

Abstract
Increased oxidative stress and abundance of reactive oxygen species (ROS) are positively correlated with a variety of pathophysiologies, including cardiovascular disease, type 2 diabetes, Alzheimer's disease, and neuroinflammation. In adipose biology, diabetic obesity is correlated with increased ROS in an age- and depot-specific manner and is mechanistically linked to mitochondrial dysfunction, endoplasmic reticulum (ER) stress, potentiated lipolysis, and insulin resistance. The cellular quality control systems that homeostatically regulate oxidative stress in the lean state are down-regulated in obesity as a consequence of inflammatory cytokine pressure leading to the accumulation of oxidized biomolecules. New findings have linked protein, DNA, and lipid oxidation at the biochemical level, and the structures and potential functions of protein adducts such as carbonylation that accumulate in stressed cells have been characterized. The sum total of such regulation and biochemical changes results in alteration of cellular metabolism and function in the obese state relative to the lean state and underlies metabolic disease progression. In this review, we discuss the molecular mechanisms and events underlying these processes and their implications for human health and disease.
Keywords: adipocyte, obesity, oxidative stress, lipid peroxidation, protein-lipid interaction, carbonylation
Introduction
Adipose oxidative stress is a major contributor to insulin resistance and cellular dysfunction (1) and is regulated in an age- and depot-specific manner. Adipose oxidative stress increases with age in both humans and experimental mice and varies between depots. For example, in contrast to younger mice (6 months), aged C57BL/6 mice (23 months) exhibit increased ROS2 in the visceral adipose depot (epididymal) as compared with the subcutaneous depot (inguinal) (2). Similar experiments on human tissue have revealed that subcutaneous adipose tissue from obese diabetic subjects exhibit increased H2O2 production compared with that from age-matched obese, nondiabetic subjects or to lean controls (3). Moreover, ROS levels are higher in epicardial fat as compared with subcutaneous adipose tissue (4). Furthermore, oxidized lipids and proteins accumulate to a greater extent in visceral depots compared with subcutaneous depots (5, 6). These observations point out the complexity of linking oxidative stress to disease and the molecular mechanisms that drive dysfunction and dysregulation. Major challenges exist in defining the specifics of reactive oxygen species–driven pathology and its connectivity to human health and disease.
Sources of adipose ROS
The origins of adipose ROS are varied and are regulated by a combination of hormonal and metabolic determinants. Indeed, although the mitochondrion is the major source of ROS and clearly represents the major regulatory node for reactive oxygen species synthesis, adipocytes express a variety of enzyme systems that generate ROS (Fig. 1). As such, a major complexity exists in defining the sources of ROS, either basally or in response to a regulatory event such as inflammation or insulin resistance. Although it is tempting to equate reactive oxygen species with negative metabolic outcomes, ROS species play an important positive regulatory role in adipose biology, for example by inactivating the dual-specificity lipid and protein phosphatase and tensin homolog protein facilitating insulin action (7) or through the oxidation of UCP1 as a component of thermogenesis and the production of beige fat cells (8).
Figure 1.
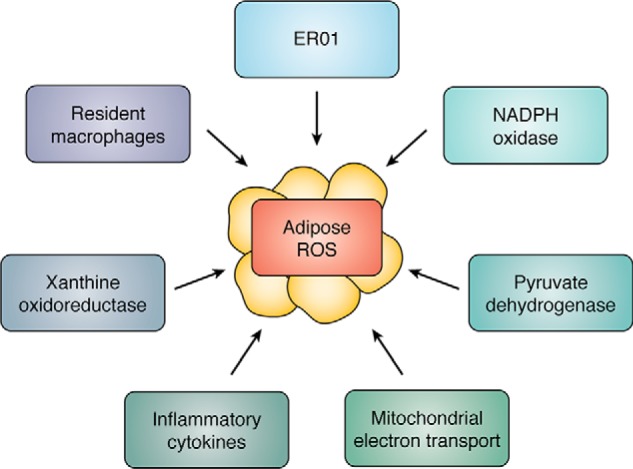
Schematic representation of the major enzymatic and/or systems that affect adipose tissue ROS levels.
