

Abstract
Background
Bakanae disease, caused by the fungus Fusarium fujikuroi, occurs widely throughout Asia and Europe and sporadically in other rice production areas. Recent changes in climate and cropping patterns have aggravated this disease. To gain a better understanding of the molecular mechanisms of rice bakanae disease resistance, we employed a 6-plex tandem mass tag approach for relative quantitative proteomic comparison of infected and uninfected rice seedlings 7 days post-inoculation with two genotypes: the resistant genotype 93–11 and the susceptible genotype Nipponbare.
Results
In total, 123 (77.2% up-regulated, 22.8% down-regulated) and 91 (94.5% up-regulated, 5.5% down-regulated) differentially expressed proteins (DEPs) accumulated in 93–11 and Nipponbare, respectively. Only 11 DEPs were both shared by the two genotypes. Clustering results showed that the protein regulation trends for the two genotypes were highly contrasting, which suggested obviously different interaction mechanisms of the host and the pathogen between 93 and 11 and Nipponbare. Further analysis showed that a noticeable aquaporin, PIP2–2, was sharply upregulated with a fold change (FC) of 109.2 in 93–11, which might be related to pathogen defense and the execution of bakanae disease resistance. Certain antifungal proteins were regulated in both 93–11 and Nipponbare with moderate FCs. These proteins might participate in protecting the cellular integrity required for basic growth of the susceptible genotype. Correlation analysis between the transcriptome and proteome revealed that Pearson correlation coefficients of R = 0.677 (P = 0.0005) and R = − 0.097 (P = 0.702) were obtained for 93–11 and Nipponbare, respectively. Our findings raised an intriguing result that a significant positive correlation only in the resistant genotype, while no correlation was found in the susceptible genotype. The differences in codon usage was hypothesized for the cause of the result.
Conclusions
Quantitative proteomic analysis of the rice genotypes 93-11and Nipponbare after F. fujikuroi infection revealed that the aquaporin protein PIP2–2 might execute bakanae disease resistance. The difference in the correlation between the transcriptome and proteome might be due to the differences in codon usage between 93-11and Nipponbare. Overall, the protein regulation trends observed under bakanae disease stress are highly contrasting, and the molecular mechanisms of disease defense are obviously different between 93 and 11 and Nipponbare. In summary, these findings deepen our understanding of the functions of proteins induced by bakanae disease and the mechanisms of rice bakanae disease resistance.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5435-5) contains supplementary material, which is available to authorized users.
Keywords: Rice, Bakanae disease, Proteomic dissection, Transcript, Correlation
Background
Rice (Oryza sativa L.) is one of the world’s most important food crops, serving as a staple for more than 50% of the world population. In China, rice is a staple food crop that feeds more than 60% of the population and contributes nearly 40% of the total calorie intake [1]. Under diverse ecological conditions, rice is frequently exposed to a variety of biotic and abiotic stresses [2]. Among biotic stresses, many diseases play an important role in determining the yield and cost of food. Rice bakanae disease, caused by the fungus Fusarium fujikuroi, is an important disease in rice. That can be soilborne or seedborne and results in characteristic symptoms of excessively elongated seedlings with chlorotic stems and leaves. Bakanae disease occurs widely throughout Asia and Europe, and sporadically in other areas of rice production [3, 4]. Recent changes in climate and in cropping patterns have aggravated the disease. Although bakanae disease can be managed to a certain extent using chemical fungicides [5] through seed treatment and soil amendment, a more efficient, sustainable and environmentally friendly solution is to explore genetic resources that are resistant to the disease and breed additional high-resistance genotypes.
Several studies have made progress in identifying quantitative trait loci (QTLs) governing resistance against bakanae in rice [2, 6, 7]. Yang et al. [6] first obtained two QTLs, qB1 and qB10, associated with more than 13% of expressed variance for resistance to bakanae disease, via artificial inoculation at the seedling stage using a japonica/indica double haploid (DH) population derived from Chunjiang 06 and TN1. Hur et al. [7] reported a major QTL for bakanae disease resistance, qBK1, which explained 65% of the total phenotypic variation in near-isogenic lines (NILs) obtained from a cross between the resistant donor YR24982–9-1 and the susceptible parent Ilpum. Fiyaz et al. [2] identified three QTLs (qBK1.1, qBK1.2 and qBK1.3, which accounted for 4.76, 24.74 and 6.49% of phenotypic variation, respectively) using a recombinant inbred line (RIL) population derived from the indica rice parents Pusa 1342 and Pusa Basmati 1121. qBK1.1 is likely the same as qBK1 identified by Hur et al. [7], but the associated phenotypic variations were extremely different because different mapping populations were used; thus, these QTLs are far from application in marker-assisted introgression of bakanae resistance in rice breeding. Recent research [8] using a GWAS approach to screen a japonica rice germplasm collection revealed two new genomic regions that were highly associated with the observed phenotypic variation for the response to bakanae infection on the short arm of chromosome 1 (designated qBK1_628091) and the long arm of chromosome 4 (designated qBK4_31750955), representing new genomic regions associated with F. fujikuroi resistance.
In addition, RNA sequencing (RNA-seq) has been used to illustrate the molecular mechanism of the interaction between F. fujikuroi and rice plants. Different expression patterns were identified for the resistant genotype 93–11 and the susceptible genotype Nipponbare through comparative transcriptome analysis [9]. The results revealed that certain WRKYs, WAK and MAP3Ks were responsible for the bakanae disease resistance of 93-11and showed that the defense-related genes (WRKYs and MARKs) on chromosome 1 that are modulated in 93–11 upon infection might play a crucial role in the rice-F. fujikuroi interaction. Matić S et al. [10] found that PR1, germin-like proteins, glycoside hydrolases, MAP kinases, and WRKY transcriptional factors were up-regulated in the resistant genotype Selenio upon infection with F. fujikuroi, whereas up-regulation of chitinases and down-regulation of MAP kinases and WRKY transcription factors were found in the susceptible cultivar Dorell. Pathways including the response to chitin, JA-dependent signalling and the hypersensitive response were found to be involved in bakanae resistance.
In addition to QTL mapping and transcriptome analysis of rice bakanae disease resistance, other technologies and methods are needed to further dissect resistance to bakanae disease. With the completion of rice genome sequencing in 2002, proteomics is becoming an increasingly powerful tool for the investigation of complex cellular processes and has also been successfully used for genetic and physiological studies [11]. Proteomic technology is also playing a key role in developing an inventory of proteins that are responsive to biotic and abiotic stresses and in dissecting rice defense pathways [12]. In rice breeding, proteomics is usually applied to detect stress-responsive proteins through comparisons among stressed plants. Further identification of candidate proteins may reveal that some of these proteins have functions that are clearly consistent with the stress-tolerance trait [13].
