

Significance
Approximately 50,000 cases of Staphylococcus aureus pneumonia occur each year. These infections are associated with high mortality rates and frequently progress to multiorgan system failure. Treatment is limited to antibiotics and supportive care. Here we show that Abl kinase inhibition enhances proliferation and differentiation of a subpopulation of lung epithelial cells following pathogen-induced lung injury. We found that a subset of secretory cells in the lower airway expands to sites of injury in the alveolar epithelium to promote regeneration. Mice treated with an Abl kinase inhibitor recovered faster than vehicle control-treated mice following pathogen-induced lung injury. Therapeutic strategies to promote lung epithelial cell regeneration after injury could profoundly improve patient outcomes when used in combination with antibiotics and supportive care.
Keywords: pneumonia, lung regeneration, Abl kinases, alveolar injury
Abstract
Current therapeutic interventions for the treatment of respiratory infections are hampered by the evolution of multidrug resistance in pathogens as well as the lack of effective cellular targets. Despite the identification of multiple region-specific lung progenitor cells, the identity of molecules that might be therapeutically targeted in response to infections to promote activation of progenitor cell types remains elusive. Here, we report that loss of Abl1 specifically in SCGB1A1-expressing cells leads to a significant increase in the proliferation and differentiation of bronchiolar epithelial cells, resulting in dramatic expansion of an SCGB1A1+ airway cell population that coexpresses SPC, a marker for type II alveolar cells that promotes alveolar regeneration following bacterial pneumonia. Furthermore, treatment with an Abl-specific allosteric inhibitor enhanced regeneration of the alveolar epithelium and promoted accelerated recovery of mice following pneumonia. These data reveal a potential actionable target that may be exploited for efficient recovery after pathogen-induced infections.
Damage to the lung epithelium in response to pathogens is a major health problem worldwide. Parenchymal lung infections disrupt lung epithelial architecture and function by eliciting destruction of airway and alveolar cell populations (1–6). Approximately 50,000 cases of lung infection by Staphylococcus aureus occur per year in the United States (7). S. aureus pneumonia has high morbidity and mortality rates, as it frequently presents in the context of hospital-acquired pneumonia and patients frequently progress to sepsis and multiorgan system failure (8–10). Currently, there are no approved drugs that directly prevent or repair epithelial cell damage following pathogen-induced lung injury. Therapeutic strategies to protect or promote lung epithelial cell regeneration following injury could profoundly improve patient outcomes when used in combination with antibiotics and supportive care, particularly in the context of infections caused by resistant bacterial strains.
Lung epithelial cells are the first line of defense against foreign agents such as pathogens and chemicals. The lung epithelium is composed of airway and alveolar cells. In the airway epithelium, elegant studies have identified both basal and secretory cells as critical cell types for regeneration during normal cell turnover and following injury (11–15). In the alveoli, type II alveolar epithelial cells (AECs) give rise to type I AECs during regeneration following injury (16). Other reports have implicated a small subpopulation of cells at the bronchioalveolar duct junction (BADJ) expressing markers of both secretory cells (SCGB1A1+) from the airway and type II AECs (SPC+, expressed by Sftpc) as progenitor cells for epithelial cells in both the distal airway and alveoli (17). However, at baseline, the contribution of this putative “bronchioalveolar stem cell” population is small, with on average <1 cell per BADJ (11). Targeting pathways that promote expansion of these or other progenitor cells after lung injury would ameliorate the morbidity and mortality associated with pneumonia and other conditions that cause lung damage.
Here we uncover a previously unknown role for the Abelson (Abl) kinases in regulating the regeneration response of the lung epithelium after pathogen-induced injury. The Abl kinases Abl1 and Abl2 are a family of nonreceptor tyrosine kinases that regulate a wide variety of cellular processes during development and normal homeostasis (18–21), but can have deleterious effects on cell survival, proliferation, and cell–cell junction adhesion upon their up-regulation following inflammation, tumorigenesis, and oxidative stress (22–28). We and others have previously identified a role for the Abl kinases in the integrity of the endothelium in the context of vascular leak syndrome (29–34). However, therapies focused on preventing vascular leak have not translated to viable therapeutic approaches for the treatment of acute lung injury in clinical trials (35). Here we report that genetic and pharmacological inactivation of Abl kinases mobilizes secretory cells from the distal airway and BADJ to promote the expansion of double-positive SCGB1A1+ SPC+ cells within 4 to 24 h after injury, leading to enhanced regeneration of the damaged alveolar epithelium following bacterial infection induced by live S. aureus or Streptococcus pneumoniae. These findings suggest that available Abl kinase-specific inhibitors, which have been used for treating leukemia, might be repurposed to treat the damaged lung following pathogen exposure.
Results
Abl Kinases Are Up-Regulated in Lung Epithelial Cells Following Injury.
To evaluate whether Abl kinases play a role in lung epithelial cell integrity during regeneration and/or repair, we first assessed Abl expression in primary human bronchial epithelial cells (HBECs) grown in air–liquid interface (ALI) cultures before and after pathogen-induced injury. HBECs were grown in ALI cultures for 28 d to induce differentiation of basal cells to a pseudostratified layer of basal, secretory, and ciliated epithelial cells (36). At day 28, HBECs were incubated with S. aureus and then harvested 1 and 5 d after bacterial inoculation to evaluate Abl kinase RNA and protein expression (SI Appendix, Fig. S1A). We observed a 20- to 30-fold increase in Abl1, and to a lesser extent Abl2, transcripts at 24 h that persisted up to 5 d following exposure to bacteria (SI Appendix, Fig. S1B). Abl protein expression was similarly enhanced after pathogen exposure (SI Appendix, Fig. S1C). Previous reports have shown that enhanced Abl kinase activity is associated with a wide variety of pathologies, including endothelial barrier dysfunction, tumorigenesis, and inflammation (19). Thus, Abl kinase inhibition may affect the response of lung epithelial cells during injury, regeneration, and/or repair.
Loss of Abl1 in SCGB1A1+ Lung Epithelial Cells Promotes Accelerated Recovery in a Mouse Model of Pneumonia.
To evaluate whether Abl has a role in regulating the response of bronchial epithelial cells to injury in vivo, we generated a conditional, secretory cell-type specific knockout of Abl1 with concomitant expression of a farnesylated GFP (i.e., membrane-bound GFP) reporter [CC10 (Scgb1a1)-CreERT; Rosa26-fGFP; Abl1fl/fl] upon administration of tamoxifen. We obtained efficient (>75%) excision of Abl1 in Scgb1a1-expressing epithelial cells following i.p. delivery of four doses of tamoxifen 2 wk before inducing injury (37). Scgb1a1, also known as CC10 or CCSP, is widely used as a marker of secretory cells in mammalian lung airways. To injure the lung epithelium, we employed a mouse model of pneumonia induced by intranasal insufflation of 5 × 108 cfu S. aureus (38) (SI Appendix, Fig. S1D). In this model, mice survive the bacterial infection despite significant lung injury accompanied by inflammation and edema. Following infection with S. aureus, we observed a >10-fold increase in Abl1 expression in isolated GFP+ (Scgb1a1 driver) cells in wild-type mice that was abrogated in Abl1fl/fl mice (SI Appendix, Fig. S1E). Abl1fl/fl mice displayed remarkable recovery from symptoms of infection compared with wild-type mice (Fig. 1 A–C). Abl1fl/fl mice were active and lacked pathological signs of infection displayed by wild-type mice after S. aureus inoculation (a 30-s video corresponding to Fig. 1B is in Movie S1; a 2-min tracing of mouse movement is in Fig. 1C). Bronchioalveolar lavage (BAL) samples from mouse lungs 3 d after exposure to S. aureus showed a significant decrease in protein (Fig. 1D), the cell-damage marker LDH (Fig. 1E), and leukocytes (SI Appendix, Fig. S2) in Abl1fl/fl compared with wild-type mice. Abl1 knockout mice also exhibited significantly diminished injury in lung tissue sections 72 h after injury (Fig. 1 F and G). Similar results were observed in Abl1fl/fl mice treated with an adenoviral vector encoding a CC10 (Scgb1a1) promoter driving the expression of Cre-recombinase (Ad5-CC10-Cre) to induce excision of the floxed locus in the Abl1fl/fl mouse (SI Appendix, Fig. S3). Of note, all wild-type mice shown were treated with tamoxifen, and evaluation of tamoxifen-treated wild-type mice versus untreated wild-type mice showed no significant differences in the degree of lung injury.
