

Abstract
Background
Significant numbers of variants detected in cancer patients are often left labeled only as variants of unknown significance (VUS). In order to expand precision medicine to a wider population, we need to extend our knowledge of pathogenicity and drug response in the context of VUS’s.
Methods
In this study, we analyzed variants from AACR Project GENIE Consortium APG (Cancer Discov 7:818-831, 2017) and compared them to the COSMIC database Forbes et al. (Nucleic Acids Res 43:D805-811, 2015) to identify recurrent variants that would merit further study. We filtered out known hotspot variants, inactivating variants in tumor suppressors, and likely benign variants by comparing with COSMIC and ExAC Lee et al. (Science 337:967-971, 2012).
Results
We have identified 45,933 novel variants with unknown significance unique to GENIE. In our analysis, we found on average six variants per patient where two could be considered as pathogenic or likely pathogenic and the majority are VUS’s. More importantly, we have discovered 730 recurrent variants that appear more than 3 times in GENIE but less than 3 in COSMIC. If we combine the recurrences of GENIE and COSMIC for all variants, 2586 are newly identified as occurring more than 3 times than when using COSMIC alone.
Conclusions
Although it would be inappropriate to blindly accept these recurrent variants as pathogenic, they may warrant higher priority than other observed VUS’s. These newly identified recurrent variants might affect the molecular profiles of approximately 1 in 6 patients. Further analysis and characterization of these variants in both research and clinical contexts will improve patient treatments and the development of new therapeutics.
Electronic supplementary material
The online version of this article (10.1186/s12885-019-5313-1) contains supplementary material, which is available to authorized users.
Keywords: Precision medicine, GENIE, Recurrent variants, Variant of unknown significance, Variant disparity
Background
In cancer genomic analysis, it is commonplace to find rare variants whose pathogenicity and contributions to various aspects of tumorigenesis are not easily evaluated. In those circumstances, such variants are labeled variants of unknown significance (VUS) and focus is shifted to pathogenic or likely pathogenic mutations. Since the bulk of variants are VUS’s, there are many efforts to characterize them by using functional cell-based assays, somatic mutation signatures [1], gene expression [2], and structure based approaches [3]. Although functional cell-based assay approaches are powerful, they are time consuming and can still fall short of capturing certain aspects of pathogenicity, particularly those of a multicellular nature, such as escape from the immune system. Somatic signature and gene expression analysis requires whole genome or exome sequencing and gene expression data that are not attainable from panel sequence assays, which are the most commonly performed assay in the clinic today. Instead by studying the characteristics of variants observed over many panel sequenced samples, it may be possible to better understand the relevancy of a particular alteration.
An oft used metric for prioritizing VUS’s is the recurrence rate. Although high recurrence is insufficient to indicate pathogenicity, it can assist doctors in hypothesizing as to the etiological cause of the tumor and highlight specific VUS’s. Databases like COSMIC [4] and cBioPortal [5] are cataloging variants observed in a wide variety of studies. By offering a comprehensive set of observed alterations, researchers and clinicians can better prioritize variants in their own samples for further study or action, particularly when the exact biological function is unclear. By studying the frequencies, distributions, and types of variants seen across many cancers and associated clinical information, it may be possible to better classify a novel variant and advance precision medicine through the development of more accurate diagnostic, prognostic, and therapeutic markers and signatures.
AACR’s GENIE project [6] is a multi-year study to advance precision oncology. By working with cancer centers around the world, GENIE has collected genomic and clinical data from tens of thousands of cancer patients. Such a project is vital to improving the identification of actionable variants, particularly in light of the high variability in detecting actionable variants found across smaller studies. A recent precision medicine study shows that only 10 % of patients are eligible for FDA-labeled targeted treatment [7]. However, approximately half of patients had actionable variants in the MOSCATO 01 trial [8]. By performing a broad variant analysis on this new resource, we hope to characterize a set of novel and potentially clinically relevant VUS’s to enable precision medicine to better address a wider patient population. Such recurrent variants would serve as new lines of research inquiry and better enable clinicians to assess and act upon the genomic profile of their own patients.
Methods
GENIE ver. 1.0, publicly released on January 5th, 2017, was used for this study. Samples in 524 tumor types from 32 tissues including both liquid and solid malignancies were sequenced at 8 participating centers using 12 cancer panels [9]. Dana Farber Cancer Institute, Memorial Sloan Kettering Cancer Center, and Vanderbilt-Ingram Cancer Center used hybridization capture whereas the remaining five centers used a PCR method. Not all panels included full genes with promoters and introns, and some only cover hotspots. Most tumor samples are not accompanied with matching normal samples except those from Memorial Sloan Kettering Cancer Center and Vanderbilt-Ingram Cancer Center; thus, it is important to remove potential germline variants. GENIE provides neither copy number alteration nor structural variants; therefore, this study focuses on recurrent SNV and small indels. The workflow of various filters to classify variants and to extract GENIE recurrent variants is illustrated (Fig. 1).
Fig. 1.
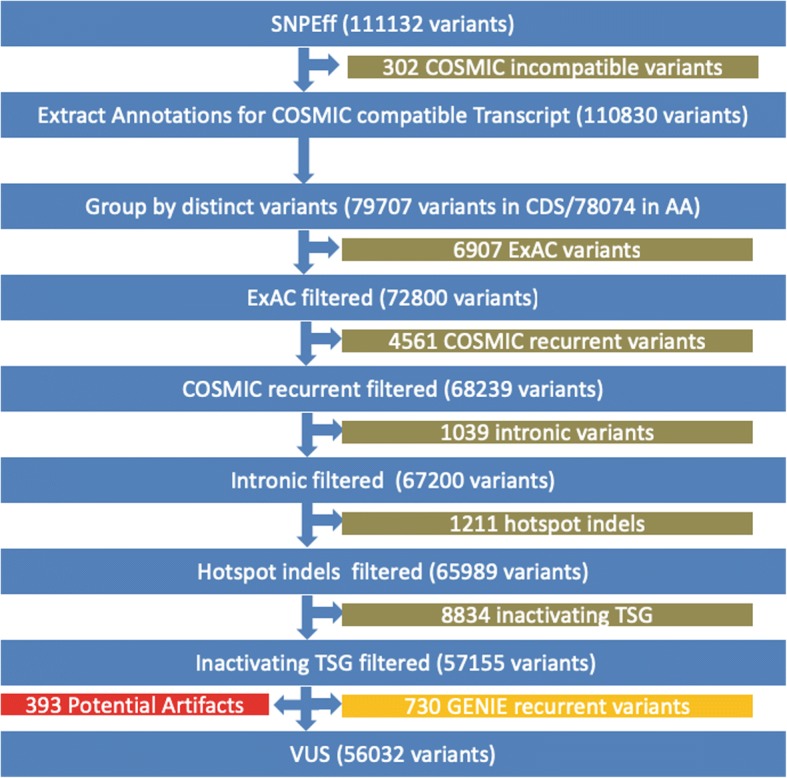
Process flow diagram of filters to remove variants
SNPEff [10] ver 4.3 using GRCh37.75 database was used to annotate variants. SNPEff annotations were extracted for COSMIC compatible transcripts. Although many COSMIC transcripts were consistent with Ensembl transcript IDs, some were provided as a RefSeq transcript ID, had been deprecated or belonged to non-human organisms. These inconsistencies were manually corrected; however, there remained several transcripts that could not be matched with any COSMIC transcripts.