A comparative proteomics study of rice genotypes that are resistant and susceptible to bakanae disease has yet not to be reported, nor has the correlation between the proteome and transcriptome under disease stress. In this study, the tandem mass tag (TMT) technique was applied to compare the differences in the proteomic pattern associated with the bakanae disease response using the resistant genotype 93–11 and the susceptible genotype Nipponbare. On the basis of our previous study of the disease response at different transcriptome levels between the two genotypes [9], correlation analysis was conducted between proteomic data and the transcriptomic data. The molecular mechanism of rice resistance to bakanae disease was thereby comprehensively dissected.
Results and discussion
Responses of 93–11 and Nipponbare after inoculation with F. Fujikuroi
Using the inoculation method applied in our previous study [9], 93–11 and Nipponbare were found to show different symptoms following inoculation with 8.8 × 106 spores/ml of F. fujikuroi. Nipponbare Seedlings were elongated while 93–11 showed no elongation (Fig. 1). The seedling height of Ne and Nck presented an extremely significant difference, whereas no difference was found between 93-11e and 93-11ck (Table 1).
Fig. 1.

Symptoms of Nipponbare and 93–11 under inoculation with 8.8 × 106 spores/ml of F. fujikuroi
Table 1.
Height of elongated seedling of 93–11 and Nipponbare treated with 8.8 × 106 spores/ml of F. fujikuroi
| Treatments | Seedling height |
|---|---|
| 93-11ck | 6.63 ± 0.40 |
| 93-11e | 7.13 ± 0.23 |
| Nipponbare-ck | 7.70 ± 0.36 |
| Nipponbare-e | 10.03 ± 0.42** |
**represents a significant difference at P = 0.01
Previous reports have revealed that indica exhibits higher resistance to rice blast (Magnaporthe grisea) and sheath blight (Rhizoctonia solani) than japonica [14–16]. Hur et al. [7] used the resistant indica variety Shingwang and the susceptible japonica variety Ilpum to generateNILs for identifying QTLs associated with bakanae disease resistance. In other studies, both resistant and susceptible indica genotypes [2] or japonica genotypes [8, 17] have been used to evaluate resistance to bakanae disease. In this study, Nipponbare (japonica) was more sensitive to F. fujikuroi, whereas 93–11 (indica) was resistant, indicating that indica 93–11 exhibited higher resistance to the bakanae disease stress than japonica Nipponbare cultivar.
Protein identification and quantification
After TMT and LC-MS/MS analysis of F. fujikuroi-infected seedlings and mock-infected seedlings collected at 7 days post-inoculation (dpi), a total of approximately 100,000 polypeptides were detected with 48,195 and 52,576 polypeptide species being identified for 93–11 and Nipponbare respectively (Table 2). After two-stage mass spectrometry and protein quantification, the identified proteins were screened in the NCBI database (https://www.ncbi.nlm.nih.gov/, file: txid39947-Oryza-sativa-28,555 s-20150412.fasta) with VennPlex software. Overall, 7, 478 and 9161 proteins were identified in the two genotypes when a false discovery rate (FDR) < 1% was applied to the dataset.
Table 2.
Results of protein identification and quantification
| Item | 93–11 | Nipponbare |
|---|---|---|
| Polypeptides identified | 100,507 | 106,997 |
| Polypeptide species identified | 48,195 | 52,576 |
| Quantitative proteins | 7478 | 9161 |
| DEPs | 123 | 91 |
| Up-regulated DEPs | 95 | 86 |
| Down-regulated DEPs | 28 | 5 |
A total of 123 and 91 DEPs accumulated in 93–11 and Nipponbare respectively. Using the criteria of P value ≤0.05 and FC ≥ 1.5 or FC ≤ 0.667, 95 (77.2%) and 28 (22.8%) DEPs in 93–11, 86 (94.5%) and 5 (5.5%) DEPs in Nipponbare were found to be up- and down-regulated, respectively. Furthermore, 11 DEPs were both shared by the two genotypes (Fig. 2).
Fig. 2.
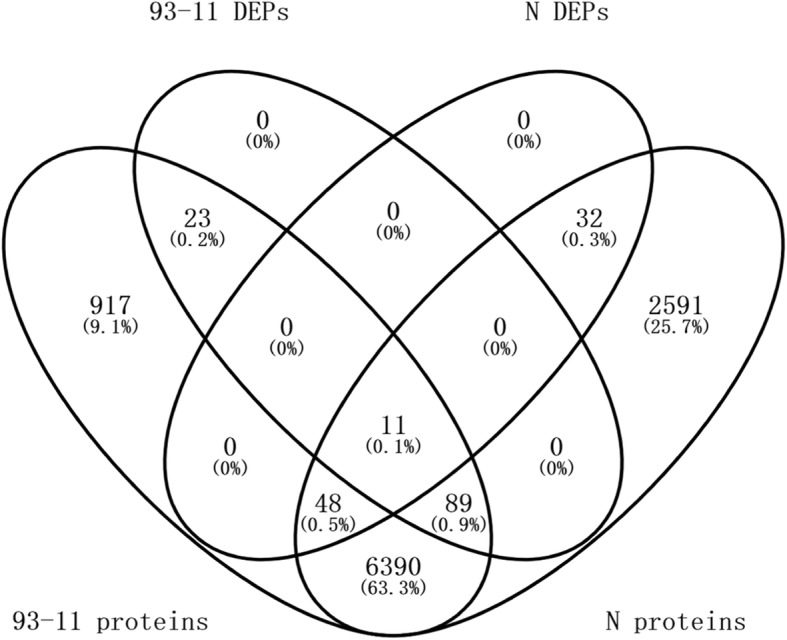
Venn diagram of total and F. fujikuroi-modulated proteins
Since only 11 DEPs were shared by the two genotypes, the difference in DEPs between 93 and 11 and Nipponbare infected by the pathogen were further investigated using cluster analysis (Fig. 3). Clustering results showed that the protein regulation trends for the two genotypes were highly contrasting, which suggested an obviously different interaction mechanism of the host and the pathogen between 93 and 11 and Nipponbare.
Fig. 3.
