

The integrated stress response reduces the cytotoxic effect of inflammation on oligodendrocytes. Chen et al. show that the small molecule Sephin1 enhances the integrated stress response, protecting oligodendrocytes against CNS inflammation and delaying disease progression in a mouse model of multiple sclerosis. Sephin1 and interferon-beta provide additive therapeutic benefits.
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, Sephin1, oligodendrocytes, integrated stress response
Abstract
Multiple sclerosis is a chronic autoimmune demyelinating disorder of the CNS. Immune-mediated oligodendrocyte cell loss contributes to multiple sclerosis pathogenesis, such that oligodendrocyte-protective strategies represent a promising therapeutic approach. The integrated stress response, which is an innate cellular protective signalling pathway, reduces the cytotoxic impact of inflammation on oligodendrocytes. This response is initiated by phosphorylation of eIF2α to diminish global protein translation and selectively allow for the synthesis of protective proteins. The integrated stress response is terminated by dephosphorylation of eIF2α. The small molecule Sephin1 inhibits eIF2α dephosphorylation, thereby prolonging the protective response. Herein, we tested the effectiveness of Sephin1 in shielding oligodendrocytes against inflammatory stress. We confirmed that Sephin1 prolonged eIF2α phosphorylation in stressed primary oligodendrocyte cultures. Moreover, by using a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis, we demonstrated that Sephin1 delayed the onset of clinical symptoms, which correlated with a prolonged integrated stress response, reduced oligodendrocyte and axon loss, as well as diminished T cell presence in the CNS. Sephin1 is reportedly a selective inhibitor of GADD34 (PPP1R15A), which is a stress-induced regulatory subunit of protein phosphatase 1 complex that dephosphorylates eIF2α. Consistent with this possibility, GADD34 mutant mice presented with a similar ameliorated experimental autoimmune encephalomyelitis phenotype as Sephin1-treated mice, and Sephin1 did not provide additional therapeutic benefit to the GADD34 mutant animals. Results presented from the adoptive transfer of encephalitogenic T cells between wild-type and GADD34 mutant mice further indicate that the beneficial effects of Sephin1 are mediated through a direct protective effect on the CNS. Of particular therapeutic relevance, Sephin1 provided additive therapeutic benefit when combined with the first line multiple sclerosis drug, interferon β. Together, our results suggest that a neuroprotective treatment based on the enhancement of the integrated stress response would likely have significant therapeutic value for multiple sclerosis patients.
Introduction
Multiple sclerosis is a chronic autoimmune neurological disease most often diagnosed in young adults (Frohman et al., 2006; Reich et al., 2018). The most common form of multiple sclerosis initially manifests with relapsing-remitting symptoms before progressing to permanent neurological damage, resulting in motor, sensory and cognitive deficits (Schaeffer et al., 2015). While the aetiology of multiple sclerosis is unknown, its pathological hallmarks include immune-mediated destruction of oligodendrocytes, the myelin-producing cells of the CNS that are highly sensitive to cellular homeostatic changes, subsequent demyelination and axonal degeneration (Frohman et al., 2006; Reich et al., 2018).
Multiple sclerosis is regarded as an immune-mediated inflammatory demyelinating disorder, and therefore considerable effort has focused on the therapeutic potential of regulators of inflammation and autoimmunity (Yadav et al., 2015). Current disease-modifying treatments, such as IFN-β, glatiramer acetate, natalizumab and terifunomide, mainly target the immune system (Cohen et al., 2001; Simpson et al., 2003; O’Connor et al., 2011; Delbue et al., 2016). Although these treatments have moderate effects on relapses, the impact of these drugs on disease progression is unknown (Hart and Bainbridge, 2016). Previous studies have demonstrated that reducing oligodendrocyte apoptosis leads to reduced demyelination and axon damage, thereby ameliorating disease severity of experimental autoimmune encephalomyelitis (EAE), a multiple sclerosis mouse model (Hisahara et al., 2000; Mc Guire et al., 2010). Therefore, it is of considerable therapeutic interest to develop alternative approaches capable of protecting oligodendrocytes and reducing demyelination that contribute to disease progression (Stone and Lin, 2015; Way et al., 2015).
We have previously demonstrated that the integrated stress response (ISR) is triggered in oligodendrocytes by CNS inflammation (Lin et al., 2005, 2007). The ISR is a cellular protective response that is activated by a variety of cytotoxic insults such as reactive nitrogen oxide species, glutamate excitotoxicity, and endoplasmic reticulum stress, all of which have been implicated in multiple sclerosis pathogenesis (Mháille et al., 2008; Cunnea et al., 2011; Way and Popko, 2016). ISR activation leads to the phosphorylation of the translation initiation factor eIF2α, which results in rapid attenuation of global protein synthesis to conserve cellular resources, while allowing the activation of a cytoprotective response coordinated by the transcription factor ATF4 (Hetz et al., 2011; Walter and Ron, 2011; Wang and Kaufman, 2016). As part of a tight feedback loop, ATF4 induces C/EBP-homologous protein (CHOP, encoded by DDIT3) expression, which in turn promotes expression of GADD34 (encoded by PPP1R15A), a cofactor of the protein phosphatase 1 catalytic subunit (PP1c). The GADD34-PP1c complex dephosphorylates phosphorylated eukaryotic translation initiation factor 2 alpha (p-eIF2α), ensuring re-initiation of translation (Walter and Ron, 2011). Importantly, when the cellular stress cannot be remedied, the accumulation of CHOP leads to apoptosis (Tabas and Ron, 2011; Li et al., 2014).
In genetic studies, the inactivation of the regulatory subunit of the p-eIF2α phosphatase GADD34 results in a prolonged ISR due to the delayed dephosphorylation of p-eIF2α (Kojima et al., 2003; Lin et al., 2008). Importantly, GADD34 mutant mice display a significant reduction in oligodendrocyte loss and demyelination in response to inflammation (Lin et al., 2008). The success of these studies attests to the potential of an enhanced ISR to confer increased oligodendrocyte and myelin protection against inflammation.
Guanabenz, an anti-hypertensive drug approved by the US Food and Drug Administration, has been shown to enhance the ISR by inhibiting the GADD34-PP1c-mediated dephosphorylation of p-eIF2α by GADD34-PP1c (Tsaytler et al., 2011). Our laboratory recently reported that treatment of EAE in mice with guanabenz delayed disease onset and dampened clinical symptoms: these effects correlated with an increase in p-eIF2α levels in oligodendrocytes and oligodendrocyte protection (Way et al., 2015). These promising results have served as the basis for a multiple sclerosis clinical trial (ClinicalTrials.Gov: NCT02423083) at the National Institutes of Health. Unfortunately, guanabenz also functions as an α2-adrenergic agonist, a feature that has been shown to cause drowsiness, lethargy and even coma upon overdose (Holmes et al., 1983). Encouragingly, a guanabenz derivative, Sephin1, was recently shown to retain GADD34-PP1c-specific inhibition activity without measurable α2-adrenergic side effects in cells or in vivo (Das et al., 2015). Here we examined the effectiveness of Sephin1 in protecting oligodendrocytes and myelin against neuroinflammation by using a chronic EAE mouse model of immune-mediated CNS demyelination. We found that Sephin1 treatment of chronic EAE mice significantly delays disease onset, which correlates with diminished oligodendrocyte, axon and myelin loss, as well as a prolonged ISR. In addition, Sephin1 reduces the number of inflammatory CD3+ T cells and inflammatory cytokine production within the CNS without affecting the peripheral immune response. Furthermore, we provide evidence using the adoptive transfer model of EAE that the beneficial effects of Sephin1 are likely due to protection of the CNS and not attributed to the infiltrating T cells. Moreover, the combination of Sephin1 and IFN-β provides additive therapeutic benefit on EAE. These data demonstrate that Sephin1 has promising therapeutic potential in the treatment of multiple sclerosis by providing protection to oligodendrocytes and myelin.
Materials and methods
Animal study
All mice used in this study were housed under pathogen-free conditions at controlled temperatures and relative humidity with a 12/12-h light/dark cycle and free access to pelleted food and water. All animal experiments were designed in accordance with ARRIVE guidelines and conducted in compliance with The University of Chicago’s Animal Care and Use Committee guidelines. For statistical validity, we used up to 12 mice for the clinical score assessment, and three to five mice for molecular biology studies and histology. All assessments were done by researchers blinded to the experimental groups. The mice were randomly assigned to the different experimental groups. Sephin1 (free base) was purchased from Apexbio. Stock solution (24 mg/ml) of Sephin1 was dissolved in DMSO (dimethyl sulphoxide, Sigma-Aldrich) and stored at −20°C. Final solutions were prepared in sterile 0.9% NaCl (DMSO concentration: 1%) for animal treatment and in culture media (50 µM) for in vitro treatment. Recombinant mouse IFN-β (2.37 × 107 units/ml) was purchased from PBL Assay Science, aliquoted and stored at −80°C. IFN-β (5000 U) in 0.9% NaCl was administered to each mouse daily.
