

The role of surface loops in encoding SH2 domain specificity has been systematically investigated by characterizing a group of loop variants obtained from screening phage-displayed SH2 domain libraries. The reported results support a general role for the EF loop (which connects the β-strands E and F) and the BG loop (which connects the α-helix B and β-strand G) in encoding SH2 specificity, add to our understanding of the mechanism of target sequence recognition by an SH2 domain in cells, and have general implications for the evolution of binding specificity of protein interaction modules.
Keywords: Cancer Biology, Chemoresistance, Breast Cancer, Mitochondria Function or Biology, Stem Cells, Autophagy, NMR, Clinical Proteomics, Antibiotics, Cancer Stem Cells, Mitochondria, NMR-metabolomic
Graphical Abstract

Highlights
Surface loops play an essential role in SH2 domain specificity.
Diverse specificities may be obtained from a single SH2 domain by combinatorial mutations in the EF and BG loops.
The specificity of a loop mutant correlates with the sequence characteristics of the bait peptide used in its isolation.
Abstract
Src homology 2 (SH2) domains play an essential role in cellular signal transduction by binding to proteins phosphorylated on Tyr residue. Although Tyr phosphorylation (pY) is a prerequisite for binding for essentially all SH2 domains characterized to date, different SH2 domains prefer specific sequence motifs C-terminal to the pY residue. Because all SH2 domains adopt the same structural fold, it is not well understood how different SH2 domains have acquired the ability to recognize distinct sequence motifs. We have shown previously that the EF and BG loops that connect the secondary structure elements on an SH2 domain dictate its specificity. In this study, we investigated if these surface loops could be engineered to encode diverse specificities. By characterizing a group of SH2 variants selected by different pY peptides from phage-displayed libraries, we show that the EF and BG loops of the Fyn SH2 domain can encode a wide spectrum of specificities, including all three major specificity classes (p + 2, p + 3 and p + 4) of the SH2 domain family. Furthermore, we found that the specificity of a given variant correlates with the sequence feature of the bait peptide used for its isolation, suggesting that an SH2 domain may acquire specificity by co-evolving with its ligand. Intriguingly, we found that the SH2 variants can employ a variety of different mechanisms to confer the same specificity, suggesting the EF and BG loops are highly flexible and adaptable. Our work provides a plausible mechanism for the SH2 domain to acquire the wide spectrum of specificity observed in nature through loop variation with minimal disturbance to the SH2 fold. It is likely that similar mechanisms may have been employed by other modular interaction domains to generate diversity in specificity.
The Src homology 2 (SH2)1 domain, originally identified in the viral oncogene product v-fps/fes, was subsequently found in numerous metazoan proteins (1, 2). It is known now that the human genome encodes ∼120 SH2 domains that are dispersed in more than 110 proteins. These include protein or lipid kinases, protein phosphatases, small GTPases, cytoskeleton regulators, and adaptor/scaffolding proteins and other regulators of signal transduction (3–4). SH2 domains exert their functions by binding to the phosphotyrosine (pY) residue embedded in specific sequence motifs, thereby enabling transduction of signals emanated from tyrosine kinases to downstream molecules (1, 5, 6). The importance of the tyrosine kinase-pY-SH2 signaling axis in normal physiology and disease pathogenesis is underscored by the fact that drugs targeting components of this axis form the largest collection of targeted therapeutics used in the clinic to treat cancer and other complex human diseases (7).
SH2 domains, related to one another by structure and function, are ∼100-residue in length and fold into a globular structure comprising a central β-sheet (with strands βA to βG) flanked by two α-helices (αA and αB) (8–10). A typical SH2 domain recognizes the pY and a specific residue C-terminal to the pY in a two-pronged plug two-holed socket mode (11, 12). Although all SH2 domains contain a pY-binding pocket and share virtually the same mode of pY recognition (8), they differ in specificity and mode of recognition for the C-terminal residue (3, 13). Based on results from a systematic structure-function analysis, we categorized the mammalian SH2 domains into three specificity classes, p + 2, p + 3 and p + 4 (13). The p + 3 class, exemplified by the Src SH2 domain, prefers a hydrophobic residue at the p + 3 position (the third residue C-terminal to the pY residue). The Grb2 and BRDG1 SH2 domains, which belong to the p + 2 and p + 4 classes respectively, prefer peptides with an Asn at the p + 2 or a hydrophobic residue at the p + 4 position (13–15).
The C-terminal specificity is mediated by a binding pocket or site—referred to herein as specificity pocket—on the surface of an SH2 domain that accommodate the p + 2, p + 3 or p + 4 residue in the peptide ligand (13). We have shown previously that two surface loops on the SH2 domain, namely the EF loop (which connects the β-strands E and F) and the BG loop (which connects the α-helix B and β-strand G), not only participate in the formation of the specificity pockets, but also control access of the peptide ligand to the pockets (14). In a typical SH2 domain, only one of the three specificity pockets is available for ligand binding whereas the remaining pockets are made inaccessible because of pocket-plugging or steric hindrance created by specific residues from the EF and BG loops. For example, in the Src SH2 domain (p + 3 class), the p + 4 pocket is plugged by a residue from the BG loop whereas in the BRDG1 SH2 domain (p + 4 class), the p + 3 pocket is blocked by an EF loop residue. In the case of the Grb2 SH2 domain (p + 2 class), both the p + 3 and p + 4 binding pockets are blocked (13). The critical role of the EF and BG loops in governing SH2 domain specificity was underscored in the observation that specificity of an SH2 domain may be altered, or even class-switched, by mutating key residues within these loops. Of note, mutating the EF1 residue in the Src SH2 domain from Thr to Trp resulted in switch of specificity from the p + 3 to p + 2 class (16). In contrast, substituting the EF2-Leu residue in the p + 4 class BRDG1 SH2 domain with an Ala caused a switch of specificity to the p + 3 class (13).