Fig. 1.

Inactivation of Abl1 in Scgb1a1+ lung epithelial cells protects mice from S. aureus-induced lung injury. (A) Timeline of experiment: Two weeks before nasal insufflation of 5 × 108 cfu S. aureus, both wild-type and Abl1fl/fl mice were treated with tamoxifen in CC10 (Scgb1a1)-CreERT2 mice. (B) Screenshot of Movie S1 of wild-type and knockout mice 24 h after nasal insufflation of S. aureus. (C) Two-minute tracing of mice performed with Adobe Premiere Pro showing a dramatic increase in mouse movement in knockout compared with wild-type mice. (D and E) Bronchioalveolar lavage showing reduced protein (D) and LDH (E) in knockout mice compared with wild-type mice. (F) H&E staining 3 d following nasal insufflation of S. aureus in wild-type and knockout mice showing increased protein and cell infiltrates in the airspace of wild-type mice compared with knockout mice. (A composite of images of mouse left lung sections using a 10× objective on the Zeiss AxioImager microscope stitched together with Zen software to recreate the entire left lung.) (G) Quantification of alveolar space infiltrates on H&E sections of wild-type vs. knockout mice normalized to a healthy, noninfected mouse. Graphs depict means and SEM of “n” mice, where n represents each individual animal used (i.e., n = 37 represents 37 individual mice). *P < 0.05, **P < 0.01, and ***P < 0.001.
Given the reduction in immune cell infiltration in the alveolar space detected in BAL fluid (SI Appendix, Fig. S2), we next evaluated whether loss of Abl1 in Scgb1a1-expressing epithelial cells could affect the immune response to S. aureus infection. We evaluated whether the observed decrease in immune cell infiltration in the BAL fluid of Abl1 knockout mice was specific to the alveolar space or whether there was also a decrease in the influx of immune cells into the lung parenchyma. Analysis of H&E sections of lungs from wild-type and Abl1 knockout mice suggested that the reduction in immune cell infiltration was specific to the alveolar space, as a neutrophilic infiltration was observed in the lung parenchyma of both wild-type and Abl1 knockout mice (SI Appendix, Fig. S2B). Fluorescence-activated cell sorting (FACS) analysis of the BAL fluid showed a decrease in the total immune cell (CD45+) and neutrophil populations (Ly6G+) in the alveolar space, without changes in the proportion of neutrophils in the BAL fluid of Abl1 knockout compared with wild-type mice (SI Appendix, Fig. S2 E–H). Moreover, we found no changes in the populations of immune cells, including neutrophils in the whole lung (non-BAL fluid component) of wild-type and Abl1 knockout mice after injury (SI Appendix, Fig. S4 C–F). The finding that loss of Abl1 in Scgb1a1+ cells leads to a decrease in leukocytes in the alveolar space only (BAL fluid component) without changing the total immune cell population in the lung (non-BAL fluid component) suggested that loss of Abl1 in Scgb1a1+ epithelial cells protects and/or promotes regeneration of epithelial barrier function following injury, thereby reducing protein and cellular infiltration into the alveolar space, which enhances gas exchange compared with wild-type mice.
To assess whether loss of Abl1 in the lung epithelium has a protective effect by blocking pathogen infection, we evaluated whether loss of Abl1 in Scgb1a1+ cells confers differential susceptibility of lung epithelial cells to bacterial infection compared with wild-type mice. We found no significant change in bacterial load in either the alveolar space (BAL fluid component) or the whole lung (non-BAL fluid component) 24 and 72 h after injury (SI Appendix, Fig. S4 A and B). We also found no significant change in cytokine production levels in the BAL fluid 24 and 72 h after injury (SI Appendix, Fig. S2 C and D).
The finding that genetic inactivation of Abl1 in Scgb1a1+ cells promotes recovery from pathogen-induced lung injury in a mouse model of pneumonia without modulating susceptibility to bacterial infection or immune responses suggests that Abl kinases modulate the regeneration response of lung epithelial cells following injury.
Genetic Inactivation of Abl1 in SCGB1A1+ Lung Epithelial Cells Promotes Regeneration of the Alveolar Epithelium After Pathogen-Induced Lung Injury.
To dissect the cellular effects of Abl kinase inactivation in lung epithelial cells that could explain the enhanced recovery observed following bacterial infection, CC10 (Scgb1a1)-CreERT; Rosa26-fGFP; Abl1fl/fl and the corresponding wild-type, control mice were killed at various times following nasal insufflation of S. aureus. Staining for lung epithelial cell populations (Fig. 2A) revealed that damage following nasal insufflation of S. aureus occurred primarily in type I alveolar epithelial cells, identified by immunostaining for the cell-surface marker RAGE (also known as AGER), without significant changes in the number of type II AECs (SPC+) and airway epithelial cell types in regions of lung injury (Fig. 2B). Damage to type I AECs is maximal at 24 to 72 h following S. aureus exposure, with wild-type mice achieving complete recovery by day 6 (Fig. 2C, Top). In contrast to wild-type mice, the alveolar epithelium of Abl1fl/fl mice exhibited complete recovery by day 3 postinfection (Fig. 2C, Bottom; quantification in Fig. 2D). These findings were surprising because Abl1 was inactivated specifically in Scgb1a1-expressing cells of the airway, and type I AECs do not express Scgb1a1.
Fig. 2.

Genetic inactivation of Abl1 promotes regeneration of the alveolar epithelium following nasal insufflation of S. aureus. (A) Schematic of distal lung epithelial cell populations in the bronchial tree. (B) CC10 (Scgb1a1)-CreER; Rosa26-fGFP; Abl1wt or Abl1fl/fl mice were given tamoxifen four times 2 wk before nasal insufflation of S. aureus to induce excision of Abl1 and/or expression of GFP in Scgb1a1+ cells. Panel of immunofluorescence stains for RAGE (type I alveolar epithelial cell marker), SPC (type II alveolar epithelial cell marker), and Hoechst 33342 (nuclear stain) shows that type I alveolar epithelial cells are the most common site of injury (yellow dotted lines) following exposure to S. aureus in wild-type mice for 3 d. (Scale bar, 100 µm.) (C) Immunofluorescence staining for the type I cell marker RAGE 4 h, 3 d, and 6 d following nasal insufflation of S. aureus shows widespread damage (loss of RAGE expression and reduced alveolar volume) to the alveolar epithelium in wild-type (WT) mice that peaks at 3 d and resolves in 1 wk compared with damage in Abl1fl/fl (KO) mice at 4 h that completely resolves by day 3. (Scale bars, 50 µm.) (D) Quantification showing reduced damage (as measured by loss of alveolar area/volume calculated across an entire section of the left lung compared with uninfected mice) in Abl1fl/fl mice compared with wild-type mice 3 d after injury. Graphs depict means and SEM of n mice, where n represents each individual animal used (i.e., n = 6 represents six individual mice). ***P < 0.001; ns, not significant.
To distinguish between protective and regenerative effects of Abl kinase inhibition, we evaluated lung tissue isolated at earlier time points after S. aureus exposure and assessed these sections for lung epithelial cell damage. At 4 h following nasal insufflation of S. aureus, type I AEC damage, as measured by alveolar volumes, was observed in both wild-type and Abl1 knockout mice (Fig. 2 C and D). These findings suggest that inactivation of Abl signaling in SCGB1A1+ cells fails to prevent pathogen-induced alveolar injury but rather promotes rapid regeneration of type I AECs, leading to accelerated recovery of Abl1fl/fl mice following pathogen exposure.
