

Abstract
Clinical conditions that result in endotoxemia, such as sepsis and alcoholic hepatitis, often are accompanied by cholestasis. Although hepatocellular changes in response to lipopolysaccharide (LPS) have been well characterized, less is known about whether and how cholangiocytes contribute to this form of cholestasis. We examined effects of endotoxin on expression and function of the type 3 inositol trisphosphate receptor (ITPR3), because this is the main intracellular Ca2+ release channel in cholangiocytes, and loss of it impairs ductular bicarbonate secretion. Bile duct cells expressed the LPS receptor TLR4, which links to activation of NF-κB. Analysis of the human ITPR3 promoter revealed five putative response elements to NF-κB, and promoter activity was inhibited by p65/p50. Nested 0.5 and 1.0 kb deletion fragments of the ITPR3 promoter were inhibited by NF-κB subunits. ChIP assay showed that NF-κB interacts with the ITPR3 promoter, with an associated increase in H3K9 methylation. LPS decreased ITPR3 mRNA and protein expression, and also decreased sensitivity of bile duct cells to calcium agonist stimuli. This reduction was reversed by inhibition of TLR4. ITPR3 expression was decreased or absent in cholangiocytes from patients with cholestasis of sepsis and from patients with severe alcoholic hepatitis.
Conclusion:
Stimulation of TLR4 via LPS activates NF-κB to downregulate ITPR3 expression in human cholangiocytes. This may contribute to the cholestasis that can be observed in conditions such as sepsis or alcoholic hepatitis.
Keywords: Sepsis, calcium signaling, cholangiocyte, cholestasis, nuclear factor-κB
INTRODUCTION
An association between endotoxin and cholestasis is well established. The clinical condition in which this has been reported most widely is sepsis. Estimates for the frequency of cholestasis in sepsis have varied from ~1% to over 50%, depending on such factors as the study population and the diagnostic criteria (1). However, several studies suggest that the frequency of cholestasis in severe sepsis is in the 20–30% range (2, 3). Endotoxin is thought to be important in the pathogenesis of alcoholic hepatitis as well (4). Although this had been considered a disease that mostly reflects inflammatory damage to hepatocytes, evolving evidence suggests that cholestasis can also be an important component of alcoholic hepatitis, and in fact the presence of cholestasis may reflect more severe disease (5–7). The molecular basis for endotoxin-induced cholestasis has been examined mostly in hepatocytes and has been attributed to decreased expression of several transporters that are important for canalicular bile formation (8–10). However, bile secretion depends not only upon formation of primary canalicular bile by hepatocytes, but also upon further conditioning of bile by cholangiocytes (11). Yet, very little is known about the effects of endotoxin on cholangiocyte secretion.
A variety of diseases affecting cholangiocytes result in cholestasis. Despite different etiologies, primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), biliary atresia and biliary obstruction each furthermore is associated with loss of the type 3 inositol 1,4,5-trisphosphate receptor (ITPR3) from cholangiocytes (12). Knockdown of ITPR3 in cholangiocytes specifically inhibits bicarbonate and fluid secretion (13), suggesting that its loss in these different cholangiopathies may represent a final common pathway to cholestasis. Certain transcription factors (14) and micro-RNA’s (15) can decrease ITPR3 expression in cholangiocytes, but the role of these may differ among the various cholangiopathies. The purpose of this study was to investigate whether and how endotoxin inhibits ITPR3 expression in human cholangiocytes, and to determine the clinical relevance of this.
MATERIALS AND METHODS
Analysis of NF-κB binding sites and transfection.
MatInspector (www.genomatix.de) was used to analyze the human ITPR3 promoter sequence, and 5 putative elements responding to nuclear factor-κB (NF-κB) were identified. For generation of fusion plasmids with firefly luciferase, 2kb, 1kb and 0.5 kb 5‘-upstream regions of the human ITPR3 gene including the proximal promoter were cloned. NHC cells were co-transfected with each of the ITPR3 promoter fragment and plasmids encoding p65 and p50 subunits of NF-κB with or without IκB-α super suppressor (IκBSR). Luciferase activity was analyzed 48 hours later using the Dual Luciferase kit (Promega) Also, NHC cells were treated with LPS (1 µg/mL) for 18 hours, after transfection with ITPR3pr 2kb. In all experiments, NHC cells were co-transfected with Renilla luciferase plasmid to normalize for transfection efficiency. Site-directed mutants of the ITPR3 promoter reporter constructs were generated using the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions. The mutated NF-κB constructs comprised five NF-κB single mutated plasmids (ITPR3-mut-NFκB1, ITPR3-mut-NFκB2, ITPR3-mut-NFκB3, ITPR3-mut-NFκB4 and ITPR3-mut-NFκB5), and the specific primers are listed in Supporting table 3. Mutated reporter constructs were verified by DNA sequencing at the W.M. Keck Biotechnology Resource Laboratory at Yale University.
Chromatin Immunoprecipitation (ChIP).
Experiments were performed using EZ-Magna ChIP HiSEns kit (Millipore-Upstate, Temecula, CA). NHC cells were fixed and cross-linked in 1% formaldehyde, inactivated with 2.5 M glycine, then nuclear lysates were prepared with isolation buffer. Chromatin samples were sheared into fragments by sonication followed by centrifugation at 10,000 rpm for 10 min at 4 °C. Sheared chromatin samples were precleared using protein G beads, then incubated overnight at 4 °C with ChIP-grade rabbit anti-NF-κB p65 monoclonal antibody (Abcam, Cambridge, MA). Anti-H3K9me or normal rabbit IgG were used as negative controls. DNA was purified and real time PCR was performed to evaluate the ChIP-enriched DNA (14).
Ca²+ measurements.
NHC cells were plated onto glass coverslips 24 hours prior to imaging. Cells were then loaded with 6 µM Fluo-4/AM (Invitrogen, Eugene, OR) for 30 minutes at 37° C. Once transferred to a custom-built perfusion chamber on the stage of a Zeiss LSM 710 confocal microscope (Carl Zeiss, Inc, Thornwood, NY), cells were perfused with HEPES buffer (16) while stimulated with 1 µM adenosine triphosphate (ATP). Ca²+ signaling was monitored in these cells by exciting at 488 nm while collecting emitted light above 505 nm. Normalized amplitudes of the ATP-induced Ca2+ signals were extracted with Image J software and plotted as previously described (17). For flash photolysis of caged InsP3, cells were transfected with a rat mCherry-ITPR3 plasmid (a gift from David Yule, University of Rochester Medical Center) and 48 hours later incubated with 1 μM caged InsP3 (Bio-Techne, Minneapolis, MN) and 6 µM Fluo-4/AM for 30 minutes at 37° C. Cells were then imaged on a Bruker Opterra II Swept field confocal microscope (Middleton, WI) using 488 nm excitation and collection above 505 nm. Uncaging was achieved by a brief pulse of 405 nm laser line on individual cells (36 µs/pixel). Changes in fluorescence were normalized by baseline levels and used to calculated the rise time of Ca2+ signals, which is the time required for the signal to increase from 20% to 80% of the maximum response.
Human liver specimens.
