

Abstract
Heme-copper oxidases (HCOs) are terminal enzymes on the mitochondrial or bacterial respiratory electron transport chain, which utilize a unique heterobinuclear active site to catalyze the 4H+/4e− reduction of dioxygen to water. This process involves a proton-coupled electron transfer (PCET) from a tyrosine (phenolic) residue and additional redox events coupled to transmembrane proton pumping and ATP synthesis. Given that HCOs are large, complex, membrane-bound enzymes, bioinspired synthetic model chemistry is a promising approach to better understand heme-Cu-mediated dioxygen reduction, including the details of proton and electron movements. This review encompasses important aspects of heme-O2 and copper–O2 (bio)chemistries as they relate to the design and interpretation of small molecule model systems and provides perspectives from fundamental coordination chemistry, which can be applied to the understanding of HCO activity. We focus on recent advancements from studies of heme–Cu models, evaluating experimental and computational results, which highlight important fundamental structure–function relationships. Finally, we provide an outlook for future potential contributions from synthetic inorganic chemistry and discuss their implications with relevance to biological O2-reduction.
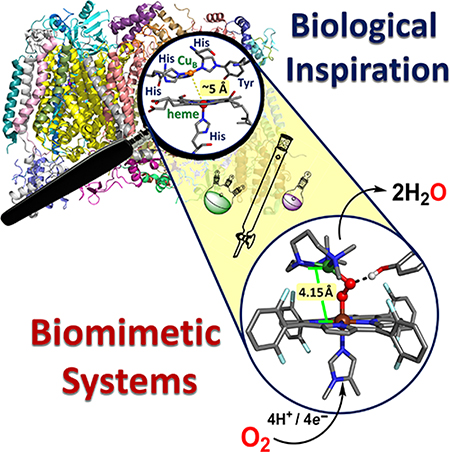
Graphical Abstract
1. INTRODUCTION
1.1. O2 Activation and Reduction
The diversity of dioxygen chemistry spans all subcategories of chemistry and is the cornerstone of aerobic life. The high oxidizing power stored in the O=O double bond requires redox “activation” in order to be accessed and utilized (i.e., for chemical transformations), as molecular oxygen is readily abundant, yet kinetically inert. Complete reduction of dioxygen to water requires 4 protons and 4 electrons (O2 + 4H+ + 4e− → 2H2O), and an electrochemical understanding of these stepwise proton transfers and/or electron transfers is well-characterized in aqueous media (see Figure 1).1,2
Figure 1.

(Top) Stepwise reduction of dioxygen. Reduction potentials are given in volts vs NHE at 25 °C, 1 bar, pH = 7. (Bottom) Frost diagrams depicting the free energy (nE) of the ROS intermediates during stepwise O2 reduction to water at (a) pH = 0 and (b) pH = 7. Adapted from ref 1. Copyright 2010 American Chemical Society.
This oxygen reduction reaction (ORR) is extremely important in biology for cellular respiration and oxidative phosphorylation, but it is also the cathodic reaction in sustainable fuel cells and, therefore, of interest for industrial applications.3–9 Incomplete reduction of O2 results in generation of (partially reduced) reactive oxygen species, (P)ROS, which can be toxic or destructive in certain biological systems and manifest in decreased efficiencies for fuel cells. Therefore, researchers have focused on developing systems to control the reactivity of dioxygen in order to better understand the favorable conditions which lead to complete, selective O2-reduction to water. Since stepwise reduction in this manner involves consecutive proton-coupled electron transfer (PCET) reactions (see Figure 1), a detailed understanding of mechanism in natural systems as well as synthetic model systems is desirable (i.e., how protons and electrons move, together or sequentially, and their relative rates and affinities).10–14 In heme-copper oxidases (HCOs), a diverse superfamily of integral membrane proteins which use a binuclear center (BNC) (Figure 2) to activate and reduce dioxygen to water, another layer of complexity stems from the coupling of the proton and electron movements associated with O2-reduction to additional transmembrane proton translocations (vide infra).
Figure 2.

Heme-copper binuclear center (BNC) active site in HCOs, as represented by X-ray crystal structures from (A) a fully reduced iron(II)∙∙∙copper(I) bovine heart cytochrome c oxidase (CcO) and (B) a reduced but Fea3 carbon monoxide (CO) bound derivative.
1.2. Dioxygen Activation and Reduction in Biology by Metalloenzymes
Partial and complete reduction of dioxygen is carried out in biological systems by a variety of metalloenzymes in which redox-active transition metal ions including iron, copper, and manganese adopt diverse coordination/ligand environments and, at times, cooperate with redox-active amino acid residues, allowing them to activate O2 (Figure 3). Despite the aqueous chemistry of dioxygen reduction being quite well-characterized, what is known about (i) how dioxygen interacts with metals, or (ii) how the protonation and/or redox chemistry of dioxygen is effected when bound in different ways to various metal ions, is very limited. Relevant to the subject reviewed herein, that is to say, the thermodynamic reduction potential of an O2-derived fragment (e.g., a peroxide group) bound to a metal ion or a bimetallic center, is certainly different from that of the free O2-derived fragment in water. Likewise, the basicity of a superoxide (O2•−) or peroxide (O22−) group bound to positively charged metal ions (however, as expressed by the pKa value of the metal-bound conjugate acids, M-(•O2H) or M-(−O2H), respectively) undoubtedly varies from the aqueous analog, and such factors substantially affect the conditions under which O–O reductive cleavage occurs, to eventually yield water. This is evidenced by the large structural variety of known metal-oxy adducts and other protein interactions which give rise to unique functionalities among metalloenzymes. Thus, for example, in cytochrome P-450 monooxygenases, the exact reduction potential and basicity of the iron(III)-hydroperoxide [i.e., FeIII(OOH) intermediate] must be understood and critically controlled so that protonation and O–O reductive cleavage and elimination of water (as an excellent leaving group) will correctly lead to the high-valent active oxygenating species Cmpd I (Figure 4A and also see section 2). The model systems which are the focus of this review are inspired by a class of enzymes which use a heme-Cu binuclear center to effect O2-reduction, although both heme-only and Cu-only enzymes are known to be active in O2-chemistry (Figure 3, Figure 4, see also section 2 and 3, respectively).15–19 A comprehensive understanding of heme-copper oxidase biochemistry therefore benefits from, and builds upon, the parallel O2-reduction chemistries of heme-only and Cu-only systems (Figure 3). For example, cytochrome P450s are a widely studied class of monooxygenases which contain a heme in their active site and catalyze two-electron reduction of O2 via a high-valent (P+•)·FeIV=O, coupled to substrate oxygenation (oxygen atom incorporation, formally) (Figure 3, Figure 4A, and section 2). Multicopper oxidases can also catalyze 4-electron reduction of O2 (in some cases, also with substrate oxygenation) by utilizing proximal Cu ions and an array of terminal and bridging [Cu]n-O2 (n = 1, 2) adducts (Figure 3, Figure 4B, and section 3). Many other heme- or copper-containing enzymes are involved in the chemistry of partial O2-reduction or ROS regulation (see Figure 3); however, perhaps the best examples we have from biology to study the complete O2 reduction reaction are HCOs (Figure 4C). Nature has evolved an electron transport chain in aerobic organisms (both prokaryotic and eukaryotic) in which multiple systems, including the terminal HCOs, work synchronously to efficiently reduce dioxygen while harnessing its thermodynamic potential energy as chemical energy to drive transmembrane proton pumping.20 Indeed, far-reaching research efforts over many decades have elucidated a great deal of the biochemistry and bioenergetics of HCOs. However, direct studies of heme-copper-oxidases are extremely complicated, especially with respect to the metal ion cofactors (including the heme–copper center where dioxygen binds and is reduced), as they are such large proteins which are embedded within a membrane and contain multiple spectroscopically active moieties. HCOs are therefore attractive targets for model chemistry, and detailed fundamental insights can be gained from systematic studies of heme- and copper-containing models as we describe below (see section 5 for details).
Figure 3.

Heme, copper, and heme/Cu enzymes involved in chemistry with dioxygen. aCatechol oxidase catalyzes four-electron reduction of dioxygen to two moles of water while oxidizing two moles of catechol substrate to two moles of quinone. bThe stoichiometry of monooxygenases (Cu-only and heme-only examples) reflects 2H+/2e− reduction of dioxygen to give one water molecule and incorporation of one O atom into a substrate (X-H → X–OH). cThe newly discovered formylglycine-generating enzyme (FGE) is a monooxygenase which is believed to utilize a mono-Cu site to oxidize cysteine to formylglycine and reduce O2 to form H2O and H2S (see section 3 for details).
Figure 4.

(A) Heme-only chemistry with O2 (section 2). (B) Mononuclear and dinuclear, copper-only chemistry with O2 (section 3). (C) O–O reductive cleavage chemistry by heme–copper oxidases, coupled to transmembrane pumping of 4 protons per mole of O2 reduced (section 4).
1.3. Synthetic Modeling Approach to Understanding O2-Reduction by HCOs
Scientific modeling can be viewed as a chemist's solution to a biologist’s problem. Certain questions regarding catalytic mechanisms and fundamental active site structure–function relationships within systems such as HCOs can likely best be or perhaps only be answered via synthetic model chemistry.21,22 Indeed, large-scale cooperative effects such as allostery and conducting channels (for moving protons, electrons, water, substrates, etc. over relatively long distances) have important roles in multifunctional enzymes such as HCOs; however, when considering the transfer of understanding to designing functional catalytic systems for industrial purposes such as fuel cells, one must identify the specific factors and conditions which lead to selective and efficient O–O bond cleavage by the local environment of the BNC. Relevant and critical aspects include knowledge of (i) O2-binding modes for O2 and its reduced derivatives (e.g., superoxo, peroxo, or oxo), (ii) protonation states of different intermediate species, and (iii) the timing and ordering of electron-transfer (ET) and proton-transfer (PT) events.
These details can be sought and probed systematically using active site models of varying complexity, and indeed the evolution of HCO model compounds has shown that there are advantages of studying both simple and elaborate systems, including small redesigned proteins (see section 5).23–26 As will be described, the essence of this approach is based on mimicry of one or multiple characteristics of the active site and evaluation of their resultant reactivities. Insights gained from heme-only and Cu-only model complexes importantly guide the design of heme-and-copper-containing models such that modifications to heme or copper ion environments (coordination number and geometry, ligand donating ability and denticity, sterics, and H-bonding factors) in these systems can be comprehensively understood in relation to O2-activation and reduction by the BNC.22 Additionally, synthetic inorganic models allow access and detailed study of inferred, yet unobserved, catalytic/reactive intermediates and give researchers control over substrate addition and properties [pKa (H+) and E0 (e−)]. Often, models can take advantage of conditions which some would consider to be irrelevant for enzyme functional mimicry (such as the use of organic solvents,27 low temperatures, or unnatural pH), in order to eliminate side reactions, solubilize metal–ligand complexes, stabilize reactive intermediates, and/or control H-bonding interactions. Recent advances in theoretical techniques and their application have led to extremely valuable contributions which supplement scientific modeling in general and relate experimental results obtained in model systems back to the understanding of HCOs. Overall, the rigorous (physical and electronic) structural characterization of HCO models, their reactivities with O2, and parallel theoretical investigations, have provided significant insight into structure–function relationships related to O2-activation and reduction chemistry by HCOs, yet it is the collaboration between enzyme studies, and these synthetic models and computations, which expands our understanding and inspires design of new systems.
2. HEME/DIOXYGEN INTERACTIONS: FROM BIOLOGY TO MODEL SYSTEMS
2.1. Hemoproteins: Structure, Function, and Mechanism
As they are ubiquitous in physiological systems within a large range of organisms, heme proteins carry out numerous pivotal functions such as gas transportation, substrate oxidation and oxygenation, and biomolecule degradation. A listing of heme-only-containing proteins and their functions is given in Table 1. It is fascinating how these protein active sites are evolutionarily fine-tuned to be suited to particular organisms and for precise function toward particular substrates, mainly by modifying the identity of their axial (i.e., proximal) ligand, the details of the amino acid composition, and/or local structure within the distal binding pocket.28,29
Table 1.
Dioxygen-Binding Hemoproteins and Their Main Catalytic Functionalities
| name | proximal ligand on heme | catalytically active speciesa | main function |
|---|---|---|---|
| hemoglobin (Hb) | histidine | N/Ab | O2 binding and transportation |
| myoglobin (Mb) | histidine | N/Ab | O2 binding and storage |
| HbNd | histidine | N/Ab | nitric oxide decomposition |
| cyanoglobind | histidine | N/Ab | O2 binding for oxidase activity |
| HbOd | histidine | N/Ab | O2 binding and transfer |
| Glb3d | histidine | N/Ab | O2 binding |
| neuroglobin | histidine | N/Ab | protection against hypoxia and oxidative stress |
| cytoglobin | histidine | N/Ab | protection against hypoxia and oxidative stress |
| erythrocruorine | histidine | N/Ab | O2 binding and transportation |
| globin E | histidine | N/Ab | O2 binding and storage |
| cytochrome P450 (Cyt-P450) | cysteine | (P•+)FeIV=Oc | monooxygenation of organic substrates |
| aromatase | cysteine | (P)FeIII–OO−c | biosynthesis of estrone from androstenedione |
| heme oxygenase (HO) | histidine | (P)FeIII–OOHc | heme degradation to biliverdin, CO(g), and free Fe2+ |
| nitric oxide synthase (NOS) | cysteine | (P)FeIII–OO•–c or (P)FeIII–OO−c |
generation of NO(g) from L-arginine |
| indoleamine 2,3-dioxygenase (IDO) | histidine | (P)FeIII–OO•–c | dioxygenation of indoleamine moieties |
| tryptophan 2,3-dioxygenase (TDO) | histidine | (P)FeIII–OO•–c | dioxygenation of tryptophan to give N-formylkynurenine |
| peroxidases | histidine | (P•+)FeIV=Oc | H2O2 activation for substrate oxidation |
| chloroperoxidase | cysteine | (P•+)FeIV=Oc | monooxygenation and halogenation of organic substrates |
| catalase | tyrosine | (P•+)FeIV=Oc | H2O2 disproportionation to H2O and O2 (g) |
| secondary amine monooxygenase | histidine | (P•+)FeIV=Oc | dealkylation of 2° amines to give 1° amines and aldehydes |
| prostaglandin synthase | histidine | (P•+)FeIV=Oc | generation of prostaglandin from arachidonic acid |
| heme sensor proteins | histidine or cysteinef | N/Ab | sensing gases (O2, NO, CO)/redox states during cell signal transduction |
The central paradigm of dioxygen binding and activation by hemoproteins follows a distinctive panel of intermediates, as the stepwise reduction of O2 occurs via O–O reductive cleavage, concomitant with the oxidation of the heme–iron moiety. A diagram depicting the generalized landscape of heme proteins with their variations, relationships, and interconversions between these key intermediates is summarized in Figure 5. Critically important realizations come from comparison of the discrete steps and partially reduced oxygen species (PROS) produced during (i) aqueous four-electron four-proton O2 reduction (see Figure 1, top) with (ii) the analogous steps and species where these partially reduced O2 derivatives are heme-bound (see Figure 4), and (iii) these same or very similar metal-bound PROS within heme enzymes wherein the identity of the proximal axial ligand is critical to function, as outlined in Figure 5. As will be noted in part in further discussions, all aspects of practical O2-utilization, that is O2 reduction in fuel cells, O2-activation for organic substrate oxidation, and water oxidation to produce dioxygen, will at a fundamental level require a complete elucidation of our understanding concerning redox potentials and pKa's for metal-bound O2-reduced and partially protonated species and their relationship to structure and reactivity.
Figure 5.

Generalized relationship among heme/dioxygen-derived oxidative species and their occurrence in biological systems.
Dioxygen transportation and storage proteins such as hemoglobin and myoglobin, respectively, possess histidine axial ligands, which lack strong electron-donating effects as in cysteinate or tyrosinate axial bases present in dioxygen-activating heme proteins such as cytochrome P450 mono-oxygenases or catalase. Furthermore, the polarity of the distal binding pocket for the latter are much greater, as compared to those of the former due to the presence of amino acid residues with much more polar side chains that aid in stabilizing charge accumulated intermediates. In fact, it has been observed that the replacement of the histidine axial base of dioxygen carrier/storage proteins with cysteinate, which is the proximal residue within dioxygen activating P450 monooxygenases, switches the protein O2-processing from reversible dioxygen binding to O2-activation via further dioxygen reduction, giving rise to oxidizing high-valent intermediates (Figure 9).39 The collective effect of these properties results in the evolution of heme local environments with the well-known “push-pull” machinery, which facilitates the O–O bond cleavage step (vide infra). On the other hand, the main oxidizing species for most heme degradation enzymes (i.e., heme oxygenases) is thought to be a (P)FeIII–OOH moiety, which effects the conversion of heme to biliverdin (a powerful physiological antioxidant) and carbon monoxide (a neurotransmitter) along with the release of FeII for its future utilization. This is to say that biological evolution has led to a (P)FeIII–OOH intermediate which does not self-destroy the porphyrinate moiety, but rather further reductive O–O cleavage occurs to give Cmpd I or II in catalases, peroxidases, and P-450 enzymes. Irregularities in this finetuning of active site chemistry may give rise to undesired heme degradation processes potentially leading to organism disease states. Structural and mechanistic details pertaining to these multifunctional heme enzymes have been meticulously reviewed elsewhere,17,40–43 and we herein mainly describe the basic differences in some of their structures and mechanisms, with the main emphasis being on properties of the key intermediates involved. The identification and understanding already in place for these long-studied enzymes which affect O2-binding, and subsequent oxidase and/or oxygenase function, will likely be relevant to O2-activation/reduction in HCOs and thus of interest for this review article.
Figure 9.

Proposed catalytic cycle for Cyt P450 enzymes. Crystallo-graphic characterization of species 1–4 has been achieved,120 while other intermediates including peroxo and hydroperoxo Cmpd I and Cmpd II have been spectroscopically observed.125,131,132 See text for details.
Upon initial binding of dioxygen to the (P)FeII (P = porphyrinate ligand supporting the iron center) center in the reduced state of the active site, a heme–iron(III)–superoxo [(P)FeIII–O2•−] species forms via single electron transfer from the FeII center to the ligated dioxygen moiety. This is widely referred to in the biological literature as the “oxy-form”, especially in the dioxygen carrier and storage proteins hemoglobin and myoglobin. These proteins contain heme in the form of protoporphyrin IX (Figure 6) and carry out reversible dioxygen association/dissociation cycles [i.e., mediate the reversible equilibrium (P)FeII + O2 ⇄ (P)FeIII–O2•−] without further activation of the bound dioxygen moiety. The most suitable electronic structure description of these oxy compounds has been under severe debate over a period of many decades, and the essential details of the current understanding best describes this intermediate as a (P)FeIII– O2•− species (and see section 2.2.2). In contrast to these dioxygen transport proteins, dioxygen-activating enzymes take things much further. That is, further reduction gives the two-electron reduced dioxygen-ligated species, an (P)iron(III)– peroxo [(P)FeIII–O22−] or hydroperoxo species [(P)FeIII– OOH] if or when protonated. This is also known as Compound 0 (Cmpd 0) and may actually exist as an FeIII– (H2O2) adduct, which is what first forms in peroxidases via resting state heme–FeIII + H2O2 chemistry (vide infra). These (P)FeIII–OOH adducts are critical intermediates in biology; while sometimes serving as the oxidant which attacks substrates (vide infra), they serve as pivotal precursors leading to the generation of much more reactive iron high-valent oxidants that can oxidize or oxygenate quite unreactive hydrocarbon substrates.44 The homolytic or heterolytic nature of the O–O bond cleavage step gives rise to either (P)FeIV= O [also known as Compound II (Cmpd II)] or (P•+)FeIV=O [also known as Compound I (Cmpd I)], respectively (vide infra).
Figure 6.

(A) Oxy- and (B) deoxy- forms of hemoglobin where the doming of the porphyrinate is observed in the deoxy- form. The iron center moves into the plane of the porphyrin as the dioxygen coordinates in the oxy-form.
The O–O reductive cleavage step is critical, and it has been the subject of many decades of critical interrogation in P-450 and peroxidase enzymes, as well as synthetic model systems.41,45–53 Still, there are aspects that require further elucidation. (P)FeIII–OOH + proton to give (P•+)FeIV=O, Cmpd I species, represents a two-electron transfer to hydroperoxide, thus the initial dioxygen molecule has been fully reduced by four-electrons, producing one molecule of water and an oxo (oxide; O2− formally) atom bound to a highly oxidized heme. In HCOs, the homolytic cleavage of a putative (P)FeIII–OOH⋯CuII intermediate takes place to give a detectable Cmpd II intermediate, (P)FeIV=O, formally also with a fully reduced oxo (O2−) ligand; formally a hydroxyl radical has been produced by the homolytic cleavage reaction but is stabilized by further reduction. Further discussion of these matters is given below.
2.1.1. Dioxygen Carrier and Storage Proteins: Hemoglobin and Myoglobin.
In aerobic organisms, dioxygen transportation and storage are carried out by two well-characterized hemoproteins, namely hemoglobin (Hb) and myoglobin (Mb), respectively. These globin proteins are highly efficient in dioxygen binding under physiological conditions, while the protein structures generally prevent further reduction of the bound dioxygen moiety leading to the release of deleterious reactive oxygen species. These hemoproteins have been a main focus of biochemical research for over 150 years, during which, they have been successfully utilized for the initial identification and characterization of heme cofactors within biological systems in the 1850s, to obtaining some of the first X-ray crystallographically characterized protein structures back in the 1950s.29,54,55
Myoglobin is a monomeric globin protein, with a single dioxygen binding protoporphyrin IX unit. In contrast, the tetrameric hemoglobin contains four Mb-like subunits, with a full binding capacity of four equivalents of dioxygen per molecule. The dioxygen binding in Hb is known to be cooperative, where the affinity is dependent on other external factors such as the partial pressure of O2(g) and pH.56 These subtle differences in affinity patterns are primarily responsible for the smooth exchange of dioxygen between Hb and Mb, where the former delivers dioxygen to muscle tissues where it is transferred to the latter over an affinity gradient for storage and future use. The dioxygen binding sites of Hb and Mb are very similar, and in the deoxy form (where dioxygen is not bound), a pentacoordinate high-spin (S = 2) ferrous center with an axially coordinated proximal histidine side-chain structure is observed. Upon dioxygen binding, an in-plane movement of the iron center is displayed, along with the shortening of all Fe-N distances, and the spin state of the FeIII center changing to low-spin (S = 1/2) (Figure 6).57–61 These fine structural modifications about the dioxygen binding heme center are transmitted to the rest of the protein by means of conformational changes throughout the protein matrix, leading to the cooperative dioxygen binding mechanism in Hb even though the distance between any two heme centers is over 25 Å. A sigmoidal (or “S-shaped”) curve is observed in a plot of Hb protein saturation with molecular oxygen as a function of the partial pressure of dioxygen, indicating that the more O2 that is bound to Hb, the easier it is for even more dioxygen to ligate, until all binding sites are saturated.56,62,63 The dioxygen affinity of these hemoproteins is also regulated by noncovalent interactions between amino acid side chains and bound dioxygen moieties. Such interactions have long been proposed64 but were definitively proved by means of spectroscopic, structural, and theoretical interrogations.65–76 This work revealed that oxy-forms of these hemoproteins contain an in-plane iron center within the heme unit with an end-on ligated dioxygen ligand that is hydrogen-bonded to a critical histidine (His) residue within the distal binding pocket. This latter interaction greatly enhances the O2-affinity for deoxy-Hb and Mb, relative to what it would be without this interaction.
Carbon monoxide (CO(g)) acts as a common inhibitor of these hemoproteins, and it inherently possesses (in a nonprotein environment) much higher affinities to bind to heme centers than does dioxygen. CO(g) is a known byproduct of biological heme degradation (vide infra). However, it is known that the globin protein matrix77,78 and the axially coordinated amino acids play major roles in decreasing the CO(g) affinity of Mb and Hb, such as by not allowing the optimal bonding which would involve a linear Fe-C-O CO-coordination due to the presence of well-placed distal histidine and valine residues.79,80 Thus, nature has evolved these heme O2-carriers to alter the intrinsic metal–ligand binding properties, enhancing Fe–O2 interactions (via the distal His H-bond) and to diminish the CO to iron(II) binding; all this being necessary in spite of the overwhelming larger amount of O2 versus CO normally present in the atmosphere.
The stability of the oxy-heme species is also of great importance for the proper functioning of these hemoproteins; however, a minor portion of Hb in most organisms (e.g., ~1 – 2% of Hb in humans)81,82 exists in oxidized ferric form lacking coordinated dioxygen; this protein form is known as met-Hb. For synthetic porphyrinates, the occurrence of this “autooxidation” reaction [formally the deoxy-heme iron(II) is oxidized to iron(III)] may take place via dimerization pathways (see section 2.2); however, such processes are disfavored within biological active sites due to the surrounding steric bulk of the protein matrix. Proposals for how/why this occurs within such systems include the suggestion of a “proton relay” mechanism facilitated by the distal histidine residue, followed by an SN2-type replacement of the coordinated superoxide by anionic nucleophiles (such as SCN−, N3−, CN−, F−, and Cl−).83,84
Although oxy-heme compounds, both biological and synthetic, have been some of the most extensively studied species in bioinorganic chemistry,85 the precise electronic structure description of the heme–Fe–O2 moiety remains as a point of discussion and research. In pioneering work, Pauling and Coryell reported the diamagnetism of carbonmonoxy-and oxy-Hb, even though deoxy-Hb was observed to exhibit paramagnetic behavior indicating the presence of unpaired electrons.86 These researchers' seminal work suggested that the electronic structure of oxy-Hb is best described as a FeII–O2 adduct (with the electronic structure of the bound dioxygen moiety undergoing major modifications in conjunction with that of the hemoglobin binding site, where two resonance forms, Fe=O–O ⇄ Fe–O=O, were introduced); here, complete electron transfer from dioxygen to iron has not occurred, meaning that the coordinated dioxygen moiety was redox innocent (Figure 7). However, dioxygen ligands are well-known to be noninnocent upon metal ligation, which led to the iron(III)–superoxo, FeIII–O2•−, model of J. J. Weiss first formulated in 1964.87 Here, the observed diamagnetism of oxy- Hb is explained by the antiferromagnetic coupling between the unpaired spin on the coordinated superoxo ligand (S = 1/2) and that on the low-spin FeIII center (S = 1/2) to result in an overall spin of S = 0 (Figure 7).
Figure 7.

Simple representations showing the three main proposals presented thus far for the explanation of the electronic structure description of oxy-Hb/Mb species. Adapted from ref 94. Copyright 2013 American Chemical Society.
In addition, the “ozone model” of McClure, Harcourt, and Goddard describes the oxy-form electronic structure as an antiferromagnetically coupled intermediate spin ferrous center (S = 1) and a triplet dioxygen ligand (S = 1) (Figure 7).88–91 This formulation recently gathered further support through a combination of variable temperature X-ray spectroscopic techniques and computations performed on a variety of Fe– O2 adducts including those of Hb, Mb, and several synthetic heme and nonheme iron complexes.92 In this way, the authors probed the molecular and electronic structure concomitantly with a comprehensive Fe–O2 sample set and deduced that the effective unpaired electron density on the Fe–O=O fragment (with 2 unpaired spins on intermediate spin ferrous iron center) aligned closely with that of the ozone radical (O3+).92 These results are presented so as to give a description of this intermediate in the context of the three models shown in Figure 7 and to clarify the nature of O2 binding to Fe relevant to understanding of protein environments and their synthetic models. Importantly, the authors also pointed out that care must be taken when making conclusions based on bond lengths and angles, as, for example, radiation from high-energy X-ray techniques can potentially induce changes in these parameters.93 An unequivocal description of the electronic structure of oxy-Hb/Mb is said to be hindered due to the highly covalent nature of the supporting porphyrinate ligand system, along within complications in accurate probing of the hydrogen-bonding interactions within the distal binding pocket, using spectroscopic and computational methods.94 Intriguingly, recent comprehensive spectroscopic and theoretical studies by Sarangi and co-workers95 reveal that the solution-state structure of oxy-Hb favor the Weiss model (FeIII-O2•−), while the solid-state crystalline oxy-Hb is more consistent with the Pauling model (FeII-O2). Thus, it was concluded that the dioxygen binding in Hb and Mb does not simply follow either of these proposed models but is a complicated representation of a combination of those proposals with subtleties involved and multiple external factors affecting covalency.92 Finally, even though the current understanding of oxy-heme intermediates is incapable of presenting a simple explanation for its electronic structure, notable recent work has exhibited the necessity of electron transfer from the iron center to the dioxygen moiety in order to form oxy-heme-type intermediates, emphasizing the unambiguous validity of the Weiss model.66,85,96–98
2.1.2. Heme Enzymes with Oxygenase Activity.
This class of heme enzymes performs extremely critical biochemical roles in a large variety of both prokaryotic and eukaryotic organisms.40 However, enzymes with oxygenase activity were only discovered in 1955 by the remarkable independent work of both Hayaishi99 (heme protein, tryptophan 2,3-dioxygenase) and Mason100 (copper protein, tyrosinase). They demonstrated that unlike heme-containing oxidases mainly known at the time for carrying out terminal electron acceptor roles using dioxygen (vide infra), these enzymes are capable of facilitating the efficient incorporation of either one or both oxygen atoms of dioxygen into organic substrates following dioxygen activation. Later on, Hayaishi named this class of enzymes as “oxygenases”,101 which are then subcategorized into mono- and dioxygenases depending on the stoichiometry of oxygen incorporation into the substrate. A majority of heme- containing oxygenases contain protoporphyrin IX as the heme prosthetic group that coordinate varying axial ligands within different enzymes, as outlined by the presentation in Figure 5. In this review, we present a brief summary and focus on the active-site structure and mechanism of action of cyt P450s, heme-oxygenases (HOs), and the dioxygenases TDO and IDO. The heme-dioxygen chemistry occurring in these enzymes well illustrates the nature of and interconversion of intermediates such as those shown in Figure 5 to help us toward a better understand of HCOs, which also possess a copper ion proximal to the heme center.
2.1.2.1. Cytochrome P450 (Cyt P450).
These widespread oxygenases are certainly one of the most comprehensively studied classes of metalloenzymes overall, emphasizing the large array of substrate oxidations they mediate, most of which are crucial for aerobic life. Notable ones include hydrocarbon (and aromatic) hydroxylation, olefin (and arene) epoxidation, alkyne oxygenation to carboxylic acids, N-/S-/O- dealkylation reactions, alcohol and aldehyde oxidations, and nitric oxide reduction.40,41 Markedly, a subclass of P450 enzymes known as aromatases also catalyze several physiological interconversions that are of crucial significance for the production of sex hormones40 (e.g., progesterone to androstenedione,102,103 androstenedione to estrogen,104–108 and pregnenolone to dehydroepiandrostenedione102,103), including those in humans. One of the most extensively studied P450s is P450cam,17,109 which catalyzes the regio- and stereospecific hydroxylation of camphor to 5-exohydroxycamphor in the first step of camphor oxidation. Within humans, the most notable transformation that P450s catalyze is xenobiotic degradation within the liver. P450s may also exert deleterious effects on important biomolecules such as DNA, where they could epoxidize critical functional groups utilizing their superior oxidative power.17
Both membrane-bound (usually mitochondrial) and soluble forms of P450s have been identified to-date, and often the overall structure of P450s exhibit higher flexibility compared to most other hemoproteins, because of the constant flux of reactants, substrates, and their products to and from the active site. Furthermore, P450s work closely with various flavin and/or iron/sulfur proteins for the supplementation of electrons during dioxygen reduction.110 The resting state of Cyt P450 contains a low-spin six coordinate ferric center with an axially coordinated thiolate group of a cysteine residue on the proximal side (Figure 8), along with a ligated water molecule at the axial distal site. The coordinated proximal thiolate residue is hydrogen-bonded to several other amino acid side chains, and these interactions have been identified as key features for fine-tuning the redox potential of the heme center.111–113 Generally in the resting state, the FeIII/FeII reduction potential lies between approximately −400 and −170 mV, and thus is inactive to reduction.41 However, upon binding of substrates which are usually hydrophobic, the water molecules (including the water molecule axially ligated to FeIII) within the binding pocket are displaced.114,115 This entropically favorable process triggers the immediate switch of the low-spin six-coordinate (6C) FeIII center to a high-spin five-coordinate state, dramatically elevating its reduction potential by ~400 mV. Now, the electron transfer becomes energetically favorable, resulting in the facile reduction of the five-coordinate FeIII center to FeII. This mechanism of generating FeII prevents spontaneous reduction of the resting state by cellular reducing agents such as NADPH, thereby thwarting the release of partially reduced, cytotoxic-reactive oxygen species.17
Figure 8.

Close view of the Cyt P450cam active site with the heme- bound dioxygen molecule, also highlighting the bound camphor substrate, the six coordinate oxy-heme center (as a formally FeIII- superoxide complex), and the critical H-bonding interactions involving the proximally ligated cysteine residue.
The FeII center readily binds dioxygen, giving rise to the initial heme-oxy intermediate, which has been characterized both by spectroscopic116–119 and structural methods,120 revealing its close reminiscence to those of Hb and Mb. Interestingly, the distal O atom of the superoxo moiety is stabilized by several hydrogen-bonding interactions, which are mediated by two new water molecules that enter the cavity.41 The ferric-bound superoxo moiety then triggers the second electron transfer generating an end-on bound iron(III)-peroxo, FeIII-O22− adduct (Figure 9), this conclusion being based on computational studies and detection via cryoenzymology studies of Davydov and Hoffman.121 A synthetic model compound of such a species was generated and characterized by Naruta and co-workers (see below).122 This species is exceedingly basic (based on computations42 and from model compound investigations122,123), in great part due to the presence of the highly donating axial cysteinate ligand. This end-on peroxo species is then rapidly protonated to generate the low-spin 6C FeIII-OOH intermediate, a Cmpd 0 analog of that known for peroxidases (vide infra) (Figure 16). The Fe–OOH moiety is now poised for heterolytic O–O bond cleavage, due to the presence of what is known as “push-pull” machinery (i.e., strong proximal thiolate donation and hydrogen-bonding by neighboring threonine (Thr252) and aspartate (Asp251) residues) (with the aid of a critically important water molecule) on the distal OH of the Fe-OOH moiety. The overall effects are strengthening of the Fe–O and O–H bonds while weakening the peroxidic O–O bond.40,124 Following another net proton transfer to the hydroperoxo distal O atom, the O–O bond is heterolytically cleaved producing the highly oxidizing Cmpd I intermediate (Figure 9). Due to its superior oxidative power, it, until recently, has only been a fleeting intermediate in P450 studies. The elegant research by Green and co-workers125–127 has led to the high yield generation of a Cmpd I enzyme intermediate from a thermophilic bacterium (CYP119) by addition of m- chloroperbenzoic acid (m-CPBA) to ferric CYP119. These studies have given credence to the long-standing mechanistic proposal describing Cmpd I as being catalytically competent toward hydroxylation of hydrocarbon substrates. Intriguingly, however, both Cmpd i108,128,129 and the ferric peroxo intermediate of P450 (5 in Figure 9)102–105,130 have been proposed as catalytically active metal oxidants for the aldehyde deformylation step (“lyase” step) involved in aromatase activity. Extremely important further insights are required pertaining to thermodynamics of the transition from Cmpd I to the next species in the catalytic cycle, formally a protonated Cmpd II. The proximal cysteinate donor makes the oxo atom of the Cmpd II species extremely basic, requiring that Cmpd I not be as strong a one-electron oxidant.
Figure 16.

Proposed catalytic cycle for peroxidases highlighting the crucial role of the distal His residue in proton translocation that induces the heterolytic O–O bond cleavage process of Cmpd 0. Adapted with permission from ref 45. Copyright 2007 University Science Books.
Although formally FeV=O, the electronic structure of Cmpd I is best described as a Fe(IV) center ferromagnetically coupled with a ligand-based π-cation radical [(P•+)FeIV=O] and has been reviewed elsewhere.133–135 The manner by which Cmpd I carries out substrate oxidations had been an active topic in the field, and in 1978 J. T. Groves proposed his now well-known and widely accepted “radical rebound mechanism” (Scheme 1).135–137 Here, in the enzyme rate-determining step (H/D KIE of >11), Cmpd I [i.e., (P•+)FeIV=O] accepts a hydrogen atom (H•) by homolytic cleavage of a substrate C–H bond, giving rise to (P)FeIV–OH and substrate radical species (R•). The latter abstracts a formally HO• radical from the (P)FeIV–OH species, giving the R–OH hydroxylated organic and ferric-heme (Figure 9). The rebound (2nd) step has to happen fast enough to avoid radical rearrangement of the substrate, in order to conserve the regio-/stereo-specificity of hydroxylation. Groves has recently summarized studies relating to aspects of this timing;135,138 Goldberg and coworkers have elegantly demonstrated the first example of the P-450 enzyme type of rebound, using an FeIV–OH model system.137 We note that alternative mechanistic proposals exist, wherein charged (instead of radical) intermediates and concerted C–H activation pathways (without the effective formation of a (P)FeIV–OH-type intermediate species) have been observed or proposed.42,43,139–143
Scheme 1.

Radical Rebound Mechanism Proposed by Groves et al.a
aSee the main text for further discussion.
Furthermore, as seen in Figure 9, O-atom surrogates such as peracids and PhIO can be used to effect catalytic hydroxylations by addition of these reagents to the substrate- FeIII form, leading directly to Cmpd I (“O-atom shunt”; as mentioned above for the work of M. Green). P450 enzymes can also undergo a productive “peroxide shunt” catalytic process,144–146 wherein H2O2 or ROOH are added to the substrate-FeIII enzyme, directly giving the FeIII–OOH(R) intermediate; that is followed by O–O cleavage, etc. We should add that just in the past few years, heme–thiolate proteins using H2O2 (and not O2) as the terminal oxidant have been or are being characterized. These “peroxygenases” are very efficient hydrocarbon (including aromatic) substrate hydroxylases and form a Cmpd I type intermediate very much like P450s.135,147,148
Interestingly, in one such recent Cmpd I example, a rare nonlinear Brønsted-Evans Polanyi relationship revealed a switch in the transition state (TS) in its hydroxylation reactivity, depending on the substrate BDE. That is, for substrates with BDE > 90 kcal/mol, a midpoint TS was observed, while for those with weaker BDEs, an early TS resulting in entropy-controlled rates was detected.149 The Cmpd II species of the same peroxygenase was later shown to oxidize unprecedentedly (for Cmpd II intermediates) strong substrate C–H bonds due to the remarkable basicity of the ferryl moiety as compared to others such as in peroxidases (vide infra).147
Unproductive branching points have been identified for the P450 catalytic cycle:150–153 (1) autoxidation of the oxy-form to release superoxide anions (or protonated superoxide), as proposed for the decay of Fe-oxy-type intermediates within model systems (see section 2.2), leaving behind the enzyme ferric state;153 (2) “oxidase uncoupling” is the two-electron/two-proton reduction/protonation of Cmpd I, producing water and the resting ferric state; (3) the reverse of the peroxide shunt is referred to as “uncoupling”, wherein productive O–O heterolytic cleavage fails, and proximal (rather than distal) O- atom protonation of the FeIII–OOH intermediate leads to the release of H2O2.
It is especially the well-understood cytochrome P-450 monooxygenase's mechanism of O–O reductive cleavage that relates/connects to HCO mechanism, in that we see that a heme–(hydro)peroxide needs to form, and appropriate machinery for H-bonding to and/or protonation of the O-atom distal to the heme, to efficiently cleave the O–O bond and produce a high-valent Fe=O species. The existence of the P450 peroxide shunt and that being a natural course in peroxygenases also supports similarities to HCOs, in that such a (non-natural) peroxide shunt also works in heme–Cu oxidases (see section 4.2). Of course, the analogy or relationship of Cyt P450 monooxygenase O–O cleavage chemistry to that occurring in HCOs is not as simple as this; in HCOs the normal reductive O–O cleavage is homolytic and CuB likely also plays roles in the enzyme proton translocation process and in setting the overall enzyme bioenergetics.
2.1.2.2. Heme Oxygenase (HO).
Heme oxygenases catalyze the regiospecific oxidative degradation of heme (6–8 mg of Hb per day, releasing ~300 mg of oxidized heme) producing biliverdin (later converted to bilirubin by bilirubin reductase),154 free ferrous irons, and CO(g); some HOs that do not produce CO(g) have been recently discovered and characterized (vide infra). Consequently, HOs are responsible for a variety of physiological functions that include heme catabolism, iron homeostasis, antioxidant defense, and cell signaling via the CO(g) produced.155–159 HO also aids in “Fe stealing” mechanisms of pathogenic organisms, where they extract Fe from the host cells under disease conditions.160,161 HO stands apart from all other heme enzymes since the heme group also behaves as the substrate in this case, mediating all three dioxygen activation steps (vide infra); HO does not require any other O2-activating cofactor during its turnover.162 The reductases that provide electrons during catalytic turnover vary among organisms. In mammals, it is NADPH-Cyt P450 reductase,163 and in some plant HOs it is thought to be NADPH-ferredoxin reductase and/or ferredoxin.164 Electron donors that support bacterial HOs are yet to be identified.
The structure of human HO165,166 demonstrates a unique active site, with a five-coordinate heme center axially ligated by histidine, with the α-meso- position of the heme porphyrinate being relatively sterically unencumbered compared to other meso- positions. Unusually close positioning of the distal and proximal helices sandwiches the heme center (Figure 10), where the latter is presumed to precisely tune the electronics of the Fe center, preventing O–O bond weakening/cleavage of the Cmpd 0 hydroperoxo intermediate. The oxy form of HO has been thoroughly characterized and is now known to possess an acute Fe–O–O angle that promotes van der Waaltype interactions, which position the distal O atom in close proximity with the α-meso carbon.167,168 Furthermore, the distal O atom also hydrogen bonds to a distal water molecule that is part of a water cluster held together by a critical aspartate residue (Figure 10).166,169–171 The mechanism of HO involves three consecutive oxidation steps (Scheme 2), each of which involves one molecule of dioxygen. Substantial details are known in the literature regarding the first oxidation step,172,173 however, the second and third steps have remained less clear. The latter has been extensively studied in the recent work by Matsui, Ikeda-Saito, and co-workers161,174,175 revealing salient mechanistic details.
Figure 10.

Active site structure of an oxy form of HO bearing the heme with axially coordinated His20, “distal” (yellow), and “proximal” (green) helices, and the critical water cluster held in place by Asp136. See the text for further discussion.
Scheme 2.

Main Mechanistic Steps Involved in the Heme-Degradation Pathway Catalyzed by HO. Adapted from ref 161. Copyright 2010 American Chemical Society.
The first step involves the regiospecific self-hydroxylation of the α-meso position of the heme porphyrinate to give α-meso-hydroxyheme (Scheme 2). The active species responsible for this step is a Cmpd 0-like FeIII–OOH moiety, which is formed by stepwise reduction/protonation events. An exceptionally rapid protonation is observed in Cmpd 0 formation, and the aforementioned water cluster is thought to relay the required protons176 similar to the above-described mechanism of Cyt-P450. Intriguingly, the Cmpd I intermediate of HO has been shown to be inactive toward the conversion of heme to α-meso-hydroxyheme.177 Furthermore, H2O2 has also been observed to successfully carry out the formation of α-meso-hydroxyheme, presumably via a peroxide shunt-type mechanism. The catalytically active FeIII-OOH intermediate of HO has been observed under cryogenic conditions179–181 and has been characterized in detail by electron paramagnetic resonance (EPR) and electron–nuclear double resonance (ENDOR) spectroscopies.179 The second proton transfer is presumed to be a concerted process (from H/D isotope-labeling)182 in that the proton transfer from the water molecule to the distal O atom of Cmpd 0 is concomitant with the C–O bond formation between the distal O atom and the α-meso-carbon of the porphyrinate. However, the involvement of an extremely reactive HO• radical (formed by homolytic O–O bond cleavage in a stepwise scenario, as described by theoretical studies)183–185 has not been completely discounted.
In the second oxidation step, the hydroxylated heme activates the second equivalent of dioxygen, which further oxidizes α-meso-hydroxyheme to give verdoheme and CO(g). This step is a spontaneous autoxidation process, where the involvement of the heme center of HO is not required.186 Interestingly, however, replacing dioxygen with H2O2 for this step does not yield desired products, unlike in the two other oxidative transformations of the HO mechanism. For this second step, two mechanistic pathways have been presented, which differ in the sequence of the dioxygen-dependent oxidation of iron(III)-α-meso-hydroxyheme, and the reduction of FeIII to FeII, ultimately yielding iron(II)–verdoheme. Given the highly feasible nature of the oxidation of iron(III)-α-meso-hydroxyheme by dioxygen,187,188 initial oxidation followed by reduction of FeIII is generally accepted as the viable pathway. Nonetheless, very little is known regarding further details of the mechanistic events involved in this step.
Finally, a verdoheme-activated third equivalent of dioxygen induces porphyrin ring-cleavage, generating the biliverdin final product, releasing the ferrous ion (Scheme 2). This ringopening step is observed to be the overall rate-limiting step under physiological conditions,189 with an anomalously slow rate of O2-binding, making this reaction amenable to be used as a physiological O2 sensor.155,174,189 Similar to the second oxidation step, this step has also been observed to occur even in the absence of HO, by either hydrolytic or redox-mediated pathways.190–192 Interestingly, iron(II)-verdoheme reacts with both O2 and H2O2 to give the biliverdin product.174 When a H2O2-mediated pathway is considered, it is proposed to bind to either the Fe center or the α-carbon on the pyrrole ring to generate hydroperoxo intermediates (Figure 10). Upon deprotonation, these would lead to biliverdin via the formation of bridged-peroxo intermediates. Interestingly, exposure of verdoheme to alkylperoxides yields alkoxy-biliverdin (Figure 11).175 This observation further strengthens the proposed formation of an Fe-based hydroperoxo adduct en route to biliverdin rather than a substrate-based hydroperoxo intermediate. In further support, similar to the first oxidation step, the proper positioning of the distal water cluster (Figure 10) was observed to be critical for this final oxidation process.174,193–195 Noticeably, verdoheme reacts rapidly with H2O2, in fact, much faster than with dioxygen, thereby playing a key role in detoxifying excess reactive oxygen species produced within the cell. Moreover, biliverdin and bilirubin also exhibit remarkable antioxidant properties,157,174,196 implicating HO in an important role in controlling the detoxification of physiologically derived reactive oxygen species (ROS).
Figure 11.

Mechanistic proposals for the transformation of iron(II)-verdoheme to iron(III)-biliverdin in the last step of HO activity. Adapted from ref 161. Copyright 2010 American Chemical Society.
Alternative heme degradation mechanisms (with no production of CO(g)) have been recently discovered in certain bacterial HOs, termed IsdG/I and MhuD,197–199 which are structurally distinct from eukaryotic HOs. These HOs have been observed to convert heme to staphylobilin (α-meso- carbon converted to and aldehyde) and mycobilin (meso-carbon liberated as formaldehyde), respectively (Figure 12).200,201 Furthermore, “heme-degrading factors” have also been identified, which are essentially proteins that produce biliverdin but with no structural homology to any known HO. The active species these proteins utilize is thought to be an Feoxy-type intermediate,178,202–204 in contrast to FeIII–OOH in traditional HOs. The exact mechanism of action these proteins follow is not known. IsdI and MhuD have been structurally characterized;199,205 heme ruffling is proposed to induce the tight regiospecificity of each step in heme oxidation, due to the distinct steric interactions observed about the active site.206 The initial oxidation step, generating meso-hydroxyheme is presumed to be mechanistically similar to that of traditional HOs; however, the events subsequent to meso-hydroxyheme formation are distinctly different. Importantly, the porphyrin ring-opening is not observed at the primarily hydroxylated site (which is either β or δ) but is rather observed at the α-carbon, where the second dioxygen attacks (Figure 12, pathways B and C). This “remote” secondary oxidation is different from conventional HOs since the dioxygen binds to the heme periphery, rather than the metal, and is the key step for suppression of CO(g) production.207 In this step, the second dioxygen molecule inserts both O atoms via a dioxetane intermediate cleaving the ring at the α-position. Thus, three O atoms are incorporated into the heme overall (dioxygenation of meso-hydroxyheme) as confirmed by 18O labeling, as compared to verdoheme or biliverdin containing only two O atoms from O2 (monooxygenation of meso-hydroxyheme). Although the precise mechanistic details are still unknown for this recently discovered heme degradation pathway, since CO(g) is not produced, any verdoheme-type intermediates can be excluded.
Figure 12.

Differences in heme degradation pathways of traditional (pathway A) versus nontraditional (pathways B and C) heme oxygenases. Adapted from ref 208. Copyright 2014 American Chemical Society.
2.1.2.3. Indoleamine 2,3-Dioxygenase (IDO) and Tryptophan 2,3-Dioxygenase (TDO).
TDOs209 and IDOs210 are two of the earliest studied oxygenases by Hayashi, and he discovered that they facilitate similar oxidations, with the latter having a broader substrate scope. The generalized reaction is the oxidative ring cleavage of indoles between the second and third carbons (eq 1). Accordingly, they catalyze the
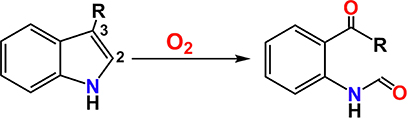 |
(1) |
O2-dependent rate-limiting transformation of L-tryptophan (l-Trp) to N-formylkynurenine (NFK), in the first and rate-limiting step of the kynurenine pathway of tryptophan metabolism.211 Importantly, IDOs are known to aid malignant cells in escaping the host immune system, making IDO inhibitors potential therapeutic targets in modern cancer therapy.212–216
The first crystallographic characterization of human-IDO was reported in 2006, revealing the presence of an axially coordinated proximal histidine (His 346) on Fe. The distal side is nonpolar, with no His residues present, in contrast with other O2-activating enzymes (Figure 13).217 The latter observation is significant, since it warranted major revisions in the prior mechanistic proposals (vide infra, Scheme 3). However, bacterial218 and human219 TDOs bear a distal His residue, but this has been shown to be nonessential for enzyme activity.220
Figure 13.

Active site structure of human-IDO. Here, a 4-phenylimidazole inhibitor molecule is bound to the active site heme (shown in green).
Scheme 3.

Previously Proposed Base (B) Catalyzed Activation of Indoles with Oxy-Heme Species Forming a Peroxyindole Intermediate, Which Reacts Further by Either Criegee (maroon) or Dioxetane (green) Pathwaysa
a Adapted from ref 211. Copyright 2011 American Chemical Society.
A striking feature of the IDO/TDO enzyme mechanism is that it only requires one reducing equivalent for turnover, in contrast to most other dioxygen-dependent hemoenzymes (vide supra). Therefore, iron(III)-superoxo-level intermediates are thought to behave as active oxidants. Initial mechanistic proposals describe a base-catalyzed pathway (Scheme 3),221 since only substrates with a proton on the indole nitrogen were observed to be active. Accordingly, a distal His residue was thought to abstract the proton from the indole N atom.221,222 However, in light of that IDO structure (showing no such distal His residue), and the observation that indoles with no proton at the 1-position are slow but still active substrates, alternative mechanistic proposals217,223 have been presented. These involve electrophilic or radical addition of the indole substrate to the ferric-superoxo moiety (Scheme 4).224 The latter is much more favored since ferric-superoxos are weak electrophiles but bear significant radical character. Furthermore, the substrate binding at the recent human TDO crystal structure219 and known chemistry of indoles all support a radical addition pathway. Nonetheless, a solid mechanistic pathway has not been explicitly established for these enzymes to-date.
Scheme 4.

Electrophilic and Radical Addition Mechanisms Proposed for IDO/TDO Enzymes Reacting via Indole Epoxide and Ferryl Intermediatesa
a As shown here, the rate-limiting steps for the two enzymes are thought to be different. Adapted from ref 225. Copyright 2016 American Chemical Society.
Importantly, formation of a ferryl-type (Cmpd II) intermediate has been proposed to occur later in the mechanism, en route to the final NFK product (Scheme 4). The evidence comes from resonance Raman experiments on an IDO turnover intermediate, with an Fe=O vibration of 799 (Δ18O2 = −37) cm −1.226 However, no such intermediate has ever been observed for TDO, suggesting disparities in their rate-limiting steps as shown in Scheme 4.225 The detection of a ferryl intermediate suggests the possibility of a stepwise transfer of the two oxygen atoms to the substrate rather than a concerted-addition mechanism. If this species were formed following the first oxygenation step, it would also require the formation of an indole epoxide intermediate (Scheme 4), a suggestion supported by recent theoretical work.227 Again, experimental evidence for latter steps of the IDO/TDO mechanism is severely lacking, either from enzymatic examples or model systems.
2.1.2.4. Nitric Oxide Synthase (NOS).
Mammalian enzyme NOS exists in several isoforms, namely endothelial- (eNOS), neuronal-(nNOS), and inducible-NOS (iNOS). Their active sites contain a heme group with an axially ligated cysteine residue like that of the Cyt P450 active site.17,228,229 Accordingly, NOS also possesses a flavin-containing electron-donating reductase and catalyzes the monooxygenation of Larginine to give L-citrulline liberating nitric oxide (NO(g)). NO(g) is an important signaling molecule that carries out an array of critical cellular functionalities. The subject of variable roles of NO(g) has been extensively reviewed elsewhere.230–237 The amino acid sequence of the heme domain of NOS lacks homology with that of P450s, which may exclude NOS from the P450 superfamily.40,238
The catalysis of NOS occurs in two distinct steps: (1) conversion of L-arginine to N-hydroxy-L-arginine and (2) oxidation of N-hydroxy-l-arginine to l-citrulline plus nitric oxide. Notably, NOS forms functional dimers by coupling its heme centers with the aid of calmodulin (a multifunctional calcium (Ca2+) binding messenger protein in eukaryotes), and a Zn2+ ion coordinated to the cysteine pair at the dimer interface, assisting the binding of the L-biopterin cofactor (BH4). This dimer facilitates interdomain electron transfer from flavin proteins to the heme center of the other monomer.239,240 While this process provides the first electron for the reduction of the Fe-bound dioxygen unit, the second electron (forming Cmpd I) is thought to rise from the l-biopterin cofactor, which then gets rereduced by intracellular reductants such as flavin reductase or ascorbic acid (vide infra).17,241 Therefore, the first oxidation step (L-arginine to N-hydroxy-L-arginine) is thought to proceed via a “traditional” P450-type mechanism.242,243 However, the details of the second step of this mechanism (N-hydroxy-L-arginine to L-citrulline) have been highly controversial (Figure 14).
Figure 14.

