

Abstract
Covalent conjugations of the SUMO-1 moiety on a target protein play important roles in the regulation of cellular protein function. SUMO-conjugation of PML is a regulatory step for PML nuclear body (PML-NB) formation, and HIPK2 is SUMO-conjugated and recruited into the PML-NBs. Although HIPK2 mutations (R861W and N951I) were found in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) patients, little is known about the underlying mechanisms by which HIPK2 mutations are associated with the pathogenesis of leukemia. Here we show that HIPK2 mutants found in AML and MDS patients are defective in SUMO-interacting motif (SIM) function. Due to defective SIM function, the HIPK2 mutants were not modified with SUMO-1, and not recruited to the PML-NBs. However, the HIPK2 mutants can normally bind to and phosphorylate AML1b. Therefore, the HIPK2 mutants can sequestrate the AML1 complex out of the PML-NBs, resulting in the disruption of AML1-mediated activation of target genes for myeloid differentiation. In addition, the differentiation of K562 blast cells was impaired by the expression of the HIPK2 SIM-defective mutants. These results suggest that HIPK2 targeting into the PML-NBs via the SIMs is crucial for HIPK2-mediated induction of myeloid differentiation, and is associated with AML pathogenesis.
Keywords: SUMOylation, HIPK2, SUMO-interacting motif, PML-NBs, myeloid differentiation
Introduction
SUMOylation, the covalent modification of the small ubiquitin-like modifier (SUMO) moiety to a target protein, is an important post-translational modification which regulates diverse cellular functions and cancer pathogenesis [1]. SUMO is classified as a ubiquitin-like protein because the three dimensional structure of SUMO is similar to that of ubiquitin [2]. SUMOylation is highly analogous to ubiquitination, and hence is executed by the sequential activity of E1 activating, E2 conjugating, and SUMO E3 ligases. SUMO E3 ligases facilitate the transfer of SUMO from the E2 conjugating enzyme Ubc9 to the target protein [3]. Attachment of the SUMO moiety to the target protein requires a conserved SUMO acceptor site, ψKxE (ψ; hydrophobic amino acid, x; any amino acid), in which a hydrophobic residue is followed by lysine and an acidic amino acid [4]. However, SUMOylation can also occur on lysine residues through the SUMO interacting motif (SIM), independent of SUMOylation acceptor sequences [5,6]. SIMs consist of hydrophobic aliphatic residues, followed by acidic or polar amino acids [5-8]. The acidic amino acids flanking the core hydrophobic residues promote electrostatic SUMO-SIM interactions [9,10]. During SIM-mediated SUMOylation to the target protein, the SUMO-loaded E2 conjugating enzyme Ubc9 is recruited to the target protein through SUMO-SIM interaction. Recruited Ubc9 promotes transfer of the SUMO moiety to nearby lysine residues. SIM forms a β-strand that can bind to the hydrophobic groove generated by the β2-strand and α1-helix of SUMO, and a negative charge imposed by a stretch of neighboring acidic amino acids determines its specificity in binding to distinct SUMO paralogues [7].
The promyelocytic leukemia protein (PML) has been identified as a fusion protein with retinoic acid receptor α (RARα) as a result of a chromosomal translocation found in acute promyelocytic leukemia patients [11,12]. PML forms multi-protein complexes to build up nuclear dot structures, which are the so-called PML nuclear bodies (PML-NBs). PML-NBs consist of constitutive resident proteins such as Sp100, Daxx as well as PML, and additional proteins are temporarily recruited to the PML-NBs, depending on the physiological condition of cells [13,14]. PML-NBs are a dynamic sub-nuclear structure which are regulated by post-translational modifications such as SUMOylation, phosphorylation, ubiquitination, and acetylation [15]. These modifications direct the interaction of PML with various binding partners. SUMOylation is essential for recruitment of PML-NB components, turnover and retention of PML-NBs as well as maintaining the proper spherical structure and functional integrity of PML-NBs. All nuclear PML isoforms contain a SIM, with the exception of PML-VI, and PML-I and PML-IV have two additional serine and acidic amino acid motifs, corresponding to amino acids encoded by exon 8a [16]. Mutation of the SIM in PML-I and PML-IV dramatically affect the structure of PML-NBs and show a reduction in mean number, loss of hollowness, and increase in size [17]. The PML gene is rearranged in acute promyelocytic leukemia (APL), an M3 subtype of acute myeloid leukemia (AML) [18,19], and the AML1 gene is the most frequent target for chromosomal translocation in leukemia [20]. The AML1 protein interacts with PML, p300/CBP, and coactivators such as MOZ and homeodomain-interacting protein kinase 2 (HIPK2) to regulate differentiation of hematopoietic cells [21,22]. In a previous study, mutations of the HIPK2 gene were identified in the screening of mutations in AML (acute myeloid leukemia) and MDS (myelodysplastic syndrome) patients using denaturing high performance liquid chromatography [23].
HIPK2 was initially identified as a co-repressor of NK-class homeoproteins and a co-activator for the androgen receptor [24-26]. The name HIPK2 was coined from the characteristics of HIPK2 to interact with various homeoproteins [26]. HIPK2 regulates diverse cellular activities ranging from cell proliferation and differentiation to apoptosis and the DNA damage response [27-30]. HIPK2 phosphorylates a wide variety of target proteins under different signaling cues, which are dynamically regulated by diverse post-translational modifications such as phosphorylation, poly-ubiquitination, acetylation, and SUMOylation [31-34]. HIPK2 was the first nuclear protein kinase identified to be covalently modified with SUMO-1 [35], and contains SIM within its speckle retention sequence (SRS) domain [36,37]. HIPK2 associates with PML-NBs through the SIMs of both PML and HIPK2 [38,39]. PML was shown to be required for HIPK2 to induce p53 phosphorylation at Ser46 and transactivation of pro-apoptotic target genes in the PML-NBs [40,41].
Abnormal SUMOylation can lead to the development of a number of diseases, including cancer [1,42,43]. Here we show that HIPK2 mutants (R861W and N951I) found in AML and MDS are defective in SIM function, and hence these HIPK2 mutants are non-functional in PML-mediated p53 activation and AML1-mediated activation of target genes. Since HIPK2 mutants show a dominant-negative function against wild-type HIPK2 in recruitment to the PML-NBs, a single allelic HIPK2 mutation is sufficient to block wild-type HIPK2 expressed from a normal allele. Differentiation of myeloid precursor cells is alleviated by the expression of HIPK2 SIM-defective mutants.
Materials and methods
Cell culture and differentiation assay
HEK293T, RKO, HeLa, H1299, and U2OS cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. For immunoblot analysis, cells were transfected with the lipofectamine 3000 reagent in six-well plates. The chronic myelogenous leukemia cell line K562 cells were grown in RPMI medium 1640 supplemented with 10% FBS, antibiotics, and L-glutamine. Differentiation of K562 cells was induced by 24-hr treatment of PMA. Cells were cytocentrifuged, and cell morphology was evaluated by Wright-Giemsa staining.
