

Abstract
Dysregulation of the ErbB family of receptor tyrosine kinases is involved in the progression of many cancers. Antibodies targeting the dimerization domains of family members EGFR and HER2 are approved cancer therapeutics, but efficacy is restricted to a subset of tumors and resistance often develops in response to treatment. A third family member, HER3, heterodimerizes with both EGFR and HER2 and has also been implicated in cancer. Consequently, there is strong interest in developing antibodies that target HER3, but to date, no therapeutics have been approved. To aid the development of anti-HER3 antibodies as cancer therapeutics, we combined antibody engineering and functional genomics screens to identify putative mechanisms of resistance or synthetic lethality with antibody-mediated anti-proliferative effects. We developed a synthetic antibody called IgG 95, which binds to HER3 and promotes ubiquitination, internalization, and receptor down-regulation. Using an shRNA library targeting enzymes in the ubiquitin proteasome system, we screened for genes that effect response to IgG 95 and uncovered the E3 ubiquitin ligase RNF41 as a driver of IgG 95 anti-proliferative activity. RNF41 has been shown previously to regulate HER3 levels under normal conditions and we now show that it is also responsible for down-regulation of HER3 upon treatment with IgG 95. Moreover, our findings suggest that down-regulation of RNF41 itself may be a mechanism for acquired resistance to treatment with IgG 95 and perhaps other anti-HER3 antibodies. Our work deepens our understanding of HER3 signaling by uncovering the mechanistic basis for the anti-proliferative effects of potential anti-HER3 antibody therapeutics.
Keywords: antibody, antibody engineering, ubiquitylation (ubiquitination), functional genomics, E3 ubiquitin ligase, cell signaling, short hairpin RNA (shRNA), cancer, protein engineering, HER3, RNF41
Introduction
The human ErbB family of receptor tyrosine kinases is comprised of four homologous, transmembrane surface proteins: EGFR (ErbB1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4), which control important oncogenic functions such as proliferation, apoptosis, migration, and differentiation (1). Under normal physiological conditions, activation of EGFR, HER3, and HER4 occurs upon ligand binding, which leads to extracellular domain reorganization from a closed form to an open form, dimerization with another family member, and signaling through the cytoplasmic kinase domains. In contrast, HER2 has no known ligand and the extracellular domain is in a permanently open conformation, permitting ligand-independent signaling via homodimerization and heterodimerization (2). In some cancers, gain-of-function mutations or gene amplifications cause hyperactivation of ErbB family signaling, and are associated with disease progression (1). As perhaps the best characterized example, ∼20% of breast cancer patients exhibit HER2 amplification (3). The tumors of these patients typically respond to anti-HER2 therapeutic antibodies such as trastuzumab or pertuzumab, but resistance and disease progression remain major challenges for HER2-amplified tumors (4, 5).
Pathophysiological signaling in HER2-amplified settings is largely dependent on HER3, which forms a potent oncogenic heterodimer with HER2 even in the absence of ligand (6, 7). The formation of HER2/HER3 heterodimers leads to phosphorylation of HER3 by HER2 at up to six phosphorylation sites that mediate phosphoinositide 3-kinase (PI3K)6 binding and Akt signaling (8–11). Given the capacity for HER3 to elicit potent signaling through PI3K/Akt, it is not surprising that the oncogenic activity of HER3 has been demonstrated in a number of models. Systems biology studies have highlighted HER3 essentiality in HER2-amplified cells (12) and in models dependent on paracrine signaling by Neuregulin 1 (NRG1) (13, 14), a HER3 ligand that promotes Akt activation by enforcing heterodimerization between HER3 and HER2 or EGFR. Furthermore, HER3 is overexpressed in response to a number of targeted therapies, such as inhibitors of HER2 (15, 16), MEK (17, 18), and BRAF (19, 20). Collectively, these data highlight the importance of HER3 in tumorigenesis and suggest value for HER3 as a potential therapeutic target.
Unlike other ErbB family members, HER3 kinase activity is weak and has no known role in signal transduction (21, 22), thereby excluding small molecule inhibitors as therapeutics (23). However, over the past decade, a number of anti-HER3 antibodies have entered pre-clinical and clinical testing (24). Most antibodies described in the literature inhibit NRG1-induced HER3 activation and subsequent PI3K/Akt pathway activation, and there is evidence that some of these antibodies directly compete with NRG1 for HER3 binding (25, 26). NRG1 expression may be a valuable prognostic marker for response to NRG1 competitive antibodies, as preclinical models with high NRG1 levels are associated with a therapeutic response (27, 28). Similarly, retrospective analysis of clinical trial samples from patients treated with NRG1-competitive antibodies have highlighted the putative benefits of using these biologics in diseases with high NRG1 expression (29–32).
The strongest evidence for anti-HER3 antibody efficacy in HER2-amplified settings that exhibit ligand-independent growth comes from studies with the anti-HER3 antibodies KTN3379 (33, 34) and LJM716 (35). These antibodies lock HER3 in the closed inactive confirmation, blocking rearrangement to the open form that binds to HER2. However, given the propensity for HER2/HER3 heterodimers to form in a ligand-independent manner upon HER2-amplification (36), we hypothesized that targeting the open form of HER3 may provide additional benefit by enabling recognition of HER3 in the open and dimerized form.
Here we report the development of a new anti-HER3 antibody, IgG 95, which binds the open, HER2-bound form of HER3, blocks ligand binding, and inhibits signaling and proliferation in ligand-dependent and ligand-independent cancer models. IgG 95 enhanced HER3 ubiquitination and reduced HER3 and phosphorylated AKT (pAKT) levels. A subsequent shRNA screen targeting the ubiquitin proteasome system (UPS) revealed that down-regulation of the mRNA for the E3 ligase RNF41 enhanced resistance to IgG 95. Our work further shows that the anti-proliferative activity of IgG 95 depends on RNF41 through a mechanism whereby antibody binding causes ubiquitination of HER3 by RNF41, which leads to receptor internalization and degradation, and consequent inhibition of signaling. We anticipate that the mechanistic insights provided by our study will aid in the development of therapeutic antibodies targeting HER3.
Results
Development of synthetic anti-HER3 antibodies
We subjected the recombinant Fc-tagged extracellular domain (ECD) of HER3 (HER3-Fc) to two rounds of binding selections with a phage-displayed synthetic Fab library (37). The phage pool from the second round was split and used for three further parallel selection rounds for binding to either HER3-Fc or an Fc-tagged heterodimer of the HER2 and HER3 ECDs (HER2/HER3-Fc). We reasoned that HER2/HER3-Fc may present epitopes not present on HER3-Fc, and therefore may yield antibody specificities not obtainable by selection on HER3-Fc alone, and it may enable the selection of anti-HER3 antibodies that can bind the HER2/HER3 heterodimer. After the final round, five clones were isolated, sequenced, and characterized using competitive phage ELISAs to sort binders based on relative affinities (Fig. 1A and data not shown). We converted the candidate clones to the IgG1 format, and found that purified IgG proteins bound to HER3-Fc but did not bind to the other ErbB family members (Fig. 1B).
Figure 1.

Characterization of anti-HER3 antibodies. A, sequences of anti-HER3 Fabs. Sequences of CDR positions randomized in the library are shown and are numbered according to the IMGT standards (72). B, IgG specificity for ErbB family members assessed by ELISA. Error bars represent the standard deviation of three independent experiments, and each point the mean of one experiment. C, Western blots of lysates from SKBR3 cells treated with the indicated IgG (5 μg/ml) for 1 h prior to treatment with NRG1 (2 nm) for 10 min. Blots were developed with the antibodies to the proteins indicated on the right. The data are representative of three independent experiments. D, cell proliferation assays with SKBR3 cells treated with the indicated antibodies. Relative confluence was measured after control cells doubled 1.5 times (∼55 h). Error bars represent the standard deviation of three independent experiments. *, p < 0.05; **, p < 0.005, one-way ANOVA (see “Experimental procedures”). E, in vivo xenograft assays. Subcutaneous BxPC3 xenografts were established in CB-17 SCID mice and treated with the indicated antibodies. Tumor size was measured at the indicated time points (n = 10 for the PBS control, and n = 9 for all other treatment groups). **, p < 0.0001; *, p = 0.0001, two-way ANOVA.
We next characterized the antibodies with cell binding and signaling assays to identify the best candidates for use as inhibitors of cell proliferation. Given the role of ligand-dependent HER3 signaling in disease models (13, 14), we tested whether the IgGs could reduce ligand-induced phosphorylation of HER3 and the downstream effector Akt. In SKBR3 cells, which exhibit HER2-amplification and high HER3 levels (38, 39), we found that NRG1 stimulation resulted in robust phosphorylation of HER3 when pretreated with or without an isotype control (Fig. 1C). Preincubation of SKBR3 cells with three of the five HER3 IgGs reduced NRG1-induced levels of phosphorylated HER3 (pHER3) and pAkt. IgGs 95, 101, and 145 bound strongly to HER3-positive SKBR3 cells transfected with control siRNA, and this staining was diminished in cells transfected with HER3 siRNA (Fig. S1).
