

Abstract
ATP synthase uses a rotary mechanism to couple transmembrane proton translocation to ATP synthesis and hydrolysis, which occur at the catalytic sites in the β subunits. In the presence of Mg2+, the three catalytic sites of ATP synthase have vastly different affinities for nucleotides, and the position of the central γ subunit determines which site has high, medium, or low affinity. Affinity differences and their changes as rotation progresses underpin the ATP synthase catalytic mechanism. Here, we used a series of variants with up to 45- and 60-residue-long truncations of the N- and C-terminal helices of the γ subunit, respectively, to identify the segment(s) responsible for the affinity differences of the catalytic sites. We found that each helix carries an affinity-determining segment of ∼10 residues. Our findings suggest that the affinity regulation by these segments is transmitted to the catalytic sites by the DELSEED loop in the C-terminal domain of the β subunits. For the N-terminal truncation variants, presence of the affinity-determining segment and therefore emergence of a high-affinity binding site resulted in WT-like catalytic activity. At the C terminus, additional residues outside of the affinity-determining segment were required for optimal enzymatic activity. Alanine substitutions revealed that the affinity changes of the catalytic sites required no specific interactions between amino acid side chains in the γ and α3β3 subunits but were caused by the presence of the helices themselves. Our findings help unravel the molecular basis for the affinity changes of the catalytic sites during ATP synthase rotation.
Keywords: ATP synthase, enzyme mechanism, ATPase, bioenergetics, enzyme catalysis, ligand-binding protein, conformational change, oxidative phosphorylation, proton translocation
Introduction
F1Fo-ATP synthase catalyzes the final step of oxidative phosphorylation and photophosphorylation, the synthesis of ATP from ADP and Pi. F1Fo-ATP synthase consists of the membrane-embedded Fo subcomplex with, in most bacteria, a subunit composition of ab2c10 and the peripheral F1 subcomplex with a subunit composition of α3β3γδϵ. The energy necessary for ATP synthesis is derived from an electrochemical transmembrane proton (or, in some organisms, sodium ion) gradient. Proton flow, down the gradient, through Fo is coupled to ATP synthesis on F1 by a unique rotary mechanism. The protons flow through (half) channels at the interface of a and c subunits, which drives rotation of the ring of c subunits. The c10 ring, together with F1 subunits γ and ϵ, forms the rotor. Rotation of γ leads to conformational changes in the catalytic nucleotide-binding sites on the β subunits where ADP and Pi are bound. The conformational changes result in formation and release of ATP. Thus, ATP synthase converts electrochemical energy, the proton gradient, into mechanical energy in the form of subunit rotation and back into chemical energy as ATP. In bacteria, under certain physiological conditions, the process runs in reverse. ATP is hydrolyzed to generate a transmembrane proton gradient, which the bacterium requires for such functions as nutrient import and locomotion (1–6).
F1 (or “F1-ATPase”) has three catalytic nucleotide-binding sites, located on the three β subunits, at the interface to the adjacent α subunit. The catalytic sites have pronounced differences in their affinity for Mg2+-nucleotide. In Escherichia coli, Kd1 for MgATP is in the nanomolar range (“high-affinity site”), Kd2 is ≈1 μm (“medium-affinity site”), and Kd3 is ≈30–100 μm (“low-affinity site”) (7, 8). The affinity of a catalytic site at any given point of time is determined by the position of the central γ subunit. This implies that during rotational catalysis the affinities change. After rotation of γ by 120°, the sites have swapped their affinities. Experimental evidence for the crucial role of γ in determining the affinities of the catalytic sites comes from observations of the dependence of substrate binding and product release on the rotational angle of γ (9–13). Further support is provided by the discovery of mutations in β at the β/γ interface that affect nucleotide binding affinities despite the fact that these mutations are located in the DELSEED loop in the C-terminal domain (14, 15), ≥30 Å away from the catalytic binding site.
The affinity differences and changes seem to be of central importance for the enzymatic mechanism with respect to coupling between rotation and catalysis as well as catalysis itself. ATP synthesis and hydrolysis occur only on the high-affinity site (16). According to most catalytic models, in ATP synthesis proton translocation–driven rotation of γ forces the high-affinity site open, thereby reducing the affinity to “low” so that the newly formed ATP can be released (2, 17–19). In ATP hydrolysis, closing of the low-affinity site around the newly bound ATP, accompanied by conversion of the site to high-affinity, is widely believed to push γ and make it rotate for the experimentally observed 80° substep. The subsequent 40° substep appears to be energetically linked to the release of Pi and/or ADP (2, 12, 15, 17, 20–22).
Although a coherent picture of the general chemomechanical coupling mechanism is emerging, many aspects of the mechanism on the molecular level are still unresolved. For one, knowledge of the interactions between γ and β (and/or α) that are responsible for the different affinities of the catalytic binding sites is fragmentary. The majority of contacts between γ and the α3β3 ring involve the long N- and C-terminal helices of γ.
Because of the large number of interactions that might possibly contribute, the first goal of the present study was to identify regions of γ that play a role in the assignment of affinities. In search of a screening method that allows looking at multiple residues at the same time, our approach was based on the observation that it is possible to generate mutants in ATP synthase from Geobacillus stearothermophilus (formerly known as Bacillus PS3) that have portions of the N- and/or C-terminal helices removed (23–26). We measured binding of MgATP to a series of N- and C-terminal truncation mutants in the G. stearothermophilus α3β3γ subcomplex. For both helices, increasing the length of the truncation converted the MgATP binding behavior from asymmetric and WT-like, with a clearly present high-affinity site, to (nearly) symmetric, as also observed in the complete absence of γ. This approach allowed us to narrow down the region of γ causing the differences in affinity of the catalytic sites to two short segments of ∼10 residues, one in each helix.
To possibly identify individual residues responsible for conferring the nucleotide binding asymmetry, we replaced the amino acids in these two segments in groups of 5–6 residues by alanine. MgATP binding experiments with the alanine-replacement mutants all gave the WT-like asymmetric binding pattern. These results suggest that there is no specific residue in γ that causes the pronounced affinity differences of the three catalytic nucleotide-binding sites. Instead, the α-helices of γ themselves in the identified region appear to be the cause.
