

Abstract
Chemically induced dimerizers (CIDs) have emerged as one of the most powerful tools to artificially regulate signaling pathways in cells; however, currently available CID systems lack the properties desired for use in regulating cellular therapies. Here, we report the development of human antibody-based chemically induced dimerizers (AbCIDs) from known small-molecule-protein complexes by selecting for synthetic antibodies that recognize the chemical epitope created by the bound small molecule. We demonstrate this concept by generating three antibodies that are highly selective for the BCL-xL/ABT-737 complex over BCL-xL alone. We show the potential of AbCIDs to be applied to regulating human cell therapies by using them to induce CRISPRa-mediated gene expression and to regulate CAR T-cell activation. We believe that the AbCIDs generated in this study will find application in regulating cell therapies, and that the general method of AbCID development may lead to the creation of many new and orthogonal CIDs.
Introduction
Chemically induced dimerizers (CIDs) are powerful tools for dose and temporal control over protein-protein interactions.1–3 CIDs have been utilized in a myriad of applications, including the development of artificial cellular circuits4, activating split-enzyme activity5, 6, and controlling protein localization. Recently, there has been a growing interest in utilizing CIDs to regulate the activity of cell therapies after they have been administered to a patient.7, 8 Of particular interest has been the utilization of CIDs as safety switches for chimeric antigen receptor T-cell (CAR T-cell) therapies, where several patient deaths have occurred in clinical trials.9 While a number of homo- and hetero-CIDs have been developed, they generally lack the properties required for use in human cell therapies.1, 3, 10–16 For example, the classical FKBP/FRB CID system utilizes the small molecule rapamycin, which is both toxic and immunosuppressant. Orthogonal “rapalogs” show reduced toxicity, but have undesirable pharmacokinetic (PK) properties. Several plant-based CID systems have been developed, but the non-human nature of these proteins makes them prone to immunogenicity issues if incorporated into a cell therapy.17 For the application of CIDs in cell therapies to reach its full potential, it is critical that new human-protein-based CIDs be developed that utilize small molecules with drug-like properties. Ideally, the small molecules should have favorable PK properties and be bioorthogonal or well-tolerated. Additionally, new CIDs should exhibit dose dependence and be easily incorporated into different cellular signaling pathways. To date, the vast majority of CID systems have been based on naturally occurring CIDs, and the ability to engineer in customized properties has been limited. While chemically linking two pharmacophores together has been employed to rationally design heteromeric CIDs not found in nature, the resulting small molecules almost universally lack drug-like properties. For these reasons, a general method to design novel CIDs with desirable properties for use in regulating human cell therapies would be of great utility.
Here, we demonstrate a strategy to generate chemical-epitope-selective antibodies that has the potential to turn many known small-molecule-protein complexes into antibody-based chemically induced dimerizers (AbCIDs) (Fig. 1a). We demonstrate this approach by engineering AbCIDs using the BCL-xL/ABT-737 complex. Furthermore, we show that AbCIDs can be used to regulate cellular processes; including CRISPRa mediated gene expression and CAR T-cell activation. We believe the broad applicability of this approach is the ability to rapidly generate CIDs from human protein-small-molecule complexes, with proteins and small molecules that meet the criteria for application in regulating human cell therapies.
Figure 1.
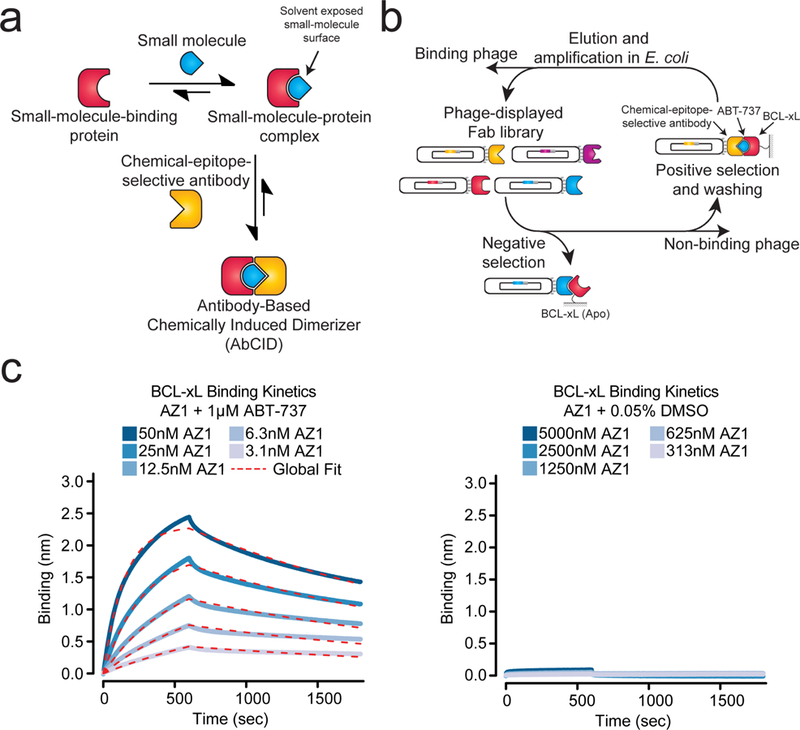
Design and characterization of antibody-based chemically induced dimerizers (AbCIDs). (a) Schematic of AbCIDs (b) Diagram of the phage selection strategy used to select ABT-737-inducible Fab binders of BCL-xL. (c) Biolayer interferometry shows potent and reversible binding of Fab AZ1 to BCL-xL in the presence of ABT-737 (left) but no significant binding was observed in the absence of ABT-737 (right). Blue curves represent measured data points and dashed red lines represent the global-fit lines used for analysis.
Results
Identification of a complex for generation of an AbCID
We reasoned that the ideal complexes to generate selective antibodies against would be those in which a large portion of the small molecule remains solvent exposed when bound. Nature has employed a similar principle in the rapamycin-FKBP12-FRB CID system, where rapamycin first binds FKBP12, generating a new binding surface that is then recognized by FRB. Several other natural products use a similar approach for artificial protein recruitment.2 Additional design principles included that the target protein be a small monomeric domain and that the small molecule inducer be commercially available with desirable pharmacokinetic properties and low toxicity, making it potentially useful for animal model applications.
After a survey of small-molecule-bound structures in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) we turned our attention to the human BCL-xL/ABT-737 complex (PDB: 2YXJ).18 BCL-xL is a member of the anti-apoptotic BCL-2 family of proteins.19 This small monomeric protein (~26 kDa) is located on the outer membrane of the mitochondria where it sequesters pro-apoptotic members of the BCL-2 family. Because of its anti-apoptotic role, a number of animal and clinically active small-molecule inhibitors have been developed against BCL-xL for the treatment of cancers.20 The crystal structure of our candidate ligand, ABT-737 (1)21, bound to BCL-xL shows that a large portion of ABT-737 is exposed to solvent (308 Å2) providing a potential chemical epitope for antibody binding. In comparison, an analysis of 866 small-molecule-bound structures in the PDB (Supplementary Results, Supplementary Fig. 1) revealed a mean solvent exposed surface area of 125 Å2, with rapamycin bound to FKBP12 being an outlier at 528 Å2 (PDB:1FKB).22 Thus, we felt that the BCL-xL/ABT-737 complex would be an ideal first target for the development of an AbCID.
