

 Storage of electrons in the Ti8O8 nodes of MOF MIL-125 is controlled by the presence of charge-balancing Na+ cations.
Storage of electrons in the Ti8O8 nodes of MOF MIL-125 is controlled by the presence of charge-balancing Na+ cations.
Abstract
Stoichiometric reduction reactions of two metal–organic frameworks (MOFs) by the solution reagents 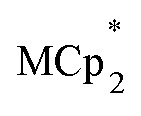 (M = Cr, Co) are described. The two MOFs contain clusters with Ti8O8 rings: Ti8O8(OH)4(bdc)6; bdc = terephthalate (MIL-125) and Ti8O8(OH)4(bdc-NH2)6; bdc-NH2 = 2-aminoterephthalate (NH2-MIL-125). The stoichiometry of the redox reactions was probed using solution NMR methods. The extent of reduction is greatly enhanced by the presence of Na+, which is incorporated into the bulk of the material. The roughly 1 : 1 stoichiometry of electrons and cations indicates that the storage of e– in the MOF is tightly coupled to a cation within the architecture, for charge balance.
(M = Cr, Co) are described. The two MOFs contain clusters with Ti8O8 rings: Ti8O8(OH)4(bdc)6; bdc = terephthalate (MIL-125) and Ti8O8(OH)4(bdc-NH2)6; bdc-NH2 = 2-aminoterephthalate (NH2-MIL-125). The stoichiometry of the redox reactions was probed using solution NMR methods. The extent of reduction is greatly enhanced by the presence of Na+, which is incorporated into the bulk of the material. The roughly 1 : 1 stoichiometry of electrons and cations indicates that the storage of e– in the MOF is tightly coupled to a cation within the architecture, for charge balance.
Introduction
Metal–organic frameworks (MOFs) are emerging as promising materials for facilitating redox reactions, including multi-e–/multi-H+ transformations. Recent studies highlight the ability of the organic linkers or the metal ions at the MOF nodes to undergo 1 – e– oxidations1–3 or reductions,4–6 for the MOFs themselves to serve as conductive materials6–12 or semi-conductors,13–16 and for the MOFs to facilitate redox reactions that are pertinent to fuel cells and energy.3,17–23 MOFs are also already commercially available in Li-ion batteries.24–27 The diversity of these studies reaffirms the promise of using MOFs in a variety of redox systems. This underscores the importance of understanding how MOFs participate in these redox processes, as well as how the change in redox state impacts the stability and reactivity of the material.28,29
The well-known Ti-based MOF MIL-125 (ref. 30) (Ti8O8(OH)4(bdc)6) and its amino-substituted analogue NH2-MIL-125 (ref. 23) (Ti8O8(OH)4(bdc-NH2)6) have recently been thoroughly investigated for their photo-activity (bdc = terephthalate); (bdc-NH2 = 2-amino-terephthalate). UV irradiation of a slurry of MIL-125 in benzyl alcohol results in oxidation of the alcohol to benzaldehyde and a color change from white to blue, due to the reduction of Ti(iv) to Ti(iii).30 It was suggested that this process reduces each MOF node (Ti8 cluster) by a net H-atom (eqn (1)). Using a simple solution NMR assay for titrating H-atom equivalents in MOFs, we have shown that this is indeed the case.5,31
| 1 |
Intrigued by the stability of these MOFs to reduction, we have turned our attention to exploring the chemical reduction of MIL-125 and NH2-MIL-125. Here, we describe a simple method to quantify the reducing equivalents added to the MOFs, and establish the importance of coupling electron transfer with cation transfer. Model studies on soluble cluster analogues suggest a specific role for Na+ in facilitating reduction. These fundamental studies provide new insights into the redox properties of MOFs, in particular the importance of internal charge balance.
Results
Chemical reduction with 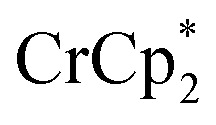
Chemical reductions of suspensions of MIL-125 and NH2-MIL-125 were explored with the outer-sphere electron donors 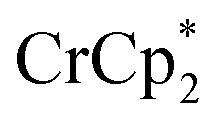 and
and 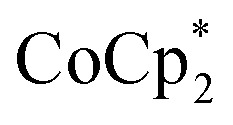 . These soluble reductants were chosen because they allow for quantification of the reduction stoichiometry (electrons transferred per Ti8 node) via solution 1H NMR spectroscopy. Both
. These soluble reductants were chosen because they allow for quantification of the reduction stoichiometry (electrons transferred per Ti8 node) via solution 1H NMR spectroscopy. Both 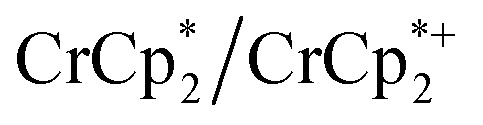 and
and 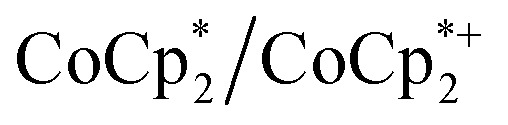 undergo fast chemical exchange on the NMR timescale. Therefore solutions containing both the oxidized and reduced forms show a single resonance for
undergo fast chemical exchange on the NMR timescale. Therefore solutions containing both the oxidized and reduced forms show a single resonance for 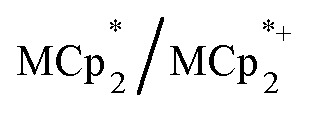 (M = Cr, Co) whose chemical shift indicates the mole fraction of each species in solution (eqn (2)),32 and hence the number of e– transferred to MIL-125.
(M = Cr, Co) whose chemical shift indicates the mole fraction of each species in solution (eqn (2)),32 and hence the number of e– transferred to MIL-125.
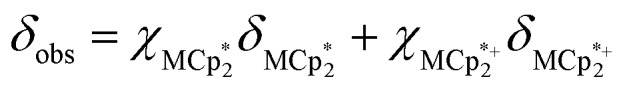 |
2 |
Thus, 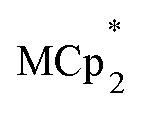 serves as both a reductant and assay for redox quantification. In these studies, suspensions of MIL-125 in solutions of
serves as both a reductant and assay for redox quantification. In these studies, suspensions of MIL-125 in solutions of 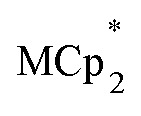 were stirred for ∼12 h, after which the reactions were filtered. NMR analysis of the filtrate yields the
were stirred for ∼12 h, after which the reactions were filtered. NMR analysis of the filtrate yields the 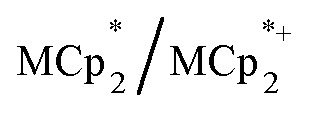 ratio. Combining this with the relative amounts of
ratio. Combining this with the relative amounts of 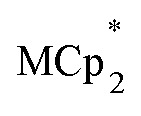 and MIL-125 quantifies the stoichiometry of the e– transferred per Ti8 node.
and MIL-125 quantifies the stoichiometry of the e– transferred per Ti8 node.
To establish whether 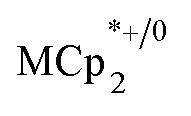 is taken up in the pores of MIL-125 (5–7 Å windows),30 which would render the aforementioned analysis inaccurate, uptake studies were conducted. For these studies,
is taken up in the pores of MIL-125 (5–7 Å windows),30 which would render the aforementioned analysis inaccurate, uptake studies were conducted. For these studies, 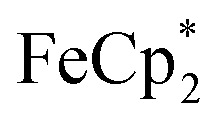 was used as an isosteric diamagnetic analog because the average Fe–Cring bond distance of 2.05 Å in
was used as an isosteric diamagnetic analog because the average Fe–Cring bond distance of 2.05 Å in 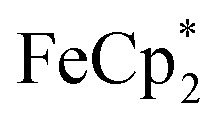 is less than those found in the
is less than those found in the 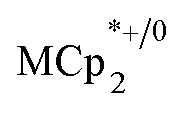 analogues used in this manuscript (for
analogues used in this manuscript (for 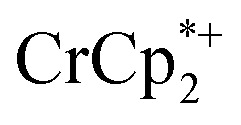 : 2.212 Å;
: 2.212 Å; 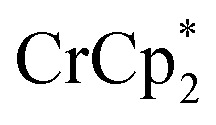 : 2.163 Å;
: 2.163 Å; 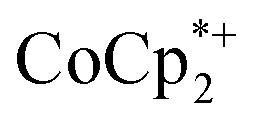 : 2.058 Å;
: 2.058 Å; 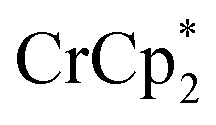 : 2.091 Å). A solution of
: 2.091 Å). A solution of 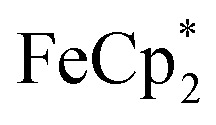 and (p-tolyl)2O as a standard in C6D6 was prepared and its 1H NMR spectrum was obtained. MIL-125 was added, the suspension stirred for 3 h, and another 1H NMR spectrum was obtained. The change in the integrals was noted. While smaller molecules such as FeCp2 were taken up into the MOF, as evidenced by a decrease in relative integration, that of
and (p-tolyl)2O as a standard in C6D6 was prepared and its 1H NMR spectrum was obtained. MIL-125 was added, the suspension stirred for 3 h, and another 1H NMR spectrum was obtained. The change in the integrals was noted. While smaller molecules such as FeCp2 were taken up into the MOF, as evidenced by a decrease in relative integration, that of 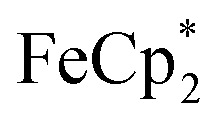 remained unchanged. Thus, we conclude that the
remained unchanged. Thus, we conclude that the 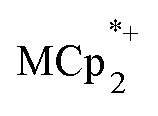 ions cannot enter the pores of the MOFs.
ions cannot enter the pores of the MOFs.
