

Abstract
Methylation of HBV cccDNA has been detected in vivo and in vitro; however, the mechanism and its effects on HBV replication remain unclear. HBx derived from a 1.2-mer HBV replicon upregulated protein levels and enzyme activities of DNA methyltransferase 1 (DNMT1), 3a, and 3b, resulting in methylation of the negative regulatory region (NRE) in cccDNA, while none of these effects were observed with an HBx-null mutant. The HBx-positive HBV cccDNA expressed higher levels of HBc and produced about 4-fold higher levels of HBV particles than those from the HBx-null counterpart. For these effects, HBx interrupted the action of NRE binding protein via methylation of the C-1619 within NRE, resulting in activation of the core promoter. Treatment with 5-Aza-2′dC or DNMT1 knock-down drastically impaired the ability of HBx to activate the core promoter and stimulate HBV replication in 1.2-mer HBV replicon and in vitro infection systems, indicating the positive role of HBx-mediated cccDNA methylation in HBV replication.
Keywords: covalently closed circular DNA, DNA methylation, HBx, hepatitis B virus, negative regulatory region binding protein
INTRODUCTION
Hepatitis B virus (HBV) is a major pathogen causing human liver diseases, ranging from acute and chronic hepatitis to liver cirrhosis and hepatocellular carcinoma (Villeneuve, 2005). It is a small enveloped virus with a 3.2-kb partially double stranded DNA genome. Upon entry into the nucleus, HBV genome is repaired to form covalently closed circular DNA (cccDNA), which serves a template for the synthesis of 3.5-kb pregenomic RNA (pgRNA) and three other transcripts (Quasdorff and Protzer, 2010). Besides serving mRNA for viral polymerase and core protein (HBc), pgRNA is also encapsidated for the replication of viral genome via reverse transcription (Quasdorff and Protzer, 2010). Therefore, transcription of pgRNA from cccDNA is now considered as a rate-limiting step in the virus life cycle (Gunther et al., 1998). Transcription of pgRNA from cccDNA is regulated by one of the four HBV promoters termed core promoter (Kramvis and Kew, 1999). Two enhancer elements, EnhI and EnhII, regulate the core promoter in a position- and orientation-dependent manner (Kramvis and Kew, 1999; Su and Yee, 1992; Yuh et al., 1992). A number of cellular transcription factors have been shown to regulate the activities of the core promoter via enhancers (Curtil et al., 2014; Ishida et al., 2000; Kramvis and Kew, 1999; Su and Yee, 1992; Yuh et al., 1992). Transcription from the core promoter is also modulated by a negative regulatory element (NRE) located between the two enhancers (Kramvis and Kew, 1999; Lo and Ting, 1994). A repressor termed NRE binding protein (NREBP) binds to a specific site on the NRE to inhibit pgRNA synthesis from the core promoter (Sun et al., 2001). Accordingly, overexpression of NREBP represses transcription of pgRNA from cccDNA, which ultimately decreases the production of HBV virions (Sun et al., 2001). The relative importance of enhancers and NRE and regulation of their activities during HBV replication are largely unknown.
Although initial studies suggested that HBx is unnecessary for virus replication in cell culture (Blum et al., 1992; Yaginuma et al., 1987), the importance of HBx during HBV infection have been demonstrated by several experimental systems, including human hepatocyte chimeric mice (Tsuge et al., 2010), HBV transgenic mice (Xu et al., 2002), tail vein-injected mice (Keasler et al., 2009), and cell culture models (Cha et al., 2009; Keasler et al., 2007; Lucifora et al., 2011). HBx can directly stimulate HBV gene expression from the cccDNA template (Keasler et al., 2007; Tang et al., 2005). For this effect, HBx interacts with cellular proteins, such as damage-specific DNA-binding protein 1 (DDB1) to target the ‘structural maintenance of chromosomes’ (Smc) complex Smc5/6 for degradation, which relieves the role of Smc5/6 as a restriction factor selectively blocking HBV DNA transcription (Decorsière et al., 2016; Hodgson et al., 2012; Leupin et al., 2005). HBx may also contribute indirectly to HBV replication by deregulating cellular signal transduction pathways, such as cytosolic calcium signaling (Bouchard et al., 2001) and phosphatidylinositol 3-kinase/Akt pathway (Rawat and Bouchard, 2015), to benefit virus replication. Accumulating evidence also suggest that epigenetic changes, including alterations in DNA methylation, histone modifications, chromatin remodeling, and noncoding RNA interference of cccDNA, contributes to HBV gene expression and replication (Belloni et al., 2009; Hong et al., 2017; Koumbi and Karayiannis, 2015; Zhang et al., 2013a). The correlation between DNA methylation and HBV replication has attracted particular interests. The HBV cccDNA genome contains three CpG islands: Island I, II, and III (Fig. 4) (Kaur et al., 2010; Vivekanandan et al., 2008; Zhang et al., 2013b). Methylation of CpG islands in HBV cccDNA has been detected in both cultured cells and human liver tissues (Guo et al., 2009; Kaur et al., 2010; Vivekanandan et al., 2008; 2010; Zhang et al., 2013b). However, our understanding of the mechanism for methylation of viral DNA and its role during HBV replication remains unclear.
Fig. 4. DNA methylation of HBV NRE by HBx.
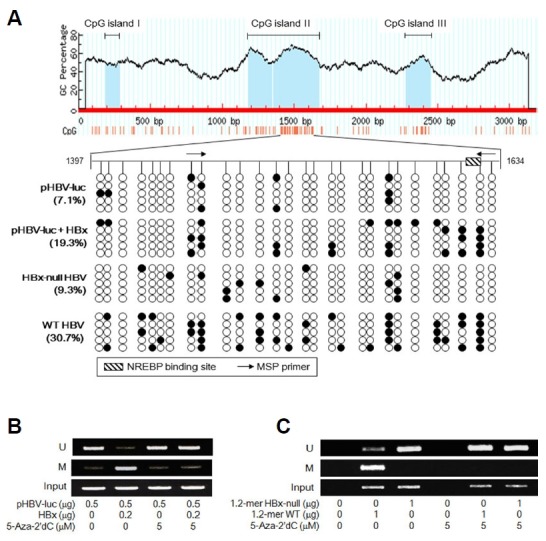
(A) The complete nucleotide sequence of a cloned HBV genome (NCBI accession number X04615.1; Okamoto et al., 1986) was analyzed for the presence of CpG islands using the MethPrimer software. Positions of three CpG islands are shown (upper panel). Bisulfite sequencing analysis was performed to determine methylation status of the NRE region from pHBV-luc and HBV cccDNA in the presence or absence of HBx (lower panels). The CpG sites in the NRE (1397 to 1634) from five different clones are shown to be either methylated (filled circles) or unmethylated (open circles). Positions of a NREBP binding site and two MSP primers are indicated. (B) HepG2 cells prepared as described in Fig. 3B were subjected to MSP to determine whether the NRE in pHBV-luc is methylated (M) or unmethylated (U). (C) HepG2 cells were transfected with an empty vector, 1.2-mer WT, or 1.2-mer HBx-null for 48 h in the presence or absence of 5 μM 5-Aza-2′dC, followed by MSP to determine the methylation status of NRE in HBV cccDNA.