Plasmid construction and site-directed mutagenesis
Expression plasmids for HA-HIPK2, Myc-HIPK2, GFP-HIPK2, GFP-HIPK2 K221R, HIPK2 SIM-VK, and HIPK2 deletion mutants were previously described [26,34,35,44]. Flag-PML isoforms (PML-I and PML-VI) were a generous gift from Keith Leppard (Warwick University, Warwick, UK). Flag-AML1b, HA-MOZ, and pMPO-luciferase reporter plasmids were a kind gift from Issay Kitabayashi (National Cancer Center Research Institute, Tokyo, Japan). To generate the mouse HIPK2 deletion mutants, PCR products amplified with specific primers were subcloned into pEntr-3C, and a Myc-tagged or GFP-fused HIPK2 deletion mutant was generated using Gateway Technology (Invitrogen). HIPK2 point mutants (HIPK2 R861W and HIPK2 N951I) were generated using the QuikChange Mutagenesis Kit (Stratagene). Mutations were verified by DNA sequencing. In order to construct pCFP-HIPK2, the 3.9 kb DNA fragment encoding HIPK2 was amplified by PCR, and inserted into the EcoRI and SalI sites of pECFP-C1 (Clontech).
Western blot analysis
Western blotting was performed as previously described [45]. Briefly, cells were lysed with RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 50 mM Tris-HCl (pH 8.0), 0.1% SDS, protease inhibitor). Whole cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were immunoblotted with various antibodies followed by detection with ECL Western detection reagents (Intron).
In vitro SUMOylation assays
In vitro SUMOylation assays were performed as described [34]. GST-SUMO-1 and GST-SAE1/2 proteins were affinity purified as described previously [8]. His-Ubc9 was purified from cell lysates by applying the mixture to Ni-NTA resin in buffer containing 20 mM Tris-HCl (pH 8.0), 350 mM NaCl, 1 mM β-mercaptoethanol, and 20 mM imidazole, and eluted from the resin in the same buffer except 400 mM imidazole. In brief, in vitro synthesized Myc-HIPK2-(800-10-49) were mixed with 500 ng of purified E1 (GST-SAE1/SAE2), 400 ng of His-Ubc9, and 2 μg of GST-SUMO-1 (GG), and incubated at 37°C for 60 min. Reactions were terminated by the addition of SDS sample buffer, and products were visualized by Western blotting with an anti-Myc antibody (Roche Molecular Biochemicals).
GST pull-down assay
In vitro pull-down assays were performed as described [38]. Briefly, assays were performed by incubating in vitro translated wild-type Myc-HIPK2 or Myc-HIPK2 mutants (R861W and N951I) with equal amounts of GST or GST-SUMO-1 fusion proteins immobilized on glutathione-Sepharose beads. The mixture was incubated for 2 hr with gentle agitation on a slowly rotating wheel at 4°C, and washed 3 times. Bound proteins were eluted by the addition of SDS sample buffer, and separated by 8% SDS-polyacrylamide gel electrophoresis, followed by Western blotting using anti-Myc antibody.
Co-immunoprecipitation analysis
Co-immunoprecipitation was performed after the lysis of 2×107 cells in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 50 mM Tris-HCl (pH 8.0), 0.1% SDS, protease inhibitor). After incubation on ice for 10 min and centrifugation at 4°C for 10 min, equal volumes of protein were diluted with lysis buffer lacking NaCl and incubated with antibody and protein A/G-Sepharose beads overnight at 4°C on a rotating wheel. The beads were washed three times with lysis buffer. The whole cell lysate and immunoprecipitates were separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were immunoblotted with various antibodies followed by detection with ECL Western detection reagents (Intron).
Immunocytochemistry
Immunocytochemistry for U2OS cells was performed as described [38]. Briefly, U2OS cells were grown on coverslips and transfected with HA-PML expression plasmid together with wild-type GFP-HIPK2 or GFP-HIPK2 mutant plasmid (HIPK2 R861W and HIPK2 N951I). Twenty-four hours after transfection, cells were fixed with 100% methanol for 5 min at -20°C and blocked with phosphate-buffered saline containing 1% bovine serum albumin. Fluorescence microscopy was performed with a Zeiss LSM-700 microscope. GFP-HIPK2 was detected using an excitation wavelength of 488 nm. HA-PML was detected using a rhodamine-conjugated secondary antibody against a mouse monoclonal anti-HA antibody. Acquired images were processed with Adobe Photoshop.
Luciferase assay
To analyze HIPK2-mediated induction of apoptosis at the transcriptional level, H1299 cells seeded into 12-well plates were transfected with the p53 expression plasmid, p53 response element-Luc reporter plasmid, and either the wild-type HIPK2 or a HIPK2 mutant (catalytically inactive K221R, HIPK2 R861W, or HIPK2 N951I) plasmid. To determine HIPK2-mediated activation of the MPO promoter, H1299 cells were transfected with the pMPO-luc reporter plasmid together with expression plasmids encoding AML1b, MOZ, and increasing amounts of wild-type HIPK2 or HIPK2 SIM mutants (HIPK2 R861W and N951I). Forty-eight hours after transfection, luciferase activity was measured using the Luciferase Reporter Assay System (Promega) and a Genios luminometer (TECAN, Austria). Transfection efficiency was normalized against β-galactosidase expression. Each experiment was repeated at least three times.
Colony formation assay
RKO cells (2×105 cells/dish) were transfected with either GFP-HIPK2 or GFP-HIPK2 mutant expression plasmid (catalytically inactive K221R mutant, HIPK2 R861W and HIPK2 N951I mutants). Cells were selected by 400 μg/ml G418 administration for two weeks. G418-resistant colonies were stained with Coomassie Brilliant Blue. Experiments were repeated at least three times.
Results
Characterization of HIPK2 mutants found in AML and MDS patients
HIPK2 mutations were identified in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) patients [23]. The sites of these HIPK2 mutations (Arg868 and Asn958) are well conserved in mouse HIPK2 (Arg861 and Asn951) and are located within the speckle retention sequence (SRS) domain, which is also known to contain the SUMO-interaction motif (SIM) (Figure 1A). HIPK2 SIM plays a crucial role in the SUMO modification of lysine residues via interaction with SUMO-Ubc9 conjugates [36]. To test whether these mutants affect the SIM functions of HIPK2, we mutated Arg861 to Trp (R861W) and Asn951 to Ile (N951I) on mouse HIPK2. GST pull-down and co-immunoprecipitation analysis showed that HIPK2 R861W and N951I mutants do not interact with SUMO-1 in vitro and in vivo, in contrast to wild-type HIPK2 (Figure 1B and 1C). Moreover, we verified that both HIPK2 R861W and N951I mutants are not conjugated with SUMO-1, whereas wild-type HIPK2-(800-1049) is conjugated to SUMO-1 in vivo and in vitro (Figure 1D and 1E). These results indicate that Arg861 and Asn951 of HIPK2 contribute to both noncovalent interactions with SUMO-1 and covalent modification of HIPK2 with SUMO-1. Next, we examined the colocalization of these HIPK2 mutants with PML-I or PML-IV by indirect immunocytochemistry of cells expressing PML-I, PML-IV, and each HIPK2 mutant (Figure 1F). The R861W and N951I mutants did not colocalize with PML-I and PML-IV, whereas wild-type HIPK2 localized to the PML-NBs containing PML-I or PML-IV (Figure 1F). Next, we performed co-immunoprecipitation assays with HA-PML-IV and Myc-HIPK2 (wild type, R861W or N951I mutant) in the presence or absence of SUMO-1. Wild-type HIPK2 interacted with SUMO-conjugated PML-IV (Figure 1G, lane 2), but the R861W and N951I mutants did not (Figure 1G, lanes 4 and 6). Taken together, the substitutions of arginine to tryptophan at amino acid 861 and of asparagine to isoleucine at amino acid 951 of HIPK2 impaired colocalization with PML-NBs and interaction with PML-IV. In addition, HIPK2 R861W and N951I mutants are defective in SIM function, and are thus functionally equivalent to the HIPK2 SIM-defective mutant (HIPK2 SIM-VK) where VSVI, the core sequence of SIM, was substituted with KSAK at amino acids 878-881 [38].