NADPH oxidase is a major regulatory enzyme in adipose tissue, generating superoxide and/or H2O2 in response to agonist stimulation. The Nox family has seven isoforms: Nox1, Nox2, Nox3, Nox4, Nox5, Duo1, and Duo2 (9). Nox4 is the major Nox isotype expressed by adipocytes and is activated by high-fat diets. Importantly, Nox4 is unique in that it generates H2O2 as opposed to superoxide anion, which is made by the other NADPH oxidase family members (10). Indeed, Shimomura and co-workers (11) have shown that the expression of gp91Phox, the catalytic subunit of the complex, plus p47Phox, p22Phox, p67Phox, and p40Phox are each up-regulated in murine obesity models such as C57BL/6 and KKAy. In murine cultured adipocytes such as 3T3-L1's, high glucose and/or saturated fatty acids increase Nox4 expression and lead to increased ROS levels. Importantly, Scherer and co-workers (12) demonstrate that maintaining 3T3-L1 cells in a low-glucose environment following differentiation attenuated ROS synthesis and diminished inflammatory cytokine secretion compared with cells maintained in a high-glucose medium, suggesting that cell autonomous factors can have a major regulatory role in defining adipose ROS.
The mitochondrion and mitochondrial sources of reactive oxygen species have long been considered to be major drivers of oxidative stress in adipose tissue. Indeed, complexes I and III of the electron transport chain are classical ROS-generating systems that produce the unstable superoxide anion that, when coupled with superoxide dismutase, generate the much more stable ROS hydrogen peroxide (13). Complex I receives electrons from NADH or the reverse reaction of complex II resulting in ROS production in the mitochondrial matrix, whereas complex III produces superoxide through the Q cycle and can release superoxide to both sides of the inner mitochondrial membrane (13). Excess mitochondrial ROS has been argued to be causative to mitochondrial dysfunction, leading to type 2 diabetes, nonalcoholic fatty liver disease, and heart failure. Other studies have suggested that mitochondrial ROS plays an immunomodulatory role inducing cardiac inflammation via the mtDNA damage pathway (14).
Fisher-Wellman and Neufer (15) have posited a novel hypothesis that under conditions of low reduced GSH, common to fat cells from insulin-resistant states, pyruvate dehydrogenase and nicotinamide nucleotide transhydrogenase form a redox circuit with the potential to be a robust generator of H2O2. Because white adipocytes require pyruvate oxidation via the dehydrogenase system to generate acetyl-CoA for citrate synthesis, pyruvate dehydrogenase-dependent production of ROS would be directly tied to de novo lipogenesis and triglyceride deposition. This consideration is consistent with experiments maintaining 3T3-L1 cells on either low or high glucose leading to either low or high levels of ROS, respectively (12). This intriguing possibility introduces the idea that whereas the mitochondrion may be a major source for ROS, pyruvate dehydrogenase, in addition to the electron transport chain, may be a critical regulator affecting reactive oxygen species flux.
An additional enzyme system often linked to adipose ROS is the xanthine dehydrogenase/oxidoreductase system. Uric acid is secreted avidly from adipocytes, potentially as a mechanism to rid the cell of excess nitrogen derived from branched chain amino acid metabolism (16). Adipocytes utilize branched chain amino acids as anapleurotic substrates for mitochondrial citrate synthesis and eventual de novo lipogenesis. Whereas the carbon atoms of branched chain amino acids are utilized for triglyceride formation, the nitrogen atoms are utilized by the purine pathway to make inosine and ultimately via xanthine oxidoreductase, uric acid. The conversion of xanthine to uric acid generates H2O2 and superoxide anion and as such potentially links de novo lipogenesis to ROS synthesis.
An underappreciated source of ROS in adipocytes is endoplasmic reticular oxidoreductin 1 (ERO1). ERO1 is a protein disulfide oxidase of the ER that produces H2O2 as a consequence of protein folding and secretion (17). Whereas protein-disulfide isomerase directly oxidizes new client proteins and is subsequently reduced, ERO1 reoxidizes and reactivates one of the two thioredoxin-like domains of protein-disulfide isomerase for a new cycle of oxidative protein folding by transferring electrons to oxygen and subsequently producing H2O2. As such, ERO1 is an important source of ER oxidative stress and may be a major source of intracellular ROS exceeding classical ROS resources such as the mitochondrion or xanthine dehydrogenase system. Given the large number of proteins secreted by fat cells, and their relative abundance, a major role for ERO1 in reactive oxygen species synthesis seems likely.
