

Abstract
Accumulating evidence indicates that neuroinflammation contributes to the pathogenesis and exacerbation of neurodegenerative disorders, such as Alzheimer disease (AD). Sphingosine-1-phosphate (S1P) is a pleiotropic bioactive lipid that regulates many pathophysiological processes including inflammation. We present evidence here that the spinster homolog 2 (Spns2), a S1P transporter, promotes microglia pro-inflammatory activation in vitro and in vivo. Spns2 knockout (Spns2KO) in primary cultured microglia resulted in significantly reduced levels of pro-inflammatory cytokines induced by lipopolysaccharide (LPS) and amyloid-beta peptide 1-42 oligomers (Aβ42) when compared to littermate controls. Fingolimod (FTY720), a S1P receptor 1 (S1PR1) functional antagonist and FDA approved drug for relapsing-remitting multiple sclerosis, partially blunted Aβ42-induced pro-inflammatory cytokine generation, suggesting that Spns2 promotes microglia pro-inflammatory activation through S1P-signaling. Spns2KO significantly reduced Aβ42-induced nuclear factor kappa B (NFκB) activity. S1P increased, while FTY720 dampened, Aβ42-induced NFκB activity, suggesting that Spns2 activates microglia inflammation through, at least partially, NFκB pathway. Spns2KO mouse brains showed significantly reduced Aβ42-induced microglia activation/accumulation and reduced levels of pro-inflammatory cytokines when compared to age-matched controls. More interestingly, Spns2KO ameliorated Aβ42-induced working memory deficit detected by Y-Maze. In summary, these results suggest that Spns2 promotes pro-inflammatory polarization of microglia and may play a crucial role in AD pathogenesis.
Keywords: Neuroinflammation, Alzheimer’s disease, Microglia, Sphingosine-1-phosphate, Spns2
Graphical Abstract
Main point: Using a mouse line deficient for the S1P transporter, Spns2, we demonstrate that Spns2 promotes Aβ42-induced pro-inflammatory activation of microglia through NFκB pathway and contributes to Aβ42-induced cognitive decline. These data suggest that Spns2 is a potential new mediator of Aβ42-induced pro-inflammatory polarization of microglia and may play a role in AD pathogenesis.
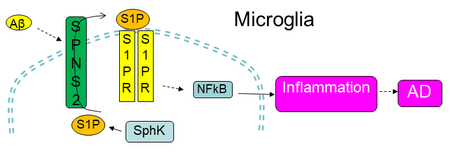
Introduction
Microglia, accounting for about 5–12% of all glial cells, are the first line defense of the central nervous system (Greter and Merad 2013; H et al. 2017; Parisi et al. 2016). Although more complicated functional heterogeneity is being discovered recently, microglia activation are mainly categorized into two states: the pro-inflammatory neurotoxic state and the anti-inflammatory neuroprotective state, (Cunningham 2013; Greter and Merad 2013). Pro-inflammatory microglia are generally identified by expression of pro-inflammatory cytokines and chemokines, such as tumor necrosis factor α (TNFα), interleukin-6 (IL6), IL-1β, and nitric oxide, while anti-inflammatory-polarized microglia identified by high expression of Arginase-1 (Arg1), Interleukin-4 (IL4), and CD206 (H et al. 2017; Parisi et al. 2016). Treatment with endotoxin LPS, amyloid beta peptide (Aβ), and other agents lead to pro-inflammatory polarization, whereas treatment with anti-inflammatory agents, such as IL4 and glatiramer acetate, result in anti-inflammatory polarization (Cao et al. 2017; Lee et al. 2008; McGeer and McGeer 2002; Minett et al. 2016). However, many aspects of the microglia biology, especially the intrinsic molecular mechanisms leading to different activation states and their corresponding effect on cellular and neuronal function in different pathophysiological context, remain elusive and demand further investigation (Cherry et al. 2014; McGeer and McGeer 2002; Minett et al. 2016; Moller et al. 2001; Ramesh et al. 2013; Wang et al. 2015b).
Sphingosine-1-Phosphate (S1P) is a major signaling lipid that plays a crucial function in inflammation in a cell type and context dependent manner (Maceyka et al. 2012; Maceyka and Spiegel 2014; Spiegel et al. 1998; Takabe and Spiegel 2014; Yu et al. 2004). S1P has been found to induce anti-inflammatory polarization of macrophages (Hughes et al. 2008) while promoting a pro-inflammatory phenotype in microglia ischemic stroke conditions (Moon et al. 2015). However, the roles of S1P in AD are unclear. On one hand, the level of S1P is reduced in postmortem AD patient brains suggesting a protective role, consistent with S1P’s often observed neuroprotective or proliferative effects in neurons and astrocytes (Ceccom et al. 2014; Couttas et al. 2014; He et al. 2010). Corroborating with this notion, the level of sphingosine kinase 1 (SphK1) is increased and that of S1P lyase (SgPL1) is decreased in these postmortem brains (Ceccom et al. 2014). On the other hand, there are also studies suggesting an opposite and disease-causative function in the initiation and/or progression of AD (Hagen et al. 2011; Lei et al. 2017; Takasugi et al. 2011). Prolonged exposure of hippocampal neurons to S1P resulted in apoptosis due to Ca2+-mobilization, depending upon the activation of protein phosphatases, possibly calcineurin and phosphatase 2A or a related phosphatase (Moore et al. 1999). Hagen et al show that accumulation of sphingosine kinase 2 (SphK2)-generated S1P induces apoptosis in SgPL1-deficient neurons, which involved dephosphorylation to sphingosine and rephosphorylation catalyzed by SphK2 (Hagen et al. 2011). Deletion of SphK2 enhanced contextual fear conditioning is enhanced in SphK2 knockout mice (Lei et al. 2017). More importantly, in a longitudinal study investigating patients with mild cognitve impairment (MCI), the level of S1P is significantly increased in the cerebral spinal fluid of those AD patients that were developed from MCI (MCI-AD), authough the S1P level eventually decreases in fully-developed AD patients, implicating S1P plays a role in the early pathogenesis of AD (Ibanez et al. 2013). Given these conflicting report and the complex roles it plays in the regulation of cellular functions, more detailed functional and mechanistic insights are needed to better understand the function of S1P and its signaling in AD (Hisano et al. 2012; Rothhammer et al. 2017).
Spns2 is an S1P transporter first discovered in zebrafish by a mutagenesis study (Kawahara et al. 2009). Mutation of Spns2 in zebrafish causes impaired migration of cardiomyocyte precursors and cardia bifida (two hearts) which can be rescued by S1P injection, indicating that Spns2 is an essential S1P transporter in vivo (Kawahara et al. 2009; Osborne et al. 2008). This S1P transportation function is validated in in vitro studies and by genetic knockout in mice, in which Spns2 deficiency results in marked reduction of lymph (or serum) S1P level. Spns2KO mice show a divergent function of this gene in mammals, exhibiting reduced circulating lymphocytes, early onset hearing loss, and cataracts (Chen et al. 2014; Kawahara et al. 2009; Mendoza et al. 2012; Mendoza et al. 2017; Nagahashi et al. 2012; Nijnik et al. 2012). Knockout of lymphatic endothelial Spns2 reduces pulmonary metastasis via a mechanism that involves induction of a lymphopenia and an increase in effector T cell and natural killer cell number to enhance tumor cell killing in the lung (van der Weyden et al. 2017). A most recent study shows that the reduced number of circulating T cells in Spns2KO mice is due to increased naive T cell death in the lymph nodes (Mendoza et al. 2017). Further, Spns2KO mice are protected in adaptive immune and in autoimmune disease models such as airway inflammation, colitis, arthritis and experimental autoimmune encephalopathy (Donoviel et al. 2015). However, Spns2’s function in microglia and neuroinflammation is not known.
In this study, we examined the function of Spns2 and S1P in microglia biology underlying AD pathology using both in vitro and in vivo approaches. Our results show that Spns2 deficiency leads to reduced pro-inflammatory polarization of primary cultured microglia in vitro. Exogenous S1P dose-dependently increased pro-inflammatory cytokine generation, and Fingolimod (FTY720), partially blunted Aβ42-induced pro-inflammatory cytokine generation, suggesting that Spns2/extracellular S1P promotes microglia inflammation. Spns2KO significantly reduced Aβ42-induced NFκB activity. S1P supplementation in both control and Spns2KO microglia increased the level of pP65, although the level was higher in Con than in Spns2KO. FTY720 treatment dampened Aβ42-induced NFκB activity. Combinative supplementation of S1P and Aβ42 exhibited a cumulative effect when compared to treatment alone, which was diminished by FTY720. Spns2 deficiency reduced microglial activation/proliferation in mice injected with Aβ42 oligomer in vivo. More interestingly, Spns2KO partially ameliorates memory loss induced by Aβ42 in vivo, suggesting Spns2/S1P plays a role in AD pathogenesis.
Materials and Methods
Materials:
The antibody against phosphorylated NFκB P65 (pP65) (Ser 536) and LPS were from Sigma Aldrich (St. Louis, MO), antibodies against GAPDH, pIκB, TNFα, IL4, IL6, and IL10 were from Santa Cruz Biotechnology (Dallas, TX). Anti-IBA1 was from Abcam (Cambridge, MA). Anti-NeuN was from EMD Millipore (Billerica, MA), Anti-P2Y12 was from Anaspec (Fremont, CA), and Aβ42 was from rPeptide (Watkinsville, GA). Huzzah® S1P (human serum albumin conjugated) and Control HAS were from Avanti Polar Lipids (Alabaster, AL).
