

Abstract
Obesity-induced adipose-tissue dysfunction is a critical contributor to the pathogenesis of nonalcoholic fatty liver disease (NAFLD). B cell-activating factor (BAFF) is an adipokine related to impaired insulin sensitivity, and the serum BAFF concentration is associated with NAFLD severity. In this study, we aimed to determine the direct in vivo role of BAFF in the development of insulin resistance, adipocyte dysfunction, and hepatic steatosis using BAFF−/− mice fed a high-fat diet (HFD). HFD-fed BAFF−/− mice exhibited significantly improved insulin sensitivity despite their increased weight gain and adiposity relative to HFD-fed wild-type mice. Moreover, inflammation, especially the accumulation of CD11c+ adipose-tissue macrophages, and fibrosis of epididymal adipose tissue were reduced, contributing to healthy adipose-tissue expansion in obese BAFF−/− mice. In line with metabolically healthy obesity, hepatic steatosis also decreased, and we observed attenuated de novo lipogenesis in both the livers and hepatocytes of BAFF−/− mice. Our data revealed that BAFF serves as a potential stimulator of unhealthy adipose-tissue expansion by triggering inflammation and fibrosis and ultimately leading to enhanced insulin resistance and NAFLD. Therefore, these results suggest that BAFF is a promising target for diabetes and NAFLD treatment.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is currently the most common subtype of chronic liver disease worldwide1 and is closely related to insulin resistance and metabolic risk factors. NAFLD represents a hepatic manifestation of metabolic syndrome2, and numerous pathogenic symptoms, such as insulin resistance, inflammation, enhanced oxidative stress, and mitochondrial dysfunction, are implicated in NAFLD development.
A fundamental function of adipose tissue involves the storage of excess energy in the form of triglycerides (TGs). Promoting healthy adipose-tissue expansion and better lipid storage in visceral adipose tissue (VAT) is crucial to maintaining glucose homeostasis and insulin sensitivity. Recent studies have suggested that increased adipose-tissue inflammation triggers lipolysis and tissue fibrosis, thereby accelerating the release of free fatty acids (FFAs) from adipose tissue3 and resulting in its ectopic accumulation in non-adipose tissue such as the liver4. The resulting lipotoxicity subsequently initiates a variety of metabolic disorders.
Obesity-associated inflammation is also accepted as a critical factor that initiates or exacerbates NAFLD5. Macrophage infiltration occurs in both adipose tissue and the liver to contribute to insulin resistance and NAFLD. Proinflammatory mediators exert direct effects on hepatocytes to increase lipid synthesis6 and/or act on both liver macrophages and hepatocytes to accelerate hepatic inflammation and promote hepatic steatosis. Recent reports indicate that de novo lipogenesis is a prominent abnormality in NAFLD and the key feature involved in progression to severe steatosis7. Combined with excess adipose FFA release, distinct increases in lipogenesis may contribute to obesity-related NAFLD.
B cell-activating factor (BAFF; CD257) belongs to the tumour necrosis factor (TNF)-ligand family and promotes B cell proliferation and survival, leading to increased serum immunoglobulin levels8. Additionally, BAFF plays an important role in the development of autoimmune diseases8. Previous studies have reported that BAFF is produced by mature adipocytes, as well as myeloid lineage cells and activated T cells9,10. Using mice with diet-induced obesity, we previously demonstrated that BAFF controls the production of adipokines and induces insulin resistance via impairment of insulin-receptor signalling in vitro9. Moreover, we found that BAFF is associated with NAFLD severity among Japanese patients11. These data suggest that BAFF is involved in glucose and lipid metabolism in obesity.
In an effort to better understand the impact of BAFF signalling on lipid metabolism in vivo, we previously investigated BAFF receptor (BAFF-R)-deficient mice12. Contrary to our expectation, hepatic steatosis was enhanced in obese BAFF-R-deficient mice, whereas glucose tolerance was improved and body weight was reduced relative to those of obese wild-type (WT) mice12. However, BAFF-R-deficient mice displayed increases in serum BAFF concentrations; therefore, there remains a lack of knowledge concerning the pathological significance of BAFF signalling in NAFLD. In this study, we aimed to determine the role of BAFF in insulin resistance, adipose-tissue dysfunction, and hepatic steatosis using high-fat diet (HFD)-fed mice. Our results demonstrated that, despite increased adiposity, BAFF deficiency ameliorated obesity-associated insulin resistance and inflammation in VAT and prevented fat accumulation in the liver. Our findings suggest that BAFF may have a potential clinically relevant impact on NAFLD pathogenesis.
Results
Altered lipid distribution and insulin sensitivity in BAFF−/− mice fed an HFD
We confirmed BAFF gene deletion in BAFF−/− mice by serum enzyme-linked immunosorbent assay (ELISA) and detection of mRNA expression in several organs (Supplementary Fig. S1). BAFF−/− mice showed splenic deficiencies and significantly reduced splenic weights as compared with those of WT mice (Fig. 1B).
Figure 1.