Selection of chemical-epitope-selective antibodies
To identify unique chemical-epitope-selective antibodies, we utilized a C-terminally truncated form of BCL-xL (residues 2–215) that lacks the mitochondrial-transmembrane domain. Biotinylated BCL-xL was immobilized on streptavidin resin and used for phage selections with a previously developed synthetic antibody-fragment library and selection stategy.23 During each round of selection, the phage library was first subjected to stringent counter selection against BCL-xL in the absence of small molecule, thereby removing any Fab-phage that was not selective for the ABT-737-bound form. Positive selections were then performed in the presence of saturating amounts of ABT-737 (1 µM), ensuring that the majority of BCL-xL was bound to ABT-737 (Fig. 1b). A total of four rounds of selection were performed. Encouragingly, we observed significant enrichment of phage titers for selections against BCL-xL in the presence of ABT-737 (Supplementary Fig. 2). After round four, individual Fab-phage clones were isolated and sequenced. A total of ten Fab-phage with unique sequences in the complementarity-determining regions (CDRs) of the Fab were identified (Supplementary Table 1).
Characterization of AbCIDs selectivity
The unique Fabs were sub-cloned into a bacterial expression vector, expressed, and purified.23 Gratifyingly, enzyme-linked immunosorbent assays (ELISA) with BCL-xL in the presence or absence of ABT-737 showed that all ten Fabs had enhanced binding in the presence of drug. Several Fabs showed excellent potency and extremely strong selectivity for binding in the presence of ABT-737 (Supplementary Fig. 3). To further profile the best three Fabs, we characterized the kinetics of BCL-xL binding in the presence or absence of ABT-737 by bio-layer interferometry (Fig. 1c and Supplementary Fig. 4).24 All three of the Fabs (AZ1, AZ2 and AZ3) were very potent binders of BCL-xL in the presence of ABT-737 (KD < 10 nM) and showed no detectable binding in the absence of ABT-737 at concentrations up to 5000 nM of Fab (Supplementary Table 2). Our most selective Fab (AZ2) showed >2000 fold selectivity for the ABT-737-bound form of BCL-xL over the apo form. Formation of the AbCID ternary complex was reversible, either through washout of the Fab (Fig. 1c and Supplementary Fig. 4) or the small molecule (Supplementary Fig. 5).
We hypothesized that the exquisite selectivity of our Fabs was the result of direct interactions of the Fab CDRs with parts of ABT-737. We reasoned that if this were the case, the Fab would bind less potently to other BCL-xL-ligand complexes. ABT-263 (2) is an analog that binds with similar potency to the same conformation of BCL-xL as ABT-737 (RMSD = 0.8) (Fig. 2a, Supplementary Fig. 6a).25 To test our hypothesis, we measured the ability of AZ1 to discriminate between ABT-737, ABT-263, and the native-ligand-derived Bak-peptide26 bound BCL-xL (Fig. 2b). As predicted, we observed dramatically weaker binding of the Fab to the BCL-xL/ABT-263 complex and no detectable binding of Fab to the Bak-peptide complex. Although we do not have a crystal structure of the AbCID complex, these data strongly suggest that AZ1 binds near if not over the small-molecule binding site.
Figure 2.
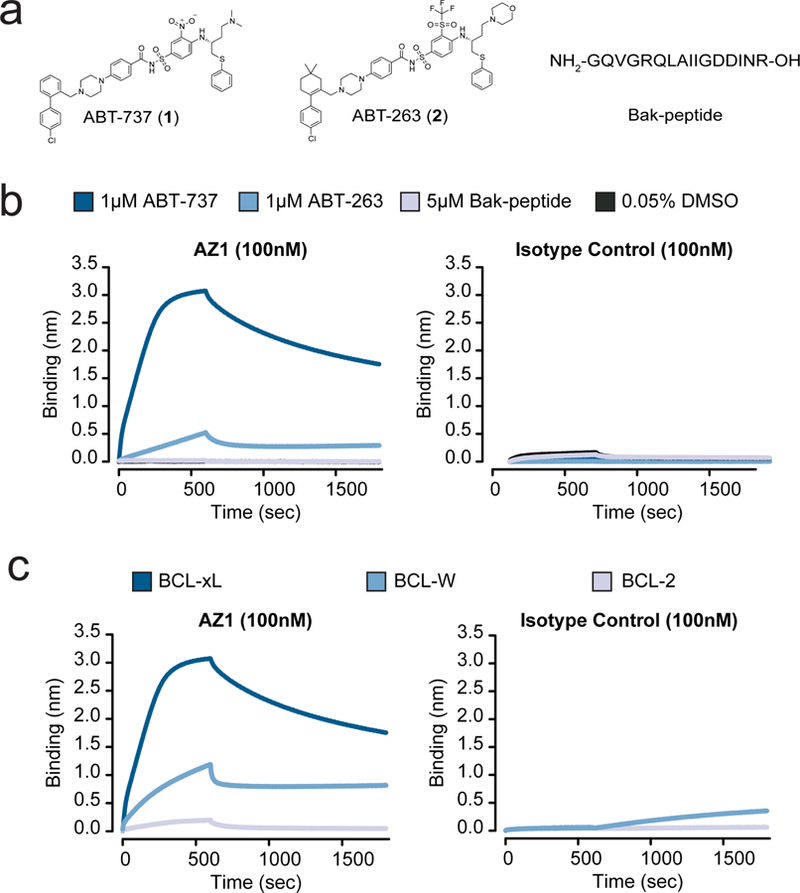
Characterization of the Fab AZ1 epitope. (a) Chemical structures and amino acid sequence of the binding ligands: ABT-737, ABT-263, and Bak-peptide. (b) Biolayer interferometry shows Fab AZ1 binds potently to BCL-xL in the presence of ABT-737, with greatly reduced potency in the presence of ABT-263, and weakly or undetectably in the presence of Bak-peptide. The data shows that AZ1 can readily discriminate between subtle structural differences in the small molecules, and supports that Fab AZ1 is chemical-epitope selective. The isotype control is a Fab selected against CD55, with an identical scaffold to AZ1 but differing CDR sequences. (c) Biolayer interferometry show Fab AZ1 binds potently to BCL-xL in the presence of ABT-737, with greatly reduced potency to BCL-W, and undetectably to BCL-2. The data shows that AZ1 can readily discriminate between subtle structural differences in the proteins, and supports that Fab AZ1 makes important contacts with BCL-xL in addition to its contacts with ABT-737. The isotype control is a Fab selected against CD55, with an identical scaffold to AZ1 but differing CDR sequences.