Several previous studies have demonstrated that overexpression of HBx upregulates expression of host DNA methyltransferases (DNMTs), resulting in DNA methylation of cellular tumor suppressor genes and subsequent downregulation of their expression in human hepatocytes (Jung et al., 2007; Lee et al., 2005; 2012; Park et al., 2007). In addition, it has been reported that HBV infection upregulates DNMTs, which in turn induces methylation of both host and viral cccDNA (Vivekanandan et al., 2010). Based on these observations, it was possible to assume that HBx is responsible for DNA methylation of HBV and this ability contributes at least in part to its role as a positive regulator of HBV replication. To prove this hypothesis, we first investigated whether HBx activates DNMTs during HBV replication via upregulation of enzyme levels and induces DNA methylation of HBV cccDNA. We then attempted to prove that the potential of HBx to induce DNA methylation of HBV cccDNA correlates with its ability to stimulate viral replication. We also tried to define a critical regulatory element(s) and factors involved in the DNA methylation-mediated activation of core promoter and to provide a detailed mechanism by which HBx stimulates HBV replication via DNA methylation. Lastly, it was investigated whether DNA methylation in HBV cccDNA positively affects virus replication using both 1.2-mer HBV replicon and in vitro infection models.
MATERIALS AND METHODS
Plasmids
The 1.2-mer-wild type (WT) replicon containing 1.2 units of the HBV genome and its HBx-null counterpart were described before (Cha et al., 2009). pCMV-3 × HA1-HBX3 encoding HBx downstream of three copies of the influenza virus heamagglutinin (HA) epitope was described before (Kwun and Jang, 2004). An HBV core promoter/enhancer reporter construct, pHBV-luc, was described before (Cha et al., 2009). pHBV-NRE-luc, pHBV-Enh-luc, and pHBV-BCP-luc were constructed by PCR-directed deletion of enhancers and/or NRE from pHBV-luc. pHBV-G1620A-luc and pHBV-G1620C-luc were also constructed from pHBV-luc by PCR-directed mutagenesis. pHBV-NRE-G1620A-luc and pHBV-NRE-G1620C-luc were similarly constructed from pHBV-NRE-luc. 1.2-mer HBV G1620A and 1.2-mer HBV G1620C were constructed from 1.2-mer WT HBV replicon by PCR-directed mutagenesis. Construction of DNMT1 shRNA plasmid using the SirenCircle RNAi system (Allele Biotechnology) was described before (Jung et al., 2007). Plasmid pCH110, encoding the Escherichia coli β-Gal gene under the control of the SV40 promoter, was obtained from Pharmacia. Scrambled (SC) shRNA plasmid and NREBP shRNA plasmid was purchased from Santa Cruz Biotechnology. The human sodium taurocholate cotransporting polypeptide (NTCP) expression plasmid, SLC10A1, was purchased from OriGene.
Cell culture and luciferase assay
HepG2 (KCLB No. 58065) was obtained from the Korean Cell Line Bank. For transient expression, 2 × 105 cells per 60-mm dish were transfected with 2 μg of appropriate plasmid( s) using WelFect-EX™ PLUS (WelGENE) following the manufacturer’s instructions. MTT assay was performed to determine cell number as described before (Lee et al., 2012). For luciferase assay, 500 ng of a reporter plasmid was co-transfected with either an empty vector or an effector plasmid under the indicated conditions. One hundred nano-grams of pCH110 were also included as an internal control. Forty-eight hours later, the luciferase assay was performed using Luciferase Reporter 1000 Assay System (Promega), and values obtained were normalized to that of the β-Gal activity measured in the corresponding cell extracts.
HBV replicon system
Cells were transiently transfected with either 1.2-mer WT or its HBx-null mutant for 48 h as described above. HBV-producing cell lines were established by stable transfection of HepG2 cells with a 1.2-mer HBV replicon and virus titers in cultured medium were determined by real-time PCR, as previously described (Yeom et al., 2018). Cells were infected with HBV as previously described (Yeom et al., 2018). Briefly, HepG2 cells were first transfected with 1 μg of NTCP expression plasmid for 24 h and then infected with HBV in the serum-free Dulbecco’s modified Eagle’s medium (DMEM, WelGENE) at a multiplicity of infection (MOI) of 1.0 genome equivalents (Geq) per cell for 2 h. Cells were then washed twice with serum-free DMEM, and maintained for additional 46 h in DMEM supplemented with 2% fetal calf serum (FCS, Gibco). Immunoprecipitation (IP)-coupled PCR was performed to measure levels of extracellular HBV particles, as previously described (Yeom et al., 2018). Briefly, HBV particles were immunoprecipitated from culture supernatant with an anti-HBV surface antigen (HBs) antibody (Santa Cruz Biotechnology) using a Classic Magnetic IP/Co-IP assay kit (Pierce). HBV genomic DNA was purified from the precipitated HBV particle-antibody complexes using the QIAamp DNA mini kit (Qiagen). For quantitation of intracellular HBV particles, total DNA including host and HBV genomes was purified from the transfected cells using the QIAamp DNA mini kit. For conventional PCR analysis of HBV DNA, the genomic DNA was amplified using the Solg 2 × Taq PCR Smartmix1 (Solgent) and a primer pair, HBV NRE 1399F and HBV NRE 1632R, which followed with numbering of the reference HBV genome (NCBI accession number X04615.1; Okamoto et al., 1986) (Table 1). As an internal control, Ecadherin DNA was amplified using a primer pair, E-cad promoter S and E-cad promoter AS (Table 1). Quantitative real-time PCR assay of HBV was carried out, as described before (Mendy et al., 2006). Briefly, HBV DNA was amplified using the SYBR premix Ex Taq II (Takara Bio) and a primer pair, HBV 379F (Table 1) in a Rotor Gene Q PCR machine (Qiagen). Enzyme-linked immunosorbent analysis (ELISA) was also performed to quantitate HBV particles using the Genedia HBsAg ELISA 3.0 (Green Cross MS) according to the manufacturer’s instructions.
Table 1.
List of primers used in this study
| Name | Sequence (5′ to 3′) | Reference |
|---|---|---|
| HBV NRE 1399F | TGG TAC CTG CGC GGG ACG TCC TT | This study |
| HBV NRE 1632R | AGC TAG CGT TCA CGG TGG TCT CC | This study |
| E-cad promoter S | TGC AGG TAC CAT AAC CCA CC | This study |
| E-cad promoter AS | TAC CGC TAG CTG GCT GAG GG | This study |
| HBV 379F | GTG TCT GCG GCG TTT TAT CA | 28 |
| HBV 476R | GAC AAA CGG GCA ACA TAC CTT | 28 |
| HBV M 1381F | TGT TAG GTT GTG TTG TTA ATT G | This study |
| HBV M 1724R | CAA ACA ATC TTT AAA ATA TAC CTC | This study |
| pGL3 Luc +81R | TAA CAT CTT CCA TAA TAA CT | This study |
| HBV 1442MF | GTT GAA TTT TGC GGA C | This study |
| HBV 1630MR | ATA TTC AAT AAA CGT TCA CG | This study |
| HBV 1442 UF | GTT GAA TTT TGT GGA T | This study |
| HBV 1630 UR | ATA TTC AAT AAA CAT TCA CA | This study |
| E-cadherin MF | TTA GGT TAG AGG GTT ATC GCG T | 29 |
| E-cadherin MR | TAA CTA AAA ATT CAC CTA CCG AC | 29 |
| E-cadherin UF | TTA TTT TAG GTT AGA GGG TTA TTG T | 29 |
| E-cadherin UR | CAC AAC CAA TCA ACA ACA CA | 29 |
| RAR-β MF | GGG ATG TCG AGA ACG CG | 30 |
| RAR-β MR | CGA CCA ATC CAA CCG AAA CGA | 30 |
| RAR-β UF | GGG ATG TTG AGA ATG TG | 30 |
| RAR-β UR | CAA CCA ATC CAA CCA AAA CA | 30 |
DNMT activity assay
Exactly 2 × 105 cells per 60-mm diameter plate were transfected with the indicated plasmids for 48 h. DNMT activity in the cell lysates was measured using EpiQuick DNMT Activity/Inhibition Assay Ultra Kit (Epigentek), following the manufacturer’s instructions.