Figure 1.
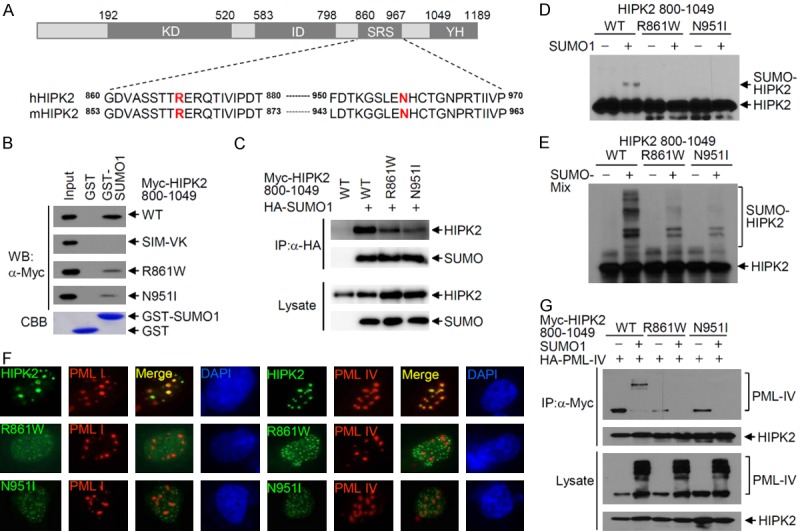
Characterization of HIPK2 mutants found in acute myeloid leukemia and myelodysplastic syndrome patients. A. Schematic of HIPK2 and position of HIPK2 mutations. KD, protein kinase domain; ID, interaction domain with homeodomain proteins; SRS, speckle retention sequence; YH, tyrosine- and histidine-rich domain. Sequence alignment of human HIPK2 and mouse HIPK2 shows conservation (hHIPK2 Arg869 and mHIPK2 Arg861; hHIPK2 Asn958 and mHIPK2 Asn951). B. GST-pull down assay showing physical interaction of HIPK2-(800-1049) with SUMO-1. In vitro translated Myc-HIPK2-(800-1049), or the R861W or N951I mutant was incubated with equal amounts of either GST protein or GST-SUMO-1. Bound proteins were eluted and resolved by 8% SDS-PAGE, followed by Western blotting using anti-Myc antibody. Affinity purified GST or GST-SUMO-1 used in this assay are shown in the lower panel. Input shows 10% of the in vitro translated Myc-HIPK2 used in the binding reaction. C. Co-immunoprecipitation of Myc-HIPK2-(800-1049) with SUMO-1. An expression plasmid coding for either Myc-HIPK2-(800-1049), the R861W or N951I mutant was transfected into HeLa cells, together with an HA-SUMO-1 expression plasmid. Transfected cells were lysed and immunoprecipitated with anti-HA antibody, followed by Western blotting using anti-Myc antibody. D. Mutation of HIPK2 at Arg861 or Asn951 abolishes SUMO-1 conjugation to HIPK2. An expression plasmid encoding either Myc-HIPK2-(800-1049), R861W or N951I was transfected into HeLa cells with or without the SUMO-1 expression plasmid. SUMO-1 conjugation to HIPK2 was determined by Western blotting using anti-Myc antibody. E. In vitro translated Myc-HIPK2-(800-1049), the R861W or N951I mutant was subjected to in vitro SUMOylation assays. Reactions were terminated by adding sample buffer, followed by Western blotting using anti-Myc antibody. F. U2OS cells were transfected with GFP-HIPK2, the R861W or N951I mutant in combination with nuclear HA-PML-I or HA-PML-IV. Cells were fixed and immunostained 24 hrs after transfection. Images were obtained with a confocal fluorescent microscope at an excitation wavelength of 488 nm and 543 nm, respectively. G. Co-immunoprecipitation of Myc-HIPK2-(800-1049) with SUMOylated PML-IV. Either Myc-HIPK2-(800-1049), the R861W or N951I mutant expression plasmid was transfected into HeLa cells with the HA-PML-IV expression plasmid in the presence or absence of the GFP-SUMO-1 expression plasmid. Total cell lysates were immunoprecipitated with anti-Myc antibody, followed by Western blotting using anti-HA antibody.
HIPK2 R861W and N951I mutants do not phosphorylate p53 at Ser46
The SIM function of HIPK2 is important in HIPK2-induced p53 phosphorylation at Ser46 and p53-mediated apoptosis [38,39]. Since the R861W and N951I mutants of HIPK2 lose SIM function (Figure 1B-E), we examined p53 Ser46 phosphorylation upon co-expression of wild-type HIPK2, or the R861W or N951I mutant. Western blot analysis revealed that phosphorylation of p53 at Ser46 was markedly reduced by expression of the R861W and N951I mutants of HIPK2, compared to strong phosphorylation by wild-type HIPK2. The catalytically inactive mutant (K221R) of HIPK2 was utilized as a positive control (Figure 2A). Moreover, these mutants did not induce p53-mediated transcription of the pro-apoptotic PUMA gene (Figure 2B). Next, we explored whether the R861W and N951I mutants affect colony formation of cancer cell and induction of apoptosis. Wild-type HIPK2 inhibited the colony formation of RKO cells, but the R861W and N951I mutants did not (Figure 2C). These results show that the R861W and N951I mutants of HIPK2 functionally mimic the HIPK2 SIM-defective mutant in HIPK2-mediated induction of apoptosis through p53 phosphorylation.
Figure 2.
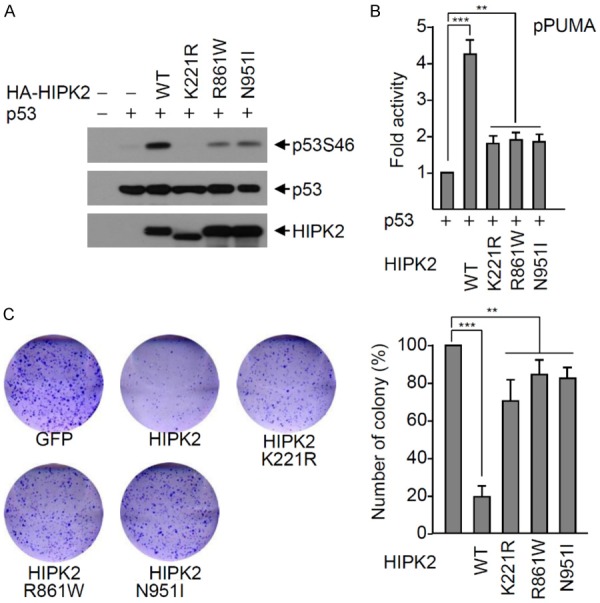
The HIPK2 R861W and N951I mutants do not phosphorylate p53 at Ser46. A. H1299 cells were transfected with p53 and either wild-type HA-HIPK2 or a HIPK2 mutant (catalytically inactive K221R mutant, R861W or N951I) expression plasmid. p53 Ser46 phosphorylation was determined with Western blotting using a p53Ser46 phospho-specific antibody. Expression levels of p53 and HIPK2 were determined by Western blotting using anti-p53 and anti-HA antibody, respectively. B. The p53 expression plasmid and each reporter plasmid (PUMA gene promoter) was transfected into H1299 cells with either wild-type HIPK2 or an HIPK2 mutant (K221R, R861W or N951I mutant). Twenty-four hours after transfection, luciferase activities were measured using the Luciferase Assay System (Promega). Transfection efficiency was normalized against β-galactosidase expression. Experiments were repeated at least three times. The data were statistically analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test (***P < 0.001, **P < 0.01). Bars represent the mean ± SD of three independent experiments. C. The HIPK2 R861W and N951I mutants are defective in p53-mediated induction of apoptosis. Wild-type GFP-HIPK2 or a GFP-HIPK2 mutant was transfected into RKO cells, and transfected cells were selected with G418 for two weeks. Colonies were visualized by staining with crystal violet and counted. The number of colonies was determined as the mean ± SD of three independent experiments, and presented as the relative percentage compared to the colony numbers of the negative control. The data were statistically analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test (***P < 0.001, **P < 0.01). A representative example is shown.