Purification and treatment of oligodendrocyte precursor cells
Isolation and immunopanning purification of oligodendrocyte precursor cells (OPCs) was performed as previously described (Dugas and Emery, 2013). Briefly, rat brain cortices from 6–7 days old pups were diced and digested with papain (Worthington) at 37°C. Cells were triturated and resuspended in a panning buffer, and then incubated at room temperature sequentially on plates coated with primary antibodies against Ran-2 and GalC for negative selection and O4 for positive selection. Ran-2 (rat neural antigen-2) is a cell surface protein of astrocytes. GalC (galactosylceramide), is a sphingolipid of myelin expressed early in the maturation of oligodendrocytes. O4 is expressed in pro-oligodendrocytes. Immunopanning sequentially with anti-Ran-2 and anti-GalC antibodies is done to remove astrocytes and oligodendrocytes from the cell mixture (negative selection), while anti-O4 is used to purify the OPCs by positive selection. OPCs were released from the final panning dish with trypsin and seeded on poly-d-lysine-coated flasks in growth medium to proliferate. For the treatment experiments, OPCs were split and plated in differentiation media overnight. Plates were randomly designated for treatment regimens: rat recombinant IFN-γ (R&D systems 200 U/ml) and thapsigargin (Sigma, 200 nM) with or without Sephin1 (50 µM).
EAE immunization and treatment
EAE was induced in 7-week-old female C57BL/6J mice (Jackson Laboratory) or GADD34 mutant (Marciniak et al., 2004) and wild-type mice by subcutaneous flank administration of 200 µg MOG35-55 peptide emulsified with complete Freund’s adjuvant (CFA) (MOG35-55/CFA) (BD Biosciences) containing inactive Mycobacterium tuberculosis H37Ra (BD Biosciences). Mice also received intraperitoneal injections of 200 ng pertussis toxin (List Biological Laboratories) in sterile phosphate-buffered saline (PBS) immediately after MOG administration and 48 h later. CFA control mice were similarly induced but lacked MOG. Mice were injected intraperitoneally with Sephin1 or the equivalent amount of vehicle (1% DMSO in 0.9% NaCl) daily beginning post-immunization Day (PID) 7. Mouse groups were randomized during the treatment. Mice were blindly scored daily for signs of EAE as follows: 0 = healthy, 1 = flaccid tail, 2 = ataxia and/or paresis, 3 = paralysis of hindlimbs and/or paresis of forelimbs, 4 = tetraparalysis, 5 = moribund or death.
Adoptive transfer of EAE induction
For the adoptive transfer EAE experiments, lymph nodes from active EAE mice were dissected on PID8. Isolated cells from lymph nodes were reactivated in the presence of MOG35-55 (20 µg/ml) and IL-12 (10 ng/ml) in HL-1 medium. After 72 h in culture, total cells were harvested and counted. A total of 4 × 106 blast cells were transferred into each recipient female mouse via tail vein injection. All recipient mice also received intraperitoneal pertussis toxin (200 ng per injection) immediately after cell transfer and 48 h later. Mice were monitored daily for the onset of EAE.
Immunofluorescence
Anaesthetized mice were transcranially perfused with 4% paraformaldehyde in PBS. Lumber spinal cords were immediately removed, embedded in O.C.T. resin, and snap frozen. The tissue was sectioned in a series of 10 µm on a cryostat. Cryosections were treated with acetone at −20°C, then blocked with PBS containing 5% goat serum and 0.1% Triton™ X-100, and incubated overnight with the primary antibodies at 4°C. Sections were incubated with secondary antibodies for 1 h at room temperature. Primary antibodies include the following: anti-MBP (Abcam, ab24567, 1:700), anti-TPPP (Thermo Fisher Scientific, PA5-19243, 1:100), anti-p-eIF2α (Abcam, ab32157, 1:100; Thermo Fisher Scientific, MA5-15133, 1:50), anti-CD3 (Santa Cruz, sc-18843, 1:200), anti-CD11b (Bio-Rad, MCA711, 1:50), anti-NG2 (Millipore, Ab5320, 1:100), anti-neurofilament (Millipore, AB5539, 1:500), anti-GFAP (Millipore, AB5541, 1:200), anti-caspase 3 (Abcam, AB2302, 1:50), anti-Ki67 (Abcam, AB15580, 1:100), anti-PDGFR-alpha (BD Biosciences, 558774, 1:100), and anti-SOX10 (R&D, AF2864, 1:100). For myelin staining, sections were incubated with FluoroMyelin™ (Thermo Fisher Scientific, F34651, 1:300) in PBS for 20 min at room temperature. The fluorescent images were acquired under a laser scanning confocal microscope system (Leica) and quantified by ImageJ. The percentage of double-positive cells was calculated by normalizing TPPP/p-eIF2α double-positive cell numbers against TPPP-positive cell numbers.
Haematoxylin and eosin stains
Fixed frozen sections were prepared as above and stained with haematoxylin and eosin using standard methods (Kluver and Barrera, 1953) to visualize leucocyte infiltration. Stained slides were imaged using a whole slide scanner (Perkin Elmer). The inflammatory scores were determined according to the size of cellular infiltration: 0 = no inflammation; 1 = infiltration only in the perivascular areas and meninges; 2 = infiltration in less than one-third of the white matter; 3 = more than one-third of the white matter; 4 = infiltration in the whole white matter (Okuda et al., 2002).
Western blot
After treatment, differentiating OPCs were rinsed twice with PBS and lysed with RIPA buffer (Sigma) containing protease inhibitors (Roche) and phosphatase inhibitors (Sigma). Anaesthetized mice were perfused with PBS. Mouse tissue was immediately isolated, snap-frozen in liquid nitrogen and homogenized in the supplemented RIPA buffer. Aliquots of 25 µg of protein extracts were separated via a 4–12% gradient gel in SDS-PAGE (Thermo Fisher Scientific) and transferred onto nitrocellulose membrane (Thermo Fisher Scientific). The blots were blocked in 5% non-fat milk in Tris-buffered saline with 1% Tween 20 (pH 8.0) for 1 h and then probed with primary antibodies at 4°C overnight. Primary antibodies include the following: anti-MBP (BioLegend, SMI-99P, 1:1000), anti-MAG (Invitrogen, 346200, 1:1000), anti-CHOP (Thermo Fisher Scientific, MA1-250, 1:500), anti-ATF4 (AVIVA, 1:500), anti-p-eIF2α (Abcam, ab32157, 1:500; Thermo Fisher Scientific, MA5-15133, 1:500), anti-T-eIF2α (Cell Signaling, 9722S, 1:500), and anti-β-actin (Sigma, A2066, 1:2000) as reference proteins to check equal loading. Following incubation with the secondary HRP-conjugated antibodies, signal detection was performed via chemiluminescence on ChemiDocTM Touch (Bio-Rad). Semiquantitative densitometric analyses of immunoblots were performed using the Image Lab software (Bio-Rad).
RNA extraction and quantitative real-time reverse transcription PCR
Total RNA was extracted from snap-frozen lumber spinal cords using Aurum™ Total RNA Fatty and Fibrous Tissue Kit (Bio-Rad). Complementary DNA was generated with iScript™ cDNA synthesis kit (Bio-Rad). Quantitative real-time reverse transcription PCR was performed on a CFX96 RT-PCR detection system (Bio-Rad) using SYBR® Green technology. Results were analysed and presented as the fold induction relative to the internal control primer for the housekeeping gene, HPRT. Quantitative real-time PCR primers (5′–3′) for mouse gene sequences were as follows: Plp1 (proteolipid protein 1)-f: CACTTACAACTTCGCCGTCCT, Plp1-r: GGGAGTTTCTATGGGAGCTCAGA; Mbp-f: TGTCACAATGTTCTTGAAGAAATGG, Mbp-r: TGTCACAATGTTCTTGAAGAAATGG; Mag-f: CTGCTCTGTGGGGCTGACAG, Mag-r: AGGTACAGGCTCTTGGCAACTG; Chop (Ddit3)-f: AGGTGCCCCCAATTTCATCT, Chop-r: TGAGTCCCTGCCTTTCACCTTGGAGA; Atf4-f: TGGATGATGGCTTGGCCAGTG, Atf4-r: GAGCTCATCTGGCATGGTTTC; Inos (Nos2)-f: GCTGGGCTGTACAAACCTTCC, Inos-r: TTGAGGTCTAAAGGCTCCGG; Tnfa (tumor necrotic factor alpha)-f: GGCAGGTTCTGTCCCTTTCA, Tnfa-r: ACCGCCTGGAGTTCTGGAA; Il (interleukin)-12-f: CTCTATGGTCAGCGTTCCAACA, Il-12-r: GGAGGTAGCGTGATTGACACAT; Il-17-f: ATGCTGTTGCTGCTGCTGAG, Il-17-r: TTTGGACACGCTGAGCTTTGAG; Ifng-f: GATATCTGGAGGAACTGGCAAAA, Ifng-r: CTTCAAAGAGTCTGAGGTAGAAAGAGATAAT; Hprt1-f: TCAGACCGCTTTTTGCCGCGA, Hprt1-r: ATCGCTAATCACGACGCTGGGAC.
