

Abstract
Bleeding heterogeneity amongst patients with immune thrombocytopenia (ITP) is poorly understood. Platelets play a role in maintaining endothelial integrity, and variable thrombocytopenia-induced endothelial changes may influence bleeding severity. Platelet-derived endothelial stabilizers and markers of endothelial integrity in ITP are largely underexplored. We hypothesized that, in a canine ITP model, thrombocytopenia would lead to alterations in endothelial ultrastructure and that von Willebrand factor (vWF) would serve as a marker of endothelial injury associated with thrombocytopenia. Thrombocytopenia was induced in healthy dogs with an anti-platelet antibody infusion; control dogs received an isotype control antibody. Cutaneous biopsies were obtained prior to thrombocytopenia induction, at platelet nadir, 24 hours after nadir, and on platelet recovery. Cutaneous capillaries were assessed by electron microscopy for vessel thickness, number of pinocytotic vesicles, number of large vacuoles, and number of gaps between cells. Pinocytotic vesicles are thought to represent an endothelial membrane reserve that can be used for repair of damaged endothelial cells. Plasma samples were assessed for vWF. ITP dogs had significantly decreased pinocytotic vesicle numbers compared to control dogs (P= 0.0357) and the increase in plasma vWF from baseline to 24 hours correlated directly with the endothelial large vacuole score (R = 0.99103; P<0.0001). This direct correlation between plasma vWF and the number of large vacuoles, representing the vesiculo-vacuolar organelle (VVO), a permeability structure, suggests that circulating vWF could serve as a biomarker for endothelial alterations and potentially a predictor of thrombocytopenic bleeding. Overall, our results indicate that endothelial damage occurs in the canine ITP model and variability in the degree of endothelial damage may account for differences in bleeding phenotype among patients with ITP.
Keywords: Endothelium, ITP, dog
Introduction
Severe thrombocytopenia can result in fatal bleeding. Surprisingly, there is great variability in bleeding manifestations in thrombocytopenic patients, suggesting that factors other than platelet count determine the phenotype. While platelets play a pivotal role in the formation of hemostatic clots at the sites of vascular injury, petechial hemorrhages and capillary leakage also occur with thrombocytopenia in the absence of vascular injury. The fact that bleeding can occur without vascular injury has led to the idea that platelets support the vascular endothelium and maintain the structural integrity of intact blood vessels [1], [2].
The ultrastructural manifestations of thrombocytopenia on the endothelium are disputed. Kitchens and colleagues reported capillary endothelial thinning in experimental thrombocytopenia in rabbits and in spontaneous severe thrombocytopenia in humans [3], [4]. However, other electron microscopic studies from a variety of thrombocytopenic animal models did not demonstrate ultrastructural changes in the microvascular endothelium [1], [5], [6], [7].
Similarly, the mechanism by which platelets support vascular integrity is incompletely understood. Platelets produce many soluble vasoactive mediators that may support their vascular-stabilizing function. Which mediator, or combination of mediators, is the most important in the maintenance of endothelial integrity remains unknown. Sphingosine 1-phoshate (S1P), a lysosphingolipid, is one such candidate platelet-derived mediator that serves to maintain inter-endothelial cell junctions [8] [9–12]. Markers of endothelial integrity that might reflect endothelial alterations in ITP, and perhaps allow clinicians to predict bleeding risk, are also unexplored. Von Willebrand factor (vWF) has served as a biomarker of endothelial dysfunction in other diseases like hemorrhagic fever and sepsis [13, 14] and it may serve a similar role in ITP.
A major unanswered clinical question centers on the factors that determine which thrombocytopenic patients will bleed in the absence of trauma. One possible explanation for the variable clinical phenotype is inter-individual differences in endothelial integrity, perhaps related to differences in the capacity of the remaining circulating platelets to maintain vascular integrity.
In the context of a previously described canine model of immune thrombocytopenia (ITP) [15], we hypothesized that thrombocytopenic bleeding is caused by ultrastructural alterations in the microvascular endothelium. We selected cutaneous endothelium because the skin is a common location for thrombocytopenic bleeding (petechiae and ecchymoses) and cutaneous sites are readily accessible for sampling at multiple time points. We also examined plasma levels of the endothelial-stabilizing factor, S1P, to determine whether S1P levels are related to endothelial ultrastructure. Finally, we evaluated plasma vWF as a potential indicator of endothelial ultrastructural changes [16].
Materials and Methods
The methods are described in more detail in the Supplementary information methods section.
Animals
Eight healthy adult (median age 2 years old; range 1–4) intact male mixed breed dogs were used in this study (28.4 ± 5.6 kg). Research dogs were loaned from Francis Owen Blood Research Laboratory at the University of North Carolina at Chapel Hill or Laboratory Animal Resources at the North Carolina State College of Veterinary Medicine (NCSU) as described [15]. All protocols were approved by the Institutional Animal Care and Use Committee of North Carolina State University.
Induction of Thrombocytopenia
As previously described, dogs (n=5) were infused with a murine monoclonal anti-GPIIb antibody (2F9) in order to model ITP and generate predictable severe thrombocytopenia (target nadir 5,000 to 30,000 platelets/μl) [15]. Control dogs (n=3) were infused with an IgG2a isotype control antibody (anti-yellow fever antibody; αYFA or CRL-1689.1). 2F9 is specific for GPIIb (CD41) as previously demonstrated by Western Blot analysis [15]. Significantly, GPIIb is absent on endothelial cells [17].
Blood sampling and preparation
S1P levels were measured at the following time points: baseline, time zero (when platelet count first fell into the target nadir range or 1 hour after control antibody administration), 2, 4, 6, 8, 12, 24 hours, and then every 24 hours until platelet count returned to baseline and new bleeding stopped, whichever came later (168–384 hours). This last time point was termed “recovery.” See Supplementary methods for blood processing details.
Bleeding quantification
At the time of each blood draw, bleeding was assessed by one author using a bleeding scale that quantifies bleeding at 8 anatomic sites. The scale was adapted from the human ITP bleeding scale as described [15, 18].
Biopsy procurement and tissue processing
Dogs were sedated for biopsy procurement at baseline, time of initial platelet nadir (time zero), 24 hours after platelet nadir, on platelet count recovery (recovery), and at the time of any observed clinical signs of spontaneous cutaneous hemorrhage (timepoint termed bleed). Sedation was achieved with intravenous dexmedetomidine dosed to effect (range 3.6–21.9 μg/kg) (Dexdomitor, Pfizer Animal Health, NY, NY). Cutaneous biopsies were collected from the region of the dorsal flank using 8 mm biopsy punches (Miltex, York, PA) and biopsy sites were closed with 3–0 PDS II (Ethicon, Inc., Somerville, NJ). Skin biopsies were also taken if dogs developed pronounced cutaneous ecchymoses, with sample collection at the border of the ecchymoses and normal skin.
Immediately following surgical collection, two 8 mm skin punch biopsy samples were processed for histopathology and transmission electron microscopy using standard methods as described in the Supplementary methods.
Histology
Histological sections were randomized (Random.org) and evaluated by a board certified veterinary pathologist (KEL) who was blinded to specimen identity. Sections were evaluated for lesions and scored for evidence of inflammatory cells, hemorrhage, edema, vessel thrombosis and endothelial cell hypertrophy. Scoring of each of these changes followed a standard lesions severity scale; 0 = no change, 1 = minimal change, 2 = mild change, 3 = moderate change and 4 = marked change.
Electron microscopy
Tissues were examined using a LEO EM910 transmission electron microscope operating at 80kV (Carl Zeiss SMT, Inc., Peabody, MA) at magnifications of 5,000, 10,000, and 20,000 as described in further detail in the supplementary methods. Initially at all time points of one 2F9-treated dog were examined to determine what time points would be the most informative. As greater thinning was demonstrated at 24 hours than at the platelet nadir, in subsequent dogs, only tissues from baseline, 24 hours, and at the time of a bleed were examined. After method development, the electron microscopist (DNL) was blinded to the time point or treatment group.
