

Abstract
In addiction, an individual’s ability to inhibit drug seeking and drug taking is thought to reflect a pathological strengthening of drug-seeking behaviors or impairments in the capacity to control maladaptive behavior. These processes are not mutually exclusive and reflect drug-induced modifications within prefrontal cortical and nucleus accumbens circuits, however unlike psychostimulants such as cocaine, far less is known about the temporal, anatomical, and cellular dynamics of these changes. We discuss what is known regarding opioid-induced adaptations in intrinsic membrane physiology and pre-/postsynaptic neurotransmission in principle pyramidal and medium spiny neurons in the medial prefrontal cortex and nucleus accumbens from electrophysiological studies and explore how circuit specific adaptations may contribute to unique facets of opioid addiction.
Keywords: addiction, opioids, prefrontal cortex, nucleus accumbens, glutamate plasticity
Graphical abstract
INTRODUCTION
Opioid-based drugs are mainstays for clinical pain management, however the misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare [1–3]. Increasing evidence suggest that neural circuit adaptations responsible for withdrawal that accompany dependence are not necessarily synonymous with those responsible for establishing drug seeking behavior, creation of relapse-driving cue/context associations and impaired control over drug intake – highlighting a need to better understand how specific aspects of opioid plasticity uniquely contribute to opioid abuse. The medial prefrontal cortex (mPFC) and nucleus accumbens (NAc) are two brain regions that are critically involved in reward, motivated behavior, and cognitive control where drugs of abuse are believed to promote pathological plasticity that leads to enduring behavioral abnormalities associated with addiction [4–10], however our understanding of this plasticity in relation to opioid abuse remains surprisingly absent. The purpose of this review is to discuss what is known and highlight important knowledge gaps regarding opioid-induced plasticity in the mPFC and NAc using primarily electrophysiology data from preclinical models, and explore potential relevance of this plasticity to unique facets of opioid addiction (e.g., reward, drug-seeking, and dependence).
MEDIAL PREFRONTAL CORTEX
Deep layer glutamatergic pyramidal neurons within the more dorsal prelimbic (PrL) and ventral infralimbic (IL) regions of the mPFC provide widespread glutamatergic input to cortical and subcortical structures [11–14]. Differences in the anatomical connectivity of these regions contribute to their dissociable (often opposing) roles in regulating limbic and cognitive functions, thus regional adaptations may have distinct implications for drug seeking behavior and impaired control of drug intake [5, 10, 15–20]. In addition to anatomical connectivity, recent evidence indicates that these neurons can be further divided into subpopulations based on morphology, molecular and physiological profile – including the expression of D1-versus D2-type dopamine receptors – as well as what type of excitatory or inhibitory input they receive [21–27]. As these sub-populations likely provide an anatomical frame work to regulate complex behavioral control and define how they undergo experience induced plasticity, a critical step towards understanding how addiction alters PFC networks will be to identify cell type and pathway-specific adaptations [26–29].
Opioid effects on mPFC pyramidal neuron activity
Preclinical studies have indeed identified a role for mPFC glutamate input to regions such as the NAc and ventral tegmental area (VTA), as well as dopaminergic neurotransmission in the PFC in numerous facets of opioid addiction, including acute reward [30–38], relapse related drug seeking following abstinence and extinction [39–43], precipitated somatic withdrawal [44], and opioid addiction memory formation [45]. Studies examining the acute effects of opioids on pyramidal neuron activity are relatively few in number, however in vivo work indicates that morphine attenuates the excitatory response of pyramidal neurons to locally applied and afferent evoked glutamate [31]. Notably, effects of morphine on transmitter release were more pronounced at mediodorsal thalamus afferents compared to input arising from the baslolateral amygdala (BLA) and hippocampus (HPC) -- highlighting the possibility of pathway- or cell-type specific regulation by mu opioid receptors [31, 46]. Moreover, these findings are in apparent contrast to in vivo studies with other drug classes (e.g., cocaine, nicotine, ethanol) where acute exposure produced a transient depolarization and subsequent activation of mPFC layer 5/6 putative pyramidal neurons [47–50].
Following more prolonged opioid exposure, reduced metabolic activity within frontal cortical regions of heroin addicts is thought to reflect impaired functionality that contributes reduced control over limiting drug intake [5, 10, 51] as well as affective withdrawal as it correlated with anhedonia in opioid dependent subjects [52]. Alternatively, drug “craving” in opioid users in response to viewing drug-associated cues is correlated with increased activation of frontal cortex regions [41, 53–58]. This dichotomy is reinforced by clinical imaging studies in cocaine addicts [53, 59] and preclinical findings in cocaine self-administering rats that exhibit a progressive reduction in basal activity of putative pyramidal neurons in the mPFC but increased activity in response to cocaine- and cocaine-related cues [58]. Similarly, exposure to environmental cues linked morphine reward is strongly associated with distinct activity in select neuron populations in the PrLC [60], and reinstatement of heroin-seeking increases neuronal markers of activity such as c-fos and zif268 [61–63].
These observations suggest that PFC neurons may undergo unique forms of plasticity that permit them to exhibit a hypo- and hyperactive state following prolonged drug use (Figure 1). In vitro electrophysiology studies in acute slices has shown that repeated, but not acute systemic cocaine exposure increases intrinsic membrane excitability in PrL, but not IL, deep layer pyramidal neurons that persists up to 45 days post drug exposure [64–68]. With more prolonged use, PrL pyramidal neurons show a reduction in intrinsic membrane excitability that is more pronounced in in aversion-resistant rats thought to reflect compulsive behavior, and hat restoring this hypoexcitability using optical stimulation of pyramidal neurons alleviates uncontrolled cocaine intake [69]. Presently, no published findings on the effect of repeated opioid exposure on mPFC pyramidal neuron excitability exist, however reductions (cell non-specific) in the activity-regulated gene, c-fos, have been reported in the orbitofrontal cortex following a two-week chronic escalating-dose regimen of non-contingent morphine (Piao et al., 2017). Further, unpublished findings from our lab have recently observed that following a 14–21 d abstinence period from two weeks of daily remifentanil self-administration, pyramidal neurons in the PrL show increased excitability and spike firing, and that these changes extend to the IL in a specific population of neurons (Anderson et al., unpublished observations). If the duration of self-administration is extended to 30–45 d, PrL neurons are less excitability (Anderson et al., unpublished observation) but exhibit an increased firing capacity at more depolarized potentials – adaptations that could significantly impact cortical information process and thus function.
Figure 1. Time- and sex-dependent adaptations in PFC pyramidal neuron physiology and underlying mechanisms.

Data indicate that in males, short-term exposure to opiods increases the intrinsic excitability PrL pyramidal neurons, in part through a downregulation in metabotropic inhibitory signaling mediated by GABABR-Girk transmission (A-B). Following prolonged exposure, the activation threshold is increased (reduced excitability) in males – a phenomenon that occurs after only two weeks of self-administration in females (A-B). Hypoexcitable states appear to reflect reductions in synaptic AMPAR transmission as well as increased GABAA transmission and coincides with an increase in action potential frequency at more depolarized potentials (B) indicating that while these neurons are more difficult to initiate firing, once activated, mechanisms normally in place to restore firing to basal levels is impaired. Although the function implications are unclear, hypoactivation states may contribute to an impaired ability of the mPFC to exert inhibitory control, that may be further exacerbated by increased (disorganized) firing that disrupts cortical information processing or may contribute to the pathological strengthening of drug-seeking/taking behavior in response to drug-associated cues.
