

Abstract
Innate immunity contributes to host defense through all cell types and relies on their shared germline genetic background, whereas adaptive immunity operates via only three main cell types, αβ T cells, γδ T cells, and B cells, and relies on their somatic genetic diversification of antigen-specific responses. Human inborn errors of innate immunity often underlie infectious diseases. The range and nature of infections depend on the mutated gene, the deleteriousness of the mutation, and other ill-defined factors. Most known inborn errors of innate immunity to infection disrupt the development or function of leukocytes other than T and B cells, but a growing number of inborn errors affect cells other than circulating and tissue leukocytes. Here, we review inborn errors of innate immunity that have been recently discovered or clarified. We highlight the immunological implications of these errors.
Keywords: Infection, inborn error of immunity, immunodeficiency, innate immunity, signaling pathway, Toll-like receptors, NF-κB, interferon, phagocytes
INTRODUCTION
Innate immunity is a germline-encoded system that enables eukaryotes to defend themselves against infection (1, 2). It consists of three main components: 1) anatomical barriers, such as the physical barrier of intact skin and the chemical barrier of low gastric pH; 2) soluble proteins secreted onto mucosal surfaces or into the bloodstream, such as complement factors; 3) a cellular compartment composed of both hematopoietic and nonhematopoietic cells. Antigen presenting cells (APC), such as dendritic cells (DCs), phagocytes, such as neutrophils, cytotoxic/cytolytic cells, such as natural killer (NK) cells and other innate lymphoid cells (ILCs), together with non-hematopoietic cells, such as fibroblasts, keratinocytes, epithelial cells, and neurons, harbor pathways that allow an adequate response to infection. Pathogens are first detected via innate immune sensors or receptors. Several classes of microbial receptors have been described: Toll-like receptors (TLRs), retinoic-acid-inducible protein 1 (RIG-I)-like receptors (RLRs), Nod-like receptors (NLR), Ctype lectin receptors (CLRs), and DNA sensors (3–5). These receptors recognize microbial structures, such as lipoproteins, lipopolysaccharide (LPS), flagellin, peptidoglycan or microbial nucleic acids. Recognition activates downstream signaling cascades, leading to a rapid (occurring within hours) but short-lived immune response. This early immune response enables the host to survive until an adaptive response is generated, but its excessive activation can trigger auto-inflammation, which is defined as damage caused by dysregulated innate immune cells to host tissues (6, 7). Cell-intrinsic immunity is a branch of innate immunity in which a set of cellular antimicrobial defenses protects individual hematopoietic and non-hematopoietic cells against microbes, especially viruses, through autocrine pathways and intracellular mechanisms, such as the production of viral restriction factors (8, 9).
The study of inborn errors of immunity was long dominated by the concept of “conventional” primary immunodeficiencies (PIDs), in which rare Mendelian traits confer susceptibility to infection with a multitude of pathogens, in a completely penetrant manner and with a measurable effect on immune cells number or function. The prototype of such PIDs is X-linked (XL) agammaglobulinemia, which is caused by deleterious mutations in the Bruton tyrosine kinase gene (BTK). BTK is expressed in hematopoietic cells, and defects of this protein render the host susceptible, for life, to recurrent life-threatening infections with various microbes and result in a detectable immunological phenotype (little or absent immunoglobulin (Ig) production and few or absent B lymphocytes). In the 1910s, Nicolle described asymptomatic infections and documented the natural variability in host susceptibility to infection. In line with these observations, “monogenic infections” were described from the 1940s onward, and their molecular basis from the 1970s onward. Increased and selective susceptibility to infections with one or a narrow range of pathogens can be referred to as “non-conventional” PIDs and include many inborn errors of innate immunity (10). An example is the susceptibility to invasive Neisseria infections in patients with late complement pathway defects (11–13). Over the last two decades, susceptibility to a narrow range of microbes, such as monogenic susceptibility to invasive pneumococcal disease (IPD), mycobacterial disease (MSMD), Herpes simplex virus 1 (HSV-1) encephalitis (HSE) and fungal disease, particularly chronic mucocutaneous candidiasis (CMC), have been ascribed to defects in single genes, often but not always encoding components of innate immunity (13, 14). However, dichotomous classifications making distinctions between adaptive and innate immunity, not to mention cell-intrinsic immunity, represent an oversimplification and prove difficult to apply to inborn errors of immunity. In fact, inborn errors of innate immunity may result in the absence or impaired production of cytokines essential for the development, survival, proliferation or function of B or T cells, and can lead to allergy and autoimmunity, indications of adaptive immune dysregulation. In this review, we therefore explore a set of inborn errors affecting “primarily” innate immunity, acknowledging their impact on the adaptive immune response. We focus on recently described gene defects in the cellular compartment of the innate immune response or on new insights into the pathogenesis of previously described defects (Table 1). So, although key to innate immunity against infections, the complement system (comprehensively reviewed elsewhere (15, 16)) will not be discussed here. For the recently described CD55 deficiency, we refer to the original publication (17).
Table 1:
Overview of recently described inborn errors of innate immunity to infections
| Protein | Gene | OMIM n. gene / disease | Inheritance and penetrance | Cells where expressed (mRNA and/or protein) | Cells where tested | Results | Immunological phenotype | Infectious phenotype | Noninfectious phenotype | Autoinfl ammation | N. of report ed patients | Therapy | Year of genetic report |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Upstream defects of the TLR and/or IL-1R signaling pathway | |||||||||||||
| IRAK1 | IRAK1 | 300283 | XL | Ubiquitous | Fibroblasts PBMCs EBV-B cells | TLR responses: impaired in fibroblasts and EBVB cells, normal in PBMCs. Normal responses to IL-1β in fibroblasts and PBMcs | NA | NA | NA (MECP2 deletion sdr) | No | 1 | NA | 2017 (24) |
| TIRAP | TIRAP | 606252 | AR - IP | Ubiquitous | Fibroblasts PBMCs Granulocytes | Impaired responses to TLR2 and TLR4 stimulation | NA | Severe staphylococcal infection | NA | No | 8 | NA | 2017 (25) |
| Inborn errors of the CBM complex | |||||||||||||
| CARD9 | CARD9 | 607212/ 212050 | AR - CP | Granulocytes | PBMCs Monocytederived dendritic cells Macrophages | Impaired T cell dependent IL-17 production. Defective cytokine and chemokine production upon fungal stimulation. Impaired neutrophil killing, impaired neutrophil recruitment to the site of infection. | Eosinophilia, high IgE | Superficial (e.g. chronic mucocutaneous candidiasis) and invasive (e.g. meningoencephalitis) fungal infections | NA | No | 58 | HSCT GM-CSF G-CSF | 2009 (50) |
| Inborn errors of LUBAC | |||||||||||||
| HOIL-1 | HOIL1 (RBCK1) | 610924 | AR - CP | Ubiquitous | Fibroblasts EBV-B cells PBMCs | Impaired NF-κB responses in fibroblasts and EBVB cells. Enhanced response to IL-1β in monocytes. | Impaired vaccine responses | Invasive bacterial infections. | Chronic autoinflammation, muscular amylopectinosis | Yes | 3 | NA | 2012 (57) |
| HOIP | HOIP (RNF31) | 612487 | AR - CP | Ubiquitous | Fibroblasts EBV-B cells PBMCs | Impaired linear ubiquitination and NF-κB activation in fibroblasts. Impaired CD40 activation in B cells. Enhanced response to IL-1β in monocytes. | Lymphopenia, antibody deficiency and impaired distribution and function of T lymphocytes | Recurrent viral and bacterial infections. | Multiorgan autoinflamma tion, chronic diarrhea, subclinical amylopectino sis, systemic lymphangiectasia, | Yes | 1 | NA | 2015 (58) |
| Defects of phagocytes | |||||||||||||
| VPS45 (SCN5) | VPS45 | 610035/615285 | AR - CP | Broadly expressed | Lymphoblasts Fibroblasts Platelets Neutrophils Bone marrow myeloid cells | Impaired cell migration, increased apoptosis, loss of lysosomes. Loss of α-granules in platelets | SCN, G-CSF treatment resistant. Platelet dysfunction, thrombocytopenia, progressive anemia | Invasive pyogenic bacterial and fungal infections of airways, skin and urinary tract; sepsis | Extramedullar hematopoiesi s, bone marrow fibrosis, nephromegaly, hepatospleno megaly, neurological involvement | No | 13 | HSCT | 2013 (69, 70) |
| JAGN1 (SCN6) | JAGN1 | 616012/ 616022 | AR - CP | Ubiquitous | Neutrophils | Ultrastructural defects, few granules, aberrant glycosylation of GCSF-R, increased apoptosis | SCN, mostly G-CSF treatment resistant | Pyogenic bacterial infections of airways and skin; sepsis | Myeloid maturation arrest, osteopenia, teeth abnormalities, facial dysmorphisms, short stature, pancreatic insufficiency | No | 16 | G-CSF HSCT | 2014 (74) |
| G-CSF receptor (SCN7) | CSF3R | 138971/ 617014 | AR - CP | Granulocyte s, placenta and other tissues | PBMCs | Impaired glycosylation, expression at the cell surface and function | SCN, G-CSF treatment resistant | Pyogenic bacterial infections of airways, skin and urinary tract | NA | No | 5 | GM-CSF | 2014 (76) |
| SMARCD2 (SGD2) | SMARCD2 | 601736/ 617475 | AR - CP | Hematopoietic progenitor cells | Hematopoieti c progenitor cells | Myeloid differentiation defects, defect of granulopoiesis and neutrophil granule scarcity | Neutropenia, neutrophil specific granule deficiency, granulocyte maturation arrest | Severe recurrent bacterial infections, sepsis and parasitic infections | Developmental delay, skeletal anomalies, dysmorphic features, delayed separation of umbilical cord, progressive myelofibrosis and MDS, chronic diarrhea | No | 4 | HSCT | 2017 (79) |
| Aip1 | WDR1 | 604734 | AR - CP | Ubiquitous | Neutrophils | Impaired neutrophil polarization, chemotaxis and survival due to impaired actin rearrangement | Mild neutropenia, herniation of neutrophil nuclear lobes, absence of neutrophil granules | Pyogenic bacterial infections of airways, skin and urinary tract; sepsis; disseminated lethal Varicella Zoster infection | Impaired wound healing, severe stomatitis with oral stenosis | Yes | 6 | HSCT | 2016 (83) |
| Defect in a lymphoid organ: isolated congenital asplenia | |||||||||||||
| Riboso mal protein SA | RPSA | 150370/ 271400 | AD - IP | Ubiquitous | PBMCs | Haploinsufficiency | Asplenia | Invasive bacterial infections | Isolated congenital asplenia | No | >30 | NA, penicilli n prophylaxis | 2013 (89) |
| Inborn errors of interferon-γ and IL-12 immunity | |||||||||||||
| TYK2 | TYK2 | 176941/611521 | AR - CP | Broadly expressed | EBV-B cells SV40-fibroblasts HVS-T cells PBMCs Fibroblasts | Impaired responses to type I IFN, IL-12/IL-23 and IL-10 | NA | Mendelian susceptibility to mycobacterial disease and infections with other intracellular organisms and viruses | Hyper-IgE syndrome reported in only one patient | No | 8 | NA | 2006 (96) 2012 (95) 2015 (94) |
| JAK1 | JAK1 | 147795 | AR - CP | Broadly expressed | PBMCs Fibroblasts | Impaired responses to type I and II IFN, IL-2, IL-4, IL-10, IL-27 | Progressive T lymphopenia, increased IgG and IgA | Mendelian susceptibility to mycobacterial disease, warts, superficial parasitic and fungal infections | Early onset bladder carcinoma | No | 1 | NA | 2016 (97) |
| ISG15 | ISG15 | 147571/ 616126 | AR - CP | Broadly expressed | PBMCs Granulocytes SV-40 fibroblasts EBV-B cells | Enhanced type I IFN response. Impaired IFN-γ production by NK and T cells | NA | Mendelian susceptibility to mycobacterial disease | Intracranial calcifications | Yes | 6 | NA | 2012 (100) |
| USP18 | USP18 | 607057/ 617397 | AR - CP | Broadly expressed | Brain tissue Fibroblasts | Enhanced STAT1 and STAT2 phosphorylation and transcription of ISGs upon type I IFN stimulation | Thrombocytopenia | NA | PseudoTORCH syndrome | Yes | 5 | NA | 2016 (104) |
| IRF8 | IRF8 | 601565/ 614894 | AR - CP | Hematopoietic cells | PBMCs Dermal APCs | Impaired IL-12/IFN-γ response, tissue macrophages and DC deficiency. Impaired NK cytotoxic function and differentiation | Monocyte and DC deficiency. NK cell deficiency with decreased CD56dim NK cells | Severe opportunistic infections, mycobacterial infections, candidiasis. Severe EBV, viral and bacterial infections | Myeloprolifer ation, lymphadenopathy, bronchiectasis | No | 4 | HSCT | 2011 (105, 107) |
| IRF8 | IRF8 | 601565/ 614893 | AD - CP | Hematopoie tic cells | PBMCs EBV-B cells | Reduced IL-12 response | Deficiency of CD11c+CD1c+ DCs | Mendelian susceptibility to mycobacterial disease | NA | No | 2 | NA | 2011 (105) |
| IRF4 | IRF4 | 601900 | AD - IP | Bone marrow, lymphoid tissues and gastrointesti nal mucosa | EBV-B cells PBMCs | Impaired Th cell responses | NA | Whipple’s disease (Tropheryma whipplei infection) | NA | No | 4 | NA | 2018 (109) |