Human liver tissue biopsies and clinical data from patients with sepsis and alcoholic hepatitis were obtained under the auspices of protocols approved by the Ethics Committee of Federal University of Minas Gerais (Belo Horizonte, Brazil) number CAAE: 06595912.2.20000.5125 and the Institutional Review Board on the Protection of the Rights of Human Subjects (Yale University, New Haven, CT) number HIC-1304011763. Sepsis specimens were from all patients who died at Hospital das Clinicas from 2015 to 2017, with sepsis listed as cause of death and with cholestasis diagnosed histologically post-mortem. The clinical characteristics of 31 consecutive patients with biopsy-proven alcoholic hepatitis at Yale-New Haven Hospital during the time period 2012 to 2014 were analyzed. Residual liver tissue used for immunohistochemistry analysis was available for 7 of those 31 patients. Histologically normal tissues were obtained from liver resections of metastatic colon cancer patients as determined by a review of pathology reports from 2004 to 2013 at Yale New Haven Hospital and 2010 to 2017 at Hospital das Clinicas (UFMG).
Statistical analysis.
Results are presented as mean ± SEM. Data were analyzed using Graphpad Prism 7 and Microsoft Excel. Differences between experimental groups were assessed for significance using Student’s t-test or one-way ANOVA followed by Bonferroni post hoc-tests. Statistical significance was defined as p<0.05.
Further methodological details.
Detailed additional Materials and Methods are available in the Supporting Information.
RESULTS
LPS decreases ITPR3 expression.
Because ITPR3 is the principal ITPR isoform in cholangiocytes and is important for bicarbonate-rich secretion (12, 13, 18), the effect of LPS on ITPR3 expression was tested in NHC cells. ITPR3 mRNA expression was reduced in cells treated with LPS concentrations of 125 (0.20 ± 0.05, p<0.001, n=5), 250 (0.30 ± 0.01, p<0.001, n=5) and 500 ng/mL (0.27 ± 0.01, p<0.001, n=5), when compared with control (Figure 1a). This decrease was not observed at lower LPS concentrations of 31 (0.86 ± .07, n=3) or 62 ng/mL (0.93 ± 0.03, n=3). Because 125 ng/mL was the lowest LPS concentration to maximally inhibit ITPR3 expression, this concentration was used for further studies. To determine whether the LPS-mediated decrease in ITPR3 expression is time-dependent, cells were treated with LPS (125 ng/mL) for 30 minutes, 6 and 18 hours. ITPR3 mRNA levels progressively decreased over time, reaching its lowest level at 18 hours (Figure 1b). ITPR3 protein levels were also reduced in NHC cells exposed to LPS for 18 hours as shown by both western blotting (control 0.34±0.03 versus LPS 0.14±0.03, p<0.01, n=3, Figure 1c) and immunofluorescence (control 38.78±5.40 versus LPS 17.74±0.77, n=3, 15 cells for each group, p<0.01, Figure 1d). Similar findings were obtained in H69 cells, in which exposure to LPS reduced both ITPR3 mRNA (control 1.00 ± 0.02; LPS 0.55 ± 0.07, p<0.05, n=3, Figure 1e) and protein expression (control 0.64 ± 0.06; LPS 0.13 ± 0.05 p<0.05, n=3, Figure 1f) when compared to untreated control cells. We also evaluated whether the expression of the two minor isoforms of the ITPR in cholangiocytes, ITPR1 and ITPR2 (13, 18), is altered by LPS treatment. Real time PCR analysis showed that treatment of NHC cells with LPS resulted in a much lesser but nonetheless significant decrease of ITPR1 mRNA (0.68 ± 0.03, p<0.001, n=5) compared with control, while no alteration in ITPR2 was observed (0.88 ± 0.08, n=5) relative to untreated control cells (Supporting Figure S1). These results are similar to what was observed in a rat model of endotoxemia (12) and together suggest that LPS treatment downregulates ITPR3 expression in human cholangiocytes as well.
FIGURE 1.
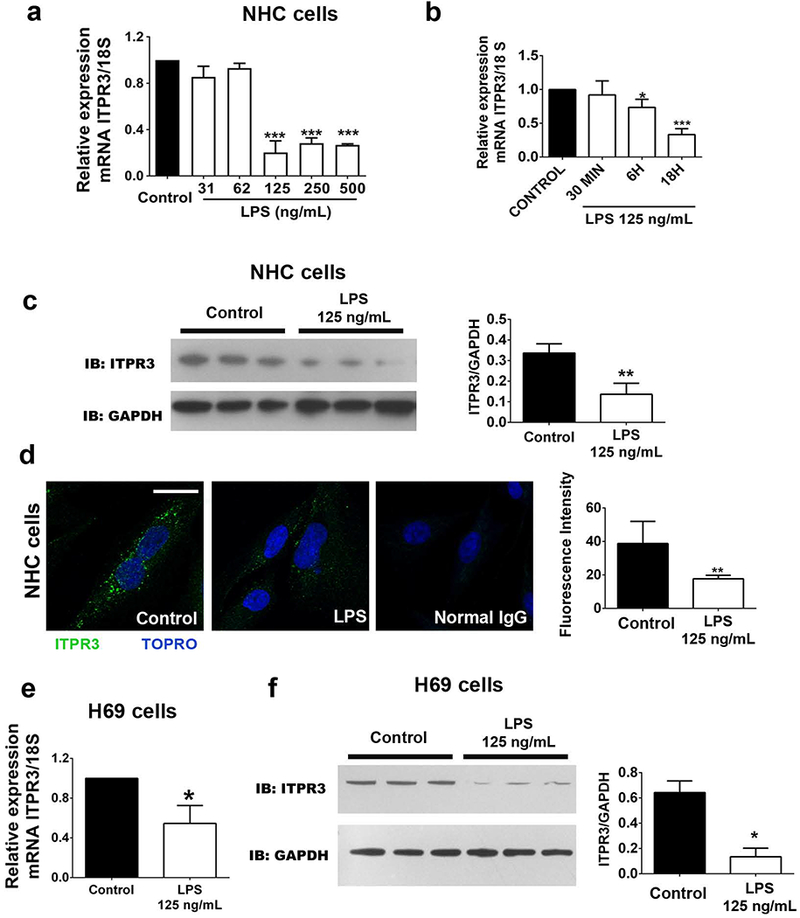
LPS decreases ITPR3 expression in cholangiocytes. (A) ITPR3 mRNA expression in NHC cells after LPS treatment for 18 hours. (B) qPCR of ITPR3 in NHC cells after LPS (125 ng/mL) treatment for different times (30 minutes, 6 and 18 hours). (C) Western blotting and (D) immunofluorescence for ITPR3 in NHC cells incubated with LPS for 18 hours. (E) ITPR3 mRNA expression and (F) western blotting in H69 cells treated with LPS 125 ng/mL for 18 hours. Scale bar=20 µm. **p<0.01 and ***p<0.001 using Student’s t test (B-E) or one-way ANOVA test, followed by Bonferroni post-hoc test (A).
NF-κB inhibits ITPR3 promoter activity.