Intrinsic and synaptic plasticity
Studies to date have identified alterations in the expression and function of numerous voltage-gated and G protein-coupled ion channel conductance as contributing factors underlying increased excitability and neuronal firing following cocaine exposure, however almost nothing is known regarding intrinsic factors (non-synaptic factors) underlying opioid-induced changes in pyramidal neuron physiology [6, 64–66, 70–77]. Our preliminary findings suggest that similar to repeated cocaine, reductions in the threshold to fire following remifentanil selfadministration align with reductions in metabotropic inhibitory signaling mediated by GABABR-dependent activation of G protein inwardly-rectifying potassium (Girk) channels in Layer 5/6 pyramidal neurons of the PrL region (Hearing et al., 2013; Anderson et al., unpublished observation). However, unlike cocaine, these effects are no longer present following 45 d abstinence – a temporal distinction that aligns with previous findings that incubating effects of cocaine on “craving” and drug-seeking persisted as long as 90 days, this phenomenon peaks at ~12 d post returned to control levels by 60 d following heroin self-administration [78–81].
A number of studies have also provided evidence for enduring changes in glutamate and GABA synaptic transmission following drug exposure. For example, cocaine increases mPFC expression of NMDA and AMPA receptor protein, while chronic alcohol upregulates postsynaptic NMDAR-mediated in layer 5/6 pyramidal cells – the latter of which was associated with elevations in spike-timing dependent plasticity [82–85]. Although opioidinduced glutamate plasticity in the mPFC has been reported, the nature of these adaptations and their functional role in opioid addiction are less clear. Electrophysiology studies indicated that excitatory synaptic strength as measured by changes in the ratio of AMPA-to-NMDA receptor currents in pyramidal neuron is not altered following withdrawal from heroin self-administration (but see [86]), however this may reflect a lack of distinguishing neurons localized to the PrL versus IL and/or plasticity produced by extinction/reinstatement in this study [87]. Alternatively, this same study indicated that short-term glutamate plasticity (in the IL) in the form of AMPAR endocytosis plays a role in reinstatement of drug-seeking [87].
During early withdrawal from an intermediate period of cocaine self-administration, reductions in GABA transmission mediated by the ionotropic, GABAA receptor, appear to contribute to augmented induction of longterm potentiation at PFC pyramidal neurons, however whether similar adaptations occur following opioid selfadministration is unclear [67, 76]. These modifications, in conjunction with observed increases in excitability of mPFC pyramidal neurons, suggest that a shift in excitation at PFC output neurons may drive initial learning of drug seeking behavior as well as relapse during early opioid use. On the other hand, published work as well as our unpublished findings indicate that hypoexcitability of pyramidal neurons may instead reflect adaptations in synaptic GABAAR transmission rather than GABAB. In support of this, increased GABAergic inhibition of PrL pyramidal neurons has been shown to facilitate relapse to heroin-seeking [87], while increases in the frequency of IPSCs were found following remifentanil self-administration (Anderson et al., unpublished findings).
Presently, almost nothing is known regarding how opioids alter plasticity in subpopulations of pyramidal neurons based on their anatomical connectivity and physiological phenotype. Effects of opioids on GABAergic interneurons have also not been directly measured, however recent optogenetic manipulation of parvalbumin expressing interneurons indicates that these neurons play a critical role in cognitive function [88, 89] as well as extinction of reward seeking [90]. As cortical information processing and thus function relies on a balance of excitation:inhibition across multiple neuron types, identifying cell-specific modifications will be an important goal moving forward.
Behavioral and clinical implications of PFC plasticity
The functional implications of the co-occurring increased responsivity to drug-associated cues and reduced basal activation of the mPFC remains a vital question towards understanding how drugs of abuse impair cognitive control. One possibility is that during early drug use, increased excitability serves to lower the threshold for induction of long-lasting plasticity in PFC circuits (i.e., mPFC-to-NAc) important for learning drug seeking and drug taking behaviors or developing drug-cue associations responsible for precipitating craving and subsequent relapse. As drug-cues retain the ability to precipitate craving and activate the PFC, functionality of PrL circuits related to motivation and initiation of drug-seeking may be maintained, with reductions in basal metabolic activity serving to allow only drug-associated cues to ably activate PFC networks and thus maintain control over behavior [58, 91].
Emergence of compulsive and uncontrollable opioid use likely reflects impaired function of the numerous frontal cortical regions as well as increasing activation of neural networks responsible for habit-like behavior [69, 92–97]. Clinical findings indicate that heroin addicts exhibit attention bias in drug-associated contexts and often display increased impulsivity, cognitive inflexibility, and impaired attention [98, 99] that are strong predictors of heroin abuse and relapse [100–102]. Although impulsivity and inflexibility are not synonymous with compulsivity or habit, impulsiveness – either intrinsic or drug-induced – appears to increase vulnerability toward compulsion and habit and may ultimately lead to substance abuse [4, 103, 104]. Moreover, perseverative responding despite its adverse consequences (cognitive inflexibility) is a significant predictor of poorer cocaine addiction treatment outcomes [105].
Preclinical studies in rodents have highlighted the PrL as a critical substrate for cognitive flexibility and attention [106–113]. Recent findings showed reduced excitability of PrL pyramidal neurons in compulsive cocaine-seeking rats, which could be rescued by in vivo optogenetic stimulation of this region [69], thus the observed hypofunction of the PrL pyramidal neurons following remifentanil may contribute to perseverative drug seeking. This is substantiated by impaired performance in mice exhibiting hypo-, but not hyperfrontal states (Anderson et al., unpublished observation). Alternatively, impulsive behavior appears to align more readily with altered functionality of the more ventral IL aspect of the mPFC as well as the OFC [114, 115]. As our unpublished observations indicate that adaptations in IL pyramidal neuron excitability are likely pathway-specific, exploring how opioids alter ILC and OFC-associated circuits will likely provide additional insight into how frontal cortical dysfunction contributes to opioid abuse.
Increasing evidence indicates that subpopulations of cortical pyramidal neurons exhibit distinctions in anatomical connectivity, molecular and physiological profile (i.e., expression of dopamine D1 vs. D2-receptors), and excitatory/inhibitory innervation that define how they undergo experience-induced plasticity and provide an anatomical frame work to regulate complex behavior related to cognitive control and reward [21, 22, 27, 28]. Given this framework, it is plausible specific PFC circuits are activated by opioids and associated stimuli, whereas concomitant impairments in other circuits – perhaps those exhibiting hypoactivation – contribute to opioid-related deficits in cognitive control and perpetuate a shift in the balance of behavioral control to more dorsal striatal (i.e., habit) circuits following cue activation
NUCLEUS ACCUMBENS
The NAc is a heterogenous structure that can be divided into a core and shell region based on anatomical connectivity and presumptive role in reward-related behavior [116–122]. GABAergic medium-spiny neurons (MSNs) make up a majority of all neurons in the NAc and receive coordinated input from glutamatergic afferents arising from cortical and limbic brain regions that elicit physiological and behavioral responses [123–126]. Canonically divided into two main subpopulations based on D1- or D2-type dopamine receptor expression and efferent regulation of basal ganglia output structures [127–132], these two subtypes display unique intrinsic physiology, differentially undergo drug-induced plasticity, and play dissociable (often opposing) roles in drug-reward-related behavior [126, 133–138].
