

Lightning in a bottle: Plasma enables selective, electrolytic production of ammonia from nitrogen and water.
Abstract
There is a growing need for scalable ammonia synthesis at ambient conditions that relies on renewable sources of energy and feedstocks to replace the Haber-Bosch process. Electrically driven approaches are an ideal strategy for the reduction of nitrogen to ammonia but, to date, have suffered from low selectivity associated with the catalyst. Here, we present a hybrid electrolytic system characterized by a gaseous plasma electrode that facilitates the study of ammonia formation in the absence of any material surface. We find record-high faradaic efficiency (up to 100%) for ammonia from nitrogen and water at atmospheric pressure and temperature with this system. Ammonia measurements under varying reaction conditions in combination with scavengers reveal that the unprecedented selectivity is achieved by solvated electrons produced at the plasma-water interface, which react favorably with protons to produce the key hydrogen radical intermediate. Our results demonstrate that limitations in selectivity can be circumvented by using catalyst-free solvated electron chemistry. In the absence of adsorption steps, the importance of controlling proton concentration and transport is also revealed.
INTRODUCTION
The artificial fixation of nitrogen (N2) has an enormous energy, environmental, and societal impact, the most important of which is the synthesis of ammonia (NH3) for fertilizers that helps support nearly half of the world’s population (1). Industrially, NH3 is currently produced via the Haber-Bosch (H-B) process by reacting N2 with hydrogen (H2) over an iron-based catalyst at high pressure (150 to 300 atm) and high temperature (400° to 500°C) (2). This heterogeneous reaction scheme consumes more energy and contributes more greenhouse gas emissions than any other process associated with the top large-volume chemicals manufactured worldwide (3). A critical reason is that the source of H2 for H-B is fossil fuels, and either coal or natural gas must be catalytically converted in multiple steps before NH3 synthesis takes place. Because of the low single-pass conversion efficiency (15%) and high temperatures and high pressures, plants that implement H-B are large and centralized to be economical, making them difficult to integrate with renewable sources of H2 such as electrolysis (4).
Strategies for large-scale NH3 synthesis at ambient conditions that use renewable sources, such as water or H2 from electrolysis, are being explored and include photochemical (5) and electrochemical (6) processes. The major drawback with these approaches has been poor selectivity (<1%) for the desired NH3 product (7) largely related to the catalyst. Photochemical reduction suffers from weak adsorption of N2 on the surface of and oxidation of the reaction products by the holes in semiconductor catalysts (8). Electrochemical reduction has been characterized by large overpotentials that are required for the relatively stable N2 to associatively or dissociatively adsorb on a metal catalyst and energetically satisfy multiple intermediates involved in the complex reaction mechanism (9, 10). Moreover, the electrocatalysts with the lowest overpotentials are metals that favor the adsorption of hydrogen species (H2, H+, etc.) over N2, making it difficult to suppress the hydrogen evolution reaction (HER), which ultimately compromises NH3 formation (11).
Plasma-based processes are capable of activating N2 without a catalyst by generating highly energetic electrons; nitrogen fixation to convert N2 and oxygen (O2) to nitrates (NOx) occurs naturally in the atmosphere from lightning (12) and was industrially developed before H-B by forming an electric arc in air known as the Birkeland-Eyde process (13). More recently, plasmas have been combined with solid catalysts to enhance the heterogeneous reaction of N2 and H2 and enable NH3 synthesis at atmospheric pressure and low temperature (14). The synthesis of NH3 from water has also been reported (15). While these studies are promising, particularly the latter to avoid a dependence on fossil fuels (16), selective reduction of N2 to NH3 from renewable sources remains elusive.
Here, we report a hybrid electrolytic approach using a gaseous plasma electrode to study NH3 formation at ambient temperature and pressure from N2 and water free of any catalytic material surface. Distinct from other plasma-based processes such as lightning and the Birkeland-Eyde process, the conditions are controlled by removing air via a gas purge and reacting solvated electrons with water containing acid to selectively produce NH3. Solvated electrons, one of the most powerful reducing agents known, are injected into water from the plasma that is formed by electrical breakdown of an N2 gas flow in contact with the liquid surface (17). The feasibility of solvated electrons reducing N2 to NH3 has been previously demonstrated using boron-doped diamond films but required ultraviolet radiation to generate the solvated electrons, and the surface termination influenced NH3 formation (18). By using only electricity and forming the solvated electrons at a gas-liquid interface, we have discovered that high selectivity (up to 100%) and large production rate (0.44 mg/hour) are possible. Measurements of NH3 at varying currents and pH and in the presence of scavengers for potential reaction intermediates provide insight that a key step in our process is the reduction of protons (H+) to hydrogen radicals (H·) by solvated electrons. Moreover, we find that by removing kinetic barriers for adsorption to a surface, NH3 formation depends primarily on the concentration and transport of H+. These results elucidate how solvated electron chemistry can be harnessed to circumvent limitations in selectivity associated with the catalyst for NH3 production from renewable energy sources.
RESULTS
NH3 formation in a plasma electrolytic system
The synthesis of NH3 from N2 and water was carried out in a plasma electrolytic system schematically depicted in Fig. 1A. The setup bears similarity to electrochemical approaches except that the metal cathode was replaced by a plasma formed in a gas gap between a stainless steel nozzle and the solution surface. Details of this general approach have been reported elsewhere (19). Here, to study NH3 formation, both argon (Ar) and N2 were investigated as the plasma supply and purge gas. We performed all the experiments with a platinum (Pt) electrode immersed in the solution, which operated as the anode. The solution contained sulfuric acid (H2SO4) in deionized water (18.2 megohm) to both supply protons (H+) for N2 reduction and trap the as-synthesized NH3.
Fig. 1. Catalyst-free, electrolytic NH.

3 production from N2 and water using a plasma electrolytic system. (A) Schematic of the plasma electrolytic system operated by a dc power supply and galvanostatically controlled using a resistor (R) in series. The direction of electron flow (e−) is indicated. (B) Total NH3 produced after 45 min at 6 mA and pH 3.5 for various gas configurations and controls. (C) Potentially important species contained in the plasma, such as vibrationally excited N2 [N2(v)], and in the water, such as solvated electrons [e−(aq)], and their involvement in reactions, such as the generation of hydrogen radicals (H·), that lead to NH3 formation. The overall reactions for N2 reduction to NH3 and H2 evolution (under acidic conditions) at the cathode are shown.