Inflammatory cells and adipose ROS
In adipose tissue, the stromal vascular fraction containing fibroblasts, preadipocytes, immune cells, and endothelial cells is the major source of reactive oxygen species. Because both adaptive cells (like T and B cells) and innate immune cells (macrophages, dendritic cells, mast cells, and eosinophils) are rich in the stroma, adipose tissue is now recognized not only as an energy storage pool for the body but also as a component of the innate immune system (18). In general, adipose tissue M1 macrophages produce pro-inflammatory cytokines and defend against infection, whereas M2 macrophages are primarily involved in anti-inflammatory processes, tumor promotion, and tissue remodeling (19). With the development and expansion of adipose tissue during obesity, M1 macrophages produce pro-inflammatory cytokines (e.g. TNFα, IL-6, and IL-1β) that attenuate adipocyte insulin signaling and lead to increased reactive oxygen species and mitochondrial dysfunction. Moreover, inflammatory cytokines down-regulate the expression of mitochondrial antioxidants, particularly GSH S-transferase A4, peroxiredoxin 3, and GSH peroxidase 4 (5, 20) in a depot-specific manner. Furthermore, some inflammatory cytokines (e.g. IL-1β) activate Nox4 expression and further increase cytosolic ROS in adipocytes.
Studies by Han and co-workers (21, 22) have revealed that Nox2-derived ROS from macrophages can influence inflammation and oxidative stress in adipocytes. Nox2 is the major NADPH oxidase expressed by adipose tissue macrophages and generates superoxide anion in response to agonists such as lipopolysaccharide or saturated fatty acids (23). Myeloid-specific deletion of Nox2 attenuates the metabolic consequences of a high-fat diet, and knockout mice exhibit a delay in inflammation and insulin resistance (24), suggesting that ROS derived from macrophages may be major contributors to total adipose reactive oxygen species signaling. Because certain ROS forms such as H2O2 are long-lived and are capable of transcellular diffusion, macrophage-derived ROS could be a major regulator of adipocyte function.
Paradoxically, M2 macrophages have also been implicated in adipose oxidative stress but from a metabolic flux perspective. The preferential utilization of fatty acids as an energy source by M2 macrophages (relative to M1 macrophages) leads to increased mitochondrial electron transport chain activity and subsequent ROS production (25).
Intracellular ROS metabolism in adipocytes
Superoxide dismutase converts the highly-reactive superoxide anion into hydrogen peroxide and is expressed at high levels in the mitochondrion to regulate O2− levels. Interestingly, adipose-specific knockout of SOD2 results in decreased weight gain in mice fed an obesogenic diet, an effect that was attributed to elevated mitochondrial biogenesis and the beiging of white adipocytes (26). These data are counterintuitive given the dogma that has largely dominated the field that considers elevated ROS as harmful. Indeed, low amounts of reactive oxygen species potentiate adipocyte differentiation, and efforts to attenuate all ROS species may be deleterious to the development of fat cells (27). However, this does demonstrate the critical emerging concept that different species and pools of ROS are functionally distinct in the cell, pointing to potentially important differences between superoxide and hydrogen peroxide in adipose biology.
In contrast to superoxide anion, H2O2 is more stable and is diffusible across membranes. This property makes it an efficient messenger to communicate changes in redox status, as it can act as a signaling molecule and/or initiate damage through the oxidation of reactive thiols. Cysteine oxidation is highly dynamic in vivo and is controlled by both the availability of ROS and the reduction of protein sulfenic acid. Accordingly, the enzymes responsible for H2O2 detoxification to O2 and H2O are ubiquitously expressed and localized to many distinct cellular sites. The catalase and peroxiredoxin families are among the many enzymes that catalyze this reaction (28). In addition, GSH peroxidase catalyzes the reduction of H2O2, in addition to lipid hydroperoxides, into water through the oxidation of GSH (29).
Protein and lipid oxidation in adipose biology
An additional fate of H2O2 is the generation of the hydroxyl radical (•OH−) under conditions that favor Fenton chemistry. In this case, H2O2 reacts with ferrous iron in a chemically driven reaction resulting in the formation of the highly reactive hydroxyl radical. The hydroxyl radical is unique because, unlike H2O2, there are no known enzymes responsible for detoxification, and as such, •OH− is only consumed through the removal of an electron from neighboring molecules, including lipids, proteins, and nucleic acids.
