

Abstract
Many normal tissues undergo age-related drift in DNA methylation, providing a quantitative measure of tissue age. Here we identify and validate 781 CpG-islands (CGI) that undergo significant methylomic drift in 232 normal colorectal tissues and show that these CGI continue to drift in neoplasia while retaining significant correlations across samples. However, compared with normal colon, this drift advanced (~3–4 fold) faster in neoplasia, consistent with increased cell proliferation during neoplastic progression. The observed drift patterns were broadly consistent with modeled adenoma-carcinoma sojourn time distributions from colorectal cancer (CRC) incidence data. These results support the hypothesis that, beginning with the founder premalignant cell, cancer precursors frequently sojourn for decades before turning into cancer, implying that the founder cell typically arises early in life. At least 77–89% of the observed drift variance in distal and rectal tumors was explained by stochastic variability associated with neoplastic progression, while only 55% of the variance was explained for proximal tumors. However, gene-CGI pairs in the proximal colon that underwent drift were significantly and primarily negatively correlated with cancer gene expression, suggesting that methylomic drift participates in the clonal evolution of CRC. Methylomic drift advanced in colorectal neoplasia consistent with extended sojourn time distributions, which accounts for a significant fraction of epigenetic heterogeneity in CRC. Importantly, these estimated long-duration premalignant sojourn times suggest that early dietary and lifestyle interventions may be more effective than later changes in reducing CRC incidence.
Keywords: epigenetic drift, tissue age, DNA methylation, CpG-islands (CGIs), gene expression, adenoma-carcinoma sojourn time, colorectal cancer (CRC), multistage clonal expansion (MSCE) model of colorectal cancer
Introduction
Colorectal cancers (CRC) arise along alternative pathways through an accumulation of mutations and epigenetic alterations accompanied by clonal expansions, along with random and selective drift (1–5). Several mutations or epigenetic changes are thought to be necessary (e.g. bi-allelic inactivation of APC, epigenetic silencing of MLH1) to initiate premalignant clonal growth (6). Occult premalignant clones that do not undergo extinction may grow into observable adenoma while accumulating (epi)genetic alterations, with some developing genomic instability, undergoing malignant transformation and invasive growth (7–9). Rates for these processes may be influenced by obesity, diet, genetics, the microbiome and other factors (3, 10–12). Although CRC genomes have been extensively profiled for somatic mutations, chromosomal abnormalities and epigenetic alterations (3, 9), little is known about the dynamics of the carcinogenic process, including the sojourn time distribution from the time when a premalignant founder cell is born to when the descendent cancer becomes clinically identifiable (13). Here we aim to better understand these dynamics and the role of epigenetic drift in the colon and rectum as an indicator of tissue aging and its potential phenotypic effects in colorectal neoplasia (14–16).
Recently, we established a key role of differential methylomic drift in the progression of Barrett’s esophagus (BE) to esophageal adenocarcinoma (EAC) by analyzing age-related differences in DNA methylation between normal esophageal epithelium, metaplastic BE tissue and EAC tissue (17, 18). Here we define methylomic, or epigenetic, drift to represent the tissue-specific and age-related increases in DNA methylation at certain CpG dinucleotides. One major finding of this earlier analysis was that epigenetic drift is widespread in BE genomes with the magnitude of drift being highly variable between individuals, suggesting significant differences in BE tissue age. We also observed a significant negative correlation of advanced methylomic drift at the CpG-island (CGI) level with the expression of 200 genes, including several genes that have recently been proposed as diagnostic markers for BE or have been implicated in esophageal carcinogenesis (19, 20).
Epigenetic drift in the colon has been previously identified at a number of genes, in particular at promoter-associated CpG island (CGI) (14, 21–23). However, methylomic tissue aging has only recently been studied more extensively in colon, using advanced statistical regression methods (24, 25) applied to data from high-throughput techniques such as reduced representation bisulfite sequencing (RRBS) and high-density DNA-methylation arrays.
Materials and methods
In this study, we used a conventional regression approach geared toward a fuller assessment of methylomic drift both at the single CpG and CGI level and, for the first time, provide a genome-wide evaluation of methylomic drift in colon (left/right) and rectum from normal and neoplastic tissue biopsies. We evaluated methylation levels at > 450,000 CpG probes using the Illumina HM450 beadchip array (HM450) in a total of 675 colorectal tissue samples. Of particular interest were site and sex differences in methylomic drift, inter-individual heterogeneity, and whether drift patterns at the probe and CGI level reflect the expected variance of tissue sojourn times in the adenoma-to-carcinoma sequence. Estimates of adenoma-to-carcinoma times for rectum, distal, and proximal colon for the two sexes were based on mathematical models developed by our group to explain age-specific incidence patterns of CRC in the US and UK (26). (See Fig. 1). Here, we derive mathematical expressions for the distribution of total sojourn times from the occurrence of the premalignant founder cell to the descendent carcinoma, with these sojourn times properly conditioned on the time (patient age) when the descendent carcinoma is diagnosed and removed for molecular analysis.
Fig. 1:
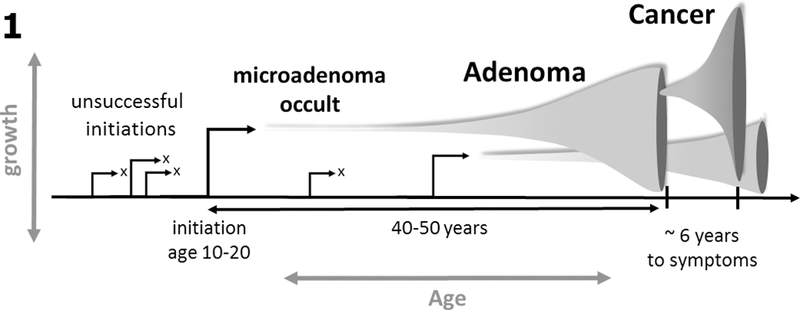
DNA methylation drift measured in a cancer tissue sample provides a measure of the sojourn time between initiation of the founder premalignant cell and the cancer that arises along this lineage. Premalignant clones may grow gradually for decades prior to generating an observable adenoma or cancer.
Of note, the premalignant sojourn times introduced here differ from clinical adenoma sojourn times (with varying estimates that range up to ~25 years (27)) as they capture the entire phase of clonal expansion including the occult phase of the adenoma, the clinical (detectable) phase, and malignant phase that leads to symptomatic cancer. Hence, the difference is that the sojourn times we estimate date back to the premalignant founder cell that undergoes slow stochastic growth and does not become extinct.
Finally, comparing CGI level methylomic drift with gene expression in CRC, we addressed the question whether methylomic drift may turn into a selective force impacting gene expression similar to our findings for EAC.
Consortia and Patient Samples
This study included normal colon and rectum samples obtained from patients participating in various studies in the Seattle-Puget Sound region, including the Luo Study (2), the Seattle ColoCare Study (28), the Screening Marker Study (SMS) (29), and GICaRes (GICR) (30). Written informed consent was obtained from all patients, the studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and the studies followed protocols approved by various Institutional Review Boards. (See Table 1).