We initially performed a series of control experiments to verify NH3 formation under the same amount of time and current of 45 min and 6 mA, respectively. Figure 1B shows the average mass of NH3 produced for the following configurations: (i) N2 both flowing into the cathode tube where the plasma is normally generated and bubbled through the solution to purge, but no electrical power applied (i.e., no plasma generated); (ii) Ar as both the supply gas in the plasma and the purge gas; (iii) Ar as the supply gas in the plasma and N2 as the purge gas; and (iv) N2 as both the supply gas in the plasma and the purge gas. The complete set of data for all trials is shown in table S1A. Without a plasma, or with only Ar in the system, no detectable amount of NH3 was found. When only Ar is present, the products are assumed to be H2 and O2 gas, the result of water electrolysis, as demonstrated in previous studies (20). In comparison, NH3 was produced (statistically nonzero; table S1B) when N2 was either the purge or the plasma supply gas, confirming that it was not coming from other sources including background contamination. The NH3 yield was significantly larger with N2 in the plasma compared with Ar in the plasma (table S1B).
A potential reaction mechanism for NH3 synthesis by solvated electron chemistry was proposed by Christianson et al. (21) based on the reduction of protons to hydrogen radicals (H·), H+ + e−(aq) ➔ H·, followed by sequential addition of H· to N2. This relatively simple picture is generally consistent with the associative pathway that has also been proposed for N2 reduction in biological and electrochemical systems (7). As shown in Fig. 1C, analogous to electrochemical synthesis of NH3, the HER is a major competing reaction, reducing selectivity, and occurs via either the second-order recombination of solvated electrons, 2e−(aq) + 2H2O ➔ H2 + 2OH−, or hydrogen radicals, 2H· ➔ H2, with the latter predominant under acidic conditions. We note that the first and rate-limiting step of this previously proposed mechanism for NH3 formation, which forms the reaction intermediate N2H·, is characterized by sluggish kinetics, and it is likely that the HER will dominate in this scenario. For this reason, while this mechanism may be relevant for our experiments with Ar in the plasma and N2 bubbled, the relatively high NH3 yield with N2 in the plasma suggests a different pathway for NH3 formation.
There are two aspects to our system that are distinct from that reported by Christianson et al. (see Fig. 1C). The first is that when N2 is supplied in the plasma, an excess of N2 is provided to the plasma-water interface. The solubility of N2 in water at atmospheric pressure and room temperature (25°C) is relatively low (6.8 × 10−4 M) and, in the case of an Ar plasma, will be even further depleted as N2 that is bubbled is converted to NH3. However, we estimate that the interfacial solvated electron concentration is ~0.5 mM at a current of 6 mA, and it is unlikely that the N2 concentration at the interface could ever be high enough to overcome the much faster HER and explain the observed higher NH3 yield. The second difference in our system is that N2 in the plasma could lead to gas-phase excitation or dissociation of N2. We point out that at atmospheric pressure, the electron energies in a plasma are dampened by collisions and the most likely mode of excitation is vibrational, which has the lowest energy threshold. In catalytic conversion of N2 and H2 to NH3, the importance of vibrationally excited N2 in lowering the activation barrier for the initial dissociative adsorption step is well known (14). The role of such excited species in plasma-water interfacial chemistry has not been reported, but we suggest, based on our results, that it is possible that vibrationally excited N2 reacts in a water vapor layer to form an intermediate that then dissolves at sufficiently high concentrations with favorable kinetics toward solvated electrons or H atoms to enhance NH3 formation.
NH3 yield and efficiency
NH3 yield and efficiency in the plasma electrolytic system were examined by measuring the amount of NH3 synthesized after different processing times and at different steady-state operating currents. Figure 2A shows the average mass of NH3 obtained as a function of time at a current of 6 mA and a pH of 3.5. The complete set of data for all trials is shown in table S2A. A linear increase in NH3 produced with time would indicate a constant production rate, but we find that the rate changes, decreasing after 10 min. The efficiency was estimated by comparing the measured NH3 mass at each time point to the amount of NH3 calculated from Faraday’s law assuming a three-electron reaction (see the Supplementary Materials for details). We note that while the set point for the current through the hybrid electrolytic system was kept constant, there were small fluctuations over the duration of the experiments arising from plasma instabilities, particularly when the plasma was ignited (fig. S1). Over the experimental period, these fluctuations did not cause substantial deviations from the average current because they occurred on very short time scales on the order of seconds. Nonetheless, to ensure that the faradaic amount of NH3 calculated was precise, the temporal current, I(t), was monitored and integrated to obtain the total charge, Q = ∫ I(t)dt. As a function of process time, the average faradaic efficiency is found to decrease from ~60% at 5 min to a statistically indistinguishable value of ~30% at 30 and 45 min (table S2B), consistent with the observed changes in the NH3 production rate. A longer time trial of 5 hours was also conducted, and the faradaic efficiency was only slightly lower than at 45 min (24.9% compared with 32.2%), confirming that the observed trends are maintained (table S2A).
Fig. 2. NH.
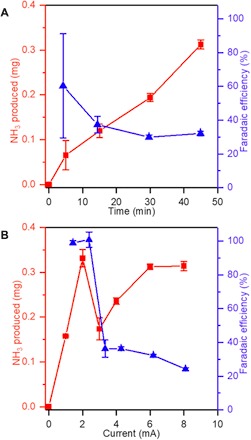
3 yield and efficiency in the plasma electrolytic system. (A) Total NH3 produced and corresponding faradaic efficiency after different processing times at 6 mA and pH 3.5. No NH3 is produced at 0 min based on the untreated electrolyte solution. (B) Total NH3 produced and corresponding faradaic efficiency as a function of current after 45 min at pH 3.5. No NH3 is produced at 0 mA based on the control experiment (see table S1A).