Results
Selection of enzyme source
The enzymes from E. coli and G. stearothermophilus (formerly Bacillus PS3) are arguably the best-characterized bacterial ATP synthases. They are sufficiently closely related so that the G. stearothermophilus F1 subcomplex can be reconstituted with E. coli Fo to give a functional ATP synthase and vice versa (27). For the present study, we chose the G. stearothermophilus enzyme because of its higher oligomeric stability. Specifically, it could be shown that the G. stearothermophilus ATP synthase can form an α3β3 complex in the absence of γ (28) that is stable enough to be crystallized (29) or monitored in high-speed atomic force microscopy (30). Under appropriate storage conditions, as ammonium sulfate precipitate at 4 °C, even after several years about two-thirds of the complex was still in α3β3 form with about 20% isolated α and β subunits and the remainder unidentified degradation products, which are absent directly after preparation of the enzyme (Fig. S1). Thus, truncations of γ should not affect the stability of the α3β3 complex.
N-truncation mutants of γ: an overview
The following N-terminal truncation mutants of the γ subunit of G. stearothermophilus ATP synthase α3(βY341W)3γ subcomplex were generated: γΔN4, missing the first 4 residues of γ; γΔN9; γΔN13; γΔN29; and γΔN45 (the numbering system assumes that E. coli, used to express the G. stearothermophilus enzyme, removes the Met encoded by the start codon as is observed for the native E. coli ATP synthase). Subcomplexes with full-length γ, and γ-less subcomplex were included as controls. The γΔN45 mutant eliminates all contacts between the N-terminal helix of γ and the α3β3 cylinder (31–33). The crystal structures of mitochondrial (31, 32) and E. coli F1 (33) indicate that the N-terminal helix of γ starts immediately at the N terminus. Secondary structure predictions suggest that the same applies to G. stearothermophilus WT γ and to the truncation mutants investigated here (Fig. 1A) with the possible exception of the γΔN9 mutant. For the γΔN9 mutant, the 5th residue is the first that reaches a probability for formation of an α-helix of ≥50%, whereas in all other cases it is the 2nd or 3rd residue; thus, for a few residues at the extreme N terminus of γΔN9, the helix might be unwound. For γΔN4, the predicted probability for formation of an α-helix by the initial residues is lower than in the other cases but still ≥50% (as compared with 90–100% for the other truncation mutants).
Figure 1.

Secondary structure predictions for the γ truncation mutants. All secondary structure predictions were obtained using the PredictProtein server (49). The amino acid sequence is given on top of the figure. The figure plots the probability of formation of an α-helix. The top panels (panels 1; black) show the prediction for the WT G. stearothermophilus enzyme with full-length γ. It should be noted that the prediction for γ of mitochondrial ATP synthase gave very similar results (not shown) with a lower probability of α-helix formation between residues γ10 and γ15 and a disruption of the helix between γ40 and γ45. The crystal structures (31, 32) show the γ10–15 segment as helical and confirm the helix break around γ40–45. A, N-terminal truncations. Panels 2–6 show the predictions for the N-terminal truncation mutants γΔN4 (green), γΔN9 (red), γΔN13 (blue), γΔN29 (pink), and γΔN45 (cyan). B, C-terminal truncations. Panels 2–6 show the predictions for the C-terminal mutations γΔC14 (green), γΔC20 (red), γΔC27 (blue), γΔC36 (pink), and γΔC60 (cyan).
C-truncation mutants of γ: an overview
The C-terminal γ truncation mutants generated for this study were as follows: γΔC14, eliminating 14 residues from the C terminus; γΔC20; γΔC27; γΔC36; and γΔC60. In the γΔC60 mutant, the C terminus is outside of the α3β3 cylinder. According to the crystal structures of the mitochondrial and E. coli enzymes (31–33), the C-terminal helix starts 73 and 76 residues, respectively, upstream of the C terminus and runs uninterrupted to the C terminus. Secondary structure predictions (Fig. 1B) suggest similar behavior for the C-terminal helix of the G. stearothermophilus enzyme, although for the terminal 6–7 residues the probability to form a helix is only about 50%. According to the predictions, also in the truncation mutants the helix reaches close to the respective C terminus.
Stability and ATPase activity of the α3β3γ subcomplexes
An α3β3γ subcomplex could be isolated from each of the five N-terminal truncation mutants and from four of five C-terminal truncation mutants. Of the N-terminal truncation mutants, based on Western blotting using an antibody against the globular portion of γ, the γΔN4 and γΔN9 mutants contained a full complement of γ. In the mutants with longer N-terminal truncations, the amount of γ was substoichiometric. Obviously, these truncations reduce the stability of the interactions of γ with the α3β3 cylinder. Still, even for the γΔN45 mutant in nearly a half of the enzyme complexes γ was still present (Table 1 and Fig. S2A). As to the C-terminal mutants, for the longest truncation, γΔC60, an α3β3γ subcomplex could not be isolated. No γ was found in the preparation; instead, an α3β3 subcomplex was obtained (data not shown). Thus, this mutant was not pursued any further. All shorter C-terminal truncations contained stoichiometric amounts of γ (Table 1 and Fig. S2B).
Table 1.
ATPase activities and ATP-binding properties of γ truncation mutants
The content of γ in the subcomplex preparations was measured by Western blotting using an antibody against the globular portion of γ. Each Western blot contained an α3β3γ control, set as 100%, and an α3β3 control, set as 0%. ATPase activities were determined in duplicate at 42 °C and pH 8.0 by the amount of Pi released. Nucleotide binding to the three catalytic sites was measured at 23 °C and pH 8.0 using the fluorescence of residue βTrp341 as signal. This technique does not allow resolving Kd values below 0.01 μm with confidence. All values given in this table represent the average from at least two independent experiments with two different enzyme preparations with standard deviations in parentheses.