Intrinsic MSN physiology
Activity of NAc MSNs is dependent on synaptic input as well as intrinsic membrane properties that directly influence the probability and pattern of neuronal firing (i.e. excitability). These intrinsic properties are dependent on factors extrinsic to the synapse, such as expression and function of voltage-gated and G protein-coupled channels and are known to be altered by drugs of abuse [6, 70–75], but often take a backseat to research focusing on adaptations in transmission mediated by synaptic receptors.
At present, relatively little is known regarding the effect of opioids on intrinsic physiology. Following 10 d of abstinence from repeated non-contingent morphine increase firing capacity of MSNs [74, 75]. While these studies did not identify MSN subpopulations based on dopamine receptor expression, morphine plasticity was examined based on membrane properties, i.e., the intrinsic excitability and spike adaptation. Morphine increased the initial firing frequency and decreased the train duration in “type I” MSNs that show high levels of intrinsic excitability basally, without altering action potential threshold (rheobase). Alternatively, morphine reduced rheobase (increased excitability) of “type II” MSNs, without altering firing frequency or train duration [74]. In contrast to the shell, repeated morphine suppressed excitability in unidentified NAc core MSNs [71]. Although the exact mechanisms underlying morphine adaptations based on neuron sub-type are unclear, increases in NAc shell MSN excitability is due in part to increased activation of extrasynaptic NMDA receptors which in turn inhibits sustained voltage-gated potassium currents, reduced excitability in the core aligns with upregulation of inwardly-rectifying potassium channels [71, 74, 139].
Interpreting the relevance of changes in the NAc core versus shell is challenging given how little is known about how the overall MSN firing is affected by the interplay between excitatory synaptic inputs projecting to MSNs (which are enhanced following long-term withdrawal of both cocaine or morphine) and MSN intrinsic excitability. Furthermore, it is unclear how alterations in MSN intrinsic activity and their inhibitory regulation of projection sites during long-term withdrawal regulate drug-seeking behaviors. Reduced intrinsic excitability of MSNs likely leaves these neurons less responsive to activation by excitatory drive, whereby only increased glutamate release driven by exposure to drug-related stimuli is sufficient to overcome this dampening of activity – thus increasing signal-to-noise ratio for drug-associated stimuli and reducing it for other stimuli [140].
Notably, intrinsic changes following morphine exposure also appear to directly contrast psychostimulant-induced adaptations. For example, in the shell, repeated non-contingent cocaine or amphetamine exposure increased various K+ currents while decreasing background Na+ and voltage-gated Ca2+ currents mediated by N− and R−type channels in unidentified MSNs that translated into suppression of MSN firing capacity in acute brain slices 1–21 d following the final drug exposure [73, 141–143]. MSNs in the NAc core on the other hand exhibit increased excitability during acute psychostimulant withdrawal, that unlike the shell, returns to control levels by day 10 of withdrawal [73]. The drug-specific discrepancies on MSN intrinsic excitability are intriguing to say the least. Particularly difficult to interpret are the opposing effects in the NAc core given its role in the in the motor activating effects of abused drugs, and that cocaine and morphine doses used in these experiments both produce prominent behavioral sensitization. However, these results appear to align with in vivo studies have previously shown differential responses in single-unit recordings during cocaine and heroin self-administration [144]. Future work using transgenic mice will be of value to address critical questions, including how these properties align based on the expression of D1 or D2Rs and how changes in excitability contribute to regulation of drug-seeking behavior.
Opioid reward and relapse-related glutamate plasticity
Ample evidence has shown that glutamate transmission at NAc MSNs contributes to opioid reward, withdrawal, and relapse [8, 9, 39, 40, 43, 108, 139, 145–152]. However neuronal “interpretation” of this glutamate release is complex, as it depends on the afferent, MSN subtype (i.e., D1- vs. D2-MSN), and efferent connectivity (i.e., VP or SN) [130, 153–156]. This complexity combined with the fact that drug-induced modifications in NAc glutamate transmission often involve a postsynaptic receptor number and/or conductance [157], presynaptic release mechanisms, and glutamate clearance mechanisms, represents a major challenge towards identifying the nature and locus of opioid-induced plasticity and how specific neural circuit modifications uniquely contribute to aspects of opioid addiction (dependence versus relapse). Below we will highlight what is known regarding changes in glutamate plasticity within the NAc at both the pre- and postsynaptic level and what circuits these modifications manifest.
AMPA- and NMDA-type ionotropic glutamate receptors are the primary mediators of rapid glutamate transmission and exert a profound influence on synaptic plasticity and cellular physiology [158]. Changes in the conductance, expression, and localization (e.g., synaptic vs. perisynaptic) of these receptors plays a key role in drug-induced and experience-related plasticity -- making these receptors a key target for studying how opioids promote persistent addicted behavior and development of future pharmacotherapies. Recent electrophysiological findings indicate that repeated non-contingent morphine increases excitatory drive at NAc shell D1-MSNs, while reduced it at D2-MSNs. The increase in synaptic strength reflects upregulation of AMPAtype receptor signaling through insertion of GluA2-lacking calcium permeable AMPARs as well as enhanced glutamate release probability [75, 134, 135]. Alternatively, weakened D2-MSN signaling reflects a reduction in presynaptic release and a time-dependent loss of AMPAR expression and dendritic spines [134, 135]. Similar to cocaine this augmented synaptic strength is not immediately present, as no changes in glutamate receptor signaling were observed 12 h following the final exposure – highlighting a need for time to develop [134–136, 149].
Unlike cocaine, increases in synaptic strength did not extend to MSNs in the NAc core [134], however recent unpublished observations indicate that AMPAR transmission (mEPSC frequency and amplitude) is increased at D1-MSNs in the core and shell following 10–14 d of withdrawal from remifentanil self-administration (Madayag et al., unpublished observations).
Dependence/Withdrawal Glutamate Plasticity
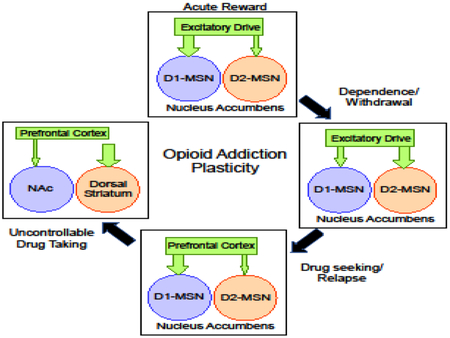
The enduring nature of adaptations within the mPFC-NAc pathway indicate that these changes likely contribute to strengthening of drug-seeking behavior and subsequent relapse in response drug-associated stimuli (Figure 2, top). However, evidence indicates that upregulation of NAc AMPAR signaling also plays a critical role in the development of dependence and expression of withdrawal symptoms. For example, chronic morphine exposure elevated GluA2-lacking AMPAR surface expression in the NAc and intra-shell infusion of a GluA2-lacking selective antagonist blocked naloxone-precipitated affective, but not somatic, withdrawal [152] [152]. Electrophysiology studies following repeated systemic morphine indicate that this upregulation may occur selectively at PVT-to-NAc shell D2-MSN synapses, and contributes to establishing withdrawal-related aversive memories [159]. Given that morphine withdrawal increases extracellular glutamate levels in the NAc (Sepulveda et al., 1998) and appears to increase release probability at D1- and D2-MSNs (pooled afferents) (Madayag et al., unpublished observation), it is possible that upregulation of excitatory signaling at D1-MSNs contribute to conditioned reward and relapse similar to those observed following intermittent non-contingent morphine [134, 135] whereas “priming” of D2-MSNs via increased AMPAR receptor activation is responsible for withdrawalrelated symptoms, with specific pathways contributing to unique aspects of this withdrawal (i.e., negative affect vs. somatic; Figure 2, bottom) that may lead individuals to increase use in attempt to mitigate these negative symptoms.