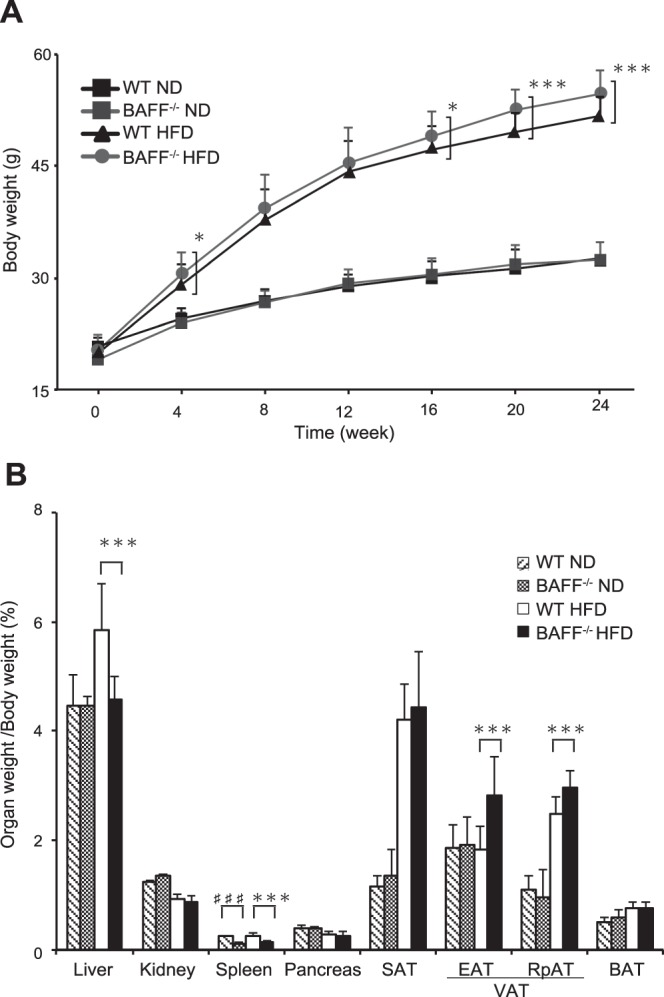
Lipid distribution in adipose tissue and liver is altered in BAFF−/− mice fed an HFD. (A) Changes in body weight of BAFF−/− and WT mice fed an ND or HFD (n = 25–55/group). (B) Organ weight [(organ weight/body weight) × 100%] in BAFF−/− and WT mice fed an ND (n = 6/group) or HFD (n = 7/group) for 24 weeks. For all bar plots shown, data are expressed as the mean ± SD, and significance was determined by two-tailed Student’s t test (A) and Mann–Whitney U test (B). ♯♯♯P < 0.001 compared to WT-ND mice. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to WT-HFD mice. BAFF, B cell-activating factor; WT, wild-type; ND, normal diet; HFD, high-fat diet; SAT, subcutaneous adipose tissue; EAT, epididymal adipose tissue; RpAT, retroperitoneal adipose tissue; VAT, visceral adipose tissue; BAT, brown adipose tissue.
There was no difference in body weight between normal chow diet (ND)-fed WT and BAFF−/− mice; however, the body weight of HFD-fed BAFF−/− mice was significantly higher than that of HFD-fed WT mice (Fig. 1A). Although VAT weight was significantly increased in HFD-fed BAFF−/− mice as compared with that in HFD-fed WT mice, liver weight was decreased (Fig. 1B). Serum TG levels did not differ between the two HFD-fed groups (WT: 56 ± 6 mg/dL; BAFF−/−: 49 ± 12 mg/dL; n = 7/group; P = 0.25).
We next examined glucose metabolism in HFD-fed BAFF−/− mice; results revealed lower fasting blood glucose and plasma insulin concentrations, as well as lower calculated homeostasis model assessment of insulin resistance indices relative to those in HFD-fed WT mice (Fig. 2A). The glucose levels at random times were also lower in HFD-fed BAFF−/− mice than in HFD-fed WT mice (WT: 271 ± 73 mg/dL; BAFF−/−: 213 ± 42 mg/dL; n = 10 /group; P < 0.05). Glucose- and insulin-tolerance tests revealed that HFD-fed BAFF−/− mice displayed better glucose tolerance and insulin sensitivity than HFD-fed WT mice (Fig. 2B,C). Glucose- and insulin-tolerance did not differ between ND-fed WT and BAFF−/− mice (Supplementary Fig. S2). These data indicated that HFD-fed BAFF−/− mice displayed significantly improved glucose tolerance as compared with HFD-fed WT mice, despite increased total body and VAT weights.
Figure 2.

BAFF−/− mice are protected against glucose intolerance and insulin resistance. (A) Fasting blood glucose, plasma insulin, and HOMA-IR values in HFD-fed BAFF−/− and WT mice over the course of 12 weeks (n = 6/group). (B) Glucose- and (C) insulin-tolerance tests (left) and corresponding area under the curve (AUC) values during the tests (right) (n = 7–14/group). For all bar plots, data are expressed as the mean ± SD, and significance was determined by the Mann–Whitney U test. *P < 0.05, **P < 0.01, and ***P < 0.001. BAFF, B cell-activating factor; WT, wild-type; ND, normal diet; HFD, high-fat diet; HOMA-IR, calculated homeostasis model assessment of insulin resistance.
Adipose-tissue inflammation is reduced in HFD-fed BAFF−/− mice
Increased adipose-tissue inflammation is considered a critical determinant of insulin resistance. We observed that the number of crown-like structures (CLSs), which are sites of M1 macrophage aggregation in obese adipose tissue, was significantly reduced in epididymal adipose tissue (EAT) from HFD-fed BAFF−/− mice as compared with that in HFD-fed WT mice (Fig. 3A). The expression of TNF-α and macrophage-specific markers, such as F4/80 and CD11c, was significantly lower in EAT from BAFF−/− mice than in WT mice after HFD feeding (Fig. 3B). However, mRNA levels of M2-like macrophage markers, such as CD206 and arginase 1, did not differ between the two groups (Fig. 3B). Flow cytometric analysis revealed that the proportion of F4/80+ CD11c+ M1-like macrophages isolated from the stromal vascular fraction (SVF) of EAT from HFD-fed BAFF−/− mice was significantly lower than that in HFD-fed WT mice (Fig. 3C). Furthermore, mRNA levels of TNF-α and resistin, which plays a role in the development of insulin resistance, were significantly lower in EAT from HFD-fed BAFF−/− mice than in EAT from HFD-fed WT mice (Fig. 3B,D), although adiponectin levels did not differ between the two groups. We also observed that serum resistin concentrations were significantly lower in HFD-fed BAFF−/− mice as compared with those in HFD-fed WT mice (Fig. 3E) and that serum TNF-α levels were undetectable in both groups. The malondialdehyde (MDA) adduct protein levels, which serve as an indicator of lipid peroxidation and oxidative stress, in the EAT were not different between two groups (Supplementary Fig. 3A). These data indicated that BAFF deficiency was associated with decreases in HFD-induced adipose-tissue inflammation via reduced proinflammatory cytokine levels.