Addition of 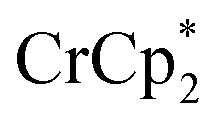 to a d8-THF + CD3CN 4 : 1 v/v suspension of MIL-125 (1/12th equiv. of
to a d8-THF + CD3CN 4 : 1 v/v suspension of MIL-125 (1/12th equiv. of 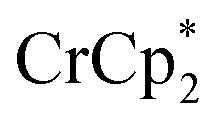 per Ti8 node) resulted in the disappearance of the 1H NMR resonance for
per Ti8 node) resulted in the disappearance of the 1H NMR resonance for 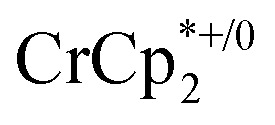 and a change in solution color from yellow to very pale yellow. The solution color suggests the formation of a very small concentration of
and a change in solution color from yellow to very pale yellow. The solution color suggests the formation of a very small concentration of 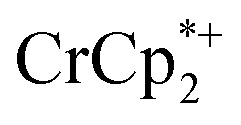 . Since neither
. Since neither 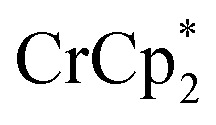 or
or 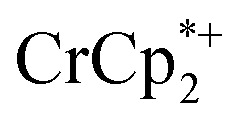 is taken up in the pores of the MOF, the absence of a
is taken up in the pores of the MOF, the absence of a 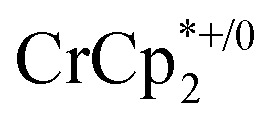 in solution suggests that
in solution suggests that 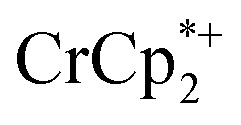 is formed and that it ion-pairs with the reduced MIL-125 (Fig. 1). The suggestion of ion pairing is supported by experiments adding 10 or 20 equiv. of nBu4N+PF6– into the 1 : 12 mixture of
is formed and that it ion-pairs with the reduced MIL-125 (Fig. 1). The suggestion of ion pairing is supported by experiments adding 10 or 20 equiv. of nBu4N+PF6– into the 1 : 12 mixture of 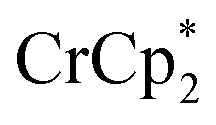 : MIL-125. This resulted in the liberation of ∼36% and 39% of
: MIL-125. This resulted in the liberation of ∼36% and 39% of 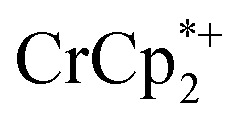 , respectively. Similar results are obtained with NH2-MIL-125 (see ESI†).
, respectively. Similar results are obtained with NH2-MIL-125 (see ESI†).
Fig. 1. Reduction of MIL-125 with 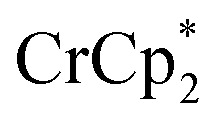 .
.

The addition of 3 equiv. of 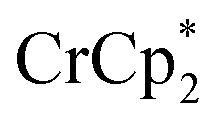 per node of MIL-125 gave solutions containing only
per node of MIL-125 gave solutions containing only 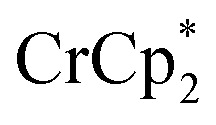 . Integration of
. Integration of 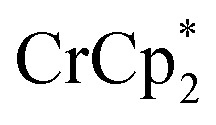 (relative to an internal standard) indicated that 0.27 equiv. of
(relative to an internal standard) indicated that 0.27 equiv. of 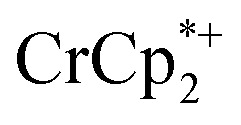 was ion-paired with the MIL-125. This represents the upper-limit for electron transfer to the MOF (batch 2, vide infra) in the absence of added Na+ cations.
was ion-paired with the MIL-125. This represents the upper-limit for electron transfer to the MOF (batch 2, vide infra) in the absence of added Na+ cations.
The effect of Na+ on chemical reductions with 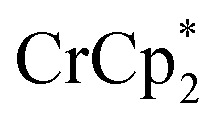
Added Na+ has a strong effect on the extent of MIL-125 reduction by 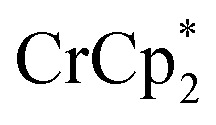 . Owing to its solubility, commercial availability, and stability of the counter anion, NaTFSI (TFSI = bis(trifluoromethane sulfonyl)imide) was used in these studies.
. Owing to its solubility, commercial availability, and stability of the counter anion, NaTFSI (TFSI = bis(trifluoromethane sulfonyl)imide) was used in these studies.
As mentioned above, addition of 3 equiv. of 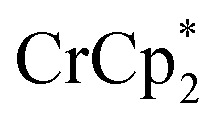 to a suspension of MIL-125 in the absence of Na+ resulted in slight reduction of MIL-125. Addition of 0.2 to 1.0 equiv. of NaTFSI into the above-mentioned mixture led to a change of the chemical shift of
to a suspension of MIL-125 in the absence of Na+ resulted in slight reduction of MIL-125. Addition of 0.2 to 1.0 equiv. of NaTFSI into the above-mentioned mixture led to a change of the chemical shift of 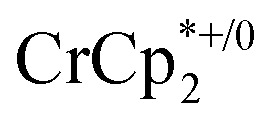 from ∼–6 ppm to ∼2 ppm, corresponding to
from ∼–6 ppm to ∼2 ppm, corresponding to 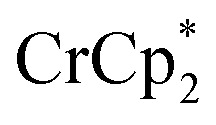 oxidation (Fig. 2). This suggests that reduction of MIL-125 is facilitated by and coupled to cation uptake, in this case, sodium, or sodium-coupled electron transfer (a version of ‘metal-ion coupled electron transfer’ coined by Nam and Fukuzumi for molecular reactions33).
oxidation (Fig. 2). This suggests that reduction of MIL-125 is facilitated by and coupled to cation uptake, in this case, sodium, or sodium-coupled electron transfer (a version of ‘metal-ion coupled electron transfer’ coined by Nam and Fukuzumi for molecular reactions33).
Fig. 2. Reduction of MIL-125 with 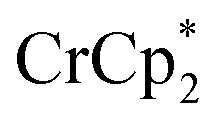 and Na+ (NaTFSI). (A) Schematic of the reaction. (B) 1H NMR spectra (d8-THF + CD3CN 4 : 1 v/v) of mixtures of MIL-125 + 3 equiv.
and Na+ (NaTFSI). (A) Schematic of the reaction. (B) 1H NMR spectra (d8-THF + CD3CN 4 : 1 v/v) of mixtures of MIL-125 + 3 equiv. 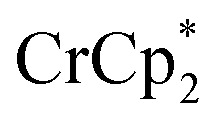 + different equiv. of Na+ per Ti8 node: 0 (red), 0.2 Na+ (yellow), 0.4 Na+ (green), 0.6 Na+ (light blue), 0.8 Na+ (dark blue), and 1.0 Na+ (purple). (C) Plot of e– transferred to MIL-125 (left axis) and fraction of
+ different equiv. of Na+ per Ti8 node: 0 (red), 0.2 Na+ (yellow), 0.4 Na+ (green), 0.6 Na+ (light blue), 0.8 Na+ (dark blue), and 1.0 Na+ (purple). (C) Plot of e– transferred to MIL-125 (left axis) and fraction of 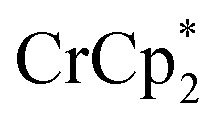 ion-paired in reduced MIL-125 (right axis) versus equiv. NaTFSI added. Orange circles: equiv. of Na+ associated with the reduced MIL-125, derived from the
ion-paired in reduced MIL-125 (right axis) versus equiv. NaTFSI added. Orange circles: equiv. of Na+ associated with the reduced MIL-125, derived from the 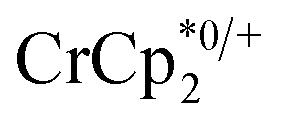 chemical shift. Blue circles: equiv.
chemical shift. Blue circles: equiv. 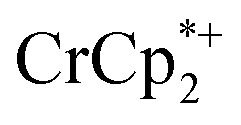 associated with the reduced MIL-125, derived from the change in integration of the
associated with the reduced MIL-125, derived from the change in integration of the 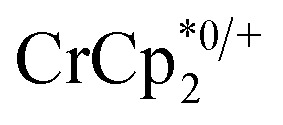 resonance upon mixing with MIL-125. Black circles: total extent of reduction (the sum of the blue and orange circles). Light blue diamonds: fraction of the reduced MIL-125 that is associated with
resonance upon mixing with MIL-125. Black circles: total extent of reduction (the sum of the blue and orange circles). Light blue diamonds: fraction of the reduced MIL-125 that is associated with 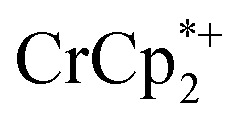 .
.

When 1 equivalent of NaTFSI was added to a mixture of MIL-125 and 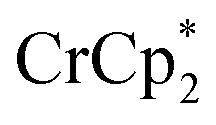 , the number of electrons in the MOF increased from 0.27 ± 0.08 (with no Na+) to 1.12 ± 0.03 per Ti8 node. Integration of the
, the number of electrons in the MOF increased from 0.27 ± 0.08 (with no Na+) to 1.12 ± 0.03 per Ti8 node. Integration of the 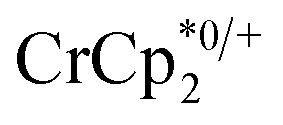 resonance showed that NaTFSI addition reduced the amount of the
resonance showed that NaTFSI addition reduced the amount of the 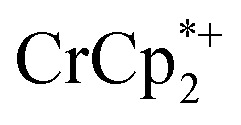 that was ion-paired with the MOF. The ion paired
that was ion-paired with the MOF. The ion paired 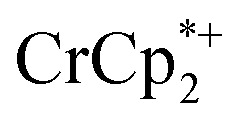 decreased from 0.25 to 0.09 equiv. per Ti8 node, which corresponds to a drop from 93% to 8% of the electrons being ion-paired with
decreased from 0.25 to 0.09 equiv. per Ti8 node, which corresponds to a drop from 93% to 8% of the electrons being ion-paired with 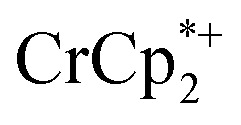 . Thus, 1 equivalent of Na+ led to roughly 1e– per TiO8 node and the charge balance for this added electron was provided by Na+, not
. Thus, 1 equivalent of Na+ led to roughly 1e– per TiO8 node and the charge balance for this added electron was provided by Na+, not 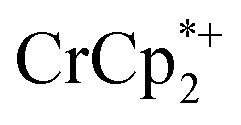 . These data suggest that all of the Na+ is associated with the reduced MIL-125. Given that there is one electron for every node in the MOF, the cation was very likely taken up into the pores, as discussed below. Similar results were obtained for NH2-MIL-125 (see ESI†). Taken together, the results show that under these conditions, MIL-125 behaves like a sodium battery, accepting electrons with sodium ions.
. These data suggest that all of the Na+ is associated with the reduced MIL-125. Given that there is one electron for every node in the MOF, the cation was very likely taken up into the pores, as discussed below. Similar results were obtained for NH2-MIL-125 (see ESI†). Taken together, the results show that under these conditions, MIL-125 behaves like a sodium battery, accepting electrons with sodium ions.
To determine how the amount of reductant impacts the extent of MOF reduction, 6 equiv. of 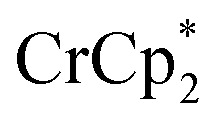 was added to a suspension of MIL-125. Under these conditions, with 0 and 1 equiv. of NaTFSI, MIL-125 is reduced by 0.28 ± 0.07 and 1.14 ± 0.04 electrons, respectively. Thus, doubling the equiv. of reductant does not impact the extent of reduction, as these values are within error of those obtained with 3 equiv. of
was added to a suspension of MIL-125. Under these conditions, with 0 and 1 equiv. of NaTFSI, MIL-125 is reduced by 0.28 ± 0.07 and 1.14 ± 0.04 electrons, respectively. Thus, doubling the equiv. of reductant does not impact the extent of reduction, as these values are within error of those obtained with 3 equiv. of 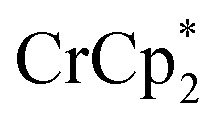 . Hence, the extent of reduction is controlled by the amount of Na+, not the amount of reductant.