Although the above studies suggest a pivotal role for the EF and BG loops in SH2-ligand binding, they also raise an important question: can these surface loops encode the wide spectrum of specificities found for the SH2 domain family in a fashion akin to the role of the complementarity-determining regions (CDR) in determining the specificity of an antibody (17)? To address this question, we generated phage-displayed libraries of the Fyn SH2 domain in which the EF and BG loops were randomized in length and residue composition. By screening the libraries with pY-peptides with diverse sequences, we identified variants that exhibited a wide range of specificities. Using peptide arrays, including Oriented Peptide Array Libraries (OPAL) and ligand peptide arrays, and in-solution binding assays, we determined the specificity and affinity for a panel of 29 variants isolated from the phage-displayed library screening. Our data shows that the EF and BG loops are highly evolvable and capable of encoding a wide spectrum of specificity found in naturally occurring SH2 domains.
MATERIALS AND METHODS
Phage Display
The Fyn-SH2 EF/BG loop library was constructed by Kunkel mutagenesis (8, 18). The library was panned against biotin-pY peptides immobilized on a Maxisorp plate (NUNC) precoated with streptavidin. After four cycles of panning, enriched phage pools were applied to infect E. coli XL1-Blue to obtain single colonies. Phage ELISA was conducted by adding the single colony phage solution into a 96-well Maxisorp plate (NUNC) precoated with streptavidin and biotin-pY peptides. Positive phages were subject to DNA sequencing to identify the sequences of the corresponding SH2 variants (8).
Peptide Synthesis
Peptides were synthesized on the Tentagel resin on an Intavis-AG MultiPep peptide synthesizer using Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry. Peptides were labeled, at the N terminus, with either biotin for printing and pulldown assays or fluorescein for binding studies by fluorescence polarization (8). A spacer containing Ahx-Ahx-Ser-Gly-Gly (Ahx, 6-aminohexanoic acid) was inserted between the biotin or fluorescein and the peptide to minimize the effect of labeling. Purity and identities of the peptides were verified by mass spectrometry.
Peptide Array Slide Preparation and Probing
Biotin-labeled peptides were incubated in PBS (phosphate buffered saline), pH 7.5 with neutravidin in a 1:1 molar ratio for 1h at room temperature. The mixture was diluted in PBS to 25 μm. SuperAB glass slides (Fisher, Pittsburgh, PA) were preactivated in 50 mm NaIO4, 0.1 m sodium acetate, pH 5.5 for 0.5h at room temperature, dried with nitrogen stream and used immediately. The peptide-neutravidin conjugates were printed onto an activated SuperAB chip (Fisher) using a Bio-Rad VersArray Chipwriter-Pro system. Before probing with a purified SH2 protein, the peptide array chip was washed three times in 3% BSA in TBST buffer (0.1 m Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% Tween 20). For probing, 1.0 μm total GST-SH2 protein was added directly to the 3%BSA/TBST buffer and incubated with the slide for 1h at RT. The slide was then washed three times in TBST and incubated with a rabbit anti-GST antibody (Abcam, Toronto, ON, Canada #ab3416). After 1h, the slide was washed 3X in TBST and incubated with a DyLight 649-labeled goat anti-rabbit IgG antibody (Pierce, Pittsburgh, PA #35565) for 1 h in the dark. The slide was washed again in TBST, dried in the dark, and scanned with a microarray laser scanner (Tecan Co., Männedorf, Switzerland). Data processing and quantification were performed using the embedded software of the scanner.
Array Data Analysis
Data processing and quantification were performed using the embedded software of the scanner. The binding signal for a variant was calculated as the average value of the quadruple repeats for each peptide. Then, the binding signals were normalized across the entire array.
The selectivity score (z-score) of a variant domain for each pY peptide is defined according to the formula
Where Bi is the average signal of binding, σ is the standard deviation of Bi. If more than one residue in the position was considered, the average of Z score for each amino acid was used.
Fluorescence Polarization Measurements
Each SH2 protein was serially diluted in a 384-well plate, followed by the addition of fluorescein-labeled peptide in PBS buffer. The mixtures were incubated in the dark for 30 min prior to fluorescent polarization measurements at RT on an EnVision Multilabel Plate Reader (PerkinElmer) with the excitation set at 480 nm and emission at 535 nm. Binding curves were generated by fitting the binding data to a hyperbolic nonlinear regression model using Prism 3.0 (GraphPad software, Inc., San Diego, CA), which also produced the corresponding dissociation constants (Kd).