Western blot analysis
Cells were lysed in buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40) supplemented with protease inhibitors. Cell extracts were separated by SDS–PAGE and transferred onto a nitrocellulose membrane (Hybond PVDF; Amersham). Membranes were then incubated with primary antibodies to DNMT1, DNMT2, DNMT3a, DNMT3b, Ecadherin, HBc, HBs, NREBP, retinoic acid receptor-β (RAR-β), and p16 (Santa Cruz Biotechnology), to HBx (Millipore) and to γ-tubulin (Sigma) and subsequently with an appropriate horseradish peroxidase-conjugated secondary antibody: anti-mouse, anti-goat, or anti-rabbit IgG (H + L)-HRP (Bio-Rad). The ECL kit (Amersham) was used to visualize protein bands via the ChemiDoc XRS imaging system (Bio-Rad).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using a ChIP assay kit (Upstate Biotechnology) according to the manufacturer’s specifications. Briefly, the sheared chromatin was immunoprecipitated with either human IgG (Sigma) or anti-NREBP antibody (Santa Cruz Biotechnology) overnight at 4°C. After adding protein A agarose/salmon sperm DNA, the mixture was incubated for additional 30 min. DNA released from the precipitated complexes was amplified by PCR using HBV NRE 1399F and HBV NRE 1632R (Table 1).
Bisulfite DNA sequencing analysis and methylation-specific PCR
Total genomic DNA was modified as described before (Lee et al., 2005). For bisulfite DNA sequencing of NRE from HBV cccDNA, modified DNA was amplified using a primer pair, HBV M 1381F and HBV M 1724R (Table 1). For the analysis of NRE from pHBV-luc, modified DNA was first amplified using a primer pair, HBV M 1381F and pGL3 Luc +81R (Table 1) and then the products were amplified again using HBV M 1381F and HBV M 1724R. For methylation-specific PCR (MSP) of E-cadherin and RAR-β, the modified DNA was amplified with Taq polymerase using both methylated and unmethylated primer pairs of E-cadherin (Herman et al., 1996) and RAR-β (House et al., 2003) as described before. For MSP of HBV NRE from cccDNA and pHBV-luc, the PCR products used for the bisulfite DNA sequencing analysis were amplified again using either a methylated primer set, HBV 1442MF and HBV 1630MR, or an unmethylated primer set, HBV 1442 UF and HBV 1630 UR (Table 1).
Statistical analysis
The values indicate means ± standard deviations from at least four independent experiments. Two-tailed student’s t-test was used for all statistical analyses; a P value of < 0.05 was considered statistically significant.
RESULTS
HBx upregulates protein levels and enzyme activities of DNMT1, 3a, and 3b during HBV replication
Initially, we investigated whether HBx activates DNMTs during HBV infection, as previously demonstrated with ectopic expression of HBx (Jung et al., 2007; Lee et al., 2005; Park et al., 2007). For this purpose, we employed a 1.2-mer HBV replicon system, mimicking the natural course of HBV infection in human hepatic cell lines (Cha et al., 2009; Melegari et al., 1998). Transient transfection of HepG2 cells with a 1.2-mer HBV replicon, 1.2-mer WT, resulted in production of HBV proteins, including HBc, HBs and HBx (Fig. 1A), releasing approximately 1.5 × 106 virus particles per ml of medium (Fig. 1B). In contrast, transfection of the cells with a 1.2-mer HBx-null HBV replicon, 1.2-mer HBx-null, led to production of lower levels of HBc and HBs but not HBx, releasing approximately 6-fold less virus particles (2.3 × 105 copies/ml), indicating the positive role of HBx in HBV replication, as previously demonstrated (Cha et al., 2009; Keasler et al., 2007; 2009; Tsuge et al., 2010). The importance of HBx in HBV replication was further confirmed by the ectopic HBx expression, which fully complemented the ability of 1.2-mer HBx-null to produce intracellular HBV proteins and release extracellular virion particles (Figs. 1A and 1B).
Fig. 1. HBx activates DNMTs during HBV infection.
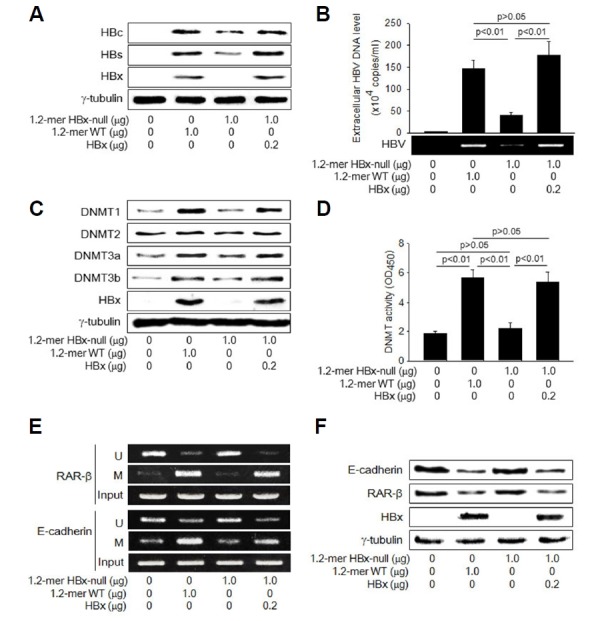
HepG2 cells were transiently transfected with either 1.2-mer WT HBV replicon or its HBx-null derivative for 48 h. For lane 4, HBx-expression plasmid was included. (A, C, F) Levels of the indicated proteins were determined by Western blotting. (B) Levels of HBV particles released from the cells were determined by IP-coupled conventional PCR and real-time PCR (n = 5). (D) DNMT activity was measured as described in Methods (n = 4). (E) MSP analysis was performed to determine whether CpG islands in the promoters of RAR-β and E-cadherin are methylated (M) or unmethylated (U) in the presence or absence of HBx.
Having established an in vitro HBV replication system, we next investigated whether HBx activates DNMTs while HBV replicates in HepG2 cells. Replication of HBV derived from 1.2-mer WT upregulated protein levels of DNMT1, 3a, and 3b (Fig. 1C), resulting in elevation of their activities to about 3-fold (Fig. 1D). In contrast, levels of DNMT2 that is indispensable for DNA methylation (Goll et al., 2006) were little affected under the condition. In addition, HBV replication induced promoter hypermethylation of cellular tumor suppressor genes, such as RAR-β and E-cadherin (Fig. 1E), leading to downregulation of their protein levels (Fig. 1F). None of these effects were observed with HBx-null HBV, indicating that HBx is responsible for these effects. The defects of HBx-null HBV in the activation of DNMTs (Figs. 1C and 1D), promoter hypermethylation of RAR-β and E-cadherin (Fig. 1E), and downregulation of their protein levels (Fig. 1F) were almost completely restored by the ectopic expression of HBx, confirming that HBx activates the host DNA methylation system to induce DNA methylation of cellular genes during HBV replication.
