

Abstract
Abnormal alternative splicing (AS) caused by alterations to splicing factors contributes to tumor progression. Serine/arginine splicing factor 1 (SRSF1) has emerged as a key oncodriver in numerous solid tumors, leaving its roles and mechanisms largely obscure in glioma. Here, we demonstrate that SRSF1 is increased in glioma tissues and cell lines. Moreover, its expression was correlated positively with tumor grade and Ki-67 index, but inversely with patient survival. Using RNA-Seq, we comprehensively screened and identified multiple SRSF1-affected AS events. Motif analysis revealed a position-dependent modulation of AS by SRSF1 in glioma. Functionally, we verified that SRSF1 promoted cell proliferation, survival, and invasion by specifically switching the AS of the myosin IB (MYO1B) gene and facilitating the expression of the oncogenic and membrane-localized isoform, MYO1B-fl. Strikingly, MYO1B splicing was dysregulated in parallel with SRSF1 expression in gliomas and predicted the poor prognosis of the patients. Further investigation revealed that SRSF1-guided AS of the MYO1B gene increased the tumorigenic potential of glioma cells through the PDK1/AKT and PAK/LIMK pathways. Taken together, we identify SRSF1 as an important oncodriver that integrates AS control of MYO1B into promotion of gliomagenesis and represents a potential prognostic biomarker and target for glioma therapy.
Keywords: Cell Biology, Oncology
Keywords: Brain cancer, RNA processing

Introduction
Alternative splicing (AS) of pre-mRNA is a vital post-transcriptional process determining the proteomic complexity of mammals (1). Generally, AS regulation depends on the strength of intrinsic cis elements, including intronic and exonic enhancers and silencers, which recruit trans-acting splicing factors to promote or suppress the utilization of adjacent splice sites (2, 3). Failure to accurately recognize spice sites due to either splice site mutation or splicing factor dysregulation ultimately leads to the generation of abnormal mature mRNA variants encoding for deleterious isoforms and contributes substantially to tumor malignancy (1, 4, 5). The “cancerous” splice variants of specific genes have turned into novel molecular biomarkers as well as therapeutic targets to outwit cancer treatment (6).
Serine/arginine-rich splicing factors (SRSFs) are well characterized for their roles in AS and are composed of 1 or 2 RNA recognition motifs (RRMs) and a serine/arginine-rich (RS) domain (7). Among these SRSFs, SRSF1 is a prototypical splicing factor that specifically binds to exonic enhancers and stimulates splicing (8). Increasing evidence demonstrates that SRSF1, predominately driven by the transcription factor MYC (9), is overexpressed in multiple human cancers and exerts oncogenic roles via control of AS of cancer-related genes (10–12). Recently, genome-wide studies have extensively identified the AS targets of SRSF1 (13, 14) and established its concentration/position-dependent splicing model (12, 15). The emerging roles of SRSF1 in AS regulation in cancer are opening up a new therapeutic avenue.
Gliomas are the most frequent primary brain tumors (16, 17). Malignant glioma, especially glioblastoma (GBM), is associated with dismal prognoses, primarily due to its infiltrating properties and the emergence of chemoresistance (18). Treatment options remain limited in part due to the still poor understanding of the basic biology of glioma. Given the complexity of splicing regulation in brain, abnormal splicing may be a significant but yet underexplored contributor to gliomagenesis. Indeed, functional studies have revealed several splicing factors as oncogenic candidates by switching the AS products toward tumor-promoting isoforms in glioma cells (19–21). However, whether SRSF1 participates in glioma onset and progression remains unknown.
In the present study, we sought to elucidate the expression, clinical relevance, biological function, and underlying mechanism of SRSF1 in gliomas, focusing on the aspect of AS control. Upregulation of SRSF1 was observed in gliomas and predicted patients’ adverse prognoses. Through RNA sequencing (RNA-Seq) and motif analyses, we systematically identified hundreds of SRSF1-affected AS events and described a position-dependent modulation of AS by SRSF1 in glioma. We further verified that SRSF1 promotes the proliferation, survival, and invasion of glioma cells by switching the AS of the myosin IB (MYO1B) gene. Taken together, our findings highlight a role for SRSF1 as a splicing regulator in glioma biology, which contributes to multiple aspects of the glioma phenotype.
Results
SRSF1 is increased in gliomas, and its higher expression predicts worse prognosis.
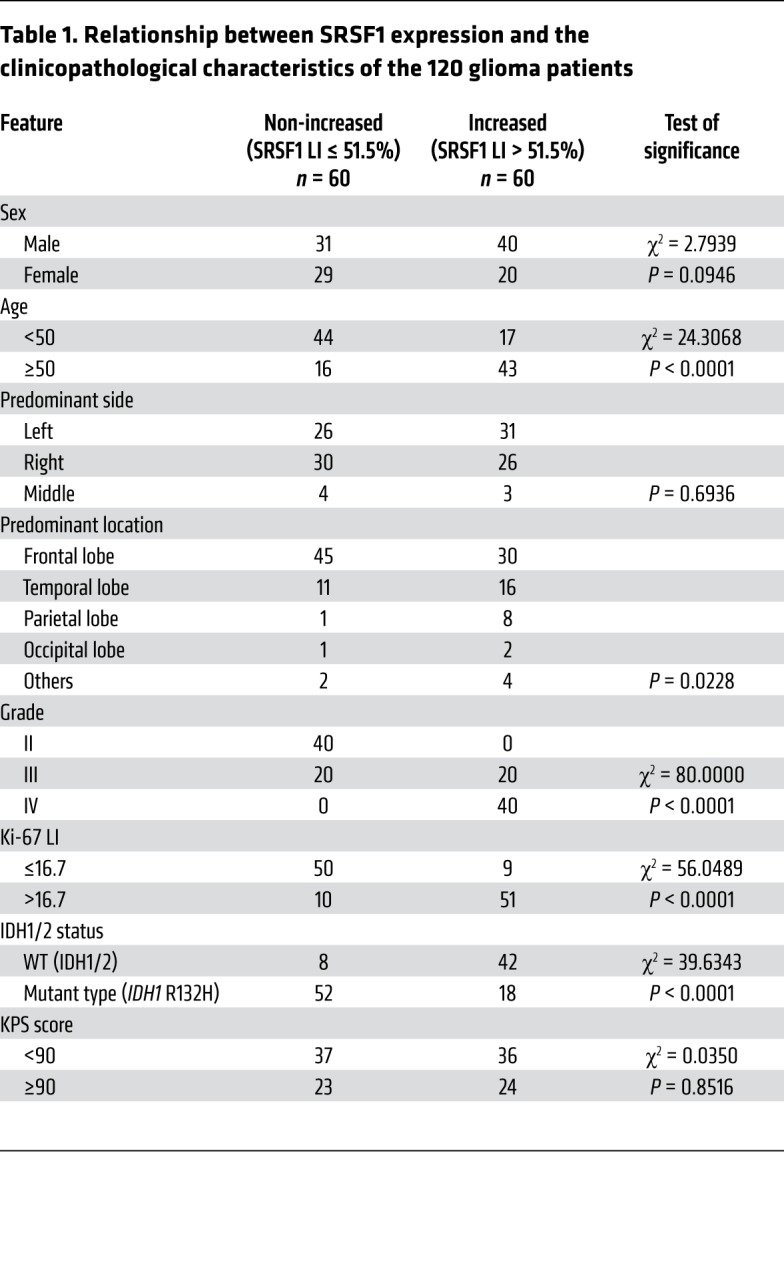
To investigate whether SRSF1 was involved in gliomagenesis, we first explored alteration of its expression in glioma tissues. In silico analysis of the 3 published datasets from Oncomine (http://www.oncomine.org) revealed a significant increase in SRSF1 mRNA content in GBM tissues as compared with normal brain (NB) tissues (P < 0.001; Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI120279DS1). This result was reinforced by the quantification of SRSF1 mRNA in 14 glioma tissues (including 6 lower-grade gliomas [LGGs; WHO grade II–III] and 8 GBMs [WHO grade IV]) and 5 NBs (Figure 1A). Western blot analysis verified that human GBM tissues and cell lines showed significantly higher levels of SRSF1 protein when compared with NBs and the human immortal astrocyte cell line UC2, respectively (Figure 1, B and C, and Supplemental Figure 1, B and C). SRSF1 IHC confirmed its nuclear localization and a progressive increase in its labeling index (LI) with the elevation of glioma grade (P < 0.001; Figure 1D). Moreover, SRSF1 expression was positively correlated with the proliferation index (Ki-67 LI; r = 0.839, P < 0.0001; Figure 1E and Supplemental Figure 1, D and E). Importantly, SRSF1 overexpression was clearly associated with older age (P < 0.0001), advanced grade (P < 0.0001), higher Ki-67 LI (P < 0.0001), and WT isocitrate dehydrogenase 1 and 2 (IDH1/2) status (P < 0.0001; Table 1). Kaplan-Meier analyses showed that patients with higher levels of SRSF1 had shorter disease-free survival (DFS; P < 0.0001) and overall survival (OS; P < 0.0001; Figure 1F). The prognostic value of SRSF1 was further verified by The Cancer Genome Atlas (TCGA) data analysis (OS: P < 0.0001; Supplemental Figure 1F). Furthermore, even within the cohort of glioma patients of similar ages (age ≥ 50, age < 50), identical IDH1/2 gene type, and similar Karnofsky Performance Status (KPS; <90, ≥90), the association between high SRSF1 expression and poor prognosis remained apparent (DFS: P < 0.01–0.0001; OS: P < 0.01–0.0001; Figure 1, G and H, and Supplemental Figure 1G). Cox regression showed that SRSF1 LI was an independent predictor of DFS and OS (Supplemental Tables 1 and 2). Taken together, these data strongly indicate that upregulation of SRSF1 is closely associated with glioma progression, and SRSF1 is a potential prognostic biomarker for glioma patients.
