

Summary
Bacteria coordinate DNA replication and cell division, ensuring a complete set of genetic material is passed onto the next generation. When bacteria encounter DNA damage, a cell cycle checkpoint is activated by expressing a cell division inhibitor. The prevailing model is that activation of the DNA damage response and protease mediated degradation of the inhibitor is sufficient to regulate the checkpoint process. Our recent genome-wide screens identified the gene ddcA as critical for surviving exposure to DNA damage. Similar to the checkpoint recovery proteases, the DNA damage sensitivity resulting from ddcA deletion depends on the checkpoint enforcement protein YneA. Using several genetic approaches, we show that DdcA function is distinct from the checkpoint recovery process. Deletion of ddcA resulted in sensitivity to yneA overexpression independent of YneA protein levels and stability, further supporting the conclusion that DdcA regulates YneA independent of proteolysis. Using a functional GFP-YneA fusion we found that DdcA prevents YneA-dependent cell elongation independent of YneA localization. Together, our results suggest that DdcA acts by helping to set a threshold of YneA required to establish the cell cycle checkpoint, uncovering a new regulatory step controlling activation of the DNA damage checkpoint in Bacillus subtilis.
Keywords: Bacillus subtilis, DNA damage response, DNA damage checkpoint, YneA, SOS response
Abbreviated Summary
Bacterial cells respond to DNA damage by expressing a protein that prevents cell division. Here, we identify a previously uncharacterized regulator, DdcA that antagonizes the checkpoint enforcement protein YneA helping to set the threshold for checkpoint activation.
Introduction
The logistics of the cell cycle are of fundamental importance in biology. All organisms need to control cell growth, DNA replication, and the process of cell division. In bacteria the initiation of DNA replication is coupled to growth rate and the cell cycle (Donachie & Blakely, 2003; Hill, Kadoya, Chattoraj, & Levin, 2012; Wang & Levin, 2009; Westfall & Levin, 2017). Bacteria also regulate cell division in response to DNA replication status through the use of DNA damage checkpoints (Kreuzer, 2013; Lenhart, Schroeder, Walsh, & Simmons, 2012). The models for the DNA damage response (SOS) were developed based on studies of Escherichia coli and subsequently extended to other bacteria. In these models, DNA damage results in perturbations to DNA replication and the accumulation of ssDNA (Friedberg et al., 2006). RecA is loaded onto ssDNA (Anderson & Kowalczykowski, 1997; Churchill, Anderson, & Kowalczykowski, 1999; Ivancic-Bace et al., 2003; Ivancic-Bace, Vlasic, Salaj-Smic, & Brcic-Kostic, 2006; Morimatsu & Kowalczykowski, 2003), and the resulting RecA/ssDNA nucleoprotein filament induces the SOS response by activating auto-cleavage of the transcriptional repressor LexA (Slilaty & Little, 1987). LexA inactivation results in increased transcription of genes involved in DNA repair and the DNA damage checkpoint (Au et al., 2005; Goranov, Kuester-Schoeck, Wang, & Grossman, 2006; Lewis, Harlow, Gregg-Jolly, & Mount, 1994; Little & Mount, 1982; Little, Mount, & Yanisch-Perron, 1981). The DNA damage checkpoint is established by relieving the LexA-dependent repression of a cell division inhibitor that enforces the checkpoint by blocking cell division (Huisman & D’Ari, 1981; Huisman, D’Ari, & Gottesman, 1984; Kawai, Moriya, & Ogasawara, 2003; Mo & Burkholder, 2010). Once the checkpoint is established, the delay in cytokinesis provides the cell with enough time to repair and complete DNA replication, thereby ensuring a complete and accurate copy of the chromosome is segregated to both daughter cells. Over several decades of study, this overarching model has been consistently demonstrated among bacteria that contain a RecA and LexA-dependent DNA damage checkpoint mechanism (Erill, Campoy, & Barbe, 2007; Kreuzer, 2013).
Where the DNA damage response varies between bacteria is in the process that enforces and alleviates the checkpoint. In E. coli and closely related Gram-negative bacteria, the checkpoint is enforced by SulA, which is a cytoplasmic protein that acts by directly inhibiting formation of the FtsZ proto-filament blocking cell division (Bi & Lutkenhaus, 1993; Huang, Cao, & Lutkenhaus, 1996; Huisman et al., 1984; Mukherjee, Cao, & Lutkenhaus, 1998; Trusca, Scott, Thompson, & Bramhill, 1998). In many other bacteria the checkpoint is enforced by a small membrane binding protein (Chauhan et al., 2006; Kawai et al., 2003; Modell, Hopkins, & Laub, 2011; Modell, Kambara, Perchuk, & Laub, 2014; Ogino, Teramoto, Inui, & Yukawa, 2008). In Caulobacter crescentus, the small membrane proteins SidA and DidA inhibit cell division through direct interactions with components of the essential cell division complex known as the divisome (Modell et al., 2011; Modell et al., 2014). In other bacteria the exact mechanism of checkpoint enforcement remains unclear. In the Gram-positive bacterium Bacillus subtilis, the checkpoint enforcement protein YneA inhibits cell division in response to DNA damage (Kawai et al., 2003). YneA is a small protein containing a transmembrane domain as well as a LysM domain (Mo & Burkholder, 2010). A previous study found that several amino acids on one side of the transmembrane alpha helix are important for function, which led the authors to speculate that YneA may also interact with a component of the divisome (Mo & Burkholder, 2010). The same study also suggested full length YneA is the active form, and that the transmembrane domain alone is not sufficient for activity (Mo & Burkholder, 2010). Although YneA is clearly involved in cell division inhibition, the role of this checkpoint in ensuring that daughter cells each receive an intact copy of the genome has not yet been firmly established, and the mechanism by which YneA enforces the checkpoint is still unknown.
The mechanism of relieving the DNA damage checkpoint has only been identified in two bacterial species, E. coli and B. subtilis. Despite the checkpoint mechanisms functioning in different cellular compartments, the strategy for checkpoint recovery is remarkably similar between these two organisms. In E. coli, Lon protease is the major protease responsible for degrading SulA (Canceill, Dervyn, & Huisman, 1990; Mizusawa & Gottesman, 1983; Sonezaki et al., 1995), and the protease ClpYQ appears to play a secondary role (Kanemori, Yanagi, & Yura, 1999; Seong, Oh, Yoo, Seol, & Chung, 1999; W. F. Wu, Zhou, & Gottesman, 1999). In B. subtilis, there are two proteases YlbL, which we rename here to DdcP (DNA damage checkpoint recovery protease) and CtpA that degrade YneA (Burby, Simmons, Schroeder, & Simmons, 2018). In the case of DdcP and CtpA, the former seems to be the primary protease in minimal media, however during chronic exposure to DNA damage in rich media both proteases are important and they can functionally replace each other when overexpressed (Burby et al., 2018). DdcP and CtpA are not regulated by DNA damage (Burby et al., 2018), suggesting that the proteases act as a buffer to YneA accumulation helping to set the threshold for checkpoint activation. Thus, in order for the checkpoint to be enforced both proteases must be saturated. Following repair of damaged DNA, LexA represses expression of YneA and the remaining YneA is cleared by DdcP and CtpA allowing cell division to proceed (Burby et al., 2018).
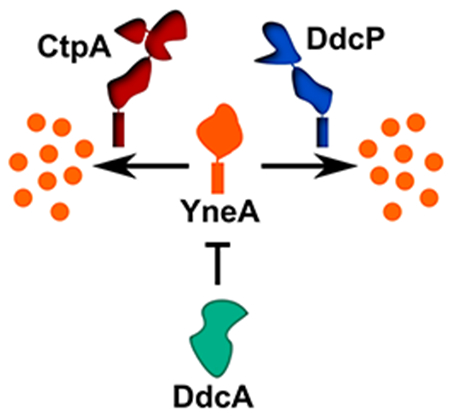
Although the DNA damage checkpoint in bacteria is well understood, it is becoming increasingly clear that establishing the checkpoint is more complex than what earlier models suggest. Work from Goranov and co-workers demonstrated that the initiation protein and transcription factor DnaA regulates ftsL levels in response to DNA replication perturbations, which contributes to cell filamentation (Goranov, Katz, Breier, Burge, & Grossman, 2005). Further, our recent report identified several genes not previously implicated in genome maintenance or cell cycle control that are critical for surviving chronic exposure to a broad spectrum of DNA damage (Burby et al., 2018). We identified genes involved in cell division and cell wall synthesis as well as genes of unknown function that rendered the deletion mutants sensitive to DNA damage (Burby et al., 2018). To understand how the DNA damage response in bacteria is regulated, we investigated the contribution of one of the unstudied genes ddcA (formerly ysoA, see below) in the DNA damage response. We report here, that DdcA antagonizes YneA action functioning to help set a threshold of DNA damage required for checkpoint activation.
Results
Deletion of ddcA (ysoA) results in sensitivity to DNA damage
We recently published a set of genome wide screens using three distinct classes of DNA damaging agents, uncovering many genes that have not been previously implicated in the DNA damage response or DNA repair (Burby et al., 2018). One gene that conferred a sensitive phenotype to all three types of DNA damage tested was ysoA, which we rename here to DNA damage checkpoint antagonist (ddcA). DdcA is a protein that is predicted to have three tetratrichopeptide repeats (Fig 1A), which are often involved in protein-protein interactions, protein complex formation, and virulence mechanisms in bacteria (Cerveny et al., 2013). In order to better understand the mechanism of the DNA damage response in B. subtilis, we investigated the contribution of DdcA. To begin, we tested the sensitivity of the ddcA deletion to DNA damage. Deletion of ddcA resulted in sensitivity to mitomycin C (MMC) an agent that causes DNA crosslinks and bulky adducts; (Iyer & Szybalski, 1963; Noll, Mason, & Miller, 2006) and phleomycin a peptide that forms double and single strand DNA breaks (Kross, Henner, Hecht, & Haseltine, 1982; Reiter, Milewskiy, & Kelley, 1972). We found that expression of Pxyl-ddcA from an ectopic locus (amyE) was sufficient to complement deletion of ddcA with or without inducing expression using xylose (Fig 1B). We conclude that deletion of ddcA results in a bonafide sensitivity to DNA damage.
Figure 1. Deletion of ddcA (ysoA) results in sensitivity to DNA damage.