Ex vivo recall and flow cytometry
Total splenocytes were collected on PID12, and two sets of cultures were setup in triplicate wells per individual mouse (n = 5–7 mice per treatment group). Cells were cultured at 5 × 105 cells/well in the presence of anti-CD3 (1 µg/ml), OVA323-339, or MOG35-55 (20 µg/ml) in HL-1 medium. For the cellular proliferation cultures, at 24 h post-culture initiation, the wells were pulsed with 1 µCi of tritiated thymidine and the cultures were harvested at 72 h and tritiated thymidine incorporation detected using a TopCount® Microplate Scintillation Counter. Results are expressed as the mean CPM (counts per minute) of triplicate cultures. For cytokine analysis replicate wells were harvested on day + 3 of culture and the level of cytokine secreted determined via multiplex Luminex® LiquiChip (Millipore).
Flow cytometric analysis and additional analysis was performed by a blinded investigator on cells from individual animals. Cells were stained with two separate panels. The first analysis panel for T cell populations contained anti-CD45 (clone 30-F11), anti-CD3 (clone 145-2C11), anti-CD4 (clone RM4-5), anti-FoxP3 (clone FJK-16 s), anti-Nrp-1 (Polyclonal Goat IgG; R&D Systems), anti-CD25 (clone PC61), and anti-CD44 (clone IM7). The second analysis panel for antigen presenting cell populations contained anti-CD3 (clone 145-2C11), anti-CD11b (clone M1/70), anti-CD11c (clone HL3), anti-CD19 (clone ID3), anti-PD-L1 (clone 1-111A), and anti-B7, i.e. anti-CD80 (clone 16-10A1) and anti-CD86 (clone GL1) (BD Bioscience). Viable cells (106 cells per tube) were analysed per individual sample using a BD Canto II cytometer (Becton Dickinson), and the data were analysed using BD FACSCanto II software (BD Bioscience).
Statistical analysis
All data were presented as mean ± SEM (standard error of mean). Multiple comparisons were carried out by one-way ANOVA followed by Tukey’s post hoc test; single comparisons were evaluated by unpaired t-test. Differences were considered statistically significant when P < 0.05.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Sephin1 prolongs eIF2α phosphorylation in oligodendrocytes under stress
It has been shown that Sephin1 prolongs eIF2α phosphorylation and delays translation recovery in HeLa cells under stress (Das et al., 2015). We therefore examined p-eIF2α levels in purified primary differentiating OPCs continuously treated with the endoplasmic reticulum (ER) stress inducer thapsigargin or the T cell-derived cytokine IFN-γ in the presence or absence of Sephin1 (Fig. 1A–D). Levels of p-eIF2α increased over time with the treatment of thapsigargin (Fig. 1A and C), which disrupts calcium homeostasis and thereby induces ER stress (Nakagawa et al., 2000). At 2 and 8 h, p-eIF2α levels started to decrease in differentiating OPCs treated only with thapsigargin but were consistently higher in cells treated with both thapsigargin and Sephin1 (P < 0.05, P < 0.001) (Fig. 1A and C). We have previously reported that IFN-γ can induce ER stress in differentiating OPCs (Lin et al., 2005). As expected, we observed increased levels of p-eIF2α in differentiating OPCs treated with IFN-γ, particularly at 4 and 12 h (Fig. 1B and D). Cells treated with both IFN-γ and Sephin1 maintained a higher level of p-eIF2α than those treated with IFN-γ alone (P < 0.01, P < 0.05) (Fig. 1B and D). Thus, Sephin1 prolongs the benefit of eIF2α phosphorylation in stressed oligodendrocytes in vitro.
Figure 1.

Sephin1 prolongs the integrated stress response in differentiating rat OPCs under ER stress and delays EAE disease onset. (A) Western blot of differentiating OPCs treated continuously with thapsigargin (Thap) alone or Thap+5 µM Sephin1. (B) Western blot of differentiating OPCs treated continuously with IFN-γ alone or IFN-γ+5 µM Sephin1. Horizontally cropped blots are displayed in the main figures. The scanned full blots are presented in Supplementary Figs 12 and 13, respectively. Immunoblot analysis is probed with p-eIF2α (phosphorylated), T-eIF2α (total) and β-actin is presented as a loading control. (C and D) Densitometry histograms of p-eIF2α after normalization to T-eIF2α with respect to blots in A and B. *P < 0.05, **P < 0.01, ***P < 0.001, significance based on unpaired t-test. Data represent an average of four biological isolations and three technique replicates for each isolation (mean ± SEM). (E) Clinical scores of C57BL/6J female mice immunized with MOG35-55/CFA to induce chronic EAE, treated with vehicle (n = 9), 4 mg/kg Sephin1 (n = 8) or 8 mg/kg Sephin1 (n = 8). (F) Histogram of average onset of disease (onset defined at score of 1), average peak of disease and average peak score of all treatment groups. Data are represented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, significance based on ANOVA and t-test.
Sephin1 treatment delays the onset of EAE
The EAE mouse model has been widely used to unravel the consequences of autoimmune CNS inflammation and to test potential treatments for multiple sclerosis (Ransohoff, 2012). Briefly, EAE was induced by injecting MOG35-55/CFA in C57BL/6 female mice (Mendel et al., 1995; Hussien et al., 2014). Chronic EAE mice display an ascending paralysis that begins in the tail and spreads to the hind limbs and forelimbs before disease plateau. Generally, in our studies EAE clinical symptoms are first detectable around Day 11 post-EAE disease induction (PID 11). The symptoms reach a peak about a week later (PID 17), and the disease course is maintained roughly for a month. To evaluate the dose range of Sephin1 capable of affecting the severity of EAE clinical disease, the average clinical scores of mice were compared among EAE groups treated daily with 4 mg/kg or 8 mg/kg of Sephin1 beginning on PID 7 (Fig. 1E). Sephin1 treatment significantly delayed clinical disease onset with both dosages, but to a greater extent with the 8 mg/kg treatment (P < 0.0001) (Fig. 1E and F). EAE mice treated with 8 mg/kg of Sephin1 reached the peak disease roughly at PID 26, which was a week later than mice treated with vehicle (P < 0.01) (Fig. 1F). Nevertheless, the peak score of EAE mice treated with 4 mg/kg (3.8) or 8 mg/kg (3.9) of Sephin1 showed no significant difference from that of vehicle-treated EAE mice (4.1) (Fig. 1F). In a separate EAE experiment, Sephin1 treatment initiated immediately after the peak of disease was reached did not confer significant benefit in terms of disease severity or duration of paralytic symptoms (Supplementary Fig. 1). Of note, no hypotensive side effects were observed with Sephin1 treatment (Supplementary Video). Sephin1-treated mice continued to remain active while mice receiving guanabenz were torpid for >2 h. These findings demonstrate that Sephin1 treatment delays the onset, but does not diminish the severity, of the EAE disease course.
Sephin1 protects oligodendrocytes and preserves myelin and axons while modulating the integrated stress response in EAE
Given that inflammatory demyelination is an important early event in the pathogenesis of multiple sclerosis and EAE (Schaeffer et al., 2015; Yadav et al., 2015), we sought to characterize the effect of Sephin1 treatment on CNS inflammation and demyelination. Lumber spinal cords were harvested from vehicle and Sephin1-treated EAE mice at PID 17, the peak of disease in vehicle-treated mice, as well as at PID 30, when disease is at a late stage in the vehicle-treated mice and soon after peak of disease in the Sephin1-treated mice. As shown in Fig. 2A, results from haematoxylin and eosin staining showed remarkable infiltration foci in the lumber spinal cords and increased inflammatory scores from the vehicle-treated EAE mice (P < 0.0001) (Fig. 2E), but not from Sephin1-treated EAE mice at PID 17. Consistent with the inflammatory cell infiltration results, significant demyelinated regions were detected by FluoroMyelin™ staining in the lumbar spinal cords only from vehicle-treated EAE mice (P < 0.0001) (Fig. 2B and F). In contrast, at PID 30, both vehicle- and Sephin1-treated EAE samples displayed cellular infiltration, compared to CFA-only control mice (P < 0.01, P < 0.001) (Fig. 2C and E). Correspondingly, extensive damage to myelin was observed in EAE mice treated either with vehicle or Sephin1 at PID 30 (P < 0.001, P < 0.0001) (Fig. 2D and F).