Dominant-negative functions of HIPK2 R861W and N951I mutants
HIPK2 was shown to be able to homo-dimerize in vitro and in vivo independent of its catalytic activity [46]. HIPK2 mutants were found in an allele of a chromosome from AML and MDS patients [23]. Thus wild-type HIPK2 was expressed from a normal allele and either HIPK2 R861W or N951I was expressed from a mutant allele, and thereby wild-type and mutant HIPK2 were expressed simultaneously. Therefore, we hypothesized that the HIPK2 R861W and N951I mutants might act in a dominant-negative manner against wild-type HIPK2. To test this idea, the effects of the HIPK2 SIM mutant on the colocalization of wild-type HIPK2 with PML-IV was explored with indirect immunocytochemistry. U2OS cells were transfected with expression plasmids encoding GFP-HIPK2 and HA-PML-IV together with either wild-type CFP-HIPK2 or the CFP-HIPK2 SIM-VK mutant. Immunostaining of transfected cells indicated that CFP-HIPK2 colocalized with GFP-HIPK2 and PML-IV, while the CFP-HIPK2 SIM-VK mutant did not colocalize with PML-IV and the localization of wild-type GFP-HIPK2 was shifted to the dot structures of the CFP-HIPK2 SIM-VK mutant. These results suggested that the HIPK2 SIM-VK mutant inhibited colocalization of wild-type HIPK2 with PML-IV, and thereby the HIPK2 SIM-VK mutant has dominant-negative functions against the colocalization of wild-type HIPK2 with PML-IV (Figure 3A). The same experiments were repeated with HIPK2 K221R, a catalytically inactive mutant, and the R861W and N951I mutants. Co-expression of the HIPK2 K221R mutant did not disrupt colocalization of wild-type GFP-HIPK2 with PML-IV, while both Myc-HIPK2 SIM-VK, R861W and N951I mutants forced relocalization of wild-type GFP-HIPK2 to small dot structures, so as not to colocalize with PML-IV (Figure 3B). These results suggest that HIPK2 R861W and N951I mutants as well as the HIPK2 SIM-VK mutant have dominant-negative effects on the partitioning of wild-type HIPK2 into the PML-NBs.
Figure 3.
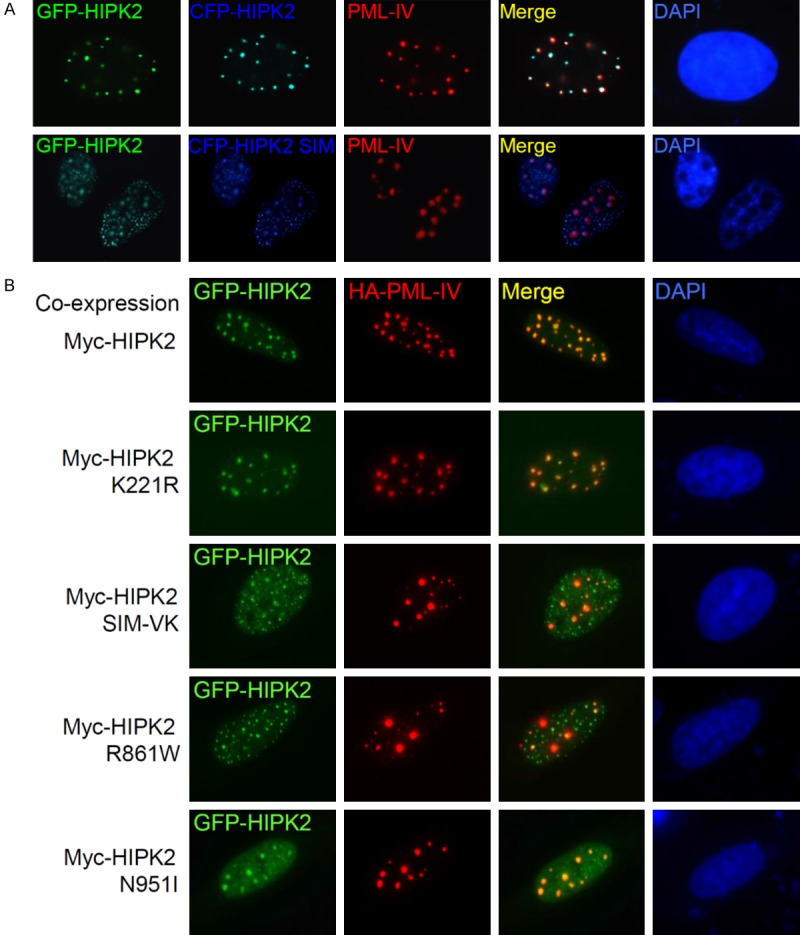
HIPK2 SIM mutants have dominant-negative function. A. U2OS cells were transfected with GFP-HIPK2, CFP-HIPK2 or a SIM-VK mutant and nuclear HA-PML-IV. Cells were fixed and immunostained 24 hrs after transfection. B. The HIPK2 R861W and N951I mutants have dominant-negative function. U2OS cells were transfected with GFP-HIPK2, Myc-HIPK2 or various mutants and nuclear HA-PML-IV. Cells were fixed and immunostained 24 hrs after transfection. Images were obtained with a confocal fluorescent microscope at an excitation wavelength of 488 nm and 543 nm, respectively.
