. (1985) Effects of exercise training on in vivo insulin action in individual tissues of the rat. J. Clin. Invest. 76, 657–666 10.1172/JCI112019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henriksen E. J., Bourey R. E., Rodnick K. J., Koranyi L., Permutt M. A., and Holloszy J. O. (1990) Glucose transporter protein content and glucose transport capacity in rat skeletal muscles. Am. J. Physiol. 259, E593–E598 [DOI] [PubMed] [Google Scholar]
- 20. Hatakeyama H., and Kanzaki M. (2017) Heterotypic endosomal fusion as an initial trigger for insulin-induced glucose transporter 4 (GLUT4) translocation in skeletal muscle. J. Physiol. 595, 5603–5621 10.1113/JP273985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koumanov F., Richardson J. D., Murrow B. A., and Holman G. D. (2011) AS160 phosphotyrosine-binding domain constructs inhibit insulin-stimulated GLUT4 vesicle fusion with the plasma membrane. J. Biol. Chem. 286, 16574–16582 10.1074/jbc.M111.226092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mafakheri S., Flörke R. R., Kanngiesser S., Hartwig S., Espelage L., De Wendt C., Schönberger T., Hamker N., Lehr S., Chadt A., and Al-Hasani H. (2018) AKT and AMPK regulate TBC1D1 through phosphorylation and its interaction with the cytosolic tail of insulin-regulated aminopeptidase IRAP. J. Biol. Chem. 293, 17853–17862 10.1074/jbc.RA118.005040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thomas E. C., Hook S. C., Gray A., Chadt A., Carling D., Al-Hasani H., Heesom K. J., Hardie D. G., and Tavaré J. M. (2018) Isoform-specific AMPK association with TBC1D1 is reduced by a mutation associated with severe obesity. Biochem. J. 475, 2969–2983 10.1042/BCJ20180475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woo J. R., Kim S. J., Kim K. Y., Jang H., Shoelson S. E., and Park S. (2017) The carboxy-terminal region of the TBC1D4 (AS160) RabGAP mediates protein homodimerization. Int. J. Biol. Macromol. 103, 965–971 10.1016/j.ijbiomac.2017.05.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kramer H. F., Taylor E. B., Witczak C. A., Fujii N., Hirshman M. F., and Goodyear L. J. (2007) Calmodulin-binding domain of AS160 regulates contraction- but not insulin-stimulated glucose uptake in skeletal muscle. Diabetes 56, 2854–2862 10.2337/db07-0681 [DOI] [PubMed] [Google Scholar]
- 26. Vendelbo M. H., Møller A. B., Treebak J. T., Gormsen L. C., Goodyear L. J., Wojtaszewski J. F., Jørgensen J. O., Møller N., and Jessen N. (2014) Sustained AS160 and TBC1D1 phosphorylations in human skeletal muscle 30 min after a single bout of exercise. J. Appl. Physiol. 117, 289–296 10.1152/japplphysiol.00044.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oki K., Arias E. B., Kanzaki M., and Cartee G. D. (2018) Prior treatment with the AMPK activator AICAR induces subsequently enhanced glucose uptake in isolated skeletal muscles from 24-month-old rats. Appl. Physiol. Nutr. Metab. 43, 795–805 10.1139/apnm-2017-0858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arias E. B., Kim J., Funai K., and Cartee G. D. (2007) Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am. J. Physiol. Endocrinol Metab 292, E1191–E1200 10.1152/ajpendo.00602.2006 [DOI] [PubMed] [Google Scholar]
- 29. Funai K., Schweitzer G. G., Sharma N., Kanzaki M., and Cartee G. D. (2009) Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 297, E242–E251 10.1152/ajpendo.00194.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Treebak J. T., Taylor E. B., Witczak C. A., An D., Toyoda T., Koh H. J., Xie J., Feener E. P., Wojtaszewski J. F., Hirshman M. F., and Goodyear L. J. (2010) Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am. J. Physiol. Cell Physiol. 298, C377–C385 10.1152/ajpcell.00297.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pehmøller C., Brandt N., Birk J. B., Høeg L. D., Sjøberg K. A., Goodyear L. J., Kiens B., Richter E. A., and Wojtaszewski J. F. (2012) Exercise alleviates lipid-induced insulin resistance in human skeletal muscle-signaling interaction at the level of TBC1 domain family member 4. Diabetes 61, 2743–2752 10.2337/db11-1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jessen N., An D., Lihn A. S., Nygren J., Hirshman M. F., Thorell A., and Goodyear L. J. (2011) Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 301, E164–E171 10.1152/ajpendo.00042.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fecchi K., Volonte D., Hezel M. P., Schmeck K., and Galbiati F. (2006) Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J. 20, 705–707 10.1096/fj.05-4661fje [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng K. K., Zhu W., Chen B., Wang Y., Wu D., Sweeney G., Wang B., Lam K. S., and Xu A. (2014) The adaptor protein APPL2 inhibits insulin-stimulated glucose uptake by interacting with TBC1D1 in skeletal muscle. Diabetes 63, 3748–3758 10.2337/db14-0337 [DOI] [PubMed] [Google Scholar]
- 35. Balsevich G., Häusl A. S., Meyer C. W., Karamihalev S., Feng X., Pöhlmann M. L., Dournes C., Uribe-Marino A., Santarelli S., Labermaier C., Hafner K., Mao T., Breitsamer M., Theodoropoulou M., Namendorf C., et al. (2017) Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat. Commun. 8, 1725 10.1038/s41467-017-01783-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., and McGraw T. E. (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2, 263–272 10.1016/j.cmet.2005.09.005 [DOI] [PubMed] [Google Scholar]
- 37. Kanzaki M., and Pessin J. E. (2001) Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J. Biol. Chem. 276, 42436–42444 10.1074/jbc.M108297200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.