Table 1:
Study, number of samples, sample location and mean patient age (range) used for this study. Note: we excluded EACs from The Cancer Genome Atlas (TCGA) with MSI and/or mucinous histology.
| Study group | Number of samples | Colorectal location | Sex | Mean age at diag. (range) | Sample histology | Patient status | ||
|---|---|---|---|---|---|---|---|---|
| Rectum | Left | Right | F/M | |||||
| SMS | 150 | 150 | 0 | 0 | 90/60 | 58.1 (31 – 79) | normal | healthy |
| GICaRes | 82 | 0 | 68 | 14 | 40/42 | 60.9 (29 – 82) | normal | healthy |
| Luo (normal) | 41 | unknown | unknown | unknown | 19/22 | 58.4 (43 – 78) | normal | matched (n=24) |
| Luo (neoplasia) | 80 (18 aden.) | 9 | 27 | 44 | 54/26 | 60.0 (23 – 89) | adv. aden. cancer | adv. aden. cancer |
| TCGA | 322 | 43 | 141 | 138 | 145/177 | 64.9 (31 – 90) | cancer | cancer |
Experimental Plan/Study Design
For discovery, we utilized SMS tissue samples (n=150) to identify significant DNA methylation drift (q-value <10−4) at the CpG probe level. Tissue samples (n=68, left colon; n=14, right colon) from the independent GICR study were used to validate the discovered drift-CpGs, including analyses of drift differentials by sex and colorectal location both at the single CpG dinucleotide and CpG-island (CGI) level (with drift-CGIs defined as containing at least 5 drift CpGs per island). Next, we obtained methylation data from endoscopic normal and cancer samples published by Luo et al. (2, 31) and The Cancer Genome Atlas (TCGA) consortium Colorectal Adenocarcinoma (COAD) and Rectum Adenocarcinoma (READ) projects (32, 33) to evaluate drift-related methylation patterns in neoplastic tissues. The TCGA data also included information on the percentage of tumor cells that we used to adjust measured drift levels in the tumors for normal, stromal and necrotic cell content. For TCGA data, we accessed the data via the Genomic Data Commons (level 1 HM450 methylation array idats) and for gene expression data via Firehose (Level 3, v2 pipeline, RSEM-normalized Illumina HiSeq 2000 gene expression counts, http://gdac.broadinstitute.org/, (34). All methylation array data were preprocessed as described in SI.
Statistical software and data metrics
Data pre-processing and most analyses were performed using the R programming language (v3.4.4) (35). The minfi Bioconductor package was used to analyze methylation data and preprocess idats as described previously (36).
Levels of DNA methylation across islands and CpGs are provided as β-values (0 < β < 1), which represents the percentage of methylation at a given site or island, or as M-values, calculated as (logit2(β)). In keeping with our previous studies of methylomic drift in Barrett’s esophagus, we preselected CpG probes that showed low levels of methylation (β < 0.5) in normal tissue samples (17). See SI for further details.
Data Availability
Methylation data used in this study is deposited on the Gene Expression Omnibus, accession GSE113904. All other data were previously published in open-access repositories.
Adenoma-to-carcinoma sojourn time distributions
We previously published estimates for the mean sojourn time of an adenoma (from the birth of its founder cell) to cancer (13, 37). However, to correlate the methylomic drift in tumor tissue samples with tissue age, it is necessary to condition the estimate on the age when the cancer tissue sample was collected, while calculating the sojourn time as beginning with the initiating event that leads to the first premalignant cell that generates an adenoma and eventually the cancer from which the tissue sample was collected. This is typically close to the patient age at the time of diagnosis. Here we provide a derivation of the sojourn time distribution conditioned on the age cancer is detected. Additional mathematical details can be found in previously published articles (38, 39).
Given the age a cancer is detected clinically, two random events are assumed to occur prior to detection: (1) Initiation of a viable (premalignant) adenoma (referred to as a p-clone) that does not become extinct by the time it transforms to cancer and (2), a malignant transformation in the clonally expanding p-clone. A third event, clinical observation of the carcinoma, coincides with the size-dependent detection of a malignant clone (m-clone) in the p-clone that forms the cancer. Let Y(t) be the (random) number of premalignant cells in a p-clone at time t and f0(u1|Y(t) > 0) be the conditional density function for the initiation of a p-clone (at time u1) that is conditioned on not becoming extinct prior to malignant transformation at time t. Further, let fp−clone(t−u1) be the conditional density function for a p-clone to undergo a first malignant transformation in time length t−u1 that leads to a first cancer. Then, as shown in the SI, we have the following expression for the conditional density function of the initiating event that leads to a first malignant clone at random time TM = t,
| (1) |
Here TM represents the time when a malignant transformation occurs that will lead to a viable malignant clone and a clinically detected cancer at a later (random) time TC = a. To account for this, we convolve the distribution in Eq. (1) over times TM=u2 for malignant transformation with the probability density for clinical detection of the malignant clone (m-clone) as a carcinoma at age TC = a.
| (2) |
Because multiple malignant transformations may occur during the lifetime of the adenoma before it turns into cancer, this formula is an approximation. However, as was shown in [13], this process can be well approximated by an effective malignant transformation in the p-clone which generates a viable m-clone with transformation rate μeff = μp∞, where μ is the rate for malignant transformations and p∞ is the asymptotic non-extinction probability (see SI for details).
Explicit formulas for f0(u1|Y(t) > 0), fp−clone(t−u1), fm−clone(t−u2), are provided in Supplemental Information (SI) (see Eqs S9, S11, and S13). The distribution of the adenoma initiation time u1 given in Eq. (2) can then be used to compute the expected adenoma-to-carcinoma sojourn times E(s) and their variance Var(s), conditioned on the carcinoma being detected at age TC = a. Since s = a−u1, we have
| (3) |
| (4) |
Regression modeling of tumor methylation data
We used a constrained non-linear regression model, corrected for the presence of normal and stromal cell fractions in the tumor samples, to fit the drift-CGI methylation levels of both TCGA and Luo tumors (excluding adenomas), separately for both sexes. The observed methylomic drift in these tumors was assumed to be the sum of an unobserved (true) neoplastic drift and drift associated with the non-tumor (normal/stromal) cell content in the sample. Specifically, we used the following model to relate the mean methylation level D across the identified 781 drift-CGI to the expected premalignant sojourn time E(s), corrected for the measured fractions of normal/stromal cells in the tumor samples, fN, and with a fixed offset ε representing the mean level of normal methylation at birth for all drift-CGI:
| (5) |
Using this model, we estimated the CGI-level drift rate αT for the tumors, while the normal drift rate αN across the 781 CGI was independently estimated using all normal tissue samples from the SMS and GICR studies. Numerical values for the parameters in Eq. (5) and estimates of the tumor drift rate αT for males and females are provided in Table S1 in the SI.
Variance of drift explained by stochastic cancer model
To assess how much of the observed variance in drift in the Luo and TCGA CRC data can be explained by the variance associated with the stochastic colon cancer model, we computed for each sample the sum of square errors SSE = Σ (Dobs - Dexp)2, where Dexp is given by Eq (5) and Dobs the observed (mean) methylomic drift for a given sample. Thus, SSE is the sum of the square residuals of the data relative to the predicted age-dependent drift, adjusted for normal/stromal cell content in the tumor samples. ‘Variance explained’ by the stochastic model is then computed as the ratio R=SSP/SSE, where SSP is the sum of square errors predicted by the stochastic model, i.e., SSP = Σ (αT)2Var(s). Thus, when R < 1 the model cannot fully explain the observed variance while for R > 1 the model yields a sojourn time variability that is inconsistent with drift data.