RESULTS
The EF and BG Loops Are Highly Evolvable
We employed the Fyn SH2 domain to test if the EF and BG loops can encode a wide range of specificity. The EF loop of the Fyn SH2 domain comprises three residues (i.e. TTR) whereas its BG loop seven residues (i.e. AAGLSSR). We generated two libraries of the Fyn SH2 domain in which the EF and BG loop residues were randomized by Kunkel mutagenesis (8, 18) (Fig. 1). The resulting libraries were displayed, respectively, on the M13 bacteriophage and screened for binding to immobilized pY peptides (8) representing the three major ligand classes (i.e. p + 2N, p + 3, and p + 4). The library screens led to the isolation of 152 unique variants (supplemental Table S1) bound by 19 bait peptides (supplemental Table S1). Based on sequence diversity of the bait peptides and the isolated variants, we selected 29 Fyn SH2 variants for further analysis (Fig. 2).
Fig. 1.
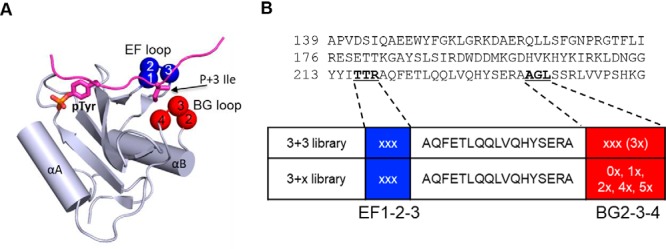
Directed evolution of the Fyn SH2 domain via systematic changes in the EF and BG loops. A, Structure of the FYN-SH2 domains in complex with a tyrosine-phosphorylated peptide (PDB ID 4U1P). The bound peptide is colored magenta, with the side chains of pTyr and p + 3 Ile residues shown as sticks. The p + 3 Ile is located between the EF and BG loops. Residues targeted for evolution via combinatorial mutagenesis are identified with blue balls for the EF loop (EF1, EF2 and EF3) and red balls for the BG loop (BG2, BG3 and BG4). B, The phage display library design. The three resides from the EF and BG loops targeted for mutagenesis were underscored in the sequence of the human Fyn-SH2 domain. The length of both loops was unchanged in the 3 + 3 library, whereas the length of BG loop was varied in the 3+x library, from two-residue shorter to three-residue longer, but not 3.
Fig. 2.

Loop sequences of the variants identified by different bait peptides. Residues in the bait peptide and EF/BG loop sequences are colored according to their chemical nature. The 29 variants selected for subcloning and further characterization are annotated with the variant numbers. A, Variants identified by bait peptides containing the pY+2N motif. B, Variants identified by bait peptides that contain a hydrophobic residue at either the pY+3 or pY+4 position.
A noteworthy feature of the variants selected by the p + 2N group of bait peptides (Fig. 2A) is the enrichment for aromatic residues within the EF loop. Indeed, 33 of the identified clones contained an aromatic residue (W, Y or F) at the EF1 position. A bulky, aromatic EF1 residue would likely block the pY+3 binding pocket in a manner like the EF1-Trp residue in the Grb2 SH2 domain (p + 2 class) (13). To encode p + 2N specificity, it is also necessary to have the p + 4 binding pocket plugged. This appeared to be accomplished in most cases by an amino acid with a long, aliphatic sidechain (L, V or I) at the BG2 position, or in some instances, the BG4 or BG3 position (Fig. 2A). Intriguingly, several variants (e.g. V10) captured by the PDGFRβ-pY716 peptide (which contained the small hydrophobic residue, Ala, at the p + 4 position in addition to an Asn at p + 2) contained a truncated BG loop in which the residues BG2–BG4 or BG3–BG4 were missing. In the same vein, selection by the L4 peptide (p + 4Leu, Fig. 2B and Table I) yielded four variants (including V17) with the BG loop either completely missing or drastically curtailed. As a shortened BG loop would leave the p + 4 binding pocket unblocked, these mutants are expected to have acquired p + 4 specificity (vide infra). As shown later (Table I), V17, but not V10, exhibited a stronger preference for the p + 4L peptide. In contrast, most variants selected by the p + 3Ile (I3) peptide featured a Leu or an Ile residue at the BG2 or BG4 position and a non-aromatic residue at the EF1 position, suggesting that these variants have retained the p + 3 specificity of the wild-type (wt) Fyn SH2 domain.
Table I. Dissociation constants and relative affinities (brackets) of a panel of Fyn SH2 variants for phosphopeptides representing different specificity classes.