A board certified veterinary pathologist (REC) who was blinded to sample identity evaluated all photomicrographs. Mean thickness of each capillary was determined using the 5,000 magnification electron micrographs. If the entire capillary was not captured in one micrograph, Gnu image manipulation program (GIMP) 2.8 was used to create a montage of micrographs to form a complete vessel. A grid with straight lines every 22.5 degrees like spokes on a wheel was randomly superimposed on the capillary as shown in Figure 1A.Intersections between grid lines and the point at which they crossed the basal lamina were used, enabling a randomized selection of points at which to measure cell thickness. Perinuclear regions were excluded because of the inherent increased thickness in size of that region. At each intersection, a line perpendicular to the basal lamina was drawn towards the lumen and its length measured. Up to 16 thickness measurements were taken of a given vessel and the mean vessel wall thickness was determined for each animal/time point.
Figure 1A. Demonstration of determination of endothelial cell thickness.
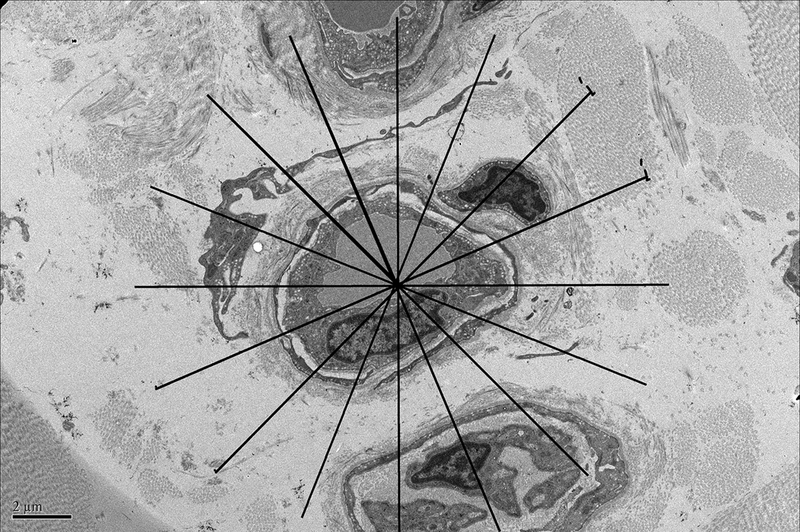
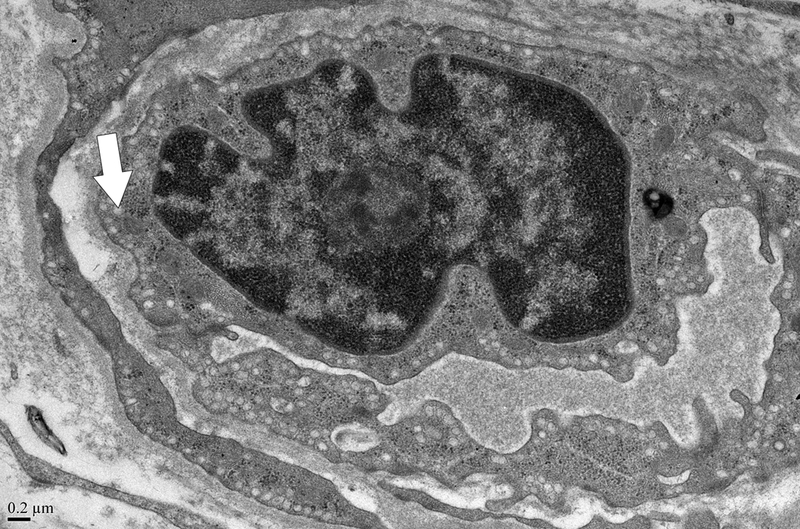
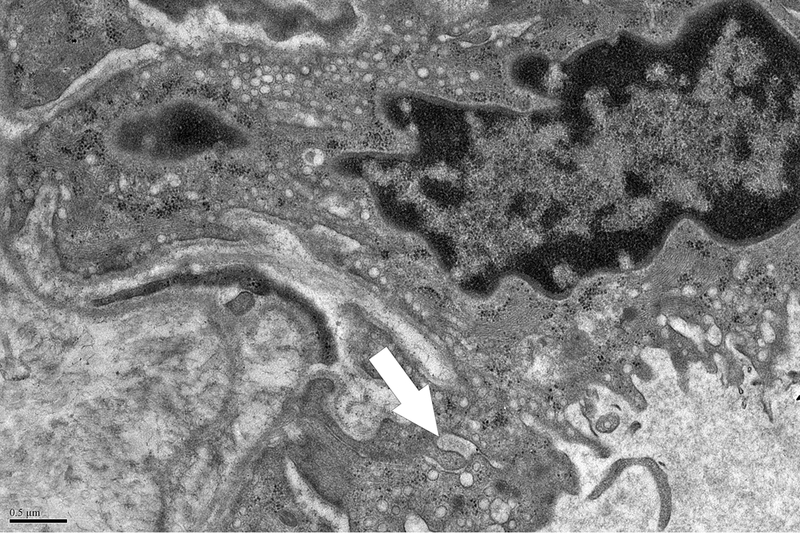
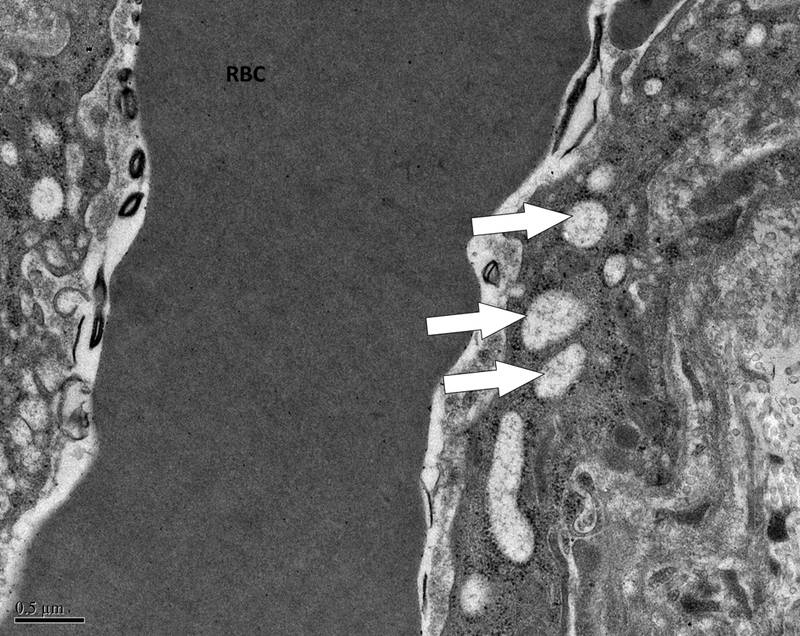
A grid was superimposed on endothelial cells and endothelial cell thickness measured each place the grid crossed the vessel. Average thickness was calculated from each individual measurement. Magnification x 5,000. Definition of ultrastructural features: B. Pinocytotic vesicles are 0.07–0.08 μm in diameter (arrow), magnification x20,000; C. Space between cells (arrow), magnification x20,000; D. Large vacuoles are > 0.1 μm (arrows); x20,000.
Evaluation of capillaries for ultrastructural features was performed at magnifications of 5,000, 10,000, and 20,000. Each capillary was examined for pinocytotic vesicles (0.07–0.08 μm), large vacuoles (>0.1 μm), and number of gaps between cells. Figure 1 demonstrates these terms. Scoring of pinocytotic vacuoles was performed as follows: 0 –absent, 1 –rare, 2 –present multifocally, 3 –present diffusely; grading of large vacuoles was 0 –absent, 1 –focal, 2 –multiple, 3 –numerous/ many. Average number of gaps between cells was determined by averaging the number of gaps per each individual capillary in a given section.
Sphingosine 1-phosphate (S1P) measurement
Platelet poor plasma samples were analyzed at the University of Kentucky Gill Heart Institute for determination of plasma S1P and dihydroS1P (dhS1P) levels. Lipids were extracted from plasma using acidified organic solvents, as previously described using 50 pmol of a C17-S1P internal standard [19],[20]. S1P levels were measured using ultra-fast liquid chromatography coupled with electrospray ionization tandem mass spectrometry as previously described [19].
Von Willebrand factor measurement
Plasma von Willebrand factor (vWF) concentration was assessed as a marker of endothelial cell damage. Platelet free plasma von Willebrand factor concentration [von Willebrand factor antigen (vWF:Ag)] was measured at the Comparative Coagulation Laboratory (Animal Health Diagnostic Center, Cornell University) using an ELISA,[21] configured with monoclonal anti-canine vWF antibodies. A canine plasma standard, with an assigned value of 100% vWF:Ag, was prepared at the Coagulation Laboratory as a pooled plasma collected from 20 healthy dogs and stored in single-use aliquots at −700 C.