Figure 3.

Adipose tissue inflammation is alleviated in BAFF−/− mice. (A) Representative H&E staining of EAT (left). The arrows point to the CLSs formed by macrophage aggregation. Quantitative measurement of CLSs (right) (scale bar, 200 μm; images from six different fields; n = 5/group). (B,D) Relative mRNA levels of the indicated genes in the EAT (n = 12/group). (C) Representative flow cytometry plots showing frequencies of CD11c+ F4/80+ macrophages among stromal cells isolated from EATs from HFD-fed BAFF−/− and WT mice (left). Quantification of Cd11c+ F4/80+ cells isolated from EAT (right) (n = 9–12/group). (E) Serum resistin concentrations (n = 14, 15/group). For all bar plots, data are expressed as the means ± SEM. *P < 0.05 and ***P < 0.001 (Mann–Whitney U test). BAFF, B cell-activating factor; WT, wild-type; H&E, haematoxylin and eosin; CLS, crown-like structure; HFD, high-fat diet; EAT, epididymal adipose tissue.
Adipose-tissue fibrosis is reduced in HFD-fed BAFF−/− mice
Adipose-tissue expansion is often associated with adipose-tissue remodelling. In mouse models of abnormal collagen accumulation, rigidity related to developmental fibrosis limits adipose-tissue expansion13. Consistent with the observed increase in VAT weight, histological analysis revealed that the adipocyte cell size in EAT was increased in BAFF−/− mice as compared with that in EAT from WT mice fed an HFD for 24 weeks (Fig. 4A). By contrast, Sirius red staining revealed extensive interstitial fibrosis of EAT from WT mice, which was significantly reduced in BAFF−/− mice (Fig. 4B,C). Additionally, we confirmed that the total collagen content in EAT from BAFF−/− mice was significantly lower than that in EAT from WT mice (Fig. 4D). The mRNA expression of transforming growth factor (TGF)-β1 was lower in EAT from BAFF−/− mice relative to levels in EAT from WT mice, whereas the α-smooth muscle actin and collagen type 1 mRNA levels did not differ between the two groups (Fig. 4E). These results indicated that a reduction in interstitial fibrosis in EAT from HFD-fed BAFF−/− mice may contribute to adipocyte expansion.
Figure 4.

Interstitial fibrosis in white adipose tissue is reduced in BAFF−/− mice. (A) The median adipocyte size (left) and the distribution of adipocyte size (right) in EAT from HFD-fed BAFF−/− and WT mice. Representative (B) Sirius red staining and (C) quantification of Sirius red-positive areas of EAT (scale bar, 100 μm; images from 10 different fields; n = 6/group). (D) Total collagen contents in EAT (n = 9–13/group). (E) Expression of genes related to fibrosis in EAT (n = 14–15/group). For all bar plots, data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (Mann-Whitney U test). BAFF, B cell-activating factor; HFD, high-fat diet; WT, wild-type; EAT, epididymal adipose tissue; TGF, transforming growth factor; SMA, smooth muscle actin; Col, Collagen.
Hepatic steatosis is attenuated in BAFF−/− mice fed an HFD
We then investigated whether BAFF deficiency influenced the development of fatty liver. Histological examination revealed that hepatic fat accumulation was dramatically lower in HFD-fed BAFF−/− mice than in HFD-fed WT mice (Fig. 5A,B), which was consistent with the lower liver weight observed in BAFF−/− mice. Furthermore, liver TG and cholesterol levels were significantly lower in HFD-fed BAFF−/− mice relative to those in HFD-fed WT mice (P < 0.01; Fig. 5C,D), although serum alanine transaminase (ALT) levels did not differ between the two groups (WT: 111 ± 54 IU/L; BAFF−/−: 111 ± 35 IU/L; n = 7/group; P = 0.81). Additionally, histological examination revealed no or mild inflammation in the livers of both groups (Fig. 5A).
Figure 5.

Hepatic steatosis is ameliorated in HFD-fed BAFF−/− mice. (A) Representative H&E staining of livers from mice after 24 weeks of ND or HFD consumption (scale bar, 500 μm). (B) Representative OsO4 staining (left) and quantification of the OsO4-positive areas (right) of livers (scale bar, 500 μm; images from eight different fields; n = 8/group). (C) Hepatic TG and (D) total cholesterol levels (n = 5/group). For all bar plots, data are expressed as the mean ± SEM, and significance was determined by the Mann–Whitney U test. *P < 0.05 and ***P < 0.001. BAFF, B cell-activating factor; WT, wild-type; ND, normal diet; HFD, high-fat diet; TG, triglyceride.