| gp130 | IL6ST | 600694 | AR - CP | Ubiquitous | PBMCs EBV-B cells T lymphoblasts | Suppressed STAT1 and STAT3 activation and signaling upon IL-6 or IL-11 stimulation, impaired upon IL-27 or OSM stimulation. Absent acute phase response after stimulation with IL6. | Increased naïve B cells, reduced nonswitched memory B cells, eosinophilia, high IgE | Recurrent severe bacterial respiratory infections with bronchiectasis, | Skeletal anomalies, craniosynosto sis, absent acute phase response | No | 1 | NA | 2017 (112) |
| Inborn errors of IL-17 immunity | |||||||||||||
| STAT1 | STAT1 | 600555/ 614162 | AD - *CP GOF *almost CP | Broadly expressed | PBMCs EBV-B cells Fibroblasts | Increased responses to type I and II IFNs, IL-6 and IL-21, | Low Th17 proportions +/− low memory B cells | Chronic mucocutaneous candidiasis, bacterial and viral infections, invasive fungal infections, mycobacterial disease | Intracranial aneurysms, autoimmunity, enteropathy, bronchiectasis | Yes | >350 | JAK inhibitor (ruxoliti nib) HSCT | 2011 (119, 196) |
| RORγ/γT | RORC | 602943/ 616622 | AR – CP | RORγ broadly expressed RORγT thymocytes and fetal lymphocytes | PBMCs | Almost absent Th17 cells. Impaired IFN-γ production by γδ T cells and CD4+CCR6+CXCR3+ αβ T cells. | Modest T lymphopenia low ILC3, absence of NKT and MAIT cells | Disseminated mycobacterial infections and CMC | NA | No | 7 | NA | 2015 (197) |
| DNA polymer ase α−1 (PDR) | POLA1 | 312040/ 301220 | XL – CP | Ubiquitous | Fibroblasts | Constitutive activation of type I IFN response and pro-inflammatory genes | NA | Recurrent pulmonary infections with bronchiectasis | Type I interferonopa thy with chronic diarrhea, failure to thrive, IBD, urethral strictures, diffuse reticulate skin hyperpigmentation, hypohidrosis, corneal scarring, characteristic facies | Yes | 21 | NA | 2016 (198) |
| Inborn errors of the TLR3 pathway | |||||||||||||
| IRF3 | IRF3 | 603734 | AD - IP | Broadly expressed – relevant for CNS cells | PBMCs Fibroblasts | Impaired IFN response to AT-rich DNA, dsDNA and HSV-1 infection | NA | Herpes simplex encephalitis | NA | No | 2 | NA | 2015 (144, 145) |
| Inborn errors of intracellular viral sensing | |||||||||||||
| DBR1 | DBR1 | 607024 | AR – CP | Ubiquitous, but strongest in the spinal cord and brainstem | Fibroblasts | Deficient lariat processing and impaired control of viral infections | Congenital neutropenia (not fully penetrant) | Viral brainstem encephalitis | Growth retardation, mental retardation, curly hair | No | 5 | NA | 2018 (146) |
| MDA5 | MDA5 (IFIH1) | 606951 | AR – CP AD – IP LOF | Broadly expressed | Respiratory epithelial cells Fibroblasts | Impaired MDA5 enzymatic and IFNinducing function with dominant negative effect. Impaired response to HRV and RSV infection. | NA | HRV and other respiratory viral infections | NA | No | 10 | NA | 2017 (150-152) |
| RNA polymerase III | POLR3A POLR3C | 614258 617454 | AD – IP Digenic or monogenic | Ubiquitous | PBMCs Monocytesmacrophages Fibroblasts Keratinocytes | Impaired IFN response to AT-rich DNA and VZV infection | NA | Acute severe VZV infection with lung and CNS involvement | NA | No | 4 | NA | 2017 (153) |
| SAMD9 | SAMD9 | 610456/617053 | AD - IP GOF | Broadly expressed | Bone marrow cells Adrenal cortex cells Fibroblasts | Structural and functional alterations of the endosome system, inhibition of cell proliferation | Variable mild lymphopenia | Severe invasive fungal and bacterial infections | MIRAGE syndrome: MDS, infection, growth failure, adrenal hypoplasia, genital anomalies, enteropathy. Developmental delay, thrombocytop enia and anemia | Yes | 19 | HSCT | 2016 (161) |
| SAMD9L | SAMD9L | 611170 | AD - IP GOF | Broadly expressed | EBV-B cells PBMCs Bone marrow cells | Inhibition of cell proliferation | Pancytopenia | Bacterial and viral infections | Ataxia/cerebe llar atrophy, pancytopenia, MDS | No | 25 | HSCT | 2016 (163) |
| Inborn errors of type I and III IFN amplification and responses | |||||||||||||
| IRF7 | IRF7 | 605047/ 616345 | AR – CP | Fibroblasts PBMCs pDCs PECs | Impaired responses to type I and III IFNs | NA | Influenza virus lifethreatening infection | NA | No | 1 | NA, influenz a vaccinat ion | 2015 (199) | |
| STAT2 | STAT2 | 600556/ 616636 | AR – IP | Broadly expressed | Fibroblasts PBMCs | Impaired type I IFN response to viral infections | NA | Severe viral infections (disseminated vaccine-strain measles) | Mitochondrial anomalies. Kawasaki-like syndrome, HLH-like syndrome. | Yes | 9 | NA, avoid MMR live vaccinat ion | 2013 (169) |
| IFNAR2 | IFNAR2 | 602376/ 616669 | AR – IP | Broadly expressed | Fibroblasts | Impaired type I IFN responses to viral infections | NA | Disseminated vaccine-strain measles and mumps | NA | No | 2 | NA, avoid MMR live vaccinat ion | 2015 (170) |
| IRF9 | IRF9 | 147574 | AR – CP | Broadly expressed | EBV-B cells SV40-fibroblasts | Impaired type I IFN response to viral infections | NA | Severe viral infections (Influenza virus life-threatening infection, complications after MMR vaccination, other respiratory viral infections) | NA | No | 1 | NA | 2018 (175) |
| IFNAR1 | IFNAR1 | 107450 | AR Digenic with IFNGR2 mutation | Broadly expressed | Fibroblasts | Impaired type I IFN responses to viral infections | NA | Disseminated CMV, Streptococcus viridans sepsis, mycobacterial disease | HLH | No | 1 | HSCT | 2017 (176) |
| Inborn errors of NK cells / with NK cells affected | |||||||||||||
| MCM4 | MCM4 | 602638/ 609981 | AR - CP | Ubiquitous | Fibroblasts EBV-B cells PBMCs | DNA replication defect | NK cell deficiency (reduction of CD56dim NK cells, reduced proliferation and excess spontaneous apoptosis of NK cells) | Recurrent viral infections, especially herpesviruses; viral pulmonary infections leading to respiratory failure | Adrenal insufficiency, severe intra- and extrauterine growth retardation, microcephaly, DNA repair disorder and susceptibility to cancer | No | 24 | Corticos teroid replace ment therapy | 2012 (185, 186, 200) |
| GINS1 | GINS1 | 610608 | AR – CP | Ubiquitous | PBMCs Fibroblasts | DNA replication defect | NK cell deficiency, neutropenia, autoimmune cytopenia, lymphopenia | Bacterial/viral respiratory and GI infections, herpesviruses infections | Intra- and extrauterine growth retardation, facial dysmorphism, eczema, hypothyroidis m, cancer, enteropathy, glaucoma | No | 5 | G-CSF | 2017 (187) |
| GATA2 | GATA2 | 137295/ 614172 | AD – CP | Hematopoietic progenitors, nonhematopoietic embryonic stem cells, lymphatic cells | PBMCs Bone marrow cells | Absent or severely deficient CD56bright NK cells. Functionally impaired CD56dim NK cells. | Deficiency of monocytes, DCs, NK cells and B lymphocytes | Susceptibility to viral infections, mycobacteria, HPV, histoplasmosis | Alveolar proteinosis, MDS/AML, lymphedema, hear loss, miscarriage | No | >400 | HSCT | 2011 (188–191) |
Legend: AML: acute myeloid leukemia; APC: antigen presenting cell; AR: autosomal recessive; AD: autosomal dominant; CBM: CARD-BCL1054 MALT1 complex; CMV: Cytomegalovirus; CNS: central nervous system; CP: complete penetrance; DC: dendritic cell; EBV-B: EBV-immortalized B cell; G-CSF: granulocyte colony-stimulating factor; GI: gastrointestinal; GM-CSF: granulocyte-macrophage colony-stimulating factor; GOF: gain of function; HLH: hemophagocytic lymphohistiocytosis; HPV: Human papillomavirus; HRV: Human rhinovirus; HSCT: hematopoietic stem cell transplantation; HVS-T: herpesvirus saimiri-immortalized T-cell; IBD: inflammatory bowel disease; IFN: interferon; Ig: immunoglobulin; IL: interleukin; ILC: innate lymphoid cell; IP: incomplete penetrance; ISG: IFN-stimulated gene; LOF: loss of function; LUBAC: Linear ubiquitination chain assembly complex; MAIT: mucosa-associated invariant T cells; MDS: myelodysplastic syndrome; MMR: measles-mumps-rubella; n: 60 number; NA: not applicable; OMIM: Online Mendelian Inheritance in Man; PBMC: peripheral blood mononuclear cell; pDC: plasmacytoid DC; PDR: reticulate pigmentary disorder; PEC: pulmonary epithelial cells ref: reference; RSV: Respiratory syncytial virus; SCN: severe congenital neutropenia; SGD: specific granule deficiency; SV40-fibroblast: simian virus 40-immortalized fibroblast; Th: T helper; TLR: Toll-like receptors; 63 TORCH: toxoplasmosis, other agents, rubella, cytomegalovirus, herpes simplex; VZV: Varicella zoster virus; XL: X-linked.
OVERVIEW OF NOVEL INBORN ERRORS ALONG MAJOR INNATE IMMUNE PATHWAYS
A. Defects in the innate immune defense against commensal pyogenic bacteria and fungi
Pyogenic bacteria such as Streptococcus pneumoniae (S. pneumoniae) and Staphylococcus aureus (S. aureus) and fungi such as Candida albicans (C. albicans) are commensals of the human skin or mucosa and rarely cause severe disease. Detailed analysis of the immune system is therefore warranted in patients with invasive or recurrent pyogenic or fungal disease (18). The canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway is key in the innate immune response to this group of pathogens (Figure 1A) (19). Notably, proximal defects in the pattern recognition receptor signaling pathway underlie susceptibility to a narrow spectrum of pyogenic infections, while defects in more distal components of the NF-κB pathway result in broader phenotypes, including susceptibility to a range of infectious agents and developmental delay. Aside from NF-κBrelated immune responses, important pathways in bacterial and fungal killing are phagocytosis and the filtration of encapsulated bacteria by the spleen.
Figure 1: A).Recognition of conserved bacterial/fungal patterns by cell surface and endosomal TLRs and/or cell surface CLRs.
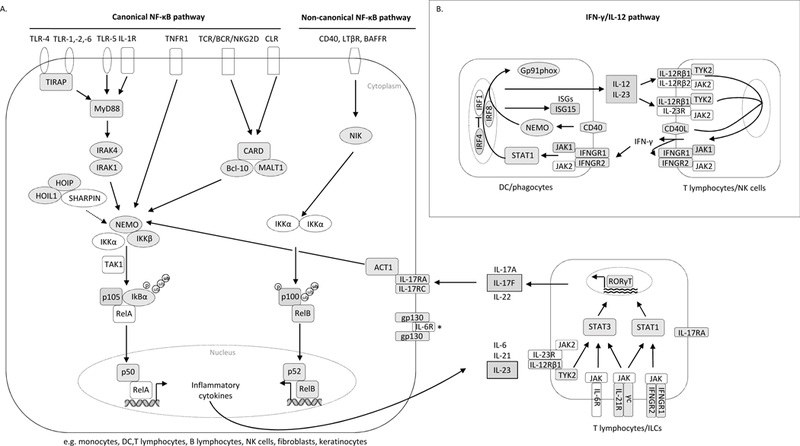
Bacterial components are recognized by TLR1/2, TLR2/6 and TLR4, triggering recruitment of the TIR domain-containing adapter molecules TIRAP and MyD88, while TLR5 and IL-1R directly activate MyD88, followed by recruitment of IRAK family members. Upon sensing of fungal infections, CLRs signal through the CARD9-BCL10-MALT1 complex. Both signaling pathways eventually lead to activation of the IKK complex (IKKα, IKKβ, and NEMO/IKKγ) and to the phosphorylation of IκBα via TAK1 (the classic MyD88-dependent pathway). After IκBα degradation, NF-κB heterodimers (p50:RelA or p50:cRel) translocate to the nucleus (canonical NF-κB pathway). By contrast, the noncanonical NF-κB pathway is induced upon activation of some TNF-R super-family members, such as CD40, BAFF-R or LT-β receptor, and involves the IKKα-mediated phosphorylation of p100 associated with RelB, leading to the generation of transcriptionally active p52:RelB complexes. IKKα activation and the phosphorylation of p100 depend on NIK activation. NFκB activation and translocation results in the induction of inflammatory cytokine genes (such as IL-21, IL-23 and IL-6). Upon binding to their receptors (expressed on T and innate lymphoid cells), the pro-inflammatory cytokines activate T cells via the transcription factor STAT3 and upregulate RORγT expression, resulting in the differentiation of these cells into IL17-producing T cells. *gp130 is a ubiquitously expressed protein scaffolding many cytokine receptors, such as IL6R, IL-11R, IL-27R, oncostatin M and other. B) IFN-γ/IL-12 response to mycobacterial infection and Salmonella. Upon mycobacterial infection, the IFN-γ/IL-12 axis is induced in APCs and T/NK cells. IL-12, IL-23 and IFN-γ are secreted in a positive loop between them, and through their receptors (IL-12Rβ1/2, IL-12Rβ1/IL-23R and IFNGR1/2) activate downstream signaling via TYK2, JAK1–2 and STAT1, finally inducing the transcription of ISGs. For all proteins shadowed in grey mutations have been described.
I. Defects in the NF-κB pathway
The NF-κB pathway consists of 1) a canonical pathway responsible for anti-microbial defense that relies on NF-κB essential modulator (NEMO)/ inhibitor of NF-κB kinase (IKK) γ-, IKKα- and IKKβ-mediated degradation of inhibitor of NF-κB (IκB) α/β/ε for the transfer of heterodimers formed by RelA or cRel and p50 to the nucleus, and 2) a non-canonical pathway that regulates lymphoid organogenesis, bone formation, B cell survival and function, and critically depends on NF-κB-inducing kinase (NIK) and IKKα-mediated processing of p100 into p52, to allow the formation of nuclear RelB:p52 dimers (figure 1). NF-κB operates downstream from TLRs/interleukin (IL)-1 receptor (IL-1R), via a TLRdependent adaptor, myeloid differentiation factor (MyD88), and the IL-1R-associated kinase (IRAK) complex, a dimer of two active kinases, IRAK-1 and IRAK-4. The canonical NF-κB pathway is also activated downstream from the B and T cell receptors (BCR, TCR), NK cell receptors (such as NKG2D), tumor necrosis factor (TNF) receptor superfamily, CLRs (dectin-1, dectin-2, Mincle), and its effects are mediated by the CBM complex, consisting of caspase recruitment domain (CARD) molecules such as CARD9 and CARD11, B-cell lymphoma 10 (BCL10) and mucosa-associated-lymphoid tissue lymphoma-translocation gene 1 (MALT1). Additionally, linear ubiquitination, catalyzed by the linear ubiquitin assembly complex [LUBAC, formed by HOIL-1 (heme-oxidized IRP2 ligase 1L), HOIP (HOIL-1-interacting protein) and SHARPIN (Shank-associated RH domain-interacting protein)], plays a crucial role in NF-κB activation.