. Hence, the extent of reduction is controlled by the amount of Na+, not the amount of reductant.
To probe the maximum amount that MIL-125 could be reduced with 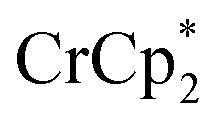 , excess NaTFSI was added to suspensions of MIL-125 with 6 equiv. of reductant. Fig. 3 shows the results of a titration: the extent of reduction versus the Na+ equiv. added. The electrons transferred to the MOF initially rises linearly with the added Na+, then levels off. With 20 equiv. of NaTFSI, MIL-125 is reduced by a 2.22 ± 0.09 electrons. The equiv. of
, excess NaTFSI was added to suspensions of MIL-125 with 6 equiv. of reductant. Fig. 3 shows the results of a titration: the extent of reduction versus the Na+ equiv. added. The electrons transferred to the MOF initially rises linearly with the added Na+, then levels off. With 20 equiv. of NaTFSI, MIL-125 is reduced by a 2.22 ± 0.09 electrons. The equiv. of 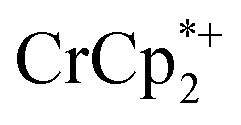 associated with reduced MIL-125 does not significantly change with increasing Na+ equiv., all being within error of one another.
associated with reduced MIL-125 does not significantly change with increasing Na+ equiv., all being within error of one another.
Fig. 3. Reduction of MIL-125 with 6 equiv. 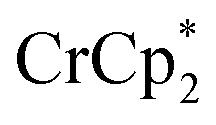 and 0–20 equiv. Na+ in d8-THF + CD3CN 4 : 1 v/v. Plot of e– transferred to MIL-125 (left axis), and fraction of
and 0–20 equiv. Na+ in d8-THF + CD3CN 4 : 1 v/v. Plot of e– transferred to MIL-125 (left axis), and fraction of 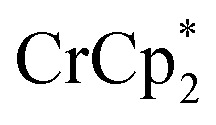 ion-paired in reduced MIL-125 (right axis), versus equiv. NaTFSI added. Orange circles: equiv. Na+ associated with the reduced MIL-125, derived from the
ion-paired in reduced MIL-125 (right axis), versus equiv. NaTFSI added. Orange circles: equiv. Na+ associated with the reduced MIL-125, derived from the 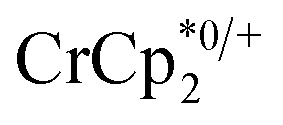 chemical shift. Blue circles: equiv.
chemical shift. Blue circles: equiv. 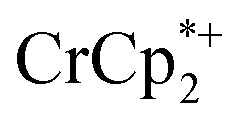 that are associated with the reduced MIL-125, derived from the change in integration of the
that are associated with the reduced MIL-125, derived from the change in integration of the 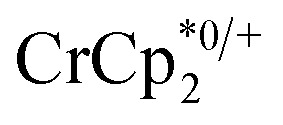 resonance upon mixing with MIL-125. Black circles: total extent of reduction (sum of blue and orange). Light blue diamonds represent the fraction of the reduced MIL-125 that is associated with
resonance upon mixing with MIL-125. Black circles: total extent of reduction (sum of blue and orange). Light blue diamonds represent the fraction of the reduced MIL-125 that is associated with 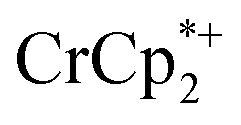 .
.

To probe the chemical oxidation of this reduced MOF (2.2e–), the reduced MIL-125 was isolated by washing with THF + MeCN (4 : 1 v/v), centrifuging and removal of the solvent layer for 5 times, followed by drying under vacuum. The reduced-MIL-125 was then reacted with 4 equiv. of 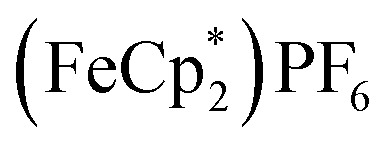 (E°′ = –0.48 V vs. Fc+/0 in MeCN), a pure 1 e–-acceptor, leading to bleaching of dark-purple colour of MIL-125. However, the colour of bleached MIL-125 was not perfectly white as the original one, which represents the reduced MIL-125 was not fully oxidized by
(E°′ = –0.48 V vs. Fc+/0 in MeCN), a pure 1 e–-acceptor, leading to bleaching of dark-purple colour of MIL-125. However, the colour of bleached MIL-125 was not perfectly white as the original one, which represents the reduced MIL-125 was not fully oxidized by 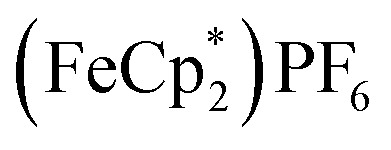 . By monitoring the change of 1H chemical shift of
. By monitoring the change of 1H chemical shift of 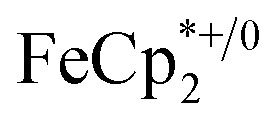 , the reaction of the reduced MIL-125 and ∼4 equiv. of
, the reaction of the reduced MIL-125 and ∼4 equiv. of 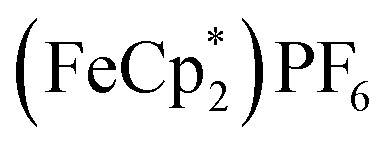 showed that the yield of
showed that the yield of 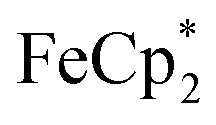 was 1.06 ± 0.19 equiv. As the reaction was further stirred for another day, the yield of
was 1.06 ± 0.19 equiv. As the reaction was further stirred for another day, the yield of 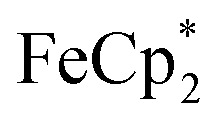 just increased less than 1% (data not shown). As shown by powder X-ray diffraction data (Fig. S8†), the structure of MIL-125 remains intact during reduction and after reoxidation.
just increased less than 1% (data not shown). As shown by powder X-ray diffraction data (Fig. S8†), the structure of MIL-125 remains intact during reduction and after reoxidation.
As addition of Na+ triggers reduction of MIL-125, we explored the converse: whether removal of Na+ would cause oxidation of the MOF. To test this, varying amounts of [2.2.2]-cryptand, a strong Na+ chelating ligand, was added to a sample of MIL-125 which was reduced with 3 equiv. 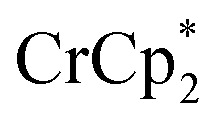 in the presence of 1 equiv. NaTFSI. Prior to addition of cryptand, NMR analysis of the solution indicated that MIL-125 was reduced by 1.07 ± 0.08 electrons, which was accompanied by uptake of 0.98 ± 0.09 equiv. of Na+ and ion-pairing of 0.09 ± 0.09 equiv. of
in the presence of 1 equiv. NaTFSI. Prior to addition of cryptand, NMR analysis of the solution indicated that MIL-125 was reduced by 1.07 ± 0.08 electrons, which was accompanied by uptake of 0.98 ± 0.09 equiv. of Na+ and ion-pairing of 0.09 ± 0.09 equiv. of 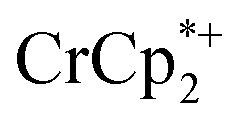 . Incremental addition of the cryptand resulted in a shift of the
. Incremental addition of the cryptand resulted in a shift of the 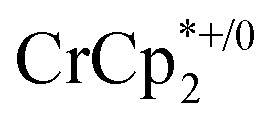 resonance, favoring more reduced
resonance, favoring more reduced 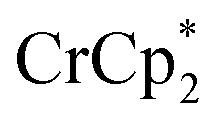 . From the peak position and integration of the
. From the peak position and integration of the 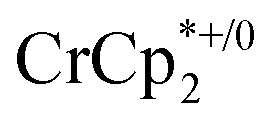 resonance, the amount of reduced MIL-125 that is associated with Na+ and
resonance, the amount of reduced MIL-125 that is associated with Na+ and 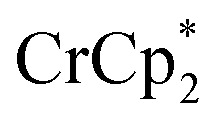 was again obtained. Fig. 4 shows that addition of cryptand results in oxidation of the MOF, with 1 equiv. of cryptand resulting in removal of 0.47 ± 0.10 electrons from MIL-125. As the cryptand simply removes Na+, the electron must go to
was again obtained. Fig. 4 shows that addition of cryptand results in oxidation of the MOF, with 1 equiv. of cryptand resulting in removal of 0.47 ± 0.10 electrons from MIL-125. As the cryptand simply removes Na+, the electron must go to 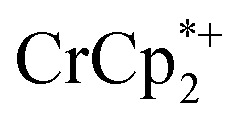 . As electrons are removed upon addition of cryptand, the equiv. Na+ associated with reduced MIL-125 decreases, as anticipated. Fig. 4 also shows that addition of cryptand increases the equiv. of
. As electrons are removed upon addition of cryptand, the equiv. Na+ associated with reduced MIL-125 decreases, as anticipated. Fig. 4 also shows that addition of cryptand increases the equiv. of 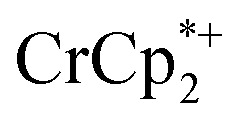 that are associated with the reduced MIL-125. In fact, with 1 equiv. of cryptand, there are 0.23 ± 0.12 equiv. of
that are associated with the reduced MIL-125. In fact, with 1 equiv. of cryptand, there are 0.23 ± 0.12 equiv. of 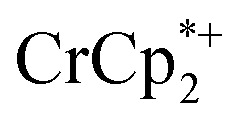 associated with the reduced MIL-125. This is within error of the maximum value obtained when 3 equiv. of
associated with the reduced MIL-125. This is within error of the maximum value obtained when 3 equiv. of 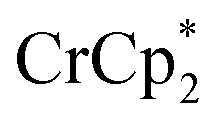 was added to MIL-125 (0.25 ± 0.08).
was added to MIL-125 (0.25 ± 0.08).