ExAC release 0.3.1 [11] was downloaded and adjusted allele counts (AC_adj) and adjusted total counts (AN_adj) were extracted for each variant. Although the GENIE dataset already had some variant filtering using ExAC, there remained alterations that appeared with higher than expected frequency in the ExAC database. After application of Hypothesis Testing for the Difference in Population Proportions with 5% (Z > = 1.645) significance level, 6907 variants are removed.
Besides transcript compatibility issues, there were other challenges in comparing variants between GENIE and COSMIC. There were slight differences in the notation of variants between COSMIC and SNPEff outputs. For instance, SNPEff duplication annotations like p.L23dup is not used in COSMIC and instead COSMIC uses ins rather than dup. Also, SNPEff promoter variants such as c.-124C > T are expressed as c.1-124C > T in COSMIC. A tandem double variant in COSMIC may be expressed as c.1798_1799GT > AA whereas SNPEff outputs it as c.1798_1799delGTinsAA. As for amino acid change notations, the SNPEff deletion p.G469del might be written as p.G469delG in COSMIC. Finally, COSMIC has many instances of “c.?”, representing an unknown coding sequence change. After resolving these issues, we successfully removed 4561 COSMIC recurrent variants with counts ≥3.
Further filtering steps included removing intronic variants, short indels in hotspots, and inactivating variants in tumor suppressor genes. Intronic variants located 2 bp outside of the exon boundary were excluded but critical splicing acceptor and donor variants were kept. Upstream and downstream variants beyond 1000 bp from start and stop codon were discarded. These steps resulted in 1039 variants being removed. Short indels in hotspots were filtered out. If we observed more than 10 overlapping indels in a region regardless of being in-frame or not, we deemed the region a hotspot. Well known regions in cancer genomics include PIK3R1 (p85alpha iSH2 domain) [12], FLT3 ITD near R595 (Y591 and Y597) in exon 14 [13], and EGFR exon 19 [14] and exon 20 [15]. 1211 hotspot indels were removed as a result.
Inactivating variants such as stop gained, start loss, frameshift, splicing acceptor, splicing donor, and stop loss were considered as likely loss of function, and when found in tumor suppressor genes, they were removed under the assumption they were likely pathogenic. We have manually annotated tumor suppressors for the 536 GENIE mutated genes. These include some unequivocal tumor suppressors accepted by many, such as TP53, RB1, PTEN, NF1, APC, and CDKN2A. Though less established, many other genes such as B2M [16], CBFB [17], CUL3 [18], FUBP1 [19], GATA3 [20], GPS2 [21],HLA-A [22], MAP3K1 [23], MGA [24], NCOR1 [25], RASA1 [26], RBM10 [27], RNF43 [28], and RYBP [29] were included based upon current evidence in the literature. The full list of tumor suppressor genes defined in this study and corresponding evidences to support their designations is provided in the supplementary material (Additional file 1: Table S1). Using this set of tumor suppressors, 8834 variants were removed by this filter.
There remains the possibility that a number of sequencing related artifacts may be present in the recurrent list. To minimize such artifacts, we removed variants found only from a single sequencing center and not listed in COSMIC. With these criteria and a frequency threshold of at least three samples, 730 recurrent variants unique to GENIE were discovered.
Results
The GENIE project contains data from 18,966 patients generated from a variety of sequence panels. A total of 111,132 variants were observed across these samples with a mean of six variants per sample. The processing of these variants is described in the methods. In brief, variants that do not lie within COSMIC gene transcripts were removed, leaving 110,830 variants. Among those, there are 79,707 coding sequence (CDS) changes and 78,074 variants leading to an amino acid change. 67,793 variants appeared only once in GENIE and 30 variants are observed over 100 times (Fig. 2).
Fig. 2.

Variant recurrence in GENIE samples. Histogram indicates the number of variants (y-axis, log-scale) that occur at a given frequency (x-axis). As the frequency of recurrence increases, the number of variants decreases. However, a sizable number of variants observed in over 100 samples, which are listed in Table 1
These highly recurrent variants are mostly found in well-established cancer genes like KRAS, TP53, and PIK3CA. KRAS G12D was the most frequently observed (711 samples) followed by BRAF V600E (615 samples) (Table 1). There are hotspot variants found for individual cancers. In NSCLC, expected recurrent variants in KRAS, TP53, and PIK3CA are observed alongside hotspot variants EGFR L858R and exon 19 deletion E746_A750del. IDH codon 132 variants are seen in various cancers [30], and AKT1 E17K is commonly observed in breast cancer [31]. FGFR3 S249C often appears in bladder cancer [32]. All the highly recurrent variants are well known to the cancer community and are part of the hall of fame list.
Table 1.