1. Upstream defects of the TLR and/or IL-1R signaling pathway
Biallelic loss-of-function mutations in IRAK-4 and MyD88 were identified in 2003 and 2008, respectively, in children with pyogenic bacterial infections (IPD, staphylococcal and Pseudomonas aeruginosa infections) and little clinical and biological inflammation (20, 21). Of note, the incidence of IPD typically decreases from adolescence onward Mendelian and the mortality of these two inborn errors of immunity declines with age (22, 23).
IRAK1
The role of IRAK-1 in the TLR/IL-1R signaling pathway was partially unveiled with the description of a boy who did not suffer from invasive pyogenic infections but who had weak inflammatory responses, like MyD88- and IRAK-4 deficient patients. He carried a de novo Xq28 microdeletion spanning Methyl CpG Binding Protein 2 (MECP2) and IRAK1 (24). IRAK-1 is expressed in lymphocytes, granulocytes and monocytes, and any deficiency of this protein would therefore be likely to affect both innate and adaptive immunity. In vitro studies of the patient’s cells showed IRAK-1 to be largely redundant for TLR/IL-1R signaling in peripheral mononuclear blood cells (PBMCs) and for the IL-1R response in fibroblasts, but essential for signaling downstream from TLR2/6, TRL4, TLR7, and TLR8 in the patient’s fibroblasts and Epstein-Barr virus-immortalized (EBV-B) cells. However, as the patient died at seven months of age due to MECP2 deficiency-related encephalopathy, it was not possible to determine the complete clinical and cellular phenotypes of IRAK-1 deficiency. The identification and study of additional patients with IRAK-1 deficiency are therefore required.
TIRAP
In a child presenting with S. aureus pneumonia and sepsis, a rare homozygous variant of TIR domain-containing adaptor protein (TIRAP), necessary for recruitment of MyD88 to TLR2 and TLR4, was found (25). The authors showed that the mutation was loss-of-function (LOF) and resulted in severely impaired responses to TLR1/2 and TLR4 stimulation, and completely abolished responses to TLR2/6 in various cell types (Simian-virus 40 (SV40)-fibroblasts, granulocytes, monocytes) in vitro (25). Intriguingly, seven healthy relatives were homozygous for the same variant but had never suffered from severe bacterial infections. Anti-staphylococcal lipoteichoic acid (LTA) antibodies (Ab) have been shown to enhance response to LTA (a ligand of TLR2), and could not be detected in the patient’s plasma, yet were present in the healthy relatives. TLR2 activation in response to LTA was rescued in vitro by addition of anti-LTA Ab-containing control plasma or anti-LTA monoclonal Ab to the patient’s whole blood or PBMCs. Hence human AR TIRAP deficiency results in invasive staphylococcal disease only in the absence of anti-LTA Ab (25). This demonstrates the redundancy of TIRAP in the presence of anti-LTA Ab, underlying the interrelatedness of the innate and adaptive immune response: human adaptive immunity can in fact rescue an inborn error of innate immunity.
2. Inborn errors of NF-κB signaling
Defects in NF-κB activation lead to a broad range of developmental manifestations and infections due to impaired signaling pathways downstream of both innate and adaptive immune system receptors, as has recently comprehensively been reviewed by Zhang et al. (26). Autosomal dominant (AD) mutations in NFKB1, encoding p105/p50, and in NFKB2, encoding p100/p52, result in a common variable immunodeficiency (CVID) phenotype with recurrent infections and autoimmunity (27–32). Null mutations in IKBKG, encoding the IKKγ, also known as NEMO, have been described in 2000 to cause incontinentia pigmenti in females and early fetal loss in males, while hypomorphic mutations classically cause XL recessive anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (33, 34). The first AD form of EDA-ID was identified in 2003, due to gain-of-function (GOF)/hypermorphic mutations in NFKBIA, encoding IκBα, in a patient with a phenotype similar to XL-EDA-ID. In these disorders, hematopoietic stem cell transplantation (HSCT) can only cure the hematopoietic defect but not the cell-intrinsic immunity of non-hematopoietic cells (35, 36). Biallelic mutations in IKBKB, encoding IKKβ, were identified as the cause of disrupted NF-κB signaling in patients with clinical characteristics of severe combined immunodeficiency (SCID), including early onset severe viral, bacterial, and fungal infections, despite normal B and T cell counts. The patients displayed impaired T and B cell activation and proliferation, particularly with a defect of TCRγδ T cells, hypogammaglobulinemia, and NK cell deficiency (37, 38). Finally, homozygous LOF mutation in mitogen-activated protein kinase 14 (MAP3K14), encoding NIK, are responsible for the disruption of non-canonical NF-κB activation and a SCID phenotype with numeric and functional defects of B, T and NK cells (39).
3. Inborn errors of the CBM complex
CARD11, MALT1 and BCL10 deficiency all result in a similar phenotype of recurrent pyogenic and severe viral infections, yet an intestinal inflammatory phenotype predominates in MALT1 deficiency (40–43) and patients with CARD11 deficiency are typically highly susceptible to Pneumocystis infection (44, 45). Immunologically, patients present with a combined immunodeficiency with normal total numbers of T and B cells, mostly displaying a naïve phenotype, and impaired TCR and BCR responses (44–49). The underlying reason for these specific cellular phenotypes remains unclear, as BCL10 has been shown to be strongly expressed in bronchial and intestinal epithelial cells and, to a lesser extent, in lymphocytes and myeloid cells (47). MALT1 is expressed primarily in lymphocytes and myeloid cells and CARD11 in lymphocytes. Moreover, Perez-De Diego et al. reported a strong impairment of NF-κB mediated fibroblast function but normal monocyte-derived macrophage and dendritic cell responses to stimulation of TLR4, TLR2/6, and Dectin-1 in patients with BCL10 deficiency (47).
CARD9
First described by Glocker et al. in 2009, biallelic mutations in CARD9 are deleterious for the innate immune defense against invasive fungal infections (50). Affected patients are particularly susceptible to superficial (CMC) or invasive (central nervous system) Candida infections, superficial or deep dermatophytosis, subcutaneous or invasive phaeohyphomycosis (caused by Dermatiaceous fungi), invasive Exophiala infections and extrapulmonary aspergillosis (50–54). Pro-inflammatory cytokine or chemokine responses to various fungal agonists were found impaired in patients’ whole blood, monocyte-derived dendritic cells and macrophages (51, 53, 55, 56). Additionally, a defect of fungal killing by neutrophils and defective Th17 immunity were reported in some patients (55, 56). A recent study of two patients with invasive extra-pulmonary aspergillosis and null mutations of CARD9 showed that CARD9 -deficient neutrophils killed Aspergillus effectively but did not accumulate at sites of infection, an observation also made in Candida infection of the central nervous system (54, 56). However, the precise mechanism of susceptibility to invasive fungal infections in patients with CARD9 deficiency remains to be defined.
4. nborn errors of LUBAC
HOIL-1 and HOIP
Three patients from two kindreds presenting with invasive pyogenic infections associated with autoinflammation and amylopectinosis-related myopathy and cardiomyopathy were found to have autosomal recessive (AR) HOIL-1 deficiency due to homozygous LOF mutations in HOIL-1 (57). A patient with similar manifestations, together with viral infections, lymphangiectasia, lymphopenia, antibody deficiency and impaired T cell function harbored biallelic LOF mutations in HOIP (57, 58). Deficiencies of HOIL-1 or HOIP result in the destabilization of LUBAC, impaired linear ubiquitination of NEMO and, ultimately, weak NF-κB activation (57–60). Linear ubiquitination by LUBAC has recently been shown to restrict the growth of bacteria invading the cytosol by activating autophagy and NF-κB (61, 62). HOIP- and HOIL-1-deficient fibroblasts display impaired NF-κB activation in response to TNF and IL-1β (57, 58). By contrast, whole blood and monocytes from both HOIP- and HOIL-1-deficient patients responded strongly to IL-1β in terms of IL-6 production, which was significantly greater than the controls. The excessive monocyte response observed in these patients may account for their clinical autoinflammation (57, 58).
1I. Defects of phagocytes
Phagocyte defects comprise disorders in phagocytes number or function (adhesion, chemotaxis and/or killing) and were recently reviewed (63–65). Defects in the number of phagocytes such as congenital neutropenia present in infancy with severe and invasive pyogenic and fungal infections as well as mucosal inflammation, and are associated with an increased risk of myelodysplastic syndrome (MDS) or leukemia. Mutations in elastase neutrophil-expressed (ELANE) are responsible for around 50% of congenital neutropenia cases (65). A second set of defects results in impaired leukocyte adhesion, impaired phagocyte chemotaxis and/or motility, manifesting as delayed healing of the umbilical cord and omphalitis, bacterial/fungal infections and poor wound healing, but also with periodontitis and syndromic features (66). Finally, mutations in the genes encoding proteins responsible for superoxide production (gp91phox, p22phox, p47phox, p67phox and p40phox), and thus for bacterial and fungal killing, cause chronic granulomatous disease (CGD), manifesting with severe infections, especially in the form of abscesses, granulomata, and an autoinflammatory phenotype (67, 68). We here discuss a number of newly described genes responsible for neutrophil defects.
1. Severe congenital neutropenia
VPS45
Vilboux et al. and Stepensky et al. independently investigated thirteen children with severe congenital neutropenia (SCN) who did not carry mutations in ELANE and who were resistant to granulocyte colony-stimulating factor (G-CSF) treatment (69, 70). The patients all presented before six months of age with severe to fatal pyogenic and fungal infections (mainly with Pseudomonas, S. aureus and other gram-positive bacteria, and Aspergillus), manifesting as omphalitis, superficial and deep abscesses, pneumonia, osteomyelitis and sepsis. Neurological involvement and myelofibrosis with extramedullary hematopoiesis, causing hepatosplenomegaly and/or nephromegaly, were also striking. Five of them succumbed to infection and six received HSCT, four of whom survived (69–73). These patients were found to carry homozygous mutations in VPS45, encoding vacuolar sortingassociated protein 45 (69, 70). VPS45 is a regulator primarily associated with Golgi, transport vesicles and endosomal membranes. It is required for intracellular trafficking and recycling of proteins through the endocytic-lysosomal pathway. The described mutations lead to accelerated neutrophil and bone marrow myeloid cell apoptosis, as well as thrombocytopenia and thrombasthenia due to impaired granule metabolism (69, 70). Other lysosome-related proteins are responsible for neutropenia (e.g. in Chédiak-Higashi syndrome, Hermansky-Pudlak syndrome type 2 and Griscelli syndrome type 2), but the exact mechanism underlying the neutropenia in defects of endosomal-lysosomal proteins is unknown.
JAGN1
Another cause of SCN unresponsive to G-CSF was reported by Boztug et al. who identified homozygous mutations in the jagunal homolog 1 (JAGN1)-encoding gene JAGN1 in fourteen patients with SCN (74). They manifested pyogenic bacterial infections, myeloid maturation arrest and bone and teeth abnormalities. Sixteen patients have been reported so far, one succumbed to sepsis and two were successfully treated with HSCT (74, 75). JAGN1 is an endoplasmic reticulum (ER) protein involved in vesicular trafficking between the ER and the Golgi. It plays a crucial role in neutrophil differentiation and survival (74). JAGN1 deficiency affects primarily neutrophils, which display scarce specific granules as well as aberrant ER structures and function, resulting in anomalous N-glycomic profiles and increased apoptosis (74). Furthermore, the defect of N-glycosylation also affects the G-CSF receptor, possibly explaining the common G-CSF resistance in these patients.
CSF3R
Biallelic defects of the G-CSF receptor-encoding gene CSF3R are responsible for another AR form of SCN, characterized by peripheral neutropenia without granulocyte maturation arrest and complete resistance to G-CSF treatment (76, 77). The expression of the mutant receptor on the cell membrane is severely reduced or completely abolished (76, 77) leading to absence of G-CSF response and neutropenia. Of note, mutations of CSF3R have been associated with acute myeloid leukemia in SCN. These acquired, somatic mutations affect the transmembrane or intracellular domain of the receptor, whereas the mutations underlying neutropenia are germline and affect the extracellular domain of the protein [reviewed in (78)].
SMARCD2
Witzel et al. recently reported four patients with delayed separation of umbilical cord, intractable diarrhea with failure to thrive, pneumonia, abscesses and recurrent sepsis, dysmorphic features and skeletal anomalies. Bone marrow analysis demonstrated scarcity of neutrophils with a granulocyte maturation arrest, neutrophil specific granule deficiency and a progression to MDS with blast excess in all patients; furthermore, one patient developed myelofibrosis (79). Two patients have undergone successful HSCT at 1 and 2 years of age respectively, and two succumbed to septic shock at one month and at five years of age (79). In these patients, homozygous mutations in SWI/SNF-related, matrix-associated, actindependent regulator of chromatin, subfamily D, member 2 (SMARCD2) were identified. SMARCD2 is part of a SWI/SNF (SWItch/Sucrose Non-Fermentable) complex, a family of chromatin-remodeling factors responsible for the unwrapping of nucleosomes, necessary for the regulation of differentiation in hematopoietic stem cells. Priam et al. have confirmed the essential role of SMARCD2 in granulopoiesis and granule production in neutrophils (80).