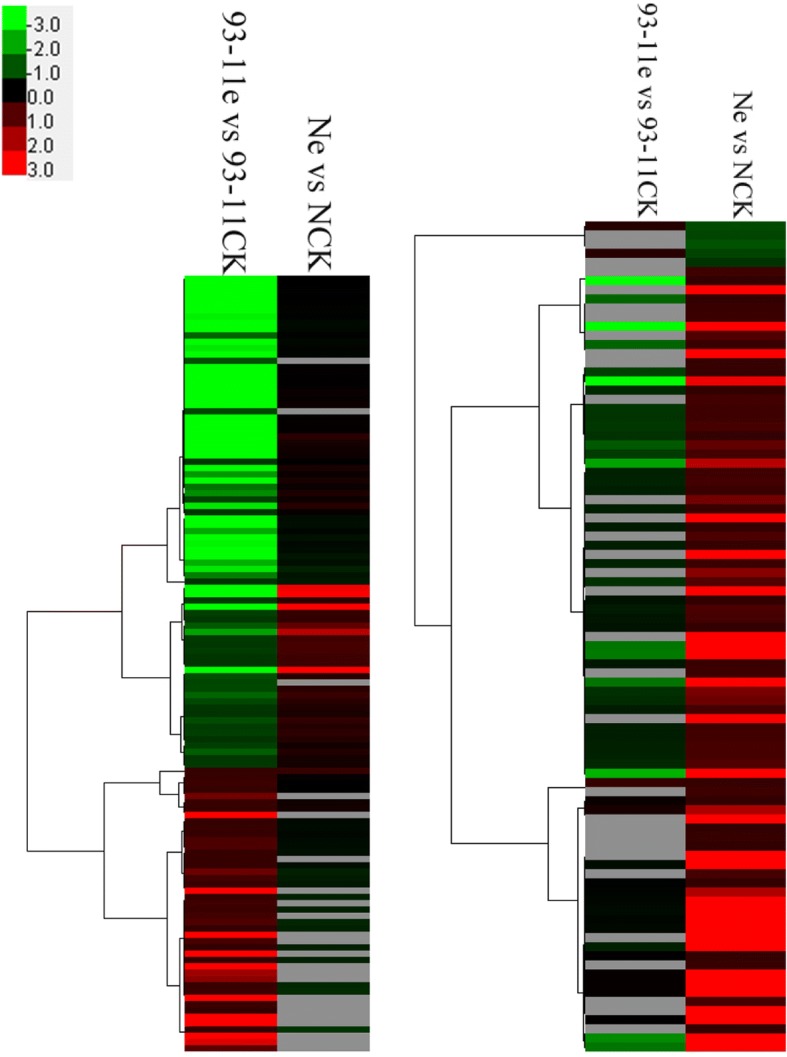
Hierarchical cluster analysis of protein expression based on FC data
Greatly up-regulated proteins might be closely related to bakanae disease resistance
To obtain target proteins to analyze the interaction mechanisms of the host and the pathogen, further criteria (≥2 unique peptides and ≥ 6 counts) were used for the DEPs in the two genotypes. With these further criteria, 29 (Table 3) and 27 (Table 4) DEPs were obtained in 93–11 and Nipponbare respectively, with 4 DEPs (Table 5) being shared. It was noted that the aquaporin (AQP) PIP2–2 was sharply up-relgulated with an FC of 109.2 in 93–11 (Table 3).
Table 3.
The 29 DEPs identified in 93–11 using the further criteria
| Accession | Gene_Symbol | Description | FC |
|---|---|---|---|
| XP_015626399.1 | LOC4330049 | probable aquaporin PIP2–2 | 109.18 |
| XP_015614532.1 | LOC4348376 | vacuolar-sorting receptor 3 | 16.01 |
| XP_015639310.1 | LOC4339191 | probable sugar phosphate/phosphate translocator At4g32390 | 7.86 |
| XP_015622682.1 | LOC4328618 | fumarylacetoacetase | 7.46 |
| XP_015646863.1 | LOC4344152 | serine carboxypeptidase II-3 | 7.23 |
| XP_015622762.1 | LOC4329858 | isocitrate dehydrogenase [NAD] regulatory subunit 1, mitochondrial isoform X2 | 3.82 |
| XP_015618186.1 | LOC4351745 | glucuronoxylan 4-O-methyltransferase 1 | 3.48 |
| XP_015634447.1 | LOC4335518 | alpha-humulene synthase | 2.08 |
| XP_015635507.1 | LOC9271676 | phenylalanine ammonia-lyase-like | 1.91 |
| XP_015638718.1 | LOC4337901 | fasciclin-like arabinogalactan protein 11 | 1.88 |
| XP_015649427.1 | LOC4345604 | alpha carbonic anhydrase 7 | 1.80 |
| XP_015622103.1 | LOC4326471 | salt stress-induced protein | 1.80 |
| XP_015626729.1 | LOC4330040 | phenylalanine ammonia-lyase | 1.67 |
| XP_015621390.1 | LOC4325129 | peroxidase 72 | 1.63 |
| XP_015641088.1 | LOC4340665 | probable peroxygenase 4 | 1.63 |
| XP_015623983.1 | LOC4329036 | salt stress root protein RS1-like | 1.62 |
| XP_015628637.1 | LOC4332648 | uncharacterized protein LOC4332648 | 1.62 |
| XP_015651336.1 | LOC4347224 | 1-aminocyclopropane-1-carboxylate oxidase 1 | 1.58 |
| XP_015621495.1 | LOC4324284 | salt stress root protein RS1 | 1.56 |
| XP_015641648.1 | LOC4340788 | UDP-glycosyltransferase 708A6 | 1.55 |
| XP_015621166.1 | LOC4325960 | scopoletin glucosyltransferase | 1.53 |
| XP_015639141.1 | LOC4339222 | peroxidase 1 | 1.53 |
| XP_015612645.1 | LOC4347709 | ribonuclease 1 | 1.51 |
| XP_015623954.1 | LOC4328586 | peroxiredoxin-2E-2, chloroplastic | 0.65 |
| XP_015619408.1 | LOC4352014 | ribulose bisphosphate carboxylase small chain A, chloroplastic | 0.64 |
| XP_015627384.1 | LOC4329821 | putative UDP-rhamnose:rhamnosyltransferase 1 | 0.63 |
| XP_015625272.1 | LOC4326593 | 50S ribosomal protein L31 | 0.62 |
| XP_015614152.1 | LOC4348979 | protein TIC110, chloroplastic | 0.61 |
| XP_015617238.1 | LOC4326583 | carbonic anhydrase, chloroplastic isoform X1 | 0.56 |
Table 4.