To investigate mechanisms underlying the attenuation of hepatic steatosis in HFD-fed BAFF−/− mice, we analysed the expression of genes related to lipid metabolism in the livers of both groups. The expression of CD36, which is related to fatty acid uptake and transport, was significantly downregulated in the livers of HFD-fed BAFF−/− mice relative to levels in HFD-fed WT mice, as were levels of sterol regulatory element-binding protein 1c (SREBP-1c) and fatty acid synthase (FAS), which are related to lipogenesis (Fig. 6A). We confirmed that the SREBP-1c and FAS protein levels were also lower in the livers of BAFF−/− mice (Fig. 6B). Additionally, levels of TNF-α, monocyte chemoattractant protein-1, and macrophage markers such as F4/80 and CD11c were significantly lower in the livers of BAFF−/− mice than in those of WT mice (Fig. 6C), although histologic analysis revealed that inflammatory cell infiltration did not differ between groups according to histologic analysis (Fig. 5A). The MDA adduct protein levels in the livers did not differ between the two groups (Supplementary Fig. 3B). These data suggested that BAFF deficiency prevented hepatic steatosis by decreasing de novo lipogenesis in the liver as well as fatty acid influx from EAT.
Figure 6.
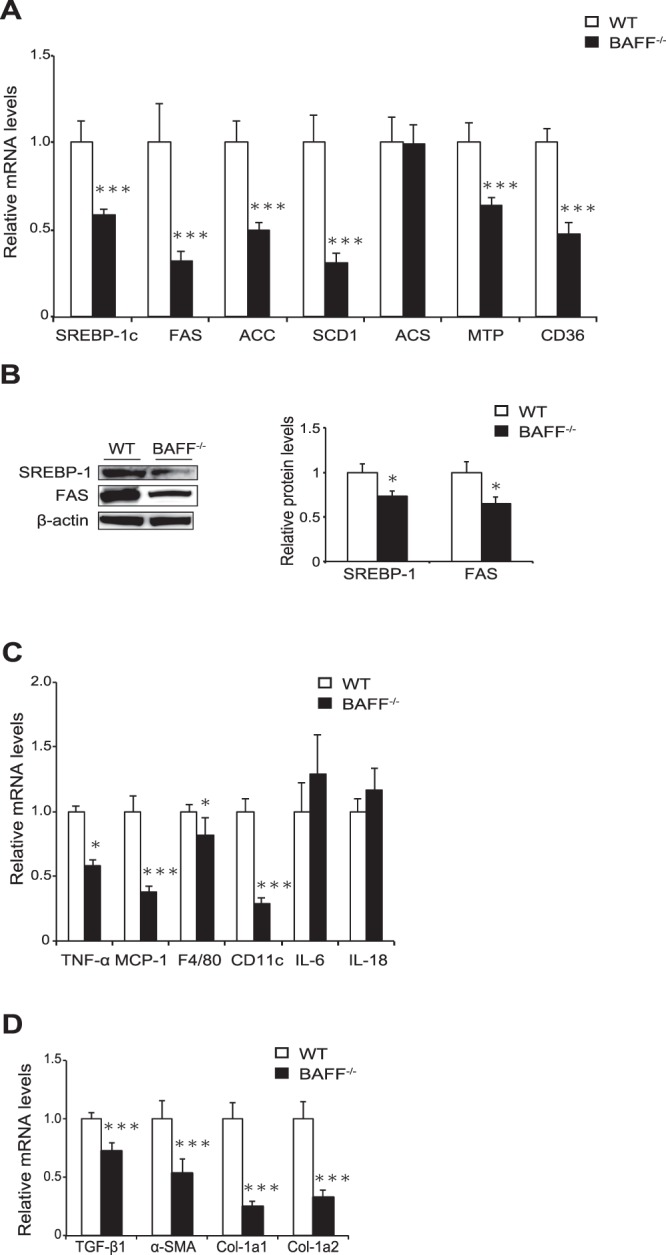
The expression of genes related to steatosis is downregulated in the livers of BAFF−/− mice. (A,C,D) The expression of genes related to lipogenesis (A), inflammation (C), and fibrosis (D) in mouse livers after 24 weeks of HFD consumption (n = 9–14/group). (B) The expression of proteins related to lipogenesis (n = 4/group). Original gel documents of western blot were shown in Supplementary Fig. S4. Protein levels were normalized to those of β-actin, and those of WT mice were set at 1. For all bar plots, data are expressed as the mean ± SEM, and significance was determined by Mann–Whitney U test. *P < 0.05 and ***P < 0.001. BAFF, B cell-activating factor; WT, wild-type; HFD, high-fat diet; FAS, fatty acid synthase; SREBP1, sterol regulatory element-binding protein; ACC, acetyl-CoA carboxylase; SCD, stearoyl-CoA desaturase; ACS, acetyl-CoA synthetase; MTP, microsomal triglyceride transfer protein; TNF, tumour necrosis factor.
BAFF deficiency decreases lipid synthesis in hepatocytes
We next evaluated the expression of genes related to lipid metabolism using primary cultured hepatocytes from HFD-fed mice. Consistent with the results obtained from whole livers, the expression of genes related to lipid synthesis was significantly downregulated in hepatocytes from BAFF−/− mice as compared with that in hepatocytes from WT mice (Fig. 7A).
Figure 7.