2. Defects in chemotaxis
WDR1
In the ‘70s, a defect of neutrophils actin polymerization and chemotaxis was described (the so-called “lazy leukocyte syndrome”), but no genetic cause could be determined (81, 82). Affected patients displayed mild neutropenia, abnormal neutrophil migration/mobilization and abnormal neutrophil morphology with nuclear lobe herniation and absence of cytosolic granules. In 2016, Kuhns et al. described five additional patients with lazy leukocyte syndrome that suffered from recurrent sinopulmonary and life-threatening systemic pyogenic infections, as well as severe chronic stomatitis, leading to oral stenosis (83). One patient succumbed to overwhelming infection and HSCT was life-saving in another patient. The group identified biallelic mutations in WD repeat-containing protein 1 (WDR1), the gene encoding actin-interacting protein 1 (Aip1), as a cause of this syndrome (83). Aip1 is required for adequate actin rearrangement and Aip1-deficient neutrophils display defects in polarization, chemotaxis and survival. Another homozygous mutation of WDR1 was recently reported as the cause of severe autoinflammatory disease, periodic fever, severe stomatitis with oral stenosis and immunodeficiency with severe recurrent infections in two girls from a consanguineous family (84). They displayed increased inflammasome activation with high serum IL-18 levels and poor T cell proliferation in vitro, but no neutropenia. One patient succumbed to the disease and the other has been successfully treated with HSCT (84). These findings underline once again the key role of the actin cytoskeleton in human immune responses (85).
III. Defect in a lymphoid organ: isolated congenital asplenia
The spleen is a secondary lymphoid organ, organized in white and red pulp separated by a marginal zone, serving critical functions both in innate and in adaptive immunity (86). Especially the marginal zone macrophages constitute the first defense against many viruses and bacteria that are filtered from the bloodstream. The adaptive immune response originating from the spleen is pivotal in the production of IgM-expressing memory B cells and their non-adherent reaction to polysaccharide antigens. Hence anatomic or functional asplenia, together with EDA-ID due to NEMO deficiency or IKBKA GOF mutations, and with IRAK4- and MyD88-deficiency, confers the highest risk to invasive infections with pyogenic, encapsulated bacteria (S. pneumoniae, Haemophilus influenzae, Neisseria meningitidis) (87).
RPSA
Isolated congenital asplenia (ICA), the absence of a spleen at birth without associated congenital anomalies, such as cardiopathy as part of heterotaxy syndromes, had been described as a hereditary trait as early as in 1968 (88). However, the genetic cause of this inborn error of immunity remained enigmatic until Bolze et al. brought together a cohort of 33 patients with ICA presenting with fulminant bacterial infections in early childhood. Hypothesizing genetic homogeneity, they identified heterozygous mutations in ribosomal protein SA (RPSA) as underlying ICA in 18 patients from 8 kindreds (89). The described mutations resulted in haploinsufficiency of RPSA, with complete penetrance. The cohort of patients has now expanded to include 28 new kindreds. Nine new heterozygous proteincoding mutations are reported. Two mutations located in the 5’-UTR and five protein-coding mutations display incomplete penetrance (90). RPSA is part of the small subunit of the ribosome, is ubiquitously expressed, and is involved in pre-ribosomal RNA processing. The pathogenesis of ICA and the role of RPSA in this context are not yet understood. The phenotype associated with RPSA deficiency is striking, since haploinsufficiency in 15 other ribosomal proteins has been associated with Diamond-Blackfan anemia and not with splenic anomalies.
B. Defects in the innate immune response to mycobacteria, Salmonella and Candida
The elucidation of the molecular mechanisms underlying MSMD has improved our understanding of the immune response to mycobacteria. All described diseases leading to MSMD affect the interferon (IFN)-γ/IL-12 axis that, through secretion of IFN-γ, IL-12 and IL23, and induction of interferon stimulated genes (ISGs), forms a loop between APCs and T/NK cells. The IFN-γ/IL-12 axis can be impaired by deleterious mutations in genes encoding pathway components, such as IFN-γ receptor 1/2 (IFNGR1/2), signal transducer and activator of transcription 1 (STAT1), tyrosine kinase 2 (TYK2) and IL-12, but also other proteins, such as cytochrome B-245 beta chain (CYBB, or gp91phox), necessary for oxidative burst in macrophages, and NEMO, which regulates the CD40-dependent production of IL-12 (Figure 1-B). Some MSMD-causing defects are also responsible for susceptibility to infection with a narrow spectrum of other pathogens, such as Salmonella, viruses and Candida. Indeed, C. albicans-induced activation of the NF-κB pathway in phagocytes results in cytokine production, such as IL-12 and IL-23, which leads to activation of T helper (Th) cells (91). This results in retinoic acid-related orphan nuclear receptor γ T (RORγT)-mediated cellular differentiation and IL-17 release by Th17 cells, connecting IL-23 function with Candida immunity (Figure 1-A). Knowledge on IL-17 immunity up to 2012 has been reviewed in Puel et al. (92). For a comprehensive review of MSMD-causing mutations published before 2013, we refer to Bustamante et al. (93).
I. Inborn errors of interferon-γ and IL-12 immunity
TYK2
Kreins et al. and Kilic et al. described a total of seven new patients from five families and four ethnicities with mycobacterial (Bacille Calmette-Guérin (BCG) and tuberculosis), intracellular bacterial (e.g. Brucella) and/or viral infections, harboring homozygous mutations in TYK2 (94, 95). None of the patients displayed any of the signs of hyper-IgE syndrome (HIES) as reported for the first patient described with AR TYK2 deficiency (96). TYK2 is expressed in T, B, and NK cells, myeloid cells and monocytes, and its scaffolding role contributes to the cell surface expression of type I IFN receptor 1 (IFNAR1), IL-12Rβ1 and IL10R2. As expected, the EBV-B cells of the TYK2-deficient patients did not express these receptors on the surface (94). The responses of both T cells and NK cells to IL-12 in these patients were impaired but not abolished, accounting for the MSMD. However, TYK2deficient patients had normal ex vivo percentages of IL-17-producing T cells and did not present a CMC phenotype, despite an impaired response to IL-23 in vitro. Their response to IFN-α/β was completely abolished in Herpesvirus saimiri-immortalized (HVS)-T cells and impaired in EBV-B cells and Simian Virus 40 (SV-40)-immortalized fibroblasts, accounting for the viral phenotype. Finally, TYK2-deficient patients’ EBV B cells showed very low basal expression of IFN-λ-inducible genes, suggesting a role of TYK2 in their regulation.
JAK1
A young man suffering from recurrent non-tuberculous mycobacterial infection, warts, onychomycosis and scabies, was found to harbor two homozygous missense mutations in janus kinase 1 (JAK1) (97). The patient succumbed to early onset bladder carcinoma at 23 years of age. Many cytokines including IL-2, IL-4, IL-7, IL-9, IL-15, IL-21, IL-27, IL-6 family and IL-10 family cytokines signal through JAK1. Together with TYK2 and JAK2, JAK1 is a scaffolding protein for type I and II IFN receptors, and acts downstream by phosphorylating STAT proteins, such as STAT1 and STAT2. The authors showed impaired phosphorylation of JAK1 and of STAT1/2 upon stimulation of type I or II IFN receptors, and reduced ISGs transcription (97). Also, they showed reduced amounts of phosphorylated TYK2 and JAK2 upon stimulation with IFN-alpha or – γ respectively. Together these broad signaling defects explain the observed susceptibility to non-tuberculous mycobacteria. Interestingly, they demonstrated that JAK1 kinase activity is needed to activate the signaling downstream from type I but not type II IFN, as IFN-γ stimulation induced STAT1 phosphorylation also in the presence of a loss-of-kinase mutation in JAK1. The occurrence of carcinomas has been previously described in patients treated with JAK1 inhibitors for immunological disorders, in line with the presentation of this patient (97, 98).
ISG15
Bogunovic et al. studied three patients from two unrelated consanguineous families presenting with MSMD and asymptomatic intracranial calcifications and three Chinese siblings with seizures and intracranial calcifications reminiscent of Aicardi-Goutières syndrome, the prototypic type I interferonopathy (reviewed in (99)). All patients harbored homozygous LOF ISG15 mutations (100, 101). The absence of BCG vaccination in some members of one family homozygous for the mutation accounted for the incomplete penetrance of the infectious phenotype (101). ISG15 is an ubiquitin-like molecule. It is induced by type I IFNs, and conversely it contributes to their downregulation. Primarily expressed by monocytes, myeloid cells, NK cells, T and B cells, it can covalently bind other proteins in a process called ISGylation. ISG15 also exists in a free form that can act intra- or extracellularly, exercising an immunomodulatory function on several immune cells, including neutrophils, NK cells, monocytes and lymphocytes, and promoting antimicrobial responses, for example by driving IFN-γ production (102). Bogunovic et al. demonstrated that granulocytes were the major source of extracellular ISG15 and that NK cells were the key ISG15-responsive leukocytes in terms of IFN-γ secretion, accounting for the MSMD phenotype observed in vivo (100). This neutrophil/NK cell loop can be seen as the innate adjunct to the IL-12/IFN-γ circuit of the adaptive immune system, which is also implicated in MSMD. The authors also showed that intracellular ISG15 deficiency prevented ubiquitin specific peptidase 18 (USP18) accumulation. USP18 inhibits IFN-α/β signaling, and its decrease is consistent with the higher levels of ISG expression in the patient’s blood (103). The observed phenotypic dichotomy of this monogenic disease, with both MSMD and autoinflammation, reflects the different roles of extracellular (secreted) and intracellular ISG15. Unsurprisingly, AR human USP18 deficiency due to biallelic LOF mutations of USP18 was recently described as a new type I interferonopathy presenting as pseudo-TORCH syndrome (neonatal neuroinflammation in the absence of typical TORCH infections) (104). Intriguingly, despite the demonstration of a role for ISG15 in antiviral defenses in mice, patients with ISG15 deficiency do not present a phenotype of susceptibility to viral infections (103).
IRF8
Hambleton et al. reported an infant presenting with disseminated BCG and CMC requiring HSCT, and two otherwise healthy subjects with a history of recurrent disseminated but curable BCG disease. All three subjects turned out to harbor mono- or biallelic mutations in IFN regulatory transcription factor 8 (IRF8) (105). IRF8 is a transcription factor expressed only in hematopoietic cells. It is a differentiation factor for granulocytes and macrophages, and is necessary for the development of dendritic cells. AR IRF8 deficiency was associated with the absence of conventional CD11c+ myeloid DCs, which secrete IL-12, and CD123+ plasmacytoid DCs (pDCs), the major producers of type I IFNs. This DC deficiency was accompanied by monocytopenia with granulocytic hyperplasia and high NK cell counts resembling hemophagocytic lymphohistiocytosis (HLH) (105, 106). T cells were normal in number, but displayed impaired IL-17 secretion in response to microbial or mitogen challenges. This finding suggests that T cells nurtured in the absence of an intact DC environment are functionally abnormal. AD IRF8 deficiency is associated with a DC deficiency limited to CD1c+CD11c+ cells (105). These findings demonstrate that the role of IRF8 in the human innate immune system extends beyond involvement in the transcriptional response of myeloid cells to IFNs and TLR agonists, to the control of DC development. Intriguingly, the reported patients did not have a phenotype of increased susceptibility to viral infections. Recently, AR IRF8 deficiency has been reported in a family with severe EBV infection and isolated NK cell deficiency of the CD56dim subset (107), and in a patient with a broad phenotype, including severe recurrent viral infections, DC and monocyte deficiency and T, B and NK cell dysfunction (108). The widespread immunological manifestations of different IRF8 LOF mutations point to a broader role for this transcription factor than previously thought.
IRF4
Guérin et al. recently described a multiplex non-consanguineous family where four individuals affected by Whipple’s disease (life-threatening intestinal and systemic infection by Tropheryma whipplei) were found to carry a heterozygous LOF mutation in IRF4 (109). There was incomplete penetrance for Whipple’s disease in the kindred. IRF4 is a lymphocyte-specific transcription factor with pleiotropic effects in T and B cells. In contrast to other IRF proteins, its expression is not regulated by IFNs, but by pathways responsible for lymphocyte activation (110). IRF4 is required for the proliferation of CD4+ T cells in response to mitogens and for the production of IL-2, IL-4, and IFN-γ, thus regulating both Th1 and Th2 responses. Moreover, it is essential to drive Th17 responses, partly through activation of RORγT. Finally, it can bind IFN-stimulated response element (ISRE) promoters and either promote or repress the transcription of ISGs, while also exercising a negative regulation on TLR signaling (110, 111). Negishi et al. have in fact shown that IRF4 is induced upon TLR activation and exercises a negative-feedback regulation on the TLR signaling, possibly by competing with IRF5 for binding to MyD88 (111). In vivo, IRF4-knock-out mice show a more potent inflammatory response and succumb more rapidly than controls upon TLR stimulation (111). In the index family with IRF4 deficiency, the authors demonstrated loss of DNA binding ability to the ISRE promoters and of the induction of transcription by the mutated IRF4, resulting in impairment of multiple signaling pathways (for instance IFNγ/STAT1, lymphotoxin-alpha (also known as TNF-β), IL-2RA). IRF4 haploinsufficiency is another example of how single-gene inborn errors of immunity can underlie life-threatening infectious disease during primary infection in otherwise healthy individuals.
IL6ST
Schwerd et al. studied a child with recurrent, early, severe bacterial respiratory and skin infections, bronchiectasis, skeletal anomalies including craniosynostosis, eczema and high IgE levels compatible with HIES, and found a homozygous LOF mutation in IL-6 signal transducer (IL6ST), encoding gp130 (112). Gp130 is a ubiquitously expressed cytokine receptor subunit involved in the recognition and signaling of multiple ligands, including IL-6, IL-11, IL-27 and oncostatin M (OSM). The authors showed that gp130 deficiency caused a complete loss of IL-6 signaling as measured by STAT3 phosphorylation in the patient’s q LOF (112, 113). This highlights the essential contribution of the IL-21/STAT3 axis, but not gp130, to Th17 subset differentiation.
II. Inborn errors of IL-17 immunity
The key role of the IL-17 pathway in the defense against C. albicans was demonstrated by the discovery of AD IL-17F and AR IL-17 receptor A (IL-17RA), IL-17RC and NF-κB activator 1 (ACT1) deficiencies underlying “isolated” CMC. Interestingly, the phenotype of IL-17RA deficiency was recently reported to be broader than initially thought (114). Indeed, Lévy et al. showed that 14 of 21 patients from 12 kindreds had staphylococcal skin disease, and that eight of these 21 patients had recurrent bacterial respiratory tract infections. These data suggest that IL-17RA is essential for mucocutaneous immunity to C. albicans in humans, and, to a lesser extent, for protection of the skin and mucosae against Staphylococcus aureus. Moreover, Conti et al(115). studied the mucosal function of the IL-17 pathway in a mouse model of oropharyngeal candidias and found neutrophil recruitment to the tongue of infected mice to be severely impaired in IL-23- and IL-17RA-deficient mice compared to wild type or IL-12-deficient mice (116). Therefore, despite belonging principally to the adaptive immunity, IL-17 pathway defects also affect the cell-intrinsic immunity of non-hematopoeietic tissue, such as fibroblasts, keratinocytes or epithelial cells.