The 27 DEPs identified in Nipponbare using the further criteria
| Accession | Gene_Symbol | Description | FC |
|---|---|---|---|
| XP_015633534.1 | LOC4335790 | beta-fructofuranosidase, insoluble isoenzyme 2 | 11.58 |
| XP_015617184.1 | LOC4349693 | G-rich sequence factor 1 isoform X1 | 10.98 |
| XP_015622762.1 | LOC4329858 | isocitrate dehydrogenase [NAD] regulatory subunit 1, mitochondrial isoform X2 | 4.50 |
| XP_015624063.1 | LOC4330051 | defensin Tm-AMP-D1.2 | 4.22 |
| XP_015644849.1 | LOC9271516 | uncharacterized protein LOC9271516 | 3.07 |
| XP_015649880.1 | LOC4344615 | cysteine-rich repeat secretory protein 55 | 2.70 |
| XP_015644657.1 | LOC9270927 | pathogenesis-related protein 1 | 2.54 |
| XP_015634447.1 | LOC4335518 | alpha-humulene synthase | 2.22 |
| XP_015643864.1 | LOC4341249 | peroxidase P7 | 1.86 |
| XP_015620386.1 | LOC4352160 | alpha-dioxygenase 1 isoform X1 | 1.79 |
| XP_015651336.1 | LOC4347224 | 1-aminocyclopropane-1-carboxylate oxidase 1 | 1.78 |
| XP_015636635.1 | LOC4334987 | subtilisin-like protease SBT3.8 isoform X2 | 1.77 |
| XP_015621390.1 | LOC4325129 | peroxidase 72 | 1.75 |
| XP_015631105.1 | LOC4332568 | pathogenesis-related protein 1 | 1.70 |
| XP_015614030.1 | LOC4348474 | uncharacterized protein LOC4348474 | 1.65 |
| XP_015651086.1 | LOC4325834 | uncharacterized protein LOC4325834 | 1.63 |
| XP_015649764.1 | LOC4345934 | tricin synthase 1 | 1.61 |
| XP_015620489.1 | LOC4352487 | major allergen Dau c 1 | 1.60 |
| XP_015615445.1 | LOC4350299 | cytochrome P450 87A3-like | 1.57 |
| XP_015629807.1 | LOC4325264 | GDSL esterase/lipase At5g45910 | 1.56 |
| XP_015618576.1 | LOC4351585 | actin-7 | 1.55 |
| XP_015625928.1 | LOC4330873 | 1-aminocyclopropane-1-carboxylate oxidase 1-like isoform X2 | 1.55 |
| XP_015649937.1 | LOC4346299 | monodehydroascorbate reductase | 1.53 |
| XP_015641256.1 | LOC4340800 | cytochrome P450 76C2 | 1.51 |
| XP_015632670.1 | LOC4325859 | salicylic acid-binding protein 2 | 1.50 |
| XP_015633866.1 | LOC4337415 | protochlorophyllide reductase A, chloroplastic | 0.57 |
| XP_015620637.1 | LOC4352505 | lipoxygenase 2.1, chloroplastic isoform X1 | 0.51 |
Table 5.
The 4 DEPs shared by the two genotypes according to the further criteria
| Accession | Gene_Symbol | Description | FC for N | FC for 93–11 |
|---|---|---|---|---|
| XP_015622762.1 | LOC4329858 | isocitrate dehydrogenase [NAD] regulatory subunit 1, mitochondrial isoform X2 | 4.50 | 3.82 |
| XP_015634447.1 | LOC4335518 | alpha-humulene synthase | 2.22 | 2.08 |
| XP_015651336.1 | LOC4347224 | 1-aminocyclopropane-1-carboxylate oxidase 1 | 1.78 | 1.58 |
| XP_015621390.1 | LOC4325129 | peroxidase 72 | 1.75 | 1.63 |
AQP belongs to a highly conserved group of membrane proteins referred to as major intrinsic proteins that facilitate water transport across biological membranes in all types of organisms. It has been reported that AQP responds to various stresses [18], including the overexpression of TaAQP8 in tobacco to increase tolerance to salt stress [19], overexpression of RWC3 in rice to increase tolerance to drought stress [20], and overexpression of BnPIP1 in tobacco to increase tolerance to water stress [21]. Zhang et al. [22] suggested that aquaporin proteins might improve plant abiotic resistance through alleviating water deficit or oxidative damage. In Arabidopsis, AQP was shown to be involved in responses to both abiotic and biotic stresses. For instance, the aquaporin PIP2–5 is up-regulated in the roots and aerial parts of Arabidopsis by both drought and cold stresses [23]. Overexpression of AtPIP1;4 enhances water flow and facilitates germination in response to cold stress [24]. Regarding biotic stress resistance, it was demonstrated that the aquaporin TaPIP1 transgenic Arabidopsis enhanced resistance to Pseudomonas syringae pv. Tomato (Pst) DC3000 infection [18]. In our study, the aquaporin protein PIP2–2 exhibited 109.18-fold up-regulation after F. fujikuroi inoculation, which is thought to be related to pathogen defense and the execution of bakanae disease resistance. Therefore, the present study provides the second report of the role of the aquaporin protein in biotic stress resistance.
Plant vacuoles are vital organelles [25]. Perturbation of the vacuolar trafficking machinery affects many cellular processes, including responses to pathogens [26]. Wang et al. [27] found that vacuolar trafficking mediated by vacuolar sorting receptor1 (VSR1) is required for osmotic stress-responsive ABA biosynthesis and osmotic stress tolerance. In this study, a vacuolar-sorting receptor 3(VSR3) was significantly up-regulated, with an FC of 16.01 in 93–11, which meant that the vacuolar-sorting protein VSR3 could improve resistance to bakanae to some extent.
According to the the susceptible genotype Nipponbare, the FCs of the genes encoding the aquaporin and VSR3 proteins were around 1 (data in the Additional file 1), which meant that no obvious change was demonstrated for the proteins between the treatment and the control plants in the susceptible genotype.