Platelet Counts
Platelet counts were determined from EDTA whole blood by the Clinical Pathology Laboratory at NCSU using an Advia 120 (Siemens Healthcare Diagnostics, Inc., Norwood, MA) or manual hemocytometer counts if platelets were <10,000 platelets/μl as previously described [15].
Assessment of platelet activation state via flow cytometry
As an alternative explanation for variable bleeding phenotypes, platelet reactivity to agonists was assessed by two different flow cytometric methods. Platelet surface expression of P-selectin in response to graded thrombin concentrations was used to determine thrombin EC50 as previously described and as detailed in supplemental methods [22]. Ability of platelets to generate coated platelets in response to dual stimulation with thrombin and convulxin was also performed as previously described and as detailed in supplemental methods [23].
Statistical analysis
For electron micrograph parameters (vessel thickness, average number of gaps between cells, average number of gaps between cells and basement membrane, large vacuole score, pinocytotic vesicle score), the Kolmogorov-Smirnov test was performed to test the changes from baseline to 24 hours between treatment and control groups. The generalized estimating equation (GEE) was applied to model the repeated measures of S1P, vWF:Ag, and dhS1P. The model included the time effect and treatment effect as covariates. It assumed an independent correlation structure to address the correlation within the repeated measures. The GEE model was also applied to test the association between S1P and platelets and the association between hematocrit and S1P. We further evaluated the association between the bleeding scores and the variables (S1P, dhS1P, vWF:Ag) by using the GEE model. The Spearman correlation between the variables (S1P, vWF:Ag, bleeding scores) and the EM parameters (vessel thickness, vessel pinocytotic score, vessel large vacuole score, space between cells) was calculated for the observations at baseline, 24 hours, and the change from baseline to 24 hours, respectively. The Spearman correlation was tested by a t-test with n-2 degrees of freedom, where n is the total sample size. Similarly, the Spearman correlations between the platelet count and the variables (vWF, vessel thickness, gaps between cells, gaps between cells and BM, large vacuole score, pinocytotic vesicle score, bleeding score) were also assessed. All analyses were performed using SAS 9.3 software (SAS Institute, Cary, NC). All the tests are evaluated at the significance level of 0.05.
Results
Platelet count in response to antibody infusion
Platelet counts are shown in Figure 2. In 2F9-treated dogs, platelet nadir was 4,000 to 11,000 platelets/μl (compared to baseline counts of 183,000 to 329,000 platelets/μl) and nadir platelet counts were on average 3% of baseline counts. Variable, but generally mild mucocutaneous bleeding was noted in 2F9 treated dogs as previously described [15]. Platelet counts in 2F9-treated dogs remained below 40,000 platelets/μl for 24 hours, after which time counts began to spontaneously recover. No changes in platelet count were observed in the dogs treated with control antibody.
Figure 2. Platelet counts in 2F9 and control antibody-treated dogs.
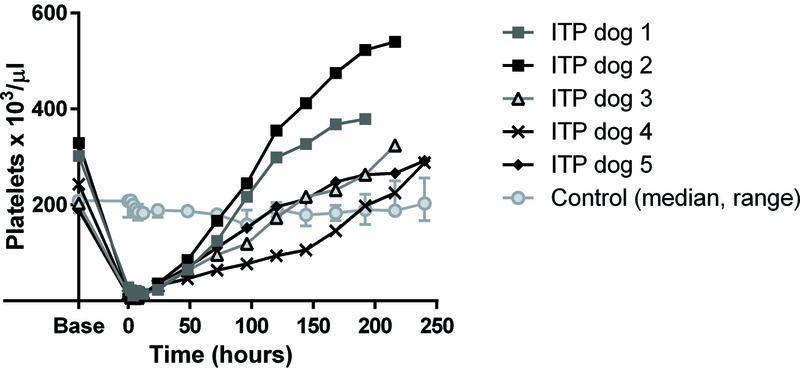
Counts are shown as for individual ITP dogs and as median and range for control dogs. Time zero is when the platelet count first fell into the target platelet range of 5,000–30,000 platelets/μl or 1 hour after control antibody infusion.
Histological observations
Supplementary Table 1 demonstrates the light microscopic data. Minimal changes were seen in most specimens.
Ultrastructural observations
Sections from the skin of the 3 control dogs and the 5 2F9-treated dogs were examined at baseline and after 24 hours of thrombocytopenia or 24 hours after control antibody administration.
Normal dogs
Figure 3 demonstrates a normal cutaneous capillary sampled from a dog at baseline with abundant pinocytotic vesicles, no endothelial cell-cell gaps, and a tightly apposed cell junction. A basement membrane surrounds the vessel and supporting pericytes are present. The vessel is composed of non-fenestrated continuous endothelium typical of normal cutaneous vasculature [24]. In the normal dogs (baseline samples), capillary wall thickness in regions away from the endothelial nuclei varied from 0.62 to 0.95 μm.
Figure 3. Normal cutaneous capillary.
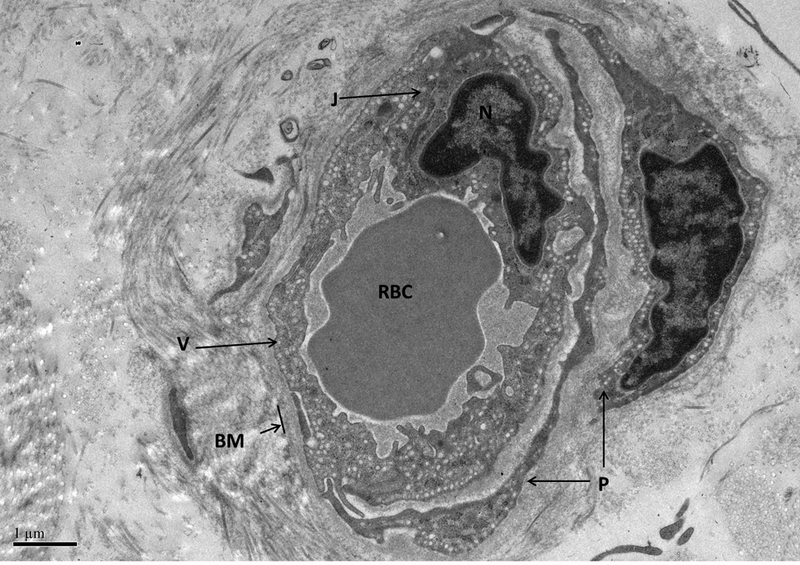
The vessel is enveloped by the basement membrane (BM). Two pericytes are visible (P). Pinocytotic vesicles are numerous (V). There are no spaces between endothelial cells and a well-apposed junction (J) is present connecting two adjacent endothelial cells adjacent to the nucleus (N). A red blood cell (RBC) is present in the vessel lumen. Magnification = 10,000 x.
Endothelial ultrastructural changes in ITP
Figure 5 and Table 1 show the ultrastructural data. After 24 hours of thrombocytopenia, thrombocytopenic vessels have significantly decreased numbers of pinocytotic vesicles from baseline when compared to changes in time-matched controls (P<0.0357) (Table 1; Figures 4–5). Thrombocytopenic microvascular endothelium tends to have increased numbers of large vacuoles and increased gaps between endothelial cells compared to baseline endothelium (Table 1; Figure 5C-D). These alterations are consistent with changes that would increase vascular permeability. However, these findings were not statistically different compared to time-matched controls (P=0.68 for change in vacuoles; P=0.29 for change in spaces between cells). There is no difference in vessel thickness in thrombocytopenic compared to control dogs (P=0.86) and the variation in vessel thickness is high in all samples. The standard deviation for vessel thickness in all baseline samples was 0.31 μm. Bleeding scores were not significantly correlated with any endothelial ultrastructural parameters.
Figure 5. Effect of ITP on endothelial ultrastructure.