Conditional Deletion of Abl1 Promotes Expansion of Double-Positive SCGB1A1+ SPC+ Cell Population Following Injury.
To dissect the role of Abl1 in Scgb1a1+ cells in the regeneration of the damaged alveolar epithelium following pathogen exposure, we performed lineage tracing experiments of the Scgb1a1+ cell population using a Rosa26-fGFP reporter in CC10 (Scgb1a1)-CreERT mice. We detected a dramatic increase in the number of GFP+ (Scgb1a1 driver) cells in the lung parenchyma of Abl1fl/fl mice compared with wild-type mice 72 h after injury (Fig. 3A and SI Appendix, Fig. S5A). Staining for the type II AEC marker surfactant protein C (SPC), encoded by Sftpc, revealed that the GFP+ cells in the alveolar space coexpress SPC (Fig. 3B). Following injury, we observed a twofold increase in the number of GFP+ SPC+ cells in Abl1 knockout compared with wild-type mice. Notably, by day 3 after injury, 30% of SPC+ cells in Abl1fl/fl mice were GFP+, and therefore lineage-derived from Scgb1a1-expressing cells (SI Appendix, Fig. S5B; quantification is in Fig. 3C). Importantly, there was no significant difference in the GFP+ SPC+ cell populations in uninfected wild-type versus Abl1fl/fl mice, 3 wk and up to 4 mo after delivery of tamoxifen to induce Abl1 excision (SI Appendix, Fig. S6).
Fig. 3.

Conditional deletion of Abl1 promotes expansion of the GFP+ (Scgb1a1 driver) SPC+ cell population in mouse lungs following injury. (A) CC10 (Scgb1a1)-CreER; Rosa26-fGFP; Abl1wt (Left) or Abl1fl/fl (Right) mice were given tamoxifen four times 2 wk before nasal insufflation of S. aureus to induce excision of Abl1 and/or expression of GFP in Scgb1a1+ cells. Three days after exposure to the live bacteria, knockout mouse lungs demonstrated a dramatic increase in the GFP+ cell population in the alveolar space extending out from the bronchioles (Br) and BADJ. (Scale bars, 70 µm.) (B) Magnified subset at the 72-h time point (A, white box) with costaining for GFP and SPC antibodies shows that the GFP+ cell population in the alveolar space expresses the type II alveolar epithelial cell marker SPC. (Scale bars, 20 µm.) (C) Quantification of the percent of SPC+ cells expressing GFP in the entire left lungs of mice infected with S. aureus shows a doubling of the double-positive GFP+ SPC+ cell population in knockout (n = 9 mice) compared with wild-type mice treated with tamoxifen (n = 16 mice) 72 h after injury. Graph represents means with SEM. *P < 0.05.
We sought to investigate whether the GFP+ (Scgb1a1 driver) SPC+ cells in the alveolar epithelium retained expression of Scgb1a1. Costaining for GFP and Scgb1a1 showed that GFP+ cells in close proximity to the BADJ retained high levels of Scgb1a1 protein, but the expression of Scgb1a1 progressively decreased in GFP+ cells localized more distally from the BADJ (SI Appendix, Fig. S5C). Henceforth, we will refer to this population of cells as double-positive SCGB1A1+ SPC+ cells, even though they demonstrate variable expression of Scgb1a1 and are likely to undergo differentiation as they expand farther away from the BADJ. These data suggest that inactivation of Abl1 in Scgb1a1-expressing cells greatly enhances a population of double-positive SCGB1A1+ SPC+ cells in response to injury, and that these cells appear to originate from the bronchioles and/or BADJ and then expand into the alveolar epithelium to promote regeneration.
Pharmacological Inhibition of Abl Kinase Promotes Recovery of Mice Following Pneumonia.
The availability of pharmacological inhibitors of the Abl kinases prompted us to evaluate whether treatment with these compounds might be a useful therapeutic strategy following S. aureus-induced lung infection. We employed the Abl kinase-specific allosteric inhibitor GNF5 to evaluate Abl kinase inhibition as a treatment modality in bacterial pneumonia. GNF5 binds specifically to the myristoyl-binding site in the kinase domain of the Abl kinases and does not inhibit other protein kinases (39). Administration of GNF5 by oral gavage twice daily (b.i.d.) results in specific inhibition of the Abl kinases without known off-target effects (39). Treatment was initiated 1 h after nasal insufflation of S. aureus, and mice were evaluated 24 and 72 h following injury (Fig. 4A). Remarkably, two doses of GNF5, one initiated 1 h and another 16 h after injury, were sufficient to promote recovery after 24 h in drug-treated mice compared with vehicle control-treated mice (a 30-s video corresponding to movement tracings in Fig. 4B can be found in Movie S2). In contrast to the immobile and sickly appearance of vehicle control-treated mice, the mice treated with GNF5 were active and exhibited a healthy appearance. Analysis of lung tissue sections at 72 h postinfection revealed a significant decrease in lung injury in the treated mice (Fig. 4 C and D).
Fig. 4.

Treatment with an Abl kinase inhibitor reverses lung injury following nasal insufflation of S. aureus. (A) Timeline of experiment: CC10 (Scgb1a1)-CreER; Rosa26-fGFP mice were treated with an Abl kinase inhibitor or vehicle control b.i.d. starting 1 h after nasal insufflation of S. aureus. Mice were evaluated 4, 24, and 72 h after injury. (B) Movement tracings of a 30-s video (Movie S2) of vehicle- and GNF5-treated mice 24 h after induction showing faster recovery in mice treated with GNF5. (C) H&E staining of the left lung of vehicle- and GNF5-treated mice 3 d after injury. (A composite of images of mouse left lung sections using a 10× objective on the Zeiss AxioImager microscope stitched together with Zen software to recreate the entire left lung.) (D) Quantification of lung injury in H&E sections in vehicle- and GNF5-treated mice. (E and F) Bronchioalveolar lavage performed 3 d after injury shows (E) a reduction in protein in the airspace in mice treated with GNF5 starting 24 h before injury (Pre) or 24 h after injury (Post) compared with control mice and (F) a reduction in protein in the airspace in mice treated with nilotinib or GNF5 24 h after injury compared with control mice. (G) Three days after injury, mice treated with GNF5 (Right) demonstrated an increase in the GFP+ cell population in the alveolar epithelium compared with vehicle-treated mice (Left). (H) Costaining for GFP and SPC antibodies showed an induction of SPC expression, a marker for type II AECs, in both the GFP+ cells of the bronchiolar and alveolar epithelium in GNF5-treated mice. (I) Quantification of the percentage of SPC+ cells from the entire left lung that were also GFP+ in mice with genetic inactivation or pharmacological inhibition of Abl kinases compared with control mice. (J) Immunofluorescence staining for the type I AEC marker RAGE 3 d after injury shows widespread damage to the alveolar epithelium (dotted yellow lines) in untreated mice that is resolved in mice treated with GNF5. Graphs depict means and SEM of n mice, where n represents each individual animal used (i.e., n = 6 represents six individual mice). *P < 0.05, **P < 0.01, and ***P < 0.001.
To evaluate the effect of delayed drug treatment that more closely represents the delay in treatment initiation following the onset of pneumonia symptoms in the clinical setting, treatment schedules were initiated for mice exposed to S. aureus 24 h after injury and compared with mice treated 24 h prior to injury. BAL samples obtained at day 3 postinfection demonstrated a significant reduction in protein concentration in both the pretreatment group and in mice treated with GNF5 starting at 24 h after injury (Fig. 4E). Thus, even delayed treatment confers a therapeutic effect. A significant reduction in lung injury was also observed in mice treated with the ATP-binding site Abl kinase inhibitor nilotinib, a Food and Drug Administration (FDA)-approved drug for treatment of chronic myelogenous leukemia driven by oncogenic BCR-ABL (Fig. 4F).