Endotoxins typically activate the transcription factor NF-κB via binding to toll-like receptor 4 (TLR4) (19), which is expressed in cholangiocytes (20). The NF-κB inhibitor BAY 11–7082 (BAY) prevented the decrease in ITPR3 that was induced by LPS in mRNA (LPS+BAY 0.80 ± 0.06 versus LPS 0.35 ± 0.04, n=3; p<0.001, Figure 2a) and protein levels (LPS+BAY 0.77 ± 0.07 versus LPS 0.18 ± 0.09, p<0.01, Figure 2b), suggesting a role for this pathway in regulating ITPR3 expression. Therefore, an in-silico analysis of the proximal 2 kb of the human ITPR3 promoter was performed to probe for putative NF-κB responsive elements. This revealed the presence of five potential NF-κB responsive elements located at −293 to −307bp (NF-κB1), −329 to −343bp (NF-κB2), −397 to −411bp (NF-κB3), −860 to −874bp (NF-κB4) and −1778 to −1792bp (NF-κB5) upstream of the transcription starting sites (Supporting Fig. S2). To validate the MatInspector software analyses, NHC were initially transfected with a 2 kb fragment of the ITPR3 promoter fused with luciferase alone or in combination with p65/p50 plasmids or else treated with LPS (1µg/mL, Supporting Fig. S3). The luciferase activity of the ITPR3 promoter construct was reduced by co-expression of NF-κB subunits (2 kb ITPR3pr 28.8 ± 2.9 versus 2 kb ITPR3pr+p50/p65 14.6 ± 0.6, p<0.05, n=6) (Figure 2c). Similar inhibition was found after LPS treatment (2 kb ITPR3pr 7.61±0.46 versus 2 kb ITPR3pr+LPS 5.78±0.49, p<0.05). Furthermore, co-transfection with IκBSR, an antagonist of NF-κB activity, rescued the p65/p50-induced inhibition of ITPR3 promoter activity (29.5 ± 4.4, n=6). Next, NHC cells were transfected with nested deletion fragments of 1.0 and 0.5 kb containing the proximal conserved NF-κB elements and alongside with p65/p50 plasmids (Figure 2d). Both nested fragments showed inhibition by p65/p50 (1.0 kb ITPR3pr 70.1 ± 1.3 versus 1.0 kb ITPR3pr+p50/p65 40.5 ± 0.9, p<0.05, n=3; and 0.5 kb ITPR3pr 48.5 ± 8.2 versus 0.5 kb ITPR3pr+p50/p65 7.9 ± 0.2, p<0.01, n=3). To determine the relative importance of each putative NF-κB binding site on ITPR3 promoter activity, NF-κB mutants of each of the 5 sites (Supporting Figure 2) were generated. Promoter activity of each of the 5 mutants was decreased by co-expression of p50/p65 (not shown). These results provide evidence that the proximal 2 kb of the human ITPR3 promoter contains functional NF-κB response elements and that none of the 5 putative binding sites is dominant, although it is alternatively possible that the effect of NF-kB on the ITPR3 promoter is indirect.
FIGURE 2.
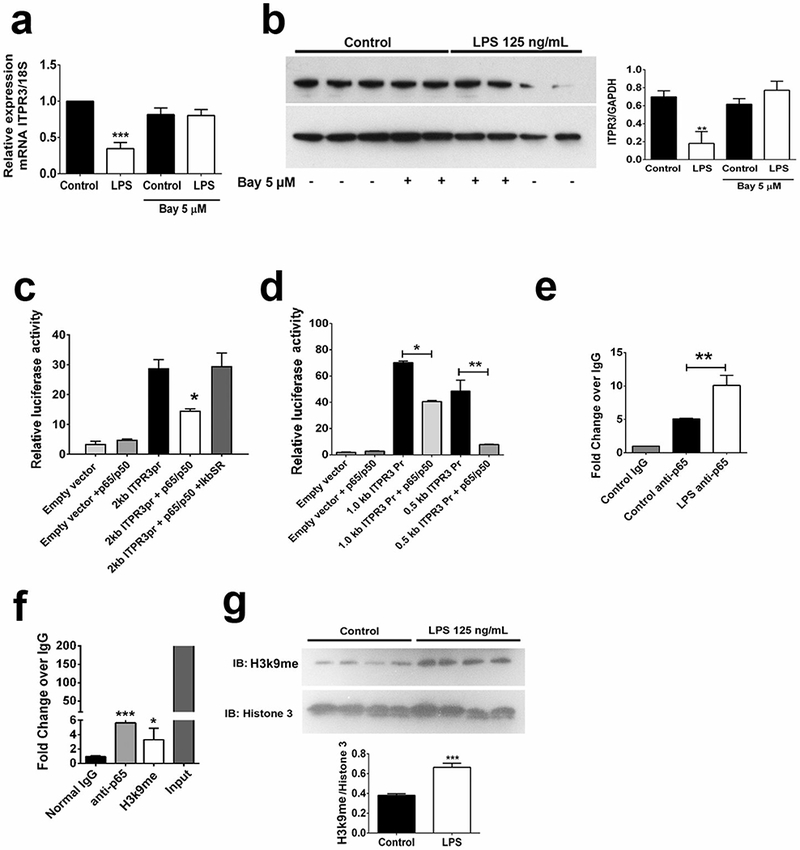
NF-κB inhibits ITPR3 expression in cholangiocytes. (A) Pharmacological inhibition of NF-κB signaling prevented the reduction of ITPR3 mRNA and (B) protein levels induced by LPS treatment. (C) Luciferase activity of the 2kb ITPR3 promoter is reduced by p65/p50 and this effect in prevented by co-expression of IκB super repressor (IkBSR). (D) Promoter activity of 0.5kb and 1.0kb ITPR3 fragments is significantly reduced by co-expression of p50/p65. (F) NHC sheared chromatin was immunoprecipitated to analyze H3K9 methylation of and p65 recruitment to the ITPR3 promoter. (F) Western blot for H3K9 methylation after LPS treatment (125 ng/mL, 18h) in NHC cells. (G) Recruitment of p65 to the ITPR3 upon treatment with LPS (125 ng/mL) for 18 hours. *p<0.05, **p<0.01, and ***p<0.001 using one-way ANOVA test, followed by Bonferroni post-hoc test.
To investigate whether the inhibitory effect of NF-κB on ITPR3 promoter activity is due to direct binding of this transcription factor to the ITPR3 promoter, a ChIP assay was performed employing a p65 antibody. p65 was recruited to the proximal NF-κB elements spanning the region between 270 to 490 bp (5.67 ± 0.28, p<0.001, n=3) and this recruitment was further induced in cells treated with LPS 125 ng/mL for 6 hours (10.1±0.9, p<0.01, n=3), compared to non-treated controls (5.1±0.1, n=3) (Figure 2e). Together these findings provide evidence that NF-κB inhibits ITPR3 expression through direct binding to the ITPR3 promoter.
Because NF-κB binding reduces ITPR3 promoter activity, we evaluated whether this binding is associated with methylation of histone H3 at lysine 9 (H3K9), a well-known repressive epigenetic signature (14). ChIP analysis showed that NHC cells co-transfected with the 2 kb ITPR3 promoter and p65/p50 plasmids displayed increased H3K9 methylation (3.30 ± 1.56, p<0.05, n=3), relative to the normal IgG control group (0.95 ± 0.60, n=3, Figure 2f). Also, LPS treatment increased overall protein levels of methylated H3K9 (0.66±0.02 versus 0.38±0.01, Figure 2g). These results suggest that p65/p50 reduces ITPR3 promoter activity via repressive epigenetic modifications at NF-κB binding sites.
TLR4 decreases ITPR3 expression via activation of NF-κB in cholangiocytes.