Animals:
The Spns2 conditional knockout mice Spns2f/f was a generous gift from Dr. Susan Schwab (NYU Langone Medical Center). These mice were bred with mice carrying Cre recombinase under the control of β-actin promoter to generate Spns2 knockout mice (Spns2−/−, Spns2KO). Spns2KO mice lack exon 3 and thus were genotyped using a pair of primers flanking the exon: upstream: ACTTGGCCTTAGCCAGGAAT, and downstream: GGAGAAGCTAAGCAGGAGCA. All animal experiments were approved by the Augusta University Institutional Animal Care and Use Committee according to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Methods
Preparation and treatment of primary microglia:
Primary glia culture were prepared from P1 to P3 pups of Spns2+/− breeding following published protocols (Ni and Aschner 2010). Microglia were collected by gently shaking the flasks and plating detached cells on dishes coated with Poly L-lysine (Invitrogen, Carlsbad, CA), following published protocols. The purity of microglia was routinely > 95% shown by labeling of CD11b (Ni and Aschner 2010). Microglia transcriptome undergoes extensive and rapid changes in culture (Gosselin et al. 2017). To minimize variations, we used primary cultures that were prepared from littermate control (Con) and Spns2KO pups prepared under the exact same conditions. The primary cultured microglia were treated with LPS (1μg/ml) and soluble human Aβ42 oligomers (1μM). The soluble Aβ42 oligomers were prepared by following previous publications with minor modifications (Ryan et al. 2013). Briefly, Aβ42 lyophilized powder was dissolved in 1% (w/v) NH4OH to prepare a stock solution of 221 mM. The stock solution was aliquoted and frozen in 80°C. The Aβ42 stock was neutralized by HCl, diluted to 100 mM using serum-free medium, and incubated in 4°C for 24 hours before experiments.
RNA extraction, RT-PCR, and quantitative PCR:
Total RNA was extracted from cells or tissues using Trizol reagent following manufacturer’s instructions (Invitrogen, Carlsbad, CA). First strand DNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR was performed on a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA). The primers were: GAPDH, 5’-CGTGTTCCTACCCCCAATGT-3’ and 5’-TGTCATCATACTTGGCAGGTTTCT-3’; TNFα, 5’- CTGAACTTCGGGGTGATCGG −3’ and 5’-TGGTGGTTTGTGAGTGTGAG-3’; IL6, 5’-GGATACCACTCCCAACAGACC-3’ and 5’-TGTTTTCTGCAAGTGCATCATCG-3’; IL-1β, 5’-TGACGGACCCCAAAAGATGA-3’ and 5’-CTTGTTGATGTGCTGCTGCG-3’; Arg1, 5’-TGTGGGGAAAGCCAATGAAG-3’ and 5’-GATGCTTCCAACTGCCAGAC-3’; CD206, 5’-GCCCTGAACAGCAACTTGAC-3’ and 5’-CTCGTCAGCACCCCAGTTAG-3’; IL4, 5’-GGTCTCAACCCCCAGCTAGT-3’ and 5’-GCCGATGATCTCTCTCAAGTGAT-3’.
ELISA:
Secreted TNFα, IL4, IL6, and IL10 proteins were measured using the TNFα Mouse ELISA kit (Invitrogen, Carlsbad, CA) and the mouse Single Analyte ELISAArray Kits Mouse (SABiosciences, Frederick, MD) for IL4, IL6, and IL10. Briefly, supernatant from primary cultured microglia were centrifuged at 2,000g for 10 min at 4°C to remove debris. Samples were added into the bottom of precoated microplate stripe wells. Mouse TNFα, IL4, IL6, or IL10 biotin conjugates were added, mixed, and incubated at room temperature for 90 min. Streptavidin-HRP were then added and incubated at room temperature for 30 min. The optical density was measured by a microplate reader after adding developer and stop solutions (BioRad, Hercules, CA).
Immunoblot analysis:
Protein concentrations were determined using the RC/DC protein assay, in accordance with the manufacturer’s (Bio-Rad) instructions. Equal amounts of protein were loaded onto a 4-20% gradient gel, and SDS-PAGE was performed using the Laemmli method. For immunoblotting, membranes were first blocked with 5% dry milk (or 3% BSA for phosphorylated proteins) in PBST (PBS containing 0.1% Tween-20) and incubated with primary antibodies in the blocking buffer overnight at 4°C. Membranes were then washed three times with PBST and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After washing, bands were detected using an Odyssey imaging system using a Li-Cor system (Li-Cor), or the ECL system (ThermoFisher Scientific).
In vivo amyloid beta injection, immunostaining and laser confocal microscopy:
Male Spns2KO 6 to 8-month-old mice were used in the experiments. Heterozygote or wild type littermates were used as controls. After ketamine-xylazine induced anesthesia, the mouse was secured onto a Kopf stereotaxic device (David Kopf Instruments, Tujunga, CA). Under aseptic conditions, the skull was exposed, a burr holes drilled, and a needed inserted into both side of the subgranular zone of hippocampus (AP, −2.0 mm; ML, ±1.3 mm; DV, −2.2 mm). Four μl of 100 μM Aβ42 was injected at a rate of 0.5μl/min. After injection, the needle was kept in place for 5 min before being withdrawn to prevent reflux of the injected material. Six weeks after injection, the mice were used for a behavioral study (Y-maze) and sample collection for further analyses with immunohistochemistry and immunoblot. For immunohistochemistry, frozen sections were immunolabeled with antibodies and images taken using a Zeiss LSM 780 inverted or a LSM 510 upright confocal laser-scanning microscope equipped with a laser at wavelength of 488 nm, 543 nm, and 633 nm. LSM 510 Meta 3.2 and Zen (blue version) software were used for image acquisition and processing.
Extraction of nuclear proteins:
Nuclear proteins were prepared using the CelLytic NuCLEAR Extraction kit following manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO). Briefly, cells were washed three times with cold PBS and collected. Lysis buffer (with DTT and protease inhibitor) and reagent IGEPAL CA-630 was added, incubated on ice for 15 min, and centrifuged at 10,000-11,000 g and 5°C. The nuclear extractor mix (1 μl 0.1M DTT+1 μl protease inhibitor cocktail+98μl extractor buffer) was then added to the pellet, mixed, incubated, and centrifuge at 20,000-21,000 g for 5min at 5°C. The nuclear fraction (pellet) was saved at −80 °C until analysis.
Y-Maze:
Working memory was tested using a Y-maze (San Diego Instruments, San Diego, CA). The Y-maze consisted of three identical beige plastic arms at 120° angle that were labeled A, B, C. Each arm was 7.5 cm in width, 38 cm in length, and 12.5 cm in height. No visual or auditory cues were present in any of the arms. The whole apparatus was under the illumination of appropriately 100 lux lighted by 3 bulbs. The mice were placed individually in the far end of one arm and their movement monitored and recorded by a webcam (C920, Logitech, Newark, CA) and analyzed using the Any-Maze software (Stoelting, Wood Dale, IL). A mouse was considered to have entered an arm if its whole body (except for the tail) entered the arm and to have exited if the whole body exited the arm. If an animal consecutively entered three different arms, it was counted as a spontaneous alternation performance (SAP), which is an indication of sound working memory. If an animal went from A to B and came back to A, it was counted as an alternate arm return (AAR). Any time a mouse went from an arm (e.g., arm A) to the center and came back to the same arm (A), it was counted as a same arm return (SAR). Both AAR and SAR indicate defected working memory. The score of alternation was calculated using the formula: Score= number of alternating triads/ (total number of triads minus 2) × 100%.
S1P quantification in culture media and cell pellet:
Primary cultured microglia were washed three times and changed into media with 10% delipidated FBS. Microglia culture media and cell pellet were collected and S1P level analyzed in the Lipidomics Core of Medical University of South Carolina and Virginia Commonwealth University (Bradley et al. 2014).
Preparation of delipidated FBS:
FBS was delipidated by charcoal according to published procedures (Obinata and Hla 2012). 2.5 g of activated charcoal was washed with distilled water three times. 25 ml FBS added and rotated overnight at 4°C. The sera were centrifuged at 1,200 g for 15 min, filtered using 0.45 μm and 0.2 μm pore size syringe filters.
Quantification and statistical analysis:
For quantification of IBA1, the total fluorescence intensity will be measured by the NIS-Element AR 3.0 Imaging system in five sections. The intensity values will be averaged for each group of animals and expressed as the mean densitometry value. The mean, SD, and significance tests of control and treatment samples were calculated using Microsoft Excel and GraphPad Prism. Student’s t-test was used to compare two groups and one-way ANOVA with Tucky post-hoc test for three or more groups. Values of p<0.05 were considered significant.