Figure 2. Proposed role of known opioid adaptations in nucleus accumbens glutamate transmission in opioid addiction.

Repeated exposure to opioids promotes a number of pre- and postsynaptic modifications in excitatory currents, most of which appears to be mediated by the ionotropic AMPA-type receptors. (Top) Enduring adaptations at mPFC-to-NAc synapses aligning with plasticity associated with establishing learned drug associations, drug-seeking/taking behavior, and relapse. Modifications include increased glutamate release probability and insertion of AMPARs that are permeable (GluA1/GluA1; blue-blue) or impermeable (GluA1/GluA2; blue-green) at PrL-to-Core (light blue) and IL-to-Shell (dark blue) synapses on D1-MSNs (red) as well as reduced excitatory drive and increased synapse numbers in D2-MSNs (green). These cell-specific adaptations promote an overall shift in excitatory drive within D1- and D2-MSN circuits that favors strengthening of D1-MSN pathways. (Bottom) The cellular, temporal, and mechanistic nuances of NAc glutamate plasticity contributing to dependence and withdrawal are less clear. Morphine withdrawal is associated with increased extracellular glutamate, which may reflect increased release probability or impaired clearance, or both. It is possible that upregulation of excitatory signaling plays distinct roles depending on the MSN sub-type, with increased signaling at D1-MSNs reflecting changes related to reward/relapse whereas signaling at D2-MSNs is responsible for withdrawal-related symptoms, with specific pathways contributing to unique aspects of this withdrawal (i.e., negative affect vs. somatic).
Pathway-specific plasticity
Increasing evidence indicates that the nature and locus of opioid-induced plasticity in the NAc indicates that this plasticity dictates the relationship to behavior. Initial examination of plasticity within mPFC-to-NAc circuits using optogenetics has shown that IL-to-Shell synapses are strengthened by increased insertion of GluA2-lacking receptors selectively at D1R-MSN synapses following 10–14 withdrawal from repeated non-contingent morphine exposure [99]. Preliminary findings from our lab have shown that similar adaptations occur at D1-MSN IL-to-Shell synapses following abstinence from remifentanil self-administration (Madayag et al., unpublished observations). While the functional implications of this plasticity following opioid self-administration are unclear, our previous work has shown that reversal of morphine-induced pathophysiology using in vivo optogenetic stimulation (10 Hz) of IL-to-Shell inputs disrupts reinstatement of morphine-evoked conditioned place preference [134]. Information regarding adaptations within the PrL-to-Core pathway is essentially non-existent, however recent examination of this pathway following remifentanil self-administration showed an increased presence of synaptic GluA2-lacking AMPAR and release probability at D1-MSNs and reduced excitatory drive at D2-MSNs, the latter of which may reflect adaptations in at the post and presynaptic level (Madayag et al., unpublished observation).
It is tempting to speculate that the emergence of plasticity in the more motor-centric core region of the NAc reflects the use of an operant- and goal-directed based model of opioid self-administration, as non-contingent exposure did not alter AMPAR transmission (or release probability) following non-contingent morphine, however it is also possible that enduring plasticity in the NAc core is primarily expressed at synapses receiving significant innervation from PrL afferents [134]. Regardless, the functional role of this plasticity requires further exploration, as work to date has primarily highlighted a role for the IL-to-Shell pathway in opioid relapse [39, 40, 148, 151, 160]. Based on previous circuit lesion studies [39, 40, 148, 160] it is possible that plasticity within the PrL-Core circuit is responsible for maintaining drug- and drug-cue associations responsible for driving relapse-related behavior, whereas context-based drug-associations are retained by plasticity in the IL-Shell circuit and that these adaptations work in concert to maintain relapse vulnerability.
Examination of circuit plasticity outside of cortico-accumbens circuits indicates that glutamate release probability is elevated at BLA- but not PrL-to-Core MSNs as early as 2 hours following an escalating regimen of morphine over 5 days [161]. Alternatively, while release probability is unchanged at BLA- and PVT-to-Shell MSNs within the first 24 h of repeated non-contingent morphine, there is an upregulation of GluA2-lacking AMPARs at PVT-to-Shell inputs selectively on D2-MSN that when reversed suppressed somatic withdrawal and aversive memories [159]. Taken together, these data highlight an intriguing possibility that enhanced transmission at thalamic transmission at D2-MSNs promote dependence/withdrawal-related negative affective states that drive continued use, whereas developing adaptations within PFC-to-NAc circuits that produce a shift in the ratio between excitatory synaptic drive at D1R- over D2R-MSN synapses represent enduring pathophysiology that drives relapse. Regardless, these data highlight a need to better understand the temporal, state-dependent, and cell-specific dynamics of opioid-induced pre- and postsynaptic plasticity, as it will provide better insight into how each may contribute to unique aspects of behavior (i.e., reward and relapse versus dependence and withdrawal).
Intrinsic and extrinsic factors influencing opioid abuse
A wealth of clinical data indicates that environmental and genetic factors contribute to an individuals susceptibility to addiction. Although outside the scope of this review (see Mistry et al., 2014 for a review), heritability of opioid abuse has been linked into part to polymorphisms in the genes encoding for dopamine d2 receptors, mu and delta opioid receptors, as well as neurotrophic and growth-related factors such as BDNF and NGF (Mistry et al., 2014). Although the co-occurrence of prolonged environmental and psychological stressors has also been linked to increased risk for opioid abuse, a critical unresolved question is how stress influences opioid use and what specific opioid- and stress-induced adaptations in cellular physiology and synaptic plasticity lead to these maladaptive behaviors. Moreover sex differences in drug use and stress responsivity is evident in humans and animal models, however a vast majority of work continues to focus primarily on males -- leaving major gaps regarding the influence of sex on drug- and stress-induced neural plasticity and behavior [162–164]. In susceptible populations, women escalate cocaine use more rapidly and show a greater tendency to relapse and experience a loss of voluntary control of intake [165, 166].
Despite a 100% increase in the number of women using heroin in recent years [167, 168], data on sex differences in opioid use are relatively lacking. Human studies have shown that women report greater craving and exhibit increased vulnerability to relapse than men [168, 169], however the underlying mechanism for this sex difference are unknown. Our preliminary findings indicate that hypoexcitability states in mPFC pyramidal neurons emerges following only 14 d of self-administration and that these changes may have significant implications for cognitive function and relapse. An important unanswered question is whether this reflects a leftward temporal shift whereby females exhibit hyperexcitability following a shorter period of drug administration (e.g., 5 days), or if this shift occurs due to intrinsic differences in pyramidal excitability (Figure 1A) across sexes [170]. Moreover, it remains unclear whether hypofrontal states reflect a shift in the corticostriatal circuits driving behavior, such as those regulating habit behavior, or if mPFC-NAc circuits are still required for initiation of this behavior.
Although difficult to causally disentangle multiple variables, identifying the locus, and temporal dynamics of corticostriatal plasticity produced by opioids and stress as well as relevant sex-specific and genetic-based differences that contribute to this plasticity and impact later opioid use in susceptible populations, will advance our understanding of how these modifications uniquely contribute to behavioral deficits in addiction and neuropsychiatric disease and aide development of more targeted and impactful therapies to treat opioid use disorders.