Hall of fame variants that appear in over 100 samples
| Gene | AA change | CDS change | Counts |
|---|---|---|---|
| KRAS | p.Gly12Asp | c.35G > A | 711 |
| BRAF | p.Val600Glu | c.1799 T > A | 615 |
| KRAS | p.Gly12Val | c.35G > T | 607 |
| PIK3CA | p.Glu545Lys | c.1633G > A | 524 |
| PIK3CA | p.His1047Arg | c.3140A > G | 500 |
| KRAS | p.Gly12Cys | c.34G > T | 449 |
| TP53 | p.Arg175His | c.524G > A | 370 |
| PIK3CA | p.Glu542Lys | c.1624G > A | 332 |
| IDH1 | p.Arg132His | c.395G > A | 323 |
| KRAS | p.Gly13Asp | c.38G > A | 260 |
| TP53 | p.Arg273His | c.818G > A | 243 |
| TP53 | p.Arg273Cys | c.817C > T | 242 |
| TP53 | p.Arg248Gln | c.743G > A | 223 |
| TP53 | p.Arg248Trp | c.742C > T | 211 |
| TP53 | p.Arg282Trp | c.844C > T | 182 |
| TP53 | p.Arg213* | c.637C > T | 179 |
| NRAS | p.Gln61Arg | c.182A > G | 173 |
| EGFR | p.Leu858Arg | c.2573 T > G | 160 |
| EGFR | p.Glu746_Ala750del | c.2236_2250delGAATTAAGAGAAGCA | 159 |
| AKT1 | p.Glu17Lys | c.49G > A | 155 |
| KRAS | p.Gly12Ala | c.35G > C | 152 |
| TP53 | p.Arg342* | c.1024C > T | 135 |
| NRAS | p.Gln61Lys | c.181C > A | 123 |
| TP53 | p.Arg196* | c.586C > T | 110 |
| IDH1 | p.Arg132Cys | c.394C > T | 109 |
| KRAS | p.Gln61His | c.183A > T | 108 |
| TP53 | p.Tyr220Cys | c.659A > G | 108 |
| APC | p.Arg1450* | c.4348C > T | 102 |
| APC | p.Arg876* | c.2626C > T | 100 |
| FGFR3 | p.Ser249Cys | c.746C > G | 100 |
Among the most frequently mutated genes, TP53 ranks highest with 8083 variants followed by KRAS with 2811 variants (Table 2). This set of highly mutated genes also contains many epigenetic regulators, such as KMT2D, ARID1A, KMT2A, ARID1B, ARID2, SMARCA4, TET2, ATRX, CREBBP, and EP300. For example, KMT2D, also known as MLL2, is a lysine methyl transferase that activates genes by methylating histone H3 at lysine 4 residue [33]. ARID1A is a SWI/SNF complex component that alters the expression of diverse genes through chromatin remodeling [34].
Table 2.
Top mutated genes in GENIE
| Gene | Variants observed |
|---|---|
| TP53 | 8083 |
| KRAS | 2811 |
| PIK3CA | 2693 |
| APC | 2674 |
| KMT2D | 1980 |
| ARID1A | 1494 |
| PTEN | 1313 |
| EGFR | 1212 |
| ATM | 1202 |
| NF1 | 1122 |
| BRAF | 1116 |
| BRCA2 | 949 |
| NOTCH1 | 924 |
| RB1 | 917 |
| ATRX | 887 |
| SETD2 | 845 |
| CREBBP | 840 |
| CDKN2A | 788 |
| ERBB4 | 740 |
| KMT2A | 739 |
| SMAD4 | 733 |
| ARID1B | 719 |
| SMARCA4 | 696 |
| ROS1 | 688 |
| FBXW7 | 687 |
| EP300 | 669 |
| ARID2 | 649 |
| PTPRD | 647 |
| TET2 | 642 |
| DNAPK | 639 |
| Others | 70,732 |
To further focus on coding VUS’s, we removed intronic variants, hotspot indels, inactivating variants in tumor suppressor genes, and variants according to their population frequencies. 6907 variants were filtered out by comparing variant frequencies between the ExAC database and GENIE (Table 3, Fig. 3) to remove variants observed in the general population at similar or higher rates than in GENIE. Following these filtering steps, 56,032 variants remained as VUS’s. Of the average six variants observed per patient, we found that approximately 1/3 are potentially significant as they are frequently mutated in cancer or are likely inactivating variants in tumor suppressor genes. Thus, with more than half of patient variants being classed as a VUS, clinical decisions or actions are often being made with fairly limited knowledge.
Table 3.
Total number of distinct variants in each classification of interest is shown
| Variant Classification | Distinct Variants | Total Counts |
|---|---|---|
| Novel | 55,192 | 59,544 |
| ExAC | 6907 | 8237 |
| Recurrent | 4561 | 25,378 |
| Intronic | 1039 | 1205 |
| Hotspot indels | 1211 | 1459 |
| Inactivating TSG | 8834 | 10,294 |
| GENIE recurrent | 730 | 2598 |
| VUS | 56,032 | 60,195 |
| Novel VUS | 45,933 | 48,203 |
| GENIE COSMIC recurrent | 2586 | 5372 |
Fig. 3.

Variants classified according to filters. The percent of variants classified by each of the following filters: ExAC – variants with similar or higher frequencies in ExAC; Recurrent – variants detected in ≥3 samples in COSMIC; Intronic – variants found in introns excluding splice junctions; Inactivating variant in TSG – likely inactivating factors that occur in tumor suppressor gene; GENIE recurrent – variants detected in ≥3 samples in GENIE and < 3 samples in in COSMIC; Potential artifacts – variants occurring only from a single sequencing center; and VUS – all remaining variants are considered variants of unknown significance. Newly retrieved recurrent variants revealed in this study accounts for 3% (GENIE recurrent)
To better characterize these recurrent variants that are observed in many patient samples (Table 4), we leveraged additional information from COSMIC. Though before beginning special care was taken to remove potential artifacts originating from a single sequencing center pipeline by only considering variants reported by at least two sequencing centers. When first looking for recurrent variants appearing in at least three GENIE samples and not reported in COSMIC, we found 730 recurrent variants unique to GENIE. These variants appear in 1932 patient samples, or 10% of patients (Additional file 2: Table S2). The number of recurrent variants grows to 2586 affecting 3288 patients when pooling COSMIC and GENIE variant frequencies and still requiring they appear in at least three samples (Additional file 3: Table S3). While the proportion of cancer patients with these recurrent variants is relatively small at 10–20%, it still translates to millions of patients. For some, this information may lead to changes in the interpretation of their molecular profile and may affect diagnosis by altering disease subgrouping or lead to different treatment options. Though there is an expected decrease in the number of recurrent variants as the observation threshold increases, we still found that 4 variants appear more than 10 times in GENIE but fewer than three in COSMIC.
Table 4.