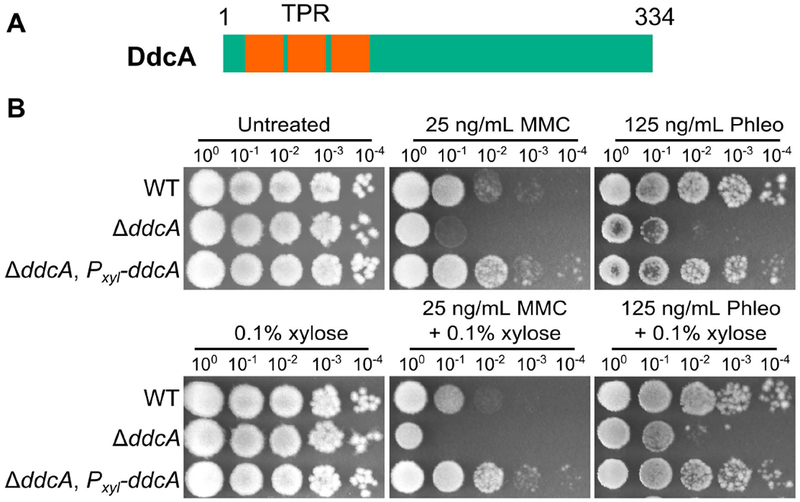
(A) A schematic of the DdcA protein. DdcA is predicted to have 334 amino acids and 3 tetratrichopeptide repeats at its N-terminus. (B) A spot titer assay in which exponentially growing cultures of B. subtilis strains WT (PY79), ΔddcA (PEB357), and ΔddcA, amyE∷Pxyl-ddcA (PEB503) were spotted on the indicated media and incubated at 30°C overnight.
DNA damage sensitivity of ddcA deletion is dependent on yneA.
We asked how DdcA functions in the DNA damage response. Our observation that a ddcA deletion allele results in sensitivity to several DNA damaging agents is similar to the result of deleting the checkpoint recovery proteases (Burby et al., 2018). Our prior study (Burby et al., 2018) showed that DNA damage phenotypes in checkpoint recovery protease mutants depend on the checkpoint enforcement protein, YneA, which is likely the result of aberrant activation of the checkpoint in the absence of YneA degradation. We asked whether deletion of yneA could rescue DNA damage sensitivity resulting from ddcA deletion. Indeed, deletion of yneA in the ddcA deletion background rescued sensitivity to MMC (Fig S1).
We also tested for a genetic interaction with nucleotide excision repair, reasoning that the absence of nucleotide excision repair would result in increased yneA expression and increased sensitivity in the ddcA deletion. Indeed, deletion of uvrAB, genes coding for components of nucleotide excision repair (Sancar, 1996), resulted in hypersensitivity to MMC (Fig S1). These data, together with the initial observation of general DNA damage sensitivity, and suppression of the sensitivity with loss of yneA function suggests that DdcA participates in regulating the DNA damage checkpoint protein YneA.
DdcA functions independent of DNA damage checkpoint recovery proteases
Based on the observation that sensitivity to DNA damage in a ΔddcA mutant was rescued by deletion of yneA, similar to our observations with the checkpoint recovery proteases (Burby et al., 2018), we hypothesized that DdcA could function within the checkpoint recovery process. For example, DdcA could inhibit CtpA and/or DdcP activity. To test this idea, we generated double mutant strains of ΔddcA with ΔctpA or ΔddcP. If DdcA functions together with CtpA or DdcP we would expect that the double mutant would have the same phenotype as the single mutant. In contrast, we observed that deletion of ddcA in a ctpA or ddcP mutant resulted in increased sensitivity to MMC (Fig 2A). These results support the hypothesis that DdcA does not function with the proteases in checkpoint recovery. To test this idea further, we detemined the effect of deletion of ddcA in a ΔddcP, ΔctpA double mutant on MMC sensitivity. We found that deletion of ddcA resulted in increased MMC sensitivity relative to the double protease mutants (Fig 2B), suggesting that DdcA functions independently of both DdcP and CtpA. We then asked if yneA was responsible for the phenotype of ΔddcA in the absence of the checkpoint recovery proteases. Strikingly, we found that the sensitivity of the triple mutant was mostly dependent on yneA, but at elevated concentrations of MMC, there was a slight but reproducible difference when ddcA was deleted in the ΔddcP, ΔctpA, ΔyneA∷loxP mutant background (Fig 2B). Taken together, these data suggest that DdcA regulation of the checkpoint is independent of the recovery proteases. Further, because the ddcA phenotype is dependent on yneA we suggest that DdcA negatively regulates the checkpoint enforcement protein YneA.
Figure 2. DdcA functions independent of the checkpoint recovery proteases.
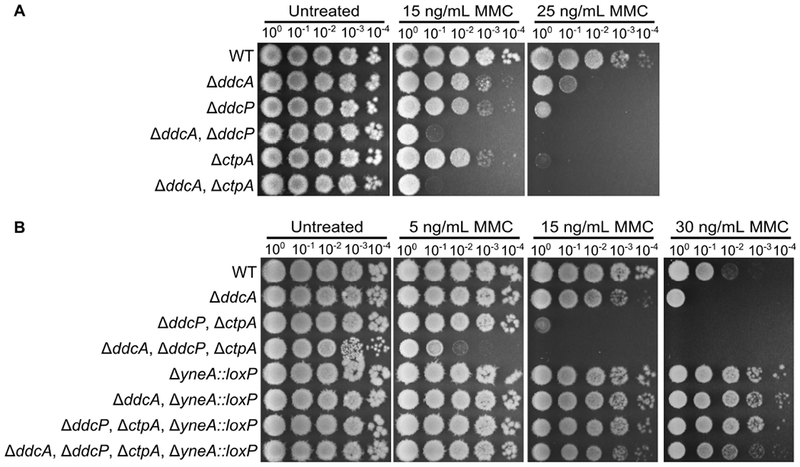
(A) Spot titer assay using B. subtilis strains WT (PY79), ΔddcA (PEB357), ΔddcP (PEB324), ΔddcA ΔddcP (PEB499), ΔctpA (PEB355), and ΔddcA ΔctpA (PEB579) spotted on the indicated media. (B) Spot titer assay using B. subtilis strains WT (PY79), ΔddcA (PEB357), ΔddcP ΔctpA (PEB555), ΔddcA ΔddcP ΔctpA (PEB639), ΔyneA∷loxP (PEB439), ΔddcA ΔyneA∷loxP (PEB587), ΔddcP ΔctpA ΔyneA∷loxP (PEB561), and ΔddcA ΔddcP ΔctpA ΔyneA∷loxP (PEB643) spotted on the indicated media.
In our previous study we found that the checkpoint recovery proteases could substitute for each other (Burby et al., 2018). Therefore to more firmly establish when DdcA regulates the checkpoint we asked if DdcA could replace the checkpoint recovery proteases or if the proteases could function in place of DdcA. To test this idea, we overexpressed ddcP and ctpA in a ΔddcA mutant and found that neither protease could rescue a ddcA deletion phenotype (Fig 3A). We also found that expression of ddcA in the double protease mutant could not rescue the MMC sensitive phenotype (Fig 3B). Further, expression of ddcP or ctpA were each able to partially complement the phenotype of the triple mutant, but expression of ddcA had no effect at higher concentrations of MMC (Fig 3B). As a control, we verified that overexpression of ddcA using high levels of xylose (0.5% xylose) could complement a ΔddcA mutant (Fig S2). We also found that at lower concentrations of MMC, expression of ddcA could rescue the ddcA deficiency of the triple mutant resulting in a phenotype indistinguishable from the double protease mutant (Fig 3C). Given that DdcA cannot substitute for DdcP and CtpA, we hypothesized that DdcA would not affect YneA protein levels following DNA damage. We tested this by monitoring YneA protein levels following MMC treatment and after recovering from MMC treatment for two hours. Deletion of ddcA alone did not result in a detectable difference in YneA protein levels compared to WT (Fig S3). Further, deletion of ddcA in the double protease mutant also did not result in an increase in YneA protein levels relative to the double protease mutant with ddcA intact (Fig S3). With these data we conclude that DdcA does not regulate YneA protein abundance.
Figure 3. DdcA cannot complement loss of checkpoint recovery proteases.
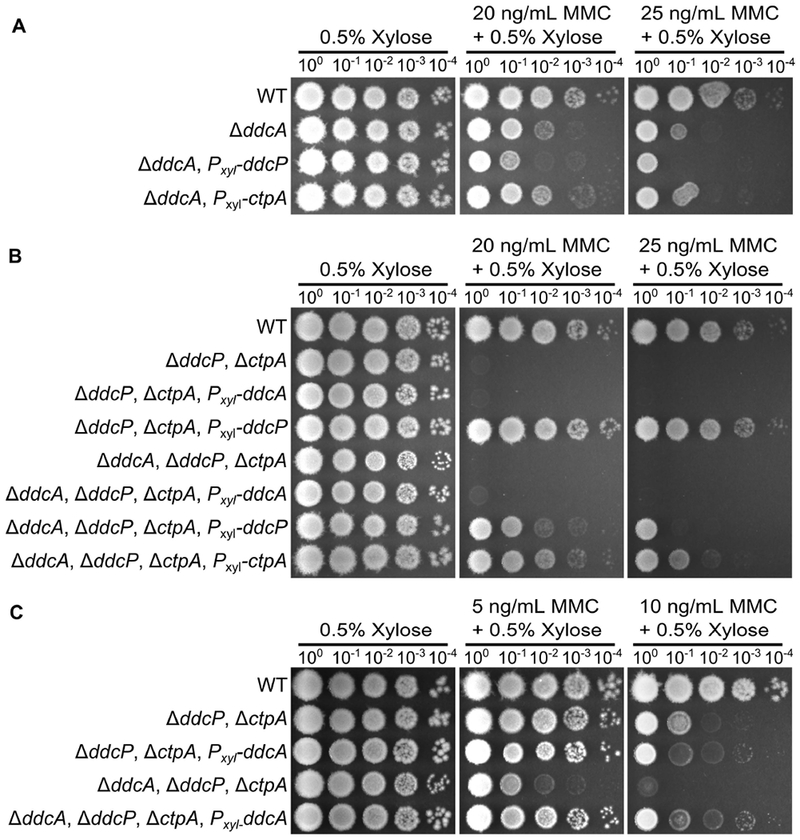
(A) Spot titer assay using B. subtilis strains WT (PY79), ΔddcA (PEB357), ΔddcA amyE∷Pxyl-ddcP (PEB836), and ΔddcA amyE∷Pxyl-ctpA (PEB837) spotted on the indicated media. (B) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔctpA (PEB555), ΔddcP, ΔctpA, amyE∷Pxyl-ddcA (PEB838), ΔddcP, ΔctpA, amyE∷Pxyl-ddcP (PEB557), ΔddcA ΔddcP ΔctpA (PEB639), ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ddcA (PEB840), ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ddcP (PEB839), and ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ctpA (PEB841) spotted on the indicated media. (C) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔctpA (PEB555), ΔddcP, ΔctpA, amyE∷Pxyl-ddcA (PEB838), ΔddcA ΔddcP ΔctpA (PEB639), ΔddcP, ΔctpA, and ΔddcA, amyE∷Pxyl-ddcA (PEB840) spotted on the indicated media.