Figure 2.

Sephin1 attenuates inflammatory infiltration and demyelination at EAE PID 17. Histological analysis of lumber spinal cord sections of PID 17 and PID 30 mice immunized with CFA only or MOG35-55/CFA (EAE), then treated daily with vehicle or Sephin1 beginning from PID7. (A and C) Haematoxylin and eosin (H&E) staining. (B and D) FluoroMyelin™ staining. Scale bars = 200 μm. Higher magnification images are taken from the boxed areas. Scale bar = 50 μm. White dashed line demonstrates the lesion border. (E) Quantification of inflammatory scores measured from haematoxylin and eosin staining. (F) Average percentage of demyelinated areas/white matter areas measured from FluoroMyelin™ staining. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, significance based on ANOVA. Data represent an average of 4–6 mice per group (mean ± SEM).
Sephin1 prolongs the ISR by inhibiting the GADD34-PP1c complex-mediated dephosphorylation of p-eIF2α (Tsaytler et al., 2011; Das et al., 2015). Therefore, we tested whether an enhancement of the ISR by Sephin1 renders oligodendrocytes resistant to inflammatory attack and prevents axon damage. Serial sections of lumber spinal cords were co-stained with p-eIF2α and TPPP, a mature oligodendrocyte marker (Skjoerringe et al., 2006). Axon loss was assessed by neurofilament immunostaining. Compared to CFA-only control mice, vehicle-treated EAE mice lost about 50% of TPPP+ oligodendrocytes (P < 0.01) and neurofilament-positive axons (P < 0.01), while Sephin1-treated EAE mice did not display a significant reduction in the numbers of either mature oligodendrocytes or axons at PID17 (Figs 3A–C, G, 4A and C). To investigate whether Sephin1 treatment affects the death or proliferation of oligodendrocytes, we stained the spinal cord sections with the apoptotic marker caspase 3 and the proliferation marker Ki67. At PID 17, the total number of caspase 3-positive apoptotic cells and the percentage of caspase3-positive mature oligodendrocytes in the lumber spinal cord section were significantly higher in vehicle-treated EAE mice than in Sephin1-treated EAE mice (Supplementary Fig. 2A, C and D). At PID 30, both groups presented with a similar percentage of caspase 3-positive apoptotic oligodendrocytes (Supplementary Fig. 2B and D). These data indicate that the apoptotic death of oligodendrocytes is delayed in the Sephin1-treated EAE mice. In addition, we observed the total number of Ki67-positive cells was significantly higher in vehicle-treated EAE mice than in the Sephin1 treated mice at PID 17, but no difference was detected at PID 30 (Supplementary Fig. 3C). When combined with antibodies to SOX10 (oligodendrocyte lineage marker) or PDGFR-alpha (OPC marker), the number of Ki67-positive, Sox10-positive cells or Ki67-positive, PDGFR-alpha-positive cells was also significantly higher in the vehicle-treated mice than the Sephin1-treated mice at PID 17, but no difference was found at PID 30 (Supplementary Fig. 3B–G). In contrast, there were very few Ki67-positive mature oligodendrocytes (TPPP-positive) in any of the groups (Supplementary Fig. 3A), which was as expected since there is little evidence that these cells have the capacity to proliferate (Keirstead and Blakemore, 1997; Carroll et al., 1998). The higher level of oligodendrocyte lineage cell proliferation in the vehicle-treated mice is likely in response to the increased oligodendrocyte cell death that occurs in these animals (Hughes et al., 2013).
Figure 3.

Sephin1 ameliorates oligodendrocyte loss at EAE PID 17, which is correlated with prolonged eIF2α phosphorylation. Lumber spinal cord sections were taken from PID 17 and PID 30 mice with CFA only or actively-induced chronic EAE treated daily with vehicle or Sephin1 beginning from PID 7. (A–F) Immunofluorescent staining for TPPP (a mature oligodendrocyte marker, green) and p-eIF2α (an ISR marker, red). Scale bar = 50 μm. Higher magnification images are taken from the boxed areas. Scale bar = 10 μm. Arrow heads point to the cells with double positive staining. (G) Quantification of cells positive for TPPP in the lesion areas. (H) The percentage of TPPP positive oligodendrocytes that are p-eIF2α positive in the whole spinal cord cross-sections. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, significance based on ANOVA. Data represent an average of 3–4 mice per group (mean ± SEM).
Figure 4.
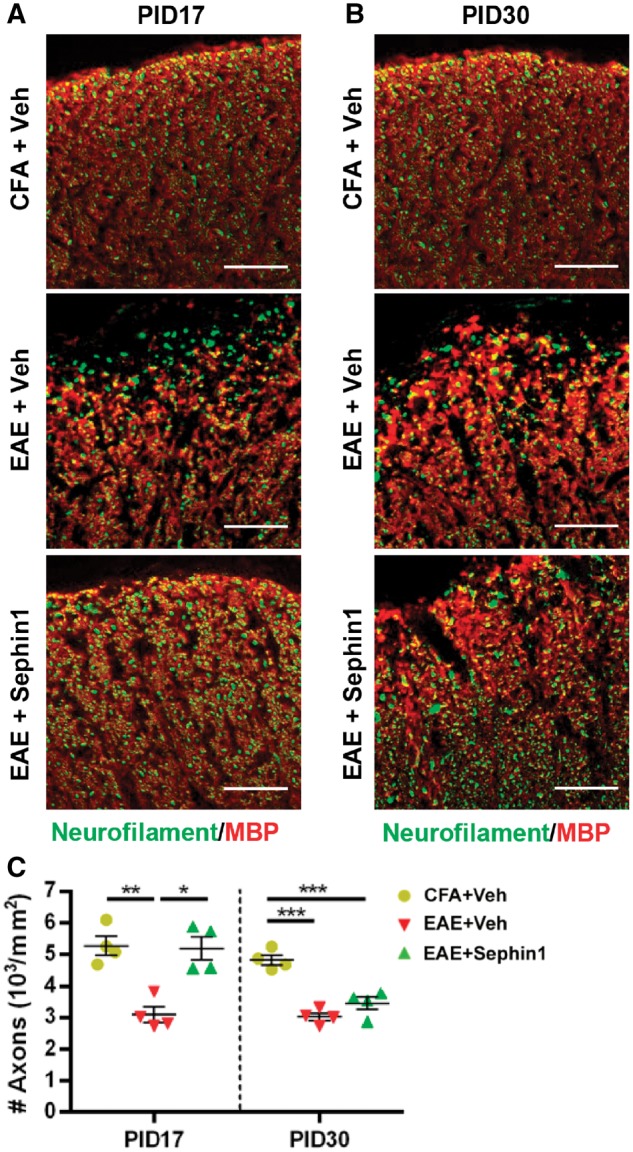
Sephin1 reduces axon loss at PID 17 in EAE. Lumber spinal cord sections were taken from PID 17 and PID 30 mice with CFA only or actively-induced chronic EAE treated daily with vehicle or Sephin1 beginning from PID 7. (A and B) Immunofluorescent staining for neurofilament (an axon marker, green) and MBP (red). Scale bar = 50 μm. Individual fluorescent channels are provided in Supplementary Fig. 10. (C) Quantification of axons positive for neurofilament in the whole spinal cord sections. *P < 0.05, **P < 0.01, ***P < 0.001, significance based on ANOVA. Data represent an average of four mice per group (mean ± SEM).