Plants have developed various mechanisms, including the production of antifungal proteins or resistance-related proteins to defend themselves against pathogenic fungi. In this study, antifungal proteins such as defensin, peroxidase and ribonuclease were regulated in both 93–11 and Nipponbare, although the FCs of these proteins were only moderately high. Furthermore, although Nipponbare was susceptible to bakanae disease, the corresponding gene encoding the beta-fructofuranosidase protein LOC4335790, also known as CIN2/GIF1, was up-regulated with an FC of 11.58. According to functional annotation with the Uniprot database (http://www.uniprot.org/uniprot/Q0JDC5), sugar homeostasis mediated by CIN2/GIF1 plays an important role in constitutive and induced physical and chemical defense against pathogens. Yuan et al. [28] reported that genes involved in plant defenses were up-regulated when senescence was induced in a susceptible rice variety after attack by the brown planthopper and that these genes might participate in protecting the cellular integrity required for the progression and completion of senescence. Therefore, antifungal or defense-related proteins in the susceptible genotype Nipponbare might participate in protecting the cellular integrity required for basic growth of the cultivar.
Correlation analysis between the proteome and transcriptome
A single protein-profiling experiment can provide only a few clues regarding the function of a protein [13]. The integration of available transcriptomic data with proteomic data is critical to add further value to the single proteomic datasets and to provide a global picture of gene regulation and metabolic networks. To better clarify potential molecular mechanisms, some studies have been performed to correlate protein and mRNA expression levels in the same sample, in an attempt to identify reliable genes for crop breeding [29–31].
In this study, correlation analysis was conducted at the differential expression level by combining the proteomic results of this study and the transcriptomic results of our previous study [9]. Correlation was considered to occur if a gene that was modulated as a DEP was also expressed as a DEG (differentially expressed gene at the transcript level) after F. fujikuroi treatment.
A total of 22 DEPs (17.9% of the total DEPs) were correlated with DEGs (2.1% of the total DEGs) in 93–11, and 18 DEPs (19.8% of the total DEPs) were correlated with DEGs (1.8% of the total DEGs) in Nipponbare (Fig. 4).
Fig. 4.
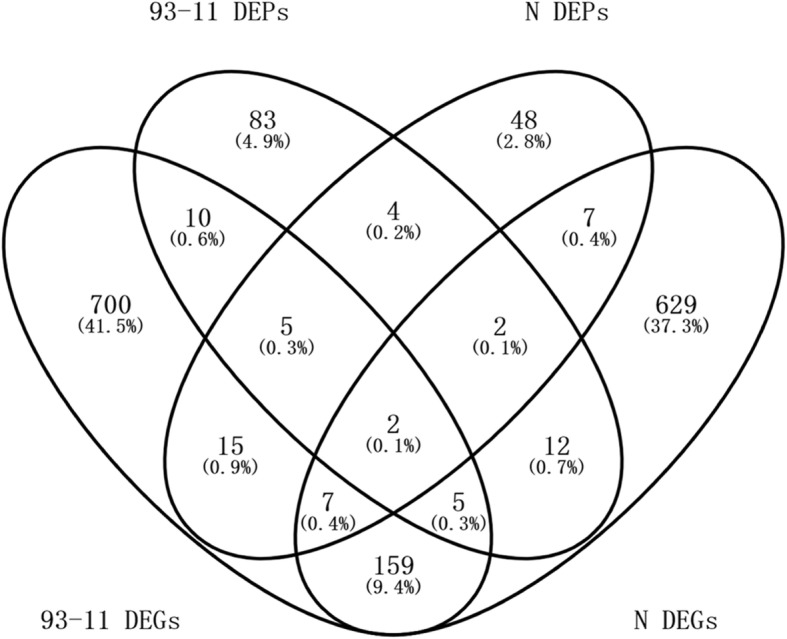
Venn diagram of DEPs and DEGs for Nipponbare and 93–11
The concordance between the changes in abundance at the transcript and protein levels was investigated using the corrgram function of the R programming language. Pearson correlation coefficients of R = 0.677 (P = 0.0005) and R = − 0.097 (P = 0.702) were obtained for 93–11 and Nipponbare, respectively, which meant that, a significant positive correlation was demonstrated in the resistant genotype at the two omics levels, whereas no such correlation was found in the susceptible genotype. Olivares-Hernández et al. [32] reported that most of the variability in the protein-transcript relationship can be explained by the variation in the codon composition, and codon usage variability therefore determines the correlation between proteome and transcriptome FCs. We hypothesize that the difference in the correlation of the two omic levels between the resistant and the susceptible genotypes might be due to the differences in the codon composition between the two genotypes under disease stress, resulting in the differences in the translation mechanism between the two genotypes. Further research is requisite to dissect the intriguing result.
Some arguments regarding this aspect have been put forth. In several reports, only a weak correlation was found between mRNA and protein abundances, due to post-transcriptional and post-translation regulation and measurement errors [33–35]. Ji et al. [31] obtained a value of R = 0.56 between the transcriptome and proteome. Certain opposite change trends were demonstrated between the two omics levels. The regulation of the process leading from mRNA to protein is generally very complex. During protein translation from mRNA, factors including mRNA splicing, altered protein turnover, protein degradation, or a combination of the above may lead to a poor correlation between the expression patterns of mRNAs and their respective proteins [36]. Komatsu [13] maintained that transcript and protein levels cannot be correlated, due to the inability of total mRNA to be translated into protein. The identification of proteomes that are not fully complete due to current limitations of proteomics [37] also affects global comparative studies of transcript and protein expression. Maier et al. [35] considered the correlation between mRNA and protein abundance to depend on various biological and technical factors. In general, with future advances in identification and quantification techniques employed in omics analyses, a more accurate picture of the organism-wide correlation between mRNA and protein will be achieved.
Conclusions
Protein expression plays an important role in the response to environmental stresses. In this study, TMT-based experiments were performed to identify proteins that were differentially accumulated between F. fujikuroi-infected and mock-inoculated seedlings of the bakanae disease-resistant and -susceptible genotypes 93-11and Nipponbare, respectively, to determine the key proteins involved in disease defense. The results revealed that the greatly up-regulated protein PIP2–2 might be related to pathogen defense and the execution of bakanae disease resistance. The vacuolar-sorting protein VSR3 can improve resistance to bakanae to some extent. Certain antifungal proteins were regulated in both the resistant and susceptible genotypes. Correlation analysis of transcriptome and proteome levels revealed a significant positive correlation only in the resistant genotype, while no such correlation was found in the susceptible genotype, which could be explained by the differences in codon usage. Overall, the molecular regulation of rice bakanae disease resistance was found to be completely different between 93-11and Nipponbare. Functional verification of the key protein PIP2–2 will be a prerequisite of our future research, which may be accomplished through transferring the related gene to the susceptible genotype or knocking down the gene in the resistant genotype.