Figure 1. SRSF1 overexpression is correlated with excessive glioma cell proliferation and predicts poor prognoses of glioma patients.

(A) Relative SRSF1 mRNA levels in glioma tissues as detected by qRT-PCR. The mean of the normal brain (NB) group was arbitrarily set to 1.0. Data are presented as mean ± SD, n = 3. (B and C) Western blot of SRSF1. The expression levels of SRSF1 were compared between GBM tissues and NBs (B), as well as among the GBM cell lines, UC2 (an immortal astrocyte cell line) and SW1088 (an anaplastic astrocytoma cell line, WHO grade III) (C). Loading control: β-actin. (D) Left: IHC staining of SRSF1 in control (nontumoral brain tissues) and glioma tissues. The negative control was established by using PBS as a substitute for the primary antibody. Scale bar: 20 μm. Right: Comparison of SRSF1 expression levels among 20 NB tissues and 120 gliomas of various grades. The expression levels are represented by labeling indexes (LIs [%]), which were calculated with Leica Image Pro Plus 5.0 software as the percentage of total cells that were positive cells. Data are presented as box plots. Boxes represent the 25th and 75th percentiles, lines represent the median, and whiskers show the minimum and maximum points. ***P < 0.001 by 1-way ANOVA with Tukey’s post test. (E) Pearson correlation analysis between the LIs of SRSF1 and Ki-67 in the glioma samples (n = 120). Pearson correlation test, r and P values are shown. (F–H) Kaplan-Meier analyses of the DFS and OS of all glioma patients (F) and patients of similar age (G) and identical IDH1/2 gene status (H). Patients were stratified into high- and low-expression groups using the medians of the SRSF1 LIs of the corresponding cohorts as cutoffs. The P values of the log-rank (Mantel-Cox) tests are presented.
Table 1. Relationship between SRSF1 expression and the clinicopathological characteristics of the 120 glioma patients.
SRSF1 increases tumorigenic potentials of glioma cells.
Prompted by the above findings, we examined whether SRSF1 exerted oncogenic functions in glioma. First, we transiently silenced endogenous SRSF1 expression with 2 independent siRNAs. Western blot analysis confirmed efficient knockdown of SRSF1 in 4 GBM cell lines, including U87MG, U251, LN229, and SNB19 (P < 0.001; Supplemental Figure 2A). Compared with the control siRNA, SRSF1 siRNAs significantly inhibited the growth of these cell lines, as gauged by Cell Counting Kit-8 (CCK-8; Dojindo) assays (P < 0.001; Supplemental Figure 2B). Using the stable sub–cell lines expressing control shRNA (sh-NC; WT) and SRSF1 shRNA (sh-SRSF1; KD), we found that SRSF1 knockdown also severely impaired cell survival and invasion as shown by colony formation (P < 0.001; Supplemental Figure 2C) and Transwell assays (P < 0.001; Supplemental Figure 2D), respectively.
To confirm whether SRSF1 is essential for SRSF1-mediated oncogenic roles in glioma, we infected the KD sub–cell line with a lentivirus coexpressing luciferase and an shRNA-resistant synonymous mutant of SRSF1 (SRSF1-mu; Supplemental Figure 2E) to restore SRSF1 expression (KD+SRSF1-mu) and infected the WT and KD sub–cell lines with a control lentivirus expressing luciferase alone (vec; WT+vec and KD+vec) as controls (Figure 2A). The 3 groups of stable sub–cell lines undergoing sequential lentivirus infection and antibiotic selection were used in the following experiments both in vitro and in vivo. Consistent with the previous results, SRSF1 knockdown severely impaired the proliferation, survival, and invasion abilities, while SRSF1 restoration significantly rescued the above defects (P < 0.001; Figure 2, B–D). In the SW1088 cell line (a grade III astrocytoma cell line with a lower level of endogenous SRSF1; Figure 1C and Supplemental Figure 1C), we also observed the promoting effects of SRSF1 on glioma cell proliferation and invasion (P < 0.001; Supplemental Figure 2, F–H). All these results demonstrate that SRSF1 is a potent promoter of glioma cell proliferation, survival, and invasion.
Figure 2. SRSF1 increases the tumorigenic abilities of GBM cells and induces reorganization of the cytoskeleton.

(A) Western blot of endogenous (endo) SRSF1 and exogenous (exo) SRSF1-mu (HA-tagged product of the SRSF1 synonymous mutant) in the extracts of cells as indicated. Loading control: β-actin. (B) Images of EdU staining (left) and the comparison of EdU-positive rates among the indicated cells (right). (C) Colony formation assay results. (D) Transwell invasion assay results. Original magnification (B and D), ×400. (E) Left: The cytoskeleton was labeled with Phalloidin (green), and cell nuclei were counterstained with DAPI. Scale bar: 20 μm. Right: Green fluorescence (Phalloidin) intensities were profiled along the red lines (upper and middle panels). Cell areas were compared between the WT and the KD sub–cell lines. Data are presented as box plots, n = 300. ***P < 0.001 by 2-tailed Student’s t test (bottom panel). Data in A–D are presented as mean ± SD, n = 3 for A and n = 5 for B–D. ***P < 0.001 by 1-way ANOVA with Dunnett’s post test. Representative images from biological triplicate experiments are shown for B–E.
In addition, we also observed changes in actin organization following SRSF1 knockdown in U87MG and U251 cells. Phalloidin labeling showed the appearance of diffused actin stress fibers with reduced fluorescence intensities in KD cells, whereas in control (WT) cells, F-actin was centralized mostly on the cell cortex or lamellae (Figure 2E). Alterations in actin organization are accompanied by the formation of focal adhesion at the edges of cells, consistent with a more spread-out cell morphology and larger KD cell area (Figure 2E).
For animal experiments, the abovementioned 3 groups of U87MG stable sub–cell lines (WT+vec, KD+vec, KD+SRSF1-mu) were transplanted into nude mice. IHC results showed that xenografts of the KD+vec group retained SRSF1 silencing, while those of the WT+vec and KD+SRSF1-mu groups expressed high levels of SRSF1 (Supplemental Figure 3A). Our results showed that SRSF1 knockdown (KD+vec) clearly inhibited the growth of the glioma xenografts and increased the OS rates of the nude mice, and these effects were almost completely reversed by replenishment of SRSF1-mu (KD+SRSF1-mu; Supplemental Figure 3, A–C). Combined with our in vitro findings, these results demonstrate that SRSF1 increases the tumorigenic potential of glioma cells by facilitating their proliferation, survival, and invasion.
Global landscape of the SRSF1-affected AS and gene expression in GBM cells.
To screen SRSF1-regulated AS events involved in gliomagenesis, we conducted high-throughput sequencing of RNA (RNA-Seq) on the WT and KD sub–cell lines of U87MG and U251. With approximately 100 million 150-nt paired-end reads, we identified a total of 1,348 and 1,332 SRSF1-regulated AS events in U87MG and U251 cells, respectively, which could be classified into 5 AS categories (Figure 3A and Supplemental Table 3). The majority of these AS events belonged to the skipped exon (SE) category.
Figure 3. Global profiles of SRSF1-affected AS in GBM cells.

(A) SRSF1-affected AS events in U87MG (left) and U251 (right) cell lines. The AS events are classified into 5 categories: skipped exon (SE), retained intron (RI), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), and mutually exclusive exon (MXE). (B) Relative fraction of AS events affected positively (activation) or negatively (repression) by SRSF1 in each category. (C) Overlapping AS events in each category of the activation/repression groups between U87MG and U251 cell lines. (D) Gene ontology of the common AS targets shared by U87MG and U251 cell lines. Fisher’s exact P values (–log2 transformed) are plotted for each enriched functional category. (E) Functional association network of the SRSF1-affected AS targets. Genes incorporated in D were analyzed using the STRING database, and the subgroups are marked according to their functions.
Subsequent analysis indicated the dual role of SRSF1 as a splicing activator and repressor, as it induced similar percentages of exon/intron inclusion (activation) and exclusion (repression; Figure 3B and Supplemental Table 3) events.
Among all the SRSF1-regulated AS events, we found multiple events shared by U87MG and U251 cells, most of which belonged to the SE category (Figure 3C and Supplemental Table 3). Importantly, these overlapping SRSF1-affected splicing targets were associated with tumor-related functions in the areas of cell cycle control, RNA splicing, cytoskeleton organization, and focal adhesion (Figure 3, D and E).
Apart from splicing control, we also observed the effects of SRSF1 on global gene expression. Heatmap analysis of the differentially expressed coding genes revealed cell type–specific variations in U87MG and U251 cells (Supplemental Figure 4A). The overlapping genes were also involved in the tumor-related functions mentioned above (Supplemental Figure 4, B and C), reiterating the roles of SRSF1 in gliomagenesis. Furthermore, we spotted the impacts of SRSF1 on the expression of numerous noncoding RNAs, which also exhibited both generality and specificity between U87MG and U251 cells (Supplemental Figure 4D). These results propose that the glioma-promoting roles of SRSF1 are closely associated with its modulating effects on global AS and gene expression.