Effects of HIPK2 mutants on AML1-mediated transcription
HIPK2 was reported to induce AML1b-dependent transcriptional activity of the myeloperoxidase (MPO) promoter through AML1b phosphorylation [23]. To know whether HIPK2 SIM-VK, R861W and N951I mutants affect the AML1b-dependent transcriptional activity, the AML1b, MOZ expression plasmids and MPO reporter plasmid were transfected into 293T cells with either wild-type HIPK2 or the HIPK2 mutants. Activation of AML1b-mediated transcription was impaired upon expression of the HIPK2 SIM-VK, R861W and N951I mutants, as measured by transcriptional activity of a natural promoter of the MPO gene. Transcriptional activity was inversely correlated to the expression levels of HIPK2 SIM-VK, R861W and N951I mutants (Figure 4A). These results suggest that HIPK2 SIM-defective mutants, including the R861W and N951I mutants, act in a dominant-negative manner against AML1-mediated transactivation of the MPO promoter. Next, to explore the repression mechanisms of AML1b-dependent transcriptional activity by HIPK2 mutants, we determined the phosphorylation of AML1b by the HIPK2 mutants, and also analyzed the interaction of AML1b with either HIPK2 or the HIPK2 mutants. Western blotting indicated that AML1b phosphorylation was not affected by the HIPK2 mutants (Figure 4B). In addition, co-IP analysis revealed that the interaction affinity of HIPK2 SIM-VK mutant, R861W and N951I mutants with AML1b were not affected (Figure 4C). These results indicate that the SIM mutations in HIPK2 do not affect AML1b phosphorylation and interaction between AML1b and HIPK2 (Figure 4B and 4C). Moreover, to characterize the colocalization between AML1b and HIPK2 SIM mutants, we examined cells expressing various GFP-HIPK2 mutants and HA-AML1b. Consistent with the results in Figure 4B and 4C, an indirect immunofluorescence assay revealed that both wild-type HIPK2 and HIPK2 SIM mutants including HIPK2 R861W and N951I mutants were colocalized with AML1b (Figure 4D). These results suggested that SIM mutations of HIPK2 do not disrupt the interaction and colocalization with AML1b.
Figure 4.
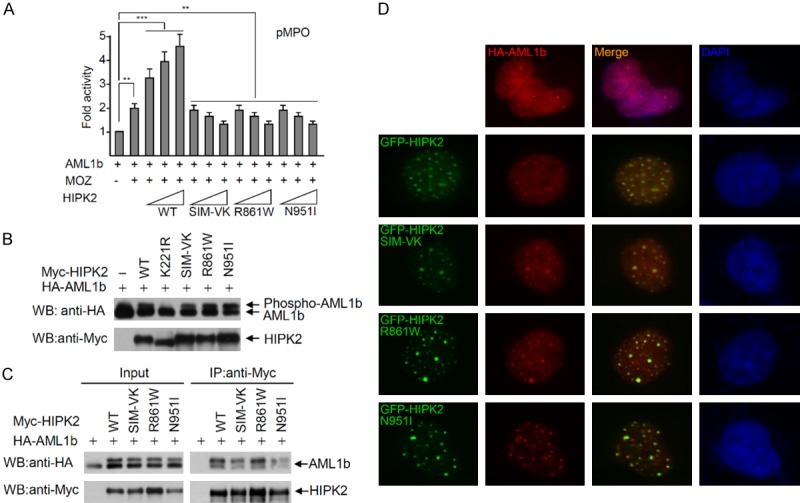
The SIM mutants of HIPK2 do not affect the phosphorylation and interaction with AML1b. A. The HIPK2 R861W and N951I mutants do not activate AML1b-dependent transcriptional activity. The AML1b, MOZ expression plasmid and MPO-reporter plasmid were transfected into 293T cells with increasing amounts of wild-type HIPK2 or a HIPK2 mutant (K221R, SIM-VK, R861W or N951I mutant). Twenty-four hours after transfection, luciferase activities were measured using the Luciferase Assay System (Promega). Transfection efficiency was normalized against β-galactosidase expression. Experiments were repeated at least three times. Error bars indicate the standard error of the means of three independent experiments. The data were statistically analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test (***P < 0.001, **P < 0.01). B. 293T cells were transfected with AML1b and either wild-type HA-HIPK2 or a HIPK2 mutant (catalytically inactive K221R mutant, SIM-VK, R861W or N951I) expression plasmid. AML1b phosphorylation was determined with Western blotting using anti-HA antibody. HIPK2 expression levels were determined by Western blotting using anti-Myc antibody. C. Co-immunoprecipitation of Myc-HIPK2 with AML1b. Either Myc-HIPK2, SIM-VK, R861W or N951I mutant expression plasmid was transfected into 293T cells with the HA-AML1b expression plasmid. Total cell lysates were immunoprecipitated with anti-Myc antibody, followed by Western blotting using anti-HA antibody. D. The SIM mutants of HIPK2 co-localize with AML1b. U2OS cells were transfected with GFP-HIPK2, or various mutants and AML1b. Cells were fixed and immunostained 24 hrs after transfection. Images were obtained with a confocal fluorescent microscope at an excitation wavelength of 488 nm and 543 nm, respectively.
The PML-I isoform specifically interacts with AML1b and facilitates functional cooperation between AML1b and p300 in transcriptional activation. Moreover, PML-I can recruit both AML1b and its cofactor p300 to the PML-NBs [21]. Since the HIPK2 SIM mutants did not colocalize with PML-I (Figure 1F), we hypothesized that the HIPK2 SIM mutations could interfere with colocalization between AML1b and PML-I. To test this idea, we examined the colocalization of the HIPK2 SIM mutant with AML1b in the presence or absence of PML-I. Immunostaining of cells indicated that wild-type HIPK2 and the SIM mutant colocalized with HA-AML1b irrespective of PML-I expression (Figure 5A). In addition, we examined the effects of HIPK2 SIM mutants including the HIPK2 R861W and N951I mutants on the colocalization of AML1b with PML-I. U2OS cells were transfected with expression plasmids encoding HA-AML1b and Flag-PML-I together with either Myc-HIPK2 or Myc-HIPK2 mutants (SIM-VK, R861W or N951I). Immunostaining of transfected cells showed that HA-AML1b colocalized with Flag-PML-I when wild-type HIPK2 was co-expressed, while HA-AML1b did not colocalize to the PML-NBs when HIPK2 SIM mutants were co-expressed (Figure 5B, arrows in zoom images). These results suggested that HIPK2 SIM mutants could sequestrate AML1b into PML-independent dot structures of HIPK2, and thereby AML1b is not localized to the PML-NBs in the presence of HIPK2 SIM mutants including the HIPK2 R861W and N951I mutants. Taken together, the HIPK2 R861W and N951I mutants disrupt AML1b-dependent transcription of target genes, which require recruitment of AML1b and its cofactors into the PML-NBs.
Figure 5.