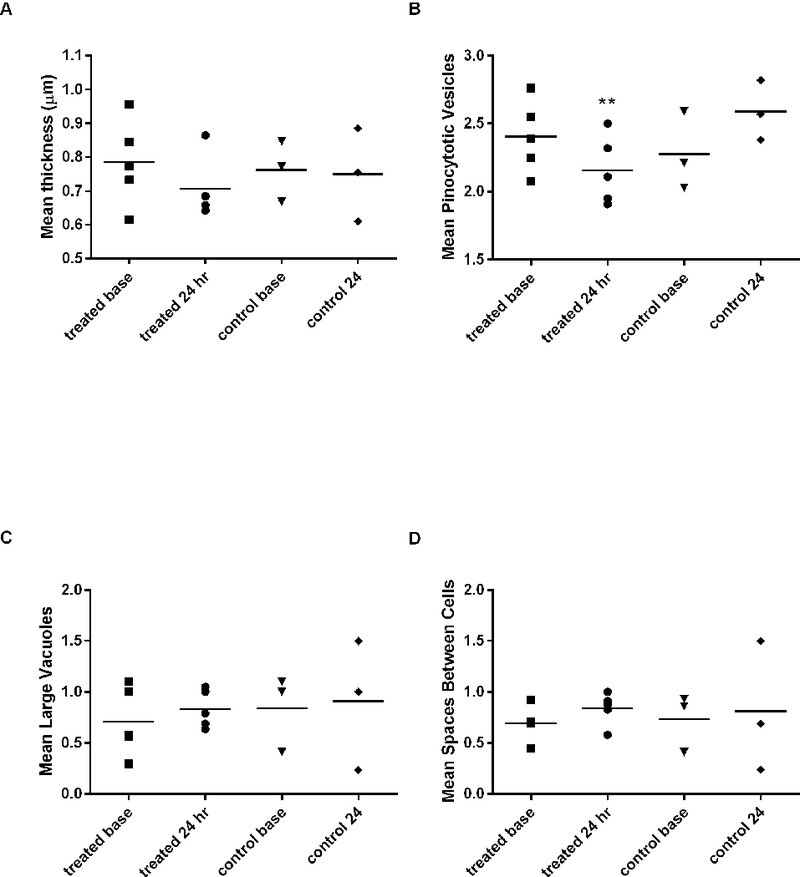
A) Vessel thickness, (B) number of endothelial pinocytotic vesicle score, (C) large vacuole score, and (D) number of spaces between cells. **There is a significant decrease in number pinocytotic vesicles from baseline to 24 hours of thrombocytopenia compared to time-matched controls (P<0.0357). See text for vacuole and vesicle scoring system. Symbols represent the mean value for all capillaries evaluated in each dog at that time point. Bars represent overall treatment group mean. n=5 2F9 treated dogs; n = 3 control dogs.
Table 1.
Ultrastructural features of endothelium in healthy dogs and dogs after 24 hours of severe thrombocytopenia.
| Treated (n=5) | Control (n=3) | |||||
|---|---|---|---|---|---|---|
| Baseline | 24 hours | Difference | Baseline | 24 hours | Difference | |
| Vessel thickness (μm) | 0.78±0.42 | 0.71±0.38 | -0.08±0.57 | 0.76±0.39 | 0.75±0.38 | -0.01±0.55 |
| Pinocytotic vesicle score | 2.41±0.48 | 2.16±0.53 | -0.25±0.71** | 2.28±0.57 | 2.59±0.51 | 0.31±0.76 |
| Large vacuole score | 0.71±0.77 | 0.83±0.86 | 0.13±1.16 | 0.84±0.78 | 0.91±0.79 | 0.07±1.11 |
| Number of spaces between cells | 0.69±0.7 | 0.84±0.79 | 0.15±1.06 | 0.73±0.6 | 0.81±0.69 | 0.08±0.92 |
See methods for vesicle and vacuole scoring system. Results are mean ± S.D.
P<0.0357 compared to change in time-matched controls.
Figure 4.
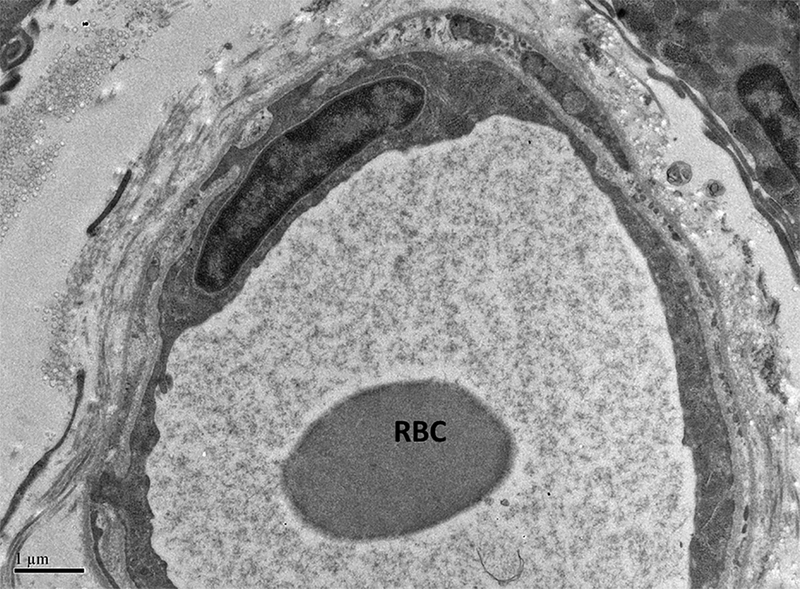
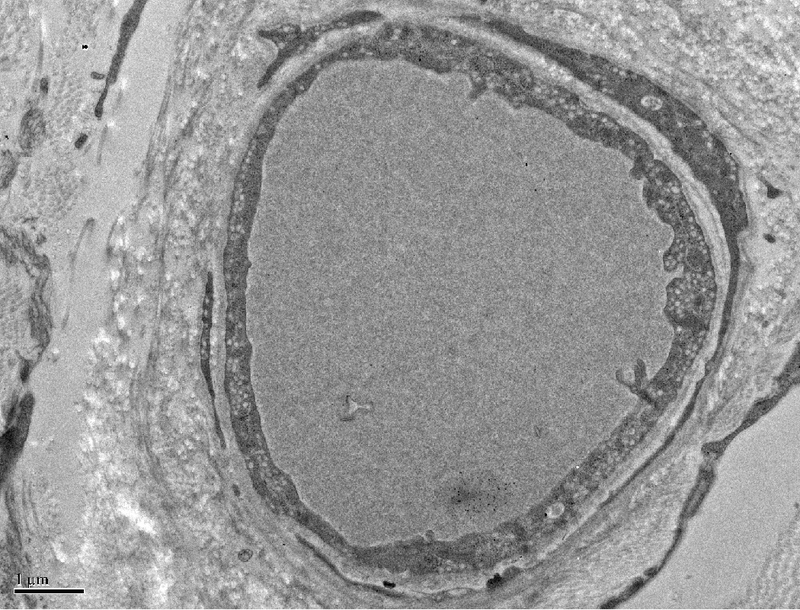
Representative micrograph demonstrating reduction in number of pinocytotic vesicles observed in thrombocytopenic dogs (A). A RBC is shown in the vessel lumen. Note that vesicles are almost absent compared to the normal vessel of comparable diameter shown in B. B; x 10,000.
Endothelial ultrastructural changes were observed in dogs with ecchymoses
Two dogs developed large ecchymoses (one on the abdominal skin and one on the axillary skin) that coincided with ultrastructural endothelial changes including pale swollen endothelial cells and gaps between adjacent endothelial cells (Figure 6A; 6C). In the dog that developed the large abdominal bruise, prominent inter-endothelial cell gaps were present after 24 hours of thrombocytopenia, which was 24 hours prior to the development of the bleed (6B). For comparison, a normal endothelial cell junction is pictured in Figure 6D. Compared to each dog’s baseline capillaries, capillaries in these bleed biopsies had increases in the average number of spaces between endothelial cells and the number of large vacuoles as well as decreases in the number of pinocytotic vesicles (Table 2). One dog’s mean vessel wall thickness at bleed was reduced from that of baseline; the other dog had a slight increase in thickness from baseline (Table 2). Histopathologic evaluation of tissues was also consistent with hemorrhage; no other explanation for the ecchymoses such as thrombosis or vasculitis was apparent (Supp Table 1).
Figure 6. Ecchymoses occurred in two dogs.