While AZ1 is able to discriminate between ABT-737 and its close analog ABT-263, suggesting that ABT-737 comprises a portion of the epitope recognized by AZ1, we hypothesized that AZ1 also makes contacts with BCL-xL. To test this, we measured the ability of AZ1 to bind to ABT-737 bound BCL-W and BCL-2. BCL-W and BCL-2 are both homologs of BCL-xL known to bind ABT-737 with similar potency to BCL-xL.21 Importantly, BCL-xL, -W, and −2 all have similar folds (Supplementary Fig. 6b). In the presence of saturating concentrations of ABT-737, AZ1 showed reduced binding to BCL-W and almost no detectable binding to BCL-2, suggesting that the epitope recognized by AZ1 encompasses specific residues on the surface of BCL-xL in addition to specific chemical epitopes on ABT-737 (Fig. 2c). This data supports the hypothesis that AZ1 makes direct contact with both the small molecule and protein portion of the BCL-xL/ABT-737 complex.
In the naturally occurring rapamycin-FKBP12-FRB CID, it is known that rapamycin potently binds FKBP12 (low nM), and has only weak affinity for FRB on its own (µM). However, FRB is able to potently bind the FKBP/rapamycin complex (low nM). As we generated our AZ1 AbCID by selecting for Fabs against the previously known BCL-xL/ABT-737 complex, we hypothesized that our CID assembled using a mechanism similar to rapamycin-FKBP12-FRB. To test this, we utilized differential scanning fluorimetry to look for changes to the Tm of AZ1 in the presence of ABT-737. As suspected, ABT-737 seemed to have no effect on the Tm of AZ1, suggesting that AZ1 does not bind ABT-737 on its own at the concentrations used in our dimerization assays (Supplementary Table 3). In comparison, BCL-xL, which is known to potently bind ABT-737, showed a ~10 °C increase in Tm in the presence of ABT-737. Together, this data supports a mechanism in which ABT-737 first binds to BCL-xL, creating a new epitope, which is then potently recognized by AZ1.
Application of AbCIDs to regulate gene expression
When developing AbCIDs, we used design principles based on a desire to utilize these tools in regulating cellular therapies. For that reason, when choosing cellular applications to demonstrate AbCIDs, we focused on cellular models of two main cell therapy modalities; regulation of gene expression and activation of immune cells. Furthermore, we attempted to incorporate our AbCIDs into technologies at the cutting edge of these fields (CRISPRa, CAR T-cell), so as to show the great promise of applying AbCIDs to next-generation cell therapies.
Current CID technologies are often used for controlling intracellular signaling pathways.2, 3 Due to the disulfide bond linking the heavy and light chains of Fabs and the reducing environment inside the cell, it is generally believed that intracellular expression of Fabs in mammalian cells would lead to an inactive species. Recently, we reported a single-chain Fab (scFab) construct in which the light and heavy chains are genetically fused as a single polypeptide.27 The scFab scaffold has a very high melting temperature (Tm = ~81 °C) so that once formed it is very stable.27 We hypothesized that conversion of our ABT-737-inducible Fabs into a scFab format may allow for their use in living cells. Indeed, transfection of the AZ1 gene in scFab format (scAZ1) into HEK293T cells resulted in robust expression as measured by immunoblotting (Supplementary Fig. 7). To test if scAZ1 was active in living cells, we constructed a genetic circuit in which scAZ1 is fused to the VPR transcriptional activation domain28 and BCL-xL is fused to dCas929 (Fig. 3a). Both constructs contain a nuclear localization sequence, which reduces the possibility of interaction with endogenous BCL-xL while simultaneously priming the system for activation in the nucleus. The dCas9-BCL-xL fusion can be targeted by addition of a specific sgRNA to a promoter that drives a luciferase reporter. If the AbCID functions in cells, addition of ABT-737 should lead to localization of AZ1-VPR to the luciferase reporter, promoting expression of luciferase, which can be readily detected. For comparison, we generated an identical circuit, but utilizing a conventional CID based on the rapamycin-FKBP12-FRB system10, as recently reported.30 Indeed, addition of ABT-737 to our engineered cells resulted in robust expression of luciferase, supporting that AZ1 and BCL-xL functioned as an ABT-737-inducible AbCID in living cells (Fig. 3b). The level of activation observed using the AbCID was comparable to that observed by the conventional CID. The induction of luciferase expression was dose dependent, with an EC50 of 8.7 ± 1.1 nM (Fig. 3c). This value was consistent with the EC50 measured by in vitro characterization of the AZ1/ABT-737/BCL-xL complex using biolayer interferometry (Supplementary Fig. 8). Importantly, addition of ABT-737 to an AbCID-gated system with a negative sgRNA resulted in no increase in luciferase expression (Supplementary Fig. 9). Together, these results support that our AbCID can be used for tunable control of biological systems in living cells.
Figure 3.
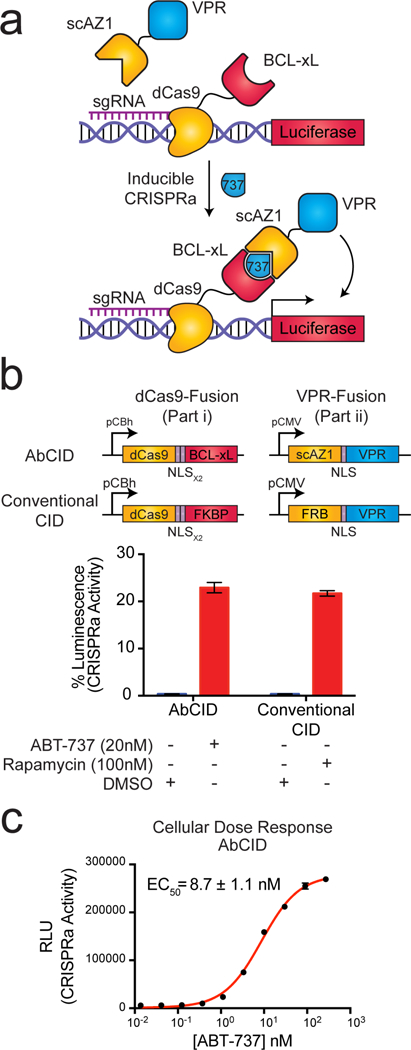
A single-chain Fab version of AZ1 can be utilized as an intracellular AbCID to regulate CRISPRa-mediated gene activation. (a) Schematic of the AbCID regulated gene activation system. Inducible recruitment of the VPR transcriptional activation domain to dCas9 results in the expression of a luciferase reporter. (b) Quantitation of luciferase activity 48 hours after addition of ABT-737 (20 nM) to the AbCID-gated system compared to the addition of rapamycin (100 nM) to the conventional CID. Values are normalized to a positive control, which is dCas9 genetically fused to VPR, and background subtracted with a negative control, which is dCas9-VPR with a negative sgRNA. Each data point represents the mean of 4 independent experiments ± s.d. (c) Dose response after 48 hour induction by addition of ABT-737 to the AbCID-gated system. Each data point represents the mean of 3 independent experiments ± s.d. The EC50 reported was calculated from the mean of 3 independent experiments using 3-parameter nonlinear regression ± s.e.m.