Results
Spns2 transports S1P in microglia
The function of Spns2 in microglia is not known. We first determined whether Spns2 is expressed in primary cultured mouse microglia. RT-PCR showed Spns2 mRNA was detected in wild type control microglia (Con), while not detectable in littermate Spns2KO microglia (Fig.1A). Spns2 expression was also detected in BV2 cells, a microglia cell line (not shown). Lipidomics analyses found that the medial level of S1P is reduced by more than 70% in Spns2KO microglia when compared to that of Con (Fig.1B), while those of dihydro-S1P (dhS1P), Sphingosine (Sph), and dihydro-Sph (dhSph) remained unchanged (Fig.1B and Fig.1C). On the other hand, the intracellular S1P level in Spns2KO microglia was not significantly altered (Fig.1D), potentially due to a small sample size or the rebalanced expression of its metabolic enzymes SphK1, SphK2, and SgPL1 (Fig.1E). Mfsd2b, a newly discovered S1P transporter (Kobayashi et al. 2018; Vu et al. 2017), was not detectable in primary cultured microglia (not shown). These data indicate that Spns2 is expressed and functions to regulate S1P transport in microglia.
Figure 1.
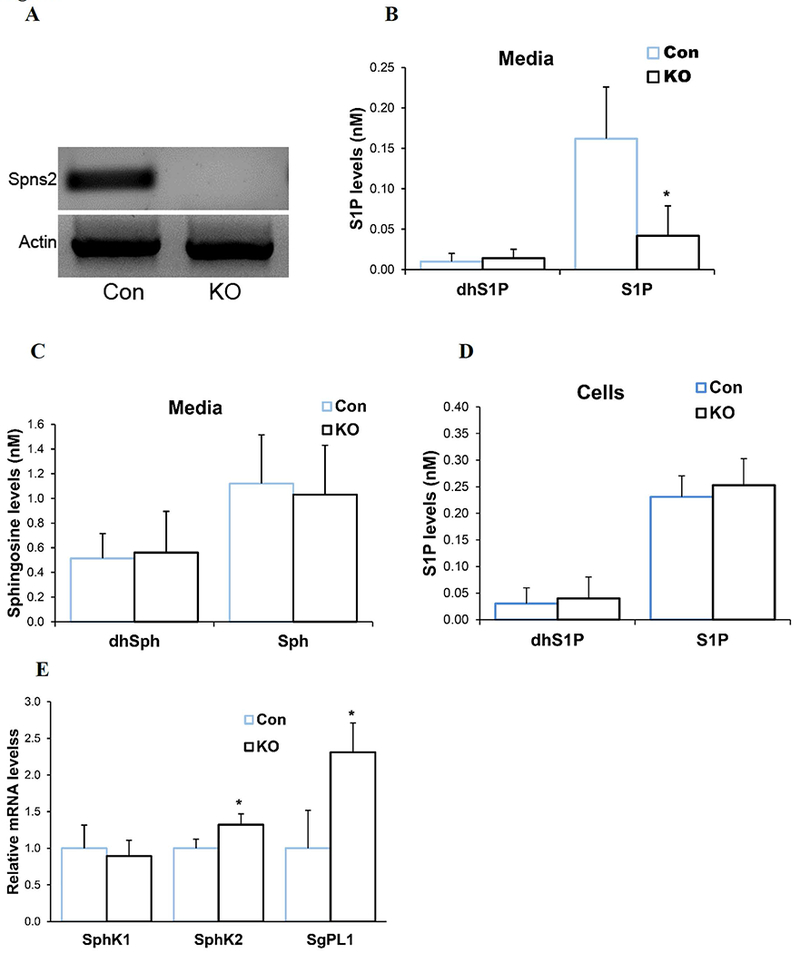
Characterization of Spns2KO microglia. A. Spns2 was expressed in littermate control primary cultured microglia (Con) while not detectable in Spns2 Knockout (Spns2KO) microglia. B. S1P was significantly reduced in the culture media of Spns2KO microglia compared to littermate Con microglia when using media with delipidated FBS, while the level of dhS1P did not change. N=3. *, p<0.05. C. The levels of Sphingosine (Sph) and dhSph were not significantly altered in Spns2KO microglia culture media. P>0.05. N=3 D. Intracellular levels of dhS1P and S1P were not significantly altered in Spns2KO microglia when compared to Con. P>0.05. N=2. E. Spns2KO differentially affected expression of S1P metabilic enzymes SphK1, SphK2, and SgPL1. *, p<0.05. N=3.
Spns2 depletion reduces pro-inflammatory activation of microglia
We next investigated the function of Spns2 in microglia activation by using primary cultured microglia. Lipopolysaccharide (LPS) is a bacterial endotoxin inducing microglia activation (Fu et al. 2017; H et al. 2017; Lall and Baloh 2017). Figure 2A shows that LPS treatment (24 hours) induced mRNA expression of TNFα, a pro-inflammatory polarization marker when compared to vehicle (Veh) treatment, in both littermate control (Con) and Spns2KO microglia. However, the level of TNFα was reduced by more than 55% in Spns2KO microglia when compared to Con (Fig.2A and 2B). Other markers of pro-inflammatory polarization, interleukin 1-β (IL-1β) and IL6, are reduced even more significantly, by more than 90% and 98%, respectively (Fig.2B). However, the effect of Spns2KO on anti-inflammatory polarization marker expression was mixed, with CD206 and IL4 being increased in Spns2KO microglia while ARG1 being reduced (Fig.2C). A time course study showed that, in both Con and Spns2KO microglia, TNFα mRNA was induced by LPS treatment within one hour and its level continued to increase until 24-48 hours at which it appeared to plateau (Fig.2D). However, the extent of TNFα mRNA increase in Spns2KO is significantly smaller than that of Con, consistent with results of Fig.2A and 2B.
Figure 2.
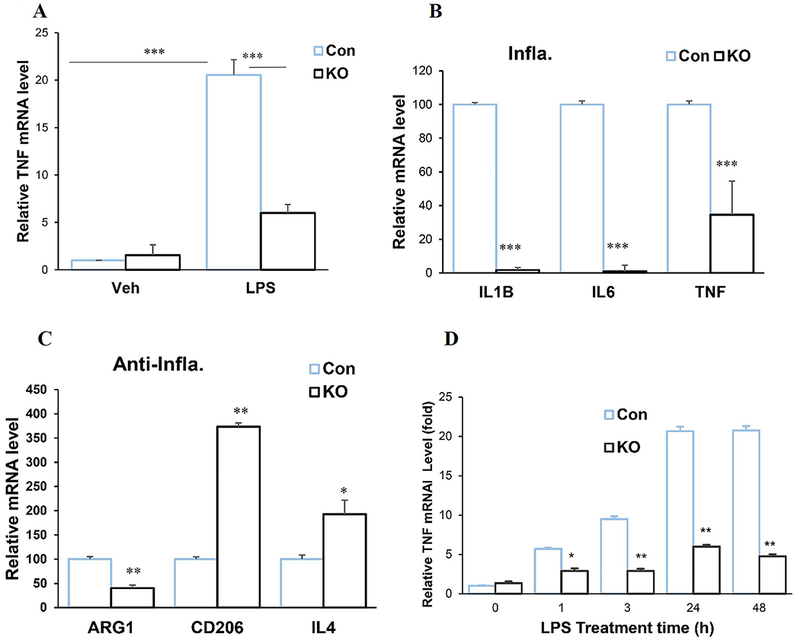
Spns2KO reduced pro-inflammatory polarization of microglia induced by LPS. Microglia were treated with LPS for 24 hours. A. LPS treatment significantly increased mRNA expression of TNFα when compared to vehicle (Veh). Spns2KO (KO) significantly reduced LPS-induced TNFα mRNA expression when compared to littermate control (Con). ***, p<0.001. N=3. B. Spns2KO significantly reduced expression of LPS-induced mRNA levels of TNFα, IL1b and IL6, when compared to littermate Con microglia. ***, p<0.001. N=3. C. Spns2KO significantly increased expression of LPS-induced mRNA levels of CD206 and IL4, but not ARG1, when compared to littermate Con microglia. *, p<0.05. **, p<0.01. N=3. B. Time response curve for LPS treatment.
Amyloid-β accumulation is one of the hallmarks of AD. We used amyloid-β peptide 1-42 oligomer (Aβ42) because it is one of the most toxic forms of amyloid peptides in inducing inflammation and neurotoxicity (Malaplate-Armand et al. 2006). Similar to LPS, the soluble Aβ42 induced microglia activation shown by drastically increased TNFα mRNA expression (Fig.3A). The dose of Aβ42 of 1μM was used based on previous publications (Clapp and Gazzaley 2012; Heurtaux et al. 2010). Spns2KO significantly reduced TNFα mRNA expression, as well as other pro-inflammatory markers IL-1β and IL6, suggesting a reduced pro-inflammatory polarization of microglia (Fig.3A and 3B). Spns2KO significantly increased the mRNA levels of all three tested anti-inflammatory markers CD206, IL4, and ARG1 (Fig.3C). The time response curve of TNFα mRNA generation after Aβ42 treatment is included in Figure 3D.
Figure 3.