Fig. 4. Effect of[ adding cryptand to reduced MIL-125 (3 equiv. 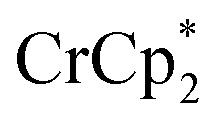 + 1 equiv of Na+), and reduction of MIL-125 with
+ 1 equiv of Na+), and reduction of MIL-125 with 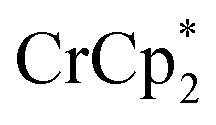 and [Na·cryptand]TFSI. (A) Schematic of this reaction. (B) 1H NMR spectra (d8-THF + CD3CN, 4 : 1 v/v) MIL-125 + 3 equiv.
and [Na·cryptand]TFSI. (A) Schematic of this reaction. (B) 1H NMR spectra (d8-THF + CD3CN, 4 : 1 v/v) MIL-125 + 3 equiv. 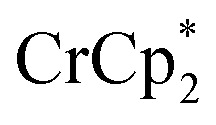 + 3 equiv. NaTFSI + 0 (red), 0.2 (yellow), 0.4 (green), 0.6 (light blue), 0.8 (dark blue), and 1.0 (purple) equiv. cryptand (per Ti8 node). The two peaks indicated by the arrow correspond to free (right) and Na-bound (left) cryptand. (C) Plot of e– in MIL-125 versus equivs of cryptand added (left axis), and equiv. of Na+ in solution (right axis). Orange circles: equivs Na+ associated with the reduced MIL-125, derived from the
+ 3 equiv. NaTFSI + 0 (red), 0.2 (yellow), 0.4 (green), 0.6 (light blue), 0.8 (dark blue), and 1.0 (purple) equiv. cryptand (per Ti8 node). The two peaks indicated by the arrow correspond to free (right) and Na-bound (left) cryptand. (C) Plot of e– in MIL-125 versus equivs of cryptand added (left axis), and equiv. of Na+ in solution (right axis). Orange circles: equivs Na+ associated with the reduced MIL-125, derived from the 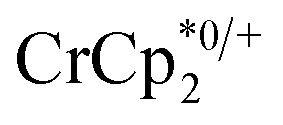 chemical shift. Blue circles: equivs
chemical shift. Blue circles: equivs 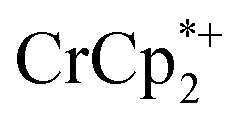 associated with the reduced MIL-125, derived from the change in integration of the
associated with the reduced MIL-125, derived from the change in integration of the 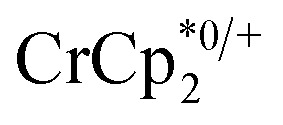 resonance. Black circles: total extent of reduction (sum of blue and orange). Light green diamonds: equiv. Na+ in solution, derived from the amount of Na+ still associated with reduced MIL-125. Dark green diamonds: equiv. of Na+ in solution, from integration of the cryptand/cryptand-sodium resonances. Open circles: number of e– in MIL-125 (per Ti8 node) for the converse reaction: addition of 3 equiv. of
resonance. Black circles: total extent of reduction (sum of blue and orange). Light green diamonds: equiv. Na+ in solution, derived from the amount of Na+ still associated with reduced MIL-125. Dark green diamonds: equiv. of Na+ in solution, from integration of the cryptand/cryptand-sodium resonances. Open circles: number of e– in MIL-125 (per Ti8 node) for the converse reaction: addition of 3 equiv. of 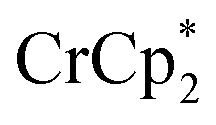 and 1 equiv. of [Na·cryptand]TFSI.
and 1 equiv. of [Na·cryptand]TFSI.

The chemical shift of the cryptand differed from that of [Na·cryptand]+, allowing for quantification of cryptand in solution that was bound to Na+. Fig. 4 shows that the equiv. of [Na·cryptand]+ from this analysis are similar to that obtained for the amount of Na+ in solution from analysis of the 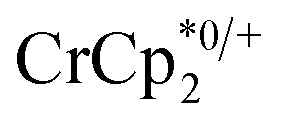 resonance.
resonance.
The converse reaction, that is, addition of 1 equiv. of [Na·cryptand]TFSI to a suspension of MIL-125 in a solution with 3 equiv. of 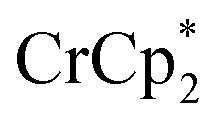 was also investigated. As in the absence of cryptand, addition of Na+ results in increased reduction of MIL-125, however, qualitatively, the reaction is slower. After ten days, MIL-125 is reduced by 0.47 ± 0.06 electrons, and the reduced material is associated with 0.37 ± 0.06 and 0.10 ± 0.08 equiv. of Na+ and
was also investigated. As in the absence of cryptand, addition of Na+ results in increased reduction of MIL-125, however, qualitatively, the reaction is slower. After ten days, MIL-125 is reduced by 0.47 ± 0.06 electrons, and the reduced material is associated with 0.37 ± 0.06 and 0.10 ± 0.08 equiv. of Na+ and 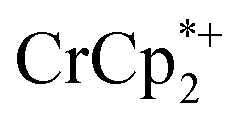 , respectively. These values are within error of those obtained when cryptand is added to the reduced MOF. Analogous results are obtained for NH2-MIL-125. Thus, the various NMR measurements are all consistent with the reactions at the top of Fig. 4, that the number of electrons and Na+ ions in the MOF change in parallel.
, respectively. These values are within error of those obtained when cryptand is added to the reduced MOF. Analogous results are obtained for NH2-MIL-125. Thus, the various NMR measurements are all consistent with the reactions at the top of Fig. 4, that the number of electrons and Na+ ions in the MOF change in parallel.
Chemical reduction with 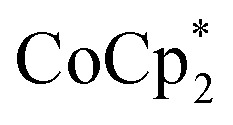 with and without Na+
with and without Na+
To test the maximum level of charging, a sample of MIL-125 was treated with 10 equiv. of NaBArF24 and 10 equiv. 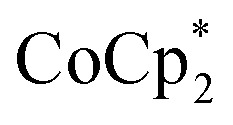 (BArF24 = tetrakis[(3,5-trifluoromethyl)phenyl]borate).
(BArF24 = tetrakis[(3,5-trifluoromethyl)phenyl]borate). 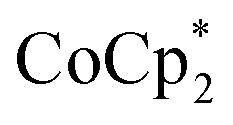 is a very strong soluble reductant, 0.45 V more reducing than
is a very strong soluble reductant, 0.45 V more reducing than 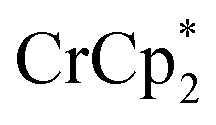 [E1/2 = –1.91 V for
[E1/2 = –1.91 V for 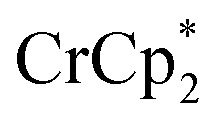 and –1.46 V for
and –1.46 V for 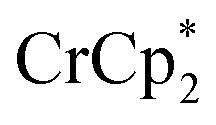 vs.
vs.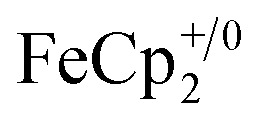 in MeCN34,35]. The NMR analysis showed 8.7 ± 0.2 equiv. of e– transferred. This roughly corresponds to every Ti center being reduced by one electron, ca. 8e– for each Ti8O8 cluster node.
in MeCN34,35]. The NMR analysis showed 8.7 ± 0.2 equiv. of e– transferred. This roughly corresponds to every Ti center being reduced by one electron, ca. 8e– for each Ti8O8 cluster node.
The reaction of MIL-125 with 10 equiv. 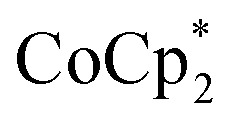 without Na+ showed very little darkening of MIL-125. From the 1H NMR
without Na+ showed very little darkening of MIL-125. From the 1H NMR 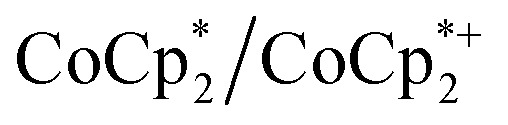 analysis, 0.05 ± 0.03 equiv. of
analysis, 0.05 ± 0.03 equiv. of 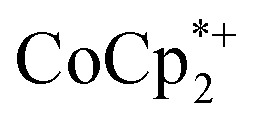 is formed per Ti8 cluster. This result is an average of 6 runs, from different MIL-125 batches (vide infra) in which the size of crystallites varied (see ESI† and Fig. 5 caption).
is formed per Ti8 cluster. This result is an average of 6 runs, from different MIL-125 batches (vide infra) in which the size of crystallites varied (see ESI† and Fig. 5 caption).
Fig. 5. (Top) Schematic of the reduction of MIL-125 with 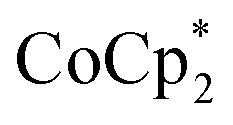 . The darkening indicates addition of electrons, and incorporation of sodium is designated by the red Na+. (Bottom) Extent of MIL-125 reduction versus equiv. of Na+ added to various batches of MIL-125. Experiments with 2 (circles) and 10 (squares) equiv. of
. The darkening indicates addition of electrons, and incorporation of sodium is designated by the red Na+. (Bottom) Extent of MIL-125 reduction versus equiv. of Na+ added to various batches of MIL-125. Experiments with 2 (circles) and 10 (squares) equiv. of 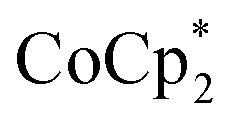 are plotted. The dashed line represents a 1 : 1 stoichiometry, and is not a fit. Description of the MIL-125 crystallites are: batch 1 has ellipsoid morphology and axes of 300 (±100)–850 (±50) nm; batch 2 has spherical morphology with axes of 85 (±17) nm; batch 3 has octahedral morphology with edges of 2650 (±1350) nm; batch 4 contains deuterated bdc linkers and has ellipsoid morphology and axes of 600 (±200)–1150 (±350) nm.
are plotted. The dashed line represents a 1 : 1 stoichiometry, and is not a fit. Description of the MIL-125 crystallites are: batch 1 has ellipsoid morphology and axes of 300 (±100)–850 (±50) nm; batch 2 has spherical morphology with axes of 85 (±17) nm; batch 3 has octahedral morphology with edges of 2650 (±1350) nm; batch 4 contains deuterated bdc linkers and has ellipsoid morphology and axes of 600 (±200)–1150 (±350) nm.

To elaborate on the role that the cation plays on reduction of the MOF, suspensions of MIL-125 with 2 equiv. of 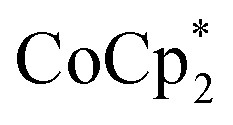 were titrated with 0–2 equiv. of NaBArF24 in THF, following the procedure described above (Fig. 5). In the presence of Na+, an immediate color change of the solids to dark purple is observed, concomitant with lightening of the solution due to conversion of
were titrated with 0–2 equiv. of NaBArF24 in THF, following the procedure described above (Fig. 5). In the presence of Na+, an immediate color change of the solids to dark purple is observed, concomitant with lightening of the solution due to conversion of 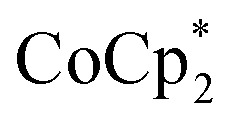 to
to 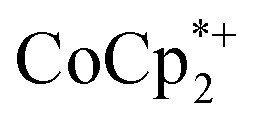 . Analysis of the
. Analysis of the 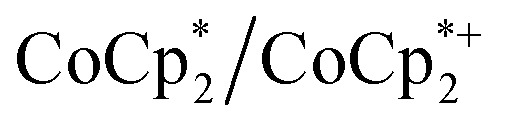 ratio indicates a good correlation between Na+ equivs and the extent of reduction, across four different batches of MIL-125 (Fig. 5). The solid MOF from reactions with 1 and 2 equiv. of Na+ per Ti8O8 cluster were also isolated, washed and analyzed by ICP. The Na : Ti ratios obtained were 0.10 ± 0.03 and 0.21 ± 0.02 (using batch 4 of MIL-125). These are in agreement with the ratios expected based on the addition stoichiometry, 0.125 and 0.25 respectively.
ratio indicates a good correlation between Na+ equivs and the extent of reduction, across four different batches of MIL-125 (Fig. 5). The solid MOF from reactions with 1 and 2 equiv. of Na+ per Ti8O8 cluster were also isolated, washed and analyzed by ICP. The Na : Ti ratios obtained were 0.10 ± 0.03 and 0.21 ± 0.02 (using batch 4 of MIL-125). These are in agreement with the ratios expected based on the addition stoichiometry, 0.125 and 0.25 respectively.