Application of AbCIDs to regulate CAR T-cells
The use of engineered T-cells for the treatment of malignancies has recently become an important paradigm in cancer therapeutics.31 One such approach, known as CAR T-cells, involves the genetic engineering of a T-cell such that it expresses a surface exposed scFv antibody fragment linked to an intracellular T-cell activation domain. The scFv is specific for a tumor antigen, and results in recruitment of the T-cell to the tumor and antigen-dependent activation of the T-cell. This technique has shown great responses in treating leukemia by targeting the CD19 antigen. However, hyperactivation of CAR T-cells has resulted in off-target cytotoxic effects and in some cases death, limiting utility of this promising modality.31 For this reason, there has been great interest in developing remote control over the activity of these cells, so as to tune the level of activation or end it should untoward toxicity develop.32–35
Previous reports of small-molecule-activated CAR T-cells utilize intracellular splitting of the CAR activation domains.32 An additional approach for control of CAR T-cell activation uses universal protein-based adaptor domains that confer antigen recognition and promote activation of the CAR T-cell when added.33–35 We hypothesized that by taking advantage of the unique antibody nature of our AbCID system we could generate a hybrid of these two paradigms, with the universal nature of an adaptor strategy but the temporal control of a small-molecule inducible system. To test this, we engineered Jurkat T-cells to express a CAR in which the scFv portion of the CAR is replaced by BCL-xL (Fig. 4a–b). This creates a T-cell that contains the machinery required for activation, but no longer binds to the antigen-presenting cells. In parallel, we generated a bispecific antibody by linking a clinically utilized αCD19 scFv36 to Fab AZ1. Upon addition of ABT-737 the bispecific antibody will be recruited to the CAR T-cell while simultaneously engaging the CD19+ cells. Such a design allows for both inducible and antigen-dependent CAR T-cell activation. To facilitate rapid quantitation of T-cell activation, we utilized a Jurkat T-cell line that had been engineered to express GFP upon activation of the NFAT pathway.37 In the presence of CD19+ K562 cells and our bispecific antibody (AZ1-αCD19), addition of ABT-737 resulted in a dose-dependent activation of the CAR T-cells as measured by expression of GFP (Fig. 4c). Activation of the T-cells was further confirmed by expression of the canonical T-cell activation markers, CD69 and secreted Interleukin-2 (Supplementary Fig. 10).38, 39 Importantly, activation of the T-cells was not observed with K562 cells lacking CD19 or if an AbCID was used that did not contain the αCD19 scFv (Fig. 4c). In addition, ABT-737 was not able to induce T-cell activation on its own. While our T-cell system showed ~65% the activation level of the conventional CAR control, the reduced activity may actually be of benefit due to the hyperactivation and toxicity observed with conventional CARs. These data demonstrate that AbCIDs can by used for extracellular regulation of cellular signaling pathways and represent a novel paradigm for small-molecule control of CAR T-cell activation.
Figure 4.
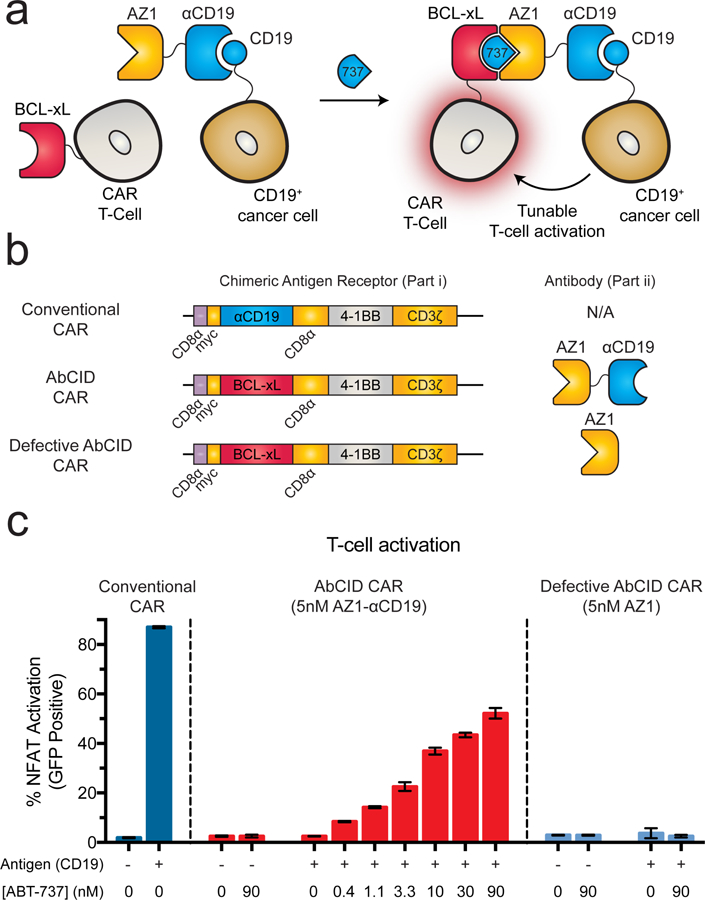
AZ1 can be utilized as an extracellular AbCID to regulate CAR T-cell activation. (a) Schematic of AbCID-regulated CAR T-cell activation where the CAR contains an extracellular BCL-xL domain in place of the typical scFv. Addition of an AZ1-αCD19 bispecific antibody and various concentrations of ABT-737 results in recruitment to CD19+ cancer cells and tunable activation of the CAR T-cells. (b) Linear diagrams of the gene constructs used to produce the CARs and schematics of corresponding antibodies for this study. (c) Quantification of NFAT-dependent GFP reporter expression 20 hours after initiation of co-culture with either CD19+ or CD19– K562 target cells and addition of antibody (5 nM) and varying concentrations of small molecule. Addition of ABT-737 in the presence of CD19+ K562 cells and bispecific antibody resulted in dose-dependent activation of the NFAT pathway, but no activation was observed in the absence of ABT-737 or when co-cultured with CD19– K562 cells. The defective AbCID CAR, which lacks the CD19-binding scFv portion of the antibody, resulted in no activation under all conditions. Each data point represents the mean of 3 independent experiments ± s.d.