| Enzyme/mutation | Content of γ | ATPase activity | MgATP binding |
||
|---|---|---|---|---|---|
| Kd1 | Kd2 | Kd3 | |||
| % | units/mg | μm | |||
| α3β3γ | 100 | 17 (3) | <0.010 | 4.4 (0.7) | 29 (6) |
| α3β3 | 0 | 0.09 (0.08) | 2.2 (0.7) | 10 (2) | 11 (2) |
| γΔN4 | 99 (5) | 25 (5) | <0.010 | 1.9 (0.6) | 54 (22) |
| γΔN9 | 103 (6) | 2.2 (0.2) | 0.41 (0.12) | 13 (2) | 13 (2) |
| γΔN13 | 67 (10) | 1.7 (0.4) | 2.2 (0.8) | 13 (2) | 13 (2) |
| γΔN29 | 53 (8) | 0.30 (0.03) | 1.4 (0.5) | 15 (3) | 16 (3) |
| γΔN45 | 46 (9) | 0.55 (0.12) | 1.2 (0.2) | 11 (2) | 11 (2) |
| γΔC14 | 95 (8) | 11 (2) | 0.015 (0.003) | 4.0 (1.0) | 23 (7) |
| γΔC20 | 104 (12) | 1.4 (0.3) | 0.012 (0.003) | 3.0 (0.8) | 16 (5) |
| γΔC27 | 114 (20) | 0.48 (0.13) | 1.1 (0.3) | 11 (2) | 11 (2) |
| γΔC36 | 110 (22) | 0.43 (0.13) | 1.8 (0.3) | 13 (2) | 13 (2) |
| γA5–10 | NDa | 13 (2) | <0.010 | 3.7 (0.9) | 16 (4) |
| γA10–15 | ND | 18 (2) | <0.010 | 4.8 (1.3) | 15 (5) |
| γA256–260 | ND | 31 (6) | <0.010 | 3.4 (0.7) | 25 (6) |
| γA260–265 | ND | 30 (6) | <0.010 | 2.8 (0.8) | 17 (6) |
a ND, not determined; as all alanine replacement mutants showed wildtype-like activities and MgATP binding pattern, measurement of the γ content seemed unnecessary.
Of the α3β3γ subcomplexes with N-terminal truncations, only γΔN4 showed an ATPase activity that was similar to that of the WT enzyme. In the case of the γΔN9 and γΔN13 α3β3γ subcomplexes, the ATPase activity was about 10% of the WT value. For γΔN29 and γΔN45 α3β3γ it was even less but still clearly higher than for a γ-less α3β3 subcomplex (Table 1). Of the C-terminal truncation mutants, again only the shortest one, γΔC14, had an ATPase activity in the same range of that of enzyme with full-length γ. The activity of γΔC20 was about 10% of WT; those of γΔC27 and γΔC36 were 2–3%. Again, even the C-terminal deletion mutants with the largest functional impairment still had ATPase activities significantly higher than the γ-less enzyme (Table 1). A general decrease of the ATPase activity with the length of the deletion had been described before for N-terminal as well as C-terminal truncations (23, 25, 26).
Effect of truncations on MgATP binding
The fluorescence of residue βTrp341 in the α3β3γ subcomplex was used to determine the MgATP binding properties of the truncation mutants. The results are compiled in Fig. 2, A (N-terminal truncations) and B (C-terminal truncations), and in Table 1. MgATP binding data for the WT α3β3γ subcomplex and the γ-less subcomplex, α3β3, are shown as controls. α3β3γ shows the well-established MgATP binding asymmetry with a Kd1 below 10 nm, a Kd2 of about 4 μm, and a Kd3 of about 30 μm. For the α3β3 subcomplex, in the absence of γ, it had been shown previously that it has a highly symmetrical structure (29) and has lost its ability to bind MgTNP-ATP5 and MgTNP-ADP with high affinity (34). Here, we confirmed the “functional symmetry” of the α3β3 subcomplex. High-affinity binding was not observed, and the three catalytic binding sites had very similar affinities for MgATP. An optimal fit was obtained using a model with three different sites; the fit suggested that one of the three sites might still have a slightly higher affinity than the remaining two sites. However, as can be seen from Fig. 2 (best visible in C), the difference between this fit (solid black line through the open circles) and the fit for a model with three identical sites (dotted black line) is minimal. In the latter case, Kd123 = 6.1 μm.
Figure 2.
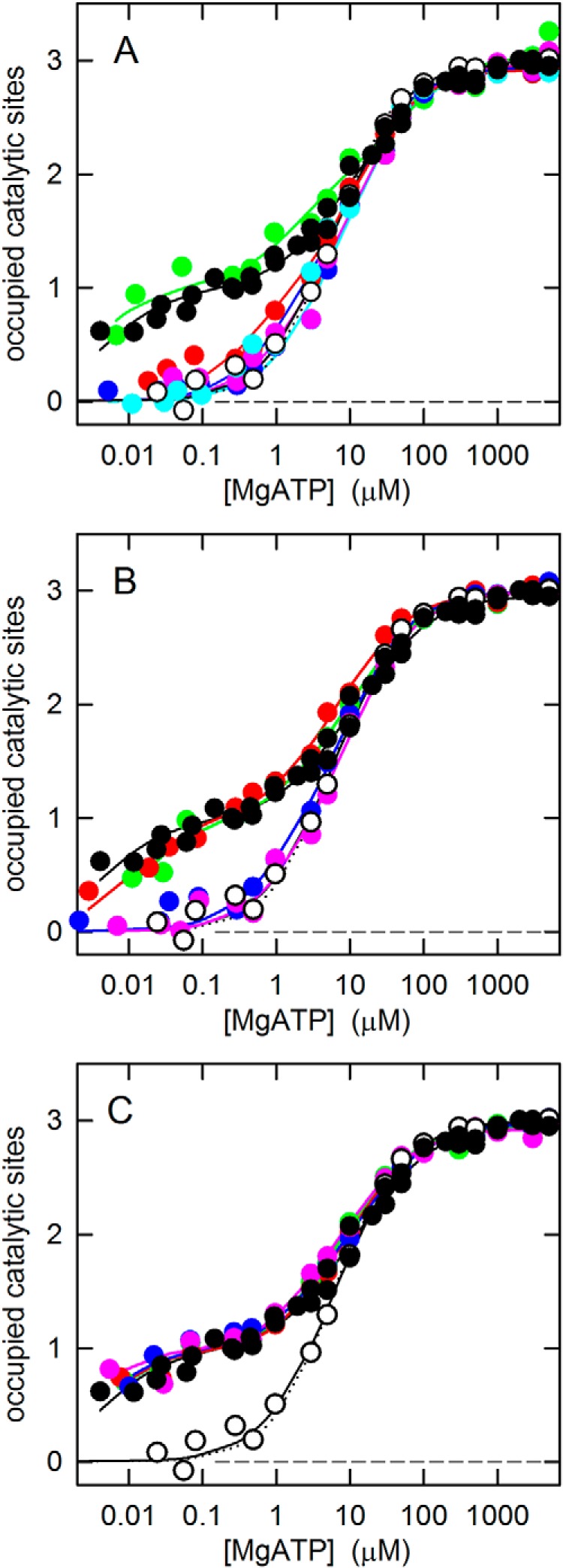
MgATP binding to the catalytic sites of the γ truncation and alanine-replacement mutants. MgATP binding to the three catalytic sites of the α3β3γ subcomplex of G. stearothermophilus ATP synthase was measured as described under “Experimental procedures.” In all panels, WT α3β3γ with full-length γ is represented by black filled circles, and α3β3 (no γ) is represented by white open circles. Each plot shows a representative experiment, combining data from four independent titrations, with the MgATP concentration increased by an order of magnitude in each step (8). The solid lines are fitted binding curves based on a model with three different, independent sites. Kd values are given in Table 1. For the γ-less α3β3 subcomplex, the fit based on a model with identical sites is also shown (dotted black lines). A, N-terminal truncation mutants: γΔN4, green; γΔN9, red; γΔN13, blue; γΔN29, pink; γΔN45, cyan. B, C-terminal truncation mutants γΔC14, green; γΔC20, red; γΔC27, blue; γΔC36, pink. C, alanine-replacement mutants: γA5–10, green; γA10–15, red; γA256–260, blue; γA260–265, pink.