Plotting the combined data (Fig. 5) shows that the ratio of e– added to MIL-125 to the number of Na+ ions is close to 1 : 1. Thus, as in the 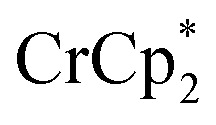 case above, each Na+ ion titrated in allows the addition of roughly one e– to the MOF.
case above, each Na+ ion titrated in allows the addition of roughly one e– to the MOF.
The close correlation of e– and Na+ stoichiometry prompted us to re-examine the observation of a small amount of electron transfer to the MOF in the absence of added cations. ICP analysis for Na and Ti was done on batch 2 of as-prepared MIL-125 and gave a Na : Ti ratio of 0.01 ± 0.02. This amount could account for the extent of MOF reduction observed in the absence of added Na+. The uncertainty in the ICP measurement is significant due to the correction for the sodium background (it is ubiquitous in the environment; see ESI†). The presence of 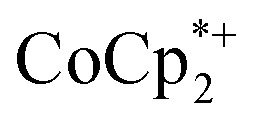 in solution in these experiments suggests the presence of a trace cation or acid (H+) impurity; if this were not the case, then all of the
in solution in these experiments suggests the presence of a trace cation or acid (H+) impurity; if this were not the case, then all of the 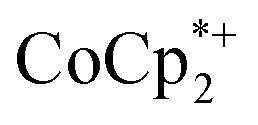 would be tightly ion-paired with the MOF and hence would not be detected in 1H NMR spectra.
would be tightly ion-paired with the MOF and hence would not be detected in 1H NMR spectra.
The EPR spectra of the 1, 2, and ∼8-electron reduced MOF were similar to that of the photoreduced MIL-125 (see ESI†). The rhombic signal broadens with increased extent of reduction, consistent with increased spin–spin interactions. Also, powder XRD patterns of the reduced materials indicate that the structure of MIL-125 is predominantly intact. Interestingly, upon reduction MIL-125's unit cell is found to slightly contract along it's c-axis, while featuring a small expansion in the ab-plane. At higher reduction levels, a few additional reflections are observed. Through Pawley fitting, these reflections were found to be forbidden for MIL-125 (Fig. S9–S11†) and may indicate some structural lability under reduction with 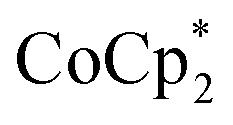 . These additional reflections are not observed in the case of reduction with
. These additional reflections are not observed in the case of reduction with 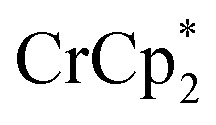 (Fig. S8†). For all chemically reduced samples described above, exposure to air restores the white color of oxidized MIL-125.
(Fig. S8†). For all chemically reduced samples described above, exposure to air restores the white color of oxidized MIL-125.
To probe whether e–/Na+ reduction occurred only at the surface or throughout the bulk of MIL-125, we examined chemical reductions using batches of the MOF that differed in their size and morphology. The smallest crystals (batch 2) were roughly spherical with a diameter of 85 ± 16.5 nm, and the largest (batch 3) were octahedral, with axes of 3650 ± 1350 nm. If reduction were limited to the surface Ti8 clusters, then these two batches should have given different extents of reduction of MIL-125, measured as the number of e– per Ti8 node in the entire sample. However, the extent of reduction does not depend on the crystal size (Fig. 5 and 6). This shows that reduction is not dependent on external surface area, that it is occurring at all of the nodes throughout the MOF. Thus, the e–/Na+ can migrate into the MOF pores. This conclusion is consistent with the very high level of reduction observed, 1–8e– per Ti8 node throughout the bulk of the material. There are simply not enough Ti8 clusters at the crystal surface to accept that many electrons, given the small ratio of surface to bulk for these sized crystals.
Fig. 6. Displacement ellipsoid (50%) representation of (top) 2-Na and (b) the anion of 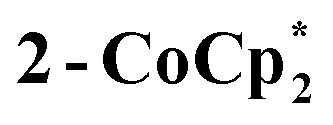 . For clarity, H-atoms are omitted and tBu groups are shown as sticks. There are two THF ligands to Na+ that are disordered, as shown (S: 24.5%, R: 75.5%); see the ESI.† Select bond distances (Å) for 2-Na: Ti1–O8 1.7796(25), Ti1–O1 1.8090(24), Ti2–O1 1.8090(25), Ti2–O2 1.7939(24), Ti3–O2 1.8285(24), Ti3–O3 1.7660(25), Ti4–O3 1.8634(25), Ti4–O4 1.7456(27), Ti5–O4 1.9075(26), Ti5–O5 1.7451(28), Ti6–O5 1.9473(27), Ti6–O6 1.9556(26), Ti7–O6 1.7346(27), Ti7–O7 1.8830(26), Ti8–O7 1.7571(25), Ti8–O8 1.8459(25). Select bond distances (Å) for
. For clarity, H-atoms are omitted and tBu groups are shown as sticks. There are two THF ligands to Na+ that are disordered, as shown (S: 24.5%, R: 75.5%); see the ESI.† Select bond distances (Å) for 2-Na: Ti1–O8 1.7796(25), Ti1–O1 1.8090(24), Ti2–O1 1.8090(25), Ti2–O2 1.7939(24), Ti3–O2 1.8285(24), Ti3–O3 1.7660(25), Ti4–O3 1.8634(25), Ti4–O4 1.7456(27), Ti5–O4 1.9075(26), Ti5–O5 1.7451(28), Ti6–O5 1.9473(27), Ti6–O6 1.9556(26), Ti7–O6 1.7346(27), Ti7–O7 1.8830(26), Ti8–O7 1.7571(25), Ti8–O8 1.8459(25). Select bond distances (Å) for 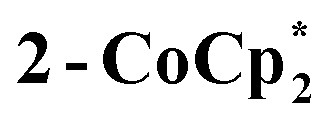 : Ti1–O2 1.8445(1), Ti1–O1 1.7990(1), Ti2–O1′ 1.8514(1), Ti2–O2 1.7666(1).
: Ti1–O2 1.8445(1), Ti1–O1 1.7990(1), Ti2–O1′ 1.8514(1), Ti2–O2 1.7666(1).

Soluble cluster analogues
To better understand the redox chemistry at a molecular level, soluble cluster analogues were prepared and studied. Ti8O8(OOCtBu)16 (1) was prepared as described36 and isolated as a white crystalline solid. Cluster 1 is comprised of eight roughly octahedral Ti(iv) centers arranged in a cyclic array, with each Ti linked to its neighbors by one oxo and two pivalate (tBuCOO–) bridging ligands. The solution 1H NMR spectrum (d8-toluene) of 1 features two sharp resonances at 1.34 and 1.22 ppm corresponding to tBu groups that are axial and equatorial with respect to the planar Ti8 ring. The ligand arrangement differs from that of the nodes in MIL-125 in that the latter has four of the carboxylates replaced by bridging hydroxo groups to give an empirical cluster formula of Ti8O8(OH)4(OOCR)12. Also, 1 features alkyl carboxylates rather than aryl carboxylates as found in MIL-125. While the molecular cluster with benzoate ligands can be prepared,36 the limited solubility and uninformative 1H NMR spectrum of Ti8O8(OOCPh)16 makes it unsuitable for these studies.
 |
3 |
 |
4 |
Syntheses of one-electron reduced congeners of 1 are readily achieved by treatment of 1 with 1 equiv. of an appropriate reductant. Thus, stirring a THF solution of 1 over 1 equiv. of Na/Hg for 6 hours results in a color change from white to blue and formation of 2-Na (eqn (3)). Addition of 1 equiv. of 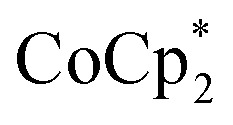 to a benzene solution of 1 also results in a color change to blue, with formation of the related 1e–-reduced congener,
to a benzene solution of 1 also results in a color change to blue, with formation of the related 1e–-reduced congener, 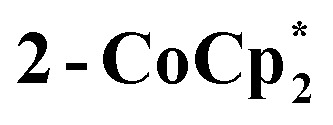 (eqn (4)). Both of these clusters have been thoroughly characterized (see ESI†), including by single-crystal X-ray diffraction (Fig. 6). Treatment of either 2-Na or
(eqn (4)). Both of these clusters have been thoroughly characterized (see ESI†), including by single-crystal X-ray diffraction (Fig. 6). Treatment of either 2-Na or 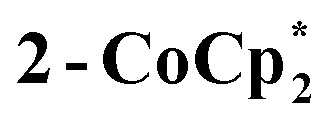 with 1 equiv. of the aminium oxidant [(p-tolyl)3N˙+]PF6– quantitatively converts them back to 1 (by 1H NMR spectroscopy in d8-THF).
with 1 equiv. of the aminium oxidant [(p-tolyl)3N˙+]PF6– quantitatively converts them back to 1 (by 1H NMR spectroscopy in d8-THF).
The X-ray crystal structure of 2-Na shows that the Ti8 ring is intact, albeit distorted, with a Na+ cation coordinated on one edge of the inside of the ring. The Na+ coordinates two THF molecules and the two μ-oxo ligands that flank Ti6. The Ti6–O distances of 1.9556(26) and 1.9473(27) Å are ∼0.12 Å elongated from the mean Ti–O distance in 1 (Ti–Oave = 1.827 Å; see Fig. 6).27 Moving around the ring from Ti6 the Ti–O distances alternate long and short. Next to Ti6 this difference is quite pronounced, with the two Ti–O distances for Ti5 and Ti7 differing by ∼0.15 Å. At Ti2 on the other side of the ring, however, the two Ti–O distances are almost the same, 1.7939(24) vs. 1.8090(25) Å, and are even shorter than the average in the fully oxidized 1. These metrical parameters indicate that the added electron is mostly localized on Ti6 in the solid-state, near the Na+, but that the influence of the added electron is clearly felt in the neighbouring octahedra. The addition of the Na+ renders the ring more elliptical, with distances between titanium atoms that are across the ring varying by 0.584 Å in 2-Na (vs. only 0.268 Å in 1).
The structure of 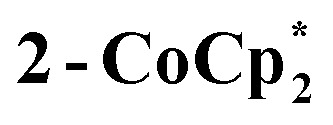 reveals no direct interaction between the anion and cation. In the cluster anion (Fig. 6 bottom), a 4-fold axis renders pairs of Ti atoms equivalent about the core. Here too, the Ti–O distances alternate between long and short, though the metrical parameters clearly show that all Ti are equivalent; the electron is delocalized. Twinning and disorder in the
reveals no direct interaction between the anion and cation. In the cluster anion (Fig. 6 bottom), a 4-fold axis renders pairs of Ti atoms equivalent about the core. Here too, the Ti–O distances alternate between long and short, though the metrical parameters clearly show that all Ti are equivalent; the electron is delocalized. Twinning and disorder in the 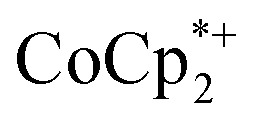 cation prevent obtaining its accurate bond metrics.
cation prevent obtaining its accurate bond metrics.