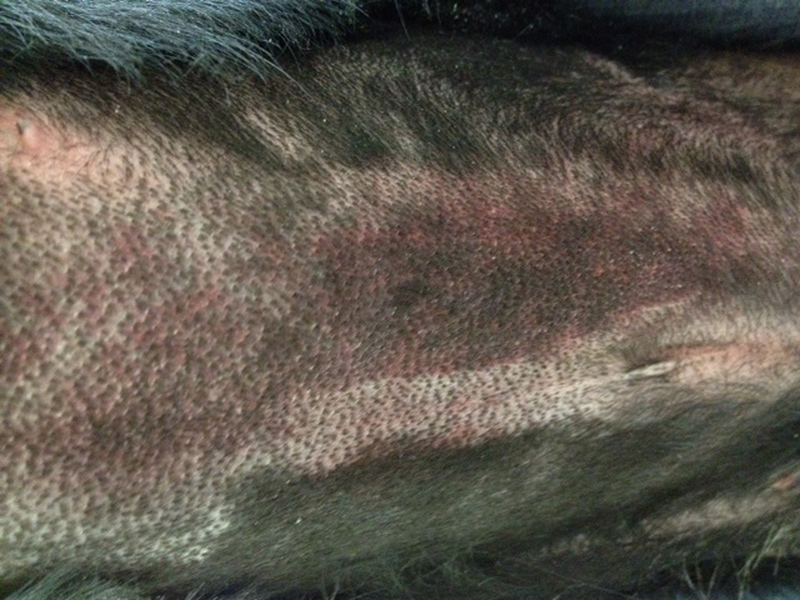
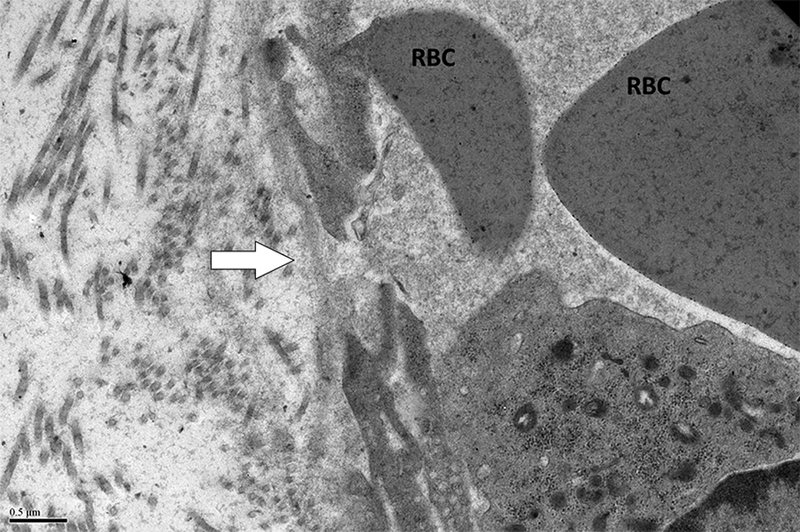
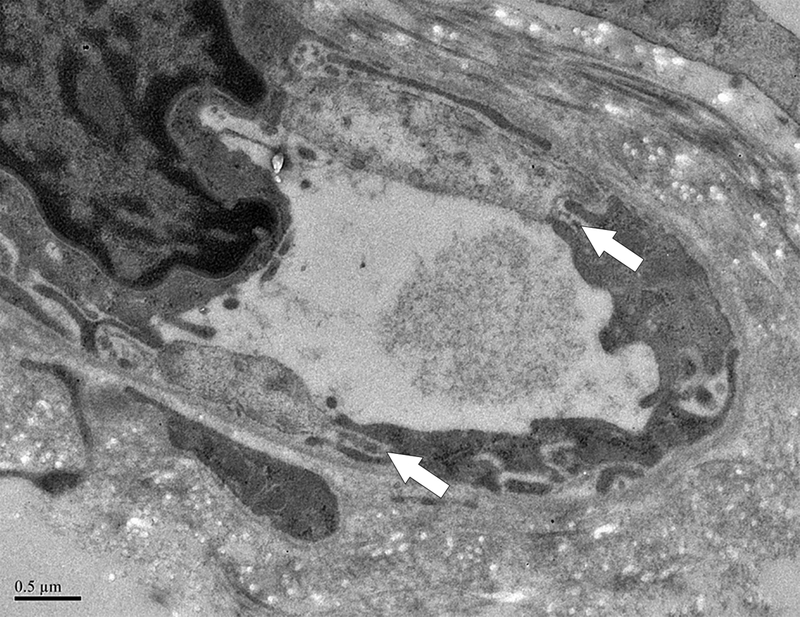
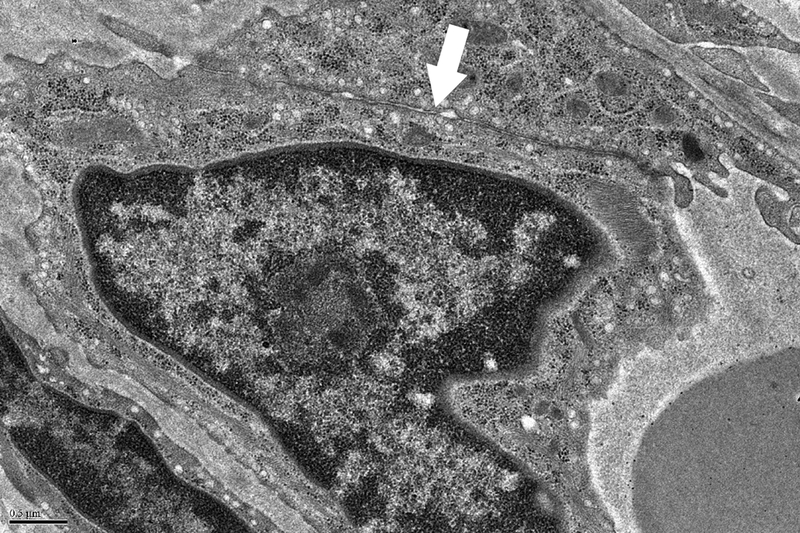
A. Photograph of the large abdominal bruise (8.5 × 13 cm) that formed at 48 hours in one dog. B. Electron micrograph from the dog in A after 24 hours of thrombocytopenia, 24 hours prior to the development of the bruise. A large gap between adjacent endothelial cells is marked with an arrow. RBCs are shown in lumen; x 20,000. C. Pale, swollen endothelial cells consistent with necrotic cells and inter-endothelial cell gaps (arrows) present in an electron micrograph from the bruised region of another dog that developed an ecchymosis; x 20,000. D. For comparison, normal closely apposed adjacent endothelial cells connected by an intact junction (arrow) in a dog at baseline are shown; x 20,000.
Table 2.
Endothelial ultrastructural features at the time of large ecchymosis formation in two dogs.
| Dog 1 | Dog 2 | |||
|---|---|---|---|---|
| Baseline | Bleed | Baseline | Bleed | |
| Vessel thickness(μm) | 0.85±0.56 | 0.68±0.43 | 0.77±0.38 | 0.8±0.45 |
| Pinocytotic vesicle score | 2.08±0.49 | 1.75±0.58 | 2.76±0.44 | 2.23±0.53 |
| Large vacuolescore | 1±1 | 1.13±0.89 | 0.29±0.47 | 0.45±0.67 |
| Number of spaces between cells | 0.69±0.75 | 1.31±0.79 | 0.71±0.69 | 1.1±0.97 |
Results are mean ± S.D of all vessels evaluated.
Platelet reactivity in relationship to bleeding
As an alternative explanation for variable bleeding phenotypes, platelet hemostatic function was also assessed throughout the study using flow cytometry. Bleeding scores were not correlated with platelet reactivity (measured by coated platelet numbers and EC50 thrombin; data not shown). In the two dogs that developed ecchymoses, platelet reactivity was equal to or greater than their baseline at the time that new bleeding occurred (Figure 7).
Figure 7. Platelet reactivity in two dogs that developed large ecchymoses when platelet hemostatic function was adequate.