| Peptide (Specificity Motif) | Fyn | Src | Grb2 | BRDG1 | V7 | V8 | V10 | V14 | V17 | V18 | V29 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EPQpYENEEE (N2, p + 2N) | 0.7 ± 0.15 | 1.7 ± 0.34 | 0.8 ± 0.25 | NB | 0.8 ± 0.23 | 0.1 ± 0.097 (7) | 1.8 ± 0.48 | 0.3 ± 0.07 (2.3) | 2.5 ± 0.29 (0.3) | 0.7 ± 0.21 | 0.2 ± 0.08 (3.5) |
| QPEpYVNQADV (ErbB2-pY1139, p + 2N) | 5.7 ± 1.36 | 4.5 ± 1.56 | 0.4 ± 0.09 | NB | 6.3 ± 0.86 | 1.7 ± 0.28 (3.3) | 7.3 ± 2.66 | 4.2 ± 0.72 (1.3) | 24.5 ± 5.00 (0.2) | 6.8 ± 1.14 | 0.3 ± 0.04 (19) |
| EPQpYEEIEE (I3, p + 3I) | 0.6 ± 0.08 | 0.7 ± 0.13 | NB | NB | NB | 4.9 ± 1.08 (0.1) | 19.9 ± 4.54 | 0.1 ± 0.02 (6) | 4.1 ± 0.47 (0.1) | 1.0 ± 0.26 | 0.5 ± 0.11 (1.2) |
| NPDpYQQDFFP (EGFR-pY1172, p + 4) | 6.5 ± 1.36 | 5.0 ± 1.43 | 10.6 ± 2.84 | 27.2 ± 13.93 | 2.3 ± 0.38 | 23.7 ± 3.36 (0.2) | 4.8 ± 1.10 | 3.6 ± 0.52 (1.8) | 2.8 ± 0.33 (2.3) | 6.3 ± 0.71 | 2.1 ± 0.29 (3.1) |
| EPQpYEEELE (L4, p + 4L) | 2.5 ± 0.46 | 4.2 ± 1.04 | NB | 4.1 ± 0.51 | NB | 6.3 ± 1.77 (0.6) | 14.4 ± 2.66 | 1.0 ± 0.13 (2.5) | 0.3 ± 0.06 (8.3) | 1.3 ± 0.57 | 1.0 ± 0.18 (2.5) |
Notes: Dissociation constants (Kd, in μm, ±standard deviation) are derived from fluorescein polarization measurements (see also supplemental Fig. S7A–S7K). NB, no binding or binding too weak to determine the Kd of. All peptides were synthesized with an N-terminal spacer containing the sequence fluorescein-Ahx-Ahx-Ser-Gly-Gly, where Ahx denotes 6-aminohexanoic acid.
Characterization of SH2 Variant Specificity by OPAL
To survey the breadth of new specificities, we selected 29 Fyn SH2 variants with distinct EF/BG loop characteristics (Fig. 2) and expressed them respectively in E. coli as GST fusion (supplemental Fig. S1). The purified GST-SH2 protein was then used to screen an Oriented Peptide Array Library (OPAL) containing the degenerated sequence x-pY-x-x-x-x-x (where x denotes a mixture of 19 natural amino acids excluding Cys) (19). We have previously employed the OPAL approach to characterize the specificity of human SH2 domains (3). A notable difference between the current and previous methods (3) is that the current OPAL sublibraries were labeled with biotin and printed onto neutravidin-coated glass slides instead of being spotted on cellulose membranes. Moreover, each sublibrary was printed in quadruplicates to control printing quality (supplemental Fig. S2).
A rabbit anti-GST antibody was used as the primary antibody and a goat anti-rabbit IgG labeled with DyLight-649 as the secondary antibody to visualize the bound GST fusion protein by far-Western. The printing buffer (red rectangle, Fig. 3A) and GST (green rectangle) were included in the OPAL slide as negative and positive controls, respectively. As shown in Fig. 3A and supplemental Fig. S3A–S3C, each variant produced a unique binding pattern on the OPAL array. For example, variant 6 (V6) showed a strong preference for an Asn (N) at the p + 2 position, suggesting that it belongs to the p + 2N class (Fig. 2A). The binding signals on an OPAL slide were subsequently quantified and the intensity of each spot was normalized against the average signal over the entire slide to derive a Z-score indicative of preference for a given amino acid residue at a specific position (Fig. 3B and supplemental Fig. S4A–S4F). This allowed for comparison of specificities for the different variants based on the corresponding Z scores on the OPAL. As shown in Fig. 3B, the variants V1, V5, V6, V8, and V11, which were selected by the p + 2N group of peptides, indeed strongly preferred an Asn residue at the p + 2 position. Exceptions were noted for a small number of variants (e.g. V10) that did not show p + 2N specificity, likely because of truncation in the BG loop (Fig. 2A). Interestingly, V19, isolated by the pYEEEL (L4) peptide, displayed a strong p + 2N selectivity. The presence of the EF1-Phe (to block the p + 3 pocket) and BG2-Leu (to plug the p + 4 pocket) makes V19 an ideal candidate for the p + 2N class.
Fig. 3.

Specificity of SH2 loop variants revealed by OPAL. A, A representative OPAL binding profile for the variant V6. Each sublibrary was printed in quadruplicate (marked by a square). Neutravidin was included as the negative control (marked by red rectangle). GST was employed as positive control (for GST fusion proteins used to probe the OPAL) and identified by a green rectangle. The Fyn-SH2 variant V6 showed p + 2N specificity. B, A heat map to show the preference of the 29 variants for residues at the p + 2 position. The Fyn, BRDG1 and PI3K-p85α SH2 domains were included as controls. The heat map was generated using the corresponding Z-scores on the OPAL.