Notably, we found a significant increase in the proportion of GFP+ (Scgb1a1 driver) SPC+ cells in GNF5-treated mice versus vehicle-treated mice, as well as enhanced SPC expression in SCGB1A1+ cells within the bronchioles of GNF5-treated mice after injury (Fig. 4 G–I). Further, the alveolar epithelium in mice treated with GNF5 recovered more rapidly by day 3 postinfection compared with control mice (Fig. 4J). These findings show that pharmacological or genetic inactivation of Abl elicits an expansion of double-positive GFP+ (Scgb1a1 driver) SPC+ cells that promote rapid regeneration of the alveolar epithelium following injury.
Genetic Deletion of Abl1 Promotes Expansion of SCGB1A1+ and SOX2+ Airway Cells That Coexpress SPC Following Pathogen-Induced Injury.
To characterize the specific cell types responsible for the marked expansion of SCGB1A1+ SPC+ cells into the damaged alveolar epithelium in Abl1-deficient mice, we employed three different mouse models: (i) CC10 (Scgb1a1)-CreERT; Rosa26-fGFP, (ii) SPC (Sftpc)-CreERT2; Rosa26-tdTomato, and (iii) SOX2-eGFP mice. Prior studies identified a small pool (<1 cell per BADJ) of SCGB1A1+ SPC+ putative bronchioalveolar stem cells within the BADJ as a potential cell type of origin for type II AECs (17). Consistent with previous reports, we found that after pathogen-induced injury, wild-type mice displayed an increase in the number of SPC+ cells that were also GFP+ around the BADJ (Fig. 3). However, the number of GFP+ SPC+ cells markedly increased from <5% up to ∼50% in the Abl1fl/fl mice after pathogen exposure (Fig. 3). The profound increase in the SCGB1A1+ SPC+ cell population in Abl1 knockout mice is unlikely to derive from the limited pool of resident double-positive SCGB1A1+ SPC+ cells within the BADJ alone.
To examine whether Abl1 inactivation in SCGB1A1+ cells affected their proliferation, we performed immunofluorescence staining for the Ki67 proliferation marker. A significant increase in proliferating SCGB1A1+ cells was observed in bronchioles of Abl1fl/fl mice compared with wild-type mice, with a maximal increase occurring at 4 h after injury (Fig. 5 A and B and SI Appendix, Fig. S7 A and B). These findings demonstrate that inactivation of Abl1 in SCGB1A1+ cells elicits an early wave of cell proliferation (∼4 h) following pathogen exposure that is not detected in wild-type controls, which show increased proliferation at 24 h postinjury compared with uninjured mice (SI Appendix, Fig. S7 A and B).
Fig. 5.
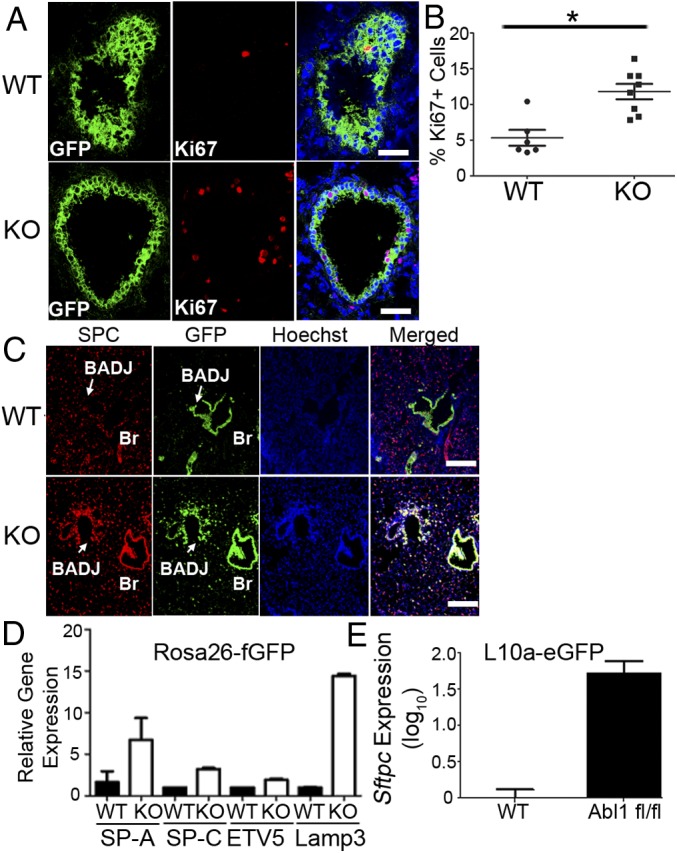
Genetic inactivation of Abl1 promotes proliferation and differentiation of Scgb1a1+ cells following injury. CC10 (Scgb1a1)-CreER; Rosa26-fGFP; Abl1wt or Abl1fl/fl mice were given tamoxifen four times 2 wk before nasal insufflation of S. aureus to induce excision of Abl1 and/or expression of GFP in Scgb1a1+ cells. (A) Four hours after exposure to S. aureus, GFP+ cells in knockout mice exhibited increased rates of proliferation as measured by Ki67 staining compared with wild-type mice. (Scale bars, 40 µm.) (B) Quantification. (C) Costaining for GFP and SPC showed a significant increase in expression of SPC within the bronchiolar secretory cell population of Abl1 knockout mice compared with wild-type mice. (Scale bars, 100 µm.) (D) GFP+ cells were isolated from the lungs of CC10 (Scgb1a1)-CreER; Rosa26-fGFP mice by FACS. RT-PCR of lysates from isolated cells showed a significant increase in expression of four genes highly expressed in type II cells in knockout mice compared with wild-type mice (n = 4 mice per group). (E) RT-PCR quantification of Sftpc mRNA expression from CC10 (Scgb1a1)-CreER; L10a-eGFP mice showing an increase in actively translated Sftpc mRNA pulled down from GFP-labeled ribosomes in knockout mice compared with wild-type control mice (n = 3 mice per group). Graphs represent means with SEM. *P < 0.05.
Next, we evaluated whether inactivation of Abl1 in SCGB1A1+ cells affected their differentiation. Costaining for GFP and SPC followed by immunofluorescence microscopy revealed a significant increase in expression of SPC within the bronchiolar secretory cell population of Abl1 knockout compared with wild-type control mice by day 3 following injury (Fig. 5C). In the absence of injury, SPC expression is specific to type II AECs and is not detected in the bronchiolar epithelium. To determine whether Abl1 inactivation elicited an increase in the expression of other type II AEC markers, we performed FACS of GFP+ cells from lungs of CC10 (Scgb1a1)-CreERT; Rosa26-fGFP; Abl1wt and Abl1fl/fl mice. RT-qPCR analysis of lysates from the isolated GFP+ cells revealed up-regulation of four of the most highly expressed genes in type II alveolar cells (Sftpa, Sftpc, Lamp3, and ETV5) (40) in Abl1 knockout compared with wild-type mice (Fig. 5D). To obtain a functional readout of altered gene transcription induced by Abl1 inactivation, we employed the translating ribosome affinity purification (TRAP) technique (41) using lungs from CC10 (Scgb1a1)-CreERT; L10a-eGFP; Abl1fl/fl and corresponding wild-type, control mice. RT-qPCR showed an increase in actively translated Sftpc mRNA that was pulled down from GFP-labeled ribosomes in Abl1 knockout compared with wild-type mice (Fig. 5E). These data show that Abl1 inactivation in a large population of SCGB1A1+ cells within the bronchioles elicits an early wave of enhanced proliferation at 4 h after alveolar epithelial damage, followed by a wave of differentiation 24 to 72 h after injury.