In the nucleotide binding experiments, both series of truncation mutants showed the same tendencies, from a WT-like asymmetrical pattern with a clearly expressed high-affinity binding site for the shorter truncations to a symmetrical binding behavior with a loss of the high-affinity site for the longer truncations. Of the N-terminal truncation mutants, only γΔN4 displayed strong asymmetric behavior (Fig. 2A); of the C-terminal truncations, both γΔC14 and γΔC20 did (Fig. 2B). In contrast, γΔN13, γΔN29, and γΔN45 exhibited the (nearly) symmetrical binding pattern of the γ-less enzyme as did γΔC27 and γΔC36. Whereas for the C-terminal truncations the binding pattern went directly from WT-like in γΔC20 to γ-less-like in γΔC27, among the N-terminal truncations γΔN9 presented a transition case. The affinity of site 1 in γΔN9 appeared to be slightly higher than in the mutants with longer truncations.
As noted above, in γΔN13, γΔN29, and γΔN45, the amount of γ was substoichiometric. Nevertheless, as these mutants display binding characteristics that are virtually indistinguishable from those of α3β3, correction of the binding curves for “contamination” by γ-less enzyme did not change the results significantly. However, this correction assumes that the amount of γ in the α3β3γ subcomplex remains constant during the binding assay. To test whether this assumption is valid, we concentrated the assay mixture after the binding assay using a Centricon centrifugal filter device with an exclusion limit of 50,000 Da, which would let isolated γ pass through. Immunoblot analysis of the concentrate indicated that for γΔN29 and γΔN45 the amount of γ in the α3β3γ subcomplex was indeed further reduced by 30–50%. However, the γΔN13 α3β3γ subcomplex showed the same amount of γ as before the binding experiment (Fig. S2C). Thus, there is no doubt that γΔN13 displays a symmetrical binding pattern, just as γ-less enzyme. Although the precise amount of γ in the α3β3γ subcomplex of γΔN29 and γΔN45 at the exact moment of the binding assay is not known and therefore a correction for the contamination by γ-less enzyme is not straightforward, it appears highly unlikely that these truncation mutants would revert to an asymmetric pattern.
The segments of γ responsible for the nucleotide binding asymmetry of the catalytic sites: N terminus
At the N terminus, truncation of just 4 residues preserved the asymmetric WT binding pattern. Truncation of 13 or more residues resulted in the symmetric pattern seen in the absence of γ. Truncation of 9 residues showed intermediate characteristics with a higher degree of asymmetry than observed for the longer truncations but no pronounced high-affinity binding site. Based on the results, one might conclude that the segment most important for determination of the affinities of the three catalytic sites consists of residues γ5–9. However, it is possible that the failure of the γΔN9 truncation mutant to give a WT-like binding pattern (like γΔN4) could be due to failure to form an α-helix immediately at its N terminus (see “Results” and Fig. 1). In contrast to γΔN9, secondary predictions for γΔN13 indicate that this mutant reaches α-helical conformation within 2–3 residues. Thus, the affinity-determining segment on the N-terminal helix should not extend beyond residue γ15, giving it the sequence γ5DIKTRINATKK15.
None of the residues of the γ5DIKTRINATKK15 segment is completely conserved; however, several display exclusively conservative substitutions (Table S1). In general, positions γ7–γ15 show an accumulation of arginine and lysine residues, between 2 and 5 in the examples shown; the net charge of this stretch is between +1 and +5. The γ5–15 segment comes close to three subunits of the α3β3 cylinder, βTP,6 βDP, and αE (Figs. 3A and S3A). In the structure of the E. coli enzyme (33), γ5–15 also has contacts with the C-terminal helix of the ϵ subunit. Possible interactions with βTP and βDP occur with the DELSEED loop of these subunits. The multiple negatively charged side chains of the DELSEED loop appear well suited for interactions with the positive charges of the γ5–15 segment. However, it has been shown that the negative charges of the DELSEED motif can be removed without affecting enzymatic activity, MgATP binding asymmetry, and rotational torque (35–38).
Figure 3.

Identification of the segments in the N- and C-terminal helices of γ responsible for the affinity differences of the catalytic sites. A, side view. The identified segment in the N-terminal helix, γ5–15, is shown in pink; the segment in the C-terminal helix, γ256–265, is shown in cyan; and the remainder of γ is shown in purple. Subunit βTP, which carries the high-affinity nucleotide-binding site, is shown in yellow; subunit βDP with the medium-affinity binding site is colored green, and subunit βE is orange. Bound nucleotides are shown as “space-filled”; the site on βE is empty in the crystal structure. For clarity, the α subunits have been removed. B, top view, looking toward the membrane. Color-coding is as in A. Bound nucleotides are shown in “stick” representation. The portion of the β subunits above the level of the nucleotide-binding sites has been removed. γ is represented just by the two segments, γ5–15 and γ256–265. Segment γ5–15 makes contact with the DELSEED loop of subunits βTP and βDP; segment γ256–265 makes contact with the catch loop of subunit βE (for further details, see “Results” and “Discussion”).
The segments of γ responsible for the nucleotide binding asymmetry of the catalytic sites: C terminus
At the C terminus, truncation of up to 20 residues gave the asymmetric, WT-like binding pattern, whereas removal of 27 or more residues resulted in the symmetric pattern, lacking a high-affinity binding site, also observed in the absence of γ. Thus, the affinity-determining segment of the C-terminal helix should, at the minimum, consist of residues γ259TLSYNRA265, which are present in γΔC20 but absent in γΔC27. Taking into account that the terminal residues of γΔC27 might not be in quite the same helical confirmation as in full-length γ, a reasonable estimate for the C-terminal affinity-determining segment appears to be γ256RTLTLSYNRA265. Of these residues, γAsn263 is conserved; several other positions contain exclusively conservative substitutions (Table S2).