Figure 2B shows the average mass of NH3 produced as a function of current after 45 min and at pH 3.5. The complete set of data for all trials is shown in table S3A. The current that drives the plasma electrolytic system is measured at the power supply and is the same as the plasma current which, like the Pt anode, is electrically connected in series. Increasing the plasma current leads to an increase in the number of gas-phase electrons, which could excite N2 or be injected from the plasma into the solution (19), and based on our proposed mechanism, should increase NH3 production. Here, however, we find that the NH3 yield exhibits a rather complicated dependence on current, significantly increasing from 1 to 2 mA, then decreasing significantly from 2 to 3 mA, and then increasing again from 3 to 6 mA before staying constant from 6 to 8 mA (table S3B). We suggest that, as the current is increased >2 mA, the injected electrons are involved in competing reactions that do not form NH3. This apparent decrease in selectivity toward NH3 is corroborated by the faradaic efficiency, which decreases significantly from 2 to 3 mA and then decreases more slowly but statistically significantly from 4 to 8 mA (table S3B). The most likely reaction pathway that competes with NH3 formation is the HER, which occurs by recombination of either solvated electrons or H· (see Fig. 1C). In general, the HER is kinetically favored at higher concentrations of solvated electrons or H· because of the second-order dependence. Assuming that the solution volume over which the solvated electrons or H· are generated is constant, increasing the current would increase their concentrations and support the observed trends in faradaic efficiency.
The high, ~100% faradaic efficiency at <2 mA indicates that the competing HER is negligible. In support, we measured H2 production by analyzing the exhaust gas from the reactor using residual gas analysis (RGA) and gas chromatography (GC). RGA measurements at 2 mA showed no detectable change in the H2 partial pressure from background at any time during plasma operation (fig. S2). GC measurements showed no discernible peak corresponding to H2 up to 30 min at 1 mA and up to 15 min at 2 mA (table S3C). A small peak was observed at 1 mA after 45 min equivalent to a concentration of 6 ppm H2, and at 2 mA after 30 and 45 min equivalent to 33 and 30 ppm, respectively (table S3C). We note that the generation of H2 in our reactor over time is not necessarily inconsistent with our faradaic efficiency estimations. In contrast to the NH3 measurements, which reflect how much is accumulated over time, the GC measures how much H2 is produced at an exact time and, assuming that the rest of the current goes toward NH3 production, provides only an instantaneous faradaic efficiency (see the Supplementary Materials for details). To obtain a comparable cumulative faradaic efficiency, we measured H2 at other times and integrated the instantaneous faradaic efficiencies by assuming two-step functions to obtain an upper and a lower bound (fig. S3). This analysis showed faradaic efficiencies after 45 min of 95.1 to 100% at 1 mA and 77.9 to 95.9% at 2 mA, which agree well with the faradaic efficiencies independently obtained from NH3 measurements (see table S3A).
NH3 stability and trapping
While a notable amount of NH3 was produced in a single-compartment cell setup, we addressed the potential decomposition of NH3 by comparing it with a split H-cell geometry where the plasma cathode was operated in one compartment, the Pt anode was contained in the other, and the solutions in the two compartments were separated by a glass frit that allowed ionic contact but prevented mixing (fig. S4). If NH3 is decomposed by oxidation at the anode in the single cell, then the measured amount of NH3 would be lower than what is actually produced at the cathode and the split cell would show a higher yield. Figure 3A compares the average masses of NH3 produced in a single and split cell at 6 mA and pH 3.5 after different processing times. The complete set of data for all trials is shown in table S4A. The NH3 yields were found to be statistically identical, indicating that NH3 is not decomposed at the anode (table S4B). This is consistent with a previous report that proposed NH3 decomposition occurs through reaction with hydroxide ions (OH−) and only becomes considerable in basic solutions (22). Another possible loss mechanism for NH3 is simply vaporization because of its relatively low solubility. We studied the effectiveness of NH3 trapping by varying the pH of our solution in the single cell and connecting the gas effluent from the cell to a second vessel containing a strongly acidic H2SO4 bath (pH 2) to ensure complete capture. Figure 3B shows the average mass of NH3 measured in the main reaction cell and the second trap vessel at different pH. The complete set of data for all trials is shown in table S5A. The results confirm that for pH ≤5.5, no detectable amount of NH3 is lost from the reaction cell. The lack of any NH3 collected in the trap under acidic conditions also shows that the NH3 measured in our electrolytic process is not formed in the gas phase, for example, by reaction in the plasma between N2 and H2 produced from the HER or water vapor, but is formed in the solution. This was corroborated by RGA measurements that showed no detectable amount of NH3 in the exhaust gas (fig. S2). We also observe that the NH3 yield and faradaic efficiency significantly increase with decreasing pH (table S5B). This is consistent with the important role that H· plays and the contribution of the competing HER pathways (see Fig. 1C). Decreasing pH should lead to an increase in the proton (H+) concentration and enhance the rate of H+ reacting with solvated electrons via mass action to produce H· relative to the second-order recombination of solvated electrons (see Fig. 1C). The former reaction step leads to either NH3, by sequential addition to some nitrogen-containing intermediate, or H2, through the second-order recombination of H·, while the latter reaction step results in H2. The pH trends for NH3 yield and faradaic efficiency support this picture of the solvated electrons reacting favorably with H+, as the pH is decreased, and also show that the second-order recombination of H· does not become important even at the lowest pH tested, pH 2, where the H+ and then the H· concentration could be high. This reaffirms that a nitrogen-containing intermediate is at sufficiently high concentrations in our process to react quickly with any available H· and form NH3. We note that the dependence of NH3 formation on H+ is independent of its trapping since all the NH3 formed is effectively captured in the reaction cell at pH ≤5.5.
Fig. 3. Stability and trapping of NH.
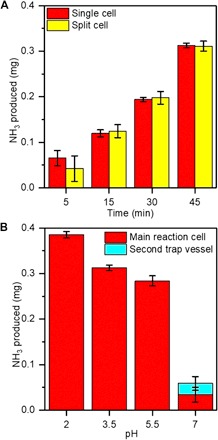
3 in the plasma electrolytic system. (A) Comparison of total NH3 produced in split-compartment versus single-compartment cells at 6 mA and pH 3.5 after different processing times. (B) Total NH3 captured in the main reaction cell and a secondary trap vessel as a function of pH in the main reaction cell. The secondary trap was a strongly acidic H2SO4 bath (pH 2).