STAT1
AD STAT1 GOF mutations were first described in 2011 (117). Recent advances in CMC genetics have shown that in over 50% of CMC cases a STAT1 GOF mutation can be found, and more than 350 patients with STAT1 GOF mutations have been described to date (117–119). The phenotype extends to bacterial, viral, mycobacterial, and invasive fungal infections (118). Forty percent of patients with STAT1 GOF mutations have autoinflammatory (innate) and autoimmune (adaptive) manifestations, especially hypothyroidism (118). The stronger IFN signaling found in STAT1 GOF mutations likely underlies these features. Other clinical features include aneurysms and increased risk of carcinoma, and recently a high prevalence of STAT1 mutations has been described in patients suffering from JC virus-induced progressive multifocal leukoencephalopathy (PML) (120). Immunologically, patients with STAT1 GOF mutations often have low proportions of circulating IL-17-producing T cells. A number of patients have also been found to have low memory B-cell and IgG2 and/or IgG4 levels (118). Based on the knowledge that janus kinase (JAK) 1 and 2 act upstream of STAT1 dimers, the JAK1/2 inhibitors ruxolitinib and more recently baricitinib, have been successfully introduced into the treatment of patients with STAT1 GOF mutations, to reverse autoimmunity and to treat CMC (121–125). However, major adverse effects, such as infection and a cytokine rebound effect, have been reported in patients treated with ruxolitinib for STAT1 GOF mutations and other conditions (126–128). Recently, an international retrospective study on a cohort of 15 patients with STAT1 GOF mutations who received HSCT showed poor overall survival (40%) and poor event-free survival (10%) (129).
RORC
Okada et al. studied seven patients from three unrelated kindreds presenting with mild CMC and severe mycobacterial infections (130). All patients developed disseminated mycobacterial infection and one died of BCG meningoencephalitis. Deleterious biallelic mutations of retinoic acid receptor-related orphan receptor C (RORC), encoding RORγ and RORγT, were identified as the underlying genetic cause. The disease showed an AR transmission and was fully penetrant. RORγT and RORγ are transcription factors, the first restricted to leukocytes and the second ubiquitously expressed. Consistent with the role of RORγT as a master transcription factor for the production of IL-17, the patients had abnormally low frequencies of circulating IL-17A, IL-17F- and IL-22-producing T cells and ILC3, accounting for their CMC (131). On the other hand, impaired antigen-specific IFN-γ production by γδ- and CD4+CCR6+ αβ T cells probably accounted for their mycobacterial disease, to which their lack of mucosa-associated invariant T cells (MAIT) and NKT cells could contribute (130).
POLA1
XL reticulate pigmentary disorder (PDR) is a type I interferonopathy characterized by autoinflammation and immunodeficiency. An intronic mutation in DNA polymerase α 1 (POLA1), affecting splicing and reducing the expression of the protein, was recently found in 21 patients belonging to 12 kindreds (132). The hemizygous males manifest reticular pigmentation abnormalities with corneal inflammation and hypohydrosis, failure to thrive due to inflammatory bowel disease, characteristic facies and recurrent severe respiratory infections, mostly pyogenic but also mycobacterial (132). An augmented type I IFN response was demonstrated in POLA-1 deficient fibroblasts, with enhanced expression of IRF- and NFκB-induced genes, such as ISGs, accounting for the autoinflammatory features of the syndrome. Moreover, the authors proved that POLA-1 was involved in the generation of cytosolic RNA:DNA, which in turn is a downregulator of the IRF pathway: a deficient POLA-1 polymerase activity in the cytoplasm leads to reduced cytosolic RNA:DNA in the patients’ cells and enhanced expression of IRF target genes. Finally, the patients showed low plasma levels of IFN-γ and IL-17A, which could explain the infectious phenotype (132).
C. Defects in the innate immune response against viruses
Host antiviral defense is based on the recognition of viral nucleic acid intermediates by membrane-bound TLRs or in the cytoplasm by intracellular viral sensors for DNA or RNA, such as RNA-sensing retinoic acid-inducible gene I (RIG-I), RIG-like receptors (RLRs), such as melanoma differentiation-associated protein 5 (MDA5), and DNA sensors. Recognition of the microbial ligand by these receptors induces the production of antiviral mediators, such as type I and III (IFN-λ) IFNs, via IRF3 and IRF7, through unique adaptors [TIR-domain-containing adaptor molecule 1 (TRIF), mitochondrial antiviral signaling protein (MAVS) and stimulator of IFN genes (STING)] (133, 134). Type I and III IFNs then activate innate cells such as NK and ILCs to eliminate virus-infected cells. The importance of the strict regulation of these pathways has been shown by the study of type I interferonopathies, a clinically heterogenic group of hereditary diseases with a constitutive activation of this pathway (Figure 2) (99).
Figure 2: Recognition of viral nucleic acids by endosomal TLRs, cytosolic RLRs and DNA sensors.
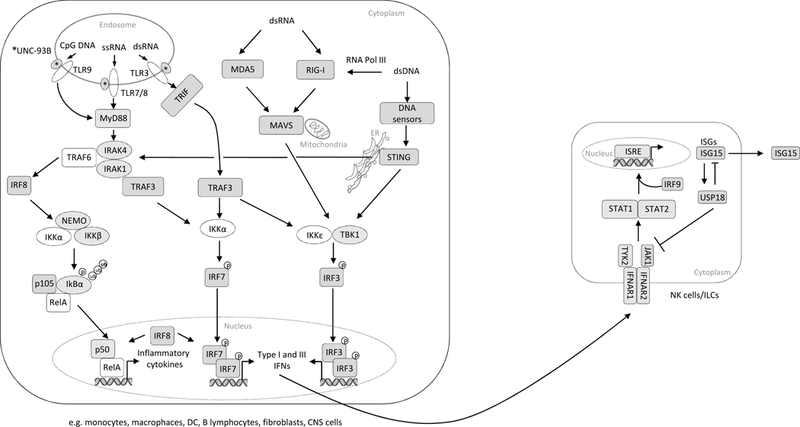
The UNC-93B chaperone protein guides TLR7/8 and TLR9 trafficking from the ER to the endosome, where these receptors recognize viral ssRNA and CpG DNA, respectively. These TLRs recruit MyD88, IRAK4 and IRAK1, which activate TRAF3 and TRAF6. TRAF6, in turn, mediates the activation of NF-κB, leading to the induction of inflammatory cytokine genes, whereas TRAF3 activates IKKα, which catalyzes the phosphorylation of IRF7 and induces type I IFN genes (the classic MyD88-dependent pathway). After recognizing the viral dsRNA, UNC-93B-dependent endosomal TLR3 recruits the adapter molecule TRIF for the activation of TBK1 and IKKε, followed by the activation of IRF3 and upregulation of IFN production (the alternative TRIF-dependent pathway). IRF8 facilitates the activation of NF-κB and IRF7. RLRs, such as MDA5 and RIG-I recruit mitochondrial MAVS adapter molecules after the recognition of cytoplasmic viral dsRNA. Various cytoplasmic dsDNA sensors signal via the ER-located adapter molecule STING. The activation of these RNA and DNA signaling pathways results in the production of inflammatory cytokines via NF-κB activation, and IFN production via TBK1-IKKε-IRF activation. Secreted type I IFNs interact with their surfaceexpressed receptor IFNAR1/2, leading to the recruitment and activation of TYK2 and JAK1, which in turn phosphorylate and activate STAT1 and STAT2. STAT1/STAT2/IRF9 form complexes that interact with ISRE promoters for the induction of ISGs expression. Intracellular ISG15 is type I IFN-inducible and is involved in the USP18-dependent regulation 47 of type I IFN and the prevention of type I IFN-dependent auto-inflammation. For all proteins 48 shadowed in grey mutations have been described.
I. Inborn errors of the TLR3 pathway
TLR3 binds double strand (ds) RNAs, leading to activation of a signaling cascade, in which the adapter molecule TRIF induces polyubiquitination of tumor necrosis factor receptor (TNFR)associated factor 3 (TRAF3) and the activation of IKKɛ and TANK-binding kinase 1 (TBK1), resulting in IRF3 phosphorylation and the induction of a type I IFN response (Figure 2) (135). Human defects in several components of this pathway (TLR3, TRIF, TRAF3, TBK1 and UNC93B1 (115, 136–140)) have already been described as genetic etiologies of HSE, often displaying incomplete penetrance. HSE is a rare (1–2 cases/500,000/year) and devastating manifestation of primary HSV-1 infection that mostly affects children between the ages of six and 36 months (141). TLR3 has proved largely redundant for responses to dsRNA in various leukocytes, but not in neurons, accounting for the neuron-specific phenotype of TLR3 deficiency. Additionally, TBK1 has been implicated in autophagy, indicating a role in the cell-intrinsic immunity of neurons (142, 143).
IRF3
An additional important player in the TLR3 pathway was identified through the genetic study of patients with sporadic HSE (144, 145). Two patients were found to carry different heterozygous mutations in IRF3, resulting in an impaired type I and III IFN response to HSV-1, as demonstrated in PBMCs and fibroblasts in vitro (144, 145). Like TRAF3 and TBK1, IRF3 encodes a downstream component of the TLR3 pathway, but deficiencies of any one of these proteins result in a narrow phenotype, primarily affecting the central nervous systemresident cells.
II. Inborn errors of intracellular viral sensing
DBR1
Zhang et al. recently described an AR and fully penetrant genetic cause of brainstem viral encephalitis in five children from three kindreds (146). Four children presented with brainstem encephalitis from different viruses (HSV-1 and Influenza B) between the age of ten months and 12 years, and two died from the infection. A fifth child manifested brainstem encephalitis from Norovirus at the age of six months, but had a broader phenotype including growth and mental retardation, curly hair and congenital neutropenia. The authors identified biallelic LOF mutations in the gene encoding the only known human RNA lariat debranching enzyme (DBR1), resulting in severely impaired expression and enzymatic activity of DBR1. DBR1 acts by hydrolyzing 2’-to-5’ branched phosphodiester bonds at the branch point of lariat intron RNA, and converting them into linear molecules and ensuring their rapid turnover. It is most represented in the peripheral and central nervous system, especially in the spinal cord and brainstem (147). In primary fibroblasts from the patients, the abundance of intronic lariat RNA was accordingly elevated, with a further marked increase after HSV-1 infection. Moreover, the authors showed impaired control of viral infections in patients’ fibroblasts, despite intact TLR3- and IFN-responsive pathways. In fact, DBR1 deficiency did not disrupt the TLR3-IFN-α/β and IFN-α/β-STAT1 responses, implying a different, at present still elusive pathogenic mechanism than those underlying HSE of the forebrain.
MDA5
Rare heterozygous GOF mutations of IFN-induced helicase C domain-containing protein 1 (IFIH1), or melanoma differentiation-associated gene 5 (MDA5), cause AD Aicardi-Goutières syndrome (148, 149), a type I interferonopathy. A child suffering from severe, recurrent viral respiratory tract infections by human rhinovirus (HRV), influenza virus, and respiratory syncytial virus (RSV), and transiently low levels of Igs, T, B and NK cells, was shown to carry a homozygous LOF mutation in MDA5 (150). The authors showed that MDA5 played a nonredundant protective role in the human airways, in sensing HRV and initiating innate immune responses against this virus, but not the other viruses, despite in vivo clinical susceptibility (150). High levels of HRV replication were observed in MDA5-silenced respiratory epithelial cells or fibroblasts and nasal epithelial cells from the MDA5-deficient patient, and the phenotype was rescued by transduction of fibroblast clones with wild-type MDA5. Additionally, Asgari et al. identified eight children carrying three putative LOF variants (homozygous in one) of MDA5 in a group of 120 previously healthy children requiring intensive care support for severe illness caused by respiratory viruses (HRV and RSV) (151). These variants were found to have a high allele frequency in the general population (1.9%), and were proved to disrupt MDA5 signaling function, enzyme activity and protein stability in vitro. Moreover, mutant MDA5 protein interfered with the function and stability of the wild-type protein, suggesting a dominant-negative effect, possibly involving prevention of the formation of normal multimeric MDA5 filaments. The authors suggested that protective effect against type I diabetes and other autoimmune diseases might account for this high frequency (151). Zaki et al. described an additional patient (152). The broad spectrum of MDA5 mutations again demonstrates the delicate balance between efficient host defense, inflammation and auto-immunity.
POLR3A and POLR3C
Ogunjimi et al. have recently identified four children, from a group of 21 patients affected by acute severe Varicella virus (VZV) infection (involving the lung or the central nervous system), harboring mutations in RNA polymerase III (POLR3) (153). Two patients displayed heterozygous mutations of either POLR3A or POLR3C, and the other two patients carried a heterozygous mutation in both genes. The penetrance of the disease is incomplete. The authors demonstrated that induction of IFN type I and III response by AT-rich DNA in human leukocytes is POL III–dependent, and that in vitro stimulation of patient-derived leukocytes with synthetic AT-rich DNA (poly(dA:dT)) or VZV genome elicits an impaired IFN response. However, the type I IFN response was normal in patients’ PBMCs upon stimulation of the RIG-I/MAVS pathway with poly(I:C), downstream from POL III (153). Finally, they showed that patients’ PBMCs were not able to restrict VZV virus replication in vitro, and that the transduction with wild type POLR3C or POLR3A rescued the control of viral replication in patients’ deficient cells (153).