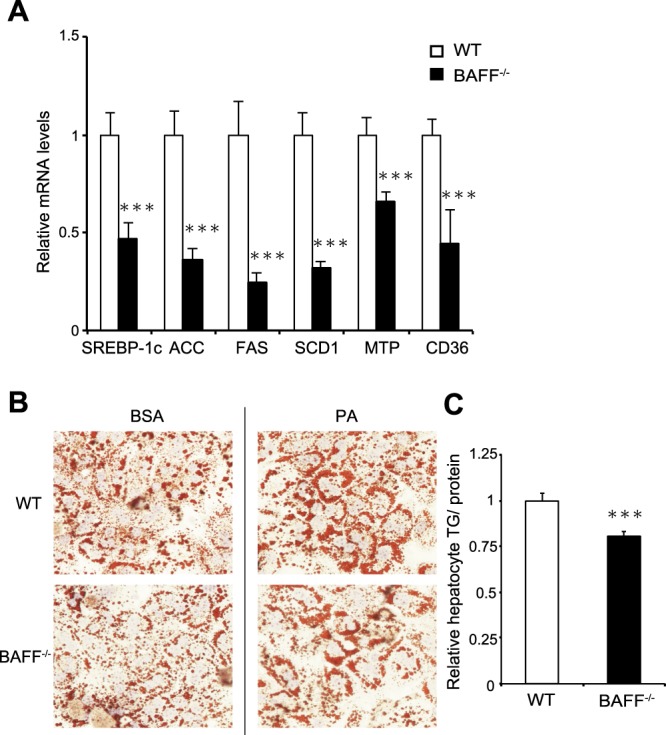
Hepatocyte fat deposition is reduced in BAFF−/− mice. (A) The expression of genes related to lipogenesis in primary hepatocytes from HFD-fed BAFF−/− and WT mice. (B) Hepatocyte fat deposition. Representative Sudan III staining of primary hepatocytes from BAFF−/− and WT mice exposed to BSA or PA (left). Quantification of TG in primary hepatocytes exposed to PA (right) (n = 8/group). For all bar plots, data are expressed as the mean ± SEM. ***P < 0.001 (Mann–Whitney U test). BAFF, B cell-activating factor; WT, wild-type; FAS, Fatty acid synthase; SREBP1, sterol regulatory element-binding protein; ACC, acetyl-CoA carboxylase; SCD, stearoyl-CoA desaturase; MTP, microsomal triglyceride transfer protein; BSA, bovine serum albumin; PA, palmitate, TG; triglyceride.
Furthermore, we analysed the role of BAFF in lipid accumulation in an in vitro model of hepatic steatosis. Primary cultured hepatocytes were exposed to palmitate in vitro. As shown in Fig. 7B, fat accumulation was attenuated to a greater degree in primary cultured hepatocytes from BAFF−/− mice than in those from WT mice. These results indicated that BAFF deficiency profoundly attenuated lipid accumulation in the hepatocytes of HFD-fed mice.
Discussion
We previously reported that BAFF impairs insulin sensitivity and is associated with NAFLD severity9,11, indicating that this molecule is an important factor in glucose and lipid metabolism. In the present study, we demonstrated that BAFF deficiency has beneficial effects on metabolic dysfunction in diet-induced obesity models. First, as expected, HFD-fed BAFF−/− mice showed lower fasting glucose levels than HFD-fed WT mice (Fig. 2A). Additionally, HFD-fed BAFF−/− mice showed better glucose tolerance and insulin sensitivity after insulin challenge than HFD-fed WT mice (Fig. 2B,C). Moreover, HFD-fed BAFF−/− mice displayed marked improvements in hepatic steatosis as compared with that observed in HFD-fed WT mice (Fig. 5), despite a lack of body weight reduction. Both insulin and glucose stimulate the expression of SREBP-1, a major transcription factor that positively regulates de novo lipogenic enzymes in the liver14. Yahagi et al.15 reported that the absence of SREBP-1 ameliorates fatty liver development, but not obesity or insulin resistance, in leptin-deficient ob/ob mice. In the present study, we observed that BAFF deficiency attenuated hyperglycaemia in HFD-fed mice and was associated with the downregulation of genes related to de novo lipogenesis, including SREBP-1c and FAS, in the liver (Fig. 6A,B). We also confirmed that the expression of genes related to lipid synthesis was significantly downregulated in primary cultured hepatocytes obtained from HFD-fed BAFF−/− mice (Fig. 7A) and that BAFF deficiency alleviated palmitate-induced fat deposition in primary cultured hepatocytes in vitro (Fig. 7B). These data indicated that a decrease in de novo lipogenesis directly contributed to the attenuation of hepatic steatosis in HFD-fed BAFF−/− mice.
In addition to de novo lipogenesis, liver fatty acids are derived from VAT. Obesity induces chronic low-grade inflammation in VAT16, which leads to morphological and functional changes, including alterations in VAT-resident immune-cell profiles, dynamic remodelling of the extracellular matrix (ECM), and altered production of adipokines17. Moreover, we previously reported that BAFF was preferentially expressed in VAT and inhibited insulin-signalling pathways in adipocytes9. As expected, in the present study, we demonstrated that BAFF deficiency reduced the accumulation of proinflammatory CD11c+ macrophages and CLS formation in VAT from HFD-fed mice (Fig. 3A,C). Additionally, levels of resistin, whose expression is induced during adipogenesis to interfere with multiple steps in the insulin-signalling cascade, were significantly lower in HFD-fed BAFF−/− mice than in HFD-fed WT mice (Fig. 3D,E). This finding is consistent with results from our previous report showing that BAFF induced resistin expression in adipocytes in vitro and in vivo9. These results suggest that BAFF may drive insulin resistance related to adipokine production, as well as suppress the infiltration of proinflammatory macrophages into VAT.