Two proposed pathways for the conversion of N-hydroxy-L-arginine to L-citrulline plus NO(g) at the active site of NOS. Adapted from ref 17. Copyright 2014 American Chemical Society.
For this subsequent oxidation/oxygenation of N-hydroxy-L-arginine, two mechanistic proposals have been put forward (Figure 14).17 Only one step requires a reducing equivalent; this process is proposed to be an iron superoxo- or peroxo-level oxidant mediating a formal H atom abstraction from the N-hydroxy-L-arginine N–H (and not the O–H) group.244 Pathway A (Figure 14) involves the one-electron reduction of the heme–oxy complex by the reduced biopterin to give a peroxo-level species, which then passes through a cyclic intermediate that collapses to produce L-citrulline and •NO. In support, formation of the BH4+•· radical has been observed during turnover;245 however, the details pertaining to back electron transfer from NO− to BH4+•· radical (final step) remain elusive. In pathway B, the heme–superoxo adduct abstracts a hydrogen atom from N-hydroxy-L-arginine giving a hydroperoxy species, which attacks the substrate as a nucleophile to give a species which decays to products. The activity of NOS is tightly regulated by calmodulin.17
In the above section of this review, we have described a few of the crucial Fe-dependent oxygenases, with the goal of highlighting the reactivity differences of divergent Fe-based dioxygen-dependent oxidants and their mechanisms of action, which helps in broadening our understanding of the mechanistic subtleties involved with the cytochrome c oxidase mechanism, at least for the heme portion of the heme–Cu active site. Most fascinatingly, the oxygenases described patently exemplify how nature utilizes Fe/dioxygen adducts with different levels of oxidation for distinct roles within biological systems. For a full set of heme oxygenases and their functionalities, the reader is referred to Table 1, and extensive literature reviews are available.17,40,41,43,212
2.1.3. Heme-Containing Oxidases: (Chloro)-peroxidases and Catalases.
Hydrogen peroxide (H2O2) serves critical roles in cellular redox signaling, and in oxidative stress,246,247 but of course it is a cosubstrate for peroxidases [and peroxygenases (vide supra)] in required enzymatic oxidation reactions. The O–O bond in H2O2 can (formally) cleave either homolytically or heterolytically. Homolytic O–O bond scission occurs in the presence of reduced iron or copper ions to give deleterious hydroxyl radicals (HO•), via the Fenton reaction. Heterolytic cleavage occurs in peroxidases or catalases, either to detoxify or utilize H2O2 for productive oxidative chemistry. Peroxidase and catalase reaction mechanisms are quite similar (vide infra), with both initially leading to Cmpd I formation. In the former, Cmpd I mediates substrate dehydrogenative oxidations, while for the latter, a second equiv of H2O2 is oxidized to dioxygen, detoxifying hydrogen peroxide via disproportionation. These enzymes contain protoporphyrin IX as the heme prosthetic group [similar to heme-containing globins and Cyt P450 (vide supra)], and the main difference among their active sites is that many peroxidases contain a proximal axially coordinated His residue while catalases accommodate a tyrosinate residue (vide infra).
Peroxidases are generally categorized into three classes: (1) intercellular [e.g., mitochondrial yeast cytochrome c peroxidase (CCP)], (2) extracellular fungal, and (3) intercellular plant [horseradish peroxidase (HRP)] peroxidases. Both peroxidases and catalases contain similar active sites, although subtle differences about their heme centers exist; the imidazole ring of this His residue in peroxidases is perpendicular to the heme, while it is parallel in catalases.45,248 Catalases are the only class of heme enzymes to possess a negatively charged tyrosinate proximal ligation. The proximal His in peroxidase is H-bonded to a conserved Asp residue (Figure 15),249,250 which is thought to induce significant anionic character at the His residue, aiding in heterolytic O–O bond cleavage (vide infra). This enhanced anionic character significantly reduces the Fe3+/Fe2+ redox potential, largely stabilizing the ferric state. This is in clear contrast with proximal His ligating globins. This phenomenon of proximal charge donation in peroxidases was first demonstrated on model systems,249,251 and then further confirmed by protein site-directed mutagenesis studies.101,252 Similarly, the anionic tyrosinate proximal ligation in catalase also results in a negatively shifted Fe3+/Fe2+ redox couple.
Figure 15.

Structure of CCP Cmpd I, where a water molecule is present in close proximity (also observed for Cmpd I of HRP). The protonated distal imidazole reveals the involvement of a proton relay mechanism in the O–O bond scission event of the corresponding Cmpd 0 species.
On the basis of a rich body of spectroscopic and structural insights, the catalytic cycle of peroxidases can be described by three distinct mechanistic transformations:253–257 (l) two-electron reduction of H2O2 by the FeIII resting state, inducing the O–O bond cleavage (via the intermediacy of a FeIII–OOH species (Cmpd 0; Figure 16) and the generation of Cmpd I [note: Cmpd I in peroxidases is unique, in that the π-cation radical can either reside on the porphyrin ring, a Trp,258,259 or a Tyr260–262 residue (vide infra)]; (2) single electron transfer from the substrate to Cmpd I, producing Cmpd II and a substrate-centered radical; and (3) second electron transfer oxidizing a second equivalent of substrate, while the enzyme shuttles back to its resting ferric state. This last step is often the slowest, and thus, the rate-determining step. Phenols, catechols, and aromatic amines often serve as substrates for peroxidases. In the first step, following the binding of H2O2 (Figure 16), the distal His residue263,264 plays a crucial role in “translocating” the proton on the proximal O atom to the distal, which triggers heterolytic O–O bond cleavage. Despite the proposed role of the distal His residue, it is too far from the proximal O atom to form a strong H-bond.265 Thus, the involvement of a water molecule has been proposed,266 which is supported by the distinct positioning of the water molecule in a crystal structure of peroxidase Cmpd I (Figure 15).267–270
Although crystal structures exist, the exact structure of Cmpd I has been a subject of much controversy. This is since spectroscopic methods such as resonance Raman271–273 and EXAFS254,274,275 (EXAFS = extended X-ray absorption fine structure) provide evidence for a FeIV=O bonding description (Fe–O = 1.64–1.67 Å), while X-ray structures256,257,276,277 reveal Fe–O distances more consistent with an FeIV–OH moiety (1.8–1.9 Å). This is now known to be due to the propensity of Cmpd I to be photoreduced upon exposure to X-rays,278,279 and more recent data with minimal X-ray dosage and neutron diffraction methods have revealed distances much more consistent with spectroscopic data [1.73 or 1.88 Å for APX (APX = ascorbate peroxidase), and 1.63 Å for CCP].280,281 Intriguingly, the distal His residue in the latest Cmpd I neutron diffraction structure by Raven and Moody (Figure 15) is protonated (but not in the resting ferric state), suggesting the existence of a proton relay mechanism that ultimately leads to the O–O bond cleavage of Cmpd 0.269,270 Water molecules are thought to be the source of these protons, resulting in a water-mediated, acid-catalyzed O–O bond heterolysis.270 The crucial role of a distal Arg residue in the stabilization and reactivity of CCP Cmpd I has also been proposed.282,283 Furthermore, another elegant recent study by Raven and Moody revealed the crystal structures for Cmpd I and Cmpd II of peroxidases, where they detailed the reasons that may contribute to the much stabilized Cmpd I in peroxidases as compared to that in P450 enzymes.280 The ligand-based radical of Cmpd I in peroxidases was found to also be distributed over a nearby tryptophan (W191; Figure 15) residue rather than just the heme in CCP.259,284 In addition, as mentioned, this radical may even migrate on to a nearby Tyr residue.261,262 By contrast, due to dissimilarities in amino acid composition, HRP Cmpd I bears the radical character mainly on the porphyrinate, generating a ligand- based π-cation radical.
As mentioned, catalase Cmpd I acts upon a second equivalent of H2O2 reducing Cmpd I in a concerted fashion, formally donating two hydrogen atoms (H•; H•· = H+ + e−) producing water and dioxygen (Figure 17). Owing to the concerted nature of this step, catalases do not accumulate Cmpd II as an observable turnover intermediate. Notably, the proton translocation of Cmpd 0, which is mediated by an Arg residue in peroxidases (vide supra), is facilitated by a critical Asn residue in catalases.257 The generally accepted mechanism for catalases is shown in Figure 17.
Figure 17.

Proposed mechanism for catalases, emphasizing the prominent role of the Asn residue. Adapted with permission from ref 45. Copyright 2007 University Science Books.
Very similar reactivities can also be ascribed to chloroper-oxidases (CPO) ( in more general terms, referred to as haloperoxidases).285–288 CPOs, as thoroughly studied highly versatile enzymes, are capable of catalyzing halogenation, dehalogenation, N-demethylation, dismutation, epoxidation, and oxidation reactions.286,288–298 CPO is named after its primary biological role, that is, to carry out chlorination of organic substrates following the C–H bond activation step. These enzymes may be described as hybrids of P450s and peroxidases, despite their structure being notably different from either. CPO contains an axially coordinated cysteinate proximal ligand; however, its distal amino acid structure resembles that of peroxidases, with the presence of a majority of polar side chains that aid heterolytic O–O bond cleavage (vide supra). In spite of the structural differences, the mechanism of formation of Cmpd I in CPO is very much similar to the aforementioned mechanism seen in peroxidases (Figure 16), with the involvement of a crucial Glu residue at the distal site.299,300 A recent theoretical study has highlighted the importance of the hydrogen-bonding network about the active site of CPO as effecting its precise regioselctivity.301 That study further suggested that the hydrogen-bonding interactions in both the proximal and distal sites work in collaboration in order to mediate fast reactivity with H2O2, while maintaining enhanced stability of Cmpd 0, yet decreased stability of Cmpd I. In fact, the Cmpd 0 intermediate of chloroperoxidase has been structurally characterized (Figure 18)302 and possesses a low-spin Fe center in its FeIII–OOH moiety, with Fe–O and O–O distances of 1.80 and 1.50 Å, respectively. Given the versatile catalytic competency of CPO, its catalytic cycle is uniquely amended to carry out multiple classes of reactions mediated by different intermediates formed during turnover (Figure 19).287,289,300,303–305
Figure 18.

Crystallographically characterized Cmpd 0 intermediate of chloroperoxidase: Fe–O = 1.80 Å, O–O = 1.50 Å, and Fe–O–O = 131°.
Figure 19.

Divergent transformations involved within the catalytic cycle of haloperoxidases. Adapted with permission from ref 287. Copyright 2006 Springer Nature Switzerland AG.
2.1.4. O–O Bond Cleavage Process in Heme Enzyme Mechanisms.
The mechanistic details involved with the O–O bond scission process of heme enzymes are of pivotal significance, not only since the subsequent formation of the high-valent species with extreme oxidizing power but also due to implications of such events in catalysis and alternative energy applications.306–308 As described above, Cmpd 0, (P)FeIII–OOH, is subjected to O–O bond cleavage, and technically, either Cmpd II, (P)FeIV=O, or Cmpd I, (P•+)-FeIV=O, can form, depending on the homolytic or heterolytic nature of O–O bond scission, respectively.309 For example, for H2O2 activation by peroxidase, it has been observed that His 42 mediates proton transfer to Cmpd 0, in order to trigger heterolytic O–O bond cleavage.310 However, in mutants where His 42 has been replaced, this proton transfer event is absent, and thus, homolytic O–O bond cleavage was observed, yielding Cmpd II. Subsequently, the Cmpd II species participates in one more electron transfer step (where a protein radical has been generated), giving Cmpd I.311 Accordingly, this two-step generation of Cmpd I was observed to be pH-independent, unlike in the wild-type enzyme. In another example, theoretical studies have proposed the possibility of homolytic O–O bond cleavage in Cyt-P450-mediated fatty acid hydroxylation, where the resultant hydroxyl radical (locked in place by a distinct hydrogen bonding network) abstracts a hydrogen atom from protonated Cmpd II, (P)FeIV–OH, yielding Cmpd I.312 Intriguingly, the necessity of an O–O bond cleavage process in the HO mechanism has also been proposed by Shaik and co-workers using theoretical studies,183–185 but supporting experimental evidence is lacking. In some cases, reversible O–O bond cleavage has also been proposed for heme enzymes.313
Careful analysis of the active site structures of O–O bond cleaving enzymes reveal that this event is almost always driven by a “push-pull”-type mechanism and, thus, is a result of a delicate structure–function relationship. Accordingly, these enzymes often possess highly electron-rich axially ligating amino acid residues such as anionic cysteinate (P450, NOS, Chloroperoxidase) or tyrosinate (catalase) residues, or neutral, but highly electron-rich (vide supra) histidine residues (peroxidases). These axial ligands are highly competent in efficiently pushing electron density into the dz2 orbital of the Fe center.314–318 Such strong axial donation (especially of the thiolate ligation in P450 and/or chloroperoxidase platforms) has been shown to significantly increase the basicity of the Cmpd II-type intermediate in rebound (the FeIV–OH intermediate resulting from the initial hydrogen-atom transfer (HAT); see Scheme 1), furthering the HAT power of the Cmpd I species, while disfavoring undesired outer-sphere electron transfer events.319,320 On the other hand, the distal binding pocket of these enzymes contains precisely positioned strongly hydrogen-bond donating amino acid residues, which interact with the distal O-atom of the Cmpd 0-type intermediate, pulling away electron density. The necessity of such structural subtleties for O–O bond cleavage is nicely demonstrated in the work of Watanabe and Morishima, where functional P450 model enzymes were generated by replacing the axial His residue of Mb with Cys.321 The O–O bond cleavage in CcO enzymes is quite different in that the oxy-form is reduced with four electrons within a single mechanistic step giving Cmpd II. Three of the four electrons in that case are donated by the binuclear heme/copper center,322 while the fourth electron (and a proton) originates from a nearby tyrosine residue (see section 4.2). In contrast, in the related cytochrome bd oxidase mechanism, all four electrons are generated from the binuclear bis-heme active site itself, resulting in the formation of Cmpd I.323 Notably, mechanistic studies of O–O bond cleavage of hemoproteins could be complicated by subsequent redox events that modify the actual products of the reaction. For example, Watanabe and coworkers have reported the rapid reduction of a Cmpd I species formed in solution (from heterolytic O–O bond cleavage) to the respective Cmpd II species (as would be expected from homolytic O–O bond cleavage) during H2O2-dependent oxidations mediated by Mb mutants.324–326 Such complications resulting from subsequent redox events have also been reported in model systems, and in fact, much detailed work has been carried out in order to understand pH and solvent effects on the O–O bond cleavage mechanism(s) and is described in section 2.4 (vide infra).49
2.1.5. Synthetic Models of Cmpd I and Cmpd II.
As described above, biological systems often utilize the superior oxidizing power of high-valent (effectively FeV or FeIV) iron oxidants in multiple substrate conversion processes. To repeat, Cmpd I’s are overall (P)FeV=O oxidants, but with electronic structures best described as a FeIV=O core magnetically coupled with a porphyrin-based π-cation radical, and thus formulated as (P•+)FeIV=O. Cmpd II’s are (P)FeIV=O species, with a neutral porphyrinate supporting ligand (Figure 20). These intermediates derive from peroxide level species’ O–O reductive cleavage, thus the understanding of such processes is relevant to the HCO reaction mechanism (see section 2.4.2). Due to their highly oxidizing nature, they have mostly been observed as fleeting intermediates in biological catalytic cycles, one clear exception being the Cmpd I of peroxidase, which has been characterized crystallographically (Figure 15).269,280 Green and co-workers more recently have extensively spectroscopically characterized the Cmpd I intermediate in cyt. P450 monooxygenases, using advanced experimental techniques to generate and handle this species; the focus has been on the study of the role of the thiolate (from cysteine) proximal axial ligand in enzyme activity, by elucidation of reaction kinetics, redox potentials, and pKa of the protonated Cmpd II.
Figure 20.

Earliest characterized high-valent oxo-FeIV=O model complexes: [(TMP•+)FeIV=O]+ (left) and [(TMP)FeIV=O] (right). See text for details.
The studies of model systems of such intermediates serve to significantly increase our understanding of their geometric and electronic structure (which delicately controls their reactivity), as well as provide insights into the scope and mechanism of substrate reactivities. Furthermore, the main C–H activation mechanism that these oxidants follow, often referred to as the “oxygen-rebound” mechanism (Scheme 1; initially proposed by Groves),136 has been extensively studied using model systems, which has shed light on pivotal factors that tightly govern the reactivity of these oxidants.135,137 In this section, rather than reporting on all Cmpd I and Cmpd II models described in the literature, we will discuss the essence of their structure and reactivity, presenting appropriate specific examples.
2.1.5.1. Cmpd I/II Models: Electronic Structure and Substituent Effects.
The pioneering work in high-valent porphyrinate models is ascribed to Groves and co-workers, when they carried out the two-electron oxidation of the FeIII precursor complex of TMP (TMP = 5,10,15,20-tetra-(mesityl)porphyrin; Figure 20), using the O-atom transferring reagent, m-CBPA, at –78 °C.327,328 The authors spectroscopically characterized the electronic structure as an overall S = 3/2 system, where an FeIV (S = 1) center is tightly ferromagnetically coupled with the porphyrin π-cation radical (S = 1/2) moiety.134 EPR spectroscopic features are consistent with this conclusion, with signals observed at g = 4.3, 3.9, and 1.99 (J > + 40 cm−1) for [(TMP•+)FeIV=O]+ at 4 K.329 In fact, this model system exhibited much stronger magnetic coupling than that observed for Cmpd I species of peroxidase enzymes (vide supra).330 In further studies, it was shown that the one-electron chemical oxidation of a Cmpd II species, obtained via thermally induced homolytic O–O bond cleavage of [{(TMP)FeIII}2(O22−)], yields Cmpd I at –70 °C in toluene (Figure 20). This connected Cmpds I and II as one-electron redox partners and illustrated that compound I species can be directly derived from dioxygen.331
Further work has also demonstrated the synthetic utility of dimethyldioxirane as an O-atom transfer agent (with TMP porphyrinate)332 and ozone (TDCPP porphyrinate; Figure 21)333 as an oxidant to successfully generate these high-valent heme models. Cmpd I model systems have been characterized using a variety of spectroscopic techniques,133 and the electronic absorption features of [(TMP•+)FeIV=O]+ bear striking similarities to the Cmpd I spectrum of catalase.334 Importantly, the combined insight from EPR and NMR spectroscopies together with theory strongly suggests that the [(TMP•+)FeIV=O]+ has an a2u ground state, where the radical is placed on a porphyrin-based orbital with an a2u symmetry. Although Cmpd I intermediates within model systems have never been crystallized, important structural information has been revealed by X-ray absorption spectroscopy, including the finding of an expected short Fe–O distance of 1.6 Å for [(TMP•+)FeIV=O]+,335 in good agreement with that observed for HRP Cmpd I.275 Resonance Raman spectroscopic data collected on the same model system definitively revealed (from 54Fe-O and Fe–18O isotope labeling) an Fe-O stretch centered at 828 cm−1 in a methanol/dichloromethane (DCM) mixture,336 which shifted to 801 cm−1 in pure DCM.337 This shift in υFe-O energy was attributed to the different properties of axially coordinated ligands (i.e., chloride anion in pure DCM versus methanol in methanol/DCM mixtures) for each case.338 In fact, these axially coordinated ligands, along with the identity of the oxidant and the solvent are known to significantly affect the formation of Cmpd I model complexes (Figure 22),133 where they govern key mechanistic features such as homolytic versus heterolytic O–O bond cleavage in synthetic precursors. For example, when [(TMP)FeIII(X)] (where X is an axially coordinated anionic Cl−, ClO4−, or benzoate ligand) is reacted with m-CPBA in DCM, the Cmpd I species, [(TMP•+)FeIV=O]+, is produced. However, when [(TMP)FeIII(X)] is oxidized with m-CPBA in toluene where X = HO−, acetate, or m-chlorobenzoate, the formation of the [(TMP)FeIII-N-oxide] complex is observed (Figure 22). The latter observation strongly suggests that under the given experimental conditions, the homolytic O–O bond cleavage of a putative Fe-m-CPBA adduct occurs, followed by N-O bond formation with one of the pyrrole N-atoms of the porphyrin ring. It is noteworthy that these major effects resulting in the differential formation and/or reactivity of high-valent model systems as a consequence of subtle experimental modifications has,135,138 and will continue to, shed light on yet unclear mechanistic details implicated with such intermediates in biology.
Figure 21.

Various porphyrin meso- and β-pyrrole derivative supporting ligands that have been utilized to prepare high-valent oxo-heme model complexes. See text for details.
Figure 22.

Synthetic methods for [(TMP•+)FeIV=O]+ and [(TMP)FeIV=O] with various axial ligands, oxidants, and solvent environment. See text for discussion. Adapted with permission from ref 133. Copyright 2016 Royal Society of Chemistry.
Apart from these aforementioned factors, differently substituted porphyrinates with varying overall electronic structure have also been utilized to generate high-valent iron compounds (Figure 21; note that these ligands could possess substituents at either meso- or pyrrole β-positions of the porphyrinate ring). In model systems, EPR spectroscopic and theoretical studies have identified two possible π-cation radical (P•+)FeIV=O states (a1u and a2u).339 The electronic structure of these (P•+)FeIV=O species are highly sensitive to the effect of substitution at the porphyrinate meso-, pyrrole β-positions.340 Incorporating electron-withdrawing groups at the porphyrin meso- position lowers the energy of the a2u state (observed in [(TMP•+)FeIV=O]+), allowing energy mixing with the fully occupied a1u molecular orbital via vibronic coupling. Concomitantly, the ferromagnetic coupling interaction between the S =1 FeIV center and S = 1/2 π-cation radical decreases as the electron-withdrawing effect increases, and the HOMO becomes increasingly rich in a1u character.340,341
Interestingly, incorporation of mesityl groups at the pyrrole β-positions (Figure 21) results in a different π-cation radical ground state, one with an a1u symmetry, as observed in the [(TMTMP•+)FeIV=O]+ model system (Figure 21).334 In pyrrole β-derivatized porphyrins, the lack of a meso-mesityl group stabilizes the a2u orbital (because, mesityl groups are electron-releasing), which makes the a1u orbital higher in energy and hence singly occupied. Unlike the a2u state, the a1u state leads to weak ferromagnetic coupling between the porphyrin S = 1/2 π-cation radical and the S =1 FeIV=O moiety due to a lack of spin density on the porphyrin nitrogen atoms. The 1H NMR and EPR spectroscopic properties of [(TMTMP•+)FeIV=O]+ are similar to those of horseradish peroxidase compound I, as well as ascorbate peroxidase and Micrococcus lysodeikticus catalase.341–343 This similarity in the strength of ferromagnetic coupling of the S =1/2 porphyrin radical with the S =1 FeIV=O is also observed in other pyrrole β-substituted synthetic models, suggesting that these represent closer approximations to biological Cmpd I species supported by protoporphyrin IX.
Cmpd I model complexes are capable of reacting with organic substrates in oxo-transfer, epoxidation, and C–H bond activation reactions (Figure 23). For phosphines (e.g., PPh3), sulfides, and olefins, oxygen atom transfer reactions are effected.133,344 Intriguingly, the reaction mechanisms of sulfoxidation and oxidative N-demethylation reactions mediated by Cmpd I model complexes have been found to be very different from those involving Cmpd I in peroxidases, for which, various crucial factors including the predominant steric effects imposed by enzyme protein matrix have been discussed.345,346 Electron-withdrawing groups increase the reactivity of these Cmpd I model complexes for cyclohexene epoxidation reactions. When the reactivity of [(TMP•+)FeIV= O]+ for cyclohexene epoxidation was compared to that of its trichloro-substituted analogue [(TTCPP•+)FeIV=O]+ (Figure 21), the latter reacts much faster.340 This is attributed to the increased redox potential of the trichloro-substituted Cmpd I species, which in turn increases the rate of olefin oxidation. The ground state (a1u or a2u) of the Cmpd I species within model systems does not appear to have a great effect on their reactivity, as both [(TMTMP•+)FeIV=O]+ and [(TMP•+)-FeIV=O]+ displayed similar activity in cyclohexene epoxidation reactions.347
Figure 23.

Organic substrate transformations carried out by synthetic compound I model species.
Cmpd I model complexes also catalyze the hydroxylation of unactivated hydrocarbon C–H bonds including aromatics, as do cytochrome P450s.126,348–350 One recent example from Groves and co-workers which incorporated a positively charged periphery in porphyrin framework, 4-TMPyP (4- TMPyP = 5,10,15,20-tetrakis(N-methyl-4-pyridinium)-porphyrin (Figure 21), displayed extraordinarily high C-H bond hydroxylation reactivity. This was attributed to the positively charged porphyrin framework, which lowered the energy of the a2u orbital, making spin state crossing more facile, while destabilizing the FeIV=O π-cation radical.351 Generally, with hydrocarbon substrates, an oxygen rebound mechanism is invoked; for instance, a H atom abstraction from the hydrocarbon substrate occurs, forming an FeIV-OH intermediate, which then rebounds with the organic substrate radical yielding the alcohol product (Scheme 1).348 Fujii and co-workers have recently investigated the factors governing hydrogen tunneling in the rate-limiting HAT step in aliphatic C-H bond activation. Kinetic studies displayed nonlinear Arrhenius plots, which could be fit with equations derived from Bell's tunneling model to prove that hydrogen tunneling contributes to the rate-limiting step. The extent of this tunneling was found to be dependent on the reaction temperature, C-H bond dissociation energy of the substrate, reactivity of the metal oxidant, [(TMP•+)FeIV=O(L)]+ (where L = NO3, Cl−, and Im), and the Fe=O bond strength.352 The role of ferryl protonation and solvent participation for oxygen atom transfer reactions of these high-valent heme models in aqueous media were also recently evaluated.353
The hydroxylation reaction of aromatic compounds is less well-understood. Several reaction mechanisms have been proposed for aryl hydroxylation by Cmpd I species, including hydrogen atom transfer (HAT), arene oxide formation, electron transfer/bond formation, and electrophilic attack (Scheme 5).354 The HAT mechanism has been largely disfavored due to several factors: (1) observation of smaller KIE values than expected, (2) the strength of aryl C–H bonds oxidized, and (3) key theoretical findings (Scheme 5, pathway A).355–357 Arene oxide formation has been ruled unlikely based on product analysis and other experimental evidence (Scheme 5, pathway b).358–360 Direct electron transfer has been observed in cytochrome P450 compound I mediated oxidation of tetramethoxybenzene, since electron transfer oxidation is exergonic for these substrates (Scheme 5, pathway C).361 In a kinetic study by Nam and co-workers, isotope labeling, inverse kinetic isotope effects, and a largely negative Hammett ρ (rho) value for the reaction of para-substituted benzenes with [(TPFPP•+)FeIV=O]+ provided evidence for an electrophilic attack type mechanism as shown in pathway D in Scheme 5.362 Very recently, Fujii and Asaka suggested a new hybrid mechanism through the generation and analyses of Marcus plots that elegantly incorporate features of both pathways C and D. In this case, the aromatic substrate and Cmpd I species participate in an initial electron transfer equilibrium (surrounded by the solvent cage), which is coupled to C–O bond formation (Scheme 5, inset). This proposal reconciles the main features of pathways C and D and provides a new potential mechanistic scenario for aromatic hydroxylation facilitated by Cmpd I species.354
Scheme 5.

Proposed Mechanistic Pathways for Aromatic Hydroxylation Reactions Mediated by Cmpd I Model Systemsa
aThe new mechanistic scenario suggested by Fuji and Asaka is shown in the inset. Adapted from ref 354. Copyright 2016 American Chemical Society.
In addition to affecting Cmpd I species formation, axial base ligands markedly influence the substrate reactivity of the (P•+)FeIV=O species, enhancing the rate of epoxidation as the donor strength is increased.363,364 For example, when imidazole and phenolate axial ligands were incorporated, the rate of substrate epoxidation increased ~100–400 fold when compared to one having an axially ligated nitrate ligand on the Fe center.364
The first example of a Cmpd II model complex was reported in 1980 by Balch and co-workers, which formed via the homolytic O–O bond cleavage of a bridging μ-peroxo ferric heme complex (see Scheme 6).365 This peroxide species was produced by oxygenating the ferrous heme complex, [(TmTP)FeII] (TmTP = meso-tetra-m-tolylporphyrin), at –80 °C in toluene. The subsequent addition of a pyridyl or imidazolyl axial base to this μ-peroxo species resulted in the homolytic O–O bond scission (Scheme 6), producing the corresponding Cmpd II species. The paramagnetic 1H NMR spectra of this model and others (prepared similarly) were compared to that of the Cmpd II species of HRP and a ferryl myoglobin derivative to confirm their assignment as an FeIV= O adduct. Specifically, the chemical shift values of the porphyrinate meso-positions366 (for β-pyrrole derivatized heme) and imidazolyl axial base protons367,368 were compared. Model Cmpd II complexes were also prepared via the iodosobenzene oxidant using a similar FeIII precursor, [(TMP)FeIII(X)], featuring strongly coordinating axial ligands such as Cl−, F−, CH3CO2−, and OH− in toluene and dichloromethane (see Figure 22). However, more weakly coordinating axial ligands such as ClO4−, CF3SO3−, and NO3− yielded the compound I analogue (Figure 22).51 In a basic methanol media, [(TMP)FeIV=O] could also be prepared via the oxidation of [(TMP)FeIII(OCH3)] with iodosylmesitylene.369
Scheme 6.

Synthetic Route to O2-Derived Cmpd II Complexes365
Other examples of porphyrinoid Cmpd II models have been prepared via chemical oxidation,370,371 ligand metathesis,372 and the reduction of compound I species.373 Interestingly in one indirect route, the oxidation of a porphyrinoid FeIII–OH species results in a π-cation radical porphyrin FeIII–OH adduct, which was then deprotonated (using tetra-N-butylammonium hydroxide) to yield the corresponding FeIV=O Cmpd II analogue.374–377 In terms of spectroscopic properties, porphyrinate FeIV=O species are EPR silent (in perpendicular mode) due to their S =1 integer spin state.365 Their FeIV=O stretching frequencies have been characterized by rR spectroscopy, and five-coordinate Fe=O stretches range from 852–863 cm−1 as determined from studies in a low-temperature (15 K) matrix134,378 Electron-withdrawing groups at the meso- positions resulted in stronger Fe=O bonds.379–381 Solution phase measurements of the Fe=O bond stretches indicated lower values ranging from 841–845 cm−1.382–384 Adding an axial imidazole ligand such as 1- methyimidazole shifted the Fe=O bond stretches to lower frequencies, ranging between 815 and 820 cm−1.385–387 However, these v(Fe = O) values are still substantially higher than those reported for Cmpd II species of peroxidases (745–790 cm−1). Notably, in a recent model system study, a [(TMP)FeIV=O] species with an axially coordinated imidazole ligand revealed its Fe=O bond stretch to occur at 792 cm−1, in good agreement with that of peroxidase Cmpd II.385
In terms of reactivity, synthetic Cmpd II complexes have been reported to carry out oxo-transfer reactions with triphenylphosphine388 and with olefins.371,372 Both solvent polarity and substrate properties are important for determining the nature of carbon-centered radical intermediates formed during the course of the reaction.372 When electron-withdrawing substituents are incorporated at the porphyrin meso-position, the rate of reaction between the porphyrin FeIV=O adduct and olefins is decreased. It was suggested that the rate-limiting step for this reaction is a disproportionation process involving the FeIV=O adduct, yielding the corresponding FeIV=O π-cation radical species (i.e., a Cmpd I species), which is in reality the active oxidant.371 Electron-withdrawing substituents would slow down this disproportionation reaction, decreasing the rate of formation of the active oxidant, and thus, the overall rate. However, Cmpd II model complexes are reportedly able to abstract hydrogen atoms from substrates with weak bond dissociation energies such as xanthene.370
Synthetic Cmpd I adducts have also been utilized in the modeling of chemistry related to two heme containing haloperoxidases, namely chloroperoxidase (CPO) and myeloperoxidase (MPO). As mentioned above, haloperoxidases are a special class of peroxidases, which carry out the two-electron oxidation of halides such as chloride and bromide to hypochlorite (−OCl), and hypobromite (−OBr), respectively. Chloroperoxidases are involved in the biosynthesis of chlorine-containing compounds, and they possess a thiolate axial base coordinated to the heme (section 2.1.3 and Figure 19).389–391 Myeloperoxidases are involved in the antimicrobial defense system in neutrophils and feature a histidine imidazole axial base. Both of these enzymes utilize H2O2 in the formation of Cmpd I, which then react with chloride to form hypochlorite, which is believed to be the key oxidant. However, details concerning oxidative halogenation by Cmpd I are not fully understood in this case; in fact, the true oxidizing agent is not known for CPO. Hence, it is a current goal to study the reactivity of small molecule Cmpd I species toward halides.389–391 Recent work has shown that although [(TMP•+)FeIV=O]+ does not react with chlorides, its perfluorinated analogue [(TPFPP•+)FeIV=O]+ does (Figure 21). Addition of tert-butylammonium chloride [(TBA)+Cl−] to [(TPFPP•+)FeIV=O]+ yielded its compound II species, [(TPFPP)FeIV=O], instead of the ferric heme, as occurs in haloperoxidases. This product could have resulted from direct oxidation of the chloride by the [(TPFPP•+)FeIV=O]+ to yield Cl2(g) and [(TPFPP)FeIV=O] or from homolytic cleavage of a Cl-O bond in a ferric hypochlorite adduct. Both of these pathways would yield chlorine radicals within the reaction mixture, which may then lead to the liberation of Cl2(g).392 Remarkably, when substrates are added to the Cmpd I/(TBA)+Cl− mixture, substrate chlorination was observed. For example, 1,3,5-trimethoxybenzene was oxidized to 1-chloro-2,4,6-trimethoxybenzene but not 1-chloromethoxyl-3,5-dimethoxybenzne. This indicated that a chlorine radical is not likely to be the active oxidant. Anisole and cyclohexene were also oxidized to p/o-chloroanisole and trans-1,2-dicyclohexene, respectively. The product ratio of p- to o-anisole was similar to that obtained from direct bubbling of Cl2 through an anisole solution.392,393 To this end, it can be postulated that Cl2 is the active chlorinating agent in these transformations, and thus, the relevance of these models to CPO and MPO is not certain.
When tert-butylammonium chloride is added to [(TPFPP•+)FeIV=O]+ in the presence of trifluoroacetic acid, a new chlorinating agent is formed, a ferric meso-chloroisoporphyrin species (Scheme 7).394 The formulation of this ferric meso-chloroisoporphyrin comes from corroborative UV-vis, NMR, and EPR spectroscopic features, when compared with those of other known isoporphyrin species. Further, electrospray ionization mass spectrometric data also validated this formulation. This species was observed to be an active chlorinating agent, producing 1-chloro-2,4,6-trimethoxybenzene from 1,3,5-trimethoxybenzene, and yielded ratios of para- and ortho-chlorinated anisole, similar to CPO. The generation of aqueous hypochlorite was also observed under acidic conditions.393,395 Even though these results do provide evidence for the chlorination competency of meso-chloroiso-porphyrinoids in model systems, the relevance of such intermediates in CPO or MPO chemistry remains unclear.
Scheme 7.

Formation of meso-Chloroisoporphyrin Complexes from [(TPFPP•+)FeIV=O]+ and Its Oxidative Reactivity with Anisole and Cyclohexenea
aAdapted from ref 394. Copyright 2012 American Chemical Society.
Although direct evidence for hypochlorite formation via a Cmpd I species was not obtained, one study showed that addition of tert-butylammonium hypochlorite to the ferric hydroxide species, [(TPFPP)FeIII-(OH)], at low temperature gives a ferric bis-hypochlorite product, [(TPFPP)- FeIII(ClO)2]−, a species that is capable of sulfoxidation, epoxidation, and chlorination chemistry.396 A predominant axial ligand effect was observed when comparing this species' reactivity to that of its monohypochlorite analogue, [(1-methylimidazole)(TPFPP)FeIII(ClO)], where the former was observed to be much more reactive. This could indicate that the ferric bis-hypochlorite adduct is a closer model of CPO, which features a more activating thiolate axial ligation on heme, as opposed to MPO, with an axially coordinating imidazole ligand.396
Synthetic models of Cmpds I and II have also been accessed through heterolytic (Cmpd I) and homolytic (Cmpd II) O–O bond cleavage of iron(III)-acylperoxide, -alkylperoxide, and -hydroperoxide species (see Scheme 8 and section 2.4). This methodology is particularly biologically relevant, considering that FeIII(−OOH) O–O cleavage is the main pathway that leads to Cmpd I- and Cmpd II-type species in catalases, peroxidases, and cytochrome P450s. Proximal axial base properties and hydrogen-bonding interactions with distal amino acid residues are critical aspects governing the O–O bond cleavage event of FeIII–OOR-type intermediates within enzymatic systems, and these factors are discussed in detail in section 2.1.
Scheme 8.

Generation of Cmpd I and Cmpd II Type Species via Respective Heterolytic and Homolytic O–O Bond Cleavage Reactions of Heme FeIII–OOR-Type Intermediates
2.2. Dioxygen Binding at Hemoprotein Models: Ferrous-Oxy/Ferric-Superoxo Systems
2.2.1. Significance and Early Studies on Biological Heme-oxy Systems and Their Models.
For the past 60 years, a major portion of bioinorganic research efforts has geared toward modeling active sites of heme-containing proteins, especially on revealing the fascinating mysteries behind biological gas carriers and storage proteins with reversible gas-binding properties. This area was majorly fueled by the elegant crystallographic work by Perutz and Kendrew on structural properties of hemoglobin and myoglobin, respectively,54,55 which earned them the Nobel Prize in 1962. Small molecule models of these hemoprotein active sites have significantly contributed to the current understanding of their biochemistry, most of which were initiated by the classic “picket-fence” porphyrin models of Collman in the early 1970s (vide infra).56,397 In this review, we will summarize heme-only model systems with different reduced derivatives of dioxygen ligated, starting from iron(III)-superoxo up to Cmpd I models (Figure 24). All of these adducts have been proposed as key intermediates for reversible dioxygen binding in O2-carrier/storage proteins and/or in enzymatic biomolecule transformations via oxidase or oxygenase activity15 (vide supra, section 2.1). In general, these intermediates are generated from heme FeII or FeIII precursor complexes through either dioxygen activation or reactivity with reduced derivatives of dioxygen [i.e., superoxide (O2−•), peroxide (O22−), or oxide (O2−)] often under specialized reaction conditions. As such models allow facile variation of electronic and steric properties surrounding the Fe center, they often provide a very useful handle on probing structure–function relationships that are crucial for the optimal function of corresponding biomolecules.
Figure 24.

Six-coordinate [i.e., with an axially ligated base (B)] heme–Fe adducts bearing different reduced derivatives of dioxygen as axial ligands (i.e., superoxo, peroxo, and oxo). Inspired by the relevance of these adducts in various enzymatic systems (see section 2.1), small molecule models have been generated and interrogated in great detail. Significant insights gained from such models are discussed in the sections that follow, highlighting their applicability toward a more thorough understanding of the CcO enzymatic system.
2.2.2. Ferric-Oxy/Ferric-Superoxo Model Systems.
Designing model systems that can accurately mimic the geometric and electronic characteristics of iron(III)-superoxo moieties that form within biological heme-containing proteins continues to be a challenging task for model chemists. This is not merely due to their impaired thermal stability but also owing to the complexity of their electronic structure,70 as well as their paramount significance in reversibility and cooperativity of dioxygen binding.56 In fact, only a handful of oxy-heme models display cooperative dioxygen binding capability to-date.71,398 Heme iron(III)-superoxo moieties can be readily generated when FeII precursor complexes are exposed to dioxygen, although their isolation and stabilization in solution often requires cryogenic temperatures. In addition, aprotic and/or sterically encumbered environments may also enhance the stability of these iron(III)-superoxo adducts, since their proposed decay pathways have been shown to involve protonation and/or dimerization processes as described in detail below.79
Two main pathways have been proposed for the irreversible oxidative decay of heme iron(III)-superoxo moieties in solution. First, the μ-peroxo pathway involves the interaction of a monomeric iron(III)-superoxo complex with a second equivalent of iron(II)-porphyrinate in solution giving a bridged peroxide ligand between two FeIII centers, (P)FeIII– (μ-O–O)– FeIII(P) (P = porphyrinate ligand; also see Scheme 9). The basis for this proposal comes from kinetic measurements of oxygenation carried out by Cohen and Caughey on 2,4-diacetyldeuteroporphyrin IX dimethyl ester iron(II) (Figure 25A), where they observed second-order and first-order rate dependencies with respect to FeII and dioxygen concentrations, respectively.399,400 This suggested that the rate-limiting step of dioxygen activation proceeds with a Fe:O2 stoichiometry of 2:1, which was also confirmed by others in later studies.401,402 In light of this work, the isolation and spectroscopic characterization of a (P)FeIII– (μ-O–O)– FeIII(P) species in cryogenic temperatures was first reported by Balch and coworkers,403,404 and propensity of such compounds to thermally decay giving a diiron(III)-monooxo-bridged “dimer”, (P)FeIII– O–FeIII(P), was later reported.367 Thus, the aforementioned μ-peroxo species was proposed to undergo homolytic O–O bond cleavage giving two equivalents of Cmpd II [or (P)FeIV=O, also see below], which then binds another equivalent of (P)FeII.71
Scheme 9.

Proposed Interconversions Involved in the μ-Peroxo Decay Pathway of Iron(III)-superoxo porphyrinate (P) Complexes; B = Axially Ligating Base, a Weak One Such As a Solvent; B’ = a Strong Axial Ligating Base Such As a Pyridyl or Imidazolyl Moiety
Figure 25.

Chemdraw depictions of iron(II) complexes of 2,4-diacetyldeuteroporphyrin IX dimethyl ester (A), protoporphyrin IX (B), octaethylporphyrin (C), and tetraphenylporphyrin (D) with substituted imidazole axial ligands; tetramesitylporphyrin (E) with axially ligated piperidine; examples of systems with tethered axial bases [F (PIm) and G (PPy)]; “picket-fence” porphyrin with substituted imidazole axial ligands (H); single-strapped (I), and double-strapped (J) “basket-handle” porphyrins; examples with steric bulk at both sides of the porphyrinate (K, M); and examples of capped porphyrinates (L, M). See the text for further discussions of these complexes.
Second, a proton-facilitated decay pathway of iron(III)- superoxo adducts has also been proposed, where protonated superoxide dissociates, leaving behind the oxidized FeIII center. This has been presented as a plausible mechanism responsible for the formation of Methemoglobin (MetHb) from oxyhemoglobin (see section 2.1), where water may play a key role as a proton donor and/or a nucleophile facilitating the release of free superoxide.83,405 Correspondingly, multiple hemoglobin and myoglobin mutant proteins have also provided evidence in favor of such a protonation-assisted decay pathway of their oxy- forms.405–408 Although probing such a mechanism using model systems is much more complicated given the possibility of acid–base chemistry involving noninnocent ligands (such as axial imidazoles or pyridines). Momenteau and co-workers have observed that the decay of oxy-heme “basket-handle” porphyrinates (Figure 25, panels K and M) display a sharp dependency on the water content in organic solvents (such as toluene and dichloromethane).79,409–411 This is an interesting observation, given the fact that “basket-handle” porphyrin complexes are incapable of dimerizing (due to their extreme steric encumbrance) and therefore cannot proceed through a μ-peroxo decay mechanism. Furthermore, a monomeric FeIII–OH moiety was observed as the final metal species following the decay process, along with the production of hydrogen peroxide (which is known to form by the disproportionation of protonated, metal-free superoxide), which led to the proposal of a second decay pathway summarized in Scheme 10.
Scheme 10.

Protonation-Assisted Decay Pathway Proposed for the Iron(III)-Superoxide Adduct of a "Basket-Handle” Porphyrin; P = Supporting Porphyrinate
In light of the insight into these mechanisms, two essential properties have been identified for model systems, in order to successfully mimic the dioxygen binding chemistry of hemoproteins: (1) a five-coordinate (5C) FeII center including one equivalent of axially ligated base (e.g., exogenous or tethered imidazole or pyridine; importantly, imidazole has been shown to exceptionally improve dioxygen binding capability compared to pyridine)251 and (2) steric bulk on either or both sides of the heme–iron center (e.g., “picket-fence”, “capped”, “strapped”, or porphyrins with “hanging base”; see Figure 25, panels A, I, J, L, and M). Generating 5C iron(II) complexes with one equivalent of coordinated axial base is often challenging, due to the strong thermodynamic driving force toward the formation of 6C iron(II) complexes with two axial bases, also known as hemochromes.399,412,413 However, fine-tuning of the steric properties has led to the successful formation and isolation of desired 5C iron(II) model complexes.
In 1973, Collman and Reed discovered the first 5C iron(II) model complex using tetraphenylporphyrinate (TPP) and axially ligated 2-methylimidazole (Figure 25D).414,415 The out-of-plane displacement of the high-spin iron(II) center in this 5C complex416 minimizes steric interactions between the porphyrin and the methylimidazole ligand, allowing its successful isolation. In contrast, the corresponding low-spin complex (with two axial imidazole ligands) would move the FeII center in-plane with the porphyrin ligand, resulting in a dramatic increase of the unfavorable steric interactions, which makes its formation an overall energetically uphill process. Since this work, numerous 5C iron(II)-porphyrinate complexes have been generated and isolated following different methodologies, some of which have been characterized structurally.56,72,79,412,417,418 Furthermore, despite being synthetically more demanding, several examples with porphyrinates with tethered axial bases have also been reported as an approach for generating stable 5C iron(II) complexes.79,419–423 Due to the intramolecular iron-base coordination, these systems produce 5C FeII complexes with greater yield and purity as compared to systems which require an exogenous base. However, in spite of the built-in steric bulk of the tethered bases, these systems have also exhibited dimer formation at cryogenic temperatures.424–426
Introducing steric bulk on single or both sides of a heme has also been utilized in order to minimize dimerization of Fe centers as proposed for the decay of iron(III)-superoxo adducts (Scheme 9). The formation of 5C iron(II) compounds of these face-hindered hemes can be more thermodynamically favorable than their 6C counterparts, which is desirable for designing models with close resemblance to the biological heme centers.79 It is noteworthy, however, that these sterically encumbered hemes had been primarily designed to provide a preferential, protected binding site for dioxygen in most cases rather than to prevent dimerization or hemochrome formation.79 Notably, the 5C FeII complexes of these sterically crowded porphyrinates serve as close models for the dioxygen binding sites of myoglobin and hemoglobin, as observed and explained using X-ray absorption spectroscopy.62,427
The geometric and electronic properties pertaining to biological oxy-heme forms have been under severe literature debate (section 2.1.1), mainly due to the lack of structural understanding until the elegant, now classic model studies carried out by Coliman and co-workers on crystallizing the oxy- form of “picket-fence” porphyrin in the 1970s (Figure 26).72,397,428–430 This porphyrinate macrocycle consists of four ortho-pivalamide phenyl groups at meso- positions, which creates an ~5.4 A deep hydrophobic pocket on one side of the iron center.397 The 5C FeII complex was initially prepared along with 1 equiv of 1-methylimidazole, which exhibited reversible dioxygen binding capability. The crystal structure of the oxygenated complex displayed an end-on bent (Fe-O–O angle ~126°) dioxygen ligand in close agreement with Pauling’s 1964 model431 for describing oxy-Hb electronic structure (vide supra). This oxygenated complex crystallized in two space groups with four molecules in each unit cell, where the Fe–O–O plane was either parallel or perpendicular to the trans- axial imidazole plane. Thus, significant distortions resulted, albeit the preliminary structure was calculated with satisfiable accuracy. The structure reveals O–O distances of 1.23(8) and 1.26(8) Å, which are in good agreement with that of coordinated superoxide.432 This and two other closely related heme iron(III)-superoxo complexes (differing only by the substituents on the axially ligated imidazole) remain the only crystallographically characterized heme–superoxo species in literature to-date.72,429,430
Figure 26.

Classic oxy-heme “picket-fence” porphyrin complex, originally designed, generated, and crystallographically characterized by Collman and co-workers,397 is here depicted in a later X-ray structure determined by Li, Schulz, and Scheidt.433 Selected bond lengths and angles are shown; these parameters are very close to those previously reported by Collman and co-workers. Also, see the text.
Finally, this complex exhibited strong resemblance in its Mössbauer spectroscopic properties to those of oxy-Hb, reaffirming its suitability as an excellent synthetic mimic.414,434 Furthermore, it has also been proposed that the pivalamide functionalities in “picket-fence” porphyrins and their derivatives could play a key role in facilitating dioxygen binding and stabilizing the oxy- form utilizing noncovalent interactions with dioxygen435 as seen with a distal histidine in the protein environment.87,431,436 However, steric properties of these bulky ligands could also significantly affect the dioxygen affinities of the ferrous center.437 Interestingly, the successive structural work on oxy-hemoglobin and oxy-myoglobin by Shaanan69 and Phillips,68 respectively, have also revealed Fe-bound end-on angular superoxide moieties similar to those in Collman’s models.
The effect of water molecules on the dioxygen affinity and selectivity at the heme center of the aforementioned O2 transport/storage proteins407,438,439 have also been modeled by Collman and co-workers. In that study they utilized several “picket-fence” derivatives with tethered axial bases to provide evidence for a negative effect of water on the dioxygen binding kinetics, possibly due to its protic properties.440 Furthermore, in addition to these oxy-heme adducts of “picket-fence” and related porphyrinates by Collman and co-workers, multiple research groups have synthesized and characterized a variety of such derivatives, as well as other sterically encumbered porphyrinates (with or without tethered axial bases; see Figure 25) with the intent of gaining more insight into the complicated dioxygen binding process of hemoproteins. These efforts have been comprehensively reviewed elsewhere.56,79,441,442
More recently, Li, Schulz, Scheidt, and co-workers have reported on detailed structural, spectroscopic, and theoretical studies on the temperature-dependency of the Mössbauer signatures of three oxy-“picket-fence” species with different coordinated axial bases.433,443 The Mössbauer spectral features of biological and synthetic oxy-forms are known to be similar; however, they exhibit atypically large quadruple splittings and line shapes that display unusual temperature-dependencies compared to other low-spin ferrous systems. The three oxy-compounds examined in this study were crystallized at variable temperatures (80–300 K), which revealed the presence of two independent orientations of the Fe-bound dioxygen unit and of the pivalamide groups. In all cases, the major dioxygen plane of the iron(III)-superoxo unit displayed a near-perpendicular orientation with respect to the axial imidazole ligand plane. Furthermore, the relative orientations of ligated dioxygen unit and pivalamide functionalities are strongly correlated with each other,444 resulting in a strong coupling between the feasibility of dioxygen rotations with relative energetics of picket positions.433 Interestingly, the previously reported iron(III) and iron(II) nitrite crystal structures of “picket-fence” porphyrinates do not exhibit such picket dynamics,445–448 revealing the strong influence of the dioxygen ligand rotations on the large disorder of these oxy-heme crystal structures. Similarly, the oxy- forms of the cobalt analogs of these ligand systems also displayed tight correlation between the dynamics of coordinated dioxygen and the pickets.449
Furthermore, this work provides detailed descriptions of ligand dynamics revealed by variable temperature solid-state structures.433 This report, for the first time, provides direct insight into the off-axis tilt (of 6.2°) of the coordinated dioxygen moiety of oxy-heme unit of “picket-fence” porphyrinates at 80 K. Such an off-axis tilt has been previously proposed via theoretical studies but lacked experimental evidence.443,450 The authors of this study have reconfirmed previous observations of Lang and co-workers on Mössbauer spectroscopic features with unusual temperature-dependence, which is common to hemoglobin, myoglobin, and their model systems.414 Moreover, the variable-temperature Mössbauer characteristics have been fitted with computational models based off of the two theoretical models introduced by Lang414 and Oldfield.434 Both models gave satisfactory fits for low temperature (<250 K) experimental data, albeit the quality of fits was observed to decrease with increasing temperature. The constant movement of the ligated dioxygen, and the complicated relationship of that with picket rotations resulting in vibronic contributions,451 was proposed to induce significant dissimilarity between experimental and theoretical Mössbauer spectroscopic characteristics at higher temperatures.
In another recent study, Hedman, Hodgson, Solomon, and co-workers have revisited the long-standing controversy over the electronic structure description of oxy-heme adducts by analyzing the L-edge X-ray absorption spectroscopic (XAS) properties of 6C “picket-fence” (TpivPP) heme-superoxo species with axially ligated 1-methylimidazole (1-MeIm; see Figure 27).94 Spectroscopic and theoretical investigations of the electronic structure of oxy-heme species have often been complicated by the highly delocalized nature of the porphyrin ligand platform. However, iron L-edge data provides a useful handle to directly analyze the electronic structure of the iron center, despite its highly covalent ligand environment. The authors in this study compared and contrasted the spectroscopic features of the 6C oxy- species with three reference complexes: the low-spin (S = 0) FeII complexes [(TpivPP)FeII(1-MeIm)2] and [(TpivPP)FeII(1-MeIm)- (cO)], the low-spin (S = 1/2) FeIII complex [(TPP)-FeIII(Im) 2](Cl),452 and the intermediate-spin FeII complex [(TpivPP)FeII] (Figure 27). These reference systems were carefully selected in order to represent the electronically different heme-Fe centers of Pauling, Weiss, and McClure- Goddard-Harcourt models, respectively, described for oxy-heme adducts within biological systems (also see section 2.1.1, Figure 7).
Figure 27.