ddcA deletion results in sensitivity to yneA overexpression independent of YneA stability
Prior work established that overexpression of yneA resulted in growth inhibition (Kawai et al., 2003; Mo & Burkholder, 2010). Previously, we demonstrated that the double checkpoint recovery protease mutant was considerably more sensitive than the WT strain or the single checkpoint protease mutants to yneA overexpression (Burby et al., 2018). Given that treatment with DNA damage has cellular consequences in addition to expression of yneA, we wanted to test whether overexpression of yneA was sufficient for enhanced growth inhibition in the absence of ddcA. Indeed, we found that the ΔddcA mutant was more sensitive to yneA overexpression than WT (Fig 4A), and that deletion of ddcA in the double protease mutant background resulted in even greater sensitivity to yneA overexpression than the double mutant or each single mutant (Fig 4A, (Burby et al., 2018)). Therefore, we asked whether YneA protein levels changed under these conditions, and again there was no detectable difference when ddcA was deleted alone or in combination with the double protease mutant (Fig 4B). We also considered the possibility that DdcA could affect the stability of YneA rather than the overall amount. To test this idea, we performed a translation shut-off experiment and monitored YneA stability over time. We induced expression of yneA in the double protease mutant with and without ddcA and blocked translation. We found that YneA protein abundance decreased at a similar rate regardless of whether ddcA was present (Fig 4C). We conclude that DdcA negatively regulates YneA independent of protein stability.
Figure 4. Deletion of ddcA results in sensitivity to yneA overexpression independent of YneA stability.
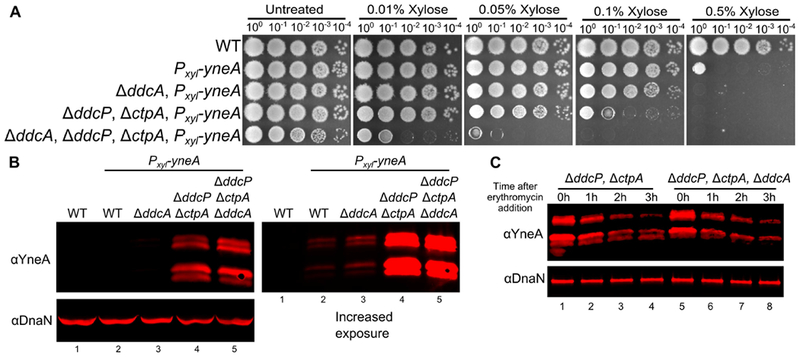
(A) Spot titer testing the effect of yneA overexpression. B. subtilis strains WT (PY79), amyE∷Pxyl-yneA (PEB846), ΔddcA amyE∷Pxyl-yneA (PEB848), ΔddcP, ΔctpA, amyE∷Pxyl-yneA (PEB850), and ΔddcA ΔddcP ΔctpA, amyE∷Pxyl-yneA (PEB852) were spotted on LB agar media containing increasing concentrations of xylose to induce yneA expression. (B) A Western blot using antisera against YneA (Upper panels), or DnaN lower panel using B. subtilis strains WT (PY79), amyE∷Pxyl-yneA (PEB846), ΔddcA amyE∷Pxyl-yneA (PEB848), ΔddcP, ΔctpA, amyE∷Pxyl-yneA (PEB850), and ΔddcA ΔddcP ΔctpA, amyE∷Pxyl-yneA (PEB852) after growing in the presence of 0.1% xylose for two hours. The panel on the left shows an increased exposure to see the faint bands of WT and ΔddcA. (C) A Western blot using antisera against YneA (upper panel) or DnaN (lower panel). Cultures of ΔddcP, ΔctpA, amyE∷Pxyl-yneA (PEB850) and ΔddcA ΔddcP ΔctpA, amyE∷Pxyl-yneA (PEB852) were grown as in panel B, except at 0 hours erythromycin was added and samples were harvest every hour for three hours.
DdcA is an intracellular protein and DdcP and CtpA are membrane anchored with extracellular protease domains
The observation that DdcA and the checkpoint recovery proteases have distinct functions led us to ask where these proteins are located within the cell to determine if there are spatial constraints on their regulation of the DNA damage checkpoint. YneA is a membrane protein with the majority of the protein located extracellularly (Mo & Burkholder, 2010). We hypothesized that proteases DdcP and CtpA should be similarly localized since YneA is a direct substrate (Burby et al., 2018). We used the transmembrane prediction software TMHMM (Krogh, Larsson, von Heijne, & Sonnhammer, 2001) and found that both DdcP and CtpA were predicted to have an N-terminal transmembrane domain, as reported previously (Tjalsma, Bolhuis, Jongbloed, Bron, & van Dijl, 2000). We tested this prediction directly using a subcellular fractionation assay (L. J. Wu & Errington, 1997). We found that DdcP and CtpA were present predominantly in the membrane fraction (Fig 5A). DdcP is predicted to have a signal peptide cleavage site (Tjalsma et al., 2000), however, we did not detect DdcP in the media (Fig 5A), suggesting that DdcP is membrane anchored and not secreted. The membrane topology of DdcP and CtpA could put the protease domains inside or outside of the cell (Fig 5B). To determine their location we used a protease sensitivity assay (Fig 5B; Wilson, Carlson, Janes, & Hanna, 2012). Cells were treated with lysozyme, followed by incubation with proteinase K. We found that DdcP and CtpA were digested by proteinase K, but that the intracellular protein DnaN was not (Fig 5C). In control reactions we added Triton X-100 to disrupt the plasma membrane, which rendered all three proteins susceptible to proteinase K (Fig 5C). To verify that the N-terminal transmembrane domain is required for DdcP and CtpA to be extracellular we created N-terminal truncations (Fig 5D), and repeated the proteinase K sensitivity assay. With these variants, DdcP and CtpA should be locked inside the cell, and indeed, both N-terminal truncations were now resistant to proteinase K similar to DnaN (Fig 5E). We conclude that DdcP and CtpA are tethered to the plasma membrane through N-terminal transmembrane domains and their protease domains are extracellular (Fig 5B, left panel).
Figure 5. DdcP and CtpA are membrane anchored with extracellular protease domains.
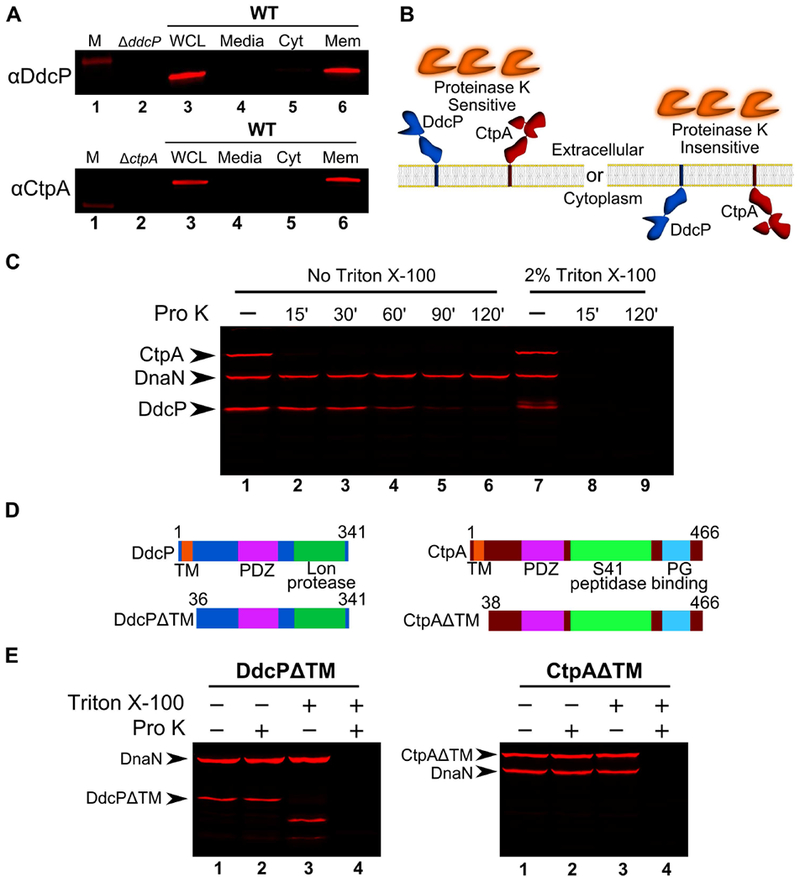
(A) Subcellular fractionation followed by Western blot analysis of WT (PY79) lysates using DdcP and CtpA antiserum (M, molecular weight standard, WCL, whole cell lysates; Media, precipitated media proteins; Cyt, cytosolic fraction; Mem, membrane fraction). (B) Competing models for membrane topology of DdcP and CtpA tested with proteinase K sensitivity assay. (C) Proteinase K sensitivity assay followed by Western blot detection of DdcP, CtpA, and DnaN with antiserum. Samples were treated with lysozyme to generate protoplasts and incubated with proteinase K for the indicated time (lanes 1-6), or the samples were incubated with lysozyme and Triton X-100 to disrupt the plasma membrane and incubated with proteinase K for the indicated time (lanes 7-9). (D) Schematics depicting the DdcPΔTM (left) and CtpAΔTM (right) in which the transmembrane domain was deleted. (E) Proteinase K sensitivity assay followed by Western blot analysis of strains expressing DdcPΔTM (left, PEB719) or CtpAΔTM (right, PEB772) performed as in panel C using a 2 hour incubation with proteinase K.