Validation and mechanistic exploration of SRSF1-guided AS in glioma.
To verify the accuracy of our RNA-Seq results on AS, we subsequently validated the top 50 SRSF1-affected AS events shared by U87MG and U251 cells (Supplemental Table 4). Representative results of 12 validated AS events are shown in Figure 4, A and B. These results confirmed that SRSF1 either activated (Figure 4A) or repressed (Figure 4B) the splicing of the target exons/introns.
Figure 4. Validation and motif discovery of SRSF1-mediated AS in GBM cells.

(A and B) RT-PCR validation of SRSF1-affected AS events. Representative images from 3 independent experiments are presented. The structure of each PCR product is indicated schematically on the right. Alternative exons/introns affected by SRSF1 are painted in orange. The 4 lanes for GOLGA4 (B) are on the same gel but noncontiguous. The percentage of total products that were exon or intron inclusion products (in%) are provided below each gel. (C) Left: Flowchart of SRSF1 motif discovery from the RNA-Seq data. Right: The sum of the log2-transformed fold change (FC) of the GA-rich 6-mers overrepresented within the 5 regions around the regulated cassette exons is compared with that around the control cassette exons. The red line represents SRSF1-mediated cassette exon activation, and the blue line represents SRSF1-mediated cassette exon repression. Potential SRSF1 motifs derived from the 6-mers overrepresented in the activated or repressed cassette exons are also given.
To determine whether the distribution of the SRSF1-binding motifs differs between SRSF1-activated and -repressed cassette exons, we performed de novo discovery of the SRSF1-binding motif using the sequences of 60 (30 activated and 30 repressed) validated SRSF1-regulated SE events shared by U87MG and U251 cells. The motifs derived from the SRSF1-activated training set indicated a predominant enrichment of GAGGGG within the cassette exons over the flanking constitutive exons and introns (Figure 4C). However, SRSF1-repressed exons showed enrichment of the putative binding motifs in the flanking constitutive exons and introns (Figure 4C).
Among the validated SRSF1-affected AS events, we focused on the myosin IB (MYO1B) gene, since its transcripts dramatically switched to the exon-skipped isoform upon SRSF1 knockdown (Figure 5A, primer set 1). The human MYO1B gene has 31 exons, of which exons 23 and 24 are subject to AS regulation. Therefore, this gene theoretically generates 4 different transcripts depending on the inclusion/exclusion of the 2 alternative exons. However, we could only distinguish 3 isoforms, because exons 23 and 24 are of the same length. Reverse transcriptase PCR (RT-PCR) using primer set 2, which could amplify all of the exon-including or -skipping isoforms, revealed that SRSF1 knockdown significantly resulted in skipping of the 2 adjacent exons and facilitated the expression of the truncated MYO1B isoform in the KD sub–cell line (MYO1B-t; Figure 5A). The splicing effect was quantified by determining the percentage of the total MYO1B transcripts that were full-length transcripts containing exons 23 and 24 (MYO1B-fl) (fl%; Figure 5A). Meanwhile, the exogenous HA-tagged SRSF1-mu dose-dependently increased the fl% and recovered the WT splicing pattern of MYO1B in the KD sub–cell line (Figure 5B and Supplemental Figure 5A). Similar results were also obtained in SW1088 cells (Supplemental Figure 5B). However, introduction of domain deletion mutants of SRSF1 (SRSF1-ΔRRM1, -ΔRRM2, -ΔRS) failed to recover or only weakly restored the WT splicing of MYO1B (Figure 5C), suggesting that all 3 domains of SRSF1 are required for efficient splicing of MYO1B pre-mRNA.
Figure 5. SRSF1 mediates inclusion of exons 23 and 24 in MYO1B pre-mRNA.

(A) Top: Diagram of the splicing variants of MYO1B mRNA and the primers for RT-PCR detection of exon 23 (primer set 1) and exons 23 and 24 (primer set 2) inclusion/exclusion. Bottom: AS of exon 23 (primer set 1) and exons 23 and 24 (primer set 2) and expression of MYO1B isoforms were examined by RT-PCR and Western blot, respectively. (B) Western blot of SRSF1 and the corresponding results of RT-PCR of MYO1B mRNA fragments (primer set 2). (C) Top: Schematic diagram of SRSF1 domains and construction of 3 SRSF1 mutants: ΔRRM1 (deleted RRM1), ΔRRM2 (deleted RRM2), ΔRS (deleted RS). All mutants were HA tagged. Bottom: Western blot of endogenous and exogenous SRSF1 with anti-HA and anti-SRSF1 antibodies. AS of exons 23 and 24 was detected by RT-PCR (primer set 2). (D) Western blot of exogenous SRSF1 and its mutants using anti–HA tag antibody. (E) Direct binding between indicated proteins and endogenous MYO1B RNA fragments was verified by CLIP. (F) Left: Schematic diagram of the MYO1B minigene with the potential SRSF1-binding sites marked in red. MYO1B splicing reporters with the indicated deletion (del1-del3) or insertion (del2in) were generated. Right: Splicing of the MYO1B minigene and the reporters was verified by RT-PCR (primer set 2). The percentages of MYO1B-fl within the total MYO1B transcripts are presented using fl% in A–C and F.
To further examine whether SRSF1 binds to MYO1B exons 23 and 24 in vivo, we overexpressed HA-tagged SRSF1-wt or its domain deletion mutants in U87MG cells (Figure 5D). To rule out experimental artifacts caused by the HA antigen, we also transfected the cells with HA-vector (vec) or HA-SRSF2-wt (SRSF2-wt) plasmids as controls (Figure 5D). In vivo crosslinking followed by immunoprecipitation (CLIP) and the subsequent RT-PCR results showed that SRSF1-wt bound to exons 23 and 24 with high affinity. However, the affinities of the 3 SRSF1 domain deletion mutants and SRSF2-wt to exons 23 and 24 were low or even negligible, similar to those to the flanking exons 22 and 25 (Figure 5E). This finding is consistent with the motif distribution feature of SRSF1-activated exons, which is characterized by the predominant enrichment of SRSF1-binding motifs within the cassette exons over the flanking constitutive exons.
To gain more mechanistic insights into SRSF1-regulated AS of the MYO1B gene, we constructed a minigene reporter spanning the genomic DNA fragment of MYO1B exons 22–25 (MYO1B-wt; Figure 5F). Splicing was assayed following transient transfection of WT and KD sub–cell lines of U87MG. In accordance with the endogenous splicing pattern, exons 23 and 24 were nearly 40% included in the WT sub–cell line (lane 1), whereas SRSF1 knockdown markedly inhibited the inclusion of the 2 exons (lane 2), indicating that the inclusion of MYO1B exons 23 and 24 is SRSF1 dependent (Figure 5F). Sequence analysis revealed several potential SRSF1-binding motifs in exons 23 and 24 of MYO1B mRNA (Supplemental Figure 5C). We then examined the role of internal binding motifs in exon inclusion in detail. To this end, we designed a series of motif deletion mutants of the MYO1B minigene, with the motif elements within exons 23 and 24 deleted individually (MYO1B-del1 and -del2) or simultaneously (MYO1B-del3; Figure 5F). Strikingly, MYO1B-del1 and -del2 displayed minor effects in exon exclusion and remained responsive to SRSF1 knockdown (Figure 5F, lanes 3–6 compared with lanes 1–2). However, simultaneous deletion of the 2 motif elements (-del3) almost abolished the inclusion of exons 23 and 24, similar to the effect of SRSF1 deprivation (Figure 5F, lanes 7–8 compared with lanes 1–2). Moreover, inserting 3 copies of GAGGGG (SRSF1-binding motif) into MYO1B-del2 (MYO1B-del2in) substantially restored the WT splicing pattern, and this effect was completely abrogated by SRSF1 knockdown (Figure 5F, lanes 9–10 compared with lanes 1–2). Collectively, these results demonstrate that enrichment of SRSF1-binding motifs within the cassette exon results in exon inclusion.
MYO1B-fl isoform increases the oncogenic capacities of GBM cells.
Given that SRSF1 facilitated the expression of full-length MYO1B protein (MYO1B-fl, containing 58 unique amino acids encoding 2 more IQ motifs in the C-terminus; Supplemental Figure 5, C and D), we next investigated whether and how MYO1B-fl contributes to gliomagenesis. We designed 2 different siRNAs targeting exons 23 and 24, and verified their isoform-specific silencing effects on MYO1B-fl by Western blot analysis (P < 0.001; Figure 6A). CCK-8 assays showed that MYO1B-fl knockdown efficiently suppressed the growth of GBM cells (P < 0.001, Figure 6B). Using the stable sub–cell lines expressing control shRNA (sh-NC) and MYO1B-fl shRNA (sh-MYO1B-fl), we found that MYO1B-fl knockdown considerably suppressed cell invasion (P < 0.001; Figure 6C). Moreover, MYO1B-fl knockdown (sh-MYO1B-fl) caused the MYO1B immunofluorescent signal at the cytomembrane to clearly fade, leaving only scattered fluorescence diffuse in the cytoplasm (Figure 6D) sh-MYO1B-fl cells also showed diffused actin stress fibers with reduced intensities (cytoskeleton disorganization) and enlarged cell areas (Figure 6D and Supplemental Figure 6A).