To test whether NF- κB is involved in the TLR4-dependent downregulation of ITPR3, NF- κB expression and localization were measured in NHC cells after LPS treatment. NF-κB mRNA (0.84 ± 0.02, n=3) and protein levels (0.85 ± 0.04, n=3) were not altered in LPS-treated cells, as compared to control proteins (0.96 ± 0.02, n=3, Figure 3a and 3b). Next, NF-κB activation was assessed in three ways. First, phosphorylation of p65 was increased in LPS-treated cells relative to controls (0.04 ± 0.004 versus. 0.02 ± 0.003, n=3, p<0.01, Figure 3b). Second, immunofluorescence of p65 in NHC cells showed that p65 translocates to the nucleus after 6 hr of treatment with LPS (101.9±8.75, 15 cells in each group; p<0.01), compared to untreated controls (41.27±78.63, n=3, 15 cells for each group, Figure 3c). Finally, protein (Figure 3d) and mRNA expression levels (Supporting Fig. S4) of NF-κB-regulated interleukins were measured. LPS treatment (125 ng/mL for 18h) increased interleukin 6 (164±15.10 versus 16.28±1.46) and IL-8 (277.3±6.41 versus 23.77±0.22) protein levels relative to controls (p<0.001). Similarly, LPS treatment increase IL-8 mRNA (2.16±0.26, p<0.01, n=4), but did not change IL-1 (0.75±0.10, n=4) or IL-6 levels (1.34±0.02, n=4). Treatment of NHC cells with the TLR4 antagonist TAK242 prevented the reduction in ITPR3 mRNA levels induced by LPS (0.90±0.03, n=3) as well as induction of the NF-κB target gene IL-8 (1.30±0.25, n=3, Supporting Fig. S4). To validate the importance of TLR4 for LPS-dependent reduction of ITPR3 expression, TLR4 was silenced with siRNA in NHC cells (Control+siTLR4 0.25±0.02 versus Control 0.97±0.24, Figure 3f). Upon TLR4 silencing, treatment of NHC cells with LPS no longer decreased ITPR3 mRNA expression (LPS+siTLR4 0.98±0.08 versus LPS 0.40±0.07, Figure 3f). Next, ITPR3 expression in bile ducts was evaluated in WT and TLR4KO mouse livers 24 hr after treatment of the mice with endotoxin. ITPR3 expression was reduced in WT mice treated with LPS relative to saline-treated controls, whereas ITPR3 expression was not decreased by LPS in TLR4KO mice (Figure 3g). Together, these results provide evidence that the reduction of ITPR3 expression induced by LPS is mediated largely if not entirely by the TLR4/NF-κB pathway.
FIGURE 3.
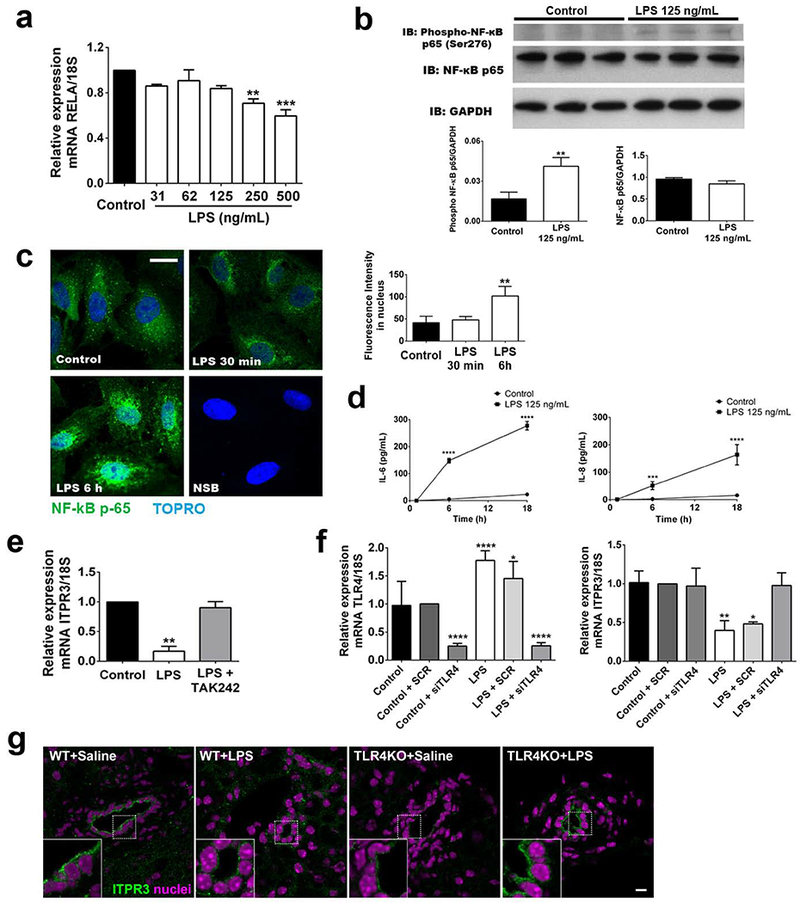
LPS reduces ITPR3 expression in cholangiocytes via TLR4 activation of NF-κB. (A) NF-κB mRNA expression in NHC cells treated multiple concentrations of LPS for 18 hours (B) Western blot and densitometry for phospho-p65 and total p65 in NHC cells incubated with LPS for 18 hours. (C) Immunofluorescence for p65 (in green) and nuclei (in blue) after 30 min and 6 hours of incubation with LPS. Scale bar=20 µm. (D) Concentration of interleukins 6 and 8 after LPS treatment in NHC cells for 6h and 18h. (E) mRNA expression of ITPR3 after LPS treatment for 18 hours in NHC cells with or without TAK242. (F) Left panel, TLR4 expression in NHC cells transfected with scrambled siRNA (SCR) and TLR4 specific siRNA (siTLR4). Right panel, ITPR3 expression after TRL4 silencing and treatment with LPS (125 ng/mL) for 18 hours. (G) Representative images of ITPR3 expression in bile ducts of WT and TLR4KO mice injected with saline or LPS (n=3–6 bile ducts from 3 animals per condition). Scale bar=10 µm. *p<0.05, **p<0.01, and ***p<0.001 using Student’s t test (B) or one-way ANOVA test, followed by Bonferroni post-hoc test (A, C and D).
LPS attenuates Ca2+ signaling in cholangiocytes.
Because ITPR3 is the main intracellular calcium release channel expressed in cholangiocytes (18), we examined whether the downregulation of ITPR3 induced by LPS affects Ca2+ signaling. ATP is an important stimulus for Ca2+-mediated bicarbonate secretion in cholangiocytes (13), and the amplitude of ATP (1µM)-induced calcium signals was similar between untreated and LPS-treated NHC cells (428 ± 33% increase in fluo-4 fluorescence in LPS-treated cells, versus. 509 ± 39% in controls, n=3 preps with 30 cells in each group, Figure 4c). However, the percentage of responding cells was decreased in the LPS-treated group (29±8%, p<0.05) relative to controls (68±7%, n=3, 30 cells in each group). Furthermore, there was an increase in the fraction of responding cells with an oscillatory pattern in the LPS-treated group relative to untreated controls (62±6% vs 33±4%, respectively; n=3 preps with 30 cells in each group; p<0.05); Ca2+ oscillations represent a submaximal signaling response compared to sustained increases in calcium (21, 22). Additionally, the rise time of Ca2+ signals induced by photo-release of caged InsP3, which is inversely correlated with expression and activity of ITPR receptors (23), was significantly faster in NHC cells overexpressing mCherry-ITPR3 that were treated with LPS when compared to non-transfected LPS-treated cells (LPS=9.75±2.02sec, n=8 versus LPS+mCherry ITPR3= 2.5±0.5 sec, n=10; p<0.01. Figure 4f and 4g). Collectively, these results provide functional evidence that LPS-induced downregulation of ITPR3 expression renders cells less responsive to Ca2+ releasing agonists and that this defect can be rescued by re-expression of ITPR3. Because cAMP is a major determinant of bicarbonate secretion by cholangiocytes and LPS may stimulate cAMP in other cells systems (24), cAMP production was compared between control and LPS-treated NHC cells. No difference between these two groups was found either under baseline (unstimulated) conditions or after treatment with Forskolin (Supporting Figure S8). These findings show that LPS-mediated decrease in ITPR3 is not accompanied by a compensatory production of cAMP.