Number of recurrent GENIE variants that are underrepresented in COSMIC (< 3 samples)
| Minimum number of recurrences | GENIE recurrent variants | GENIE and COSMIC combined recurrent variants |
|---|---|---|
| 10 | 4 | 5 |
| 9 | 4 | 7 |
| 8 | 5 | 15 |
| 7 | 15 | 44 |
| 6 | 40 | 100 |
| 5 | 87 | 264 |
| 4 | 236 | 740 |
| 3 | 730 | 2586 |
Discussion
COSMIC compatibility
With the intent of discovering new cancer-relevant variants from the GENIE data, we leveraged COSMIC as a point of reference for the current state of variant observation. A necessary consideration in such a comparison is the ability to map genes and variants between both resources. Across the 12 sequencing panels that comprise the GENIE dataset, 536 genes are mutated in GENIE samples. There was agreement between COSMIC and GENIE on most of the gene names and transcript ids with a few exceptions. For instance, PRKDC is the HGNC approved symbol [35]; however, COSMIC instead uses DNAPK. Additionally, CDK1’s canonical transcript is not defined in COSMIC. There were transcript compatibility issues for RUNX1T1, GNAS, DMD, and several other genes. For example, COSMIC picked ENST00000371085 (GNAS-015) with 394 amino acid residues as the canonical transcript whereas ENST00000371100 (GNAS-001) has 1037 amino acid residues. As a result, many variants can fall outside of COSMIC’s canonical transcript. Thus, we tried to rescue those variants by adapting the ENST00000371100 transcript as well. While in the GNAS case most variants could be rescued, 302 still fell outside of the COSMIC transcript. Recognizing the purpose of this study is to compare the GENIE variants with the standard COSMIC database, we opted not to rescue further variants.
Unusual variants
Our analysis revealed a number of notable variants that had not previously been reported or were not observed at the same frequency in COSMIC. The frameshift variant EGFR L747 fs was found 13 times in GENIE but not once in COSMIC or ExAC. Although this particular variant was removed by the hotspot indels filter, we deemed it noteworthy because both its observed frequency is significantly higher than in COSMIC and it is an inactivating variant in a well-established oncogene. Indeed, as the variant occurs in the kinase domain, it would likely contribute to the truncation of that domain and the inactivation of the gene. Interestingly, it has been reported in literature that a patient harboring this variant has shown intermediate response to gefitinib (progression within 12 months) [36]. While at this point there remains the possibility that these are sequencing artifacts or the result of structural variants, such as amplification, the frequency with which they occur and the genes they fall within suggests their mechanisms warrant further study.
We also found several cases of variants likely leading to exon skip events. 13 variants were observed in the splice donor of MET exon 14 (c.3082 + 1 or c.3082 + 2). These variants are known to lead to MET exon 14 skipping events creating a constitutively active form of MET, and such patients were found to generally respond well to MET inhibitors, crizotinib and cabozantinib [37]. In addition to those splicing donor variants, we discovered an additional 17 variants in the coding region of the splicing donor. MET D1028H, MET D1028Y, and MET D1028N might also yield abnormal splicing similar to the exon 14 skipping variants. All D1028 variants were from NSCLC samples. These events should be confirmed with PCR or other methods before treatment with MET inhibitors.
Highly recurrent variants
There are 40 novel, highly recurrent variants that are defined as appearing in more than 6 samples in the GENIE dataset and fewer than three in COSMIC (Table 5). The most frequent among them is MET A179T, which is found 19 times in GENIE and once in COSMIC. This variant has been reported in a chronic myelomonocytic leukemia patient but with no mention of its pathogenicity [38]. In GENIE the majority of samples in which it was detected were from NSCLC patients; although, all such samples were from a single sequencing center raising the possibility this particular variant is an artifact. Though as MET is already known to be frequently mutated in lung adenocarcinoma [39], study of this variant should likely be given priority.
Table 5.
List of highly recurrent GENIE variants (≥ 6 samples) that are underrepresented in COSMIC (< 3 samples)
| Gene | AA change | CDS change | GENIE count | COSMIC count | Cancer types |
|---|---|---|---|---|---|
| MET | p.A179T | c.535G > A | 19 | 1 | Non-Small Cell Lung Cancer(13);Melanoma(2);Colorectal Cancer(4) |
| ERBB3 | p.E928G | c.2783A > G | 14 | 2 | Melanoma(1);Colorectal Cancer(2);Bladder Cancer(2);Hepatobiliary Cancer(1);Small Bowel Cancer(1);Breast Cancer(6);Esophagogastric Cancer(1) |
| CDKN2A | p.P75S | c.223C > T | 13 | 1 | Prostate Cancer(1);Colorectal Cancer(2);Ovarian Cancer(5);Non-Small Cell Lung Cancer(1);Endometrial Cancer(2);Breast Cancer(1);Cervical Cancer(1) |
| SMO | p.L23dup | c.67_69dupCTG | 11 | 2 | Thyroid Cancer(1);Melanoma(1);Glioma(2);Leukemia(2);Non-Small Cell Lung Cancer(2);Cancer of Unknown Primary(1);Endometrial Cancer(1);Breast Cancer(1) |