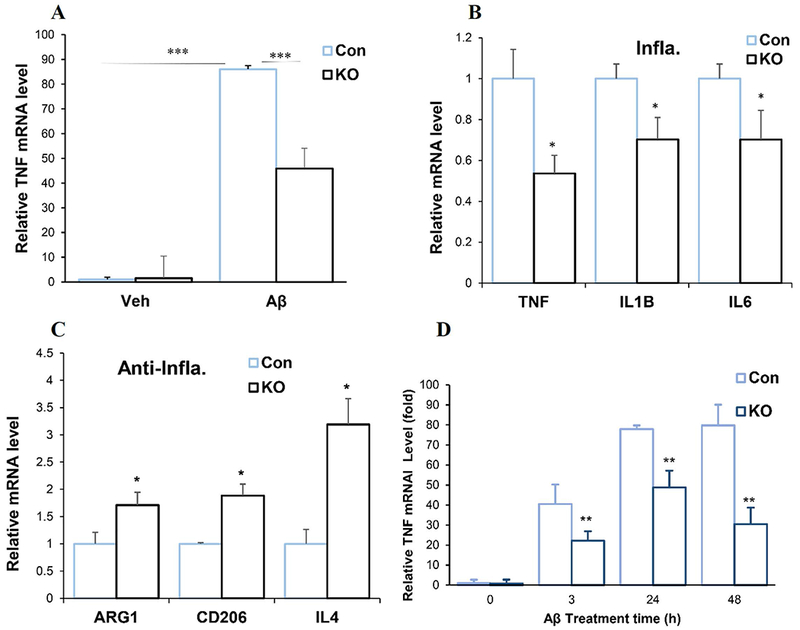
Spns2KO reduced pro-inflammatory polarization and increased anti-inflammatory polarization induced by Aβ42 Oligomer (Aβ42). Microglia were treated with Aβ42 for 24 hours. A. Aβ42 treatment significantly increased mRNA expression of TNFα when compared to vehicle (Veh). Spns2KO (KO) significantly reduced Aβ42-induced TNFα mRNA expression. ***, p<0.001. N=3. B. Spns2KO significantly reduced expression of Aβ42-induced mRNA levels of TNFα, IL1b and IL6, when compared to littermate Con microglia. *, p<0.05. N=3. C. Spns2KO significantly increased expression of Aβ42-induced mRNA levels of ARG1, CD206, and IL4, when compared to littermate Con microglia. *, p<0.05. N=3.
To further validate that Spns2KO reduces production of Aβ42-induced pro-inflammatory cytokines and increases production of anti-inflammatory cytokines, we measured the protein levels of TNFα, IL6, IL4 and IL10 using ELISA in the media of primary cultured microglia treated with Aβ42. Figures 4A shows that the secreted protein levels of TNFα and IL6 were significantly reduced in Spns2KO microglia induced by Aβ42, while those of the IL4 and IL10 increased when compared to littermate Con
Figure 4.
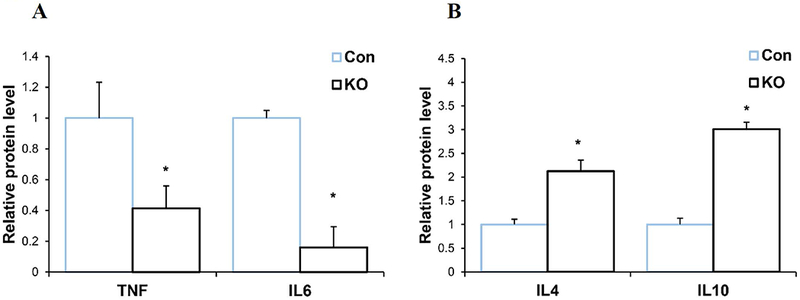
Spns2KO reduced secreted pro-inflammatory and increased anti-inflammatory protein levels from microglia induced by Aβ42. A. Secreted protein levels of TNFα and IL6 were reduced in Spns2KO microglia detected by ELISA. p<0.05. N=3. B. Secreted protein levels of IL4 and IL10 were increased in Spns2KO microglia detected by ELISA. p<0.05. N=3.
Spns2 regulates microglia activation partly through the S1P/S1PR axis
The above data indicate that Spns2 deficiency reduces Aβ42-induced pro-inflammatory polarization in microglia in vitro. Since Spns2 is a major S1P transporter in microglia (Fig.1B), we added S1P (human serum albumin conjugated) into the culture media to determine S1P’s role in this process. S1P supplementation increased gene expression of pro-inflammatory cytokine IL1β and IL6 and suppressed that of anti-inflammatory cytokines IL4 and CD206 (Fig.5A). These S1P dosages were chosen based on previous publications (Lv et al. 2016).
Figure 5.
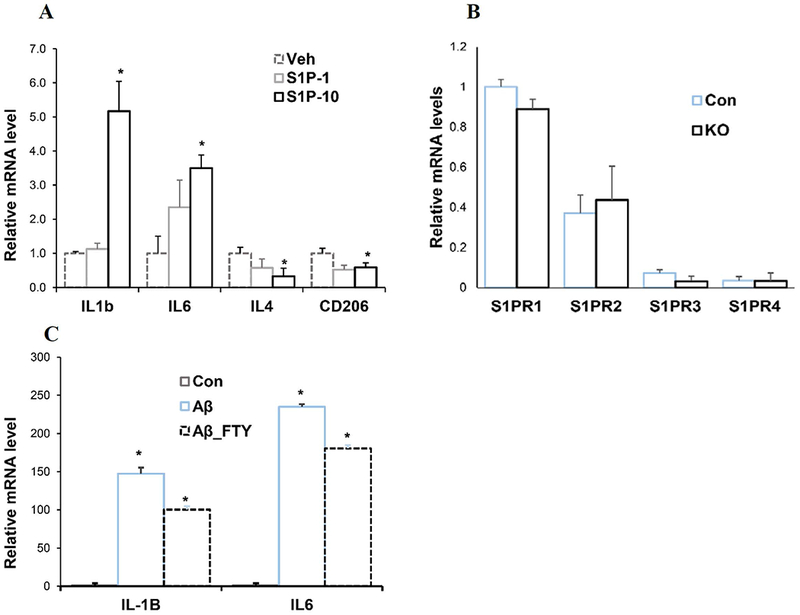
Spns2 enhanced microglia pro-inflammatory polarization through S1P signaling. A. S1P augmented pro-inflammatory cytokine production. 1nM (S1P-1) and 10nM (S1P-10) S1P were supplemented into microglia culture. Samples were collected 1 hour later for qPCR. p<0.05. N=3. B. S1PR mRNA levels in untreated microglia. N=6-8. C. Control microglia were treated with 1μM Aβ42 oligomer for 1 hour with or without FTY-720. mRNA was extracted and IL-1β and IL6 assayed by qPCR. *, p<0.05. N=3.
Extracellular S1P functions through five S1P receptors (S1PR) (Kim et al. 2009; Maceyka et al. 2012; Maceyka and Spiegel 2014; Takabe and Spiegel 2014). To further dissect the mechanism underlying Spns2KO-mediated reduction of pro-inflammatory cytokine generation, we assayed the levels of S1PRs. qPCR from mRNA of untreated control microglia detected all S1PRs except S1PR5, among which S1PR1 was the most abundant (Fig. 5B). S1PR5 was below detection limit in contrary to a previous report, possibly due to different culture conditions, treatment, or genetic background (Blaho and Hla 2014). Thus, we reasoned that Spns2KO reduces pro-inflammatory response mainly through S1PR1 in microglia. This notion was supported by treatment with FTY-720, a functional antagonist for S1PR1 (Montalban et al. 2011; Noda et al. 2013), which significantly attenuated Aβ42-induced IL-1β and IL6 expression in control microglia by 32.0±4.3% and 23.4±2.4% (Ab_FTY) (Fig. 5C). These data suggest that Spns2 promotes microglial pro-inflammatory activation, likely through extracellular S1P.
Spns2 regulates microglia activation through NFκB signaling
NFκB is a major downstream effector of the S1PR signaling (Blaho and Hla 2014; Hisano et al. 2012). The most frequently activated form of NFκB is a heterodimer composed of P65 (RelA) and p50. It is kept in the cytoplasm as a latent and inactive form by interaction with IkB proteins in unstimulated cells (Blaho and Hla 2014; Liang et al. 2013). Upon stimulation, IκB is rapidly phosphorylated (p- IκB) and degraded, allowing phosphorylation of P65 (pP65) and nuclear translocation of the heterodimer which activates transcription activation of pro-inflammatory genes (Blaho and Hla 2014; Liang et al. 2013). To define NFκB activity in Spns2KO microglia in AD, we measured the levels pP65 in Aβ42-treated microglia. The nuclear level of pP65 was greatly increased by Aβ42 (Fig.6A), consistent with previous reports that amyloid activates the NFκB pathway (Bonaiuto et al. 1997; Cramer et al. 2012). Spns2KO significantly reduced the nuclear level of pP65 to 40.8±9.0% of control when normalized to Histone H3 (HisH3) (Fig. 6A). Accordingly, the level of pIκB in Spns2KO microglia was significantly reduced to 42.8±8.9% of Con (Fig. 6B).
Figure 6.