In adipose tissue, unsaturated fatty acids are a significant target of oxidation by •OH−. Polyunsaturated fatty acids (PUFAs) in particular are reactive due to the instability caused by the double bonds. Because PUFAs are abundant in membrane structures, one major consequence of lipid peroxidation is membrane damage–a mechanism shown to play a causal role in mitochondrial dysfunction (30). In addition, peroxidation of PUFAs ultimately results in the release of diffusible reactive lipid aldehydes. Among the wide variety of reactive lipids formed through this mechanism, 4-hydroxynonenal (4-HNE) derived from oxidation of n-6 fatty acids and 4-hydroxyhexenal (4-HHE) from n-3 fatty acid oxidation are the most widely studied in the context of adipose biology. Such reactive lipid aldehydes are detoxified by aldehyde dehydrogenase 2-dependent oxidation, aldoketoreductase reduction, and glutathionylation by GSH S-transferase a3 and a4. In the latter case, glutathionylated lipids are secreted by fat cells and signal to macrophages to potentiate inflammation (31). Cytokine-dependent down-regulation of GSH S-transferase a4 and aldehyde dehydrogenase 2 mRNA expression in visceral adipose tissue potentiates lipid aldehyde accumulation by adipocytes (5). Increased oxidizing pressure leads to reduced GSH pools that in turn affect ER stress and the secretion of adipokines.
Lipid aldehydes are highly electrophilic and are prone to nucleophilic attack by the side chains of lysine, histidine, and cysteine residues of proteins, resulting in a covalent lipid–protein adduct in a process termed protein carbonylation (32). Such alkylation is not believed to be reversible, and there are currently no known enzymes that can remove the lipid adducts. Furthermore, Lys, His, and Cys are often found within active sites of enzymes or within critical structural motifs, so their stable modification by 9- or 6-carbon lipids generally leads to inhibition or deactivation of protein function.
Protein carbonylation is elevated in many tissues as a consequence of acute or prolonged oxidative stress. In the context of obesity, 4-HNE and 4-HHE levels are significantly increased in visceral but not subcutaneous adipose depots in mice and increase with body mass index in adipose tissue of humans (33). In addition, elevated protein carbonylation has been shown in cultured cells and in vivo as a result of a variety of metabolic challenges, including obesogenic diet, antioxidant depletion, inflammation, ER stress, and aging (32, 34, 35). Together, these observations lead to the hypothesis that protein carbonylation is a causal link between oxidative stress and metabolic dysfunction.
Proteomic analysis of 4-HNE and 4-HHE protein adducts has revealed a variety of cytoplasmic, mitochondrial, and nuclear targets (6, 36, 37). Cytoplasmic protein carbonylation has been implicated in the control of insulin signaling as well as glucose and lipid metabolism. Indeed, many reports have demonstrated that glycolytic enzymes are highly carbonylated in vivo where the modification largely leads to enzyme inactivation (37, 38). In cultured fat cells, silencing of mitochondrial GSH S-transferase leads to increased carbonylation of enzymes and proteins linked to tricarboxylic acid cycle metabolism, electron transport chain function, and branched chain amino acid metabolism. The outcomes of such modification led to diminished membrane potential and respiration (37) and potentiated ROS synthesis implying a potential feed-forward loop where ROS leads to carbonylation of mitochondrial proteins that in turn lead to elevated ROS (Fig. 2).
Figure 2.