FIGURE 4.
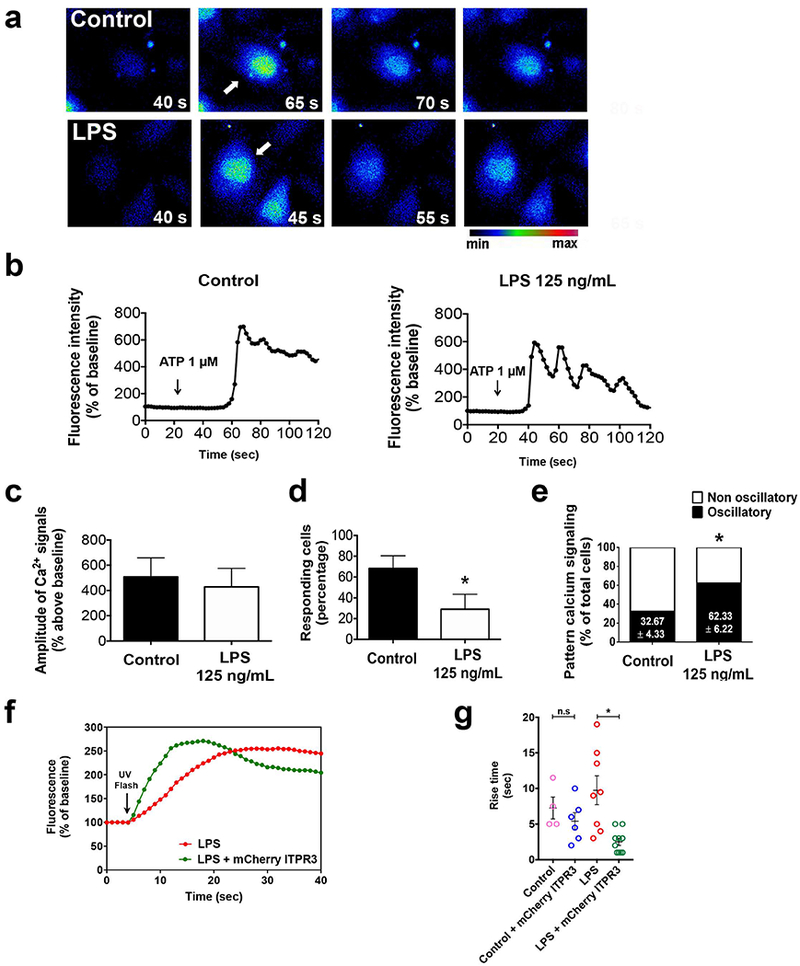
LPS reduces the responsiveness of cholangiocytes to calcium agonists. (A) Representative images and (B) representative time lapse fluorescence changes in NHC cells (indicated by the arrow) loaded with the fluorescent calcium dye Fluo-4/AM (6 µM) and stimulated with ATP (1 µM). (C) Amplitude of calcium signaling is not affected by treatment with LPS. (D) Percentage of cells responding to ATP stimulation is decreased in LPS-treated cells. (E) Percentage of cells with an oscillatory rather than a sustained calcium signaling pattern is increased in the LPS group, *p<0.05 by Student’s t test. (F) Representative Ca2+ signaling changes over time upon photolysis of caged InsP3 in LPS-treated cells with or without overexpression of mCherry-ITPR3. Arrow indicates UV photolysis. (G) Rise time of Ca2+ signals are reduced in LPS-treated cells expressing mCherry-ITPR3 when compared to LPS-treated cells alone.
ITPR3 expression is decreased in bile ducts of patients with clinical conditions associated with endotoxemia.
To investigate the clinical relevance of these findings, immunohistochemistry was performed in liver samples from patients with sepsis or alcoholic hepatitis, two conditions associated with endotoxemia. Liver samples from autopsies of patients with cholestasis-associated sepsis (Supporting Table S1) were examined for p65, ITPR3 and TLR4 labeling (Figures 5). TLR4 expression appeared increased in sepsis samples and labeling of p65 was detected in the nucleus of 27% of cholangiocytes in sepsis specimens (n=226 cells), as compared to 7% of cholangiocytes in histologically normal liver tissues (n=222 cells; p<0.0001 by Chi-squared test), suggesting that NF-κB is frequently activated in cholangiocytes in patients with sepsis (11 ducts in 6 sepsis specimens, 8 ducts in 5 control specimens). Conversely, the apical ITPR3 staining observed in cholangiocytes in normal human liver (12, 18) was reduced in cholangiocytes of patients with sepsis (309±52, n=5, vs. 94±20, n=6, respectively; p<0.01). Moreover, apical ITPR3 labeling inversely correlated with the percentage of NF-κB in the nucleus (R2=0.8693, ***p<0.001).
FIGURE 5.