HBx stimulates HBV replication via activation of the host DNA methylation system
According to Fig. 1, HBx activated viral gene expression and stimulated HBV replication, as previously demonstrated (Cha et al., 2009; Keasler et al., 2007; 2009; Tsuge et al., 2010). In addition, HBx activated DNMTs to repress cellular gene expression via DNA methylation during HBV replication. Therefore, it was intriguing to investigate whether the potential of HBx to activate DNMTs is implicated in the stimulation of HBV replication by HBx. For this purpose, we employed a universal DNMT inhibitor, 5-Aza-2′dC, which could abolish the potential of HBx to repress cellular gene expression via DNA methylation (Jung et al., 2007; Lee et al., 2005). Interestingly, treatment with 5-Aza-2′dC decreased the amount of HBV particles released from the cells transfected with 1.2-mer WT HBV in a dose-dependent manner (Fig. 2A), while it little affected the cell proliferation rate, as demonstrated with MTT assay (Fig. 2A). In addition, knockdown of DNMT1 downregulated both HBV gene expression and replication, while neither was affected by transfection with SC shRNA plasmid (Figs. 2E and 2F). Inhibition of DNA methylation by either treatment with 5-Aza-2′dC or DNMT1 knock-down significantly impaired the replication of both WT HBV and HBx-null mutant but the effect was more drastic in the case of WT HBV whose HBx activated DNMTs (Fig. 2B to 2F). Accordingly, the potential of HBx to stimulate HBV replication was weakened as DNA methylation of cccDNA was inhibited. However, neither 5-Aza-2′dC nor DNMT1 shRNA could completely abolish the potential of HBx to stimulate HBV replication, implying the presence of another mechanism(s) involved in the stimulation of HBV replication by HBx.
Fig. 2. HBx stimulates HBV replication via DNA methylation.
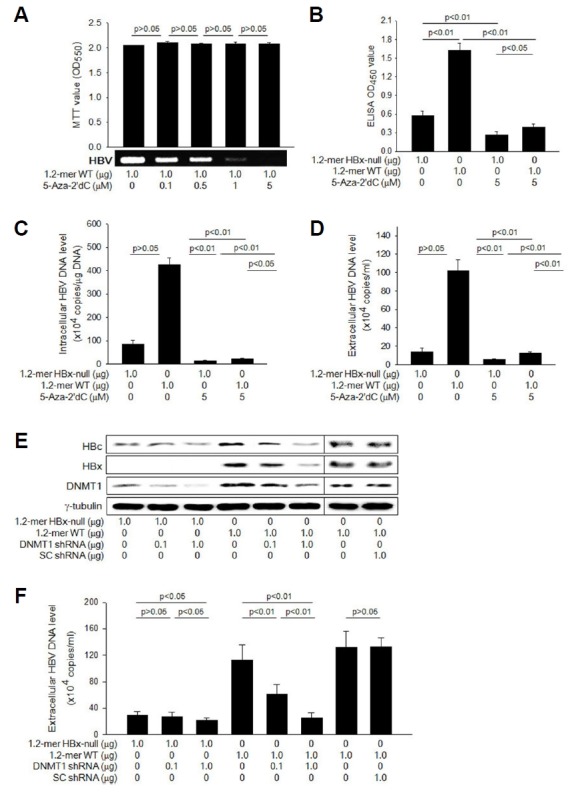
(A) HepG2 cells were transfected with 1.2-mer WT as described in Fig. 1 in the presence of an increasing concentration of 5-Aza-2′dC. For the determination of viable cell number, MTT assay was performed as described in Methods (n = 4). Levels of extracellular HBV particles were determined by IP-coupled conventional PCR. (B) HepG2 cells were transfected with either 1.2-mer WT or its HBx-null derivative for 48 h in the presence or absence of 5 μM 5-Aza-2′dC. Levels of extracellular HBV particles were determined by HBsAg ELISA (n = 5). (C) Levels of intracellular HBV DNA and (D) extracellular virus particles from HepG2 cells prepared as in (B) were determined by IP-coupled real-time PCR (n = 6). (E) HepG2 cells were transfected with either 1.2-mer WT or its HBx-null derivative along with the indicated amount of DNMT1 shRNA plasmid or SC shRNA plasmid for 48 h, followed by Western blotting. (F) Levels of extracellular HBV particles released from HepG2 cells prepared in (E) were determined by IP-coupled real-time PCR (n = 6).
HBx activates HBV core promoter via DNA methylation of NRE
Previous reports have demonstrated that HBx stimulates HBV replication via activation of the core promoter, increasing transcription of pgRNA from cccDNA (Quasdorff and Protzer, 2010; Tang et al., 2005). To investigate the mechanism by which HBx activates the HBV core promoter, we generated three deletion reporter constructs from pHBV-luc containing the full-length HBV core promoter (Cha et al., 2009). The basal luciferase activities of pHBV-luc and its derivatives in the absence of HBx were significantly higher compared to that of an empty reporter pGL2-luc (Fig. 3A). HBx increased the luciferase activity from pHBV-luc, up to approximately 4-fold (Fig. 3B). Treatment with 5-Aza-2′dC significantly decreased the basal core promoter activity and its activation by HBx (Fig. 3B). Similarly, DNMT1 knock-down significantly decreased the effect of HBx on the core promoter (Fig. 3C), indicating that DNA methylation serves an essential mechanism by which HBx activates the HBV core promoter. To define a region(s) responsible for the DNA methylation-mediated activation of core promoter by HBx, we constructed three deletion reporter derivatives from pHBV-luc (Fig. 3A). The luciferase activity from pHBV-Enh-luc, containing all the control elements of pHBV-luc except NRE, was significantly upregulated by HBx (Fig. 3D), indicating that HBx activates the core promoter by elevating the transcriptional activities of positive regulators acting on the enhancers, as previously demonstrated (Quasdorff and Protzer, 2010; Tang et al., 2005). In addition, treatment with 5-Aza-2′dC upregulated the luciferase activity from pHBV-Enh-luc in the presence and absence of HBx (Fig. 3D), possibly by facilitating the binding of positive regulators through the demethylation of the regulatory elements located on the enhancers. This result is consistent with the HBx-mediated activation of pHBV-luc and stimulation of HBV replication in the presence of 5-Aza-2′dC (Figs. 2 and 3B). HBx also upregulated the luciferase activity from pHBV-NRE-luc (Fig. 3E). Unlike in the case of pHBV-Enh-luc, the potential of HBx to upregulate luciferase activity from pHBV-NRE-luc was almost completely abolished by treatment with 5-Aza-2′dC (Fig. 3E) or DNMT1 knock-down (Fig. 3F), indicating that the NRE-driven reporter activation by HBx exclusively depends on DNA methylation. Neither HBx nor 5-Aza-2′dC affected the luciferase activity from pHBV-BCP-luc that contains the basic core promoter (Fig. 3G).
Fig. 3. HBx activates HBV core promoter via DNA methylation of NRE.
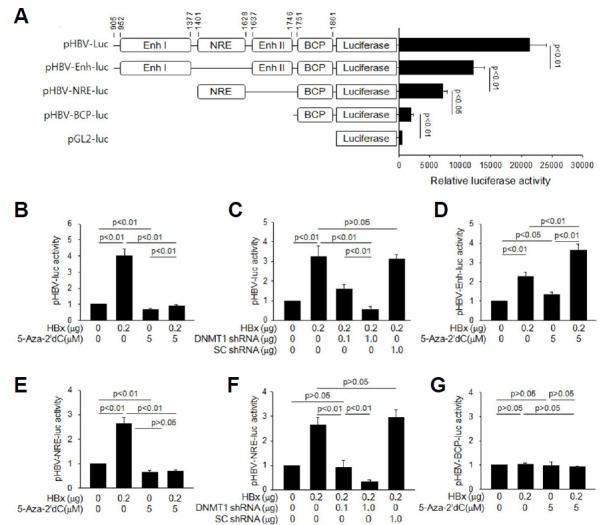
(A) Schematic representation of pHBV-luc and its derivatives. The basal luciferase activities of these constructs along with an empty vector pGL2-luc (Promega) were compared in HepG2 cells (n = 4). (B to G) Either an empty vector or HBx-expression plasmid was co-transfected with the indicated reporter construct into HepG2 cells for 48 h, followed by luciferase assay. The values indicate the relative luciferase activity compared to the basal level of the control (n = 4). (B, D, E, and G) Cells were either mock-treated or treated with 5 μM 5-Aza-2′dC. (C and F) The indicated amount of DNMT1 shRNA plasmid or SC shRNA plasmid was included in the transfection mixtures.