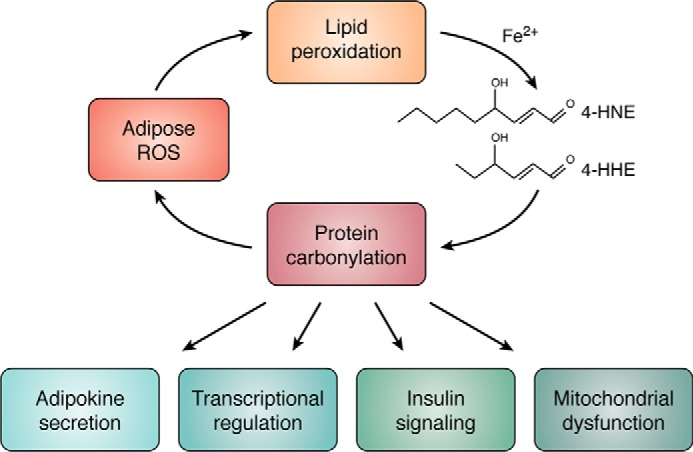
ROS–carbonylation cycle. Increased adipocyte ROS leads to lipid hydroperoxide product and the generation of reactive lipid electrophiles such as 4-HNE and 4-HHE. Reactive lipid aldehydes in turn covalently modify proteins of the adipocyte, particularly those in the mitochondrion and nucleus to regulate major metabolic processes, including gene expression, protein secretion, and insulin action.
Strikingly, recent work indicates that carbonylated proteins accumulate preferentially in the nucleus of epididymal adipose depot from mice fed an obesogenic diet (6). This observation is important for two reasons, First, zinc finger proteins and histones were among the most highly enriched for protein carbonylation (6). These proteins, including transcription factors, chromatin-modifying enzymes, and critical regulatory residues on histones, are nodes for transcriptional regulation (Fig. 2). Their modification by reactive lipids represent a new redox-dependent signaling paradigm by which oxidative stress may signal in a retrograde manner from the mitochondrion to the nucleus to affect transcriptional outcomes. Second, although the mitochondrion is the most significant source of ROS, and it has largely been assumed that 4-HNE and 4-HHE were also produced from mitochondrial membranes, discovery of lipid–protein adducts in the nucleus of adipocytes suggests either a different source of ROS contributes to lipid peroxidation or a mechanism exists to sequester and shuttle reactive aldehydes to specific subcellular localizations.
The link between oxidative stress and metabolic disorders has been investigated in both a whole-body and tissue-specific context tissue using murine models. Knockout mouse models of antioxidant enzymes have provided important insights into the causal nature of oxidative stress, but also to the tissue-specific roles of antioxidant enzymes (44). Conversely, overexpression models show promising evidence that tissue-specific amelioration of oxidative stress can prevent the development of metabolic disease phenotypes, although the importance of tissue-specific targeting of such strategies is clear and remains an important and active area in translational research (44).
Importantly, there are a number of examples of polymorphisms among the critical antioxidants described here that are linked to metabolic disease in humans. One well-characterized polymorphism in aldehyde dehydrogenase 2 (ALDH2) has been shown to be associated with type 2 diabetes in several studies (39). In this case, the rs671 SNP (G-to-A) results in a Glu-to-Lys substitution at residue 504, ultimately causing decreased enzyme activity of ALDH2 (40). Other examples include polymorphisms in members of the catalase (41, 42) and GSH S-transferase families (43). Furthermore, studies in humans show a positive correlation between lipid peroxidation and body mass index (11) and adipose protein carbonylation (33). Together, these data demonstrate the necessity for further study in humans to address the role of oxidative stress and resultant lipid peroxidation in the etiology of metabolic disease.
To parallel molecular analysis of genes linked to oxidative stress, several groups have approached redox control and protein carbonylation using small molecule antioxidants (45). In general, although clinical trials focused on small molecule antioxidant therapy have been disappointing, newer findings suggest that some molecules may be effective. Indeed, Anderson et al. (45) have shown that the dipeptide l-carnosine (β-alanyl histidine) can attenuate the metabolic effects of high-fat feeding (including reduced 4-HNE) when given to experimental mice. Consistent with this, l-carnosine has been used for years as a clinically useful component of lubricant eye drops given to patients with oxidation-linked ocular disease (46).
Summary
Depot and age-regulated accumulation of ROS underlies adipocyte biology and plays a fundamental role in signaling, fat cell metabolism, and dysfunction potentially affecting clinical outcomes such as type 2 diabetes and obesity. Potentiated accumulation of ROS in the visceral adipose depot of obese insulin-resistant mice compared with insulin-sensitive mice and humans implies that inflammation plays a major determining role in oxidative stress. Whether this observation implies that inflammatory cells themselves are the source of ROS or that cytokine-dependent down-regulation of adipocyte anti-oxidants underlies metabolic deficiency is unknown. Furthermore, it remains unclear what the intracellular sources of ROS are that contribute to oxidative stress phenotypes and organelle-specific ROS pools. Moreover, in many cases increased oxidative stress produces a “feed-forward” increase in additional ROS synthesis suggesting oxidative stress cascades with temporal and spatial regulation. Finally, the mechanistic studies focusing on the molecules targeted by ROS and how oxidation affects their function are critical goals for understanding the effects of oxidative stress and their linkage to metabolic disease. The availability of new technology in terms of animal models, analytical methodologies, and genome engineering tools now make approaches toward a broader understanding of the role(s) of oxidative stress in health and disease attainable.