Furthermore, co-localization of TPPP with p-eIF2α showed that vehicle-treated EAE mice (82.2%, P < 0.0001) at PID 17 showed a significantly higher percentage of p-eIF2α positive mature oligodendrocytes than CFA mice (66.5%) (Fig. 3A, B and H). This result is in accordance with the evidence that several components of the ISR are upregulated in multiple sclerosis and EAE lesions (Stone and Lin, 2015; Way and Popko, 2016). Notably, we observed a significant increase in p-eIF2α positive mature oligodendrocytes in Sephin1-treated EAE mice (74.9%) compared to CFA control mice (P < 0.05), even though CNS lesions and inflammation were absent at the PID 17 time point (Figs 2A and 3A, C and H). At PID 17, both vehicle- and Sephin1-treated EAE mice also exhibited significantly higher percentages of p-eIF2α positive astrocytes labelled by GFAP (Supplementary Fig. 4A–C), indicating that Sephin1 does not solely target the ISR in oligodendrocytes. We also examined the presence of p-eIF2α in CD3-positive infiltrating T cells, but there was no significant difference in the percentage of p-eIF2α-positive T cells between vehicle- and Sephin1-treated mice at PID 30 (Supplementary Fig. 4D and E). More than 80% of mature oligodendrocytes were p-eIF2α positive in both vehicle- and Sephin1-treated groups at PID 30, when EAE clinical disease had progressed in both groups (Fig. 3D–F and H). At this stage, there was no significant reduction in the number of mature oligodendrocytes in either vehicle- or Sephin1-treated EAE mice, likely because the NG2-positive OPCs replenished the mature oligodendrocytes (Supplementary Fig. 5). OPCs migrate to the demyelinated lesion and subsequently differentiate to mature oligodendrocytes to remyelinate the damaged axons (Hughes et al., 2013). Nevertheless, this process is often incomplete in multiple sclerosis (Fancy et al., 2010). Of note, Sephin1-treated EAE mice at PID 30 displayed a larger number of OPCs than vehicle-treated EAE mice at PID 17 (P < 0.01), suggesting that Sephin1 may promote OPC survival as well (Supplementary Fig. 5). Despite the evidence that precursor cells responded well in EAE treated with Sephin1, axon damage accumulated at the end of the disease course (P < 0.001) (Fig. 4B and C).
Activation of the ISR pathway was further confirmed by immunoblotting of lumber spinal cord lysates from EAE mice. Compared to CFA mice, the expression of p-eIF2α was significantly increased in vehicle-treated EAE samples (P < 0.05) at PID 17 (Fig. 5A and B). Although there was an increasing trend, we did not observe significant changes in p-eIF2α levels in Sephin1-treated EAE tissues compared to CFA controls. Moreover, in vehicle-treated EAE samples the ISR markers CHOP and ATF4 were highly expressed and MBP and MAG levels were notably reduced. Compared to vehicle-treated EAE samples, Sephin1 treatment not only significantly reduced the expression of CHOP/DDIT3 and ATF4 at both protein and mRNA levels, but also increased the levels of myelin proteins (Fig. 5A–C, Supplementary Fig. 6A and B). This result is consistent with a previous study, which showed that Sephin1 treatment attenuated the expression of pro-apoptotic CHOP, an indicator of cell loss (Das et al., 2015). These observations suggest that Sephin1 treatment preserves oligodendrocyte viability in the early stage of EAE by modulating the ISR pathway.
Figure 5.

Sephin1 reduces demyelination at EAE PID 17 and modulates ISR activity. Lysates of lumber spinal cords were analysed from PID 17 and PID 30 CFA or EAE mice treated with vehicle or Sephin1 beginning from PID 7. (A and B) Immunoblot analysis of MBP and MAG, and the key ISR components CHOP, ATF4, and p-eIF2α. T- eIF2α and β-actin is presented as a loading control. Images cropped horizontally are displayed in A and images cropped horizontally and vertically displayed in B. The scanned full blots are presented in Supplementary Figs 14 and 15, respectively. (C and D) Densitometry histograms of the ISR component proteins after normalization to β-actin or T-eIF2α, respectively. (E and F) Densitometry histograms of myelin proteins after normalization to β-actin. *P < 0.05, **P < 0.01, significance based on ANOVA. Data represent an average of 3–4 mice per group (mean ± SEM).
At PID 30, both vehicle- and Sephin1-treated EAE groups displayed a significant reduction in the levels of myelin proteins compared to CFA controls (Fig. 5D, F and Supplementary Fig. 6D). Despite higher percentages of mature oligodendrocytes with positive p-eIF2α staining (Fig. 3D–H), no obvious difference in the expression of p-eIF2α was found between groups at this stage as determined by western blots from the lumbar spinal cord lysate samples (Fig. 5D and E). This discrepancy could be due to the loss of oligodendrocytes at this later time point. Interestingly, we noticed that the difference in CHOP and ATF4 expression between vehicle- and Sephin1-treated EAE tissues was abrogated (Fig. 5D and E). This can be explained by changes in the mRNA for these specific transcripts. Results from quantitative real-time reverse transcription PCR indicated that Atf4 mRNA was downregulated in vehicle-treated EAE groups (P < 0.05), while Chop/Ddit3 message levels were significantly increased in Sephin1-treated EAE mice (P < 0.05) (Supplementary Fig. 6C).
Sephin1 alters the immune cell response and cytokine profiles in the CNS of EAE mice
CNS inflammation in EAE is characterized by an increase in T cell numbers and activation of microglia/macrophages (Pierson et al., 2012). Haematoxylin and eosin staining showed that Sephin1 treatment reduced the inflammatory cell infiltration into the spinal cords at PID 17, but not at PID 30 (Fig. 2). To examine the infiltration of T cells and activation of microglia/macrophages, we immunostained the lumber spinal cords for CD3 (T cells) and CD11b (microglia and macrophages) (Fig. 6). We detected very few CD3+ T cells in the CFA mouse spinal cords at both PID 17 and PID 30 (Fig. 6A). Meanwhile, there were ∼330/mm2 CD3+ cells in the vehicle-treated EAE mice at PID 17, but no detectable cells in Sephin1-treated groups at the same time point (Fig. 6A and B). Similarly, EAE mice treated with vehicle but not Sephin1 exhibited a significant increase in the number of CD11b+ cells (P < 0.0001) at PID 17 compared to CFA mice (Fig. 6C and D). While ISR-mediated protection of oligodendrocytes via Sephin1 may indirectly limit inflammatory cellular infiltration into the CNS by reducing myelin antigen presentation, it is also possible that Sephin1 directly affects the activation and/or trafficking of immune cells. Nevertheless, Sephin1-treated EAE mice at PID 30 have a significantly higher number of CD3+ cells than CFA (P < 0.0001) or vehicle-treated EAE mice (P < 0.01) at this time point (Fig. 6A and C), demonstrating robust immune cell infiltration in the presence of Sephin1.
Figure 6.

Sephin1 reduces the number of T cells and activated microglia and suppresses cytokine production in the CNS at EAE PID 17 but not PID 30. All analyses conducted using lumber spinal cords of PID 17 or PID 30 mice immunized with CFA only or MOG35-55/CFA (EAE), then treated with vehicle or Sephin1 daily from PID 7. (A) Immunofluorescent staining for CD3 (a T cell marker, green) and MBP (red). (B) Immunofluorescent staining for CD11b (a microglial marker, green) and MBP (red). Scale bar = 100 μm. (C and D) Quantification of cells positive for CD3 or CD11b, respectively. Real-time reverse transcription PCR analysis in E–H using lysates of lumber spinal cords. (E and G) Relative mRNA levels at PID 17 and PID 30 of Th1 and Th17 cytokines IFN-γ and IL-17, respectively. (F and H) Relative mRNA levels of macrophage/microglia-derived cytokines Tnfa, Nos2 and Il12 (TNF-α, iNOS and IL-12). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, significance based on ANOVA. Hash symbol represents significance when comparing values from PID 17 and PID 30 of the same treatment group. Data represent an average of 3–4 mice per group (mean ± SEM).
In addition, we assessed the effect of Sephin1treatment on the cytokine response of EAE mice at PIDs 17 and 30. At PID 17, mRNA levels of Ifng (IFN-γ), Tnfa (TNF-α) and Nos2 (iNOS) were significantly higher in vehicle-treated EAE mice than in CFA control mice or Sephin1-treated EAE mice (Fig. 6E and F). Interestingly, no significant difference was detected in T cell cytokine IL-17 levels among the three groups (Fig. 6E and F). Generally, the levels of cytokines in vehicle-treated EAE mice were reduced by PID 30 and were not significantly different from those in Sephin1-treated EAE mice (Fig. 6G and H). Taken together, these findings show that Sephin1 treatment reduces the presence of pro-inflammatory cytokines in the CNS during the earlier phases of EAE, which correlates with its effect on T cell infiltration.