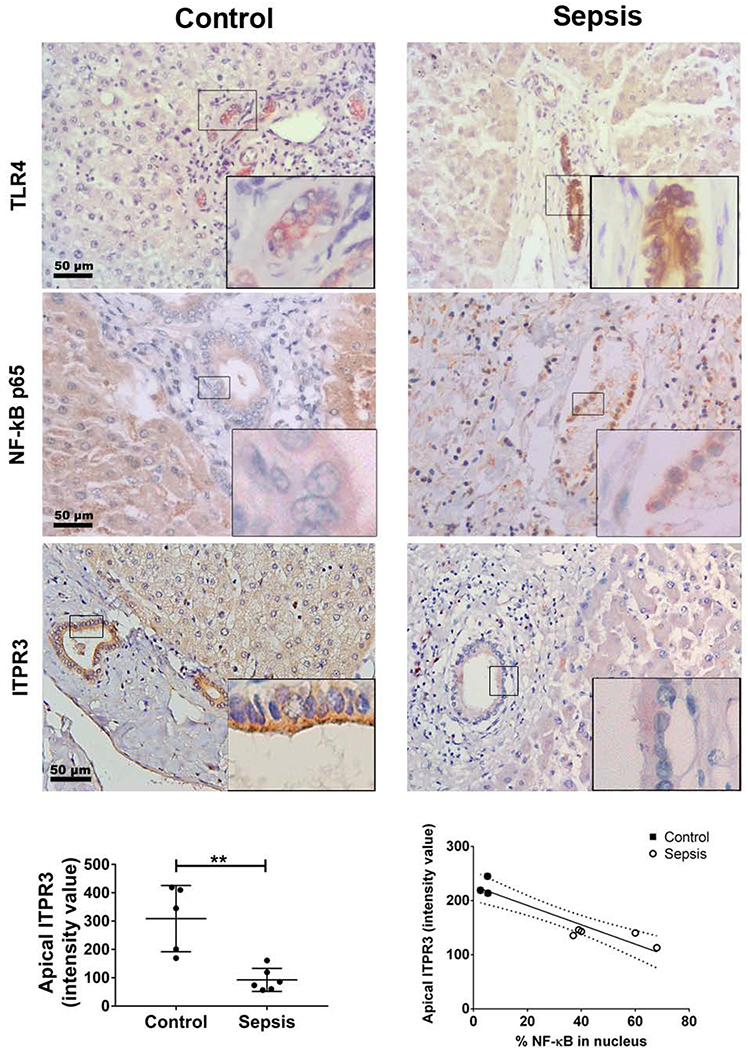
NF-κB is found in the nucleus of cholangiocytes more frequently in patients with sepsis-associated cholestasis. Immunohistochemistry for TLR4, NF-KB p65 and ITPR3 (brown) are shown. Control specimens are on the left and sepsis specimens on the right (20× magnification). Insets show enlarged view of the selected regions. Scale bar 50 µm. TLR4 expression is increased in the sepsis samples (first row). Bile duct cells positive for p65 in the nucleus were counted in blinded fashion (middle row). In sepsis, 27% of cholangiocytes were positive for nuclear p65 (n=226 cholangiocytes) as opposed to 7% in control specimens (n=222 cholangiocytes). Apical ITPR3 expression is reduced in cholangiocytes of patients with sepsis-associated cholestasis (bottom row). Left graph shows average intensity of apical ITPR3 staining in bile ducts from control and sepsis livers (n=6 sepsis patients and 5 controls; **p<0.01 by Student’s t test). Right graph shows the correlation between apical ITPR3 (intensity value) and percentage of p65-postivie nuclei (control=3 and sepsis=5). The linear regression curve (R2=0.8693, ***p<0.001) is indicated by the solid line with 95% CI indicated by the dashed lines.
Liver specimens from alcoholic hepatitis (AH) were evaluated as an additional clinical condition of increased endotoxemia in which cholestasis is a manifestation. Analysis of laboratory data from 31 consecutive AH patients at Yale-New Haven Hospital that underwent liver biopsy to confirm the diagnosis showed that 23 of these individuals had elevated alkaline phosphatase (ALP) levels (Supplementary Table S2), consistent with cholestasis. ITPR3, p65 and TLR4 immunolocalization was performed in the liver samples that were available from these patients. Similar to the findings in sepsis samples, TLR4 expression was increased and NF-кB was translocated to the nucleus in AH liver specimens (p65 positive nuclei in 3.5%, n=301 cells from 4 control patients versus 73.5%, n=156 cells from 2 AH patients). Apical ITPR3 staining in cholangiocytes of 7 AH livers was decreased relative to controls (121±16 versus 186±20, respectively; p<0.05, Figure 6) and was inversely correlated with the percent of p65-positive nuclei (R2=0.6645, *p<0.05). Notably, no increase in nuclear factor, erythroid 2-like 2 (Nrf2) staining was detected in either AH or sepsis cholangiocytes (Supporting Fig. S5 and S6). Similarly, there was no significant change in Nrf2 mRNA in NHC cells after LPS treatment (Supporting Fig. S7). This transcription factor has been linked to decreased ITPR3 expression in other, more classical cholestatic disorders (14). Taken together, these results support the idea that endotoxin downregulates ITPR3 expression in human cholangiocytes. This may contribute to the mechanism by which conditions of endotoxemia result in cholestasis.
FIGURE 6.
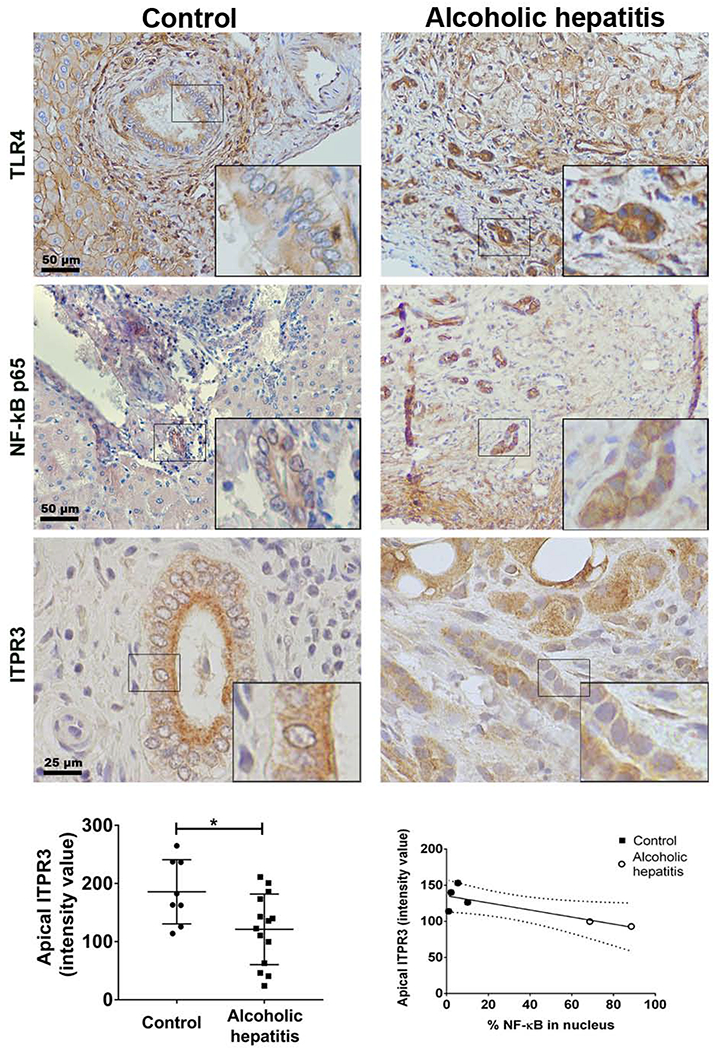
Apical ITPR3 expression is decreased in cholangiocytes of patients with alcoholic hepatitis. Immunohistochemistry for TLR4, NF-кB p65 and ITPR3 are shown (brown). Insets show 40× zoom of representative cholangiocytes. Scale bar, 50 μm. Left graph shows average intensity of apical ITPR3 staining in bile ducts from control and alcoholic hepatitis livers (n=14 AH patients and 8 controls; *p<0.05 by Student’s t test). Scale bar, 50 µm. TLR4 expression is increased in alcoholic hepatitis compared to controls. The percentage of bile duct cells with NF-kB p65 in the nucleus was counted in blinded fashion and correlated with the intensity of the apical ITPR3 signal (control=4 patients and alcoholic hepatitis=2 patients). Right graph shows the linear regression curve (R2=0.6645, ***p<0.05) with 95% CI indicated by the dashed lines.