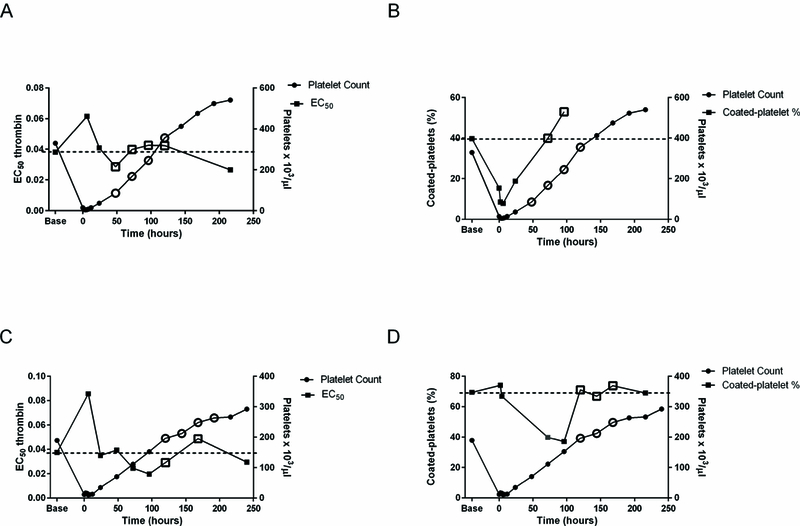
Unfilled symbols show time at which new bleeding was occurring. A. EC50 thrombin for P-selectin expression and platelet count in one dog. Dashed line indicates baseline EC50 in that dog. B. Coated-platelet levels and platelet count in the same dog. Dashed line indicates baseline coated-platelets in that dog. C. EC50 thrombin and platelet count in the second dog. D. Coated-platelet levels and platelet count in the second dog.
ITP is associated with a reduction in plasma S1P
Plasma S1P had a linear association with platelet count in treated and control dogs over all time points (P <0.0356) (Supplementary Figure 2). Hematocrit was evaluated as a potential confounder. As has been previously reported in humans, hematocrit was significantly correlated with plasma S1P (P<0.0001) [19], but was not significantly correlated with platelet count. Thus, it cannot be concluded that hematocrit is a confounder in the correlation between S1P and platelet count. Despite the relationship between platelet count and plasma S1P, there were no correlations between plasma S1P and endothelial thickness or the evaluated endothelial ultrastructural parameters at baseline and 24 hours. Similarly, there was no significant correlation between plasma S1P and bleeding score at baseline or at 24 hours.
Plasma Von Willebrand factor correlates with endothelial large vacuole score
In both 2F9-treated dogs and in control dogs, plasma vWF:Ag increased similarly over time (difference over time P <0.0001; difference between treatment groups P =0.78; Figure 8). The change in plasma vWF:Ag from baseline to 24 hours correlated directly with the change in endothelial large vacuole score (R= 0.99103, P<0.0001). Plasma vWF:Ag was also directly correlated with pinocytotic vessel score (R = 0.93415; P<0.0007) at 24 hours. There were no correlations between plasma vWF:Ag and endothelial thickness, the other evaluated endothelial ultrastructural parameters, or bleeding score.
Figure 8. Alterations of von Willebrand factor over time in thrombocytopenic and control dogs.
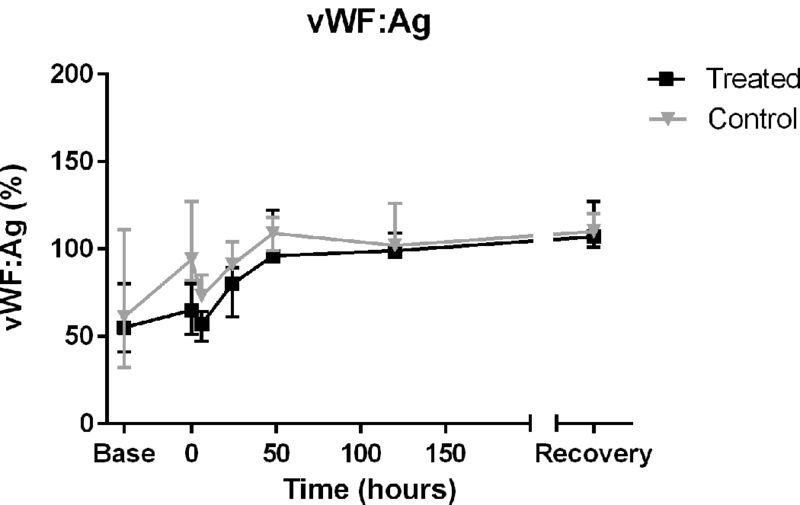
Similar increases in vWF:Ag in both groups of dogs likely reflect vWF’s induction as an acute phase reactant secondary to biopsies (P<0.0001 for change in vWF:Ag over time). However, vWF may also play a role as a marker of thrombocytopenic endothelial damage as demonstrated by its correlation with large vacuole score. Plasma vWF:Ag concentration is reported as percent compared to a normal canine standard (median, range).
Discussion
ITP patients have varied bleeding phenotypes, and it is not possible to accurately predict which patients will bleed based on severity of thrombocytopenia alone. We hypothesized that variations in endothelial integrity may influence bleeding. In a canine model of ITP, we observed changes in the endothelial ultrastructure of cutaneous vessels during severe thrombocytopenia. Most notably, pinocytotic vesicles decreased significantly in the thrombocytopenic endothelium. The exact mechanism of vesicle decrease is unknown but vesicle reduction is observed in several other examples of endothelial damage including methotrexate toxicity, hypoxia, and snake bite envenomation [25],[26], [27]. Pinocytotic vesicles provide a reserve of plasma membrane and can be translocated to the endothelial cell surface by exocytosis when there is a demand for increased membrane either due to cellular distention or membrane damage [25]. This is believed to be the reason for vesicle decrease following snake bite envomenation [25]. Formation of pinocytotic vesicles is an energy dependent process [28]. An alternate explanation for their observed decrease is an endothelial metabolic derangement and a perturbance in overall endothelial cell health. The exact mechanisms of platelet support of endothelial metabolism are unknown, but many platelet-derived growth factors such as vascular endothelial growth factor (VEGF) promote endothelial cell survival [29],[30]. VEGF can also directly induce vesicle formation in endothelial cells [31]. Reduction of VEGF and other growth factors in the face of thrombocytopenia likely result in altered endothelial metabolism [32]. Regardless of mechanism, the observed vesicle decrease is evidence of a thrombocytopenia-induced endothelial cell abnormality.
We also observed some lesions in biopsies from thrombocytopenic dogs that would result in increased vascular permeability, including increased frequency of gaps between endothelial cells, and increased number of large vacuoles. However, the magnitude of these changes was not statistically significant. The large vacuoles likely represent part of the vesiculo-vacuolar organelle (VVO), an endothelial cell permeability structure that provides a major route of extravasation of macromolecules at sites of augmented vascular permeability [33]. VVOs are a series of interconnecting vacuoles (on average 100 nm in diameter) that provide a transcellular pathway from vascular lumen to ablumen [33].
The thrombocytopenic trends we observed - increases in intercellular spaces and increases in large vacuoles – were expected based on others’ observations and knowledge of how endothelial permeability is controlled [34]; [35]. Pro-integrity factors like S1P stabilize endothelial cell junctions [36] while pro-permeability factors disrupt junctions and promote VVO transport [33], [30]. In contrast, endothelial thinning, although described by Kitchens [37], is not a predicted feature of thrombocytopenic vascular fragility and was not observed in our study nor in other small animal models [7].
The breaches in the endothelial walls and widened endothelial junctions observed in the dogs that developed ecchymoses are noteworthy (Figure 7). There are controversial data regarding the integrity of endothelial junctions in the face of thrombocytopenia. Kitchens and colleagues observed normal endothelial junctions and predicted that increased vesicular transport accounts for alterations in endothelial permeability during thrombocytopenia [37]. Others have reported normal endothelial junctions in thrombocytopenic guinea pigs, rats, and rabbits [7]. Contrary to these reports and similar to our findings in 2 dogs with eccchymoses, widened intercellular junctions, sometimes containing erythrocytes, have been previously described in thrombocytopenic rats and guinea pigs [34]; [38]. The junctions of thrombocytopenic dogs that did not develop cutaneous hemorrhage appeared normal. Our findings, in conjunction with previously reported data, suggest that in thrombocytopenic bleeding, erythrocytes extravasate through altered endothelial cell junctions or damaged endothelial cell walls [38].