| SMARCA4 | p.R1189Q | c.3566G > A | 8 | 2 | Glioma(1);Bladder Cancer(3);Renal Cell Carcinoma(1);Cancer of Unknown Primary(1);Breast Cancer(1);Esophagogastric Cancer(1) |
| CDKN2A | p.E69G | c.206A > G | 7 | 0 | Non-Small Cell Lung Cancer(6);Colorectal Cancer(1) |
| ERBB3 | p.M91I | c.273G > A | 7 | 2 | Bladder Cancer(6);Endometrial Cancer(1) |
| ERBB3 | p.K329E | c.985A > G | 7 | 0 | Bladder Cancer(4);Breast Cancer(1);Colorectal Cancer(1);Esophagogastric Cancer(1) |
| ERCC2 | p.N238S | c.713A > G | 7 | 0 | Bladder Cancer(5);Breast Cancer(2) |
| FANCA | p.K1283R | c.3848A > G | 7 | 0 | Bladder Cancer(1);Endometrial Cancer(1);Hepatobiliary Cancer(1);Breast Cancer(3);Leukemia(1) |
| FLT1 | p.R501K | c.1502G > A | 7 | 0 | Melanoma(1);Ovarian Cancer(1);Non-Small Cell Lung Cancer(2);Cancer of Unknown Primary(1);Breast Cancer(1);Cervical Cancer(1) |
| FLT4 | p.P30fs | c.88delC | 7 | 2 | Non-Small Cell Lung Cancer(1);Colorectal Cancer(6) |
| IKZF1 | p.D22N | c.64G > A | 7 | 0 | Colorectal Cancer(1);Non-Small Cell Lung Cancer(1);Melanoma(3);Glioma(1);Embryonal Tumor(1) |
| KDR | p.S265 L | c.794C > T | 7 | 0 | Myelodysplasia(1);Melanoma(1);Colorectal Cancer(1);Sellar Tumor(1);Non-Small Cell Lung Cancer(2);“Skin Cancer, Non-Melanoma”(1) |
| PIK3CB | p.R604fs | c.1809dupC | 7 | 1 | Bladder Cancer(1);“Skin Cancer, Non-Melanoma”(1);Breast Cancer(3);Endometrial Cancer(1);Esophagogastric Cancer(1) |
| APC | p.T1160K | c.3479C > A | 6 | 0 | Ovarian Cancer(1);Salivary Gland Cancer(1);Non-Small Cell Lung Cancer(1);Melanoma(1);Colorectal Cancer(2) |
| ARID1A | p.S735 N | c.2204G > A | 6 | 0 | Bladder Cancer(1);Hepatobiliary Cancer(1);Leukemia(1);Non-Small Cell Lung Cancer(1);Breast Cancer(1);Cervical Cancer(1) |
| ASXL1 | p.S1028 L | c.3083C > T | 6 | 1 | Thyroid Cancer(1);Non-Small Cell Lung Cancer(2);Melanoma(1);Small Cell Lung Cancer(1);Prostate Cancer(1) |
| ATRX | p.G1071R | c.3211G > A | 6 | 0 | Skin Cancer, Non-Melanoma(1);Non-Small Cell Lung Cancer(2);Breast Cancer(1);Colorectal Cancer(1);Myeloproliferative Neoplasm(1) |
| BRCA1 | p.E597K | c.1789G > A | 6 | 1 | Bladder Cancer(2);“Skin Cancer, Non-Melanoma”(1);Non-Small Cell Lung Cancer(1);Breast Cancer(1);Colorectal Cancer(1) |
| CARD11 | p.R377Q | c.1130G > A | 6 | 2 | Non-Hodgkin Lymphoma(1);Colorectal Cancer(1);Uterine Sarcoma(1);Ovarian Cancer(1);Small Bowel Cancer(1);“Skin Cancer, Non-Melanoma”(1) |
| CDKN2A | p.V106 V | c.318G > A | 6 | 2 | Melanoma(1);Non-Small Cell Lung Cancer(2);Breast Cancer(1);Colorectal Cancer(1);Gastrointestinal Stromal Tumor(1) |
| ERBB4 | p.E452K | c.1354G > A | 6 | 2 | Skin Cancer, Non-Melanoma(2);Melanoma(4) |
| ERCC3 | p.R742W | c.2224C > T | 6 | 1 | Glioma(1);Sex Cord Stromal Tumor(1);Renal Cell Carcinoma(1);Non-Small Cell Lung Cancer(1);Leukemia(1);Colorectal Cancer(1) |
| FBXW7 | p.R441W | c.1321C > T | 6 | 1 | Colorectal Cancer(3);Endometrial Cancer(1);Renal Cell Carcinoma(1);Esophagogastric Cancer(1) |
| IGF1R | p.R1246C | c.3736C > T | 6 | 1 | CNS Cancer(1);Endometrial Cancer(2);“Skin Cancer, Non-Melanoma”(1);Breast Cancer(1);Glioma(1) |
| IKZF1 | p.E304K | c.910G > A | 6 | 0 | Melanoma(6) |
| KMT2D | p.K4832 N | c.14496G > T | 6 | 0 | Ovarian Cancer(1);Breast Cancer(1);Colorectal Cancer(2);Head and Neck Cancer(1);Thymic Tumor(1) |
| MET | p.D1028H | c.3082G > C | 6 | 2 | Non-Small Cell Lung Cancer(6) |
| MET | p.D1028Y | c.3082G > T | 6 | 0 | Non-Small Cell Lung Cancer(6) |
| MYC | p.S161 L | c.482C > T | 6 | 0 | Ovarian Cancer(1);Endometrial Cancer(2);Colorectal Cancer(2);Cervical Cancer(1) |
| PDCD1 | p.T36 fs | c.104delC | 6 | 2 | Endometrial Cancer(2);Non-Small Cell Lung Cancer(1);Glioma(1);Colorectal Cancer(2) |
| PDCD1LG2 | p.P81S | c.241C > T | 6 | 0 | Colorectal Cancer(1);Head and Neck Cancer(1);Appendiceal Cancer(1);Mesothelioma(1);Leukemia(1);Pancreatic Cancer(1) |
| PIK3C2B | p.G1435R | c.4303G > C | 6 | 0 | Head and Neck Cancer(1);Ovarian Cancer(1);Sellar Tumor(1);Appendiceal Cancer(1);Non-Small Cell Lung Cancer(1);Breast Cancer(1) |
| PMS2 | p.K651R | c.1952A > G | 6 | 1 | Bladder Cancer(2);Uterine Sarcoma(1);Endometrial Cancer(1);Colorectal Cancer(2) |
| RAF1 | p.S259F | c.776C > T | 6 | 1 | Small Bowel Cancer(1);Bladder Cancer(1);Non-Small Cell Lung Cancer(1);Melanoma(3) |
| ROS1 | p.G1915R | c.5743G > A | 6 | 0 | Melanoma(1);“Skin Cancer, Non-Melanoma”(1);Non-Small Cell Lung Cancer(1);Breast Cancer(2);Cancer of Unknown Primary(1) |
| SMAD4 | p.D351N | c.1051G > A | 6 | 2 | Appendiceal Cancer(1);Bladder Cancer(1);Hepatobiliary Cancer(1);Breast Cancer(1);Colorectal Cancer(2) |
| SMAD4 | p.R361S | c.1081C > A | 6 | 2 | Pancreatic Cancer(1);Hepatobiliary Cancer(1);Colorectal Cancer(4) |
| SMAD4 | p.G419 W | c.1255G > T | 6 | 1 | Colorectal Cancer(6) |
The next most frequent variant is ERBB3 E928G. This particular variant has been experimentally confirmed to have higher activity and appears to activate EGFR allosterically upon heterodimerization [40, 41]. ERBB3 has two additional highly recurrent variants. The M91I variant appeared primarily in bladder cancer (6 of 7 samples), where it has been previously reported though its pathogenicity remains unknown. K329E variant was observed in seven samples, and four were endometrial cancer. Another ERBB family member variant, ERBB4 E452K, appeared mainly in skin cancers and has been confirmed to increase activity [42].