Having established that HBx activates the core promoter via DNA methylation of NRE, we next investigated whether HBx actually induces DNA methylation of NRE. When the complete nucleotide sequence of a cloned HBV genome (NCBI accession number X04615.1; Okamoto et al., 1986) was analyzed for the presence of clustered CpG sequences using MethPrimer software (http://www.urogene.org/methprimer/index1.html), three CpG islands were located in the HBV genome (Fig. 4A), as demonstrated in previous reports (Vivekanandan et al., 2008; Zhang et al., 2013b). According to the data from bisulfite DNA sequencing analysis shown in Fig. 4A, the NRE from both pHBV-luc and HBV cccDNA exhibited DNA methylation in a few CpG dinulceotides in the absence of HBx (7.1% and 9.3%, respectively), whilst their DNA methylation frequencies became about 3-fold higher in the presence of HBx (19.3% and 30.7%, respectively). In addition, according to the MSP data, the NRE in the pHBV-luc and HBV genome was mostly unmethylated in the absence of HBx, whilst it became hypermethylated in the presence of HBx (Figs. 4B and 4C). The potential of HBx to induce DNA methylation of the HBV NRE was almost completely abolished by treatment with 5-Aza-2′dC. Based on these observations, it was possible to conclude that HBx induces DNA methylation of CpG sites within the NRE of cccDNA during HBV replication.
HBx stimulates HBV replication via DNA methylation of NRE
We next explored the detailed mechanism by which HBx activates the core promoter via DNA methylation of NRE during HBV replication. As a potent negative regulator, NREBP is known to inhibit the core promoter activity via NRE, repress pgRNA transcription, and ultimately decrease HBV particle production (Sun et al., 2001). Consistently, knockdown of NREBP using a specific siRNA significantly elevated the luciferase activity from both pHBV-luc and NRE-luc (Figs. 5A and 5B), while it little affected the luciferase activity from pHBV-Enh-luc (Fig. 5C), resulting in upregulation of HBV DNA levels of intracellular HBV and Ecadherin were measured by PCR (middle panels). Protein levels of NREBP, HBx, and γ-tubulin were determined by Western blotting (lower panels). DNA and proteins, including HBc and HBx, in the cells and elevation of extracellular HBV particles (Fig. 5D). The potential of NREBP silencing to induce these effects was much weaker in the presence of HBx, possibly due to the impaired action of NREBP in the presence of HBx. In addition, the effects of HBx on the core promoter activity, viral gene expression, and HBV replication were almost completely abolished by NREBP knock-down, suggesting that HBx executes its potential by inhibiting the role of NREBP as a negative regulator of the core promoter.
Fig. 5. DNA methylation of NRE interferes with NREBP binding to increase HBV replication.
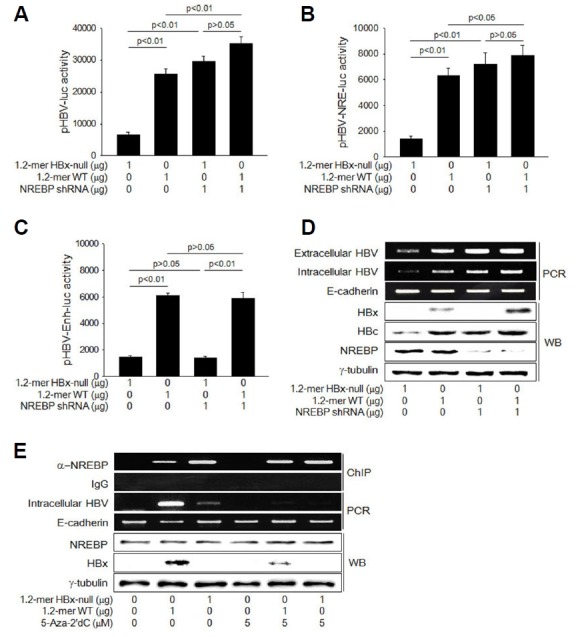
(A–C) HepG2 cells were co-transfected with pHBV-luc (A), pHBV-NRE-luc (B), or pHBV-Enh-luc (C) with either 1.2-mer WT or 1.2-mer HBx-null in the presence or absence of NREBP shRNA plasmid for 48 h, followed by luciferase assay (n = 5). (D) HepG2 cells were transfected with either 1.2-mer WT or 1.2-mer HBx-null with or without NREBP shRNA plasmid for 48 h. Levels of extracellular and intracellular HBV DNA and E-cadherin DNA as an internal control were determined by PCR (upper panels). Levels of HBx, HBc, and NREBP were determined by Western blotting (lower panels). (E) HepG2 cells were transfected with either 1.2-mer WT or 1.2-mer HBx-null for 48 h for 48 h in the presence or absence of 5 μM 5-Aza-2′dC. ChIP assay was performed to determine levels of NREBP bound on the NRE of HBV cccDNA (upper panels).
Based on the data obtained so far, it is possible to speculate that NREBP freely acts on the NRE in the absence of HBx, whilst its action is disrupted by HBx that induces DNA methylation of its binding site. To test this hypothesis, we examined whether HBx interferes with the binding of NREBP on the NRE during HBV replication. Neither HBx nor 5-Aza-2′dC altered protein levels of NREBP in HepG2 cells (Fig. 5E). According to the data from ChIP assay, the amount of NREBP bound on the NRE of WT HBV cccDNA was much lower compared to that from the HBx-null mutant (Fig. 5E). In addition, the ability of HBx to interrupt NREBP binding was almost completely abolished by treatment with 5-Aza-2′dC (Fig. 5E), suggesting that the methylated NRE in the WT HBV cccDNA is not appropriate for NREBP binding. Taken together, we conclude that HBx activates the HBV core promoter by preventing NREBP from binding on the NRE via DNA methylation.
DNA methylation of C-1619 in the NREBP binding site is critical for the stimulation of HBV replication by HBx
According to Fig. 4A, the 27th CpG site located within the NREBP binding site was totally unmethylated in the absence of HBx, whereas it was mostly methylated in the presence of HBx. In addition, the C-1619 is known to be a highly conserved nucleotide in the HBV NREBP binding site (Li et al., 2011). Therefore, we investigated whether DNA methylation of this CpG site is associated with the HBx-mediated inhibition of NREBP binding and subsequent activation of the core promoter. For this purpose, we constructed two pHBV-luc derivatives, pHBV-G1620A-luc and pHBV-G1620C-luc, in which the G-1620 was substituted with A and C, respectively, to prevent methylation of the C-1619 from taking place. The basal luciferase activity from pHBV-G1620A-luc and pHBV-G1620C-luc was lower than that from pHBV-luc (Fig. 6A), suggesting that methylation of C-1619 prevents NREBP from interacting with its binding site and thereby inhibiting the core promoter. Although HBx still significantly activated the core promoter from pHBV-G1620A-luc and pHBV-G1620C-luc, possibly due to its effect on the enhancers, as shown in Fig. 3D, the potential was much lower compared to that obtained with pHBV-luc. Moreover, it was impossible to observe the stimulatory effect of HBx on the luciferase activity from pHBV-NRE-G1620A-luc and pHBV-NRE-G1620C-luc (Fig. 6B). These results suggest that HBx activates the HBV core promoter via DNA methylation of the C-1619 located in the NREBP binding site.
Fig. 6. DNA methylation of C-1619 is critical for the stimulation of HBV replication by HBx.