Acknowledgments
Relevant Funding Source: R00-DA038706–01 (NIDA)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Florence CS, et al. , The Economic Burden of Prescription Opioid Overdose, Abuse, and Dependence in the United States, 2013. Med Care, 2016. 54(10): p. 901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirson NY, et al. , The Economic Burden of Opioid Abuse: Updated Findings. J Manag Care Spec Pharm, 2017. 23(4): p. 427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scarpati LM, et al. , Opioid Abuse: A Detailed Examination of Cost Drivers over a 24-Month Follow-up Period. J Manag Care Spec Pharm, 2017. 23(11): p. 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everitt BJ and Robbins TW, Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci, 2005. 8(11): p. 1481–9. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein RZ and Volkow ND, Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci, 2011. 12(11): p. 652–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hearing MC, Zink AN, and Wickman K, Cocaine-induced adaptations in metabotropic inhibitory signaling in the mesocorticolimbic system. Rev Neurosci, 2012. 23(4): p. 325–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalivas PW and Volkow ND, The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry, 2005. 162(8): p. 1403–13. [DOI] [PubMed] [Google Scholar]
- 8.Peters J, Pattij T, and De Vries TJ, Targeting cocaine versus heroin memories: divergent roles within ventromedial prefrontal cortex. Trends Pharmacol Sci, 2013. 34(12): p. 689–95. [DOI] [PubMed] [Google Scholar]
- 9.Peters J, et al. , Opposing roles for the ventral prefrontal cortex and the basolateral amygdala on the spontaneous recovery of cocaine-seeking in rats. Psychopharmacology (Berl), 2008. 197(2): p. 319–26. [DOI] [PubMed] [Google Scholar]
- 10.Van den Oever MC, et al. , Prefrontal cortex plasticity mechanisms in drug seeking and relapse. Neurosci Biobehav Rev, 2010. 35(2): p. 276–84. [DOI] [PubMed] [Google Scholar]
- 11.Sesack SR, et al. , Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol, 1989. 290(2): p. 213–42. [DOI] [PubMed] [Google Scholar]
- 12.Sesack SR and Pickel VM, Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol, 1992. 320(2): p. 145–60. [DOI] [PubMed] [Google Scholar]
- 13.Taber MT, Das S, and Fibiger HC, Cortical regulation of subcortical dopamine release: mediation via the ventral tegmental area. J Neurochem, 1995. 65(3): p. 1407–10. [DOI] [PubMed] [Google Scholar]
- 14.Taber MT and Fibiger HC, Electrical stimulation of the prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. J Neurosci, 1995. 15(5 Pt 2): p. 3896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Killcross S and Coutureau E, Coordination of actions and habits in the medial prefrontal cortex of rats. Cereb Cortex, 2003. 13(4): p. 400–8. [DOI] [PubMed] [Google Scholar]
- 16.Sierra-Mercado D, Padilla-Coreano N, and Quirk GJ, Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology, 2011. 36(2): p. 529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vertes RP, Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse, 2004. 51(1): p. 32–58. [DOI] [PubMed] [Google Scholar]
- 18.Hoover WB and Vertes RP, Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct, 2007. 212(2): p. 149–79. [DOI] [PubMed] [Google Scholar]
- 19.Miller EK and Cohen JD, An integrative theory of prefrontal cortex function. Annu Rev Neurosci, 2001. 24: p. 167–202. [DOI] [PubMed] [Google Scholar]
- 20.O’Doherty JP, Contributions of the ventromedial prefrontal cortex to goal-directed action selection. Ann N Y Acad Sci, 2011. 1239: p. 118–29. [DOI] [PubMed] [Google Scholar]
- 21.Gee S, et al. , Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci, 2012. 32(14): p. 4959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santana N, Mengod G, and Artigas F, Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex, 2009. 19(4): p. 849–60. [DOI] [PubMed] [Google Scholar]
- 23.Gaspar P, Bloch B, and Le Moine C, D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci, 1995. 7(5): p. 1050–63. [DOI] [PubMed] [Google Scholar]
- 24.Vincent SL, Khan Y, and Benes FM, Cellular distribution of dopamine D1 and D2 receptors in rat medial prefrontal cortex. J Neurosci, 1993. 13(6): p. 2551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dembrow NC, Chitwood RA, and Johnston D, Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci, 2010. 30(50): p. 16922–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee AT, et al. , Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron, 2014. 81(1): p. 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seong HJ and Carter AG, D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci, 2012. 32(31): p. 10516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenni NL, Larkin JD, and Floresco SB, Prefrontal Dopamine D1 and D2 Receptors Regulate Dissociable Aspects of Decision Making via Distinct Ventral Striatal and Amygdalar Circuits. J Neurosci, 2017. 37(26): p. 6200–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Land BB, et al. , Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci, 2014. 17(2): p. 248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, et al. , Morphine disinhibits glutamatergic input to VTA dopamine neurons and promotes dopamine neuron excitation. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giacchino JL and Henriksen SJ, Opioid effects on activation of neurons in the medial prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry, 1998. 22(7): p. 1157–78. [DOI] [PubMed] [Google Scholar]
- 32.Heijna MH, et al. , Opioid receptor-mediated inhibition of dopamine and acetylcholine release from slices of rat nucleus accumbens, olfactory tubercle and frontal cortex. Eur J Pharmacol, 1990. 181(3): p. 267–78. [DOI] [PubMed] [Google Scholar]
- 33.Rossetti ZL, et al. , Extraneuronal noradrenaline in the prefrontal cortex of morphine-dependent rats: tolerance and withdrawal mechanisms. Brain Res, 1993. 609(1–2): p. 316–20. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka E and North RA, Opioid actions on rat anterior cingulate cortex neurons in vitro. J Neurosci, 1994. 14(3 Pt 1): p. 1106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vezina P, et al. , Nicotine and morphine differentially activate brain dopamine in prefrontocortical and subcortical terminal fields: effects of acute and repeated injections. J Pharmacol Exp Ther, 1992. 261(2): p. 48490. [PubMed] [Google Scholar]
- 36.Williams JT and Zieglgansberger W, Neurons in the frontal cortex of the rat carry multiple opiate receptors. Brain Res, 1981. 226(1–2): p. 304–8. [DOI] [PubMed] [Google Scholar]
- 37.Wood PL and Rao TS, Morphine stimulation of mesolimbic and mesocortical but not nigrostriatal dopamine release in the rat as reflected by changes in 3-methoxytyramine levels. Neuropharmacology, 1991. 30(4): p. 399401. [DOI] [PubMed] [Google Scholar]
- 38.Yuan K, et al. , Morphine treatment enhances glutamatergic input onto neurons of the nucleus accumbens via both disinhibitory and stimulating effect. Addict Biol, 2017. 22(6): p. 1756–1767. [DOI] [PubMed] [Google Scholar]
- 39.Bossert JM, et al. , Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci, 2011. 14(4): p. 420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bossert JM, et al. , Role of projections from ventral medial prefrontal cortex to nucleus accumbens shell in context-induced reinstatement of heroin seeking. J Neurosci, 2012. 32(14): p. 4982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daglish MR, et al. , Changes in regional cerebral blood flow elicited by craving memories in abstinent opiatedependent subjects. Am J Psychiatry, 2001. 158(10): p. 1680–6. [DOI] [PubMed] [Google Scholar]
- 42.Glick SD and Cox RD, Changes in morphine self-administration after tel-diencephalic lesions in rats. Psychopharmacology (Berl), 1978. 57(3): p. 283–8. [DOI] [PubMed] [Google Scholar]