The cell cycle regulating protein, CDKN2A, is frequently inactivated in various cancer types. While COSMIC there are several variants occurring at CDKN2A P75 residue, such as P75L and P75S, that are reported only once, we observed them 13 times in GENIE. CDKN2A P75L has been functionally studied and concluded to be benign [43]. Another CDKN2A variant, E69G, takes places mostly in NSCLC. Although E69G is never observed in COSMIC, other codon E69 variants have been reported there. E69G was observed in GENIE as belonging mostly to NSCLC samples. There have been reports of CDKN2A E69G in familial melanoma patients with 30% decreased binding to CDK4 compared with its wild type [44]. The CDKN2A variant, V106 V, is a synonymous mutation for CDKN2A; however, the same locus is used for protein p14 (ARF), which is a tumor suppressor. This mutation translates to p14(ARF) A162T.
SMO L23dup (or L23_G24insL in COSMIC notation) was found 11 times in GENIE but only twice in COSMIC. This variant, along with two other detected variants (L23_G23insLL and L23_G23insA), resides in a signal peptide domain found in the first 27 residues. SMO L23dup was previously reported in a mesothelioma cell line LO68 and two gastric cancer patients; however, no functional significance was observed but it might affect processing of SMO precursor [45]. Though this alteration was detected in GENIE in a diverse array of cancers, there is potential for it to be a sequencing artifact because it originated from only a single sequencing center.
Variants in the SWI/SNF components, ARID1A S735 N and SMARCA4 R1189Q, were also found to be highly recurrent. SMARCA4 R1189Q has been reported in 2 COSMIC samples, and in GENIE, 3 of 7 samples were bladder cancer. There are not yet reports on pathogenicity regarding these two variants. It may be possible to assess whether these variants in SWI/SNF genes contribute to tumorigenesis by studying epigenetic signatures using techniques like ATAC-seq [46].
FBXW7 is a ubiquitin ligase and known to function as a tumor suppressor regulating NOTCH, MYC, and other oncogenes [47, 48]. FBXW7 is frequently mutated in colorectal cancer. FBXW7 R441W appears 3 times out of 6 in colorectal cancer and is located near R465, R479, and R505 hotspots. There are currently no reports in literature for this particular variant. Although the FBXW7 variant is not generally considered actionable, FBXW7 is one of the most mutated genes in cancer and developing sensitivity or resistance information related the variant would be beneficial.
DNA repair genes BRCA1, ERCC2, ERCC3, and FANCA are known to affect responses to chemotherapeutic agents and PARP1 inhibitors. ERCC2 N238S was observed seven times in GENIE and five of those samples were bladder cancer. ERCC2 variants are also known to improve response to platinum agents [49]. These ERCC2 variants could prove informative for changing the outcome of certain patients by serving as a therapeutic biomarker. FANCA K1283R appeared three times in breast cancer out of seven cases. FANCA variants have been reported in non-BRCA1/2 familial breast cancer patients [50]. FANCA’s role in homologous recombination suggests that patients with loss of function variants might be susceptible to PARP1 inhibitor treatment [51]. While BRCA1 is obviously an important cancer gene, the clinical significance of recurrent BRCA1 E597K variant is not yet known.
FLT4 frameshift variant P30fs was observed in colorectal cancers in 6 of the 7 samples it appeared, and the 2 COSMIC reported cases were also colorectal cancer. Given FLT4’s believed function as an oncogene playing a role in invasion and metastasis [52], further investigation should be made as to the relevancy of this variant or FLT4’s role in pathogenicity in colorectal cancer. This might also indicate a potential tumor suppressor role for FLT4 gene in colorectal cancer. Another FLT family member variant FLT1 R501K was found to be highly recurrent. FLT1 is a VEGF receptor along with KDR (VEGFR2), which also had a highly recurrent variant in GENIE, S265 L. Neither FLT1 R501K nor KDR S265 L have confirmed pathogenicity.
SMAD4 has three recurrent variants D351N, R361S, and G419 W. SMAD4 is one of the most mutated genes in colorectal cancer. Considering its high occurrence in colorectal cancer, these variants may reduce activity of SMAD4 and contribute to the development of colorectal cancer. Along with SMAD4, APC is another important gene in colorectal cancer. APC T1160K appeared in colorectal cancer for 2 out of 6 samples it was found. At this point, none of these variants have confirmed pathogenicity.
Many variants in IKZF1 are observed in melanoma. Three of the 7 samples D22N was found and all 6 samples where E304K was detected originated from melanoma samples. The relationship between IKZF1 and melanoma is not yet well established. However, it was recently reported that IKZF1 expressing cells respond better to PD-1/CTLA-4 [53]. These variants in IKZF1 along with PDCD1 (PD-L1) T36 fs and PDCD1LG2 (PD-L2) P81S should be investigated for response to PD-1/CTLA-4 inhibitors.
There are various kinases – IFI1R, PIK3C2B, ROS1, and RAF1 – in the set of highly recurrent variants. Although BRAF has gained more attention in melanoma, RAF1 plays an important role in MAPK signaling. The RAF1 S259 residue is critical to bind the inhibitory 14–3-3 protein [54]. Since 3 of the 6 samples that possessed RAF1 S259F were melanoma, this variant may contribute to melanoma development.
Conclusions
While our variant analysis of the GENIE dataset focusing on VUS’s is only beginning to scratch the surface, it does provide a more comprehensive assessment of the landscape of cancer variants. Many of these VUS’s require additional study to disentangle their roles in cancer formation and progression. Yet, using the frequencies with which they occur and how they are distributed among cancer types, this analysis can already aid clinicians working to develop a course of treatment. Currently, there are significant disparities in the reporting of variants. For instance, there are thousands of papers concerning BRAF V600E and EGFR L858R, but many of the most frequent variants registered in COSMIC are not published in a journal article. COSMIC contains 2 million unique coding variants, and it is not practical to publish articles on all equally. However, the recurrent variants revealed in this study are good candidates for further research. There exist several reasons, both technical and biological, for the differences between our findings in GENIE and that of COSMIC. The technical reasons include differences in platforms, reagents, and data processing pipelines. The biological differences may be partly attributable to ethnic and regional sampling differences. For instance, chemical and microbial exposure can vary greatly region to region. Well-coordinated strategies to cover these variants must be developed to mitigate such differences, to efficiently deploy scientific resources, and to overcome the lack of coverage in the published literature. Only with these persistent efforts will the clinical utility of precision medicine be fully demonstrated.