Is it possible to identify the residue(s) responsible for the nucleotide binding asymmetry?
To answer this question, all residues in the two identified segments were replaced by alanine. We made two alanine mutants per segment by replacing 5–6 residues at a time by alanine because making longer alanine mutants was challenging. Thus, we generated the mutations γA5–10, γA10–15, γA256–260, and γA260–265 for N- and C-terminal segments, respectively.
The results of the functional assays are shown in Table 1 and Fig. 2C. All four mutant enzymes had WT-like ATPase activity and a WT-like asymmetric MgATP binding pattern. Thus, it seems that no specific interaction between amino acid side chains in γ and β (or α) causes the binding asymmetry. Instead, it appears as if the α-helices themselves in this region are responsible.
Discussion
One major goal of the research on ATP synthase in our lab at this time is to explore the molecular basis for the changes in affinity of the catalytic sites during rotation and to analyze the role of these affinity changes in coupling of catalysis and rotation. The affinity of a catalytic site for substrate(s) and product(s) at any given point of time is determined by the position of the central γ subunit. In the absence of γ, all three sites have the same, rather low affinity. Several residues at the β/γ interface that affect binding affinities of the catalytic sites have been described before (14, 15, 39). In the present study, we aimed to identify the part(s) of γ that included all residues that are responsible for the affinity differences. For this purpose, we measured nucleotide binding to the catalytic sites of two series of γ truncation mutants, one that had between 4 and 45 residues from the N terminus of γ removed and the other between 14 and 60 residues from the C terminus. The truncations were done using the enzyme from G. stearothermophilus because of its superior oligomeric stability. In the absence of γ, it can form a stable α3β3 complex (28), which has not yet been described for the E. coli enzyme. However, both enzymes are functionally so similar, specifically in their nucleotide binding pattern (for comparison, see the Kd(MgATP) values for E. coli given in the Introduction and the G. stearothermophilus values in Table 1), that there is no reason to assume that these patterns were achieved by different mechanisms.
The truncation approach yielded two segments of about 10 residues, one on the N-terminal helix, γ5–15, and the other on the C-terminal helix, γ256–265. Both segments are required to give the asymmetric, WT-like nucleotide binding pattern with pronounced affinity differences and a clearly expressed high-affinity site. If one of them is missing, the enzyme shows the symmetric binding behavior with a relatively low overall affinity that is also observed in the complete absence of γ.
Over both segments, about half of the residues are conserved or conservatively substituted (see “Results” and Tables S1 and S2), and a number of these residues are able to form hydrogen bonds and/or salt bridges with residues on the α or β subunits (Fig. S3). However, experiments where the residues in both segments were replaced by alanine gave a WT-like asymmetric nucleotide binding pattern and WT-like catalytic activity. Thus, no individual amino acid side chains appear to be responsible for asymmetry and activity. This finding is reminiscent of the βDELSEED loop, which is essential for driving γ rotation and enzymatic activity due to its overall bulk shape, without individual interactions between amino acid side chains required for its function (35–38).
The affinity-determining segment of the N-terminal helix, γ5–15, makes contact with the DELSEED loop of the βTP and βDP subunits, which carry the high- and medium-affinity catalytic site, respectively (18). This observation confirms the importance of the DELSEED loop in affinity regulation of the catalytic sites as suggested by previous studies (14, 15). Absence of the γ5–15 segment apparently fails to bring the respective β subunit into the high- or medium-affinity conformation. The affinity-determining segment of the C-terminal helix, γ256–265, is located at a level similar to its N-terminal counterpart, γ5–15, offset by approximately one helical turn in direction away from the membrane (Fig. 3). A portion of the γ256–265 segment approaches the “catch loop” (31, 40) of the βE subunit, residues β309–316. The γΔC27 truncation removes any interaction of the C-terminal helix with the catch loop. Considering that the affinity of the low-affinity catalytic site on βE is not affected by γ, it seems possible that the role of this segment in determination of the affinity of the catalytic sites on βTP and βDP is indirect, by stabilizing the N-terminal γ5–15 segment and its interactions with α3β3. Theoretically, it would also be possible that lack of this portion of the C-terminal helix might cause failure to form the N-terminal helix (41). However, we could show that a γ subunit consisting just of the N-terminal 35 or 42 residues was able to sustain a certain degree of ATP synthesis activity (19), which would be highly unlikely if the segment were unstructured. Furthermore, it has been shown that truncation of the N terminus by 50 residues (26) or of the C terminus by 36 residues (25) still allows production of torque close to 50% of WT, whereas the torque generated by an enzyme without both helices is significantly lower (24). Taken together, these findings suggest that each of the helices can exist without support of the other. Thus, the structural integrity of γ is preserved in the truncation mutants.
Interestingly, despite the presence of a high-affinity site, the γΔC20 truncation mutant showed only low ATPase activity; adding back 6 more residues, in the γΔC14 mutant, restored WT-like activities. This finding is in contrast to the situation at the N terminus where the presence of a high-affinity site went hand in hand with normal ATPase activities as observed with γΔN4; the γΔN9 mutant, which had largely lost high-affinity binding capability, also had substantially reduced enzymatic activity. Thus, as far as the mutants under investigation here are concerned, the presence of a high-affinity site appears to be a necessary, but not sufficient, condition for high activity.
The 2 residues immediately downstream of the identified segment, γArg266 and γGln267 are strictly conserved. However, from the results presented here, it is clear that they have no influence on the binding affinity of the catalytic sites as these residues are missing in γΔC20, which nevertheless has a WT-like binding pattern. Given the importance of γArg266 and γGln267 for the catalytic function (40, 42),7 it is likely that their absence in γΔC20 is responsible for the low enzymatic activity of this truncation mutant. The lack of residue γThr271 in γΔC20 (which is present in γΔC14) could be another factor for its catalytic impairment (42, 43). It is interesting to note that loss of hydrogen-bonding capability of γThr271 resulted in reduced nucleotide-binding affinity of the catalytic site(s) in the transition state (measured as rate of formation of the transition-state analog MgADP–fluoroaluminate complex (43)). However, as the results presented here show, γThr271 has no role in determining the affinity in the ground state.