We tested whether IgGs 95 and 101 could inhibit proliferation of cell lines reliant on HER3 signaling. Both IgGs exhibited significant anti-proliferative activity in vitro with HER2-amplified, ligand-independent SKBR3 cells (Fig. 1D). Using BxPC3 cells as a model for NRG1-reliant tumor growth in mouse xenografts (28), we found that IgG 95 inhibited tumor growth (at 35 days, TGI30 mg/kg = 64.6%, p < 0.0001, TGI10 mg/kg = 68.3%, p < 0.0001) sooner and more efficaciously than IgG 101 (at 35 days, TGI10 mg/kg = 30.7%, p = 0.0001) (Fig. 1E). Together, these data show that IgG 95 is a promising anti-proliferative antibody for cancer cells that rely on HER3 signaling.
We next assessed the specificity of IgG 95 by immunoprecipitation MS (IP-MS) experiments with lysates from HER2-amplified BT474 cells, which are also known to express HER3 (39). HER3 was the major protein isolated from IgG 95 immunoprecipitates, as measured by spectral counts, but was never observed in the IgG control immunoprecipitates (Table S1). The IgG 95 immunoprecipitates were also enriched for HER3 interactors and mediators of HER3 signaling, including SHC1, GRB2, and PI3K catalytic (PIK3CA and PIK3CB) and regulatory (PIK3R1, PIK3R2, and PIK3R3) subunits. Notably, HER2 peptides were also found in anti-HER3 immunoprecipitates. These findings are consistent with IgG 95 being derived from selections for binding to the HER2/HER3-Fc heterodimer, and suggest that IgG95 can bind to HER3 in the HER2-bound state. Furthermore, optical biolayer interferometry measurements of binding kinetics found that IgG 95 bound similarly to monomeric HER3 and to an engineered HER2/HER3-Fc heterodimer (Table 1). Taken together, the IP-MS and affinity data suggest that the presence of HER2 does not alter the affinity of IgG 95 for HER3.
Table 1.
Kinetic constants for IgG 95 binding monomeric or heterodimeric HER3
Values are representative of 3 independent experiments.
| Antigen | Kon | Koff | KD |
|---|---|---|---|
| 104m−1 s−1 | 10−3 s−1 | nm | |
| HER3-His | 9.5 ± 0.1 | 1.0 ± 0.01 | 10 ± 0.02 |
| HER3/HER2-Fc | 6.0 ± 0.1 | 0.9 ± 0.01 | 15 ± 0.03 |
To further investigate this hypothesis, we performed co-immunoprecipitation experiments followed by Western blotting for HER2 and HER3. For comparison, we also analyzed IgG MOR09825, which locks HER3 in a closed conformation that cannot bind to HER2 (35), and a commercial antibody (IgG CST) that binds to the intracellular domain of HER3. All three antibodies precipitated HER3, but we could not detect co-precipitation of HER2 beyond what was observed with a negative control IgG (Fig. 2A). However, pretreatment of cells with the membrane impermeable reagent 3,3′-dithiobis(sulfosuccinimidyl propionate) (DTSSP), which stabilizes protein complexes through covalent cross-linking, resulted in a clear increase in co-precipitated HER2 by IgG 95 compared with the negative control IgG. In contrast, cross-linking abolished precipitation of HER3 by IgG MOR09825 but did not affect precipitation by IgG CST. Taken together, these results suggest that the DTSSP cross-linker stabilizes HER2/HER3 heterodimers, which are precipitated by IgG 95 but are not recognized by IgG MOR09825.
Figure 2.

Comparison of IgGs 95 and MOR09825. A, Western blots of BT474 cell lysates immunoprecipitated with the antibodies indicated above the lanes. Cells were untreated (−DTSSP) or treated with a protein cross-linking agent (+DTSPP) prior to lysis and immunoprecipitation, and blots were developed with antibodies to the proteins indicated to the right. HER2 band intensities are indicated below the lanes. Representatives of three independent experiments are shown. B, ELISA for detection of NRG1 binding to immobilized HER3-Fc preblocked with the indicated IgG. Error bars represent the standard deviation of three independent experiments, and each point the mean of one experiment. *, p < 0.005, one-way ANOVA. C, proliferation assays for BT474 cells treated with 15 μg/ml of the indicated IgG or mixture (7.5 μg/ml of each antibody) for 7 days. Error bars represent the standard deviation of three independent experiments, and each point the mean of one experiment. *, p < 0.005, one-way ANOVA.
Given that IgG 95 apparently bound to HER3 in an open conformation and did not compete with HER2 binding, we investigated whether the antibody could compete with NRG1. We assessed binding of soluble NRG1 to immobilized HER3-Fc by ELISA and found that pretreatment of the HER3-Fc with IgG 95 greatly reduced binding of NRG1 (Fig. 2B). Thus, IgG 95 competes with NRG1 for binding to HER3, but it does not compete with HER2, suggesting that the antibody binds to the open form of HER3 at a site that may overlap with the NRG1-binding site.
Based on the evidence for different binding modes of IgGs 95 and MOR09825, we reasoned that ligand-independent heterodimers on the BT474 cell surface would present HER3 epitopes accessible to IgG 95, but not IgG MOR09825, and that IgG MOR09825 would block heterodimer formation, but IgG 95 would not. Therefore, we hypothesized that together they may exhibit a stronger anti-proliferative effect on HER2-amplified cells than either antibody alone. Indeed, proliferation of BT474 cells was more potently inhibited by treatment with a mixture of IgGs 95 and MOR09825 than with an equal concentration of either antibody alone (Fig. 2C), suggesting that the two antibodies recognize distinct forms of HER3 on cells.
IgG 95 induces HER3 ubiquitination and degradation
Given that IgG 95 binds the HER2-bound form of HER3, and that the antibody is anti-proliferative in HER2-amplified cell lines BT474 and SKBR3, we assessed changes in the activation of signaling pathways downstream of HER3 to uncover how the antibody might function in this ligand-independent setting. In the absence of ligand, IgG 95 caused a dose-dependent reduction in pHER3 and pAkt, and did so comparably to the anti-HER2 IgG trastuzumab (Fig. 3A). The effect on phosphorylated ERK (pERK) was minimal in the absence of ligand, consistent with the role of HER3 in controlling activation of the Akt pathway (40). IgG 95 treatment led to a comparable inhibition of pHER3 in the presence or absence of ligand, although, in agreement with the literature (36, 41), trastuzumab did not. As well, pAkt levels were higher in IgG 95-treated cells that were also stimulated with NRG1 compared with unstimulated cells, suggesting that IgG 95 cannot completely inhibit ligand-induced signaling in the presence of excess NRG1. Interestingly, IgG 95 also caused down-regulation of total HER3 in a dose-dependent manner, pointing to a putative mechanism by which the antibody might function to reduce ligand-independent HER3 activation.
Figure 3.

Effects of IgG 95 on HER3 internalization and ubiquitination. A, Western blots of lysates from SKBR3 cells treated with the indicated IgGs for 90 min. The following concentrations were used for IgG 95: 0.04, 0.2, 1, or 5 μg/ml, and 5 μg/ml was used for IgG control and trastuzumab. The indicated cells were stimulated with 2 nm NRG1 for 10 min before harvest. Blots were developed with the antibodies indicated to the right. The data are representative of 3 independent experiments. B, Western blots of lysates from SKBR3 cells treated with 100 μm chloroquine (chlor) or 50 nm bortezomib (bort) for 1 h prior to treatment with 1 μg/ml of IgG 95 for 2 h, as indicated. Blots were developed with antibodies to the proteins indicated to the right. Representative of three independent experiments. C, internalization of IgG 95. SKBR3 cells were treated with 100 μm chloroquine or 50 nm bortezomib for 1 h prior to treatment with 1 μg/ml of IgG 95 for 2 h, as indicated. The cells were then processed for flow cytometry by staining with IgG 95. Error bars represent the S.D. of 4–5 independent experiments, and each point the relative median fluorescent intensity of a single experiment. *, p < 0.005, one-way ANOVA. D, Western blots for assessment of ubiquitination of HER3. The indicated cells were treated with 100 μm chloroquine for 30 min followed by treatment with 1 μg/ml of IgG 95 for 30 min, as indicated. Lysates were subjected to immunoprecipitation with IgG 95, followed by development with antibodies to the proteins indicated to the right. The data are representative of three independent experiments.
To identify the cellular processes that drive IgG 95-mediated HER3 down-regulation, we assessed the effect of chloroquine, a lysosomal protease inhibitor, and bortezomib, a proteasome inhibitor, on HER3 levels after IgG 95 treatment. HER3 down-regulation was partially restored in the presence of chloroquine, suggesting that IgG 95 promotes the lysosomal degradation of HER3 (Fig. 3B). The dependence of antibody-mediated HER3 degradation on lysosomal proteases suggested that IgG 95-induced internalization, a hypothesis consistent with the observation that treatment of cultured cells with IgG 95 led to reduced IgG 95 staining (Fig. 3C).