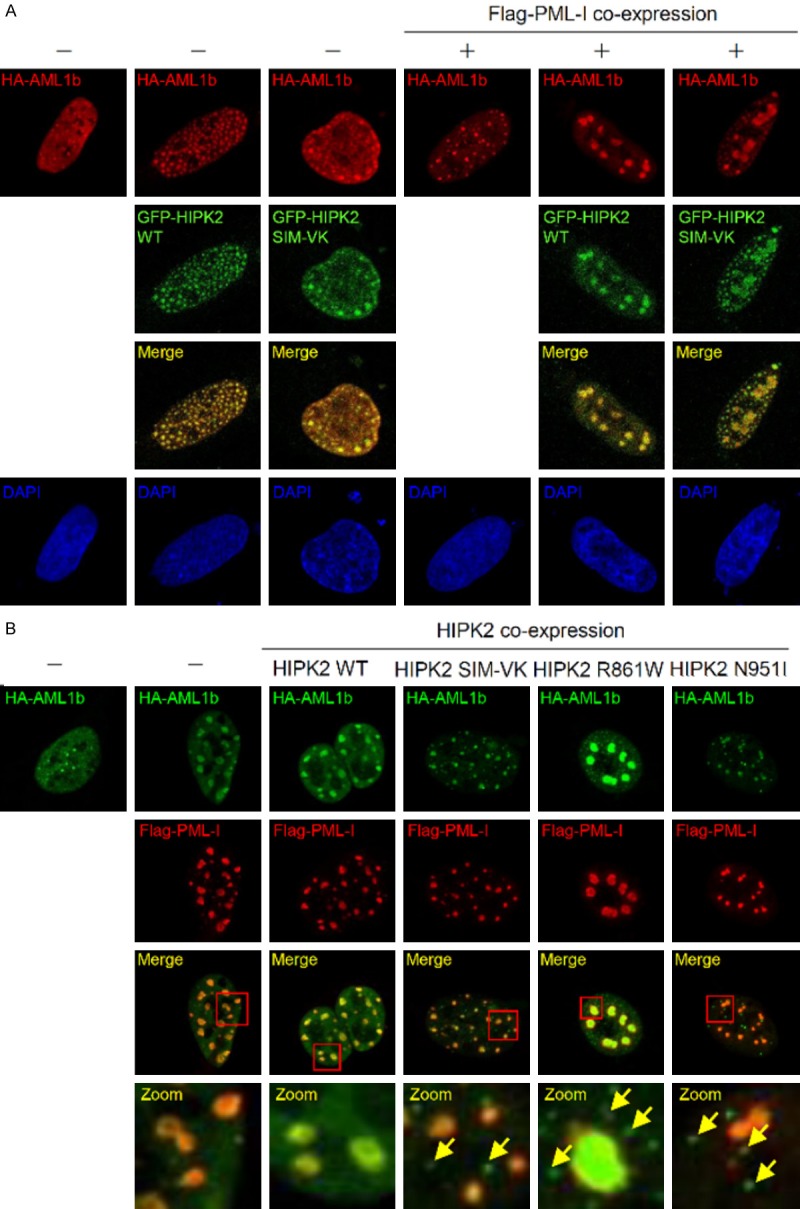
HIPK2 SIM mutants impair co-localization between AML1b and PML-I. A. U2OS cells were transfected with HA-AML1b in the presence or absence of Flag-PML-I in combination with GFP-HIPK2 or GFP-HIPK2 SIM mutant. Cells were fixed and immunostained 24 hrs after transfection. B. U2OS cells were transfected with HA-AML1b and Flag-PML-I in combination with Myc-HIPK2 or Myc-HIPK2 mutants (HIPK2 SIM-VK, R861W or N951I). Cells were fixed and immunostained 24 hrs after transfection. Zoom image is 10 times magnification of inset. Arrows indicate the AML1b-containing puncta independent of PML-I. Images were obtained with a confocal fluorescent microscope at an excitation wavelength of 488 nm and 543 nm, respectively.
HIPK2 function on myeloid blast cell differentiation
AML1b was reported to be a nuclear matrix-associated transcription factor, and essential for hematopoiesis [47]. To test whether the HIPK2 SIM mutation affects the differentiation of myeloid precursor cells, differentiation of K562 cells (chronic myelogenous leukemia cell line) were induced by the administration of PMA, and differentiation was determined by staining the cells with Wright-Giemsa. Differentiation of K562 was observed 1 day after cell culture with induction medium containing PMA. Expression of HIPK2 SIM mutants including the R861W and N951I HIPK2 mutants impaired differentiation of the K562 cells, while wild-type HIPK2 did not affect differentiation (Figure 6A and 6B). These results showed that the recruitment of HIPK2 into PML-NBs is very important for AML1b-mediated transcriptional activation and the differentiation of K562 cells, and thereby HIPK2-dependent recruitment of AML1b into the PML-I NBs is required for the differentiation of hematopoietic precursor cells.
Figure 6.
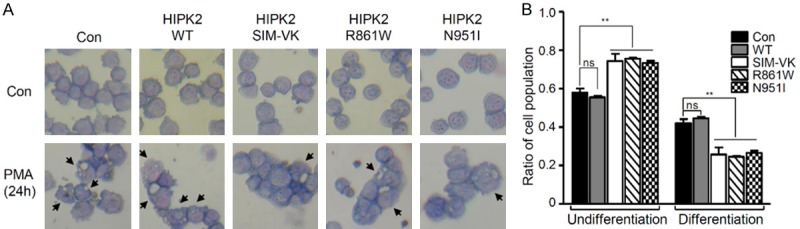
HIPK2 SIM mutants impair AML1b-dependent differentiation of K562 cells. A. Morphological features of K562 cells in which differentiation was induced by PMA treatment for 24 hrs. K562 cells were transfected with GFP-HIPK2, or GFP-HIPK2 mutant (HIPK2 SIM-VK mutant, R861W or N951I). Cytospins were stained with Wright-Giemsa. Original amplification, ×400. Arrows indicate the differentiated K562 cells. B. The number of differentiated or undifferentiated K562 cells were counted, and are shown on the graph. The data were statistically analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test (**P < 0.01). Data are presented as the mean ± SEM.
Discussion
Post-translational modifications of HIPK2 such as phosphorylation, acetylation, poly-ubiquitination, and SUMOylation, are crucial for HIPK2 stability, catalytic activities and selection of binding partners [31-33]. SUMO-modification of HIPK2 plays multiple roles in the regulation of HIPK2 stability and partitioning to specific sub-cellular structures [38,39,48,49]. SUMO-modification of a target protein occurs at a SUMOylation acceptor site such as ψKxE. HIPK2 Lys25 is a major SUMOylation site which matches well with the SUMOylation acceptor site consensus sequence. However, the C-terminus of HIPK2 is also SUMOylated by a different molecular mechanism in which Ubc9-loaded SUMO interacted with SIM [36], and the SUMO moiety is transferred to the nearby lysine residues by association with the E2 conjugating enzyme Ubc9. SIM-mediated SUMOylation of HIPK2 at lysine residues near SIM play a role in HIPK2 targeting into PML-NBs [38,39]. N-terminal SUMOylation of HIPK2 at Lys25 was associated with stability and selection of binding partners, but not partitioning to the PML-NBs which require C-terminal SUMOylation at a lysine nearby the HIPK2 SIM [34,48,49].