This work was supported by National Institutes of Health Grant DK053189 (to D. A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- Nox
- NADPH oxidase
- mtDNA
- mitochondrial DNA
- ERO
- endoplasmic reticular oxidoreductin
- PUFA
- polyunsaturated fatty acid
- 4-HNE
- 4-hydroxy-2,3-trans-nonenal
- 4-HHE
- 4-hydroxy-2,3-trans-hexenal
- ER
- endoplasmic reticulum.
References
- 1. Houstis N., Rosen E. D., and Lander E. S. (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948 10.1038/nature04634 [DOI] [PubMed] [Google Scholar]
- 2. Zhang L., Ebenezer P. J., Dasuri K., Fernandez-Kim S. O., Francis J., Mariappan N., Gao Z., Ye J., Bruce-Keller A. J., and Keller J. N. (2011) Aging is associated with hypoxia and oxidative stress in adipose tissue: implications for adipose function. Am. J. Physiol. Endocrinol. Metab. 301, E599–E607 10.1152/ajpendo.00059.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chattopadhyay M., Khemka V. K., Chatterjee G., Ganguly A., Mukhopadhyay S., and Chakrabarti S. (2015) Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell. Biochem. 399, 95–103 10.1007/s11010-014-2236-7 [DOI] [PubMed] [Google Scholar]
- 4. Salgado-Somoza A., Teijeira-Fernández E., Fernández A. L., González-Juanatey J. R., and Eiras S. (2010) Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 299, H202–H209 10.1152/ajpheart.00120.2010 [DOI] [PubMed] [Google Scholar]
- 5. Long E. K., Olson D. M., and Bernlohr D. A. (2013) High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic. Biol. Med. 63, 390–398 10.1016/j.freeradbiomed.2013.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hauck A. K., Zhou T., Hahn W., Petegrosso R., Kuang R., Chen Y., and Bernlohr D. A. (2018) Obesity-induced protein carbonylation in murine adipose tissue regulates the DNA-binding domain of nuclear zinc finger proteins. J. Biol. Chem. 293, 13464–13476 10.1074/jbc.RA118.003469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwon J., Lee S.-R., Yang K.-S., Ahn Y., Kim Y. J., Stadtman E. R., and Rhee S. G. (2004) Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. U.S.A. 101, 16419–16424 10.1073/pnas.0407396101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chouchani E. T., Kazak L., Jedrychowski M. P., Lu G. Z., Erickson B. K., Szpyt J., Pierce K. A., Laznik-Bogoslavski D., Vetrivelan R., Clish C. B., Robinson A. J., Gygi S. P., and Spiegelman B. M. (2016) Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116 10.1038/nature17399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bedard K., and Krause K.-H. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- 10. Han C. Y., Umemoto T., Omer M., Den Hartigh L. J., Chiba T., LeBoeuf R., Buller C. L., Sweet I. R., Pennathur S., Abel E. D., and Chait A. (2012) NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 287, 10379–10393 10.1074/jbc.M111.304998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., and Shimomura I. (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin Y., Berg A. H., Iyengar P., Lam T. K., Giacca A., Combs T. P., Rajala M. W., Du X., Rollman B., Li W., Hawkins M., Barzilai N., Rhodes C. J., Fantus I. G., Brownlee M., and Scherer P. E. (2005) The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J. Biol. Chem. 280, 4617–4626 10.1074/jbc.M411863200 [DOI] [PubMed] [Google Scholar]
- 13. Cadenas S. (2018) Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1859, 940–950 10.1016/j.bbabio.2018.05.019 [DOI] [PubMed] [Google Scholar]
- 14. Yao X., Carlson D., Sun Y., Ma L., Wolf S. E., Minei J. P., and Zang Q. S. (2015) Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 10, e0139416 10.1371/journal.pone.0139416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fisher-Wellman K. H., and Neufer P. D. (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153 10.1016/j.tem.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsushima Y., Nishizawa H., Tochino Y., Nakatsuji H., Sekimoto R., Nagao H., Shirakura T., Kato K., Imaizumi K., Takahashi H., Tamura M., Maeda N., Funahashi T., and Shimomura I. (2013) Uric acid secretion from adipose tissue and its increase in obesity. J. Biol. Chem. 288, 27138–27149 10.1074/jbc.M113.485094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zito E. (2015) ERO1: a protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 83, 299–304 10.1016/j.freeradbiomed.2015.01.011 [DOI] [PubMed] [Google Scholar]