SAMD9 and SAMD9L
Very recently, sterile alpha motif domain-containing protein 9 (SAMD9) was shown to be a host innate antiviral factor against poxvirus (e.g. vaccinia and myxoma virus), Japanese encephalitis virus and low-risk Papillomavirus in humans (154–159). Mutations in SAMD9 and its paralog SAMD9-like (SAMD9L) are associated with a heterogeneous clinical phenotype. SAMD9 bears 59% amino acid similarity to SAMD9L. Both are located on 7q21 and are tumor suppressors, primarily expressed in hematopoietic cells and induced by TNF-α, IFN-α and IFN-γ. Upon viral infection, SAMD9 and SAMD9L downregulate IFN-driven hematopoiesis. Recently, two groups have identified heterozygous GOF mutations in SAMD9 in children with growth failure, adrenal hypoplasia, genital anomalies, severe enteropathy, thrombocytopenia and anemia (160, 161). Most patients succumbed in infancy due to the hematological manifestations or due to infection with fungi (Candida) or bacteria (mostly Staphylococcus). Thymic size was small. Patients with SAMD9L GOF mutations instead develop ataxia-pancytopenia syndrome, with variable severity, and some experience viral infections, such as Cytomegalovirus (CMV) (162, 163). A subset of patients showed hematopoietic monosomy 7 (by uniparental disomy), deletion of 7q (so-called adaptationby-aneuploidy) or neutralization of the GOF mutation by LOF mutations in SAMD9 or SAMD9L (160–162, 164, 165). These adaptations were dynamic, progressive and often multiple and associated with increased survival. However, a subset of patients with monosomy 7 and 7q- developed MDS, probably due to haploinsufficiency of other 7q21 genes (162–165).
III. Inborn errors of type I and III IFN amplification and responses
IRF7
In an otherwise healthy child suffering from life-threatening acute respiratory distress syndrome during primary Influenza A virus (IAV) infection at the age of 2.5 years, compound heterozygous null mutations of IRF7 were identified (166). IRF7 is a transcription factor activated downstream of RIG-I, TLR3, TLR7, TLR9. It plays a crucial role in and amplifying IFN type I and III production upon detection of virus (167, 168). It is expressed in abundance by pDCs, the main producers of type I IFNs. The authors showed a complete lack of IFN-α2b and decreased IFN-β and IFN-λ1 production in the patient’s pDCs and PBMCs upon exposure to different viruses or TLR stimulation. Moreover, IAV replication was significantly enhanced in the patient’s inducible pluripotent stem cell-derived pulmonary epithelial cells (PECs) and fibroblasts, and this phenotype was rescued by treatment with IFN-α2b, IFN-β or IFN-λ1. The absence of IRF7-dependent type I and III IFN amplification by pDCs, PECs, and possibly other cell types, probably accounts for the severe phenotype of this patient. By the time at which this case was reported, the child had not suffered from other severe infections, implying that human IRF7 is largely redundant in host defense against most common viruses (166).
IFNAR2 and STAT2
The key role of type I IFN in host antiviral defense is also highlighted by reports on human AR IFNAR2 and STAT2 deficiencies, displaying a similar phenotype and incomplete penetrance (169–172). These patients typically present with complicated vaccine-strain measles after immunization with the live vaccine, but can also suffer from life-threatening infections with other viruses. Duncan et al. identified a homozygous LOF mutation in IFNAR2 in a previously healthy child, who died from encephalitis following the administration of live measles vaccine, and his newborn sister (170). The authors demonstrated that the mutation resulted in a loss of the IFN-α response in patient’s fibroblasts in vitro (170). Nine patients with STAT2 deficiency have been described to date: two died of viral infections, six developed severe measles after vaccination, and excessive clinical inflammation has been reported in at least four patients (Kawasaki-like syndrome, HLH-like syndrome) (169, 171, 172). The recent finding that STAT2 is essential for the USP-18-mediated inhibition of type I IFN signaling, at least in the conditions studied in vitro, provides an illustration of the tight regulation embedded in the innate immune system (173).
IRF9
Downstream from IFNAR1/2, IRF9 combines with STAT1/STAT2 heterodimers to form the ISGF3 transcription factor. This translocates to the nucleus and initiates transcription after binding to an ISRE promoter. AR IRF9 deficiency was recently characterized in a child who presented with complications after measles-mumps-rubella vaccine, recurrent viral infections and severe IAV infection requiring intensive care treatment, a clinical picture overlapping with both IRF7 and STAT2 deficiency (174). The authors demonstrated absent activation of ISGF3, and absent transcription of factors driven by the ISRE promoter, which consequently impairs ISGs expression and anti-viral immunity in patient’s fibroblasts. The impairment in transcriptional response to IFN stimulation was found to be as broad as that of STAT1- or STAT2-deficient patients.
IFNAR1
Hoyos-Bachiloglu et al. have reported a child, born from consanguineous parents, who at the age of 2 months suffered from Streptococcus viridans sepsis, severe CMV infection and mycobacteriosis, and went on to develop HLH. Of note, he later received attenuated measles, mumps and rubella virus vaccines without any adverse effect. The group identified homozygous mutations in both IFNGR2 and IFNAR1 (175). The mutation in IFNGR2 is LOF and considered responsible for the mycobacterial susceptibility (175). The mutation in IFNAR1 is a deletion replacing a stop codon with 46 new codons, resulting in a slightly longer protein. The effect is predicted to be deleterious based on decreased STAT1 and STAT2 phosphorylation, partial impairment of p-STAT1 nuclear translocation and failure to induce an appropriate antiviral response after IFN-α stimulation of the patient’s fibroblasts, that was rescued by transduction with wild type IFNAR1 (175). However, the simultaneous presence of homozygous mutations in both type I and II IFN pathways renders difficult the attribution of the clinical infectious phenotype to one or the other gene defect.
IV. Inborn errors of NK cells and innate lymphoid cells
ILCs were recently described as a subfamily of lymphocytes differing from adaptive T cells in their absence of a clonotypic TCR and their independence of gene rearrangement (176). They are the “innate” counterparts of differentiated adaptive T cells and are able to respond to threats within hours of activation. ILCs are classified into three groups based on their phenotype and functional resemblance to T cytotoxic or helper cells: ILC1, ILC2 and ILC3 are driven by T-bet, GATA-3 and RORγt, respectively, and secrete IFN-γ, IL-5/IL-13 and IL-17/IL22, respectively (177). ILCs are abundant at mucosal surfaces, where they maintain the delicate balance between pathogen containment and avoidance of auto-inflammation, particularly in mice (176, 177). Research into human ILC development and function is in its infancy. Vély et al. showed that peripheral blood ILCs other than NK cells were more abundant in umbilical cord blood and in children than in adults (178). They also showed that T-B+NK- SCID patients with mutations in IL-2 receptor subunit gamma (IL2RG) or JAK3 had no circulating ILCs, consistent with the dependence of these cells on IL-7 and IL-15 (178). Moreover, in the absence of myeloablative conditioning, ILC deficiency persists in blood and tissue after HSCT, with no major detectable clinical impact, even after long-term follow-up, at least in conditions in which functional T and B cells are present, suggesting that ILCs are redundant for host defense in humans. The ILC population is reconstituted after myeloablative HSCT, indicating that ILC precursors are present in human bone marrow and cord blood (178). Long-term observations in a larger number of patients are required to assess the potential increase in cancer risk and to determine whether the observed redundancy persists with aging. However, recent studies have shown that NK cells, ILC1, ILC2 and IL-22+ ILC3 but not IL-17A+ ILC3s can develop from ILC precursors in RORC-deficient patients (131). It is therefore possible that the lack of ILC3s in humans with biallelic LOF RORC mutations contributes to the susceptibility of these individuals to infection with Candida and mycobacteria. The role of NK cells in human immune defense remains a matter of debate and is accurately reviewed in several newly published papers (107, 179, 180). The patient described by Biron et al. in 1989 with severe varicella, recurrent viral infections, NK cell deficiency and lack of NK cytotoxic function was later found to harbor a mutation in GATA2 (181, 182). Mace et al. went on to characterize the NK cell defect of GATA2-deficient patients (see paragraph on GATA2) (182). In another child with fatal varicella infection and NK cell deficiency with abolished NK cytotoxic function, Hanna et al. found a homozygous mutation in RTEL1, responsible for a form of dyskeratosis congenita with bone marrow failure (Hoyeraal-Hreidarsson syndrome) (183). These reports highlight how NK cells can be first affected in a variety of inborn errors, preceding the development of defects in other hematopoietic cell lines, and therefore deserve to be accurately studied. Novel findings in NK defects include the discovery of defects in mini-chromosome maintenance 4 (MCM4), Go-Ichi-Ni-San (GINS) complex subunit 1 gene (GINS1), IRF8 and the extended phenotype of GATA2 deficiency.
MCM4
Two independent groups recently identified a homozygous mutation in MCM4 in 14 patients from five kindreds with NK cell deficiency, severe viral infection (EBV, CMV and other viruses), growth failure and adrenal failure (184, 185). These patients displayed a selective loss of mature CD56dim NK cells, due to blocking of the differentiation of CD56bright into CD56dim, lack of proliferation, and excess of spontaneous apoptosis of both CD56bright and CD56dim NK cells. Moreover, the patients showed signs of increased DNA breakage, highlighting the essential role of MCM4 in DNA replication during cell division.
GINS1
NK cell deficiency associated with neutropenia and intrauterine growth retardation has been reported in five patients from four kindreds displaying enhanced susceptibility to viral (CMV, VZV, RSV, Adenovirus), bacterial and fungal infections. All the affected patients had compound heterozygous mutations of GINS1 (186). The GINS complex, like the MCM2–7 complex containing MCM4, is part of the CMG helicase (Cdc45, Mcm2–7, GINS) essential for eukaryotic DNA replication. Unlike patients lacking MCM4, patients with GINS1 deficiency have abnormally low numbers of both CD56bright and CD56dim NK cells. Finally, these patients have an excess of promyelocytes with reduced metamyelocyte and mature neutrophil levels, which can be corrected by G-CSF treatment.
GATA2
Heterozygous mutations in GATA2, resulting in haploinsufficiency of GATA2, are responsible for AD MonoMAC syndrome, Emberger syndrome and familial MDS/acute myeloid leukemia (AML) (187–190). The immunophenotype is characterized by monocytopenia, B and NK cell lymphopenia, clinically translating into mycobacterial, fungal and viral infections. The clinical spectrum has been extended in the last five years and involves susceptibility to disseminated nontuberculous mycobacterial infections, papillomavirus infections, opportunistic fungal infections, and pulmonary alveolar proteinosis; lymphedema, hearing loss and risk of miscarriage are also reported (191). In 2013, Mace et al. showed that the CD56bright NK cells of GATA2 deficient patients are absent or severely reduced in numbers, and that CD56dim cells are functionally impaired (182). Other characteristics are B cell, monocyte and DC deficiency, bone marrow hypocellularity and myeloid dysplasia, often with karyotypic abnormalities leading to MDS and leukemia (187, 191, 192). The only available treatment is HSCT, but the timing of this intervention is a matter of debate (193).
FUTURE DIRECTIONS & CONCLUSION
We have reviewed here a set of recently described inborn errors of innate immunity conferring susceptibility to infection. The studies we described offer insights on the close interplay between innate and adaptive immunity and on the redundancy of some innate immune pathways and factors that can be traced back to the evolution of adaptive immunity. The innate immune system is more conserved through the species than the adaptive, nonetheless a great degree of variation has characterized the development of human innate immunity. Patin et al. have recently used flow cytometry and a genome-wide association approach to study 1000 healthy individuals, and have found that natural variation in the parameters of innate cells, such as the cell-surface expression of protein markers, is more strongly driven by genetic factors than in adaptive cells (194). In most of the inborn errors of innate immunity discussed here, adaptive immunity is also affected to some extent, and conversely defects manifesting as T and B cell dysfunction often display some innate immunity involvement, as is the case in defects of NF-κB and the CBM complex. Some defects, such as those in the TLR3 pathway or in the type I/III IFN pathway, illustrate the redundancy of the pathway for host defense in general: the phenotype is in fact restricted to impaired cell-intrinsic immunity against one or a limited number of pathogens and in a single non-hematopoietic cell line. TIRAP deficiency, that only causes an infectious susceptibility in the absence of anti-LTA Ab, is a fine example of both the redundancy of an innate pathway and the mutual influence of innate and adaptive immunity. Finally, we illustrate that tight regulation of the immune pathways is crucial to confer protection from infections while avoiding auto-inflammation. In many conditions discussed here, the increased susceptibility to infection is associated with auto-inflammation, and defects in the same gene can cause either immunodeficiency or autoinflammation depending on their detrimental or enhancing effect on protein function, such as in STAT1 and MDA5 mutations. Infection and auto-inflammation can be allelic. From a clinical perspective, the inborn errors of innate immunity also teach us that the absence of a measurable immunological defect (e.g. hypogammaglobulinemia) does not exclude an immunodeficiency and should not result in a delay in treatment. Moreover, many of the described clinical entities present with hematological manifestations that can dominate the phenotype and result in fatal outcome (e.g. MDS/AML in GATA2 deficiency and SAMD9 GOF mutation). The intertwinement of these manifestations also shows the importance of regulation of hematopoiesis upon infection. The genetic theory of infectious diseases (13) suggests that many inborn errors of immunity in general and of innate immunity in particular are yet to be characterized by the study of patients with severe infections. Explaining the underlying mechanisms of increased resistance to disease after infection with a pathogenic microbe is another challenge. Ultimately, these experiments of nature will improve our knowledge and understanding of human (innate) immunity to infection, and should enable us to provide better treatment to individual patients and entire populations.
Acknowledgments
Declaration of all sources of funding: GB is supported by the FWO Vlaanderen (project G0C8517N). LM is supported by the CSL Behring Chair in Primary Immunodeficiencies, by the KID-FONDS charity of KU Leuven and by the Jeffrey Modell Foundation. BB is supported by the Boehringer Ingelheim Fonds PhD fellowship. XB is supported by Jeffrey Modell Foundation Translational Research Program, by a grant from the KU Leuven (GOA/13/013)and by a FWO Vlaanderen TBM grant. JLC is funded by the Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, National Institute of Allergy and Infectious Diseases (R01AI088364, R37AI095983, P01AI061093, R01AI127564, U19AI111143), National Institute of Neurological Disorders and Stroke (R01NS072381); AP is funded by Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), French National Research Agency (ANR)under the “Investments for the future” program (ANR-10-IAHU-01), ANR HGDIFD (ANR-14- CE15-0006-01) and eRARE EURO-CMC (ANR-14-RARE-0005-02), an AP-HP interface contract and Jeffrey Modell Foundation Translational Research Program. IM is supported by the Jeffrey Modell Foundation and by the FWO Vlaanderen (project G0C8517N).