Figure 6. MYO1B-fl promotes GBM malignancy, and MYO1B isoforms differ in subcellular localization.

(A) Endogenous MYO1B-fl was efficiently knocked down in U87MG and U251 cells by MYO1B-fl siRNA (si-MYO1B-fl-1#, si-MYO1B-fl-2#) transfection, as verified by Western blot. Loading control: β-actin. (B) Growth curves of U87MG and U251 cells transfected with the siRNAs as indicated. (C) Transwell invasion assay results. Original magnification, ×400. (D) Fluorescence images of the cells as indicated. MYO1B was stained by immunofluorescence (red) and the cytoskeleton was labeled by phalloidin (green). Cell nuclei were counterstained with DAPI (blue). Scale bars: 20 μm. (E) Subcellular distribution of EGFP-fused MYO1B. Cell nuclei were counterstained with DAPI (blue). Scale bars: 20 μm. Data in A–C are presented as mean ± SD, n = 3 for A and n = 5 for B and C. ***P < 0.001 by 1-way ANOVA with Dunnett’s post test for A and B, 2-tailed Student’s t test for C. Representative images from triplicate biological experiments are shown for C and D.
Prompted by the above results, we questioned whether the 2 MYO1B isoforms (MYO1B-fl and -t) differ in their subcellular localization. To this end, we expressed EGFP-fused MYO1B-fl or -t in U87MG and U251 cells (Supplemental Figure 6B) and found that MYO1B-fl-EGFP localized mainly on the cytomembrane, while MYO1B-t-EGFP was dispersed in cytoplasm (Figure 6E and Supplemental Figure 6, C and D). Furthermore, we found that SRSF1 knockdown induced a switch of MYO1B protein from cytomembrane aggregation to cytoplasmic dispersion (Supplemental Figure 6E). All of these results strongly indicate a discrepancy in the subcellular localization among MYO1B isoforms.
To investigate whether subcellular localization determines the biologic functions of MYO1B isoforms, we designed 2 shRNAs targeting the 3′-UTR of MYO1B mRNA (sh-MYO1B-total 1# and 2#) to simultaneously knock down all the MYO1B isoforms (Supplemental Figure 7A). We then established the sh-MYO1B-total sub–cell line using the more efficient shRNA (Figure 7A). Thereafter, we infected the sub–cell line with the control lentivirus expressing luciferase alone (vec; sh-MYO1B-total+vec) or the lentivirus coexpressing luciferase plus the fusion proteins MYO1B-fl-EGFP (MYO1B-fl; sh-MYO1B-total+MYO1B-fl) or MYO1B-t-EGFP (MYO1B-t; sh-MYO1B-total+MYO1B-t) to investigate the individual functions of MYO1B isoforms (Figure 7A). Notably, knockdown of all MYO1B isoforms suppressed the proliferation, survival, and invasion abilities of U87MG and U251 cells. While MYO1B-fl markedly reversed the above defects, MYO1B-t exerted almost no rescue effects (Figure 7, B and C, and Supplemental Figure 7B). Accordingly, MYO1B-fl overexpression significantly increased the colony-forming efficiency of U251 cells, while MYO1B-t had no obvious effect (Supplemental Figure 7C). In animal experiments, glioma xenografts of the MYO1B-fl overexpression group exhibited higher growth rates as compared with the control group (Supplemental Figure 7, D and E). These results demonstrate that MYO1B-fl strongly promotes the proliferation, survival, and invasion of GBM cells, whereas MYO1B-t lacks the above oncogenic properties.
Figure 7. MYO1B isoforms differ in their biological functions.

(A) Western blot of endogenous MYO1B and exogenous MYO1B-fl/t expression. MYO1B was knocked down by the specific shRNA (sh-MYO1B-total) in U87MG and U251 cells, and the EGFP-fused full-length and truncated isoforms were overexpressed individually. Loading control: β-actin. (B and C) Images of EdU staining and Transwell invasion assays (B) and the statistical analysis results (C). Representative images from triplicate biological experiments are shown for B. Data in C are presented as mean ± SD, n = 5. **P < 0.01; ***P < 0.001 by 1-way ANOVA with Dunnett’s post test. Original magnification, ×400.
Restoration of MYO1B-fl reverses the antiglioma effects of SRSF1 knockdown.
To provide more evidence that SRSF1 promotes gliomagenesis by inducing MYO1B-fl expression, we infected the U87MG WT and KD sub–cell lines with the control lentivirus expressing luciferase alone (vec; WT+vec, KD+vec) and infected the KD sub–cell line with the lentivirus coexpressing luciferase plus the fusion proteins SRSF1-mu-HA (SRSF1-mu; KD+SRSF1-mu), MYO1B-fl-EGFP (MYO1B-fl; KD+MYO1B-fl), or MYO1B-t-EGFP (MYO1B-t; KD+MYO1B-t) to overexpress the corresponding protein (Figure 8A). Using the 5 groups of stable sub–cell lines, we found that restoration of SRSF1-mu (KD+SRSF1-mu) and MYO1B-fl (KD+MYO1B-fl), but not MYO1B-t (KD+MYO1B-t), efficiently reversed the adverse effects of SRSF1 knockdown (KD+vec) on GBM cell proliferation, survival, and invasion (Figure 8B and Supplemental Figure 8, A and B), underscoring the importance of SRSF1-regulated MYO1B splicing in gliomagenesis.
Figure 8. MYO1B-fl partially recapitulates the SRSF1-mediated tumor-promoting phenotypes in GBM cells.

(A) Western blot of endogenous and exogenous MYO1B and SRSF1 in U87MG cells. Loading control: β-actin. (B) EdU staining and Transwell invasion assays of U87MG cells as indicated. Representative images from triplicate biological experiments are shown. Original magnification, ×400. (C) Bioluminescence images of the intracranial glioma xenografts formed by the indicated U87MG cells. Images of representative mice are shown. (D) Bioluminescence quantification results at days 4, 11, 18, and 25 after implantation (n = 8 for each group). Data are presented as mean ± SD. **P < 0.01; ***P < 0.001 by 1-way ANOVA with Dunnett’s post test. (E) Kaplan-Meier analysis of the OS of the glioma-bearing nude mice. **P < 0.01 for the difference of WT+vec vs. KD+vec, KD+SRSF1-mu vs. KD+vec, WT+vec vs. KD+MYO1B-t, and KD+SRSF1-mu vs. KD+MYO1B-t; *P < 0.05 for the difference of KD+MYO1B-fl vs. KD+vec and KD+MYO1B-fl vs. KD+MYO1B-t by the log-rank (Mantel-Cox) test. (F) IHC of SRSF1 and Ki-67 in outgrowing tumor slices and H&E staining images showing the junctions between glioma xenografts and surrounding brain tissues. Red dotted lines outline the boundaries of the tumors, and red double-sided arrows indicate invasion distances. Scale bars for IHC: 20 μm. Scale bars for H&E: 100 μm (×100) and 50 μm (×400). Images of representative tumors are shown.
We then investigated the functional significance of MYO1B-fl in mediating the oncogenic effects of SRSF1 in vivo. The abovementioned 5 groups of U87MG stable sub–cell lines (WT+vec, KD+vec, KD+SRSF1-mu, KD+MYO1B-fl, KD+MYO1B-t) were transplanted into nude mice. Consistent with the in vitro results, both SRSF1-mu and MYO1B-fl abrogated the suppressive effects of SRSF1 silencing on xenograft growth and tumor cell proliferation (as assessed by Ki-67 index), and the 2 groups of nude mice showed lower survival rates (Figure 8, C–F, and Supplemental Figure 8C). SRSF1 silencing and restoration were confirmed by IHC (Figure 8F). Most prominently, in contrast to the WT+vec controls, xenograft tumors formed by SRSF1-silenced cells (KD+vec) showed no sign of invasion (Figure 8F). Although SRSF1 reexpression (KD+SRSF1-mu) completely restored the growth and invasion of the xenografts, MYO1B-fl exerted only partial effects (Figure 8, C–F), suggesting the existence of other SRSF1-regulated AS targets. However, unlike MYO1B-fl, MYO1B-t failed to exert any “rescue” effects on tumor growth and invasion (Figure 8, C–F, and Supplemental Figure 8C). Collectively, these data indicate that SRSF1 facilitates GBM cell proliferation, survival, and invasion at least partially by switching the MYO1B splicing pattern and favoring the expression of the full-length MYO1B isoform.
Increased MYO1B-fl levels correlate with SRSF1 expression and predict poor prognoses of glioma patients.
We next examined the splicing pattern of MYO1B exons 23 and 24 in 14 gliomas and 5 NBs, as mentioned above. Much in agreement with the oncogenic potentials of MYO1B-fl, inclusion of exons 23 and 24 was more frequent in gliomas than in NBs and more frequent in GBMs than in LGGs (Figure 9A). A positive correlation was observed between the SRSF1 mRNA level and MYO1B-fl% in gliomas (P < 0.01; Figure 9B). These findings were further reinforced by analyzing RNA-Seq data of a large cohort of glioma patients obtained from the TCGA database: MYO1B-fl% was higher in GBMs than in LGGs (P < 0.001; Figure 9C) and positively correlated with SRSF1 level (P < 0.0001; Figure 9D). Furthermore, higher MYO1B-fl% was closely associated with worse OS of the patients (P < 0.0001; Figure 9E). Together, these results validate the mechanistic link between MYO1B-fl and SRSF1 overexpression in gliomas and suggest that MYO1B splicing can be used as a novel independent prognosis factor for glioma patients.