Computer code
R-code used to derive the following results is available on https://github.com/gluebeck/Epigenetic-Drift-in-Colon
Results
In this analysis, we: 1) identified and validated CpG probes that drift significantly in normal colorectal tissues; 2) examined the variability of drift in neoplastic tissues vs normal tissues (Luo data); 3) determined corresponding drift rates at the CpG island (CGI) level defining drift-CGIs as CpG islands that contain at least 5 drift-CpGs; 4) compared drift rates by sex and colorectal location (proximal, distal, and rectum); 5) obtained island-level drift distributions in CRCs; and 6) computed the expected variability of drift observable in CRCs associated with the modeled distributions of premalignant (adenoma-to-carcinoma) sojourn times, defined by the time the ancestral premalignant progenitor cell is born until cancer diagnosis.
Identification of methylomic drift at the CpG probe-level in normal colorectal tissue
To identify age-related methylomic drift across a population of normal tissue samples, we performed probe-wise linear regressions using all 150 samples (both sexes) from the SMS study. Only probes with β < 0.5 across all samples were included in the discovery to select for positive drift, i.e. gradual increases of DNA methylation levels with age. This resulted in a total of 182,498 CpG probes being tested by regressing age (at the time of biopsy) on the methylation level (M-value) measured. Among these, we identified 13,525 probes with highly significant (mostly upward) drift (q-value <10−4) as shown in Fig. 2.
Fig. 2.

Estimated CpG drift rates of 182,498 CpG probes vs the (log2) ratio of methylation variance in tumor samples relative to the corresponding variance in normal tissue samples from the Luo study [11]. Variances and drift rates were computed using M-values. The drift rates were estimated using linear regression of methylation vs patient age (in years). CpGs in dark grey undergo significant methylomic drift (q-value <10−4), CpGs in medium grey are considered static, i.e., do not show significant linear trends with patient age. The subset of CpGs marked in light grey serves as a control group for the analysis of gene expression and methylomic drift (see Results).
Furthermore, when these drift probes were evaluated separately in 41 normal tissue samples and 80 neoplastic tissue samples of the Luo study (2), we found that the methylomic drift was mostly associated with an increased variance in neoplastic tissues compared with normal tissues (Fig. 2) suggesting a high level of tissue-age related heterogeneity in the neoplasia.
Validation of methylomic drift in GICR study
We used 68 additional normal (left colon) samples from the GICR study (30) to validate the set of drift-CpGs we identified in the SMS study (29). Although the SMS samples were exclusively collected in rectum, we found that out of the 13,525 drift-CpGs identified in SMS 12,700 could be validated as positively (drift rate > 0) and significantly drifting in the GICR study (p-value < 0.05) using Pearson’s correlation.
Drift at the CpG-island (CGI) level
Motivated by our recent findings of widespread epigenetic drift involving > 1,000 CGI in Barrett’s esophagus (18), we also evaluated age-related drift at the CGI level in colon and rectum. Among the 12,700 CpG probes that exhibited significant positive drift in both SMS and GICR data sets, we identified 871 CGI with at least 5 drift-probes per island (we will refer to such CGI as drift-CGI). As expected, island-level methylation was also highly correlated between the drift-CGI in normal tissue (mean Pearson r=0.68), however it was attenuated in cancers (mean Pearson r=0.42 for left colon and rectum; r=0.55 for right colon in TCGA).
To boost the overall correlations between drift-CGI, we selected a subset of 781 CGI that were consistently and significantly correlated with one another across TCGA cancers in both left and right colon. This filtering improved the mean drift-CGI correlations to 0.71 for normal colon, 0.46 for left colon and rectum, and 0.6 for right colon. However, we obtained similar results with the full set of 871 CGI.
For the subset of 781 drift-CGIs, we list the genomic location, associated genes, proximity to transcription start sites (TSS200 or TSS1500), the number of array probes and number of identified drift probes and the island-level drift rate (regression slopes) in Table S2. For comparison, we also identified > 1000 CGI that do not appear to undergo methylomic drift in normal colon but that may or may not drift differentially in colorectal neoplasia. We refer to this comparison group as ‘static-CGI’.
Drift at the CpG-probe vs CGI-level
While >90% of drift-CpGs identified are located within or near CGI, only 60% of all probes were associated with CGI on the array (i.e., are situated on an island, shore, or shelf), which shows that methylomic drift in normal colorectum (as defined here) occurs predominantly at islands. Furthermore, drift rates appear to be more uniform at the island level compared with estimated drift rates at the single probe level (shown as drift-rate distributions by dashed and solid lines in Fig. 3 at the probe- and island-level, respectively).
Fig. 3.
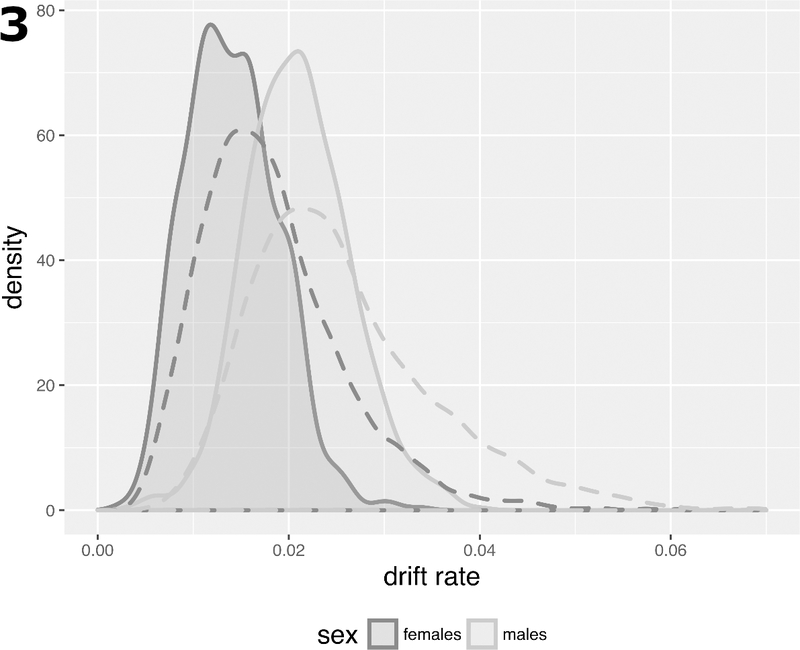
Drift rate distributions in SMS for 781 CGI with a minimum of 5 identified drift-CpGs by sex (solid curves) versus analogous distributions at the probe-level comprising 12,700 CpGs (dashed curves).
Next, to adjust for systemic differences in methylomic drift between the sexes, we performed an analysis of covariance (ANCOVA) allowing for differences in drift rates by gender (SMS and GICR left colon data, comparing 127 females and 91 males). Incremental differences in drift rates between males and females were statistically significant for 759 of 781 drift islands tested (q-value < 0.05) and are shown by their distinct distributions for males and females in Fig. 3.