Due to the role of ubiquitin in directing receptors for lysosomal-mediated degradation (42), we tested whether IgG 95 promotes HER3 ubiquitination in HER2-amplified SKBR3 and BT474 cells, as well as the ligand-dependent BxPC3 cell line. Cells were pre-treated with chloroquine to inhibit lysosomal degradation and were then treated for a short time with IgG 95. The cell lysates were precipitated with IgG 95 and blotted with anti-HER3 or anti-ubiquitin antibodies. Treatment with IgG 95 caused a substantial increase in ubiquitinated HER3 in SKBR3, BT474, and BxPC3 cells (Fig. 3D). Collectively, these data define a putative mechanism whereby IgG 95 inhibits HER3 signaling by promoting HER3 ubiquitination and down-regulation via proteolytic degradation in the lysosome.
shRNA screens reveal that RNF41 depletion generates resistance to IgG 95
Having shown that IgG 95 induces ubiquitination, internalization, and degradation of HER3, we designed an shRNA screen to determine which UPS enzymes are involved and whether this process is required for the anti-proliferative effects induced by IgG 95. Thus, we generated a custom lentiviral-based pooled shRNA library, named the UPS library, which targets 500 UPS enzymes, including E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, E3 ubiquitin ligases, and deubiquitinases, with approximately 5 shRNAs for each gene (Table S2). We introduced the library into SKBR3 cells, because this model exhibited the highest levels of antibody-induced ubiquitination, by transduction, and performed a rescue screen to identify shRNAs that enhance cell proliferation in the presence of IgG 95 (Fig. 4A). Thus, shRNAs that provided a selective growth advantage under the pressure of IgG 95 would become enriched in the pooled cell population. Relative shRNA abundances were quantified by deep sequencing and, for each shRNA, relative abundance in the untreated and treated pools was calculated. Principal component analysis indicated that replicates of different time points and treatments clustered together (Fig. S2A), providing confidence in the quality of the screening data.
Figure 4.

Functional genomic screen for mediators of IgG 95 anti-proliferative effects. A, schematic of the shRNA dropout screen performed to identify IgG 95 desensitizers in the ubiquitin pathway. B, normalized RNF41 hairpin reads from the screen. Counts from control IgG or IgG 95-treated cell lines are illustrated with a solid or dashed line, respectively. C, effects of shRNAs on RNF41 mRNA levels. qPCR was performed using reverse-transcribed mRNA from cells stably expressing the indicated shRNAs. Error bars represent the S.D. of three independent experiments, and each point the mean of one experiment. *, p < 0.005, one-way ANOVA. D, proliferation assays for SKBR3 cells expressing the indicated shRNA treated with IgG 95 for 5 days. Error bars represent the S.D. of three independent experiments. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 when comparing RNF41 hairpins to the corresponding luc (56) control, one-way ANOVA.
We identified MARCH10, UBR4, and RNF41 as candidate genes with three or more shRNAs that had significantly different normalized hairpin counts between control samples and samples treated with IgG 95. Analysis of MARCH10 hairpin plots indicated that it is likely a false-positive, as some shRNAs appear to desensitize, whereas others sensitize (Fig. S2B). The UBR4 E3 ubiquitin ligase has been reported to interact with the ErbB family member EGFR upon ligand stimulation (43) (Fig. S2C). The RNF41 E3 ubiquitin ligase was implicated in the ubiquitination and down-regulation of HER3 (44) and was the top desensitizer to IgG 95 treatment. RNF41 shRNAs were significantly enriched in cell pools treated with IgG 95, implying a role for RNF41 in the anti-proliferative response to IgG 95 (Fig. 4B).
Assessment of on-target activity in SKBR3 cells by RT-PCR assays showed that RNF41 shRNAs 49 and 50 (see Fig. S2D for TRCN numbers) depleted RNF41 mRNA by >50% compared with controls (Fig. 4C). To validate RNF41 knockdown as an IgG 95 desensitizer, we performed proliferation assays of SKBR3 cells transduced with either RNF41 shRNA 49 or 50, or with a control luciferase shRNA 56, in the presence or absence of IgG 95. SKBR3 cells stably transduced with RNF41 shRNAs exhibited decreased sensitivity across an order of magnitude of IgG 95 concentration compared with cells transduced with the luciferase shRNA (Fig. 4D). We note that RNF41 shRNAs 51 and 53 caused anti-proliferative effects in isolation, which complicated attempts to measure synergy with IgG 95 treatment (Fig. 4B and data not shown). Together, these results support the observations made from the genetic screens and validate RNF41 down-regulation as a desensitizer of the anti-proliferative effects of IgG 95.
RNF41 depletion reduces ubiquitination and down-regulation of HER3 caused by IgG 95
Because RNF41 shRNAs attenuated the anti-proliferative activity of IgG 95, we tested whether they also attenuated the effects of IgG 95 on the ubiquitination and down-regulation of HER3. In SKBR3 cells expressing shRNAs targeting RNF41 or control proteins (RFP or luciferase), we assessed HER3 levels after treatment with a negative control IgG or IgG 95. In the presence of the negative control IgG, cells expressing RNF41 shRNA exhibited higher HER3 levels than those expressing control shRNAs (Fig. 5A), which is consistent with previous reports (45, 46) and with on-target shRNA activity. Moreover, down-regulation of HER3 levels was attenuated in cells expressing RNF41 shRNA in response to IgG 95 compared with a control IgG (Fig. 5B). Furthermore, whereas IgG 95 enhanced ubiquitination of HER3 in all cells tested, the effect was dampened by the expression of RNF41 shRNAs (Fig. 5C). Consistent with cell proliferation data (Fig. 4D), which showed that RNF41 shRNA expression reduced the anti-proliferative effect of IgG 95, neither IgG 95 nor trastuzumab could reduce pAkt in cells expressing RNF41 shRNA compared with those expressing a control shRNA (Fig. 5D). Taken together, these results suggest that binding of IgG 95 to the HER3 ECD stimulates RNF41-mediated ubiquitination of the HER3 intracellular domain.
Figure 5.
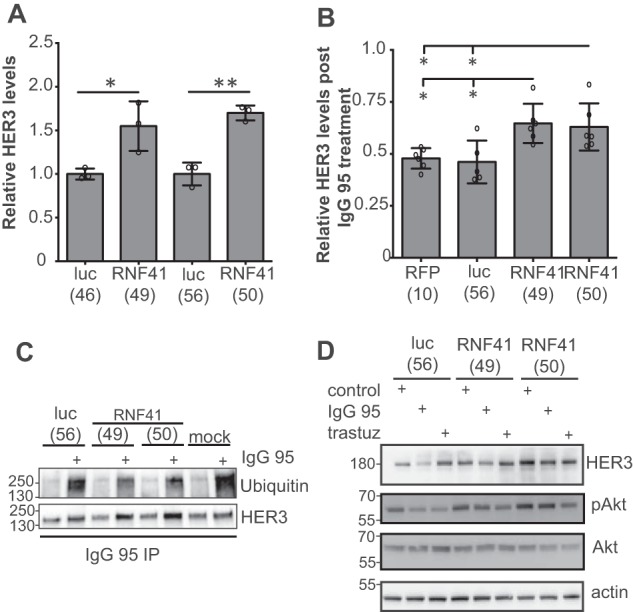
RNF41 depletion protects HER3 protein from degradation induced by IgG 95. A, quantification by Western blotting of HER3 in lysates from SKBR3 cells stably expressing the indicated shRNA. Error bars represent the S.D. of three independent experiments, and each point the value of one experiment. *, p < 0.05; **, p < 0.005, t test. B, quantification by Western blotting of HER3 in lysates from SKBR3 cells stably expressing the indicated shRNA and treated with 1 μg/ml of IgG 95 or control IgG for 2.5 h. Error bars represent the S.D. of at least five independent experiments, and each point the value of one experiment. *, p < 0.05, one-way ANOVA. C, Western blots of IgG 95 immunoprecipitates from SKBR3 cells stably expressing the indicated shRNA and pretreated with 100 μm chloroquine for 30 min, and then with 1 μg/ml of control or IgG 95 for 30 min. Representative data from one of three independent experiments are shown. D, Western blots of lysates from SKBR3 cells stably expressing the indicated shRNA and treated with 5 μg/ml of the indicated IgG for 2.5 h. The data are representative of three independent experiments.
In summary, our data highlight a functional contribution of RNF41 to the anti-proliferative activity of IgG 95 in SKBR3 cells and support a dual role for this E3 ligase in the regulation of HER3. First, as also described previously (44, 45, 47), RNF41 regulates resting HER3 levels, as knockdown of RNF41 results in an increase in total HER3. Second, as shown here for the first time, RNF41 is required for HER3 ubiquitination and down-regulation in response to an anti-HER3 antibody, IgG 95, which in turn is necessary for the reduction of pAkt and the anti-proliferative response induced by IgG 95.
RNF41 disruption with CRISPR/Cas9 causes a resistor phenotype in cancer models
It has been shown previously that RNF41 depletion does not alter the down-regulation of HER3 in BxPC3 cells treated with the anti-HER3 IgG 9F7-F11 (48). Together with our findings, these results suggest that the effect of RNF41 on the response to anti-HER3 antibodies may depend on the antibody and/or the cancer cell line. Therefore, to further refine our understanding of the role of RNF41 in the cellular response to IgG 95, we investigated how the RNF41 gene disruption affects the activity of IgG 95 in different genetic backgrounds, including other HER2-amplified cell models (AU565 and BT474), which exhibit ligand-independent HER3 activation, and the aforementioned ligand-dependent BxPC3 cell line.