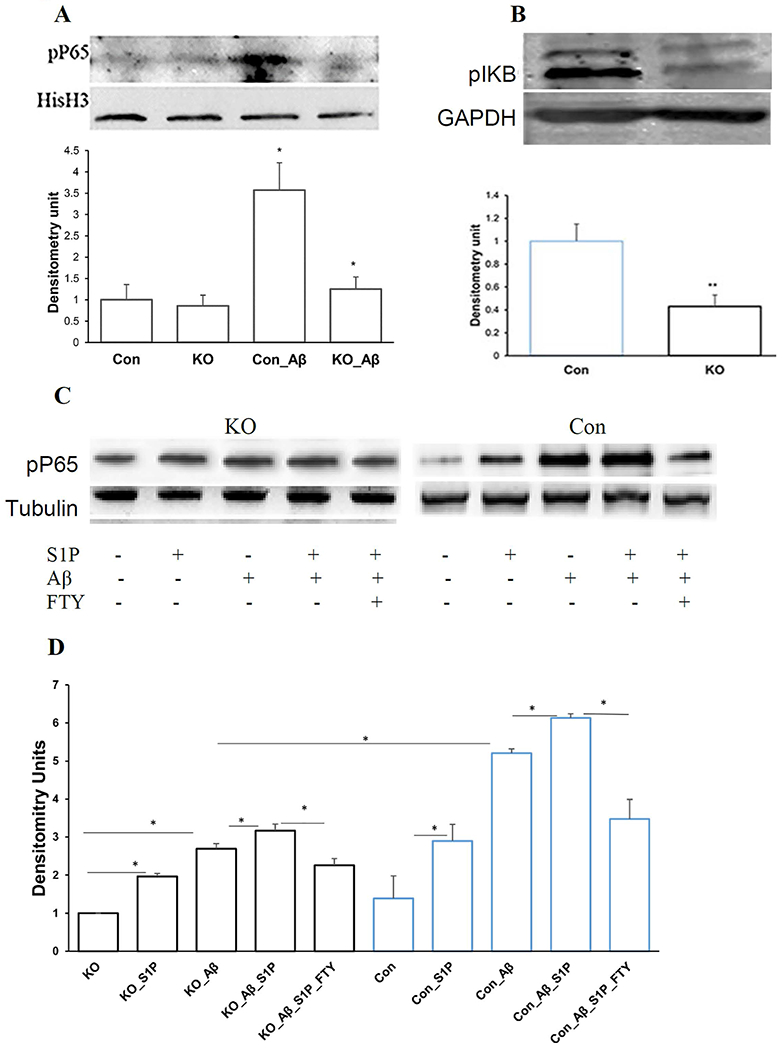
Spns2 enhanced microglia pro-inflammatory polarization partly through NFκB signaling. A. Spns2KO reduced Aβ42-induced nuclear pP65. *, p<0.05. N=3. The bands were quantified by densitometry using ImageJ and normalized to Histone H3 (HisH3). B. Spns2KO significantly reduced Aβ42-induced pIκB level. The bands were quantified using ImageJ and normalized to GAPDH. **, p<0.01. N=3. C and D, S1P coordinated with Aβ42 to induce NFκB activity determined by Western blot (C) and densitometry analysis (D). *, p<0.05. N=3.
To investigate whether the reduced NFκB activity in Spns2KO microglia is caused by decreased extracellular S1P level, we added S1P (human serum albumin conjugated) into the culture media of microglia. S1P supplementation in both Con and Spns2KO microglia increased the level of pP65, although the level was higher in Con than in Spns2KO (Fig.6C and 6D). FTY720 treatment dampened Aβ42-induced NFκB activity. Combinative supplementation of S1P and Aβ42 exhibited a cumulative effect when compared to treatment alone, which was diminished by FTY720 (Fig.6C and 6D). These data suggest that S1P coordinates with or contributes to Aβ42-induced NFκB activation.
Spns2 deletion reduces microglia activity/acumulation and improves cognition in vivo
Neuroinflammation contributes to impaired cognitive function (Lee et al. 2008; Qin et al. 2007; Shamim and Laskowski 2017). To investigate the role of Spns2 in microglia activation in vivo, we stereotaxically injected Aβ42 oligomers into the subgranular zone of Spns2KO mouse brains (AP:−2mm; ML: ±1.3mm; DL:−2mm) (Jean et al. 2015). Aβ42 oligomer has been found to induce microglia activation in vitro and in vivo (Jean et al. 2015; Sasaki et al. 1997; Wu et al. 2000). After 6 weeks, we carried out a Y-maze behavioral test to assess spatial working memory affected by Spns2KO. Alternation of arm entries in a Y-maze is driven by an instinct to visit a novel place and requires the mouse to remember which arms it entered in its immediately previous exploration (for review, see [54]). During the 3 min test session in the Y-maze, Aβ42-treatment significantly reduced the probability of spontaneous alternation performance (SAP) to 51% (Con_Aβ) from 74% in vehicle-treated control mice (Con) (Fig.7A). Spns2KO prevented the decline of SAP caused by Aβ administration (KO_Aβ) (Fig.7A). Correspondingly, Aβ42 significantly increased the probability of alternative arm return (AAR) and same arm return (SAR), while Spns2KO reversed the phenotype (Fig.7B and 7C). Treatment with scrambled Aβ42 did not produce a difference between Con and Spns2KO in the Y-Maze test (not shown). These results suggest that Spns2KO prevents/rescues the spatial working memory loss caused by Aβ42 administration.
Figure 7.
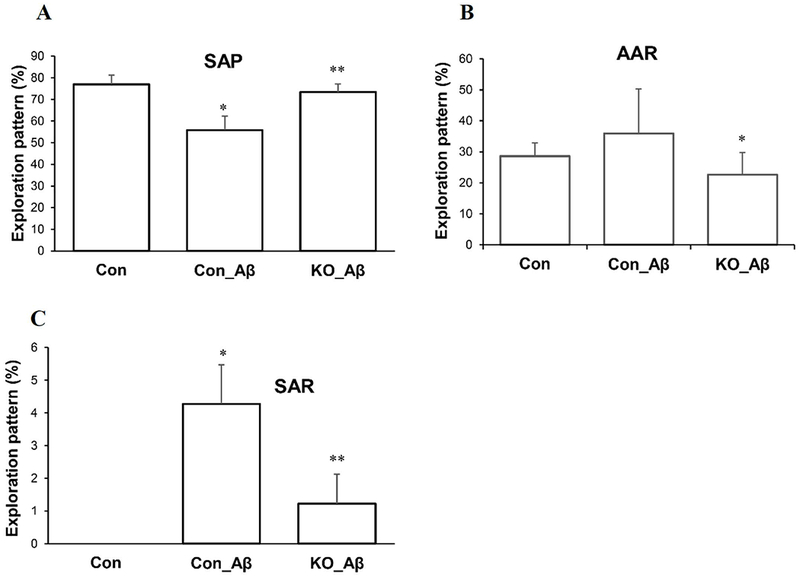
Spns2KO ameliorated Aβ42-induced cognition in vivo. Four μl of 5nM Aβ42 oligomers were injected stereotactically into the subgranular zone of 6 to 8-month-old male mouse brains. Six weeks later, the animals were subjected to Y-maze to test spatial working memory. Spns2KO ameliorated the memory decline caused by Aβ42 shown by: A, improved Spontaneous alternation performance (SAP), B, reduced alternative arm return (AAR), and C, reduced same arm return (SAR). *, p<0.05; **, p<0.01. N=4. SAP is defined as the percentage of triads that an animal goes into three different arms of the Y-maze in a triad entry; AAR as the percentage of an animal going into alternative arms in a triad entry, and SAR as the percentage of an animal returning to the same arm in any consecutive entries in a triad entry.
After behavioral studies, the mice were sacrificed and brain sections were collected to detect microglia activation/proliferation by using IBA1 and P2Y12, marker proteins of microglia (Asai et al. 2015; Butovsky et al. 2014; Cao et al. 2017). Immunostaining showed that Aβ42 induced profound activation/proliferation of IBA1 (+) cells in control brain (Con_Aβ) when compared to vehicle treated brains (Con_Veh), many of which exhibited an activated phenotype characterized by enhanced IBA1-immunoreactivity, retracted processes, perikaryal hypertrophy, and amoeboid appearance (Fig.8A and 8B). In contrast, Spns2KO brains exhibited a markedly reduced number of IBA1 (+) cells and IBA1 immunoreactivity (Fig.8A and 8B). Microglia labeling with another microglia exclusive marker, P2Y12 (Ahlers et al. 2015; Bedard et al. 2007; Butovsky et al. 2014; Clark et al. 2010; Valdearcos et al. 2017), showed a similar phenotype (Fig.8C). Staining with secondary antibody-only did not show any discernible signal (Fig. S1). Next, we detected the level of cytokines in these brain samples. Figures 8D and 8E show that in the Spns2KO brains, the levels of brain pro-inflammatory cytokines TNFα and IL6 were reduced, while those of anti-inflammatory cytokines IL4 and IL10 increased, when compared to Con, consistent with the results of our in vitro experiments (Fig. 2–4). Treatment with vehicle or scrambled Aβ42 did not induce microglia activation/proliferation and cytokine production (not shown). Collectively, these data indicate that Spns2 and extracellular S1P promote microglia pro-inflammatory activation and Spns2 deficiency ameliorates Aβ42-mediated inflammation and cognition in vivo.
Figure 8.