Validating CGI level drift by gender and site
Using ANCOVA regression with sex as a categorical variable and age as the continuous independent variable, we were able to validate island-level drift first identified in the SMS study. The GICR study comprised a total of 68 normal tissue samples from the left colon (31 males, 37 females) and 14 normal tissue samples from the right colon (11 males, 3 females).
All GICR samples were from cancer-free patients at the time of biopsy. Fig. 4 shows the estimated drift rates for the two studies by gender. The drift rate distributions for males and females in the left colon are clearly distinct for the two sexes with males showing 40% (SMS) and 65% (GICR) higher mean drift rates compared with females. Due to small sample size, only results for males and females combined are shown in Fig. 4 for methylomic drift in right (proximal) colon (gray symbols).
Fig. 4.
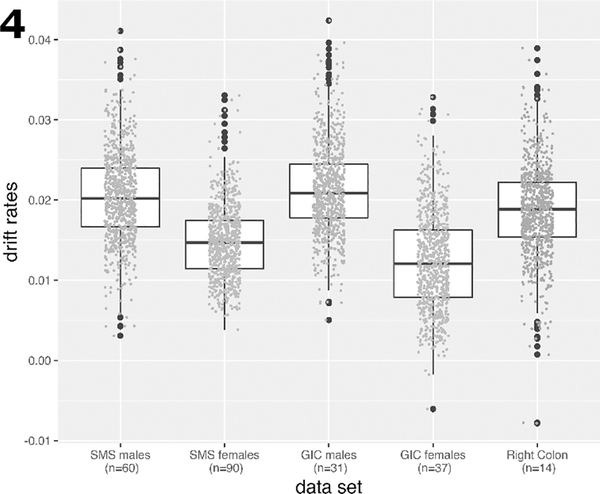
Boxplots of the drift rate distributions for the same CGI as in Fig.(2), but validated in samples from the GICR study for left (distal) and right (proximal) colon samples. For each group the individual drift rate estimates are shown as data points. Due to small sample sizes for males and females in right colon, drift rates were determined for both sexes combined in right colon.
Methylomic drift in neoplastic tissues
The expected sojourn times E(s) of the parental adenoma that led to the clinically detected cancers and measured methylation drift rates (adjusted for tissue composition) are shown for males in Fig. 5A and 5B, respectively, by age and anatomical site together with their 95% confidence bands. Similarly, Fig. 5C and 5D show female expected parental adenoma sojourn times and adjusted methylation drift rates, respectively. The parameters used for these predictions are taken from Meza et al. (26) who fitted 3-stage clonal expansion models to colorectal incidence data in the US and the UK. Although the model parameters (adjusted for secular trends) were similar for the US and UK, we chose to use the model parameters obtained for the UK population, which historically had lower colorectal screening utilization than the US, therefore better reflect the natural history of CRC. Note, the computed age-specific sojourn times do not differ significantly between males and females for neoplasia in right (proximal) colon and rectum. For neoplasia in left (distal) colon we obtained sojourn times that are between those for rectal and proximal colon among males (Fig. 5A) but are more similar to the sojourn times in rectal colon among females (Fig. 5C). However, for all sites and the two sexes, our predictions suggest that adenoma bound for cancer started early in life, most likely before the age of 20. See Fig. 5A and 5C and Fig. S1 in SI.
Fig. 5.
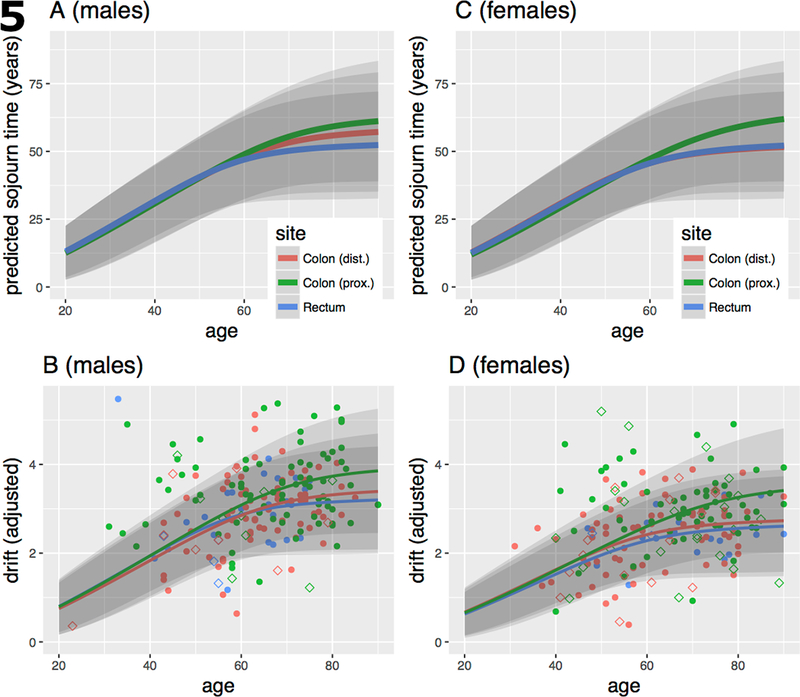
A) Expected (mean) premalignant sojourn times (in years) for males by age of cancer diagnosis and anatomical site with 95% confidence bands, based on the model fits described in Meza et al. [10] for UK males. B) Sojourn time-dependent drift curves fitted to normal/stromal cell content corrected TCGA (solid symbols) and Luo (empty symbols) methylomic CGI-level drift in tumors by sex and anatomical site. Regression model described in Material and methods. C, D) same as A and B, respectively, but for females.
To see whether our sojourn time estimates are consistent with methylomic drift patterns in neoplasia, we assumed constant drift rates and fitted them by regressing drift-related methylation levels (at the island-level) on patient age using the computed age-, sex- and site-specific premalignant sojourn times (see Material and Methods). The estimated neoplastic drift rates (M-value/year), although similar for the 3 anatomical sites, were about 12–22% higher in males than females (proximal colon – females/males: 0.056/0.065, distal colon: 0.054/0.061, rectum: 0.051/0.061). This difference is not unexpected since we found much stronger drift rate differences between the sexes in normal colorectum (Fig. 3). Of note, (1) although the corrected drift patterns shown in panels B (males) and D (females) of Fig. 5 still exhibit a high degree of variability compared with the expected variance, especially for proximal (right) colon, we observe that the ordering of the drift patterns and their fits closely follow the predicted ordering of premalignant sojourn times in panel Fig. 5A and 5C. (2) the estimated island-level drift rates that best fit the methylomic patterns shown in panel 5B (males) and panel 5D (females) are similar for the 3 sites and are approximately 3 to 4-fold higher than the corresponding drift rates for normal colon. (3) our estimates of a roughly 3 to 4-fold acceleration of methylomic drift in colorectal neoplasia is consistent with various cell proliferation measurements (discussed below) in normal colorectal epithelium vs adenoma and carcinoma, suggesting that the epigenetic drift (as defined here) is likely correlated with mitotic activity (23, 40–42).