Complexes utilized by Hedman, Hodgson, and Solomon94 for electronic structure studies on “picket-fence” oxy-heme adduct: (A) the “picket-fence” oxy heme complex with axially coordinated 1-MeIm, (B) [(TpivPP)FeII(1-MeIm)2], (C) [(TpivPP)FeII(1-MeIm)(CO)], (D) [(TPP)FeIII(Im)2](Cl), and (E) [(TpivPP)FeII]. See text for further discussion.
The L-edge X-ray absorption spectroscopic signatures reveal that the oxy species, [(TpivPP)(1-MeIm)FeIII(O2•−)], bears an electronic structure that is closely reminiscent of that of the low-spin ferrous reference complex, [(TpivPP)FeII(1-MeIm)2], albeit with some differences in the high-energy region. These differences arise from the higher Zeff of the oxy-complex due to strong σ and π interactions of the Fe-O2 unit. Both other reference complexes, ([(TPP)FeIII(Im)2](Cl) and [(TpivPP)-FeII]), exhibit strikingly different XAS features. These observations are intriguing, since they indicate that none of the three aforementioned electronic structural models are capable of perfectly describing the electronic properties of the highly covalent Fe-O2 unit. In-detail spectroscopic investigations presented in this study, together with theoretical computations point out the fact that the oxidation state of the iron center in oxy-heme is between ferric and ferrous forms, predominantly due to the strong electronic perturbations exerted by the remarkably covalent heme ferric-superoxo moiety.
2.2.2.1. Heme-Superoxo Model Systems with Potential Implications in Heme/Cu/O2 Chemistry.
Apart from “picket-fence” examples, other more recent examples of porphyrinates possessing both built-in steric bulk and functionalities that mediate secondary coordination sphere interactions are scarce but do exist. The “single-coronet” and “twin-coronet” ligand platforms from Naruta and co-workers are such examples (vide infra), which consist of highly sterically bulky substituted dinaphthalene moieties that create a hydrophobic local environment around the iron center, while offering hydroxide groups for secondary coordination sphere interactions (Figure 29). This unique ligand architecture has been shown to alter the iron-dioxygen and iron-carbon monoxide affinities,453–455 with lowered and enhanced stabilities of iron-carbonyl and iron-oxy adducts, respectively, as observed for biological heme centers.407,456 This is intriguing given that the CO affinity for free heme centers are ≈20000 times greater than that for dioxygen.457 This remarkable change in affinity is brought about by suppressing back-bonding interactions that stabilize heme-carbonyl adducts, while exerting stability-enhancing hydrogen-bonding interactions on heme-dioxygen moieties (see Figure 28).453,458
Figure 29.

(A) “Single-coronet” and (B and C) “twin-coronet” model systems designed by Naruta and co-workers. The “twin-coronet” systems incorporate (B) thiobenzyloxy and (C) thioglycolate axial tethers as mimics for the cytochrome P450 active site. See text for further discussion.
Figure 28.

Destabilization of carbonyl complexes through reduced back-bonding (left), and stabilization oxy-heme species through hydrogen-bonding interactions (right) resulting from hydroxylated naphthyl functionalities in “coronet” model systems from Naruta and co-workers. Adapted from ref 455. Copyright 2003 Wiley-VCH.
The “twin-coronet” systems were initially synthesized as cytochrome P450 mimics bearing axially tethered thiolate groups with varying electron-donating capabilities (Figure 29). These systems have been later adopted as globin protein mimics and will be discussed below. The oxy-heme forms of cytochrome P450 models in general are noteworthy, since they’re often severely destabilized by the strongly electron- donating anionic thiolate axial ligation. These thiolate ligands primarily aid in modeling the “push-pull” machinery, which predominantly contributes to the eventual O–O bond heterolysis in the enzymatic active site (see section 2.1). Although several 6C oxy-heme model systems with axial thiolate ligands have previously been reported in litera- ture,459,460 the first fully characterized heme-superoxo complex of this class (including vibrational spectroscopic characterization) was reported by Naruta, utilizing his unique “twin-coronet” ligand platform.458 The built-in steric bulk on both sides of the “twin-coronet” not only prevents unfavorable autoxidation decay pathways of the superoxo complexes (vide supra) but also assists in protecting the tethered thiolate group from oxidative degradation via disulfide formation. These ligand systems are capable of unique modeling of the hydrogen-bonding interactions between the heme-superoxo moiety and a threonine residue (Thr252) at the cytochrome P450cam active site (see section 2.1.2.1),120 by aforementioned noncovalent interactions between their heme-superoxo adducts and hydroxyl groups on the ligand coronets (see Figure 29).
These heme-superoxo complexes were generated from ferric starting complexes and one equiv of added potassium superoxide (KO2) under a dioxygen atmosphere in tetrahydrofuran (THF) at −80 °C. The complexes exhibited reversible dioxygen binding, with dioxygen ligand being readily replaced by carbon monoxide (CO). These superoxo complexes displayed fairly high thermal stabilities with halflives of 1 h at −20 °C and 5 h at 0 °C for the thiobenzyloxy and thioglycolate ligated adducts, respectively (see Figure 29). These differences in stabilities correlate with the reduction potentials of the corresponding Fe centers (thiobenzyloxy and thioglycolate ligated adducts displayed −1.35 V and −1.12 V vs Fc+/Fc for the FeIII/FeII pair, respectively), where the system with the less electron-donating thioglycolate axial tether gives rise to the better-stabilized superoxo adduct. The rR spectra for these complexes exhibited sharp features centered at 1138 cm−1 (for the thiobenzyloxy ligated) and 1137 cm−1 (for the thioglycolate ligated) with Soret excitation, which shifted to 1074 and 1073 cm−1 upon 18O-isotope substitution, respectively. Furthermore, these observed isotope shifts matched well with the calculated values for v(O–O) stretching frequencies, providing further evidence for the oxy-heme complexes of these new porphyrin systems. Intriguingly, these rR features and their isotopic shifts are also in good agreement with those of oxy-cytochrome P450cam [1139 (Δ 18O2 - 66) cm−1],118,461,463 which is known to possess a 6C low-spin ferric-superoxo adduct. These two heme-superoxo intermediates were also observed to possess hydrogen-bonding interactions between the hydroxyl groups of the dinaphthalene moieties of the “twin-coronet” ligand (see Figure 29) and the iron-bound superoxide ligand. Evidence for such an interaction originate from their rR spectra, where the observed v(O–O) frequencies were up-shifted by 2 cm−1 upon deuterium exchange of the ligand hydroxyl functionalities. Similar shifts in v(O–O) frequencies upon hydrogen/deuterium exchange of hydrogen-bonding functionalities were also observed in previous work on oxy-cobalt hemoglobin and myoglobin.464–466
In a later study, Naruta and co-workers also synthesized hemoglobin/myoglobin models from the same “twin-coronet” ligand platform, by introducing a tethered imidazole or pyridine on to the porphyrinate periphery (see Figure 30).455 These ligand systems again exhibit decreased CO affinity, thereby favoring dioxygen binding under competitive conditions. Thus, these mimics utilize secondary coordination sphere noncovalent interactions in order to lower CO-affinity at the Fe center, as seen in heme-containing binding pockets of Mb and Hb.69,438,467–476 Specifically, the ligand-originated polar interactions about the nonpolar Fe-CO unit were spectroscopically observed to facilitate the decrease of backbonding between the iron center and CO ligand [unusually low v(Fe-O) = 470 cm−1 (rR) and high v(C-O) = 1994 cm−1 (IR), as well as significant shielding of 13C NMR features], making it destabilized with respect to the more polar Fe-O2 unit. The tunability of dioxygen affinity at the heme center by altering polar properties within the distal binding site has been demonstrated in both biological and model systems: Momenteau had observed that the dioxygen affinity of “basket- handle” porphyrin (Figure 25) can be increased ≈10-fold by replacing its ether linkages by amide functionalities,477 while replacing the distal histidine residue of Mb with glycine was observed to decrease its O2-affinity by ≈10 times.407 In similar work reported very recently, Boitrel and Hayashi have seen the increment of dioxygen affinity of a new strapped porphyrinate system change by 2 orders of magnitude just by replacing a distal ester functionality by a carboxylate group.478
Figure 30.

Amended “twin-coronet” model platforms of hemoglobin and myoglobin active sites with tethered axial N-based ligands. See text for discussion.
The dioxygen adducts of these globin models of Naruta (Figure 30) displayed remarkable stability at room temperature in toluene, with half-lives up to several days. Furthermore, it has been previously observed that the dioxygen affinity at the iron center of “picket-fence” systems479 and “basket-handle” porphyrinates477 decreased upon the replacement of axial imidazole ligand by pyridine. Such an affinity shift is rather anticipated, given the enhanced π-acidity of the imidazole ligand compared to that of pyridine, which results in lowering of the dioxygen dissociation rate for the oxy-heme adduct. However, the “twin-coronet” ligand system showed a reverse dioxygen affinity, where the higher oxophilicity was observed for the one with axial pyridine ligated.455 This reversal is proposed to result from large distortions in the oxy-species of the imidazole-ligated system, enhancing its O2 dissociation rate with respect to the pyridine-ligated. The axial imidazole and pyridine ligated heme-superoxo adducts exhibited rR features for v(Fe-O) stretching centered at 586 (Δ18O2 = −26) cm−1 and 583 (Δ18O2 = −25) cm−1, respectively. The upshift in energy of these features with respect to those of other Fe-oxy species in model systems (“basket-handle”,480 “picket- fence”,481 OEP, TPP, TMP, and protoporphyrin IX482 porphyrinates) as well as biological examples (Hb483 and Mb484) was attributed to hydrogen-bonding interactions between the superoxide moiety and the hydroxyl groups of the coronet ligand. The v(O–O) stretching frequency could not be assigned unambiguously, possibly due to vibrational coupling of internal modes as observed in globin proteins.465,466 In order to further confirm the intramolecular hydrogen-bonding interactions of the oxy- forms, the oxygenated species of “twin-coronet” systems were analyzed by rR spectroscopy. Here, the v(Fe-O) stretching frequencies displayed a unique temperature-dependency, where the v(Fe-O) features moved to higher energy [586 (Δ18O2 = −26) to 599 (Δ18O2 = −29) and 583 (Δ18O2 = −25) to 590 (Δ18O2 = −27) cm−1 for axial imidazole and pyridine complexes, respectively] upon cooling down the sample from room temperature to −45 °C.455 This behavior was attributed to increased back-bonding upon cooling down, strengthening the Fe-O bond, during which, the weakening of the O–O bond is anticipated. Furthermore, the O–H IR stretching frequency of the ligand hydroxyl groups significantly lost intensity in the oxy- species compared to deoxy- and carbonyl- adducts, providing further evidence for hydrogen bonding. Therefore, the “twin-coronet” ligand systems of Naruta and coworkers were capable of successful modification of both O2 and CO affinities providing insight into how Mb and/or Hb would carry out highly selective physiological O2-binding despite the exceptionally large driving force to produce carbonyl complexes.
Interestingly, in a heme/Cu containing CcO model system of Naruta and co-workers, it was observed that the heme/Cu μ-peroxo complex, FeIII–O22−–CuII, formed upon oxygenation of the fully reduced compound (FeII/CuI), readily transforms into the heme–superoxo-CuI species in 20% MeCN/THF solution at −70 °C (also see section 5).485 This model consists of a porphyrin-linked copper chelate bearing a covalently linked imidazole-phenol cross-link, that mimics the histidine- tyrosine cofactor at the CcO active site (Figure 31).485,486 The tethered axial imidazole generates the 5C ferrous center for initial O2 binding.
Figure 31.

Formation and decay of the heme/Cu μ-peroxo species giving heme-superoxo and reduced CuI as observed by Naruta. Since the rate of formation of the superoxo is increased upon added water, a water-mediated hydrogen-bonding interaction between the tethered phenol and the superoxo ligand is postulated. R = H, MOM. Adapted with permission from ref 485. Copyright 2005 Wiley-VCH.
The entire transformation from the fully reduced parent compound, FeII…CuI, to the heme/Cu μ-peroxo adduct, FeIII– O22−–CuII, leading to the final heme–superoxo/Cu species, FeIII–O2•−…CuI, has been fully analyzed by rR spectroscopy, and only the v(Fe–O) stretching frequency of the heme– superoxo adduct was observed at 574 (Δ18O2 = −26) cm−1. This assignment was reconfirmed by analyzing rR signatures of an authentic heme–superoxo complex prepared with the same ligand platform in the absence of Cu metal. Notably, in the absence of CuI, the dioxygen binding to FeII was observed to be reversible; however, with the inclusion of CuI, the reversibility was lost. Thus, the CuI ion in this case stabilizes the heme–superoxo adduct presumably through a Lewis acid- type interaction, preventing its reversal to FeII. The formation of the bridged peroxo species, FeIII– (O22−)–CuII, and its subsequent transformation into FeIII–O2•−---CuI is quite unique, and the authors propose that the initial oxygenation of the reduced metal complex gives rise to a cupric superoxo complex, CuII–O2•− (for more details on Cu/dioxygen intermediates see section 3), which further reacts to give the FeIII–(O22−)– CuII adduct. The strongly electron-donating axial imidazole ligand most likely destabilizes this heme/Cu μ-peroxo complex, causing its disruption even at low temperatures giving the more thermodynamically favorable FeIII– O2•−…CuI combination. The redox potentials of the FeIII– O2•−/FeIII–O22− and CuII/CuI couples are thought to play a central role in the observed chemistry, where a large negative potential for the former couple than the latter would facilitate the formation of the observed product. Furthermore, a water-mediated hydrogen-bonding network involving the tethered phenol and the oxy-ligand may further facilitate the formation of the final heme–superoxo complex (Figure 31). In support, upon added water, the faster decay of the heme/Cu μ-peroxo complex to the heme–superoxo was observed, and upon replacing the hydroxyl functionality of the phenol with a methoxymethyl group, a drastic switch in the chemistry was observed, where the decay of the bridged-peroxo produced a heme/Cu μ-oxo (i.e., O2−) complex, FeIII– (O2−)– CuII. The latter decay pathway is commonly observed for thermal decomposition of heme/Cu μ-peroxo complexes, as described in detail in section 5.
Recently, Naruta and co-workers have also reported the formation of a ferrous-superoxo complex by cryo-reduction of the corresponding ferric superoxide adduct.487 Early studies on cryo-reduction of ferric-superoxo adducts, FeIII–O2•−, of both biological and model heme centers488,489 with γ-ray radiation in aprotic solvents, have been shown to generate ferrous superoxo adducts, FeII–O2•−. These are highly reduced oxygenated complexes with facile O–O bond cleavage capability, which could serve as important intermediates in catalytic dioxygen reduction reactions. These complexes are unique in the fact that the added electron on to the ferric-superoxo moiety is localized at the Fe center rather than the bound superoxo ligand, thus the electronic structure is best described as a ferrous-superoxo species, rather than a ferric-peroxo adduct. Typically, upon annealing at higher temperatures, these complexes produce ferric-peroxo or ferric- hydroperoxo complexes reaffirming their initial assignment as ferrous-superoxo complexes. As mentioned above, Naruta and co-workers have recently reported such a system, where the ferric superoxo complex of a system bearing a tethered imidazole moiety has been cryo-reduced using γ-ray radiation at 77 K in 20% MeCN/2-MeTHF (Figure 32).487 Further evidence comes from EPR spectroscopy of this FeII-O2•– adduct, exhibiting a g = 2.10 signal, which is indicative of the uncoupled superoxo radical but not the FeIII-O2•– or FeIII-O22– complexes. Furthermore, upon annealing at 173 K, the g = 2.10 EPR signal disappeared, upon concomitant emergence of new features corresponding to low-spin FeIII-OOH species (as previously observed by Naruta,490 see section 2.4 for detailed discussions).
Figure 32.

Cryo-reduction of heme ferric-superoxo species using γ-ray radiation at 77 K in frozen solution reported by Naruta and co-workers and its subsequent reactivity to produce the ferric-hydroperoxo adduct.
More importantly, the authors were able to carry out low-temperature rR characterization of the ferrous-superoxo complex, with the v(Fe-O) band detected at 459 (Δ 18O2 – 26) cm−1. The v(O–O) stretching feature was not observed. It is interesting to note that the Fe-O stretching frequency in this case has been significantly down-shifted with respect to that of the corresponding FeIII-O2•– (579 cm−1) and is directly comparable to the respective band in nonheme NiII- O2•– adducts [v(Ni-O) = 437 (Δ 18O2 – 21) cm−1].491 Moreover, the oxidation state sensitive v4 band of the porphyrin shifts from 1367 to 1357 cm−1 upon γ-ray irradiation, indicating the reduction of the Fe center. This study marks the only example in literature where the low-temperature rR characterization of a heme ferrous-superoxo complex has been reported. In addition, density functional theory (DFT) computations were also carried out in order to gain more insight into the chemical events involving the ferrous-superoxo species. With optimized geometry, Fe-O and O–O distances of the iron(II)-superoxo were calculated to be 1.93 and 1.37 Å, displaying a significant elongation of the FeO bond upon reduction, consistent with the down-shift of the v(Fe-O) rR feature by ≈120 cm−1 (vide supra).487
With an added proton, the ferrous-superoxo was observed to produce the ferric-hydroperoxo species as observed by EPR spectroscopy. The basicity of the ferrous-superoxo adduct was calculated to be much larger (pKa ≈ 39) than its ferric counterpart (pKa ≈ 0), corroborating the protonation of the former during annealing even in the absence of a known proton source. Authors suspect the proton source in this case as either adventitious water or solvent-based cation radical species resulting from γ-ray irradiation. Finally, the calculated reduction potential for the protonated ferric superoxo species was found to be +600 mV, indicating that the usually large thermodynamic barrier for reduction of ferric-superoxo into ferric-(hydro)peroxo is minimized with the presence of protons, suggesting a more PCET-like machinery. This detail is crucial in understanding biological enzymatic transformations, as well as in targeting efficient porphyrin-based oxygen-reduction catalysts and designing better biological mimics.
Another example of an iron(II)-superoxo adduct was characterized by Ivanović-Burmazović and co-workers, where an iron(III)-peroxide complex supported by a porphyrin-crown ether conjugate was observed to coexist in equilibrium with the corresponding iron(II)-superoxo species in DMSO solution (Figure 33).492 The formulation of the latter was confirmed by IR, EPR, and Mössbauer spectroscopies, along with DFT computations, and the detailed solution behavior of this system has been characterized by high-pressure studies.493 They propose that the iron(III)-peroxo ⇌ iron(II)-superoxo equilibrium favors the formation of iron(II)-superoxo upon added acid or coordinating solvent, giving FeII decay products rather than FeIII (also see section 2.3).493,494 Notably, an equilibrium of the aforementioned nature, and the dissociation of protonated-superoxide ions from the reduced metal center, may have implications in the proposed proton-assisted heme- superoxo decay pathways relevant to both biological and model systems (vide supra, Scheme 10).79
Figure 33.

Solution equilibrium between an FeIII-peroxo and an FeII-superoxo species of a porphyrin-crown ether conjugate as observed by Ivanović-Burmazović and co-workers.492
Several other examples of heme-superoxo complexes have also been reported by Karlin and co-workers, most of which have been subsequently utilized in the synthesis of their heme/Cu μ-peroxo adducts, FeIII-O22−-CuII, by several different methodologies (see section 5 for more information).495–497 Their initial report on the [(F8)FeIII(O2•–)] adduct [F8 = tetrakis(2,6-difluorophenyl)porphyrin; see Figure 34] appeared in 1999,496 which was detected as a transient intermediate en route to the heme/Cu μ-oxo species, FeIII- O2−-CuII, by stopped-flow experiments at low temperatures. In a subsequent study, the one-electron reduction of this superoxo complex with CoCp2 to give the FeIII-peroxo adduct, [(F8)FeIII(O22−)]− (side-on bound), was reported, which then led to the formation of the heme/Cu μ-peroxo adduct, FeIII-O22−CuII, upon addition of the copper(II) complex under cryogenic conditions.495 The detailed characterization of the ferric-superoxo adduct, [(F8)FeIII(O2•–)], was later reported in 2001, along with its entire decay pathway via rigorous spectroscopic analysis (Figure 34).498 This information is critical in designing logical CcO heme/Cu models for dioxygen binding that eventually leads to O–O bond cleavage giving water. The [(F8)FeIII(O2•–)] adduct was generated in THF solvent at −80 °C, exhibiting complete reversibility of dioxygen binding. Its formulation was confirmed by manometric measurements of dioxygen uptake, where an Fe/O2 stoichiometry of 1:1 was observed. In a later report, the 6C low-spin ferric-superoxo complex, [(THF)(F8)FeIII(O2•–)], was further characterized by rR spectroscopy where v(O–O) and v(Fe–O) vibrations were identified at 1178 (Δ18O2 = −64) and 568 (Δ18O2 = −24), respectively.
Figure 34.

Reversible dioxygen binding to [(F8)FeII] to form [(THF)(F8)FeIII(O2•–)] and its thermal decay giving [(F8)-FeIII(OH)]. Adapted from ref 498. Copyright 2001 American Chemical Society.
Another useful technique that has been extensively used to gain insight into the properties of these fleeting intermediates is 2H NMR analysis of metal species supported by porphyrinates with deuterated pyrrolic positions. This allows direct visualization of the deuterated pyrrolic positions, unobstructed by multiple other resonances of the macrocycle. Accordingly, the 2H NMR spectrum of the superoxo adduct (with deuterated β-pyrrolic positions) provided evidence of a diamagnetic low-spin iron center (δpyrrole = 8.9 ppm), indicating the presence of a 6C FeIII center, [(THF)(F8)-FeIII(O2•−)], in the superoxo complex. The diamagnetism of FeIII–O2•− complexes arise from the antiferromagnetic coupling between the unpaired electrons on the low-spin FeIII center and the radical anionic superoxo ligand (see section 2.1.1 for diamagnetism of oxy-Mb).79,499 Upon warming to RT, the complete formation of the mononuclear high-spin [(F8)FeIII(OH)] complex is observed (Figure 34), with a paramagnetic 2H NMR shift at δpyrrole = 125 ppm (recorded at –80 °C). Furthermore, the characteristic 2H NMR shift corresponding to the [(THF)(F8)FeIII-(O2•−)] complex enabled its unambiguous formulation against a FeIII/FeIII μ-peroxo dimer, [{(F8)FeIII}2(O22−)],367,403,498,500 a highvalent ferryl species, [(F8)FeIV=O],365,367,498 a mononuclear ferric peroxo adduct, [(F8)FeIII(O22−)]−,495,498,501 or a FeIII/FeIII μ-oxo dimer, [{(F8)FeIII}2(O2−)].502 Moreover, the 19F-NMR spectrum of (THF) [(F8)FeIII(O2•−)] complex shows two fluorine resonances at –111 and –113 ppm with near identical integrations, indicating two chemically different environments for the 2,6-difluoro substituents of the porphyrinate, presumably due to the two chemically inequivalent sides of the porphyrinate, with one bearing the superoxide ligand.367
Intriguingly, when the parent FeII complex, [(F8)FeII], was oxygenated in noncoordinating solvents such as toluene or CH2Cl2, the immediate formation of an FeIII/FeIII μ-peroxo dimer, [{(F8)FeIII}2(O22–)], was observed at −80 °C (see Figure 35), with complete reversibility in its formation observed in toluene as in the case of [(THF)(F8)FeIII(O2•–) in coordinating solvents (vide supra).498 The formulation of FeIII/FeIII μ-peroxo dimer, [{(F8)FeIII}2(O22–)], was further corroborated by dioxygen titration of [(F8)FeII] in CH2Cl2 at –80 °C, where a stoichiometry of 0.5–0.55 equiv of O2 per equiv of [(F8)FeII] was established. Interestingly, the dioxygenbinding in CH2Cl2 was observed to be irreversible. 2H NMR experiments carried out in CH2Cl2 at −80 °C provided evidence for the formation of an initial mixture of FeIII/FeIII μ-peroxo dimer, [{(F8)FeIII}2(O22–)], (δpyrrole = 17.5 ppm)367,403,498,500 and 5C FeIII-superoxo adduct, [(F8)- FeIII(O2•–)] (δpyrrole = 8.9 ppm),367,384,498 which upon warming up to −50 °C resulted in the full formation of the peroxo-bridged dimer in the expense of the superoxo species. Thus, the ferric superoxo complex was still an intermediate en route to the observed FeIII/FeIII μ-peroxo dimer in noncoordinating solvents. Upon warming up to room temperature, the complete transformation of [{(F8)FeIII}2(O22–)] to [(F8)FeIII(OH)] was observed as expected. This dioxygen reactivity pattern was also followed by 19F-NMR spectroscopy, providing further evidence for the aforementioned mechanism for the stepwise conversion of [(F8)FeII] into [(F8)FeIII(OH)].
Figure 35.

Stepwise transformation of [{(F8)FeIII}2(O22−)] to [(F8)FeIII(OH)] with the intermediacy of the ferryl species ([(B)(F8)FeIV=O]) in the presence of a coordinating base. Adapted from ref 498. Copyright 2001 American Chemical Society.
In accordance with the proposed μ-peroxo decay pathway of heme–superoxo intermediates (vide supra; Scheme 9), the conversion of FeIII/FeIII μ-peroxo dimer to the final FeIII–OH species should proceed with the intermediacy of a ferryl species (FeIV=O or compound II) resulting from the homolytic O–O bond scission (Figure 35).79,367 In order to interrogate the existence of such a high-valent ferryl species in this system, Karlin and co-workers carried out 2H NMR experiments in CH2Cl2, whereupon the addition of 1 equiv of 4-(dimethylamino)pyridine (DMAP, a coordinating base) to the FeIII/FeIII μ-peroxo dimer triggered the immediate formation of a new species with δpyrrole = 3.5 ppm, consistent with a 6C ferryl species, [(DMAP)(F8)FeIV=O].365,367,498,503 In support, the conversion of a FeIII/FeIII μ-peroxo dimer to the ferryl species upon the addition of 1-methylimidazole was previously observed by Balch and co-workers for tetraarylporphyrin systems.365–368,388 The [(DMAP)(F8)FeIV=O] species was observed to be indefinitely stable within the temperature range from −50 °C to −80 °C, whereupon further warming up, the final decay product [(F8)FeIII(OH)] is observed (Figure 35).498 Finally, the addition of an excess of a coordinating solvent such as THF effected the direct conversion of the FeIII/FeIII μ-peroxo dimer to the [(F8)-FeIII(OH)] with the absence of any observable intermediate. Therefore, this work and Kitagawa's studies on tetramesityl-porphyrinate (TMP)384 are two excellent examples where the stepwise decay of the initial ferric-superoxo complex has been clearly observed with the intermediacy of the corresponding ferryl species, providing further evidence for the μ-peroxo decay model of metal-superoxo adducts described by Balch and co-workers (Scheme 9).367
Karlin and co-workers have also presented several other reports on heme-superoxo complexes based on the F8 (vide supra) platform bearing tethered axial nitrogenous bases (such as imidazole or pyridine moieties) and/or tethered tri- or tetradentate copper chelates. The first of this class, an F8-derived ligand platform with the tethered tetradentate copper chelate, TMPA (TMPA = tris(2-pyridylmethyl)amine; often referred to in the literature as TPA), known as 6L, was shown to produce a clean ferric-superoxo complex at −80 °C regardless of the solvent properties.504 By 2H NMR studies (of the deuterated β-pyrrolic positions of the porphyrin), this solvent-independency was found to be due to the coordination of a pyridyl arm of the copper chelate (with no copper metal ion present; see Figure 36) to the Fe center giving a 6C ferric-superoxo complex in contrast to the case of F8 (vide supra). In THF solvent at −80 °C, the parent FeII complex, [(X)(6L)- FeII], was found to be 5C, existing as a mixture of species where X = THF (δpyrrole = 95 ppm) or X = pyridyl arm from the TMPA tether (δpyrrole = 79 ppm). However, upon oxygenation, an immediate shift in the pyrrole NMR signals to δpyrrole = 8.9 ppm was observed, which upon warming up moves to δpyrrole = 121 ppm, indicating the formation of the final decay product [(6L)FeIII(OH)].505 The pyrrole shift of the ferric-superoxo complex is evident of a 6C low-spin FeIII species (vide supra), [(Y)6FeIII(O2•−)], where Y could be either a THF solvate or a pyridyl arm from the TMPA tether (Figure 36), the identity of which could not be unambiguously determined by 2H NMR studies alone.
Figure 36.

Two structural possibilities for a 6C low-spin [(6L)-FeIII(O2•−)] complex formed via the oxygenation of the parent FeII complex in THF at −380 °C. The sixth ligand on the FeIII center of the superoxide could either be a THF solvate (right) or a pyridyl arm from the TMPA tether (left) as shown here. Adapted from ref 504. Copyright 2002 American Chemical Society.
In DCM solvent, the [(X)(6L)FeII] species could again be observed at −80 °C with δpyrrole = 75 ppm, where in the absence of a coordinating solvent, X had to be a pyridyl arm from the TMPA tether. Upon dioxygen binding, two pyrrole shifts are observed at 9.3 and 17 ppm, where the former is attributed to a 6C low-spin [(Y)(6L)FeIII-(O2•−)] species where Y = a pyridyl arm from the TMPA tether, while the latter is a minor impurity of a FeIII/FeIII μ-peroxo dimer, [{(6L)FeIII}2(O22−)],498 which immediately rearranges to the more thermally stable 6C superoxo species (δpyrrole = 9.3 ppm) upon warming up to −50 °C. As anticipated, upon warming up to room temperature, the superoxo adduct displayed the full conversion to the high-spin [(6L)FeIII(OH)] species with a pyrrole shift of 122 ppm. Nevertheless, if the coordinating pyridyl arm from the TMPA tether in the case of 6C low-spin superoxo adduct is intramolecular or intermolecular in nature, it could not be explicitly assigned by 2H NMR interrogations carried out in this study. Finally, this 6C low-spin superoxo adduct displayed reversible O2 binding both in THF and DCM, albeit with reduced stability in the latter.504
In a later study, Karlin and co-workers have described another F8-derived ligand with an appended MePY2 (MePY2 = bis[2-(2-pyridyl)ethyl]methylamine)]) fragment, 2L (see Figure 37A), of which the Fe-only dioxygen binding chemistry to form a 6C low-spin superoxo adduct, and its implications in heme/Cu/O2 chemistry have been investigated.506,507 In other work, the Cu-only dioxygen chemistry of MePY2 ligand platform has been well-established (see section 3).508–512 The (2L)FeII complex (with empty copper chelate) exhibited a 5C high-spin FeII center in both solid-state (X-ray crystal structure) and solution (1H NMR studies) with one of the pyridine arms of the MePY2 moiety coordinating to the Fe center. This complex, upon dioxygen binding at −80 °C in DCM, gave the 6C low-spin ferric-superoxo species, [(X)- (2L)FeIII(O2•−)] (where X = a pyridine arm from the MePY2 tether), with a 2H NMR (on porphyrinate bearing deuterated β-pyrrolic positions) chemical shift of 9 ppm. Clean formation of this stable (under the aforementioned conditions) ferric-superoxide complex further establishes the fact that a 5C FeII center is necessary for its formation, especially within noncoordinating solvents such as DCM (vide supra). Similar to F8 and 6L platforms, 2L ligand also displayed reversible dioxygen binding of its FeII complex, and upon warming up of the superoxide complex to room temperature, the high-spin [(2L)FeIII(OH)] species (δpyrrole = 80 ppm) was produced.
Figure 37.

(A) The dioxygen binding of the 5C [(2L)FeII] giving the 6C low-spin [(2L)FeIII(O2•−)] complex. Note that in both structures one of the pyridine arms of the MePY2 tether is coordinated to the iron center. (B) The reaction landscape observed when the fully reduced [(2L)FeII/CuI]+ complex is oxygenated in EtCN at −80 °C.
Following the characterization of the heme-only dioxygen chemistry, CuI was added to the MePY2 chelate of 2L, effectively detaching the pyridine arm from the FeII center in solution. Now, the spin-state of the FeII center was observed to be highly solvent-dependent, where in strongly coordinating (such as pyridine), weakly coordinating [such as THF, acetone, EtCN (EtCN = propionitrile) or MeCN], and noncoordinating (such as DCM) solvents, low-spin (S = 0), high-spin (S = 2), and intermediate-spin (S = 1) iron centers were found by 2H NMR, respectively.506 The dioxygen binding/reduction in the formation of the heme/Cu μ-peroxo adduct, [(2L)FeIII(O22−)CuII]+, starting from the fully reduced FeII/CuI complex was analyzed by stopped-flow spectroscopic techniques in EtCN at −105 °C. In this case, the transient formation of the FeIII-superoxo/CuI adduct, [(EtCN)(2L)-FeIII(O2•−)…CuI(NCEt)]+,498,513 was observed within 0.03 s of mixing, which rapidly transformed into the heme/Cu μ-peroxo species after ≈0.3 s of mixing (Figure 37B). However, two indistinguishable isomeric forms of the heme-superoxide could be present in solution as highlighted in Figure 37, while independent Cu-only dioxygen activation studies (see section 3)508 rule against the formation of a cupric superoxide species under these conditions. Furthermore, when the heme/Cu μ-peroxo species, [(2L)FeIII(O22−)CuII]+, is reacted with 1 equiv of PPh3 (PPh3 = triphenylphosphine) in THF at −75 °C, the heme-superoxide to heme/Cu-peroxide chemistry is essentially reversed, and the 6C ferric superoxide (δprole = 9 ppm) and the cuprous-phosphine complex, [(THF)(2L)-FeIII(O2•−)····CuI(PPh3)]+, are formed. The large thermodynamic driving force to form cuprous-phosphine complexes within a tridentate ligand environment is well-precedented in literature.514,515 The heme-superoxo species thus formed thermally decayed into the high-spin [(2L)FeIII(OH)] (vide supra) generating the final decomposition product, [(2L)-FeIII(OH)····CuI(PPh3)]+, in 47% yield.506,516
As very well-reviewed by Momenteau and Reed, Collman, and others, porphyrinates with tethered axial bases have been used in numerous studies to generate 5C FeII centers for dioxygen binding, which are known to produce better- stabilized ferric superoxo species (vide supra).56,79 Karlin and co-workers have also reported the reversible dioxygen binding chemistry of two such porphyrinates with either tethered imidazole (PIm)517 or pyridine (PPy) (Figure 38).420 The solution spin-states of the FeII complexes of these porphyrinates vary in the same pattern as observed for F8 ligand (vide supra), the only exception being that PIm possessing a 6C low-spin FeII center (with a strongly interacting axially bound solvate) in THF resulted in diamagnetic pyrrole shifts (for deuterated β-pyrrolic positions) centered at 8.4, 10.9, and 12.6 ppm in its 2H NMR spectrum. Both PIm and PPy FeII starting complexes at −80 °C in THF gave 6C low-spin ferric superoxo species, [(PIm)FeIII-(O2•−)] and [(PPy)FeIII(O2•−)], with their 2H NMR pyrrole chemical shifts centered at 9.8 and 9.1 ppm, respectively. The former was also recently characterized by rR spectroscopy, where the Fe-O and O–O stretches were observed at 575 (Δ 18O2 - 23) and 1180 (Δ 18O2 - 56) cm−1, respectively.518 [(PPy)FeIII(O2•−)] has been utilized for the formation of a heme/Cu μ-peroxo species, [(PPy)FeIII(O22−)CuII(AN)]+ (AN = bis[3-(dimethylamino)propyl]amine; also see section 5), where the tethered axial pyridine was found to be dissociated.421 In addition, both these superoxo complexes exhibited reversible dioxygen binding, where the iron(III)-superoxo complex fully converted to the FeII parent compound upon Ar bubbling and warming up to room temperature.420 Moreover, the carbonyl complexes of the ferrous species, [(PIm)FeII(CO)] and [(PPy)FeII(CO)], were also generated, exhibiting a v(C-O) IR stretch of 1985 cm−1 for both examples. This was somewhat surprising in that the v(C-O) stretch of the carbonyl moiety does not seem to correlate with the electron-donating ability of the axial ligand (imidazolyl > pyridyl;455,477,479 one would expect that with strong electron donation, the v(C–O) IR stretch would be reduced in energy due to back bonding,519 however, a similar lack of correlation of electronic effects has been previously observed for other heme–carbonyl systems.519–521
Figure 38.

Schematic diagram indicating the formation of the 6C low- spin ferric-superoxo adducts of [(PPy)FeII] (top) and [(PIm)FeII] (bottom) at −380 °C in THF. Adapted with permission from ref 420. Copyright 2013 Elsevier Ltd.
Several examples of double-phase hindered porphyrinates522 have also been investigated in dioxygen binding chemistry by Karlin and co-workers, where the top phase of the porphyrinate bears a tethered tridentate copper chelate similar to that in 2L (Figure 37), while the bottom phase possesses a tethered nitrogenous base as in the case of PIm (Figure 38) for axial binding. [(3L)FeII] (with empty copper chelate; see Figure 39) upon oxygenation in THF, toluene, or DCM produces the 6C low-spin ferric-superoxo complex, [(3L)-FeIII(O2•−)], at –80 °C as characterized by 1H NMR spectroscopy and spectrophotometric titrations show a Fe:O2 stoichiometry of 1:1. [(3L)FeIII(O2•−)] binds dioxygen reversibly, exhibiting the complete reversal to [(3L)FeII] upon warming up to room temperature. The analogous complex [(4L)FeII] (Figure 39) displayed similar binding of dioxygen, albeit the reversibility was not observed. Such drastic differences in reversibility upon subtle alterations of the ligand architecture have also been previously observed in heme/Cu μ-oxo complexes (see section 5).523 In the presence of CuI, both of these FeIII–O2•− adducts were shown to form the heme/Cu μ-peroxo species at low-temperatures; however, only the 4L derivative gave the expected heme/Cu μ-oxo complex, [(4L)FeIII(O)CuII]+,502 while the 3L derivative gave [(3L)-FeIIIOH···CuII]+ as the product of thermal decay.
Figure 39.

Dioxygen reactivity patterns of 3L and 4L ligand platforms forming ferric-superoxo complexes. Only 3L (top) exhibits reversible dioxygen binding.
Similar to the aforementioned examples where heme–superoxo adducts have been observed within heme/Cu platforms (vide supra), Collman and co-workers have reported a system where “stable” heme–superoxide have been observed even in close proximity to CuI (Figure 40).524,525 Such models are of great significance since the Oxy intermediate of CcO (referred to as A) is thought to bear a heme–superoxide moiety, prior to CuI-mediated reduction to peroxide (see sections 4 and 6). Collman and co-workers exhibited the dioxygen binding chemistry of the heme/Cu complex, [(NMePr)FeII/CuI]+, in 5% MeCN/THF at room temperature with a stoichiometry of 1:1, yielding a diamagnetic product as observed by 1H NMR, 19F-NMR, and EPR spectroscopies (Figure 40). Unfortunately, rR experiments failed to reveal an O–O stretching frequency; nevertheless, an v(Fe–O) stretch was observed at 570 (Δ 18O2 = −26) cm−1 (also see Table 3). This was attributed to a ferric superoxo species based on the v(Fe–O) stretching frequency of an authentic superoxo sample of the same system [575 (Δ 18O2 = −21) cm−1].524 Thus, this new species was formulated as the ferric-superoxo species bearing a proximal CuI center, [(NMePr)FeIII(O2•−)···CuI]+ (Figure 40). It is also noteworthy that [(NMePr)FeIII(O2•−)···CuI]+ is stable for up to few hours at room temperature and does not exhibit reversibility in its dioxygen binding chemistry. On the other hand, the copper-free [(NMePr)FeII] complex does not bind dioxygen in coordinating solvents (as above) at room temperature but completely converts to the authentic ferric- superoxo complex when cooled down to −60 °C as observed by 1H NMR spectroscopy. The dioxygen binding is fully reversible in this case, giving the FeII starting complex back either upon warming up to room temperature or applying reduced pressure. Thus, the presence of the CuI ion remarkably increases the dioxygen affinity of the heme center, as well as the half-life of the heme-superoxo complex presumably due to increased H-bonding interactions between the oxy ligand and the pickets in the Fe/Cu system as previously observed for Co/Cu systems of the same ligand.526 The CuI center may exert Lewis acid like interactions that significantly affect the affinity and reversibility of dioxygen binding (as previously seen in Naruta's systems; see Figure 31), as well as the reactivity of the superoxo moiety (vide infra).
Figure 40.

Heme-superoxo/copper(I) adduct, [(NMePr)FeIII– (O2•−)····CuI]+, reported by Collman and co-workers (R = methyl; R’ = propyl).525 The reactivity of this species with phenols, as biomimetic substrates, is described in section 5.
Table 3.
Selected Resonance Raman Spectroscopic Data: Heme-Peroxo-Copper Complex Core Vibrations
| system | vO–O (Δv)a | vFe–O (Δv)a | vCu–O (Δv)a | figure number, [reference] |
|---|---|---|---|---|
| tetradentate Cu ligands | ||||
| [(DCHIm)(TACNAcr)FeIII-(O22−)-CuII] + | 758 (−18) | n/r | n/r | 100933 |
| [(TPP)FeIII-(O22−)-CuII(tpa)] + | 803 (−44) | n/r | n/r | 95 A934 |
| [(TPP)FeIII-(O22−)-CuII(5-Metpa)]+ | 793 (−42) | n/r | n/r | 95 B930 |
| [(TMP)FeIII-(O22−)-CuII(5-Metpa)] + | 790 (−44) | n/r | n/r | 95C930 |
| [(TMP)FeIII-(O22−)-CuII(LOH)] + | 799 (−47) | n/r | n/r | 106 A486,935 |
| [(TMP)FeIII-(O22−)-CuII(LOMOM)] + | 801 (−46) | n/r | n/r | 935 |
| [(TMPIm)FeIII-(O22−)-CuII(LN4−OH)] + | 803/787 (−52) | n/r | n/r | 106 B485 |
| [(TMPIm)FeIII-(O2)2−-CuII(LN4−OMOM)] + | 804/752 (−52) | n/r | n/r | 935 |
| [(6L)FeIII-(O22−)-CuII]+ | 787 (−43) | 533 | n/r | 101 A936 |
| [(5L)FeIII-(O22−)-CuII]+ | 809 (−53) | n/r | n/r | 101 B936 |
| [(F8)FeIII-(O22−)-CuII(TMPA)]+ | 808 (−46) | 538 (−22) | 516 (−24) | 94498 |
| [(DCHIm)(F8)FeIII-(O22−)-CuII(TMPA)] + | 812 (−50) | 623 (−27) | 533 (−24) | 98497 |
| [(F8)FeIII-(O22−)-CuII(LN4OH)]+ | 813 (−44) | 529 (−21) | n/r | 104 B937 |
| [(F8)FeIII-(O22−)-CuII(LN4OMe)]+ | 815 (−46) | 528 (−22) | n/r | 104937 |
| [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)4] + | 876, 863 (−56) | 591, 585 (−27) | n/r | 108497 |
| tridentate copper ligands | ||||
| [(NMePr)FeIII(O2• −) CuI]+b | n/r | 570 (−26) | n/r | 40524 |
| [(2L)FeIII-(O2)2−-CuII]+ | 747 (−49) | n/r | n/r | 102 A938 |
| [(TMP)FeIII-(O22−)-CuII(LN3−OH)]+ | 810 (−46) | n/r | n/r | 105 A935 |
| [(TMP)FeIII-(O22−)-CuII(LN3−ONa)] + | 812 (−50) | n/r | 491(−28) | 105 B935 |
| [(F8)FeIII-(O22−)-CuII(AN)]+ | 746 (−48) | n/r | n/r | 94516 |
| [(DCHIm)(F8)FeIII-(O22−)-CuII(AN)]+ | 796 (−42) | 586 (−22) | n/r | 98928 |
| [(F8)FeIII-(O22−)-CuII(LMe2N)]+ | 752 (−43) | n/r | n/r | 103513 |
| 767 (−40) | ||||
| [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)3]+ | 876 (−55) | 594 (−28) | n/r | 108497 |
| [(F8)FeIII-(O22−)-CuII(MeTHF)3]+ | 737 (−41) | 555 (−27) | n/r | 107497 |
Values given in cm−1; values in parentheses are ∆18O2 shifts.
Characterized as a heme-FeIII-superoxide⋯CuI complex, see Section 2.2.2; n/r = not reported.
In a later study, Collman and co-workers have reported on the reactivity of this putative [(NMePr)FeIII(O2•−)···CuI]+ species with exogenous phenolic substrates introduced as tyrosine mimic compounds.525 As mentioned above, such oxidative reactivity may be suggestive of Lewis acid like interactions between the superoxo moiety and the distal CuI center, especially given that (heme)iron(III)-superoxo adducts are generally not strong oxidants toward organic substrates (compared to other reduced O2-derived species. When [(NMePr)FeIII(O2•−)···CuI]+ was reacted with 3–12 equiv of substituted phenol in DCM, the generation of 2 equiv of phenoxyl radical was observed and quantified using EPR standards. Kinetic analysis of this reaction reveals a bimolecular process, with a second-order rate constant of 0.012 M−1 min−1, resulting in formation of a heme-hydroperoxo species, [(NMePr)FeIII(O2•−)···CuI]+. This was then proposed to undergo CuI-mediated intramolecular reduction leading to homolytic O–O bond rupture giving the ferryl, [(NMePr)-FeIV=O···(HO)-CuII]+. In evidence, when an excess of PPh3 was added into the reaction mixture following the formation of 1 equiv of phenoxyl radical, the rapid generation of O=PPh3 was observed, indirectly indicating the formation of the ferryl species upon reacting with 1 equiv of substrate. The second equivalent of the phenoxyl radical is proposed to rise from the reaction between the phenol and the ferryl species, presumably giving the highly thermodynamically favored FeIII-hydroxide end product, [(NMePr)FeIV=O···(HO)-CuII]+.
Such systems with gradual reduction of dioxygen in the presence of an exogenous substrate are excellent models of the enzymatic transformation occurring within the CcO active site (see sections 4 and 5). However, the stepwise reduction of oxygenated species in the absence of substrates technically allows the detailed analysis of each intermediate leading to O–O bond cleavage. With this intent, chemical reduction of heme-superoxo species have been previously reported by Naruta and Karlin leading all the way up to hydroperoxo species in some studies (vide supra; see sections 2.3 and 2.4). In contrast, Anxolabehere-Mallart and co-workers have recently reported the electrochemical reduction of a ferric- superoxo species supported by meso-tetrakis- (pentafluorophenyl)porphyrinate (F20) in DMF at room temperature (Figure 41).527 The formation of this superoxide adduct was achieved by reacting the FeII complex with dioxygen in solution, which was subsequently reduced by cyclic voltammetry to give the ferric-peroxo species. This peroxide complex was then protonated with (CF3)3COH in the presence of an axially coordinating base 1-MeIm, to generate the hydroperoxo complex, [(F20)FeIII(OOH)]. In further evidence, the authors have followed the entire proposed reactivity landscape using other techniques such as spectroe-lectrochemistry, bulk electrolysis, and EPR spectroscopy. Interestingly, the authors have observed the formation of significant amounts of FeII upon the addition of the 1-MeIm and (CF3)3COH to the peroxo complex, thus formulating the final decay product of the FeIII-OOH species as [(F20)- FeII(OH)] (Figure 41).492–494,528 In support, other studies have shown instances where FeIII-peroxo adducts are reduced to FeII in the presence of 1-MeIm.529
Figure 41.

Electrochemical reduction of heme-superoxo systems studied by Anxolabehere-Mallart and co-workers. See text for discussion.
2.2.2.2. Heme Superoxos with Five-Coordinate (5C) Iron or Side-On-Bound Superoxide.
It is well-established that most heme protein active sites that carry out dioxygen binding/activation consist of 5C oxophilic FeII centers, which upon O2 binding, produce low-spin 6C heme-superoxo species.17 A majority of these adducts have been thoroughly characterized following numerous spectroscopic and theoretical analyses to possess an end-on bound superoxo ligand on the ferric center (vide supra; section 2.1). Accordingly, a majority of small molecule heme model systems have been designed to possess heme centers that mimic the same geometries as well as superoxide binding modes. However, a handful of model systems have been reported with structural alterations of the Fe-superoxo unit when compared with the traditional examples described in detail above. In 1982, Nakamoto and co-workers have investigated the dioxygen binding chemistry of [(TPP)-FeII] using matrix co-condensation techniques, where they generated the “base-free” FeII complex, [(TPP)FeII], from [(TPP)FeII(pip)2] (pip = piperidine) under reduced pressure at 97 °C.530 In this work, the [(TPP)FeII(pip)2] complex was vaporized at 197 °C and co-condensed with cold argon gas or a dioxygen/argon gaseous mixture on a CsI window under helium gas, of which the IR signatures were measured at −258 °C. Upon condensation with dioxygen/argon mixture, they observed two new IR signatures, centered at 1195 (Δ 18O2 = −68) and 1106 (Δ 18O2 = −63) cm−1, where the former exhibited greater intensity (×6) with respect to the latter. These frequencies fall within the typical v(O–O) range of heme iron(III)-superoxo species, and thus were attributed to two isomeric forms of the 5C ferric-superoxo species, [(TPP)FeIII-(O2•−)], formed by condensation. In support, variable concentration experiments revealed a Fe:O2 stoichiometry of 1:1. Intriguingly, when this mixture was warmed up to −173 °C, the complete disappearance of the 1106 cm−1 band was observed, and in its expense, the band at 1195 cm−1 was observed to grow in. Intriguingly, cooling back down to −258 °C recovered the full intensity of the band at 1106 cm−1. This behavior indicated the thermal equilibrium present between the two aforementioned isomeric forms of [(TPP)-FeIII(O2•−)] within the given environment. Although the identities of those intermediates were unclear in this initial work, in a later study they probed the same system with isotope scrambled dioxygen (16O18O) and normal coordinate computations.531 This work, together with their previous studies on different Fe, Co, and Mn systems,532 revealed that the isomer with the 1106 cm−1 IR feature corresponds to a ferric-superoxo species with a side-on bound dioxygen ligand, while that at 1195 cm−1 corresponds to the end-on isomer of the ferric-superoxide (Figure 42). Although side-on Fe- superoxo species have previously been proposed for ligandfree iron-dioxygen systems,533 this work by Nakamoto and coworkers marks the only example of a biomimetic side-on heme-superoxo adduct to-date.
Figure 42.

Thermal equilibrium present between the end-on and the side-on bound heme-superoxo isomers as observed by Nakamoto and co-workers. See text for further discussion.531
Recently, another example of a 5C low-spin (end-on) heme superoxide (with a square pyramidal geometry) was observed by Harris and co-workers, which was isolated within the porphyrinic metal–organic framework (MOF) PCN-224 FeII.534 The MOF ficantly contributed to the isolation of this fleeting intermediate by preventing its undesirable dimerization reactions (vide supra). This species was also structurally characterized (Figure 43), revealing an end-on bound superoxo moiety with Fe–O and O–O distances of 1.79(l) and 1.15(4) Å, respectively, and an Fe– O–O angle of 118° reported. This work is therefore the only example with a structurally characterized “base-free” 5C heme–superoxide adduct. In this study, the ferrous heme complex, [(TPP)FeII] was incorporated into the MOF giving an overall four-coordinate (4C) FeII center, which upon exposure to dioxygen at –78 °C, generated the superoxo adduct. Surprisingly, this FeII parent compound did not react with dioxygen at room temperature but required the lowering of temperature in order for the reaction to proceed. Accordingly, upon warming up of the ferric-superoxo sample to room temperature, the complete reversal of dioxygen binding was observed with the generation of stoichiometric amounts of the parent FeII complex. Variable temperature experiments reveal an Fe:O2 stoichiometry of 1:1. In addition, these temperature-dependent measurements also provide insight into the energetics of dioxygen binding, where a binding enthalpy of –34(4) kJ/mol was found. This value is significantly lower than that of a 6C FeII center (–63 to –65 kJ/mol), which is attributed to the differences in redox potentials (≈ 250 mV) of their FeII centers. These findings point out to the critical significance of the axially coordinated base on strong binding of dioxygen at the Fe center of hemes, justifying the 5C FeII complexes nature has chosen for dioxygen binding/storage and or reduction (see sections 2.1 and 4).
Figure 43.