- 18. Makki K., Froguel P., and Wolowczuk I. (2013) Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013, 139239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atri C., Guerfali F. Z., and Laouini D. (2018) Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 19, E1801 10.3390/ijms19061801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Curtis J. M., Grimsrud P. A., Wright W. S., Xu X., Foncea R. E., Graham D. W., Brestoff J. R., Wiczer B. M., Ilkayeva O., Cianflone K., Muoio D. E., Arriaga E. A., and Bernlohr D. A. (2010) Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59, 1132–1142 10.2337/db09-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han C. Y. (2016) Roles of reactive oxygen species on insulin resistance in adipose tissue. Diabetes Metab. J. 40, 272–279 10.4093/dmj.2016.40.4.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Den Hartigh L. J., Omer M., Goodspeed L., Wang S., Wietecha T., O'Brien K. D., and Han C. Y. (2017) Adipocyte-specific deficiency of NADPH oxidase 4 delays the onset of insulin resistance and attenuates adipose tissue inflammation in obesity. Arterioscler. Thromb. Vasc. Biol. 37, 466–475 10.1161/ATVBAHA.116.308749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan H.-Y., Wang N., Li S., Hong M., Wang X., and Feng Y. (2016) The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid. Med. Cell Longev. 2016, 2795090 10.1155/2016/2795090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pepping J. K., Vandanmagsar B., Fernandez-Kim S. O., Zhang J., Mynatt R. L., and Bruce-Keller A. J. (2017) Myeloid-specific deletion of NOX2 prevents the metabolic and neurologic consequences of high fat diet. PLoS ONE 12, e0181500 10.1371/journal.pone.0181500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nomura M., Liu J., Rovira I. I., Gonzalez-Hurtado E., Lee J., Wolfgang M. J., and Finkel T. (2016) Fatty acid oxidation in macrophage polarization. Nat. Immunol. 17, 216–217 10.1038/ni.3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han Y. H., Buffolo M., Pires K. M., Pei S., Scherer P. E., and Boudina S. (2016) Adipocyte-specific deletion of manganese superoxide dismutase protects from diet-induced obesity through increased mitochondrial uncoupling and biogenesis. Diabetes 65, 2639–2651 10.2337/db16-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee H., Lee Y. J., Choi H., Ko E. H., and Kim J.-W. (2009) Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 284, 10601–10609 10.1074/jbc.M808742200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Netto L. E., and Antunes F. (2016) The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 39, 65–71 10.14348/molcells.2016.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brigelius-Flohé R. (2006) Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 387, 1329–1335 [DOI] [PubMed] [Google Scholar]
- 30. Nomura K., Imai H., Koumura T., Kobayashi T., and Nakagawa Y. (2000) Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem. J. 351, 183–193 10.1042/bj3510183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frohnert B. I., Long E. K., Hahn W. S., and Bernlohr D. A. (2014) Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 63, 89–100 10.2337/db13-0777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hauck A. K., and Bernlohr D. A. (2016) Oxidative stress and lipotoxicity. J. Lipid Res. 57, 1976–1986 10.1194/jlr.R066597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frohnert B. I., Sinaiko A. R., Serrot F. J., Foncea R. E., Moran A., Ikramuddin S., Choudry U., and Bernlohr D. A. (2011) Increased adipose protein carbonylation in human obesity. Obesity 19, 1735–1741 10.1038/oby.2011.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanase M., Urbanska A. M., Zolla V., Clement C. C., Huang L., Morozova K., Follo C., Goldberg M., Roda B., Reschiglian P., and Santambrogio L. (2016) Role of carbonyl modifications on aging-associated protein aggregation. Sci. Rep. 6, 19311 10.1038/srep19311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng J., Mutcherson R. 2nd, and Helfand S. L. (2005) Calorie restriction delays lipid oxidative damage in Drosophila melanogaster. Aging Cell 4, 209–216 10.1111/j.1474-9726.2005.00159.x [DOI] [PubMed] [Google Scholar]