To determine whether Abl kinase inhibition specifically in type II alveolar epithelial cells contributes to the observed regenerative phenotype in mice after injury, we used SPC (Sftpc)-CreERT2; Rosa26-tdTomato mice (37) that were Abl1wt or Abl1fl/fl. After exposure to S. aureus, damage to the lung alveolar epithelium was detected in untreated SPC (Sftpc)-CreERT2; Rosa26-tdTomato; Ablwt and Abl1fl/fl mice compared with SPC (Sftpc)-CreERT2; Rosa26-tdTomato mice treated with the Abl kinase inhibitor GNF5 (SI Appendix, Fig. S8 A and B). Notably, we detected tdTomato+, RAGE+ (type I) cells derived from tdTomato+, SPC+, RAGE− (type II) cells at sites of damage only in mice treated with GNF5 3 d after injury (SI Appendix, Fig. S8C). While it has been well-established that type II AECs contribute to type I AEC regeneration after injury, our findings are consistent with published data that, in wild-type mice, type I AECs lineage-derived from type II AECs are only observed 5 to 7 d after injury (16, 42). These data suggest that knockout of Abl1 in type II AECs (SPC-CreERT2 model) alone is unlikely to contribute to the accelerated regeneration of type I AECs observed 72 h after knockout of Abl1 in Scgb1a1+ cells (CC10-CreERT model).
To further ascertain whether the expansion of SCGB1A1+ SPC+ cells arises from the bronchioles and BADJ versus the small fraction (∼10%) of SCGB1A1+ type II AECs (12), we employed a SOX2eGFP knockin/knockout mouse model (43). Within the lung, SOX2 is expressed specifically in airway cells but not in alveolar cells (44). Experiments performed in heterozygous mice in which the ORF of SOX2 is replaced by eGFP allow for identification of cells actively expressing SOX2, and not just lineage-derived cells from the airway cell population. Remarkably, we found a dramatic increase in the proportion of SOX2+ SPC+ cells expanding from the bronchioles into the lung parenchyma in mice treated with the Abl kinase allosteric inhibitor GNF5 compared with vehicle control-treated mice (Fig. 6). Thus, Abl kinase inhibition promotes expansion of airway cells that coexpresses SPC following injury.
Fig. 6.

Inhibition of Abl1 promotes expansion of the SOX2+ SPC+ cell population in mouse lungs following injury. SOX2eGFP mice were pretreated with vehicle or the Abl kinase inhibitor GNF5 (100 mg/kg, b.i.d. via oral gavage) 24 h before nasal insufflation of S. aureus. Three days after injury, lungs were harvested, paraffin-embedded, sectioned, and stained with antibodies to GFP and SPC. (A) Immunofluorescence staining showed a significant increase in the number of double-positive GFP+ SPC+ cells in GNF5-treated mice compared with vehicle-treated mice 72 h after injury. (Scale bars, 50 µm.) (B) Quantification of the proportion of double-positive cells was performed over 5-µm sections of the entire left lungs of mice (n = 5 mice per group). *P < 0.05. Bar graphs represent means with SEs of measurement.
Taken together, data from these three different mouse models suggest that inactivation of Abl kinases specifically in SCGB1A1+ and SOX2+ airway epithelial cells, but not type II AECs, leads to expansion of double-positive SCGB1A1+ SPC+ or SOX2+ SPC+ cell populations that precede regeneration of the damaged alveolar epithelium following injury.
Mobilization of SCGB1A1+ SPC+ Cells to Sites of Injury Promotes Alveolar Regeneration.
To track the mobilization of the double-positive SCGB1A1+ SPC+ cell population during regeneration of the alveolar epithelium, we performed immunofluorescence staining for GFP (Scgb1a1 driver), SPC (type II AEC marker), and RAGE (type I AEC marker) proteins in wild-type and Abl1 knockout mice at various times after exposure to S. aureus. In wild-type mice, we found an increase in the number of SPC+ but GFP− type II AECs at sites of injury in the alveolar epithelium at day 3 postinfection (Fig. 7A, Top). By contrast, Abl1 knockout in CC10 (Scgb1a1)-CreERT; Rosa26-fGFP; Abl1fl/fl mice exposed to S. aureus displayed increased numbers of GFP+ (Scgb1a1 driver) SPC+ cells at sites of alveolar repair following damage (Fig. 7A, Bottom). The architecture of the damaged alveolar epithelium was largely restored (as evaluated by immunostaining for RAGE) in the Abl1 knockout mice by day 3 postinfection with areas of hypercellularity and decreased alveolar volume, suggestive of damaged epithelium undergoing repair. In Abl1 knockout mice, GFP+ (Scgb1a1 driver) SPC+ cells expand from the bronchioles and/or BADJ to sites of damage, which precedes enhanced alveolar epithelial cell regeneration, and these phenotypes are not observed in wild-type mice (Fig. 7 B and C).
Fig. 7.

Scgb1a1+ SPC+ cells expand to sites of injury and promote regeneration. (A) Three days following exposure to S. aureus, CC10 (Scgb1a1)-CreER; Rosa26-fGFP; Abl1wt mouse lungs (Top) exhibit large areas of alveolar epithelial damage shown by staining for the type I AEC marker RAGE. There is an accumulation of type II AECs, indicated by staining for SPC, at sites of damage (purple dashed lines). Knockout mouse lungs (Bottom) exhibit regenerated alveolar epithelium with regions of hypercellularity and reduced area of alveolar loss/volume. In areas of repair (purple dashed lines), a dramatic increase in double-positive GFP+ SPC+ cells was observed. (Scale bars, 25 μm.) (B) Lower-magnification images of knockout (Right) mouse lungs 3 d after exposure to S. aureus demonstrate the enhanced proliferation and differentiation of Scgb1a1+ cells to Scgb1a1+ SPC+ cells compared with wild-type (Left) mouse lungs. (Scale bars, 50 µm.) (C) Higher-magnification images of cells expanded from the BADJ to sites of damage (boxes in B). (Scale bars, 20 µm.) (D) Model. Following injury to the alveolar epithelium, wild-type mice (Left) initiate regeneration through expansion of a rare population of double-positive Scgb1a1+ (CC10+) SPC+ cells at the BADJ and local differentiation of type II AECs. By contrast, knockout of Abl1 in Scgb1a1-expressing cells in the bronchioles and BADJ promotes proliferation and differentiation into double-positive Scgb1a1+ SPC+ cells, which in turn mobilize to sites of injury to indirectly promote regeneration of type I AECs from resident type II AECs.
To evaluate whether SCGB1A1+ SPC+ cells promote alveolar epithelium regeneration directly by differentiating into type I AECs, mouse lung sections were stained at various times following injury to detect the presence of triple-positive GFP+ (Scgb1a1 driver), RAGE+, SPC+ versus GFP+, RAGE+ but SPC− cells at sites of injury. Small clusters of triple-positive cells as well as GFP+ (Scgb1a1 driver) RAGE+ cells were detected in lung sections of both wild-type and Abl1 knockout mice 30 d following injury but not at earlier time points (SI Appendix, Fig. S9). Because Abl1 knockout mice exhibited accelerated recovery from infection 24 to 72 h after injury, it is unlikely that differentiation of GFP+ (Scgb1a1 driver) SPC+ cells into GFP+ RAGE+ cells at day 30 postinjury significantly contributed to the observed early alveolar regeneration phenotypes. It is more likely that double-positive SCGB1A1+ SPC+ cells mobilize to sites of injury and promote local type II AECs to regenerate type I AECs, leading to repair of the damaged lung epithelium (model, Fig. 7D). Consistent with this possibility is the observation that SPC (Sftpc)-CreERT2; Rosa26-tdTomato mice treated with GNF5 displayed an increase in the proportion of tdTomato+ (Sftpc driver) RAGE+ type I AECs compared with untreated wild-type and Abl1 knockout mice in SPC+ type II AECs (SI Appendix, Fig. S8).