Methods
Plant materials and inoculation with F. Fujikuroi
The indica rice genotype 93–11 and the japonica genotype Nipponbare (sourced from Qian Qian’s laboratory at China National Rice Research Institute) were inoculated with F. fujikuroi. 93–11 is resistant and Nipponbare is susceptible to bakanae disease. The details of the inoculation method were the same as in our previous study [9]. For inoculum production, an isolate of F. fujikuroi xj1105 isolated from Chinese bakanae-infected rice plants was used. The fungus was grown for 7 days on potato dextrose agar (PDA) solid medium at 28 °C in the dark, after which microconidia were harvested using sterile distilled water and the concentration was adjusted to 8.8 × 106 spores/ml. Rice seeds were surface sterilized in 70% ethanol for 1 min, then in 5% sodium hypochlorite for 5 min and subsequently were rinsed 5 times in sterile water. Surface sterilized seeds were germinated for three days to uniform size and vigor and were then subjected to F. fujikuroi stress treatment. Germinated seeds were soaked in the spore suspension and shaken for 24 h at 30 °C, while the control seeds for each genotype were soaked in sterile distilled water. The germinated seeds were further transferred to wet test paper to grow to seedlings at 28–30 °C, with a 12 h photoperiod. At the time point of 7 dpi, the seedling length of the treated plants (designated 93-11e and Ne) and the corresponding controls (designated 93-11ck and Nck) was calculated. The seedlings were further frozen in liquid nitrogen immediately after collection and stored at − 80 °C. Three biological replicates were performed.
Protein extraction
Samples were ground in liquid nitrogen with a mortar and pestle. The sample powder was then dissolved (vortex blending) with trichloroacetic acid (TCA)-acetone and left to stand for at least 12 h at − 20 °C in precooling tubes. The mixture was then centrifuged in a Allegra 64R rotor (BeckMAN, USA) at 14,000 g at 4 °C for 15 min to remove the supernatant, and the precipitate was suspended in acetone at − 20 °C for 2 h. The suspension was centrifuged at 14,000 g at 4 °C for 10 min to remove the supernatant, and the precipitate was washed with chilled acetone and 90% acetone before vacuum drying. The lyophilized precipitate was cracked into smaller molecules with a protein breaker (diagenode, Belgium) at 4 °C for 1 h using protein extraction buffer (8 M urea, 0.1% SDS) containing additional 1 mM phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Roche, USA), and followed by centrifugation at 14,000 g for 10 min. The supernatant was carefully collected, and the protein quality was tested with a bicinchoninic acid (BCA) protein assay kit. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was also carried out to test protein quality with a 12% cross-linked polyacrylamide gel. Protein bands on the gel were visualized with silver staining. The gels was scanned with a Molecular Imager (Bio-Rad Laboratories, USA).The protein samples were stored at − 80 °C until further processing.
TMT labeling
The tandem mass tags TMT6 (Pierce, USA) with different reporter ions (126–131 Da) were applied as isobaric tags for relative quantification, and TMT labeling was performed according to the manufacturer’s instructions. Briefly, 100 μg per condition was transferred to a new tube, and 100 mM triethyl ammonium bicarbonate (TEAB) buffer was added to the protein solution to a final volume of 100 μL. Then, 5 μL of 200 mM Tris (2-carboxyethyl) phosphine (TCEP) was added, and the sample was incubated at 55 °C for 1 h. Next, 5 μL of 375 mM iodoacetamide was added to the sample, followed by incubation for 30 min at room temperature while protected from light. The proteins were precipitated with pre-chilled (− 20 °C) acetone. After resuspension with 100 μL of 100 mM TEAB, the proteins were digested overnight at 37 °C with 2.5 μg trypsin (Sigma, USA). The digested samples were individually labeled at room temperature for 1 h as follows: TMT6 reagents were used for 93–11, where the controls, with three replicates, were labeled with TMT6–126, 127 and 128, and the treatments were labeled with TMT6–129, 130 and 131; TMT10 reagents were used for Nipponbare, where the controls, with three replicates, were labeled with TMT10–126, 127C, and 127 N, the treatments were labeled with TMT10-128C, 128 N and 129 N, respectively. The labeling reaction was quenched by adding 8 μL of 5% hydroxylamine. Finally, the labeled peptide aliquots were combined for subsequent fractionation.
Fractionation of labeled peptides
For the fractionation of labeled peptides, samples were first lyophilized and reconstituted in solvent A (2% ACN, pH 10). Then, the samples were loaded onto an Xbridge PST C18 Column (130 Å, 5 μm particle size, 250 × 4.6 mm; Waters, USA) and resolved via a basic RPLC method using a gradient of 5 to 95% solvent B (90% ACN, pH 10) in 40 min. A total of 40 fractions were collected, which were then concatenated into 20 fractions, vacuum dried and stored at − 80 °C until further LC-MS/MS analysis.
LC-MS/MS analysis
LC-MS/MS analysis was performed at CapitalBio Technology using a Q Exactive mass spectrometer (Thermo Scientific, CA). The peptide mixture was separated through reversed-phase chromatography in a DIONEX nano-UPLC system using an Acclaim C18 PepMap100 nano-Trap column (75 μm × 2 cm, 2 μm particle size) (Thermo Scientific, USA) connected to an Acclaim PepMap RSLC C18 analytical column (75 μm × 25 cm, 2 μm particle size) (Thermo Scientific, USA). Before loading, the sample was dissolved in sample buffer containing 4% acetonitrile and 0.1% formic acid. A linear gradient from 3 to 30% mobile phase B (0.1% formic acid in 99.9% acetonitrile) over 48 min followed by a steep increase to 80% mobile phase B in 1 min was used at a flow rate of 300 nL/min. The nano-LC was coupled online to the Q Exactive mass spectrometer using a stainless steel emitter coupled to a nanospray ion source. Mass spectrometry analysis was performed in a data-dependent manner with full scans (350–1600 m/z), which were acquired using an Orbitrap mass analyzer at a mass resolution of 70,000 at 400 m/z in Q Exactive. The twenty most intense precursor ions from a survey scan were selected for MS/MS from each duty cycle and detected at a mass resolution of 35,000 at an m/z of 400 in an Orbitrap analyzer. All the tandem mass spectra were produced by the higher-energy collision dissociation (HCD) method. Dynamic exclusion was set at 18 s.