Methylomic tissue age (mAge) vs drift-based sojourn time predictions
Several ‘universal’ methylomic clocks have been introduced recently to predict biological tissue age using regularized regression methods (24, 25). In contrast to the drift-based clock used for this study, which scans individual CpG probes for significant correlations with age, these multi-tissue-type clocks primarily rely on elastic net regressions to predict chronological age from a selection of CpG probes. To compare sojourn time estimates for the TCGA samples included in this study (using Eq (5)) with estimates of mAge provided by others, we computed mAge for two published clocks by Horvath (Horvath 1: 353 CpGs; Horvath 2: 110 CpGs) and a clock developed by Hannum et al. (71 CpGs) (24, 25). All estimates were adjusted for normal cell content assuming that the normal cell fraction in the tumors contributes a term proportional to patient age. Table S3 shows the correlations between our predicted sojourn times and mAge for the 3 models, as well as their means and p-values for difference in mean mAge and mean E(s) = 61 years of the 322 TCGA colorectal samples used for this comparison. We find significant correlations of mAge with our predictions (r = 0.45, p-value < 2.2 · 10−16 for the Hannum et al. clock). Moreover, upon a simple recalibration of the unadjusted mAge estimates of normal tissue to closely fit patient age, this clock also predicts excessively long sojourn times. In contrast, while the Horvath 110 CpG clock still provides good correlations with our sojourn time predictions (r = 0.32, p-value = 6.9 ·10−9), the 353 CpG clock correlates only poorly (r=0.07, p-value = 0.2).
Degree of methylomic drift in cancers shows strong correlation with methylation levels in normal tissue
To compare drift patterns at the CGI-level between normal and cancer tissue, we ordered the samples by their mean methylation levels across the drift-CGI in normal colon tissue (Fig. 6). The resulting heatmaps (for left colon in Fig. 6, for right colon in Fig. S2) show that the base-level of methylation in normal colon is predictive of the amount of drift occurring in cancers. CGI that are static (first 300 top rows) in normal colon do not drift discernibly in cancers (although they can be altered). In contrast, CGI that drift in normal colon show accelerated drift in cancers with increasing levels of methylation in normal tissue (Fig. S3). The correlation between mean drift in normal tissue and mean drift in the cancer tissues across the 781 drift-related islands is 0.81 (p-value < 2.210−16). In contrast, static CGI, defined here as islands that comprise at least 5 CpG probes found not to undergo drift in normal colon (drift rates < 0.002/year, shown as light grey data points in Fig. 2), do not drift in the cancers (Fig. S4).
Fig. 6.
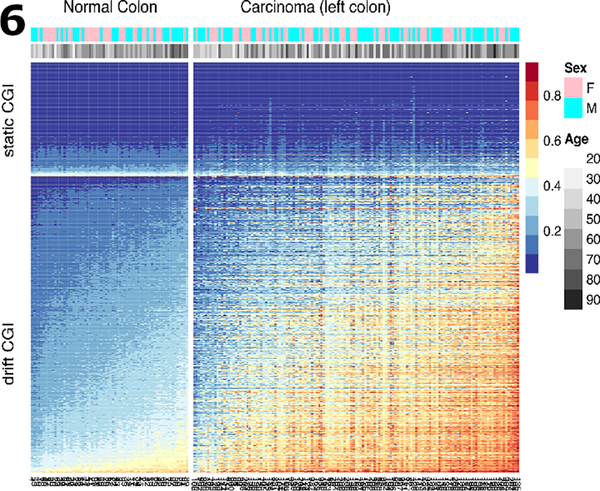
Methylation heat map of 300 static CGI (top rows) and 781 drift-related CGI (bottom rows) for 68 normal samples and 141 TCGA colon cancer samples (left colon only). Sample groups (normal, cancers) are shown ordered by their mean island level methylation.
Methylomic drift and gene expression
We previously found that advanced methylomic drift on some CGI associated with actively transcribed genes in EAC are significantly associated with reduced gene expression and possibly gene silencing (18). Thus, here we investigated whether methylomic drift in CRC is similarly associated with widespread transcriptional repression. To this end, we computed the Pearson correlation between methylation and gene expression (RNA-seq) and its statistical significance for all gene-CGI pairs for left colon (n = 184, including rectum) and right colon cancers (n = 138) from the TCGA. Out of a total of 668 identified gene-island pairs in right colon we found 373 (56%) of pairs that show a significant negative Pearson correlation, while only 34 (5%) of pairs show a significant positive correlation (q-value < 0.01). In contrast, left colon cancers show fewer pairs being correlated. Out of a total of 663 identified gene-CGI pairs in left colon we found only 170 (26%) pairs that show a significant negative correlation, while 46 (7%) pairs show a significant positive correlation (q-value < 0.01, see Table S4).
Although the overall number of correlated gene-CGI pairs in right colon is almost twice the number in left colon, 78% (169/216) of the pairs in left colon are also found to be significantly correlated in right colon. Furthermore, we find no significant difference in the fractions of repressed vs over-expressed genes affected by drift in the right vs left colon (p-value = 0.24, Fisher’s exact test). For comparison, we identified >1000 islands that appear ‘static’ in normal colon tissue (see Fig. 2) and do not show discernible age-related drift among cancers (Fig. 6 and Figs. S2 and S3). Surprisingly, 16% (19%) of these static islands in left (right) colon also show strong correlations between methylation and gene expression suggesting that they, albeit under stronger epigenetic control in normal tissue, can also be altered in neoplasia and may participate in the clonal evolution of a cancer (see Table S5). However, compared to gene-CGI pairs that are associated with methylomic drift and exhibit significant methylation-gene expression correlations in the cancers, fold-changes (> 2-fold up or down) in expression are less common among static pairs (<25%) compared with drift pairs (> 62%) in left and right colon.
Discussion
Methylomic drift appears to be widespread in normal colon and rectum, involving at least 7% of CpG probes tested (q-value < 10−4). Over 90% of these probes are found on (or near) CGI, while only 64% of HM450 probes are located on or near CGI. However, among probes with β < 0.5 in normal colorectum, the fraction of HM450 probes on or near islands is about 88%, similar to the fraction of drift-CpGs we identified. In contrast, a study by Irizarry et al. (43) of the human colon cancer methylome showed that aberrant methylation predominantly occurred at conserved tissue-specific CGI shores, with hypermethylation typically enriched closer to the associated CGI and hypomethylation enriched further from the associated CGI. While this finding appears in conflict with our findings of drift occurring mainly on (or near) CGI, we point out that our definition of CGI includes the shore regions which extend 2kb from the island boundaries. Thus, the island-level drift (including shore regions) in neoplastic tissue observed in our study, is not inconsistent with the tissue-specific methylation changes in cancers observed by Irizarry et al. (43) and may well play an important role in the phenotypic evolution of cancer.
Several important conclusions can be drawn from our findings:
Methylomic drift in normal colon continues unabated at an increased rate in neoplastic tissue (about 3–4 fold faster compared with normal colon), with drift-associated methylation in proximal colon showing the highest gains across the older aged (age >60) cancer population, followed by distal colon and with rectum showing the lowest gains. However, the estimated drift rates for neoplasia in these sites are similar which suggests that, on average, neoplastic lesions in the proximal colon sojourn longer than lesions in the distal colon and rectum. This conclusion is consistent with the findings from independent mathematical modeling of age-specific incidence curves of CRC in the US and UK (26) that suggested significantly slower growth rates of proximal adenoma compared with distal and rectal adenoma.