In this study, we observed that HFD-fed BAFF−/− mice displayed significant increases in EAT weight and adipocyte size (Figs 1 and 4) but decreases in liver weight and hepatic fat accumulation as compared with HFD-fed WT mice (Figs 1 and 5). A previous study demonstrated that hepatic steatosis is associated with the loss of epididymal fat mass in HFD-fed mice18. Moreover, in humans, loss of adipocyte mass (lipodystrophy) is associated with hepatomegaly due to steatosis19. These observations are consistent with our findings. In addition to insulin resistance and proinflammatory conditions, adipose-tissue fibrosis may be involved in this process. A previous study suggested that excessive accumulation of ECM proteins (i.e., collagen) is a key feature of adipose-tissue dysfunction during obesity20. Further, several groups have reported that fibrosis of VAT limits lipid-storage capacity by inhibiting adipocyte hypertrophy, which plays a role in ectopic lipid accumulation in the liver21,22. Divoux et al.23 reported that adipose-tissue fibrosis is negatively correlated with adipocyte diameter in human adipose tissue. Moreover, increased interstitial fibrosis decreases ECM flexibility, reduces tissue plasticity, and limits adipose-tissue expansion24, leading to an increased frequency of adipocyte cell death and enhanced inflammatory response. TGF-β signalling contributes to ECM deposition, specifically in VAT25. The reduced TGF-β1 expression in VAT from HFD-fed BAFF−/− mice (Fig. 4), as observed in the present study, may support this process. VAT expansion in the relative absence of fibrosis and inflammation maintains metabolic homeostasis in response to an energy surfeit26 and represents a hallmark of metabolically “healthy” obesity27. These data suggest that BAFF−/− mice are protected against obesity-induced adipose-tissue fibrosis and exhibit increased lipid-storage capacity, which may reduce FFA efflux from the VAT to the liver. Recent studies indicate that adipose oxidative stress in the adipose tissue is associated with metabolic disorders and obesity, including healthy adipose tissue expansion and ectopic lipid accumulation28. However, oxidative stress levels, as estimated by the levels of MDA adducts, in the VAT were not different between HFD-fed BAFF−/− mice and HFD-fed WT mice (Supplementary Fig. 3A).The reasons for this discrepancy are not known; further studies are necessary in this regard.
Here, we demonstrated for the first time a role for BAFF in hepatic steatosis using BAFF−/− mice. Previously, Kim et al.29,30 reported that BAFF depletion ameliorated glucose intolerance and resulted in increased body weight in HFD-fed mice, which is consistent with our observations. However, they30 showed that BAFF−/− mice did not exhibit an elevated fat mass in the EAT as compared with WT mice fed with an HFD for 10 weeks. Additionally, liver weight did not differ between the two groups, although they did not investigate liver histology. This discrepancy may be caused by the different protocols used in the two studies. Strissel et al.18 reported that EAT weight and hepatic steatosis were inversely correlated after 12 weeks of HFD feeding, which may be related to adipocyte death and adipose-tissue remodelling.
The phenotype of BAFF-R-deficient mice is similar to that of BAFF−/− mice31. Both types of mice exhibit a reduced number of mature B cells and impaired antigen-dependent antibody responses. Additionally, both BAFF-R−/− and BAFF−/− mice exhibit attenuated inflammation in VAT and improved insulin resistance12. However, HFD-fed BAFF-R−/− mice displayed exacerbated hepatic steatosis along with elevated expression of genes related to de novo lipogenesis12. The reasons for this discrepancy between BAFF-R−/− and BAFF−/− mice are unclear; however, the serum BAFF concentrations in BAFF-R−/− mice were approximately threefold higher than those in WT mice (data not shown). In our preliminary study, we measured serum BAFF concentrations in patients with histologically diagnosed NAFLD (n = 65) and found that the prevalence of severe steatosis (>66%) was significantly higher among patients with higher BAFF concentrations than among those with lower BAFF concentrations (data not shown). This may support the idea that BAFF plays a role in exacerbating hepatic steatosis. We analysed the expression levels of three previously identified BAFF receptors (BAFF-R, B cell-maturation antigen, and transmembrane activator and calcium modulating cyclophilin-ligand interactor) and found that BAFF-R may represent the main functional receptor in the mouse liver12. However, the roles of other receptors, including those that remain unidentified, cannot be disregarded. Furthermore, recent evidence has indicated that B cells contribute to the development and promotion of insulin resistance32, adipose-tissue dysfunction33, and NAFLD34. In the present study, we primarily focused on the metabolic aspects of BAFF in VAT and the liver; however, BAFF also plays an important role in maintaining a healthy immune system8. Therefore, future studies may be necessary to investigate the immunological role(s) of BAFF and B-cell compartments.
In summary, we demonstrated that BAFF depletion improved HFD-induced insulin resistance and liver steatosis via suppression of VAT inflammation and fibrosis, subsequently leading to beneficial VAT expansion. Our data suggested that targeting BAFF might be beneficial for treating obesity-related NAFLD and diabetes.
Methods
Animals
All methods were carried out in accordance with the guidelines and regulations of Ehime University (Ehime, Japan), and all experimental protocol was approved by Ehime University Animal Research (No. 05TI70-16).
Male C57BL/6J WT mice and B6.129S2-Tnfsf13btm1Msc/J (BAFF−/−) mice in C57BL/6J background were purchased from CLEA Japan (Tokyo, Japan) and the Jackson Laboratory (Bar Harbor, ME, USA), respectively. They were maintained in a temperature-, humidity-, and light-controlled room (12-h light/dark cycles), allowed free access to water, and fed an ND (13% fat, 26% protein, and 60% carbohydrates; 360 kcal/100 g; Oriental Yeast, Tokyo, Japan). Six-week-old animals were fed ad libitum with ND or HFD (D12492; 60% fat, 20% protein, and 20% carbohydrates; 520 kcal/100 g; Research Diets, New Brunswick, NJ, USA).
Serum was extracted after 15 h of fasting and stored at −80 °C. In some experiments, serum was extracted at random times. Serum TG and ALT levels were measured using a Hitachi 7180 Autoanalyzer (Hitachi, Ltd., Tokyo, Japan). The liver and EAT were harvested, submerged in RNA-later (Life Technologies, Carlsbad, CA, USA) overnight, and stored at −20 °C until use.