The solution 1H NMR spectrum of 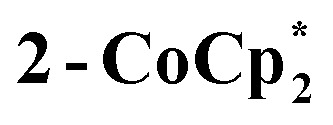 in d8-THF consists of two paramagnetically shifted and broadened resonances at 1.373 and 1.217 ppm, which correspond to the tBu protons of the cluster. The 1H NMR spectrum of 2-Na (d8-THF) likewise features two broadened resonances at 1.415 and 1.234 ppm. To determine whether these resonances correspond to all 144 pivalate protons of the cluster or just to only protons from tBu protons that are distant from the odd electron, an NMR sample of 2-Na in d8-THF was oxidized with 1 equiv. of [(p-tolyl)3N˙+]PF6. The integrals of the tBu resonances were the same versus an internal standard before and after oxidation, indicating that all tBu groups are observed in the 1H NMR spectrum of 2-Na. These results show that for both 2-Na and
in d8-THF consists of two paramagnetically shifted and broadened resonances at 1.373 and 1.217 ppm, which correspond to the tBu protons of the cluster. The 1H NMR spectrum of 2-Na (d8-THF) likewise features two broadened resonances at 1.415 and 1.234 ppm. To determine whether these resonances correspond to all 144 pivalate protons of the cluster or just to only protons from tBu protons that are distant from the odd electron, an NMR sample of 2-Na in d8-THF was oxidized with 1 equiv. of [(p-tolyl)3N˙+]PF6. The integrals of the tBu resonances were the same versus an internal standard before and after oxidation, indicating that all tBu groups are observed in the 1H NMR spectrum of 2-Na. These results show that for both 2-Na and 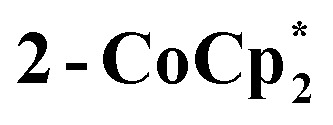 , the added electron is delocalized around the ring on the NMR timescale in solution. For 2-Na in C6D6, where the Na+ must be closely associated with the anionic Ti8O8 ring, this requires that there be rapid migration of the Na+ around the cluster ring. This contrasts with the localization of the Na(THF)2+ unit in the solid-state, as discussed above.
, the added electron is delocalized around the ring on the NMR timescale in solution. For 2-Na in C6D6, where the Na+ must be closely associated with the anionic Ti8O8 ring, this requires that there be rapid migration of the Na+ around the cluster ring. This contrasts with the localization of the Na(THF)2+ unit in the solid-state, as discussed above.
Discussion
MIL-125 and NH2-MIL-125 have previously been shown to be redox-active, like other titanium-oxo containing MOFs.23,30,37,38 UV irradiation (or visible for the amino derivative) in the presence of an alcohol causes reduction of the MOF, as indicated by the blue colour of the reduced material. EPR spectroscopy suggests that the reduction is Ti-based.5,23,30 We have recently shown that this reduction corresponds to a maximum of ∼2e– per Ti8O8 node. That reduction is a proton-coupled electron transfer (PCET) process, so each Ti8 cluster is reduced by (e– + H+), a net H-atom.31 Similar chemistry was shown for the COK-69 MOF that contains a [Ti3IV(μ3-O)(O)2(RCOO)6] cluster at each node.5
The studies reported here provide a detailed look at the cation-coupled reduction of MIL-125 and NH2-MIL-125. Using organometallic, soluble reductants and Na+ salts, and with the preparation of molecular versions of the reduced Ti8O8 nodes, this work provides an unusually detailed and quantitative view of the reduction of a MOF.
A simple 1H NMR assay to measure the redox stoichiometry in MOF redox reactions
A 1H NMR spectroscopic assay was first developed to determine the number of electrons added to a known weight of the MOF. By using bulky soluble reductants, 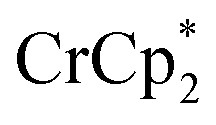 and
and 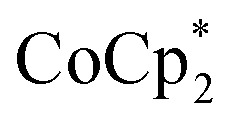 , it was shown that the reductant is not taken up into the pores of MIL-125. Then 1H NMR spectra reveal the concentrations of reduced and oxidized forms,
, it was shown that the reductant is not taken up into the pores of MIL-125. Then 1H NMR spectra reveal the concentrations of reduced and oxidized forms, 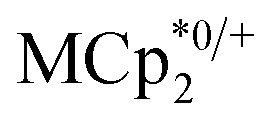 . In this case, both
. In this case, both 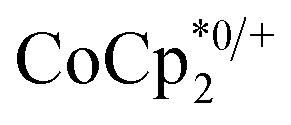 and
and 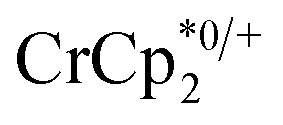 undergo fast chemical exchange, so a single resonance is observed, with the resonance position indicative of the mole fraction of each species (eqn (2)). The 1H NMR spectra showed no additional resonances, indicating the absence of decomposition of the MIL-125 or
undergo fast chemical exchange, so a single resonance is observed, with the resonance position indicative of the mole fraction of each species (eqn (2)). The 1H NMR spectra showed no additional resonances, indicating the absence of decomposition of the MIL-125 or 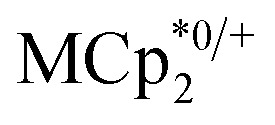 (M = Cr, Co). This assay should be widely applicable to redox reactions of materials as long as the reagent has a well-defined and well-positioned redox couple that undergoes clean reactivity and whose components do not all adsorb strongly on or in the material. Though there are now quite a few examples of redox-active MOFs, the extent of oxidation/reduction is often not quantified. An early exception was the work of de Combarieu et al. showing electrochemically that an iron MOF could absorb 0.6 equiv. of Li with reduction of Fe3+ to Fe2+.27 Other studies of MOF battery materials have followed.251H NMR spectroscopy has been used to quantify the oxidation of linkers in Mn-MOF-74 by the Dincă group.1 Such 1H NMR assays are typically only applicable when there is a substantial amount of redox change; they would challenging for 0.1% doping of a semiconducting MOF, for example.
(M = Cr, Co). This assay should be widely applicable to redox reactions of materials as long as the reagent has a well-defined and well-positioned redox couple that undergoes clean reactivity and whose components do not all adsorb strongly on or in the material. Though there are now quite a few examples of redox-active MOFs, the extent of oxidation/reduction is often not quantified. An early exception was the work of de Combarieu et al. showing electrochemically that an iron MOF could absorb 0.6 equiv. of Li with reduction of Fe3+ to Fe2+.27 Other studies of MOF battery materials have followed.251H NMR spectroscopy has been used to quantify the oxidation of linkers in Mn-MOF-74 by the Dincă group.1 Such 1H NMR assays are typically only applicable when there is a substantial amount of redox change; they would challenging for 0.1% doping of a semiconducting MOF, for example.
Reduction occurs throughout the MOF, not just at the surface
Treatment of MIL-125 with 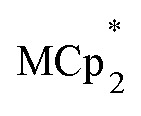 and a soluble Na+ source gave substantial charging of the MOF. With 1 equivalent of Na+ per Ti8O8 node, 1H NMR studies indicated roughly one electron per Ti8O8 cluster had been transferred to the MOF. With additional Na+, the MIL-125 may be reduced by up to 2 electrons per Ti8 node with
and a soluble Na+ source gave substantial charging of the MOF. With 1 equivalent of Na+ per Ti8O8 node, 1H NMR studies indicated roughly one electron per Ti8O8 cluster had been transferred to the MOF. With additional Na+, the MIL-125 may be reduced by up to 2 electrons per Ti8 node with 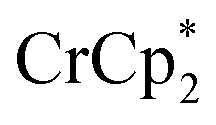 and up to a remarkable 8 electrons per node with
and up to a remarkable 8 electrons per node with 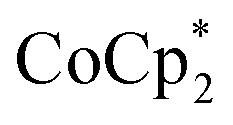 .
.
These very high charging levels are consistent only with reduction throughout the MIL-125 crystallites. There are simply not enough titanium ions in the surface layer of the MOF to accommodate all of these electrons (in the limit of 8e– per node, formally every titanium has been reduced to Ti(iii)). In addition, different batches of MIL-125 with different sizes and morphologies behave very similarly, showing that reduction is not simply a surface property. This reduction throughout the material, as discussed in the next section, is possible because the Na+ ions migrate into the pores to provide local charge balance for the added electrons.
Roughly 1 : 1 Na+ to e– stoichiometry of MIL-125 reduction
Titration experiments adding aliquots of Na+ to suspensions of MIL-125 with excess 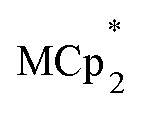 showed an equimolar increase in the number of electrons transferred to the MOF (a few electrons were transferred in the absence of Na+; see below). For instance, Fig. 5 and 6 show that the extent of reduction of MIL-125 with
showed an equimolar increase in the number of electrons transferred to the MOF (a few electrons were transferred in the absence of Na+; see below). For instance, Fig. 5 and 6 show that the extent of reduction of MIL-125 with 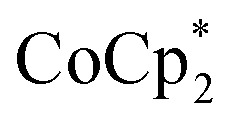 is equal to the amount of Na+ up to 2 equiv. of Na+. This is true when 2 or 10 equiv. of
is equal to the amount of Na+ up to 2 equiv. of Na+. This is true when 2 or 10 equiv. of 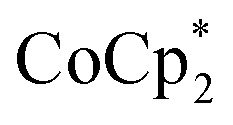 is used, and with any of four distinct batches of MIL-125 are used. These batches differ in their size, morphology, and in one case, feature deuterated bdc linkers. Thus, the effect of Na+ on extent of reduction is not an artefact of one batch. The data are clear that the Na+ cation controls the extent of reduction. This 1 : 1 stoichiometry based on the total amount of Na+ added and the total number of Ti8O8 nodes is consistent with the conclusion that the electrons and Na+ ions penetrate throughout the MOF. The
is used, and with any of four distinct batches of MIL-125 are used. These batches differ in their size, morphology, and in one case, feature deuterated bdc linkers. Thus, the effect of Na+ on extent of reduction is not an artefact of one batch. The data are clear that the Na+ cation controls the extent of reduction. This 1 : 1 stoichiometry based on the total amount of Na+ added and the total number of Ti8O8 nodes is consistent with the conclusion that the electrons and Na+ ions penetrate throughout the MOF. The 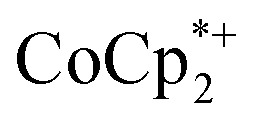 cation is too large to enter MIL-125 and therefore cannot provide coulombic stabilization proximal to each node as Na+ can. Therefore
cation is too large to enter MIL-125 and therefore cannot provide coulombic stabilization proximal to each node as Na+ can. Therefore 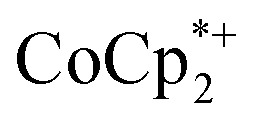 cation can only stabilize a small amount of reduction of the MOF.
cation can only stabilize a small amount of reduction of the MOF.