YneA has a transmembrane domain and has previously been shown to be localized to the plasma membrane (Mo & Burkholder, 2010), and we now show that DdcP and CtpA are membrane anchored as well. To better understand how DdcA limits YneA activity, we asked where DdcA is located. We were unable to find DdcA detected in any previous proteomic experiments that interrogated cytosolic or extracellular proteins (Buttner et al., 2001; Eymann et al., 2004; Hirose et al., 2000). Also, the secretome of B. subtilis was analyzed using bioinformatics and did not report DdcA as a secreted protein (Tjalsma et al., 2000). Therefore, we used several programs to predict the subcellular location of DdcA (Bendtsen, Kiemer, Fausboll, & Brunak, 2005; Hofmann, 1993; Krogh et al., 2001; Yu et al., 2010), all of which suggested that DdcA is cytosolic.
In order to experimentally determine the location of DdcA, we generated GFP fusions to the N- and C-termini of DdcA. We tested whether GFP-DdcA and DdcA-GFP were functional by assaying for the ability to complement a ddcA deletion. We found that GFP-DdcA was able to complement a ddcA deletion in the presence or absence of xylose for induced expression (Fig 6A), similar to that observed with untagged DdcA (Fig 1). In contrast, DdcA-GFP was partially functional, because complete complementation was only observed when expression of ddcA-gfp was induced using xylose, but not in the absence of xylose (Fig 6A). As a control we asked if we could detect free GFP via Western blotting using GFP specific antiserum. We did not detect the fusion proteins in lysates if expression was not induced using xylose. We found that both DdcA fusions were detectable at their approximate molecular weight of 67.6 kDa when induced with 0.05% xylose (Fig 6B), though we did see that the C-terminal fusion had a slight increase in mobility (Figure 6B, arrowhead). Importantly, we did not detect a significant band near 25 kDa, the approximate size of GFP (Fig 6B), suggesting that GFP is not cleaved from DdcA. We did detect a very faint proteolytic fragment (Fig 6B, arrow) that seemed to occur during the lysis procedure. After establishing the functionality and integrity of the GFP-DdcA fusion we chose to visualize DdcA localization via fluorescence microscopy.
Figure 6. GFP-DdcA is an intracellular protein and is present in the cytosolic and membrane fractions.
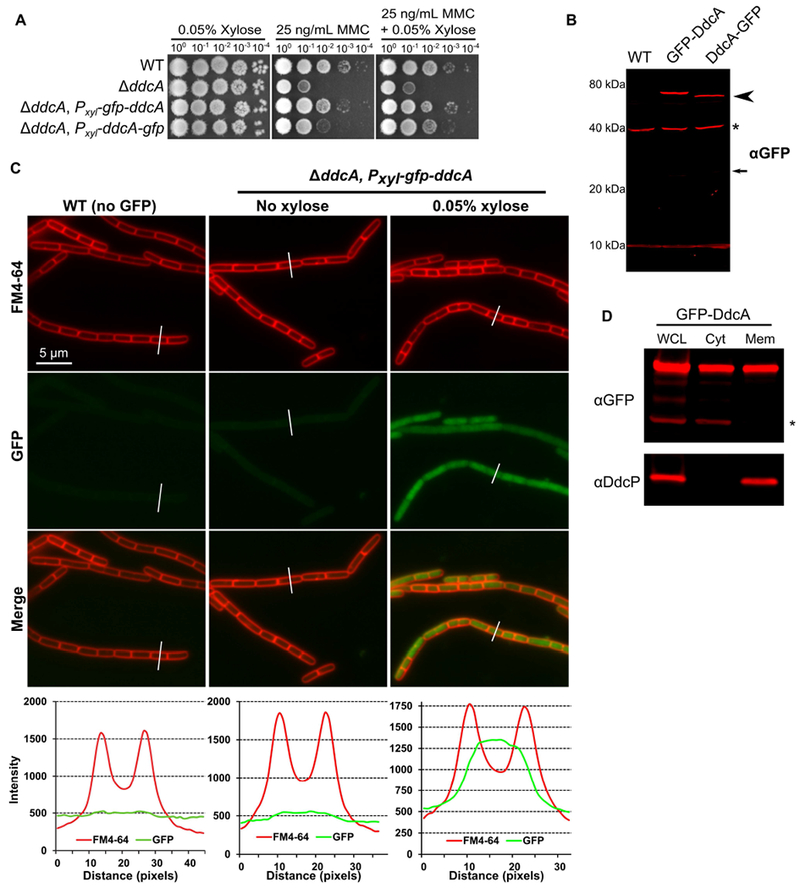
(A) Spot titer assay using B. subtilis strains WT (PY79), ΔddcA (PEB357), ΔddcA amyE∷Pxyl-gfp-ddcA (PEB854), and ΔddcA amyE∷Pxyl-ddcA-gfp (PEB856) spotted on the indicated media. (B) Western blot of cell extracts from B. subtilis strains WT (PY79), ΔddcA amyE∷Pxyl-gfp-ddcA (PEB854), and ΔddcA amyE∷Pxyl-ddcA-gfp (PEB856) using antiserum against GFP. The arrowhead highlights the slightly increased mobility of DdcA-GFP, and the asterisk denotes a cross-reacting species detected by the GFP antiserum. The smaller arrow indicates the expected migration of free GFP. (C) Micrographs from WT (PY79) and ΔddcA amyE∷Pxyl-gfp-ddcA (PEB854) cultures grown in S750 minimal media containing 1% arabinose with (far left and right panels) or without (middle panels) 0.05% xylose. Images in red are the membrane stain FM4-64, green are GFP fluorescence and the bottom images are a merge of FM4-64 and GFP fluorescence. The white lines through cells in the images are a representation of the line scans of fluorescence intensity generated in ImageJ and plotted below the micrographs. Scale bar is 5 μm. (D) Western blot of whole cell lysate (WCL), cytosolic fraction (Cyt), and membrane fraction (Mem) from ΔddcA amyE∷Pxyl-gfp-ddcA (PEB854) cell extracts using antisera against GFP (upper panel) or DdcP (lower panel). The asterisk denotes a cross-reacting species detected by the GFP antiserum.
To compare the background fluorescence of B. subtilis cells, we imaged WT (PY79) cells under the same conditions as the GFP-DdcA fusion strain. We found a low level of background fluorescence in WT cells, and when a line scan of fluorescence intensity through a cell was plotted there was a very slight increase in signal intensity in the span between the fluorescent membrane peaks (Fig 6C). The GFP-DdcA fusion was detectable throughout the cell at very low levels in the absence of xylose induction, with the intensity being slightly greater than WT cells (Fig 6C). We then imaged cells under conditions in which gfp-ddcA expression was induced with 0.05% xylose. This experiment shows that GFP-DdcA was found throughout the cytosol, and the scan of fluorescence intensity was significantly greater than WT (Fig 6C). We observed that the partially functional DdcA-GFP fusion was also present diffusely throughout the cytosol (Fig S3A). Finally, we tested DdcA localization using subcellular fractionation. We found that GFP-DdcA was detectable in the membrane and cytosolic fractions (Fig 6D), and similar results were obtained with DdcA-GFP (Fig S4B). As controls, we found that DdcP was found in the membrane fraction and not the cytosolic fraction (Fig 6D), and a cross-reacting protein detected by our GFP antiserum was found in the cytosol and not the membrane fractions (Fig 6D). Taken together, DdcA appears to be an intracellular protein that is primarily located in the cytosol with some molecules localized to the membrane. Importantly we now show that DdcA and the checkpoint recovery proteases are separated in space by the plasma membrane, demonstrating that YneA regulators are present in the cytosol (DdcA) and in the extracellular space (DdcP and CtpA). Further, the demonstration of DdcA occupying a different subcellular location from DdcP and CtpA explains their distinct roles in regulating YneA.
YneA-dependent cell elongation is enhanced in cells lacking DdcA and the recovery proteases.
DdcA appears to regulate YneA activity independent of protein abundance and stability. We initially hypothesized that DdcA could interact directly with YneA to inhibit its activity. To test this hypothesis, we assayed for a protein-protein interaction using a bacterial two-hybrid, but did not detect an interaction (Fig S5). We then asked whether DdcA affected the localization of YneA, hypothesizing that DdcA could prevent YneA from reaching the plasma membrane. To address this question, we built a strain in which GFP was fused to the N-terminus of YneA, and placed gfp-yneA under the control of the xylose-inducible promoter Pxyl. We expressed both YneA and GFP-YneA in strains lacking ddcA, the checkpoint recovery proteases, or the triple mutant and found that GFP-YneA is able to inhibit growth to a similar extent as YneA (Fig 7A), suggesting that the GFP fusion is functional. We visualized GFP-YneA following induction with 0.1% xylose for 30 minutes. We found that GFP-YneA localized to the mid-cell, while also demonstrating diffuse intracellular fluorescence (Fig 7B), which we suggest is free GFP generated by the checkpoint recovery proteases after YneA cleavage. Deletion of ddcA alone did not affect GFP-YneA localization, with both WT and ΔddcA strains having similar mid-cell localization frequencies (Fig 7B). The absence of both checkpoint recovery proteases resulted in puncta throughout the plasma membrane (Fig 7B).
Figure 7. DdcA inhibits YneA activity.