Another notable feature of ecchymoses in this model is that we found no evidence of a platelet function defect that might have contributed to inadequate hemostasis at this time (Figure 7). Given the endothelial abnormalities we observed in biopsies taken from those sites of bleeding (Figure 6), we speculate that the bruising may result from progressive endothelial destabilization.
With the goal of identifying a mechanism by which platelets maintain vascular integrity, we monitored plasma S1P in treated and control dogs. Platelet count correlated significantly with plasma S1P (Supplementary Figure 2). To our knowledge this platelet count-plasma S1P correlation has not been previously reported [39–42]. Although we did not find any correlations between plasma S1P and endothelial ultrastructure or bleeding score, this does not exclude the possibility that platelet-derived S1P may have important local effects on preserving the vasculature. In the future, local S1P concentrations could be assessed by immunostaining endothelial biopsies. Additionally, our ITP model is one of platelet destruction. However, in naturally occurring ITP, platelet production may also be inadequate [43]. When platelet production is decreased, plasma S1P might be more impacted than during peripheral platelet destruction that may maintain the pool of circulating S1P. Alternatively, other platelet-derived vascular-stabilizing mediators like endostatin and angiopoietin-1 might be more important [30, 44]. Reliable canine-reactive assays for these mediators are unfortunately not available.
We investigated plasma vWF:Ag as a potential biomarker of ITP-induced endothelial damage. vWF is produced by endothelial cells and stored within endothelial organelles known as Weibel-Palade bodies. Unlike in other species, minimal vWF is found in canine platelets [45]. vWF is released from damaged or dysfunctional endothelial cells and high circulating vWF levels may result from increased synthesis in the acute phase response to an inflammatory stimulus [45–47]. vWF increased over time in both ITP and control dogs, likely as a sequel to repeated biopsies. Of note however, we did observe that changes in plasma vWF from time 0 to 24 hours were positively correlated with changes in vessel large vacuole score (R = 0.99103; P<0.0001). As large vacuoles are thought to represent the VVO, an endothelial cell permeability structure, the observed corresponding increases in vWF suggest that plasma vWF could serve as a circulating biomarker of endothelial dysfunction in ITP. Interestingly, vWF has been demonstrated to promote vascular permeability in animal models of stroke and peritonitis and could even serve as a direct mediator of endothelial damage [48, 49].
Plasma vWF:Ag was also positively correlated with vessel pinocytotic score after 24 hours of thrombocytopenia (R = 0.97008; P <0.0001). Decreases in the number of pinocytotic vesicles indicate membrane damage and repair, and thus we would have expected an inverse relationship between vWF and pinocytotic vesicles. However, increases in plasma vWF concentration reflect a combination of factors including endothelial cell injury, stimulated synthesis, and rate of macrophage-mediated plasma clearance [50, 51]. Further exploration is required to clarify the relationship between ultrastructural changes and circulating VWF levels.
Our ultrastructural studies have several limitations. We were evaluating structural lesions when the endothelial-stabilizing effect of platelets may be more of a functional one. Rodent models often use Evans Blue dye leakage to assess vascular integrity; however, because canine skin is so much thicker this would not be a reliable marker in dogs, nor could infusion of this toxic dye be employed in survival studies. In the future, we plan to conduct functional studies utilizing the ITP dog model such as monitoring lymphatic volume as an assessment of the effect of thrombocytopenia on vascular permeability.
A major limitation of the study was the small sample size. Given the variation in endothelial ultrastructural features that we observed even within a given dog and time point, more dogs may have been needed to identify significant changes in ultrastructural features other than pinocytotic vesicles. That we did not replicate the thinning of endothelium noted by Kitchens and colleagues more likely represents a difference in methodology or species than a sample size issue given that other studies similarly failed to identify thrombocytopenia-induced vessel thinning [7]. Additionally, our study may have been limited by the short duration of severe thrombocytopenia. By 24 hours, platelet counts had started to recover (>30,000 platelet/μl in 4 of 5 of the 2F9-treated dogs) which may be above the threshold for maintenance of vascular integrity. In murine models of thrombocytopenic hemorrhage, only five percent of the normal platelet count was needed to achieve the vascular protective effect of platelets [52], [2]. It may be that longer duration of severe thrombocytopenia in dogs is necessary to accentuate relevant lesions. We plan to achieve this in future studies with repeated 2F9 administration.
Recent work by Goerge and colleagues suggests one possible explanation for the relatively mild bleeding and the overall minor changes in endothelial ultrastructure observed in the model dogs [52]. This group demonstrated that thrombocytopenic hemorrhage only occurs in the face of inflammation [52]. In order for bleeding to occur, leukocyte diapedesis may be necessary in combination with the vascular destabilizing effects of thrombocytopenia [53, 54]. Ultrastructural studies have not been performed to look at the combined effects of inflammation and hemorrhage. As our dogs had minimal to no cutaneous inflammation (Supplementary Table 1), they may have lacked a second “hit” necessary to create sufficient vascular damage for significant bleeding to occur. We suspect mild bleeding is also the reason we did not identify a correlation between bleed score and endothelial structural changes.
Conclusion
In summary, this study suggests that there is an association between thrombocytopenia and ultrastructural endothelial abnormalities in a canine model of ITP. The significant decrease in pinocytotic vesicles in thrombocytopenic endothelium shows that endothelial damage occurs in this model and warrants further study in model ITP dogs and in dogs with spontaneous disease. Variability in the degree of alterations of vascular ultrastructure may account for differences in bleeding phenotype in immune thrombocytopenia. Plasma vWF could serve as a biomarker for endothelial alterations and should be investigated as a predictor of thrombocytopenic bleeding. Ultimately, measurement of markers of endothelial dysfunction or platelet vascular stabilizing mediators could enable us to predict which ITP patients are at risk of bleeding and tailor patient management accordingly. Furthermore, better understanding of the mechanism of thrombocytopenic bleeding will allow for generation of new therapeutics to prevent and treat bleeding in thrombocytopenia.
Supplementary Material
Acknowledgements
The authors wish to thank Victoria Madden, Steven Ray, and Robert Bagnell for their invaluable technical support and expertise with electron microscopy.
Declaration of Interests
This work was supported in part by research funding from the North Carolina State University Center for Comparative Medicine and Translational Research (DNL, SKN, THF, MBB, NSK). DNL and MM were supported by NIH grant T32 HL007149.
Footnotes
The authors declare that they have no conflicts of interest.