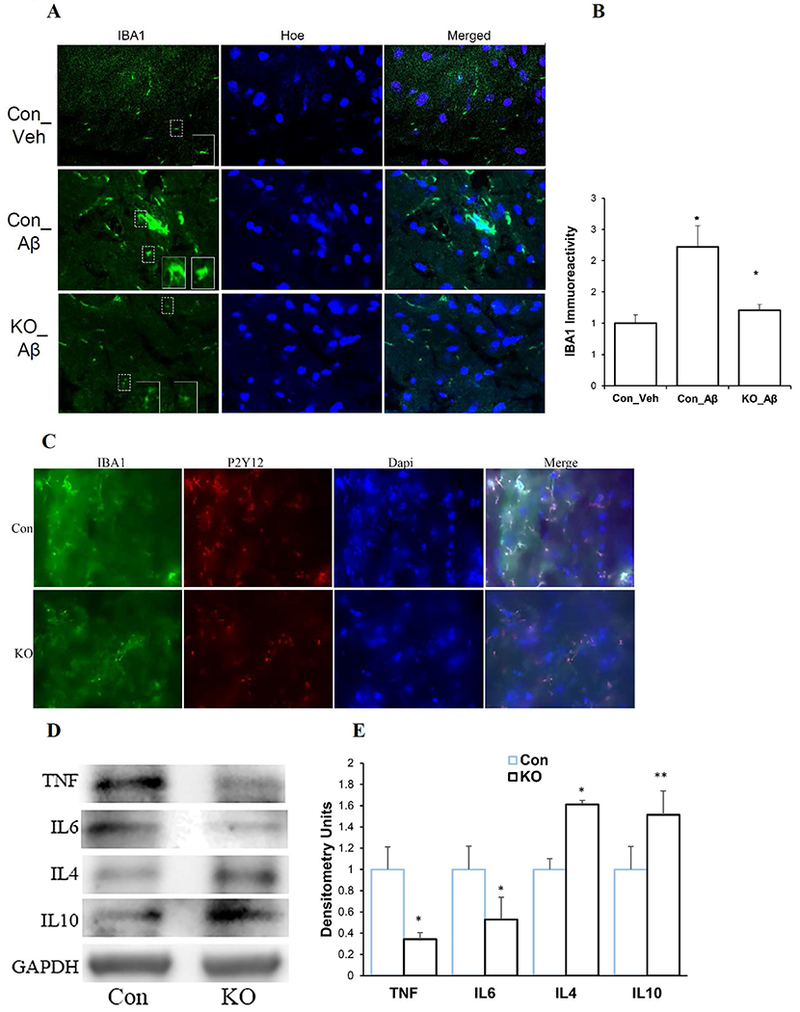
Spns2KO reduced Aβ42-induced inflammation in vivo. Four μl of 5nM Aβ42 oligomers were injected as in Figure 7. Brain samples were collected for immunofluorescence and Western Blot analyses. A. Representative images of microglia labeling of microglia in control (Con) and Spns2KO (KO) brains injected with Aβ42 and Vehicle (Veh) for IBA1 (microglia), NeuN (neuron), and Hoechst (Hoe, nuclei). B. Quantification of immunoreactivity of IBA1 in A. *, p<0.05. N=4. C. Co-labeling of microglia with IBA1 and P2Y12 in brain sections injected with Aβ42. D. Western blot to detect TNFα, IL6, IL4, and IL10 protein levels in brains injected with Aβ42. E. Densitometry quantification of the bands in D. *, p<0.05; **, p<0.01. N=3-5.
Discussion
Chronic activation and production of pro-inflammatory cytokines by microglia is a contributing factor to neurodegenerative disease (Hollingworth et al. 2011; McGeer and McGeer 2015; Naj et al. 2011; Sims et al. 2017; Wes et al. 2016; Zhang et al. 2012). Long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs) protects against subsequent development of AD and Parkinson’s disease (PD), although randomized controlled trials using NSAIDs do not reveal statistically significant outcomes in treating AD after disease onset (Bornebroek et al. 2007; Gagne and Power 2010; Vlad et al. 2008; Wang et al. 2015a). Inhibiting TNFα mediated inflammation signaling pathway leads to improved cognition in AD (Shamim and Laskowski 2017). Microglia depletion/inhibition in animal models of AD ameliorates memory loss in adult mice (Asai et al. 2015). In addition, Genomic wide association studies (GWAS) have identified variants of several immune genes are risk factors for late onset AD including CD33 and TREM2 (Abduljaleel et al. 2014; Hollingworth et al. 2011; Jonsson et al. 2013; Naj et al. 2011; Painter et al. 2015; Wang et al. 2015c). A most recent three-stage case-controlled study of 85,133 subjects identified three new genome-wide significant nonsynonymous variants in PLCG2, ABI3, TREM2 (Sims et al. 2017). All of these genes are highly expressed in microglia, have limited expression in other neural cells, and play essential roles in inflammatory response. These studies provide clear evidence that microglia-mediated innate immune response associates with the development of neurodegenerative diseases. However, recent studies have also found that the functional effects of microglia activation is much more complex than assumed, and are age and context dependent (Chen et al. 2012; Stetler et al. 2014). In aging or pathological conditions where chronic inflammation is predominant, microglia tend to polarize to a more destructive phenotype as exhibited in AD and other situations (Cherry et al. 2014; H et al. 2017; Parisi et al. 2016; Wang et al. 2015b). Thus, strategies to boost anti-inflammatory biased phenotype while inhibiting the pro-inflammatory phenotype may have a profound potential for effective treatment (Cherry et al. 2014; McGeer and McGeer 2015; Wes et al. 2016).
In AD, multiple approaches have been employed to redirect/reintroduce anti-inflammatory phenotype to aid in Aβ clearance and cognition improvement, including pharmacological methods, cell transplantation, and direct use of anti-inflammatory cytokines (Butovsky et al. 2006; Cherry et al. 2014; Cramer et al. 2012; Kiyota et al. 2010; Mandrekar-Colucci et al. 2012; Neumann and Takahashi 2007). Some of these approaches target PPARγ signaling and TGFβ signaling that are shown to favor anti-inflammatory polarization (Mandrekar-Colucci et al. 2012; Wyss-Coray et al. 2001). Overexpression of a homeobox gene msh-like homeobox −3 (MSX3) promotes anti-inflammatory but impedes pro-inflammatory polarization (Yu et al. 2015). MSX3 is dynamically regulated in experimental autoimmune encephalomyelitis but its role in AD or other neuroinflammatory diseases is unclear (Yu et al. 2015). Therefore, the intrinsic molecular mechanisms controlling pro-inflammatory vs anti-inflammatory dynamics are largely debatable and remain one of the most challenging questions in microglia research (Parisi et al. 2016).
Sphingosine-1-Phosphate (S1P) is a potent pleotropic bioactive sphingolipid that plays diverse roles in cellular function, including cell survival, proliferation, migration, and most remarkably, inflammation (Maceyka et al. 2012; Maceyka and Spiegel 2014; Spiegel et al. 1998). The multi-functional characteristics of S1P and its signaling make it an attractive target for therapies of many diseases, e.g. cancer and neurological disorders. The recently approved immunosuppressant drug for multiple sclerosis, Fingolimod (FTY720), functions through S1P metabolism and signaling (Brinkmann et al. 2010; Doggrell 2007; Kappos et al. 2006; Montalban et al. 2011). FTY720 treatment decreases Aβ peptide production in cultured neurons (Takasugi et al. 2013). However, 6-days treatment of APP transgenic mice with FTY720 resulted in a decrease in Aβ40, but an increase in Aβ42 levels in brains (Takasugi et al. 2013). Another study showed that, in 3-month-old 5xFAD mice, FTY720 decreased plaque density, soluble and insoluble Aβ, GFAP staining, and the number of activated microglia (Aytan et al. 2016). These studies suggest that FTY720 or S1P modulators present a great potential for AD.
Using the Spns2 deficient mouse, we found that the lipid transporter plays key roles in microglia activation transition. We demonstrated that Spns2 is expressed and exports S1P in microglia (Fig.1), consistent with previous reports (Bradley et al. 2014; Donoviel et al. 2015; Kawahara et al. 2009; Koso et al. 2016; Mendoza et al. 2017; Nagahashi et al. 2012). Spns2 deficiency in primary cultured microglia led to reduced levels of pro-inflammatory cytokines TNFα, IL-1β, and IL6 in response to LPS and Aβ42 oligomer treatment, indicating a reduced pro-inflammatory phenotype (Fig.2, 3, and 4). On the other hand, the levels of anti-inflammatory cytokines were mostly reduced in Spns2KO microglia, suggestive of anti-inflammatory phenotype (Fig.2, 3, and 4). These data indicate that Spns2 plays a key role in enhancing pro-inflammatory polarization of microglia in vitro.