Glucose- and insulin-tolerance tests
Glucose-tolerance tests were performed after a 16-h fast. Blood glucose concentrations were measured by a blood glucose test meter (Antisense III; HORIBA Medical, Kyoto, Japan) at 0, 15, 30, 60, 90, and 120 min after intraperitoneal injection of glucose (1.5 mg/g body weight). Insulin sensitivity was assessed using an insulin-tolerance test. After 6 h of fasting, insulin (1 U/kg body weight; Eli Lilly, Indianapolis, IN, USA) was administered intraperitoneally, and blood samples were drawn from the tail vein at 0, 30, 60, 90, and 120 min after administration. Plasma insulin levels were measured with an ELISA kit (Morinaga Institute of Biological Science, Kanagawa, Japan).
Histological and morphometric analysis
Liver tissue and EAT were fixed with neutral-buffered formalin and embedded in paraffin. Sections (3-µm-thick) were stained with haematoxylin and eosin (H&E) or Sirius red, and adipocyte size and number in the EAT were measured digitally in H&E sections (10×) using ImageJ software (6 sections per animal, n = 5 animals/group; National Institutes of Health, Bethesda, MD, USA). The CLS density of EAT was obtained by counting the total numbers of CLSs and adipocytes per section. For the measurement of fibrosis, Sirius red-positive areas were measured digitally in histological light-microscopy images (10×; 10 sections per animal, n = 6 animals/group). To evaluate the degree of fat accumulation, livers were stained with OsO4. Briefly, fixed livers were fixed again with buffer containing 2% OsO4, 5% potassium dichromate, and acetic acid for 24 h, and OsO4-positive areas were measured digitally using ImageJ software (6 sections per animal, n = 8 animals/group; National Institutes of Health).
Isolation of SVF and flow cytometric analysis
EAT was collected and cut into small pieces. After filtering through a 40-μm strainer, tissues were centrifuged and resuspended in RPMI(−) medium. The dissociated SVF was washed twice with PBS, incubated for 10 min in erythrocyte-lysing buffer, and washed again with PBS. Cells were stained with the following antibodies and incubated for 30 min at 4 °C in the dark: CD11c (BioLegend, San Diego, CA, USA) and F4/80 (BD Biosciences, Franklin Lakes, NJ, USA). Staining was analysed by fluorescence-assisted cell sorting (Galios; Beckman Coulter, Tokyo, Japan) using FlowJo 7.6.3. software (Tree Star, Ashland, OR, USA).
Quantitative real-time PCR
RNA was extracted from livers and EAT using an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and an RNeasy Plus Lipid Kit (Qiagen), respectively. Reverse-transcription reactions were performed using SYBR Green I (Roche Diagnostics, Basel, Switzerland) on a LightCycler 480II (Roche Diagnostics). Primer sequences and annealing temperatures are provided in Supplementary Table S1. Gene-expression data were normalized with the housekeeping gene encoding hypoxanthine phosphoribosyltransferase 1 and expressed as a ratio to values obtained for WT mice.
ELISA
Serum concentrations of BAFF and resistin were determined using ELISA (MBLYS0 for BAFF and MRSN00 for resistin; R&D Systems, Minneapolis, MN, USA). The MDA adduct levels in the liver and EAT were estimated using a commercially available ELISA kit (Oxiselect MDA Adduct Competitive ELISA kit; Cell Biolabs, San Diego, CA, USA), according to the manufacturer’s protocol.
Collagen content in adipose tissue
Total collagen content in adipose tissue was measured using a commercially available kit (QuickZyme total collagen assay; QuickZyme Biosciences, Leiden, Netherlands).
Measurement of hepatic TG and cholesterol
Hepatic TG and cholesterol levels were measured at Skylight Biotech (Akita, Japan) using the Folch technique with Cholestest TG and Cholestest CHO kits (Sekisui Medical, Tokyo, Japan), respectively.
Western blotting analysis
Total liver and isolated primary hepatocyte lysates were prepared using radioimmunoprecipitation assay buffer with a protease-inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Protein (30 μg) was placed in the wells of 4–12% Bis-Tris Gels (Invitrogen, Carlsbad, CA, USA) and resolved. The products were then blotted onto a polyvinylidene difluoride membrane using NuPage transfer buffer and XCell SureLock (both from Invitrogen) and incubated with the following specific antibodies at 4 °C overnight: FAS (1:1000; #3180; Cell Signaling Technology, Danvers, MA, USA), SREBP-1 (1:1000; AB28481; Abcam, Cambridge, UK), and β-actin (1:3000; MAB1501; Millipore, Billerica, MA, USA).
Isolation of primary cultured hepatocytes
Primary cultured hepatocytes were isolated by liver perfusion. Briefly, after digestion with Liver Digest Medium (Life Technologies), the liver was excised, minced, filtered, and centrifuged. We washed the samples twice with Williams’ Medium E (Gibco; Life Technologies, Gaithersburg, MD, USA) supplemented with 10% foetal bovine serum (FBS), 1% penicillin–streptomycin, insulin, 1% l-glutamine, and 1.5% HEPES. For mRNA-expression analysis, hepatocytes were used before attachment. To assess fat deposition, after attachment, primary cultured hepatocytes were cultured with Williams’ Medium E supplemented with 10% FBS, 1% penicillin–streptomycin, 1% l-glutamine, and 1.5% HEPES added to bovine serum albumin (BSA) or palmitate (0.3 mM) for 18 h.
Measurement of hepatocyte fat deposition
Cells were stained with Sudan III, and additional cells were harvested and subjected to a chemical assay to quantify intracellular levels of TGs using a Triglyceride E-Test (Wako Pure Chemical Company, Osaka, Japan) using the Bligh–Dyer procedure.
Statistical analysis
Data were analysed using JMP version 11.2.0 software (SAS Institute, Cary, NC, USA), and values are presented as the mean ± SEM or SD. Normally distributed and skewed data were analysed using Student’s t tests and Mann–Whitney U tests, respectively. Differences were considered statistically significant at P < 0.05.