HIPK2 mutants with Arg868 substitution to Trp and Asn958 to Ile were found in AML and MDS patients [23]. In this study, we demonstrated that mouse HIPK2 mutations at Arg861 and Asn951, which are equivalent to human Arg868 and Asn958, disrupted HIPK2 SIM function, and thereby the HIPK2 C-terminus was not modified with SUMO-1 (Figure 1). As a result, HIPK2 was not recruited to the PML-NBs, which is crucial for the p53-mediated DNA damage response (Figure 2) and AML1b-dependent differentiation of hematopoietic cell lineage (Figure 6). Mouse HIPK2 SIM (VSVITISSDTDEEEE) spans amino acids 878 to 892, and is located at the center of the SRS domain (aa 860-967). The SRS domain was identified by serial deletion analysis of N- or C-terminal HIPK2, and indicates the minimal motif required for HIPK2 to form the nuclear dot structure which colocalizes with the PML-NBs [35]. The HIPK2 mutations found in AML and MDS patients, Arg868 and Asn951, are located near the N- and C-terminal end points of the HIPK2 SRS domain, respectively. Previously, we demonstrated that the HIPK2 K966R mutant also shows defective SIM function [38]. In addition, serial deletion analysis of Pc2 SIM and mutational analysis of the amino acids surrounding the Pc2 SIM indicated that several conserved amino acids surrounding the Pc2 SIM are essential in SUMO binding [50]. Taken together, it is plausible that HIPK2 SRS domain may be a minimal motif to maintain the three dimensional structure of SIM to associate with the hydrophobic groove generated by the SUMO β2-strand and α1-helix. The HIPK2 mutations found in AML and MDS patients may disrupt the tertiary conformation of SIM and the integrity of the SRS domain to associate with SUMO. AML and MDS are associated with the AML1 protein, and formation of the AML1 complex including HIPK2 and p300/CBP in the PML-NBs plays a role in AML1-mediated transcriptional activation of target genes for cell differentiation [21,22]. The association of HIPK2 with the PML-NBs requires both SIM function and C-terminal SUMOylation. SUMOylated HIPK2 recognizes the SIM of PML-I and PML-IV, and HIPK2 SIM binds to SUMOylated PML. Therefore, SIM-defective HIPK2 mutants such as R861W and N951I mutants, cannot bind to SUMOylated PML, and the PML SIM cannot recognize these HIPK2 mutants due to the lack of SUMOylation at the HIPK2 C-terminus, resulting in a loss of association of the HIPK2 mutants with the PML-NBs (Figures 3 and 5). On the other hand, HIPK2 SUMOylation is not required for binding to and phosphorylating AML1 (Figure 4). Therefore, HIPK2 SIM mutants sequestrate AML1 out of PML-NBs, and hence HIPK2 SIM mutants act in a dominant negative manner against wild-type HIPK2 (Figures 3 and 5).
We demonstrated an additional role of HIPK2 and PML-NBs during the differentiation of myeloid precursor cells. AML1b associates with the MOZ coactivator and PML-I to induce transcriptional activation of AML1b target genes [21]. During normal differentiation of myeloid precursor cells, HIPK2 participated in the AML1-mediated transcriptional activation of downstream target genes by phosphorylation of both AML1b and MOZ [22]. PML activated transcription of AML1 target genes by protecting HIPK2 and p300 from proteasomal degradation [51]. However, the HIPK2 SIM-VK mutant and other HIPK2 mutants defective in SIM function such as HIPK2 R861W and N951I mutants bind to AML1b and MOZ but do not associated with the PML-NBs. The outcome of the differential association of HIPK2 mutants to PML, AML1 and MOZ is the sequestration of AML1 and MOZ to other nuclear dots independent of the PML-NBs. Consequently, AML1b and MOZ could not induce the transcription program for differentiation of myeloid precursor cells in the presence of the HIPK2 SIM mutant. These results indicated that recruitment of HIPK2 into PML-NBs via SIM is crucial for the appropriate differentiation of hematopoietic precursor cells. Furthermore, since HIPK2 targeting into PML-NBs is also critical for p53-mediated induction of apoptosis [38,39], cells expressing HIPK2 SIM-defective mutants are resistant to DNA damage response and cell death. Therefore, partitioning of HIPK2 into the nuclear dot structures independent of PML-NBs plays dual roles in inhibition of cell death and myeloid differentiation, resulting in the development of cancers such as AML.
Acknowledgements
This work was supported by the National Research Foundation of Korea grant (2017R1A2B2009818 and SRC-2017R1A5A1014-560 to C.Y.C.) funded by the Korea Ministry of Science and ICT.
Disclosure of conflict of interest
None.
References
- 1.Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184–197. doi: 10.1038/nrc.2016.143. [DOI] [PubMed] [Google Scholar]
- 2.Bayer P, Arndt A, Metzger S, Mahajan R, Melchior F, Jaenicke R, Becker J. Structure determination of the small ubiquitin-related modifier SUMO-1. J Mol Biol. 1998;280:275–286. doi: 10.1006/jmbi.1998.1839. [DOI] [PubMed] [Google Scholar]
- 3.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 4.Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- 5.Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275:36316–36323. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 6.Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci U S A. 2004;101:14373–14378. doi: 10.1073/pnas.0403498101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem. 2006;281:16117–16127. doi: 10.1074/jbc.M512757200. [DOI] [PubMed] [Google Scholar]
- 8.Song J, Zhang Z, Hu W, Chen Y. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J Biol Chem. 2005;280:40122–40129. doi: 10.1074/jbc.M507059200. [DOI] [PubMed] [Google Scholar]
- 9.Sekiyama N, Ikegami T, Yamane T, Ikeguchi M, Uchimura Y, Baba D, Ariyoshi M, Tochio H, Saitoh H, Shirakawa M. Structure of the small ubiquitin-like modifier (SUMO)-interacting motif of MBD1-containing chromatin-associated factor 1 bound to SUMO-3. J Biol Chem. 2008;283:35966–35975. doi: 10.1074/jbc.M802528200. [DOI] [PubMed] [Google Scholar]
- 10.Cappadocia L, Mascle XH, Bourdeau V, Tremblay-Belzile S, Chaker-Margot M, Lussier-Price M, Wada J, Sakaguchi K, Aubry M, Ferbeyre G, Omichinski JG. Structural and functional characterization of the phosphorylation-dependent interaction between PML and SUMO1. Structure. 2015;23:126–138. doi: 10.1016/j.str.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 11.de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–684. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- 12.Kakizuka A, Miller WH Jr, Umesono K, Warrell RP Jr, Frankel SR, Murty VV, Dmitrovsky E, Evans RM. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66:663–674. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- 13.Weidtkamp-Peters S, Lenser T, Negorev D, Gerstner N, Hofmann TG, Schwanitz G, Hoischen C, Maul G, Dittrich P, Hemmerich P. Dynamics of component exchange at PML nuclear bodies. J Cell Sci. 2008;121:2731–2743. doi: 10.1242/jcs.031922. [DOI] [PubMed] [Google Scholar]
- 14.Brand P, Lenser T, Hemmerich P. Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys. 2010;3:3. doi: 10.1186/1757-5036-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng X, Kao HY. Post-translational modifications of PML: consequences and implications. Front Oncol. 2012;2:210. doi: 10.3389/fonc.2012.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nisole S, Maroui MA, Mascle XH, Aubry M, Chelbi-Alix MK. Differential roles of PML Isoforms. Front Oncol. 2013;3:125. doi: 10.3389/fonc.2013.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Peng Q, Wan X, Sun H, Tang J. C-terminal motifs in promyelocytic leukemia protein isoforms critically regulate PML nuclear body formation. J Cell Sci. 2017;130:3496–3506. doi: 10.1242/jcs.202879. [DOI] [PubMed] [Google Scholar]