Data analysis and TMT quantification
Proteome Discoverer software (version 1.4) (Thermo Scientific, USA) was used for database searching against the NCBI Oryza sativa Japonica Group database. The following criteria were applied: precursor mass tolerance of 10 ppm and fragment mass tolerance of 20 mmu. Trypsin was specified as the digesting enzyme, and 2 missed cleavages were allowed. Cysteine carbamidomethylation and TMT modifications (N-terminus and lysine residues) were defined as fixed modifications and methionine oxidation and phosphorylation as variable modifications. The results were filtered using the following settings: only high-confidence peptides with a global FDR < 1% based on a target-decoy approach were included in the results. In the TMT quantitation workflow, the most confident centroid method was used with an integration window of 20 ppm. For protein quantitation, only unique peptides were employed. Proteins with a P value ≤0.05 and FC ≥ 1.5 or ≤ 0.5 between the treatment and the control plants were recognized as significant DEPs.
Protein hierarchical cluster analysis
Cluster analysis was performed for all of the DEPs identified in the 93-11ck vs. 93-11e and Nck vs. Ne comparisons on May 5th, 2018. The FCs for each DEP were clustered with Cluster 3.0 software (http://bioservices.capitalbio.com/xzzq/rj/3885.shtml) using the hierarchical method, and the results were visualized using Treeview software [38].
Transcriptome study and correlation analysis the proteome and transcriptome
Transcriptome study was conducted in our previous report [9] via RNA-Seq and bioinformatics. Correlation was considered to occur if a gene that was modulated as a DEP was also expressed as a DEG after F. fujikuroi treatment. Pearson correlation coefficients were calculated using the R programming language (R version 2.15.3) to evaluate the concordance of the changes in abundance at the transcript and protein levels.
Additional file
The total proteins and DEPs of 93-11 and Nipponbare. (XLSX 9906 kb)
Acknowledgements
We thank Dr. Dawei Xue of Hangzhou Normal University for his advice on the project design. We thank the anonymous reviewers and Dr. Zhou Zhongjing of Zhejiang Academy of Agricultural Sciences for their suggestion and assistance on revision of the manuscript. We also thank the CapitalBio Corporation at Beijing for its assistance in original data processing.
Funding
This work was supported by funds from the Science Technology Department of Zhejiang Province to the Zhejiang Agricultural Key Breeding Project (2016C02050–4), the National Key Research and Development Program of China (2016YFD0101104, 2017YFD0100301), Major Project for Breeding New Varieties of Transgenic Organisms of the Ministry of Agriculture of China (2016ZX08001–002), the Zhejiang Provincial Natural Science Foundation (LQ17C130005), and an Open Fund of Key Laboratory of Integrated Crop Pest Management in Southwest China (XNYH2016-1).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the additional supporting files.
Abbreviations
- AQP
Aquaporin
- DEGs
Differentially expressed genes at the transcript level
- DEPs
Differentially expressed proteins
- dpi
days post-inoculation
- FC
Fold change
- FDR
False discovery rate
- PDA
Potato dextrose agar
- QTLs
Quantitative trait loci
- RIL
Recombinant inbred line
- RNA-seq
RNA sequencing
- TMT
Tandem mass tag
Authors’ contributions
CY and QQ designed the study. ZJ, YZ and YL conducted the experiments and analyzed the data. JZ wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zhijuan Ji, Email: zhijuanji@126.com.
Yuxiang Zeng, Email: zyxyes1@gmail.com.
Yan Liang, Email: ly318318@126.com.
Qian Qian, Email: qianqian188@hotmail.com.
Changdeng Yang, Email: yangchangdeng@126.com.
References
- 1.Cheng SH, Zhuang JY, Fan YY, Du JH, Cao LY. Progress in Research and Development on hybrid rice: a super-domesticate in China. Ann Bot. 2007;100:959–966. doi: 10.1093/aob/mcm121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiyaz RA, Yadav AK, Krishnan SG, Ellur RK, Bashyal BM, Grover N, et al. Mapping quantitative trait loci responsible for resistance to Bakanae disease in rice. Rice. 2016;9:45. doi: 10.1186/s12284-016-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun S, Snyder W. The Bakanae disease of the rice plant. In: Nelson PE, Toussoun TA, Cook RJ, editors. Fusarium: diseases, biology, and taxonomy. University Park: Pennsylvania State University Press; 1981. pp. 104–113. [Google Scholar]
- 4.Webster RK, Gunnell PS. Compendium of rice diseases. St. Paul: APS Press; 1992. [Google Scholar]
- 5.Iqbal M, Javed N, Sahi ST, Cheema NM. Genetic management of Bakanae disease of rice and evaluation of various fungicides against Fusarium moniliforme in vitro. Pakistan J Phytopathol. 2011;23:103–107. [Google Scholar]
- 6.Yang CD, Guo LB, Li XM, Ji ZJ, Ma LY, Qian Q. Analysis of QTLs for resistance to rice Bakanae disease. Chin J Rice Sci. 2006;6:657–659. [Google Scholar]
- 7.Hur Y-J, Lee SB, Kim TH, Kwon T, Lee J-H, Shin D-J, et al. Mapping of qBK1, a major QTL for Bakanae disease resistance in rice. Mol Breed. 2015;35:78. doi: 10.1007/s11032-015-0281-x. [DOI] [Google Scholar]
- 8.Volante A, Tondelli A, Aragona M, Valente MT, Biselli C, Desiderio F, et al. Identification of bakanae disease resistance loci in japonica rice through genome wide association study. Rice. 2017;10:29. doi: 10.1186/s12284-017-0168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji ZJ, Zeng YX, Liang Y, Qian Q, Yang CD. Transcriptomic dissection of the rice–Fusarium fujikuroi interaction by RNA-Seq. Euphytica. 2016;211:123–137. doi: 10.1007/s10681-016-1748-5. [DOI] [Google Scholar]
- 10.Matić S, Bagnaresi P, Biselli C, Orru L, Carneiro GA, Siciliano I, et al. Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen Fusarium fujikuroi. BMC Genomics. 2016;17:608. doi: 10.1186/s12864-016-2925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal GK, Rakwal R. Rice proteomics: a move toward expanded proteome coverage to comparative and functional proteomics uncovers the mysteries of rice and plant biology. Proteomics. 2011;11:1630–1649. doi: 10.1002/pmic.201000696. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Nie Y, Zhang Z, Ye Z, Zou X, Zhang L, et al. Comparative proteomic analysis of methyl jasmonate-induced defense responses in different rice cultivars. Proteomics. 2014;14:1088–1101. doi: 10.1002/pmic.201300104. [DOI] [PubMed] [Google Scholar]