Porphyrinic MOF, PCN-224FeII, synthesized by Harris and co-workers (left) that reversibly binds dioxygen to give the first structurally characterized 5C low-spin heme–superoxo complex (right). Adapted from ref 534. Copyright 2014 American Chemical Society.
2.3. Models of Heme Ferric-Peroxo Intermediates
Heme ferric-peroxo species are crucial intermediates within the catalytic cycles of heme-containing oxygenases and oxidases, especially in the “peroxide shunt”-type pathways described in detail in section 2.1. Within biological systems, ferric-peroxo adducts typically possess a six-coordinate FeIII center, with an end-on bound peroxo ligand. Such species are highly destabilized, due to the electron-rich iron(III)-peroxo moiety, with a charged distal Operoxo-atom that is not coordinated to the metal center. Thus, until recently, it has been a challenging endeavor to design such end-on heme–peroxo intermediates within model systems, whereas most of the early model systems contained a side-on bound peroxo on the FeIII center. Another significant challenge faced in modeling iron(III)- peroxo systems starting from FeII and dioxygen is that an exogenous electron has to be delivered to the iron(III)-superoxo adduct to be reduced to the iron(III)-peroxo level, while preventing the reduction of the FeIII center and/or any decomposition or dimerization of the metal adducts. Carrying out this second dioxygen reduction step has now been successfully achieved by both electrochemical and chemical reduction methodologies (vide infra). Such end-on heme–peroxo models may also reveal insightful information implicated with the CcO mechanism, where a peroxo-bridged FeIII/CuII intermediate has been proposed during turnover (see section 4.2).535
2.3.1. Binuclear FeIII/FeIII μ-Peroxo Species.
As mentioned above in section 2.2, these intermediates have been observed to form during the thermal decay of a variety of monomeric heme FeIII–superoxo intermediates, upon their interaction with a second equiv of the FeII parent compound (Scheme 9).536 These [{(porphyrinate)FeIII}2(μ-O22−)] species are typically generated by the oxygenation of ferrous forms of unhindered porphyrinates under cryogenic (~ –70 °C) conditions but decay rapidly when warmed up to ambient temperatures.404,500 These complexes, similar to μ-oxo FeIII/FeIII binuclear compounds, are typically diamagnetic due to the antiferromagnetic coupling of the unpaired spins on FeIII centers. Notably, unlike monomeric iron(III)–peroxo complexes (vide infra), these binuclear forms are highly inert toward substrate reactivity, presumably due to the steric hindrance experienced by the substrate while approaching the peroxo moiety, sandwiched between the two porphyrinates.388 For example, [{(porphyrinate)FeIII}2(μ-O22−)] species were found to be inert toward triphenylphosphine; however, triphenylphosphine oxide formation was observed during their thermal decay. The rationale behind this observation is that a ferryl intermediate is formed following homolytic O–O bond cleavage (Scheme 9; section 2.2).
However, as described in section 2.2, the [{(porphyrinate)- FeIII}2(μ-O22−)] species readily react with nitrogenous bases (such as piperidine, pyridine, or imidazole)335,365–367,498 and SO2537 even at cryogenic temperatures (eqs 2 and 3). The
| (2) |
| (3) |
former reaction (eq 2) intriguingly yields the six-coordinate low-spin FeIV=O species (Cmpd II), upon homolytic O–O bond cleavage induced by axial ligation of the base, while the latter (eq 3) produces sulfate anions (either free or bridging between two ferric porphyrinates). Finally, these [{(porphyrinate)FeIII}2(μ-O22−)] species could be extremely photosensitive,384 although some have been characterized by rR spectroscopy, possessing an Fe-O stretch centered within the range of 570–577 cm−1.538
2.3.2. Monomeric Iron(III)-Peroxo Adducts.
The first example of a synthetic iron(III)-peroxo complex was reported by Valentine and co-workers, where they subjected FeIII starting complexes of two porphyrinates (OEP and TPP, where OEP = octaethylporphyrinate and TPP = tetraphenyl- porphyrinate; see Figure 44) to excess KO2 in DMSO or
 |
(4) |
 |
(5) |
MeCN solvent at RT.539 At least two equiv of KO2 were required, where the first equiv reduces the FeIII complex to FeII, concomitantly producing O2; in the subsequent reaction, FeII + O2•− would yield the FeIII side-on bound η2-peroxide complex (eqs 4 and 5, Figure 44). The presence or absence of an axially coordinated solvent molecule in the FeIII-O22− species could not be unequivocally determined. This pattern of reactivity was observed to be highly dependent on multiple experimental conditions such as the solvent, moisture content, concentration of the iron solution, and reaction temperature. For example, in strongly coordinating (such as pyridine), or noncoordinating solvents (such as toluene or dichloromethane), the formation of a stable six-coordinate low-spin FeII complex, or the μ-oxo-bridged FeIII–(O)–FeIII species, were observed following the reaction with KO2, respectively, with no trace of the ferric-peroxo intermediate. Furthermore, in this work, the isolation of the iron(III)-peroxo complex from solution in trying to obtain X-ray quality crystals was unsuccessful, presumably due to its decay through a dimerization pathway. Therefore, spectroscopic characterization (UV-vis, EPR, IR, and rR) of solution samples was reported.539 The IR and rR O–O stretching frequencies were detected at 806 and 780 cm−1, respectively.538 The combination of spectroscopic insights led to the formulation of these complexes as high-spin iron(III)-peroxo species with a rhombic symmetry, possessing a side-on bound peroxo ligand on the FeIII center.
Figure 44.

Methodology utilized by Valentine and co-workers in the generation of side-on bound heme-iron(III)-peroxo complexes. Two equiv of KO2 were observed to be essential, where the first equiv would reduce the starting FeIII complex to FeII (eq 4) and the second equiv reacts with the FeII complex, producing the iron(III)-peroxo adduct (eq 5).
In a later report, the tetramethylammonium salt of the corresponding iron(III)-peroxo complex of the OEP porphyrinate, [(Me)4N][(OEP)FeIII-(O22−)], was isolated as a microcrystalline solid from the reaction between [(OEP)-FeIII(Cl)] and [(Me)4N](O2•−) in MeCN at −330 °C.540 Solution magnetic susceptibility measurements, Mössbauer isomer shifts, and zero-field splitting parameters (calculated from temperature-dependent EPR measurements) were all consistent with a high-spin FeIII complex within a rhombic symmetry, in agreement with the proposed side-on peroxo binding to FeIII. Following Valentine's initial report, Dolphin, James, and co-workers reported on the synthesis of [(OEP)-FeIII-(O22−)]− via the reduction of the corresponding superoxo complex (i.e., the oxy-heme), [(OEP)FeIII- (O2•−)], by electrochemical methods.541 The synthesis involved a fully reversible one-electron redox event at −30.24 V (vs Ag+/Ag0) in 1:1 MeCN/DMSO at −25 °C, as confirmed by spectroelectrochemical experiments. Other characterizations confirmed the peroxo species to also be a rhombic, highspin species.539 In another report, Momenteau carried out structural characterization of [(TPP)FeIII(O22−)]− using EXAFS spectroscopy, where a side-on bound iron(III)-peroxo unit was again detected, with an Fe-O distance of 1.80(3) Å.542
The reactivities of metal-peroxo adducts are known to be either electrophilic543 or nucleophilic,544–546 for which multiple factors could weigh in, including the identity of the metal, supporting ligand/solvent properties, and conditions of the experiment. Valentine and co-workers interrogated the epoxidation reactivity of two ferric-peroxo heme complexes, [(TPP)FeIII(O22−)]− and [(TMP)FeIII(O22−)]−,547 which only reacted with electron-deficient olefins, displaying nucleophilic reactivity. In a related study it was observed that by having strong electron-withdrawing substituents on the porphyrin ring [such as in TPFPP (TPFPP = 5,10,15,20-[tetrakis-(pentafluorophenyl)]porphyrin); see Figure 41; section 2.2.2], the nucleophilic epoxidation reactivity of the peroxo complexes could be turned off, while significantly increasing the stability of the peroxo complex.548 However, even with electron-withdrawing substituents, the peroxo moiety could not be influenced to become electrophilic. Notably, with the coordination of an axial ligand (iron(III)-peroxo complexes of TPFPP were observed to readily bind axial ligands such as PPh3 and DMSO), the nucleophilic reactivity of this previously unreactive iron(III)-peroxo complex could be turned on, exhibiting dramatic rates of epoxidation.529,548 For example, [(TPFPP)FeIII(O22−)]− instantaneously reacted with menadione in DMSO, producing 60–70% yield of menadione epoxide. This distinct increase in the nucleophilic reactivity of [(DMSO)(TPFPP)FeIII(O22−)]− as compared to [(TPFPP)-FeIII(O22−)]− was attributed to a more end-on like peroxo moiety in the former, with the charge-accumulated O atom more readily available as a nucleophile (Scheme 11).
Scheme 11.

Proposed “Turn-On” Mechanism of Nucleophilic Activity of [(TPFPP)FeIII(O22−)]− upon Axial Coordination of the DMSO Solvatea
aAdapted from ref 529. Copyright 1998 American Chemical Society. See text for discussion.
In comparative studies, Valentine and co-workers demonstrated heme iron(III)-peroxo adducts to be much more nucleophilic than many other metalloperoxo species (such as the peroxo adducts of MnIII, PtII, and TiIV),549 which may have been the reason for nature to select Fe as the active site metal of numerous dioxygen-dependent oxidation/oxygenation enzymes. In light of these findings, it was also suggested that biological iron(III)-peroxo intermediates could be the active catalytic species responsible for O2-dependent olefin epox- idation reactivities of Cyt-P450. In support, they showed that the [(protoporphyrin(IX))FeIII-peroxo]− adduct displayed superior reactivity over several other synthetic porphyrin systems.548 Moreover, Valentine also reported on a aromatase model utilizing the heme iron(III)-peroxo complex, [(TMP)- FeIII(O22−)]−, where it was concluded that the enolization of the steroid substrate at the 3-keto group is critical for its deformylation by the enzymatic heme-peroxo active oxi-dant.550 Watanabe and co-workers also carried out in-detail analysis of aldehyde deformylation reactivity of iron(III)- peroxo models using derivatives of the TPP ligand platform.551 Those reactions yielded both the deformylated and nitrilated products of the aldehyde substrate (Scheme 12 and Table 2), the latter as a result of substrate condensation with the solvent.
Scheme 12.

Reactivity of Cyclohexanecarboxyaldehyde with Heme Iron(III)-Peroxo Adducts Giving Both the Deformylated and Nitrilated Products As Observed by Watanabe and Co-workersa
aAdapted with permission from ref 551. Copyright 1998 Elsevier.
Table 2.
Product Distribution of Cyclohexanecarboxyaldehyde Deformylation Reactivity Mediated by Various Heme Iron(III)-peroxo Adducts As Reported by Watanabe and Co-workersa
| percent yields of products [%]a |
|||
|---|---|---|---|
| Entry | Compound |  |
 |
| 1 | [(TPP)FeIII-(O2)]− | 15 | 20 |
| 2 | [(TPP)FeIII-(O2)]−/PBNb | 8 | 15 |
| 3 | [(TMP)FeIII-(O2)]− | trace | 50 |
| 4 | [(TDCPP)FeIII-(O2)]− | 18 | 5 |
| 5 | KO2 | trace | 40 |
Adapted with permission from ref 551. Copyright 1998 Elsevier.
Yield were determined by GC/MS based on peroxoiron(III) prophyrin complex used.
[PBN] = 10 mM (PBN: phenyl-tert- butylnitrone).
Given the biological significance of O–O bond cleavage of peroxoiron(III) species (vide supra), the further activation of the O–O bond in these peroxo adducts has long been of interest. Such reactions involving dimeric μ-peroxo FeIII/FeIII complexes were described above (section 2.3.1). For monomeric iron(III)–peroxo adducts, Khenkin and Shteinman demonstrated that upon the addition of acylating agents (such as anhydrides or acyl chlorides), iron(III)-peroxo complexes (of TPP and TMP porphyrinates) convert into the corresponding Cmpd I species (Scheme 13 A), probably with the intermediacy of an acylperoxoiron(III) intermediate that exhibits excellent hydroxylation capability of unactivated hydrocarbons.552 Moreover, Weiss and Trautwein have reported the conversion of the iron(III)-peroxo species of the “picket-fence” porphyrin to the corresponding Cmpd II species upon reacting with CO2 in THF at −370 °C (Scheme 13 B).503 This Cmpd II product was characterized by UV-vis, 1H NMR, EPR, and Mössbauer spectroscopies. This study is also notable since the authors prepared the iron(III)-peroxo adduct by chemical reduction of the iron(III)-superoxo species using one equiv of sodium bis(2-methoxyethoxy)aluminum hydride. However, the stoichiometry of the CO2 interaction, and the mechanism of O–O bond cleavage in that study remain elusive, although the formation of a peroxymonocarbonate intermediate was proposed (Scheme 13B). In a later study, these authors reported an even more intriguing result where they could generate the Cmpd I species in noncoordinating solvents (such as DCM or toluene) from the same iron(III)-peroxo species, upon its protonation (Scheme 13 C).553 This serves as a direct mimic of the proposed mechanism for P450 enzymes, where O–O bond cleavage is triggered upon the protonation of the iron(III)-peroxo species at the distal O atom (vide supra). This protonation reaction displayed interesting solvent-dependency, where in coordinating solvents, such as THF, the generation of Cmpd II instead of Cmpd I was observed. The cyclohexene oxidation reactivities of the resulting Cmpd I and Cmpd II species were also investigated, where the former reacted readily to give cyclohexene oxide, while the latter was observed to be inert toward cyclohexene. It is of great interest how the identity of the added species (acylating agents, CO2, or protons) in the aforementioned studies could either lead to homolytic or heterolytic O–O bond cleavage of the ferric-peroxo moiety. Such variations in O–O bond cleavage patterns of iron(III)- peroxo/hydroperoxo (end-on bound in most cases) adducts have biological implications, given the differences in mechanistic details of CcO versus Cyt P450 or peroxidases (see sections 2.1 and 4.2).17
Scheme 13.

O–O Bond Cleavage Reactivity of Heme Iron(III)-Peroxo Intermediates upon the Addition of Either (A) Acylating Agents, (B) CO2, or (C) Protons
In more recent work, Karlin and co-workers demonstrated that the strong one-electron reductant CoCp2 could be used to reduce the iron(III)–superoxo species of the F8 porphyrinate (see Figures 34 and 35, section 2.2) to generate the corresponding monomeric side-on bound iron(III)-peroxo species, [(F8)FeIII(η2-O22−)]−, in THF at –80 °C (see section 5 for more detailed descriptions).495 [(F8)FeIII(η2−O22–)]– was characterized by UV–vis, EPR, and 1H NMR spectroscopies and was shown to be spectroscopically analogous to Valentine's complexes (vide supra). [(F8)FeIII(η2−O22–)]– was subsequently utilized to assemble [(heme)FeIII(O22–)-CuII(L)]+ complexes, taking advantage of the nucleophilic behavior of the ferric-peroxo adduct by reacting it with a ligand (L)-CuII compound.
2.4. Generation and Characterization of Heme FeIII–OOH Models, and O–O Bond Cleavage of Heme FeIII–OOR-type Intermediates
2.4.1. Generation of Heme Ferric-Hydroperoxo Models.
Heme ferric-hydroperoxo adducts (FeIII–OOH; also often referred to as Cmpd 0 in Cyt-P450 literature) are commonly proposed (or observed) intermediates in dioxygen activation by a series of hemoproteins, including heme monooxygenases such as Cyt-P450, peroxidases, and catalases, as well as in heme oxygenases (HOs); in the latter, as discussed, the FeIII–OOH species is thought to be the catalytically active oxidant (see sections 2.1). Within enzymatic O2-activation steps, FeIII–OOH intermediates originate from the reduction/protonation of the initial oxy-heme (i.e., ferric-superoxo) adducts and thus are comprised of a low-spin six-coordinate FeIII center with an axially bound end-on hydroperoxo ligand.
Heterolytic or homolytic O–O bond cleavage of FeIII–OOH moieties give rise to the high-valent Cmpd I or Cmpd II, respectively. Formation of a bridged FeIII–(OOH)–CuII moiety at the CcO heme/Cu active site may precede homolytic O–O bond cleavage (with the aid/involvement of the nearby Tyr residue) producing the spectroscopically observable Cmpd II intermediate, PM (see section 4.2). Nature has carefully tuned the reactivity of these heme FeIII–OOH intermediates within different enzymatic active sites, also evolving distal hydrogen-bonding amino acids or networks which may also support the placement of key water molecules which play a crucial role in hetero- or homolytic O–O reductive bond cleavage. In light of these biological implications, it has been a long-standing interest in the field to generate, and unequivocally characterize, FeIII–OOH intermediates within model systems and to carry out detailed investigations into their O– O bond cleavage chemistry. However, it has been a challenging task, mostly due to the highly destabilized nature of low-spin 6C heme FeIII–OOH intermediates when compared to their alkyl- or acyl-peroxo counterparts.146,347,554–557
FeIII–OOH intermediates were proposed in early studies involving the pH-dependent decomposition of H2O2 by [(TPP)FeIII]+ (TPP = tetraphenylporphyrin; see Figure 25, section 2.2)558 but were neither observed nor characterized experimentally. To this end, Tajima and co-workers utilized rapid freeze techniques coupled with electronic absorption and EPR spectroscopies to study the first example of a heme FeIII– OOH model compound during the H2O2 activation by [(TPP)FeIII(Cl)] at –45 °C in DMF solvent.559 Notably, [(TPP)FeIII(OOH)] was only observed under basic conditions, where either tripropylamine or potassium hydroxide was used as the base. Success in the observation of FeIII–OOH species also depended on initial reaction times (before freezing) and the presence of ~5% water. The development of an intense red color was characteristic of ferric hydroperoxo complex formation, and its EPR spectrum implied the presence of a low-spin ferric center, with characteristic peaks centered at g ≈ 1.962, 2.157, and 2.264.559 The axially ligating base was presumed to be −OH (Figure 45). In support, the related tert-butyl hydroperoxo ferric adduct had been previously characterized in methanol media and was found to bind methoxide as the axial base, having been formulated as [(OCH3)(TPP)FeIII(OOtBu)].557 Interestingly, when [(TPP)-FeIII(Cl)] was reacted with H2O2 at 25 °C, key spectroscopic changes (resulting in very weak electronic absorption features along with an intense g = 4.3 EPR signal) accompanied by a change in color was observed. These spectroscopic signatures were reminiscent of high-spin ferric centers of nonheme iron systems,560–562 suggesting that porphyrinate ring destruction under these conditions may have occurred. Thus, this observation lent credence to the proposal that a six-coordinate FeIII-OOH intermediate plays the role of the key active oxidant in heme degradation catalyzed by heme oxygenase (vide supra). In support, when a solution of [(OH)(TPP)-FeIII(OOH)]−was warmed and then refrozen, spectroscopic features inherent to the final product mixture resulting from the room temperature experiments could be observed.559
Figure 45.

First FeIII-OOH model system, [(TPP)(OH)- FeIII(OOH)]−, generated and characterized by Tajima and coworkers.559
Later work by Tajima and co-workers included the characterization of FeIII-OOH species derived from OEP and TMP porphyrinates (see Figure 25, section 2.2), carried out using the same rapid-freeze/UV-vis/EPR coupled methodology.563,564 In the OEP system, the authors observed that the six-coordinate iron(III)-superoxo complex, [(py)- (OEP)FeIII(O2•−)] (py = pyridine), readily reacts with sodium ascorbate or ascorbic acid in basic media to produce a new species with UV-vis and EPR features similar to those of [(OH)(TPP)FeIII(OOH)]− (Figure 46).563 Ascorbate was observed to carry out the one-electron reduction of the ferric superoxo moiety, and the resulting ascorbate radical was observed in a frozen solution (-195 °C) EPR spectrum (g = 2.006). This reaction was again observed to be tightly solvent- dependent and was only productive in the DMF/water mixture at −340 °C. This ferric-hydroperoxo adduct was formulated as [(OH)(OEP)FeIII(OOH)]−, and in support, [(OEP)FeIII(Cl)] reacted with H2O2 in basic media (similar to the TPP work; vide supra) to give the same compound (Figure 46).563
Figure 46.

Two synthetic routes for generating the low-spin six-coordinate FeIII-OOH adduct of OEP porphyrinate, [(OH)(OEP)FeIII(OOH)]−, as reported by Tajima and co-workers.563
In a later study, authors demonstrated the preparation of low-spin six-coordinate FeIII-OOH adducts of TPP, TMP, and OEP porphyrinates (see Figure 25, section 2.2) with imidazole as the axially coordinating sixth ligand.564 Initially, the [(OH)(P)FeIII(OOH)]− (P = supporting porphyrinates mentioned above) complexes were prepared at −60 °C in a DMF/MeOH/toluene/water mixture according to their previously published procedures (vide supra).557,563 The frozen [(OH)(P)FeIII(OOH)]− samples were then thawed at −60 °C, and upon addition of a ~ 40 molar excess of imidazole, the formation of a new low-spin ferric species occurred for the TMP system, as the EPR g values changed from 1.963, 2.156, and 2.257 to 1.943, 2.191, and 2.320. Concomitant changes in the electronic absorption spectra were also reported.564 In sharp contrast, for TPP and OEP porphyrins, heme degradation was observed upon the addition of ~25 molar excess of imidazole. The new ferric species formed in the case of TMP system was formulated as [(Im)(TMP)FeIII(OOH)] after careful analysis of EPR and crystal field parameters. This work exemplifies the much more robust nature of the FeIII-OOH species supported by the somewhat more sterically encumbered TMP ligand system when compared to TPP- and OEP-based ferric-hydroperoxo adducts.
Catalase model systems reported by Nocera and co-workers supported by the “hangman” porphyrinates are proposed to operate with the intermediacy of FeIII-OOH adducts (Figure 47), which are readily formed by the reactivity between FeIII starting complexes and H2O2.565 Importantly, the “hanging” carboxylic acid functionality was proposed to facilitate the O- O bond cleavage process by both hydrogen bond donation and proton transfer on to the distal O atom of the FeIII-OOH species. Accordingly, when the carboxylic acid was replaced by an ester functionality, the activity of the porphyrin complex reduced significantly. Interestingly, the addition of the same carboxylic acid exogenously (untethered) or changing the distance between the H-bonding functionality and the metal center (using a dibenzofuran-based tether to replace the xanthene-based tether) did not give the same efficiency as the initial tethered acid systems, suggesting the importance of the precise positioning of the protic/hydrogen-bond donating functionality in O–O bond cleavage.565 A later study also revealed that the electronic properties pertaining to this H- bonding group within the secondary coordination sphere can significantly affect the catalase activity.566 While these “Hangman” porphyrinates have been demonstrated to mediate both homolytic and heterolytic O–O cleavage depending on the reaction conditions, the carboxylate functionality has been observed to exclusively facilitate the heterolytic cleavage giving Cmpd I, as opposed to the ester group.567 These points are directly related to heme enzyme active sites, where the exact positioning of some distal amino acids (often hydrogen bond donors) has been shown to be crucial for proper functioning, including crucial mechanistic events such as O–O cleavage (see section 2.1). Furthermore, this work suggests that the delicate balance between hydrogen bond or proton-donating properties of these distal amino acid functionalities are critical for optimal turnover frequency.
Figure 47.

“Hangman” porphyrinate that catalyzes the disproportionation of H2O2 with the proposed intermediacy of an FeIII– OOH species that mediates proton-assisted O–O heterolysis. Adapted from ref 565. Copyright 2003 American Chemical Society.
The first fully characterized series of FeIII–OOH model systems accompanied by critical vibrational spectroscopic characterization came from the more recent work by Naruta and co-workers.122,490,568 Their first report of a low-spin six-coordinate FeIII–OOH species (Figure 48) supported by a TMP-derived porphyrinate with a tethered axial base, [(TMPIm)FeIII(OOH)],490 exhibited a versatile synthetic profile: (l) an FeII complex with addition of KO2 produced the side-on bound FeIII-peroxo species, which could then be protonated with MeOH, (2) carrying out the FeII + KO2 direct reaction in MeOH, and (3) 1e− reduction of the FeIII-superoxo adduct, and subsequent addition of MeOH, all produced [(TMPIm)FeIII(OOH)]. This work has marked a significant advancement in the field,44,569 which successfully mimicked the protonation of a heme peroxo FeIII–O22− species to generate the respective heme FeIII–OOH species, as has been proposed for hemoproteins (see section 2.1). This new FeIII–OOH intermediate was thoroughly characterized by UV–vis, EPR, rR, and Mössbauer spectroscopies, where the rR O–O and Fe–O stretches were –47) and 570 (Δ18O2 = −26) cm−1, respectively.490 Furthermore, upon H/D substitution of the FeIII–OOH moiety, the O–O and Fe–O stretching frequencies shifted 4 cm−1 units higher and lower in energy, respectively, providing further strong evidence for the formulation of [(TMPIm)FeIII(OOH)]. The side-on peroxo complex of this system (Figure 48) was also characterized by rR spectroscopy, with O–O and Fe–O stretches observed at 807 (Δ18O2 = −49) and 475 (Δ18O2 = −20) cm−1, respectively. The relatively lower-energy O–O and higher-energy Fe–O stretches of [(TMPIm)FeIII(O22−)]− with respect to the parent [(TMP)FeIII(o22−)]− compound [v(O–O) = 809 and v(Fe–O) 470 cm−1]490 indicates larger backbonding to the peroxo moiety in the case of the former. In support, the respective rR features of [(DMSO)(TMP)-FeIII(O22−)]−are virtually identical to those of [(TMPIm)-FeIII(O22−)]−.
Figure 48.

Versatile synthetic approaches utilized by Naruta in order to generate the FeIII–OOH species supported by the TMPIm ligand platform. Adapted with permission from ref 490. Copyright 2009 Wiley-VCH.
Naruta and co-workers have also interrogated the dioxygen binding of a modified “hangman” ligand platform with an axially tethered imidazole moiety, where they generate the first example of an end-on low-spin heme–peroxo complex by one-electron reduction of the respective superoxo complex (Figure 49).122 The scarcity of end-on heme–peroxo complexes presumably rise from their tendency to dimerize, or form side-on peroxo adducts, and/or undergo protonation to produce heme–hydroperoxo complexes (vide infra). The oxy-heme complex in this study was generated from ferrous starting complex and dioxygen in 20% MeCN/THF at –30 °C (MeCN = acetonitrile). The superoxo complex only exhibited an isotope sensitive rR band at 582 (Δ18O2 = −26) cm−1, which was attributed to the v(Fe–O) stretching frequency.
Figure 49.

Summary of work carried out by Naruta and co-workers on the modified “hangman” platform (left, Mes = mesityl).The heme-superoxo adduct which was initially generated could then be reduced to give the end-on low-spin ferric peroxo complex, which is then protonated to generate the corresponding ferric-hydroperoxo species (right). Adapted from ref 122. Copyright 2010 American Chemical Society.
The reduction of this superoxo complex with cobaltocene (CoCp2) resulted in a novel ferric peroxo complex, of which, the optical absorption and EPR features closely matched with those of peroxymyoglobin,570–572 formulated as a 6C low-spin end-on iron(III)-peroxo system. The rR spectrum of this new peroxo intermediate exhibits two sets of peaks that are shifted as expected with 18O-isotope labeling: 808 (Δ18O2 = −37) and 585 (Δ18O2 = −25) cm−1, as the v(O–O) and v(Fe–O) stretching frequencies, respectively. Although this v(O–O) stretching frequency is quite similar to that of heme side-on peroxo adducts (807 cm−1),490 the v(Fe–O) feature is significantly up-shifted (475 cm−1 for side-on peroxide) in the case of this end-on peroxide. This peroxo adduct could be readily converted to the hydroperoxo complex by reacting with ≈400 equiv of MeOH in 20% MeCN/THF at –70 °C. Further characterization of the hydroperoxo complex was carried out by vibrational spectroscopy, where the v(O–O) and v(Fe–O) stretching frequencies appeared at 807 (Δ18O2 = −31) and 575 (Δ18O2 = −25) cm−1, respectively. These features were also sensitive to H/D substitution at the Fe-OOH moiety, where the v(O–O) band upshifted by 2 cm−1, while the v(Fe-O) band down-shifted by 3 cm−1. Such behavior of rR features is revealing of hydrogen-bonding interactions involving the Fe-OOH unit.573,574 Authors attribute this observation along with the 10 cm−1 shift in the v(Fe-O) frequency upon protonation to possible interactions between added excess of MeOH solvent with both the Fe-OOH unit and the axial imidazole ligand, which could decrease the electron back-donation from the imidazole to the π* orbital of the hydroperoxo fragment.122
In a much more recent study, Naruta has reported the formation of another heme-hydroperoxo adduct (Figure 50, 3a), from the reaction between the corresponding iron(III)- superoxo complex (2a) and the ferrous species (as the reductant; 1a in Figure 50) in 80% EtCN/CH2Cl2 at −65 °C, utilizing a “hangman”-like ligand system bearing an in situ carboxylic acid group as the proton source.568 In this study, EPR spectroscopy provided evidence for the presence of a small low-spin impurity along with the high-spin iron(III)-superoxide complex upon oxygenation of the ferrous starting complex. Thus, a proton-coupled electron transfer (PCET) event from the starting ferrous complex (1a) to the ferric-superoxo species (2a) was predicted, which would account for the small low-spin impurity of a FeIII-OOH adduct (3a) in solution. This proposal was further interrogated by the addition of 1 equiv of ferrous complex into a ferric-superoxo sample, where the formation of a 1:1 molar ratio of the low-spin FeIII-OOH species (3a) and the high-spin “naked” FeIII complex were confirmed by EPR spectroscopy. In further evidence, the same hydroperoxo complex could be generated by reducing the superoxo complex with the exogenous reductant CoCp2. Intriguingly, when the carboxylate functionality of the secondary coordination sphere was replaced by an ester group, the clean formation of the corresponding superoxo complex (2b) was observed, which produced the FeIII-OOH adduct (3b) upon reduction-protonation (Figure 50). The aforementioned Fe-oxygen adducts were also characterized by rR spectroscopy: (1) FeIII-O2•− species, 2a and 2b exhibited Fe–O stretches at 576 (Δ18O2 = −26) cm−1 and 577 cm−1, respectively. The O–O stretching frequencies were not observed; (2) the FeIII-OOH complexes revealed their O–O and the Fe-O stretches centered at 807 (Δ18O2 = −40) and 811 cm−1 for 3a and 579 (Δ18O2 = −28) cm−1 and 576 cm−1 for 3b, respectively. Upon H/D exchange of 3a, a 7 cm−1 upshift of the O–O stretching frequency was observed, along with a 2 cm−1 downshift of the Fe-O stretch, supporting its formulation as a FeIII-OOH adduct.
Figure 50.

Derivatized “hangman”-type porphyrin systems recently reported by Naruta, which possess carboxylic (top) and ester (bottom) groups in the secondary coordination sphere. Under cryogenic conditions, the heme-superoxo complex of the former (2a) was observed to undergo reduction-protonation with the ferrous parent complex (1a), giving the heme-hydroperoxo adduct (3a). When the secondary coordination sphere's carboxylic acid group was replaced by an ester functionality, a stable superoxo complex was observed (2b), which could be converted to the corresponding ferric-hydroperoxo species (3b) upon reduction-protonation with exogenous reagents. Adapted with permission from ref 568. Copyright 2016 The Royal Society of Chemistry.
2.4.2. O–O Bond Cleavage Process in FeIII-OOR (R = H, Alkyl, or Acyl) Adducts.
Reactivity studies of well-defined FeIII-OOH model systems are extremely scarce and, consequently, are often only discussed within the events en route to high-valent oxidants within enzymatic catalytic cycles. In fact, previous computational42,575–577 and experimental578–580 studies have suggested that these intermediates may very well be sluggish oxidants, when compared to highvalent Cmpd I- or Cmpd II-type species. In contrast, some theoretical studies have suggested that FeIII-OOH-type species can be very efficient oxidants in Cyt-P450-mediated sulfoxidation reactions.581 Furthermore, the efficiency and accuracy of FeIII–OOH active species in “traditional” heme oxygenases clearly demonstrates the fact that these intermediates could be strong oxidants upon proper tuning of their steric and electronic properties. Unfortunately, it is difficult to arrive at definitive conclusions regarding key factors affecting their reactivity, given the severely limited set of synthetic examples found in the current literature.
As in the case of Fe-oxo intermediates (see section 2.1.5),363,582 the nature of the axial ligation can significantly modulate the properties of FeIII–OOR (R = H, alkyl, or acyl) intermediates, thus, significantly affecting their O–O cleavage chemistry (i.e., heterolytic versus homolytic nature of O–O bond scission).49–52,328,583–585 In early studies, Groves and Watanabe observed that [(TMP)FeIII(Cl)] (see Figure 25, section 2.2) plus m-CPBA would yield Cmpd I upon O–O bond heterolysis in polar solvents, while exhibiting O–O bond homolysis giving [(TMP)Fe-N-oxide] in nonpolar solvents (Scheme 14).50,52 However, Krik and co-workers later reported that Cmpd I of this system could in fact be generated in nonpolar solvents under the same experimental conditions, in the presence of axially coordinating anions (such as triflate).586 This work clearly reveals the prominent effect of axial ligation on O–O bond scission of the FeIII–m-CPBA adduct of TMP (Scheme 14). Furthermore, Nam and co-workers have reported on the disparities of [(X)(TPFPP)FeIII]-facilitated (also see Figure 41, section 2.2; X = axial ligand) oxygenation of hydrocarbons using H2O2 as the oxidant, where the feasibility and/or the yields of epoxidation and hydroxylation reactivities were observed to be strongly affected depending on the identity of X.583
Scheme 14.

Homolytic versus Heterolytic O–O Bond Cleavage of the m-CPBA Adduct of [(TMP)FeIII]+
Recently, van Eldik and co-workers carried out in-depth investigations into axial ligand influences on O–O bond scission of [(TPFPP)FeIII(OOH)] (see Figure 41, section 2.2) in the presence of organic substrates.585 The TPFPP-supported FeIII–OOH adduct exhibited enhanced stability with respect to TMP-derived systems, presumably due to the electron-withdrawing nature of the former. In this work, the FeIII–OOH compounds were prepared following a similar methodology to that of Tajima (FeIII starting complexes plus H2O2 in the presence of a base)559,563,564 in MeCN at –15 °C. Intriguingly, the researchers were able to prepare both the five- coordinate, [(TPFPP)FeIII(OOH], and the six-coordinate complexes, [(OH)(TPFPP)FeIII(OOH)]−, where the latter displayed superior stability compared to its five-coordinate counterpart. [(OH)(TPFPP)FeIII(OOH)]− was observed to undergo heterolytic O–O bond cleavage giving the corresponding Cmpd I species, while homolytic O–O bond scission was observed for [(TPFPP)FeIII(OOH)] producing Cmpd II (Scheme 15).585 The reactivities of FeIII–OOH complexes were investigated against electron-poor olefins (for epoxidation) and triphenylphosphine (PPh3; for possible O atom transfer). However, they did not react with any of the substrates chosen, displaying a similarly inert nature as the side-on iron(III)-peroxo adduct of the same ligand platform, [(TPFPP)FeIII(O22−)]−, as previously reported by Valentine and co-workers (see section 2.3).529,548 This inertness to oxidative reactivity is presumably due to the strong electron-withdrawing properties of TPFPP. Interestingly, this unreactive nature of the FeIII–OOH adduct is in contrast to the conclusions drawn by Newcomb, Hollenberg, Coon, and co-workers,53,587,588 suggesting the Cyt-P450 Cmpd 0 as an alternative electrophilic oxidant in alkane hydroxylation reactions. However, it should be noted that these differences in reactivity profiles should be viewed within the caveat of the large differences between the geometric and electronic structures of the model system when compared with those of the P450 active site.
Scheme 15.

Differences in Formation, and Reactivity Landscapes of Five-Coordinate [(TPFPP)FeIII(OOH)] and Six-Coordinate [(OH)(TPFPP)FeIII(OOH]− Ferric Hydroperoxo Complexes
The O–O bond cleavage chemistry of FeIII–OOR intermediates are known to depend on a number of crucial parameters that are inherent to the Fe-porphyrinate, the HOOR-type oxidant, and the reaction conditions/medium. The classical work by Bruice47,585–595 and Traylor48,593,594 has presented contradictory conclusions on the O–O bond cleavage process in heme iron(III)-alkylperoxo adducts, where Bruice favored a homolytic bond cleavage, while Traylor reported evidence favoring a heterolytic mechanism. Bruice demonstrated that the FeIII–OOR adducts [where R = C(CH3)3 or C(CH3)2Ph] of meso-tetrakis(2,6-dimethyl-3- sulfonatophenyl)porphyrinate undergo pH-independent homolytic O–O bond scission, with the concomitant formation of the expected R-based radical organic products.595,596 On the other hand, the highly efficient alkene (cyclooctene and norbornene) epoxidation with the TPFPP porphyrinate and C(CH3)3OOH in methanol led to the proposal of O–O heterolysis, which was shown to give Cmpd I by Traylor.48 However, Traylor's observations were put into question in light of the protic nature of the methanol solvent used. In the aforementioned intriguing report by Bruice and co-workers, the authors demonstrated that the meso-tetrakis(2,6-dimethyl- 3-sulfonatophenyl)porphyrinate can promote either heterolytic or homolytic O–O bond scission of its FeIII–OOR species depending on the identity of the R group.595,596 In this work, iron(III)-acylperoxo and −3alkylperoxo complexes underwent heterolytic and homolytic O–O bond cleavage, respectively, in aqueous media (Figure 51). It was also clearly observed that the homolytic O–O bond cleavage of the alkylperoxo adducts was pH-independent. However, Nam and co-workers have reported that some water-soluble porphyrinates facilitate pH-dependent epoxidation of stilbene utilizing alkylperoxides [HOOR, where R = C(CH3)3 or 2-methyl-1-phenylpropan- 2-yl] as oxidants, where the O–O cleavage mechanism was proposed to switch over from heterolytic to homolytic as the pH of the medium is elevated.597,598 Accordingly, the yields of stilbene epoxide were observed to decrease with increasing pH. The latter observation was attributed to the inferior oxidative power of Cmpd II (compared to Cmpd I) resulting from the homolytic O–O cleavage that occurs at high pH values. Moreover, these researchers also observed that the electronic properties of the supporting porphyrinate could also significantly affect the O–O bond cleavage mechanism. That is, in agreement with the previous studies,599–601 electron-withdrawing properties of the supporting ligand were observed to favor heterolytic O–O bond cleavage, while electron- donating substituents favored a homolytic pathway. The identity of the axial ligand was also observed to exert such effects; coordination of 5-chloro-1-methylimidazole and 1-phenylimidazole significantly increased the yield of stilbene epoxide, as compared to 1-methylimidazole or 1,2-dimethyli-midazole, which did not significantly modify the epoxidation efficiency, presumably due to their strong electron-donating nature.
Figure 51.

Iron(III)-acylperoxo (top) and iron(III)-alkylperoxo (bottom) complexes that lead to heterolytic and homolytic O–O bond cleavage, respectively (in aqueous media), as observed by Bruice and co-workers. See text for discussion.
The aforementioned pH dependence of O–O bond cleavage of FeIII–OOR adducts was further investigated by van Eldik and co-workers using the ferric complex of highly water-soluble, non-dimer-forming porphyrin TMPS (Figure 52a) along with H2O2, m-CPBA, and PhIO as oxidants.602 When H2O2 is used as the oxidant in MeOH:H2O (4:1) mixture at –35 °C, the formation of Cmpd I as the sole oxidant was observed at pH < 5.5, while at pH > 7.5 Cmpd II dominates in solution. Within the pH range of 5.5–7.5, both Cmpd I and Cmpd II were observed to coexist. Although differences in axial ligation were observed at different pH values, the O–O bond cleavage patterns were concluded to be primarily governed by the pH-dependence of redox properties of the iron center (see the Pourbaix diagram shown in Figure 52b).49,602,603 Thus, the speciation of the heme was found to be tunable with respect to the pH of the medium, where the most thermodynamically stable form is different for various ranges in pH/E°’. The identity of the resulting Fe-based oxidant could also strongly depend on the redox properties of the oxidant used, where a strong two-electron oxidant (such as m-CPBA) could still produce Cmpd I at pH > 7.5, although it is rapidly transformed to the thermodynamically favorable Cmpd II form, emphasizing the fact that the genuine O–O bond cleavage product may not always be observable in solution, especially in the presence of subsequent energetically favorable redox processes. In a similar situation, Watanabe and co-workers reported that for O–O bond heterolysis of H2O2 by Mb (peroxidase model), the Cmpd I species is only a fleeting intermediate and was found to rapidly get reduced to Cmpd II due to a subsequent energetically downhill redox event involving the nearby His64 residue.324–326
Figure 52.

(a) Water-soluble TMPS porphyrin utilized by van Eldik and co-workers in their pH-dependent studies of O–O bond scission, and (b) Pourbaix diagram describing pH-dependent speciation of redox forms of [(TMPS)FeIII]+. Adapted with permission from ref 602. Copyright 2007 Wiley-VCH.
Further studies were carried out with the same porphyrinate and the m-CPBA oxidant (in H2O/MeOH or H2O/MeCN solvent mixtures), where the immediate oxidation products were analyzed at low temperatures using rapid-scan stopped- flow techniques.604 This work in H2O/MeOH revealed that the six-coordinate Cmpd II species, [(HOR)(TMPS)FeIV(O)] (R = H or CH3), that dominates in solution at pH > 7.5 originates from the homolytic O–O bond cleavage of a FeIII–OOR species that results from the reaction between the Cmpd I species and hydroxide/methoxide anions in solution (Scheme 16). In addition, the generation of FeIII–OR was also observed at high pH conditions (pH > 10), presumably following ligand exchange between the FeIII–OOR species and hydroxide/methoxide anions present in high concentrations under these experimental conditions (Scheme 16). When a large excess of m-CPBA is added under these highly basic conditions, the rapid, clean formation of Cmpd II, [(TMPS)FeIV(O)(HOR)], was observed and is proposed to be the product of comproportionation between Cmpd I and FeIII–OR species. Notably, the switch in reaction solvent to H2O/MeCN alters the reaction landscape remarkably, where the clean formation of Cmpd II is observed at high pH, which is the product of comproportionation between Cmpd I and FeIII starting complex. Unlike when MeOH was present, there was no trace of the ligand substitution pathway that leads to FeIII–OR in this case. Overall, this study revealed that with m-CPBA, FeIII starting complexes would always produce Cmpd I; however, depending on its stability under the given conditions, either Cmpd I or a further-reduced species may accumulate in solution.
Scheme 16.

Proposed Reaction Pathways Surrounding the Oxidation of [(TMPS)FeIII(OR)2] with m-CPBA in a Water/MeOH Mixture under Basic Conditions
The effects of an axially coordinated imidazole ligand in the formation of the same Cmpd II (starting from FeIII–OH and H2O2) has also been investigated in water,605 where biphasic kinetics have been observed. In that, the rate of formation of Cmpd II initially increased linearly with the N-methylimidazole concentration, followed by a sharp decrement, finally reaching a constant value. This unique kinetic profile was attributed to the initial formation of the five-coordinate monoimidazole ferric species in solution, which is much more reactive than the starting FeIII-OH complex. As described above, axially ligated imidazolyl groups will “push” electron density on to the π- antibonding orbitals of the iron(III)-peroxo moiety in this case,317,318,594 which presumably leads to the rapid formation of Cmpd II. As the imidazole concentration increases, the formation of the kinetically inert low-spin six coordinate bisimidazole FeIII species results, effectively lowering the rate of Cmpd II formation. Mechanistically, this second process was proposed to proceed via the initial protonation of the bound imidazole by H2O2, followed by the exchange of protonated imidazole with the HOO− anion. This proposal is supported by thermodynamic studies that indicate an overall associative process.605,606 Similar studies for the formation and stabilization of Cmpd I intermediates in aqueous media are much rarer; however, recent work by Groves and van Eldik have revealed that the successful preparation of Cmpd I intermediates can be carried out in moderately acidic (pH = 4.5–5.0) aqueous media at 5–10 °c.351,607 A detailed discussion on the formation and reactivity of Cmpd I/II models is found in section 2.1.5 of this review.
In summary, the subtleties involved in O–O bond cleavage are known to depend on a number of critical factors involving the identity and properties related to (1) the ROOH-type oxidant (including the redox properties and the identity of the R group), (2) the electronic properties of the supporting porphyrinate, (3) electron-donating power of axial ligands, and (4) the reaction medium and experimental conditions (such as the pH, temperature, etc.). Despite its crucial importance within biological systems including CcO, the understanding of the O–O bond cleavage process of FeIII-OOH species is still in its infancy. Such studies on discrete FeIII–OOH examples, such as those reported by Naruta (vide supra) could reveal critical factors pertaining to axial ligand effects, pH influences, and/or solvent effects that choreograph the O–O bond cleavage mechanism of these pivotal intermediates.
3. COPPER CHEMISTRY WITH DIOXYGEN: BIOLOGY AND MODEL SYSTEMS
The undeniably rich chemistry of copper with dioxygen has motivated biochemical and synthetic inorganic studies, which have shed light on the important aspects of metal ion coordination environment and electronic structure that control the outcomes and mechanisms of various chemical reactions. Copper-containing metalloenzymes employ mononuclear or binuclear copper active sites to carry out diverse functions, including partially (by 2e−) or completely (by 4e−) reducing dioxygen; a process which is often coupled to substrate oxidation or oxygenation (vide supra, section 1, Figures 3 and 4). Importantly, as relevant to the focus of this review, a thorough understanding of copper-O2 interactions is valuable in order to comprehensively analyze heme–Cu systems’ reactivities with dioxygen. Therefore, in this section, we outline important O2-activating enzymes containing copper and discuss the variety of proposed intermediate structures, of the general form, (Cu)n–O2(H) (where n = 1, 2). Additionally, we highlight the bioinspired, synthetic inorganic studies which have elucidated important structure–function relationships based on systematic ligand modifications and rigorous spectroscopic analyses. Many results from these studies have informed the design of heme- and copper-containing HCO models and have aided in the interpretation of their structure-directed reactivities and the O–O reductive cleavage mechanism. As the field is rapidly advancing, several recent reviews have thoroughly covered the latest important advances;18,19,608–612 therefore, here we will provide a brief introduction into copper metalloenzyme biochemistry but focus on the aspects of Cu–O2 chemistry which have been or can be directly applied to the design of heme–copper model complexes and the overall understanding of heme–copper oxidase (bio)chemistry.
3.1. Single Copper Site Enzymes
Metalloenzymes which contain a single copper ion in their active site can be classified, based on their active-site copper coordination environment and function, as “Type I” blue copper proteins (or cupredoxins) or “Type II” (nonblue copper proteins). Type I blue copper enzymes, such as plastocyanin and azurin, have (NHis)2-(SCys,Met)2 coordination and function as electron transfer proteins rather than interacting with dioxygen or other small molecules; therefore, we will not focus on their properties or reactivity here, and instead the interested reader is directed to several recent reviews.16,19,613,614
In contrast, Type II copper enzymes have an active site with a single Cu ion possessing (His)2 or (His)3 imidazole coordination and partially (by 2e−) or fully (by 4e−) reduce dioxygen. Enzymes in this family include amine oxidase and galactose oxidase (Figure 53),615–621 along with lysyl oxidase, the latter being a potential target in cancer treatment.622
Figure 53.

Active site structures and O2-reduction/substrate oxidation reactions of mononuclear copper oxidases (A) amine oxidase and (B) galactose oxidase. See text for discussion.
Several so-called uncoupled dicopper monooxygenase enzymes exhibit similar reactivity, wherein dioxygen chemistry (i.e., O2 binding and reduction, and substrate oxygenation) occurs at a single copper site (termed CuM) and the other copper ion (CuH) regulates electron transfer. Interestingly, in these cases, the copper ion controlling electron transfers (CuH) is a Type II-like center, and the O2-binding site CuM is ligated by two His and one Met residue (Type I-like). Such enzymes include dopamine β-monooxygenase (DβM), peptidylglycine α-hydroxylating monooxygenase (PHM), and tyramine-β-monooxygenase (TβM) (Figure 54A).19,623 It is now generally agreed upon that the initial copper(I)-dioxygen adduct forms at CuM, a CuII(O2•−) complex; the substrate also binds to the protein in close proximity to CuM. An X-ray crystal structure in an O2 bound (oxy) state has been reported by the Amzel research group (Figure 54A).624
Figure 54.

Monocopper monooxygenase active site structures for particular protein examples (PDB codes given) and their O2-reduction/substrate oxygenation reactions: (A) peptidylglycine α-hydroxylating monooxygenase (PHM), (B) lytic polysaccharide monooxygenase (LPMO), (C) particulate methane monooxygenase (pMMO) with either a di- or monocopper active site, and (D) formylglycine-generating enzyme (FGE), crystallized with Ag in the site proposed to normally contain copper. See text for further discussions.
In the proposed mechanism of action, the cupric-superoxide fragment effects a substrate hydrogen-atom transfer (HAT) reaction to give a product hydroperoxo-copper(II) complex plus substrate radical species (Figure 55).625 In fact, Amzel's group more recently characterized a PHM derivative with a side-on peroxo or hydroperoxo entity bound to CuM (Figure 56), giving credence to the possibility of such an intermediate structure forming in the catalytic cycle.626 Such entities are known in copper coordination chemistry (vide infra) and may be critical in the PHM enzymatic pathway.625 In previously discussed mechanisms,612,627 O–O reductive cleavage is said to occur with substrate rebound giving the product alcohol and CuII-oxyl (a “cupryl”, CuII–O· high-valent species, to be further discussed, vide infra) as shown in Figure 55, pathway 1; the latter abstracts an electron from CuH and with proton transfer gives a CuII-hydroxide species. Solomon's most recent calculations625 provide an advanced mechanistic model, where the substrate radical formed attacks (rebounds) the non-protonated O atom of the side-on bound hydroperoxide ligand, and with O–O cleavage, a strong oxidant containing a CuII-substrate radical forms, “pulling” the CuH electron, to give the same products (Figure 55, pathway 2). Critical to the analysis of this pathway, is the discussion of the importance and relevance of unusual properties ascribed to the CuH site, which possesses an unusual coordination geometry. CuH is most often observed with a distorted T-shaped geometry (His)3 (see discussion below) and does not seem to bind typical anions (e.g., azide, nitrite) unless exposed to very large excesses.625,628 This electron-transfer center possesses a large Marcus theory reorganization energy (λ), that being important to the overall PHM mechanism, as it allows “time” for substrate radical rebound, prior to electron-transfer reduction.625
Figure 55.

Postulated steps in the mechanism of action of PHM and related monooxygenases including key steps where the cupric-superoxide species initially formed upon O2 binding carries out a hydrogen atom abstraction (HAA) on substrate yielding either an end-on (pathway 1) or side-on (pathway 2) cupric-hydroperoxide. Subsequent rebound of the substrate radical to the Cu-hydroperoxide gives a highly oxidizing Cu – oxyl (i.e., CuII–O•) or CuII-substrate radical intermediate, respectively (highlighted in yellow). An electron, drawn from the enzyme (CuH)site (Figure 54A), and a proton react with the radical intermediate to release the hydroxylated alcohol product leaving a CuII–OH moiety which can be reduced to copper(l) to restart the catalytic cycle.
Figure 56.

PHM active site with side-on bound (hydro)peroxo ligand coordinated to Cum.
Further discussion of the coordination chemistry of the (His)3-ligated CuH center is warranted (Figure 54A). Here, we see a very infrequent ligation of adjacent His residues, H107 and H108. Such coordination is nonexistent in iron, manganese, and zinc biochemistry, and it is unusual in copper biochemistry, except for PHM and DβM and the CuB site in cytochrome oxidases (i.e., HCOs), see Figure 57 and further discussion in section 6. Histidine-rich peptides are found to be important in biological contexts, and in particular when possessing adjacent “bis-His” residues, a high affinity for cuprous ions is observed, and linear two-coordinate imidazole- Cu(I)-imidazole complexes readily form, usually with short (Cu–NIm = 1.86–1.88 Å) strong bonds.629–632 Interestingly, adjacent bis-His residues are also found within the metal binding region of the 40–42 amino-acid long Alzheimer’s disease Aβ peptide.629,633,634
Figure 57.

(A) Reduced active site of CcO featuring the tris-His ligation for CuB; the overall geometry is distorted T- or Y-shaped, and for H290 and H291, binding occurs to the Nε atom of the imidazole groups. (B) A PHM derivative in the oxidized state. Here, CuH is also found in a distorted T-shaped geometry, where H107 and H108 form a near-linear coordination environment (∠ H107-Cu-H108 = 164°). By contrast, these two His residues bind the copper ion through their imidazole Nδ atoms in a very favorable strong binding interaction (as does H172). See text for further discussion.
Many PHM X-ray structures show CuH is coordinated in a T- or Y-shaped geometry, where adjacent residues H107 and H108 chelate copper in a near linear fashion, and a third histidine, His172, is weakly coordinated to the metal center (Figure 57). Blackburn and co-workers635–637 conducted EXAFS spectroscopic studies of reduced wild-type and a H172A PHM mutant and found similar average Cu–NHis bond lengths (1.92 Å for WT and 1.88 Å for H172A), despite the mutant having one fewer histidine available to coordinate to CuH. These results are consistent with the notion that adjacent histidine residues H107 and H108 impart a near two-coordinate linear geometry on reduced CuH, as one would expect linear coordination of CuH to exhibit short Cu–NHis bond lengths.
Model studies by Karlin and co-workers, and others, using “histidylhistidine” ligand frameworks, have further corroborated these coordination chemistry concepts.633 Through regioselective synthetic derivatization at the imidazole N atoms, well-defined cuprous compounds with synthetic peptides (Figure 58; Lδ, Lε) coordinated through either the Νδ or Nε of histidine derivatives could be generated and characterized. From EXAFS spectroscopy, the complexes obtained show both model complexes have very similar Cu–N distances (1.876 and 1.863 Å, respectively) as compared to the reduced PHM mutant H172A. A complex with Lε was determined to be dimeric, [{(Lε)Cu}2]2+, while with Lδ a single-Cu complex formed, [(Lδ)]+ (Figure 58). The results highlight that adjacent bis-histidine ligation strongly favors intramolecular, linear, two-coordinate copper(I) geometries but only when the Nδ atoms of histidine are available. These coordination concepts may apply to enzyme systems; following the discussion above, we restate that CuH in reduced PHM exhibits a tautomeric preference to bind the Nδ atoms of H107/H108, giving a large Hisn–Cu–Hisn+1 angle (143.0° in PDB 3PHM; 164° in PDB 1PHM; Figure 57), while CuB in CcO binds to the Ne atoms of H290/H291 giving a smaller Hisn–Cu–Hisn+1 angle (91.4° for PDB IV54; 100.4° for PDB 5B1B). For how these differences in copper coordination may be involved in the catalytic mechanism of CcO, see section 6.3.5.2.
Figure 58.