Figure 9. AS of MYO1B is correlated with SRSF1 levels and predicts poor prognoses.

(A) Splicing pattern of MYO1B in glioma tissues as detected by RT-PCR (primer set 2). MYO1B-fl% is presented as mean ± SD, n = 3. (B) Pearson correlation analysis between SRSF1 mRNA levels and MYO1B-fl% in tissue samples (n = 19) as indicated in (A); r and P values by Pearson correlation test are presented. (C) Comparison of MYO1B-fl% between LGGs (n = 120) and GBMs (n = 120) using TCGA RNA-Seq data. ***P < 0.001 by 2-tailed Student’s t test. (D) Pearson correlation analysis between SRSF1 mRNA levels and MYO1B-fl% using the above TCGA data (n = 240). Pearson correlation test, r and P values are presented. (E) Kaplan-Meier analysis of the OS of the above patients in TCGA database. Patients were stratified into high and low expression subgroups using the median of MYO1B-fl% as the cutoff. P < 0.0001 by log-rank (Mantel-Cox) test.
SRSF1-guided MYO1B splicing determines cell fate through the PDK1/AKT and PAK/LIMK pathways.
To better understand the intracellular signaling network underlying the antiglioma effects due to SRSF1 knockdown, we surveyed potential cancer-related signaling pathways using phospho-antibody microarrays. We identified a spectrum of proteins whose phosphorylation levels were more than 15% increased (upregulation) or decreased (downregulation) in SRSF1-silenced U87MG cells (Figure 10A). Many of these proteins in the phosphorylated form are of great importance for tumor cell proliferation and invasion. Analysis of the array revealed a reduction in phosphorylation of several key components crucial for AKT signaling, actin organization, and MAPK signaling upon SRSF1 knockdown (Figure 10B). Among these proteins, several could be rephosphorylated by MYO1B-fl overexpression (Supplemental Figure 9A), illustrating the importance of these molecules in mediating the oncogenic roles of the SRSF1/MYO1B-fl axis in GBM cells.
Figure 10. SRSF1-guided MYO1B splicing determines cell fate through the PDK1/AKT and PAK/LIMK pathways.

(A and B) Phosphoproteome array analysis of the expression changes of phosphoproteins upon SRSF1 knockdown in U87MG cells. The levels of the individual proteins were normalized to total protein levels. Phosphoproteins whose levels increased or decreased by more than 15% were labeled red and blue, respectively. (C) Western blot of the indicated proteins in the extracts of U87MG cells. (D) Co-IP confirmation of the interaction between EGFP-fused MYO1B proteins (MYO1B-fl and MYO1B-t) and endogenous p85 PI3K in U87MG and U251 cells. (E) Subcellular distribution of exogenous MYO1B-fl or MYO1B-t (green) and endogenous p85 PI3K (red) in U87MG cells. Scale bar: 20 μm. Representative images from triplicate biological experiments are shown. (F) Western blot of the indicated proteins in U87MG cells. The lanes for MYO1B were on the same gel but noncontiguous. (G) Representative images of EdU staining from triplicate biological experiments (left) and quantification (right). Original magnification, ×400. Data are presented as mean ± SD, n = 5. ***P < 0.001 by 1-way ANOVA with Tukey’s post test.
Using Western blot analysis, we screened 2 groups of phosphoproteins whose levels were obviously impacted by SRSF1 knockdown and rescued by MYO1B-fl but not MYO1B-t. The first group included the key components and downstream effectors of the pyruvate dehydrogenase kinase 1 (PDK1)/AKT pathway. Within this group, the levels of phospho-PDK1 (Ser241), phospho-AKT (Ser473), cyclin-dependent kinase 2 (CDK2), and cyclin E2 were decreased, while that of cyclin-dependent kinase inhibitor 1A (p21WAF1) was increased upon SRSF1 knockdown and rescued by MYO1B-fl overexpression (Figure 10C). These changes were perfectly simulated by wortmannin, a specific PI3K inhibitor, which completely abrogated the rescue effects of MYO1B-fl as well (Supplemental Figure 9B, top).
The second group comprised phospho-p21 (RAC1) activated kinase 1/2/3 (PAK1/2/3; Thr423/402/421), phospho-LIM domain kinase 1/2 (LIMK1/2; Thr508/505), and phospho-cofilin (Ser3). All of these belong to the PAK/LIMK pathway, and their levels were decreased upon SRSF1 silencing and rescued by MYO1B-fl overexpression (Figure 10C). The PAK inhibitor IPA-3 efficiently mimicked the effects of SRSF1 silencing and abolished the rescue effects of MYO1B-fl (Supplemental Figure 9B, bottom). We also observed that MYO1B-t overexpression failed to reverse the activities of the PDK1/AKT and PAK/LIMK pathways (Figure 10C). All of these findings clearly verify that PDK1/AKT and PAK/LIMK are key pathways mediating the oncogenic functions of SRSF1 and its splicing target, MYO1B-fl, in glioma.
Membrane-localized PI3K is known to catalyze the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 then recruits effectors (e.g., PDK1, AKT) and induces their activation (22). Based on the above finding of MYO1B-fl cytomembrane localization, we wondered whether MYO1B-fl directly recruits PI3K to the cytomembrane to activate PDK1/AKT and PAK/LIMK signaling in glioma cells. Coimmunoprecipitation (Co-IP) assays demonstrated that exogenous MYO1B-fl bound to endogenous p85, a regulatory subunit of PI3K, while MYO1B-t showed only very weak binding to p85 (Figure 10D). Immunofluorescence detection also verified that MYO1B-fl and p85 colocalized on the cytomembrane in U87MG cells overexpressing MYO1B-fl-EGFP, while MYO1B-t and p85 scattered separately in the cytoplasm in U87MG cells overexpressing MYO1B-t-EGFP (Figure 10E and Supplemental Figure 9C). The membrane recruitment of PI3K by MYO1B-fl ultimately resulted in the activation of PDK1/AKT and PAK/LIMK signaling (Supplemental Figure 9D).
To understand the role of AKT activation in the tumor-promoting effects of MYO1B-fl, we introduced constitutively active myristoylated AKT (myr-AKT) into the sh-MYO1B-fl and sh-MYO1B-total sub–cell lines. Strikingly, overexpression of myr-AKT effectively rescued the expression of the downstream effectors (p21WAF1, CDK2) as well as the proliferation and survival of GBM cells (Figure 10, F and G, and Supplemental Figure 9, E and F). Therefore, AKT activation is crucial for the functions of the tumor promoter MYO1B-fl. To conclude, all of these results demonstrate that PDK1/AKT and PAK/LIMK are important pathways linking SRSF1-regulated AS of MYO1B and glioma progression.
Discussion
Our present work represents a comprehensive study of the splicing factor SRSF1 and its downstream AS landscape in glioma. During this study, we identified a key AS target, MYO1B, whose full-length isoform (MYO1B-fl) was closely associated with the onset and progression of glioma and the outcome of the patients. We concluded that SRSF1 promotes gliomagenesis by controlling the AS of tumor-associated genes, further highlighting the importance of AS as a crucial contributor to tumorigenesis.
The human SRSF1 gene is located on chromosome 17q23. Amplification of this locus is frequently seen in various kinds of tumors and is correlated with poor prognoses (23, 24). Additionally, insightful reports from other scholars have demonstrated that SRSF1 is overexpressed in extracranial tumors and plays oncogenic roles via control of the AS of several tumor-related genes (10–12). Here, we provide evidence that SRSF1 is overexpressed in gliomas, and its overexpression is associated with a higher malignancy grade and poorer survival. The subsequent functional study verified that SRSF1 knockdown inhibits the proliferation, survival, and invasion of GBM cells in vitro and suppresses the growth and infiltration of intracranial glioma xenografts in vivo. All of these defects could be rescued by SRSF1 restoration. Additionally, SRSF1 overexpression promoted the proliferation and invasion of a lower-grade glioma cell line (SW1088). These findings indicate that SRSF1 is an important promoter of gliomagenesis and suggest its potential value as a prognostic biomarker in glioma patients.
Recently, genome-wide studies have extensively identified the endogenous AS targets of SRSF1 (13, 14) and established a concentration/position-dependent splicing model (12, 15). However, partly due to the context-dependent nature of SRSF1 target selection, the key SRSF1-governed AS networks responsible for tumorigenesis usually differ greatly among different tumor types (13–15), and unfortunately, little is known about SRSF1-affected AS in glioma up to now. In the present study, taking advantage of RNA-Seq and other validating methods, we identified hundreds of SRSF1-affected AS events in U87MG and U251, 2 GBM cell lines. Most of these events belong to the skipped exon category, and they participate in a wide range of tumor-related functions in the areas of cell cycle control, RNA splicing, cytoskeleton organization, and focal adhesion. Among these events, the common events shared by U87MG and U251 represent only a relatively small fraction, and this could be explained by the discrepancies in the genomic and proteomic backgrounds of the 2 cell lines (25, 26). Nevertheless, the “common” events still deserve further study, for they may reflect the generality of GBMs. In the following mechanistic investigations, including motif analyses, CLIP, and minigene reporter assays, we found that enrichment of SRSF1-binding motifs within the cassette exons always led to exon inclusion, while enrichment of the motifs in the flanking constitutive exons and introns always led to exon exclusion. These findings strongly indicate that, like several other splicing factors (27, 28), SRSF1 determines exon inclusion/exclusion in a position-dependent manner.