NOx scavenging and measurement
The crucial role of solvated electrons and H· in plasma electrolytic synthesis of NH3 was verified by scavenger experiments. The high reactivity of these species allows a controlled impurity or scavenger to be added to the solution, which rapidly reacts to prevent their formation or convert them, potentially impeding their subsequent reaction. This approach is well known in radiation chemistry to elucidate reaction pathways involving solvated electron formation and their reaction by-products (23). Relevant to our study, scavengers have also been shown to reduce the concentration of solvated electrons generated by a plasma (17). We initially studied nitrate (NO3−), which has a high reactivity for presolvated electrons (24), the precursor to solvated electrons, and solvated electrons with a measured rate constant for plasma-injected solvated electrons, k = 7.0 ± 2.6 × 109 M−1 s−1 (17), and would be expected to rapidly lower the solvated electron concentration, as depicted in Fig. 4A. Figure 4B shows that the NH3 yield is slightly reduced by the addition of 10 mM NO3− and more substantially reduced (~50% reduction), but not completely suppressed, at 1 M. The complete set of data for all trials is shown in table S6A. We also studied nitrite (NO2−), which has been found to have a similar reactivity as NO3− for plasma-injected solvated electrons, k = 5.2 ± 2.6 × 109 M−1 s−1, but reacts close to 500 times faster with H· (25) (see Fig. 4A). At the same concentrations as NO3−, NO2− more effectively reduced NH3 production, with almost complete suppression at 1 M (see Fig. 4B and table S6B). We believe that both NO3− and NO2− do not completely suppress NH3 formation at 10 mM because the scavenger concentration is actually much lower at the plasma-water interface, and the reactions involving the scavenger become mass transport limited. Previous measurements of the solvated electron concentration at the plasma-water interface by optical absorbance indicate that the scavenger bulk concentration must be higher than ~0.1 M to completely attenuate the absorption signal (17). The NO2− may be more effective at reducing NH3 formation at this bulk concentration because of its enhanced reactivity with H·. At 1 M bulk concentrations, the interfacial scavenger concentrations are sufficiently high to completely quench the solvated electrons and, in the case of NO2−, stop NH3 formation. The unexpected continued formation of NH3 in the case of NO3−, even at these high concentrations, suggests a more complicated picture involving the formation of a new reducing radical species such as NO32− (26) in our system and a different reaction pathway independent of solvated electrons. Nonetheless, the scavenger experiments, particularly NO2−, substantiate that the formation of NH3 occurs through solvated electron chemistry and that one of the key intermediates is H·.
Fig. 4. Influence of NO.
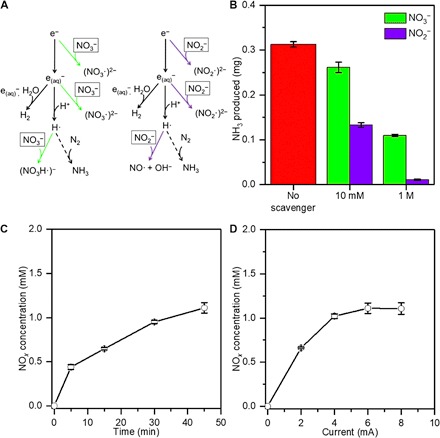
x on NH3 formation and in situ NOx production in the plasma electrolytic system. (A) Potential scavenging reaction pathways of NO3− (green) and NO2− (purple) before and after solvation of electrons in water. (B) Comparison of total NH3 produced after 45 min at 6 mA and pH 3.5 in the presence of NO3− and NO2− at 10 mM and 1 M concentrations. The total NH3 produced for the same conditions in the absence of any scavenger is included for reference. (C) NOx concentration measured after different processing times at 6 mA and pH 3.5. (D) NOx concentration measured as a function of current after 45 min and pH 3.5.
Although the single-compartment cell was closed and purged with Ar or N2 to remove background ambient air, the oxidation process at the Pt anode in our system evolves O2 gas, which could serve as an unintended impurity during the experiments in several ways. The presence of O2 in the plasma could lead to reactions with gas-phase electrons and reduce their flux to the solution surface, thus decreasing the concentration of solvated electrons produced (27). In addition, O2 could react with N2 in the plasma to produce NOx in the gas phase, as historically demonstrated by the Birkeland-Eyde process (13), and its subsequent dissolution forms NO2− and NO3− in the solution (28), which would affect solvated electron chemistry via the aforementioned scavenging reactions (see Fig. 4A). The relative similarity of NH3 yields in the split and single cells shows that the former is not an issue. To address the latter, we measured the NOx generated in the solution for our plasma electrolytic system. Figure 4 (C and D) shows the NOx concentration after different processing times at 6 mA and as a function of current after 45 min, respectively. The complete set of data for all trials is shown in table S7A. The NOx concentrations were not found to be large enough to scavenge and quench the solvated electrons (see Fig. 4B). We verified that the NOx formation was from O2 gas evolution at the Pt anode by also carrying out these measurements in the split cell, which showed no detectable amount of NOx. This was further corroborated by comparing the measured NOx with an expected amount from the faradaic yield of O2 and assuming that all of it reacts with N2 (in the plasma) to form either NO2 or NO3 (fig. S5).
DISCUSSION
The two key results that emerge from our study are the high, 100% selectivity toward NH3 formation at low currents (1 and 2 mA) and the marked decrease in selectivity as the current is increased to 3 mA and beyond. Given that O2 gas evolution and subsequent NOx formation are not sizeable, it is not likely that scavenging of solvated electrons and/or H· is responsible for the current-dependent decrease in NH3 production. Alternatively, we propose that there is a combination of factors related to the solvated electron, H+, and H· concentrations, which collectively have been found to play a critical role in NH3 formation in our system, and their spatiotemporal evolution. Initially, before the plasma is ignited and reactions begin to occur, the solution concentrations of solvated electrons and H· are zero, and the concentration of H+ at the plasma-water interface is the same as its bulk concentration. After the plasma is ignited, the solvated electron concentration immediately increases as plasma electrons are injected into the solution, and H+ is depleted and H· is generated as a result of the reduction of H+ by solvated electrons. All of this occurs primarily in an interfacial region where the solvated electrons are formed and react, and the interfacial H+ concentration becomes disparate from (lower than) its bulk concentration. As the steady-state current is increased, the production rate of solvated electrons concomitantly increases, depleting the interfacial concentration of H+ even faster. At sufficiently high currents, the H+ may be depleted, slowing down or stopping NH3 formation. While H+ may be present in the bulk from the initial acid and water oxidation at the Pt anode [2H2O(l) ➔ O2(g) + 4H+ + 4e−], mass transport limitations could prevent the bulk H+ from reaching the plasma-water interface. The high concentrations of solvated electrons and the low concentrations of H+ would result in higher rates of HER by both decreasing the reaction rate of the H+ reduction and increasing the reaction rate of the second-order recombination of solvated electrons. Thus, at higher currents, the HER becomes more important and reduces the selectivity toward NH3 formation. We note that the decrease in NH3 production rate and faradaic efficiency with time also supports a similar picture with H+ being depleted at the plasma-water interface and transport-limited (see Fig. 2A). A similar interplay between reaction and transport in a plasma electrolytic system has been observed for the reduction of silver ions to silver nanoparticles (29). There, the actual concentration of silver ions needed to achieve 100% faradaic efficiency was found to be 10 times higher than predicted by reaction modeling because of transport limitations. Transport issues may also be present in other electrolytic approaches to NH3 production but are relatively less important because of the kinetic limitations related to adsorption on the catalytic surface. Our findings show that by removing the adsorption step, transport limitations become a challenge but can be overcome to enable high selectivity.