Several studies have carefully measured cell proliferation in both normal and neoplastic (adenoma) colon mucosae. The study by Kikuchi et al. (44) evaluated the Ki-67 (MIB-1) cell proliferation marker in normal colon, adenoma of various histology, and carcinoma. Although Ki-67 labeling is strongly dependent on cell position within crypts, Ki-67 labeling in normal colon was 14% ± 5% while in adenoma Ki-67 was 26.5% ± 9% for low grade adenoma and 35% ± 6% for high grade adenoma in the Kikuchi et al. study, suggesting a 2–3-fold increase in cell proliferation between normal and neoplastic colon tissue. In carcinoma, Ki-67 labeling is about 3-fold higher than normal tissue (53% ± 5%). Similarly, the study by Baker et al. (45) found 3-fold increase in the number of Ki-67+ cells at the crypt base in adenomatous colon tissue that lost APC, compared with wild-type normal colon.
The computed age-dependent sojourn time distributions for proximal colon, distal colon, and rectum indicate that the first premalignant cell that generates a cancer-forming adenoma typically arises early in life and may take decades before developing into cancer. This conjecture is supported by our analysis of methylomic drift in colorectal neoplasia relative to normal colon tissue which shows that drift rates in neoplastic colon are increased similarly to independently measured rates of cell proliferation in adenoma and carcinoma compared with those in normal colon (44). Furthermore, an independent application of 3 universal (multi-tissue-type) clocks yields similarly long time scales (~60 years) for the TCGA samples analyzed in this study (Table S3) with 2 of the 3 clocks showing significant correlations with the computed drift-based sojourn times.
Although methylomic drift appears highly variable in the tumors (even after correction for normal/stromal cell content), 55–89% of the total (island-level) variance in DNA methylation observed in the tumor samples can be attributed to the stochasticity of the tumor growth process and random events that lead to a cancer and its detection. Note, this range of variability explained by the model does not account for any variability present in normal tissue prior to adenoma initiation. However, a variance analysis of methylation levels of static CGIs in normal tissues compared with variances of drift-CGI in normal and cancer tissues indicates that the normal sample population has a constant (non-drift) variance of about 0.018, which increases >3-fold for drift-CGI to 0.07, while in cancers this variance increases to about 0.36. Thus, assuming static methylomic variability across samples approximates the variability at adenoma initiation, only ~5% (0.018/0.36) of the observed drift variance across cancers may be attributed to pre-existing methylomic variability in normal tissue.
Consistent with findings of a recent analysis of methylomic drift in Barrett’s esophagus and in esophageal adenocarcinoma (18), we found that advanced methylomic drift at the islands-level is frequently (> 50% in right colon, > 25% in left colon) associated with significant reductions in gene expression. We identified only a small number of drift-related CGI-gene pairs for which drift correlated positively with gene expression (e.g., SIM2, TBX5, see Table S4). Although our analysis does not demonstrate causality, the fact that epigenetic drift of CGI in normal colon is more prominently associated with transcriptional changes in colorectal neoplasia than static CGI that undergo little or no drift in normal colorectum suggests a potential role of methylomic drift in the clonal evolution to cancer.
Our study has several limitations related to the nature of the available data, their clinical annotation and the methylation array platform used. In particular, the HM450 platform only covers a small fraction (~1.6%) of CpGs in the human genome and has an uneven distribution of CpGs at the island level. Thus, our selection of drift-CpGs is biased toward islands with a larger number of array probes likely resulting in an underestimation of genome-wide methylomic drift. Furthermore, we lack gene expression data for our normal tissue samples (GICR, SMS and Luo study) preventing a comparative study of drift-related phenotypic changes in normal vs neoplastic tissue. Although, we are able to explain up to 77–89% of the observed methylomic variance of drift-CGI in distal colon and rectum, we only explain a much smaller fraction (55%) of this variance in proximal colon. It is conceivable that other unaccounted factors contribute differentially to the observed variance including environmental exposures, diet, microbiome, immune status, cancer (epi)genetics and measurement errors. Unfortunately, most of these covariates are unavailable in our data and TCGA and have not been included in the modeling of the adenoma sojourn times. In spite of the limitations of the modeling and lack of further data that more fully explain the observed inter-individual heterogeneity in methylomic drift in these samples, our results support the hypothesis that adenoma that lead to cancer arise early in life, even for CRCs that occur at advanced ages. Thus, starting chemoprevention and lifestyle interventions early in life (rather than later in life) may be more effective in reducing the cancer burden given our findings that cancer precursors likely sojourn for decades before turning into cancer.
In summary, our analysis shows that age-related methylomic drift is a genome-wide phenomenon that occurs in normal colon and continues at an accelerated pace in colorectal neoplasia. Furthermore, we show that differences in age-related drift between normal and neoplastic tissues are broadly consistent with predicted long-duration but individually variable total adenoma-carcinoma sojourn times that capture approximately 55–89% of the variance of drift-CGI heterogeneity in CRCs. Other factors, including those related to genetics, obesity, diet, lifestyle and environmental factors and use of chemo-preventative agents such as use of non-steroidal anti-inflammatory drugs (NSAIDS) may account for much of the remaining heterogeneity. Importantly, the estimated long duration of premalignant sojourn times suggests that CRC incidence may be reduced through early (and ideally lifelong) dietary and lifestyle interventions.
Supplementary Material
Statement of Significance.
Findings present age-related methylomic drift in colorectal neoplasia as evidence that premalignant cells can persist for decades before becoming cancerous.
Acknowledgements
We gratefully acknowledge the Luo, Seattle ColoCare, and SMS study teams, the GICR biorepository team, and the study participants.
Financial Support: This research was supported by the following grants: NIH grants U01CA182940 (G.E. Luebeck, W.D. Hazelton, W.M. Grady, S.K. Madden, K. Curtius), U01CA199336 (G.E. Luebeck, W.D. Hazelton); Barts Charity grant 472–2300, London (K. Curtius) and UK Medical Research Council Rutherford fellowship (K. Curtius); and NIH grants (P30CA15704, U01CA152756, R01CA194663, R01CA220004, U54CA143862, P01CA077852), R.A.C.E. Charities, Cottrell Family Fund, R03CA165153, Listwin Family Foundation, Seattle Translational Tumor Research program, Fred Hutchinson Cancer Research Center (S.K. Madden, M. Yu, K.T. Carter, and W.M. Grady), R01CA189184 (C. Lee, C.M. Ulrich, S.K. Madden, M. Yu, K.T. Carter, and W.M. Grady), R01CA112516, R01CA114467, R01CA120523 (C.M. Ulrich, S.K. Madden, M. Yu, K.T. Carter, and W.M. Grady), Huntsman Cancer Foundation, U01 CA206110, R01CA189184 R01CA 207371 and P30CACA042014 (C.M. Ulrich). U24CA074794 (P.A. Newcomb, S.K. Madden, M. Yu, K.T. Carter, and W.M. Grady). This material is the result of work supported in part by resources from the VA Puget Sound Health Care System and the ColoCare Study. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Footnotes
Conflict of interest statement. The authors declare no potential conflicts of interest.