Loss of Abl1 in SCGB1A1+ Lung Epithelial Cells Promotes Recovery in Mice Following S. pneumoniae-Induced Injury.
To assess whether conditional knockout of Abl1 promoted lung epithelial regeneration in mice following exposure to other pathogens, we used S. pneumoniae. We evaluated recovery in wild-type and Abl1 knockout mice following nasal insufflation of S. pneumoniae (45), a more virulent bacterial strain than the strain of S. aureus used. We found that while both wild-type and Abl1 conditional knockout mice (Scgb1a1 driver) lost the same amount of body weight within 12 h after injury, the Abl1 knockout mice recovered body weight more quickly compared with wild-type, control mice (SI Appendix, Fig. S10A). Consistent with these findings, we found decreased damage to the lung parenchyma, as measured by an increase in average alveolar area/volume, in Abl1 knockout compared with wild-type mice (SI Appendix, Fig. S10B). We also found a significant increase in the proportion of GFP+ (Scgb1a1 driver) SPC+ cells in Abl1 knockout versus wild-type mice. Interestingly, in the S. pneumoniae injury model, we found a dramatic expansion of the double-positive cells as early as 6 h after injury (SI Appendix, Fig. S10 C and D). Thus, data from both S. aureus- and S. pneumoniae-induced injury models demonstrate that conditional knockout of Abl1 (Scgb1a1 driver) leads to expansion of a subpopulation of SCGB1A1+ SPC+ cells, promotes lung regeneration, and accelerates mouse recovery after injury.
Discussion
Respiratory pathologies are the third-leading cause of death in the industrialized world and, with the emergence of resistant bacterial strains, there is an urgent need to identify novel therapies for use in combination with antibiotics and supportive care. Here we identify the Abl kinases as a promising therapeutic target to promote alveolar epithelial regeneration following lung injury. Previous studies showed that distal lung epithelium, including type I and type II AECs, can be derived from a population of SCGB1A1+ SPC+ cells. During lung development, these cells have been identified as a putative cell type of origin for both type I and type II AECs (40). There is evidence that a rare population of these putative bronchioalveolar stem cells exists in the adult lung and resides at the BADJ (17). Accordingly, we found that in wild type- and/or vehicle control-treated mice, normal regeneration of lung alveolar epithelium involves expansion (from <5% to 10 to 15%) of a small pool (<1 cell per BADJ) of SCGB1A1+ SPC+ cells originating at the BADJ, with complete recovery taking about 1 wk. Another recent study showed expansion of this population of cells 11 to 17 d after bleomycin- or influenza-induced lung injury (46). Surprisingly, we found that genetic or pharmacological inactivation of Abl kinases promotes a dramatic expansion (<5% to 25 to 50%) of airway cells positive for Scgb1a1 or SOX2 that coexpress the SPC type II alveolar marker by 24 h following S. aureus-induced injury and as early as 6 h following S. pneumoniae-induced injury. Further, Abl1 genetic inactivation specifically in Scgb1a1+-expressing cells mobilizes a large pool of Scgb1a1+ cells from the bronchioles that are not appreciably involved in regeneration of the alveolar epithelium in wild-type mice. We detected cell proliferation within 4 h after pathogen-induced injury in Abl1-deficient mice, followed by a wave of differentiation (24 to 72 h) of an expanded pool of SCGB1A1+ secretory cells within the bronchiole, leading to dramatic expansion of double-positive SCGB1A1+ SPC+ cells at sites of injury, followed by enhanced alveolar regeneration compared with wild-type mice. The rapid timeline of regeneration observed in Abl1-deficient mice is relevant because S. aureus and S. pneumoniae infections are associated with progression to sepsis and multiorgan system failure, and early intervention is key in reducing mortality in these patients. In fact, early antibiotic treatment within the first 1 to 6 h is the only proven intervention to reduce mortality, highlighting the importance of early intervention (47, 48). We found no effect of Abl1 inactivation in SCGB1A1 cells on immune cell responses and susceptibility to bacterial infection compared with wild-type mice. Both genetic and pharmacological inactivation of Abl kinases elicited rapid recovery following pathogen-induced injury and enhanced regeneration of the alveolar epithelium.
Genetic inactivation of Abl1 in Scgb1a1+ cells was found to not only promote expansion of SCGB1A1+ SPC+ cells but also to enhance regeneration of RAGE+ type I alveolar epithelium within 3 d after injury. Our data suggest that inactivation of Abl kinases promotes expansion of SCGB1A1+ SPC+ cells, which indirectly leads to regeneration of type I AECs following injury. The absence of GFP+ (Scgb1a1 driver) RAGE+ cells at day 3 post S. aureus-induced injury suggests that the double-positive SCGB1A1+ SPC+ cells are not likely to directly differentiate into type I AECs to regenerate the alveolar epithelium. While it is not possible to rule out epigenetic silencing at the Rosa26 locus as a potential mechanism for the loss of GFP expression in type I AECs following differentiation, the fact that small clusters of GFP+ RAGE+ cells were detected at day 30 suggested that epigenetic silencing was unlikely the reason. It is more likely that type I AEC regeneration occurs through the differentiation of type II AECs locally at sites of injury induced by the expansion and mobilization of SCGB1A1+ SPC+ cells. Alternatively, the double-positive SCGB1A1+ SPC+ cells may function to activate stromal cells at sites of injury to promote regeneration of type I AECS from type II alveolar cells. In this regard, signaling by bone morphogenetic protein 4 was shown to regulate alveolar progenitor cell proliferation and differentiation, in part by targeting stromal cells in the alveolar stem cell niche (49).
The finding that inactivation of Abl kinases promotes regeneration of the alveolar epithelium following pathogen-induced injury uncovers a previously unappreciated deleterious role for activation of Abl kinases following lung injury. The activity of Abl kinases is required for normal mouse development, as genetic inactivation of Abl1 results in perinatal lethality (18, 21). Inhibition of Abl kinases with pharmacological agents in healthy adult mice does not produce deleterious effects, thereby suggesting that endogenous Abl kinases are not required for normal cellular homeostasis in the adult (22). Abl kinases are hyperactive in BCR-ABL–positive leukemia and some solid tumors, as well as in response to inflammation, DNA damage, and oxidative stress, and in various pathologies (19, 22, 27). Abl kinase inhibitors have beneficial therapeutic effects for the treatment of human leukemia, metastatic tumors in mice, as well as pathologies linked to inflammation and neurodegeneration (19, 27, 29–31, 50). The consequences of Abl activation for cell proliferation are cell context-dependent. Whereas activation of the Abl kinases promotes cell proliferation in leukemia cells, some solid tumors, and multiple cell types stimulated with growth factors, Abl1 activation is antiproliferative in the response to DNA damage (51), and Abl2 negatively regulates myoblast proliferation in mice (52).
Abl kinases regulate various downstream targets, some of which have been shown to play a role in the regulation of lung epithelial cells following injury. These include transcriptional coactivators of the Hippo (Yap1, Taz) and Wnt (β-catenin) signaling pathways, both of which have been implicated in alveolar regeneration (53–55). The Hippo pathway regulates organ development, and Yap1 activation regulates mechanical tension-induced pulmonary alveolar regeneration (55) as well as airway regeneration following exposure to naphthalene (54). Functional interactions among Abl1, Yap1, and Taz have been reported in development, cancer, and the cellular response to damage. Activation of Abl1 in response to DNA damage was reported to promote activation of a proapoptotic program mediated by Yap1 (56). Abl kinases were recently shown to regulate osteoblast differentiation and lung adenocarcinoma metastasis by regulating stabilization of the Taz transcriptional coactivator (50, 57). Previously, we reported that Abl kinases regulate the Wnt-β-catenin pathway in lung cancer cells (50). Wnt signaling was shown to regulate cell proliferation and differentiation during alveologenesis (53). Future studies will seek to identify the molecular targets downstream of activated Abl kinases in lung epithelial cells that regulate cell proliferation and differentiation pathways in response to injury induced by S. aureus and other agents.