AbCIDs activate in a non-toxic concentration regime
ABT-737 is a soluble, cell-permeable, bioavailable, potent, and commercially available compound, making it an excellent molecule for use in a CID, both in cells and potentially in animals. However, it is known that ABT-737 induces apoptosis in some cells types, particularly hematopoietic cells that have high expression levels of BCL-2 family members.21 We thus tested the concentration of ABT-737 necessary to induce apoptosis in Jurkat, K562, and HEK293T cells. Importantly, the concentration ranges used to induce AbCID CAR (<100 nM) and CRISPRa (<270 nM) activity were below the concentrations at which cell death was observed (Jurkat IC50 ~2 µM, K562 IC50 >10 µM, and HEK293T IC50 ~10 µM) (Supplementary Fig. 11). ABT-737 has been used extensively in mouse cancer models and is generally well tolerated by mice, except for platelet toxicity.21 However, the concentrations used to activate AbCIDs in our cellular assays (<100 nM) are far below the concentration observed to be toxic to platelets (low µM).40 Additionally, others have also shown that ABT-737 can be applied to activate engineered proteins in live-cell experiments with little observed cytotoxicity.41 Collectively these data support the feasibility of using ABT-737 activated AbCIDs in cellular and animal applications with minimal effect on the viability of these model organisms. Moreover, while ABT-737’s lack of bioorthogonality may be a caveat for research applications, it may actually be of benefit from a therapeutic perspective if the AbCID CAR approach described here were to be applied to the treatment of ABT-737-sensitive B-cell malignancies.
Discussion
Here we described a novel method to rapidly generate chemically induced dimerizers using known small-molecule-protein complexes and synthetic antibody libraries. We demonstrated this method by generating AbCIDs from the BCL-xL/ABT-737 complex. Additionally, we showed that these AbCIDs could be applied to regulate a diverse range of biological processes in living cells, including CRISPRa mediated gene expression and CAR T-cell activation. Finally, we showed that the concentration range of ABT-737 used to activate AbCIDs was far below the concentration at which toxicity was observed in cells.
One of the inspirations for developing AbCIDs came from previous work showing it possible to use phage display to generate antibodies that could specifically bind to protein conformations “trapped” by binding of small molecules.42–45 In these cases, the antibody shows an increased affinity for the small-molecule-bound form of the protein, similar to a CID. However, the antibody is often able to bind the protein in the trapped conformation, independent of small molecule. For this reason, the selectivity of conformation-selective antibodies for the bound form over the apo form is limited, reducing their utility as CIDs. With the development of AbCIDs, we generated antibodies that target a small-molecule-protein complex but utilize the small-molecule as part of the binding epitope. This provided higher selectivity for the bound form of the protein, and in turn, the desired properties for use as CIDs. This solution is reminiscent of several naturally occurring CID systems, including the rapamycin-FKBP12-FRB system, in which binding of rapamycin to FKBP12 creates a novel-binding surface necessary for recognition by FRB.
While rapamycin has favorable PK properties in humans, its toxicity and immunosuppressant properties make it incompatible with regulating CAR T-cell therapy. In cellular CID assays, researchers typically use rapamycin in a concentration range of 30–100 nM, despite rapamycin toxicity being observed in cell lines at concentrations of 100–300 nM, a difference of only 3-fold.12, 46, 47 In comparison, the EC50 for activation of our AbCID CAR with ABT-737 is ~6 nM and the IC50 for cell killing is ~2 μM, a >330-fold difference. While the commonly used rapamycin analog AP21967 lacks the toxicity and immunosuppressive properties of rapamycin, its short half-life in mouse plasma (<4 hr) greatly reduces its utility in activating cell therapies in vivo. In fact, previous studies demonstrating small-molecule activation of CAR T-cells in mice have been limited due to the PK liabilities of AP21967.32 Fortunately, ABT-737 has been shown to have a half-life in mouse plasma of 14–18 hr, which should greatly facilitate the use of our AbCIDs to activate CAR T-cells in mouse models of cancer.48
To our knowledge, AbCIDs represent the first demonstration of a general strategy to engineer CIDs from existing small-molecule-protein interaction pairs. While in this study we have utilized synthetic antibody fragment libraries, we envision that diversity libraries built upon alternative binding scaffolds could be applied to this technique, including but not limited to, DARPins, FNIII, ubiquitin, knottins, and nucleic acid aptamer libraries.49, 50 We envision that much of the power of our strategy will come from the ability to rapidly generate new AbCIDs from different small-molecule-protein pairs in which a significant portion of the small-molecule is solvent accessible. We believe that AbCIDs represent a novel and promising approach to develop next-generation CID tools with the properties necessary for application in human cell therapies.
Online Methods
Small molecule and peptide reagents.
ABT-737 (>99%, ChemieTek CT-A737), ABT-263 (>99%, Selleckchem S1001), and Bak-peptide (>95%, Anaspec AS-61616) were used without further purification. For use, ABT-737, ABT-263, and Bak-peptide were each dissolved in DMSO as 10 mM stocks. Stocks were stored at –80 °C until used.
Analysis of ligand solvent exposure.
Small-molecule-protein complexes were identified in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) using their in-house advanced search feature. Search parameters used were: Molecular Weight Search: Min Molecular Weight=100.0 Max Molecular Weight=50000.0 and Binding Affinity: Binding affinity min is 0.001 and Binding affinity max is 1000 and Affinity Type is Ki and TAXONOMY is just Homo sapiens (human) and TAXONOMY is only just Homo sapiens (human). The list generated was then curated by hand so as to remove complexes in which the ligand was not an organic small molecule, resulting in a final list of 866 structures. Solvent accessible surface area for bound ligands was calculated using Naccess V2.1.1 with default parameters and hydrogen and heteroatoms considered in the calculation. The plot of solvent exposed surface area was generated using the ggplot2 package in R-studio.
Expression and biotinylation of BCL-xL, BCL-2, and BCL-W.
The genes encoding C-terminally truncated BCL-xL (residues 2–215), BCL-2 (residues 2–207), and BCL-W (residues, 2–164) with an N-terminal AviTag were purchased as gBlocks™ (IDT). The genes were cloned into the pMCSG7 vector51 using Gibson cloning. For BCL-xL a Tabaco Etch Virus (TEV) cut site was then introduced between the AviTag and BCL-xL domain using sight directed mutagenesis. The genes for BCL-2 and BCL-W were purchased with a TEV-cut site already incorporated. The sequence of the final constructs were confirmed by sequencing of the entire gene. The plasmids were transformed into BL21(DE3) E. coli cells and a single colony was used to inoculate 1.5 L of 2xYT media containing carbenicillin (100 µg/mL). The culture was grown at 37 °C to an OD600 of 1–1.2, cooled to 18 °C for 1 h and then induced at 18 °C overnight with 0.5 mM IPTG. Cells were harvested by centrifugation and the pellet were stored at –80 °C.
For each protein purification, the pellet was thawed at 0 °C and then re-suspended in 10 mL of lysis buffer (50 mM Tris, pH 8.0, 200 mM NaCl, 20 mM imidazole) supplemented with PMSF (100 µg/mL). The cells were lysed using a micro-fludizer and the lysate was cleared by centrifugation at 4 °C. The cleared lysate was added to 400 µL of Ni-NTA Superflow resin (Qiagen) and rotated at 4 °C for 1 h. The resin was washed (3x) with lysis buffer and then transferred to a spin column. The purified protein was eluted with elution buffer (50 mM Tris, pH 8.0, 200 mM NaCl, 600 mM imidazole). Fractions were analyzed by SDS-PAGE and those that were found to be >95% pure were pooled, exchanged into storage buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT) and concentrated.