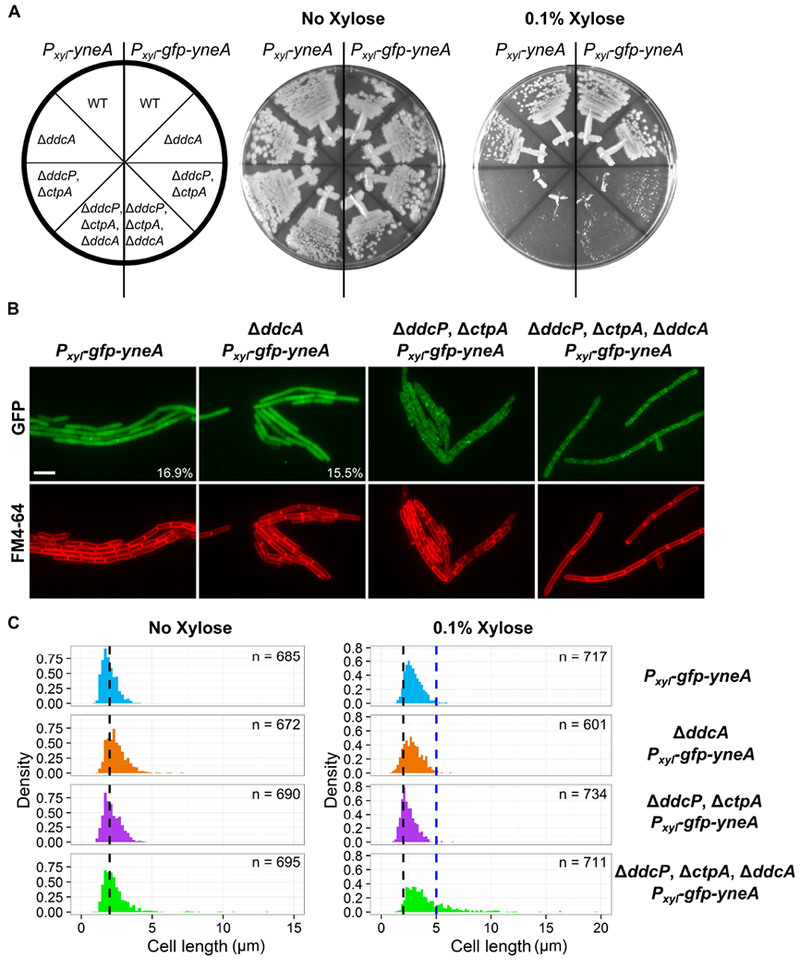
(A) B. subtilis strains amyE∷Pxyl-yneA (PEB846), ΔddcA amyE∷Pxyl-yneA (PEB848), ΔddcP, ΔctpA, amyE∷Pxyl-yneA (PEB850), and ΔddcA ΔddcP ΔctpA, amyE∷Pxyl-yneA (PEB852), amyE∷Pxyl-gfp-yneA (PEB876), ΔddcA amyE∷Pxyl-gfp-yneA (PEB882), ΔddcP, ΔctpA, amyE∷Pxyl-gfp-yneA (PEB888), and ΔddcA ΔddcP ΔctpA, amyE∷Pxyl-gfp-yneA (PEB894) were struck onto LB or LB + 0.1% xylose and incubated at 30°C overnight. (B) Micrographs from the indicated strains from Panel A, grown in minimal media and treated with 0.1% xylose for 30 minutes. Green images are GFP fluorescence and red images are FM4-64 membrane stain. The percentage of septal localization is shown for PEB876 (n=591) and PEB882 (n=542). The p-value of a two-tailed z-test was 0.516. (C) Cell length distributions of strains grown with (right) or without (left) 0.1% xylose. The number of cells measured (n) for each condition is indicated.
Intriguingly, deletion of ddcA in addition to the checkpoint recovery proteases resulted in severe cell elongation, however, GFP-YneA localization was not affected (Fig 7B). The difference in cell length was quantified by measuring the cell length of at least 600 cells following growth in the presence of 0.1% xylose for 30 minutes. The cell length distributions of strains lacking ddcA or ddcP and ctpA were similar to the WT control (Fig 7C). The distribution for the strain lacking ddcA, ddcP, and ctpA had a significant skew to the right indicating greater cell lengths (Fig 7C). The percentage of cells greater than 5 μm in length was approximately 22% for the triple mutant and significantly greater than the other three strains in which approximately 1% of cells were greater than 5 μm (Table 1). As a control, we determined the cell length distributions prior to xylose addition and found all four strains to have similar cell length distributions in the absence of xylose (Fig 7C). With these data, we conclude that DdcA prevents YneA from inhibiting cell division.
Table 1. Over-expression of GFP-YneA results in a significant increase in cells greater than 5 μm in length in cells lacking ddcP, ctpA, and ddcA.
Data are from expression of GFP-YneA using 0.1% xylose for 30 minutes. The mean cell length ± the standard deviation is listed. The percent of cells greater than 5 μm (number/total cells scored), with the p-value from a two-tailed z-test are listed.
| No Xylose | 0.1% Xylose | ||||
|---|---|---|---|---|---|
| Strain | Genotype | Cell length (mean ± sd) | Cell length (mean ± sd) | % ≥ 5 μm | p-value |
| PEB876 | amyE∷Pxyl-gfp-yneA | 1.98 ± 0.51 (n = 685) | 2.91 ± 0.75 | 0.84% (6/717) | N/A |
| PEB882 | ΔddcA, amyE∷Pxyl-gfp-yneA | 2.48 ± 0.73 (n = 672) | 2.86 ± 0.85 | 1.16% (7/601) | 0.55 |
| PEB888 | ΔddcP, ΔctpA, amyE∷Pxyl-gfp-yneA | 2.18 ± 0.60 (n = 690) | 2.49 ± 0.70 | 0.68% (5/734) | 0.73 |
| PEB894 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-gfp-yneA | 2.39 ± 1.10 (n = 695) | 4.09 ± 2.09 | 22.4% (159/711) | <0.00001 |
Discussion
A model for DNA damage checkpoint activation and recovery
The DNA damage checkpoint in bacteria was discovered through seminal work using E. coli as a model organism (Friedberg et al., 2006). An underlying assumption in the models is that the input signal of RecA coated ssDNA and the affinity of LexA for its binding site is sufficient to control the rate of cell division in response to DNA damage. A finding that the initiator protein, DnaA, controls the transcription of ftsL, and as a result the rate of cell division, in response to replication stress, gave a hint that coordination of cell division and DNA replication may be more complex (Goranov et al., 2005). Here, we elaborate on the complexity of regulating cell division in response to DNA damage by uncovering a DNA damage checkpoint antagonist, DdcA (Fig 8). In response to DNA damage, the repressor LexA is inactivated, which results in expression of yneA. Accumulation of YneA must saturate two proteases, DdcP and CtpA, and overcome DdcA-dependent inhibition in order to block cell division. We previously reported that DdcP and CtpA are not induced by DNA damage (Burby et al., 2018), and a previous study reported that transcripts of ddcA, ddcP, and ctpA are not induced by DNA damage or inhibition of DNA replication (Goranov et al., 2006). Thus, we model all three proteins functioning to set a threshold of YneA required for checkpoint activation with DdcA located in the cytosol and DdcP and CtpA protease domains located extracellularly. These regulators require that YneA expression overcomes a cytosolic regulator and then two extracellular regulators before the checkpoint can be activated. After the checkpoint is established, DNA repair occurs and the integrity of the DNA is restored the SOS response is turned off, LexA represses yneA expression, and the checkpoint recovery proteases degrade the remaining YneA. The genetic experiments attempting to substitute the checkpoint proteases for DdcA and vice versa strongly suggest that DdcA does not function in the checkpoint recovery process (Fig 3). Together, our results uncover a unique strategy in regulating a bacterial DNA damage checkpoint by identifying a proteolysis independent mechanism of setting a threshold for DNA damage checkpoint activation.
Figure 8. DdcA inhibits enforcement of the DNA damage checkpoint.
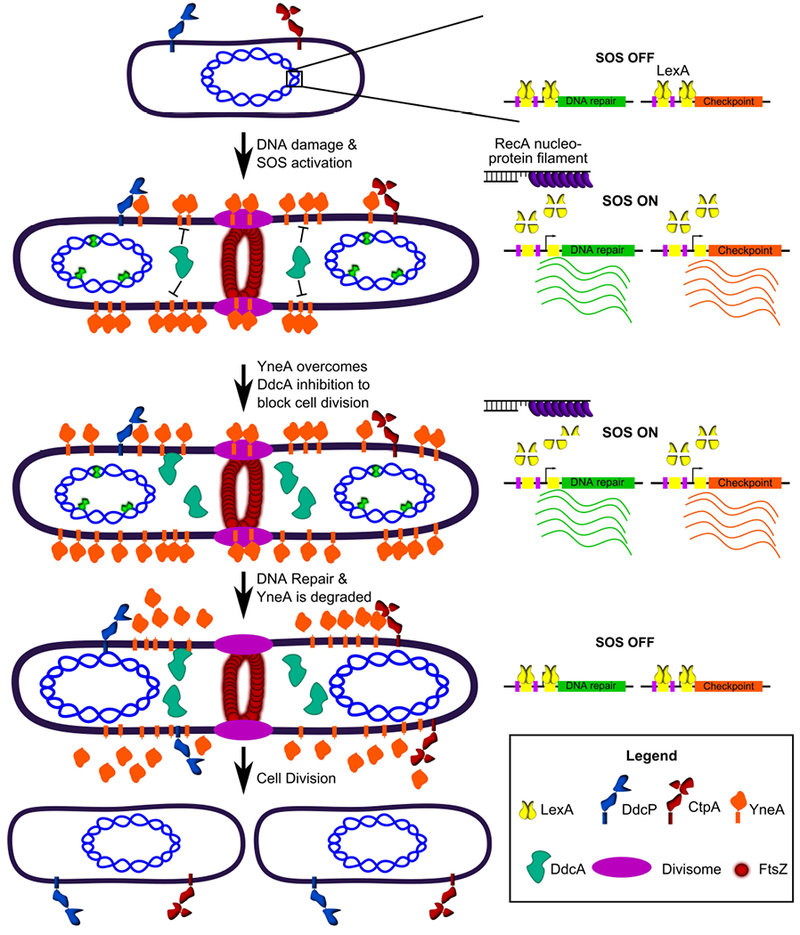
A working model for how DdcA inhibits the activity of YneA. DdcA prevents access to the target of YneA, however, when the SOS response has been activated for a prolonged period of time, YneA is able to overcome DdcA dependent inhibition to prevent cell division. Following DNA repair and completion of DNA replication the SOS response is turned off and the checkpoint recovery proteases degrade YneA allowing cell division to resume.
How does DdcA inhibit YneA?
Our results are most supportive of DdcA acting as an antagonist to YneA, rather than functioning in checkpoint recovery. Two lines of evidence support this model. First, DdcA does not affect YneA protein levels, stability, or localization (Figs S3 & 4). Second, if DdcA was involved in checkpoint recovery, we would predict that expression of one of the checkpoint proteases would be able to compensate for deletion of ddcA. Instead, we found that the checkpoint recovery proteases and DdcA cannot replace each other (Fig 3). As a result, we hypothesized that DdcA acts by preventing YneA from accessing its target. We tested for an interaction between YneA and DdcA using a bacterial two-hybrid assay and we were unable to identify an interaction with full length or a cytoplasmic “locked” YneA mutant lacking its transmembrane domain (Fig S5). We also ruled out the hypothesis that DdcA affects the subcellular localization of YneA using a GFP-YneA fusion, which had similar localization patterns with and without ddcA (Fig 7B). Taken together, all these results support a model where DdcA prevents YneA from inhibiting cell division, which could occur through preventing access to the target of YneA or through an indirect mechanism.