While determination of the detailed reaction mechanism and transport parameters is subject to future studies, we have shown that the plasma electrolytic system allows the study of NH3 formation without the need for a catalyst and is capable of highly efficient production. Table 1 compares the overall performance of our system with recently reported results for similar aqueous (containing water) electrically driven demonstrations at ambient conditions. We note that only studies that used a non-nitrogen control gas are included, and the highest faradaic efficiency in each study is highlighted. The approach described in this study is found to have the highest faradaic efficiency while maintaining high production rate, over an order of magnitude higher than other electrochemical methods at similar reaction geometric areas. Our system is characterized by a power consumption of 2270 kWh/kg at 2 mA for 45 min based on a plasma voltage of ~500 V (table S8). The high energy consumption is a result of the electrical power required to produce sufficiently large electric fields in the plasma to generate and sustain electrons. Only a very small fraction of the energy is dissipated as heat, which is why these plasmas are referred to as nonthermal (30). In support, we measured the temperature profile of the solution surface during a process run at 6 mA and found a maximum temperature of 65°C after 45 min (fig. S6).
Table 1. Comparison of electrically driven N.
2 reduction to NH3 demonstrations at ambient temperature and pressure.
The power consumption of our plasma electrolytic system is considerably larger than that of H-B, which requires about 9 to 13 kWh/kg NH3 using natural gas, coal, or fuel oil as feedstocks (31). Electrochemical synthesis of NH3 has the potential to be competitive with H-B if the faradaic efficiency could be improved, but catalyst selectivity for NH3 production over H2 remains a challenge. Thus, the development and integration of a successful electrocatalyst into a full-cell device is the intense focus of current research. A relevant recent study provides a point of comparison for full electrochemical cell operation: At 65°C, 1.6 V, and 4.5 μA/cm2 NH3 current, the faradaic efficiency is 0.044% for an energy consumption of ~17,000 kWh/kg (32). Notably, higher faradaic efficiencies have been reported in half-cell experiments (see Table 1). Simulations suggest that an electrochemical NH3 cell has the potential to be competitive with H-B if a catalyst can be developed to have >50% faradaic efficiency (33). Similar to electrochemical approaches, the plasma electrolytic process does not require high pressures or temperatures and can operate at a small scale but does not require a catalyst. Considering the overall cost of NH3 that is associated with production, capital, shipping, and storage costs, our technology could be economically attractive by enabling smaller-scale distributed networks. Future studies should be aimed at lowering the energy consumption by enhancing electron generation in plasmas by, for example, modifying the electrode geometry or exploring other electrode materials (34). Even with the power requirements reported here, it is possible for this system to be integrated with renewable energy, which is continually decreasing in cost. Similar plasma systems have already been integrated with solar power for portable applications (35). With further exploration and optimization, the approach presented here could become a potentially promising technology for green, economical NH3 production.
In summary, we demonstrate that a hybrid plasma electrolytic system can serve as a tool to study NH3 formation without a catalytic material surface and achieve high faradaic efficiencies (up to 100%) at ambient temperature and pressure for NH3 production from N2 and water. Experiments conducted at different pH values and with scavengers show that solvated electrons play a key role in the reaction chemistry, particularly the reduction of H+, and NH3 formation occurs through a mechanism involving H·. Our analysis reveals that in the absence of adsorption barriers, the study and optimization of transport are required to further improve performance. In addition, a comparison of this technology with other similar electrolytic ammonia generation alternatives suggests that it may be a promising approach for distributed, renewable NH3 production.
MATERIALS AND METHODS
NH3 synthesis from N2 and water in a plasma electrolytic system
The synthesis of NH3 from N2 and water was carried out in a quartz reaction cell sealed with a polytetrafluoroethylene (PTFE) lid. The PTFE lid contained feedthroughs for the electrodes that consisted of a stainless steel capillary tube (outer diameter, 1.59 mm; inner diameter, 0.508 mm; and length, 10 cm; Restek Inc.) held 1 mm above the electrolyte solution surface and a Pt foil (purity, 99.9%; thickness, 0.0254 mm; Alfa Aesar) immersed in the electrolyte solution. A plasma was ignited in the gas gap between the tube and solution surface in a flow of either Ar (airgas, 99.5%) or N2 (airgas, 99.99%+), using a negative high-voltage power supply (RR15-10R, Gamma High Voltage Research) that was ballasted (0.25 to 1 megohm). The plasma served as the cathode and the Pt foil as the anode. Two additional feedthroughs were used to bubble the solutions before each experiment and exhaust the gas. The H-shaped split cell was similar except that the plasma cathode and Pt anode were in separate compartments, each with its own PTFE lid and feedthroughs for bubbling and exhaust, connected by a glass frit. The electrolyte solution was a mixture of 18.2 megohm water and sulfuric acid (purity, 99.999%; Sigma-Aldrich), typically at pH 3.5. The solution was bubbled with either Ar or N2 for 30 min before igniting the plasma to ensure the removal of any dissolved gases and to purge the headspace of the vessel.
The voltage and current were measured by a previously reported electrical circuit (19). Briefly, the voltage was measured between the capillary tube and the Pt anode by using a digital multimeter, and the current was obtained from the voltage drop across a resistor in series between the Pt anode and the power supply. The current was measured in real time using LabVIEW. Temperature measurements of the solution surface as a function of position and time during a process run were performed using an FLIR i3 infrared camera with the PTFE lid removed to allow optical access. An emissivity, ε, of 0.6 was assumed, and the accuracy of the temperatures was verified by a thermometer to be ±2°C.