Additional files
Table S1. List of Genes in GENIE. -The file contains genes in GENIE with transcripts used both in GENIE and COSMIC. Each tumor suppressor gene is marked with evidences in Pubmed ID. This information was used to identify inactivating variants in tumor suppressor genes. (XLSX 34 kb)
Table S2. Variants recurring more than 3 times in GENIE samples only. Variants are ordered by the recurrence counts in GENIE. For each variant, AA change and CDS change are shown with COSMIC and GENIE sample counts. Z-value was computed for each variant using ExAC information (ExAC_AC, ExAC_AN, sample number). The table also contains sample IDs and cancer types for each variant. (XLSX 83 kb)
Table S3. Variants recurring more than 3 times in GENIE and COSMIC samples combined. Variants are ordered by the combined recurrence counts in GENIE and COSMIC. . For each variant, AA change and CDS change are shown with COSMIC and GENIE sample counts. Z-value was computed for each variant using ExAC information (ExAC_AC, ExAC_AN, sample number). The table also contains sample IDs and cancer types from both GENIE and COSMIC. (XLSX 287 kb)
Acknowledgements
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of study authors.
Funding
The authors received no specific funding for this work.
Availability of data and materials
GENIE ver 1.0 is available from https://synapse.org/genie
COSMIC V79 is available from https://cancer.sanger.ac.uk/cosmic/download?genome=37
ExAC ver 0.3.1 is available from http://exac.broadinstitute.org/downloads
SNPEFF v 4.3 is available from http://snpeff.sourceforge.net/
Supplementary Materials (supplementary_1.xls, supplementary_2.xls, and supplementary_3.xls) are provided.
Abbreviations
- AACR
American Association for Cancer Research
- CDS
coding sequence
- COSMIC
Catalogue Of Somatic Mutations In Cancer
- ExAC
Exome Aggregation Consortium
- FDA
Food and Drug Administration
- GENIE
Genomics Evidence Neoplasia Information Exchange
- NSCLC
non-small cell lung cancer
- VUS
variant of unknown significance
Authors’ contributions
TK prepared data, analyzed recurrent variants, and drafted manuscript. KR analyzed recurrent variants and revised the manuscript. LP supervised the study and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All the three authors are employees of IBM T J Watson Research Center and declare no conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Takahiko Koyama, Email: tkoyama@us.ibm.com.
Kahn Rhrissorrakrai, Email: krhriss@us.ibm.com.
Laxmi Parida, Email: parida@us.ibm.com.
References
- 1.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C, Yang X, Lubonja R, Tran N, Nguyen C, et al. Systematic functional interrogation of rare Cancer variants identifies oncogenic alleles. Cancer Discov. 2016;6(7):714–726. doi: 10.1158/2159-8290.CD-16-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang WW, Zhang Q, McLellan MD, et al. Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat Genet. 2016;48(8):827–837. doi: 10.1038/ng.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium APG AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryce AH, Egan JB, Borad MJ, Stewart AK, Nowakowski GS, Chanan-Khan A, Patnaik MM, Ansell SM, Banck MS, Robinson SI, et al. Experience with precision genomics and tumor board, indicates frequent target identification, but barriers to delivery. Oncotarget. 2017:27145–54. [DOI] [PMC free article] [PubMed]
- 8.Massard C, Michiels S, Ferte C, Le Deley MC, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S, Ammari S, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7(6):586–595. doi: 10.1158/2159-8290.CD-16-1396. [DOI] [PubMed] [Google Scholar]
- 9.AACR GENIE Data Guide version 1.0. In.; 2017.
- 10.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1(2):170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vempati S, Reindl C, Kaza SK, Kern R, Malamoussi T, Dugas M, Mellert G, Schnittger S, Hiddemann W, Spiekermann K. Arginine 595 is duplicated in patients with acute leukemias carrying internal tandem duplications of FLT3 and modulates its transforming potential. Blood. 2007;110(2):686–694. doi: 10.1182/blood-2006-10-053181. [DOI] [PubMed] [Google Scholar]
- 14.D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS, Zakowski MF, Rusch VW, Ladanyi M, Kris MG. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29(15):2066–2070. doi: 10.1200/JCO.2010.32.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, Huberman MS, Cohen DW, Nakayama S, Ishioka K, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5(216):216ra177. doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, Dalla-Favera R. Combined genetic inactivation of beta2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20(6):728–740. doi: 10.1016/j.ccr.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniuchi I, Osato M, Ito Y. Runx1: no longer just for leukemia. EMBO J. 2012;31(21):4098–4099. doi: 10.1038/emboj.2012.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorr C, Janik C, Weg M, Been RA, Bader J, Kang R, Ng B, Foran L, Landman SR, O'Sullivan MG, et al. Transposon mutagenesis screen identifies potential lung Cancer drivers and CUL3 as a tumor suppressor. Mol Cancer Res. 2015;13(8):1238–1247. doi: 10.1158/1541-7786.MCR-14-0674-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333(6048):1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dydensborg AB, Rose AA, Wilson BJ, Grote D, Paquet M, Giguere V, Siegel PM, Bouchard M. GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene. 2009;28(29):2634–2642. doi: 10.1038/onc.2009.126. [DOI] [PubMed] [Google Scholar]
- 21.Huang XD, Xiao FJ, Wang SX, Yin RH, Lu CR, Li QF, Liu N, Zhang Y, Wang LS, Li PY. G protein pathway suppressor 2 (GPS2) acts as a tumor suppressor in liposarcoma. Tumour Biol. 2016;37(10):13333–13343. doi: 10.1007/s13277-016-5220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrido C, Paco L, Romero I, Berruguilla E, Stefansky J, Collado A, Algarra I, Garrido F, Garcia-Lora AM. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis. 2012;33(3):687–693. doi: 10.1093/carcin/bgr318. [DOI] [PubMed] [Google Scholar]
- 23.Pham TT, Angus SP, Johnson GL. MAP3K1: genomic alterations in Cancer and function in promoting cell survival or apoptosis. Genes Cancer. 2013;4(11–12):419–426. doi: 10.1177/1947601913513950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jo YS, Kim MS, Yoo NJ, Lee SH. Somatic mutation of a candidate tumour suppressor MGA gene and its mutational heterogeneity in colorectal cancers. Pathology. 2016;48(5):525–527. doi: 10.1016/j.pathol.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 25.Wang W, Song XW, Bu XM, Zhang N, Zhao CH. PDCD2 and NCoR1 as putative tumor suppressors in gastric gastrointestinal stromal tumors. Cell Oncol (Dordr) 2016;39(2):129–137. doi: 10.1007/s13402-015-0258-0. [DOI] [PubMed] [Google Scholar]