Synthetically designed “histidylhistidine” ligands and their calculated (truncated) cuprous compounds. Selective functionalization of either the Nδ or Nε of the imidazole moiety allows for generation of discrete tautomeric compounds featuring either (A) Nδ–Cu–Nδ or (B) Nε–Cu–Nε ligand coordination.633
Lytic polysaccharide monooxygenases (LPMOs), only characterized starting about 10 years ago, contain a unique copper ligand environment in which two His residues are bound; however, one acts as a bidentate ligand, aptly termed, a “histidine brace” (Figure 54B).638–642 There is great interest in LPMOs as they have biotechnological significance and effect a HAT reaction from substrates which have exceedingly strong (>100 kcal/mol) C–H bonds.642 Further, with the most recent information, the presence of a “histidine brace” at the single copper ion site in LPMOs exists in common with new findings on particulate methane monooxygenases (pMMOs, vide infra). Also, some LPMO actives sites have a key neighboring Tyr residue (as in HCOs), which is critical for the O2 chemistry.643
Recent reactivity and spectroscopic studies of LPMOs have emphasized the importance of nearby secondary coordination sphere amino acid residues and the effect they may have on the mechanism of substrate hydroxylation.641,644,645 Marletta and co-workers have demonstrated the importance of two H-bonding residues (His and Glu) and their role in stabilizing bound dioxygen as well as increasing the donating ability of the nearby Tyr residue to Cu, possibly stabilizing a CuII-oxyl reactive intermediate.644 A recent structure reported by Chen and co-workers shows a copper-bound peroxide moiety with partial deprotonation of the –NH2 group of the His-brace (Figure 59A).641 This may stabilize a CuIII intermediate in the enzymatic active site, as has been observed in synthetic model systems employing anionic ligands (see section 3.3.1.3).609,646,647 Multiple high-resolution X-ray and neutron crystal structures involving either peroxide bound to copper or dioxygen bound adjacent to the active site of LPMO were recently published by Meilleur and co-workers (see Figure 59B for one of these structures).645 The authors postulated that a nearby nonligated and doubly protonated His residue may play a role in O2 binding and activation. Together, these enzymatic studies display the importance of secondary coordination sphere interactions in the activation of dioxygen at the copper active site of LPMOs.
Figure 59.

Crystal structures showing peroxidic O2 fragments bound to the static structures formed at the active site of LPMOs.
Several computational studies on copper active-site mediated polysaccharide oxidative degradation have included discussions of reductive activation of dioxygen eventually leading to a CuII–oxyl oxidant and subsequent substrate H atom abstraction following O–O cleavage.642,648,649 A recent report650 demonstrates that LPMO activity can be observed through bypassing dioxygen and reducing equivalents; H2O2 serves to effect enzyme activity, this being a peroxide-shunt analogous to what is known to occur in cytochrome P-450 enzymes. If this work is correct, it implies that substrate C–H abstraction must follow O–O cleavage, supporting the supposition that a CuII–oxyl active intermediate is very likely relevant to initial polysaccharide substrate H atom abstraction. Very recent QM/MM calculations by Rovira, Walton, and coworkers support the hypothesis that a CuII–O• species is the intermediate responsible for substrate C–H HAT in LPMOs.651 Starting with CuI and H2O2 in the enzyme active site, the O–O bond of H2O2 is cleaved in a homolytic fashion (facilitated by ET from the CuI ion) to give a CuII–OH species and •OH (hydroxyl radical). This radical species (normally known to be highly reactive) is held in place through secondary coordination sphere H-bonding interactions, stabilizing it long enough to abstract the H atom from the CuII–OH compound.651 The CuII–O• intermediate thus formed is positioned for selective polysaccharide substrate hydroxylation.
In fact, several research groups have published that LPMOs are in fact peroxygenases (i.e., normally using H2O2 as cosubstrate).650,652 However, Marletta and co-workers reported on the comparison of substrate oxidation by LPMOs (using both soluble and insoluble cellulose substrates) using either O2 or H2O2 as the cosubstrate oxidant, starting with reduced enzyme (cuprous state).653 This study showed that LPMOs are able to act as an oxygenase or peroxygenase, with faster reactions occurring with the use of H2O2 as cosubstrate. However, regioselective substrate hydroxylation, one of the key interesting aspects of LPMOs, was only achieved when O2 was the cosubstrate. This, along with the extensive enzyme oxidative damage when H2O2 is employed, points to O2 as the native cosubstrate oxidant for LPMOs.
One can imagine that even if O2 is the cosubstrate utilized by the enzyme, the reaction mechanism discussed above (starting from CuI and H2O2) may still be applicable. A CuII–OOH intermediate (formed from reduction and protonation of a cupric superoxide species made from CuI/O2 chemistry) may cleave via protonation and reduction to give CuI and H2O2, which would then start the proposed cycle.651 Clearly, future studies are needed to decipher the full detailed mechanism of LPMOs.
Particulate methane monooxygenase performs the difficult oxidation of methane to methanol and yields one H2O molecule per O2 consumed (Figure 54C). The active site contains a similar “histidine brace” as that found in LPMOs. However, the unresolved nature of the active site of pMMO has been a somewhat controversial topic in the literature, as the active site can be crystallized with one or two copper ions (PDB IDs: 3RFR and 1YEW, respectively, see Figure 54C).654,655 Starting from the coordinates of these crystal structures, the mechanism of methane oxidation has been thoroughly investigated using DFT calculations. Multiple copper-oxy species have been suggested as the possible reactive intermediate, including (μ-oxo)(μ-hydroxo)CuIICuIII, (μ-oxo)CuII2, and CuII-O•.655 However, a very recent study involving quantum refinement of a library of existing pMMO crystal structures has claimed that the combined analysis using these methods rules out a dicopper active site, which of course will have an effect on mechanistic studies and interpretations, since many previous studies implicated a dicopper active site.656 Indeed, if a monocopper active site with histidine brace is able to convert methane to methanol, then, as for LPMOs, a CuII-oxyl intermediate that attacks substrate will highlight near-future investigations. Gas-phase generated CuII-oxyl species can readily attack and oxygenate methane (vide infra). While the soluble methane monooxygenase, sMMO, has a more well-understood diiron structure,655,657,658 studies involving the structure and activity of the transmembrane, Cu-containing pMMO, remain to be of high importance, as multiple aspects of the reaction (i.e., methane activation/strong C–H bond cleavage as well as O–O reductive cleavage) are relevant for global energy concerns.
Very recently, a small and quite unique family of monooxygenase enzymes was discovered, called formylgly-cine-generating enzymes (FGEs), which utilize a copper cofactor to carry out O2-dependent cysteine oxidation to formylglycine (see Figure 54D). In these enzymes, the copper ion is ligated by two Cys residues, leaving open coordination sites for both the specific Cys substrate and dioxygen to bind. The proposed mechanism currently implicates a cupric superoxide as the active species, which undergoes HAT and ET from the substrate to yield a CuI–OOH, releasing H2S and the formylglycine product. A critical redox balance governs this catalysis, as disulfide bridge formation between active site Cys residues can result in copper release and an inactive enzyme. Currently, these mechanistic studies are supported largely by EPR spectroscopy, product characterization, and crystallographic studies.659–661
3.2. Metalloenzymes with Dicopper Active Sites
Several so-called Type III copper enzymes utilize a bimetallic active site comprised of two magnetically coupled copper ions which cooperate to carry out either O2-transporting or O2-activating functionality (Figure 60).19,662,663 Hemocyanin (Hc) and (poly)phenol oxidases (Tyrosinase, Ty; catechol oxidase, COx) both have dicopper active sites in which the copper ions are ligated by 3 histidine residues each (see Figure 60) and are held ~4–5 Å apart in their deoxygenated forms.664 Both of these types of enzymes efficiently bind dioxygen as a symmetric, μ-η2:η2 peroxo moiety (based on rR data and X-ray crystal structure determination),665 which induces geometrical changes in the active site. Hc acts only as an O2-carrier, binding dioxygen reversibly (KO2 on the order of 105, relative to ~104 for myoglobin),56,666,667 whereas the O2-activating enzymes reductively cleave the O–O bond and direct the energy released in that reaction into substrate oxidations. So, what gives rise to this distinction in the reactivity of enzymes with such structurally similar active sites? It has been suggested that substrate access to the Cu2–O2 core contributes to this distinction, and indeed, when blocking residues are removed by mutagenesis in Hc, this enzyme can hydroxylate phenolic substrates.668–670 On a molecular level, model chemistry represents a useful approach to addressing such issues (vide infra); however, biochemical research over the past several decades has elucidated many important details of the O2 reactivity of these enzymes.
Figure 60.

(A) Overlaid active site crystal structures of octopus hemocyanin (green), Streptomyces castaneoglobisporus tyrosinase (red), Aspergillus oryzae catechol oxidase (blue), with O2 in the active sites highlighting their structurally similar active sites (copper and oxygen atoms are shown as large and small spheres, respectively). A representation of (B) the oxygenated active site, and (C) the substrate reactivity of these dicopper enzymes, including that for NspF.
The reactivity of (poly)phenol oxidase pairs O–O reductive cleavage with substrate oxidation (catechol to quinone) or oxygenation (phenol to catechol), wherein one of the O2-derived oxygen atoms is incorporated into the substrate (see Figure 60). In phenol oxidases such as catechol oxidase, the catecholic substrate is oxidized by 2e− to the quinone which can then be further oxidized by other processes into various polyphenols, or polymerized into higher molecular weight compounds such as melanin, a well-known pigment in skin and a culprit in enzymatic food browning.671,672 Stoichiometrically, these enzymes oxidize two moles of catechol substrate and produce two moles of H2O per mole of O2 reduced. The monooxygenase activity of tyrosinase reduces dioxygen, simultaneously converting a tyrosine (phenolic) residue to the corresponding catechol and releasing one molecule of H2O.673 Within the past decade, a new family of hydroxyanilinase enzymes (here referred to as the most well studied, NspF) was discovered674 and has been shown to be especially selective for its native substrate, 3-amino-4-hydroxybenzamide, with an impressive kcat of 260 s−1 and Km = 0.72 mM.674–676 Interestingly, a closely related enzyme with a nearly identical dicopper active site, GriF natively performs two-electron oxidation of 3-amino-4-hydroxybenzaldehyde to yield the iminoquinone; however, it is also active toward para-substituted phenols in vitro.675,677 This difference in selectivity by two closely related enzymes was attributed based on computational analysis to the effects of the secondary coordination sphere on substrate positioning/approach and substrate–protein interactions which overall effect the reaction landscape surface as opposed to some difference in the Cu2–O2 core structure.674 While model chemistry tends (at least initially) to focus on the immediate/proximal geometry and structure around the metal ion centers, the effects of the second coordination sphere interactions cannot be overlooked.
The nature of certain reactive copper-oxy intermediates formed during turnover, including their electronic properties and the mechanisms by which protons and electrons are delivered in order to effect O–O reductive cleavage, can inform design of model systems as well as contribute to understanding of structure–function relationships in general. Of course, several enzymes utilize copper in other unique fashions such as trinuclear copper clusters (i.e., in laccase)19 or copper ions paired with other transition metal ions, like zinc in Cu/Zn superoxide dismutase,678 and iron in heme–copper oxidases. The two former cases interact with dioxygen (or its reduced form, superoxide); however, they are structurally and functionally quite far removed from the focus of this review, and therefore we will not detail their properties or reactivities here.
3.3. Biomimetic Copper-O2 Adducts
The structural variety of active sites and diverse interactions with dioxygen make copper-containing enzymes appealing targets for bioinspired model chemistry.679,680 In synthetic inorganic systems, the chemist can control aspects of the copper ion coordination sphere, such as ligand denticity and donating ability, coordination geometry, and second coordination sphere interactions like hydrogen-bonding. In doing so, it is possible to investigate the reactivity of such systems with dioxygen, as different types of Cun-O2 adducts display unique/characteristic spectroscopic features (UV–vis, EPR, and rR spectroscopy, vide infra). Then, those interactions and O2 reduction reactivity can be evaluated in terms of the structural and/or electronic properties of the copper environment.680 In many cases, model studies are carried out in nonbiological conditions (i.e., low temperatures, organic solvents); however this allows for mechanistic investigations ofrapid reactions and isolation or in situ characterization of interesting reactive intermediates which might not otherwise be feasible. Therefore, it is important to remember that the goals of the scientific modeling studies discussed in this review include incorporation of only certain aspects of an enzyme active site in order to maintain the chemist's ability to systematically study particular cause-effect relationships. Indeed, insights garnered from small molecule model systems have been invaluable to the understanding of the fundamental, molecular-level chemistry which occurs in various Cu enzymes. By extension, many of the (L)Cu structure–function relationships have informed the design of heme–copper systems and aided in assessing their reactivities with O2 (vide infra, section 5)
3.3.1. Characterization of Mononuclear Cu–O(2) Adducts and Ligand Effects.
Structure–function relationships of Cun-O2 model compounds can directly inspire the design of heme–copper models as well as aid in the interpretation of their spectroscopic properties and reactivity trends. Many research programs have been prominent in detailing the chemistry of small molecule (L)Cun-O2 systems using computational or synthetic chemistry, including the laboratories of Karlin, Solomon, Zuberbuhler, Fujisawa, Stack, Tolman, Cramer, Schindler, Sundermeyer, Itoh, Reglier, Masuda, Suzuki, Kodera, Fukuzumi, and others. In combination, they have characterized various potential enzyme or chemical system intermediates, some of which we describe below. The details of the properties of these and other formulations are highly dependent on the ligand structure (identity, denticity, and donating ability), which also controls ligand-Cu redox properties; in many cases, the Cun-O2 species can be interconverted by changing reaction conditions such as solvent or temperature, or by addition of specific substrates (vide infra).610,650,651 Indeed, in order to isolate or even characterize certain intermediates along various biologically relevant pathways, the synthetic inorganic chemist can employ such strategies as low temperatures, organic solvent, and/or anaerobic atmospheric conditions.682
3.3.1.1. Structural and Electronic Considerations Regarding Copper(II)-Superoxide Adducts.
Oxygenation of cuprous complexes possessing nitrogen-containing tetradentate tripodal ligands results in the formation of primary dioxygen adducts, often an end-on η1 cupric superoxide (CuII–O2•−) species (Figure 61a), which typically have optical spectra displaying a stronger peak around 410–430 nm plus broad but distinctive low-energy absorption features (~600–800 nm) and rR stretching frequencies of v(O–O) = 1110–1130 cm−1 and v(Cu–O) = 460–480 cm−1.18
Figure 61.

Types of monocopper-oxy complexes.
As first deduced by Roth and co-workers utilizing the TMG3tren ligand (Chart 1), such species display paramagnetic, S =1 triplet electronic structures,683 an observation which was later corroborated by MCD, rR, and 2H NMR spectroscopic studies, along with DFT investigations.684,685 In general, copper(II) superoxide complexes rapidly react, even at low temperatures, with another equivalent of ligand-copper(I) complex to form peroxide-bridged dicopper(II) species (vide infra). There are now a good number of synthetic model studies having used rational ligand design and cryogenic solution conditions to allow for the stabilization and characterization of cupric-superoxide complexes. The strategies employed include modifying the electronic or structural (steric) properties of the copper coordination environment, most often using tripodal tetradentate (N4) or tridentate (N3) ligands.
Chart 1.

Primary Copper(I)-Dioxygen Adducts, [(L)CuII(O2•−)]+ Complexesa
aSee text for further details.
For one case, from Itoh and co-workers, a neutral tridentate ligand (PEDACO, Chart 1) is able to support a cupric-superoxide structure; this complex succumbs to biomimetic and highly interesting ligand hydroxylation at the benzyl position.686,687 In studies exploring the effects of electronic structural variations, TMPA derivatives (as in this review; see Chart 1) have been employed, systematically varying electron-donating substituents on the pyridyl rings [dimethylamino (dmaTMPA) or dimethyl-methoxy (dmmTMPA)], or by substituting a ligand pyridyl arm so as to incorporate a thioether donor (dmaN3S). Such ligands (Chart 1) have been shown to allow for low-temperature stabilization of copper(II)- superoxide species which have been characterized by UV-vis and rR spectroscopies.688–691 Until just recently,692 the parent complex, [(TMPA)CuII(O2•−)]+, could only be observed using low-temperature stopped-flow spectroscopic monitoring693 (or via time-resolved laser spectroscopic studies; see section 5.2.3.2), confirming the profound influence of the ligand electronic properties, and therefore, very donating ligands most stabilize these intermediate copper(II)-superoxide complexes. Although all of these [(L)CuII(O2•−)]+ species possess similar optical spectra (vide supra), ligand effects are best correlated to (ligand)CuII/I complex reduction potentials: TMPA (–420 mV) > dmaN3S (–470 mV) > DMMTMPA (–570 mV) > DMATMPA (–700 mV), measured by cyclic voltammetry (E1/2 values vs Fc+/0 at RT in MeCN solvent).690,691 No distinct trend is observed with respect to the rR spectroscopic data and the vibrational properties of the varying complexes (Chart 1). There are no differences in ligand steric demands when using pyridyl 4-substituted substituent changes; however, we find that the copper-superoxide complex with DMAN3S ligand is more reactive toward hydrogen-atom abstraction reactions with phenol (O-H, to give a phenoxyl radical product) and N-methyl-9,10-dihydroacridine (C-H) substrates, than the complexes with DMMTMPA or DMATMPA.689,690 This may be due to the lesser donating ability of the thioether-containing ligand, conferring greater electrophilicity to the resulting [(DMAN3S)CuII(O2•−)]+ complex. Thus, we can cautiously posit that changes in ligand donor ability affects both complex stability and substrate reactivity. This result highlights the potential role of the Met ligand in PHM and DβM active sites in modulating the reduction potential of the Cu center and therefore its ability to activate dioxygen.
Copper(II)-superoxide complexes can also be formed utilizing chelates which provide stabilization against reaction with a second copper(I) complex, through imposition of ligand steric properties. Such examples include the (X3tren)Cu derivatives, where sterically bulky tetramethylguanidine (X = TMG), hexaisopropylterphenyl (X = HIPT), and tetraisopropylterphenyl (X = TIFT) groups are employed to prevent formation of dicopper(II) peroxide species (Chart 1).685,694–696 A recent update on the latter compounds indicates that a mixture of CuII-O2•− and CuIII-O22– forms.696 Changing sterics can also change the complexes’ coordination geometry, and it has been observed that with reference to their abilities to perform HAT reactions, trigonal bipyramidal end-on CuII-superoxides are less reactive than tetrahedral end-on CuII-superoxides.686,690,697,698 (Note: a similar caveat has been defined for the C-H activation capabilities of CuII-hydroperoxide complexes, vide infra.)
Tolman and co-workers have reported a superoxide supported by a dianionic pyridine dicarboxamide ligand via addition of KO2 to the LCuII compound.699 X-band EPR and 1H NMR spectroscopies, along with DFT calculations of LCuII-(O2•−) are consistent with a S =1 ground state where the superoxide coordinates in an end-on (η1) fashion to the cupric center. Resonance Raman spectroscopy shows an isotope sensitive peak at 1104 cm−1 (Δ18O = −60 cm−1), an O–O stretching frequency that is just a bit lower than those found in typical η1-superoxides (Chart 2).
Chart 2.

Ligand Design Strategies to Effect Stability of Side-On 1:1 Copper Dioxygen Adducts through the Implementation of Steric Effectsa
aSee text for further details.
A complementary method initiated by Masuda and coworkers is the incorporation of intramolecular hydrogenbonding groups on TMPA chelate arms to stabilize a [(L)CuII(OOH)]+ complex700,701 and binuclear trans-peroxo-dicopper(II) complexes (vide infra). Karlin and co-workers used one of these hydrogen-bonding ligands to generate the stabilized dioxygen adduct [(PVTMPA)CuII (O2•−)]+ (Chart 1) at −125 °C. This was characterized by rR spectroscopy and DFT calculations, the latter indicating H-bonding to either the proximal or the distal O atom of the superoxide moiety.702 The H-bonding interaction interestingly caused an upshift (of ~10 cm−1) in both the Cu-O and O–O stretching frequencies relative to TMPA derivatives with no H-bond capable pendants and was more reactive in H atom abstraction reactions from weak C–H702 and O–H692 bond substrates. A structural study on a family of TMPA derivatives bearing one or multiple H-bonding pendant arms [primary amine(s), secondary amine(s), and amide(s)] revealed that these pendant arms impact the physical and electronic structure around the Cu ion as well703 (a concept which may be extended to His-Tyr cross-link mimics in HCO model systems). It was found via cyclic voltammetry studies of [(L)CuII(Cl)]Cl complexes that increasing the number of N–H containing arms from 0 to 3 increased the reduction potential of the copper complex up to 500 mV; in other words, the Cu(I) state is more stable with greater H-bonding in these cases (and the H-bond interaction itself is stronger in the Cu(I) complex than the Cu(II) complex).703 In fact these differences were rationalized based on structural data (increasing the number of substituents increased Cu-Nligand and Cu-Cl bond lengths) as well as IR and NMR spectroscopic data assessing the overall effects of H-bonding. The total H-bonding effect is greater when multiple H-bonding interactions are involved (due to combined weaker H-bonds); the strongest single H-bonding interaction (between the N-H and Cu-bound Cl ligand) occurs when only one H-bonding moiety is present.703 Such observations may in the future be extended to other (heme and) Cu-containing models with dioxygen and our understanding of the active site H-bonding network, which is critical for O2-binding and reduction by HCOs.
Although it is not often implicated in copper enzymes' catalytic turnover, it is still interesting to consider η2 “side-on” bound 1:1 copper dioxygen complexes (Figure 61b and 62d) using model systems, as they are still plausible reactive species. Relatively few examples of such intermediates have been characterized to date; they feature tridentate or bidentate ligand coordination (Chart 2).697,704–709 The electronic nature of these adducts has been thoroughly investigated, and they fall into a spectrum of electronic structures somewhere between CuII–O2•− and CuIII–O22− formulations. Interestingly, unlike the triplet end-on superoxides, the mononuclear side-on cupric superoxide species tend to have a diamagnetic (S = 0) ground state electronic structure (i.e., antiferromagnetic coupling between the S =1/2 spins on the CuII ion and the superoxide radical anion ligand), as determined by optical, vibrational, magnetic susceptibility, and computational methods.683,705,710 To differentiate between the two possible electronic descriptions (CuII–O2•− vs CuIII–O22−), X-ray absorption spectroscopy (XAS) experiments were conducted on TpAd,iPr and β-diketiminate (R = Me) copper-ligand complexes (Chart 2),707,708 confirming the nature of these dioxygen adducts as [(TpAd,iPr)CuII–O2•−] and [(β-diketiminate)CuIII–O22–].707 In cases where η2-dioxygen binding is synonymous with side-on CuII(O2•−) formulation, v(O–O) stretching frequencies 10–77 cm−1 lower than the end-on analogues are observed. In the cases where η2-dioxygen binding gives a CuIII–(O22−) species, v(O–O) values of up to 159 cm−1 lower than for η1-CuII(O2•−) complexes are reported. Initial experiments by Tolman and co-workers using a β-diketiminate ligand framework showed that the resulting 1:1 copper dioxygen adducts [having significant CuIII–O22− character and possessing an elongated O–O bond (1.392 Å)] react with LCuI complexes to form asymmetric dicopper(III) bis-μ-oxide compounds.710
Figure 62.

Cationic (not indicated) complexes from Castillo and co-workers. A side-on bound cupric-superoxide species and its C–H activation reactivity.697
A recent study indicates that interesting substrate reactivity can occur for side-on 1:1 copper dioxygen species.697,706 Castillo and co-workers reported on a dibenzimidazole N3S ligand which provides an interesting case in which the ligand is said to impose a square pyramidal geometry for the superoxide-bound form of the complex and the thioether arm comes unbound.697 The benzimidazole donors support relatively electrophilic copper complexes [E1/2 ~ 300 mV more positive than analogous (pyridine)N3S systems], and the authors postulate that this fact as well as the imposed geometry leads to an unusual triplet ground state wherein the superoxide moiety is bound in a side-on fashion as shown in Figure 62 (the assignment being made upon agreement of optical spectroscopy and DFT calculations, although no v(O–O) or v(Cu–O) stretching frequencies could be observed via rR spectroscopy).697 This cupric superoxide species apparently also has higher activity toward C-H abstraction than the singlet side-on superoxides mentioned above (see Figure 62).
Betley and co-workers recently reported on the generation of a room temperature stable η2-superoxide complex using a dipyrromethene supporting ligand [ArLCuII(O2•–)].706 The X- ray crystal structure reveals an O–O bond length of 1.383 Å, and preliminary DFT investigations suggest an S = 0 ground state. Substrate reactivity studies show that this complex exhibits both nucleophilic and electrophilic reactivity (Scheme 17).
Scheme 17.

Reactivity of a Side-On Superoxide, [ArLCuII(O2•−)], Showing Both Nucleophilic (Acid/Base Chemistry) As Well As Electrophilic [O Atom Transfer and Hydrogen Atom Abstraction (HAA)] Charactera
aAdapted with permission from ref 706. Copyright 2017 The Royal Society of Chemistry.
Expanding on Tolman's dianionic pyridine dicarboxamide supported CuII(O2•–) chemistry, McDonald and co-workers found that this tridentate η1-superoxide complex exhibits nucleophilic behavior, for example converting benzoyl chloride and benzaldehyde derivatives to the corresponding benzoic acid (Figure 63).711 This reactivity contrasts with those of other η1-superoxides, which exhibit electrophilic behavior, such as HAT reactions with phenols (vide supra).
Figure 63.

McDonald and co-workers reported nucleophilic reactivity for a LCuII–(O2•–) complex. Adapted with permission from ref 611. Copyright 2016 Wiley-VCH.
3.3.1.2. Formation, Stabilization, and Reactivity of Copper(II) Hydroperoxide Complexes.
Net hydrogen atom transfer to a cupric superoxide results in the formation of hydroperoxide species, (L)CuII–OOH (Figure 61c), which have characteristic UV–vis bands around 330–380 nm and decidedly lower rR stretching frequencies [v(O–O) = 830–900 cm–1] than their superoxide analogs,18,687,698,712 as expected from the O–O bond-order lowering. Cupric hydroperoxide species can also be generated following addition of H2O2 and a base to a (L)CuII complex.18 Interestingly, a pair of formally copper(III) alkylperoxo complexes, (L)CuIII– OOR (R = tBu, cumyl), have recently been characterized following their low-temperature formation via addition of excess ROOH to a (L)CuII–OH species, followed by chemical oxidation using a ferrocenium salt.713 While CuII/III-OOR species have been shown to be active in substrate oxidation reactions, they are less relevant to the focus of this review, and therefore, we direct the interested reader to other recent reviews on the subject.18,610,679 Instead, here we will discuss synthetic copper-hydroperoxides, which are proposed to be reactive intermediates in various O2-activating mononuclear copper enzymes (vide supra) and since a heme–Cu bridging hydroperoxide has been considered computationally as a potential fleeting but very important intermediate in the catalytic cycle of HCOs. As with other Cu-oxy species, the ligand structure (coordination geometry, ligand denticity, type, and sterics), electronics (reduction potential), and secondary coordination sphere (H-bonding) have profound effects on the formation, stability, and reactivity of copper(II) hydroperoxides.
Most often in enzymatic systems, to aid in proton delivery to a metal-bound oxy species, there exists a hydrogen-bonding network implementing amino acid residues and/or water molecules. Indeed, there have been general efforts to study the chemistry of dioxygen with many synthetic (L)Cu complexes employing appended H-bonding moieties in the second coordination sphere. There, H-bonding interactions either serve to promote O–O reductive cleavage or stabilize CuII–OO(H) species (there are also many similar examples for synthetic heme systems, vide supra, section 2.4).714 Additional details about the nature of the interaction, including the number and strength of the H-bond donors and the particular O atom which acts as an H-bond acceptor, have profound effects on the stability or reactivity of the cupric hydroperoxo species.
In fact, the first example of an isolated CuII–OOH species, supported by a tripodal, tetradentate amino-pyridyl ligand framework (parent = TMPA, also sometimes called TPA) having two bulky pivalamide N–H donors appended in the 6- pyridyl position (called BPPA in the original work701 and subsequently referred to as LPiv2)703 (see Figure 64A) came from Masuda and co-workers.701 X-ray crystallographic characterization of this cupric hydroperoxo complex (Figure 64B) revealed that both N–H groups of the pivalamide moieties on the ligand are able to H-bond to the proximal (copper-bound) O atom of the hydroperoxide moiety, as determined by their short N–O distances (2.78 and 2.79 Å); these stabilizing interactions allow an usually thermally unstable species to survive at room temperature for up to a month. The EPR spectrum is consistent with a trigonal bipyramidal mononuclear copper hydroperoxide species, with rR spectroscopy confirming the peroxidic ligand [v(O–O) = 863 cm−1; g∥ = 2.00, g⊥ = 2.20].701 This ligand design strategy has been used to make a series of TMPA-derived ligands with H-bonding moieties (see Figure 64A),692,703 most of which have been investigated in terms of copper(l) dioxygen chemistry and utilized in the formation of cupric hydroperoxides, highlighting the effect of H-bonding on the stability of the complexes generated.712,715–719
Figure 64.

(A) Strategy of Masuda and co-workers for systematic introduction of H-bonding groups of varying strengths into the TMPA ligand framework.700,701,722,723 (B) Crystal structure (50% probability) of the stable (LPiv2)CuII-OOH species showing two H-bonds to the proximal O atom.701 (C) Spectroscopically characterized [(BA)CuII–OOH]+ species stable at low temperature.720 (D and E) Ligands with and without substituents positioned to H-bond to the distal O atom of a CuII-OOH species.700 (F) Overall conclusions regarding the stability or reactivity imposed by H-bonding to proximal vs distal O atoms for LCuII-OOH complexes.721
Another complex employing a ligand with a single benzylamine substituent on a pyridyl arm of the parent TMPA was also shown to be capable of stabilizing a hydroperoxo moiety via an H-bonding interaction between the N–H of the secondary amine and the proximal O atom (Figure 64C).720 A unique property of this complex is that only one equiv H2O2(aq) is needed to fully form [(BA)CuII– OOH]+ (Figure 64C). However, in this case, the hydroperoxo species was stable at –90 °C but decayed to a trans-μ-1,2- peroxodicopper [(BA)CuII–O–O–Cuii(bA)]2+, even if just warming to –50 °C in acetone. These examples highlight the stabilizing nature of an H-bond interacting with the proximal peroxide O atom (next to and bound directly to the copper ion). In an elegant new work, Borovik and co-workers extended this concept to a small artificial copper protein, “synthetically” placing a CuII–OOH moiety into a protein matrix, thus implementing aqueous and ambient conditions. The study utilized a biotinylated bis(2-pyridylethyl)amine ligand cupric complex inserted into a streptavidin host (Figure 65).721 Site-directed mutagenesis could then be utilized to systematically probe H-bonding effects in the local environment of a Cu active site designed to support the monocopper hydroperoxo complex (generated from the addition of hydrogen peroxide).
Figure 65.

Engineered protein active site designed by Borovik and coworkers and crystallographic determination at 1.62 Å resolution.721 Multiple H-bonding interactions are observed with the hydroperoxide moiety in the CuII–OOH structure: 01···03 = 3.2 Å and 04···Ν(Ν49) = 3.0 Å. Further, Cu–O3 = 1.9 Å; O3–O4 = 1.5 Å; and Cu–O2 = 2.6 Å.
Similar to the small-molecule systems, it was found that H-bonding can have drastic effects on the stability and reactivity of CuII–OOH species, and specifically, in this macromolecular model there exist active site residues capable of H-bonding with either the proximal or distal O atom of the −OOH moiety (residues S112, which does not directly H-bond to the hydroperoxide ligand but positions a water molecule instead, or N49, respectively, see Figure 65). With both H-bonding residues in place, the CuII–OOH species was stable for over 24 h, and interestingly, a mutation of N49 to Ala revealed that alone, an H-bond to the proximal O atom is equally as stabilizing as having both H-bonds and the hydroperoxo species is then unreactive toward weak C–H substrates. However, a S112A mutation showed that a single H-bond to the distal O atom destabilizes the hydroperoxo intermediate (reduces the half-life to 20 min), and the hydroperoxo species is capable of C–H activation of an exogenously added 4- chlorobenzylamine substrate.721 Earlier work involving a comparative study of two N4 ligands, one of which contains a moiety capable of H-bonding with the distal O atom,700 demonstrated that such an interaction results in destabilization of the cupric hydroperoxide, effectively activating the hydroperoxide moiety, as has been suggested in DβM, and also seen in the artificial copper enzyme work from Borovik. The destabilizing/activating effect was observed in acetonitrile based on the differing rates of decomposition of the two hydroperoxo species (Figure 64, panels D and E) as spectroscopically monitored at –30 °C. Complex D decays an order of magnitude faster than complex E, when there is no H bonding [kobs = 2.4(2) × 10−2 min−1 vs kobs = 7.3(6) × 10−3 min−1]. Additionally, the H-bonding interaction results in a larger |A∥| in the EPR spectrum (82 vs 75 G) and longer wavelength LMCT transition in the optical spectrum (381 vs 372 nm), both of which support a weakened coordination of the hydroperoxo moiety to the copper ion.700 From the Masuda and Borovik studies, the overall effect of positioning of H-bonding interactions in CuII–OOH species is summarized by Figure 64F.
With ligand design for synthetic model systems, it is important to remember that the imposed structural changes (in the primary or secondary coordination sphere) are likely not mutually exclusive, as it was shown that the ligand set depicted in Figure 64A indeed had various appended H-bond moieties (of different strengths and numbers), however, the reduction potentials of the CuII/I complexes were also significantly different.703 The conclusions based on IR monitoring of the N–H stretching frequency of (LPiv-n)- CuII/ICl complexes and CV studies of the (L)CuII/ICl showed that hydrogen-bonding strength for the pivalamide-containing ligands follows the trend, LPiv-1 > LPiv-2 > LPiv-3 (for each individual pivalamide group; LPiv-3 has the greatest overall H-bonding effece), and the reduction potentials in acetonitrile follow the trend, TPA < LAm-n < LNp-n < LPiv-n (where in each case, a greater effect was observed with increasing values of n).703 These structural and electronic effects highlight a challenge in assessing ligand effects in such complexes; however, this study has clear implications for the O2 chemistry of these ligand-Cu complexes. Oxygenation of LCuI has been reported for TMPA (vide supra) and most of the ligands containing H-bonding moieties. In many cases μ-1,2-trans- peroxodicopper(II) species are formed (LAm-n, LNp2, and LPivl),715,716,719 but in some cases cupric superoxides were directly implicated or observed (LNp2 and LPiv-n) 692,701,702,718,719,722,723
Rational ligand design has also allowed for control of the ligand denticity and sterics within a given framework and evaluation of the subsequent changes in stability and/or reactivity. One such comparative study involved the reactivity of (L)CuII complexes (where L = the pyridylethylamine ligands depicted in Figure 66) with H2O2/NEt3 and resulting (L)CuII–OOH species (Figure 66 for L1 and L2).724 (TEPA)CuII–OOH decayed quickly to a Cu(I) analogue (probably via Cu–OOH homolytic cleavage, also forming •OOH), whereas the tridentate (L1)CuII–OOH yielded a putative short-lived mononuclear cupric side-on peroxide species (the first of its kind) based on UV–vis, EPR, and kinetic analysis. Interestingly, the less sterically encumbered tridentate L2 ligand complex reacted so quickly that no hydroperoxo intermediate could be detected; the product was the side-on bound peroxide dicopper(II) species. These results along with DFT analysis of the process highlight the significance of both sterics and ligand denticity in supporting cupric (hydro)peroxides, as (i) the additional bulk of the L1 ligand allowed for slower rearrangement/reorganization to a side-on peroxo complex and thus relative stabilization of the hydroperoxide as compared to that with L2, but (ii) the lack of an extra coordination site with the tetradentate TEPA complex disallowed rearrangement to the side-on peroxide.724–726
Figure 66.

Supporting ligands used by Itoh and co-workers to generate CuII–OOH complexes from H2O2 and base. Hydroperoxides generated from tridentate ligands (L1 and L2) decay to dicopper(II) μ-η2:η2 peroxides through a proposed mononuclear side-on peroxide, while the tetradentate TEPA complex decays to a reduced CuI form.724
Karlin and co-workers have published multiple reports on the oxidative capabilities, including aryl hydroxylation and N-dealkylation reactions of CuII-OOH species.727–729 Mainly, TMPA-derivatives containing a substitutent appended to the 6-pyridyl position of one arm were utitlized, with the substituent acting as an internal substrate. These reactions were proposed to be initiated by O–O cleavage, with a putative CuII-O• species acting as the oxidizing intermediate (which was supported via similar reactions with CuI/PhIO).729 As mentioned above, LCuII–OOH complexes are generally formed by addition of excess H2O2/NEt3 to an LCuII compound at cryogenic temperatures. However, an important reinvestigation, including experimental and DFT analyses, of the system where oxidative N-dealkylation of a ligand appended dibenzylamine moiety occurred, showed that in fact the LCuII–OOH complex is not capable of this C–H activation reaction.730 Instead, Cu–O bond homolysis of the CuII–OOH intermediate was proposed to occur, allowing for the well-known Fenton reaction to occur between this ligand-CuI ion complex and excess H2O2 present. The hydroxyl radical (•OH) formed was said to be the true entity responsible for substrate C–H attack and the observed N-dealkylation. This proposed reaction mechanism is reminiscent of that put forth for LPMOs,651 that is discussed above (see section 3.1), wherein the reaction of CuI and H2O2 forms •OH, which then back-abstracts a H atom from the CuII–OH moiety to give the CuII–O• substrate oxidant. From computations, such a mechanism was proposed in a nonheme iron case, where a FeIII–OH species formed from an FeII + H2O2 reaction itself was further attacked by the •OH formed, giving the final FeIV=O oxidant.731
Combining computational methods plus analysis of electronic structure and oxidase activity of various other cupric hydroperoxide species (with no H-bonding stabilization/destabilization effects), it has been found that not only sterics but also the Cu ion coordination geometry can influence the activity of these species. Generally, tetradentate ligand frameworks which impose square pyramidal or trigonal bipyramidal CuII–OOH geometries are far less reactive than certain tridentate ligands which adopt tetrahedral or square planar cupric-hydroperoxide geometries.732–735 Although this trend across several examples in the literature is thought-provoking as to how local coordination geometry and ligand fields can effect stabilization or reactivity of CuII–OOH intermediates, a more systematic approach to address this concept is necessary. In that way, it will be possible to draw more definitive conclusions relevant for understanding enzyme activities or to design and construct novel catalysts.
3.3.1.3. High-Valent Copper Complexes: Theory and Implications.
Homolytic O–O bond cleavage of (L)CuII– OOH can in principle lead to the generation of a mononuclear CuII-oxyl species (i.e., “cupryl”), (L)CuII–O•, a reactive species that has been considered in computational studies as a potentially powerful oxidant (in H atom abstraction) in the catalytic cycle of copper-containing monooxygenases (see section 3.1) like DβM,736,737 PHM,738 and LPMOs642,648,649 Predicted reaction barriers for substrate hydroxylation (via initial hydrogen atom abstraction) are significantly lower than other possible intermediates such as superoxo or hydroperoxo ligand-copper(II) species. Recent studies that evaluate the role of a hydrogen-bonding network in the secondary coordination sphere of a fungal LPMO (AA9)644 similarly support the formation of (L)CuII–O• as a relevant active intermediate (Figure 67A).651 And finally, a just published work supports a mononuclear nature for the copper active site in pMMO;656,739 this makes (L)CuII-O• a good candidate for the reactive intermediate that breaks the strong C–H bond of methane during the catalytic cycle (Figure 67B).
Figure 67.

Copper-oxyl [(L)CuII-O•] and its conjugate acid [(L)CuIII–OH] as reactive intermediates in the active site of (A) LPMO and (B) pMMO enzymes.
However, copper(II)-oxyl species have not yet been observed experimentally in solution, neither in the synthetic models nor in enzymes. Only what might be referred to as its conjugate acid, (L)CuIII-OH (Figure 67), has been studied and characterized in recent works from Tolman and co-workers (vide infra).646,647,740–742 Cupryl entities have been observed only in the gas phase, where it has been shown to be very capable in attacking the strong C–H bonds in methane (BDE = 104 kcal/mol).743,744
There have been several hints or pieces of indirect evidence pointing to the existence of (L)CuII–O• species in some synthetic systems. Karlin and co-workers for example, suggested that the intramolecular hydroxylation of a methyl group in a copper ligand (TMG3tren; see section 3.3.1) was effected by a copper-oxyl intermediate, generated either through homolysis of the O–O bond of the (L)CuII–OOH or through the reaction of the [(TMG3tren)CuI]+ precursor and the O atom donor iodosobenzene (Figure 68A). In both cases, ligand oxidation was observed.698 Itoh and co-workers, on the other hand, suggested that a copper(II)-oxyl intermediate was generated from the O–O bond homolysis of a cumylperoxo copper(II) complex. In the presence of the radical trap agent 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), the putative (L)CuII–O• forms a complex that could be characterized by ESI-MS and EPR spectroscopy (Figure 68B). In absence of the radical trap, oxidations of exogenous substrates were carried out.745
Figure 68.

Chemical systems which have provided indirect evidence for the formation of copper-oxyl (CuII–O•) species.698,746 See text for further discussion.
Protonation of the elusive (L)CuII-O• would generate a (L)CuIII-OH species, which, unlike its conjugate base, has been studied and characterized. Using anionic ligands (Figure 69), Tolman and co-workers have been able to obtain (L)CuIII-OH complexes from one electron oxidation of its (L)CuII-OH precursor.646,740 Key spectroscopic features of (L)CuIII-OH species include: EPR silent spectrum (X-band) with a strong UV-vis absorption band at 500–580 nm (with ε ~ 10000 to 15000 M−1 cm−1; L, NO2L, PIPL2; Figure 69) assigned as a ligand aryl π to Cu dx2-y2 charge transfer (LMCT) transition. Also, through resonance Raman spectroscopy a v(Cu-O) value of 633 cm−1 (Δ18O = 26 cm−1) was obtained for several ligand complexes (628 cm−1 for NO2L and PIPL). Finally, using a normalized version of Badger’s rule, Cu–O bond distances were estimated to be 1.80 Å(L) or 1.81 Å (NO2L and PIPL); this represents a Cu–O distance 0.05 Å(L), 0.05 Å(NO2L), and 0.08 Å (PIPL) shorter than found in their (L)CuII–OH precursors, further supporting the increased oxidation state of Cu.741
Figure 69.

Copper(III) hydroxide complexes: (A) L, (B) NO2L, and (C) PIPL.
Reactivity studies of (L)CuIII–OH complexes have shown the ability of this moiety to attack substrates with C–H bond enthalpies ranging from 76 kcal/mol (9,10-dihydroanthracene) to 99 kcal/mol (cyclohexane),647 which further supports a high valent CuIII–OH species as a viable intermediate in the catalytic cycle of enzymes like LPMO or pMMO (Figure 54, panels B and C).
The topic of CuIII-hydroxide or CuII–O• species requires considerable future research. This is because, as mentioned, it could be relevant to the mechanism of action of LPMOs and pMMOs (Figure 54, panels B and C). From a broader perspective, such a reactive species likely would derive from the homolytic O–O reductive cleavage of a CuII–OOH that arose originally from copper(I)-dioxygen chemistry. The intimate details of the O–O reductive cleavage process require elucidation. (i) What is the timing of electron-transfer and/or protonation steps? (ii) What are the required site(s) of reduction and/or protonation; for example, at which O atom within a peroxo copper(II)–O–OH species does protonation occur giving homolytic versus heterolytic cleavage (if either are possible)? (iii) What strength of reductant (E° value) or pKa of proton is required for cleavage? Further, such details are also likely pertinent to heme–copper oxidase (bio)chemistry. As will be described, we have been able to design and carry out research on a synthetic system wherein a heme–FeIII– (O– O)2−–CuII(ligand) complex undergoes reductive O–O cleavage which is initiated by H-bonding at the Operoxo atom bound to the copper(II) ion. Mechanistic insights derived from experiments and theoretical calculations suggest that homolytic cleavage occurs and a species with CuII–oxyl character forms.7 The research results and discussions are given in sections 5.2.6, 5.2.7, and 6.
3.3.2. Characterization of Dinuclear Cu2–O2 Adducts and Ligand Effects.
3.3.2.1. Dicopper trans, Side-On Peroxo, and Bis-μ-Οxο Isomers.
Binuclear copper dioxygen species are known to be critical active intermediates in the function of many copper-based metalloproteins. The roles of these proteins are diverse and include dioxygen transport, tyrosine hydroxylation, catechol oxidation, and C–H bond activation. As outlined above, small molecule synthetic model systems have been used as proxies for enzyme intermediates to model their spectroscopic, electronic, and reactivity properties. More fundamentally, this approach expands the inorganic chemists' understanding of copper’s capacity to activate dioxygen for substrate oxidation or reduction to water. For model systems incorporating binucleating ligand frameworks, or mononucleating ligands where it is sterically and electronically allowed, the reaction of a CuII–O2•− adduct with another equivalent of CuI in solution leads to the generation of dicopper peroxo complexes. These dicopper peroxo species have been shown to adopt several different structural isomers, as shown in Figure 70.18 The two most well-characterized dicopper peroxide examples include end-on cis/trans-μ-1,2-peroxo (C/TP) species and side-on binding peroxo species in a μ-η2:η2 conformation (SP) (Figure 70). Side-on (and certain rare cases end-on748) dicopper peroxide species are known to exist in equilibrium with bis-μ-oxo dicopper(III) (O) species, where the O–O bond is reductively cleaved. The position of this equilibrium is sensitive to solvent, counterion, and ligand effects, which we will focus on here. Generally speaking, tetradentate copper chelates favor the end-on μ-1,2-peroxide isomer, whereas tridentate ligands or ligands of lower denticity favor the side-on μ-η2:η2-peroxodicopper/bis-μ-oxo equilibrium, although certain ligands can result in exclusively one or the other.749 In terms of reactivity properties, trans-μ-1,2- peroxo dicopper species are generally nucleophilic in character,750,751 whereas the μ-η2:η2-peroxo dicopper species and bis-μ-oxo dicopper(III) species are generally electrophilic,750,752 and any discussion of side-on-peroxo or bis-μ-oxo reactivity toward substrates must consider the participation of the other isomer. Our coverage of this topic here is not comprehensive, and the reader is directed to several recent very thorough reviews for in-depth coverage of each variable.18,750,753–756
Figure 70.

Binuclear copper peroxo, and bis-μ-oxo species which have been characterized or proposed, featuring ligand chelates which bind either one or two copper ions.
3.3.2.2. General Spectroscopic Properties of trans-μ-1,2-Peroxo, Side-On μ-η2:η2-Peroxo and Bis-μ-Oxo Cores.
The three main types of binuclear copper species can be readily distinguished based on their distinctive electronic (UV–vis), vibrational (rR) spectroscopies, as well as structural parameters such as their Cu–O, O–O, and Cu∙∙∙Cu distances [X-ray diffraction (XRD), EXAFS]. These distinctions are informative for both understanding metal–oxygen interactions on a fundamental/molecular level, as well as how these properties may vary in mixed-metal binuclear systems.757 In terms of their electronic spectra, the reaction of Cu' complexes with dioxygen gives intense ligand-to-metal charge transfer (LMCT) bands which are generally attributed to peroxide-to-copper charge transfer transitions. The general electronic spectra of trans-μ-1,2 peroxides, side-on μ-η2:η2 peroxides, and bis-μ-oxo species are distinctive, although each can vary depending on ligand properties (Figure 71).608 Dicopper end-on trans peroxide species tend to have Cu∙∙∙Cu distances of 4.4–4.5 Å and rR stretching frequencies of v(Cu–O) = 530–561 cm−1 (Δ18O2 = −25 cm−1) and v(O–O) = 805–830 cm−1 (Δ18O2 = −45 cm−1).753 The Cu–O bond lengths determined via X-ray crystallography are ~1.85 Å (see also section 6.2 and Tables 5 and 6), intermediate between those of the side-on peroxo and bis-μ-oxo isomers.758,759 Side-on peroxo species display substantially more activated (or weaker) O–O bond stretching frequencies in the range of v(O–O) = 716–760 cm−1 (Δ18O2= –40 cm−1), with smaller Cu∙∙∙Cu distances (3.5–3.6 Å) and slightly longer Cu–O bonds (1.9 Å) .753,760
Figure 71.