The purified BCL-xL, BCL-2, and BCL-w proteins were biotinylated on their AviTags using the standard protocol provided by Avidity. Biotinylation was monitored by intact protein mass spectrometry on a Xevo G2-XS Mass Spectrometer (Waters) and found to be quantitative. The biotinylated proteins were then purified on Ni-NTA as described above, separated into aliquots, analyzed by SDS-PAGE (Supplementary Fig. 12), snap-frozen, and stored at –80 °C for later use.
Phage display selections and phage titering.
All phage selections were done according to previously established protocols.23 Briefly, selections with antibody phage library F were performed using biotinylated BCL-xL captured with streptavidin-coated magnetic beads (Promega). Prior to each selection, the phage pool was incubated with 1 µM of BCL-xL immobilized on streptavidin beads in the absence of ABT-737 in order to deplete the library of any binders to the apo form of BCL-xL. Subsequently, the beads were removed and ABT-737 was added to the phage pool at a concentration of 1 µM. In total, four rounds of selection were performed with decreasing amounts of BCL-xL antigen (100 nM, 50 nM, 10 nM and 10 nM). To reduce the deleterious effects of nonspecific binding phage, we employed a “catch and release” strategy, where specific BCL-xL binding Fab-phage were selectively eluted from the magnetic beads by the addition of 2 µg/mL TEV protease. Individual phage clones from the fourth round of selection were analyzed for sequencing.
Phage titers were performed according to standard protocols. Briefly, TEV eluted phage were used to infect log-phase XL1-Blue E. coli cells (Stratagene). Infected cells were incubated at room temperature for 20 minutes on an orbital shaker. Cells were then serially diluted and spotted on LB agar-plates with carbenicillin (50µg/mL) and incubated overnight at 37 °C. Phage titers were measured for each round of selections against both the BCL-xL/ABT-737 complex and against apo BCL-xL.
Expression of Fabs.
Fabs were expressed according to a previously described protocol.23 Briefly, C43 (DE3) Pro+ E. coli containing expression plasmids were grown in 2xYT at 37 °C to an OD600 of 0.6–0.8 and then Fab expression was induced by the addition of 1 mM IPTG. Incubation temperature was subsequently reduced to 30 °C and the cultures were allowed to shake for 16–18 h. Cells were harvested by centrifugation and Fabs were purified by Protein A affinity chromatography. Fab purity and integrity was assessed by SDS-PAGE (Supplementary Fig. 13) and intact protein mass spectrometry using a Xevo G2-XS Mass Spectrometer (Waters).
Fab ELISAs.
ELISAs were performed according to standard protocols.23 Briefly, 96-well Maxisorp plates were coated with NeutrAvidin (10 μg/ml) overnight at 4 °C and subsequently blocked with BSA (2% w/v) for 1 h at 20 °C. 20 nM of biotinylated BCL-xL was captured on the NeutrAvidin-coated wells for 30 minutes followed by the addition of various concentrations of Fab with either 1 µM ABT-737 or 0.05% DMSO for 30 minutes. The bound Fabs were then detected using a horseradish peroxidase (HRP)-conjugated anti-Fab monoclonal antibody (Jackson ImmunoResearch 109–036-097).
Binding kinetics analysis.
Biolayer interferometery data were measured using an Octet RED384 instrument (ForteBio). Biotinylated BCL-xL, BCL-2, or BCL-W were immobilized on a Streptavidin (SA) biosensor using a 200 nM solution. Serial dilutions of Fabs in kinetics buffer (PBS, pH 7.4, 0.05% Tween-20, 0.2% BSA, 10 µM biotin) with small molecule (1 µM), peptide (5 µM), or vehicle (0.05% DMSO) were used as analyte. Affinity (KD) and kinetic parameters (kon and koff) were calculated from a global fit (1:1) of the data using the Octet RED384 software. For the ABT-737 titration experiment, the concentration of AZ1 was held constant (100 nM) with serially diluted concentrations of ABT-737. For the ABT-737 washout experiment, the disassociation step was performed in the presence of AZ1 (50 nM) but absence of ABT-737.
Differential Scanning Fluoremetry.
DSF was conducted on a LC480 Lightcycler Instrument II (Roche). Briefly, purified recombinant protein was diluted to 5 µM in DSF buffer (PBS, pH 7.4, Sypro Orange 5X) with small molecule (20 µM ABT-737) or vehicle (0.05% DMSO) and then subjected to a temperature gradient (0.01 °C/s) from 25 to 95 °C. Data were continuously acquired at ∼465 nm (excitation) and ∼580 nm (emission). Data was processed to generate first derivative curves where the curve maximum was reported as the melting temperature of the protein.
Vector generation for cellular assays.
Fab AZ1 was converted into a previously described single-chain Fab construct using Gibson cloning.27
A gene encoding the Conventional CAR construct (CD8 Signal Sequence-Myc Tag-αCD19scFv-CD8 Hinge Domain-CD8 Transmembrane Domain-4IBB Co-stimulatory Region-CD3ζ Domain) was purchased as a gBlock™ (IDT). The gene was amplified by PCR and cloned into the pLX302 vector (Addgene plasmid #25896) using Gibson cloning. The sequence of the final construct was confirmed by sequencing of the entire gene. The AbCID CAR construct was generated by replacing the αCD19scFv portion of the Conventional CAR vector with the BCL-xL gene (residues 2–215) by Gibson cloning, followed by conversion of BCL-xL to BCL-xL(M159P) by site directed mutagenesis. The M159P mutation has previously been shown to prevent BCL-xL from forming a domain-swapped dimer.52 We feared that the two-dimensional confinement of the AbCID CAR on the cell membrane would promote dimer formation in BCL-xL(WT), and lead to antigen-independent activation of the CAR T-cells. The M159P mutation did not affect ABT-737 or AZ1 binding (data not shown). The sequence of the final construct was confirmed by sequencing of the entire gene.
The gene for CD19 was obtained from the ORFeome53 and fused to a P2A-mCherry gene by overlap extension PCR. The gene was cloned into the pLX302 vector using Gibson cloning. The sequence of the final construct was confirmed by sequencing of the entire gene.
Culturing of cell lines.
The NFAT reporter Jurkat cells utilized were a generous gift from Arthur Weiss. The K562 and HEK293T cells utilized were from frozen stocks maintained by the Wells lab. The cell lines were not authenticated before use. No test for mycoplasma contamination was performed. Unless otherwise noted all Jurkat and K562 cells lines were cultured in RPMI supplemented with 10% FBS and 1X Pen/Strep. All Jurkat NFAT reporter cells were maintained in G418 (2 mg/mL). All CAR containing Jurkat cell lines were maintained in puromycin (2 µg/mL) in addition to G418. CD19+ K562 cells were maintained in puromycin (2 µg/mL). HEK293T cells containing the Gal4-UAS-Fluc operon were maintained in High Glucose DMEM supplemented with 10% FBS, 1X Pen/Strep, and puromycin (2 µg/mL). All cell lines were cultured at 37 °C under 5% CO2.