Our findings revealed that inactivation of Abl kinases promotes lung epithelial cell regeneration in mouse models of pneumonia induced by S. aureus and S. pneumoniae. The data suggest that FDA-approved inhibitors for the treatment of BCR-ABL+ leukemias might be repurposed for the treatment of lung injury. In fact, several case reports have demonstrated that treatment of leukemia patients with imatinib (which inhibits not only ABL kinases but the PDGFR and other kinases) resulted in resolution of pulmonary symptoms such as acute interstitial pneumonia or drug-induced pneumonitis (58–62). Our studies have focused on lung injury induced by S. pneumoniae and S. aureus, which target the alveolar epithelium resulting in necrotizing pneumonias, and future studies are warranted to evaluate whether inactivation of Abl kinases might be exploited as a therapeutic strategy to promote lung regeneration in response to injury induced by a variety of toxins and other pathogens. These include destructive agents such as chlorine gas and influenza virus, as well as nondestructive lung injury such as drug-induced pneumonitis or atypical pneumonia. Our findings of regeneration in mice with Abl-specific inactivation following pathogen-induced injury, together with case reports of patients treated with imatinib showing improved pulmonary function following drug-induced pneumonitis, suggest that Abl inhibitors might be used to promote repair and/or regeneration of the lung in a wide variety of lung pathologies.
Materials and Methods
Additional experimental details are provided in SI Appendix, including bacteria and reagents used, as well as procedures for real-time PCR analysis, immunoblotting, bronchioalveolar lavage, immunohistochemistry, immunofluorescence, FACS, and mouse experiments.
Cell Culture.
Primary human bronchial epithelial cells were isolated from normal donors as previously described (36), and were provided by the University of North Carolina at Chapel Hill Cystic Fibrosis Center Tissue Procurement and Cell Culture Core. Passage 1 cells were derived from human lungs unsuitable for transplantation from nonsmoker organ donors without preexisting lung disease under protocol 03-1396 approved by the University of North Carolina at Chapel Hill Biomedical Institutional Review Board. Informed consent was obtained from authorized representatives of all organ donors.
Mice.
CC10 (Scgb1a1)-CreERT; Rosa26R-CAG-farnesylated GFP (Rosa26-fGFP) and SPC (Sftpc)-CreERT2; Rosa26-tdTomato mice were provided by Mark Onaitis, University of California, San Diego, La Jolla, CA and generated by Brigid Hogan at Duke University and have been previously described (12, 37). These mice were crossed with Abl1flox/flox mice (63) into a C57BL/6 genetic background. Sox2tmHoch/J mice (referred to as SOX2-eGFP mice) were purchased from the Jackson Laboratory. Gt(Rosa)26Sortm9(EGFP/Rpl10z)Amc/J (referred to as L10-eGFP) mice were purchased from the Jackson Laboratory and crossed to CC10-CreERT; Abl1flox/flox mice into a C57BL/6 background for the TRAP experiments. To induce expression of Cre-recombinase, 8- to 20-wk-old male and female mice were injected intraperitoneally four times every other day with 0.25 mg/g body weight tamoxifen (Sigma; T5648) in corn oil (Spectrum; CO136). Exposure to S. aureus was initiated 15 d after the last dose of tamoxifen. All experiments were performed under Duke University Institutional Animal Care and Use Committee-approved protocols A098-16-04 and A130-16-06. Additional experimental details are provided in SI Appendix.
Statistical Analysis.
The number of animals in each group is indicated in the figures and/or figure legends. Differences among groups for immunofluorescence and H&E experiments were assessed using one-way ANOVA followed by post hoc Tukey tests. All quantification for these experiments was performed using stitched images for the entire left lung (>100,000 cells counted per image). Representative high-resolution images from each animal group are indicated in the figures. Results are presented as means ± SE of measurement. BAL experiments were analyzed using a repeated-measure multivariate analysis of variance (MANOVA) followed by post hoc Fisher’s protected least significant difference testing to account for difference in variance between cell count, protein, and LDH data collected from each animal. Individual group testing was performed only when statistical significance was achieved from the ANOVA and MANOVA. All quantification for these experiments was performed using fluid from both the right and left lungs from each animal. For the human bronchial epithelial cell experiments, cells from three nonsmoker donors were used. Real-time RT-qPCR experiments were all performed in duplicate or triplicate in two or three separate mice or human patient samples. *, **, and *** indicate P values, <0.05, <0.01, and <0.001, respectively. Bar graphs represent means with SEs of measurement.
Supplementary Material
Acknowledgments
We thank Dr. Claire Doerschuk (University of North Carolina) for providing the glycerol stock and advice on the growth of the S. pneumoniae bacteria. We thank Dr. Emily Riggs (Duke University) for breeding and maintaining the mouse colonies. We thank Craig Marshall and Martha Salinas (Duke University) for preparation of live S. aureus and technical assistance. We thank Dr. Brigid Hogan (Duke University) and Dr. Mark Onaitis (UCSD) for generating the CC10-CreER mouse line and graciously providing these mice. Biomarker profiling was performed under the management of Dr. Andrew N. Macintyre and direction of Dr. Gregory D. Sempowski in the Immunology Unit of the Duke Regional Biocontainment Laboratory, which received partial support for construction from the National Institute of Allergy and Infectious Diseases (Grant UC6-AI058607). A.K. receives support from National Heart, Lung, and Blood Institute Grant F30HL126448 and Training Grant T32GM007171. S.H.R. receives support from National Institutes of Health Grant DK065988 and Cystic Fibrosis Foundation Grant BOUCHE15R0. P.R.T. receives support from National Heart, Lung, and Blood Institute Grant 4R00HL127181, and is a Whitehead Scholar. C.A.P. receives support from National Institutes of Health Grant 1R01HL135239-01A1. B.D.K. receives support from National Institutes of Health Grant 1K08HL130557-01A1. This work was supported by National Institutes of Health Grants R01CA195549 and R01AI056266 (to A.M.P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1816030116/-/DCSupplemental.