Experimental procedures
Bacterial strains and plasmids
For generation of the γ truncation mutants in G. stearothermophilus α3β3γ and the γ-less α3β3 subcomplexes, the background plasmid was pNM2. Plasmid pNM2 is a derivative of plasmid pKAGB1 (44). pKAGB1 is used to express a Cys- and Trp-less form of the α3β3γ subcomplex of G. stearothermophilus ATP synthase. pNM2 contains an additional mutation to generate an α3(βY341W)3γ subcomplex, which allows monitoring nucleotide binding to the three catalytic sites, and a His10 tag at the N terminus of the β subunits to facilitate purification. Site-directed mutagenesis was performed using the QuikChange II XL kit. To generate the N-terminal truncations, downstream of the γ start codon ATG, the codons for the next 4–45 amino acids were eliminated. To obtain the C-terminal truncations, stop codons were inserted at the desired positions. For generation of the γ-less enzyme, a stop codon was introduced at the γ7 position. The insertion of an NheI site downstream of this stop codon allowed us to remove the remainder of the gene for γ on an NheI-NheI fragment as there is a natural NheI site downstream of the γ gene. Removal of segments was confirmed by DNA sequencing of the plasmid product. For expression of the mutant proteins, the plasmids were transformed into E. coli strain JM103Δ(uncB-uncD).
Isolation of α3β3(γ) subcomplex and quantification of truncated γ subunit
The purification method of α3β3γ or α3β3 subcomplex was modified from a previously described procedure (45). Cells were grown aerobically at 37 °C in terrific broth medium containing 100 μg/ml ampicillin. After cell lysis by French press, the cell debris was removed by centrifugation at 35,000 rpm for 30 min. The supernatant containing the complex was applied to a Ni2+-nitrilotriacetic acid column (Qiagen) equilibrated with 20 mm imidazole and 100 mm NaCl, pH 7.0. The column was washed with 50 mm imidazole and 100 mm NaCl, pH 7.0, and the enzyme was eluted with 500 mm imidazole and 100 mm NaCl, pH 7.0. The subcomplex was stored as precipitate in 70% saturated ammonium sulfate at 4 °C. The amount of truncated γ subunit was determined via Western blotting using antibodies raised against a peptide corresponding to a part of the globular portion of γ; the antibodies were a kind gift from Drs. Toshiharu Suzuki and Masasuke Yoshida (Japan Science and Technology Agency, Tokyo, Japan). α3β3γ subcomplex with full-length γ served as a standard. Blots were then developed with enhanced chemoluminescence substrate (Thermo Scientific). The expression level of γ subunit was quantified by measuring the integrated density of bands using a Photodyne imaging system and NIH ImageJ acquisition software. Control experiments with an anti-β antibody (Agrisera, Vännäs, Sweden) showed that the amount of β was not affected by the mutations (Fig. S2, A and B).
The oligomeric state of α3β3γ and α3β3 samples was assessed by size exclusion chromatography. A Bio-Sil SEC 250 7.8 × 80-mm column, equilibrated with 100 mm sodium phosphate buffer, pH 6.8, was used. The eluate was monitored by UV absorbance at 280 nm. The calculated molecular masses of α3β3γ and α3β3 complexes and α and β individual subunits, based on their amino acid sequences, are 352, 320, 55, and 52 kDa, respectively. BSA (66 kDa) and bovine heart lactate dehydrogenase (137 kDa) were used as controls.
Functional analysis of mutant enzymes
ATPase activities were assayed in a buffer containing 50 mm Tris/H2SO4, 10 mm ATP, and 4 mm MgSO4, pH 8.0, at 42 °C. The reaction was started by addition of 10–20 μg/ml enzyme and stopped after 1 or 2 min (depending on the activity) by addition of SDS (final concentration, 5%, w/v). The released Pi was measured as described (46). 1 unit of enzymatic activity corresponds to 1 μmol of ATP hydrolyzed (equivalent to 1 μmol of Pi produced)/min.
Binding of MgATP to the catalytic sites of the purified α3β3 or α3β3γ subcomplex was measured using the fluorescence of the inserted Trp residue βTrp341 (7, 8). Before use, the α3β3γ ammonium sulfate precipitate was pelleted by centrifugation and redissolved in a buffer containing 50 mm Tris/HCl and 10 mm CDTA, pH 8.0. After 1-h incubation at 23 °C, the α3β3γ subcomplex was passed through two subsequent centrifuge columns containing Sephadex G-50 equilibrated with 50 mm Tris/HCl and 0.1 mm EDTA, pH 8.0. After this treatment, the enzyme subcomplex is essentially nucleotide-free (47); using the luciferin/luciferase method after heat denaturation of the enzyme complex, we found <0.1 mol of nucleotide (ATP plus ADP)/mol of subcomplex. To measure MgATP binding, fluorescence titrations were performed in a buffer containing 50 mm Tris/H2SO4 and 2.5 mm MgSO4, pH 8.0, with ATP added to the desired concentration. Kd values were determined by fitting of theoretical curves to the experimental data points by nonlinear least-squares analysis. All functional assays were performed within 2–3 days after preparation of the enzyme.
Miscellaneous
Protein concentrations were determined by the method of Bradford (48) using BSA as a standard. Secondary structure predictions were performed using the PredictProtein server (49). Figs. 3 and S3 were created using PyMOL (Schrödinger, Portland, OR) using Protein Data Bank code 1E79 (32) as starting material.
Author contributions
N. M. and Y. L. data curation; N. M., Y. L., and J. W. formal analysis; N. M. and Y. L. investigation; N. M., Y. L., and J. W. writing-review and editing; J. W. conceptualization; J. W. supervision; J. W. funding acquisition; J. W. writing-original draft; J. W. project administration.
Supplementary Material
Acknowledgment
We thank Cristina Pinal for valuable technical assistance.
Part of this work was supported by National Institutes of Health Grant GM071462 (including American Recovery and Reinvestment Act Administrative Supplement) (to J. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Tables S1 and S2.
The nomenclature of α and β subunits is based on the nucleotide occupancy of the catalytic sites in the original structure of the mitochondrial enzyme (31). Subunits αTP and βTP contribute to formation of the catalytic site occupied by a non-hydrolyzable ATP analog, AMP-PNP (5′-adenylyl-β,γ-imidodiphosphate); αDP and βDP contribute to formation of the catalytic site occupied by ADP; and αE and βE contribute to formation of the empty catalytic site.