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. The New England journal of medicine. 1988;319(9):525–32. Epub 1988/09/01. doi: 10.1056/NEJM198809013190901. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 2.Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, Wang J, Willis JE, Makar KW, Ulrich CM, Lutterbaugh JD, Shrubsole MJ, Zheng W, Markowitz SD, Grady WM. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147(2):418–29 e8. Epub 2014/05/06. doi: 10.1053/j.gastro.2014.04.039. PubMed PMID: ; PMCID: PMC4107146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borras E, San Lucas FA, Chang K, Zhou R, Masand G, Fowler J, Mork ME, You YN, Taggart MW, McAllister F, Jones DA, Davies GE, Edelmann W, Ehli EA, Lynch PM, Hawk ET, Capella G, Scheet P, Vilar E. Genomic Landscape of Colorectal Mucosa and Adenomas. Cancer Prev Res (Phila). 2016;9(6):417–27. doi: 10.1158/1940-6207.CAPR-16-0081. PubMed PMID: 27221540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62(3):367–86. Epub 2013/01/24. doi: 10.1111/his.12055. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 5.Shibata D Inferring human stem cell behaviour from epigenetic drift. The Journal of pathology. 2009;217(2):199–205. Epub 2008/11/26. doi: 10.1002/path.2461. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang D, Zhang M, Gold B. Origin of Somatic Mutations in beta-Catenin versus Adenomatous Polyposis Coli in Colon Cancer: Random Mutagenesis in Animal Models versus Nonrandom Mutagenesis in Humans. Chem Res Toxicol. 2017;30(7):1369–75. doi: 10.1021/acs.chemrestox.7b00092. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 7.Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SAC, Wright NA, Simons BD, Jansen M, Graham TA. Quantification of Crypt and Stem Cell Evolution in the Normal and Neoplastic Human Colon. Cell Rep. 2014;8(4):940–7. doi: 10.1016/j.celrep.2014.07.019. PubMed PMID: WOS:000341573500003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. P Natl Acad Sci USA. 2015;112(1):118–23. doi: 10.1073/pnas.1421839112. PubMed PMID: WOS:000347447100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. Epub 2012/07/20. doi: 10.1038/nature11252. PubMed PMID: ; PMCID: 3401966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comstock SS, Hortos K, Kovan B, McCaskey S, Pathak DR, Fenton JI. Adipokines and obesity are associated with colorectal polyps in adult males: a cross-sectional study. PloS one. 2014;9(1):e85939 Epub 2014/01/28. doi: 10.1371/journal.pone.0085939. PubMed PMID: ; PMCID: 3895019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bastide N, Morois S, Cadeau C, Kangas S, Serafini M, Gusto G, Dossus L, Pierre FH, Clavel-Chapelon F, Boutron-Ruault MC. Heme Iron Intake, Dietary Antioxidant Capacity, and Risk of Colorectal Adenomas in a Large Cohort Study of French Women. Cancer Epidemiol Biomarkers Prev. 2016;25(4):640–7. doi: 10.1158/1055-9965.EPI-15-0724. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 12.Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A, Moris F, Rodrigo L, Mira A, Collado MC. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. Journal of gastroenterology. 2014. Epub 2014/05/09. doi: 10.1007/s00535-014-0963-x. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 13.Luebeck EG, Curtius K, Jeon J, Hazelton WD. Impact of tumor progression on cancer incidence curves. Cancer research. 2013;73(3):1086–96. Epub 2012/10/12. doi: 10.1158/0008-5472.CAN-12-2198. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issa JPJ, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the Estrogen-Receptor Cpg Island Links Aging and Neoplasia in Human Colon. Nature Genetics. 1994;7(4):536–40. doi: Doi 10.1038/Ng0894-536. PubMed PMID: WOS:A1994PA83200023. [DOI] [PubMed] [Google Scholar]
- 15.Galamb O, Kalmar A, Bartak BK, Patai AV, Leiszter K, Peterfia B, Wichmann B, Valcz G, Veres G, Tulassay Z, Molnar B. Aging related methylation influences the gene expression of key control genes in colorectal cancer and adenoma. World J Gastroentero. 2016;22(47):10325–40. doi: 10.3748/wjg.v22.i47.10325. PubMed PMID: WOS:000390172600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124(1):24–9. doi: 10.1172/Jci69735. PubMed PMID: WOS:000329333500006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtius K, Wong CJ, Hazelton WD, Kaz AM, Chak A, Willis JE, Grady WM, Luebeck EG. A Molecular Clock Infers Heterogeneous Tissue Age Among Patients with Barrett’s Esophagus. PLoS Comput Biol. 2016;12(5):e1004919. doi: 10.1371/journal.pcbi.1004919. PubMed PMID: ; PMCID: PMC4864310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luebeck EG, Curtius K, Hazelton WD, Maden S, Yu M, Thota PN, Patil DT, Chak A, Willis JE, Grady WM. Identification of a key role of widespread epigenetic drift in Barrett’s esophagus and esophageal adenocarcinoma. Clin Epigenetics. 2017;9:113 Epub 2017/10/20. doi: 10.1186/s13148-017-0409-4. PubMed PMID: ; PMCID: PMC5644061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chettouh H, Mowforth O, Galeano-Dalmau N, Bezawada N, Ross-Innes C, Ma/cRae S, Debiram-Beecham I, O’Donovan M, Fitzgerald RC. Methylation panel is a diagnostic biomarker for Barrett’s oesophagus in endoscopic biopsies and non-endoscopic cytology specimens. Gut. 2017. Epub 2017/11/01. doi: 10.1136/gutjnl-2017-314026. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research N, Analysis Working Group: Asan U, Agency BCC, Brigham, Women’s H, Broad I, Brown U, Case Western Reserve U, Dana-Farber Cancer I, Duke U, Greater Poland Cancer C, Harvard Medical S, Institute for Systems B, Leuven KU, Mayo C, Memorial Sloan Kettering Cancer C, National Cancer I, Nationwide Children’s H, Stanford U, University of A, University of M, University of North C, University of P, University of R, University of Southern C, University of Texas MDACC, University of W, Van Andel Research I, Vanderbilt U, Washington U, Genome Sequencing Center: Broad I, Washington University in St L, Genome Characterization Centers BCCA, Broad I, Harvard Medical S, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins U, University of North C, University of Southern California Epigenome C, University of Texas MDACC, Van Andel Research I, Genome Data Analysis Centers: Broad I, Brown U, Harvard Medical S, Institute for Systems B, Memorial Sloan Kettering Cancer C, University of California Santa C, University of Texas MDACC, Biospecimen Core Resource: International Genomics C, Research Institute at Nationwide Children’s H, Tissue Source Sites: Analytic Biologic S, Asan Medical C, Asterand B, Barretos Cancer H, BioreclamationIvt, Botkin Municipal C, Chonnam National University Medical S, Christiana Care Health S, Cureline, Duke U, Emory U, Erasmus U, Indiana University School of M, Institute of Oncology of M, International Genomics C, Invidumed, Israelitisches Krankenhaus H, Keimyung University School of M, Memorial Sloan Kettering Cancer C, National Cancer Center G, Ontario Tumour B, Peter MacCallum Cancer C, Pusan National University Medical S, Ribeirao Preto Medical S, St. Joseph’s H, Medical C, St. Petersburg Academic U, Tayside Tissue B, University of D, University of Kansas Medical C, University of M, University of North Carolina at Chapel H, University of Pittsburgh School of M, University of Texas MDACC, Disease Working Group: Duke U, Memorial Sloan Kettering Cancer C, National Cancer I, University of Texas MDACC, Yonsei University College of M, Data Coordination Center CI, Project Team: National Institutes of H. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017;541(7636):169–75. doi: 10.1038/nature20805. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyota M, Issa JP. CpG island methylator phenotypes in aging and cancer. Seminars in cancer biology. 1999;9(5):349–57. Epub 1999/11/05. doi: 10.1006/scbi.1999.0135. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 22.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer research. 2001;61(9):3573–7. Epub 2001/04/28. PubMed PMID: . [PubMed] [Google Scholar]