Supplementation of S1P increased pro-inflammatory cytokine production and reduced anti-inflammatory cytokine production in a dose-dependent manner, indicating that extracellular S1P promotes pro-inflammatory polarization (Fig.5A). This notion is supported by treatment with FTY720, a functional inhibitor of S1PR1 signaling, the major S1PR in microglia (Fig.5C). For signaling transduction, Aβ42 activated NFκB signaling control microglia, which was significantly attenuated in Spns2KO microglia. Supplementation of S1P and Aβ42 in both Con and Spns2KO microglia increased NFκB activity (Fig.6C and 6D). Combinatory supplementation exhibited a cumulative effect, which was attenuated by FTY720 the (Fig.6C and 6D), suggesting that S1P contributes to Aβ42-induced NFκB signaling. S1P receptors have been found to interact with TLR4 functionally to enhance pro-inflammatory cytokine production in human gingival epithelial cells and lung cells (Eskan et al. 2008; Fernandez-Pisonero et al. 2014; Roviezzo et al. 2017). Aβ42 is also found binds to TLR4/TLR2/CD14 to activate microglia (Doens and Fernandez 2014; Frank et al. 2008; Saijo et al. 2013). Thus, it is possible that S1P receptors coordinate with amyloid receptors (e.g., TLR4) in stimulating NFκB signaling and microglia activation. Another interesting observation is that the levels of pP65 were generally higher in Con microglia that those of Spns2KO under the same treatment (Fig.6C and 6D), suggesting that there are potentially other mechanisms involved in the process.
To test the in vivo neuroinflammation-regulatory function of Spns2, we injected Aβ42 oligomers into the subgranular zone. Spns2KO mouse brains showed markedly less IBA1 (+) and P2Y12 (+) cells, suggesting that Spns2 regulates microglia activity/proliferation in vivo (Fig.8A-C). Western blot analyses revealed that the protein levels of pro-inflammatory cytokines TNFα and IL6 were reduced while those of anti-inflammatory cytokines IL4 and IL10 increased in Spns2KO mouse brains (Fig. 8C and 8D). More interestingly, Spns2 deficiency in mice showed improved working memory in a Y-Maze test and reduced neuronal cell death after Aβ42 treatment in vivo (Fig.7). This is consistent with previous reports that Aβ42 oligomer infusion induces neurodegeneration (Jean et al. 2015; Takuma et al. 2004; Wan et al. 2010).
Our data suggest that Spns2/S1P plays a pro-inflammatory role in microglia in Aβ42-induced neuroinflammation. However, we could not exclude the potential effect of Spns2/S1P on other neural cell types, such as astrocytes and neurons, and infiltrating macrophages, in the in vivo anti-inflammatory effects (Fig. 7 and 8). It should also be noted that although the intracellular S1P level in Spns2KO microglia was not significantly altered (Fig.1D), we could not rule out the potential role of local or acute changes of intracellular S1P or its intracellular interacting proteins (Alvarez et al. 2010).
In conclusion, our data demonstrate that Spns2 promotes Aβ42-induced pro-inflammatory activation of microglia and contributes to Aβ42-induced cognitive decline. These data suggest that Spns2 could be a new mediator of Aβ42-induced pro-inflammatory polarization of microglia and may play a crucial role in AD pathogenesis.
Supplementary Material
Acknowledgment:
We thank Dr. Lin Mei and Hongsheng Wang for technical assistance in the Y-Maze experiment. We appreciate Dr. Susan Schwab (NYU Langone Medical Center) for providing the Spns2f/f mice. We thank the institutional support from the Department of Neuroscience and Regenerative Medicine, Augusta University (Chaired by Dr. Lin Mei), and the Department of Physiology, University of Kentucky (Chaired by Dr. Alan Daugherty). The project is supported by funding from American Lung Association RG-351396 to GW and NIH grants R01AG034389 and R01NS095215 to EB.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- Abduljaleel Z, Al-Allaf FA, Khan W, Athar M, Shahzad N, Taher MM, Elrobh M, Alanazi MS, El-Huneidi W. 2014. Evidence of trem2 variant associated with triple risk of Alzheimer’s disease. PLoS One 9:e92648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlers KE, Karacay B, Fuller L, Bonthius DJ, Dailey ME. 2015. Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX-dependent neurodegeneration. Glia 63:1694–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T and others. 2010. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T. 2015. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytan N, Choi JK, Carreras I, Brinkmann V, Kowall NW, Jenkins BG, Dedeoglu A. 2016. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci Rep 6:24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard A, Tremblay P, Chernomoretz A, Vallieres L. 2007. Identification of genes preferentially expressed by microglia and upregulated during cuprizone-induced inflammation. Glia 55:777–89. [DOI] [PubMed] [Google Scholar]
- Blaho VA, Hla T. 2014. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res 55:1596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaiuto C, McDonald PP, Rossi F, Cassatella MA. 1997. Activation of nuclear factor-kappa B by beta-amyloid peptides and interferon-gamma in murine microglia. J Neuroimmunol 77:51–6. [DOI] [PubMed] [Google Scholar]
- Bornebroek M, de Lau LM, Haag MD, Koudstaal PJ, Hofman A, Stricker BH, Breteler MM. 2007. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Neuroepidemiology 28:193–6. [DOI] [PubMed] [Google Scholar]
- Bradley E, Dasgupta S, Jiang X, Zhao X, Zhu G, He Q, Dinkins M, Bieberich E, Wang G. 2014. Critical role of spns2, a sphingosine-1-phosphate transporter, in lung cancer cell survival and migration. PLoS One 9:e110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. 2010. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9:883–97. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE and others. 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. 2006. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A 103:11784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Karthikeyan A, Dheen ST, Kaur C, Ling EA. 2017. Production of proinflammatory mediators in activated microglia is synergistically regulated by Notch-1, glycogen synthase kinase (GSK-3beta) and NF-kappaB/p65 signalling. PLoS One 12:e0186764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccom J, Loukh N, Lauwers-Cances V, Touriol C, Nicaise Y, Gentil C, Uro-Coste E, Pitson S, Maurage CA, Duyckaerts C and others. 2014. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol Commun 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ingham N, Kelly J, Jadeja S, Goulding D, Pass J, Mahajan VB, Tsang SH, Nijnik A, Jackson IJ and others. 2014. Spinster homolog 2 (spns2) deficiency causes early onset progressive hearing loss. PLoS Genet 10:e1004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. 2012. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32:11706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O’Banion MK. 2014. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp WC, Gazzaley A. 2012. Distinct mechanisms for the impact of distraction and interruption on working memory in aging. Neurobiol Aging 33:134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. 2010. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia 58:1710–26. [DOI] [PubMed] [Google Scholar]
- Couttas TA, Kain N, Daniels B, Lim XY, Shepherd C, Kril J, Pickford R, Li H, Garner B, Don AS. 2014. Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol Commun 2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ and others. 2012. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335:1503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C 2013. Microglia and neurodegeneration: the role of systemic inflammation. Glia 61:71–90. [DOI] [PubMed] [Google Scholar]
- Doens D, Fernandez PL. 2014. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J Neuroinflammation 11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggrell SA. 2007. Is fingolimod an advancement in the treatment of multiple sclerosis? KAPPOS L, ANTEL J, COMI G. et al.: Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. (2006) 355:1124-1140. Expert Opin Pharmacother 8:383–6. [DOI] [PubMed] [Google Scholar]
- Donoviel MS, Hait NC, Ramachandran S, Maceyka M, Takabe K, Milstien S, Oravecz T, Spiegel S. 2015. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J 29:5018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskan MA, Rose BG, Benakanakere MR, Zeng Q, Fujioka D, Martin MH, Lee MJ, Kinane DF. 2008. TLR4 and S1P receptors cooperate to enhance inflammatory cytokine production in human gingival epithelial cells. Eur J Immunol 38:1138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Pisonero I, Lopez J, Onecha E, Duenas AI, Maeso P, Crespo MS, San Roman JA, Garcia-Rodriguez C. 2014. Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS One 9:e109081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T. 2008. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56:1438–47. [DOI] [PubMed] [Google Scholar]
- Fu Y, Xin Z, Liu B, Wang J, Wang J, Zhang X, Wang Y, Li F. 2017. Platycodin D Inhibits Inflammatory Response in LPS-Stimulated Primary Rat Microglia Cells through Activating LXRalpha-ABCA1 Signaling Pathway. Front Immunol 8:1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne JJ, Power MC. 2010. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology 74:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP and others. 2017. An environment-dependent transcriptional network specifies human microglia identity. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Merad M. 2013. Regulation of microglia development and homeostasis. Glia 61:121–7. [DOI] [PubMed] [Google Scholar]
- H EH, Noristani HN, Perrin FE. 2017. Microglia Responses in Acute and Chronic Neurological Diseases: What Microglia-Specific Transcriptomic Studies Taught (and did Not Teach) Us. Front Aging Neurosci 9:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen N, Hans M, Hartmann D, Swandulla D, van Echten-Deckert G. 2011. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ 18:1356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. 2010. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 31:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurtaux T, Michelucci A, Losciuto S, Gallotti C, Felten P, Dorban G, Grandbarbe L, Morga E, Heuschling P. 2010. Microglial activation depends on beta-amyloid conformation: role of the formylpeptide receptor 2. J Neurochem 114:576–86. [DOI] [PubMed] [Google Scholar]
- Hisano Y, Nishi T, Kawahara A. 2012. The functional roles of S1P in immunity. J Biochem 152:305–11. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V and others. 2011. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. 2008. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res 102:950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez C, Simo C, Barupal DK, Fiehn O, Kivipelto M, Cedazo-Minguez A, Cifuentes A. 2013. A new metabolomic workflow for early detection of Alzheimer’s disease. J Chromatogr A 1302:65–71. [DOI] [PubMed] [Google Scholar]