We generated a mixed population of RNF41 gene-edited cells using CRISPR/Cas9, so that we could monitor the relative abundance of genomic insertions and deletions (INDELs) at the RNF41 locus after an outgrowth period in the presence or absence of IgG 95 (Fig. 6A). Our model suggests that RNF41 is required for the anti-proliferative activity of IgG 95, so we reasoned that RNF41 INDELs would be selected for upon extended treatment with IgG 95. On-target single guide RNA activity for two RNF41 guides was confirmed using the TIDE webtool (49), which identified single bp frameshift mutations within the RNF41 gene (Fig. 6B). We cultured the mixed population of RNF41 gene-edited SKBR3 cells in the presence of IgG 95 or an isotype control IgG for 13 days. TIDE analysis revealed specific enrichment of RNF41 INDELs in cells treated with IgG 95, suggesting that RNF41 disruption decreases sensitivity to IgG 95 and supporting the RNF41 shRNA knockdown experiments. Other HER2-amplified breast cancer cell lines, including BT474 and AU565, also exhibited RNF41 INDEL enrichment upon treatment with IgG 95, thus indicating that the anti-proliferative activity of IgG 95 is modulated by RNF41 disruption in multiple HER2-amplified breast cancer cell lines (Fig. 6C). We also tested RNF41 INDEL enrichment upon IgG 95 treatment in HER2 nonamplified BxPC3 cells, shown previously to exhibit HER3 down-regulation upon treatment with IgG 9F7-F11 in an RNF41-independent manner (48). IgG 95 has an anti-proliferative effect on BxPC3 cells only in low serum conditions (Fig. S3), suggesting that HER3 down-regulation by IgG 95 in these cells may depend on RNF41 only under these conditions. Indeed, mixed populations of BxPC3 RNF41 gene-edited cells exhibited INDEL enrichment upon IgG 95 treatment only when grown in low serum (Fig. 6C). Taken together with the previous finding that IgG 9F7-F11 did not require RNF41 for antibody-mediated down-regulation (48), our results suggest that different antibodies promote the activity of different ubiquitin ligases.
Figure 6.

Treatment of cells with IgG 95 enriches for CRISPR/Cas9-induced RNF41 INDELs. A, experimental strategy used to measure the effect of RNF41 deletion on IgG 95 sensitivity. CRISPR/Cas9 was used to edit the RNF41 gene, and the relative abundance of RNF41 INDELs was measured after a period of outgrowth in the presence of IgG 95. B, quantification of RNF41 INDELs by TIDE (49) after transfecting SKBR3 cells with RNF41 sgRNA. C and D, quantification of RNF41 INDELs by TIDE (49) after cells were edited with the indicated RNF41 sgRNA and grown in the absence or presence of 1 μg/ml of IgG 95 (C) or higher doses of antibody (D) as indicated for 2–3 weeks. SKBR3 cells were assayed in D. Error bars represent the S.D. of measurements made from at least three biological replicates grown in parallel (except SKBR3/sgRNA 2/control (C) and 10 μg/ml of MOR09825 (D), where two biological replicates were measured). *, p < 0.05; **, p < 0.005, t test (C), one-way ANOVA (D).
To explore whether RNF41 may influence the antiproliferative response of anti-HER3 antibodies that bind differently than IgG 95, we compared IgG 95 to IgG MOR09825, which locks HER3 in the closed confirmation (35). Treatment of a mixed population of RNF41 gene-edited SKBR3 cells with either IgG 95 or MOR09825 resulted in INDEL enrichment, providing support that RNF41 may be involved in the anti-proliferative activity of other anti-HER3 antibodies (Fig. 6D). Given that RNF41 appears to regulate basal HER3 levels (Fig. 5A), we tested whether higher concentrations of antibodies could overcome the resistance to treatment caused by RNF41 disruption, but RNF41 INDELs were enriched significantly even in the presence of 10-fold greater concentrations of either IgG 95 or IgG MOR09825 (Fig. 6D). Thus, these data suggest that increasing the IgG dose is insufficient to overcome the increased HER3 levels that result from RNF41 disruption. Collectively, our results show that RNF41 ubiquitinates HER3 in response to anti-HER3 antibody treatment, which leads to HER3 degradation and consequent anti-proliferative effects.
Discussion
Many targeted cancer therapies rely on the disruption of effector proteins in signaling networks responsible for malignant cell growth. Delineating mechanisms of action and genetic factors that influence therapeutic efficacy will facilitate the maximal utility of these drugs. HER3 has been intensely studied as a therapeutic target, yet there are currently no approved therapies that inhibit this growth factor receptor directly. Because heterodimerization of HER3 with HER2 is thought to be the most potent ErbB oncogenic signaling unit (6, 7), and is especially prominent in HER2-amplified cancers, we hypothesized that targeting the HER3 arm of this protein complex would benefit the treatment of this cancer subclass.
Our data suggest that IgG 95 can bind the HER2-bound form of HER3 and blocks NRG1 binding, characteristics consistent with binding to the open form of HER3 at an epitope that may overlap with the NRG1-binding site. Unlike NRG1, however, IgG 95 does not appear to induce HER3 activation, suggesting that binding of IgG 95 alters the HER2–HER3 heterodimer from the active conformation enforced by NRG1 binding. Based on the structural homology of HER3 (50) with EGFR (51) and HER4 (52), NRG1 binding to HER3 likely traps the receptor in an open conformation that exposes the dimerization arm, as observed with EGFR (53, 54) and HER4 (55). Interestingly, structural studies with high and low affinity EGFR ligands have highlighted how asymmetric dimers made up of subtly different open forms of EGFR correlate with the durability of downstream signaling (56), as well as rates of ubiquitination, endocytosis, and recycling (57). Therefore, it is possible that differences in the open form of HER3 upon IgG 95 binding versus NRG1 binding account for different signaling outcomes. Alternatively, or in addition, the bivalent nature of the IgG, as opposed to monomeric NRG1, may induce further clustering of receptor dimers, which may in turn induce ubiquitination and degradation. It is enticing to speculate that the additive antiproliferative effects of IgG 95 and MOR09825 result from combined targeting of the open and closed forms.
Based on our observations that IgG 95 induces ubiquitination and down-regulation of HER3, we furthered explored the mechanism of IgG 95 function by performing a UPS-targeted functional genomic screen and found that the E3 ubiquitin ligase RNF41 is at least partially responsible for the anti-proliferative effects of IgG 95. Previous studies have found that overexpression of RNF41 leads to ubiquitination and down-regulation of HER3 (44–47) and that NRG1 promotes RNF41-mediated HER3 ubiquitination (58). However, our work is the first to uncover a role for RNF41 in mediating a cellular response to an anti-HER3 antibody, which may be important for understanding the mechanisms of action and resistance for other anti-HER3, including clinical candidates. Our data supports a model whereby antibody-mediated HER3 ubiquitination and down-regulation is driven by RNF41, and HER3 down-regulation is in turn responsible for reduced levels of pAkt. Interestingly, RNF41 does not regulate HER3 levels in response to all anti-HER3 antibodies. IgG 9F7-F11, which does not block NRG1 binding (59), also did not promote RNF41-mediated ubiquitination. Rather HER3 ubiquitination and degradation in BxPC3 cells treated with IgG 9F7-F11 depended on the E3 ligase ITCH (48). Whether ITCH drives the anti-proliferative response of BxPC3 cells to IgG 9F7-F11 remains untested. We observed RNF41 INDEL enrichment in BxPC3 cells treated with IgG 95, suggesting a functional role for RNF41 in regulating IgG 95 anti-proliferative effects. Collectively, these data suggest that exposure to different antibodies can lead to engagement of different ubiquitination pathways. It will be interesting to see if combinations of anti-HER3 antibodies that recruit multiple ubiquitination pathways result in more potent HER3 shutdown.
In HER2-amplified SKBR3 cells, RNF41 plays the dual role of controlling both total HER3 levels and antibody-mediated degradation. We predict that these two functions may render RNF41-depleted cancer cell lines more aggressive as a result of higher HER3 levels, as seen elsewhere (45, 46), and less sensitive to antibodies like IgG 95 that depend on RNF41 for their efficacy, especially in the context of HER2 amplification where HER3 signaling is driven in a ligand-independent manner. Conversely, given that RNF41 knockdown increases the proliferative response to NRG1 (46), cancers exhibiting low RNF41 and ligand-dependent signaling would likely respond to NRG1 competitive anti-HER3 antibodies. An analogous finding was observed for cells with knockdown of NEDD4, another E3 ubiquitin ligase that negatively regulates total HER3 levels (60). That is, NEDD4 knockdown resulted in a higher proliferative response to NRG1, and a greater anti-proliferative effect for the anti-HER3 antibody U1–59. Collectively, our work highlights a potential benefit to targeting the open form of HER3 to inhibit cancer proliferation, underscores the importance of HER3 ubiquitination in the anti-proliferative effect of IgG 95, and suggests that HER3 down-regulation may be an important marker of response to IgG 95 and other antibodies as potential anti-cancer agents.