- 23.Issa JP. Aging and epigenetic drift: a vicious cycle. The Journal of clinical investigation. 2014;124(1):24–9. Epub 2014/01/03. doi: 10.1172/JCI69735. PubMed PMID: ; PMCID: 3871228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell. 2013;49(2):359–67. Epub 2012/11/28. doi: 10.1016/j.molcel.2012.10.016. PubMed PMID: ; PMCID: 3780611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10). doi: Artn R115 Doi 10.1186/Gb-2013-14-10-R115. PubMed PMID: ISI:000329387500008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meza R, Jeon J, Renehan AG, Luebeck EG. Colorectal cancer incidence trends in the United States and United kingdom: evidence of right- to left-sided biological gradients with implications for screening. Cancer research. 2010;70(13):5419–29. Epub 2010/06/10. doi: 10.1158/0008-5472.CAN-09-4417. PubMed PMID: ; PMCID: 2914859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutter CM, Knudsen AB, Marsh TL, Doria-Rose VP, Johnson E, Pabiniak C, Kuntz KM, van Ballegooijen M, Zauber AG, Lansdorp-Vogelaar I. Validation of Models Used to Inform Colorectal Cancer Screening Guidelines: Accuracy and Implications. Med Decis Making. 2016;36(5):604–14. doi: 10.1177/0272989X15622642. PubMed PMID: 26746432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liesenfeld DB, Grapov D, Fahrmann JF, Salou M, Scherer D, Toth R, Habermann N, Bohm J, Schrotz-King P, Gigic B, Schneider M, Ulrich A, Herpel E, Schirmacher P, Fiehn O, Lampe JW, Ulrich CM. Metabolomics and transcriptomics identify pathway differences between visceral and subcutaneous adipose tissue in colorectal cancer patients: the ColoCare study. Am J Clin Nutr. 2015;102(2):433–43. Epub 2015/07/15. doi: 10.3945/ajcn.114.103804. PubMed PMID: ; PMCID: PMC4515859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams SV, Newcomb PA, Burnett-Hartman AN, Wurscher MA, Mandelson M, Upton MP, Zhu LC, Potter JD, Makar KW. Rare Circulating MicroRNAs as Biomarkers of Colorectal Neoplasia. PloS one. 2014;9(10). doi: ARTN e108668 10.1371/journal.pone.0108668. PubMed PMID: WOS:000345743700027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, Mussolin B, Falcomata C, Binder AM, Cristiano C, Oddo D, Guarrera S, Cancelliere C, Bustreo S, Bencardino K, Maden S, Vanzati A, Zavattari P, Matullo G, Truini M, Grady WM, Racca P, Michels KB, Siena S, Esteller M, Bardelli A, Sartore-Bianchi A, Di Nicolantonio F. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut. 2017. Epub 2017/10/07. doi: 10.1136/gutjnl-2016-313372. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gene Expression Omnibus (GEO) GSE48684. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48684 Global DNA methylation alterations reveal multiple pathways in the initiation and progression of colorectal cancer [Internet]. National Center for Biotechnology Information (NCBI) 2014. [cited Mar 26, 2018]. [Google Scholar]
- 32.TCGA. TCGA Research Network. See: http://cancergenome.nih.gov/ and https://portal.gdc.cancer.gov/projects/TCGA-COAD. Accessed 6/16/2017.
- 33.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM. Toward a Shared Vision for Cancer Genomic Data. New Engl J Med. 2016;375(12):1109–12. doi: 10.1056/NEJMp1607591. PubMed PMID: WOS:000383537100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 2011;12. doi: Artn 323 10.1186/1471-2105-12-323. PubMed PMID: WOS:000294361700001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R Core Team. R: A language and environment for statistical computing. R. Foundation for Statistical Computing, Vienna, Austria: 2013. Available from: http://www.R-project.org/. [Google Scholar]
- 36.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9. doi: DOI 10.1093/bioinformatics/btu049. PubMed PMID: WOS:000336530000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meza R, Jeon J, Moolgavkar SH, Luebeck EG. Age-specific incidence of cancer: Phases, transitions, and biological implications. Proc Natl Acad Sci U S A. 2008;105(42):16284–9. Epub 2008/10/22. doi: 10.1073/pnas.0801151105. PubMed PMID: ; PMCID: 2570975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeon J, Meza R, Moolgavkar SH, Luebeck EG. Evaluation of screening strategies for pre-malignant lesions using a biomathematical approach. Mathematical biosciences. 2008;213(1):56–70. Epub 2008/04/01. doi: 10.1016/j.mbs.2008.02.006. PubMed PMID: ; PMCID: 2442130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dewanji A, Jeon J, Meza R, Luebeck EG. Number and size distribution of colorectal adenomas under the multistage clonal expansion model of cancer. PLoS Comput Biol. 2011;7(10):e1002213 Epub 2011/10/25. doi: 10.1371/journal.pcbi.1002213. PubMed PMID: ; PMCID: PMC3192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beerman I, Bock C, Garrison BS, Smith ZD, Gu HC, Meissner A, Rossi DJ. Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Cell Stem Cell. 2013;12(4):413–25. doi: 10.1016/j.stem.2013.01.017. PubMed PMID: WOS:000329569500009. [DOI] [PubMed] [Google Scholar]
- 41.Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Human molecular genetics. 2013;22(R1):R7–R15. Epub 2013/08/07. doi: 10.1093/hmg/ddt375. PubMed PMID: ; PMCID: PMC3782071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng SC, Widschwendter M, Teschendorff AE. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics. 2016;8(5):705–19. doi: 10.2217/epi-2015-0017. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 43.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash J, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–86. Epub 2009/01/20. doi: 10.1038/ng.298. PubMed PMID: ; PMCID: PMC2729128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kikuchi Y, Dinjens WN, Bosman FT. Proliferation and apoptosis in proliferative lesions of the colon and rectum. Virchows Archiv : an international journal of pathology. 1997;431(2):111–7. Epub 1997/08/01. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 45.Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014;8(4):940–7. doi: 10.1016/j.celrep.2014.07.019. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation data used in this study is deposited on the Gene Expression Omnibus, accession GSE113904. All other data were previously published in open-access repositories.