- Jean YY, Baleriola J, Fa M, Hengst U, Troy CM. 2015. Stereotaxic Infusion of Oligomeric Amyloid-beta into the Mouse Hippocampus. J Vis Exp:e52805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ and others. 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW and others. 2006. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 355:1124–40. [DOI] [PubMed] [Google Scholar]
- Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A, Mochizuki N. 2009. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323:524–7. [DOI] [PubMed] [Google Scholar]
- Kim RH, Takabe K, Milstien S, Spiegel S. 2009. Export and functions of sphingosine-1-phosphate. Biochim Biophys Acta 1791:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. 2010. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J 24:3093–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Kawasaki-Nishi S, Otsuka M, Hisano Y, Yamaguchi A, Nishi T. 2018. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci Rep 8:4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koso H, Tsuhako A, Lai CY, Baba Y, Otsu M, Ueno K, Nagasaki M, Suzuki Y, Watanabe S. 2016. Conditional rod photoreceptor ablation reveals Sall1 as a microglial marker and regulator of microglial morphology in the retina. Glia 64:2005–24. [DOI] [PubMed] [Google Scholar]
- Lall D, Baloh RH. 2017. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. 2008. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Shafique A, Shang K, Couttas TA, Zhao H, Don AS, Karl T. 2017. Contextual fear conditioning is enhanced in mice lacking functional sphingosine kinase 2. Behav Brain Res 333:9–16. [DOI] [PubMed] [Google Scholar]
- Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D and others. 2013. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23:107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv M, Zhang D, Dai D, Zhang W, Zhang L. 2016. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm Res 65:551–62. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Harikumar KB, Milstien S, Spiegel S. 2012. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 22:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M, Spiegel S. 2014. Sphingolipid metabolites in inflammatory disease. Nature 510:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaplate-Armand C, Florent-Bechard S, Youssef I, Koziel V, Sponne I, Kriem B, Leininger-Muller B, Olivier JL, Oster T, Pillot T. 2006. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis 23:178–89. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. 2012. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci 32:10117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. 2002. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol 8:529–38. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. 2015. Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets 19:497–506. [DOI] [PubMed] [Google Scholar]
- Mendoza A, Breart B, Ramos-Perez WD, Pitt LA, Gobert M, Sunkara M, Lafaille JJ, Morris AJ, Schwab SR. 2012. The transporter spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep 2:1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza A, Fang V, Chen C, Serasinghe M, Verma A, Muller J, Chaluvadi VS, Dustin ML, Hla T, Elemento O and others. 2017. Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature 546:158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C, Ince PG, Nicoll JA, Boche D, Mrc C. 2016. Microglial immunophenotype in dementia with Alzheimer’s pathology. J Neuroinflammation 13:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller T, Contos JJ, Musante DB, Chun J, Ransom BR. 2001. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem 276:25946–52. [DOI] [PubMed] [Google Scholar]
- Montalban X, Comi G, O’Connor P, Gold S, de Vera A, Eckert B, Kappos L. 2011. Oral fingolimod (FTY720) in relapsing multiple sclerosis: impact on health-related quality of life in a phase II study. Mult Scler 17:1341–50. [DOI] [PubMed] [Google Scholar]
- Moon E, Han JE, Jeon S, Ryu JH, Choi JW, Chun J. 2015. Exogenous S1P Exposure Potentiates Ischemic Stroke Damage That Is Reduced Possibly by Inhibiting S1P Receptor Signaling. Mediators Inflamm 2015:492659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AN, Kampfl AW, Zhao X, Hayes RL, Dash PK. 1999. Sphingosine-1-phosphate induces apoptosis of cultured hippocampal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 94:405–15. [DOI] [PubMed] [Google Scholar]
- Nagahashi M, Kim EY, Yamada A, Ramachandran S, Allegood JC, Hait NC, Maceyka M, Milstien S, Takabe K, Spiegel S. 2012. Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels and the lymphatic network. FASEB J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK and others. 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Takahashi K. 2007. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 184:92–9. [DOI] [PubMed] [Google Scholar]
- Ni M, Aschner M. 2010. Neonatal rat primary microglia: isolation, culturing, and selected applications. Curr Protoc Toxicol Chapter 12:Unit 12 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijnik A, Clare S, Hale C, Chen J, Raisen C, Mottram L, Lucas M, Estabel J, Ryder E, Adissu H and others. 2012. The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J Immunol 189:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda H, Takeuchi H, Mizuno T, Suzumura A. 2013. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol 256:13–8. [DOI] [PubMed] [Google Scholar]
- Obinata H, Hla T. 2012. Assessment of sphingosine-1-phosphate activity in biological samples by receptor internalization and adherens junction formation. Methods Mol Biol 874:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne N, Brand-Arzamendi K, Ober EA, Jin SW, Verkade H, Holtzman NG, Yelon D, Stainier DY. 2008. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr Biol 18:1882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, Bu G. 2015. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener 10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi C, Napoli G, Pelegrin P, Volonte C. 2016. M1 and M2 Functional Imprinting of Primary Microglia: Role of P2X7 Activation and miR-125b. Mediators Inflamm 2016:2989548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. 2007. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh G, MacLean AG, Philipp MT. 2013. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm 2013:480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V, Kenison JE, Tjon E, Takenaka MC, de Lima KA, Borucki DM, Chao CC, Wilz A, Blain M, Healy L and others. 2017. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc Natl Acad Sci U S A 114:2012–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roviezzo F, Sorrentino R, Terlizzi M, Riemma MA, Iacono VM, Rossi A, Spaziano G, Pinto A, D’Agostino B, Cirino G. 2017. Toll-Like Receptor 4 Is Essential for the Expression of Sphingosine-1-Phosphate-Dependent Asthma-Like Disease in Mice. Front Immunol 8:1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TM, Caine J, Mertens HD, Kirby N, Nigro J, Breheney K, Waddington LJ, Streltsov VA, Curtain C, Masters CL and others. 2013. Ammonium hydroxide treatment of Abeta produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ 1:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Crotti A, Glass CK. 2013. Regulation of microglia activation and deactivation by nuclear receptors. Glia 61:104–11. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y. 1997. Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94:316–22. [DOI] [PubMed] [Google Scholar]
- Shamim D, Laskowski M. 2017. Inhibition of Inflammation Mediated Through the Tumor Necrosis Factor alpha Biochemical Pathway Can Lead to Favorable Outcomes in Alzheimer Disease. J Cent Nerv Syst Dis 9:1179573517722512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC and others. 2017. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Cuvillier O, Edsall L, Kohama T, Menzeleev R, Olivera A, Thomas D, Tu Z, Van Brocklyn J, Wang F. 1998. Roles of sphingosine-1-phosphate in cell growth, differentiation, and death. Biochemistry (Mosc) 63:69–73. [PubMed] [Google Scholar]
- Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, Jing Z, Chen J, Zigmond MJ, Gao Y. 2014. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol 114:58–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Spiegel S. 2014. Export of Sphingosine-1-Phosphate and Cancer Progression. J Lipid Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Sasaki T, Ebinuma I, Osawa S, Isshiki H, Takeo K, Tomita T, Iwatsubo T. 2013. FTY720/fingolimod, a sphingosine analogue, reduces amyloid-beta production in neurons. PLoS One 8:e64050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Sasaki T, Suzuki K, Osawa S, Isshiki H, Hori Y, Shimada N, Higo T, Yokoshima S, Fukuyama T and others. 2011. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J Neurosci 31:6850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Tomiyama T, Kuida K, Mori H. 2004. Amyloid beta peptide-induced cerebral neuronal loss is mediated by caspase-3 in vivo. J Neuropathol Exp Neurol 63:255–61. [DOI] [PubMed] [Google Scholar]
- Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP and others. 2017. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab 26:185–197 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden L, Arends MJ, Campbell AD, Bald T, Wardle-Jones H, Griggs N, Velasco-Herrera MD, Tuting T, Sansom OJ, Karp NA and others. 2017. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 541:233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. 2008. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70:1672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TM, Ishizu AN, Foo JC, Toh XR, Zhang F, Whee DM, Torta F, Cazenave-Gassiot A, Matsumura T, Kim S and others. 2017. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 550:524–528. [DOI] [PubMed] [Google Scholar]
- Wan J, Fu AK, Ip FC, Ng HK, Hugon J, Page G, Wang JH, Lai KO, Wu Z, Ip NY. 2010. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J Neurosci 30:6873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tan L, Wang HF, Tan CC, Meng XF, Wang C, Tang SW, Yu JT. 2015a. Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis 44:385–96. [DOI] [PubMed] [Google Scholar]
- Wang WY, Tan MS, Yu JT, Tan L. 2015b. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH and others. 2015c. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160:1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, Gan L. 2016. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 64:1710–32. [DOI] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. 2000. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging 21:797–806. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. 2001. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–8. [DOI] [PubMed] [Google Scholar]
- Yu N, Lariosa-Willingham KD, Lin FF, Webb M, Rao TS. 2004. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 45:17–27. [DOI] [PubMed] [Google Scholar]
- Yu Z, Sun D, Feng J, Tan W, Fang X, Zhao M, Zhao X, Pu Y, Huang A, Xiang Z and others. 2015. MSX3 Switches Microglia Polarization and Protects from Inflammation-Induced Demyelination. J Neurosci 35:6350–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK and others. 2012. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44:852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.