Experimental procedures
Isolation, characterization, and production of anti-HER3 antibodies
Anti-HER3 Fabs were selected from the phage-displayed synthetic human Fab library F (37). Binding selections, phage ELISAs, and Fab protein purification were performed as described (61, 62). Briefly, phage from Library F were cycled through successive rounds of binding selections with antigen adsorbed onto 96-well MaxiSorp Immunoplates (Fisher Scientific). The first two rounds were performed with the recombinant, Fc-tagged extracellular domain of HER3 (HER3-Fc), and the final three rounds on either HER3-Fc or HER2/HER3-Fc heterodimers generated using knobs-into-holes technology (63)(kindly provided by Mark Walter). Fab-phage were produced for individual clones, and phage ELISAs were performed to detect binding to HER3-Fc. Clones exhibiting antigen-binding signals more than 2-fold over background binding to bovine serum albumin were sequenced. Competitive phage ELISAs were performed for unique clones by incubating Fab-phage with 17 nm HER3-Fc prior to capture with immobilized antigen, and then detecting bound Fab-phage with anti-M13–HRP antibody conjugate. To produce IgGs, the genes encoding light and heavy chains were cloned into vectors designed for expression in 293F cells (Invivogen). IgG proteins were affinity-purified using protein A-Sepharose (GE Healthcare). The sequence for the HER3 antibody MOR09825 was derived from a published structure (35), and codon-optimized DNA fragments encoding for the antibody light and heavy chains were synthesized and cloned into IgG expression vectors. ELISAs were performed with IgG proteins to assess binding to ErbB family members (R&D Technologies) utilized an anti-human κ-HRP conjugate (Jackson) as described (64). To assess binding of NRG1 to HER3 in the presence of IgG 95, HER3-Fc (Sino Biological) was coated on MaxiSorp Immunoplates followed by incubation with saturating (133 nm) amounts of IgG 95, then 800 nm NRG1-His (R&D Systems, 5898-NR), followed by detection of NRG1 with an anti-His–HRP antibody conjugate (R&D Systems, MAB050H).
Cells and Western blotting
SKBR3, BxPC3, BT474, and AU565 cells were grown in McCoy's 5A (Gibco), RPMI 1640 (Gibco), DMEM/F-12 (Gibco), or RPMI 1640 (Gibco) media, respectively, supplemented with 10% FBS (Gibco) and 1× NormocinTM (Invivogen). Cells were cultured in a 37 °C incubator with a humidified atmosphere of 5% CO2 in air. Cell treatments were performed with NRG1β (R&D Systems, 396-HB), chloroquine (BioShop), or bortezomib (SelleckChem). For Western blotting, cells were harvested on ice in 1% Triton X-100 in 50 mm Tris, pH 7.5, 150 mm NaCl and supplemented with a protease/phosphatase inhibitor mixture (Cell Signaling Technology (CST) or HALT (ThermoFisher). Lysates were incubated on ice for 30 min, and the extract was clarified by centrifugation at 13,000 × g for 15 min at 4 °C. Protein concentration was determined using a bicinchoninic acid assay (Pierce). For cross-linking experiments, cells were washed twice with wash buffer (50 mm HEPES, pH 7.4, 150 mm NaCl) prior to treatment on ice for 1 h with 2 mm DTSSP (ThermoFisher) prepared in wash buffer just before use. Cross-linking was stopped by treating cells with 50 mm Tris, pH 7.4, 150 mm NaCl. Lysates were processed as above, and equal volumes of samples were immunoprecipitated with 2 μg of antibody and a slurry of protein A-Sepharose (GE Healthcare). Primary antibodies binding to pHER3(Tyr-1289) (CST 4791), HER3 (CST 4754), pAkt(Ser-473) (CST 4060), Akt (CST 2920), ubiquitin (CST 3936), pERK1/2(Thr-202/Tyr-204) (CST 4370), ERK1/2 (CST 9102), GAPDH (CST 2118), and actin (Abcam 3280) were utilized for Western blotting. Appropriate HRP-conjugated secondary antibodies were used for detection. Immunoprecipitations for ubiquitin blots were performed using 1 μg of IgG 95/mg of cell extract and a slurry of protein A-Sepharose (GE Healthcare).
Flow cytometry
Primary staining of cells was performed with 15 μg/ml of an anti-HER3 IgG. Alexa Fluor 488 AffiniPur F(ab′)2 goat anti-human was used as a secondary antibody (Jackson, 109-546-097). Dead cells were excluded using 7-aminoactinomycin D. Flow cytometry was performed on either a BD FACSCanto II (BD Biosciences) or a BD LSR Forstessa (BD Biosciences). Acquired data were analyzed with FlowJo software (FlowJo, LLC). For experiments utilizing siRNA, cells were reverse transfected according to the manufacturer's recommendations using Lipofectamine RNAiMAX (ThermoFisher) and siRNA nontargeting pool #1 (Dharmacon) or HER3 siRNA SMARTpool (Dharmacon).
Biolayer interferometry
Binding kinetics were determined by BLI using a ForteBio Octet HTX system (Pall Corporation) with IgG 95 immobilized on amine-reactive generation-2 (AR2G) biosensors (ForteBio) according to the manufacturer's instructions. Sensor-captured IgG 95 was exposed to 200 nm HER3/HER2-Fc, 200 nm HER3-His (Sino Biological), or 3-fold serial dilutions in PBS, 1% bovine serum albumin, 0.05% Tween 20. Association and dissociation were each monitored for 600 s with 1000 rpm shake speed at 25 °C. Analysis was performed using a 1:1 Langmuir model and globally fit to determine kon and koff values using Octet Software (ForteBio). KD was calculated as the ratio of koff/kon. To produce HER3/HER2-Fc for BLI studies, we generated a fragment of domains 1–4 of HER3 using PCR (F, 5′-GATTGAATTGTCTAGAATGAGGGCGAACGACGCT-3′; R, 5′-TGTGTGAGTTTTGTCTGTCAGATGGGTTTTGCCG-3′) suitable for in-frame cloning using Gibson Assembly (New England Biolabs) with the CH2 and CH3 domains (accession number 1605217A, beginning at hinge IMGT positions “DKTH” (positions 6–9)) containing the following “knob” mutations (IMGT numbering) in CH3: Q3M, Y5F, T6D, T22W, L24M. We used a similar strategy to clone domains 1–4 of HER2 (F, 5′-GATTGAATTGTCTAGAATGGAGCTGGCGGCCTTG; R, 5′-TGTGTGAGTTTTGTCCGTCAGAGGGCTGGCTC-3′) in-frame with the CH2 and CH3 domains (accession number 1605217A, beginning at hinge IMGT positions DKTH (positions 6–9)) containing the following “hole” mutations (IMGT numbering) in CH3: S10I, E13L, K15.1S, T22S, L24A, Y86V. HER2/HER3-Fc was produced similarly to IgG proteins (described above).
IP-MS
BT474 cells were grown to ∼70% confluence and lysed in lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 1.5 mm MgCl2, 1 mm EGTA, 10 mm NaPPi, 10 mm NaF, 1 mm sodium orthovanadate, 10 mg/ml of leupeptin, 2 mm benzamidine and protein protease/phosphatase inhibitor mixture (CST)). Samples were clarified by centrifugation for 10 min at 20,000 × g, followed by a second spin at 20,000 × g for 2 h. Protein concentration was determined using a bicinchoninic acid assay. Total lysate (4 mg) was tumbled overnight at 4 °C with 10 μg of capture IgG 95, followed by addition of protein A-Sepharose and an additional 3 h of mixing. Beads were washed 3 times with lysis buffer and twice with water. Bound proteins were eluted 3 times with 0.15% TFA. 1 m Ammonium bicarbonate, 45 mm DTT, and 100 mm iodoacetamide were added to eluants in a stepwise manner. l-1-tosylamido-2-phenylethyl chloromethyl ketone-trypsin (1 μg/ml) was added and samples were incubated overnight at 25 °C. Samples were acidified with 20% TFA to a final concentration of 1% and peptide mixtures were purified on C18 Stage Tip columns (ThermoScientific) followed by LC tandem MS analysis on a Q-Exactive hybrid quadrupole Orbitrap hybrid mass spectrometer. Abundant ions were selected for collision-activated dissociation. Data were acquired using Xcalibur software and analyzed on SEQUEST and X!Tandem search engines, using the human Uniprot database as a reference. Results were imported onto Scaffold Viewer to determine spectral counts.
Xenograft assays
Xenograft assays were conducted following the guidelines of the Canadian Council on Animal Care (CCAC), and the experiments were performed under a protocol approved by the University Animal Care Committee (UACC) at the University of Toronto. The experimental procedures were similar to those described previously (65). Briefly, CB-17 SCID mice (7 weeks old, female) were purchased from Taconic Biosciences Inc. (Hudson, NY) and housed in a pathogen-free environment at the animal facility at the University of Toronto. To generate tumor xenografts, BxPC3 cells (5 × 106) in Dulbecco's PBS were injected subcutaneously into the right flank of each mouse. Tumors were measured using vernier calipers and mice were weighed twice weekly. Tumor volume was calculated using the formula: ½ (length × width2). When tumor volumes reached an average of ∼160 mm3, mice were randomly divided into four groups (9 or 10 mice per group) such that the mean tumor volumes of each group were similar. Each group received one of the following treatments, twice weekly via intraperitoneal injection for 4 weeks: vehicle buffer (5% d-(+)-trehalose dihydrate (BioShop Canada Inc.) in Dulbecco's PBS), IgG 101 (10 mg/kg), IgG 95 (10 mg/kg), or IgG-95 (30 mg/kg). Tumor growth inhibition (TGI) of the antibodies was calculated as % TGI = [(MTVcontrol − MTVAb-treated)/MTVcontrol] × 100, where MTV = median tumor volume.