GADD34 deletion delays the onset of EAE and abolishes the protection of Sephin1
We previously reported that the genetic inactivation of GADD34 diminished oligodendrocyte loss and hypomyelination in mice that ectopically expressed IFN-γ in the CNS (Lin et al., 2008). Given that Sephin1 selectively inhibits the GADD34-PP1c complex under ER stress (Das et al., 2015; Carrara et al., 2017), we sought to determine whether GADD34 deficiency provides protection from EAE in a similar manner as Sephin1 treatment. EAE was induced in both GADD34 mutant mice (Marciniak et al., 2004) and wild-type littermates. Disease progression of GADD34 mutant mice was significantly delayed compared to wild-type mice (Fig. 7A), which resembled what we observed in Sephin1-treated EAE mice (Fig. 1E). Similar to Sephin1, GADD34 inactivation reduced CD3+ T cell infiltration into lumber spinal cords at PID 17 (P < 0.01) (Fig. 7B and C). We next examined the myelin integrity by FluoroMyelin™ and the survival of TPPP+ oligodendrocyte in these animals. Compared to wild-type mice, GADD34 mutant mice preserved more oligodendrocytes and myelin (P < 0.05, P < 0.01) at PID 17 (Fig. 7D, F and G). Similarly, GADD34 deficiency also reduced axon loss (P < 0.05) (Fig. 7E and H). Of note, there is no difference in the numbers of oligodendrocytes and axons between GADD34 mutants and wild-type mice immunized with CFA only. These results indicate that GADD34 inactivation recapitulates the protective effect of Sephin1 in EAE. To determine if the protective effect of Sephin1 is consistent with blocking GADD34-PP1c complex activity, we treated GADD34 mutant and wild-type EAE mice, starting from PID 7. Notably, Sephin1 treatment did not change the disease course of GADD34 mutant mice (Fig. 7I), which is in accord with the protective effects of Sephin1 in EAE being mediated by GADD34 inhibition.
Figure 7.

EAE in GADD34 mutant mice. (A) Clinical scores of GADD34 wild-type (WT) (n = 8) and GADD34 mutant (Mut) (n = 12) female mice immunized with MOG35-55/CFA to induce chronic EAE. Data are represented as mean ± SEM. *P < 0.05, ***P < 0.01, ****P < 0.0001, significance based on unpaired t-test. (B) Immunofluorescent staining for CD3 (green) and MBP (red) of lumbar spinal cord sections from PID 17. Scale bar = 100 μm. (C) Quantification of cells positive for CD3 of wild-type and mutant mice in EAE. (D) Immunofluorescent staining for FluoroMyelin™ (green) and TPPP (red). Scale bar = 100 μm. (E) Immunofluorescent staining for neurofilament (green) and MBP (red). Scale bar = 50 μm. Individual fluorescent channels are provided in Supplementary Fig. 11. (F–H) Average percentage of demyelination areas/white matter areas, the number of TPPP in the lesions and the number of neurofilament positive axons from all groups are quantified, respectively. Data represent an average of three mice per group for histology analysis (mean ± SEM). *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001, significance based on unpaired t-test and ANOVA. (I) Clinical scores of GADD34 wild-type and mutant female immunized to induce chronic EAE, treated with vehicle (wild-type: n = 8 and mutant: n = 5), or 8 mg/kg Sephin1 (wild-type: n = 7 and mutant: n = 5). Data are represented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, significance based on ANOVA. (J) Clinical scores of GADD34 wild-type and mutant female mice after adoptive transfer of MOG-specific lymph node T cells. Lymph node T cells were harvested from the GADD34 wild-type or mutant immunized 8 days previously and re-stimulated in vitro with MOG35-55 (10 ng/ml) and IL-12 (20 µg/ml) for 72 h. #P < 0.05, **P < 0.01, significance based on ANOVA. Hash symbol represents significance when comparing scores from wild-type→wild-type (n = 7) and mutant→mutant (n = 5) and asterisk represents significance when comparing scores from mutant→wild-type (n = 10) and wild-type→mutant (n = 10). Data are represented as mean ± SEM.
Sephin1 does not alter the peripheral immune response in EAE
Enhanced ISR activity has been linked to a diminished inflammatory response (Zhang and Kaufman, 2008; Hasnain et al., 2012; Cláudio et al., 2013). To determine whether Sephin1 treatment may have a direct immunomodulatory effect, we isolated the splenocytes from EAE mice at PID 12, immediately after vehicle-treated EAE mice exhibited symptoms. Compared to vehicle-treated EAE mice, Sephin1-treated mice showed no difference in the numbers of total immune cells, CD4+ T cells and B cells in the spleen (Supplementary Fig. 7A). In particular, the numbers of Tregs (regulatory T cells), activated/suppressive Tregs (Nrp-1+), and Teff cells (effector T cells) were not altered with Sephin1 treatment (Supplementary Fig. 7B). In addition, splenocytes were reactivated ex vivo in the presence of a pan T cell activator (anti-CD3), a negative control antigen (OVA323-339) or the immunizing antigen (MOG35-55). As shown in Supplementary Fig. 7C, no significant difference between treatments was found in cell proliferation, as measured by tritiated thymidine incorporation. Also, the levels of IFN-γ, IL-17, GM-CSF, IL-4, and IL-10 cytokines produced by splenocytes were not significantly different in the presence of Sephin1 (Supplementary Fig. 7D–H). These findings indicate that Sephin1 treatment has no detectable immunosuppressive effect either on the initial activation or the proliferation and cytokine production of peripheral immune cells. Moreover, when infiltrating T cell numbers were examined in the lumber spinal cords of EAE mice at PID 9, 2 days after the initiation of Sephin1 treatment, no difference was observed between the vehicle and Sephin1 treated animals, suggesting that the drug does not impact the initial T cells infiltration into the CNS of EAE mice (Supplementary Fig. 8).
GADD34 inactivation in MOG-primed T cells does not alter the disease progression of an adoptive transfer model of EAE
To explore whether the therapeutic effects of Sephin1 on the EAE disease course are mediated by a CNS or peripheral immune response we used an adoptive transfer (passive) EAE model exploiting the GADD34 mutant mice. Lymph node T cells from either GADD34 wild-type or mutant mice collected at PID 8 of EAE were reactivated in vitro by MOG35-55 and IL-12 for 72 h and then transferred into naïve GADD34 wild-type or mutant mice. All recipient mice exhibited a mild but consistent EAE phenotype (Fig. 7J). Symptoms of wild-type mice that received wild-type MOG35-55 activated T cells peaked at Day 20 after cell transfer. Mutant mice that received MOG35-55 activated mutant T cells displayed a delayed disease course, peaking at Day 28 (Fig. 7J), which is consistent with active EAE in the GADD34 mutant animals (Fig. 7A). Strikingly, wild-type mice that received mutant T cells presented a similar disease course to wild-type mice receiving wild-type cells; whereas GADD34 mutant mice that received wild-type T cells displayed a delayed disease course (Fig. 7J). These data indicate that the beneficial effects of GADD34 inactivation on the EAE disease course are mediated by an effect on the CNS not infiltrating T cells.
Combination treatment has additive benefits in alleviating EAE symptoms and lessening demyelination
IFN-β is a frequently used first-line therapy for relapsing-remitting multiple sclerosis (La Mantia et al., 2016; Comi et al., 2017). Although IFN-β has been shown to reduce the relapse severity and disease activity of relapsing-remitting multiple sclerosis, it does not slow the progression of disease. We hypothesize that in combination with ISR-mediated protective therapies, the immunomodulatory IFN-β may achieve a better therapeutic outcome. To test this in mice, we initiated treatment of chronic EAE mice with vehicle, IFN-β alone, or a combined treatment of Sephin1 with IFN-β at PID 7 before the onset of clinical disease. Vehicle-treated EAE mice developed a typical disease course, while IFN-β treatment dampened disease severity beginning at PID 19 (Fig. 8A). Interestingly, a more dramatic decrease in clinical disease development was shown in the combination treatment (Sephin1+IFN-β), starting from PID 11 to the end of the study (Fig. 8A). As shown in Fig. 8B, IFN-β treatment did not affect the onset of the disease by itself, whereas when it was combined with Sephin1 we found a significant delay of the onset of disease from PID 11 to PID 17 (P < 0.001). Moreover, EAE mice treated with both Sephin1 and IFN-β, but not IFN-β alone, reached the peak of disease much later than vehicle-treated EAE mice (P < 0.05) (Fig. 8B). Importantly, the average peak score of clinical symptoms in EAE mice with the combined treatment was also significantly lower than vehicle-treated EAE mice (P < 0.05) (Fig. 8B). Histological analysis of lumber spinal cords from PID 17 showed that all treatment groups reduced oligodendrocyte loss and demyelination (Fig. 8C, E and F). Moreover, less demyelination was observed in the Sephin1 and combination treatment groups than in IFN-β group at this stage (P < 0.001 and P < 0.05). At PID 30, there were still significantly less demyelinated areas in the white matter of mice with combination treatment than with vehicle (P < 0.05) (Fig. 8D and F). Compared to the combination treatment, Sephin1 alone did not prolong its protection on the survival of oligodendrocytes and myelin, nor did Sephin1 limit the T cells infiltration at this late stage (P < 0.05, P < 0.01 and P < 0.05) (Fig. 8D–F and Supplementary Fig. 9). These data indicate that combining Sephin1 and IFN-β provides an additive therapeutic benefit in ameliorating and delaying the symptoms of EAE.