Immunoblotting.
HEK293T cells were plated at approximately 0.5×106 cells/well in a 6-well plate and cultured overnight at 37 °C under 5% CO2 before transfection. The cells were transfected with a plasmid encoding scAZ1-avitag using TransIT-293 (Mirus Bio) following the manufacturer’s procedure. The cells were further incubated at 37 °C for 48 h. The cells were washed with PBS and lysed with M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with Complete™ protease inhibitor cocktail (Roche) at 4 °C for 10 minutes. Immunoblotting was performed using an anti-AviTag antibody (GenScript mouse mAb, A01738).
CRISPRa-mediated luciferase assay.
For CRISPRa-mediated transcriptional activation, the reporter HEK293T cell line containing the Gal4-UAS-Fluc operon54 was seeded at ~0.5×106 cells/well in 6-well plates and cultured under 5% CO2 at 37 °C overnight. The cells were transfected with a plasmid encoding scAZ1-VPR and another plasmid encoding dCas9-BCL-xL and Gal4 sgRNA at a 1:1 ratio. The transfected cells were trypsinized and resuspended in fresh DMEM supplemented with 10% FBS 24 h after transfection. Cells were then aliquoted into a 96-well poly-D-lysine coated plate (Corning) and allowed to adhere for 24 h before 20 nM ABT-737 was added to induce CRISPRa activity. Cells were then further incubated for 48 h before evaluation of luciferase gene expression. To determine luciferase activity, cells were lysed with Bright-Glo Luciferase Assay substrate (Promega) and analyzed using an Infinite M200 PRO plate reader (Tecan). The luciferase activities were background-subtracted with a negative control (cells expressing full-length dCas9-VPR and PHOX2B negative-sgRNA), and normalized against a positive control (cells expressing full-length dCas9-VPR and Gal4 sgRNA). For investigation of cellular dose response, different concentrations of ABT-737 (0.014 nM, 0.041 nM, 0.12 nM, 0.37 nM, 1.1 nM, 3.3 nM, 10 nM, 30 nM, 90 nM, 270 nM) were added to the cells after cells were transfected and aliquoted to a 96-well plate.
Amino Acid Sequence of scAZ1.
Light Chain:
MASDIQMTQSPSSLSASVGDRVTITCRASQSVSSAVAWYQQKPGKAPKLLIYSASSLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQYYWGFPSLFTFGQGTKVEIKRTVAAPSVFIFPPSDSQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Linker:
GGSSGSGSGSTGTSSSGTGTSAGTTGTSASTSGSGSGGGGGSGGGGSAGGTATAGASSGS
Heavy Chain:
EVQLVESGGGLVQPGGSLRLSCAASGFNLSYSSMHWVRQAPGKGLEWVASISPYSSYTSYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCARGWVGMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT
Expression of bispecific antibodies.
Expi293 (Life Technologies) cells were transiently co-transfected with two pFUSE (InvivoGen) vectors harboring the AZ1 heavy chain and the AZ1 light chain genetically fused to the αCD19 scFv at a ratio of 1:1. The ExpiFectamine 293 transfection kit (Life Technologies) was used for transfections as per manufacturer’s instructions. Cells were incubated for 7 days at 37 °C in a 5% CO2 environment before the supernatants were harvested by centrifugation. Protein was purified by Protein A affinity chromatography and assessed for quality and integrity by SDS-PAGE (Supplementary Fig. 14).
Generation of cell lines.
All CAR containing Jurkat cells and CD19+ K562 cells used for the T-cell activation experiments were generated by lentiviral transduction. To produce virus, HEK293T cells were transfected with a mixture of second-generation lentiviral packaging plasmids at ~80% confluence. FuGene HD (Promega) was used for transfection of the plasmids using 3 µg DNA (1.35 µg pCMV delta8.91, 0.15 µg pMD2-G, 1.5 µg pLX302) and 7.5 µL of FuGene HD per well of a six-well plate. Media was changed to complete DMEM after 6 h of incubation with transfection mixture. The supernatant was harvested and cleared by passing through a 0.2 µm filter 72 h post transfection. Cleared supernatant was added to target Jurkat NFAT reporter cells and K562 cells (~1 million cells per mL) with 8 µg/mL polybrene and cells were centrifuged at 1000 g at 33 °C for 2 h. Cells were then incubated with viral supernatant mixture overnight before the media was changed to fresh complete RPMI. Cells were expanded for a minimum of 48 h before they were grown in drug selection media. Drug selection for stable cell lines was started by the addition of 2 µg/mL puromycin. Following at least 72 h of incubation in puromycin containing media, cells were analyzed by flow cytometry for expression of the CAR or CD19. High expressing populations of CD19+ K562 cells were enriched by flow cytometry by gating for expression of an intracellular mCherry marker genetically linked to CD19 by a P2A sequence. Jurkat cells displaying high levels of CARs were enriched by flow cytometry by gating for Myc tag antibody staining using a Myc-Tag Mouse mAb Alexa Fluor647 conjugate (Cell Signaling 2233S). All flow cytometry cell sorting was performed using an AriaII (BD Biosciences).
Quantification of CAR-T cell activation.
Jurkat cells expressing CARs were mixed with antigen positive (CD19+) or antigen negative (CD19−) K562 target cells at a ratio of 1:2. Bispecifc antibody (AZ1-αCD19) or Fab (AZ1) and ABT-737 or DMSO was diluted in media and added to cell mixtures. After overnight incubation at 37 °C, cells were pelleted by centrifugation. NFAT-dependent GFP reporter expression was quantified by flow cytometry using a FACSCanto II (BD Biosciences). CD69 expression was quantified by immunofluorescence flow cytometry using a FACSCanto II (BD Biosciences) using an APC anti-human CD69 Antibody (Biolegend 310910) (Supplementary Fig. 15). IL-2 secretion was quantified by collection of supernatants and analysis by ELISA using the BD Human IL-2 ELISA set as per manufacturer’s protocol. All flow cytometry data analysis was performed using FlowJo software and all plots were generated using Prism software (GraphPad).
Assaying cellular toxicity of ABT-737.
WT Jurkat, AbCID CAR Jurkat, Conventional CAR Jurkat, WT K562, CD19+ K562, and HEK293T cells were plated in 96-well plates at ~5000 cells per well. Each cell line was incubated with varying concentrations of ABT-737 (10 µM initial, 3-fold serial-dilutions, 8 times) or DMSO alone (0.1%). After 24 h, cell viability was measured using a CellTiter-Glo® Luminescent Cell Viability Assay (Promega) and the manufacturer’s standard protocol. The percent viability relative to DMSO treatment was plotted and analyzed for each cell line using Prism software (GraphPad).
Statistical Analysis.