The cited references use the E. coli enzyme. Although at the N terminus of γ the amino acid numbers for G. stearothermophilus and E. coli are the same, at the C terminus adding 2 to the G. stearothermophilus numbers gives those for E. coli.
- TNP
- trinitrophenyl
- CDTA
- trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid.
References
- 1. Weber J., and Senior A. E. (2003) ATP synthesis driven by proton transport in F1Fo-ATP synthase. FEBS Lett. 545, 61–70 10.1016/S0014-5793(03)00394-6 [DOI] [PubMed] [Google Scholar]
- 2. Nakamoto R. K., Baylis Scanlon J. A., and Al-Shawi M. K. (2008) The rotary mechanism of the ATP synthase. Arch. Biochem. Biophys. 476, 43–50 10.1016/j.abb.2008.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von Ballmoos C., Wiedenmann A., and Dimroth P. (2009) Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Rev. Biochem. 78, 649–672 10.1146/annurev.biochem.78.081307.104803 [DOI] [PubMed] [Google Scholar]
- 4. Watanabe R., and Noji H. (2013) Chemomechanical coupling mechanism of F1-ATPase: catalysis and torque generation. FEBS Lett. 587, 1030–1035 10.1016/j.febslet.2013.01.063 [DOI] [PubMed] [Google Scholar]
- 5. Junge W., and Nelson N. (2015) ATP synthase. Annu. Rev. Biochem. 84, 631–657 10.1146/annurev-biochem-060614-034124 [DOI] [PubMed] [Google Scholar]
- 6. Nakanishi-Matsui M., Sekiya M., and Futai M. (2016) ATP synthase from Escherichia coli: mechanism of rotational catalysis, and inhibition with the ϵ subunit and phytopolyphenols. Biochim. Biophys. Acta 1857, 129–140 10.1016/j.bbabio.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 7. Weber J., Wilke-Mounts S., Lee R. S., Grell E., and Senior A. E. (1993) Specific placement of tryptophan in the catalytic sites of Escherichia coli F1-ATPase provides a direct probe of nucleotide binding: maximal ATP hydrolysis occurs with three sites occupied. J. Biol. Chem. 268, 20126–20133 [PubMed] [Google Scholar]
- 8. Weber J., and Senior A. E. (2004) Fluorescent probes applied to catalytic cooperativity in ATP synthase. Methods Enzymol. 380, 132–152 10.1016/S0076-6879(04)80006-5 [DOI] [PubMed] [Google Scholar]
- 9. Nishizaka T., Oiwa K., Noji H., Kimura S., Muneyuki E., Yoshida M., and Kinosita K. Jr. (2004) Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation. Nat. Struct. Mol. Biol. 11, 142–148 10.1038/nsmb721 [DOI] [PubMed] [Google Scholar]
- 10. Adachi K., Oiwa K., Nishizaka T., Furuike S., Noji H., Itoh H., Yoshida M., and Kinosita K. Jr. (2007) Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321 10.1016/j.cell.2007.05.020 [DOI] [PubMed] [Google Scholar]
- 11. Watanabe R., Iino R., and Noji H. (2010) Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nat. Chem. Biol. 6, 814–820 10.1038/nchembio.443 [DOI] [PubMed] [Google Scholar]
- 12. Watanabe R., and Noji H. (2014) Timing of inorganic phosphate release modulates the catalytic activity of ATP-driven rotary motor protein. Nat. Commun. 5, 3486 10.1038/ncomms4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weber J. (2010) Structural biology: Toward the ATP synthase mechanism. Nat. Chem. Biol. 6, 794–795 10.1038/nchembio.458 [DOI] [PubMed] [Google Scholar]
- 14. Scanlon J. A., Al-Shawi M. K., and Nakamoto R. K. (2008) A rotor-stator cross-link in the F1-ATPase blocks the rate-limiting step of rotational catalysis. J. Biol. Chem. 283, 26228–26240 10.1074/jbc.M804858200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mnatsakanyan N., Krishnakumar A. M., Suzuki T., and Weber J. (2009) The role of the βDELSEED-loop of ATP synthase. J. Biol. Chem. 284, 11336–11345 10.1074/jbc.M900374200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scanlon J. A., Al-Shawi M. K., Le N. P., and Nakamoto R. K. (2007) Determination of the partial reactions of rotational catalysis in F1-ATPase. Biochemistry 46, 8785–8797 10.1021/bi700610m [DOI] [PubMed] [Google Scholar]
- 17. Gao Y. Q., Yang W., and Karplus M. (2005) A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase. Cell 123, 195–205 10.1016/j.cell.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 18. Mao H. Z., and Weber J. (2007) Identification of the βTP site in the x-ray structure of F1-ATPase as the high-affinity catalytic site. Proc. Natl. Acad. Sci. U.S.A. 104, 18478–18483 10.1073/pnas.0709322104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mnatsakanyan N., Hook J. A., Quisenberry L., and Weber J. (2009) ATP synthase with its γ subunit reduced to the N-terminal helix can still catalyze ATP synthesis. J. Biol. Chem. 284, 26519–26525 10.1074/jbc.M109.030528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pu J., and Karplus M. (2008) How subunit coupling produces the γ-subunit rotary motion in F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1192–1197 10.1073/pnas.0708746105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yasuda R., Noji H., Yoshida M., Kinosita K. Jr, and Itoh H. (2001) Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410, 898–904 10.1038/35073513 [DOI] [PubMed] [Google Scholar]
- 22. Shimabukuro K., Yasuda R., Muneyuki E., Hara K. Y., Kinosita K. Jr, and Yoshida M. (2003) Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40° substep rotation. Proc. Natl. Acad. Sci. U.S.A. 100, 14731–14736 10.1073/pnas.2434983100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hossain M. D., Furuike S., Maki Y., Adachi K., Ali M. Y., Huq M., Itoh H., Yoshida M., and Kinosita K. Jr. (2006) The rotor tip inside a bearing of a thermophilic F1-ATPase is dispensable for torque generation. Biophys. J. 90, 4195–4203 10.1529/biophysj.105.079087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furuike S., Hossain M. D., Maki Y., Adachi K., Suzuki T., Kohori A., Itoh H., Yoshida M., and Kinosita K. Jr. (2008) Axle-less F1-ATPase rotates in the correct direction. Science 319, 955–958 10.1126/science.1151343 [DOI] [PubMed] [Google Scholar]