Among the numerous AS targets of SRSF1 identified in glioma, we focused on the MYO1B gene, as its mature mRNA products were dramatically switched to the exon-skipped isoform upon SRSF1 knockdown. In the following experiments, we found that SRSF1 participated in the AS of MYO1B pre-mRNA by facilitating the inclusion of exons 23 and 24, and thereby induced the expression of the full-length MYO1B isoform (MYO1B-fl; Figure 11). During this process, the 2 RRM domains and the RS domain of SRSF1 were all required, since the corresponding deletion mutants showed decreased affinities to the core elements within exons 23 and 24 and failed to recover the splicing pattern after SRSF1 knockdown.
Figure 11. Schematic illustration of the molecular pathways by which SRSF1 promotes gliomagenesis by regulating the AS of MYO1B pre-mRNA.

MYO1B, a member of the class I myosin family, is a widely expressed, single-headed, actin-associated molecular motor that is associated with organelle trafficking, membrane tethering, Golgi organization, actin organization, and actin polymerization (29). Previous studies have shown that MYO1B is highly expressed in metastatic prostate cancer and head and neck squamous cell carcinoma (30, 31). Importantly, knockdown of MYO1B significantly inhibited the migratory and invasive abilities of the tumor cells by influencing actin organization (30, 31). In agreement with these reports, we found that the full-length isoform of MYO1B (MYO1B-fl) displayed glioma-promoting activities by enhancing the proliferation, survival, and invasion of GBM cells. We also found that exogenous MYO1B-fl, but not MYO1B-t (the isoform preferentially expressed after SRSF1 knockdown), partially rescued the malignant phenotypes and the tumorigenic abilities of the SRSF1-silenced GBM cell lines. Although these results cannot exclude the existence of other mediators, they are still adequate to demonstrate that MYO1B-fl is a crucial “bridge” molecule that mediates the oncogenic effects of SRSF1 in glioma. Since we have also shown in human glioma samples that the level of MYO1B-fl mRNA was increased in parallel with that of SRSF1 and the tumor grade and occupied a considerable proportion of the total MYO1B mRNA in GBM, we can safely conclude that SRSF1 participates in glioma formation and malignant progression by inducing the expression of the full-length MYO1B isoform.
Furthermore, we observed that the distribution of MYO1B changed from cytomembrane aggregation to cytoplasmic dispersion upon SRSF1 knockdown. This is an interesting phenomenon, since the localization of MYO1B is strictly controlled in mammalian cells. Previous studies reported that MYO1B-fl concentrates mainly within the dynamic areas of the actin cytoskeleton, most notably in membrane ruffles, and the artificial truncated mutants containing the motor or IQ tail domains show a partial overlapping cytoplasmic localization with MYO1B-fl but never concentrate in membrane ruffles (30, 32). Using EGFP fusion proteins, we provided evidence that EGFP-fused MYO1B-fl localized mainly on the cytomembrane, while EGFP-fused MYO1B-t was dispersed in the cytoplasm. Thus, the redistribution of MYO1B upon SRSF1 knockdown could be perfectly explained by the switch in RNA splicing and the decrease of the MYO1B-fl isoform. Our results provide an interpretation of the diversity of MYO1B localization naturally seen in normal and tumor cells and lead us to postulate that, although encoded by the same gene, different isoforms of MYO1B differ greatly in their molecular features and biologic functions. Indeed, cellular experiments demonstrated that exogenous MYO1B-fl, but not MYO1B-t, could fully reverse the antineoplastic effects of MYO1B-total knockdown on GBM cells.
In the identification of the downstream pathways mediating the oncogenic effects of SRSF1 and MYO1B-fl, we adopted the criteria that the phosphorylation profiles of the key components are changed by SRSF1 knockdown and reversed by the replenishment of MYO1B-fl, but not MYO1B-t, as this criteria helped to guarantee the specificity of the cause and effect. Two pathways, PDK1/AKT and PAK/LIMK, attracted our attention. Activated through a series of phosphorylation events, including the ones mediated by PDK1, the AKT signaling pathway plays an important role in glioma progression and aggressiveness (33). Different from the previous finding that SRSF1 bypasses the upstream AKT pathway to activate mTOR complex 1 (mTORC1) (34), we found that SRSF1 knockdown severely repressed the phosphorylation of PDK1 and AKT, led to the increase of p21WAF1 and the decrease of CDK2, and thereby inhibited the growth of glioma cells.
Downstream members of the Rho-GTPase pathway are critical intracellular mediators of actin modeling. These factors control directional cell migration and are frequently dysregulated in GBM (35). PAK-phosphorylated LIMK functions as a key promoter of mesenchymal and amoeboid migration, largely through phosphorylating cofilin at the serine-3 (Ser3) residue and blocking its actin binding ability (36). Screening of the invasion-associated proteins found upregulated LIMKs and phospho-cofilin accumulating in the periphery of GBM (37). In the present study, we observed that SRSF1 knockdown markedly repressed PAK/LIMK phosphorylation and thereby blocked the phosphorylation of cofilin. Moreover, MYO1B-fl could rescue the activity of the PDK1/AKT and PAK/LIMK signaling pathways in SRSF1-knockdown GBM cells, while MYO1B-t exerted no rescue effects. Combined with the observation that specific inhibitors of the PI3K and PAK pathways abrogated the rescue effects of MYO1B-fl, our findings confirmed that these 2 pathways are the key pathways that mediate the promoting effects of SRSF1 and MYO1B-fl on glioma cell growth and invasion.
As is known, PI3K is activated via membrane translocation induced by RTK, and then activated PI3K triggers AKT and PAK signaling by catalyzing PIP2 phosphorylation to produce PIP3 in the cytomembrane (38). Our co-IP and immunofluorescence results identified that MYO1B-fl, but not MYO1B-t, bound to p85 with high affinity, recruited p85 to the cytomembrane, and subsequently activated PDK1/AKT and PAK/LIMK signaling (Figure 11). Moreover, myr-AKT overexpression effectively abolished the suppression by MYO1B-fl or MYO1B-total knockdown on the proliferation and survival of GBM cells, further demonstrating the importance of AKT activation in mediating the oncogenic functions of MYO1B-fl in glioma. These findings indicate that SRSF1-upregulated MYO1B-fl can activate the PDK1/AKT and PAK/LIMK signaling pathways by directly binding and recruiting PI3K to the cytomembrane, therefore revealing a new mechanism by which the SRSF1/MYO1B axis promotes gliomagenesis and highlighting the importance of PI3K in mediating the above MYO1B-fl oncogenic effects.
In summary, in the present study, we demonstrated that unlike the truncated isoform of MYO1B, full-length MYO1B serves as an oncogenic factor for glioma by increasing the proliferation, survival, and invasion of glioma cells. The splicing factor SRSF1 promotes gliomagenesis and malignant progression by facilitating the inclusion of exons 23 and 24 in MYO1B pre-mRNA and inducing the expression of the full-length isoform. SRSF1 and MYO1B-fl represent prognostic biomarkers and potential targets for the therapy of malignant gliomas.
Methods
Tissue samples and clinicopathological data.
The surgical specimens of 120 astrocytic gliomas and 20 nontumoral brain tissues (control) were collected from Tianjin Medical University General Hospital (TMUGH) with written consent. After surgical excision, specimens were fixed immediately in 3.7% buffered formaldehyde solution and embedded in paraffin. Then, 5 μm continuous sections were prepared for IHC of SRSF1 and Ki-67. Histopathological diagnoses were made independently by 2 neuropathologists according to the 2016 WHO classification of central nervous system tumors (39). Sanger sequencing was adopted to verify the gene type of IDH1/2 (39). The WHO grades, IDH1/2 statuses, KPS scores, and patients’ clinical features are summarized in Supplemental Table 5. All 120 glioma patients had complete information and were followed from the date of operation until December 31, 2013, with the follow-up time ranging from 4.5 to 89 months.
Oncomine data and TCGA RNA-Seq data analysis.
Oncomine data analysis was performed as previously described (28). RNA-Seq data from a total of 240 human glioma samples (120 cases of GBM and 120 cases of LGG) were downloaded from TCGA (https://cancergenome.nih.gov/). The percentage of all MYO1B transcripts that were MYO1B-fl transcripts (MYO1B-fl%) was calculated based on the number of reads supporting inclusion and exclusion events. SRSF1 gene expression was calculated by counting the number of reads falling into the SRSF1 gene. Linear analysis was performed between the 2 variables.
IHC.
IHC was performed with mouse anti–human SRSF1 (sc-33652, Santa Cruz Biotechnology Inc.) and mouse anti–human Ki-67 primary antibodies (sc-23900, Santa Cruz Biotechnology Inc.) as previously described (40). PBS was used as a substitute for the primary antibody as a negative control (only with bio-IgG and the ABC complex). IHC images were acquired under a DM6000B microscope (Leica).
Cell culture and reagents.