ABBREVIATIONS
- Ab
Antibody
- ACT1
NF-κB activator 1
- AD
Autosomal dominant
- Aip1
Actin-interacting protein 1
- AML
Acute myeloid leukemia
- APC
Antigen presenting cell
- AR
Autosomal recessive
- BAFFR
B cell-activating factor receptor
- BCG
Bacillus Calmette-Guérin
- BCL10
B-cell lymphoma/leukemia 10
- BCR
B cell receptor
- BTK
Bruton tyrosine kinase
- CARD
Caspase recruitment domain
- CBM complex
CARD-BCL10-MALT1 complex
- CGD
Chronic granulomatous disease
- CLR
C-type lectin receptor
- CMC
Chronic mucocutaneous candidiasis
- CMG
CDC45, MCM2–7, GINS
- CMV
Cytomegalovirus
- CSF3R
G-CSF receptor
- CYBB
Cytochrome B-245 beta chain
- DBR1
Debranching enzyme 1
- DC
Dendritic cell
- ds
Double-stranded
- EBV
Epstein-Barr virus
- EDA-ID
Anhidrotic ectodermal dysplasia with immunodeficiency
- ELANE
Elastase, neutrophil-expressed
- ER
Endoplasmic reticulum
- GATA2
GATA-binding protein 2
- G-CSF
Granulocyte colony-stimulating factor
- GINS1
Go-Ichi-Ni-San (GINS) complex subunit 1
- GOF
Gain of function
- HIES
Hyper-IgE syndrome
- HLH
Hemophagocytic lymphohistiocytosis
- HOIL-1
Heme-oxidized IRP2 ubiquitin ligase 1
- HOIP
HOIL-1-interacting protein
- HSCT
Hematopoietic stem cell transplantation
- HSE
Herpes simplex virus 1 encephalitis
- HSV-1
Herpes simplex virus-1
- HRV
Human rhinovirus
- HVS-T cells
Herpesvirus saimiri-immortalized T-cells
- IAV
Influenza A virus
- ICA
Isolated congenital asplenia
- IFIH1
IFN-induced helicase C domain-containing protein 1
- IFN
Interferon
- IFNAR
Type I interferon receptor
- IFNGR
Type II interferon receptor
- Ig
Immunoglobulin
- IκB
inhibitor of NF-κB
- IKK
IκB kinase
- IL
Interleukin
- IL2RG
IL-2 receptor subunit gamma
- IL6ST
IL-6 signal transducer
- IL-1R
Interleukin 1 receptor
- IL-17R
Interleukin 17 receptor
- ILC
Innate lymphoid cell
- IPD
Invasive pneumococcal disease
- IRAK
IL-1R-associated kinase
- IRF
IFN regulatory transcription factor
- ISG
IFN stimulated genes
- ISRE
IFN-stimulated response element
- JAGN1
jagunal homolog 1
- JAK
Janus kinase
- LAD
Leukocyte adhesion deficiency
- LOF
Loss of function
- LPS
Lipopolysaccharide
- LTA
Lipoteichoic acid
- LT-βR
receptor for lymphotoxin-β
- LUBAC
Linear ubiquitination chain assembly complex
- MAIT
Mucosa-associated invariant T cells
- MALT1
Mucosa-associated lymphoid tissue lymphoma translocation protein 1
- MAP3K14
Mitogen-activated protein kinase 14
- MAVS
Mitochondrial antiviral signaling protein
- MCM2–7
Mini-chromosome maintenance protein 2–7
- MCM4
Mini-chromosome maintenance protein 4
- MDA5
Melanoma differentiation associated gene 5
- MDS
Myelodysplastic syndrome
- MECP2
Methyl CpG-binding protein 2
- MonoMAC
Monocytopenia and Mycobacterium Avium Complex/Dendritic Cell, Monocyte
- B and NK
Lymphocyte deficiency
- MSMD
Mendelian susceptibility to mycobacterial disease
- MyD88
Myeloid differentiation primary response gene 88
- NEMO
NF-kappa-B essential modulator
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NIK
NF-κB-inducing kinase
- NK
Natural killer
- NLR
Nod-like receptor OSM: oncostatin M pDC: Plasmacytoid DC
- PBMC
Peripheral mononuclear blood cell
- PEC
Pulmonary epithelial cells
- PID
Primary immunodeficiency
- PML
Progressive multifocal leukoencephalopathy
- POLA1
DNA polymerase α 1
- POLR3
RNA polymerase III
- RIG-I
Retinoic-acid-inducible protein 1
- RLR
RIG-I-like receptor
- RORC
Retinoic acid receptor-related orphan receptor C
- RPSA
Ribosomal protein SA
- RSV
Respiratory syncytial virus
- SAMD9
Sterile alpha motif domain-containing protein 9
- SAMD9L
Sterile alpha motif domain-containing protein 9-like
- SCID
Severe combined immunodeficiency
- SCN
Severe congenital neutropenia
- SMARCD2
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily D, member 2
- SHARPIN
Shank-associated RH domain-interacting protein ss: Single-stranded
- STAT
Signal transducer and activator of transcription
- STING
Stimulator of IFN genes
- SV40
Simian virus 40
- TAK1
Transforming growth factor beta-activated kinase 1
- TBK1
TANK-binding kinase 1
- TCR
T cell receptor
- Th
T helper
- TIR
Toll/IL-1 receptor
- TIRAP
TIR domain-containing adaptor
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- TNFR
Tumor necrosis factor receptor
- TORCH
Toxoplasmosis, other agents, rubella, cytomegalovirus, herpes simplex
- TRAF
TNF receptor-associated factor
- TRIF
TIR domain-containing adaptor molecule 1
- TYK2
Tyrosine kinase 2
- USP18
Ubiquitin-specific peptidase 18
- VPS45
vacuolar sorting-associated protein 45
- VZV
Varicella zoster virus
- WDR1
WD repeat domain 1
- XL
X-linked
Footnotes
The Authors do not have a Conflict of Interest.
REFERENCES
- 1.Beutler BA. TLRs and innate immunity. Blood. 2009;113(7):1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ronald P, Beutler B. Plant and animal sensors of conserved microbial signatures. Science. 2010;330(6007):1061–4. [DOI] [PubMed] [Google Scholar]
- 3.Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol 2006;117(5):979–87. [DOI] [PubMed] [Google Scholar]
- 4.Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL. Inborn errors of anti-viral interferon immunity in humans. Current Opinion in Virology. 2011;1(6):487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Neill L, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–60. [DOI] [PubMed] [Google Scholar]
- 6.Masters S, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009;27:621–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tosi MF. Innate immune responses to infection. J Allergy Clin Immunol 2005;116(2):241–9. [DOI] [PubMed] [Google Scholar]
- 8.Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol 2012;13(3):214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol 2004;5(11):1109–15. [DOI] [PubMed] [Google Scholar]
- 10.Casanova JL. Human genetic basis of interindividual variability in the course of infection. PNAS 2015;112(51):E7118–E27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim D, Gewurz A, Lint TF, Ghaze M, Sepheri B, Gewurz H. Absence of the sixth component of complement in a patient with repeated episodes of meningococcal meningitis. J Pediatr. 1976;89(1):42–7. [DOI] [PubMed] [Google Scholar]
- 12.Petersen BH, Graham JA, Brooks GF. Human deficiency of the eighth component of complement. The requirement of C8 for serum Neisseria gonorrhoeae bactericidal activity. J Clin Invest. 1976;57(2):283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casanova J Severe infectious diseases of childhood as monogenic inborn errors of immunity. PNAS. 2015;112(52):E7128–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer A, Rausell A. Primary immunodeficiencies suggest redundancy within the human immune system. Sci Immunol 2016;1(6). [DOI] [PubMed] [Google Scholar]
- 15.Freeley S, Kemper C, Le Friec G. The “ins and outs” of complement-driven immune responses. Immunol Rev. 2016;274(1):16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liszewski MK, Java A, Schramm EC, Atkinson JP. Complement Dysregulation and Disease: Insights from Contemporary Genetics. Annu Rev Pathol 2017;12:25–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozen A, Comrie WA, Ardy RC, Dominguez Conde C, Dalgic B, Beser OF, et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N Engl J Med. 2017;377(1):52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis. 2014;59(2):244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. [DOI] [PubMed] [Google Scholar]
- 20.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299(5615):2076–9. [DOI] [PubMed] [Google Scholar]
- 21.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321(5889):691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore). 2010;89(6):403–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frans G, Moens L, Schrijvers R, Wuyts G, Bouckaert B, Schaballie H, et al. PID in Disguise: Molecular Diagnosis of IRAK-4 Deficiency in an Adult Previously Misdiagnosed With Autosomal Dominant Hyper IgE Syndrome. J Clin Immunol. 2015;35(8):739–44. [DOI] [PubMed] [Google Scholar]
- 24.Della Mina E, Borghesi A, Zhou H, Bougarn S, Boughorbel S, Israel L, et al. Inherited human IRAK-1 deficiency selectively impairs TLR signaling in fibroblasts. Proc Natl Acad Sci U S A 2017;114(4):E514–E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Israel L, Wang Y, Bulek K, Della Mina E, Zhang Z, Pedergnana V, et al. Human Adaptive Immunity Rescues an Inborn Error of Innate Immunity. Cell. 2017;168(5):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. ․Cell. 2017;168(1–2):37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 Subunit p50 in Common Variable Immunodeficiency. Am J Hum Genet 2015;97(3):389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-κB1 Haploinsufficiency Causing Immunodeficiency and EBV-Driven Lymphoproliferation. J Clin Immunol 2016;36(6):533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol. 2017;pii: S0091-6749(17):30053–2. [DOI] [PubMed] [Google Scholar]
- 30.Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet 2013. November 7;93(5):812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindsley A, Qian Y, Valencia CA, Shah K, Zhang K, Assa’ad A. Combined immune deficiency in a patient with a novel NFKB2 mutation. J Clin Immunol. 2014;34(8):910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA. Novel NFKB2 mutation in early-onset CVID. J Clin Immunol 2014;34(6):686–90. [DOI] [PubMed] [Google Scholar]
- 33.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappa-B signaling. ․ Nature Genet 2001;27:277–85. [DOI] [PubMed] [Google Scholar]
- 34.Hubeau M, Ngadjeua F, Puel A, Israel L, Feinberg J, Chrabieh M, et al. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood 2011;118(4):926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miot C, Imai K, Imai C, Mancini AJ, Kucuk ZY, Kawai T, et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG / NEMO mutations. Blood. 2017. September 21;130(12):1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boisson B, Puel A, Picard C, Casanova JL. Human IκBα Gain of Function: a Severe and Syndromic Immunodeficiency. J Clin Immunol. 2017. July;37(5):397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med 2013;369(26):2504–14. [DOI] [PubMed] [Google Scholar]
- 38.Mousallem T, Yang J, Urban TJ, Wang H, Adeli M, Parrott RE, et al. A nonsens mutation in IKBKB causes combined immunodeficiency. Blood. 2014;25(13):2046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willmann K, Klaver S, Doğu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic lossof-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun 2014;5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. 2013;132(1):151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol. 2015;35(2):135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rozmus J, McDonald R, Fung SY, Del Bel KL, Roden J, Senger C, et al. Successful clinical treatment and functional immunological normalization of human MALT1 deficiency following hematopoietic stem cell transplantation. Clin Immunol. 2016;168:1–5. [DOI] [PubMed] [Google Scholar]
- 43.Charbit-Henrion F, Jeverica AK, Begue B, Markelj G, Parlato M, Avcin SL, et al. Deficiency in Mucosa-associated Lymphoid Tissue Lymphoma Translocation 1: A Novel Cause of IPEX-Like Syndrome. J Pediatr Gastroenterol Nutr. 2017;64(3):378–84. [DOI] [PubMed] [Google Scholar]
- 44.Stepensky P, Keller B, Buchta M, Kienzler A, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immun. 2013;131(477–485). [DOI] [PubMed] [Google Scholar]
- 45.Greil J, Rausch T, Giese T, Bandapalli O, Daniel V, Bekeredjian-Ding I, et al. Wholeexome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immun 2013;131:1376–83. [DOI] [PubMed] [Google Scholar]
- 46.Torres J, Martinez-Barricarte R, García-Gómez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest 2014;124(12):5239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pérez de Diego R, Sánchez-Ramón S, López-Collazo E, Martínez-Barricarte R, CubillosZapata C, Ferreira Cerdán A, et al. Genetic errors of the human caspase recruitment domainB-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: Molecular, immunologic, and clinical heterogeneity. J Allergy Clin Immunol 2015;136(5):1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKinnon M, Rozmus J, Fung S, Hirschfeld A, Del Bel K, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immun 2014;133(1458–1462). [DOI] [PubMed] [Google Scholar]
- 49.Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immun 2013;132:151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glocker E, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009;361(18):1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida speciesinduced meningoencephalitis, colitis, or both. J Allergy Clin Immunol 2015;135(6):1558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lanternier F, Barbati E, Meinzer U, Liu L, Pedergnana V, Migaud M, et al. Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J Infect Dis 2015;211(8):1241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med 2013. October 31;369(18):1704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight 2016;1(17):e89890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Wang W, Lin Z, Wang X, Li T, Yu J, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol. 2014;133(3):905–8 e3. [DOI] [PubMed] [Google Scholar]
- 56.Drummond R, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog 2015;11(12):e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13(12):1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med 2015;212:939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ombrello M, Kastner DL, Milner JD. HOIL and water: the two faces of HOIL-1 deficiency. Nat Immunol. 2012;13(12):1133–5. [DOI] [PubMed] [Google Scholar]
- 60.Nilsson J, Schoser B, Laforet P, Kalev O, Lindberg C, Romero N, et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann Neurol. 2013;74:914–9. [DOI] [PubMed] [Google Scholar]
- 61.Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F. LUBACsynthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat Microbiol 2017;2:17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hailfinger S, Schmitt A, Schulze-Osthoff K. The paracaspase MALT1 dampens NF-κB signalling by cleaving the LUBAC subunit HOIL-1. FEBS J 2016;283(3):400–2. [DOI] [PubMed] [Google Scholar]
- 63.Dinauer MC. Primary immune deficiencies with defects in neutrophil function. Hematology Am Soc Hematol Educ Program. 2016;2016(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primers. 2017;3:17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. 2012;1250:50–5. [DOI] [PubMed] [Google Scholar]
- 67.Roos D Chronic granulomatous disease. Br Med Bull. 2016;118(1):50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):89–99, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vilboux T, Lev A, Malicdan MC, Simon AJ, Jarvinen P, Racek T, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N Engl J Med. 2013;369(1):54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stepensky P, Saada A, Cowan M, Tabib A, Fischer U, Berkun Y, et al. The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy. Blood. 2013;121(25):5078–87. [DOI] [PubMed] [Google Scholar]
- 71.Stepensky P, Simanovsky N, Averbuch D, Gross M, Yanir A, Mevorach D, et al. VPS 45associated primary infantile myelofibrosis--successful treatment with hematopoietic stem cell transplantation. Pediatr Transplant. 2013;17(8):820–5. [DOI] [PubMed] [Google Scholar]
- 72.Shah RK, Munson M, Wierenga KJ, Pokala HR, Newburger PE, Crawford D. A novel homozygous VPS45 p.P468L mutation leading to severe congenital neutropenia with myelofibrosis. Pediatr Blood Cancer. 2017. September;64(9). [DOI] [PubMed] [Google Scholar]
- 73.Meerschaut I, Bordon V, Dhooge C, Delbeke P, Vanlander AV, Simon A, et al. Severe congenital neutropenia with neurological impairment due to a homozygous VPS45 p.E238K mutation: A case report suggesting a genotype-phenotype correlation. Am J Med Genet A. 2015;167A(12):3214–8. [DOI] [PubMed] [Google Scholar]
- 74.Boztug K, Jarvinen PM, Salzer E, Racek T, Monch S, Garncarz W, et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat Genet. 2014;46(9):1021–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baris S, Karakoc-Aydiner E, Ozen A, Delil K, Kiykim A, Ogulur I, et al. JAGN1 Deficient Severe Congenital Neutropenia: Two Cases from the Same Family. J Clin Immunol. 2015;35(4):339–43. [DOI] [PubMed] [Google Scholar]
- 76.Triot A, Jarvinen PM, Arostegui JI, Murugan D, Kohistani N, Dapena Diaz JL, et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood. 2014;123(24):3811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klimiankou M, Klimenkova O, Uenalan M, Zeidler A, Mellor-Heineke S, Kandabarau S, et al. GM-CSF stimulates granulopoiesis in a congenital neutropenia patient with loss-offunction biallelic heterozygous CSF3R mutations. Blood. 2015;126(15):1865–7. [DOI] [PubMed] [Google Scholar]