Control by the sodium cation is further evident in the experiment where Na+ was removed from the MOF. A sample of MIL-125 was equilibrated with 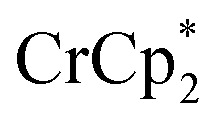 and NaTFSI, resulting in substantial electron transfer to the MOF (Fig. 4). Addition of cryptand to this mixture resulted in removal of some of the Na+ from the reduced MIL-125, as evidenced by the observation of [Na·cryptand]+ in NMR spectra. Removal of the Na+ resulted in back electron transfer to the
and NaTFSI, resulting in substantial electron transfer to the MOF (Fig. 4). Addition of cryptand to this mixture resulted in removal of some of the Na+ from the reduced MIL-125, as evidenced by the observation of [Na·cryptand]+ in NMR spectra. Removal of the Na+ resulted in back electron transfer to the 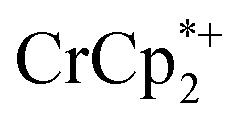 present in the solution, with oxidation of the reduced MOF. Cation control of electron transfer works in both directions, as adding Na+ leads to MOF reduction and removing the cation causes MOF oxidation. Despite the emphasis on the electron in the language used (“oxidation and reduction”), the cation is the key actor.
present in the solution, with oxidation of the reduced MOF. Cation control of electron transfer works in both directions, as adding Na+ leads to MOF reduction and removing the cation causes MOF oxidation. Despite the emphasis on the electron in the language used (“oxidation and reduction”), the cation is the key actor.
The cryptand does not remove all of the Na+, likely due this being an equilibrium process. Some insight into the binding of Na+ in MIL-125 is provided by the synthesis and characterization of molecular analogues of the Ti8O8 MOF nodes. The crystal structure of 2-Na shows the sodium ion chelated by two oxide ligands to a single titanium ion, which is likely to have the highest Ti(iii) character. However, NMR spectra of the molecular clusters in solution show that the sodium is rapidly moving around the Ti8O8 ring such that the high symmetry of the oxidized form is retained. This molecular material likely gives a detailed sense of what is likely occurring inside MIL-125 + (Na+ + e–), including the low barrier to Na+ migration around the Ti8O8 ring. A similar mode of cation coordination has been suggested to occur in the electrochemical lithiation of UiO-66 (Zr6O4(OH)4(bdc)6).39
The structure of the molecular anion with a decamethylcobaltocenium anion, 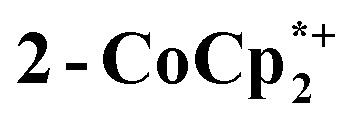 , shows no close contacts between the anion and cation, and a more delocalized anion. The solid is an ionic solid, a 3D lattice of [2–] and
, shows no close contacts between the anion and cation, and a more delocalized anion. The solid is an ionic solid, a 3D lattice of [2–] and 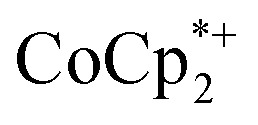 . These are of course in a 1 : 1 ratio in the solid, as the unit cell of all ionic compounds must have no net charge. The same thermodynamic preference for charge balance likely drives the 1 : 1 stoichiometry of Na+ to electrons. This is especially important at the high charging levels reached here, where essentially every Ti8O8 unit has been reduced by at least one electron. These MOFs are behaving just like the oxide cathodes of lithium ion batteries, which have a 1 : 1 stoichiometry of e–:Li+, for the same reason.
. These are of course in a 1 : 1 ratio in the solid, as the unit cell of all ionic compounds must have no net charge. The same thermodynamic preference for charge balance likely drives the 1 : 1 stoichiometry of Na+ to electrons. This is especially important at the high charging levels reached here, where essentially every Ti8O8 unit has been reduced by at least one electron. These MOFs are behaving just like the oxide cathodes of lithium ion batteries, which have a 1 : 1 stoichiometry of e–:Li+, for the same reason.
MIL-125 reduction in the absence of added Na+
Some electron transfer to MIL-125 is observed upon its reaction with 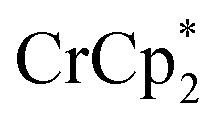 in the absence of Na+. With 3 equiv. of
in the absence of Na+. With 3 equiv. of 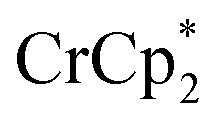 , roughly ∼0.25 equiv. of electrons were transferred. Reaction with a small amount of
, roughly ∼0.25 equiv. of electrons were transferred. Reaction with a small amount of 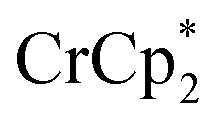 results in loss of the
results in loss of the 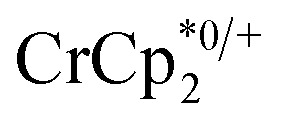 1H NMR signal, because of ion-pairing to the reduced MOF. The
1H NMR signal, because of ion-pairing to the reduced MOF. The 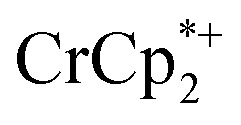 can be in part displaced by addition of the inert salt nBu4N+PF6– which can also engage in ion pairing. Since the
can be in part displaced by addition of the inert salt nBu4N+PF6– which can also engage in ion pairing. Since the 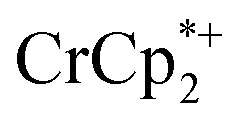 is too large to fit in the pores, electrons can be accommodated only in surface layers of the MOF, and only a limited number of electrons can be transferred.
is too large to fit in the pores, electrons can be accommodated only in surface layers of the MOF, and only a limited number of electrons can be transferred.
Studies done with 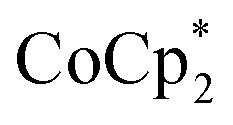 as a reductant in most ways closely paralleled those done with
as a reductant in most ways closely paralleled those done with 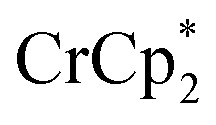 . In some cases, however, some
. In some cases, however, some 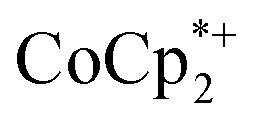 could be observed when small amounts of
could be observed when small amounts of 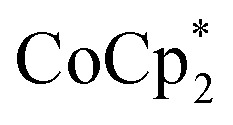 were used in the absence of Na+. This may be due to the higher sensitivity of the 1H NMR experiment for the sharp diamagnetic signal for
were used in the absence of Na+. This may be due to the higher sensitivity of the 1H NMR experiment for the sharp diamagnetic signal for 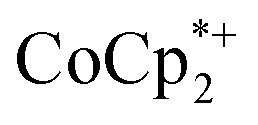 versus the broad paramagnetic signal of the chromium analogue. In one of these solutions, ICP analysis provided evidence for a small amount of sodium in the reduced MIL-125. Sodium is ubiquitous in the environment, for example on glass surfaces, and could be present in sufficient trace amounts to account for the
versus the broad paramagnetic signal of the chromium analogue. In one of these solutions, ICP analysis provided evidence for a small amount of sodium in the reduced MIL-125. Sodium is ubiquitous in the environment, for example on glass surfaces, and could be present in sufficient trace amounts to account for the 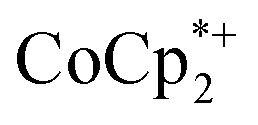 observed. Particularly at low concentrations or low charging levels, we caution other researchers to be wary of the large influence of trace Na+ or trace acid (H+).
observed. Particularly at low concentrations or low charging levels, we caution other researchers to be wary of the large influence of trace Na+ or trace acid (H+).
Effect of the reductant; stability of the reduced MOF
As emphasized above, both 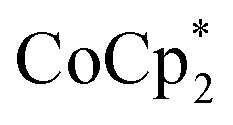 and
and 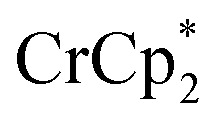 transfer electrons to MIL-125 in a roughly 1 : 1 stoichiometry with the amount of Na+ present. The fact that
transfer electrons to MIL-125 in a roughly 1 : 1 stoichiometry with the amount of Na+ present. The fact that 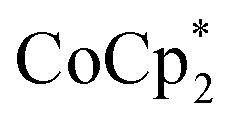 is a much stronger reducing agent, by about –0.45 V, has no effect on this stoichiometry. However, the higher reducing power of
is a much stronger reducing agent, by about –0.45 V, has no effect on this stoichiometry. However, the higher reducing power of 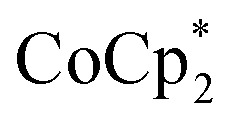 does change the overall extent to which MIL-125 can be reduced. Reductions with
does change the overall extent to which MIL-125 can be reduced. Reductions with 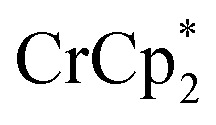 proceed only to ca. 2 electrons per Ti8O8 node, even the presence of excess reductant and excess Na+ (Fig. 3). Using
proceed only to ca. 2 electrons per Ti8O8 node, even the presence of excess reductant and excess Na+ (Fig. 3). Using 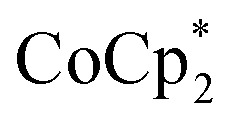 , in contrast, allows the addition of ∼8 electrons per node (perhaps with a small amount of decay of the MOF). This suggests that (e– + Na+) can be added into MIL-125 up to a certain concentration that is set by the thermodynamic reducing power of the reductant.
, in contrast, allows the addition of ∼8 electrons per node (perhaps with a small amount of decay of the MOF). This suggests that (e– + Na+) can be added into MIL-125 up to a certain concentration that is set by the thermodynamic reducing power of the reductant.
Conclusions
The stoichiometric addition of electrons and sodium ions to the MOFs MIL-125 and NH2-MIL-125 has been examined in detail. A simple solution NMR assay was developed, in concert with titration experiments, to separately quantify the e– and Na+ stoichiometries. This assay should be widely applicable to other MOFs and heterogeneous solids. MIL-125 show little reactivity with the metallocene reductants 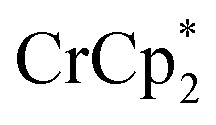 and
and 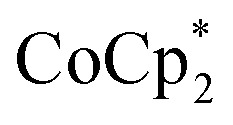 alone. Addition of Na+ cations to the suspensions, however, led to substantial reduction of the MOF. Clean and reversible reduction of the MIL-125 has been observed up to 1 (e– + Na+) per Ti8O8 node. This high charging amount, and studies with MIL-125 crystallites of different sizes, show that reduction occurs at each node throughout the MOF. Molecular analogues of the reduced Ti8O8 nodes have been prepared and their X-ray crystal structures show that Na+ binds inside these clusters while
alone. Addition of Na+ cations to the suspensions, however, led to substantial reduction of the MOF. Clean and reversible reduction of the MIL-125 has been observed up to 1 (e– + Na+) per Ti8O8 node. This high charging amount, and studies with MIL-125 crystallites of different sizes, show that reduction occurs at each node throughout the MOF. Molecular analogues of the reduced Ti8O8 nodes have been prepared and their X-ray crystal structures show that Na+ binds inside these clusters while 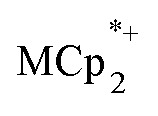 does not.
does not.