Measurement of NH3 produced
The production of NH3 in the plasma electrolytic system was measured by the o-phthalaldehyde method (36). First, 2 ml of the processed solution was removed and refrigerated in an N2-purged, sealed vial until analysis. Ten-microliter aliquots were then pipetted into a 96-well analysis tray, and 90 μl of an assay solution containing buffer solution and active reagents (QuantiFluo DNH3200, BioAssay Systems) was added. After a 15-min incubation period with no light exposure, the concentration of NH3 was determined from fluorescence measurements at an excitation wavelength of 360 nm and emission wavelength of 450 nm using a Molecular Devices Spectramax M2 microplate reader. Calibration of the method was carried out with four known concentrations of aqueous ammonium hydroxide solutions as standards: 0, 0.25, 0.50, and 0.75 mM (fig. S7). The fluorescence intensity had a lower detection limit of 12 μM NH3 and was found to increase linearly with NH3 concentration.
Measurement of H2 gas produced
The production of H2 in the plasma electrolytic system was measured using a Shimadzu GC-2014 gas chromatograph with a thermal conductivity detector and a Restek ShinCarbon ST 80/100 mesh 2 m by 2 mm column. Ar was used as the carrier gas to allow identification of the N2 from the reactor and to enhance measurement sensitivity for H2. The gas analysis was performed by connecting the reactor exhaust directly into a six-way valve, filling a 100-μl sample loop, and injecting at different times during the process run into the gas chromatograph. Confirmation of the retention times for the H2 and N2 peaks and calibration of the peak areas to obtain H2 concentration were carried out by analyzing gas mixtures of H2 and N2 controlled by two separate digital mass flow controllers. The reactor exhaust was also simultaneously analyzed using a Stanford Research Systems RGA100 atmospheric pressure sampling mass spectrometer.
Measurement of NOx produced
The production of NOx in the plasma electrolytic system was measured by the Griess method (37) with reagents from Sigma-Aldrich (23479-1KT-F). First, 2 ml of the process solution was removed and refrigerated, similar to the NH3 measurements. Ten-microliter aliquots were then pipetted into a 96-well tray, and 70 μl of buffer along with 10 μl of nitrate reductase and 10 μl of the provided Griess coenzymes were added. After a 2-hour incubation period, 50 μl of Griess reagent A was added and incubated for 5 min before addition of 50 μl of Griess reagent B and incubation for an additional 10 min. The absorbance was then measured at 540 nm using a Molecular Devices Spectramax M2 microplate reader to determine the total moles of NOx, and the concentration was obtained from the initial 10-μl volume. Calibration of the method was carried out with four known molar quantities of sodium nitrate at 0, 2, 4, and 8 nmol in a 10-μl volume (fig. S8).
Statistical analysis
All data are represented as the mean of a dataset ± SE, which was calculated from the variance in the raw data within two SDs of the mean (approximately 95% confidence interval). Statistical differences between datasets were determined using a two-sample t test. To determine whether a dataset was nonzero, a one-sample t test was used. All t tests were performed using the Minitab 2017 Statistical Software. The results of all of the t tests and the sample size for each dataset are tabulated in the Supplementary Materials. For all statistical tests, a threshold value of α = 0.05 was chosen, and a P value at or less than 0.05 indicated significance.
Supplementary Material
Acknowledgments
We thank J. Toth for assistance with GC and D. Matthiesen for assistance with RGA. We are grateful to D. Go and D. Bartels for the useful discussions. Funding: This work was supported by the U.S. Army Research Office under grant no. W911NF-17-1-0119 and the CWRU Faculty Investment Fund (FIF). R.H. and R.M.S. also acknowledge the CWRU SOURCE program for support. This work was also partially supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program, under grant no. DE-SC0016529. Specifically, J.N.R. was a supported by award DE-SC0016529 during some of the experimental work and analysis performed for this publication. Author contributions: R.M.S. and J.N.R. designed the experiments. R.H. and S.G. set up the plasma electrolytic system and performed the ammonia synthesis. R.H. and C.X. carried out the ammonia measurements. R.H. and E.G. carried out the hydrogen measurements. R.H., R.M.S., and J.N.R. discussed the results and analyzed the data. R.H. and J.N.R. did the statistical analysis. R.M.S. and J.N.R. wrote the manuscript. All authors contributed to the editing of the manuscript. Competing interests: R.M.S. and J.N.R. are inventors on a provisional patent application related to this work filed by Case Western Reserve University (no. 62/647,021; filed on 23 March 2018). All the other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat5778/DC1
Equation for faradaic efficiency calculation for NH3 from NH3 measurements
Equation for instantaneous faradaic efficiency calculation for NH3 from H2 measurements
Equation for faradaic NH3 concentration
Equation for energy consumption
Fig. S1. Representative current waveforms measured in the plasma electrolytic system during NH3 synthesis.
Fig. S2. RGA measurements of mass/charge ratio (m/z) = 2 and 17, corresponding to H2 and NH3 partial pressure, at 2 mA in a plasma electrolytic reactor by analyzing exhaust gas from cell as a function of time.
Fig. S3. H2 production in the plasma electrolytic system.
Fig. S4. Schematic of the hybrid plasma electrolytic system in a split H-cell geometry.
Fig. S5. Comparison of NOx produced with that predicted from O2 gas evolution in plasma electrolytic system.
Fig S6. Heating of solution in the plasma electrolytic system.
Fig. S7. Representative fluorescence assay calibration used to determine NH3 produced.
Fig. S8. Representative fluorescence assay calibration used to determine NOx produced.
Table S1A. Summary of NH3 produced by plasma electrolytic synthesis for the following configurations: N2 both flowing through the cathode tube where the plasma is normally generated and bubbled through the solution to purge, but no electrical power applied, Ar as both the supply gas in the plasma and purge gas, Ar as the supply gas in the plasma and N2 as the purge gas, and N2 as both the supply gas in the plasma and purge gas.
Table S1B. Summary of one-sample and two-sample t tests carried out on datasets in table S1A.
Table S2A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis after different processing times.
Table S2B. Summary of one-sample and two-sample t tests carried out on datasets in table S2A.
Table S3A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis at different currents.
Table S3B. Summary of one-sample and two-sample t tests carried out on datasets in table S3A.
Table S3C. Summary of H2 produced in plasma electrolytic reactor by analyzing exhaust gas using GC.
Table S4A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis in a split cell.
Table S4B. Summary of two-sample t tests carried out on datasets in table S4A (and tables S2A and S3A).
Table S5A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis at different pH values.
Table S5B. Summary of one-sample and two-sample t tests carried out on datasets in table S5A.
Table S6A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis in the presence of NO3 and NO2 scavengers.