- 43.Rogers JL, Ghee S, and See RE, The neural circuitry underlying reinstatement of heroin-seeking behavior in an animal model of relapse. Neuroscience, 2008. 151(2): p. 579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bassareo V, Tanda G, and Di Chiara G, Increase of extracellular dopamine in the medial prefrontal cortex during spontaneous and naloxone-precipitated opiate abstinence. Psychopharmacology (Berl), 1995. 122(2): p. 202–5. [DOI] [PubMed] [Google Scholar]
- 45.Rosen LG, et al. , Molecular and neuronal plasticity mechanisms in the amygdala-prefrontal cortical circuit: implications for opiate addiction memory formation. Front Neurosci, 2015. 9: p. 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.James AS, et al. , Opioid self-administration results in cell-type specific adaptations of striatal medium spiny neurons. Behav Brain Res, 2013. 256: p. 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trantham-Davidson H, et al. , Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci, 2012. 32(36): p. 12406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodward JJ and Pava MJ, Effects of ethanol on persistent activity and up-States in excitatory and inhibitory neurons in prefrontal cortex. Alcohol Clin Exp Res, 2009. 33(12): p. 2134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein EA, et al. , Nicotine-induced limbic cortical activation in the human brain: a functional MRI study. Am J Psychiatry, 1998. 155(8): p. 1009–15. [DOI] [PubMed] [Google Scholar]
- 50.Lambe EK, Picciotto MR, and Aghajanian GK, Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology, 2003. 28(2): p. 216–25. [DOI] [PubMed] [Google Scholar]
- 51.Botelho MF, et al. , Brain blood flow SPET imaging in heroin abusers. Ann N Y Acad Sci, 2006. 1074: p. 466–77. [DOI] [PubMed] [Google Scholar]
- 52.Zijlstra F, et al. , Neurobiological substrates of cue-elicited craving and anhedonia in recently abstinent opioiddependent males. Drug Alcohol Depend, 2009. 99(1–3): p. 183–92. [DOI] [PubMed] [Google Scholar]
- 53.Childress AR, et al. , Limbic activation during cue-induced cocaine craving. Am J Psychiatry, 1999. 156(1): p. 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldstein RZ, et al. , Severity of neuropsychological impairment in cocaine and alcohol addiction: association with metabolism in the prefrontal cortex. Neuropsychologia, 2004. 42(11): p. 1447–58. [DOI] [PubMed] [Google Scholar]
- 55.Langleben DD, et al. , Reduced prefrontal and temporal processing and recall of high “sensation value” ads. Neuroimage, 2009. 46(1): p. 219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langleben DD, et al. , Acute effect of methadone maintenance dose on brain FMRI response to heroin-related cues. Am J Psychiatry, 2008. 165(3): p. 390–4. [DOI] [PubMed] [Google Scholar]
- 57.Suh JJ, et al. , Low prefrontal perfusion linked to depression symptoms in methadone-maintained opiatedependent patients. Drug Alcohol Depend, 2009. 99(1–3): p. 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun W and Rebec GV, Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci, 2006. 26(30): p. 8004–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldstein RZ and Volkow ND, Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry, 2002. 159(10): p. 1642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun N, et al. , Acquisition, extinction, and recall of opiate reward memory are signaled by dynamic neuronal activity patterns in the prefrontal cortex. Cereb Cortex, 2011. 21(12): p. 2665–80. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt ED, et al. , Differential involvement of the prelimbic cortex and striatum in conditioned heroin and sucrose seeking following long-term extinction. Eur J Neurosci, 2005. 22(9): p. 2347–56. [DOI] [PubMed] [Google Scholar]
- 62.Shaham Y, et al. , The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl), 2003. 168(1–2): p. 3–20. [DOI] [PubMed] [Google Scholar]
- 63.Shalev U, et al. , Selective induction of c-Fos immunoreactivity in the prelimbic cortex during reinstatement of heroin seeking induced by acute food deprivation in rats. Behav Brain Res, 2003. 145(1–2): p. 79–88. [DOI] [PubMed] [Google Scholar]
- 64.Dong Y, et al. , Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci, 2005. 25(4): p. 936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hearing M, et al. , Repeated cocaine weakens GABA(B)-Girk signaling in layer 5/6 pyramidal neurons in the prelimbic cortex. Neuron, 2013. 80(1): p. 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nasif FJ, Hu XT, and White FJ, Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci, 2005. 25(14): p. 3674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang CC, Lin HJ, and Hsu KS, Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex, 2007. 17(8): p. 1877–88. [DOI] [PubMed] [Google Scholar]
- 68.Huang CC, et al. , Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci, 2007. 27(11): p. 2958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen BT, et al. , Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature, 2013. 496(7445): p. 359–62. [DOI] [PubMed] [Google Scholar]
- 70.Kourrich S, Calu DJ, and Bonci A, Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci, 2015. 16(3): p. 173–84. [DOI] [PubMed] [Google Scholar]
- 71.Heng LJ, et al. , Repeated morphine exposure decreased the nucleus accumbens excitability during short-term withdrawal. Synapse, 2008. 62(10): p. 775–82. [DOI] [PubMed] [Google Scholar]
- 72.Kourrich S, et al. , Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell, 2013. 152(1–2): p. 236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kourrich S and Thomas MJ, Similar neurons, opposite adaptations: psychostimulant experience differentially alters firing properties in accumbens core versus shell. J Neurosci, 2009. 29(39): p. 12275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu X, et al. , Effects of morphine withdrawal on the membrane properties of medium spiny neurons in the nucleus accumbens shell. Brain Res Bull, 2013. 90: p. 92–9. [DOI] [PubMed] [Google Scholar]
- 75.Wu X, et al. , Potentiation of synaptic strength and intrinsic excitability in the nucleus accumbens after 10 days of morphine withdrawal. J Neurosci Res, 2012. 90(6): p. 1270–83. [DOI] [PubMed] [Google Scholar]
- 76.Lu H, et al. , Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron, 2010. 67(5): p. 821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nasif FJ, et al. , Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther, 2005. 312(3): p. 1305–13. [DOI] [PubMed] [Google Scholar]
- 78.Freeman WM, et al. , Persistent alterations in mesolimbic gene expression with abstinence from cocaine selfadministration. Neuropsychopharmacology, 2008. 33(8): p. 1807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grimm JW, et al. , Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci, 2003. 23(3): p. 742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu L, et al. , Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. Journal of Neurochemistry, 2003. 85(6): p. 16041613. [DOI] [PubMed] [Google Scholar]
- 81.Shalev U, et al. , Time-dependent changes in extinction behavior and stress-induced reinstatement of drug seeking following withdrawal from heroin in rats. Psychopharmacology (Berl), 2001. 156(1): p. 98–107. [DOI] [PubMed] [Google Scholar]
- 82.Ben-Shahar O, et al. , Extended daily access to cocaine results in distinct alterations in Homer 1b/c and NMDA receptor subunit expression within the medial prefrontal cortex. Synapse, 2009. 63(7): p. 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kroener S, et al. , Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One, 2012. 7(5): p. e37541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klenowski PM, et al. , Increased Synaptic Excitation and Abnormal Dendritic Structure of Prefrontal Cortex Layer V Pyramidal Neurons following Prolonged Binge-Like Consumption of Ethanol. eNeuro, 2016. 3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang W, et al. , Alterations in ionotropic glutamate receptor subunits during binge cocaine self-administration and withdrawal in rats. J Neurochem, 2004. 89(4): p. 1021–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mickiewicz AL and Napier TC, Repeated exposure to morphine alters surface expression of AMPA receptors in the rat medial prefrontal cortex. Eur J Neurosci, 2011. 33(2): p. 259–65. [DOI] [PubMed] [Google Scholar]