The OPAL analysis indicates that the specificities of the bait peptide and the isolated variants are closely related (Fig. 4A). To facilitate analysis, we set 1.0 as the minimum Z score required for a variant to qualify for a specificity class. Based on this criterium, 82% (9/11) of the variants selected by peptides containing the pYxN motif could be assigned to the p + 2N class. In contrast, only 50% (9/18) of the variants captured by bait peptides without this motif could be assigned to the p + 2N class by OPAL. Similarly, the percentage of variants with the p + 3 specificity increased from 12% (2/17) to 50% when the bait peptides contained the pYxx[I/L/V] motif. Based on the corresponding Z scores for p + 2N and p + 3[I/V/L], we clustered the variants into four groups (I-IV). Group II variants, to which V6, V11, V8, and V1 belonged, exhibited greater specificity for p + 2N than p + 3[I/L/V]. These variants were all selected by peptides containing the pYxN motif. In contrast, Group IV variants, composed of V24, V14, V26 and V23 and selected by the pYxx[I/V/L] motif-containing peptides, had the opposite specificity preference to the previous group. Intriguingly, the Group I variants V27, V29, V21, and V20 (selected by bait peptides with no apparent motif) and the Fyn and p85α-N terminal (PI3K regulatory subunit) SH2 domains showed moderate specificity for both p + 2N and p + 3[I/L/V] (Fig. 4B). Group III variants, on the contrary, displayed a low propensity of binding to peptides with either the pYxN or pYxx[I/L/V] motif. Together, these data suggest that the EF/BG loops in the Fyn SH2 domain can evolve a wide spectrum of specificities that match grossly those of the bait peptides.
Fig. 4.

Bait peptides affect specificity of the isolated loop variants. A, Pie charts showing the number of variants belonging to the specificity group p + 2N or p + 3[I/L/V] based on the corresponding OPAL binding profiles. B, The variants in (A) are divided into four groups based on their Z scores.
Determination of Variant Specificity by Ligand Peptide Array
To complement the OPAL assay, we determined the specificity of the loop variants by peptide ligand array. The same phosphopeptides used in the SH2 library screening were individually synthesized, purified and printed onto a glass slide. The resulting peptide ligand array was probed for binding to different variants (supplemental Fig. S5A and S5B). The binding signals were quantified and normalized to generate the corresponding Z score in the same manner as for the OPAL data. We found that, on average, the larger the Z score, the higher the affinity for a variant-ligand peptide pair (supplemental Fig. S6). Consistent with the OPAL results, the SH2 variants formed distinct clusters with the bait peptides containing the pYxN or pYxx[I/L/V] motif in the heat maps generated from the corresponding Z scores (Fig. 5). For example, the majority of the Group II and some of the Group I variants (Fig. 4B), including V1–8, V11, V12, V15, V16, and V28, clustered with the pYxN peptides (Fig. 5, rectangle a). In contrast, the Group IV variants and some of the Group I variants (Fig. 4B), including V14, V20, V21, V23, V24, V26, V27, and V29, showed a stronger preference for the pYxx[I/L/V] peptides than the pYxN peptides (Fig. 5, rectangle b). Intriguingly, the Fyn and p85α SH2 domains bound to both types of ligands, suggesting that these naturally occurring SH2 domains have broad specificities.
Fig. 5.

Characterization of variant specificity by peptide ligand array. A heat map depicting the binding specificity of the loop variants (top) for the different peptides (right) included in the ligand array. Rectangle “a” identifies variants with strong binding for pY+2N peptides, in agreement with the OPAL data. Similarly, variants showing p + 3 [ILV] preference on the OPAL exhibited strong binding to peptides containing these residues at pY+3 position (rectangle “b”).
Specificity-determining Residues in the EF and BG Loops
The OPAL-derived Z scores allowed us to rank the 29 variants for proclivity to bind the pYxN, pYxxψ, or pYxxxψ motif. In turn, this enabled us to identify residues within the EF and BG loops that likely play an important role in conferring specificity (13). For the p + 2 class of SH2 domain, it has been shown that the peptide ligand must adopt a β-turn conformation to avoid steric clash with the bulky EF1 residue (14). Indeed, we found that the 7 variants with the strongest preference for the pYxN motif (Z>2.0) contained a bulky aromatic residue (Trp, Tyr or Phe) at the EF1 position (Fig. 6A). Furthermore, the same variants contained an aliphatic residue (Ile, Leu, or Val) at the BG2 position, which likely functions to plug the p + 4 binding pocket (Fig. 6A). Intriguingly, the next 11 variants that ranked immediately after the above group with a moderate p + 2N selectivity (with 1.0<Z<2.0) contained one or more charged (R, K, D, or E) or hydrophilic (T, S, N, or Q) residues within the EF loop. Curiously, the Fyn and p85α SH2 domains also belonged to this group. It is possible that the charged or hydrophilic residues in these variants facilitate the formation of hydrogen bonds with the sidechain of the p + 2 Asn residue or the backbone amide in the ligand peptide. Thus, the EF loop may encode p + 2N specificity using a variety of different mechanisms.
Fig. 6.

The specificity defining residues in the loop variants. Ranking of variants for p + 2N (A), p + 3[I/V/L] (B) and p + 4[L/F] (C) specificity based on the corresponding Z scores on OPAL.