The number of e– added to the MOF closely parallels the number of Na+ added. Addition of cryptand to remove the Na+ from the MOF results in the corresponding transfer of e– back to 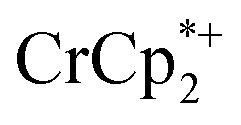 . In the presence of excess
. In the presence of excess 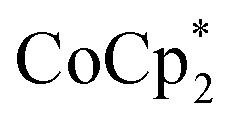 and Na+, MIL-125 can be reduced up to 8e– per Ti8 node. These data show that reduction of the MOF is controlled by the number of Na+. Only limited reduction occurs in the absence of Na+ since the
and Na+, MIL-125 can be reduced up to 8e– per Ti8 node. These data show that reduction of the MOF is controlled by the number of Na+. Only limited reduction occurs in the absence of Na+ since the 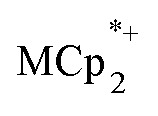 cations are too large to fit in the pores of the MOF. This study thus shows the critical role of charge balance by the cation that accompanies the electron, a principle that is likely to be general for MOFs and many other redox-active materials.
cations are too large to fit in the pores of the MOF. This study thus shows the critical role of charge balance by the cation that accompanies the electron, a principle that is likely to be general for MOFs and many other redox-active materials.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
C. T. S. gratefully acknowledges financial support from the U.S. National Institute of Health, postdoctoral fellowship 1F32GM099316. J. M. M. thanks the U.S. National Science Foundation for support via awards CHE-1151726 and CHE-1609434. S. S., B. B. and D. D. V. gratefully acknowledge the FWO for funding (Aspirant, post-doctoral grant and project funding).
Footnotes
†Electronic supplementary information (ESI) available: Additional experimental descriptions and data. CCDC 1868235 and 1868236. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04138e
References
- Cozzolino A. F., Brozek C. K., Palmer R. D., Yano J., Li M., Dincă M. J. Am. Chem. Soc. 2014;136:3334–3337. doi: 10.1021/ja411808r. [DOI] [PubMed] [Google Scholar]
- Tulchinsky Y., Hendon C. H., Lomachenko K. A., Borfecchia E., Melot B. C., Hudson M. R., Tarver J. D., Korzyński M. D., Stubbs A. W., Kagan J. J., Lamberti C., Brown C. M., Dincă M. J. Am. Chem. Soc. 2017;139:5992–5997. doi: 10.1021/jacs.7b02161. [DOI] [PubMed] [Google Scholar]
- Aubrey M. L., Long J. R. J. Am. Chem. Soc. 2015;137:13594–13602. doi: 10.1021/jacs.5b08022. [DOI] [PubMed] [Google Scholar]
- Smolders S., Lomachenko K. A., Bueken B., Struyf A., Bugaev A. L., Atzori C., Stock N., Lamberti C., Roeffaers M. B. J., De Vos D. E. ChemPhysChem. 2017;19:373–378. doi: 10.1002/cphc.201700967. [DOI] [PubMed] [Google Scholar]
- Bueken B., Vermoortele F., Vanpoucke D. E. P., Reinsch H., Tsou C.-C., Valvekens P., De Baerdemaeker T., Ameloot R., Kirschhock C. E. A., Van Speybroeck V., Mayer J. M., De Vos D. Angew. Chem., Int. Ed. 2015;54:13912–13917. doi: 10.1002/anie.201505512. [DOI] [PubMed] [Google Scholar]
- Feng D., Lei T., Lukatskaya M. R., Park J., Huang Z., Lee M., Shaw L., Chen S., Yakovenko A. A., Kulkarni A., Xiao J., Fredrickson K., Tok J. B., Zou X., Cui Y., Bao Z. Nat. Energy. 2018;3:30–36. [Google Scholar]
- Morozan A., Jaouen F. Energy Environ. Sci. 2012;5:9269. [Google Scholar]
- Aubrey M. L., Wiers B. M., Andrews S. C., Sakurai T., Reyes-Lillo S. E., Hamed S. M., Yu C.-J., Darago L. E., Mason J. A., Baeg J.-O., Grandjean F., Long G. J., Seki S., Neaton J. B., Yang P., Long J. R. Nat. Mater. 2018;17:625–632. doi: 10.1038/s41563-018-0098-1. [DOI] [PubMed] [Google Scholar]
- DeGayner J. A., Jeon I.-R., Sun L., Dincă M., Harris T. D. J. Am. Chem. Soc. 2017;139:4175–4184. doi: 10.1021/jacs.7b00705. [DOI] [PubMed] [Google Scholar]
- Xie L. S., Sun L., Wan R., Park S. S., DeGayner J. A., Hendon C. H., Dincă M. J. Am. Chem. Soc. 2018;140:7411–7414. doi: 10.1021/jacs.8b03604. [DOI] [PubMed] [Google Scholar]
- Sun L., Campbell M. G., Dincă M. Angew. Chem., Int. Ed. 2016;55:3566–3579. doi: 10.1002/anie.201506219. [DOI] [PubMed] [Google Scholar]
- Sun L., Hendon C. H., Minier M. A., Walsh A., Dincă M. J. Am. Chem. Soc. 2015;137:6164–6167. doi: 10.1021/jacs.5b02897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheberla D., Sun L., Blood-Forsythe M. A., Er S., Wade C. R., Brozek C. K., Aspuru-Guzik A., Dincă M. J. Am. Chem. Soc. 2014;136:8859–8862. doi: 10.1021/ja502765n. [DOI] [PubMed] [Google Scholar]
- Alvaro M., Carbonell E., Ferrer B., Llabrés i Xamena F. X., Garcia H. Chemistry. 2007;13:5106–5112. doi: 10.1002/chem.200601003. [DOI] [PubMed] [Google Scholar]
- Usman M., Mendiratta S., Lu K.-L. Adv. Mater. 2016;29:1605071. doi: 10.1002/adma.201605071. [DOI] [PubMed] [Google Scholar]
- Stassen I., Burtch N., Talin A., Falcaro P., Allendorf M., Ameloot R. Chem. Soc. Rev. 2017;46:3185–3241. doi: 10.1039/c7cs00122c. [DOI] [PubMed] [Google Scholar]
- Kornienko N., Zhao Y., Kley C. S., Zhu C., Kim D., Lin S., Chang C. J., Yaghi O. M., Yang P. J. Am. Chem. Soc. 2015;137:14129–14135. doi: 10.1021/jacs.5b08212. [DOI] [PubMed] [Google Scholar]
- Fang Y., Ma Y., Zheng M., Yang P., Asiri A. M., Wang X. Coord. Chem. Rev. 2018;373:83–115. [Google Scholar]
- Zhou J., Wang B. Chem. Soc. Rev. 2017;46:6927–6945. doi: 10.1039/c7cs00283a. [DOI] [PubMed] [Google Scholar]
- Mahmood A., Guo W., Tabassum H., Zou R. Adv. Energy Mater. 2016;6:1600423. [Google Scholar]
- Wang H., Zhu Q.-L., Zou R., Xu Q. Chem. 2017;2:52–80. [Google Scholar]
- Zhao Y., Song Z., Li X., Sun Q., Cheng N., Lawes S., Sun X. Energy Storage Materials. 2016;2:35–62. [Google Scholar]
- Fu Y., Sun D., Chen Y., Huang R., Ding Z., Fu X., Li Z. Angew. Chem., Int. Ed. 2012;51:3364–3367. doi: 10.1002/anie.201108357. [DOI] [PubMed] [Google Scholar]
- Peng Z., Yi X., Liu Z., Shang J., Wang D. ACS Appl. Mater. Interfaces. 2016;8:14578–14585. doi: 10.1021/acsami.6b03418. [DOI] [PubMed] [Google Scholar]
- Wang L., Han Y., Feng X., Zhou J., Qi P., Wang B. Coord. Chem. Rev. 2016;307:361–381. [Google Scholar]
- Lou X., Ning Y., Li C., Shen M., Hu B., Hu X., Hu B. Inorg. Chem. 2018;57:3126–3132. doi: 10.1021/acs.inorgchem.7b02939. [DOI] [PubMed] [Google Scholar]
- de Combarieu G., Morcrette M., Millange F., Guillou N., Cabana J., Grey C. P., Margiolaki I., Férey G., Tarascon J.-M. Chem. Mater. 2009;21:1602–1611. [Google Scholar]
- D'Alessandro D. M. Chem. Commun. 2016;52:8957–8971. doi: 10.1039/c6cc00805d. [DOI] [PubMed] [Google Scholar]
- Lin S., Usov P. M., Morris A. J. Chem. Commun. 2018;54:6965–6974. doi: 10.1039/c8cc01664j. [DOI] [PubMed] [Google Scholar]
- Dan-Hardi M., Serre C., Frot T., Rozes L., Maurin G., Sanchez C., Férey G., Maurín G. J. Am. Chem. Soc. 2009;131:10857–10859. doi: 10.1021/ja903726m. [DOI] [PubMed] [Google Scholar]
- Saouma C. T., Richard S., Smolders S., Delley M. F., Ameloot R., Vermoortele F., De Vos D. E., Mayer J. M. J. Am. Chem. Soc. 2018;140(47):16184–16189. doi: 10.1021/jacs.8b09120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandström J., Dynamic NMR Spectroscopy, 1982. [Google Scholar]
- Morimoto Y., Kotani H., Park J., Lee Y.-M., Nam W., Fukuzumi S. J. Am. Chem. Soc. 2011;133:403–405. doi: 10.1021/ja109056x. [DOI] [PubMed] [Google Scholar]
- Connelly N. G., Geiger W. E. Chem. Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- Valdez C. N., Schimpf A. M., Gamelin D. R., Mayer J. M. J. Am. Chem. Soc. 2016;138:1377–1385. doi: 10.1021/jacs.5b12182. [DOI] [PubMed] [Google Scholar]
- Frot T., Cochet S., Laurent G., Sassoye C., Popall M., Sanchez C., Rozes L. Eur. J. Inorg. Chem. 2010;8:5650–5659. [Google Scholar]
- Nguyen H. L., Gándara F., Furukawa H., Doan T. L. H., Cordova K. E., Yaghi O. M. J. Am. Chem. Soc. 2016;138(13):4330. doi: 10.1021/jacs.6b01233. [DOI] [PubMed] [Google Scholar]
- Castells-Gil J., Padial N. M., Almora-Barrios N., Albero J., Ruiz-Salvador A. R., González-Platas J., García H., Martí-Gastaldo C. Angew. Chem. 2018;130:8589–8593. doi: 10.1002/anie.201802089. [DOI] [PubMed] [Google Scholar]
- Tang B., Huang S., Fang Y., Hu J., Malonzo C., Truhlar D. G., Stein A. J. Chem. Phys. 2016;144:194702. doi: 10.1063/1.4948706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.