Unless otherwise noted all error bars represent the mean of 3 independent experiments ± s.d. All IC50/EC50 values reported were calculated from the mean of 3 independent experiments using 3-parameter nonlinear regression in Prism7 (GraphPad) ± s.e.m.
Data Availability.
All data generated and analyzed during the study are included either in this article or the associated supplementary information. All data and research resources, including protocols and plasmids are available upon reasonable request. The data utilized to generate Supplementary Fig. 1 is publically available in the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do).
Supplementary Material
Acknowledgments
We thank Sachdev Sidhu (University of Toronto) for providing the phage-displayed Fab library. We thank Arthur Weiss (UCSF) and Theresa Kadlecek (UCSF) for kindly providing the NFAT-dependent GFP reporter Jurkat cell line. Funding was provided by R01 grants from the NIH (CA191018 and GM097316). Z.B.H. was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation and HHMI, as well as a Pathway to Independence Award from the NIH-NCI (K99CA203002). A.J.M. was supported by a predoctoral fellowship from the NSF GRFP. D.P.N. is the Connie and Bob Lurie Fellow of the Damon Runyon Cancer Research Foundation (DRG-2204–14).
Footnotes
Competing Financial Interest
Z.B.H., A.J.M., J.A.W., and the University of California San Francisco have filed a patent application related to the technology described in this manuscript. The value of this patent application may be affected by publication of this manuscript.
References
- 1.Spencer DM, Wandless TJ, Schreiber SL & Crabtree GR Controlling signal transduction with synthetic ligands. Science 262, 1019–1024 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Fegan A, White B, Carlson JC & Wagner CR Chemically controlled protein assembly: techniques and applications. Chem. Rev 110, 3315–3336 (2010). [DOI] [PubMed] [Google Scholar]
- 3.DeRose R, Miyamoto T & Inoue T Manipulating signaling at will: chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflugers Arch 465, 409–417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lienert F, Lohmueller JJ, Garg A & Silver PA Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol 15, 95–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shekhawat SS & Ghosh I Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol 15, 789–797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen DP et al. Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity. Nat. Commun 7, 12009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straathof KC et al. An inducible caspase 9 safety switch for T-cell therapy. Blood 105, 4247–4254 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Stasi A et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N.Engl. J. Med 365, 1673–1683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeFrancesco L CAR-T’s forge ahead, despite Juno deaths. Nat. Biotechnol 35, 6–7 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Rivera VM et al. A humanized system for pharmacologic control of gene expression. Nat. Med 2, 1028–1032 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Farrar MA, Alberol-Ila J & Perlmutter RM Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 383, 178–181 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto T et al. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat. Chem. Biol 8, 465–470 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erhart D et al. Chemical development of intracellular protein heterodimerizers. Chem. Biol 20, 549–557 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kopytek SJ, Standaert RF, Dyer JC & Hu JC Chemically induced dimerization of dihydrofolate reductase by a homobifunctional dimer of methotrexate. Chem. Biol 7, 313–321 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Liang FS, Ho WQ & Crabtree GR Engineering the ABA plant stress pathway for regulation of induced proximity. Sci. Signal 4, rs2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czlapinski JL et al. Conditional glycosylation in eukaryotic cells using a biocompatible chemical inducer of dimerization. J. Am. Chem. Soc 130, 13186–13187 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schellekens H Factors influencing the immunogenicity of therapeutic proteins. Nephrol. Dial. Transplant 20 Suppl 6, vi3–9 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Lee EF et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ 14, 1711–1713 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Czabotar PE, Lessene G, Strasser A & Adams JM Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol 15, 49–63 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Besbes S, Mirshahi M, Pocard M & Billard C New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 6, 12862–12871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oltersdorf T et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL & Clardy J Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol 229, 105–124 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Hornsby M et al. A High Through-put Platform for Recombinant Antibodies to Folded Proteins. Mol. Cell. Proteomics 14, 2833–2847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah NB & Duncan TM Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J. Vis. Exp, e51383 (2014). [DOI] [PMC free article] [PubMed]
- 25.Tse C et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68, 3421–3428 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Sattler M et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Koerber JT, Hornsby MJ & Wells JA An improved single-chain Fab platform for efficient display and recombinant expression. J. Mol. Biol 427, 576–586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavez A et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi LS et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y et al. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 13, 1043–1049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fesnak AD, June CH & Levine BL Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CY, Roybal KT, Puchner EM, Onuffer J & Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Y et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed 55, 7520–7524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodgers DT et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 113, E459–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma JS et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 113, E450–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.June CH, Levine BL, Porter DL, Kalos MD & Michael MC Compositions and methods for treatment of cancer. US Patent 9,540,445 (2017). [Google Scholar]
- 37.Wei P et al. Bacterial virulence proteins as tools to rewire kinase pathways in yeast and immune cells. Nature 488, 384–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegler SF, Ramsdell F & Alderson MR The activation antigen CD69. Stem Cells 12, 456–465 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Smith-Garvin JE, Koretzky GA & Jordan MS T cell activation. Annu. Rev. Immunol 27, 591–619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 14, 943–951 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Goreshnik I & Maly DJ A small molecule-regulated guanine nucleotide exchange factor. J. Am. Chem. Soc 132, 938–940 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Gao J, Sidhu SS & Wells JA Two-state selection of conformation-specific antibodies. Proc. Natl. Acad. Sci. USA 106, 3071–3076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizk SS et al. Allosteric control of ligand-binding affinity using engineered conformation-specific effector proteins. Nat. Struct. Mol. Biol 18, 437–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staus DP et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomsen ND, Koerber JT & Wells JA Structural snapshots reveal distinct mechanisms of procaspase-3 and −7 activation. Proc. Natl. Acad. Sci. USA 110, 8477–8482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barlow AD et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia 55, 1355–1365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B et al. Rapamycin inhibiting Jurkat T cells viability through changing mRNA expression of serine/threonine protein phosphatase 2A. Transpl. Immunol 26, 50–54 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Kelly PN, Grabow S, Delbridge AR, Adams JM & Strasser A Prophylactic treatment with the BH3 mimetic ABT-737 impedes Myc-driven lymphomagenesis in mice. Cell Death Differ 20, 57–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiedler M & Skerra A in Handbook of Therapeutic Antibodies Vol. 1 (eds. Dubel S & Reichert JM) 435–474 (Wiley-VCH Verlag GmbH & Co. KGaA, 2014). [Google Scholar]
- 50.Kong HY & Byun J Nucleic Acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol. Ther 21, 423–434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seiler CY et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res 42, D1253–1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajan S et al. Structural transition in Bcl-xL and its potential association with mitochondrial calcium ion transport. Sci. Rep 5, 10609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collaboration O.R. The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat. Methods 13, 191–192 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analyzed during the study are included either in this article or the associated supplementary information. All data and research resources, including protocols and plasmids are available upon reasonable request. The data utilized to generate Supplementary Fig. 1 is publically available in the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do).