Table S6B. Summary of one-sample and two-sample t tests carried out on datasets in table S6A.
Table S7A. Summary of NOx produced by plasma electrolytic synthesis after different processing times and at different currents.
Table S7B. Summary of one-sample and two-sample t tests carried out on datasets in table S7A.
Table S8. Summary of voltages measured between the plasma cathode and the Pt anode as a function of the plasma current in the plasma electrolytic system.
REFERENCES AND NOTES
- 1.Erisman J. W., Sutton M. A., Galloway J., Klimont Z., Winiwarter W., How a century of ammonia synthesis changed the world. Nat. Geosci. 1, 636–639 (2008). [Google Scholar]
- 2.M. Appl, Ammonia, 1. Introduction. In Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH Verlag GmbH, 2012).
- 3.International Energy Agency, Technology Roadmap Energy and GHG reductions in the chemical industry via catalytic processes (IEA, DECHEMA, ICCA, 2013); www.iea.org/publications/freepublications/publication/TechnologyRoadmapEnergyandGHGReductionsintheChemicalIndustryviaCatalyticProcesses.pdf
- 4.Tunå P., Hulteberg C., Ahlgren S., Techno-economic assessment of nonfossil ammonia production. Environ. Prog. Sustain. Energy 33, 1290–1297 (2014). [Google Scholar]
- 5.Medford A. J., Hatzell M. C., Photon-driven nitrogen fixation: Current progress, thermodynamic considerations, and future outlook. ACS Catal. 7, 2624–2643 (2017). [Google Scholar]
- 6.Kyriakou V., Garagounis I., Vasileiou E., Vourros A., Stoukides M., Progress in the electrochemical synthesis of ammonia. Catal. Today 286, 2–13 (2017). [Google Scholar]
- 7.van der Ham C. J. M., Koper M. T. M., Hetterscheid D. G. H., Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 43, 5183–5191 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Mozzanega H., Herrmann J. M., Pichat P., Ammonia oxidation over UV-irradiated titanium dioxide at room temperature. J. Phys. Chem. 83, 2251–2255 (1979). [Google Scholar]
- 9.Skúlason E., Bligaard T., Gudmundsd S., Studt F., Rossmeisl J., Abild-Pedersen F., Vegge T., Jónsson H., Nørskov J. K., A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 14, 1235–1245 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Montoya J. H., Tsai C., Vojvodić A., Nørskov J. K., The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 8, 2180–2186 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Singh A. R., Rohr B. A., Schwalbe J. A., Cargnello M., Chan K., Jaramillo T. F., Chorkendorff I., Nørskov J. K., Electrochemical ammonia synthesis—The selectivity challenge. ACS Catal. 7, 706–709 (2017). [Google Scholar]
- 12.von Liebig J., Une note sur la nitrification. Ann. Chem. Phys. 35, 329–333 (1827). [Google Scholar]
- 13.Eyde H. S., The manufacture of nitrates from the atmosphere by the electric arc—Birkeland-Eyde process. J. R. Soc. Arts 57, 568–576 (1909). [Google Scholar]
- 14.Mehta P., Barboun P., Herrera F. A., Kim J., Rumbach P., Go D. B., Hicks J. C., Schneider W. F., Overcoming ammonia synthesis scaling relations with plasma-enabled catalysis. Nat. Catal. 1, 269–275 (2018). [Google Scholar]
- 15.Haruyama T., Namise T., Shimoshimizu N., Uemura S., Takatsuji Y., Hino M., Yamasaki R., Kamachi T., Kohno M., Non-catalyzed one-step synthesis of ammonia from atmospheric air and water. Green Chem. 18, 4536–4541 (2016). [Google Scholar]
- 16.Tremel A., Wasserscheid P., Baldauf M., Hammer T., Techno-economic analysis for the synthesis of liquid and gaseous fuels based on hydrogen production via electrolysis. Int. J. Hydrogen Energy 40, 11457–11464 (2015). [Google Scholar]
- 17.Rumbach P., Bartels D. M., Sankaran R. M., Go D. B., The solvation of electrons by an atmospheric-pressure plasma. Nat. Commun. 6, 7248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu D., Zhang L., Ruther R. E., Hamers R. J., Photo-illuminated diamond as a solid-state source of solvated electrons in water for nitrogen reduction. Nat. Mater. 12, 836–841 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Richmonds C., Witzke M., Bartling B., Lee S. W., Wainright J., Liu C.-C., Sankaran R. M., Electron-transfer reactions at the plasma–liquid interface. J. Am. Chem. Soc. 133, 17582–17585 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Witzke M., Rumbach P., Go D. B., Sankaran R. M., Evidence for the electrolysis of water by atmospheric-pressure plasmas formed at the surface of aqueous solutions. J. Phys. D Appl. Phys. 45, 442001 (2012). [Google Scholar]
- 21.Christianson J. R., Zhu D., Hamers R. J., Schmidt J. R., Mechanism of N2 reduction to NH3 by aqueous solvated electrons. J. Phys. Chem. B 118, 195–203 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Kim K.-W., Kim Y.-J., Kim I.-T., Park G.-I., Lee E.-H., The electrolytic decomposition mechanism of ammonia to nitrogen at an IrO2 anode. Electrochim. Acta 50, 4356–4364 (2005). [Google Scholar]
- 23.Wolff R. K., Bronskill M. J., Hunt J. W., Picosecond pulse radiolysis studies. II. Reactions of electrons with concentrated scavengers. J. Chem. Phys. 53, 4211 (1970). [Google Scholar]
- 24.Nguyen J., Ma Y., Luo T., Bristow R. G., Jaffray D. A., Lu Q.-B., Direct observation of ultrafast-electron-transfer reactions unravels high effectiveness of reductive DNA damage. Proc. Nat. Acad Sci. U.S.A. 108, 11778–11783 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madden K. P., Mezyk S. P., Critical review of aqueous solution reaction rate constants for hydrogen atoms. J. Phys. Chem. Ref. Data Monogr. 40, 023103 (2011). [Google Scholar]
- 26.Cook A. R., Dimitrijevic N., Dreyfus B. W., Meisel D., Curtiss L. A., Camaioni D. M., Reducing radicals in nitrate solutions. The NO32- system revisited. J. Phys. Chem. A 105, 3658–3666 (2001). [Google Scholar]
- 27.Rumbach P., Bartels D. M., Sankaran R. M., Go D. B., The effect of air on solvated electron chemistry at a plasma/liquid interface. J. Phys. D Appl. Phys. 48, 424001 (2015). [Google Scholar]