DISCUSSION
A principal finding of the current study is that LPS binds to TLR4 on cholangiocytes to cause an NF-κB-mediated decrease in ITPR3 expression. This has several clinical implications. First, ITPR3 plays an important role in regulating bicarbonate secretion in cholangiocytes. It is most concentrated in a specialized region of the endoplasmic reticulum (ER) beneath the apical membrane, where it is positioned to release calcium to activate the calcium-dependent chloride channel transmembrane member 16a (TMEM16a), which in turn couples with the chloride-bicarbonate exchanger AE2 to result in net bicarbonate secretion (25). This ITPR3 pool is effectively released by nucleotides in bile, which activate apical P2Y receptors, resulting in local formation of InsP3 (13). Although hepatocytes can secrete ATP into bile (26, 27), activation of the cystic fibrosis transmembrane conductance regulator (CFTR) in cholangiocytes may be an additional route by which this occurs (28). Furthermore, ITPR3 expression is lost in a variety of human cholangiopathies (12), and selective knockdown of ITPR3 inhibits biliary bicarbonate secretion as well (13). Therefore, LPS-induced loss of ITPR3 from cholangiocytes may represent a previously unappreciated mechanism by which sepsis induces cholestasis. Second, in cells that co-express multiple ITPR isoforms, ITPR3 is the isoform that preferentially couples to mitochondria and transmits apoptotic calcium signals into them (29). Selective loss of ITPR3 reduces apoptosis, including in response to apoptotic stimuli such as bile acids (29). Small, proliferating bile ductules are a common histological finding in obstructive cholestasis as well as in certain other cholangiopathies (30), and loss of ITPR3 may permit these ductules to proliferate despite increased exposure to bile acids because of their decreased sensitivity to apoptotic stimuli. Moreover, ITPR3 is concentrated apically in both small and large ducts (14), and we found that the loss of ITPR3 was similar in both types of ducts in severe alcoholic hepatitis (not shown). Third, it has long been appreciated that chronic biliary infections or inflammation increases the risk of developing cholangiocarcinoma (31). The current work is consistent with the idea that such conditions may also lead to chronic loss of ITPR3 in cholangiocytes. However, several investigators have reported that loss of ITPR3 can be a premalignant condition in cancers such as melanoma, mesothelioma, and prostate cancer (32, 33). This effect also has been attributed to impaired mitochondrial calcium signaling and a resulting decrease in apoptosis. Further work will be needed to determine whether loss of ITPR3 from cholangiocytes due to LPS or other inflammatory conditions contributes not only to cholestasis, but also to ductular proliferation and ultimately to the development of cholangiocarcinoma.
The current study also provides evidence that ITPR3 is lost in cholangiocytes of patients with several conditions of endotoxemia; sepsis and severe alcoholic hepatitis. The most immediate clinical effect of this would be cholestasis. Cholestasis has long been recognized as a complication of sepsis (1), but most previous studies have focused on the effects of LPS on secretion by hepatocytes rather than cholangiocytes (8, 9). Several studies have carefully delineated the loss of transporters such as Na+-taurocholate cotransporting polypeptide (NTCP) (10, 34) and multidrug resistance-associated protein 2 (MRP2) (8) that occurs in response to LPS. There is also evidence that LPS reduces expression of ITPR2 in hepatocytes (35), which plays an analogous role to ITPR3 in cholangiocytes. ITPR2 is the principal intracellular calcium release channel in hepatocytes and it is most concentrated in a specialized region of the ER beneath the canalicular membrane (36, 37). It participates in targeting and insertion of MRP2 and bile salt export pump (BSEP) into the canalicular membrane and its loss may participate in the pathogenesis of cholestasis (23, 35). LPS also causes transient increases in NF-κB and activator protein 1 (AP-1) in hepatocytes (38). Although there is little to no ITPR3 in normal hepatocytes (36) and it is not known whether NF-κB inhibits ITPR2 expression, there is recent evidence that AP-1 inhibits expression of ITPR2 (39). It had been reported that LPS decreases ITPR3 in cholangiocytes in a rodent model (12), although the mechanism for this and the relevance in patients with cholestasis had not been examined. It also has been shown that inflammatory cytokines inhibit ductular secretion (40). It is not known whether and to what extent this is also due to decreased expression of ITPR3, and if so, whether cytokines mediate this effect through NF-κB, or else by the oxidant stress sensor Nrf2 or miR-506, each of which have been shown to inhibit ITPR3 expression in other types of cholangiopathies (14, 15).
It is now widely accepted that endotoxin plays a major role in the development of alcoholic hepatitis. Its role is attributed mostly to immune activation and consequent damage to hepatocytes (41). There is an evolving appreciation that cholestasis can be an important component of alcoholic hepatitis as well. For example, a retrospective study of over 4,000 patients hospitalized for liver disease in Shanghai found that 16% of alcoholic hepatitis patients had cholestasis (42). In another study, elevated alkaline phosphatase was found to be an independent predictor of increased mortality in patients with alcoholic hepatitis (5). In a study that reported a histological scoring system to determine severity of alcoholic hepatitis, the presence of cholestasis was found to be an independent predictor of increased mortality (6). In another study, the micro-RNA profile of alcoholic hepatitis patients was analyzed and found to reflect changes seen in cholestatic liver diseases (43). These findings collectively suggest that cholestasis is a frequent complication of alcoholic hepatitis and may even portend a worse outcome. As is the case in the cholestasis of sepsis, it is possible and perhaps likely that at least some of the cholestasis that occurs in alcoholic hepatitis is due to impaired secretion by hepatocytes. However, by demonstrating a loss of ITPR3 in cholangiocytes from these patients, our findings provide evidence that bile duct abnormalities may contribute to this complication. The transcription factor Nrf2 appears to be responsible for decreased cholangiocyte ITPR3 expression in a number of cholangiopathies (14), so it is notable that Nrf2 expression was not increased in bile ducts of patients with alcoholic hepatitis. This suggests that multiple, distinct signaling pathways may converge to decrease ITPR3 expression in cholangiocytes. Future work will be needed to understand the range of mechanisms by which ITPR3 expression is regulated in cholangiocytes, and to fully define the downstream effects that result from this.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the support of the UFMG Liver Center. We also thank Emma Kruglov and Romina Fiorotto (Section of Digestive Diseases, Yale School of Medicine) for technical assistance and David Yule (University of Rochester Medical Center) for providing the rat mCherry-ITPR3 plasmid.
Financial Support: National Institutes of Health (P01-DK57751, P30-DK34989, R56-DK99470, R01-DK45710, R01-DK114041, R01-DK112797, S10-OD023598, and T32-DK07356), CNPq, INCT, FAPEMIG and CAPES.