- 18.Pandolfi PP, Alcalay M, Fagioli M, Zangrilli D, Mencarelli A, Diverio D, Biondi A, Lo Coco F, Rambaldi A, Grignani F, et al. Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J. 1992;11:1397–1407. doi: 10.1002/j.1460-2075.1992.tb05185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the acute leukaemias. french-american-british (FAB) co-operative group. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 20.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen LA, Pandolfi PP, Aikawa Y, Tagata Y, Ohki M, Kitabayashi I. Physical and functional link of the leukemia-associated factors AML1 and PML. Blood. 2005;105:292–300. doi: 10.1182/blood-2004-03-1185. [DOI] [PubMed] [Google Scholar]
- 22.Aikawa Y, Nguyen LA, Isono K, Takakura N, Tagata Y, Schmitz ML, Koseki H, Kitabayashi I. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 2006;25:3955–3965. doi: 10.1038/sj.emboj.7601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li XL, Arai Y, Harada H, Shima Y, Yoshida H, Rokudai S, Aikawa Y, Kimura A, Kitabayashi I. Mutations of the HIPK2 gene in acute myeloid leukemia and myelodysplastic syndrome impair AML1- and p53-mediated transcription. Oncogene. 2007;26:7231–7239. doi: 10.1038/sj.onc.1210523. [DOI] [PubMed] [Google Scholar]
- 24.Moilanen AM, Karvonen U, Poukka H, Janne OA, Palvimo JJ. Activation of androgen receptor function by a novel nuclear protein kinase. Mol Biol Cell. 1998;9:2527–2543. doi: 10.1091/mbc.9.9.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trost M, Kochs G, Haller O. Characterization of a novel serine/threonine kinase associated with nuclear bodies. J Biol Chem. 2000;275:7373–7377. doi: 10.1074/jbc.275.10.7373. [DOI] [PubMed] [Google Scholar]
- 26.Kim YH, Choi CY, Lee SJ, Conti MA, Kim Y. Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J Biol Chem. 1998;273:25875–25879. doi: 10.1074/jbc.273.40.25875. [DOI] [PubMed] [Google Scholar]
- 27.Kuwano Y, Nishida K, Akaike Y, Kurokawa K, Nishikawa T, Masuda K, Rokutan K. Homeodomain-interacting protein kinase-2: a critical regulator of the DNA damage response and the epigenome. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73:2829–2850. doi: 10.1007/s00018-016-2130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann TG, Glas C, Bitomsky N. HIPK2: a tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays. 2013;35:55–64. doi: 10.1002/bies.201200060. [DOI] [PubMed] [Google Scholar]
- 30.Puca R, Nardinocchi L, Givol D, D’Orazi G. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene. 2010;29:4378–4387. doi: 10.1038/onc.2010.183. [DOI] [PubMed] [Google Scholar]
- 31.Choi DW, Choi CY. HIPK2 modification code for cell death and survival. Mol Cell Oncol. 2014;1:e955999. doi: 10.1080/23723548.2014.955999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saul VV, Schmitz ML. Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J Mol Med (Berl) 2013;91:1051–1058. doi: 10.1007/s00109-013-1042-0. [DOI] [PubMed] [Google Scholar]
- 33.de la Vega L, Grishina I, Moreno R, Kruger M, Braun T, Schmitz ML. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol Cell. 2012;46:472–483. doi: 10.1016/j.molcel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Sung KS, Go YY, Ahn JH, Kim YH, Kim Y, Choi CY. Differential interactions of the homeodomain-interacting protein kinase 2 (HIPK2) by phosphorylation-dependent sumoylation. FEBS Lett. 2005;579:3001–3008. doi: 10.1016/j.febslet.2005.04.053. [DOI] [PubMed] [Google Scholar]
- 35.Kim YH, Choi CY, Kim Y. Covalent modification of the homeodomain-interacting protein kinase 2 (HIPK2) by the ubiquitin-like protein SUMO-1. Proc Natl Acad Sci U S A. 1999;96:12350–12355. doi: 10.1073/pnas.96.22.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Zhu S, Guzzo CM, Ellis NA, Sung KS, Choi CY, Matunis MJ. Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J Biol Chem. 2008;283:29405–29415. doi: 10.1074/jbc.M803632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelhardt OG, Boutell C, Orr A, Ullrich E, Haller O, Everett RD. The homeodomain-interacting kinase PKM (HIPK-2) modifies ND10 through both its kinase domain and a SUMO-1 interaction motif and alters the posttranslational modification of PML. Exp Cell Res. 2003;283:36–50. doi: 10.1016/s0014-4827(02)00025-3. [DOI] [PubMed] [Google Scholar]
- 38.Sung KS, Lee YA, Kim ET, Lee SR, Ahn JH, Choi CY. Role of the SUMO-interacting motif in HIPK2 targeting to the PML nuclear bodies and regulation of p53. Exp Cell Res. 2011;317:1060–1070. doi: 10.1016/j.yexcr.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 39.de la Vega L, Frobius K, Moreno R, Calzado MA, Geng H, Schmitz ML. Control of nuclear HIPK2 localization and function by a SUMO interaction motif. Biochim Biophys Acta. 2011;1813:283–297. doi: 10.1016/j.bbamcr.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 40.D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4:1–10. doi: 10.1038/ncb715. [DOI] [PubMed] [Google Scholar]
- 42.Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The post-translational modification, SUMOylation, and cancer (Review) Int J Oncol. 2018;52:1081–1094. doi: 10.3892/ijo.2018.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, He Y, Wang X, Liang Z, He G, Zhang P, Zhu H, Xu N, Liang S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017;7 doi: 10.1098/rsob.170167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi DW, Seo YM, Kim EA, Sung KS, Ahn JW, Park SJ, Lee SR, Choi CY. Ubiquitination and degradation of homeodomain-interacting protein kinase 2 by WD40 repeat/SOCS box protein WSB-1. J Biol Chem. 2008;283:4682–4689. doi: 10.1074/jbc.M708873200. [DOI] [PubMed] [Google Scholar]
- 45.Choi DW, Na W, Kabir MH, Yi E, Kwon S, Yeom J, Ahn JW, Choi HH, Lee Y, Seo KW, Shin MK, Park SH, Yoo HY, Isono K, Koseki H, Kim ST, Lee C, Kwon YK, Choi CY. WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol Cell. 2013;51:374–385. doi: 10.1016/j.molcel.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 46.Bitomsky N, Conrad E, Moritz C, Polonio-Vallon T, Sombroek D, Schultheiss K, Glas C, Greiner V, Herbel C, Mantovani F, del Sal G, Peri F, Hofmann TG. Autophosphorylation and Pin1 binding coordinate DNA damage-induced HIPK2 activation and cell death. Proc Natl Acad Sci U S A. 2013;110:E4203–4212. doi: 10.1073/pnas.1310001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shima Y, Kitabayashi I. Deregulated transcription factors in leukemia. Int J Hematol. 2011;94:134–141. doi: 10.1007/s12185-011-0905-9. [DOI] [PubMed] [Google Scholar]
- 48.Hofmann TG, Jaffray E, Stollberg N, Hay RT, Will H. Regulation of homeodomain-interacting protein kinase 2 (HIPK2) effector function through dynamic small ubiquitin-related modifier-1 (SUMO-1) modification. J Biol Chem. 2005;280:29224–29232. doi: 10.1074/jbc.M503921200. [DOI] [PubMed] [Google Scholar]
- 49.Gresko E, Moller A, Roscic A, Schmitz ML. Covalent modification of human homeodomain interacting protein kinase 2 by SUMO-1 at lysine 25 affects its stability. Biochem Biophys Res Commun. 2005;329:1293–1299. doi: 10.1016/j.bbrc.2005.02.113. [DOI] [PubMed] [Google Scholar]
- 50.Merrill JC, Melhuish TA, Kagey MH, Yang SH, Sharrocks AD, Wotton D. A role for non-covalent SUMO interaction motifs in Pc2/CBX4 E3 activity. PLoS One. 2010;5:e8794. doi: 10.1371/journal.pone.0008794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shima Y, Shima T, Chiba T, Irimura T, Pandolfi PP, Kitabayashi I. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol Cell Biol. 2008;28:7126–7138. doi: 10.1128/MCB.00897-08. [DOI] [PMC free article] [PubMed] [Google Scholar]