- 36. Grimsrud P. A., Xie H., Griffin T. J., and Bernlohr D. A. (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 283, 21837–21841 10.1074/jbc.R700019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Curtis J. M., Hahn W. S., Stone M. D., Inda J. J., Droullard D. J., Kuzmicic J. P., Donoghue M. A., Long E. K., Armien A. G., Lavandero S., Arriaga E., Griffin T. J., and Bernlohr D. A. (2012) Protein carbonylation and adipocyte mitochondrial function. J. Biol. Chem. 287, 32967–32980 10.1074/jbc.M112.400663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. England K., O'Driscoll C., and Cotter T. G. (2004) Carbonylation of glycolytic proteins is a key response to drug-induced oxidative stress and apoptosis. Cell Death Differ. 11, 252–260 10.1038/sj.cdd.4401338 [DOI] [PubMed] [Google Scholar]
- 39. Li G. Y., Li Z. B., Li F., Dong L. P., Tang L., Xiang J., Li J. M., and Bao M. H. (2017) Meta-analysis on the association of ALDH2 polymorphisms and type 2 diabetic mellitus, diabetic retinopathy. Int. J. Environ. Res. Public Health 14, 165 10.3390/ijerph14020165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshida A., Huang I. Y., Ikawa M. (1984) Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in orientals. Proc. Natl. Acad. Sci. U.S.A. 81, 258–261 10.1073/pnas.81.1.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen H., Yu M., Li M., Zhao R., Zhu Q., Zhou W., Lu M., Lu Y., Zheng T., Jiang J., Zhao W., Xiang K., Jia W., and Liu L. (2012) Polymorphic variations in manganese superoxide dismutase (MnSOD), glutathione peroxidase-1 (GPX1), and catalase (CAT) contribute to elevated plasma triglyceride levels in Chinese patients with type 2 diabetes or diabetic cardiovascular disease. Mol. Cell. Biochem. 363, 85–91 10.1007/s11010-011-1160-3 [DOI] [PubMed] [Google Scholar]
- 42. dos Santos K. G., Canani L. H., Gross J. L., Tschiedel B., Souto K. E., and Roisenberg I. (2006) The catalase-262C/T promoter polymorphism and diabetic complications in Caucasians with type 2 diabetes. Dis. Markers 22, 355–359 10.1155/2006/983408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amer M. A., Ghattas M. H., Abo-Elmatty D. M., and Abou-El-Ela S. H. (2011) Influence of glutathione S-transferase polymorphisms on type-2 diabetes mellitus risk. Genet. Mol. Res. 10, 3722–3730 10.4238/2011.October.31.14 [DOI] [PubMed] [Google Scholar]
- 44. Lei X. G., Zhu J. H., Cheng W. H., Bao Y., Ho Y. S., Reddi A. R., Holmgren A., and Arnér E. S. (2016) Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 96, 307–364 10.1152/physrev.00010.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson E. J., Vistoli G., Katunga L. A., Funai K., Regazzoni L., Monroe T. B., Gilardoni E., Cannizzaro L., Colzani M., De Maddis D., Rossoni G., Canevotti R., Gagliardi S., Carini M., and Aldini G. (2018) A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Invest. 128, 5280–5293 10.1172/JCI94307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Babizhayev M. A., Micans P., Guiotto A., and Kasus-Jacobi A. (2009) N-Acetylcarnosine lubricant eyedrops possess all-in-one universal antioxidant protective effects of l-carnosine in aqueous and lipid membrane environments, aldehyde scavenging, and transglycation activities inherent to cataracts: a clinical study of the new vision-saving drug N-acetylcarnosine eyedrop therapy in a database population of over 50,500 patients. Am. J. Ther. 16, 517–533 10.1097/MJT.0b013e318195e327 [DOI] [PubMed] [Google Scholar]