- 78.Klimiankou M, Mellor-Heineke S, Zeidler C, Welte K, Skokowa J. Role of CSF3R mutations in the pathomechanism of congenital neutropenia and secondary acute myeloid leukemia. Ann N Y Acad Sci. 2016;1370(1):119–25. [DOI] [PubMed] [Google Scholar]
- 79.Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, et al. Chromatinremodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat Genet. 2017;49(5):742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Priam P, Krasteva V, Rousseau P, D’Angelo G, Gaboury L, Sauvageau G, et al. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPvarepsilon dependent mechanism. Nat Genet. 2017;49(5):753–64. [DOI] [PubMed] [Google Scholar]
- 81.Miller M, Oski FA, Harris MB. Lazy-leucocyte syndrome. A new disorder of neutrophil function. Lancet 1971;1(7701):665–9. [DOI] [PubMed] [Google Scholar]
- 82.Gallin J, Malech HL, Wright DG, Whisnant JK, Kirkpatrick CH. Recurrent severe infections in a child with abnormal leukocyte function: possible relationship to increased microtubule assembly. Blood 1978;51(5):919–33. [PubMed] [Google Scholar]
- 83.Kuhns D, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, Riva D, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood. 2016;128(17):2135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Standing A, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med 2017;214(1):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moulding D, Record J, Malinova D, Thrasher AJ. Actin cytoskeletal defects in immunodeficiency. Immunol Rev 2013;256(1):282–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5(8):606–16. [DOI] [PubMed] [Google Scholar]
- 87.Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kevy SV, Tefft M, Vawier GF, Rosen FS. Hereditary splenic hypoplasia. Pediatrics. 1968;42(5):752–7. [PubMed] [Google Scholar]
- 89.Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340(6135):976–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bolze A, Boisson B, Bosch B, Antipenko A, Bouaziz M, Sackstein P, et al. Incomplete penetrance for isolated congenital asplenia in humans with mutations in translated and untranslated RPSA exons. PNAS 2018;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Richardson JP, Moyes DL. Adaptive immune responses to Candida albicans infection. Virulence. 2015;6(4):327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 2012;12(6):616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol 2014;26(6):454–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kreins A, Ciancanelli MJ, Okada S, Kong XF, Ramírez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. 2015;212(10):1641–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160(6):1055–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745–55. [DOI] [PubMed] [Google Scholar]
- 97.Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun 2016;7:13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol 2018;9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodero M, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med 2016;213(12):2527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 2012. September 28;337(6102):1684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and autoinflammation. Nature. 2015;517(7532):89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dos Santos PF, Mansur DS. Beyond ISGlylation: Functions of Free Intracellular and Extracellular ISG15. J Interferon Cytokine Res. 2017;37(6):246–53. [DOI] [PubMed] [Google Scholar]
- 103.Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7:11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meuwissen M, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 2016;213(7):1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011;365(2):127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salem S, Langlais D, Lefebvre F, Bourque G, Bigley V, Haniffa M, et al. Functional characterization of the human dendritic cell immunodeficiency associated with the IRF8(K108E) mutation. Blood 2014;124(12):1894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mace E, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest 2017;127(1):306–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bigley V, Maisuria S, Cytlak U, Jardine L, Care MA, Green K, et al. Biallelic interferon regulatory factor 8 mutation: A complex immunodeficiency syndrome with dendritic cell deficiency, monocytopenia, and immune dysregulation. J Allergy Clin Immunol. 2018. June;141(6):2234–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guerin A, Kerner G, Marr N, Markle JG, Fenollar F, Wong N, et al. IRF4 haploinsufficiency in a family with Whipple’s disease. Elife 2018. March 14;7 pii: e32340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Biswas PS, Gupta S, Stirzaker RA, Kumar V, Jessberger R, Lu TT, et al. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J Exp Med. 2012;209(3):581–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102(44):15989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017;214(9):2547–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205(7):1551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lévy R, Okada S, Béziat V, Moriya K, Liu C, Chai LY, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Nat Acad Sci. 2016;113:E8277–E85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–12. [DOI] [PubMed] [Google Scholar]
- 116.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206(2):299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JCet al. International STAT1 Gain-of-Function Study Group. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Van de Veerdonk F, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011;365(1):54–61. [DOI] [PubMed] [Google Scholar]
- 120.Zerbe CS, Marciano BE, Katial RK, Santos CB, Adamo N, Hsu AP, et al. Progressive Multifocal Leukoencephalopathy in Primary Immune Deficiencies: Stat1 Gain of Function and Review of the Literature. Clin Infect Dis. 2016;62(8):986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Weinacht K, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol 2017;139(5):1629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vargas-Hernandez A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. 2018. June;141(6):2142–2155.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mössner R, Diering N, Bader O, Forkel S, Overbeck T, Gross U, et al. Ruxolitinib Induces Interleukin 17 and Ameliorates Chronic Mucocutaneous Candidiasis Caused by STAT1 Gain-of-Function Mutation. Clin Infect Dis. 2016;62(7):951–3. [DOI] [PubMed] [Google Scholar]
- 124.Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-offunction signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol 2015;135(2):551–3. [DOI] [PubMed] [Google Scholar]
- 125.Meesilpavikkai K, Dik WA, Schrijver B, Nagtzaam NMA, Posthumus-van Sluijs SJ, van Hagen PM, et al. Baricitinib treatment in a patient with a gain-of-function mutation in signal transducer and activator of transcription 1 (STAT1). J Allergy Clin Immunol. 2018. July;142(1):328–330.e2. [DOI] [PubMed] [Google Scholar]
- 126.Zimmerman O, Rosler B, Zerbe CS, Rosen LB, Hsu AP, Uzel G, et al. Risks of Ruxolitinib in STAT1 Gain-of-Function-Associated Severe Fungal Disease. Open Forum Infect Dis 2017;4(4):ofx202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eyal O, Flaschner M, Ben Yehuda A, Rund D. Varicella-zoster virus meningoencephalitis in a patient treated with ruxolitinib. Am J Hematol 2017;92(5):E74–E5. [DOI] [PubMed] [Google Scholar]
- 128.Coltro G, Mannelli F, Guglielmelli P, Pacilli A, Bosi A, Vannucchi AM. A life-threatening ruxolitinib discontinuation syndrome. Am J Hematol. 2017. August;92(8):833–838. [DOI] [PubMed] [Google Scholar]
- 129.Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. 2018;141(2):704–17 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349(6248):606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lim A, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell. 2017;168(6):1086–100. [DOI] [PubMed] [Google Scholar]
- 132.Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerasealpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17(5):495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hoving J, Wilson GJ, Brown GD. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol. 2014;16(2):185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL. Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol 2011;1(6):487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Häcker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 2011;11(7):457–68. [DOI] [PubMed] [Google Scholar]
- 136.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208(10):2083–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang S, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–7. [DOI] [PubMed] [Google Scholar]
- 138.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33(3):400_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. 2011;121(12):4889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 2012;209(9):1567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Whitley R, Kimberlin DW. Herpes simplex encephalitis: children and adolescents. Semin Pediatr Infect Dis 2005;16(1):17–23. [DOI] [PubMed] [Google Scholar]
- 142.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011;8(6039):228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012;37(2):223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Andersen L, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 2015;212(9):1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mork N, Kofod-Olsen E, Sorensen KB, Bach E, Orntoft TF, Ostergaard L, et al. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015;16(8):552–66. [DOI] [PubMed] [Google Scholar]
- 146.Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, et al. Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell. 2018;172(5):952–65 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Garrey SM, Katolik A, Prekeris M, Li X, York K, Bernards S, et al. A homolog of lariatdebranching enzyme modulates turnover of branched RNA. RNA. 2014;20(8):1337–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rice GI, del Toro Duany Y, Jenkinson E, Forte G, Anderson B, Ariaudo G, et al. Gain-offunction mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nature Genet 2014;46:503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006;441:101–5. [DOI] [PubMed] [Google Scholar]
- 150.Lamborn I, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med 2017. July 3;214(7):1949–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci U S A. 2017;114(31):8342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zaki M, Thoenes M, Kawalia A, Nurnberg P, Kaiser R, Heller R, et al. Recurrent and Prolonged Infections in a Child with a Homozygous IFIH1 Nonsense Mutation. Front Genet 2017;8:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ogunjimi B, Zhang SY, Sorensen KB, Skipper KA, Carter-Timofte M, Kerner G, et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest. 2017;127(9):3543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang LK, Chai F, Li HY, Xiao G, Guo L. Identification of host proteins involved in Japanese encephalitis virus infection by quantitative proteomics analysis. J Proteome Res. 2013;12(6):2666–78. [DOI] [PubMed] [Google Scholar]
- 155.Wang J, Dupuis C, Tyring SK, Underbrink MP. Sterile alpha Motif Domain Containing 9 Is a Novel Cellular Interacting Partner to Low-Risk Type Human Papillomavirus E6 Proteins. PLoS One. 2016;11(2):e0149859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. Identification of Restriction Factors by Human Genome-Wide RNA Interference Screening of Viral Host Range Mutants Exemplified by Discovery of SAMD9 and WDR6 as Inhibitors of the Vaccinia Virus K1L-C7L- Mutant. MBio 2015;6(4):e01122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meng X, Zhang F, Yan B, Si C, Honda H, Nagamachi A, et al. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog. 2018;14(2):e1006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu J, McFadden G. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol. 2015;89(3):1925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nounamo B, Li Y, O’Byrne P, Kearney AM, Khan A, Liu J. An interaction domain in human SAMD9 is essential for myxoma virus host-range determinant M062 antagonism of host anti-viral function. Virology. 2017;503:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. 2017;127(5):1700–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48(7):792–7. [DOI] [PubMed] [Google Scholar]
- 162.Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-offunction SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. 2017;129(16):2266–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, et al. AtaxiaPancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet. 2016;98(6):1146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31(8):1827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Davidsson J, Puschmann A, Tedgard U, Bryder D, Nilsson L, Cammenga J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia. 2018;32(5):1106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dittmann M, Hoffmann HH, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, et al. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell 2015;160(4):631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, et al. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol 2003;170(9):4465–74. [DOI] [PubMed] [Google Scholar]
- 169.Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci U S A 2013;110(8):3053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Duncan C, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci Transl Med. 2015;7(307):307 ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol. 2017. June;139(6):1995–1997.e9. [DOI] [PubMed] [Google Scholar]
- 172.Shahni R, Cale CM, Anderson G, Osellame LD, Hambleton S, Jacques TS, et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain 2015;138:2834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Arimoto K, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol 2017;24(3):279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hernandez N, Melki I, Jing H, Danielson J, Habib T, Huang S, et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. Journal of Experimental Medicine. 2018;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hoyos-Bachiloglu R, Chou J, Sodroski CN, Beano A, Bainter W, Angelova M, et al. A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J Clin Invest. 2017;127(12):4415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol. 2015;15(7):415–28. [DOI] [PubMed] [Google Scholar]
- 177.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–9. [DOI] [PubMed] [Google Scholar]
- 178.Vély F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17(11):1291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jouanguy E, Gineau L, Cottineau J, Béziat V, Vivier E, Casanova JL. Inborn errors of the development of human natural killer cells. Curr Opin Allergy Clin Immunol. 2013;13(6):589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mace E, Orange JS. Genetic Causes of Human NK Cell Deficiency and Their Effect on NK Cell Subsets. Front Immunol 2016;7:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320(26):1731–5. [DOI] [PubMed] [Google Scholar]
- 182.Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. 2013;121(14):2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hanna S, Beziat V, Jouanguy E, Casanova JL, Etzioni A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J Allergy Clin Immunol. 2015;136(4):1113–4. [DOI] [PubMed] [Google Scholar]
- 184.Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest 2012;122(3):821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hughes C, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest 2012;122(3):814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cottineau J, Kottemann MC, Lach FP, Kang YH, Vély F, Deenick EK, et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J Clin Invest 2017;127(5):1991–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43(10):929–31. [DOI] [PubMed] [Google Scholar]
- 190.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123(6):863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tholouli E, Sturgess K, Dickinson RE, Gennery A, Cant AJ, Jackson G, et al. In vivo Tdepleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood. 2018;131(12):1383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol. 2018;19(3):302–14. [DOI] [PubMed] [Google Scholar]