Characteristic UV-vis, rRaman, and structural parameters for trans-μ-1,2-peroxo, side-on μ-η2:η2-peroxo, and bis-μ-oxo isomers.753.
Table 5.
Selected Bond Lengths and Angles of Synthetic and Enzymatic Metal-“Oxy” Speciesa
| entry no. | complex | “oxy” species | 0–0 (Å) | Fe-O (Å) | M2–O (Å) | Ml-M2 (Å) | Ml–O–O M2–O–O | Ml–O–O–M2 |
|---|---|---|---|---|---|---|---|---|
| O2 | NA | 1.21 | NA | NA | NA | NA | NA | |
| KO2 | NA | 1.28 | NA | NA | NA | NA | NA | |
| H2O2 | NA | 1.47 | NA | NA | NA | 95° | 120° | |
| Superoxides | ||||||||
| (1) | sperm whale Mb | ES1092 | 1.24 | 1.81 | NA | NA | 122.1° | NA |
| (2) | sperm whale Mb | ES1131 | 1.23 | 1.84 | NA | NA | 115.5° | NA |
| (3) | human Hb | Es69 | 1.24 | 1.87 | NA | NA | 159.1° | NA |
| (4) | [CuII2,(UN-O)(O2)]2+c | μ-1, 1-S783 | 1.29 | NA | 2.07 | 3.29 | 127.3° | 103.4° |
| 2.12 | 128.4° | (Cu-O-Cu) | ||||||
| (5) | [CuII2,(UN-O)(O2)]2+c | μ-1, 2-S783 | 1.29 | NA | 2.02 | 3.49 | 121.4° | 14.4° |
| 2.04 | 123.1° | |||||||
| (hydro)peroxides | ||||||||
| (6) | CPO-OOH | HP302 | 1.50 | 1.80 | NA | NA | 131° | NA |
| (7) | Mb-O2d | P570 | 1.33 | 1.85 | NA | NA | 120° | NA |
| (8) | Mb-O2d | P571 | 1.26 | 1.84 | NA | NA | 119° | NA |
| (9) | [CuII(BPPA)(OOH)]+ | HP701 | 1.46 | NA | 1.89 | NA | 114.5° | NA |
| (10) | [CuII(biot-et-dpea)(OH2)(OOH)]+ | HP721 | 1.52 | NA | 1.94 | NA | 138.2° | NA |
| (11) | [CuII2(LEt)(OOH)]2+ | HP789 | 1.46 | NA | 1.98 | 3.53 | 114.0° | 125.6° |
| 1.99 | 120.3° | (Cu-O-Cu) | ||||||
| (12) | [CuII2(XYL-O)(mCPBA)]2+ | AcP786 | 1.46 | NA | 1.97 | 3.20 | 122.4° | 108.3° |
| 1.97 | 123.3° | (Cu-O-Cu) | ||||||
| (13) | [ZnII (Ts3Tren)(H2O2)]− | H2P1132 | 1.45 | NA | 2.17 | NA | 129.8° | NA |
| dicopper(II) trans-peroxides | ||||||||
| (14) | [{CuII(TMPA)}2 (O2)]2+ | TP766 | 1.43 | NA | 1.85 | 4.36 | 107.7° | 180° |
| (15) | [{CuII(Me6Tren)}2(O2)]2+ | TP758 | 1.37 | NA | 1.90 | 4.59 | 116.4° | 180° |
| (16) | [{CuII(Bz3Tren)}2(O2)]2+ | TP767 | 1.45 | NA | 1.87 | 4.48 | 111.5° | 180° |
| (17) | [{CuII(Tet b)}2(O2)]2+ | TP768 | 1.50 | NA | 1.92 | 4.57 | 114.9° | 143.7° |
| 1.94 | 115.2° | |||||||
| (18) | [{CuII(TPANHPh)}2(O2)]2+ | TP769 | 1.48 | NA | 1.90 | 4.69 | 118.5° | 180° |
| (19) | [CuII4(1L−)2(O2)(OH)2(ClO4)]+ | TP1133 | 1.45 | NA | 1.96 | 4.18 | 113.7° | 107.0° |
| (20) | [CuII4(1L−)2(O2)(OH)2(ClO4)]+ | TP1133 | 1.45 | NA | 1.94 | 4.32 | 113.8° | 120.9° |
| (21) | [CuII4(1L−)2(O2)(OH)2]2+ | TP1134 | 1.37 | NA | 1.95 | 4.82 | 137.7° | 119.8° |
| (22) | [CuII4(1L−)2(O2)(OH)2]2+ | TP1134 | 1.37 | NA | 2.02 | 4.85 | 119.6° | 177.4° |
| (23) | Cu-SSZ-13c | TP805 | 1.35 | NA | 1.82 | 3.98 | NR | NR |
| 1.83 | ||||||||
| dicopper(II) cis-peroxides | ||||||||
| (24) | [CuII2(LMe)(O2)] + | CP790 | 1.50 | NA | 1.90 | 3.80 | 119.2° | 65.2° |
| 1.91 | 118.9° | |||||||
| (25) | [CuII2(LEt)(O2)] + | CP791 | 1.46 | NA | 1.87 | 3.68 | 101.9° | 104.2° |
| 1.88 | 103.6° | |||||||
| (26) | [CuII2(UN-O)(O2)]+c | CP783 | 1.36 | NA | 1.99 | 3.44 | 112.7° | 71.9° |
| 1.97 | 105.4° | |||||||
| (27) | [CuII4(L−)2(O2)(OH)2(ClO4)]+ | CP1133 | 1.45 | NA | 1.96 | 3.03 | 113.7° | 7.0° |
| 1.94 | 113.8° | |||||||
| (28) | [CuII4(L−)2(O2)(OH)2]2+ | CP1134 | 1.37 | NA | 1.95 | 3.90 | 137.7° | 28.8° |
| 2.02 | 119.6° | |||||||
| dicobalt (III) trans-peroxides (29) | [{CoIII(Tren)}2(O2)(CN)]2+ | TP1135 | 1.47 | NA | 1.87 | 4.53 | 113.2° | 180° |
| (30) | [{CoIII(Pytacn)}2(O2)(NCCH3)]4+c | TP1136 | 1.40 | NA | 1.90 | 4.50 | 121.6° | 131.9° |
| (31) | [CoIII2(L4-)(O2)] | TP1137 | 1.36 | NA | 1.93 | 4.15 | 119.4° | 101.9° |
| 1.94 | 117.4° | |||||||
| dibacolt (III) cis-peroxides | ||||||||
| (32) | [{CoIII(tmpa)}2(O2)(OH)]3+ | CP1138 | 1.41 | NA | 1.84 | 3.26 | 110.8° | 59.8° |
| 1.85 | 112.7° | |||||||
| (33) | [CoIII2(Oxapyme2−)(O2)(NO2)] + | CP1139 | 1.41 | NA | 1.86 | 3.77 | 111.1° | 96.8° |
| 1.85 | 110.8° | |||||||
| (34) | [CoIII2(LO−)(O2)(PO2Ph2)] + | CP1140 | 1.41 | NA | 1.86 | 3.23 | 112.8° | 57.8° |
| 1.86 | 109.2° | |||||||
| heme-peroxo-copperb,c (35) |
[(DCHIm)F8FeIII-O2-CuII(AN)] + | TP497 | 1.42 | 1.82 | 1.90 | 4.19 | 121.0° | 163.4° |
| 92.4° | ||||||||
| (36) | [(DCHIm)F8FeIII-O2-CuII(AN)]+ (ArOH) | TP747 | 1.43 | 1.82 | 1.95 | 4.16 | 124.6 | 130.9° |
| 98.4 | ||||||||
| (37) | [(DCHIm)F8FeIII-O2-CuII(TMPA)] + | TP497 | 1.40 | 1.82 | 1.89 | 4.49 | 119.9° | 165.2° |
| 113.6° | ||||||||
| (38) | [(DCHIm)F8FeIII-O2-CuII(DCHIm)3] + | TP919 | 1.37 | 1.83 | 1.96 | 4.16 | 122.7° | 131.5° |
| 100.1° | ||||||||
| (39) | [(DCHIm)F8FeIII-O2-CuII(DCHIm)4]+ | TP919 | 1.37 | 1.84 | 1.99 | 4.46 | 121.3° | 142.1° |
| 112.2° | ||||||||
| (40) | [(DCHIm)F8FeIII-O2-CuII(DCHIm)4]+ (ArOH) | TP919 | 1.41 | 1.83 | 2.00 | 4.37 | 123.0° | 111.7° |
| 121.0° | ||||||||
| (41) | bovine CcO (1V54) | TP747 | 1.41 | 1.83 | 1.90 | 4.26 | 120.4° lina | 128.6° |
| 110.0° |
Abbreviations used: ES = end-on superoxide; μ-1,1-S = superoxide bridging through one O-atom; μ-1,2-S = superoxide bridging through both O-atoms; HP = hydroperoxide; P = peroxide; AcP = acylperoxide; H2P = hydrogen peroxide bound; TP = trans-peroxide; CP = cis-peroxide
DFT calculated structure.
Nonprotonated peroxide, H-bonding to the N–H of a nearby His residue.
Table 6.
Average of Selected Bond Lengths and Angles of Synthetic and Enzymatic Metal-“Oxy” Species and Structural Parameters for “As-Isolated” CcO X-ray Structures Claimed to Possess Heme-Cu BNCs with a Bridging Peroxo Dianionic Liganda,b
| entry no. | complex | “oxy” species | O–O (Å) | Fe–O (Å) | M2–O (Å) | M1–M2 (Å) | M1–O–O M2–O–O | M1–O–O–M2 |
|---|---|---|---|---|---|---|---|---|
| heme (average of 3) | ES | 1.24 | 1.84 | NA | NA | 132.2° | NA | |
| heme (average of 3) | HP or P | 1.36 | 1.83 | NA | NA | 123.3° | NA | |
| (di)copper (average of 4) | HP or AcP | 1.48 | NA | 1.96 | NA | 122.1° | NA | |
| dicopper (average of 10) | TP | 1.42 | NA | 1.91 | 4.48 | 116.9° | 154.3° | |
| dicopper (average of 5) | CP | 1.43 | NA | 1.94 | 3.57 | 114.7° | 55.4° | |
| dicobalt (average of 14) | TP | 1.40 | NA | 1.90 | 4.47 | 115.3° | 158.1° | |
| dicobalt (average of 14) | CP | 1.42 | NA | 1.86 | 3.40 | 111.7° | 67.9° | |
| heme–Cu (average of 7) | TP | 1.40 | 1.83 | 1.94 | 4.30 | 121.8° | 139.1° | |
| 106.9° | ||||||||
| (42) | bovine CcO (5B1A) | TP842 | 1.55 | 2.32 | 2.16 | 4.83 | 140.8° | 146.7° |
| 90.1° | ||||||||
| (43) | bovine CcO (3WG7) | TP1141 | 1.55 | 2.35 | 2.14 | 4.90 | 149.1° | 141.5° |
| 94.0° | ||||||||
| (44) | ba3 CcO (3S8F) | TP988 | 1.54 | 2.25 | 2.30 | 4.87 | 144.4° | 144.8° |
| 91.7° | ||||||||
| (45) | ba3 CcO (3S8G) | TP911 | 1.52 | 2.39 | 2.25 | 4.92 | 140.6° | 146.6° |
| 89.4° | ||||||||
| (46) | bovine CcO (2ZXW) | TP911 | 1.70 | 2.24 | 2.19 | 4.80 | 153.9° | 141.5° |
| 96.1° | ||||||||
| (47) | bovine CcO (3ABL) | TP911 | 1.70 | 2.24 | 2.17 | 4.87 | 144.1° | 146.9° |
| 90.5° | ||||||||
| (48) | bovine CcO (3ABM) | TP911 | 1.70 | 2.19 | 2.16 | 4.89 | 139.3° | 150.8° |
| 92.2° | ||||||||
| (49) | bovine CcO (2Y69) | TP1142 | 1.11 | 2.24 | 2.49 | 4.89 | 128.7° | 156.8° |
| 91.6° | ||||||||
| (50) | P. denitrificans CcO (3HB3) | TP985 | 1.49 | 1.93 | 1.92 | 4.62 | 108.2° | 175.0° |
| 117.5° | ||||||||
| average CcO valuesc | 1.61d | 2.28 | 2.23 | 4.87 | 142.6° | 147.0° | ||
| 92.0° |
Abbreviations used: ES = end-on superoxide; HP = hydroperoxide; P = peroxide; AcP = acylperoxide; TP = trans-peroxide; and CP = cis-peroxide.
For the Heme-Cu and CcO entries, M1 = Fe and M2 = Cu.
Excluding data from entry 50 (PDB: 3HB3)
Excluding data from entry 49 (PDB: 2Y69)
These experimental observations are supported by theoretical investigations into the nature of the frontier molecular orbitals involved in the Cu–O2 bonding, Scheme 18. The interaction between the copper d-orbitals and the peroxide π and σ antibonding orbitals provide insight into the observed trends in peroxide O–O bond stretching frequency. For endon trans-peroxides, there is no back-bonding from the copper d-manifold into the peroxide σ* orbital, as occurs in its side-on analogue and, as a result, trans-peroxides display less activated O–O bonds.761–764 Finally, the bis-μ-oxo isomer has a characteristic core vibrational feature in its resonance Raman spectrum at v(Cu–O) = 580–647 cm−1 (Δ18O2 = −(20–30) cm−1] and the shortest Cu∙∙∙Cu and Cu–O distances of the three isomers at 2.8 and 1.8 Å, respectively.753,765 As mentioned above, tetradentate copper chelates generally favor the end-on trans-μ-1,2-peroxo, whereas ligands of lower denticity (1–3) favor either the side-on μ-η2:η2-peroxo, bis-μ-oxo, or an equilibrium mixture containing both (vide infra).
Scheme 18.

Frontier Molecular Orbital Interactions for End-On and Side-On Isomers of Peroxodicopper(II) Species
3.3.2.3. Trans- or Cis-μ-1,2-Peroxo Species: Ligand Effects and General Reactivity.
The first example of a trans-μ-1,2- peroxo species was characterized by the Karlin group in 1988, by adding dioxygen to a cuprous complex bearing TMPA.766 Reversible dioxygen and CO binding was demonstrated in propionitrile and dichloromethane. The crystal structure of [{(TMPA)CuII}2(O22−)]2+ was obtained, confirming its formulation as an end-on trans-μ-1,2-peroxo dicopper species (Figure 72A). The copper coordination geometry is trigonal bipyramidal, with the ligand amine nitrogen and peroxo oxygen occupying axial positions, while the pyridyl nitrogens of TMPA occupied equatorial positions. The Cu∙∙∙Cu distance is 4.359 Å and an O–O bond of 1.432(6) Å was obtained, consistent with its formulation as a peroxo-type species. This landmark report, although not immediately biologically relevant, showed that unexpected, new, highly sensitive copper oxygen species could be stabilized and characterized via XRD.759,766 system has also provided a useful benchmark with which other derivatives can be compared, in terms of their ligand properties. Since then, many trans-μ-1,2-peroxodicopper(II) species have been spectroscopically characterized.18,7S3 There are also additional structurally characterized end-on trans- peroxo dicopper species featuring tris(aminoethyl)amine (tren) type ligand frameworks, as well as a macrocyclic ligand and H-bonding TMPA-derivatives (Figure 72).758,767
Figure 72.

Depiction of crystal structures of synthetically derived trans-μ-1.2-peroxod\copper species with listings of relevant structural parameters. Created using data from references 758 and 766–769.
As mentioned previously, trans-peroxide cores are generally favored by tetradentate ligands, making the copper ion 5-coordinate upon oxygenation, which disfavors formation of a side-on peroxide isomer or bis-μ-oxide species. Ligand donor strength is also an important factor in determining the structural and electronic properties of trans-μ-1,2-peroxodicopper(II) species. Karlin and co-workers evaluated the effect of sulfur and oxygen ligation on peroxo-dicopper(II) complex properties by replacing a pyridyl arm of TMPA with an alkyl thioether [ESE; (2-ethylthio-N,N-bis((pyridin-2-yl)-methyl)ethanamine)], thioanisole [ASM; (2-(methylthio)-N,N-bis((pyridin-2-yl)methyl)benzenamine)], and alkyl ether [EOE; (2-ethoxy-N,N-bis((pyridin-2-yl)methyl)ethanamine)] ligands (Figure 73).770,771 The trans-μ-1,2-peroxodicopper(II) species bearing the ASM and ESE ligands both displayed distinctly different UV-vis/rRaman spectra when compared to the parent TMPA. The UV-vis spectra of both [{(ASM)-CuII}2(O22−)]2+ and [{(ESE)CuII}2(O22−)]2+ display an inversion in intensity of the π*σ /π*v peroxo-to-copper LMCT bands, and rR spectroscopy showed lower O–O bond stretching values (relative to TMPA) of 828 and 817 cm−1 for ASM and ESE, respectively. These changes demonstrated a strongly coordinating thioether ligand and a distortion of the CuII geometry from trigonal bipyramidal toward square pyramidal. This distortion was observed in the crystal structures of the mononuclear CuII complexes in each case. The alkyl ether analogue did not form a trans-μ-1,2-peroxodicopper(II) species upon oxygenation of its CuI complex but instead formed a bis-μ-oxodicopper(III) species, consistent with tridentate copper chelation, where the alkyl ether arm is not bound to the resulting CuIII ion. This demonstrated the stronger donor properties of thioether ligands relative to alkyl ether ligands.771
Figure 73.

Effects of ligand donor strength on copper complex speciation for tetradentate copper chelates, as followed by UV-vis spectroscopy, including those with heteroatoms such as sulfur and oxygen. Adapted from ref 771. Copyright 2010 American Chemical Society.
Steric factors can be instrumental in determining the types of copper species formed after oxygenation of copper(I) complexes in aprotic solutions. In order to evaluate the role of pyridyl arm sterics on dicopper peroxo complex speciation, the Karlin group modified the 6 position of one pyridyl arm in TMPA to include either −3XHR (where X = N or C, and R = CH3 or benzyl) or −3X(R)2 moieties (Figure 74).772 When −3XHR moieties are incorporated, the trans-μ-1,2-peroxodicopper(II) isomer is formed, similar to other tetradentate copper chelates. However, when the steric bulk of the ligand arm is increased by incorporating an −3XR2 moiety, pyridyl binding to copper is weakened and a bis-μ- oxodicopper(III) species is formed instead, which is capable of monooxygenase chemistry. Bis-μ-oxo complexes formed with the ligand 6-TBP [(6-tBu-phenyl-2-pyridylmethyl)bis(2-pyridylmethyl)amine] could be converted to [{(TMPA)-CuII)2(O22−)]2+ via ligand exchange when excess TMPA is added. Formation of H2O2 and [(6TBP)CuII(Cl)]+ following addition of hydrochloric acid to [{(6TBP)CuIII}2(O2−)2]2+ was suggestive of a bis-μ-oxo/trans-μ-1,2-peroxo equilibrium.
Figure 74.

Ligand arm steric bulk can be influential for the copper species formed upon oxygenation of copper(I) complexes (THF solvent, −380 °C). The higher steric bulk of −3XR2 disfavors pyridyl ligand binding to Cu upon dimerization, thereby giving tridentate copper coordination, which favors the bis-μ-oxo isomer. See the text.
Chelate ring size is also a critical factor in determining the structural properties and reduction potential of the bound copper ion.773 When the methylene linkers of the parent TMPA chelate are systematically extended by one to ethylene linkers, the resulting series of copper(I) complexes have progressively increasing (more positive) CuII/CuI redox potentials, as well as distinct dioxygen reactivities (Figure 75). This change in redox properties is concomitant with a structural distortion from trigonal bipyramidal in TMPA to square pyramidal in TEPA (tris[2-(2-pyridyl)ethyl]amine). The decrease in reduction potential as the chelate ring size for each pyridine ligand is decreased from 6 to 5 is a result of the favorability of copper(II) for smaller chelate ring sizes.774–776 The bite angle of the ligand is also influential in determining the coordination number around the copper(I) complex in solution, with TMPA being 5-coordinate with a solvent ligand bound, as favored by the distortion of copper toward the trigonal plane. This contrasts to PMEA (bis[(2-pyridyl)methyl]-2-(2-pyridyl)ethylamine), PMAP (bis[2-(2-pyridyl)ethyl]-(2-pyridyl)methylamine), and TEPA (Figure 75), which are all 4-coordinate and do not bind solvent. Copper(II)-superoxide and trans-μ-1,2-peroxodicopper(II) species could only be detected with TMPA. Stopped flow studies revealed a transiently stable trans-μ-1,2-peroxodicopper(II) species for PMEA, which decomposed rapidly; no superoxide species could be detected in this reaction. With PMAP, no peroxidic intermediates could be detected upon oxygenation, and TEPA is unreactive toward dioxygen. This work demonstrated that at parity of ligand donor type, 5-membered rings preferentially stabilize peroxodicopper(II) species.773
Figure 75.

Chelate ring size modulates the coordination geometry around copper, as well as the copper(II/I) redox potential.773 Cuprous chelate reactivity toward dioxygen is also strongly influenced by ligand structure. See text for further discussion.
In addition to causing structural distortions that alter the CuII/I redox potential of the copper(I) chelate, ring size can have an effect on how strongly a given ligand type binds to copper. To explore this effect, and the relative donor strength of imidazole compared to pyridine, two TMPA derivatives were synthesized featuring imidazole ligands attached to the amine nitrogen via methylene (LMIm; (lH-imidazol-4-yl)-N,N- bis((pyridin-2-yl)methyl)methanamine) and ethylene (LEIm; 2-(lH-imidazol-4-yl)-N,N-bis((pyridin-2-yl)methyl)ethanamine) linkers (Figure 76).777 Infrared spectroscopy of the stretching frequencies of (L)CuI-CO complexes of TMPA, LMIm, and LEIm demonstrated that although the imidazole is a stronger donor than pyridine, chelate ring size is still highly deterministic of solution phase behavior. Both TMPA, and LMIm have four- and five-coordinate states, with the imidazole coming unbound in LMIm. This ligand exchange was not observed in LEIm, where only one CO stretching frequency is observed. The CuII/I redox potentials of TMPA, LMIm, and LEIm are consistent with the ligand donor strength and chelate ring size trends discussed above. Namely, CuI(LMIm) has a redox potential of −3620 mV (vs FeCp2) in DMF, slightly lower than that of CuI(TMPA) (-610 mV); this could be attributed to the slightly stronger donating properties of the imidazole. Consistent with distortion toward a square pyramidal geometry as chelate ring size is increased from 5 to 6, CuI(LEIm) has a higher redox potential at −3570 mV.773 The imidazole lone pair donor orbital of LEIm is also better aligned to donate to CuII acceptor orbitals (Figure 76). This is borne out in its red-shifted LMCT bands (raised π*σJπ*v orbital energies) and lowered O–O bond stretching frequencies (decreased peroxo-to-copper donation) of the trans-μ-1,2-peroxodicopper(II) species formed upon oxygenation of [(LEIm)CuI](BArF) (BArF = (B(C6F5)4−).777 This work provides an example of how chelate ring size not only effects the reduction potential of copper complexes, but also how strongly a given ligand type can bind and its effect on the resultant spectroscopic properties of copper dioxygen species.
Figure 76.

In addition to altering copper chelate coordination geometry and redox potential, chelate ring size can influence how strongly an imidazole ligand binds to copper, the resulting complex coordination geometry, and the O2 reactivity and spectroscopic properties. See text.
Binucleating ligand frameworks have also been utilized to show the impact of strain on copper(I) O2 binding kinetics and trans-μ-1,2-peroxodicopper(II) complex stability.778 A binucleating ligand framework with two TMPA chelates attached via an ethylene linker at their 5 positions (D1, Figure 77) formed transient mixed-valent (CuICuII) 2:1 Cu:O2 superoxide and stable trans-μ-1,2-peroxodicopper(II) species upon oxygenation of its dicopper(I) complex at low temperatures (-80 °C). Although dioxygen binding and peroxo formation by [(D1)CuI2]2+ is complicated by isomerization between open and closed forms of [(D1)CuI2(O2)(EtCN)]2+, subsequent formation of a trans-μ-1,2-peroxodicopper(II) species is entropically favored because reaction with a second equivalent of CuI is intramolecular. Despite this entropically favored intramolecular reaction, strain in the ethylene linker between the TMPA moieties of [(D1)CuII2(O22-)]2+ enthalpically disfavors peroxo formation relative to its untethered analogue TMPA at low temperatures. In fact, at higher temperatures, formation of intermolecular trans-μ-1,2-peroxodicopper species with D1 are observed, as well as higher order {Cu2-O2}n oligomers, although the nature of these species was not explicitly confirmed.778 This comparison of binucleating and mononucleating ligands demonstrated that intramolecular reactions of dicopper complexes with O2 can sometimes be superseded by intermolecular reactions if ligand strain is a factor. Subsequent modification of D1 by incorporating an ether link yielded the ligand DO (Figure 77). This ligand design alleviated the ligand strain that accompanied intramolecular trans-μ-1,2-peroxodicopper(II) formation in D1.779 As a result, reaction of [(DO)CuI2]2+ with O2 yielded [(D°)CuII2(O22-)]2+ with no enthalpic impedance due to strain and maximized entropic favorability due to the reaction's intramolecular nature. This dicopper(II) peroxide species was also stable at ambient temperatures, demonstrating a rare example of such thermal stability in a synthetic system. No superoxide species could be observed in the reaction of [(DO)CuI2]2+ with O2. This comparison further illustrated the importance of ligand design in copper oxygen chemistry.
Figure 77.

Binucleating ligand analogs to TMPA, and a schematic of the trans-μ-1,2-peroxo-dicopper(II) complexes that are formed with these ligands, show modified kinetic or thermodynamic behavior. With both D1 and DO, the peroxo complexes form with greater kinetic facility. The peroxo-dicopper(II) complex with DO is thermodynamically more stable than that with TMPA; however, the binuclear peroxo-dicopper(II) complex with D1 is thermodynamically unstable and slowly rearranges to a different structure.778,779
The Karlin group has also utilized symmetric and asymmetric binucleating ligand frameworks with a phenolate moiety to hold two copper ions in close proximity to access biomimetic peroxo and hydroperoxo dicopper species. Dicopper(I) complexes [(XYL-O−)CuI2]+ and [(UN-O−)-CuI2]+ both react with dioxygen at low temperature to yield peroxide species with O–O bond stretching frequencies of 803 and 818 cm−1 for XYL-O− and UN-O−, respectively (Figure 78).780–782 Mixed-isotope labeling with 16O18O indicated that in [(XYL-O−)CuII2(O22−)]+ the peroxide is terminally and/or asymmetrically bound.782 More recently, DFT studies indicated that the peroxide binding mode for [(UN-O−)-CuII2(O22−)]+ is most likely asymmetric but possessing a μ-1,2geometry.783
Figure 78.

Generation of binuclear peroxo-dicopper(II) species with the symmetrical XYL-O− (top) and the unsymmetrical UN-O− (bottom) binucleating ligand frameworks. See text for details.
The protonated forms of both XYL−O− and UN−O− are denoted XYL−OH and UN−OH, respectively. Unlike their deprotonated forms discussed above, both dicopper(I) complexes [(XYL-OH)CuI2]2+ and [(UN-OH)CuI2]2+ yield μ-1,1-hydroperoxo dicopper(II) species, [(XYL-O−)CuII2(OOH)]2+ and [(UN-O−)CuII2(OOH)]2+, upon reaction with dioxygen at low temperatures (Figure 79).784,785 In both cases, the similarity of the EXAFS properties of these hydroperoxo species to their crystallographically characterized hydroxyl-bridged dicopper(II) analogues [(XYL-O−)−CuII2(OH)]2+ and [(UN-O−)CuII2(OH)]2+ as well as the m-CPBA adduct [(XYL-O−)CuII2(m-CPBA)]2+ (Figure 80A)786 led to their assignment as μ-1,1-hydroperoxo dicopper(II) species. These hydroperoxo species could also be attained by addition of hydrogen peroxide to the fully oxidized dicopper-(II) complexes [(XYL-O−)Cu2II(OH)]2+ and [(UN-O−) Cu2II(OH)]2+, or through direct protonation of the peroxodicopper species discussed above.787 Another similar unsymmetrical binucleating ligand framework UN2-OH, which features one bidentate copper chelate (Figure 79), was also used to obtain a μ-1,1-hydroperoxo dicopper(II) species.788
Figure 79.

Generation of μ-1,1 binuclear (hydro)peroxo-dicopper(II) species via the XYL-OH (top), UN-OH (middle), and UN2-OH (bottom) binucleating ligand frameworks. See text for details.
Figure 80.

Depiction of X-ray crystal structures of μ-1,1-bound peroxidic dicopper complexes: (A) acylperoxo [CuII2(XYL-O)(m-CPBA)]2+ 786 and (B) hydroperoxo [CuII2(LEt)(OOH)]2+.789.
Recently, the Karlin and Meyer groups have reported new examples and more in-depth analysis of previous cis-μ-1,2-peroxodicopper(II) species in binuclear ligand systems (Figure 81). Using the unsymmetrical binuclear ligand framework, UN-O−, Karlin and co-workers characterized an equilibrium between μ-1,1- and μ-1,2-superoxidedicopper(II) species confirmed by mixed isotope dioxygen labeling and rRaman spectroscopy. The μ-1,2-superoxide species could be reduced to the previously discussed μ-1,2-peroxide species with a cis- conformation as determined by DFT analysis. Computationally calculated structures for all three of these species have been reported (Figure 82). The observed redox potential for this one-electron reduction was in the range of biologically relevant redox processes, in spite of its organic media and small molecule ligand.783
Figure 81.

Binuclear phenolato and pyrazolato cupric superoxo, cis-peroxo (cP), and hydroperoxo species characterized by the Karlin (top) and Meyer (bottom) groups.783,789,790.
Figure 82.

(A and B) Depiction of X-ray crystal and (C, D, and E) computationally calculated structures of synthetic binuclear peroxo and superoxo dicopper(II) complexes. Created with data from references 783,790, and 791.
The Meyer group also reported a cis-μ-1,2-peroxodicopper-(II) species using a binuclear copper ligand with a pyrazolate- bridging ligand between the copper ions (Figure 81). The formulation of this peroxide species was confirmed by UV-vis, infrared, and resonance Raman spectroscopies and also a crystal structure with sodium also bound to the peroxo ligand, the first crystallographically characterized cis-μ-1,2-peroxodicopper complex (Figure 82A).790 In a related ligand system, a similar cis-μ-1,2 peroxodicopper species was characterized, and it displayed distinct electronic structure from previously published trans-μ-1,2-peroxodicopper(II) analogues. Namely, the overall spin state of the new cis-μ-1,2-peroxodicopper species was S =1 (i.e., with copper ions ferromagnetic coupling;791 this stands in contrast to the commonly observed antiferromagnetic coupling in trans-peroxodicopper(II) complexes). This peroxo species can be reversibly protonated (pKa ~ 22.3 in CH3CN) to yield a μ-1,1-hydroperoxide species with high ambient temperature stability (Figure 81), and a crystal structure was obtained for this new hydroperoxide species (Figure 80B).789 A very recent investigation enabled a determination of the redox potential of the cis-μ-1,2-peroxo dicopper species and superoxide redox partner to be determined (E0 = −0.59 V vs Fc/Fc+, in MeCN), and from this data, the CuOO-H bond-dissociation free-energy (BDFE) of the μ-1,1-hydroperoxide complex could be approximated at ~72 kcal/mol.792
In addition to chelate ring size, ligand flexibility, and ligand donor strength, hydrogen-bonding substituents attached to the ligand can have an effect on trans-μ-1,2-peroxodicopper(II) complex stability. The Masuda group derivatized the TMPA ligand framework with methyl (originally reported by Suzuki, Uehara, and co-workers),793 pivalamido, and amino groups at the 6-position of one pyridine ring (Figure 83, left).715 The crystal structure of [CuII(MPPA)(N3)]ClO4, where a stable azido containing complex was generated and characterized in order to help with an understanding of the details of the binding of the peroxo ligand in [{CuII(MPPA)}2(O22−)]2+, revealed an intramolecular hydrogen-bonding interaction between the pivalamido N-H and the proximal (bound to copper) nitrogen atom of the axially bound azide anion. This observation indicates that hydrogen-bonding interactions from the copper chelate to the peroxidic oxygen atom can occur in peroxodicopper(II) species, like similar monocopper superoxo and hydroperoxo compounds previously discussed (vide supra). Following oxygenation of cuprous complexes with ligands in Figure 83, trans-μ-1,2-peroxodicopper(II) species formed in all cases, and their peroxo-to-copper LMCT bands (π*σ → dσ and σ*v→ dσ) indicated that hydrogen-bonding effects the relative energies of the peroxo π*σ and σ*v orbitals. Namely π*σ is stabilized, blue-shifting its LMCT band, and the σ*→ dσ transition is broadened because of a restricted Cu-O bond rotation, which disfavors overlap between the σ*v and dσ. orbitals. Hydrogen-bonding moieties also increase the thermal stability of these trans-μ-1,2-peroxodicopper(II) species, despite their added steric bulk due to attachment at the pyridyl 6-position, which for non-hydrogen bonding groups decreases trans-peroxide stability.715
Figure 83.

Tetradentate ligand frameworks featuring hydrogen-bonding moieties studied by the Masuda group. See text for further discussion.
In another systematic study, the Masuda group added amino substituents at the 6-position of each pyridyl ligand successively to yield MAPA, BAPA, and TAPA (Figure 83, right).716 The crystal structures obtained for the [(L)-CuII(N3−)]+ complexes bearing these ligands displayed an end-on binding mode of the azide ligand, with clear hydrogen-bonding interactions between the Cu-bound Nazido-atom with the amino N–H bonds, with Nazido…H-N distances ranging from 2.84–3.05 Å, indicating that hydrogen-bonding type interactions do occur to ligands bound to copper(II) in these systems. The increase in the number of amino groups coincides with a structural distortion from trigonal bipyramidal toward square pyramidal and also with a higher CuII/CuI redox potential. This distortion was attributed to intramolecular hydrogen-bonding and steric interactions. Upon reaction with dioxygen at low temperature, all the cuprous chelates of TMPA, MAPA, BAPA, and TAPA formed end-on trans-μ-1,2-peroxodicopper(II) species, and the thermal stability of these peroxo complexes increased as more amino groups are incorporated. The steric constraints imposed by the amino groups, as well as their hydrogen-bonding interactions with the peroxo π* orbitals are implicated in the higher-energy O–O bond stretches due to electron withdrawal from the peroxo antibonding orbitals (Figure 84, left).716 Another trans-μ-1,2 peroxodicopper(II) complex from the Masuda group with the BNPA ligand framework (Figure 83, right) was characterized as well, via oxygenation of its cuprous salt. Oxygenation of [CuI(BNPA)]+ in the presence of substrates with weak X–H bonds (BDE < 72.6 kcal mol−1) yielded a cupric hydroperoxide species instead, implicating a cupric superoxide in hydrogen atom abstraction, which was supported by the use of a spin-trap reagent.719 A very recent example of a trans-μ-1,2-peroxodicopper species featuring intramolecular hydrogenbonding interactions was structurally characterized by the Szymczak group (Figure 84, right).769 Interestingly, the H bonding found in this complex differs from that previously proposed by Masuda and co-workers in that it contains both proximal and distal hydrogen bonds (Figure 84). In this work, other trans-μ-1,2-peroxodicopper(II) species with electronically tunable 6-substituted tris(2-pyridylmethyl)amine ligands featuring NH(para-R-C6H4) hydrogen bond donating substituents were also spectroscopically characterized.769
Figure 84.

Trans-μ-1,2-peroxodicopper species with (A) hydrogen-bonding interactions proposed by Masuda and co-workers716 and (B) crystallized by Szymczak and co-workers.769.
3.3.2.4. Side-On μ-η2:η2-Peroxo and Bis-μ-Οxο Dicopper Species.
A very large number of side-on peroxo dicopper(II) species and bis-μ-oxo dicopper(III) species have been characterized, and the two isomers are known to interconvert, with the position of this equilibrium depending on many factors including ligand steric and electronic properties, solvent, counterion, and temperature.18,750,753 Here we present a very brief overview of some steric and electronic effects on the side-on peroxo/bis-μ-oxo species formed and their equilibrium position. Readers are pointed to recent in-depth reviews for a fuller understanding of these topics.18,608
The first example of a side-on μ-η2:η2-peroxodicopper(Π) species was structurally characterized by Kitajima and Fujisawa in 1989 from oxygenation of a tris-pyrazolyl borate complex, [CuI(HB(3,5-iPr2pz)3)], at –78 °C in acetone to yield [{(HB(3,5-iPr2pz)3)CuII}2(O22–)]2+ (Figure 85A).760 The core structure of this bridging peroxide species is similar to that of oxy-hemocyanin and oxy-tyrosinase (section 3.2), both of which were published after the initial finding by Kitajima. The O–O bond length was reported as 1.41 Å, consistent with its formulation as a peroxo bridge. The Cu…Cu distance was reported as 3.56 Å, which is shorter than the analogous trans-μ-1,2-peroxodicopper(II) species [4.359(1)Å] and comparable to those of oxy-Hc and oxy-Ty (3.58–3.66 and 3.63 Å, respectively).760 Such side-on μ-η2:η2-peroxodicopper(II) species were also observed with binuclear copper ligands, and their UV–vis spectroscopic properties were similar to those of oxy-hemocyanin, although they were not structurally characterized.512,794 Soon afterward, the Tolman group successfully isolated a crystal of a bis-μ-oxodicopper(III) species using a triazacyclononane ligand framework, [{(LBn3)-CuIII}2(O2–)2]2+ 765 (Figure 85B). The Cu–O and Cu…Cu bond distances (1.81 and 2.798 Å, respectively) obtained from a crystal structure were distinct from previously characterized μ-η2:η2-peroxodicopper(II) species795 and similar in magnitude to those of other Mn and Fe M–(O2–)2–M species.796,797 This work also provided key insights into the role of the solvent environment in determining copper-dioxygen complex speciation. Dichloromethane solution favored the side-on μ-η2:η2-peroxodicopper(II) isomer, whereas tetrahydrofuran favored the bis-μ-oxodicopper(III) species. With the use of acetone as the solvent, a mixture of both species was obtained. This was the first observation of the equilibrium between μ-η2:η2-peroxodicopper(II) species and their bis-μ-oxo isomers. Soon after, the Stack group798 characterized a bis-μ-oxo dicopper(III) complex bearing bidentate ligands with similar structural characteristics to those first observed by Tolman (Figure 85C).765
Figure 85.

(A) The first structurally characterized μ-η2:η2- peroxodicopper(II) species. (B and C) Two of the earliest structurally characterized bis-μ-oxo dicopper(III) complexes formed from ligand-copper(I) dioxygen reactions. Created with data from references 760, 765, and 798.
Ligand electronic properties have also been shown to affect not only the side-on peroxo/bis-μ-oxo equilibrium but also the spectroscopic properties of the resultant dicopper-dioxygen species. The Karlin group evaluated the role of ligand electronic properties by derivatizing the R-MePY2 [(MePY2: (bis[2-(4-R-2-pyridn-2-yl)ethyl]methylamine))] ligand framework, containing groups with varying electron-donating or −3withdrawing capabilities (Figure 86, top row).799 Electron-donating groups such as methoxy and N,N-dimethylamino substituents increased the N-donor strength of the pyridine ligands. Oxygenation reactions of [(R-MePY2)CuI]+ complexes in this study were carried out in tetrahydrofuran at –80 °C. Resonance Raman spectroscopic studies revealed that the additional Npy-donor strength weakened the observed CuIII–O bonds in the bis-μ-oxide isomer; however, there was little effect on the O–O bond stretches of the side on μ-η2:η2- peroxodicopper(II) isomer. The absence of a shift in the O– O bond stretching frequency was attributed to greater electron density on the CuII ion, which decreases backbonding from the peroxo π*σ orbital to CuII. The resultant extra negative charge on the peroxo ligand makes back-donation from the CuII d-orbitals into the peroxo σ* orbital less favored, which is necessary to decrease the O–O bond strength to “activate” it for reductive cleavage. The percentage of bis-μ-oxo isomer formed also increased as the donor strength of the R-MePY2 ligand increased due to thermodynamic stabilization of the CuIII oxidation state in the bis-μ-oxide species, although in each case the product remained a mixture that favored the side-on peroxide. This provided an example of how electronic effects can alter the equilibrium between the side-on peroxide and bis-μ-oxo isomers but not necessarily the spectroscopic properties of each.799 Another systematic study of the role of ligand electronic effects on the position of the side-on peroxo/bis-μ-oxo equilibrium was carried out using the R-PYAN [PYAN: (N-[2-(pyridin-2-yl)ethyl]-N,N',N'-trimethylpropane-1,3-diamine)] ligand framework by the Karlin group (Figure 86, bottom row).770 Here though, the CuII/I redox potentials and C–O stretches (determined by IR spectroscopy of the [(R-PYAN)CuI(CO)]+ complexes) in each ligand system displayed only minor changes depending on the pyridyl ligand substitution. However, the position of the equilibrium between side-on peroxo and bis-μ-oxo isomers was highly sensitive to substituent effects at the para position of the pyridyl group. Namely the parent ligand (PYAN), and its p-chloro substituted analogue (Cl-PYAN), yielded the side-on peroxo isomer almost exclusively. The O–O bond stretching frequencies of these side-on peroxo species shifted by up to 10 cm−1, depending on the para-substitution. This is particularly interesting because O–O bond activation is observed in conjunction with an increase in bis-μ-oxo formation. In contrast to electron-withdrawing groups, when using the N,N-dimethylamino derivatized ligand (NMe2-PYAN), the bis-μ-oxo isomer is obtained almost exclusively. The methoxy- derivatized ligand showed a mixture of the two isomers. This increased sensitivity with the PYAN framework compared to the MePy2 ligand may be due to the presence of two aliphatic amine ligands in the former, which have greater donor strength than the pyridine and as such electronic effects are more tunable. This work further demonstrated that the side-on peroxo/bis-μ-oxo equilibrium could be shifted to either extreme using ligand electronic effects alone and that this process corresponds to O–O reductive cleavage.770
Figure 86.

Ligand electronic effects on the speciation of CuI:O2 binuclear reaction products. See text for details. E1/2 values were determined in DMF as solvent and are reported vs Fe(Cp)2+/0.
Ligand sterics are also a major determinant of the type of copper oxygen species obtained from reacting copper(I) chelates with dioxygen at low temperature. The Stack group has extensively investigated the copper-dioxygen species formed with peralkylated-diamine-type ligands.749,798 Generally speaking, as the steric demands of the ligand are increased, the CuI:O2 reaction stoichiometry decreases, with the most sterically demanding ligands being unreactive toward O2 (Figure 87). As the alkyl substituents of the diamine ligands are increased in steric bulk (Me to Et to tBu), the main isomer formed in solution goes from trinuclear oxide species (T), to bis-μ-oxo species (O), to side-on peroxide type species (SP) (Figure 87). A recent example from the Stack group using the tBu3tacn ligand framework showed the highest reported stability for a side-on peroxide species, which is catalytically competent for catechol oxidation to quinone, phenol coupling, and alcohol oxidation.800
Figure 87.

Ligand steric continuum depicting predominant copper–oxygen species formed as ligand steric demands increase. Adapted with permission from ref 749. Copyright 2003 The Royal Society of Chemistry.
In order to better understand the dioxygen binding and release processes for mononuclear tetradentate and binuclear tridentate copper-oxygen species, the Karlin group carried out laser-induced photoexcitation studies on the copper-dioxygen species [{(TMPA)CuII}2(O22−)]2+ and [(Nn)Cu2II(O22−)]2+ (n = 3 or 5) which form trans-μ-1,2- and side-on μ-η2:η2-peroxodicopper(II) species, respectively, following oxygenation of their cuprous salts.801 Photoexcitation of [{(TMPA)CuII}2(O22−)]2+ yielded a mixed-valent cupric superoxide intermediate, [(TMPA)CuII-(O2•−)-CuI(TMPA)]2+, which rapidly proceeded with another intramolecular electron transfer reaction to form an “O2-caged” dicopper(I) species [{(TMPA)CuI}2(O2)]2+. This O2-caged species then rapidly reformed the trans-μ-1,2-peroxodicopper(II) species, [{(TMPA)CuII}2(O22−)]2+, with no O2 release (Figure 88, top). With [(Nn)Cu2II(O2)]2+ (n = 3 or 5), a mixed-valent superoxide species was also formed, however, instead of forming an O2 caged species, electron transfer back to the second CuII from superoxide resulted in O2 release and two CuI ions (Figure 88, bottom). The difference in reaction pathways for these tetradentate and tridentate ligand frameworks can be understood in terms of the relative redox potentials of their cuprous chelates. Cuprous complexes with tetradentate chelates have more negative redox potentials than those of their tridentate analogues, which makes the reformation of peroxo species more favorable for the O2 caged [{(TMPA)CuI}2(O2)]2+. Both of the Nn ligands are tridentate, which favor copper(I), making photoejection of O2 facile.801
Figure 88.

Photoejection chemistry for (top) [{(TMPA)CuII}2(O22–)]2+ and (bottom) [(Nn)Cu2II(O22–)]2+, where n = 3 or 5.801.
3.3.2.5. Dicopper(II) μ-Mono-oxo Complexes and Methane Oxidizing Zeolites.
A very recent reinvestigation into the previously reported crystal structures of pMMO has determined that the enzymatic active site may only contain a single copper ion;656 original reports were ambiguous in the number of copper ions (see section 3.1).655,802–804 As has been suggested as possibly relevant, copper ion loaded zeolites (Cu-ZSM-5), and others (vide infra),805 are known to readily oxidize methane to methanol in air, and spectroscopic interrogation of this material by Solomon and co-workers led these researchers to suggest that it is a dicopper(II) complex bearing one bridging oxo ligand (Cu-O-Cu or [Cu2O]2+) that is the active species which hydroxylates methane.806–808 Thus, great interest has developed to learn how such species form and the mechanism by which they oxygenate methane. In the past, some synthetic compounds with a [Cu2O]2+ core have been synthesized using mono- and binucleating ligands. Such species can be formed through several pathways starting from CuI complex precursors, including addition of dioxygen, iodosobenzene (PhIO, a well-known O atom donor), or nitric oxide (NO) (Scheme 19, top).18,807 Initial studies by Karlin and co-workers revealed that certain [(R-PY2)CuI]+ complexes could be reacted with dioxygen at −380 °C giving μ:η2:η2- peroxo-dicopper(II) complexes (Cu/O2 = 2:1) (Scheme 19, and see section 3.3.2) which, upon warming, decayed into a( μ-oxo)dicopper(II) species.511,809,810 The same [Cu2O]2+ complex could be formed directly upon oxygenation of the initial cuprous species at 0 °C, with a reaction stoichiometry of Cu/O2 = 4:1. Reactivity studies showed that these complexes can oxidize triphenylphosphine to triphenylphosphine oxide. Limberg and co-workers807 have studied the characterization and reactivity of [Cu2O]2+ complexes bearing unique binucleating ligands (RXanthdim and FurNeu, Scheme 19). Unfortunately, neither the Karlin nor Limberg [Cu2O]2+ complexes show exceptional reactivity toward substrates; with the FurNeu ligand complex, oxidative coupling of phenols was observed.
Scheme 19.

Formation of (μ-Oxo)dicopper(II) Complexes Bearing Chelates for Single-Cu Species or Utilizing Binucleating Ligands511,807,809,810
Most recently, Kieber-Emmons and co-workers811 published on the thermodynamic parameters of hydrogen-atom-transfer reactivity by a model [Cu2O]2+ complex. Titration of a structurally characterized (μ-hydroxo)dicopper(II) complex bearing the TMPA ligand with the base 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) allowed for the determination of the pKa of the bound hydroxide ligand (pKa = 24.3 ± 1.9 in MeCN). Cyclic voltammetry on the same bridging hydroxo complex gave the CuII2/CuIICuI reduction potential as –0.48 V versus Fc0/+ in MeCN. A thermodynamic square scheme could thus be constructed giving the BDFE of the O– H bond in a (μ-hydroxo)CuIICuI species as 77.2 kcal mol−1 (Scheme 20, left). Experiments found to be consistent with this result were that the complex could react with substrates containing weak C–H and O–H bonds (cyclohexadiene and TEMPO-H), giving the products benzene and TEMPO, respectively (Scheme 20). 811 While very strong C–H bonds, such as methane, could not be oxidized, this is the first indepth study obtaining thermodynamic parameters relevant to oxidation by a [Cu2O]2+ species.
Scheme 20.

Thermodynamic Square Scheme for the (μ-Oxo)dicopper(II) Complex Bearing the TMPA Ligand, and Reactivity of the Same [Cu2O]2+ Species with Substrates Containing Weak C–H and O–H Bondsa
aAdapted from ref 811. Copyright 2017 American Chemical Society.
While methane hydroxylation has not yet been attained in synthetic model systems, as mentioned a great deal of information has been obtained for the involvement of the [Cu2O]2+ moiety in Cu–ZSM-5.808 Resonance Raman and optical spectroscopic interrogation showed that the active CuII–O–CuII species possesses a Cu–O–Cu angle of 140°, while temperature-dependent studies showed there is an activation barrier of 15.7 kcal mol−1 for methane to MeOH conversion.806 Further investigations have since identified two different Cu2O2 precursors (derived from O2-reaction with reduced CuI–zeolite material) leading to the active [Cu2O]2+ oxidant (Scheme 21).808 For Cu–ZSM-5, the initial formation of a dicopper(II) side-on peroxide complex was confirmed by UV–vis and resonance Raman spectroscopies.812 In a more recent investigation on other types of Cu-loaded zeolites, for example, Cu–SSZ-13, the O2-adduct formed initially has been verified using rR spectroscopy, to be a dicopper(II) trans-peroxide species.805 The resulting [Cu2O]2+ species in Cu– SSZ-13 is said to have a smaller ∠Cu–O–Cu bond angle (~100°) compared to that found in Cu–ZSM-5 (140°). Future studies may show how this change in geometry may affect the reactivity of [Cu2O]2+ complexes.
Scheme 21.

Proposed Activation of O2 in Copper-Loaded Zeolites, Resulting in the [Cu2O]2+ Reactive Intermediatea
aAdapted from ref 812. Copyright 2010 American Chemical Society.
Two electrons (and the loss of an oxide) are needed in order for these dicopper(II) peroxide species to convert into the reactive [Cu2O]2+ intermediate. It is postulated that two spectator Cu(I) ions donate the electrons needed, while one of the resulting oxo ligands can enter the zeolite lattice, giving [Cu2O]2+ (Scheme 21).808,812 The finding of a key oxo-dicopper(II) complex in Cu-loaded zeolites which can convert CH4 to MeOH may suggest that a dicopper site, perhaps also a two-copper-ion containing [Cu2O]2+ intermediate, is responsible for the pMMO enzyme activity.
The related reaction which might occur in enzymes is that a Cu2O2 species picks up two electrons and two protons giving an active [Cu2O]2+ species plus H2O (eq 6).
| (6) |
But exactly how does such a reaction occur mechanistically, with respect to the O–O cleavage, and by analogy to cytochrome P-450 Cmpd I formation for a heme–FeIII– OOH complex (vide supra)? One can hypothesize that double-protonation of a peroxidic Cu2O2 species followed by heterolytic cleavage could possibly give a high-valent, CuIII–O–CuIII, a thus far unknown entity in copper (bio)chemistry. For a single-copper hydroperoxide complex CuII–OOH (section 3.3.1.2), addition of one-electron and one proton could give an active high-valent CuII–O•·oxidant plus water. In copper coordination chemistry and copper-enzymes, the detailed mechanisms by which such O–O reductive cleavage events occur are poorly understood.
4. HEME-COPPER ENZYMATIC ACTIVE SITES FOR EFFICIENT, SELECTIVE O2-REDUCTION
An estimated 2.4 billion years ago, the so-called “Great Oxidation Event”, which is believed to be linked to the activity of photosynthetic cyanobacteria, raised atmospheric O2 levels over 4 orders of magnitude (to ~10% of current levels) and drastically shifted the evolutionary course for life on earth.813–815 To harness the high oxidizing power of dioxygen, nature has developed metalloproteins for aerobic respiration, completing the oxygen cycle and securing the value of O2 in biology.816 Selective and complete reduction of O2 to water by mitochondria and aerobic bacteria is accomplished by heme–copper oxidases (HCOs). These metalloenzymes have deservedly garnered extensive research efforts, as a detailed understanding of the HCO four-electron four-proton oxygen reduction reaction (ORR) gives vital fundamental biochemical insights [O2-reduction is coupled to energy production (i.e., ATP synthesis)];817 insufficient oxidase activity has been linked to neurodegenerative diseases818–821 including Alzheimer's822–824 and Leigh syndrome,825 as well as oxidative stress and aging in general.826–829 A detailed understanding of the HCO reaction mechanism utilizing heme- and copper-based catalysis also has practical industrial potential (i.e., in fuel cell development, where the cathodic ORR normally requires a precious platinum catalyst).4,7
4.1. Characteristics of the HCO Superfamily and Discussion of Important Redox Cofactors
Heme-copper oxidases (oxygen reductases) comprise a large superfamily of integral membrane proteins (Figure 89A) with diversity in heme-type (Figure 89C), electron donor, and number of subunits; these metalloenzymes exist in eukaryotic mitochondria and some bacteria.830–833 As their name suggests, HCOs use a binuclear center (BNC) containing a heme and copper cofactor to activate and reduce dioxygen. Structural and functional similarities to nitric oxide reductases (NORs) suggest that the two families share a common ancestor (see section 6.5.5),834,835 yet oxygen reductases contain key features which make them unique, including the catalytically active Tyr residue at the BNC and transmembrane proton pumping channels. As the terminal electron acceptors in the mitochondrial respiratory electron transport chain (“Complex IV”), HCOs catalyze O2-reduction to water, following the equation: O2 + 8H+in + 4e− → 2H2O + 4H+out. The proton subscripts in this equation denote the redox-coupled proton-pumping (4 protons per turnover) via a Grotthuss-type mechanism836,837 across the inner mitochondrial membrane, which contributes to the electrochemical proton gradient utilized by ATP synthase for generating the cellular energy carrier, ATP, from ADP and inorganic phosphate.836,838 The electrons can be funneled sequentially to the BNC from either a cytochrome c electron transfer hemeprotein via the nearby, mixed-valent, fully delocalized, formally di-Cu1.5 site (CuA) in subunit II which gets reduced to a CuI…CuI state, as in Cytochrome c oxidases (CcOs) (Figure 89B), or from a ubiquinol substrate embedded in the protein subunit I, as in Quinol Oxidases (QOs).839 In the case of QOs, the two ubiquinol protons freed by the double proton-coupled electron transfer (PCET) reaction, which converts ubiquinol to ubiquinone, are pumped to the P-side of the membrane, while the electrons are shuttled to the BNC (Figure 89D).840,841 In both subcategories of HCOs, electrons then travel to a low-spin six-coordinate heme a which is axially tethered to the high-spin heme a3 iron of the BNC where dioxygen binds as the sixth ligand. The copper ion (CuB site of the BNC) sits 5.1 Å from the high-spin heme a3 iron when the enzyme is in the fully reduced state (PDB: 5B1B, Figure 90)842 and is ligated by three histidine residues. Unless otherwise noted, the amino acid numbering scheme used in this review corresponds to the Bos taurus (bovine heart) cytochrome c oxidase (see Figure 90), as it is the most widely studied in the family.
Figure 89.

(A) Crystal structure of membranous bovine heart CcO at 1.6 Å resolution (PDB ID: 5B1B), (B) redox cofactors and H+ and e− pathways via the heme-Cu active site, (C) heme types found in HCOs, (D) redox cofactors active in quinol oxidase catalyzed O2-reduction and the distinct proton pumping mechanism from CcOs. (B) Adapted with permission from ref 843. Copyright 2010 National Academy of Sciences. (C) Adapted from ref 535. Copyright 2004 American Chemical Society. (D) Adapted with permission from ref 840. Copyright 2000 Nature America Inc.
Figure 90.

Active site of fully reduced FeII…CuI bovine heart CcO.
4.1.1. Unique Histidine-Tyrosine Cross-Link.
A highly conserved heme-Cu active site post-translational modification consists of a covalent cross-link of one of the His ligands to a Tyr (His-Nε-Tyr-Cε2, see Figure 90), and this cofactor has long been known to be necessary for maintaining oxidase function (vide infra).844–849 The covalent C–N linkage is estimated to decrease the pKa of the tyrosine–OH by ≥1.1 pKa units and increase the redox potential by approximately 66 mV, based on model studies,850–853 consistent with its proposed involvement as a proton and electron donor during O2 activation and reduction. From another perspective, the cross-link also affects the electron-donating ability of the histidine as a ligand for CuB, which undoubtedly fluctuates throughout turnover due to the participation of Tyr–OH in the reaction.844 Organization of a hydrogen-bonding network consisting of one or more water molecules linking the Tyr–OH and the bound O2 moiety (either the superoxide and/or proposed peroxide forms, vide infra) could aid in shuttling in the proton(s) necessary for O–O cleavage.854–857 Additionally, X-ray crystallographic data has shown that the active-site Tyr residue hydrogen bonds to one of the heme farnesyl chains possibly linking its role to substrate/proton gating.858,859 The biogenesis of this unique cofactor is not well-understood, although mechanisms have been proposed, both where the cross-link is formed during the first catalytic turnover (presumably following formation of the TyrO•· radical) or via some other anaerobic pathway (Scheme 22, panels A and B, respectively).22,612,848,860 The reaction to form the cross-link is formally a two-electron oxidative C(phenol) –N(imidazole) coupling of the amino-acid side-chains of a Tyr and a His residue which reside close to each other (in space), that is in a i/i+4 relationship on an α helix in a pro-enzyme form.
Scheme 22.

Supposed Mechanisms of His-Tyr Crosslink Biogenesis in the (A) Presence or (B) Absence of O2a
aAdapted with permission from ref 612. Copyright 2016 Springer. Other reaction sequences to effect oxidative cross-coupling of His and Tyr residues can be envisioned.
4.1.2. Proton-Pumping Function of HCOs.
The HCO proton-pumping function is coupled to the O2-reduction chemistry (vide infra, section 4.2), but is, itself, an area which deserves and has attracted considerable attention from physical biochemists, bioenergeticists, and theoretical-computational researchers due to the importance of the formed membrane electrochemical gradient which drives ATP synthesis.861 Subunit I, which houses the important redox cofactors, also contains two proton-pumping channels lined with polar residues.19 The so named, K- and D-channels, are responsible for supplying chemical protons to the BNC and transporting both chemical and pumped protons, respectively; these conclusions come from extensive mutational studies (and recently molecular dynamics simulations).843,862–865 Protons are shuttled (from the opposite side of the membrane as electrons) to the BNC in a controlled fashion which does not allow back-leakage but does make use of loading sites (“LS” in Figure 91) such as a conserved glutamic acid (E242, Figure 91) residue near the edge of heme a, and a Mg-water cluster adjacent to heme-a3, both of which are responsive to and/or cause redox changes related to the chemistry of the BNC (reduced vs oxidized metal centers induce conformational changes, and increased introduction of positive charges at the loading sites increases the electron affinity of the BNC).19,842,866–868 Uptake of protons to the BNC is necessary for O–O bond scission; however, all four protons pumped per catalytic turnover are proposed to be translocated into the intermembrane space after the O–O bond cleavage event, utilizing the energy released in these steps. Coupling to the O2-reduction mechanism implies participation of active site moieties in either gating of the pumping pathways or causing structural (allosteric) changes in the protein. Indeed, it has been suggested by time-resolved IR data that the initial binding of O2 to CuB induces proton collection and results in a conformational change which increases the O2-affinity of heme-a3, and the subsequent binding of O2 to the heme creates a bulge at Serine-382, closing the proton uptake channel to prevent back-leakage.854,869,870 Finally, the H-bond that exists between the cross-linked Tyr244 and heme-a3 farnesyl group (see below, Scheme 23, R) acts as a gate for the K-channel, with the O(H)…O(H) distance ranging between 2.6 and 4.1 Å depending on the oxidation state of the BNC.871
Figure 91.