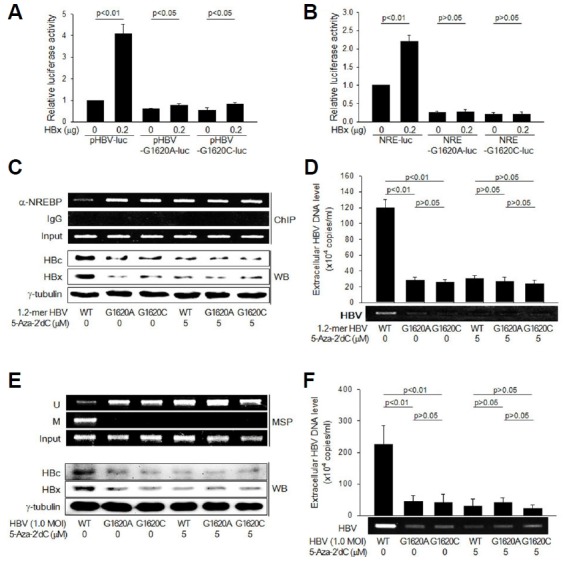
(A) pHBV-luc and its derivatives, pHBV-G1620A-luc and pHBV-G1620C-luc, were co-transfected with either an empty vector or HBx expression plasmid into HepG2 cells, followed by luciferase assay (n = 4). (B) pHBV-NRE-luc and its derivatives, pHBV-NRE-G1620A-luc and pHBV-NRE-G1620C-luc, were co-transfected with either an empty vector or HBx expression plasmid into HepG2 cells, followed by luciferase assay (n = 4). (C) HepG2 cells were transfected with 1.2-mer WT or its derivatives, 1.2-mer HBV G1620A and 1.2-mer HBV G1620C for 48 h in the presence or absence of 5 μM 5-Aza-2′dC. Levels of NREBP bound on the NRE in HBV cccDNA were determined by ChIP assay (upper panels). Levels of HBc and HBx were determined by Western blotting (lower panels). (D) Levels of extracellular HBV particles released from cells prepared as in (C) were determined by IP-coupled conventional PCR and real-time PCR (n = 4). (E) HepG2 cells transfected with the NTCP expression plasmid were infected with the indicated HBV at an MOI of 1.0 for 48 h, followed by Western blotting. (F) Levels of extracellular HBV particles released from cells prepared as in (E) were determined by IP-coupled conventional PCR and real-time PCR (n = 4).
To further demonstrate that DNA methylation of the C-1619 is critical for the stimulation of HBV replication by HBx, we generated two 1.2-mer WT derivatives named 1.2-mer HBV G1620A and 1.2-mer HBV G1620C, in which the G-1620 in 1.2-mer WT was substituted with A and C, respectively. The amount of NREBP bound on the cccDNA derived from 1.2-mer HBV G1620A and 1.2-mer HBV G1620C was higher compared to that obtained with 1.2-mer HBV WT (Fig. 6C), which led to impaired HBV replication, as demonstrated by lower levels of intracellular HBx and HBc proteins (Fig. 6C) and reduced production of HBV particles (Fig. 6D). Interestingly, treatment with 5-Aza-2′dC dramatically increased the amount of NREBP bound on the cccDNA derived from 1.2-mer HBV WT and inhibited HBV replication, whilst none of these effects were observed with HBV G1620A and HBV G1620C (Figs. 6C and 6D). As a consequence, it was impossible to observe the differences between WT and mutant versions of HBV in the presence of 5-Aza-2′dC (Figs. 6C and 6D). Lastly, it was attempted to evaluate the importance of C-1619 methylation in the HBx-mediated stimulation of HBV replication using an in vitro HBV infection system. Consistent to the data obtained with the 1.2-mer HBV replicon system (Figs. 6C and 6D), HBV G1620A and HBV G1620C replicated less effectively, compared to HBV WT, in NTCP-expressing HepG2 cells, as evidenced by lower levels of intracellular HBx and HBc proteins (Fig. 6E) and reduced production of HBV particles (Fig. 6F). The negative effect of 5-Aza-2′dC on the replication of WT HBV was also exactly reproduced in the HBV infection system (Figs, 6E and 6F). As the NRE from HBV WT, HBV G1620A and HBV G1620C recruited the same amount of NREBP in the presence of 5-Aza-2′dC (Fig. 6C), it is unlikely that the substitution itself affected NREBP binding on the cccDNA. Therefore, DNA methylation of the C-1619 can be considered to be critical for the stimulation of HBV replication by HBx.
DISCUSSION
Several experimental systems, including cell culture (Belloni et al., 2009; Cha et al., 2009; Keasler et al., 2007; 2009; Leupin et al., 2005), chimeric mice with humanized livers (Tsuge et al., 2010) and transgenic mice models (Xu et al., 2002) have demonstrated that HBx acts as a positive regulator of HBV replication. Consistently, the present study showed that HBx stimulates HBV replication in cultured human hepatocytes, as demonstrated with the higher replication rate of HBV derived from a 1.2-mer WT HBV replicon compared to its HBx-null counterpart (Figs. 1A and 1B). Although several different functions have been attributed to HBx regarding HBV replication (Bouchard et al., 2001; Leupin et al., 2005; Melegari et al., 2005), the transactivation function of HBx is likely to be critical for its role during HBV replication. It has been demonstrated that transcription of pgRNA from HBV cccDNA is positively regulated by HBx and several cellular transcription factors acting on the two enhancer elements located upstream of the core promoter (Ishida et al., 2000; Kramvis and Kew, 1999; Su and Yee, 1992; Yuh et al., 1992). Consistently, the present study showed that HBx upregulates the enhancer-driven core promoter activity approximately 4-fold (Fig. 3B). In addition, the present study provides several lines of evidence that HBx stimulates HBV replication by interfering with the function of NREBP. First, the effects of NREBP silencing on the HBV gene expression and replication were lower in the presence of HBx (Figs. 5A and 5D). Second, the stimulatory effects of HBx on the core promoter and virus replication were lower when NREBP was knocked-down (Figs. 5A and 5D). Third, HBx reduced the binding affinity of NREBP without affecting its protein level (Fig. 5E). Fourth, the effects of HBx on the core promoter and HBV replication were almost completely abolished by a single nucleotide substitution at the NREBP binding site, which facilitated NREBP binding on this site in the presence of HBx (Fig. 6).
Recent studies have identified DNA methylation of HBV DNA as an important mechanism for the regulation of viral gene expression and HBV replication (Hong et al., 2017; Koumbi and Karayiannis, 2015; Zhang et al., 2013a). Both protein levels and enzyme activities of DNMTs were upregulated in human hepatocytes infected with HBV, resulting in methylation of viral cccDNA (Guo et al., 2009; Kaur et al., 2010; Vivekanandan et al., 2008; 2010). A previous report suggested that DNMTs are upregulated in hepatocytes as a defense mechanism in response to chronic HBV infection (Vivekanandan et al., 2010). However, the causative agent responsible for this event is still unclear. According to the present study, both the enzyme activity and protein levels of DNMTs were upregulated by WT HBV, resulting in DNA methylation of cellular genes and HBV cccDNA, whilst none of these effects were observed in cells with HBx-null HBV. In addition, ectopic expression of HBx normally induced DNA methylation of cccDNA derived from HBx-null mutant (data not shown). Therefore, it is more likely that HBx is responsible for the methylation of cccDNA during HBV replication.