The human GBM cell line U87MG and the anaplastic astrocytoma (WHO grade III) cell line SW1088 were obtained from the American Type Culture Collection (ATCC). LN229, U251, U343, and SNB19 cells were purchased from the China Academia Sinica Cell Repository. The GBM cell line TJ905, from a Chinese patient, was established and maintained in house. The human immortalized astrocyte cell line UC2 was maintained by our lab and was used as the nontumoral control. All cells were cultured in DMEM (Gibco) containing 10% FBS (Gibco) at 37°C in a humidified incubator with 5% CO2. Wortmannin (PI3K inhibitor) and IPA-3 (PAK inhibitor) were ordered from Selleck (China).
siRNAs, plasmids and lentiviruses.
The siRNAs targeting SRSF1 or MYO1B-fl were synthesized by Gene Pharma and transfected with Lipofectamine RNAiMAX (Invitrogen). Their sequences are listed in Supplemental Table 6.
The plasmids expressing HA-tagged WT SRSF1 (SRSF1-wt), synonymous mutated SRSF1 (SRSF1-mu), and SRSF1 domain deletion mutants (SRSF1-ΔRRM1, -ΔRRM2, -ΔRS) were constructed using the pCDNA3.0-HA vector. The plasmids expressing EGFP-fused full-length MYO1B (MYO1B-fl) and truncated MYO1B (MYO1B-t) were constructed using the pEGFP-C1 vector. The MYO1B minigene was constructed by amplifying the genomic sequence spanning exons 22–25 of the human MYO1B gene and cloning into the pCMV-Tag2b vector, and mutants in which a specific sequence was deleted or inserted were made based on the primary minigene. All cloning primers are listed in Supplemental Table 7. The expression plasmid for constitutively active AKT (myr-AKT) was obtained from Addgene (plasmid 9008).
The knockdown lentiviruses expressing negative control shRNA (sh-NC) and shRNAs targeting SRSF1 (sh-SRSF1), full-length MYO1B (sh-MYO1B-fl), or all isoforms of MYO1B (sh-MYO1B-total) were constructed using the PLKO.1-puro lentiviral vector and packaged in 293T cells (puromycin resistance). Their sequences are listed in Supplemental Table 8. The overexpression lentiviruses expressing luciferase alone (vec) as well as those coexpressing luciferase plus fusion proteins SRSF1-mu-HA (SRSF1-mu), MYO1B-fl-EGFP (MYO1B-fl), or MYO1B-t-EGFP (MYO1B-t) were constructed and packaged by Applied Biological Materials (Canada; blasticidin resistance).
Stable sub–cell line.
First, 4 kinds of stable sub–cell lines were individually established from GBM cell lines by infecting with knockdown lentivirus expressing sh-NC, sh-SRSF1, sh-MYO1B-fl, or sh-MYO1B-total and selecting with puromycin. They included a control group (WT/sh-NC) and the knockdown groups of SRSF1 (KD), MYO1B-fl (sh-MYO1B-fl), or MYO1B-total (sh-MYO1B-total). Their pooled cultures were infected again with the corresponding lentiviruses in order to overexpress the indicated proteins and were selected with blasticidin. The stable sub–cell lines infected by dual lentiviruses included the control group (WT+vec), knockdown groups (KD+vec, sh-MYO1B-total+vec), rescue groups (KD+SRSF1-mu, KD+MYO1B-fl, KD+MYO1B-t, sh-MYO1B-total+MYO1B-fl, sh-MYO1B-total+MYO1B-t), and the overexpression group (WT+MYO1B-fl), and their pooled cultures were used for the present study.
Cell growth, proliferation, survival, and invasion assays.
Cell growth was quantified with CCK-8, and the proliferating cells were labeled by 5-ethynyl-2′-deoxyuridine (EdU) staining as described previously (39). The ability of anchorage-dependent growth (cell survival) was assessed by colony formation assays as described previously (39). Transwell invasion assays were performed as described previously (41).
Immunofluorescence staining.
Cells grown on glass coverslips were fixed with 4% paraformaldehyde (PFA, Sigma-Aldrich), permeabilized with 0.2% Triton X-100, and stained following standard procedures (42). Rabbit anti-human MYO1B antibody (catalog HPA013607) was purchased from Sigma-Aldrich. Fluorescent Alexa Fluor 488 Phalloidin (catalog A-12379), rabbit anti-human p85 (PIK3R1) antibody (catalog 710400), and TRITC-labeled goat anti-rabbit IgG antibody (catalog A-16101) were all purchased from Thermo Fisher Scientific. DAPI reagent was used to stain cell nuclei. Cells were imaged using an FV-1000 laser-scanning confocal microscope (Olympus).
Measurements of fluorescent labeling, cell area, and protein colocalization.
To accurately identify the effects of SRSF1 and MYO1B-fl knockdowns on F-actin distribution in the cytoskeleton and on cell morphology, we measured the line profile intensity of fluorescence labeled by phalloidin and cell area of the sub–cell lines in the WT/sh-NC, KD, and sh-MYO1B-fl groups. To detect differences between the subcellular localizations of MYO1B-fl and MYO1B-t, we expressed the exogenous EGFP fusion proteins, MYO1B-fl-EGFP or MYO1B-t-EGFP, in U251 cells, measured the fluorescence density (pixels) of the border (membrane location) and total in single cells of each group, and calculated the percentage ratio (membrane index) of the border pixels to total pixels. All measurement analyses were completed using ImageJ software (NIH) as described previously (43, 44). The above experiments were repeated independently 6 times. The mean ± SD of the cell area and membrane index was identified using 50 cells randomly selected from each of the 6 replicates (300 cells in total). The correlation of the subcellular localization of endogenous p85 (red) and exogenous MYO1B-fl-EGFP or MYO1B-t-EGFP (green) was detected by the colocalization finder plugin in ImageJ software.
Intracranial xenograft assay in nude mice.
Female BALB/C athymic nude mice at 4 weeks of age were purchased from the animal center of the Cancer Institute of Chinese Academy of Medical Science (Beijing, China). The pooled cultures of stable sub–cell lines sequentially infected with dual lentiviruses and selected with puromycin and blasticidin (i.e., control group [WT+vec], SRSF1-KD group [KD+vec], and rescue groups [KD+SRSF1-mu, +MYO1B-fl, +MYO1B-t]) were transplanted intracranially (3 × 105 cells per mouse) under the guidance of a stereotactic instrument, as described previously (41). Bioluminescent imaging was used to monitor intracranial tumor growth, and the survival of the animals was recorded every day. After sacrifice, the mouse brains were collected and subjected to IHC and H&E staining.
RNA isolation, quantitative reverse transcriptase PCR (qRT-PCR), RT-PCR, and in vivo CLIP.
Total RNA was isolated using the TRIzol reagent (Invitrogen) following the standard protocol. Quantitative reverse transcriptase PCR (qRT-PCR) of the mRNAs under study was performed using the SYBR Green PCR kit (Takara Bio). The specific primers were synthesized by Beijing Genomics Institute (BGI), and their sequences are listed in Supplemental Table 9. The fold changes of the mRNA levels were calculated by the 2–ΔΔCt method. To discriminate the mRNA variants of SRSF1-guided AS, RT-PCR was performed as described previously (27). In vivo CLIP assays were performed as described previously (42). In brief, the plasmids coding for HA-tagged SRSF1 and its domain deletion mutants as well as the empty vector were transiently transfected into U87MG cells. After ultraviolet crosslinking, IP was performed using the Magna RIP kit (Millipore). RNA enrichment was measured by RT-PCR. Specific primers for RT-PCR were synthesized by BGI (Supplemental Table 10).
RNA-Seq and data analysis.
Total RNA isolated from the stable WT and KD groups of U87MG and U251 cells were subjected to paired-end RNA-Seq using the Illumina HiSeq 2000 system according to the manufacturer’s instructions. Read mapping and data analysis for differentially spliced exons/introns were carried out as described previously (27). All raw RNA-Seq data were deposited in the NIH Gene Expression Omnibus database (accession 109436).
Western blot analysis and Co-IP.
Western blot analysis was carried out as described previously (39). The detailed information regarding the commercially available primary antibodies is summarized in Supplemental Table 11. For co-IPs, cell extracts were incubated successively with GFP antibody (T0005, Affinity Biosciences) for 4–6 hours and with protein A/G agarose beads (sc-2003; Santa Cruz Biotechnology) for 12 to 16 hours. Bound proteins were then washed in lysis buffer (P0013J, Beyotime Biotechnology), resuspended in protein sample buffer, separated by SDS-PAGE, and detected by immunoblot.
Motif analysis of SRSF1-regulated cassette exons.
Based on the previously CLIP-identified (15) and RNA-Seq–predicted (13) consensus sequences for SRSF1, and also taking into account the degenerate nature of RNA-binding sequences, we defined GA-rich 6-mers that include at least 1 G and 1 A with GA content ≥50% as potential SRSF1-binding sites. Motif analysis for SRSF1 in U87MG and U251 cells was performed as described previously (27).
Cancer signaling phosphoprotein profiling using an antibody array.
Cell lysates were applied to the Phospho Explorer Antibody Array, which was designed and manufactured by Full Moon Biosystems. Data were collected and analyzed by Wayen Biotechnologies. A ratio computation was used to measure the extent of protein phosphorylation: phosphorylation ratio = phosphorylated value/unphosphorylated value.
Statistics.