Proliferation assays
SKBR3 (2000 cells/well) or BT474 (7000 cells/well) cells were seeded into a 96-well dish and incubated overnight at 37 °C with 5% CO2 in humidified air. Cells were treated with IgG 95, 101, or isotype control (Jackson Labs), and proliferation was assessed after 2–7 days, either by measuring cell confluence with an IncuCyte ZOOM (Essen BioScience) or by counting the cells. For counting, cells were either stained with Hoechst 33342 (Thermo Scientific) and imaged with an IN Cell Analyzer 2200 (GE Healthcare), and the nuclei were counted using CellProfiler2.0 software (66), or stained with Vybrant DyeCycle green and imaged and counted using an IncuCyte ZOOM (Essen BioScience).
UPS shRNA functional genomic screen
An shRNA library directed against 500 UPS genes (3–5 shRNAs per gene) was assembled as a subset of a previously described genome-wide library (67), and lentivirus containing the library was produced as described (68). SKBR3 cells were treated with 4 μg/ml of Polybrene and infected with the lentivirus shRNA library at a multiplicity of infection of ∼0.45. The media was changed 24 h after infection, and cells were treated with 4 μg/ml of puromycin for 48 h to select for infected cells. Triplicate T0 samples were taken at a representation of 1000 cells per shRNA and also seeded in triplicate at this same representation, followed by treatment with 1 μg/ml of control IgG (Jackson Labs) or IgG 95. Cells were passaged every 3–4 days and sampled at a representation of 1000 cells per shRNA. Genomic DNA was isolated using a DNA blood and cell culture midi kit (Qiagen) according to the manufacturer's instructions. shRNA inserts were amplified from genomic DNA by PCR with primers harboring Illumina TruSeq adapters with i5 and i7 barcodes, and the resulting pools were sequenced on a Illumina HiSeq2500. The differential expression of shRNA between control samples and samples treated with IgG 95 was determined using DESeq (69).
shRNA validation assays
Polyclonal pools of SKBR3 cells expressing an shRNA hairpin were generated by treating cells with 4 μg/ml of Polybrene and infecting cells with lentivirus encoding a specific shRNA. After selection with 4 μg/ml of puromycin for 48 h, cells were maintained in culture and used for downstream assays.
qPCR
The RNEasy Plus Mini kit (Qiagen) was used to isolate RNA from cells according to the manufacturer's instructions. RNA was converted to cDNA using the Superscript III First Strand cDNA synthesis system for RT-PCR (Invitrogen). RT-PCR was performed using the POWER SYBR Green PCR Mastermix (Applied Biosystems) on a Bio-Rad CFX96 Real-Time system with the following primers: RNF41-F, 5′-TGCATTAAGCACCTGCGC-3′; RNF41-R, 5′-AGCAGGACAGCCACTCTCC-3′; GAPDH-F, 5′-CTCCTGCACCACCAACTGCT-3′; GAPDH-R, 5′-GGGCCATCCACAGTCTTCTG-3′. Analysis was done using the comparative cycle threshold method (70) with RNF41 expression normalized to GAPDH expression.
INDEL enrichment assay
Single guide RNAs (sgRNAs) targeting RN41 (RNF41 guide 1, 5′-GCAACGCCTGCATCACCCAG-3′; RNF41 guide 2, 5′-CCGAGGTACTGGGCGCAGAT-3′), or LacZ (LacZ guide, 5′-CCCGAATCTCTATCGTGCGG-3′) were cloned into pLCKO (Addgene catalogue number 73311) or lentiCRISPR v2 (a gift from Feng Zhang, Addgene catalogue number 52961 (71)) plasmids. X-tremeGENE 9 transfection reagent (Sigma) was used to co-transfect SKBR3 cells with a pLCKO plasmid expressing an sgRNA targeting RNF41 or LacZ and plasmid Lenti-Cas9–2A-blast (Addgene catalogue number 73310). The following day, cells were treated with 1 μg/ml of puromycin for 48 h and allowed to recover for 12 days. Cells were sampled, seeded in triplicate populations, further grown out in the presence or absence of IgG 95 (1 μg/ml) for 13 days, and sampled again. Crude gDNA extracts were generated from the sampled cell pellets using QuickExtract DNA Extraction Solution (Epicenter) according to the manufacturer's instructions. The second exon of RNF41, which contains the target site for both sgRNAs, was amplified by the PCR using the following primers: 5′-CTTTGCTGTGACCCAGTGGA-3′ and 5′-GCCAACATGCCAAAGAGACC-3′. Primers and dNTPs were removed from the reaction using ExoI (MJS BioLynx) and shrimp alkaline phosphatase (MJS BioLynx) and the amplified DNA was subjected to DNA sequencing by the Sanger method. Quantification of insertions/deletions was performed as described (49) using TIDE (Tracking of INDELs by DEcomposition). For AU565, BT474, and BxPC3 cells, RNF41 editing was induced by infecting cells with Cas9–2A-puro/RNF41 sgRNA co-expressing virus followed by selection with puromycin and cell recovery. Cells were then split into triplicate populations, challenged with IgG 95 or control IgG, and reseeded every 3–4 days. BxPC3 cells in 1% FBS did not grow fast enough to require reseeding, so the media was changed every 4 days. Sampling occurred after 15 (AU565), 18 (BT474), 14 (BxPC3 + 10% FBS), or 12 (BxPC3 + 1% FBS) days. BxPC3 cells in 1% FBS were allowed to recover for 4 days after treatment in full growth media. To measure the effect of IgG MOR09825 and high dose IgG 95 on RNF41 INDELs, SKBR3 cells were infected with lentivirus co-expressing Cas9–2A-puro/RNF41 sgRNA or Cas9–2A-puro/LacZ sgRNA followed by selection with puromycin and cell recovery. Cells expressing RNF41 or LacZ sgRNA were mixed 1:1, seeded in triplicate, and grown out in the presence or absence of antibodies, followed by sample processing and data analysis as outlined above.
Statistics
Statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA). For all statistical analyses, populations were assumed to be normally distributed and of equal variance. A two-tailed, unpaired t test was used to analyze Figs. 5A and 6C. A one-way ANOVA followed by Tukey's multiple comparisons test was used for Figs. 2, B and C, 3C, 4C, 5B, and 6D. For Figs. 1D and 4D, a one-way ANOVA was performed followed by Sidak's multiple comparisons test. For Fig. 1D, means were compared between the 10 μg/ml of IgG control and the 10 or 1 μg/ml of IgG 95 or 101 groups, the 0.1 μg/ml of IgG control group, and the 0.1 or 0.01 μg/ml of 95 or 101 experimental groups, and the 0.001 μg/ml of control and 0.001 μg/ml of 95- or 101-treated groups. For Fig. 4D, means from a given IgG 95 dosage were compared between the 56 (luc) group and the corresponding 49 (RNF41) or 50 (RNF41) groups. For Fig. 1E, a two-way ANOVA followed by Tukey's multiple comparison test was used to calculate p values for each time point.
Author contributions
J. P. T., E. W. T. L., X. W., K. R. B., F. A. F., and K. K. J. data curation; J. P. T., E. W. T. L., X. W., K. R. B., F. A. F., and K. K. J. formal analysis; J. P. T., E. W. T. L., X. W., K. R. B., F. A. F., and K. K. J. investigation; J. P. T., E. W. T. L., X. W., K. R. B., F. A. F., and K. K. J. methodology; J. P. T. writing-original draft; E. W. T. L., X. W., K. R. B., F. A. F., K. K. J., J. P., J. M., and S. S. writing-review and editing; J. P., J. M., and S. S. conceptualization; J. P., J. M., and S. S. supervision; J. P., J. M., and S. S. project administration; S. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank Nick Jarvik and Levi Blazer for BLI studies and helpful discussions, and Leroi DeSouza and Mike Moran for IP-MS studies.
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-136944 (to S. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs S1–S3 and Tables S1 and S2.
- PI3K
- phosphoinositide 3-kinase
- NRG1
- Neuregulin 1
- MEK
- mitogen-activated protein kinase
- EGFR
- epidermal growth factor receptor
- UPS
- ubiquitin proteasome system
- ECD
- extracellular domain
- DTSSP
- 3,3′-dithiobis(sulfosuccinimidyl propionate)
- shRNA
- short hairpin RNA
- IP
- immunoprecipitation
- INDEL
- insertions and deletion
- TGI
- tumor growth inhibition
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- sgRNA
- single guide RNA
- ANOVA
- analysis of variance.