Supplementary information
Acknowledgements
We thank Mr. Kenji Tanimoto, Ms. Takako Muneta, Ms. Sakiko Sugawara, Ms. Yuki Kokubun, and Ms. Takana Fujino for their valuable contributions to this study. This research was supported in part by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (JSPS KAKENHI 15K09007 and 18K07911).
Author Contributions
Y.N. designed and performed the experiments, analysed data, and wrote the manuscript; M.A. designed and supervised the experiments and revised the manuscript. K.K. and T.M. advised on and conducted the animal experiments. T.W., O.Y., M.H. and B.M. provided technical advice on molecular and immunological experiments and data analysis. H.Y.supervised the experiments and critically revised the manuscript.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37403-y.
References
- 1.Rinella ME, Sanyal AJ. NAFLD in 2014. Genetics, diagnostics, and therapeutic advances in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2015;12:65–66. doi: 10.1038/nrgastro.2014.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010;53:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J. Clin. Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuel VT, Shulman GI. Mechanism for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cay Y, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology. 2018;68:48–61. doi: 10.1002/hep.29777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C, et al. Targeting arginase-II protects mice from high-fat-diet-induced hepatic steatosis through suppression of macrophage inflammation. Sci. Rep. 2016;6:20405. doi: 10.1038/srep20405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackay F, Silveira PA, Brink R. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr. Opin. Immunol. 2007;19:327–336. doi: 10.1016/j.coi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Hamada M, et al. B cell-activating factor controls the production of adipokines and induces insulin resistance. Obesity. 2011;19:1915–1922. doi: 10.1038/oby.2011.165. [DOI] [PubMed] [Google Scholar]
- 10.Alexaki VI, et al. Adipocytes as immune cells: differential expression of TWEAK, BAFF, and APRIL and their receptors (Fn14, BAFF-R, TACI, and BCMA) at different stages of normal and pathological adipose tissue development. J. Immunol. 2009;183:5948–5956. doi: 10.4049/jimmunol.0901186. [DOI] [PubMed] [Google Scholar]
- 11.Miyake T, et al. B cell-activating factor is associated with the histological severity of nonalcoholic fatty liver disease. Hepatol. Int. 2013;7:539–547. doi: 10.1007/s12072-012-9345-8. [DOI] [PubMed] [Google Scholar]
- 12.Kawasaki K, et al. Blockade of B cell-activating factor signaling enhances hepatic steatosis induced by a high-fat diet and improves insulin sensitivity. Lab. Invest. 2013;93:311–321. doi: 10.1038/labinvest.2012.176. [DOI] [PubMed] [Google Scholar]
- 13.Vila IK, et al. Immune cell toll-like receptor 4 mediates the development of obesity- and endotoxemia- associated adipose tissue fibrosis. Cell Rep. 2014;7:1116–1129. doi: 10.1016/j.celrep.2014.03.062. [DOI] [PubMed] [Google Scholar]
- 14.Hasty AH, et al. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J. Biol. Chem. 2000;275:31069–31077. doi: 10.1074/jbc.M003335200. [DOI] [PubMed] [Google Scholar]
- 15.Yahagi N, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J. Biol. Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 16.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 17.Lawler HM, et al. Adipose tissue hypoxia, inflammation, and fibrosis in obese insulin-sensitive and obese insulin-resistant subjects. J. Clin. Endocrinol. Metab. 2016;101:1422–1428. doi: 10.1210/jc.2015-4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strissel KJ, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 19.Garg A. Acquired and inherited lipodystrophies. N. Engl. J. Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa Y, et al. Repression of adipose tissue fibrosis through a PRDM16-GTF2IRD1 complex improves systemic glucose homeostasis. Cell Metab. 2018;27:180–194. doi: 10.1016/j.cmet.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan T, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol. Cell. Biol. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka M, et al. Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nat. Commun. 2014;5:4982. doi: 10.1038/ncomms5982. [DOI] [PubMed] [Google Scholar]
- 23.Divoux A, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chun TH, et al. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591. doi: 10.1016/j.cell.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 25.Lee MJ. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018;1864:1160–1171. doi: 10.1016/j.bbadis.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 26.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity, and the metabolic syndrome–an allostatic perspective. Biochim. Biophys. Acta. 2010;1801:338–349. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Denis GV, Obin MS. ‘Metabolically healthy obesity’: origins and implications. Mol. Aspects Med. 2013;34:59–70. doi: 10.1016/j.mam.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okuno Y, et al. Oxidative stress inhibits healthy adipose expansion through suppression of SREBF1-mediated lipogenic pathway. Diabetes. 2018;67:1113–1127. doi: 10.2337/db17-1032. [DOI] [PubMed] [Google Scholar]
- 29.Kim B, Do MS, Hyun CK. B-cell-activating factor deficiency attenuates high-fat diet-induced glucose intolerance by potentiating adipose tissue function. Biochem. Biophys. Res. Commun. 2015;464:1171–1177. doi: 10.1016/j.bbrc.2015.07.099. [DOI] [PubMed] [Google Scholar]
- 30.Kim DH, Do MS. BAFF knockout improves systemic inflammation via regulating adipose tissue distribution in high-fat diet-induced obesity. Exp. Mol. Med. 2015;47:e129. doi: 10.1038/emm.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J. Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 32.Shen L, et al. B-1a lymphocytes attenuate insulin resistance. Diabetes. 2015;64:593–603. doi: 10.2337/db14-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duffaut C, Galitzky J, Lafontan M, Bouluomie A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem. Biophys. Res. Commun. 2009;384:482–485. doi: 10.1016/j.bbrc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Bigorgne AE, et al. Obesity-induced lymphocyte hyperresponsiveness to chemokines: a new mechanism of fatty liver inflammation in obese mice. Gastroenterology. 2008;134:1459–1469. doi: 10.1053/j.gastro.2008.02.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.