Proton pumping channels (K and D) in bovine CcO with key residues labeled and the location of the MgH2O cluster loading site noted. Reproduced with permission from ref 872. Copyright 2015 Elsevier.
Scheme 23.

Catalytic Mechanism of O2-Reduction by HCOsa
aReduction potential values obtained from refs 875–878 shown as a range of values obtained using various methods or conditions. Heme-a3 peripheral R group = 15-C farnesyl group, see Figure 89C. See text, both here and in section 6, for clarification or further alternative descriptions of the intermediates depicted here, or for unobserved putative intermediates.
4.2. Catalytic O2-Reduction Mechanism
In coupling the O2-reduction and proton-pumping functions of CcO, intricate mechanisms of cooperative allostery exist so as to (i) prevent release of partially reduced reactive oxygen species (PROS), (ii) allow gating of water and proton-pumping channels while preventing back-leakage of these species, and of course (iii) energetically couple the redox chemistry to these events.865 Many of the details of these interrelated steps are not yet entirely understood and will require studies on the complete enzyme and its mutants. However, the mechanism of O2-reduction by the binuclear center of CcO (which has recently been reviewed at length;19,20 also see the review article from M. Wikstrom and co-workers873 in this special themed Chemical Reviews issue, along with the following valuable recent reviews and perspectives),11,817,874 has seen immense contributions from heme-copper model systems (which are the focus of this review, vide infra and see section 5) toward the fundamental understanding of this important reaction and the tightly regulated transfer of protons and/or electrons in directing efficient reactivity/(bio)catalysis in general. Although certain details are still debated (especially with the recent rise in computational capabilities), spectroscopic studies of reactivity paired with X-ray crystallographic structural data, together over the last few decades, have culminated in the currently accepted mechanism shown in Scheme 23.
“…[CcO’s] behavior reflects a mechanism in which conditions that allow efficient dioxygen bond cleavage are not inherent to the active site but are only established as the reaction proceeds. This catalytic strategy provides an effective means by which to couple the free energy available in late intermediates in the reduction reaction to the proton-pumping function of the enzyme.” Quoted with permission from ref 979. Copyright 1993 National Academy of Sciences.
4.2.1. O–O Bond Reductive Cleavage (Oxidative Phase of the Catalytic Mechanism).
Coupling of time-resolved IR, rR, and X-ray techniques following flash photolysis of heme-CO-bound CcO879 have recently confirmed a longstanding proposal that upon reaching the BNC, O2 first transiently binds to CuB; a phenomenon which induces conformational changes on the distal side of heme-a3 related to gating of the water channel and increasing the O2-affinity of heme-a3.869,879 Nonetheless, it is typically considered that introduction of dioxygen to the fully reduced state of the enzyme, R, {Fea32+/CuB+; Fea2+; CuA+} begins the oxidative phase of the catalytic cycle with the formation of the A/Oxy species, v(Fe-O) = 571 cm−1 (∆18O2 = −27 cm−1) (Scheme 23),880 which is spectroscopically similar to the O2-adducts, oxy-hemoglobin and oxy-myoglobin [bent, “end-on” ferrous- oxy (FeII-O2) or ferric-superoxo (FeIII-O2·•−); see section 2.1.1], although proof that this species is not a peroxide level intermediate (e.g., such as IP) does not exist since an O–O stretching vibration has not been detected. (Note: see further discussions, section 6.3.1.) Rapid cleavage of the O–O bond has so-far prevented spectroscopic isolation or characterization of a bridging (hydro)peroxide species (i.e., the proposed IP, see Scheme 23, and below, sections 4.2.3 and 6), although the Fea3 and CuB may lie in close enough proximity to allow for such a formulation and indeed it is a chemically plausible intermediate to precede O–O cleavage. Furthermore, the presence of an additional proton in the active site is calculated to decrease the overall energetic barrier for O–O cleavage,747,855 and protonation of a nearby Mg-water cluster, believed to function as a proton loading site, increases the redox potential of the active site.867 Indeed the decay of the A species reveals a measurable hydrogen/deuterium kinetic isotope effect (KIE) of κH/κD = 1.8 ± 0.2 for the Rhodobacter sphaeroides bacterial enzyme881 or κH/κD = 1.9 ± 0.4 for bovine heart CcO, when the enzyme was studied in H2O or D2O media, respectively.882
Nonetheless, the next observable state by rR (still termed “P” as it was originally believed to be a peroxo species, though this was eventually ruled out by mixed isotope 16O18O experiments)322 is the O–O cleaved ferryl-oxo species, v(FeIV-16O) = 804 cm−1 (∆(18O) = −40 cm−1] with an optical absorption band observed at 607 nm, which is analogous to the so-called Compound II species (FeIV= O2−) characterized in many hemoproteins (see section 2.1). Notably, a P intermediate, which additionally contains a CuBII- OH species can also be generated during reduction of O2 by the mixed-valent CcO where only the BNC metals begin in the reduced state or by reaction of H2O2 with the fully oxidized CcO (both result in oxy-ferryl species formation within the BNC structure, PM in Scheme 23; however, the latter is sometimes referred to as PH).883–885 Dioxygen reduction by the fully reduced CcO where extra reducing equivalents are available as compared to the mixed-valent enzyme has opened discussion as to whether a more reduced P state (PR, in which the neutral Tyr radical in PM has undergone one electron reduction from heme a to yield tyrosinate, TyrO−, see Scheme 23) may also exist; however, it is likely that the energy necessary for this electron transfer may instead manifest in protein structural changes possibly related to proton movements (vide infra).881–886 The involvement of Tyr244 in the catalytic cycle has been postulated since the first crystal structure was solved; however, support for its role as an electron-donor came later from radioactive iodide labeling/peptide mapping,849 time-resolved spectroscopic methods,850 and most recently by freeze-quench X-band and D-band EPR spectroscopy on PM generated with excess H2O2 confirming the Tyr radical character (gx = 2.0059, gy = 2.0051, and gz = 2.00 1 7).887 A conserved tryptophan residue (Trp236) which π-stacks with one of the CuB histidine ligands was shown to be necessary for oxidase function as well; therefore, originally it was considered as a potential donor for the fourth electron. However, the substantial evidence supporting the Tyr244 radical suggests that this Trp is instead involved in a radical- transfer process related to the O2-reduction chemistry, which is not yet fully understood.886,888 The redox activity and protonation/deprotonation of other highly conserved tyrosine (129, 280) and tryptophan (272) residues in CcO during catalytic turnover has been shown to be involved in regulating proton translocation functionality or to serve as electron relay sites. 884,887,889–891
The timing of electron transfers to cleave the O–O bond is important to prevent release of ROS, so a “Compound I”-type intermediate, (P+•)FeIV=O, like those known to form in heme peroxidases, catalases, and Cyt P450s (vide supra, section 2), has also been considered as a possible intermediate in HCO chemistry, as the porphyrin is a local cofactor (besides the Tyr and Trp residues) that could provide the fourth necessary electron (the others derived from the iron, copper or Tyr cofactor). A highly reactive and transiently lived Cmpd I intermediate would precede PM (Scheme 23) and has been detected by EPR and rR spectroscopies in experiments where CcO (oxidized forms) are exposed to H2O2. A Cmpd I species in CcO has been considered computationally, though never observed in the native catalytic cycle (i.e., reduced enzyme plus O2).883,884,892–894 it has been ruled out in the reaction of CcO with dioxygen, based on vibrational data and recent calculations which state that it is energetically less favorable to oxidize the porphyrin than nearby residues, specifically Tyr244. 322,747,887,891,895
4.2.2. Reductive Phase of the Catalytic Cycle Including Proton-Pumping.
The analysis above shows that all four reducing equivalents required for O2-reduction are provided by the redox active cofactors in the BNC [one from copper (CuBI → CuBII), two from iron (Fea3II → Fea3IV) and one from tyrosine-244 (TyrOH → TyrO•)]; however, regeneration of the reduced state of the enzyme is of interest as well because those proton and/or electron transfer events are importantly coupled to the structurally complex proton-pumping function of CcO.882,896 Both rR spectroscopy and site-directed mutagenesis along the proton pumping channels have been integral in elucidating how O2-reduction drives or is coupled to proton translocation; however, the relationship between these processes still requires further investigation. Following formation of the P intermediate, electron transfer from heme a results in a new (still ferryl-oxo) species with λmax = 580 nm and v(Fe–16O) = 785 (Δ 18O2 = −35) cm−1,897 assignable to the F state (Scheme 23), and a three-coordinate CuB. In the time-resolved rR spectroscopic data, the features associated with F overlap significantly with those of the P intermediate, suggesting that the electrons from CuA/heme a preferably go toward a proton-coupled reduction of PM → F rather than only an electron transfer, PM → PR.886 It has been unequivocally shown that this PM → F transition is coupled to proton uptake from the D-channel and proton pumping across the membrane based on mutation studies and, moreover, that the proton uptake occurring in this step controls/drives electron transfer from the CuA site to heme a (and not vice versa; Scheme 23), with additional support from studies on a quinol oxidase.896,898 Finally, FTIR experiments indicate that Tyr is deprotonated in F, so it is expected that the proton taken up transfers to a transient CuB-OH moiety, releasing H2O and giving the F structure shown in Scheme 23 with a CuB/TyrO−.899
Additional H+/e− transfer to the BNC results in formation of the OH (“fully oxidized” relevant to turnover) intermediate (and additional proton translocation), which has a resonance Raman band at v(Fe–16O) = 450 (∆18O2 = −25) cm−1 and, with support from H2O/D2O exchange studies [wherein the rRband is shifted to v(Fe–16O) = 443 (∆18O2 = −28) cm−1 in D2O], has been assigned to an Fea3III-OH…CuIIB species.895 The rR feature of this species is significantly lower than that of an analogous state in other hemeproteins, which leads to the assignment of OH as a high-spin and strongly hydrogen-bonded ferric-hydroxy species present along with a three-coordinate (and thus high potential) CuB site; alternatively, a strained metal-bridging hydroxide structure as shown in Scheme 23 can describe this intermediate, though the exact oxidation state of Cu is debated (i.e., OH = FeIII-OH…CuI/TyrO• ↔·FeIII-OH-CuII/TyrO−). Either of these formulations may account for the reduction/protonation and proton pumping driven by the nature of the structure and bonding/electronic properties of this turnover intermediate OH to give Eh (Scheme 23).846,859,900,901 Aspects of the OH structure and coordination chemistry relevant to these properties are further discussed in section 6.3. Here, one of the first proposed computational structures for OH, a strained μ-hydroxo heme– Cu species, is shown below (Figure 92).
Figure 92.

A calculated/optimized structural possibility for the OH intermediate in the reductive phase of the catalytic mechanism of O2- reduction in HCOs (Scheme 23). Φ is the displacement of the iron above the plane of the porphyrin. Figure created with data taken from ref 901.
The F and OH states as well as the F → OH transition have been a recent focus for extensive biophysical and computational analysis in which the role of water molecules and CuB redox potential are hotly debated, as comprehensive spectroscopic/structural data has not yet elucidated mechanistic details.859,875,901–904 Two additional unresolved proton-coupled electron-transfers regenerate the TyrOH (forming the EH state, which has only been observed in electron injection experiments)905 and finally the reduced state of the enzyme once again, completing the catalytic cycle and releasing the two water molecules as well as pumping two additional protons (Scheme 23).906–908
4.2.3. Metal-Bridging Peroxo Intermediate and the Importance of Its H-Bonding or Protonation.
A putative (hydro)peroxide species in the catalytic cycle prior to O–O reductive cleavage has never been observed spectroscopically during turnover, although chemical intuition invokes a formal peroxide on the reduction pathway from dioxygen to water. Such Mn+-(−OO(H)) species are known to exist in other heme proteins (such as cyt P450s and peroxidases), multicopper oxidases,19 and model systems (also see sections 2, 3, and 5), where these intermediates immediately precede the O–O reductive cleavage step. For heme enzymes, nature goes to great effort to set up structures so that H-bonding and/or proton donation to a heme–peroxo distal O atom (that further from the iron) helps to mediate O–O cleavage (also see section 2).46,138 Noodleman calculates that a pure peroxo-bridged Fea3-O–O-CuB species (E°calcd = −0.73 vs SHE) would not undergo any reduction by cytochrome c or heme b.909 As will be illustrated in section 5, synthetic (heme)FeIII-peroxo-CuII-ligand complexes which have been tested are also unreactive even to moderately strong chemical reductants (as only electron-donor agents). In terms of electronics, a bridging peroxide avoids buildup of a large overpotential, and it could allow for ferromagnetic coupling between the Fea3 and CuB centers so that electron transfer from the Tyr would not necessitate spin-surface crossing for the reductive O–O bond rupture.909,910
In fact, the Fea3 and CuB ions in HCOs lie in close enough proximity to support a diatomic ligand, and several, including a peroxide as a bridging ligand (O22−) have been isolated in multiple CcO crystal structures [PDB IDs: 2ZXW, 5B1A, 3HB3 (O22−) and many others (see section 6.2); 1OCO, 3AG1, 3AG2 (CO); 3AG3 (NO); 3AG4 (CN−)].842,870,911 The fact that many O22−-bridged structures have been described in publications (however, see discussions in section 6.2), and that they are stable enough to be crystallized, perhaps contradicts the proposal that such a structure is a catalytically active intermediate,19 and it has been suggested that a bridging peroxide might uncouple the O2-reduction and proton-pumping functions.907,911 Nonetheless, a short-lived912 (bridging) peroxide intermediate now seems quite certain (vide infra); it was originally suggested by Proshlyakov, Pressler, and Babcock,913 and many theoretical-computational studies (Blomberg, Siegbahn, Wikström, Noodleman and co-workers) have supported this important assertion.20,850,855,861,875,901–903,909,914–918
Further, it has been shown in some or many of these theoretical-computation studies and deduced from very recent investigations on model systems (which include computational results; see section 5),747,919 that in order to reductively cleave the peroxidic O–O bond, it is energetically required to first transfer a proton to the O22– moiety,868,917,918,920 in fact to the O atom furthest from Fea3, generating a putative FeIII– (−OOH)…CuBIII hydroperoxo adduct which may or may not also be bound also to CuBII. Noodleman and co-workers909,917 go further and propose that the −OOH group is both iron(III) and copper(II) bound, with the proton located on the O-atom next to CuB, Fea33+–O–O(H)–CuB2+ (see Figure 93); this very much looks like a cytochrome P-450 heme–hydroperoxide ready to undergo heterolytic O–O cleavage giving Cmpd I. However, in HCOs, this FeIII–OOH moiety is now also bound to the copper ion via the distal O atom (with respect to Fe) and poised for homolytic O–O reductive cleavage. As discussed, only one electron will further derive from heme a3 (to transform to an FerV=O species), while the second electron comes from the proximal tyrosine, giving product, P (PM, Scheme 23, vide supra), a binuclear center formulated as FerV=O..HO–CuII YO·. From spectroscopic studies, Wikström and co-workers850 have been able to demonstrate that the proton on the hydroxide bound to CuB derives from the active-site tyrosine.
Figure 93.

(A) Noodleman and co-workers,909,917 computational structure depicting the key hydroperoxo bridged hemea3-CuB complex (with a proton derived from Y237 on the heme distal O-atom) which will now undergo O–O reductive cleavage following electron-transfer from the deprotonated Y237 phenol. This could be intermediate IP (Scheme 23). (B) Computational structure from Karlin and co-workers919 of a synthetic (porphryinate)FeIII-(μ-O22–)-CuII construct where the heme distal peroxo O atom is H-bonded to an exogenous phenol (p-nitrophenol), poised to undergo O–O reductive cleavage, as described in sections 5.2.6.
A most exciting development, a confirmation, comes from new experiments from Brzezinski and co-workers,918 carried out on Thermus thermophilus ba3 oxidase. They were able to show that at 10 °C (pH 7) electron transfer from the low-spin heme b to the heme–Cu binuclear active site is ~10 times (t ~ 11 ms) faster than formation of the FeIV=O heme ferryl PR intermediate (t ~ 110 ms). This is a clear indication that dioxygen is reduced prior to O–O reductive cleavage. In other words, O2-binding to give intermediate A (an iron(III)-superoxo species, FeIII(O2•–), similar to that which forms in hemoglobin, known as “oxy-Hb”, although in the biocommunity this is not spoken of as an O2-reduction) must be followed by formation of another intermediate prior to when the O–O bond splits. Further, corresponding experiments, carried out at pH 10, showed the rates of ET from heme b and PR formation to be very close to each other. Accompanied by DFT analysis (from coauthor Blomberg),918 intermediate A is concluded to pick up an electron either from CuB or the active site tyrosine (Y237), while the latter also contributes its proton. Thus, these researchers state that the binuclear active site peroxo intermediate IP exists as {Fea33+-(O–OH)− Cu1+ YO· μ Fea33+-(O–OH) − Cu2+ YO−}; these are resonance structures. This species is depicted in Scheme 23 as an intermediate form which includes H-bonding from the active site Tyr. The electron transfer observed by Brzezinski and coworkers was proposed to occur from heme b to this IP intermediate, resulting in a species the authors call IP− [i.e., Fea33+–(O–OH)− Cu1+ YO−].918 This ET prior to O–O cleavage lowers the activation barrier for formation of PR an O–O bond cleaved intermediate (Scheme 23).
Detailed investigations of the chemistry of synthetic heme–Cu analogs have led to many insights relevant to HCO active-site structure and chemistry,15,19,20,56,535,921 such as studies involving (i) binding molecules such as carbon monoxide, nitric oxide (NO), and cyanide, with perspectives relevant to kinetics, structure, and spectroscopy, (ii) reactivity of reduced (heme)FeII–CuI constructs with molecular oxygen, giving peroxo-bridged complexes, which in certain cases lead to O–O cleavage in the presence of electron and proton sources or where peroxo O-atom H-bond interactions are present, cf. Figure 93B, and (iii) electrocatalytic O2-reduction. In section 5, we detail advances made especially in the last 10 or more years, while in section 6 we provide discussions and insights specific to certain aspects of HCO biochemistry, as well as points of consideration or debate arising from an inorganic/coordination chemistry point of view.
5. SMALL MOLECULE SYNTHETIC MODELS OF HEME-COPPER OXIDASES
5.1. General Considerations for Modeling HCOs
In this section, we overview the work of research groups who employ synthetically derived small-molecule “biomimics” to model the heterobinuclear active site of heme-copper oxidases in order to evaluate aspects of the metal ion environment (i.e., the ligand and its exact nature) which give rise to dioxygen reactivity with reduced heme-Cu assemblies, determination of their structures and electronic properties (i.e., bonding), and their reactivity toward electrons and protons, to effect reductive O–O cleavage chemistry. Given that the reactivity of a metal center in a metalloprotein active site generally adheres to the fundamental chemistry of the metal ion itself, rational design of appropriate model complexes allows for interrogation of the structural, magnetic, and electronic properties of both stable species and transient intermediates proposed to form during enzymatic turnover.922 Such biomimetic studies can provide insights into the roles of metal ion coordination number, coordination geometry, ligand donor type, metal ion redox properties, local environmental factors, and hydrogen bond donor/acceptor moieties on the stability and/or reactivity of inorganic reaction intermediates. Additionally, they provide access to species which are not observed or easily isolated in protein studies. Most certainly, only by using synthetic models (or computational methods) can one break down such an intricate overall process into its specific elementary steps, so that profound basic understandings can be realized. While such small molecule systems do not and cannot duplicate the exact structural and mechanistic characteristics of enzymatic systems, nor can they in any absolute way prove enzyme mechanism, they can sharpen questions that need to be asked and allow for systematic studies to answer critical aspects. Much of the chemistry described below illustrates this type of research leading to the answering of some questions of great interest.
There have been two main approaches utilized in synthetic modeling of cytochrome c oxidase. One involves synthesis of discrete heme-copper assemblies, and examination of structures, and research into the question of symbiotic behavior of iron and copper into complex formation, especially O2-derived adducts. At an initial level of inquiry, one wishes to deduce how, in a heme-Cu binuclear assembly, the ligand-Cu ion influences dioxygen binding to iron(II)-porphyrinates, and equally, how hemes alter or control O2-binding to copper(I). Heme-superoxo- or heme-peroxo-copper complexes have generally required the utilization of cryogenic conditions and the use of aprotic solvents. Metal-coordinated O2-derived reduced fragments such as superoxide or peroxide are susceptible to deligation (via protonation) especially in protic solvents. Iron, and especially copper ions, exhibit significant ligand lability in their complexes, and low-temperature handling helps to overcome dissociation/exchange of ligands such as superoxide or peroxide.
The other primary research approach has been to examine the use of heme-copper assemblies as supported electrocatalysts for O2 reduction, preferably to give water, as in HCOs, avoiding the generation of partially reduced oxygen species (PROS). In some sense, the requirements for electrocatalysis are less rigid, in that an air stable compound, for example an oxidized FeIII…CuII assembly, can be used as the electrocatalytic starting material. Extensive work by the groups of Collman, Boitrel, Karlin, Naruta, and others (vide infra) has thus yielded detailed correlations between the structural, spectroscopic, electronic, and magnetic properties of the catalyst and possibly otherwise characterized discrete heme-O2-copper assemblies, under conditions where electrons derive from the electrode and protons from the medium used. In addition to spectroscopic studies, the stoichiometric reactivity of these species toward exogenous reductants, acid sources, and hydrogen atom donors (H+ + e−) has been evaluated to gauge their reactivity properties. The electrocatalytic O2-reduction with heme-Cu catalysts in fact has proven to be very successful, especially from the systematic studies carried out by Collman and co-workers.56,921,923,925,1044 Insights can and have probed aspects such as the role of the Cu ion, pH effects, solvent polarity influences, and how rates of electron flux (from an electrode) govern O2-reduction efficiency.
5.1.1. Dioxygen Derived Heme-Peroxo-Copper Assemblies as HCO Synthetic Models:
Generation, Structures, and Spectroscopic Properties. The study of O2 reactivity with heme and copper complexes, or preorganized synthetic reduced heme-Cu assemblies, is critical in the modeling of HCOs, and this strategy has been extensively employed by the Karlin, Naruta, Collman, and others. In these model compound systems, the Fe…Cu distance is determined in part by the bonding interactions of the Fe and Cu to the bridging ligand, which derives from dioxygen reactivity with reduced iron and copper ions. Many of the studies, must be carried out at low temperatures (~ −380 °C), under dry conditions due to the thermal instability of the resulting oxygenated adducts, susceptibility of generated heme- peroxo-copper complexes to protonate, releasing (usually) hydrogen peroxide, and/or their tendency to form bridging oxo species (i.e., by disproportionation, vide infra), and hydroxides (i.e., via further reaction with water). This methodology has enabled the systematic variation of the coordination environment around the Cu as well as the Fe ion and characterization of interesting biorelevant intermediates. Additionally, these adducts can and have been studied by spectroscopic techniques which help correlate aspects of heme/Cu O2-adduct molecular and electronic structure to their reactivity toward substrates (e.g., H+, e−, and phenols).
Before going in depth into heme-peroxo-copper model system reported in the literature, it is instructive to cover a few pivotal aspects of this chemistry, utilizing seminal examples from by the Karlin and Naruta groups, in order to give better context for other systems described, especially the more recent ones. To this end, in this section we detail the synthesis, typical geometric and electronic properties, and key spectroscopic signatures of high-spin and low-spin heme-peroxo-copper model systems, and their relationship with similar/relevant intermediates of CcO. Our discussion includes peroxo complexes supported by tethered (i.e., potential copper ion chelates covalently appended to the synthetic porphyrinate) ligand architectures, as well as those bearing appended phenol moieties as a mimic for the His-Tyr cross-link at CcO active site.
The study of [(F8)FeIII-(O22−)-CuII(TMPA)]+ (HS-TMPA) and [(F8)FeIII-(O22−)-CuII(AN)]+ (HS-AN) (note: both as perchlorate and/or BArF salts) (Figure 94) have helped to illustrate the influence of copper chelate denticity and axial base binding on heme-peroxo-copper bonding interactions. HS-TMPA was generated via the addition of O2 to a 1:1 mixture of [(TMPA)CuI(CH3CN)]+ and (F8)FeII in THF at −380 °c.926,927 Its classification as a heme peroxo copper complex was based on rR spectroscopy, which showed an 18/16O2 isotope sensitive O–O and Fe-O stretching frequencies at 808 (∆18O2 = −46) and 533 (∆18O2 = −22) cm−1, respectively. The oxygenated adduct was also characterized by MALDI-TOF-MS, which showed a parent peak at m/z = 1239 corresponding to [(HS-TMPA) −3ClO4 + CH3CN] and showed an increase by 4 mass units upon oxygenation with 18O2. Dioxygen uptake measurements confirmed a 1:1:1 stoichiometry for FeII:CuI:O2. The para- magnetically shifted 1H, 19F, and 2H (via the pyrrole deuterated derivative d8-F8) NMR spectra along with the Evans method experiments confirmed an overall S = 2 spin state. Mössbauer spectroscopy established the iron as a high spin ferric ion, bound to an electron-rich peroxide ligand.926,927 This data can be reconciled by considering that the d9 CuII is antiferro-magnetically coupled to the unpaired electrons on the highspin (d5) FeIII ion through the peroxidic bridging ligand. In order to better understand the basic coordination chemistry of this bridging peroxo species, extended X-ray absorption fine structure (EXAFS) spectroscopy was used in conjunction with DFT calculations. Iron and copper K-edge EXAFS data conformed to the proposed structure (but see just below) and were fit with a Cu-Fe distance of ~3.72 Å.
Figure 94.

Generation of high-spin heme-peroxo-copper adducts (top) [(F8)FeIII-(O22–)CuII(TMPA)]+ (HS-TMPA) and (bottom) [(Fg)FeIII- (O22−)CuII(AN)]+ (HS-AN). Adapted from ref 928. Copyright 2010 American Chemical Society.
Initially, the structure/coordination around the peroxo ligand in HS-TMPA was not known, but the critically important synthetic and structural work of Naruta and co-workers (see Figure 95, compounds A-C) allowed the formulation of the η2:η1-peroxo ligation in HS-TMPA.927,929 Naruta's contributions consisted of, in fact, very similar ligand donors to those used by Karlin 930,931 Naruta however utilized derivatives of tetraphenylporphyrinate (TPP) or tetramesityl-porphyrinate (TMP) where a TMPA or its methylated derivative is covalently tethered to the porphryinate periphery. Complexes (A-C) (Figure 95) are all FeIII-peroxo-CuII complexes, and the X-ray structure shown was obtained for compound (C) (Figure 95, right). Extensive physical measurements and reactivity studies were carried out for this crystalline material, (C), [(TMP)FeIII-(O22−)-(5MeTPA)CuII]+ (as a BPh4− salt). This heme-peroxo-copper complex, (C), displayed similar spectroscopic properties to analogous compounds created by Karlin and co-workers. Specifically, (C) is an S = 2 species with antiferromagnetic coupling between the FeIII and CuII, with a corresponding magnetic moment of 4.65 μΒ, EPR silent (in perpendicular mode), and has a peroxidic rR feature, v(O–O) = 790 (∆18O2 −3 44) cm−1. Some structural parameters of interest are that of d(O–O) = 1.460(6) Å and d(Fe…Cu) = 3.916 Å.
Figure 95.

Heme-peroxo-copper adducts (A-C) synthesized and spectroscopically characterized by the Naruta group. Complex (C) includes the crystal structure for [(TMP)FeIII-(O22−)-(5MeTPA)CuII](BPh4). (C) was created using data taken from ref 931.
To expand the scope of chelates used in heme-peroxo-copper complexes, the Karlin group utilized several tridentate copper chelates, the justification and interest in this being that (i) the HCO binuclear active site contains a tridentate copper ion (CuB) coordination and (ii) the redox properties and O2-chemistry vastly differs for tridentate versus tetradentate copper(I) chelates, as detailed above in section 3.3. In one such example, oxygenation of a solution containing [(AN)-CuI](BArF) and (F8)FeIII results high-spin heme peroxo copper adduct, [(F8)FeIII-(O22−)-CuII(AN)]+ (Figure 94B).926 The previously established UV-vis, 2H NMR, and rR spectroscopic properties of [(F8)FeIII-(O22−)-CuII(TMPA)]+ (Figure 94A) provided precedence for the classification of [(F8)FeIII-(O22−)- CuII(AN)]+ (Figure 94B) as a high spin heme-peroxo-copper adduct, which is supported by its paramagnetic pyrrole 2H chemical shift centered at 96 ppm (-80 °C). The rR spectrum for [(F8)FeIII-(O22−)-CuII(AN)]+ also displays an isotope sensitive peroxide O–O stretching vibration at 756 (∆18O2 = −48) cm−1.516 This O–O bond stretch is similar to that found for an analogous dicopper complex with μ-η2:η2- peroxo binding, [{(MeAN)CuII}2-(O22−)]2+, at 721 (18O2 −3 38) cm−1.1206 The formation of these copper-only complexes in the heterobimetallic reactions is ruled out by the presence of a single Soret band (indicating a single chemically equivalent heme Fe environment) and the pyrrole chemical shift found for [(F8)FeIII-(O22−)-CuII(AN)]+.516
Invested readers will note that there is a substantial difference between the value of the rR O–O bond stretching frequencies observed for heme-O22−-copper adducts having tetradentate [v(O–O) = 788–808cm−1 as opposed to tridentate [v(O–O) = 747–767 cm−1] copper coordination (Table 3). It is well-established in dicopper oxygen chemistry that the nature of the copper ligand and minor changes in chelate denticity can greatly affect the properties of the resultant copper-oxygen adduct (see section 3); i.e., tetradentate copper chelates induce formation of end-on μ-1,2-peroxo dicopper(II) structures having high (≥800 cm−1) O–O bond stretching frequencies, whereas tridentate ligands generate side-on μ-η2:η2-peroxo dicopper(II) species with lower O–O bond-stretching frequencies (<760 cm−1) (Figure 96).18,680,681,753 The distinctly different v(O–O) stretch values have been associated with back-bonding interactions from the copper d orbitals into the antibonding σ* orbital of the bridging peroxide moiety, resulting in a measurably weaker O–O bond,761 as also detailed in section 3.2.
Figure 96.

Dicopper dioxygen binding modes comparing specific structural and physical properties. Adapted from ref 928. Copyright 2010 American Chemical Society.
In order to try to understand the origin of these differences among heme-peroxo-copper complexes featuring tri- versus tetradentate copper chelates, detailed spectroscopic analysis have been carried out along with complementary DFT calculations (Figure 97).910,928 As mentioned earlier, the crystal structure and physical properties of [(TMP)FeIII-(O22−)-(5MeTPA)CuII]+ (Figure 95, C) obtained by Naruta and co-workers were important precursors to this work given the structural and spectroscopic similarities to HS-TMPA. When comparing the Fe-Cu distances for HS-TMPA (Figure 97, A) and HS-AN (Figure 97, B), it is noteworthy that HS-TMPA has a ~ 0.4 Å greater Fe…Cu distance than HS-AN, which can be rationalized by the different binding modes. Additionally, similar Fe pre-edge XAS intensities for both HS-TMPA and HS-AN suggest that the binding to Fe is similar in both complexes; that is, the Fe is pulled out of the heme plane due to peroxide binding. Further rR spectroscopic studies comparing HS-TMPA and HS-AN, showed that the spin state marker bands for both complexes are similar, ~1362 cm−1, and distinct from an authentically generated end-on O2-bound heme (1370 cm–1), an observation that is consistent with the assignment of the peroxide as a (side-on bound) bidentate ligand to Fe in both cases. The optimized structures of both HS-TMPA and HS-AN calculated using density functional theory (DFT) are in good agreement with the EXAFS data discussed above. An updated DFT model of HS-TMPA essentially reproduced previously reported core properties and provided additional data on the ruffling of the porphyrin macrocycles. The optimized DFT structure of HS-AN shows that the peroxide binding mode is η2 to the copper ion, whereas that of HS-TMPA is instead η1 (Figure 97).
Figure 97.

High-spin heme iron(lIl)-peroxo-copper(II) complexes, utilizing either the (A) tetradentate ligand TMPA or the (B) tridentate chelate AN. Structural details are derived from DFT calculations. Adapted from ref 928. Copyright 2010 American Chemical Society.
Electronic structure calculations on HS-TMPA and HS-AN provide a rationalization of the distinct rR spectral results while accounting for their identical spin states which is facilitated by antiferromagnetic coupling. In both cases, the d9 CuII, and high spin d5 FeIII ions are strongly antiferromagnetically coupled through the peroxide bridge, via the σ-bonding framework (Figure 97, right). However, the nature of the copper ligand (tridentate vs tetradentate) results in different singly unoccupied net bonding orbitals on the copper ion. In the case of HS-TMPA, the TMPA ligand favors a five-coordinate, trigonal bipyramidal geometry for Cu, with the singly occupied orbital on copper having 3dz2 character, which favors end-on binding by the peroxide to copper. In contrast, for HS-AN, the more flexible AN ligand allows for a square pyramidal binding geometry, with a bidentate peroxide ligand, where the singly unoccupied orbital on Cu is of 3dx2-y2 character. The antiferromagnetic coupling in both cases is mediated though the πσ* orbital of the peroxide. The iron porphyrin fragments in both HS-TMPA and HS-AN were found to be essentially identical with σ interactions occurring between the Fe 3dxz orbital and the peroxide πσ* and δ bonding interactions occurring between the Fe 3dxy and the peroxo πv* orbital. These Fe-peroxide bonding interactions are effectively the same in both HS-TMPA and HS-AN, and therefore, these σ-bonding interactions do not account for the ~50 cm−1 shift in the O–O bond stretching frequency observed between HS-TMPA (v(O–O) = 804 cm−1) and HS-AN (v(O–O) = 747 cm−1). Analysis of the occupied valence orbitals of HS-AN revealed that they embody much greater σ* character than those of HS-TMPA, indicating that in HS-AN, the peroxo σ* orbital acts as a π-acceptor from the 3d manifold of the CuII. Due to the highly antibonding nature of the peroxo σ* orbital, its contribution to any bonding orbitals would greatly impact the observed O–O stretch.928
For these high-spin complexes, the iron spin state of the peroxo species can be changed upon the addition of an exogenous axially ligating (to heme) base moiety [1,5-dicyclohexylimidazole, (DCHIm)] to form the corresponding low-spin heme–peroxo–copper species, [(DCHIm)(F8)FeIII-(O22−)-CuII(TMPA)]+, and [(DCHIm)(F8)FeIII-(O22–)-CuII(AN)]+ shown in Figure 98, labeled LS-TMPA and LS-AN, respectively.497,932 In order to better understand the structures and bonding and confirm the peroxidic nature of LS-TMPA and LS-AN, rR spectroscopy, XAS (for LS-AN) and DFT studies were used to establish optimized structures. A higher peroxidic stretching frequency was observed for LS-AN (Table 3), consistent with an end-on, end-on (i.e., μ-η:η1-peroxo) coordination, resulting in decreased back-donation from the CuII d-orbitals to the peroxo σ*, and hence a stronger O–O bond.910 At the same time, the heme Fem recedes into the plane of the porphyrin, decreasing back bonding contribution from the FeIII d orbitals. This coordination mode was also reproduced from EXAFS spectroscopy, which is consistent with the expected optimal geometry produced by DFT calculations. The fact that this low-spin peroxo is end-on to both the CuII and FeIII is significant because this was the first confirmed example of this type of binding mode for heme–copper models. Electronic structure calculations provided additional insight into the HS to LS conversion of the FeIII center. Upon the addition of the axially ligating base 1,5-dicyclohexylimidazole to HS-AN conversion to LS-AN occurs causing the FeIII to recede into the plane of the porphyrin. The strong ligand field contributed by the porphyrinate thus helps account for the HS to LS conversion. The antiferromagneti- cally coupled ground state of the LS adduct is also favored by its acute Fe–O–O–Cu dihedral angle.910 This change from high-spin to low-spin may have implications for O–O bond cleavage in CcO, as a precursor to further proton coupled electron transfer, producing the FeIV=O and Cu–OH/OH2-containing PM species (Scheme 23; also see sections 5.2.6 and 5.2.7).
Figure 98.

Formation of (top) end-on μ-1,2 low-spin heme–peroxo–copper adducts [(DCHIm)(F8)FeIII-(O22–)-CuII(TMPA)]+ (LS-TMPA) and (bottom) [(DCHIm)(F8)FeIII-(O22–)-Cuii(An)]+ (LS-AN) from their high spin precursors. Adapted from ref 497. Copyright 2015 American Chemical Society. Adapted from ref 910. Copyright 2011 American Chemical Society.
With the precedents set concerning structure and spectroscopies for heme–peroxo–copper species, generated primarily by the Naruta and Karlin groups, the binding motif of the bridging peroxo species can be inferred based on its O–O bond stretching frequency and the denticity of the copper ligand structure. Three general modes have been observed in these systems, as schematically depicted in Figure 99; O–O bond stretching frequencies for these and other heme–peroxo–copper adducts are given in Table 3.
Figure 99.

Distinct heme-peroxo-copper binding motifs characterized by Karlin928 and Naruta.931 See text for details.
The first example of a heme–peroxo–copper assembly was reported by Collman and co-workers in 1994933 based on the 1,4,7-triazacyclononane-capped picket fence porphyrinate (TACNAcr). This ligand was metalated to yield a cytochrome c oxidase model complex with FeII and CuI ions in close proximity. The precursor iron(II)–copper(I) complex reacts irreversibly with dioxygen in the presence of excess 1,5-dicyclohexylimidazole (DCHIm) to yield a proposed peroxide adduct, [(DCHIm)(TACNAcr)FeIII(O22– )-CuII]+ (Figure 100A). The Fe only version of this 1,4,7-triazacyclononane capped porphryinate reacted reversibly with O2, and at a much slower rate, implicating the copper ion as essential to rapid dioxygen reactivity in this system. Further evidence for the peroxidic nature of the oxygenated form, that depicted in Figure 100, was provided by its rR spectra where an isotope sensitive peak was observed at 758 cm−1 (∆18O2 = −18) with 16O2, said to be consistent with the presence of a peroxide species, most likely bridged between the Fe and Cu ions. Although the coordination environment of the Fe center was concluded to be six-coordinate in this case (with an axially ligating DCHIm ligand), the low-energy O–O rR stretch could be indicative of a μ-η2:η2 bridging peroxo moiety and thus bearing a high-spin FeIII center and a tridentate copper chelate, similar to systems reported by Karlin (Figure 96). The dioxygen adduct was also reported to react with four equiv of cobaltocene to reduce one mol of dioxygen, per mol of catalyst, and was capable of performing this chemistry for multiple cycles.
Figure 100.

First example of a heme-peroxo–copper adduct, reported by Collman and co-workers.933 [(DCHIm)(TACNAcr)-FeIII(O22–)-CuII]+. L = solvent or 1,5-dicyclohexylimidazole.
The first example of a heme-peroxo-copper adduct from the Karlin group was reported in 1999 (Figure 101A).936 This work employed the heme–copper binucleating ligand 6L, consisting of a fluorinated-tetraarylporphyrinate with a TMPA copper chelate appended via an ether linkage at the 6' position of one of its pyridyl arms. The fully reduced complex [(6L)FeIICuI]+ reacted with O2 in acetonitrile and acetone to give a metastable peroxo species [(6L)FeIII(O22–)CuII]+ (A). An analogous model system was also developed where the cuprous chelate was attached to the parent heme at the 5′ position instead, [(5L)FeIICuI]+. The rR spectra of these peroxo adducts exhibited O–O bond stretching vibrations centered at 787 (∆18O2 = −43) and 809 (∆18O2 = −53) cm−1 for (A) and (B), respectively. (Figure 101). The EPR and NMR signatures of [(6L)FeIII-(O22–)CuII]+ (A), indicated an overall S = 2 complex, resulting from the antiferromagnetic coupling between the high spin iron(III) (S = 5/2) and the copper(II) (S = 1/2) spin manifolds through the peroxidic bridge. MALDI-TOF-MS of the oxygenated form of (A) gave evidence for a dioxygen adduct (m/z 1191,16O2), which shifted by four mass units with 18O2 to m/z 1195.936,939 EXAFS spectroscopic interrogation of (A) led to some structural insights, Fe–O (1.83 Å), Cu–O (1.882 Å), and Fe…Cu [3.35(2) Å]. Stopped flow kinetic studies on the oxygenation reaction of (A) shows that it forms a transient intermediate within the first 1 ms of oxygenation, which could be either an [(6L)FeIII–(O2•−)–CuI]+ (superoxide) or a bis-superoxide adduct [(6L)FeIII(O2•−)-CuII(O2•−)]+, either of which would subsequently transform to the bridging peroxo species, [(6L)FeIII–(O22–)– CuII]+ (A). While the formulated structure of this complex likely has an end-on μ-η1:η1-peroxo bridging ligation,939 the low frequency O–O stretching vibration (787 cm−1) and close similarity of v(O–O) and binucleating ligand architecture to those of Naruta's group (Figure 95) would indicate an η2:η1-peroxo structure like that of [[(TMP)FeIII-(O22–)-(5MeTPA)CuII]+ (Figure 95A) is a better assignment. An explanation for why the v(O–O) for [(6L)FeIII– (O22– )– CuII]+ (A) is 20 cm−1 lower than for the closely related complex [(F8)FeIII(O22–)-CuII(TMPA)]+ (Figure 94A) is not clear but is interesting. One difference in reactivity is apparent; [(F8)FeIII(O22– )-CuII(TMPA)]+ is unreactive toward exposure to excess CO(g) while [(6L)FeIII– (O22–)–Cun]+ rapidly loses O2 and gives a bis-carbonyl complex [(6L)FeII–CO··· CuI–CO]+, suggesting ligand strain in [(6L)FeIII– (O22–)–CuII]+ due to its tethered chelate for Cu leads to an overall less stable peroxo complex.516 Essentially, this is a process involving the reverse of O2-binding to give back the fully reduced complex [(6L)FeIIμCuI]+, which subsequently binds CO, indicating that as for proteins, CO binds very or more strongly than O2 to reduced hemes (and to CuI-species).940 For the 5L ligand platform, a dioxygen adduct forms, [(5L)FeIII(O22−)-CuII]+ (Figure 101, B), as evidenced by the peroxo stretching frequency observed (Table 3); however, the reaction stoichiometry was found to be 2:1 (B: O2) not 1:1 as expected, and 1H NMR spectroscopy indicated that the product is diamagnetic. An understanding of the chemistry occurring here is lacking.
Figure 101.

Heme-peroxo-copper adducts characterized by the Karlin group936,939 featuring tetradentate binucleating ligand frameworks: [(6L)FeIII-(O22–)CuII]+ (A) and [(5L)FeIII-(O22–)CuII]+ (B). See text for details.
The Karlin group has also studied several binuclear systems with tridentate copper chelates, including those depicted in Figure 102, A–C. The most extensively studied of these was [(2L)FeIICuI]+, which employed the ligand 2L, featuring a bis(2-(2-pyridyl)ethyl) amine copper chelate attached to the parent F8 heme via an amide linkage. The dioxygen reactivity of metalated [(2L)FeIICuI]+ was monitored at low-temperature (CH2Cl2/EtCN) by UV–vis spectroscopy and observed to form a side-on (μ-η2:η2) heme–peroxo–copper adduct, [(2L)FeIII– (O22–)– CuII]+ (Figure 102, A), confirmed by 1H and 2H NMR and EPR spectroscopic studies. Resonance Raman spectroscopic studies corroborated a high-spin ferric heme and a μ-η2:η2 binding mode for the bridging peroxide moiety, which has an O–O stretching frequency of 747 (∆18O2 = −49) cm−1.938
Figure 102.

Heme-peroxo-copper and heme–superoxo adducts characterized by the Karlin group featuring heme plus tridentate binuclear ligand frameworks: (A) [(2L)FeIII–(O22–)–CuII]+,938 (B) [(3L)FeIII(O2•–)], and (C) [(4L)FeIII–(O22–)–CuII]+.522 See text for details.
Stopped flow kinetics were utilized to monitor the O2 binding chemistry of [(2L)FeIICuI]+ in a mixture of CH2Cl2/EtCN at low temperature. From this work, a transiently stable, six-coordinate, ferric superoxide species, [(2L)FeIII–(O2•–)–CuI]+ could be identified and assigned based on its first-order decay to yield [(2L)FeIII–(O22–)–CuII]+ (Figure 102, A). Given the tendency of tridentate pyridyl cuprous complexes to strongly bind to nitrile solvate molecules, the formation of a bis-superoxide adduct, [(2L)FeIII-(O2•–)-CuII(O2•–)]+, is unlikely. The FeII/CuI complex of 2L ([(2L)FeIICuI]+) also reacts with carbon monoxide to form a bis-CO adduct in MeCN, [(2L)FeII(CO)-CuI(CO)]+, based on the solution IR spectroscopic bands at 1980 and 2089 cm−1 for FeII–CO and CuI–CO moieties,509 respectively. The CO stretch of the Fe-bound CO molecule shifts to 1968 cm−1 when THF is used as a solvent, suggesting some axial ligation by the solvent and a resultant transeffect on the Fe-bound CO.519,941 This chemistry is valuable in that it provides a small molecule model which can, in future studies, be interrogated using flash photolysis studies to evaluate the effect of CO binding on the O2 reactivity of tridentate monotethered heme–copper models and, by extension, similar situations within the cytochrome c oxidase active site.
Another pair of tridentate binuclear complexes prepared by the Karlin group feature a similar cuprous chelate with the addition of a tethered axial pyridine moiety on the distal side of the porphyrin. The cuprous chelate, and axial pyridine linkers for 3L, and 4L are attached to the parent heme via methylene and ethylene linkers, respectively (Figure 102, B and C). The different linker lengths were intended to evaluate the subtle effect of ligand environment changes on dioxygen reactivity. The Fe-only versions of ([(3L)FeIII]) (B) and ([(4L)FeII]) (C) both bind dioxygen to give the corresponding FeIIIO2•– species, although only [(3L)FeIII(O2•–)]+ (B) was shown to form reversibly (Figure 102, B; also see section 2.2). The inclusion of both FeII and CuI within the binucleating site of 3L and 4L yielded the corresponding reduced binuclear species, [(3L)FeIICuI]+ and [(4L)FeIICuI]+. Upon reaction with dioxygen, both species form dioxygen adducts, with spectroscopic properties similar to those discussed above (see Figures 94 and 101). However, only the species [(4L)FeIII(O22–)– CuII]+ (Figure 102, C) was explicitly characterized as a heme- peroxo-copper adduct. This assignment was predominantly based on the similarity in the UV-vis, EPR, and 1H NMR spectrum of (C) to other previously characterized examples from Collman933 and Karlin.938
In addition to the previously discussed high spin heme– peroxo–copper adduct generated from (F8)Fe° and [CuI(AN)](BArF),516 another bipodal cuprous chelate was employed to evaluate the effect of pyridyl ligands in a tridentate copper chelate.513 By using a cuprous chelate with a dimethylaminopyridine derivatized ligand, LMe2N, a new heme-peroxo–copper adduct was synthesized and characterized (Figure 103). The dioxygen reactivity of a 1:1 mixture of (F8)FeII and [CuI(LMe2N)](BArF) was evaluated in CH2Cl2/6% EtCN at –80 °C. Similar to other heme copper systems, the formation of [(F8)FeIII(O22–)-CuII(LMe2N)]+ (Figure 103) is preceded by the formation of an FeIII–O2•– species, which subsequently reacts with [CuI(LMe2N)]+. Dioxygen reactivity with [CuI(LMe2N)]+ is impeded by the nitrile solvent, precluding the formation of copper–oxygen species, in favor of binding to iron(II). The UV–vis, 2H NMR, EPR, and rR spectroscopic properties of [(F8)FeIII(O22–)-CuII(LMe2N)] + are consistent with those of other μ-η2:η2 heme-peroxo copper adducts (Table 3).513
Figure 103.

Heme-peroxo-copper complex [(F8)FeIII-(O22–)-CuII(LMe2N)]+ featuring a tridentate copper chelate with electron-donating N,N-dimethylamino substituents.513.
5.1.2. Heme-Peroxo-Copper Adducts Containing an Appended Phenol Moiety at the Copper Chelate.
The active site of cytochrome c oxidase contains a cross-linked imidazole-phenol moiety that is critical to enzyme function and selectivity for the four proton, four electron reduction of dioxygen to water.942 To emulate this functionality in model systems the copper chelate, LN4OH, and its anisole analogue LN4OMe (Figure 104, A) were prepared in order to study the effect of an appended phenol on dioxygen reactivity in copper and heme-copper systems. Oxygenation of an equimolar mixture of (F8)Fen and [(LN4OH)CuI]+ leads immediately to the formation of a heme–peroxo–copper species, [(F8)FeIII (O22−)-CuII(LN4OH)]+ (Figure 104, B).937 The UV–vis, 2H NMR, EPR, and rR spectroscopic properties were consistent with a high-spin (S = 2) heme–peroxo–copper adduct with antiferromagnetically coupled CuII and FeIII metal centers via a peroxide bridging ligand. The resonance Raman spectra for [(F8)FeIII-(O22–)-CuII(LN4OH)]+ (B) show an O–O bond stretching frequency of 813 (∆18O2 – 44) cm−1 and an Fe–O bond stretch of 529 (∆18O2 = −21) cm−1. Mixed isotope labeling of the bound O2 molecule (16O18O) indicates a symmetric binding mode for the peroxide ligand, although this complex has similar spectroscopic properties to those of other μ-η2:η1 heme–peroxo–copper complexes. Thus, its coordination mode has not been precisely defined. The anisole analogue of (B), [(F8)FeIII-(O22–)-CuII(LN4OMe)]+, also displays a similar peroxide stretching frequency, at 815 (∆18O2 = −46) cm−1 and an Fe–O stretch at 528 (∆18O2 = −22) cm−1.
Figure 104.

Heme-peroxo-copper adducts characterized by the Karlin group featuring a covalently appended tyrosine mimic on the copper chelate. (A) LN4OR ligand where R = H or OMe. (B) [(Fg)FeIII(O22−)-CuII(LN4OH)]+.937.
Naruta’s investigations of heme–copper binuclear model systems also involved tridentate cuprous chelates with an appended phenol moiety. These model systems displayed different dioxygen reactivity compared to their tetradentate analogues. For system [(TMP)FeIII-(O22–)-CuII(LN3-OH)] + (Figure 105, A), oxygenation of the FeII/CuI complex in nitrile solvents yields a ferric superoxide species, with an oxidized CuII ion. However, when a solvent mixture consisting of dichloromethane and propionitrile is employed, a “roughly” equal parts mixture of the superoxide, plus a heme-peroxo-copper adduct is obtained. This solvent dependence of dioxygen reactivity is distinct from the more robust heme-peroxo–copper adduct generated in the tetradentate systems, where the formation of heme–peroxo–copper species is not solvent-dependent. When the phenol moiety is deprotonated to yield [[(TMP)FeIII-(O22–)-CuII(LN3-ONa)]+ (Figure 105, B), the proportion of the heme-peroxo-copper adduct in the product mixture increases from ~50% to ~70%, as a result of increased reactivity of the CuI toward O2 imparted by the enhanced electron-donating ability of the supporting ligand. It is noteworthy that the O–O bond stretching frequency of this heme–peroxo–copper adduct is 812 cm−1, which is substantially higher than those of other μ-η2:η2 heme–peroxo–copper adducts containing tridentate copper chelates (Table 3 and Figures 94, 100, 102, and 103). In fact, this high-energy O–O stretching frequency may imply the presence of the μ-η2:η1 peroxo binding mode, as observed in analogous complexes with tetradentate copper chelates discussed above.935
Figure 105.

Binuclear heme–copper model systems investigated by Naruta featuring tridentate copper chelates and covalently appended phenol groups. (A) [(TMP)FeIII-(O22–)-CuII(LN3OH)]+ and (B) [(TMP)FeIII-(O22–)-Cun(LN3-ONa)]+.935.
In the case of [(LOH)FeII–CuI]+ (Figure 106, A) also reported by Naruta and co-workers, the complex reacts to form a low-temperature stable heme-peroxo-copper species, which has an isotope sensitive O–O bond stretching frequency of 799 (∆18O2 = −47) cm−1 based on rR spectroscopy. The ESI-MS spectrum of [(LOH)FeIII–(O22–)–CuII]+ is also consistent with an oxygenated adduct, and the parent peak shifts by 4 mass units when 18O2 is utilized rather than 18O2. An analogue of [(LOH)FeIII(O22–)-CuI]+ with a methoxymethyl-protected phenol moiety, [(LOMOM)FeIII–CuII]+, displayed similar dioxygen reactivity to its deprotected analogue LOH, and its rR data are included in Table 3.486 Upon incorporation of an imidazolyl heme axial base ligand, the nature of the oxygenated intermediates appear to change. On oxygenation of [(TMPIm)FeIII(O22–)-Cun(LN4-OH)]+ (Figure 106, B), a bridging peroxo species forms. In this case, its 16O2 O–O stretching frequency is split due to vibrational Fermi coupling between the bound O2 molecule and an internal vibrational mode of the porphyrin, resulting in two features at 787 and 803 cm−1 in the rR spectrum; however, oxygenation with 18O2 results in a single O–O bond stretch at 751 cm−1, ruling out a second bound oxygen species. These O–O stretching bands also appear simultaneously with Fe–O stretches at 611 (∆18O2 = −27) cm−1. This peroxo intermediate, [(TMPIm)FeIII-(O22–)-CuII(LN4-OH)]+ (B), is not stable and rapidly decays to form a low-temperature stable heme–superoxo–copper(I) complex, as evidenced by an isotope sensitive superoxide Fe–O stretch at 574 (Δ18O2 = −26) cm−1. It was proposed that this peroxo-to-superoxide transformation occurs due to a “push effect” by the axial imidazole, causing the Cu–O bond to break yielding a ferric superoxo CuI species, a transformation which could be thermodynamically favored by a relatively low FeIII–(O2•–)/FeIII–(O22–) redox couple relative to that of [(LN4OH)-CuII/I]+.485
Figure 106.