- 87.Van den Oever MC, et al. , Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci, 2008. 11(9): p. 1053–8. [DOI] [PubMed] [Google Scholar]
- 88.Kim H, et al. , Prefrontal Parvalbumin Neurons in Control of Attention. Cell, 2016. 164(1–2): p. 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riga D, et al. , Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci, 2014. 8: p. 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sparta DR, et al. , Activation of prefrontal cortical parvalbumin interneurons facilitates extinction of rewardseeking behavior. J Neurosci, 2014. 34(10): p. 3699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kalivas PW, The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci, 2009. 10(8): p. 561–72. [DOI] [PubMed] [Google Scholar]
- 92.Jog MS, Building Neural Representations of Habits. Science, 1999. 286(5445): p. 1745–1749. [DOI] [PubMed] [Google Scholar]
- 93.Packard MG and Knowlton BJ, Learning and memory functions of the Basal Ganglia. Annu Rev Neurosci, 2002. 25: p. 563–93. [DOI] [PubMed] [Google Scholar]
- 94.Balleine BW, Delgado MR, and Hikosaka O, The role of the dorsal striatum in reward and decision-making. J Neurosci, 2007. 27(31): p. 8161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Porrino LJ, et al. , The expanding effects of cocaine: studies in a nonhuman primate model of cocaine selfadministration. Neurosci Biobehav Rev, 2004. 27(8): p. 813–20. [DOI] [PubMed] [Google Scholar]
- 96.Porrino LJ, et al. , Cocaine self-administration produces a progressive involvement of limbic, association, and sensorimotor striatal domains. J Neurosci, 2004. 24(14): p. 3554–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kasanetz F, et al. , Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science, 2010. 328(5986): p. 1709–12. [DOI] [PubMed] [Google Scholar]
- 98.Zeng H, et al. , Impulsivity, cognitive function, and their relationship in heroin-dependent individuals. J Clin Exp Neuropsychol, 2013. 35(9): p. 897–905. [DOI] [PubMed] [Google Scholar]
- 99.Lee TM, et al. , Neural activity associated with cognitive regulation in heroin users: A fMRI study. Neurosci Lett, 2005. 382(3): p. 211–6. [DOI] [PubMed] [Google Scholar]
- 100.Jones JD, et al. , The effects of heroin administration and drug cues on impulsivity. J Clin Exp Neuropsychol, 2016. 38(6): p. 709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schippers MC, et al. , Unidirectional relationship between heroin self-administration and impulsive decisionmaking in rats. Psychopharmacology (Berl), 2012. 219(2): p. 443–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao Q, et al. , Neural Correlates of Drug-Related Attentional Bias in Heroin Dependence. Front Hum Neurosci, 2017. 11: p. 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diergaarde L, et al. , Impulsive choice and impulsive action predict vulnerability to distinct stages of nicotine seeking in rats. Biol Psychiatry, 2008. 63(3): p. 301–8. [DOI] [PubMed] [Google Scholar]
- 104.Schoenbaum G and Shaham Y, The role of orbitofrontal cortex in drug addiction: a review of preclinical studies. Biol Psychiatry, 2008. 63(3): p. 256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Turner TH, et al. , Measures of cognitive functioning as predictors of treatment outcome for cocaine dependence. J Subst Abuse Treat, 2009. 37(4): p. 328–34. [DOI] [PubMed] [Google Scholar]
- 106.Di Ciano P and Everitt BJ, Conditioned reinforcing properties of stimuli paired with self-administered cocaine, heroin or sucrose: implications for the persistence of addictive behaviour. Neuropharmacology, 2004. 47 Suppl 1: p. 202–13. [DOI] [PubMed] [Google Scholar]
- 107.Peters J, LaLumiere RT, and Kalivas PW, Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci, 2008. 28(23): p. 6046–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.LaLumiere RT, Smith KC, and Kalivas PW, Neural circuit competition in cocaine-seeking: roles of the infralimbic cortex and nucleus accumbens shell. Eur J Neurosci, 2012. 35(4): p. 614–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalivas PW and McFarland K, Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology (Berl), 2003. 168(1–2): p. 44–56. [DOI] [PubMed] [Google Scholar]
- 110.Stefanik MT and Kalivas PW, Optogenetic dissection of basolateral amygdala projections during cue-induced reinstatement of cocaine seeking. Front Behav Neurosci, 2013. 7: p. 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Birrell JM and Brown VJ, Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci, 2000. 20(11): p. 4320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dias R, Robbins TW, and Roberts AC, Dissociation in prefrontal cortex of affective and attentional shifts. Nature, 1996. 380(6569): p. 69–72. [DOI] [PubMed] [Google Scholar]
- 113.Floresco SB, Block AE, and Tse MT, Inactivation of the medial prefrontal cortex of the rat impairs strategy set-shifting, but not reversal learning, using a novel, automated procedure. Behav Brain Res, 2008. 190(1): p. 8596. [DOI] [PubMed] [Google Scholar]
- 114.Dalley JW, Everitt BJ, and Robbins TW, Impulsivity, compulsivity, and top-down cognitive control. Neuron, 2011. 69(4): p. 680–94. [DOI] [PubMed] [Google Scholar]
- 115.Torregrossa MM, Quinn JJ, and Taylor JR, Impulsivity, compulsivity, and habit: the role of orbitofrontal cortex revisited. Biol Psychiatry, 2008. 63(3): p. 253–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sesack SR and Grace AA, Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology, 2010. 35(1): p. 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shiflett MW and Balleine BW, Molecular substrates of action control in cortico-striatal circuits. Prog Neurobiol, 2011. 95(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Voorn P, et al. , Putting a spin on the dorsal–ventral divide of the striatum. Trends in Neurosciences, 2004. 27(8): p. 468–474. [DOI] [PubMed] [Google Scholar]
- 119.Heimer L, et al. , The accumbens: beyond the core-shell dichotomy. J Neuropsychiatry Clin Neurosci, 1997. 9(3): p. 354–81. [DOI] [PubMed] [Google Scholar]
- 120.Everitt BJ, et al. , Associative processes in addiction and reward. The role of amygdala-ventral striatal subsystems. Ann N Y Acad Sci, 1999. 877: p. 412–38. [DOI] [PubMed] [Google Scholar]
- 121.Heimer L, et al. , Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience, 1991. 41(1): p. 89–125. [DOI] [PubMed] [Google Scholar]
- 122.Zahm DS and Brog JS, On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience, 1992. 50(4): p. 751–67. [DOI] [PubMed] [Google Scholar]
- 123.Silva AJ, et al. , Molecular and cellular approaches to memory allocation in neural circuits. Science, 2009. 326(5951): p. 391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hogarth L, et al. , Associative learning mechanisms underpinning the transition from recreational drug use to addiction. Ann N Y Acad Sci, 2013. 1282: p. 12–24. [DOI] [PubMed] [Google Scholar]
- 125.Goto Y and Grace AA, Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goaldirected behavior. Nat Neurosci, 2005. 8(6): p. 805–12. [DOI] [PubMed] [Google Scholar]
- 126.Pascoli V, et al. , Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature, 2014. 509(7501): p. 459–64. [DOI] [PubMed] [Google Scholar]
- 127.Smith RJ, et al. , Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol, 2013. 23(4): p. 546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Le Moine C and Bloch B, D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol, 1995. 355(3): p. 418–26. [DOI] [PubMed] [Google Scholar]