The YneA target that results in the inhibition of cell division is unknown. YneA is a membrane bound cell division inhibitor. This class of inhibitor in bacteria is typified as being a small protein that contains an N-terminal transmembrane domain, and they have been identified in several species (Bojer et al., 2018; Chauhan et al., 2006; Kawai et al., 2003; Modell et al., 2011; Modell et al., 2014; Ogino et al., 2008). In Caulobacter crescentus, the cell division inhibitors SidA and DidA inhibit the activity of FtsW/N, which are components of the divisome. A recent study in Staphylococcus aureus identified a small membrane division inhibitor, SosA, and its target appears to be PBP1 (Bojer et al., 2018), which is involved in peptidoglycan synthesis at the septum (Claessen et al., 2008; Scheffers & Errington, 2004). It is tempting to speculate that YneA could target an essential component of the cell division machinery, in particular because previous work found a conserved face of the transmembrane domain that was required for activity (Mo & Burkholder, 2010). Prior studies of C. crescentus and S. aureus were able to detect interactions between the cell division inhibitors and their targets using the bacterial two-hybrid assay (Bojer et al., 2018; Modell et al., 2011; Modell et al., 2014). We reasoned that we might be able to identify an interacting partner of YneA or DdcA using this approach. We used DdcA and YneA in a bacterial two-hybrid assay using several proteins involved in cell division and cell wall synthesis, many of which had phenotypes in our previous Tn-seq genetic screens (Burby et al., 2018), but we were unable to identify a positive interaction (data not shown). Still, there are fundamental differences between YneA and other membrane bound cell division inhibitors. YneA has two major predicted features: an N-terminal transmembrane domain and a C-terminal LysM domain, and both have been found to be required for full activity (Mo & Burkholder, 2010). The other cell division inhibitors SidA, DidA, and SosA do not have a LysM domain (Bojer et al., 2018; Modell et al., 2011; Modell et al., 2014). LysM domains bind to peptidoglycan (PG) and many proteins containing LysM domains have cell wall hydrolase activity (Buist, Steen, Kok, & Kuipers, 2008). Thus, another possibility is that YneA acts directly on the cell wall to inhibit cell division instead of or in addition to targeting a membrane protein.
Intriguingly, the cell division inhibitor of Mycobacterium tuberculosis, Rv2719c, also contains a LysM domain and was shown to have cell wall hydrolase activity in vitro (Chauhan et al., 2006). The localization of GFP-YneA is also similar to previous reports of fluorescent vancomycin labeling of nascent peptidoglycan synthesis (Fig 7B; Daniel & Errington, 2003; Tiyanont et al., 2006). The difficulty with the model of targeting cell wall synthesis directly is that it is not clear how DdcA would prevent YneA activity given that these proteins are separated by the plasma membrane. One explanation is that DdcA directly or indirectly affects the folding of YneA as it is transported across the membrane, resulting in a form of YneA that is not competent for PG binding. DdcA contains a TPR domain and proteins containing TPR domains have been found to have chaperone activity and act as co-chaperones (Smith, 2004). It is intriguing that ddcA is just upstream of the chaperone trigger factor (tig) in the B. subtilis genome, and this organization is conserved in some bacterial species.
Negative regulation of YneA occurs through three distinct mechanisms
The checkpoint recovery proteases and DdcA utilize multiple strategies to inhibit YneA. Although both DdcP and CtpA degrade YneA, they are very different proteases. DdcP has a Lon peptidase domain and a PDZ domain, whereas CtpA has an S41 peptidase domain and a PDZ domain. The PDZ domains of DdcP and CtpA have different functions in vivo and show homology to different classes of PDZ domains found in proteases in E. coli (Fig. S6, see supporting results). Thus, it appears that the proteases utilize different strategies to degrade YneA. DdcA is unique, because it acts as an antagonist without affecting protein abundance, stability, or localization. Also, DdcA appears to function prior to checkpoint establishment and not in recovery, whereas the proteases perform both functions. Together, DdcA, DdcP, and CtpA provide a buffer to expression of YneA, thereby setting a threshold of YneA for checkpoint enforcement.
The discovery of a specific DNA damage checkpoint antagonist brings the total known proteins to negatively regulate YneA to three, which begs the question: why isn’t a single protein sufficient? One explanation is that the process can be fine-tuned. By utilizing several proteins, the process has more nodes for regulation, which is advantageous at least for B. subtilis. A second explanation is that this strategy evolved in response to more efficient DNA repair. The SOS-regulon is highly conserved in bacteria and yet the checkpoint strategies vary significantly (Erill et al., 2007). If an organism evolves a more efficient DNA repair system in which DNA repair could be completed faster, the same level of checkpoint protein will no longer be required, because the checkpoint would delay cell division longer than necessary to complete DNA repair. This could be the explanation for the highly divergent nature of cell division inhibitors in bacteria as well as the explanation for the complex control over YneA found in B. subtilis.
Materials and Methods
Bacteriological and molecular methods
All B. subtilis strains are derivatives of PY79 (Youngman, Perkins, & Losick, 1984), and are listed in Table 2. Construction of individual strains is detailed in the supporting methods using double cross-over recombination or CRISPR/Cas9 genome editing as previously described (Burby & Simmons, 2017; Burby et al., 2018). B. subtilis strains were grown in LB (10 g/L NaCl, 10 g/L tryptone, 5 g/L yeast extract) or S750 media [1× S750 salts (diluted from 10× S750 salts: 104.7g/L MOPS, 13.2 g/L, ammonium sulfate, 6.8 g/L monobasic potassium phosphate, pH 7.0 adjusted with potassium hydroxide), 1× metals (diluted from 100× metals: 0.2 M MgCl2, 70 mM CaCl2, 5 mM MnCl2, 0.1 mM ZnCl2, 100 μg/mL thiamine-HCl, 2 mM HCl, 0.5 mM FeCl3), 0.1% potassium glutamate, 40 μg/mL phenylalanine, 40 μg/mL tryptophan] containing either 2% glucose or 1% arabinose as indicated in each method. Plasmids used in this study are listed in Table S1. Individual plasmids were constructed using Gibson assembly as described previously (Burby et al., 2018; Gibson, 2011). The details of plasmid construction are described in the supporting methods. Oligonucleotides used in this study are listed in Table S2 and were obtained from Integrated DNA technologies (IDT). Antibiotics for selection in B. subtilis were used at the following concentrations: 100 μg/mL spectinomycin, 5 μg/mL chloramphenicol, and 0.5 μg/mL erythromycin. Antibiotics used for selection in Escherichia coli were used at the following concentrations: 100 μg/mL spectinomycin, 100 μg/mL ampicillin, and 50 μg/mL kanamycin. Mitomycin C (Fisher bioreagents) and phleomycin (Sigma) were used at the concentrations indicated in the figures and legends.
Table 2.
Strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| PY79 | PY79 | (Youngman et al., 1984) |
| PEB309 | ΔuvrAB | This study |
| PEB324 | ΔddcP (ylbL) | (Burby et al., 2018) |
| PEB355 | ΔctpA | (Burby et al., 2018) |
| PEB357 | ΔddcA (ysoA) | (Burby et al., 2018) |
| PEB433 | ΔyneA∷erm | (Burby et al., 2018) |
| PEB439 | ΔyneA∷loxP | (Burby et al., 2018) |
| PEB495 | ΔddcA, ΔyneA∷erm | This study |
| PEB497 | ΔuvrAB, ΔddcA | This study |
| PEB499 | ΔddcP, ΔddcA | This study |
| PEB503 | ΔddcA, amyE∷Pxyl-ddcA | This study |
| PEB555 | ΔddcP, ΔctpA | (Burby et al., 2018) |
| PEB557 | ΔddcP, ΔctpA, amyE∷Pxyl-ddcP | (Burby et al., 2018) |
| PEB561 | ΔddcP, ΔctpA, ΔyneA∷loxP | (Burby et al., 2018) |
| PEB579 | ΔctpA, ΔddcA | This study |
| PEB587 | ΔddcA, ΔyneA∷loxP | This study |
| PEB619 | ΔddcP, ΔctpA, amyE∷Pxyl-ctpA | (Burby et al., 2018) |
| PEB639 | ΔddcP, ΔctpA, ΔddcA | This study |
| PEB643 | ΔddcP, ΔctpA, ΔddcA, ΔyneA∷loxP | This study |
| PEB719 | ΔddcP, amyE∷Pxyl-ddcPΔTM | This study |
| PEB772 | ΔctpA, amyE∷Pxyl-ctpAΔTM | This study |
| PEB774 | ddcPΔPDZ | This study |
| PEB776 | ctpAΔPDZ | This study |
| PEB836 | ΔddcA, amyE∷Pxyl-ddcP | This study |
| PEB837 | ΔddcA, amyE∷Pxyl-ctpA | This study |
| PEB838 | ΔddcP, ΔctpA, amyE∷Pxyl-ddcA | This study |
| PEB839 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ddcP | This study |
| PEB840 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ddcA | This study |
| PEB841 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-ctpA | This study |
| PEB846 | amyE∷Pxyl-yneA | This study |
| PEB848 | ΔddcA, amyE∷Pxyl-yneA | This study |
| PEB850 | ΔddcP, ΔctpA, amyE∷Pxyl-yneA | This study |
| PEB852 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-yneA | This study |
| PEB854 | ΔddcA, amyE∷Pxyl-gfp-ddcA | This study |
| PEB856 | ΔddcA, amyE∷Pxyl-ddcA-gfp | This study |
| PEB876 | amyE∷Pxyl-gfp-yneA | This study |
| PEB882 | ΔddcA, amyE∷Pxyl-gfp-yneA | This study |
| PEB888 | ΔddcP, ΔctpA, amyE∷Pxyl-gfp-yneA | This study |
| PEB894 | ΔddcP, ΔctpA, ΔddcA, amyE∷Pxyl-gfp-yneA | This study |
Spot titer assays
Spot titer assays were performed as previously described (Burby et al., 2018). Briefly, B. subtilis strains were grown on an LB agar plate at 30°C overnight and a single colony was used to inoculate a liquid LB culture. The cultures were grown at 37°C to an OD600 between 0.5 and 1. Cultures were normalized to an OD600 = 0.5, and serial dilutions were spotted on to LB agar media containing the drugs as indicated in the figures. Plates were grown at 30°C overnight (16-20 hours). All spot titer assays were performed at least twice.