DNA methylation in host DNA has been shown to be the main mechanism inactivating tumor suppressor genes in HCC, suggesting a potential role of DNMT inhibitors in the treatment of HCC (Claus et al., 2005; Tong et al., 2009). However, the role of DNA methylation in HBV cccDNA during virus replication is controversial. DNA methylation in general provides a negative regulatory mechanism for gene expression (Bestor, 2000). Early studies thus have suggested that methylation of HBV DNA leads to decreased viral gene expression and replication. For example, co-transfection experiments with full-length HBV genome and DNMT3 led to the downregulation of viral protein and pgRNA production (Vivekanandan et al., 2010). In addition, transfection of artificially methylated HBV DNA led to reduced HBV mRNA levels, decreased HBs and HBc protein expression, and decreased secretion of HBV proteins (Vivekanandan et al., 2009). Moreover, methylation of HBV cccDNA in patient samples is associated low serum HBs titers (Zhang et al., 2014). Consistently, the present study showed that the enhancer-driven core promoter activity was upregulated by treatment with 5-Aza-2′dC (Fig. 3D), confirming DNA methylation as a negative regulatory mechanism of HBV replication. However, the present study also provides several lines of evidence that DNA methylation of the NRE region in HBV cccDNA stimulates virus replication. First, the WT HBV cccDNA with hypermethylated CpG island II replicated more effectively compared to the HBx-null mutant cccDNA with hypomethylated CpG island II. Second, inhibition of DNA methylation by either 5-Aza-2′dC treatment or DNMT1 knock-down dramatically decreased the core promoter activity, viral gene expression, and replication of HBV. These effects were more dramatic in the presence of HBx that induced DNA methylation in HBV cccDNA. Third, HBx interfered with the action of NREBP during HBV replication via DNA methylation of the C-1619 located within its binding site. The methylation frequencies of the C-1619 and CpG island II region were closely correlated, both of which were affected by the HBx-mediated activation of DNMTs. Fourth, substitution of the G-1620 with A or C to block methylation at the C-1619 facilitated NREBP binding and thereby decreased HBV replication, which almost completely abolished the potential of HBx to stimulate HBV replication. It is not easy to explain the discrepant roles of DNA methylation in HBV replication. The relative importance of enhancers and NRE in the regulation of core promoter activity by HBx can be considered. If the positive regulation via enhancers and transactivators is dominant over the negative regulation involving NRE and NERBP in the regulation of the core promoter by HBx, DNA methylation in HBV cccDNA may suppress HBV replication, as demonstrated in the previous reports (Guo et al., 2009; Vivekanandan et al., 2009; Zhang et al., 2013a). In contrast, an opposite outcome can be obtained if NRE and NERBP play more crucial roles in the HBx-mediated activation of the core promoter, as demonstrated in the present study. The relative importance of enhancers and NRE in the regulation of the core promoter activity can be affected by several factors, including HBV genotype, HBeAg positivity, patient age and liver disease stage, and other experimental conditions, which are also relevant to DNA methylation status in cccDNA (Zhang et al., 2014). The present study may provide theoretical basis for the development of a target-specific drug from DNMT inhibitors, which can inhibit HBV replication and also interfere with the progression of hepatocellular carcinogenesis.
ACKNOWLEDGMENTS
We thank W.-S Ryu for providing the HBV replicon system and pHBV-luc plasmid used in this study. This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry for Health & Welfare, Republic of Korea (grant number: HI12C1315).
REFERENCES
- Belloni L., Pollicino T., De Nicola F., Guerrieri F., Raffa G., Fanciulli M., Raimondo G., Levrero M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA. 2009;106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor T.H. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Blum H.E., Zhang Z.S., Galun E., von Weizsacker F., Garner B., Liang T.J., Wands J.R. Hepatitis B virus X protein is not central to the viral life cycle in vitro. J Virol. 1992;66:1223–1227. doi: 10.1128/jvi.66.2.1223-1227.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M.J., Wang L.H., Schneider R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–2378. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- Cha M.Y., Ryu D.K., Jung H.S., Chang H.E., Ryu W.S. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J Gen Virol. 2009;90:978–986. doi: 10.1099/vir.0.009928-0. [DOI] [PubMed] [Google Scholar]
- Claus R., Almstedt M., Lubbert M. Epigenetic treatment of hematopoietic malignancies: in vivo targets of demethylating agents. Semin Oncol. 2005;32:511–520. doi: 10.1053/j.seminoncol.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Curtil C., Enache L.S., Radreau P., Dron A.G., Scholtes C., Deloire A., Roche D., Lotteau V., Andre P., Ramiere C. The metabolic sensors FXRalpha, PGC-1alpha, and SIRT1 cooperatively regulate hepatitis B virus transcription. FASEB J. 2014;28:1454–1463. doi: 10.1096/fj.13-236372. [DOI] [PubMed] [Google Scholar]
- Decorsière A., Mueller H., van Breugel P.C., Abdul F., Gerossier L., Beran R.K., Livingston C.M., Niu C., Fletcher S.P., Hantz O., Strubin M. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–389. doi: 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- Goll M.G., Kirpekar F., Maggert K.A., Yoder J.A., Hsieh C.L., Zhang X., Golic K.G., Jacobsen S.E., Bestor T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- Gunther S., Piwon N., Will H. Wild-type levels of pregenomic RNA and replication but reduced pre-C RNA and eantigen synthesis of hepatitis B virus with C(1653) --> T, A(1762) --> T and G(1764) --> A mutations in the core promoter. J Gen Virol. 1998;79:375–380. doi: 10.1099/0022-1317-79-2-375. [DOI] [PubMed] [Google Scholar]
- Guo Y., Li Y., Mu S., Zhang J., Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol. 2009;81:1177–1183. doi: 10.1002/jmv.21525. [DOI] [PubMed] [Google Scholar]
- Herman J.G., Graff J.R., Myohanen S., Nelkin B.D., Baylin S.B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson A.J., Hyser J.M., Keasler V.V., Cang Y., Slagle B.L. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology. 2012;426:73–82. doi: 10.1016/j.virol.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X., Kim E.S., Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology. 2017;66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House M.G., Guo M., Iacobuzio-Donahue C., Herman J.G. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis. 2003;24:193–198. doi: 10.1093/carcin/24.2.193. [DOI] [PubMed] [Google Scholar]
- Ishida H., Ueda K., Ohkawa K., Kanazawa Y., Hosui A., Nakanishi F., Mita E., Kasahara A., Sasaki Y., Hori M., et al. Identification of multiple transcription factors, HLF, FTF, and E4BP4, controlling hepatitis B virus enhancer II. J Virol. 2000;74:1241–1251. doi: 10.1128/jvi.74.3.1241-1251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.K., Arora P., Pagano J.S., Jang K.L. Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res. 2007;67:5771–5778. doi: 10.1158/0008-5472.CAN-07-0529. [DOI] [PubMed] [Google Scholar]
- Kaur P., Paliwal A., Durantel D., Hainaut P., Scoazec J.Y., Zoulim F., Chemin I., Herceg Z. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J Infect Dis. 2010;202:700–704. doi: 10.1086/655398. [DOI] [PubMed] [Google Scholar]