In contrast to the identification of numerous variants with strong p + 2N selectivity, few variants showed a stronger preference for the pYxx[I/V/L] motif than the Fyn SH2 domain (Fig. 6B). This suggests that the wt Fyn SH2 domain is optimized for p + 3 binding. We noted that the 9 lowest ranked variants for the p + 3[I/L/V] specificity all contained an aromatic residue at the EF1 position, suggesting that these variants would favor the pYxN motif. Indeed, 6 of these variants (V6, V11, V8, V1, V5, and V7) were ranked with the strongest p + 2N selectivity. Although the Fyn SH2 domain is not known to possess p + 4 specificity, it showed a moderate preference for bait peptides containing the pYxxx[L/F] motif. Intriguingly, several variants, including V17 and V29, exhibited a greater preference for this motif than the Fyn SH2 domain. As shown below, the V17 and V29 variants bound to pYxxx[L/F] peptides with markedly greater affinities than the Fyn SH2 domain. Collectively, these data suggest that SH2 variants with distinct specificities can be evolved through combinatorial mutations in the EF and BG loops.
Identification of Variants with Distinct Specificities From the Parent SH2 Domain
Although the OPAL and ligand peptide arrays enabled us to identify variants with specificities that are different from that of the parent domain, it is necessary to confirm the predicted binding specificities/affinities in solution. To this end, we measured the dissociation constants of several SH2 variants for peptides containing the pYxN, pYxxψ or pYxxxψ motifs by fluorescence polarization with purified proteins and fluorescein-labeled peptides. We included the Fyn SH2 domain for comparison and the Grb2, Src and BRDG1 SH2 domains as representatives of the p + 2, p + 3, and p + 4 specificity classes, respectively. The change in affinity for a variant relative to the Fyn SH2 domain was used as a measure of specificity toward the same peptide ligand.
In line with the peptide array results (Figs. 4 and 5), we found that the Fyn SH2 domain was capable of binding to all three types of peptides with submicromolar to micromolar affinities (Table I, supplemental Fig. S6). However, the strongest affinity was observed for the I3 peptide, in agreement with the Fyn SH2 domain belonging to the p + 3 specificity class. Curiously, the Fyn SH2 domain bound much more tightly to the N2 than the ErbB2-pY1139 peptide despite both containing the pYxN motif. Similarly, it displayed a significantly greater affinity for the L4 than the EGFR-pY1172 peptide although both peptides contained a hydrophobic residue at the p + 4 position (Table I). This suggests that the negatively charged Glu residues in the N2, I3 and L4 peptides play a significant role in binding the Fyn SH2 domain. Nevertheless, because these three peptides differ only in the residue at the p + 2, p + 3 or p + 4 position, they are ideal for gauging the specificity changes for the variants.
Compared with the wt Fyn SH2 domain, the variant V8 displayed a 7-fold increase in affinity for the N2 peptide, but a 10-fold decreased affinity for the I3 peptide. This indicates that V8, which showed a strong preference for an Asn at the p + 2 position in the OPAL screen (Fig. 6A), has indeed acquired a dominant p + 2N specificity. Similarly, variant V14, which was predicted to possess a stronger p + 3[I/L/V] specificity than Fyn SH2 domain (Fig. 6B) indeed showed 6-fold increased affinity for the I3 peptide. Variant V17, which was predicted to prefer p + 4 over other specificity (Fig. 6C), displayed 2–8-fold increased affinity for the L4 (p + 4L) and EGFR-pY1172 (p + 4F) peptides and simultaneous 2.5–10-fold decreased affinity for the N2 and I3 peptides. This suggests that V17 has acquired a dominant p + 4 specificity compared with the parent Fyn SH2 domain. Because the BG loop in V17 is completely truncated, this would leave the p + 4 pocket open for peptide binding (Fig. 7A). In comparison, variant V29, which was predicted to have a greater preference for the p + 2N and p + 4[L/F] motifs than the Fyn SH2 domain (Fig. 6), indeed showed stronger binding to the corresponding peptides (Table I). The EF1 position in V29 is occupied by a Trp, which could be used to block the p + 3 binding pocket and thereby engendering p + 2N specificity for the variant. Intriguingly, the BG loop of V29 comprises a triad of bulky aromatic residues (W-Y-W) that would not fit the p + 4 binding pocket, thereby leaving the p + 4 pocket accessible for ligand binding. Therefore, depending on the peptide, V29 may deploy either the p + 2N or p + 4 binding mode for ligand recognition (Fig. 7B and 7C). Intriguingly, V10, which features a truncated BG loop, exhibited marked lower affinities for the N2, I3 and L4 peptides than the wt Fyn SH2 domain. Compared with V17 that is also characterized with a truncated BG loop, V10 contains a bulky Trp residue at the EF1 position, which would render it to favor p + 2N specificity rather than p + 4 even though the p + 4 binding pocket is open. Indeed, V10 displayed a greater affinity for the N2 than L4 peptide. In contrast, V17 preferred L4 to N2 because the variant contains a small Gly residue at the EF1 position (Table I).
Fig. 7.
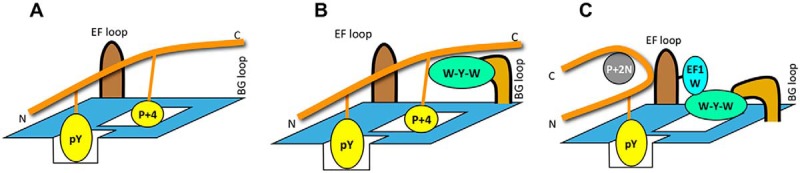
A cartoon model depicting the mechanism of p + 4 recognition by V17 (A) and p + 4 and p + 2N recognition by V29 (B, C). The peptide ligand is shown in orange with specificity residues shown. Specificity-determining residues in the EF and BG loops are shown.