Western blotting
Western blotting experiments for YneA were performed essentially as described (Burby et al., 2018). Briefly, for the MMC recovery assay, samples of an OD600 = 10 were harvested via centrifugation and washed twice with 1× PBS pH 7.4 and re-suspended in 400 μL of sonication buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 20% glycerol, 2× Roche protease inhibitors, and 5 mM PMSF) and lysed via sonication. SDS sample buffer was added to 2× and samples (10 μL) were incubated at 100°C and separated using 10% SDS-PAGE (DnaN) or 16.5% Tris-Tricine SDS-PAGE (YneA). Proteins were transferred to a nitrocellulose membrane using the BioRad transblot-turbo following the manufacturer’s instructions. Membranes were blocked in 5% milk in TBST for 1 hour at room temperature. Membranes were incubated with YneA antiserum at a 1:3000 dilution in 2% milk in TBST for two hours at room temperature or at 4°C overnight. Membranes were washed three times with TBST for five minutes each and secondary antibodies (LiCor goat anti-Rabbit-680LT; 1:15000) were added and incubated for one hour at room temperature. Membranes were washed three times with TBST for five minutes each. Images of membranes were captured using the LiCor Odyssey.
For overexpression of YneA, cultures of LB were inoculated at an OD600 = 0.05 and incubated at 30°C until an OD600 of about 0.2 (about 90 minutes). Xylose was added to 0.1% and cultures were incubated at 30°C for 2 hours. Samples of an OD600 = 25 were harvested and re-suspended in 500 μL sonication buffer as above. All subsequent steps were performed as described above.
For GFP-DdcA and DdcA-GFP, samples of an OD600 = 1 were harvested from LB + 0.05% xylose cultures via centrifugation and washed twice with 1× PBS pH 7.4. Samples were re-suspended in 100 μL 1× SMM buffer (0.5 M sucrose, 0.02 M maleic acid, 0.02 M MgCl2, adjusted to pH 6.5) containing 1 mg/mL lysozyme and 2× Roche protease inhibitors. Samples were incubated at room temperature for one hour and SDS sample buffer was added to 1× and incubated at 100°C for 7 minutes. Samples (10 μL) were separated via 10% or 4-20% SDS-PAGE. All subsequent steps were as described above, except GFP antisera (lot 1360-ex) was used at a 1:5000 dilution at 4°C overnight.
YneA stability assay
Cultures of LB were inoculated at an OD600 = 0.05 and incubated at 30°C until an OD600 of about 0.2 (about 90 minutes). Xylose was added to 0.1% and cultures were incubated at 30°C for 2 hours. To stop translation, erythromycin was added to 50 μg/mL and samples (OD600 = 10) were taken at 0, 60, 120, and 180 minutes (the strains for this experiment contain the chloramphenicol resistant gene, cat, which prevents chloramphenicol from being used). Western blotting was performed as described above.
Subcellular fractionation
Fractionation experiments were performed as described previously (L. J. Wu & Errington, 1997). A cell pellet equivalent to 1 mL OD600 = 1 was harvested via centrifugation (10,000 g for 5 minutes at room temperature), and washed with 250 μL 1× PBS. Protoplasts were generated by resuspension in 100 μL 1× SMM buffer (0.5 M sucrose, 0.02 M maleic acid, 0.02 M MgCl2, adjusted to pH 6.5) containing 1 mg/mL lysozyme and 1× Roche protease inhibitors at room temperature for 2 hours. Protoplasts were pelleted via centrifugation: 5,000 g for 6 minutes at room temperature. Protoplasts were re-suspended in 100 μL TM buffer (20 mM Tris, pH 8.0, 5 mM MgCl2, 40 units/mL DNase I (NEB), 200 μg/mL RNase A (Sigma), 0.5 mM CaCl2, and 1× Roche protease inhibitors) and left at room temperature for 30 minutes. The membrane fraction was pelleted via centrifugation: 20,800 g for 30 minutes at 4°C. The cytosolic fraction (supernatant) was transferred to a new tube and placed on ice, and the pellet was washed with 100 μL of TM buffer and pelleted via centrifugation as above. The supernatant was discarded and the pellet was re-suspended in 120 μL of 1× SDS dye. SDS loading dye was added to 1× to the cytosolic fraction and 12 μL of each fraction were used for Western blot analysis.
Culture supernatant protein precipitation
Culture supernatants were concentrated by TCA precipitation as described previously with minor modifications (Link & LaBaer, 2011). A culture was grown at 30°C until OD600 about 1, and the cells were pelleted via centrifugation: 7,000 g for 10 minutes at room temperature. The culture supernatant (30 mL) was filtered using a 0.22 μm filter and placed on ice. Proteins were precipitated by addition of 6 mL ice-cold 100% TCA (6.1N), and left on ice for 30 minutes. Precipitated proteins were pelleted via centrifugation: 18,000 rpm (Sorvall SS-34 rotor) for 30 minutes at 4°C. Pellets were washed with 1 mL ice-cold acetone and pelleted again via centrifugation: 20,000 g for 15 minutes at 4°C. The supernatant was discarded, and the residual acetone was evaporated by placing tubes in 100°C heat block for 1-2 minutes. Protein pellets were re-suspended in 120 μL 6× SDS-loading dye and 12 μL were used in Western blot analysis.
Proteinase K sensitivity assay
Proteinase K sensitivity assays were performed similar to previous reports (Navarre & Schneewind, 1994; Wilson et al., 2012). A cell pellet from 0.5 mL OD600 = 1 equivalent was harvested and washed as in “subcellular fractionation.” Protoplasts were generated by resuspension in 36 μL 1× SMM buffer (0.5 M sucrose, 0.02 M maleic acid, 0.02 M MgCl2, adjusted to pH 6.5) containing 1 mg/mL lysozyme at room temperature for 1 hour. Either 9 μL of 1× SMM buffer or 0.5 mg/mL proteinase K (dissolved in 1× SMM buffer) was added (final proteinase K concentration of 100 μg/mL) and incubated at 37°C for the time indicated in the figures. Reactions were stopped by the addition of 5 μL 50 mM PMSF (final concentration of 5 mM) and 25 μL 6× SDS-dye (final concentration of 2×). For Western blot analysis, 12 μL were used.
Microscopy
Strains were grown on LB agar plates containing 5 μg/mL chloramphenicol at 30°C overnight. For GFP-DdcA and DdcA-GFP, LB agar plates were washed with S750 media containing 1% arabinose and cultures of S750 media containing 1% arabinose and 0.05% xylose were inoculated at an OD600 = 0.1 and incubated at 30°C until an OD600 of about 0.4. Samples were taken and incubated with 2 μg/mL FM4-64 for 5 minutes and transferred to pads of 1× Spizizen salts and 1% agarose. Images were captured with an Olympus BX61 microscope using 250 ms and 1000 ms exposure times for FM4-64 (membranes) and GFP, respectively. The brightness and contrast were adjusted for FM4-64 images with adjustments applied to the entire image. Strains with GFP-YneA were grown on LB agar plates containing 5 μg/mL chloramphenicol overnight at 30°C. Plates were washed with S750 minimal media containing 1% arabinose and cultures started at an OD600 = 0.1. Cultures were grown at 30°C until an OD600 of about 0.3 and xylose was added to 0.1%. Cultures were grown for 30 minutes at 30°C and imaged as for GFP-DdcA with exposure times of 300 ms for FM4-64 and 500 ms for GFP.
Supplementary Material
Acknowledgements
This work was supported by NIH grant R01 GM107312 to LAS and NSF predoctoral fellowship #DGE1256260 to PEB.
Footnotes
The authors have no conflict of interest to declare.