Statistical analyses were performed using SPSS 21.0 software (International Business Machines Corporation). Two-tailed Student’s t tests, Mann-Whitney U tests, 1-way ANOVAs, Pearson correlation analyses, Kaplan-Meier analyses, log-rank tests, Cox’s proportional hazards regression model, and χ2 tests were used to analyze the corresponding data. The medians of the SRSF1 LIs, SRSF1 mRNA levels, and MYO1B-fl% were used as the cutoffs in the survival analyses of the corresponding cohort of glioma patients. A P value of less than 0.05 was considered statistically significant. All experiments on cell lines were performed at least in triplicate. All quantitative data are presented as mean ± SD.
Study approval.
Collection of human surgical specimens from TMUGH was approved by the Ethics Committee of TMUGH, and written informed consent was received from participants. qRT-PCR, Western blot analysis, and IHC on human samples were carried out in accordance with the principles of the Helsinki Declaration and approved by the Ethics Committee of TMUGH. The animal experiments were conducted strictly in accordance with a protocol approved by the Institutional Animal Care and Use Committee of TMUGH.
Author contributions
Conception and design were performed by XZ, RW, XL, and SY. Data acquisition was performed by XZ, RW, XL, LY, DH, C Sun, C Shi, WL, CR, and ZJ. Data analysis was performed by XZ, RW, XL, QW, and SY. Drafting of the manuscript was performed by XZ, YF, QW, and SY.
Supplementary Material
Acknowledgments
This project was supported by grants from the National Natural Science Foundation of China (nos. 81302002, 81402050, 81502166, 81672592, and 81872061), the Programs of Science and Technology Commission Foundation of Tianjin Municipality (14JCQNJC12300, 15JCZDJC34600, 15JCYBJC49900, 16JCQNJC13400, and 17JCYBJC27100), the Program of Tianjin Municipal Health Bureau (15KJ147), the Foundation of Tianjin Medical University and General Hospital (2015KYZQ11 and ZYYFY2015032), and the “New Century” Talent Training Project of TMUGH (2016, to X. Zhou). We thank Liqun He (Tianjin Neurological Institute, TMUGH, Tianjin, China) for providing help with the bioinformatics analysis.
Version 1. 11/27/2018
In-Press Preview
Version 2. 01/14/2019
Electronic publication
Version 3. 01/16/2019
Corrected values in figure 3A.
Version 4. 02/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(2):676–693.https://doi.org/10.1172/JCI120279.
Contributor Information
Xuexia Zhou, Email: sjs_xxzhou@126.com.
Run Wang, Email: 15378999678@163.com.
Xuebing Li, Email: xbli@tmu.edu.cn.
Lin Yu, Email: onoblivion@tmu.edu.cn.
Dan Hua, Email: kila1126@hotmail.com.
Cuiyun Sun, Email: suncy548@163.com.
Cuijuan Shi, Email: scjuan148@163.com.
Wenjun Luo, Email: luowenjun609@163.com.
Chun Rao, Email: rao_c11@163.com.
Zhendong Jiang, Email: 758590883@qq.com.
Ying Feng, Email: fengying@sibs.ac.cn.
Qian Wang, Email: wangqiantni@163.com.
Shizhu Yu, Email: tjyushizhu@163.com.
References
- 1.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24(21):2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6(5):386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14(5):802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 5.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119(pt 13):2635–2641. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 6.Dehm SM. mRNA splicing variants: exploiting modularity to outwit cancer therapy. Cancer Res. 2013;73(17):5309–5314. doi: 10.1158/0008-5472.CAN-13-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417(1):15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 8.Graveley BR, Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell. 1998;1(5):765–771. doi: 10.1016/S1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 9.Das S, Anczuków O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1(2):110–117. doi: 10.1016/j.celrep.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang L, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12(4):e1005895. doi: 10.1371/journal.pgen.1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14(3):185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghigna C, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20(6):881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Anczuków O, et al. SRSF1-regulated alternative splicing in breast cancer. Mol Cell. 2015;60(1):105–117. doi: 10.1016/j.molcel.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Miguel FJ, Sharma RD, Pajares MJ, Montuenga LM, Rubio A, Pio R. Identification of alternative splicing events regulated by the oncogenic factor SRSF1 in lung cancer. Cancer Res. 2014;74(4):1105–1115. doi: 10.1158/0008-5472.CAN-13-1481. [DOI] [PubMed] [Google Scholar]
- 15.Pandit S, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50(2):223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louis DN, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 17.Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre JY. Primary brain tumours in adults. Lancet. 2012;379(9830):1984–1996. doi: 10.1016/S0140-6736(11)61346-9. [DOI] [PubMed] [Google Scholar]
- 18.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 19.Correa BR, et al. Functional genomics analyses of RNA-binding proteins reveal the splicing regulator SNRPB as an oncogenic candidate in glioblastoma. Genome Biol. 2016;17(1):125. doi: 10.1186/s13059-016-0990-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golan-Gerstl R, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 21.Lefave CV, et al. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011;30(19):4084–4097. doi: 10.1038/emboj.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62–71. doi: 10.1016/j.ceb.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersen CL, et al. High-throughput copy number analysis of 17q23 in 3520 tissue specimens by fluorescence in situ hybridization to tissue microarrays. Am J Pathol. 2002;161(1):73–79. doi: 10.1016/S0002-9440(10)64158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinclair CS, Rowley M, Naderi A, Couch FJ. The 17q23 amplicon and breast cancer. Breast Cancer Res Treat. 2003;78(3):313–322. doi: 10.1023/A:1023081624133. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Lochmüller H, Yong VW, Karpati G, Nalbantoglu J. Adenovirus-mediated wild-type p53 gene transfer and overexpression induces apoptosis of human glioma cells independent of endogenous p53 status. J Neuropathol Exp Neurol. 1997;56(8):872–878. doi: 10.1097/00005072-199708000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Li H, et al. Differences in protein expression between the U251 and U87 Cell Lines. Turk Neurosurg. 2017;27(6):894–903. doi: 10.5137/1019-5149.JTN.17746-16.1. [DOI] [PubMed] [Google Scholar]
- 27.Zhou X, et al. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 2014;42(6):4019–4030. doi: 10.1093/nar/gkt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo C, et al. SRSF2 regulates alternative splicing to drive hepatocellular carcinoma development. Cancer Res. 2017;77(5):1168–1178. doi: 10.1158/0008-5472.CAN-16-1919. [DOI] [PubMed] [Google Scholar]
- 29.Hartman MA, Spudich JA. The myosin superfamily at a glance. J Cell Sci. 2012;125(pt 7):1627–1632. doi: 10.1242/jcs.094300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makowska KA, Hughes RE, White KJ, Wells CM, Peckham M. Specific myosins control actin organization, cell morphology, and migration in prostate cancer cells. Cell Rep. 2015;13(10):2118–2125. doi: 10.1016/j.celrep.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohmura G, et al. Aberrant myosin 1b expression promotes cell migration and lymph node metastasis of HNSCC. Mol Cancer Res. 2015;13(4):721–731. doi: 10.1158/1541-7786.MCR-14-0410. [DOI] [PubMed] [Google Scholar]
- 32.Tang N, Ostap EM. Motor domain-dependent localization of myo1b (myr-1) Curr Biol. 2001;11(14):1131–1135. doi: 10.1016/S0960-9822(01)00320-7. [DOI] [PubMed] [Google Scholar]
- 33.Chautard E, Ouédraogo ZG, Biau J, Verrelle P. Role of Akt in human malignant glioma: from oncogenesis to tumor aggressiveness. J Neurooncol. 2014;117(2):205–215. doi: 10.1007/s11060-014-1382-9. [DOI] [PubMed] [Google Scholar]
- 34.Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105(40):15323–15327. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol. 2013;3:241. doi: 10.3389/fonc.2013.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi K. Roles of cofilin in development and its mechanisms of regulation. Dev Growth Differ. 2015;57(4):275–290. doi: 10.1111/dgd.12213. [DOI] [PubMed] [Google Scholar]
- 37.Park JB, et al. Transcriptional profiling of GBM invasion genes identifies effective inhibitors of the LIM kinase-Cofilin pathway. Oncotarget. 2014;5(19):9382–9395. doi: 10.18632/oncotarget.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 39.Li H, et al. miR-320a functions as a suppressor for gliomas by targeting SND1 and β-catenin, and predicts the prognosis of patients. Oncotarget. 2017;8(12):19723–19737. doi: 10.18632/oncotarget.14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, et al. miR-146b-5p functions as a tumor suppressor by targeting TRAF6 and predicts the prognosis of human gliomas. Oncotarget. 2015;6(30):29129–29142. doi: 10.18632/oncotarget.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi C, et al. miR-29a/b/c function as invasion suppressors for gliomas by targeting CDC42 and predict the prognosis of patients. Br J Cancer. 2017;117(7):1036–1047. doi: 10.1038/bjc.2017.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou X, et al. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun. 2014;5:4581. doi: 10.1038/ncomms5581. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, et al. Molecular determinants differentiating photocurrent properties of two channelrhodopsins from chlamydomonas. J Biol Chem. 2009;284(9):5685–5696. doi: 10.1074/jbc.M807632200. [DOI] [PubMed] [Google Scholar]
- 44.Asrani K, et al. mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation. J Clin Invest. 2017;127(11):4001–4017. doi: 10.1172/JCI92893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.