DISCUSSION
Despite having the same protein fold, different antibodies can recognize different antigens. The remarkable ability of antibodies to recognize a vast array of antigens is dependent, in a large part, on the versatility of the six hypervariable loops within the variable domains of antibodies, commonly termed complementarity determining regions (CDRs) (20). These loops connect the β-strands of the antibody and are different from one antibody to another.
The principle of antibody-antigen recognition has been exploited in monobodies, antibody mimetics engineered from modular domains of much smaller size than a typical antibody. For example, the fibronectin type III domain (FN3), a molecular scaffold containing ∼100 residues, has been engineered to create novel target-binding variants, including those that can function as an SH2 domain inhibitor, by modifying the loops connecting the β-strands (21–24).
As shown in this work, the same principle of loop-mediated ligand recognition applies to the SH2 domain. Specifically, we showed that the EF and BG loops in the Fyn SH2 domain are highly adaptable and evolvable. The extreme versatility of the EF and BG loops afford them the ability to encode the broad spectrum of specificity found in naturally occurring SH2 domains. That the EF and BG loops of a single SH2 domain may be evolved to acquire specificities distinct from the parent domain is remarkable. Indeed, our comprehensive analysis of 29 loop variants selected by different bait peptides led to the identification of Fyn SH2 mutants that had switched specificity class from p + 3 to p + 2 or from p + 3 to p + 4. Furthermore, we demonstrated that the finer specificity and affinity of a variant is determined by the characteristics of the selection peptide, suggesting that the EF and BG loops not only control the major specificity of the SH2 domain but may also fine-tune specificity and affinity. Although our study was focused on the Fyn SH2 domain, it is likely that other SH2 domains are also capable of evolving variants with a wide spectrum of specificity through loop diversification. This unique property of the EF and BG loops provides an explanation for how different SH2 domains with the same globular structure may recognize different pY targets in cells.
SH2 variants with tailored specificity may provide a unique collection of tools with potential applications in research and cancer therapeutics. Naturally occurring SH2 domains such as the Fyn SH2 domain can bind to multiple pY targets in the cell, making it difficult to dissect the functions of specific SH2-pY pairs. To increase specificity, Yasui et al. developed pY-clamps by fusing a mutated SH2 domain to an FN3 loop variant that has evolved the ability to recognize the sequences flanking the pY site (14). Our work suggests that SH2 variants with tailored specificity for a given pY site may be evolved directly on the SH2 scaffold by EF/BG loop engineering. These variants would afford a class of pY sensors by which to dissect tyrosine kinase signaling in vivo. Because the specificity pocket and the pY-binding pocket are separate on an SH2 domain (8, 13, 25), we may also be able to create a panel of SH2 variants with desired specificity and affinity. It should be noted that an SH2 domain may also select a p + 1 residue (26, 27) and in certain cases, residues N-terminal to the pTyr (11) or C-terminal to the p + 4 site (28), which may not necessarily involve the EF or BG loop. Nevertheless, it can be envisioned that simultaneous in vitro evolution of the pTyr-binding pocket and the specificity pocket in an SH2 domain may yield a new class of SH2 variants with tailored affinity and specificity for research and potential therapeutic applications.
Supplementary Material
Footnotes
* This work was supported, in part, by grants (to SSCL or SS) from the Canadian Institute of Health Research (CIHR). SSCL held a Canada Research Chair in Functional Genomics and Cellular Proteomics.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SH2
- Src homology 2
- CDR
- complementarity-determining loops
- OPAL
- oriented peptide array library.