References
- Anderson DG, & Kowalczykowski SC (1997). The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell, 90(1), 77–86. [DOI] [PubMed] [Google Scholar]
- Au N, Kuester-Schoeck E, Mandava V, Bothwell LE, Canny SP, Chachu K, Lovett CM (2005). Genetic composition of the Bacillus subtilis SOS system. J Bacteriol, 187(22), 7655–7666. doi: 10.1128/jb.187.22.7655-7666.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Kiemer L, Fausboll A, & Brunak S (2005). Non-classical protein secretion in bacteria. BMC Microbiol, 5, 58. doi: 10.1186/1471-2180-5-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, & Lutkenhaus J (1993). Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J Bacteriol, 175(4), 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojer MS, Wacnik K, Kjelgaard P, Gallay C, Bottomley AL, Cohn MT, Ingmer H (2018). SosA inhibits cell division in Staphylococcus aureus in response to DNA damage. bioRxiv. doi: 10.1101/364299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buist G, Steen A, Kok J, & Kuipers OP (2008). LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol Microbiol, 68(4), 838–847. doi: 10.1111/j.1365-2958.2008.06211.x [DOI] [PubMed] [Google Scholar]
- Burby PE, & Simmons LA (2017). MutS2 Promotes Homologous Recombination in Bacillus subtilis. J Bacteriol, 199(2). doi: 10.1128/jb.00682-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE, Simmons ZW, Schroeder JW, & Simmons LA (2018). Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery. PLoS Genet, 14(7), e1007512. doi: 10.1371/journal.pgen.1007512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner K, Bernhardt J, Scharf C, Schmid R, Mader U, Eymann C, Hecker M (2001). A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilis. Electrophoresis, 22(14), 2908–2935. doi: [DOI] [PubMed] [Google Scholar]
- Canceill D, Dervyn E, & Huisman O (1990). Proteolysis and modulation of the activity of the cell division inhibitor SulA in Escherichia coli lon mutants. J Bacteriol, 172(12), 7297–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, & Stulik J (2013). Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun, 81(3), 629–635. doi: 10.1128/iai.01035-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Lofton H, Maloney E, Moore J, Fol M, Madiraju MV, & Rajagopalan M (2006). Interference of Mycobacterium tuberculosis cell division by Rv2719c, a cell wall hydrolase. Mol Microbiol, 62(1), 132–147. doi: 10.1111/j.1365-2958.2006.05333.x [DOI] [PubMed] [Google Scholar]
- Churchill JJ, Anderson DG, & Kowalczykowski SC (1999). The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of chi, resulting in constitutive recombination activation. Genes Dev, 13(7), 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, & Edwards DH (2008). Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol Microbiol, 68(4), 1029–1046. doi: 10.1111/j.1365-2958.2008.06210.x [DOI] [PubMed] [Google Scholar]
- Daniel RA, & Errington J (2003). Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell, 113(6), 767–776. [DOI] [PubMed] [Google Scholar]
- Donachie WD, & Blakely GW (2003). Coupling the initiation of chromosome replication to cell size in Escherichia coli. Curr Opin Microbiol, 6(2), 146–150. [DOI] [PubMed] [Google Scholar]
- Erill I, Campoy S, & Barbe J (2007). Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev, 31(6), 637–656. doi: 10.1111/j.1574-6976.2007.00082.x [DOI] [PubMed] [Google Scholar]
- Eymann C, Dreisbach A, Albrecht D, Bernhardt J, Becher D, Gentner S, Hecker M (2004). A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics, 4(10), 2849–2876. doi: 10.1002/pmic.200400907 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, & Ellenberger T (2006). DNA Repair and Mutagenesis (2nd ed.). Washington, D.C.: ASM Press. [Google Scholar]
- Gibson DG (2011). Enzymatic assembly of overlapping DNA fragments In Voigt C (Ed.), Synthetic Biology, Pt B: Computer Aided Design and DNA Assembly (Vol. 498, pp. 349–361). San Diego: Elsevier Academic Press Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goranov AI, Katz L, Breier AM, Burge CB, & Grossman AD (2005). A transcriptional response to replication status mediated by the conserved bacterial replication protein DnaA. Proc Natl Acad Sci U S A, 102(36), 12932–12937. doi: 10.1073/pnas.0506174102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goranov AI, Kuester-Schoeck E, Wang JD, & Grossman AD (2006). Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol, 188(15), 5595–5605. doi: 10.1128/jb.00342-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill NS, Kadoya R, Chattoraj DK, & Levin PA (2012). Cell size and the initiation of DNA replication in bacteria. PLoS Genet, 8(3), e1002549. doi: 10.1371/journal.pgen.1002549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose I, Sano K, Shioda I, Kumano M, Nakamura K, & Yamane K (2000). Proteome analysis of Bacillus subtilis extracellular proteins: a two-dimensional protein electrophoretic study. Microbiology, 146 (Pt 1), 65–75. doi: 10.1099/00221287-146-1-65 [DOI] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W (1993). TMBASE - A database of membrane spanning protein segments.
- Huang J, Cao C, & Lutkenhaus J (1996). Interaction between FtsZ and inhibitors of cell division. J Bacteriol, 178(17), 5080–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman O, & D’Ari R (1981). An inducible DNA replication-cell division coupling mechanism in E. coli. Nature, 290(5809), 797–799. [DOI] [PubMed] [Google Scholar]
- Huisman O, D’Ari R, & Gottesman S (1984). Cell-division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proc Natl Acad Sci U S A, 81(14), 4490–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivancic-Bace I, Peharec P, Moslavac S, Skrobot N, Salaj-Smic E, & Brcic-Kostic K (2003). RecFOR function is required for DNA repair and recombination in a RecA loading-deficient recB mutant of Escherichia coli. Genetics, 163(2), 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivancic-Bace I, Vlasic I, Salaj-Smic E, & Brcic-Kostic K (2006). Genetic evidence for the requirement of RecA loading activity in SOS induction after UV irradiation in Escherichia coli. J Bacteriol, 188(14), 5024–5032. doi: 10.1128/jb.00130-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VN, & Szybalski W (1963). A molecular mechanism of mitomycin action: Linking of complementary DNA strands. Proc Natl Acad Sci U S A, 50, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori M, Yanagi H, & Yura T (1999). The ATP-dependent HslVU/ClpQY protease participates in turnover of cell division inhibitor SulA in Escherichia coli. J Bacteriol, 181(12), 3674–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y, Moriya S, & Ogasawara N (2003). Identification of a protein, YneA, responsible for cell division suppression during the SOS response in Bacillus subtilis. Mol Microbiol, 47(4), 1113–1122. [DOI] [PubMed] [Google Scholar]
- Kreuzer KN (2013). DNA damage responses in prokaryotes: regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb Perspect Biol, 5(11), a012674. doi: 10.1101/cshperspect.a012674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, & Sonnhammer EL (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol, 305(3), 567–580. doi: 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- Kross J, Henner WD, Hecht SM, & Haseltine WA (1982). Specificity of deoxyribonucleic acid cleavage by bleomycin, phleomycin, and tallysomycin. Biochemistry, 21(18), 4310–4318. [DOI] [PubMed] [Google Scholar]
- Lenhart JS, Schroeder JW, Walsh BW, & Simmons LA (2012). DNA repair and genome maintenance in Bacillus subtilis. Microbiol Mol Biol Rev, 76(3), 530–564. doi: 10.1128/mmbr.05020-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LK, Harlow GR, Gregg-Jolly LA, & Mount DW (1994). Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J Mol Biol, 241(4), 507–523. doi: 10.1006/jmbi.1994.1528 [DOI] [PubMed] [Google Scholar]
- Link AJ, & LaBaer J (2011). Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb Protoc, 2011(8), 993–994. doi: 10.1101/pdb.prot5651 [DOI] [PubMed] [Google Scholar]
- Little JW, & Mount DW (1982). The SOS regulatory system of Escherichia coli. Cell, 29(1), 11–22. [DOI] [PubMed] [Google Scholar]
- Little JW, Mount DW, & Yanisch-Perron CR (1981). Purified lexA protein is a repressor of the recA and lexA genes. Proc Natl Acad Sci U S A, 78(7), 4199–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusawa S, & Gottesman S (1983). Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci U S A, 80(2), 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo AH, & Burkholder WF (2010). YneA, an SOS-induced inhibitor of cell division in Bacillus subtilis, is regulated posttranslationally and requires the transmembrane region for activity. J Bacteriol, 192(12), 3159–3173. doi: 10.1128/jb.00027-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Hopkins AC, & Laub MT (2011). A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev, 25(12), 1328–1343. doi: 10.1101/gad.2038911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Kambara TK, Perchuk BS, & Laub MT (2014). A DNA damage-induced, SOS-independent checkpoint regulates cell division in Caulobacter crescentus. PLoS Biol, 12(10), e1001977. doi: 10.1371/journal.pbio.1001977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimatsu K, & Kowalczykowski SC (2003). RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell, 11(5), 1337–1347. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Cao C, & Lutkenhaus J (1998). Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli. Proc Natl Acad Sci U S A, 95(6), 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, & Schneewind O (1994). Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol Microbiol, 14(1), 115–121. [DOI] [PubMed] [Google Scholar]
- Noll DM, Mason TM, & Miller PS (2006). Formation and repair of interstrand cross-links in DNA. Chem Rev, 106(2), 277–301. doi: 10.1021/cr040478b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H, Teramoto H, Inui M, & Yukawa H (2008). DivS, a novel SOS-inducible cell-division suppressor in Corynebacterium glutamicum. Mol Microbiol, 67(3), 597–608. doi: 10.1111/j.1365-2958.2007.06069.x [DOI] [PubMed] [Google Scholar]
- Reiter H, Milewskiy M, & Kelley P (1972). Mode of action of phleomycin on Bacillus subtilis. J Bacteriol, 111(2), 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A (1996). DNA excision repair. Annu Rev Biochem, 65, 43–81. doi: 10.1146/annurev.bi.65.070196.000355 [DOI] [PubMed] [Google Scholar]
- Scheffers DJ, & Errington J (2004). PBP1 is a component of the Bacillus subtilis cell division machinery. J Bacteriol, 186(15), 5153–5156. doi: 10.1128/jb.186.15.5153-5156.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong IS, Oh JY, Yoo SJ, Seol JH, & Chung CH (1999). ATP-dependent degradation of SulA, a cell division inhibitor, by the HslVU protease in Escherichia coli. FEBS Lett, 456(1), 211–214. [DOI] [PubMed] [Google Scholar]
- Slilaty SN, & Little JW (1987). Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc Natl Acad Sci U S A, 84(12), 3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DF (2004). Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones, 9(2), 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonezaki S, Ishii Y, Okita K, Sugino T, Kondo A, & Kato Y (1995). Overproduction and purification of SulA fusion protein in Escherichia coli and its degradation by Lon protease in vitro. Appl Microbiol Biotechnol, 43(2), 304–309. [DOI] [PubMed] [Google Scholar]
- Tiyanont K, Doan T, Lazarus MB, Fang X, Rudner DZ, & Walker S (2006). Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc Natl Acad Sci U S A, 103(29), 11033–11038. doi: 10.1073/pnas.0600829103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Bolhuis A, Jongbloed JD, Bron S, & van Dijl JM (2000). Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev, 64(3), 515–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusca D, Scott S, Thompson C, & Bramhill D (1998). Bacterial SOS checkpoint protein SulA inhibits polymerization of purified FtsZ cell division protein. J Bacteriol, 180(15), 3946–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JD, & Levin PA (2009). Metabolism, cell growth and the bacterial cell cycle. Nat Rev Microbiol, 7(11), 822–827. doi: 10.1038/nrmicro2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall CS, & Levin PA (2017). Bacterial Cell Size: Multifactorial and Multifaceted. Annu Rev Microbiol, 71, 499–517. doi: 10.1146/annurev-micro-090816-093803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MJ, Carlson PE, Janes BK, & Hanna PC (2012). Membrane topology of the Bacillus anthracis GerH germinant receptor proteins. J Bacteriol, 194(6), 1369–1377. doi: 10.1128/jb.06538-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, & Errington J (1997). Septal localization of the SpoIIIE chromosome partitioning protein in Bacillus subtilis. Embo j, 16(8), 2161–2169. doi: 10.1093/emboj/16.8.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WF, Zhou Y, & Gottesman S (1999). Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J Bacteriol, 181(12), 3681–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman P, Perkins JB, & Losick R (1984). Construction of a cloning site near one end of TN917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-bourne ERM gene. Plasmid, 12(1), 1–9. doi: 10.1016/0147-619x(84)90061-1 [DOI] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Brinkman FS (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26(13), 1608–1615. doi: 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.