References
- 1.Gillet Y, et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359:753–759. doi: 10.1016/S0140-6736(02)07877-7. [DOI] [PubMed] [Google Scholar]
- 2.Garnier F, et al. Pneumonia and new methicillin-resistant Staphylococcus aureus clone. Emerg Infect Dis. 2006;12:498–500. doi: 10.3201/eid1203.051040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gillespie SH, Balakrishnan I. Pathogenesis of pneumococcal infection. J Med Microbiol. 2000;49:1057–1067. doi: 10.1099/0022-1317-49-12-1057. [DOI] [PubMed] [Google Scholar]
- 4.Zaas DW, Duncan MJ, Li G, Wright JR, Abraham SN. Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. J Biol Chem. 2005;280:4864–4872. doi: 10.1074/jbc.M411702200. [DOI] [PubMed] [Google Scholar]
- 5.Inoshima I, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17:1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci USA. 2010;107:13473–13478. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs DM, Shaver A. Prevalence of and outcomes from Staphylococcus aureus pneumonia among hospitalized patients in the United States, 2009-2012. Am J Infect Control. 2017;45:404–409. doi: 10.1016/j.ajic.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Hageman JC, et al. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003-04 influenza season. Emerg Infect Dis. 2006;12:894–899. doi: 10.3201/eid1206.051141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finelli L, et al. Influenza-associated pediatric mortality in the United States: Increase of Staphylococcus aureus coinfection. Pediatrics. 2008;122:805–811. doi: 10.1542/peds.2008-1336. [DOI] [PubMed] [Google Scholar]
- 10.Mulcahy ME, McLoughlin RM. Staphylococcus aureus and influenza A virus: Partners in coinfection. MBio. 2016;7:e02068-16. doi: 10.1128/mBio.02068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogan BL, et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawlins EL, et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4:525–534. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tata PR, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–223. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rock JR, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotton DN, Morrisey EE. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat Med. 2014;20:822–832. doi: 10.1038/nm.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barkauskas CE, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim CF, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 18.Koleske AJ, et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 19.Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129:9–16. doi: 10.1242/jcs.175521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. Defective T cell development and function in the absence of Abelson kinases. J Immunol. 2007;179:7334–7343. doi: 10.4049/jimmunol.179.11.7334. [DOI] [PubMed] [Google Scholar]
- 21.Chislock EM, Ring C, Pendergast AM. Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1-mediated survival. Proc Natl Acad Sci USA. 2013;110:12432–12437. doi: 10.1073/pnas.1304188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat Rev Cancer. 2013;13:559–571. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wetzel DM, McMahon-Pratt D, Koleske AJ. The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal Leishmania infection. Mol Cell Biol. 2012;32:3176–3186. doi: 10.1128/MCB.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton EA, Plattner R, Pendergast AM. Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 2003;22:5471–5479. doi: 10.1093/emboj/cdg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tammer I, Brandt S, Hartig R, König W, Backert S. Activation of Abl by Helicobacter pylori: A novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology. 2007;132:1309–1319. doi: 10.1053/j.gastro.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 26.Sun X, et al. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J Biol Chem. 2000;275:17237–17240. doi: 10.1074/jbc.C000099200. [DOI] [PubMed] [Google Scholar]
- 27.Schlatterer SD, Acker CM, Davies P. c-Abl in neurodegenerative disease. J Mol Neurosci. 2011;45:445–452. doi: 10.1007/s12031-011-9588-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B. c-Abl in oxidative stress, aging and cancer. Cell Cycle. 2005;4:246–248. [PubMed] [Google Scholar]
- 29.Chislock EM, Pendergast AM. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS One. 2013;8:e85231. doi: 10.1371/journal.pone.0085231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rizzo AN, et al. Imatinib attenuates inflammation and vascular leak in a clinically relevant two-hit model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1294–L1304. doi: 10.1152/ajplung.00031.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizzo AN, Aman J, van Nieuw Amerongen GP, Dudek SM. Targeting Abl kinases to regulate vascular leak during sepsis and acute respiratory distress syndrome. Arterioscler Thromb Vasc Biol. 2015;35:1071–1079. doi: 10.1161/ATVBAHA.115.305085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aman J, et al. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation. 2012;126:2728–2738. doi: 10.1161/CIRCULATIONAHA.112.134304. [DOI] [PubMed] [Google Scholar]
- 33.Aman J, Peters MJ, Weenink C, van Nieuw Amerongen GP, Vonk Noordegraaf A. Reversal of vascular leak with imatinib. Am J Respir Crit Care Med. 2013;188:1171–1173. doi: 10.1164/rccm.201301-0136LE. [DOI] [PubMed] [Google Scholar]
- 34.Dudek SM, et al. Abl tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21:4042–4056. doi: 10.1091/mbc.E09-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfson RK, Lang G, Jacobson J, Garcia JGN. Therapeutic strategies to limit lung endothelial cell permeability. In: Voelkel N, Rounds S, editors. The Pulmonary Endothelium: Function in Health and Disease. John Wiley & Sons; Hoboken, NJ: 2009. pp. 337–354. [Google Scholar]
- 36.Fulcher ML, Randell SH. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol. 2013;945:109–121. doi: 10.1007/978-1-62703-125-7_8. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci USA. 2012;109:4910–4915. doi: 10.1073/pnas.1112499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Athale J, et al. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53:1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP) Nat Protoc. 2014;9:1282–1291. doi: 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Kumar VS, Zhang W, Rehman J, Malik AB. Activation of type II cells into regenerative stem cell antigen-1(+) cells during alveolar repair. Am J Respir Cell Mol Biol. 2015;53:113–124. doi: 10.1165/rcmb.2013-0497OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnold K, et al. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell. 2011;9:317–329. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tompkins DH, et al. Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS One. 2009;4:e8248. doi: 10.1371/journal.pone.0008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez JC, Dang H, Martin JR, Doerschuk CM. Nrf2 modulates host defense during Streptococcus pneumoniae pneumonia in mice. J Immunol. 2016;197:2864–2879. doi: 10.4049/jimmunol.1600043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng D, et al. Regeneration of alveolar type I and II cells from Scgb1a1-expressing cells following severe pulmonary damage induced by bleomycin and influenza. PLoS One. 2012;7:e48451. doi: 10.1371/journal.pone.0048451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sterling SA, Miller WR, Pryor J, Puskarich MA, Jones AE. The impact of timing of antibiotics on outcomes in severe sepsis and septic shock: A systematic review and meta-analysis. Crit Care Med. 2015;43:1907–1915. doi: 10.1097/CCM.0000000000001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrer R, et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: Results from a guideline-based performance improvement program. Crit Care Med. 2014;42:1749–1755. doi: 10.1097/CCM.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 49.Chung MI, Bujnis M, Barkauskas CE, Kobayashi Y, Hogan BLM. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 2018;145:dev163014. doi: 10.1242/dev.163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu JJ, et al. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight. 2016;1:e89647. doi: 10.1172/jci.insight.89647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keshet R, et al. c-Abl antagonizes the YAP oncogenic function. Cell Death Differ. 2015;22:935–945. doi: 10.1038/cdd.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JK, Hallock PT, Burden SJ. Abelson tyrosine-protein kinase 2 regulates myoblast proliferation and controls muscle fiber length. eLife. 2017;6:e29905. doi: 10.7554/eLife.29905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frank DB, et al. Emergence of a wave of Wnt signaling that regulates lung alveologenesis by controlling epithelial self-renewal and differentiation. Cell Rep. 2016;17:2312–2325. doi: 10.1016/j.celrep.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lange AW, et al. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J Mol Cell Biol. 2015;7:35–47. doi: 10.1093/jmcb/mju046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Z, et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 2016;16:1810–1819. doi: 10.1016/j.celrep.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 56.Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29:350–361. doi: 10.1016/j.molcel.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 57.Matsumoto Y, et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J Clin Invest. 2016;126:4482–4496. doi: 10.1172/JCI87802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carnevale-Schianca F, et al. Complete resolution of life-threatening bleomycin-induced pneumonitis after treatment with imatinib mesylate in a patient with Hodgkin’s lymphoma: Hope for severe chemotherapy-induced toxicity? J Clin Oncol. 2011;29:e691–e693. doi: 10.1200/JCO.2011.35.6733. [DOI] [PubMed] [Google Scholar]
- 59.Fenocchio E, et al. Successful treatment of gemcitabine-induced acute interstitial pneumonia with imatinib mesylate: A case report. BMC Cancer. 2016;16:793. doi: 10.1186/s12885-016-2833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langberg MK, et al. Imatinib may reduce chemotherapy-induced pneumonitis. A report on four cases from the SWENOTECA. Acta Oncol. 2018;57:1401–1406. doi: 10.1080/0284186X.2018.1479072. [DOI] [PubMed] [Google Scholar]
- 61.Majhail NS, Schiffer CA, Weisdorf DJ. Improvement of pulmonary function with imatinib mesylate in bronchiolitis obliterans following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2006;12:789–791. doi: 10.1016/j.bbmt.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 62.Distler JH, Manger B, Spriewald BM, Schett G, Distler O. Treatment of pulmonary fibrosis for twenty weeks with imatinib mesylate in a patient with mixed connective tissue disease. Arthritis Rheum. 2008;58:2538–2542. doi: 10.1002/art.23694. [DOI] [PubMed] [Google Scholar]
- 63.Moresco EM, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25:6105–6118. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.