- 28.Rumbach P., Witzke M., Sankaran R. M., Go D. B., Decoupling interfacial reactions between plasmas and liquids: Charge transfer vs. plasma neutral reactions. J. Am. Chem. Soc. 135, 16264–16267 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Ghosh S., Hawtof R., Rumbach P., Go D. B., Akolkar R., Sankaran R. M., Quantitative study of electrochemical reduction of Ag+ to Ag nanoparticles in aqueous solutions by a plasma cathode. J. Electrochem. Soc. 164, D818–D824 (2017). [Google Scholar]
- 30.Mariotti D., Sankaran R. M., Microplasmas for nanomaterials synthesis. J. Phys. D Appl. Phys. 43, 323001 (2010). [Google Scholar]
- 31.Giddy S., Badwal S. P. S., Munnings C., Dolan M., . Ammonia as a renewable energy transportation media. ACS Sustainable Chem. Eng. 5, 10231–10239 (2017). [Google Scholar]
- 32.Kong J., Lim A., Yoon C., Jang J.-H., Ham H. C., Han J., Nam S., Kim D., Sung Y.-E., Choi J., Park H. S., Electrochemical synthesis of NH3 at low temperature and atmospheric pressure using a γ-Fe2O3 catalyst. ACS Sustainable Chem. Eng. 5, 10986–10995 (2017). [Google Scholar]
- 33.Kugler K., Ohs B., Scholz M., Wessling M., Towards a carbon independent and CO2-free electrochemical membrane process for NH3 synthesis. Phys. Chem. Chem. Phys. 16, 6129–6138 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Bilici M. A., Haase J. R., Boyle C. R., Go D. B., Sankaran R. M., The smooth transition from field emission to a self-sustained plasma in microscale electrode gaps at atmospheric pressure. J. Appl. Phys. 119, 223301 (2016). [Google Scholar]
- 35.Ni Y., Lynch M. J., Modic M., Whalley R. D., Walsh J. L., A solar powered handheld plasma source for microbial decontamination applications. J. Phys. D Appl. Phys. 49, 355203 (2016). [Google Scholar]
- 36.Goyal S. S., Rains D. W., Huffaker R. C., Determination of ammonium ion by fluorometry or spectrophotometry after on-line derivatization with o-phthalaldehyde. Anal. Chem. 60, 175–179 (1988). [DOI] [PubMed] [Google Scholar]
- 37.Ridnour L. A., Sim J. E., Hayward M. A., Wink D. A., Martin S. M., Buettner G. R., Spitz D. R., A spectrophometric method for the direct detection of quantification of nitric oxide, nitrite, and nitrate in cell culture media. Anal. Biochem. 281, 223–229 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Shi M.-M., Bao D. C., Wulan B.-R., Li Y.-H., Zhang Y.-F., Yan J.-M., Jiang Q., Au sub-nanoclusters on TiO2 toward highly efficient and selective electrocatalyst for N2 conversion to NH3 at ambient conditions. Adv. Mater. 29, 1606550 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Zhou F., Azofra L. M., Ali M., Kar M., Simonov A. N., McDonnell-Worth C., Sun C., Zhang X., MacFarlane D. R., Electro-synthesis of ammonia from nitrogen at ambient temperature and pressure in ionic liquids. Energy Environ. Sci. 10, 2516–2520 (2017). [Google Scholar]
- 40.Bao D., Zhang Q., Meng F. L., Zhong H.-X., Shi M.-M., Zhang Y., Yan J.-M., Jiang Q., Zhang X.-B., Electrochemical reduction of N2 under ambient conditions for artificial N2 fixation and renewable energy storage using N2/NH3 cycle. Adv. Mater. 29, 1604799 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat5778/DC1
Equation for faradaic efficiency calculation for NH3 from NH3 measurements
Equation for instantaneous faradaic efficiency calculation for NH3 from H2 measurements
Equation for faradaic NH3 concentration
Equation for energy consumption
Fig. S1. Representative current waveforms measured in the plasma electrolytic system during NH3 synthesis.
Fig. S2. RGA measurements of mass/charge ratio (m/z) = 2 and 17, corresponding to H2 and NH3 partial pressure, at 2 mA in a plasma electrolytic reactor by analyzing exhaust gas from cell as a function of time.
Fig. S3. H2 production in the plasma electrolytic system.
Fig. S4. Schematic of the hybrid plasma electrolytic system in a split H-cell geometry.
Fig. S5. Comparison of NOx produced with that predicted from O2 gas evolution in plasma electrolytic system.
Fig S6. Heating of solution in the plasma electrolytic system.
Fig. S7. Representative fluorescence assay calibration used to determine NH3 produced.
Fig. S8. Representative fluorescence assay calibration used to determine NOx produced.
Table S1A. Summary of NH3 produced by plasma electrolytic synthesis for the following configurations: N2 both flowing through the cathode tube where the plasma is normally generated and bubbled through the solution to purge, but no electrical power applied, Ar as both the supply gas in the plasma and purge gas, Ar as the supply gas in the plasma and N2 as the purge gas, and N2 as both the supply gas in the plasma and purge gas.
Table S1B. Summary of one-sample and two-sample t tests carried out on datasets in table S1A.
Table S2A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis after different processing times.
Table S2B. Summary of one-sample and two-sample t tests carried out on datasets in table S2A.
Table S3A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis at different currents.
Table S3B. Summary of one-sample and two-sample t tests carried out on datasets in table S3A.
Table S3C. Summary of H2 produced in plasma electrolytic reactor by analyzing exhaust gas using GC.
Table S4A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis in a split cell.
Table S4B. Summary of two-sample t tests carried out on datasets in table S4A (and tables S2A and S3A).
Table S5A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis at different pH values.
Table S5B. Summary of one-sample and two-sample t tests carried out on datasets in table S5A.
Table S6A. Summary of NH3 produced and faradaic efficiencies by plasma electrolytic synthesis in the presence of NO3 and NO2 scavengers.
Table S6B. Summary of one-sample and two-sample t tests carried out on datasets in table S6A.
Table S7A. Summary of NOx produced by plasma electrolytic synthesis after different processing times and at different currents.
Table S7B. Summary of one-sample and two-sample t tests carried out on datasets in table S7A.
Table S8. Summary of voltages measured between the plasma cathode and the Pt anode as a function of the plasma current in the plasma electrolytic system.