- 26.Sung H, Kanchi KL, Wang X, Hill KS, Messina JL, Lee JH, Kim Y, Dees ND, Ding L, Teer JK, et al. Inactivation of RASA1 promotes melanoma tumorigenesis via R-Ras activation. Oncotarget. 2016;7(17):23885–23896. doi: 10.18632/oncotarget.8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13(4):466–472. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488(7413):665–669. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 29.Chen D, Zhang J, Li M, Rayburn ER, Wang H, Zhang R. RYBP stabilizes p53 by modulating MDM2. EMBO Rep. 2009;10(2):166–172. doi: 10.1038/embor.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. 2011;17(3):291–293. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 31.Rudolph M, Anzeneder T, Schulz A, Beckmann G, Byrne AT, Jeffers M, Pena C, Politz O, Kochert K, Vonk R, et al. AKT1 (E17K) mutation profiling in breast cancer: prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer. 2016;16:622. doi: 10.1186/s12885-016-2626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.di Martino E, Tomlinson DC, Knowles MA. A decade of FGF receptor research in bladder Cancer: past, present, and future challenges. Adv Urol. 2012;2012:429213. doi: 10.1155/2012/429213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–1208. doi: 10.1038/nm.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15(6):655–664. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wain HM, Bruford EA, Lovering RC, Lush MJ, Wright MW, Povey S. Guidelines for human gene nomenclature. Genomics. 2002;79(4):464–470. doi: 10.1006/geno.2002.6748. [DOI] [PubMed] [Google Scholar]
- 36.Bria E, Pilotto S, Amato E, Fassan M, Novello S, Peretti U, Vavala T, Kinspergher S, Righi L, Santo A, et al. Molecular heterogeneity assessment by next-generation sequencing and response to gefitinib of EGFR mutant advanced lung adenocarcinoma. Oncotarget. 2015;6(14):12783–12795. doi: 10.18632/oncotarget.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reungwetwattana T, Ou SH. MET exon 14 deletion (METex14): finally, a frequent-enough actionable oncogenic driver mutation in non-small cell lung cancer to lead MET inhibitors out of "40 years of wilderness" and into a clear path of regulatory approval. Transl Lung Cancer Res. 2015;4(6):820–824. doi: 10.3978/j.issn.2218-6751.2015.12.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A et al: Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122(22):3616–3627; quiz 3699. [DOI] [PMC free article] [PubMed]
- 39.Cancer Genome Atlas Research N: comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23(5):603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 41.Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal. 2014;7(354):ra114. doi: 10.1126/scisignal.2005786. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 42.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41(10):1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang G, Rajadurai A, Tsao H. Recurrent patterns of dual RB and p53 pathway inactivation in melanoma. J Invest Dermatol. 2005;125(6):1242–1251. doi: 10.1111/j.0022-202X.2005.23931.x. [DOI] [PubMed] [Google Scholar]
- 44.Kannengiesser C, Brookes S, del Arroyo AG, Pham D, Bombled J, Barrois M, Mauffret O, Avril MF, Chompret A, Lenoir GM, et al. Functional, structural, and genetic evaluation of 20 CDKN2A germ line mutations identified in melanoma-prone families or patients. Hum Mutat. 2009;30(4):564–574. doi: 10.1002/humu.20845. [DOI] [PubMed] [Google Scholar]
- 45.Lim CB, Prele CM, Cheah HM, Cheng YY, Klebe S, Reid G, Watkins DN, Baltic S, Thompson PJ, Mutsaers SE. Mutational analysis of hedgehog signaling pathway genes in human malignant mesothelioma. PLoS One. 2013;8(6):e66685. doi: 10.1371/journal.pone.0066685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, Oscier D, Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938. doi: 10.1038/ncomms11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato M, Rodriguez-Barrueco R, Yu J, Do C, Silva JM, Gautier J. MYC is a critical target of FBXW7. Oncotarget. 2015;6(5):3292–3305. doi: 10.18632/oncotarget.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeishi S, Nakayama KI. Role of Fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br J Cancer. 2014;111(6):1054–1059. doi: 10.1038/bjc.2014.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O'Connor KW, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4(10):1140–1153. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Litim N, Labrie Y, Desjardins S, Ouellette G, Plourde K, Belleau P, BRCAs I, Durocher F. Polymorphic variations in the FANCA gene in high-risk non-BRCA1/2 breast cancer individuals from the French Canadian population. Mol Oncol. 2013;7(1):85–100. doi: 10.1016/j.molonc.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front Oncol. 2014;4:42. doi: 10.3389/fonc.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9(3):209–223. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 53.Chen JC, Perez-Lorenzo R, Saenger YM, Drake CG, Christiano AM: IKZF1 enhances immune infiltrate recruitment in solid tumors and susceptibility to immunotherapy. Cell Syst 2018, 7(1):92–103 e104. [DOI] [PubMed]
- 54.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Genes in GENIE. -The file contains genes in GENIE with transcripts used both in GENIE and COSMIC. Each tumor suppressor gene is marked with evidences in Pubmed ID. This information was used to identify inactivating variants in tumor suppressor genes. (XLSX 34 kb)
Table S2. Variants recurring more than 3 times in GENIE samples only. Variants are ordered by the recurrence counts in GENIE. For each variant, AA change and CDS change are shown with COSMIC and GENIE sample counts. Z-value was computed for each variant using ExAC information (ExAC_AC, ExAC_AN, sample number). The table also contains sample IDs and cancer types for each variant. (XLSX 83 kb)
Table S3. Variants recurring more than 3 times in GENIE and COSMIC samples combined. Variants are ordered by the combined recurrence counts in GENIE and COSMIC. . For each variant, AA change and CDS change are shown with COSMIC and GENIE sample counts. Z-value was computed for each variant using ExAC information (ExAC_AC, ExAC_AN, sample number). The table also contains sample IDs and cancer types from both GENIE and COSMIC. (XLSX 287 kb)
Data Availability Statement
GENIE ver 1.0 is available from https://synapse.org/genie
COSMIC V79 is available from https://cancer.sanger.ac.uk/cosmic/download?genome=37
ExAC ver 0.3.1 is available from http://exac.broadinstitute.org/downloads
SNPEFF v 4.3 is available from http://snpeff.sourceforge.net/
Supplementary Materials (supplementary_1.xls, supplementary_2.xls, and supplementary_3.xls) are provided.