List of Abbreviation:
- LPS
lipopolysaccharide
- ITPR3
type 3 inositol trisphosphate receptor
- Ca2+
ion calcium
- TLR4
toll like receptor 4
- NF-κB
nuclear factor-κB
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- NHC
normal human cholangiocyte
- H69
non-tumor SV-40 immortalized human cholangiocytes
- IκBSR
IκB-α super suppressor
- ChIP
chromatin immunoprecipitation
- DAB
3,3’-diaminobenzidine
- IL
interleukins
- ALP
alkaline phosphatase
- WBC
white blood cells
- Nrf2
nuclear factor, erythroid 2-like 2
- TMEM16a
transmembrane member 16A
- CFTR
cystic fibrosis transmembrane conductance regulator
- NTCP
Na+-taurocholate cotransporting polypeptide
- MRP2
multidrug resistance-associated protein 2
- BSEP
bile salt export pump
- AP-1
activator protein 1
- miR-506
micro-RNA 506
Footnotes
Denotes equal contributions
Denotes equal contributions
REFERENCES
- 1.Chand N, Sanyal AJ. Sepsis-induced cholestasis. Hepatology 2007;45:230–241. [DOI] [PubMed] [Google Scholar]
- 2.Nesseler N, Launey Y, Aninat C, Morel F, Malledant Y, Seguin P. Clinical review: The liver in sepsis. Crit Care 2012;16:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan HB, Yang DL, Chen AS, Li Z, Xu LT, Ma XJ, Zhou H, et al. Sepsis-associated cholestasis in adult patients: a prospective study. Am J Med Sci 2013;346:462–466. [DOI] [PubMed] [Google Scholar]
- 4.Szabo G Gut-liver axis in alcoholic liver disease. Gastroenterology 2015;148:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasztelan-Szczerbinska B, Slomka M, Celinski K, Szczerbinski M. Alkaline phosphatase: the next independent predictor of the poor 90-day outcome in alcoholic hepatitis. Biomed Res Int 2013;2013:614081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, Augustin S, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014;146:1231–1239 e1231–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura-Cots M, Coulter S, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trauner M, Arrese M, Soroka CJ, Ananthanarayanan M, Koeppel TA, Schlosser SF, Suchy FJ, et al. The rat canalicular conjugate export pump (mrp2) is down-regulated in intrahepatic and obstructive cholestasis. Gastroenterology 1997;113:255–264. [DOI] [PubMed] [Google Scholar]
- 9.Bolder U, Ton-Nu HT, Schteingart CD, Frick E, Hofmann AF. Hepatocyte transport of bile acids and organic anions in endotoxemic rats: impaired uptake and secretion. Gastroenterology 1997;112:214–225. [DOI] [PubMed] [Google Scholar]
- 10.Trauner M, Arrese M, Lee H, Boyer JL, Karpen SJ. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J Clin Invest 1998;101:2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata K, Nathanson MH. Bile duct epithelia regulate biliary bicarbonate excretion in normal rat liver. Gastroenterology 2001;121:396–406. [DOI] [PubMed] [Google Scholar]
- 12.Shibao K, Hirata K, Robert ME, Nathanson MH. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 2003;125:1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minagawa N, Nagata J, Shibao K, Masyuk AI, Gomes DA, Rodrigues MA, Lesage G, et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes through release of ATP into bile. Gastroenterology 2007;133:1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weerachayaphorn J, Amaya MJ, Spirli C, Chansela P, Mitchell-Richards KA, Ananthanarayanan M, Nathanson MH. Nuclear Factor, Erythroid 2-Like 2 Regulates Expression of Type 3 Inositol 1,4,5-Trisphosphate Receptor and Calcium Signaling in Cholangiocytes. Gastroenterology 2015;149:211–222 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ananthanarayanan M, Banales JM, Guerra MT, Spirli C, Munoz-Garrido P, Mitchell-Richards K, Tafur D, et al. Post-translational Regulation of the Type III Inositol 1,4,5-Trisphosphate Receptor by miRNA-506. J Biol Chem 2015;290:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlosser SF, Burgstahler AD, Nathanson MH. Isolated rat hepatocytes can signal to other hepatocytes and bile duct cells by release of nucleotides. Proc.Natl.Acad.Sci.USA 1996;93:9948–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, et al. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A 2003;100:2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirata K, Dufour JF, Shibao K, Knickelbein R, O’Neill AF, Bode HP, Cassio D, et al. Regulation of Ca(2+) signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology 2002;36:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology 2006;44:287–298. [DOI] [PubMed] [Google Scholar]
- 20.Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, Strazzabosco M. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology 2011;141:1498–1508, 1508 e1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rooney TA, Sass EJ, Thomas AP. Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single Fura-2 loaded hepatocytes. J.Biol.Chem. 1989;264:17131–17141. [PubMed] [Google Scholar]
- 22.Nathanson MH, Burgstahler AD, Mennone A, Boyer JL. Characterization of cytosolic Ca2+ signaling in rat bile duct epithelia. Am.J.Physiol.Gastrointest.Liver Physiol. 1996;271:G86–G96. [DOI] [PubMed] [Google Scholar]
- 23.Cruz LN, Guerra MT, Kruglov E, Mennone A, Garcia CR, Chen J, Nathanson MH. Regulation of multidrug resistance-associated protein 2 by calcium signaling in mouse liver. Hepatology 2010;52:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin SL, Lan L, Zoudilova M, Conti M. Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol 2005;175:1523–1531. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Dutta A, Kresge C, Bugde A, Feranchak AP. Bile acids stimulate cholangiocyte fluid secretion by activation of membraneTMEM16A Cl(−) channels. Hepatology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlosser SF, Burgstahler AD, Nathanson MH. Isolated rat hepatocytes can signal to nearby hepatocytes and bile duct cells by secretion of nucleotides. Gastroenterology 1996;110:A1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nathanson MH, Burgstahler AD, Masyuk A, Larusso NF. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J 2001;358:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiorotto R, Spirli C, Fabris L, Cadamuro M, Okolicsanyi L, Strazzabosco M. Ursodeoxycholic acid stimulates cholangiocyte fluid secretion in mice via CFTR-dependent ATP secretion. Gastroenterology 2007;133:1603–1613. [DOI] [PubMed] [Google Scholar]
- 29.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005;280:40892–40900. [DOI] [PubMed] [Google Scholar]
- 30.Nakanuma Y, Ohta G. Immunohistochemical study on bile ductular proliferation in various hepatobiliary diseases. Liver 1986;6:205–211. [DOI] [PubMed] [Google Scholar]
- 31.Braconi C, Patel T. Cholangiocarcinoma: new insights into disease pathogenesis and biology. Infect Dis Clin North Am 2010;24:871–884, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, Pellegrini L, et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017;546:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017;546:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green RM, Beier D, Gollan JL. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology 1996;111:193–198. [DOI] [PubMed] [Google Scholar]
- 35.Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5-trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology 2011;54:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirata K, Pusl T, O’Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002;122:1088–1100. [DOI] [PubMed] [Google Scholar]
- 37.Nagata J, Guerra MT, Shugrue CA, Gomes DA, Nagata N, Nathanson MH. Lipid rafts establish calcium waves in hepatocytes. Gastroenterology 2007;133:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48:322–335. [DOI] [PubMed] [Google Scholar]
- 39.Khamphaya T, Chukijrungroat N, Saengsirisuwan V, Mitchell-Richards KA, Robert ME, Mennone A, Ananthanarayanan M, et al. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spirli C, Nathanson MH, Fiorotto R, Duner E, Denson LA, Sanz JM, Di Virgilio F, et al. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 2001;121:156–169. [DOI] [PubMed] [Google Scholar]
- 41.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol 2010;16:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao X, Gao Y, Zhang W, Xu P, Fu Q, Chen C, Li C, et al. [Cholestasis morbidity rate in first-hospitalized patients with chronic liver disease in Shanghai]. Zhonghua Gan Zang Bing Za Zhi 2015;23:569–573. [DOI] [PubMed] [Google Scholar]
- 43.Blaya D, Coll M, Rodrigo-Torres D, Vila-Casadesus M, Altamirano J, Llopis M, Graupera I, et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut 2016;65:1535–1545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.