- 25. Hossain M. D., Furuike S., Maki Y., Adachi K., Suzuki T., Kohori A., Itoh H., Yoshida M., and Kinosita K. Jr. (2008) Neither helix in the coiled coil region of the axle of F1-ATPase plays a significant role in torque production. Biophys. J. 95, 4837–4844 10.1529/biophysj.108.140061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohori A., Chiwata R., Hossain M. D., Furuike S., Shiroguchi K., Adachi K., Yoshida M., and Kinosita K. Jr. (2011) Torque generation in F1-ATPase devoid of the entire amino-terminal helix of the rotor that fills half of the stator orifice. Biophys J 101, 188–195 10.1016/j.bpj.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steffens K., Di Gioia A., Deckers-Hebestreit G., and Altendorf K. (1987) Structural and functional relationship of ATP synthases (F1Fo) from Escherichia coli and the thermophilic bacterium PS3. J. Biol. Chem. 262, 6334–6338 [PubMed] [Google Scholar]
- 28. Miwa K., and Yoshida M. (1989) The α3β3 complex, the catalytic core of F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 86, 6484–6487 10.1073/pnas.86.17.6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shirakihara Y., Leslie A. G., Abrahams J. P., Walker J. E., Ueda T., Sekimoto Y., Kambara M., Saika K., Kagawa Y., and Yoshida M. (1997) The crystal structure of the nucleotide-free α3β3 subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5, 825–836 10.1016/S0969-2126(97)00236-0 [DOI] [PubMed] [Google Scholar]
- 30. Uchihashi T., Iino R., Ando T., and Noji H. (2011) High-speed atomic force microscopy reveals rotary catalysis of rotorless F1-ATPase. Science 333, 755–758 10.1126/science.1205510 [DOI] [PubMed] [Google Scholar]
- 31. Abrahams J. P., Leslie A. G., Lutter R., and Walker J. E. (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 10.1038/370621a0 [DOI] [PubMed] [Google Scholar]
- 32. Gibbons C., Montgomery M. G., Leslie A. G., and Walker J. E. (2000) The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat. Struct. Biol. 7, 1055–1061 10.1038/80981 [DOI] [PubMed] [Google Scholar]
- 33. Cingolani G., and Duncan T. M. (2011) Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nat Struct Mol Biol 18, 701–707 10.1038/nsmb.2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaibara C., Matsui T., Hisabori T., and Yoshida M. (1996) Structural asymmetry of F1-ATPase caused by the γ subunit generates a high affinity nucleotide binding site. J. Biol. Chem. 271, 2433–2438 10.1074/jbc.271.5.2433 [DOI] [PubMed] [Google Scholar]
- 35. Hara K. Y., Noji H., Bald D., Yasuda R., Kinosita K. Jr, and Yoshida M. (2000) The role of the DELSEED motif of the β subunit in rotation of F1-ATPase. J. Biol. Chem. 275, 14260–14263 10.1074/jbc.275.19.14260 [DOI] [PubMed] [Google Scholar]
- 36. Mnatsakanyan N., Kemboi S. K., Salas J., and Weber J. (2011) The β subunit loop that couples catalysis and rotation in ATP synthase has a critical length. J. Biol. Chem. 286, 29788–29796 10.1074/jbc.M111.254730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanigawara M., Tabata K. V., Ito Y., Ito J., Watanabe R., Ueno H., Ikeguchi M., and Noji H. (2012) Role of the DELSEED loop in torque transmission of F1-ATPase. Biophys. J. 103, 970–978 10.1016/j.bpj.2012.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watanabe R., Koyasu K., You H., Tanigawara M., and Noji H. (2015) Torque transmission mechanism via DELSEED loop of F1-ATPase. Biophys. J. 108, 1144–1152 10.1016/j.bpj.2015.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boltz K. W., and Frasch W. D. (2006) Hydrogen bonds between the α and β subunits of the F1-ATPase allow communication between the catalytic site and the interface of the β catch loop and the γ subunit. Biochemistry 45, 11190–11199 10.1021/bi052592+ [DOI] [PubMed] [Google Scholar]
- 40. Greene M. D., and Frasch W. D. (2003) Interactions among γ R268, γ Q269, and the β subunit catch loop of Escherichia coli F1-ATPase are important for catalytic activity. J. Biol. Chem. 278, 51594–51598 10.1074/jbc.M309948200 [DOI] [PubMed] [Google Scholar]
- 41. Lumb K. J., Carr C. M., and Kim P. S. (1994) Subdomain folding of the coiled coil leucine zipper from the bZIP transcriptional activator GCN4. Biochemistry 33, 7361–7367 10.1021/bi00189a042 [DOI] [PubMed] [Google Scholar]
- 42. Iwamoto A., Miki J., Maeda M., and Futai M. (1990) H+-ATPase γ subunit of Escherichia coli. Role of the conserved carboxyl-terminal region. J. Biol. Chem. 265, 5043–5048 [PubMed] [Google Scholar]
- 43. Boltz K. W., and Frasch W. D. (2005) Interactions of γ T273 and γ E275 with the β subunit PSAV segment that links the γ subunit to the catalytic site Walker homology B aspartate are important to the function of Escherichia coli F1Fo ATP synthase. Biochemistry 44, 9497–9506 10.1021/bi050070o [DOI] [PubMed] [Google Scholar]
- 44. Matsui T., and Yoshida M. (1995) Expression of the wild-type and the Cys-/Trp-less α3β3γ complex of thermophilic F1-ATPase in Escherichia coli. Biochim. Biophys. Acta 1231, 139–146 10.1016/0005-2728(95)00070-Y [DOI] [PubMed] [Google Scholar]
- 45. Adachi K., Noji H., and Kinosita K. Jr. (2003) Single-molecule imaging of rotation of F1-ATPase. Methods Enzymol. 361, 211–227 10.1016/S0076-6879(03)61013-X [DOI] [PubMed] [Google Scholar]
- 46. Taussky H. H., and Shorr E. (1953) A microcolorimetric method for the determination of inorganic phosphorus. J. Biol. Chem. 202, 675–685 [PubMed] [Google Scholar]
- 47. Ren H., Bandyopadhyay S., and Allison W. S. (2006) The α3(βMet222Ser/Tyr345Trp)3γ subcomplex of the TF1-ATPase does not hydrolyze ATP at a significant rate until the substrate binds to the catalytic site of the lowest affinity. Biochemistry 45, 6222–6230 10.1021/bi060232w [DOI] [PubMed] [Google Scholar]
- 48. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 49. Rost B., Yachdav G., and Liu J. (2004) The PredictProtein server. Nucleic Acids Res. 32, W321–W326 10.1093/nar/gkh377 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.