References
- 1. Arteaga C. L., and Engelman J. A. (2014) Erbb receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 10.1016/j.ccr.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Kofler M., Jorissen R. N., Nice E. C., Burgess A. W., and Ward C. W. (2003) The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 11, 495–505 10.1016/S1097-2765(03)00048-0 [DOI] [PubMed] [Google Scholar]
- 3. Slamon D. J., Clark G. M., Wong S. G., Levin W. J., Ullrich A., and McGuire W. L. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 10.1126/science.3798106 [DOI] [PubMed] [Google Scholar]
- 4. Nahta R., and Esteva F. J. (2007) Trastuzumab: triumphs and tribulations. Oncogene 26, 3637–3643 10.1038/sj.onc.1210379 [DOI] [PubMed] [Google Scholar]
- 5. Parakh S., Gan H. K., Parslow A. C., Burvenich I. J. G., Burgess A. W., and Scott A. M. (2017) Evolution of anti-HER2 therapies for cancer treatment. Cancer Treat. Rev. 59, 1–21 10.1016/j.ctrv.2017.06.005 [DOI] [PubMed] [Google Scholar]
- 6. Alimandi M., Romano A., Curia M. C., Muraro R., Fedi P., Aaronson S. A., Di Fiore P. P., and Kraus M. H. (1995) Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 10, 1813–1821 [PubMed] [Google Scholar]
- 7. PinkasKramarski R., Soussan L., Waterman H., Levkowitz G., Alroy I., Klapper L., Lavi S., Seger R., Ratzkin B. J., Sela M., and Yarden Y. (1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 15, 2452–2467 10.1002/j.1460-2075.1996.tb00603.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schulze W. X., Deng L., and Mann M. (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 1, E1–E13 10.1038/msb4100012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prigent S. A., and Gullick W. J. (1994) Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 13, 2831–2841 10.1002/j.1460-2075.1994.tb06577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soltoff S. P., Carraway K. L. 3rd, Prigent S. A., Gullick W. G., and Cantley L. C. (1994) Erbb3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth-factor. Mol. Cell Biol. 14, 3550–3558 10.1128/MCB.14.6.3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hellyer N. J., Cheng K., and Koland J. G. (1998) ErbB3 (HER3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem. J. 333, 757–763 10.1042/bj3330757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcotte R., Brown K. R., Suarez F., Sayad A., Karamboulas K., Krzyzanowski P. M., Sircoulomb F., Medrano M., Fedyshyn Y., Koh J. L. Y., van Dyk D., Fedyshyn B., Luhova M., Brito G. C., Vizeacoumar F. J., et al. (2012) Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2, 172–189 10.1158/2159-8290.CD-11-0224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheng Q., Liu X., Fleming E., Yuan K., Piao H., Chen J., Moustafa Z., Thomas R. K., Greulich H., Schinzel A., Zaghlul S., Batt D., Ettenberg S., Meyerson M., Schoeberl B., et al. (2010) An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 17, 298–310 10.1016/j.ccr.2009.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson T. R., Lee D. Y., Berry L., Shames D. S., and Settleman J. (2011) Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 20, 158–172 10.1016/j.ccr.2011.07.011 [DOI] [PubMed] [Google Scholar]
- 15. Sergina N. V., Rausch M., Wang D., Blair J., Hann B., Shokat K. M., and Moasser M. M. (2007) Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445, 437–441 10.1038/nature05474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gijsen M., King P., Perera T., Parker P. J., Harris A. L., Larijani B., and Kong A. (2010) HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLos Biol. 8, e1000563 10.1371/journal.pbio.1000563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turke A. B., Song Y., Costa C., Cook R., Arteaga C. L., Asara J. M., and Engelman J. A. (2012) MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 72, 3228–3237 10.1158/1538-7445.AM2012-3228,10.1158/0008-5472.CAN-11-3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun C., Hobor S., Bertotti A., Zecchin D., Huang S., Galimi F., Cottino F., Prahallad A., Grernrum W., Tzani A., Schlicker A., Wessels L. F. A., Smit E. F., Thunnissen E., Halonen P., et al. (2014) Intrinsic resistance to MEK inhibitionin KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 7, 86–93 [DOI] [PubMed] [Google Scholar]
- 19. Abel E. V., Basile K. J., Kugel C. H. 3rd, Witkiewicz A. K., Le K., Amaravadi R. K., Karakousis G. C., Xu X., Xu W., Schuchter L. M., Lee J. B., Ertel A., Fortina P., and Aplin A. E. (2013) Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Invest. 123, 2155–2168 10.1172/JCI65780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Montero-Conde C., Ruiz-Llorente S., Dominguez J. M., Knauf J. A., Viale A., Sherman E. J., Ryder M., Ghossein R. A., Rosen N., and Fagin J. A. (2013) Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 3, 520–533 10.1158/2159-8290.CD-12-0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi F., Telesco S. E., Liu Y., Radhakrishnan R., and Lemmon M. A. (2010) ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 7692–7697 10.1073/pnas.1002753107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinkamp M. P., Low-Nam S. T., Yang S., Lidke K. A., Lidke D. S., and Wilson B. S. (2014) erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Mol. Cell Biol. 34, 965–977 10.1128/MCB.01605-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang N., Chang Y., Rios A., and An Z. (2016) HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 48, 39–48 [DOI] [PubMed] [Google Scholar]
- 24. Malm M., Frejd F. Y., Ståhl S., and Löfblom J. (2016) Targeting HER3 using mono- and bispecific antibodies or alternative scaffolds. MAbs 8, 1195–1209 10.1080/19420862.2016.1212147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaborit N., Lindzen M., and Yarden Y. (2016) Emerging anti-cancer antibodies and combination therapies targeting HER3/ERBB3. Hum. Vaccin. Immunother. 12, 576–592 10.1080/21645515.2015.1102809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Clorennec C., Bazin H., Dubreuil O., Larbouret C., Ogier C., Lazrek Y., Garambois V., Poul M.-A., Mondon P., Barret J.-M., Mathis G., Prost J.-F., Pèlegrin A., and Chardès T. (2017) Neuregulin 1 allosterically enhances the antitumor effects of the noncompeting anti-HER3 antibody 9F7-F11 by increasing its binding to HER3. Mol. Cancer Therap. 16, 1312–1323 10.1158/1535-7163.MCT-16-0886 [DOI] [PubMed] [Google Scholar]
- 27. Schoeberl B., Faber A. C., Li D., Liang M. C., Crosby K., Onsum M., Burenkova O., Pace E., Walton Z., Nie L., Fulgham A., Song Y., Nielsen U. B., Engelman J. A., and Wong K. K. (2010) An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 70, 2485–2494 10.1158/0008-5472.CAN-09-3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meetze K., Vincent S., Tyler S., Mazsa E. K., Delpero A. R., Bottega S., McIntosh D., Nicoletti R., Winston W. M., Weiler S., Feng B., Gyuris J., and Weng Z. (2015) Neuregulin 1 expression is a predictive biomarker for response to AV-203, an ERBB3 inhibitory antibody, in human tumor models. Clin. Cancer Res. 21, 1106–1114 10.1158/1078-0432.CCR-14-2407 [DOI] [PubMed] [Google Scholar]
- 29. Yonesaka K., Hirotani K., von Pawel J., Dediu M., Chen S., Copigneaux C., and Nakagawa K. (2017) Circulating heregulin level is associated with the efficacy of patritumab combined with erlotinib in patients with non-small cell lung cancer. Lung Cancer 105, 1–6 10.1016/j.lungcan.2016.12.018 [DOI] [PubMed] [Google Scholar]
- 30. Liu J. F., Ray-Coquard I., Selle F., Poveda A. M., Cibula D., Hirte H., Hilpert F., Raspagliesi F., Gladieff L., Harter P., Siena S., Del Campo J. M., Tabah-Fisch I., Pearlberg J., et al. (2016) Randomized phase II trial of seribantumab in combination with paclitaxel in patients with advanced platinum-resistant or -refractory ovarian cancer. J. Clin. Oncol. 34, 4345–4353 10.1200/JCO.2016.67.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meulendijks D., Jacob W., Martinez-Garcia M., Taus A., Lolkema M. P., Voest E. E., Langenberg M. H. G., Fleitas Kanonnikoff T., Cervantes A., De Jonge M. J., Sleijfer S., Soerensen M. M., Thomas M., Ceppi M., Meneses-Lorente G., et al. (2016) First-in-human phase I study of lumretuzumab, a glycoengineered humanized anti-HER3 monoclonal antibody, in patients with metastatic or advanced HER3-positive solid tumors. Clin. Cancer Res. 22, 877–885 10.1158/1078-0432.CCR-15-1683 [DOI] [PubMed] [Google Scholar]
- 32. Mendell J., Freeman D. J., Feng W., Hettmann T., Schneider M., Blum S., Ruhe J., Bange J., Nakamaru K., Chen S., Tsuchihashi Z., von Pawel, Copigneaux J. C., and Beckman R. A. (2015) Clinical translation and validation of a predictive biomarker for patritumab, an anti-human epidermal growth factor receptor 3 (HER3) monoclonal antibody, in patients with advanced non-small cell lung cancer. EBioMedicine 2, 264–271 10.1016/j.ebiom.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiao Z., Carrasco R. A., Schifferli K., Kinneer K., Tammali R., Chen H., Rothstein R., Wetzel L., Yang C., Chowdhury P., Tsui P., Steiner P., Jallal B., Herbst R., Hollingsworth R. E., and Tice D. A. (2016) A potent HER3 monoclonal antibody that blocks both ligand-dependent and -independent activities: differential impacts of PTEN status on tumor response. Mol. Cancer Ther. 15, 689–701 10.1158/1535-7163.MCT-15-0555 [DOI] [PubMed] [Google Scholar]