Figure 8.

Combination treatment with Sephin1 and IFN-β alleviates and delays clinical symptoms of EAE as well as reduces the oligodendrocytes and myelin loss during EAE course. (A) Clinical scores of C57BL/6 female mice immunized with MOG35-55/CFA to induce chronic EAE, treated with vehicle (n = 12), 5000 U of IFN-β (n = 12), 8 mg/kg of Sephin1 or Sephin1 combined with 5000 U of IFNβ (n = 10) daily from PID 7 to the end of the study. (B) Average onset of disease, peak of disease and average peak score of all treatment groups. (C and D) Immunofluorescent staining for TPPP (green) and MBP (red) of lumbar spinal cord sections from PID 17 (C) and PID 30 (D). Scale bar = 100 μm. (E) Quantification of cells positive for TPPP in the lesion areas. (F) Average percentage of demyelinated areas/white matter areas measured from MBP staining. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, significance based on ANOVA. Data represent an average of 3–4 mice per group (mean ± SEM).
Discussion
In this study we demonstrate that Sephin1, which inhibits the dephosphorylation of the ISR target eIF2α, effectively delays the onset of EAE, while a combination treatment with IFN-β exerts additive beneficial effects in attenuating EAE disease progression. Additional experiments show that Sephin1 protects oligodendrocytes, axons and myelin in EAE mice, which is correlated with a prolonged ISR, as well as reduced CNS inflammation. Our data further indicate that the protective effect of Sephin1 is not attributed to altered T cell priming in the peripheral immune system, and our adoptive transfer studies indicate that the therapeutic target of GADD34 inhibition resides within the CNS. Together, these results support the idea that the modulation of the ISR may be an effective neuroprotective therapy for multiple sclerosis patients.
Although the control of eIF2α phosphorylation and dephosphorylation is complex, the therapeutic potential of its modulation in the treatment of neurological disorders is great, such that significant focus has recently been placed on this pathway (Fullwood et al., 2012; Moreno et al., 2012; Ma et al., 2013; Das et al., 2015; Way et al., 2015). Salubrinal, an inhibitor of eIF2α dephosphorylation (Boyce et al., 2005), has been shown to confer remarkable protective effects in a number of animal models of neurodegenerative diseases, such as Alzheimer’s disease (Hoozemans et al., 2009) and Huntington’s disease (Reijonen et al., 2008). Nonetheless, a major concern with salubrinal is that this drug not only inhibits stress-induced GADD34-PP1c binding, but also disrupts the non-stress related phosphatase CReP-PP1c complex (Boyce et al., 2005). Disruption of this latter complex results in prolonged eIF2α phosphorylation and sustained inhibition of protein synthesis, which may lead to cell death (Teng et al., 2014). The discovery that guanabenz is an inhibitor of the GADD34-PP1c protein phosphatase complex that does not target the CReP-PP1c complex, therefore, provided a considerable advance (Tsaytler et al., 2011). Nevertheless, guanabenz is also an α2 adrenergic receptor agonist, which causes a number of undesirable side effects, including hypotension, dry mouth, drowsiness and fatigue (Holmes et al., 1983). Recently, Sephin1 has been shown to be devoid of α2 adrenergic pathway activity while preserving the ISR-modifying functions of guanabenz, suggesting that Sephin1 has greater therapeutic potential than guanabenz (Das et al., 2015; Way et al., 2015). Indeed, in agreement with our observations, Das et al. (2015) demonstrated that chronic treatment with Sephin1 has no measurable adverse effects on mice.
Recently, the molecular mechanism of action of guanabenz and Sephin1 on the ISR has been under intense investigation (Carrara et al., 2017; Crespillo-Casado et al., 2017, 2018). Regardless of the exact manner by which these small molecules act, their presence clearly leads to prolonged, elevated levels of phosphorylated eIF2α in stressed cells (Tsaytler et al., 2011; Das et al., 2015; Way et al., 2015), thereby enhancing the ISR protective response. In addition, our studies have demonstrated that oligodendrocytes under inflammatory stress display prolonged and elevated levels of eIF2α phosphorylation in the presence of either guanabenz (Way et al., 2015) or Sephin1 (Fig. 1). Moreover, the ameliorated EAE disease course is very similar in GADD34 mutant mice and Sephin1-treated wild-type EAE mice, and we did not observe any additive beneficial effects of Sephin1 on EAE progression in GADD34 mutant mice, suggesting that the therapeutic effects of Sephin1 on EAE are dependent on GADD34. Nonetheless, the molecular details of the protective effects of guanabenz and Sephin1 on oligodendrocytes in the presence of mediators of inflammatory cytotoxicity deserves deeper investigation.
Our results demonstrate that when administered before mice exhibit EAE clinical symptoms, Sephin1 protects mature oligodendrocytes while enhancing eIF2α phosphorylation in these cells. Meanwhile, Sephin1 does not increase CHOP expression at this early stage, suggesting that transiently enhanced eIF2α phosphorylation by Sephin1 with resultant inhibition of translation is beneficial to the survival of oligodendrocytes under inflammatory attack. Nonetheless, in contrast to its effects early in the disease course, Sephin1 treatment neither protects oligodendrocytes nor ameliorates EAE severity later in the disease, when extensive cellular stress is triggered by accumulated inflammation. Excessive eIF2α phosphorylation maintained by Sephin1 at this late stage of disease further upregulated pro-apoptotic CHOP expression, which suggests that Sephin1 cannot halt the divergence from the protective ISR to cell death when inflammation is overwhelming.
Our current study does not directly address the potential effects of ISR enhancement on the remyelination process. Following oligodendrocyte loss and demyelination, remyelinating oligodendrocytes develop from OPCs that are abundantly present throughout the adult CNS (Hughes et al., 2013). In multiple sclerosis patients, these OPCs are required to mature and myelinate axons in the adverse inflammatory environment of the demyelinated lesions. We are currently examining the potential of Sephin1 to provide protection to remyelinating oligodendrocytes against inflammation, which may result in an enhanced remyelination response.
A growing body of evidence suggests that the ISR and inflammation share extensive cross-talk (Zhang and Kaufman, 2008; Hasnain et al., 2012; Cláudio et al., 2013). Our previous report on guanabenz inferred that the protective effect of guanabenz on EAE depends on both ISR-mediated oligodendrocyte protection and moderate immunomodulatory activities, perhaps as a result of its adrenergic receptor activity (Way et al., 2015). In our current study, we observed that Sephin1 suppressed the mRNA levels of cytokines and the presence of inflammatory cells in the spinal cords of EAE mice at PID 17. Nevertheless, Sephin1 did not affect the activation of immune cells in the spleen nor did it have a significant effect on the proliferation or cytokine production of splenic T cells. Our adoptive transfer EAE studies with the GADD34 mutant mice provided further evidence that the inactivation of GADD34 in peripheral T cells did not provide therapeutic benefit. It is likely that Sephin1-mediated protection of oligodendrocytes dampens CNS inflammation by limiting apoptotic oligodendrocyte cell death and the subsequent release of myelin antigens, thus dampening epitope spreading. When the treated mice developed severe clinical symptoms at PID 30, Sephin1 did not halt CNS inflammation, which is thought to be secondary to an underlying CNS disease process (Frischer et al., 2009). In support of this, a recent study from our groups demonstrated that oligodendrocyte death can trigger myelin autoimmunity and inflammatory damage in the CNS (Traka et al., 2016). Collectively, our data strongly suggest that Sephin1 has oligodendrocyte-protective capabilities not related to direct immunomodulation.
It is also worth noting that IFN-β treatment delayed the onset of clinical disease when combined with Sephin1, while IFN-β treatment alone merely dampened the severity of EAE. IFN-β therapy, which is one of the most commonly used disease-modifying treatments in multiple sclerosis, reduces the number of attacks, the frequency of relapses and number of brain lesions in relapsing-remitting multiple sclerosis (Haji Abdolvahab et al., 2016). Although its mechanism of action remains unclear, IFN-β is thought to modulate the immune response by impairing the trafficking of inflammatory cells into the CNS (Yong, 2002; Haji Abdolvahab et al., 2016). The additive effect of combination treatment further supports the suggestion that Sephin1 works on mechanisms distinct from the immunomodulation mediated by IFN-β. The neuroprotective effects of Sephin1 when used in combination with the immunomodulatory drugs currently available might provide added benefit to multiple sclerosis patients.
Importantly, the inhibition of GADD34-mediated eIF2α dephosphorylation should have minimal impact on healthy, non-stressed cells. In support of this, GADD34 mutant cells and guanabenz-treated cells display normal p-eIF2α levels and translational activity in the absence of stress (Tsaytler et al., 2011; Das et al., 2015; Way et al., 2015). Therefore, Sephin1, which selectively disrupts eIF2α dephosphorylation, should primarily affect cells under cytotoxic stress, thereby minimizing the concerns of deleterious effects on unstressed cells.