- 13.Komatsu S. Research on the rice proteome: the contribution of proteomics technology in the creation of abiotic stress-tolerant plants. Rice. 2008;1:154–165. doi: 10.1007/s12284-008-9013-8. [DOI] [Google Scholar]
- 14.Sato H, Ideta O, Ando I, Kunihiro Y, Hirabayashi H, Iwano M, et al. Mapping QTLs for sheath blight resistance in the rice line WSS2. Breed Sci. 2004;54:265–271. doi: 10.1270/jsbbs.54.265. [DOI] [Google Scholar]
- 15.Shang J, Tao Y, Chen X, Zou Y, Lei C, Wang J, et al. Identification of a new rice blast resistance gene, Pid3, by genome wide comparison of paired nucleotide-binding site-leucine-rich repeat genes and their pseudogene alleles between the two sequenced rice genomes. Genetics. 2009;182:1303–1311. doi: 10.1534/genetics.109.102871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willocquet L, Noel M, Sackville Hamilton R, Savary S. Susceptibility of rice to sheath blight: an assessment of the diversity of rice germplasm according to genetic groups and morphological traits. Euphytica. 2012;183:227–241. doi: 10.1007/s10681-011-0451-9. [DOI] [Google Scholar]
- 17.Lee SB, Hur YJ, Cho JH, Lee JH, Kim TH, Cho SM, et al. Molecular mapping of qBK1WD, a major QTL for bakanae disease resistance in rice. Rice. 2018;11:3. doi: 10.1186/s12284-017-0197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu PP, Yu TF, Zheng WJ, Chen M, Zhou YB, Chen J, et al. The wheat Bax Inhibitor-1 protein interacts with an aquaporin TaPIP1 and enhances disease resistance in Arabidopsis. Front Plant Sci. 2018;9:20. doi: 10.3389/fpls.2018.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu W, Yuan Q, Wang Y, Cai R, Deng X, Wang J, et al. Overexpression of a wheat aquaporin gene, TaAQP8, enhances salt stress tolerance in transgenic tobacco. Plant Cell Physiol. 2012;53:2127–2141. doi: 10.1093/pcp/pcs154. [DOI] [PubMed] [Google Scholar]
- 20.Lian HL, Yu X, Ye Q, Ding X, Kitagawa Y, Kwak SS, et al. The role of aquaporin RWC3 in drought avoidance in rice. Plant Cell Physiol. 2004;45:481–489. doi: 10.1093/pcp/pch058. [DOI] [PubMed] [Google Scholar]
- 21.Yu Q, Hu Y, Li J, Wu Q, Lin Z. Sense and antisense expression of plasma membrane aquaporin BnPIP1 from Brassica napus in tobacco and its effects on plant drought resistance. Plant Sci. 2005;169:647–656. doi: 10.1016/j.plantsci.2005.04.013. [DOI] [Google Scholar]
- 22.Zhang Y, Wang Z, Chai T, Wen Z, Zhang H. Indian mustard aquaporin improves drought and heavy-metal resistance in tobacco. Mol Biotechnol. 2008;40:280–292. doi: 10.1007/s12033-008-9084-1. [DOI] [PubMed] [Google Scholar]
- 23.Jang JY, Kim DG, Kim YO, Kim JS, Kang H. An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana. Plant Mol Biol. 2004;54:713–725. doi: 10.1023/B:PLAN.0000040900.61345.a6. [DOI] [PubMed] [Google Scholar]
- 24.Jang JY, Lee SH, Rhee JY, Chung GC, Ahn SJ, Kang H. Transgenic Arabidopsis and tobacco plants overexpressing an aquaporin respond differently to various abiotic stresses. Plant Mol Biol. 2007;64:621–632. doi: 10.1007/s11103-007-9181-8. [DOI] [PubMed] [Google Scholar]
- 25.Marty F. Plant vacuoles. Plant Cell. 1999;11:587–600. doi: 10.1105/tpc.11.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surpin M, Raikhel N. Traffic jams affect plant development and signal transduction. Nat Rev Mol Cell Biol. 2004;5:100–109. doi: 10.1038/nrm1311. [DOI] [PubMed] [Google Scholar]
- 27.Wang ZY, Gehring C, Zhu JH, Li FM, Zhu JK, Xiong LM. The Arabidopsis vacuolar sorting Receptor1 is required for osmotic stress-induced abscisic acid biosynthesis. Plant Physiol. 2015;167:137–152. doi: 10.1104/pp.114.249268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan H, Chen X, Zhu L, He G. Identification of genes responsive to brown planthopper Nilaparvata lugens Sta° l (Homoptera: Delphacidae) feeding in rice. Planta. 2005;221:105–112. doi: 10.1007/s00425-004-1422-3. [DOI] [PubMed] [Google Scholar]
- 29.Wu HX, Jia HM, Ma XW, Wang SB, Yao QS, Xu WT, et al. Transcriptome and proteomic analysis of mango (Mangifera indica Linn) fruits. J Proteome. 2014;105:19–30. doi: 10.1016/j.jprot.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Xu Z, Zhang Y, Chai L, Yi H, Deng X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J Exp Bot. 2014;65:1651–1671. doi: 10.1093/jxb/eru044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji JL, Yang LM, Fang ZY, Zhuang M, Zhang YY, Lv HH, et al. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. Journal of Proteomics. 2018;179:80–91. doi: 10.1016/j.jprot.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Olivares-Hernández R, Bordel S, Nielsen J. Codon usage variability determines the correlation between proteome and transcriptome fold changes. BMC Syst Biol. 2011;5:33. doi: 10.1186/1752-0509-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan ZY, Zeng YL, An JY, Ye JL, Xu Q, Deng XX. An integrative analysis of transcriptome and proteome provides new insights into carotenoid biosynthesis and regulation in sweet orange fruits. Journal of protemics. 2012;75:2670–2684. doi: 10.1016/j.jprot.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 34.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol BioSyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier T, Güell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Zhang HY, Lei G, Zhou HW, He C, Liao JL, Huang YJ. Quantitative iTRAQ-based proteomic analysis of rice grains to assess high night temperature stress. Proteomics. 2017. 10.1002/pmic.201600365. [DOI] [PMC free article] [PubMed]
- 37.Bitton DA, Okoniewski MJ, Connolly Y, Miller CJ. Exon level integration of proteomics and microarray data. BMC Bioinformatics. 2008;9:118. doi: 10.1186/1471-2105-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The total proteins and DEPs of 93-11 and Nipponbare. (XLSX 9906 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the additional supporting files.