References
- 1.Shepro D, Sweetman HE, and Hechtman HB, Experimental thrombocytopenia and capillary ultrastructure. Blood, 1980. 56(5): p. 937–9. [PubMed] [Google Scholar]
- 2.Ho-Tin-Noe B, Demers M, and Wagner DD, How platelets safeguard vascular integrity. Journal of Thrombosis and Haemostasis, 2011. 9 Suppl 1: p. 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitchens CS and Pendergast JF, Human thrombocytopenia is associated with structural abnormalities of the endothelium that are ameliorated by glucocorticosteroid administration. Blood, 1986. 67(1): p. 203–6. [PubMed] [Google Scholar]
- 4.Kitchens CS, Amelioration of endothelial abnormalities by prednisone in experimental thrombocytopenia in the rabbit. J Clin Invest, 1977. 60(5): p. 1129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Horn DL and Johnson SA, The escape of carbon from intact capillaires in experimental thrombocytopenia. J Lab Clin Med, 1968. 71(2): p. 301–311. [Google Scholar]
- 6.Van Horn DL and Johnson SA, The mechanism of thrombocytopenic bleeding. Am J Clin Pathol, 1966. 46(2): p. 204–13. [DOI] [PubMed] [Google Scholar]
- 7.Miles RG and Hurley JV, The effect of thrombocytopenia on the ultrastructure and reaction to injury of vascular endothelium. Microvasc Res, 1983. 26(3): p. 273–90. [DOI] [PubMed] [Google Scholar]
- 8.Takuwa N, et al. , Tumor-suppressive sphingosine-1-phosphate receptor-2 counteracting tumor-promoting sphingosine-1-phosphate receptor-1 and sphingosine kinase 1 - Jekyll Hidden behind Hyde. Am J Cancer Res, 2011. 1(4): p. 460–81. [PMC free article] [PubMed] [Google Scholar]
- 9.Camerer E, et al. , Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest, 2009. 119(7): p. 1871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.chaphorst KL, et al. , Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol, 2003. 285(1): p. L258–67. [DOI] [PubMed] [Google Scholar]
- 11.Polzin A, et al. , Sphingosine-1-phosphate is thromboxane-dependently released from platelets and modulates chemotaxis of human monocytes, in American Heart Association; 2010: Chicago, IL. [Google Scholar]
- 12.Jonnalagadda D, et al. , Granule-mediated release of sphingosine-1-phosphate by activated platelets. Biochim Biophys Acta, 2014. 1841(11): p. 1581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arslan M, et al. , Importance of Endothelial Dysfunction Biomarkers in Patients with Crimean-Congo Hemorrhagic Fever. J Med Virol, 2017. [DOI] [PubMed] [Google Scholar]
- 14.Paulus P, Jennewein C, and Zacharowski K, Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarkers, 2011. 16 Suppl 1: p. S11–21. [DOI] [PubMed] [Google Scholar]
- 15.LeVine DN, et al. , A novel canine model of immune thrombocytopenia: has immune thrombocytopenia (ITP) gone to the dogs? Br J Haematol, 2014. 167(1): p. 110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson SJ, et al. , Endothelial cell damage in heparin-induced thrombocytopenia. Blood Coagul Fibrinolysis, 2007. 18(4): p. 317–20. [DOI] [PubMed] [Google Scholar]
- 17.Mikkola HK, et al. , Expression of CD41 marks the initiation of definitive hematopoiesis in the mouse embryo. Blood, 2003. 101(2): p. 508–16. [DOI] [PubMed] [Google Scholar]
- 18.Page LK, et al. , The immune thrombocytopenic purpura (ITP) bleeding score: assessment of bleeding in patients with ITP. British Journal of Haematology, 2007. 138(2): p. 245–8. [DOI] [PubMed] [Google Scholar]
- 19.Selim S, et al. , Plasma levels of sphingosine 1-phosphate are strongly correlated with haematocrit, but variably restored by red blood cell transfusions. Clin Sci (Lond), 2011. 121(12): p. 565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews TP, et al. , Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem, 2010. 53(7): p. 2766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Catalfamo JL, et al. , Canine platelets are virtually devoid of von Willebrand factor. Blood, 1991. 78: p. 261a. [Google Scholar]
- 22.Peng J, et al. , Alteration of platelet function in dogs mediated by interleukin-6. Blood, 1994. 83(2): p. 398–403. [PubMed] [Google Scholar]
- 23.Brooks MB, et al. , Scott syndrome dogs have impaired coated-platelet formation and calcein-release but normal mitochondrial depolarization. J Thromb Haemost, 2007. 5(9): p. 1972–4. [DOI] [PubMed] [Google Scholar]
- 24.Aird WC, Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res, 2007. 100(2): p. 158–73. [DOI] [PubMed] [Google Scholar]
- 25.Gutierrez JM, et al. , Blood flow is required for rapid endothelial cell damage induced by a snake venom hemorrhagic metalloproteinase. Microvasc Res, 2006. 71(1): p. 55–63. [DOI] [PubMed] [Google Scholar]
- 26.Fitzl G, et al. , Protective effects of Gingko biloba extract EGb 761 on myocardium of experimentally diabetic rats. I: ultrastructural and biochemical investigation on cardiomyocytes. Exp Toxicol Pathol, 1999. 51(3): p. 189–98. [DOI] [PubMed] [Google Scholar]
- 27.Fuskevag OM, et al. , Microvascular perturbations in rats receiving the maximum tolerated dose of methotrexate or its major metabolite 7-hydroxymethotrexate. Ultrastruct Pathol, 2000. 24(5): p. 325–32. [DOI] [PubMed] [Google Scholar]
- 28.Chavez A, Smith M, and Mehta D, New insights into the regulation of vascular permeability. Int Rev Cell Mol Biol, 2011. 290: p. 205–48. [DOI] [PubMed] [Google Scholar]
- 29.Mohle R, et al. , Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A, 1997. 94(2): p. 663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nachman RL and Rafii S, Platelets, petechiae, and preservation of the vascular wall. N Engl J Med, 2008. 359(12): p. 1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, et al. , VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol, 2002. 282(5): p. C1053–63. [DOI] [PubMed] [Google Scholar]
- 32.Verheul HM, et al. , Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res, 1997. 3(12 Pt 1): p. 2187–90. [PubMed] [Google Scholar]
- 33.Dvorak AM and Feng D, The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem, 2001. 49(4): p. 419–32. [DOI] [PubMed] [Google Scholar]
- 34.Gore I, Takada M, and Austin J, Ultrastructural basis of experimental thrombocytopenic purpura. Arch Pathol, 1970. 90(3): p. 197–205. [PubMed] [Google Scholar]
- 35.Aursnes I and Pedersen OO, Petechial hemorrhage in the ciliary processes of thrombocytopenic rabbits. An electron microscopic study. Microvasc Res, 1979. 17(1): p. 12–21. [DOI] [PubMed] [Google Scholar]
- 36.McVerry BJ and Garcia JG, In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal, 2005. 17(2): p. 131–9. [DOI] [PubMed] [Google Scholar]
- 37.Kitchens CS and Weiss L, Ultrastructural changes of endothelium associated with thrombocytopenia. Blood, 1975. 46(4): p. 567–78. [PubMed] [Google Scholar]
- 38.Dale C and Hurley JV, An electron-microscope study of the mechanism of bleeding in experimental thrombocytopenia. J Pathol, 1977. 121(4): p. 193–204. [DOI] [PubMed] [Google Scholar]
- 39.Venkataraman K, et al. , Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res, 2008. 102(6): p. 669–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pappu R, et al. , Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science, 2007. 316(5822): p. 295–8. [DOI] [PubMed] [Google Scholar]
- 41.Dahm F, et al. , Distribution and dynamic changes of sphingolipids in blood in response to platelet activation. J Thromb Haemost, 2006. 4(12): p. 2704–9. [DOI] [PubMed] [Google Scholar]
- 42.Bode C, et al. , Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem. 109(6): p. 1232–43. [DOI] [PubMed] [Google Scholar]
- 43.Aledort LM, et al. , Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am J Hematol, 2004. 76(3): p. 205–13. [DOI] [PubMed] [Google Scholar]
- 44.Brankin B, et al. , Endostatin modulates VEGF-mediated barrier dysfunction in the retinal microvascular endothelium. Exp Eye Res, 2005. 81(1): p. 22–31. [DOI] [PubMed] [Google Scholar]
- 45.Brott DA, et al. , Evaluation of von Willebrand factor and von Willebrand factor propeptide in models of vascular endothelial cell activation, perturbation, and/or injury. Toxicol Pathol, 2014. 42(4): p. 672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lip GY and Blann A, von Willebrand factor: a marker of endothelial dysfunction in vascular disorders? Cardiovasc Res, 1997. 34(2): p. 255–65. [DOI] [PubMed] [Google Scholar]
- 47.Zezos P, et al. , Elevated plasma von Willebrand factor levels in patients with active ulcerative colitis reflect endothelial perturbation due to systemic inflammation. World J Gastroenterol, 2005. 11(48): p. 7639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, et al. , Recombinant ADAMTS13 reduces tissue plasminogen activator-induced hemorrhage after stroke in mice. Ann Neurol, 2013. 73(2): p. 189–98. [DOI] [PubMed] [Google Scholar]
- 49.Petri B, et al. , von Willebrand factor promotes leukocyte extravasation. Blood, 2010. 116(22): p. 4712–9. [DOI] [PubMed] [Google Scholar]
- 50.van Schooten CJ, et al. , Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood, 2008. 112(5): p. 1704–12. [DOI] [PubMed] [Google Scholar]
- 51.Xiang Y and Hwa J, Regulation of VWF expression, and secretion in health and disease. Curr Opin Hematol, 2016. 23(3): p. 288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goerge T, et al. , Inflammation induces hemorrhage in thrombocytopenia. Blood, 2008. 111(10): p. 4958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.<j/>Ho-Tin-Noe, B., et al. , Innate immune cells induce hemorrhage in tumors during thrombocytopenia. Am J Pathol, 2009. 175(4): p. 1699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hillgruber C, et al. , Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med, 2015. 212(8): p. 1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.