- Keasler V.V., Hodgson A.J., Madden C.R., Slagle B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol. 2007;81:2656–2662. doi: 10.1128/JVI.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasler V.V., Hodgson A.J., Madden C.R., Slagle B.L. Hepatitis B virus HBx protein localized to the nucleus restores HBx-deficient virus replication in HepG2 cells and in vivo in hydrodynamically-injected mice. Virology. 2009;390:122–129. doi: 10.1016/j.virol.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumbi L., Karayiannis P. The Epigenetic Control of Hepatitis B Virus Modulates the Outcome of Infection. Front Microbiol. 2015;6:1491. doi: 10.3389/fmicb.2015.01491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramvis A., Kew M.C. The core promoter of hepatitis B virus. J Viral Hepat. 1999;6:415–427. doi: 10.1046/j.1365-2893.1999.00189.x. [DOI] [PubMed] [Google Scholar]
- Kwun H.J., Jang K.L. Natural variants of hepatitis B virus X protein have differential effects on the expression of cyclin-dependent kinase inhibitor p21 gene. Nucleic Acids Res. 2004;32:2202–2213. doi: 10.1093/nar/gkh553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Seo S.Y., Tiwari I., Jang K.L. Epstein-Barr Virus latent membrane protein 1 overcomes all-trans retinoic acid-induced apoptosis by inhibiting retinoic acid receptor-beta(2) expression. Biochem Biophys Res Commun. 2012;423:313–318. doi: 10.1016/j.bbrc.2012.05.118. [DOI] [PubMed] [Google Scholar]
- Lee J.O., Kwun H.J., Jung J.K., Choi K.H., Min D.S., Jang K.L. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24:6617–6625. doi: 10.1038/sj.onc.1208827. [DOI] [PubMed] [Google Scholar]
- Lee M.H., Na H., Na T.Y., Shin Y.K., Seong J.K., Lee M.O. Epigenetic control of metastasis-associated protein 1 gene expression by hepatitis B virus X protein during hepatocarcinogenesis. Oncogenesis. 2012;1:e25. doi: 10.1038/oncsis.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leupin O., Bontron S., Schaeffer C., Strubin M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J Virol. 2005;79:4238–4245. doi: 10.1128/JVI.79.7.4238-4245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.S., Lau T.C., Chan S.K., Wong C.H., Ng P.K., Sung J.J., Chan H.L., Tsui S.K. The G1613A mutation in the HBV genome affects HBeAg expression and viral replication through altered core promoter activity. PLoS One. 2011;6:e21856. doi: 10.1371/journal.pone.0021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W.Y., Ting L.P. Repression of enhancer II activity by a negative regulatory element in the hepatitis B virus genome. J Virol. 1994;68:1758–1764. doi: 10.1128/jvi.68.3.1758-1764.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora J., Arzberger S., Durantel D., Belloni L., Strubin M., Levrero M., Zoulim F., Hantz O., Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Melegari M., Scaglioni P.P., Wands J.R. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol. 1998;72:1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melegari M., Wolf S.K., Schneider R.J. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol. 2005;79:9810–9820. doi: 10.1128/JVI.79.15.9810-9820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendy M.E., Kaye S., van der Sande M., Rayco-Solon P., Waight P.A., Shipton D., Awi D., Snell P., Whittle H., McConkey S.J. Application of real-time PCR to quantify hepatitis B virus DNA in chronic carriers in The Gambia. Virol J. 2006;3:23. doi: 10.1186/1743-422X-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H., Imai M., Shimozaki M., Hoshi Y., Iizuka H., Gotanda T., Tsuda F., Miyakawa Y., Mayumi M. Nucleotide sequence of a cloned hepatitis B virus genome, subtype ayr: comparison with genomes of the other three subtypes. J Gen Virol. 1986;67:2305–2314. doi: 10.1099/0022-1317-67-11-2305. [DOI] [PubMed] [Google Scholar]
- Park I.Y., Sohn B.H., Yu E., Suh D.J., Chung Y.H., Lee J.H., Surzycki S.J., Lee Y.I. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology. 2007;132:1476–1494. doi: 10.1053/j.gastro.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Quasdorff M., Protzer U. Control of hepatitis B virus at the level of transcription. J Viral Hepat. 2010;17:527–536. doi: 10.1111/j.1365-2893.2010.01315.x. [DOI] [PubMed] [Google Scholar]
- Rawat S., Bouchard M.J. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J Virol. 2015;89:999–1012. doi: 10.1128/JVI.02440-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Yee J.K. Regulation of hepatitis B virus gene expression by its two enhancers. Proc Natl Acad Sci USA. 1992;89:2708–2712. doi: 10.1073/pnas.89.7.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.T., Lo W.Y., Wang I.H., Lo Y.H., Shiou S.R., Lai C.K., Ting L.P. Transcription repression of human hepatitis B virus genes by negative regulatory element-binding protein/SON. J Biol Chem. 2001;276:24059–24067. doi: 10.1074/jbc.M101330200. [DOI] [PubMed] [Google Scholar]
- Tang H., Delgermaa L., Huang F., Oishi N., Liu L., He F., Zhao L., Murakami S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol. 2005;79:5548–5556. doi: 10.1128/JVI.79.9.5548-5556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A., Gou L., Lau Q.C., Chen B., Zhao X., Li J., Tang H., Chen L., Tang M., Huang C., et al. Proteomic profiling identifies aberrant epigenetic modifications induced by hepatitis B virus X protein. J Proteome Res. 2009;8:1037–1046. doi: 10.1021/pr8008622. [DOI] [PubMed] [Google Scholar]
- Tsuge M., Hiraga N., Akiyama R., Tanaka S., Matsushita M., Mitsui F., Abe H., Kitamura S., Hatakeyama T., Kimura T., et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol. 2010;91:1854–1864. doi: 10.1099/vir.0.019224-0. [DOI] [PubMed] [Google Scholar]
- Villeneuve J.P. The natural history of chronic hepatitis B virus infection. J Clin Virol. 2005;34(Suppl 1):S139–142. doi: 10.1016/s1386-6532(05)80024-1. [DOI] [PubMed] [Google Scholar]
- Vivekanandan P., Daniel H.D., Kannangai R., Martinez-Murillo F., Torbenson M. Hepatitis B virus replication induces methylation of both host and viral DNA. J Virol. 2010;84:4321–4329. doi: 10.1128/JVI.02280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan P., Thomas D., Torbenson M. Hepatitis B viral DNA is methylated in liver tissues. J Viral Hepat. 2008;15:103–107. doi: 10.1111/j.1365-2893.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- Vivekanandan P., Thomas D., Torbenson M. Methylation regulates hepatitis B viral protein expression. J Infect Dis. 2009;199:1286–1291. doi: 10.1086/597614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Yen T.S., Wu L., Madden C.R., Tan W., Slagle B.L., Ou J.H. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J Virol. 2002;76:2579–2584. doi: 10.1128/jvi.76.5.2579-2584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaginuma K., Shirakata Y., Kobayashi M., Koike K. Hepatitis B virus (HBV) particles are produced in a cell culture system by transient expression of transfected HBV DNA. Proc Natl Acad Sci USA. 1987;84:2678–2682. doi: 10.1073/pnas.84.9.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom S., Jeong H., Kim S.S., Jang K.L. Hepatitis B virus X protein activates proteasomal activator 28 gamma expression via upregulation of p53 levels to stimulate virus replication. J Gen Virol. 2018;99:655–666. doi: 10.1099/jgv.0.001054. [DOI] [PubMed] [Google Scholar]
- Yuh C.H., Chang Y.L., Ting L.P. Transcriptional regulation of precore and pregenomic RNAs of hepatitis B virus. J Virol. 1992;66:4073–4084. doi: 10.1128/jvi.66.7.4073-4084.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Hou J., Lu M. Regulation of hepatitis B virus replication by epigenetic mechanisms and microRNAs. Front Genet. 2013a;4:202. doi: 10.3389/fgene.2013.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Li C., Zhang Y., Zhu H., Kang Y., Liu H., Wang J., Qin Y., Mao R., Xie Y., et al. Comparative analysis of CpG islands among HBV genotypes. PLoS One. 2013b;8:e56711. doi: 10.1371/journal.pone.0056711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Mao R., Yan R., Cai D., Zhang Y., Zhu H., Kang Y., Liu H., Wang J., Qin Y., et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS One. 2014;9:e110442. doi: 10.1371/journal.pone.0110442. [DOI] [PMC free article] [PubMed] [Google Scholar]