- 34. Lee S., Greenlee E. B., Amick J. R., Ligon G. F., Lillquist J. S., Natoli E. J. Jr., Hadari Y., Alvarado D., and Schlessinger J. (2015) Inhibition of ErbB3 by a monoclonal antibody that locks the extracellular domain in an inactive configuration. Proc. Natl. Acad. Sci. U.S.A. 112, 13225–13230 10.1073/pnas.1518361112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garner A. P., Bialucha C. U., Sprague E. R., Garrett J. T., Sheng Q., Li S., Sineshchekova O., Saxena P., Sutton C. R., Chen D., Chen Y., Wang H., Liang J., Das R., Mosher R., et al. (2013) An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res. 73, 6024–6035 10.1158/0008-5472.CAN-13-1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Junttila T. T., Akita R. W., Parsons K., Fields C., Lewis Phillips G. D., Friedman L. S., Sampath D., and Sliwkowski M. X. (2009) Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440 10.1016/j.ccr.2009.03.020 [DOI] [PubMed] [Google Scholar]
- 37. Persson H., Ye W., Wernimont A., Adams J. J., Koide A., Koide S., Lam R., and Sidhu S. S. (2013) CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol. 425, 803–811 10.1016/j.jmb.2012.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hynes N. E., Gerber H. A., Saurer S., and Groner B. (1989) Overexpression of the c-erbB-2 protein in human breast tumor cell lines. J. Cell Biochem. 39, 167–173 10.1002/jcb.240390208 [DOI] [PubMed] [Google Scholar]
- 39. Chen X., Yeung T. K., and Wang Z. (2000) Enhanced drug resistance in cells coexpressing ErbB2 with EGF receptor or ErbB3. Biochem. Biophys. Res. Commun. 277, 757–763 10.1006/bbrc.2000.3731 [DOI] [PubMed] [Google Scholar]
- 40. Hellyer N. J., Kim M.-S., and Koland J. G. (2001) Heregulin-dependent activation of phosphoinositide 3-kinase and Akt via the ErbB2/ErbB3 co-receptor. J. Biol. Chem. 276, 42153–42161 10.1074/jbc.M102079200 [DOI] [PubMed] [Google Scholar]
- 41. Agus D. B., Akita R. W., Fox W. D., Lewis G. D., Higgins B., Pisacane P. I., Lofgren J. A., Tindell C., Evans D. P., Maiese K., Scher H. I., and Sliwkowski M. X. (2002) Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2, 127–137 10.1016/S1535-6108(02)00097-1 [DOI] [PubMed] [Google Scholar]
- 42. Clague M. J., and Urbé S. (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143, 682–685 10.1016/j.cell.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 43. Tong J., Taylor P., and Moran M. F. (2014) Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol. Cell Proteomics 13, 1644–1658 10.1074/mcp.M114.038596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qiu X.-B., and Goldberg A. L. (2002) Nrdp1/FLRF is a ubiquitin ligase promoting ubiquitination and degradation of the epidermal growth factor receptor family member, ErbB3. Proc. Natl. Acad. Sci. U.S.A. 99, 14843–14848 10.1073/pnas.232580999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu H., Li H., Mao D., Zhu Z., and Sun H. (2014) Nrdp1 inhibits growth of colorectal cancer cells by nuclear retention of p27. Tumor Biol. 35, 8639–8643 10.1007/s13277-014-2132-5 [DOI] [PubMed] [Google Scholar]
- 46. Yen L., Cao Z., Wu X., Ingalla E. R., Baron C., Young L. J., Gregg J. P., Cardiff R. D., Borowsky A. D., Sweeney C., and Carraway K. L. 3rd (2006) Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 66, 11279–11286 10.1158/0008-5472.CAN-06-2319 [DOI] [PubMed] [Google Scholar]
- 47. Diamonti A. J., Guy P. M., Ivanof C., Wong K., Sweeney C., and Carraway K. L. 3rd (2002) An RBCC protein implicated in maintenance of steady-state neuregulin receptor levels. Proc. Natl. Acad. Sci. U.S.A. 99, 2866–2871 10.1073/pnas.052709799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le Clorennec C., Lazrek Y., Dubreuil O., Larbouret C., Poul M.-A., Mondon P., Melino G., Pèlegrin A., and Chardès T. (2016) The anti-HER3 (ErbB3) therapeutic antibody 9F7-F11 induces HER3 ubiquitination and degradation in tumors through JNK1/2-dependent ITCH/AIP4 activation. Oncotarget 7, 37013–37029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brinkman E. K., Chen T., Amendola M., and van Steensel B. (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cho H.-S., and Leahy D. J. (2002) Structure of the extracellular region of HER3 reveals an interdomain tether. Science 297, 1330–1333 10.1126/science.1074611 [DOI] [PubMed] [Google Scholar]
- 51. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H. S., Leahy D. J., and Lemmon M. A. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 10.1016/S1097-2765(03)00047-9 [DOI] [PubMed] [Google Scholar]
- 52. Bouyain S., Longo P. A., Li S., Ferguson K. M., and Leahy D. J. (2005) The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. U.S.A. 102, 15024–15029 10.1073/pnas.0507591102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J.-H., Saito K., Sakamoto A., Inoue M., Shirouzu M., and Yokoyama S. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 10.1016/S0092-8674(02)00963-7 [DOI] [PubMed] [Google Scholar]
- 54. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Zhu H.-J., Walker F., Frenkel M. J., Hoyne P. A., Jorissen R. N., Nice E. C., Burgess A. W., and Ward C. W. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α. Cell 110, 763–773 10.1016/S0092-8674(02)00940-6 [DOI] [PubMed] [Google Scholar]
- 55. Liu P., Cleveland T. E. 4th, Bouyain S., Byrne P. O., Longo P. A., and Leahy D. J. (2012) A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. U.S.A. 109, 10861–10866 10.1073/pnas.1201114109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Freed D. M., Bessman N. J., Kiyatkin A., Salazar-Cavazos E., Byrne P. O., Moore J. O., Valley C. C., Ferguson K. M., Leahy D. J., Lidke D. S., and Lemmon M. A. (2017) EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell 171, 683–695.e18 10.1016/j.cell.2017.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roepstorff K., Grandal M. V., Henriksen L., Knudsen S. L., Lerdrup M., Grøvdal L., Willumsen B. M., and van Deurs B. (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10, 1115–1127 10.1111/j.1600-0854.2009.00943.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cao Z., Wu X., Yen L., Sweeney C., and Carraway K. L. 3rd (2007) Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol. Cell Biol. 27, 2180–2188 10.1128/MCB.01245-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lazrek Y., Dubreuil O., Garambois V., Gaborit N., Larbouret C., Le Clorennec C., Thomas G., Leconet W., Jarlier M., Pugnière M., Vié N., Robert B., Monnet C., Bouayadi K., Kharrat H., Mondon P., Pèlegrin A., and Chardès T. (2013) Anti-HER3 domain 1 and 3 antibodies reduce tumor growth by hindering HER2/HER3 dimerization and AKT-induced MDM2, XIAP, and FoxO1 phosphorylation. Neoplasia 15, 335–347 10.1593/neo.121960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang Z., Choi B.-K., Mujoo K., Fan X., Fa M., Mukherjee S., Owiti N., Zhang N., and An Z. (2015) The E3 ubiquitin ligase NEDD4 negatively regulates HER3/ErbB3 level and signaling. Oncogene 34, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 61. Fellouse F. A., Esaki K., Birtalan S., Raptis D., Cancasci V. J., Koide A., Jhurani P., Vasser M., Wiesmann C., Kossiakoff A. A., Koide S., and Sidhu S. S. (2007) High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 373, 924–940 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 62. Rajan S., and Sidhu S. S. (2012) Simplified synthetic antibody libraries. Methods Enzymol. 502, 3–23 10.1016/B978-0-12-416039-2.00001-X [DOI] [PubMed] [Google Scholar]
- 63. Ridgway J. B., Presta L. G., and Carter P. (1996) “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9, 617–621 10.1093/protein/9.7.617 [DOI] [PubMed] [Google Scholar]
- 64. Sidhu S. S., Li B., Chen Y., Fellouse F. A., Eigenbrot C., and Fuh G. (2004) Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J. Mol. Biol. 338, 299–310 10.1016/j.jmb.2004.02.050 [DOI] [PubMed] [Google Scholar]
- 65. Steinhart Z., Pavlovic Z., Chandrashekhar M., Hart T., Wang X., Zhang X., Robitaille M., Brown K. R., Jaksani S., Overmeer R., Boj S. F., Adams J., Pan J., Clevers H., Sidhu S., Moffat J., and Angers S. (2017) Genome-wide CRISPR screens reveal a Wnt–FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 23, 60–68 [DOI] [PubMed] [Google Scholar]
- 66. Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., and Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., Grenier J. K., Carpenter A. E., Foo S. Y., Stewart S. A., Stockwell B. R., Hacohen N., et al. (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 10.1016/j.cell.2006.01.040 [DOI] [PubMed] [Google Scholar]
- 68. Blakely K., Ketela T., and Moffat J. (2011) Pooled lentiviral shRNA screening for functional genomics in mammalian cells. Methods Mol. Biol. 781, 161–182 10.1007/978-1-61779-276-2_9 [DOI] [PubMed] [Google Scholar]
- 69. Anders S., and Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lefranc M.-P., Pommié C., Ruiz M., Giudicelli V., Foulquier E., Truong L., Thouvenin-Contet V., and Lefranc G. (2003) IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 27, 55–77 10.1016/S0145-305X(02)00039-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.