In summary, our study indicates that Sephin1 treatment asserts a significant delay on disease progression in chronic EAE, a mouse model of multiple sclerosis, by enhancing the ISR-mediated neuroprotection against CNS inflammation. Current therapeutic choices for multiple sclerosis are largely driven by the risk-benefit evaluation. Therefore, we believe that Sephin1, free from adrenergic agonist side effects, could lead to a better clinical outcome in multiple sclerosis patients as a safe neuroprotective drug, perhaps when used in combination with immune-modulatory therapies.
Supplementary Material
Acknowledgements
We thank S. W. Way for experimental advice and critically reading manuscript, G. Wright for help preparing the figures, A. Solanki and W. Wang for technical assistance, and S. Bond from the University of Chicago Integrated Light Microscopy Core Facility for help with whole slide scanning.
Glossary
Abbreviations
- CFA
complete Freund’s adjuvant
- CHOP
C/EBP-homologous protein
- EAE
experimental autoimmune encephalomyelitis
- GADD34
growth arrest and DNA damage 34
- ISR
integrated stress response
- IFN
interferon
- OPC
oligodendrocyte precursor cell
- PID
post-immunization day
- PLP
proteolipid protein
- PP1c
protein phosphatase 1 catalytic subunit
- P/T-eIF2α
phosphorylated/total eukaryotic translation initiation factor 2 alpha
Funding
The work was supported by NIH/NINDS (R01 NS034939 and R01 NS099334) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Competing interests
The authors report no competing interests.
References
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D et al. . A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005; 307: 935–9. [DOI] [PubMed] [Google Scholar]
- Carrara M, Sigurdardottir A, Bertolotti A. Decoding the selectivity of eIF2α holophosphatases and PPP1R15A inhibitors. Nat Struct Mol Biol 2017; 24: 708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll WM, Jennings AR, Ironside LJ. Identification of the adult resting progenitor cell by autoradiographic tracking of oligodendrocyte precursors in experimental CNS demyelination. Brain 1998; 121 (Pt 2): 293–302. [DOI] [PubMed] [Google Scholar]
- Cláudio N, Dalet A, Gatti E, Pierre P. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J 2013; 32: 1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Cutter GR, Fischer JS, Goodman AD, Heidenreich FR, Jak AJ et al. . Use of the multiple sclerosis functional composite as an outcome measure in a phase 3 clinical trial. Arch Neurol 2001; 58: 961–7. [DOI] [PubMed] [Google Scholar]
- Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet 2017; 389: 1347–56. [DOI] [PubMed] [Google Scholar]
- Crespillo-Casado A, Chambers JE, Fischer PM, Marciniak SJ, Ron D. PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. Elife 2017; 6: e26109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespillo-Casado A, Claes Z, Choy MS, Peti W, Bollen M, Ron D. A Sephin1-insensitive tripartite holophosphatase dephosphorylates translation initiation factor 2α. J Biol Chem 2018; 293: 7766–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnea P, Mháille AN, McQuaid S, Farrell M, McMahon J, FitzGerald U. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult Scler 2011; 17: 808–18. [DOI] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N et al. . Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015; 348: 239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbue S, Comar M, Ferrante P. Natalizumab treatment of multiple sclerosis: new insights. Immunotherapy 2016; 9: 157–71. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Emery B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb Protoc 2013; 2013: 745–58. [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Kotter MR, Harrington EP, Huang JK, Zhao C, Rowitch DH et al. . Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp Neurol 2010; 225: 18–23. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al. . The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009; 132: 1175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med 2006; 354: 942–55. [DOI] [PubMed] [Google Scholar]
- Fullwood MJ, Zhou W, Shenolikar S. Targeting phosphorylation of eukaryotic initiation factor-2α to treat human disease. Prog Mol Biol Transl Sci 2012; 106: 75–106. [DOI] [PubMed] [Google Scholar]
- Haji Abdolvahab M, Mofrad MRK, Schellekens H. Interferon beta: from molecular level to therapeutic effects. Int Rev Cell Mol Biol 2016; 326: 343–72. [DOI] [PubMed] [Google Scholar]
- Hart FM, Bainbridge J. Current and emerging treatment of multiple sclerosis. Am J Manag Care 2016; 22: s159–70. [PubMed] [Google Scholar]
- Hasnain SZ, Lourie R, Das I, Chen AC-H, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol 2012; 90: 260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev 2011; 91: 1219–43. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Araki T, Sugiyama F, Yagami K, Suzuki M, Abe K et al. . Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J 2000; 19: 341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS. Guanabenz. A review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs 1983; 26: 212–29. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJM, van Haastert ES, Nijholt DAT, Rozemuller AJM, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 2009; 174: 1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 2013; 16: 668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussien Y, Cavener DR, Popko B. Genetic inactivation of PERK signaling in mouse oligodendrocytes: normal developmental myelination with increased susceptibility to inflammatory demyelination. Glia 2014; 62: 680–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Blakemore WF. Identification of post-mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J Neuropathol Exp Neurol 1997; 56: 1191–201. [DOI] [PubMed] [Google Scholar]
- Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol 1953; 12: 400–3. [DOI] [PubMed] [Google Scholar]
- Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K et al. . The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J 2003; 17: 1573–75. [DOI] [PubMed] [Google Scholar]
- Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) 2014; 46: 629–40. [DOI] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD et al. . The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest 2007; 117: 448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol 2005; 169: 603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol 2008; 173: 1508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P et al. . Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 2013; 16: 1299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mantia L, Di Pietrantonj C, Rovaris M, Rigon G, Frau S, Berardo F et al. . Interferons-beta versus glatiramer acetate for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev 2016; 11: CD009333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R et al. . CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Guire C, Volckaert T, Wolke U, Sze M, de Rycke R, Waisman A et al. . Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J Immunol 2010; 185: 7646–53. [DOI] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol 1995; 25: 1951–59. [DOI] [PubMed] [Google Scholar]
- Mháille AN, McQuaid S, Windebank A, Cunnea P, McMahon J, Samali A et al. . Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J Neuropathol Exp Neurol 2008; 67: 200–11. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG et al. . Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012; 485: 507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA et al. . Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98–103. [DOI] [PubMed] [Google Scholar]
- O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP et al. . Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011; 365: 1293–303. [DOI] [PubMed] [Google Scholar]
- Okuda Y, Okuda M, Bernard CCA. The suppression of T cell apoptosis influences the severity of disease during the chronic phase but not the recovery from the acute phase of experimental autoimmune encephalomyelitis in mice. J Neuroimmunol 2002; 131: 115–25. [DOI] [PubMed] [Google Scholar]
- Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev 2012; 248: 205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci 2012; 15: 1074–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med 2018; 378: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijonen S, Putkonen N, Nørremølle A, Lindholm D, Korhonen L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 2008; 314: 950–60. [DOI] [PubMed] [Google Scholar]
- Schaeffer J, Cossetti C, Mallucci G, Pluchino S. Multiple sclerosis In: Zigmond MJ, Rowland LP, Coyle JT, editors. Neurobiology of Brain disorders. Cambridge, MA: Elsevier; 2015. p. 497–520. [Google Scholar]
- Simpson D, Noble S, Perry C. Spotlight on glatiramer acetate in relapsing-remitting multiple sclerosis. BioDrugs 2003; 17: 207–10. [DOI] [PubMed] [Google Scholar]
- Skjoerringe T, Lundvig DMS, Jensen PH, Moos T. P25alpha/Tubulin polymerization promoting protein expression by myelinating oligodendrocytes of the developing rat brain. J Neurochem 2006; 99: 333–42. [DOI] [PubMed] [Google Scholar]
- Stone S, Lin W. The unfolded protein response in multiple sclerosis. Front Neurosci 2015;9:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13: 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Gao M, Wang J, Kong Q, Hua H, Luo T et al. . Inhibition of eIF2α dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis 2014; 5: e1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci 2016; 19: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011; 332: 91–4. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–86. [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016; 529: 326–35. [DOI] [PubMed] [Google Scholar]
- Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB et al. . Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun 2015; 6: 6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, Popko B. Harnessing the integrated stress response for the treatment of multiple sclerosis. Lancet Neurol 2016; 15: 434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav SK, Mindur JE, Ito K, Dhib-Jalbut S. Advances in the immunopathogenesis of multiple sclerosis. Curr Opin Neurol 2015; 28: 206–19. [DOI] [PubMed] [Google Scholar]
- Yong VW. Differential mechanisms of action of interferon-beta and glatiramer aetate in MS. Neurology 2002; 59: 802–8. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454: 455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.