- 129.Lobo MK, et al. , FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci, 2006. 9(3): p. 443–52. [DOI] [PubMed] [Google Scholar]
- 130.Kupchik YM, et al. , Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci, 2015. 18(9): p. 1230–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thibault D, et al. , Evaluation of D1 and D2 dopamine receptor segregation in the developing striatum using BAC transgenic mice. PLoS One, 2013. 8(7): p. e67219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bertran-Gonzalez J, et al. , Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci, 2008. 28(22): p. 5671–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pascoli V, Turiault M, and Luscher C, Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature, 2011. 481(7379): p. 71–5. [DOI] [PubMed] [Google Scholar]
- 134.Hearing MC, et al. , Reversal of morphine-induced cell-type-specific synaptic plasticity in the nucleus accumbens shell blocks reinstatement. Proc Natl Acad Sci U S A, 2016. 113(3): p. 757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Graziane NM, et al. , Opposing mechanisms mediate morphine- and cocaine-induced generation of silent synapses. Nat Neurosci, 2016. 19(7): p. 915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Terrier J, Luscher C, and Pascoli V, Cell-Type Specific Insertion of GluA2-Lacking AMPARs with Cocaine Exposure Leading to Sensitization, Cue-Induced Seeking, and Incubation of Craving. Neuropsychopharmacology, 2016. 41(7): p. 1779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Matamales M, et al. , Striatal medium-sized spiny neurons: identification by nuclear staining and study of neuronal subpopulations in BAC transgenic mice. PLoS One, 2009. 4(3): p. e4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Valjent E, et al. , Looking BAC at striatal signaling: cell-specific analysis in new transgenic mice. Trends Neurosci, 2009. 32(10): p. 538–47. [DOI] [PubMed] [Google Scholar]
- 139.Shen H, et al. , Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci U S A, 2011. 108(48): p. 19407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kalivas PW and Hu XT, Exciting inhibition in psychostimulant addiction. Trends Neurosci, 2006. 29(11): p. 6106. [DOI] [PubMed] [Google Scholar]
- 141.Huang YH, Schluter OM, and Dong Y, Cocaine-induced homeostatic regulation and dysregulation of nucleus accumbens neurons. Behav Brain Res, 2011. 216(1): p. 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ishikawa M, et al. , Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci, 2009. 29(18): p. 5820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mu P, et al. , Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J Neurosci, 2010. 30(10): p. 3689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chang JY, Janak PH, and Woodward DJ, Comparison of mesocorticolimbic neuronal responses during cocaine and heroin self-administration in freely moving rats. J Neurosci, 1998. 18(8): p. 3098–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dworkin SI, et al. , Kainic acid lesions of the nucleus accumbens selectively attenuate morphine selfadministration. Pharmacol Biochem Behav, 1988. 29(1): p. 175–81. [DOI] [PubMed] [Google Scholar]
- 146.van der Kooy D, et al. , Reinforcing effects of brain microinjections of morphine revealed by conditioned place preference. Brain Res, 1982. 243(1): p. 107–17. [DOI] [PubMed] [Google Scholar]
- 147.Olds ME, Reinforcing effects of morphine in the nucleus accumbens. Brain Res, 1982. 237(2): p. 429–40. [DOI] [PubMed] [Google Scholar]
- 148.Bossert JM, et al. , Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology, 2006. 31(10): p. 2197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hearing M, et al. , Opioid and Psychostimulant Plasticity: Targeting Overlap in Nucleus Accumbens Glutamate Signaling. Trends Pharmacol Sci, 2018. 39(3): p. 276–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Peters J and De Vries TJ, Glutamate mechanisms underlying opiate memories. Cold Spring Harb Perspect Med, 2012. 2(9): p. a012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Badiani A, et al. , Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci, 2011. 12(11): p. 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Russell SE, et al. , Nucleus Accumbens AMPA Receptors Are Necessary for Morphine-Withdrawal-Induced Negative-Affective States in Rats. J Neurosci, 2016. 36(21): p. 5748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hikida T, et al. , Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron, 2010. 66(6): p. 896–907. [DOI] [PubMed] [Google Scholar]
- 154.Lobo MK, et al. , Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science, 2010. 330(6002): p. 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bock R, et al. , Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci, 2013. 16(5): p. 632–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Stefanik MT, et al. , Optogenetic evidence that pallidal projections, not nigral projections, from the nucleus accumbens core are necessary for reinstating cocaine seeking. J Neurosci, 2013. 33(34): p. 13654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Manabe T, et al. , Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol, 1993. 70(4): p. 1451–9. [DOI] [PubMed] [Google Scholar]
- 158.Kauer JA and Malenka RC, Synaptic plasticity and addiction. Nat Rev Neurosci, 2007. 8(11): p. 844–58. [DOI] [PubMed] [Google Scholar]
- 159.Zhu Y, et al. , A thalamic input to the nucleus accumbens mediates opiate dependence. Nature, 2016. 530(7589): p. 219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bossert JM, et al. , Differential effects of blockade of dopamine D1-family receptors in nucleus accumbens core or shell on reinstatement of heroin seeking induced by contextual and discrete cues. J Neurosci, 2007. 27(46): p. 12655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yuan K, et al. , Morphine treatment enhances glutamatergic input onto neurons of the nucleus accumbens via both disinhibitory and stimulating effect. Addict Biol, 2016. [DOI] [PubMed] [Google Scholar]
- 162.Cahill SP, et al. , Dissemination of exposure therapy in the treatment of posttraumatic stress disorder. J Trauma Stress, 2006. 19(5): p. 597–610. [DOI] [PubMed] [Google Scholar]
- 163.Gutierrez-Lobos K, et al. , The influence of age on the female/male ratio of treated incidence rates in depression. BMC Psychiatry, 2002. 2: p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Keers R and Aitchison KJ, Gender differences in antidepressant drug response. Int Rev Psychiatry, 2010. 22(5): p. 485–500. [DOI] [PubMed] [Google Scholar]
- 165.Becker JB and Hu M, Sex differences in drug abuse. Front Neuroendocrinol, 2008. 29(1): p. 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Becker JB, Perry AN, and Westenbroek C, Sex differences in the neural mechanisms mediating addiction: a new synthesis and hypothesis. Biol Sex Differ, 2012. 3(1): p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Marsh JC, et al. , Gender differences in trends for heroin use and nonmedical prescription opioid use, 2007–2014. J Subst Abuse Treat, 2018. 87: p. 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jones CM, et al. , Vital Signs: Demographic and Substance Use Trends Among Heroin Users - United States, 2002–2013. MMWR Morb Mortal Wkly Rep, 2015. 64(26): p. 719–25. [PMC free article] [PubMed] [Google Scholar]
- 169.Kennedy AP, et al. , Sex differences in cocaine/heroin users: drug-use triggers and craving in daily life. Drug Alcohol Depend, 2013. 132(1–2): p. 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Marron Fernandez de Velasco E, et al. , Sex differences in GABA(B)R-GIRK signaling in layer 5/6 pyramidal neurons of the mouse prelimbic cortex. Neuropharmacology, 2015. 95: p. 353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]