REFERENCES
- 1. Pawson T., and Scott J. D. (2005) Protein phosphorylation in signaling–50 years and counting. Trends Biochem. Sci. 30, 286–290 [DOI] [PubMed] [Google Scholar]
- 2. Sadowski I., Stone J. C., and Pawson T. (1986) A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol. Cell. Biol. 6, 4396–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang H., Li L., Wu C., Schibli D., Colwill K., Ma S., Li C., Roy P., Ho K., Songyang Z., Pawson T., Gao Y., and Li S.S. (2008) Defining the specificity space of the human SRC homology 2 domain. Mol. Cell. Proteomics 7, 768–784 [DOI] [PubMed] [Google Scholar]
- 4. Liu B. A., Jablonowski K., Raina M., Arcé M., Pawson T., and Nash P.D. (2006) The human and mouse complement of SH2 domain proteins establishing the boundaries of phosphotyrosine signaling. Mol. Cell 22, 851–868 [DOI] [PubMed] [Google Scholar]
- 5. Pawson T. (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 [DOI] [PubMed] [Google Scholar]
- 6. Scott J.D., and Pawson T. (2000) Cell communication: the inside story. Sci Am 282, 72–79 [DOI] [PubMed] [Google Scholar]
- 7. Wu P., Nielsen T. E., and Clausen M. H. (2015) FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 36, 422–439 [DOI] [PubMed] [Google Scholar]
- 8. Kaneko T., Huang H., Cao X., Li X., Li C., Voss C., Sidhu S.S., and Li S.S. (2012) Superbinder SH2 domains act as antagonists of cell signaling. Sci Signal 5, ra68. [DOI] [PubMed] [Google Scholar]
- 9. Waksman G., Kominos D., Robertson S.C., Pant N., Baltimore D., Birge R.B., Cowburn D., Hanafusa H., Mayer B.J., Overduin M., Resh M.D., Rios C.B., Silverman L., and Kuriyan J. (1992) Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature 358, 646–653 [DOI] [PubMed] [Google Scholar]
- 10. Waksman G., Shoelson S. E., Pant N., Cowburn D., and Kuriyan J. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72, 779–790 [DOI] [PubMed] [Google Scholar]
- 11. Hwang P. M., Li C., Morra M., Lillywhite J., Muhandiram D.R., Gertler F., Terhorst C., Kay L.E., Pawson T., Forman-Kay J.D., and Li S.C. (2002) A “three-pronged” binding mechanism for the SAP/SH2D1A SH2 domain: structural basis and relevance to the XLP syndrome. EMBO J. 21, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bradshaw J. M., Grucza R. A., Ladbury J. E., and Waksman G. (1998) Probing the “two-pronged plug two-holed socket” model for the mechanism of binding of the Src SH2 domain to phosphotyrosyl peptides: a thermodynamic study. Biochemistry 37, 9083–9090 [DOI] [PubMed] [Google Scholar]
- 13. Kaneko T., Huang H., Zhao B., Li L., Liu H., Voss C.K., Wu C., Schiller M.R., and Li S.S. (2010) Loops govern SH2 domain specificity by controlling access to binding pockets. Sci Signal 3(120):ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yasui N., Findlay G.M., Gish G.D., Hsiung M.S., Huang J., Tucholska M., Taylor L., Smith L., Boldridge W.C., Koide A., Pawson T., and Koide S. (2014) Directed network wiring identifies a key protein interaction in embryonic stem cell differentiation. Mol. Cell 54, 1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu B. A., Engelmann B. W., and Nash P. D. (2012) The language of SH2 domain interactions defines phosphotyrosine-mediated signal transduction. FEBS Letters 586, 2597–2605 [DOI] [PubMed] [Google Scholar]
- 16. Marengere L. E., Songyang Z., Gish G.D., Schaller M.D., Parsons J.T., Stern M.J., Cantley L.C., Pawson T. (1994) SH2 domain specificity and activity modified by a single residue. Nature 369, 502–505 [DOI] [PubMed] [Google Scholar]
- 17. Bajorath J., Harris L., and Novotny J. (1995) Conformational similarity and systematic displacement of complementarity determining region loops in high resolution antibody x-ray structures. J. Biol. Chem. 270, 22081–22084 [DOI] [PubMed] [Google Scholar]
- 18. Huang R., Fang P., and Kay B. K. (2012) Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods 58, 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodriguez M., Li S. S., Harper J. W., and Songyang Z. (2004) An oriented peptide array library (OPAL) strategy to study protein-protein interactions. J. Biol. Chem. 279, 8802–8807 [DOI] [PubMed] [Google Scholar]
- 20. Sela-Culang I., Kunik V., and Ofran Y. (2013) The structural basis of antibody-antigen recognition. Frontiers Immunol. 4, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koide S., Koide A., and Lipovsek D. (2012) Target-binding proteins based on the 10th human fibronectin type III domain ((1)(0)Fn3). Methods Enzymol. 503, 135–156 [DOI] [PubMed] [Google Scholar]
- 22. Koide A., Wojcik J., Gilbreth R. N., Hoey R. J., and Koide S. (2012) Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 415, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wojcik J., Hantschel O., Grebien F., Kaupe I., Bennett K.L., Barkinge J., Jones R.B., Koide A., Superti-Furga G., and Koide S. (2010) A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 17, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karatan E., Merguerian M., Han Z., Scholle M.D., Koide S., and Kay B.K. (2004) Molecular recognition properties of FN3 monobodies that bind the Src SH3 domain. Chem Biol 11, 835–844 [DOI] [PubMed] [Google Scholar]
- 25. Kaneko T., Sidhu S. S., and Li S. S. (2011) Evolving specificity from variability for protein interaction domains. Trends Biochem. Sci. 36, 183–190 [DOI] [PubMed] [Google Scholar]
- 26. Tinti M., Kiemer L., Costa S., Miller M.L., Sacco F., Olsen J.V., Carducci M., Paoluzi S., Langone F., Workman C.T., Blom N., Machida K., Thompson C.M., Schutkowski M., Brunak S., Mann M., Mayer B.J., Castagnoli L., and Cesareni G. (2013) The SH2 domain interaction landscape. Cell Rep 3, 1293–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Virdee S., Macmillan D., and Waksman G. (2010) Semisynthetic Src SH2 domains demonstrate altered phosphopeptide specificity induced by incorporation of unnatural lysine derivatives. Chem. Biol. 17, 274–284 [DOI] [PubMed] [Google Scholar]
- 28. Zadjali F., Pike A.C., Vesterlund M., Sun J., Wu C., Li S.S., Rönnstrand L., Knapp S., Bullock A.N., and Flores-Morales A. (2011) Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J. Biol. Chem. 286, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.