Binuclear heme–peroxo–copper model systems investigated by Naruta and co-workers featuring appended phenol moieties and imidazolyl axial base: (A) [(LOH)FeIII(O22–)-CuII]+ and (B) [(TMPIm)FeIII(O22–)-CuII(LN4-OH)]+.485,486.
5.1.3. Novel Synthetic Approach for Generation of Heme-Peroxo-Cu Models.
One overarching goal of scientific enzyme modeling is to incorporate one or more specific active site attributes into a synthetic construct so as to maintain the ability to systematically modify and measure certain aspects of interest in order to determine structure–function relationships. In HCO modeling, copper ligand chelate design specifically has afforded a vast library of mono- or binuclear copper ion complexes which react with dioxygen alone or in the presence of hemes (vide supra). In these cases, incorporation of aspects such as ligand denticity, donor type or strength, and sterically influential or H-bonding features in turn impact factors such as metal ion coordination geometry and O2-binding mode, CuII/I redox potential, and overall O2-activation/reduction reactiv- ity.507,928,935 However, the (L)CuI reagent complexes employed are often extremely unstable and/or O2-sensitive, unjustifiably complicating their use. While it is certainly a challenge to balance mimicry and manipulability, we have recently devised a new strategy which both simplifies synthesis and expands the (Cu-environment) scope of heme–peroxo– copper model complexes, essentially O2-adducts of FeIII–CuI constructs.
5.1.3.1. Generation of Synthon and Proof of Concept.
This advantageous method builds upon a simple synthetic template (Figure 107) which is generated by bubbling dry O2 through a cryogenic solution of ferrous heme in 2-methyltetrahydrofuran (MeTHF) to first give an oxy-heme Fe(III)-end-on-superoxide species, which is followed by addition of one equivalent of a chelate free CuI salt [in this case, CuI(CH3CN)4(BArF)]. The synthon, HS-3MeTHF,497 has been characterized as a “high-spin” (SCu(H) = −1/2 and SFe(III) = +5/2), where with antiferromagnetic coupling through the peroxo bridging ligand, an overall S = 2, EPR-silent (perpendicular mode) complex is formed. It possesses a paramagnetically shifted 2H NMR signal (Figure 107), obtained by using a pyrrole-deuterated F8-d8 porphyrin in this heme–(O22–)–copper complex.496 The peroxide ligand is bound in a bridging, “side-on” manner (η2 to both Fe and Cu), based on rR507,516 spectroscopic data [v(Fe–O) = 555 cm−1, v(O–O) = 737 cm−1] and correlation to previously well- characterized analogous compounds;507 importantly, it contains a “naked” (solvent-ligated) copper site (Figure 107). As an initial justification for use as a synthetic foundation, the HS-3MeTHF complex was shown to be a suitable starting point from which to generate previously characterized, high-spin and low-spin complexes at low-temperature by simple addition of a Cu-ligand chelate or Cu-ligand/heme plus axial base, respectively (see Figure 108 and discussions above).927,928,932,943 This methodology is particularly useful moving forward in the area of rational ligand design because no chelate is inoperative irrespective of how unstable its (L)CuI complex might be (for example, if the (L)CuI itself would generally undergo disproportionation and/or is extremely airsensitive).
Figure 107.

Generation and spectroscopic properties of the HS-3MeTHF synthon. Adapted from ref 497. Copyright 2015 American Chemical Society.
Figure 108.

Scheme showing the utility of the synthon, HS-3MeTHF, for generating new and formerly characterized high-spin and low-spin heme-peroxo-copper complexes (left) with different spectroscopic features (right). Adapted from ref 497. Copyright 2015 American Chemical Society. Adapted from ref 516. Copyright 2007 American Chemical Society. Adapted from ref 910. Copyright 2011 American Chemical Society.
5.1.3.2. Characterization of New Complexes with Monodentate imidazole Donors.
Perhaps of greater interest is the fact that HS-3MeTHF is open to an entirely new ligand set: monodentate donors, such as (substituted) pyridines and imidazoles, as well as chelating multidentate ligands. Employing monodentate ligands at the copper site in heme-peroxo-copper complexes has the potential to impart a high degree of tunability and flexibility (vide infra). In fact, it has been shown in enzyme studies that upon binding of a (peroxo-like) CN− ligand at the active site of CcO, a nontrivial elongation of the Cu-N(His290bovine) bond (from 2.1 Å in the reduced CcO structure to 2.8 Å) occurs despite the complex protein tertiary structure constraints. In that study, the authors also propose that this movement of His290, or the analogous His ligand in bacterial CcO, may be connected to the proton pumping/gating function (proton pumping events during CcO activity have not yet been incorporated into model studies, but this is certainly of utmost interest).854,870,871 Such examples of primary coordination sphere elasticity highlight the value of ligand structure for a model compound which does not need to conform to chelation (i.e., where minor rearrangements can occur to accommodate substrates for reactivity). Of course, the monodentate ligand scope with HS-3MeTHF still retains the chemist's ability to modify systems in the same ways as in chelate design (i.e., donor type, and denticity/coordination geometry, etc.).944 In fact, cryogenic spectrophotometric titration of 1,5-dicyclohexylimidazole (DCHIm) into a solution of HS-3MeTHF affords two new, spectroscopically distinct complexes depending on temperature (Figure 108). At −125 °C, full formation of the tridentate (on copper), LS-3DCHIm, complex occurs after addition of four equiv imidazole, whereas at −390 °C, full formation of the tetradentate (on copper), LS-4DCHIm, complex occurs following addition of five equiv imidazole (the fourth and fifth DCHIm ligands, respectively, are bound as the heme axial base). Interestingly, warming the LS-3DCHIm species to −395 °C in the presence of one excess equiv DCHIm, resulted in full formation of the LS-4DCHIm species; however, cooling the LS-4DCHIm below −120 °C does not lead to the formation of LS-3DCHIm.497
Both LS-3DCHIm and LS-4DCHIm have UV-visible signatures typical of other low-spin heme-peroxo-copper complexes (Figure 108) in which the low-energy absorption features arise due to O22− to FeIII charge-transfer transitions.932 This is substantiated by the strong resonance enhancement of the Fe-O stretch when exciting at these lower energies (>800 nm) to obtain rR data. Although, these two complexes have uncharacteristically high Fe-O and O–O stretching frequencies as determined by rR spectroscopy: v(Fe-O) = 594 cm−1, v(O–O) = 876 cm−1 for LS-3DCHIm, and v(Fe-O) = 591, 585 cm−1, v(O–O) = 876, 863 cm−1 for LS-4DCHIm. (Note: the splitting of the Fe-O and O–O vibrations is due to the occurrence of a Fermi resonance.) These vibrational insights classify them as “end-on” peroxo complexes, which is corroborated by their EPR-silent nature, owing to the antiferromagnetic coupling of the S = +1/2 CuII and S = −1/2 low-spin FeIII through the peroxo bridge in each case. Use of a pyrrole-deuterated F8 porphyrin (d8) provides further support for the low-spin-iron formulations in that the 2H NMR pyrrole shift appears in the diamagnetic region of the spectrum (δ = 9.3 ppm for LS-3DCHIm and δ = 10.6 ppm for LS-4DCHIm) indicating that overall, S = 0. For a heme and/or heme-Cu complex with overall S ≥ 1/2, the pyrrole resonances are paramagnetically shifted out of the diamagnetic region.
Although the LS-3DCHIm and LS-4DCHIm complexes are remarkably similar in their spectroscopic features, density functional theory provides important structural insights on these low-temperature stable species. On the basis of DFT-optimized structures (note: cyclohexyl substituents on DCHIm ligands were truncated as isopropyl groups to lower the computational cost, yet still capture some of the inherent steric effects), the lowest energy configuration of LS-3DCHIm has a compressed metal-metal distance and a distorted square planar copper geometry (τ = 0.4, where 0 = square planar, and 1 = tetrahedral), whereas LS-4DCHIm has a more open peroxo core and a distorted square pyramidal copper geometry (τ = 0.1)945 in which the fourth DCHIm ligand occupies the axial position. These two complexes are therefore a useful set to interrogate how structural/copper-ligation differences can translate to function/reactivity (vide infra, section 6.5.2). Importantly, the ability to fine-tune the heme-peroxo-copper constructs in general, beginning from the HS-3MeTHF, shows promise for facile probing of a variety of factors which can contribute in different ways to their O2-activation and reduction chemistries. (Additionally, although no such cases yet exist, it is plausible to imagine that different monodentate ligands can be incorporated into the same complex simply using a ratio of different donors, and in those cases, perhaps the heme axial base could even be distinct from the Cu ligands.) As the synthetic diversity and undoubtedly wide-ranging chemical reactivity of such compounds is still only beginning to be explored, we look forward to gaining detailed fundamental insights into heme-Cu dioxygen reactivity with such models and thereby possibly shedding further light on aspects of actual enzyme HCO reactivity. The vastly different substrate reactivities of LS-3DCHIm and LS-4DCHIm testify to this fact (vide infra).
5.2. Reactivity of Selected Heme-Copper Complexes
5.2.1. Reactivity of Heme-Peroxo-Copper Complexes with π-acids, Brønsted Acids, and Chemical Reductants.
The chemical reactivity properties of several heme–peroxo–copper adducts synthesized by the Karlin group can be compared based on the copper chelate denticity and covalent attachment of the copper ligand. The generation and spectroscopic properties of the species shown below in Figure 109 have been described earlier and display a systematic variation along these lines.
Figure 109.

Heme-peroxo-copper complexes from the Karlin group utilized to study CO binding and reactivity toward triphenylphos- phine, electron, and proton sources. See text for details.
Carbon monoxide has played a very important role in enzyme studies of cytochrome c oxidase. Its tendency to bind to ferrous hemes has been exploited in laser photoejection studies to track CO and O2 binding to CuB.946–948 Additionally, carbon monoxide is known to give CO2 when reacted with reductively activated O2.949 For this reason, investigation of the nature of interactions of CO with peroxidic intermediates of model systems has been of great interest, in order to determine if CO can displace peroxide as O2 or undergo activation/reaction chemistry. Concomitant with the CO reactivity with heme-peroxo-copper adducts, A and C (Figure 109), the peroxide bridge is liberated as molecular O2, resulting in the ferrous and cuprous carbonyl complexes as shown in Figure 110. It is interesting how in these examples the thermodynamic driving force for the formation of carbonyl complexes possessing the reduced metal centers mediate the back electron transfer from the peroxidic moiety to FeIII and CuII generating the observed products.941 Such divergent reactivity patterns of tridentate versus tetradentate copper chelate-containing heme-peroxo-copper adducts were also observed with triphenylphosphine (PPh3); another π-acid also known to bind strongly to low valent metal ions such as Fen and CuI.759,950 In the case of both of the tridentate chelates (Figure 109, panels C and D), the triphenylphosphine displaces oxygen from the copper, but not the iron, yielding CuI-PPh3 adducts along with the corresponding ferric superoxide species (Figure 110). In contrast, neither of the systems utilizing a tetradentate chelate reacted with triphenylphosphine.
Figure 110.

Schematic overview of the distinct reactivity properties of (A-D) heme-peroxo-copper species bearing tetradentate or tridentate copper chelates. See text for discussion. Adapted from ref 928. Copyright 2010 American Chemical Society.
The interaction of these heme-peroxo-copper adducts with protons and electrons is also of utmost interest, in light of the enzymatic considerations involving protons/electrons in the reduction of dioxygen to water (see section 4). All four of the heme-peroxo–copper adducts in Figure 109 were observed to react with HCl, to yield the corresponding FeIIICl and/or CuII-X (X = Cl, OH, and OH2) species, upon protonation of the peroxide bridge to produce H2O2 as confirmed spectrophotometrically (Figure 110).928 Further, species A–C (Figure 109) react with 2 or more equivalents of cobaltocene to produce the μ-oxo derivative of the heme- copper complex, and in the case of (C), a μ-hydroxide species was detected. Intriguingly, these μ-oxo species were stable even in the presence of excess CoCp2, rather than further reduction/protonation, as previously reported by Collman. The origin of this difference may be a result of the different spin states of the heme in Collman's versus Karlin's systems (i.e., the axial base in Collman's systems results in a low-spin Fem center favoring its reduction to FeII, while in Karlin's systems, it is high-spin).
5.2.2. Hydrogen Peroxide Activation in Heme-Cu Species.
The Casella group has explored the cooperative effects of a copper ion in enhancing H2O2 activation by a natural protoporphyrin IX derivative with an imidazole axial base attached using a flexible Gly-L-His-OMe linker, denoted HMGH (Figure 111). Further covalent attachment of a bidentate poly(benzimidazolate) copper chelate at the second heme-propionate side chain residue yielded the small molecule CcO model (A) (Figure 111).951 The oxidized FeIII/CuII form of this synthetic construct displayed a greater azide binding constant than its iron-only analogue. Azide binding resulted in a change in the iron spin state, as observed from EPR spectroscopy. Namely, a high and low spin mixture of starting iron species fully transformed to low-spin, and the CuII signal shifted and decreased in intensity upon azide binding. This provided evidence that the azide is bridging between the FeIII and CuII metal centers, allowing magnetic communication between the paramagnetic metal centers. The authors suggested that the FeIII and CuII centers are spatially oriented to form a binuclear pocket analogous to that of the binuclear active site of cytochrome c oxidase. Addition of H2O2 to the FeIII/CuII and heme iron-only complexes (with the latter containing no second metal in the binuclear site) of (A) at low temperature indicated the formation of a peroxide adduct in both cases. Kinetic studies revealed that in acidic media, the copper ion enhances the peroxidase activity of (A), resulting in the phenol coupling reaction of para-cresol. In basic media, this reactivity was impeded due to the formation of a stable FeIII/Cun bridging hydroxide species. The authors further demonstrated the utility of this complex as a CcO model by oxygenation of its CO-bound FeII/CuI form at low temperature, which yielded a peroxide adduct. This model demonstrated the cooperative effect of copper for not only O2 binding but also H2O2 binding and activation by natural hemes.951 It is yet to be investigated if in this case the copper ion plays a similar role to that of a distal His residue at the peroxidase active site, which assists in proton translocation in intermediates such as (C) leading to heterolytic O–O bond scission.
Figure 111.

Examples relevant to heme-copper-mediated hydrogen peroxide activation from Casella and co-workers.951,952
In another study, Casella and co-workers utilized a free tridentate amino-bis(benzimidazole) (BBH) metal chelate with the same protoporphyrin IX derivative to evaluate the role of other metals on the peroxidase reactivity of this natural heme derivative HMGH (Figure 111B).952 Iron and copper were utilized in the nonheme site as NOR and CcO biomimics, respectively. Non-HCO relevant metals Co, Mn, and Zn were used to compare the cooperative role those metal ions could play in promoting the heme oxidative reactivity. Using pH spectrophotometric titrations the authors were able to show that ancillary metal ions lower the pKa of water bound to the natural heme, with the largest effects coming from FeIII, CuII, and ZnII due to their greater Lewis acidity. Interaction of the heme and nonheme metal centers was said to be facilitated by a hydroxo bridge (Figure 111B). Kinetic studies on the oxidative reactivity of HMGH in the presence of nonheme metal ions showed that redox active metal centers (Fem, Cun, and CoII) serve to enhance the catalytic reactivity of the heme by favoring H2O2 binding to the heme Fem (via the formation of adducts formulated as C in Figure 111), and its heterolytic cleavage to form (HMGHμ+)Ferv=O/[M(BBH)]"+ as the active oxidant. This work demonstrated the utility of ancillary nonheme metal complexes to stabilize catalytically active heme-iron-oxygen species across different oxidation states of iron, with the greatest effects observed for redox active metals, Fem, Cun, and Con.952
5.2.3. Heme-Copper Carbonyl Complexes and Relevant CuI–O2 Kinetic Studies.
5.2.3.1. Structural Properties and Photodissociation Chemistry of Heme-Copper Carbonyl Complexes.
Photoinduced CO dissociation studies have been widely utilized in detailed investigations of O2 binding kinetics, as well as in investigating competitive binding of CO/O2 of heme-containing globin proteins,953–956 and this approach has been extended to CcO, revealing some intriguing observations.900,957–960 These studies have demonstrated that in the presence of CO and O2 at the reduced CcO active site, the FeII-CO adduct readily forms, which upon photoejection, results in the rapid transfer of the CO ligand to the nearby CuB center (~4.5 Å away).961–963 Two distinct decay pathways have been identified for this CuB-CO adduct involving (1) the back-transfer of the CO ligand to the heme a3 (FeII) center, or (2) the departure of CO, abandoning the fully reduced active site, allowing O2 binding (Figure 112). These observations, along with previous work suggesting the formation of a CuI-O2 adduct (i.e., a CuII-superoxo species) in the initial binding of dioxygen at the CcO active site, suggest that the CuB center serves as a “doorway” for small molecules entering and exiting the active site. The CuII–O2•− intermediate in that case is further reduced by the FeII center, generating a heme– peroxo–copper adduct (see sections 4 and 6 for further discussion on such species).
Figure 112.

Reversible photodissociation of the heme-carbonyl complex of the CcO active site that involves an unstable CuI – CO adduct. The latter may also produce the naked reduced enzyme upon the departure of the CO. Note, the Fea3⋯CuB separation in PDB ID: 1OCO is 5.18 Å.
In light of this biological precedence, the Fe-only, and the Fe–Cu containing forms of 5/6L model systems were subjected to CO reactivity, and the resultant adducts were characterized by UV–vis, IR, and 1H/13C NMR spectroscopies in coordinating and noncoordinating solvents.941 The overall spectroscopic properties of [(5/6L)FeII-(CO)] and [(5/6L)-FeII–CO---CuI(CO)]+ (Figure 113) complexes were similar to those of their parent compounds, [(F8TPP)FeII(CO)] and [(TMPA)CuI(CO)]+.415,964–967 13C-NMR experiments suggested the presence of two distinct isomeric forms of the Fe- only carbonyl complex of 5/6L platforms, where the CO could either be at the same or the opposite side of the tethered copper chelate (Figure 113). Furthermore, infrared spectroscopic insights revealed the presence of two isomeric forms of [(5/6L)FeII–CO⋯CuI(CO)]+ complexes as well, where the CuI–CO unit is supported by either a tri- (major isomer; IR v(CO)Cu 2070 cm−1) or tetradentate (minor isomer IR v(co)Cu 2091–2093 cm−1) fashion, with the presence of a dangling, dissociated pyridine in the case of the former (Figure 113). IR studies also exhibited that the Cu-bound CO ligands could be readily displaced upon Ar purging, yielding the [(5/6L)FeII-CO⋯CuI]+ species.
Figure 113.

Heme-Cu carbonyl complex assemblies utilizing 5L and 6L binucleating ligands.941.
The photoinduced CO dissociation chemistry of the monocarbonylated compound [(6L)FeII–CO⋯CuI]+ was investigated in a subsequent study using time-resolved IR spectroscopic methods.968 In this work, the room-temperature dissociation of FeII-bound CO (in MeCN) was accompanied by the immediate bleaching of its IR signature centered at 1975 cm−1, with the concomitant formation of a new IR feature at 2091 cm−1. This indicated that upon photodissociation, the FeII-bound CO migrates to the CuI center giving [(6L)FeII⋯CuI–CO]+, at a rate of 1.59 X 105 s−1 (Figure 114). This slower rate of CO transfer to the CuI center compared to the enzyme was attributed to comparative solvent binding at the CuI center, and the flexibility of the tethered copper chelate of the 6L ligand, which may locate the CuI center away from the heme, resulting in inefficient CO capture (Figure 113). Upon thermal equilibration, the CO was observed to back-transfer to the FeII center at an observed rate of 1600 s−1, giving back the starting monocarbonylated complex, [(6L)FeII–CO⋯CuI]+. This Cu-to-Fe CO-transfer rate is within the range observed for CO transfer from CuB to heme in the HCO enzymatic examples and indicates that the ferrous heme–CO adduct is the thermodynamic product.968 Interestingly, the enthalpic contribution to the activation energy for this last CO transfer step was found to be remarkably similar to that of the parent copper complex, [(TMPA)CuI(CO)]+. This reveals the substantial influence of CuI–CO dissociation energetics on small molecule interactions with the heme center of both model systems and enzymatic examples. When CO is photoejected from the iron in the bis-carbonyl complex, [(6L)FeII–CO⋯CuI-CO]+, the rebinding to the heme, is much more rapid (kco = 1.0 × 107 s−1), due to the unavailability of the copper binding site. This rate constant is comparable to other reported values for CO recombination to ferrous hemes, in the absence of other metals.947
Figure 114.

Photoexcitation studies carried out on the heme–Cu 6L (tethered) ligand platform.968.
Photoinduced CO dissociation of the six-coordinate heme FeII–CO adduct, [(DCHIm)(F8)FeII(CO)], was also studied in the presence and absence of exogenously added CuI complexes with either tri- or tetradentate supporting ligands (Figure 115).969 Similar to the above-mentioned, the photo-ejected CO in this case was also observed to rapidly react with the CuI complex in solution, which then slowly returns the CO back to the FeII center. Consequently, the FeII-CO rebinding rate decreased from 65 to 10 s−1 in the presence of a tetradentate CuI chelate and to 7 s−1 with a tridentate chelate in THF solvent. Intriguingly, in nitrile solvents, such effects of CuI complexes were absent, presumably due to the solvent hindrance in the binding of CO to CuI (vide supra). Similar observations were reported in acetone solvent for the tetradentate CuI system; however, for the case of the tridentate-CuI complex, thermal CO transfer from [(DCHIm)- (F8)FeII(CO)] to the copper center occurred (without photoexcitation), resulting in the formation of an equimolar mixture of [(F8)FeII(DCHIm)2] and [(F8)FeII(solvent)2].
Figure 115.

Photodissociation chemistry of six-coordinate heme-CO/NO complexes in the presence of CuI systems with either tetra- or tridentate supporting ligands. See text for details. Adapted from ref 969. Copyright 2009 American Chemical Society.
5.2.3.2. Copper(I)-O2 Kinetic Studies: Relevance to Binuclear Center CO- and O2-Binding.
As discussed, details concerning the ligand-copper(I)-dioxygen interaction are needed if we are to deeply understand the reductive activation of O2 by heme-copper assemblies occurring in HCOs. As is now well-known, the coordination mode, metal center geometry, electronic-structure/bonding, and the resulting reactivity of primary copper(I)-O2 adducts critically depend on the exact nature of the cuprous ion ligand environment (see section 3).18,611,612,680 In fact there is a direct connection between such chemistry and that occurring at HCO binuclear active sites, since, as will be described below, strong evidence exists that binding of O2 initially proceeds through a CuB-O2 adduct and this is related to the findings that point to CuB as regulating the trafficking (being a “gateway” or “doorway”) of incoming small molecule ligands (O2, CO, NO, and others).
As mentioned in the previous section, such conclusions were determined from utilizing time-resolved spectroscopic methods (optical UV-vis, IR, or rR), initiated by CO photodissociation (i.e., “flow-flash”)970 from the thermodynamically stable ferrous heme-CO adduct, followed by “immediate” CO movement to CuB, followed by either recombination (i.e., rebinding to Fea3) or active-site exit into solution (Figure 116). [Note: CO (and NO) binding to all heme proteins has historically and is still being studied where these diatomic gases are utilized as O2-surrogates for research purposes.] A classic study from Einarsdottir, Woodruff, and co-workers962 revealed details of this process; earlier, Alben and co-workers971 were able to cryogenically catch the CuI-CO intermediate, characterizing it by infrared spectroscopy, v(C-O) = 2062 cm−1 (whereas for the ferrous Fea3 CO-adduct, v(C-O) = 1963 cm−1). Later on it was shown that the Fea3-CO to CuB-CO conversion could occur in less than one picosecond,972 while Yoshikawa and co-workers870 were able to characterize such a CuB-CO adduct by X-ray crystallography, revealing an interesting side-on weak binding (Figure 117A) (Note: Avery similar CuB-NO with side-on nitric oxide binding was also characterized crystallographically).973 Examination of CO binding or rebinding shows that it first binds to CuB before transferring back to the heme (i.e., the reverse of the (photo)dissociation chemistry). A related X-ray structural and IR spectroscopic study on Thermus thermophilus (tT) ba3 oxidase also revealed photolysis of an iron-bound CO, leading to CO transfer/binding to CuB [v(C–O) = ~ 2050 cm−1 for the CuI–CO moiety].974
Figure 116.

Scheme depicting the dioxygen binding to bovine CcO for most kinetic-spectroscopic studies where a heme a3-CO adduct is utilized for experiments. Laser flash photolysis results in picosecond transfer to give a Cubi-CO species, whereupon CO either rebinds to the heme a3 or it escapes into solution. When the latter occurs, in the presence of dissolved O2(g), the fully reduced enzyme (PDB ID: 5B1B, center) can react with O2, giving a transient Cub-O2 adduct before it transfers to heme a3. In this scheme, that latter species is depicted as a peroxo-bridged Fea3III-CubII complex (PDB ID: 5B1A), many of which have been structurally characterized. It is generally not considered that this exact peroxide-bridged form (in its details) is an intermediate during enzyme turnover. Also, see the text.
Figure 117.

(A) Structure of the bovine aa3 enzyme binuclear active site showing CO bound side-on to CuB; the species forming following low-temperature photolysis of the more stable heme a3 carbonyl derivative. (B) Depiction of a portion of the CcO O2-reduction mechanism proposed by Yoshikawa and co-workers870 based in some part on enzyme derivative X-ray structural investigations. Dioxygen initially binds to the “gatekeeper” CubI ion, depicted as a CuI(O2) adduct II [but normally best described as a CuII(O2•−) species] then transfers to heme a3, whereupon the iron(III)-superoxo intermediate (III) (the “Oxy” or A intermediate) undergoes electron- and proton-transfer reduction, cleaving the O–O bond leading to intermediate V [for details on intermediates P, (PM or PH) see section 4.2].
Thus, in earlier studies, and still currently, CO-photo-dissociation was/is most often required in order to study O2- binding kinetics (Figure 116), as formation of O2-adducts is too fast to study by stopped-flow (mixing) methods. The HCO + CO(g) studies are consistent with the labeling of CuB as a “gatekeeper” for ligand binding, and in fact, early studies by flow-flash and optical monitoring led to suggestions that CuB first binds O2, before it transfers to heme a3;975,976 at that time, Malmström975 cited a synthetic model CuI/O2 kinetics study977 in support of their proposal. Some of such studies are described below, in fact demonstrating that O2-binding to ligand-copper complexes may be extremely fast, consistent with the notion that in HCO function, dioxygen initially binds to CuBI. With the active site hemea3–CO and CuB–CO X-ray crystal structures,870 and the information about the initial O2- binding, Yoshikawa proposed an O2-binding and reduction mechanism which incorporates this special CuB-O2 adduct formation prior to its transfer (or bridging) to the heme; an as yet unobservable key intermediate is the heme–FeIII– (O2•–)··· CuI species (Figure 117B, intermediate IV),20,87° while as discussed we prefer the bridged heme–FeIII–(O22–)–CuII description. In fact, most researchers acknowledge that this O2-interaction with CuB first occurs,879,942,978–980 although that fact is most often left out of primary publications and review articles (as we have also done, Scheme 23). Rousseau and co-workers,980 in studies on the tT ba3 enzyme, deduce that CuB plays a critical role in the regulation of O2-reaction kinetics with the O2-to-heme a3 transfer being rate-limiting; the role is said to be less prominent in aa3 enzymes. Kitagawa and co-workers,879 utilizing advanced time-resolved infrared spectroscopic techniques in experiments with the bovine enzyme, recently proposed that this CuB-interaction leads to a structural change in a neighboring protein α-helix, opening a “gate” to a channel letting in water (as a proton source), which when present in sufficient concentrations leads to the O2- derived fragment transferring (or bridging) to heme a3. CuB thus plays a critical “sensing” role and detects water present in a nearby protein channel and binds O2 only when the enzyme is “ready”. This conclusion is further corroborated from advanced radiation damage free enzyme, in time-resolved (i.e., nano- to microsecond) crystallographic studies (utilizing XFEL and SF-ROX methods) on CO (as O2-surrogate)-bound protein.981 It is said that when proton saturation is achieved at the key Mg2+ water cluster,842 the CuB structure is perturbed, increasing O2-affinity by moving this metal ion out of the CuI(His)3 trigonal planar environment. Thus, CuB- O2 formation induces water channel closure, leading to O2- transfer to heme a3 (likely as a peroxidic moiety bound to both Fea3 and CuB, vide infra), followed by steps involving O–O bond reductive cleavage and proton pumping.
As mentioned in section 4, HCOs derive from many sources and aerobic organisms, from mammals to bacteria. Over the last 20 years, it has been clearly documented that there are both subtle and major differences in HCOs from varying sources, in aspects of the structures of the binuclear active sites and in differences in ligand binding and ligand dynamics, conformational changes, etc. For example, v(C–O) varies for CuB-COs from varying sources and kinetics of ligand binding or transfer between metal centers vary, etc. Derivatives and X- ray structures of various HCO derivatives are available from bovine heart (aa3 HCOs)20,842,982,983 or bacterial forms (aa3 from Paracoccus denitrificans984,985 and bacterial Rhodobacter sphaeroides)986 or the rather different tT ba3 oxidse987–989 and the bo3 oxidase from Escherichia coli.840
There has always been a concern as to if studies of O2- binding kinetics and dynamics in HCOs were influenced or inhibited by the presence of the CO utilized to initiate Fea3- CO photodissociation. Einarsdottir and co-workers990,991 have carried out an extensive research program addressing this problem, first developing tools and reagents to photodissociate O2 from solution reagents (e.g., cobalt coordination complexes), so as to study O2-binding to reduced HCO active sites on a very fast time scale and in the absence of carbon monoxide. Readers are referred to both primary and recent review articles from key research groups, summarizing a wide variety of observations and results concerning HCO ligand (CO, NO, and O2) binding dynamics and the O2-reduction mechanism across a range of HCOs (and see section 4).879,963,979,991–999 Some major conclusions are (i) the widely employed method of studying O2-binding kinetics by initially photodissociating CO bound to the heme at the heme–Cu binuclear active site is found to be inhibitory in tT ba3 CcO but not in bovine aa3 CcO; in other words, the CO flow-flash method does not accurately reflect the O2 and NO binding kinetics in tT ba3 under physiological conditions, namely, in the absence of CO. (ii) tT ba3 CcO possesses a unique Y- shaped extended hydrophobic tunnel as a very low-barrier passage for O2 or other ligands entering the heme–Cu active site. Here, in tT ba3, the binding of dioxygen (and NO(g)) is “superfast”; they both occur with rate constants of ~109 M−1 s−1, 912,990 near the diffusion limit (and about 100 times greater than that for myoglobin; also see Table 4). These authors argue that O2(g) bypasses CuB and directly binds to the heme.991 (iii) This O2-binding rate is ~10 times greater than that observed for bovine aa3 CcO.1001 (iv) The CuB in ba3 has a significantly higher affinity for CO compared to the bovine enzyme, and the crystal structure of the CuB–CO complex shows that the CuB–CO bond is significantly shorter (1.88 Å)974 and therefore stronger than the corresponding bond in the bovine enzyme (2.43 Å).870 (v) The obligatory path of CO, to and from the high-spin heme in the heme–copper oxidases most likely involves the transient binding of CO to CuBI.
Table 4.
Comparison of CO and O2 Bimolecular Rates and Binding Constants for Select (Ligand)copper(I) Complexes, Hemocyanins (Hc), and Selected Hemesa
 | ||||||
|---|---|---|---|---|---|---|
| compound or protein (solvent) | KCO (M−1) | KO2 (M−1) | kCO (M−1 s−1) | kO2 (M−1 s−1) | k−CO (s−1) | k−O2 (s−1) |
| NMe2LCuI (THF) | 4.9 × 103 | 100 | 2.5 × 109 | 2.3 × 1011 | 5.0 × 105 | 2.3 × 109 |
| ImLCuI (THF) | 2.4 × 103 | 17.4 | 2.8 × 109 | 3.4 × 1010 | 1.1 × 106 | 1.9 × 109 |
| (TMPA)CuI (THF) | 1.2 × 104 | 15.4 | 1.9 × 109 | 1.3 × 109 | 1.6 × 105 | 1.3 × 108 |
| (TMPA)CuI (EtCN) | 220 | 0.38 | 5.9 × 107 | 5.8 × 107 | 2.7 × 105 | 1.5 × 108 |
| (tBuTMPA)CuI (THF)c | 1.7 | 1.6 × 108 | 9 × 107 | |||
| (TMPA)CuI (EtCN) | 15.5 | 1.2 × 107 | ||||
| (PV-TMPA)CuI (MeTHF) | 4.8 × 107 | |||||
| (TMG3tren)CuI (MeTHF) | ~ 1 | 2.7 × 107 | 1.5 × 107 | |||
| (LiPr)CuI (THF)d | 6.3 | 5.9 × 102 | 1.5 × 106 | |||
| Cyt aa3 oxidase (CuB)e | 7 × 103 | 3.5 × 108 | 5 × 104 | |||
| Cyt bo3 oxidase (cyt o3)f | 3.8 × 107 | |||||
| Tt ba3g | 1 × 109 | |||||
| myoglobin (human) | 2.6 × 107 | (0.74–117) × 104 | 7.6 × 105 | (1.4–25) × 107 | 0.022 | 22 |
| hemoglobin (human) | 4.6 × 108 | (2.9–48) × 105 | 4.6 × 106 | (2.9–22) × 107 | 0.009 | 13.1 |
| FeII-porphyrinatesh | 105 – 106 | 106 – 108 | ||||
| Limulus Hc (arthropod) | (2.7–11.2) × 103 | (2.6–5.4) × 105 | (2–4.3) × 105 | (1.3–1.9) × 106 | 38–75 | 2.4–7.5 |
| Busycon Hc (mollusk) | 220 × 103 | 1.8 × 105 | 7.7 × 105 | (1.1–2.2) × 106 | 3–4 | 6.5–11.5 |
Values correspond to 298 K, and for synthetic copper complexes, they have been extrapolated to this temperature from cryogenic measurements. See the following citations and references cited therein.691,940,977,1002,1003 Also see sections 2 and 3 in this review for further discussions regarding many of the synthetic complexes mentioned in this table.
MeTHF = 2-methyl tetrahydrofuran.
A TMPA ligand with 4-substituted (t-butyl) pyridyl donors.691
Extrapolated to 298 K from data in reference.1004
Binding to the o3 heme, E. coli bo3 ubiquinol oxidase.1005
Includes hemes such as picket fence, pocket, durene capped, chelated proto- or meso-, deutero- or tetraphenyl- porphyrin.
Fundamental information concerning Cu(I)-dioxygen interactions is required to fully understand copper-dioxygen coordination chemistry and copper-mediated oxidative catalysis, and as it became apparent that such insights may be relevant to HCO dioxygen-reactivity, our research group has carried out copper(I)-O2 kinetics studies from the time of the late 1980s. These studies involved cryogenic, stopped-flow kinetic/thermodynamic investigations on O2-binding to synthetically derived, well-characterized mono- or dicopper(I) complexes which had already been proven to form O2-adducts, formally superoxo CuII (or peroxo CuII2(O22−) complexes. In relation to the binding of dioxygen to CuBI in HCOs, our investigations with [(TMPA)CuI]+ and analogs have led to considerable insights and have provided kinetic and thermodynamic parameters of great interest. Following the discovery that O2-binding to cuprous chelates can occur on a time-scale faster than that which can be measured by low- temperature stopped-flow experiments, we collaborated to carry out transient-absorption studies, requiring laser excitation induced CO-ejection of copper(I)–carbonyl complexes in the presence of O2 (vide infra).
Figure 118 shows schematically how the fast O2 and CO binding events were experimentally determined for the parent compound [(TMPA)CuI]+ where at low temperature (203 K), the second-order rate constant for O2 binding was found to be kO2 = 2.18 × 108 M−1 s−1. With temperature-dependent studies carried out and activation parameters determined accordingly, the extrapolated room temperature (298 K) rate constant was determined to be kO2 = 2.9 × 109 M−1 s−1 (Table 4);940 these studies were carried out in tetrahydrofuran (THF) as solvent, that being a very weak if not nonexistent ligand for copper(I). When nitrile solvents such as MeCN or propionitrile are used, the rate constants are depressed by about 2 orders of magnitude (Table 4); this is explained by the fact the RCN molecules are strong ligands for cuprous ion, stabilizing its reduced state, thus limiting or precluding fast electron transfer from copper(I) to O2 to give cupric-superoxide adducts. Figure 119 reveals the broader kinetic scheme utilized to determine both CO and O2 binding properties to a range of [(ligand)- CuI]+ complexes, and Table 4 lists the main findings in comparison to data obtained from copper proteins, heme proteins, and some of their synthetic model compounds. It is evident that the binding of O2 to these types of cuprous ion copper(I) complexes often proceeds with a very low activation barrier.
Figure 118.

(Top) Scheme depicting the laser “flash-and-trap” experiment, wherein [(TMPA)CuI(CO)]+ in the presence of various mixtures of dissolved CO(g) and O2(g) in THF is laser flashed, releasing CO and forming a solvate complex, which then reacts with either CO or O2 (but at differing rates). (Bottom left): Difference spectra {[(TMPA)Cu(X)] + – [(TMPA)CuI(CO)]+} recorded after pulsed 355 nm excitation of [(TMPA)CuI(CO)] + in THF at 198 K under a 1 atm O2:CO (7:3) mixture. The spectra were recorded at various delay times: 0 to 2 μs where [(TMPA)CuI(THF)]+ (λmax, 333 nm) converts to a mixture of [(TMPA)CuI(CO)]+ and [(TMPA)CuII(O2•–)]+ (λmax = 425 nm): ■ (black spectrum), 0 μs; red •· (red spectrum), 0.5 μs; blue ♦ (blue spectrum), 2.0 μs. The inset is an absorption transient monitored at 425 nm (corresponding with [(TMPA)CuII(O2•–)]+) with a superimposed first-order fit (in red), kobs = 3.0 × 106 s−1 (Bottom right): Plot of kfast – kCO [CO] vs [O2], where the slope corresponds to the rate constant for the O2-reaction. Adapted from ref 940. Copyright 2003 American Chemical Society. Adapted from ref 1002. Copyright 2010 American Chemical Society.
Figure 119.

Schematic employed to experimentally determine reaction kinetics [i.e., the 1:1 primary interaction of O2 and CO with various [(ligand)CuI] complexes (ligand = dL)], using transient absorption spectroscopy and the “flash-and trap” approach.940,1002 Experiments are conducted in the presence of precisely determined concentrations of O2 and CO (as mixtures) within the temperature range of interest and on nanosecond and longer time scales. As depicted here, initial laser photolysis of DLCuI-CO leads to CO photoejection, giving solvated dLCuI in a solution of both CO and O2. The initial “fast” process (kfast) involves the competitive binding of both CO and O2 with DLCuI, either regenerating DLCuI-CO or forming DLCuII-O2•−; thus, kfast is a combination of two rate constants, kO2 and kco. The latter is independently determined from transient absorption experiments with DLCuI–CO with only CO(g) present (i.e., in the absence of dioxygen). A complementary determination of the key parameters comes from observations that binding of carbon monoxide is thermodynamically favored compared to dioxygen, as is well-known for heme proteins and is also the case in copper-dioxygen proteins and complexes, Kco ≫ Ko2 (where K is the binding constant). Thus, CO subsequently displaces coordinated O2 from DLCuII-O2•− to reform the initial DLCuI–CO species (kslow; millisecond time scale), according to the equation for 1/kslow shown in the graphic, as originally deduced by Antonini and Burnori1255 in studies of heme protein O2-carriers. Adapted from ref 1002. Copyright 2010 American Chemical Society.
What do these results mean with respect to copper-dioxygen reactivity, comparison to hemes, and in the context of HCO O2-reactivity? (1) These tripodal tetradentate ligand complexes like [(TMPA)CuI]+ and its many analogs such as those given in Table 4, can react with O2 at exceedingly fast rates. However, there are many more examples in the literature such as (LiPr)CuI (Table 4), and many others not included in the table, which react very slowly with dioxygen. Such reactivity greatly depends on the exact nature of the ligand, its denticity and imposed geometry, identity of donor atoms present, and the geometric and electronic structure of the ligand-copper- dioxygen adduct, with these factors contributing to an overall characteristic (ligand)CuII/I reduction potential. As classically elucidated in studies of (ligand)cobalt(II) complex reactions with O2, the rates (and equilibrium constants) for O2 adduct formation are primarily driven by electron-transfer from metal to O2, thus ligands which are better donors [i.e., and stabilizing the higher oxidized state (implying more favorable CoII-to- CoIII conversion, i.e., less favorable more negative CoIII/II reduction potentials (E°)] result in larger rate constants; thus, clear linear-free-energy-relationships exist.1006,1007 (2) [(Ligand)CuI]+ complexes bind O2 at rates greater than hemes, perhaps because iron(II)-porphyrinates that bind O2 are normally high-spin species, and a conversion to low-spin with some kinetic barrier occurs upon O2-binding (however, this supposition requires further research). But, these 1:1 Cu/O2 adducts release dioxygen also at very large rates; k-O2 values are as great or greater than those of hemes (Table 4), revealing the fact that copper(I) and copper(II) complexes are much more labile. Thus, equilibrium constants (KO2) at room temperature are much greater for O2-adducts of heme proteins and iron-porphryinate complexes compared to those of copper-dioxygen complexes (Table 4). For hemes, k-O2 values are small; the low-spin six-coordinate iron(III)-superoxide (i.e., oxy-heme) products are not highly labile and do not readily release dioxygen compared to adducts of copper. Of course, the deeper chemical principles and reasons behind the observations made here are not as simple as the statements made. Another factor that surely comes into play are the overall charge differences; we are comparing properties for the formation of higher oxidation state FeII/FeIII(O2•−) versus CuI/CuII(O2•−) species, although the porphyrinate on iron is a (2-) ligand while the ligand sets of imidazole/pyridine donors for copper are neutral. (3) With respect to fast O2-binding to copper(I) ions, Table 4 reveals that four of the complexes with listed kO2 values (when extrapolated to room temperature from low-temperature measurements) match or exceed that of the rate constant measured and published for bovine aa3 CuB, kO2 = 3.5 × 108 M−1 s−1. Clearly, coordination chemistry copper(i)-dioxygen investigations, as revealed here (and in section 3), indicate that observations and conclusions drawn concerning the O2-chemistry of CuB centers (i.e., very rapid dioxygen binding for bovine aa3 and other HCOs; see Table 4) have a clear synthetic model compound chemical basis. in other words, copper(I) complexes can very well bind to dioxygen at rates greater than found for most or all hemes, indicating the conclusion or supposition that O2 binding to reduced CuB occurs first is well-founded (but see above the suggestion from Einarsdottir and co-workers).990,991
5.2.4. Electrocatalytic Systems and Heme-Peroxo- Copper Assemblies.
The earlier developments of “picket fence” porphyrin systems by Collman, Reed, Ibers et al.,56,79,442,1008 and other models for O2-binding mimicking hemoglobin/myoglobin function were critical to their first design, synthesis, and characterization of structurally relevant heterobinuclear model systems for cytochrome c oxidase. The synthetic chemistry of the earliest heme-based heterobinuclear systems was published in the late 1970s by several groups including Reed, Momenteau, Collman, and Boitrel, and is discussed elsewhere in this review (vide supra), and over the past 30 years, Collman, Boitrel, Karlin, and others have evaluated the utility of these species as electrocatalysts for O2 reduction.1009,1010 in heme copper model systems which grew out of these studies, the Fe⋯Cu distance (a parameter which may be useful to judge biological relevance) is determined by the ligand properties and is typically ~4.5–5 Å Classic work in this field has been covered previously, and the reader is referred to several in-depth reviews of this work.3,56,924 Generally, here we separately discuss prominent examples to highlight accomplishments and the different approaches in ligand design.
Among the earliest electrocatalytic CcO models from Collman and Boitrel were two “arbor” porphyrin inspired systems (Figure 120) where 1,4,7-triazacyclononane (TACN) (Figure 120A), or N,Nˊ,N˝-tribenzyltris(aminoethyl) amine (TBTren) (Figure 120B) as a ligand-for-copper attached to a “picket fence” heme moiety, with an imidazole or pyridine axial base ligand tethered to the other (proximal) side of the porphyrinate. An earlier version of the complex (A) shown in Figure 120 lacking a tethered axial base (see Figure 123, bottom right) was characterized and reported to bind dioxygen irreversibly to generate a bridging FeIII(O22−)-CuII “peroxo” intermediate, as evidenced rR spectroscopy, which exhibited an isotope sensitive O–O stretching frequency at 758 cm−1 (this only shifted to 740 cm−1 upon 18O2 incorporation; the small magnitude of this isotope shift was explained in terms of vibronic coupling).933 This species represented the first characterized discrete heme-peroxo-copper adduct. Interestingly, O2-reduction chemistry occurs on a graphite electrode for both (A) and (B). The former exhibits a 2e− reduction yielding hydrogen peroxide, whereas the latter catalyzes the 4e− reduction of dioxygen to water. This difference was explained to come about as a result of the relative reduction potentials of the FeIII and CuII centers.925,1012,1013
Figure 120.

“Arbor” porphyrin inspired heme-copper models featuring (A) TACN and (B) TBTren from Collman and Boitrel. See text for details. Adapted with permission from ref 1011. Copyright 2011 John Wiley & Sons, Inc.
Figure 123.

Binucleating heme-copper model systems developed by the Coutsolelos group,1026–1028 to study the effect of covalently attached copper ligands and phenol moieties. See text for discussion.
Electrochemical studies of these complexes in which the iron is replaced by cobalt, highlight the influence of metal ion redox properties on reaction selectivity. Specifically, the more positive reduction potential of CuII in (A) (Fe/Cu) enables CuII to be reduced before iron in the catalytic cycle, which leads to O2 binding and 2e− reduction mediated by copper independently. In contrast, the chemistry of the Co/Cu analogue of (A) shows that CoIII is reduced first, leading to O2 binding and reduction in the binding pocket, as the distal position is blocked by an axial base. This allows subsequent PROS to be quickly reduced in an overall four electron process and thereby minimizing leakage.1014 These studies demonstrate the importance of ligand design for electrocatalytic systems and metal ion identity in modulating the redox properties of the complex overall and the selectivity of the O2- reduction chemistry via variations in binuclear site accessibility and axial base ligation properties.
Subsequent work by Boitrel and co-workers shows similar results in electrocatalytic studies with a 3-quinolinoyl “picket fence” porphyrin system (Figure 121A.i-ii). It is worth noting that these studies were carried out utilizing a graphite electrode, which provides electrons to the catalyst at a much greater rate than they are biologically obtained from cytochrome c, so the necessity of CuI as a redox active site is relaxed because catalyst turnover is not limited by internal electron transfer processes, as is the case in CcO. During electrocatalytic studies of these complexes, it is apparent that the copper ion is not essential to the catalyst's ability to reduce dioxygen to water at physiological pH.924 These systems are, in fact, more selective for the four electron reduction of dioxygen in the absence of copper. This result is proposed to be due to side reactions of copper to yield PROS such as H2O2.
Figure 121.

Several binucleating heme-copper model systems developed for use in electrocatalytic O2-reduction. See text for discussion.
With respect to ligand design, Boitrel, Weiss, and others have addressed the role of flexibility in the copper binding site, as well as the necessity of a built-in axial sixth ligand. Several binucleating porphyrinate ligands with phenanthroline, N3O, and tren-binding moieties for copper were evaluated as CcO models (Figure 121, B-E).1015,1016 To determine the proximity of the iron and copper centers in the solution structure of these models, CO binding and photolysis studies can be utilized to determine if photoexcitation results in transfer of CO between the iron and copper centers. Carbon monoxide transfer from Fe to Cu was verified by attenuated total reflection in mid-infrared spectroscopy (ATR-MIR) immediately after photolysis. Detection of CuI bound CO stretches between 2030 and 2100 cm−1 indicated Fe-to-Cu CO transfer. For (B) (Figure 121), upon photolysis, the bound CO molecule transfers from the iron to the copper atom, which indicates that the Fe⋯Cu distance is such that both metal centers could potentially interact with small diatomic ligands. This is consistent with the predicted Cu⋯Zn distance of 5.06 Å observed in the crystal structure of Zn/Cu analogue of (B).1017 With series (C.i-iii), the absence of an observed CuI-CO stretch on photolysis revealed that the copper ion is not well-positioned to interact with CO photoejected from Fe, and here a 2e− reduction of O2 to H2O2 predominates.1018 Series (D.i-iii) suffers from steric constraints, which limit the access of small molecules to the active site and, in addition, lacks an axial base and is largely insoluble in organic solvents. Nevertheless, (D.i) is competent for the electrocatalytic 4- electron reduction of dioxygen.1019
Complex (E), which is a modified version of complex (B), allows for relevant comparisons. Both FeII/CuI complexes bind dioxygen reversibly, and the binding could be monitored with 1H NMR spectroscopy to track the spin-state of the Fe/Cu complex. In these studies, a binding event is inferred based on the change from a paramagnetic (H.S. FeII, S = 2) to diamagnetic (S = 0) 1H NMR spectrum, marked by the disappearance of paramagnetically shifted resonances corresponding to the ß-pyrrolic protons in the range from 40 to 50 ppm. However, rR spectroscopic characterization of the Fe-O, O–O, and possibly Cu-O bonds have thus far remained elusive. Interestingly, by changing the auxiliary ligand in (B) to a picoline moiety in (E), the accessibility of the binding pocket is assured for O2, making (E) a better dioxygen binding ligand than (B). Complex (E) is also less efficient as an O2-reduction catalyst in terms of the overpotential required for catalysis but yields fewer PROS overall.1020 These systems provide insight into how drastically the catalytic activity of CcO model systems can change as a result of minor changes in axial ligand basicity and pendant arm flexibility.
In very recent work, H. Kitagishi et al.1021 prepared a new porphyrin/cyclodextrin supramolecular complex that serves as a CcO model in aqueous media. The design of this model system was facilitated by the propensity of per-o-methylated β- cyclodextrins to form stable 1:2 complexes with porphyrin molecules such as 5,10,15,20-tetrakis(4-sulfonatophenyl)-porphyrinato iron(III) [(TPPS)FeIII]+.1021,1022 With this in mind, two per-o-methylated β-cyclodextrin molecules were connected via a copper(II) terpyridyl complex linker in order to mimic the distal tris-histidine ligated CuB site of CcO (Figure 122). This new ligand complex framework denoted [(TerpyCD2)CuII]2+ was then titrated with [(TPPS)FeIII]+ to yield a new 1:1 supramolecular complex [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+, which features a heterobinuclear Fe/Cu active site in the hydrophobic pocket of the β-cyclodextrin dimer. Characterization by ESI-MS and optical absorption spectroscopy revealed that [(TPPS)FeIII]+/[(TerpyCD2)- CuII]2+ could form a μ-oxo (FeIII-O–CuII) core structure with red-shifted Soret band (λmax = 453 nm). These types of bridging FeIII–O–CuII structures are described below in detail in section 5.3. The pKa of this new μ-oxo species was assessed with pH titration and found to be 8.8 (Figure 122), similar to previously reported values for μ-oxo and μ-hydroxo-bridged heme copper oxidase model compounds (section 5.3).1023 Reduction of [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+ with sodium dithionite yielded the reduced analogue [(TPPS)FeII]0/[(TerpyCD2)CuI]+, which reacted with O2 to yield a ferric superoxide species formulated as a (P)FeIII–(O2•−)–CuI(L3) complex. Resonance Raman spectroscopy revealed an Fe–O stretching frequency of 578 cm−1, consistent with previously reported iron(III)-superoxide Fe – O stretches;485,524,894,1024,1025 a superoxide O–O stretching frequency was not reported (but see discussions in section 6.3.1). There was no indication that a μ-peroxo FeIII– (O22–)–CuII complex ever forms (vide supra).
Figure 122.

(Top) Formation of the supramolecular oxidized heme-copper complex [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+ or depending on the solution pH, μ-oxo, or μ-hydroxo bridged species, formed by titration of [(TerpyCD2)CuII]2+ with one equiv of [(TPPS)FeIII]+. (Bottom) Dioxygen adduct formation on reaction of the reduced form [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+ with O2 to yield ferric superoxide cuprous complex PFeIII(O2•−)-CuIL3.
The superoxide complex was metastable' and it gradually converted to the oxidized analogue [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+ with a pH-dependent rate constant. At lower pH values, in the range of ~7–10, the superoxide decayed more rapidly, suggesting that some proton coupled process is involved in the conversion of the superoxide complex to [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+. Electrochemical technique analyses of O2-reduction by [(TPPS)- FeIII]+/[(TerpyCD2)CuII]2+ and its reference samples, [(TPPS)FeIII]+ and [(TPPS)FeIII]+/[(TerpyCD2)CuII]2+, showed that under the conditions employed, diffusion limited catalytic O2-reduction (below –1.0 V vs Ag/AgCl) was enhanced for the heme–Cu complex (n ~ 3.0 electrons per O2) compared to that for the “reference” compounds (n ~ 1.6), interpreted by the authors to mean that the Cu(I) ion functioned as an electron storage site (Figure 122).
Another recent addition to the field of electrocatalytic models for cytochrome c oxidase has come from the Coutsolelos group (Figure 123, A–E), where the copper ligand is covalently attached to the heme which can be further functionalized with a phenolic moiety (as a model for the HCO active site tyrosine).1026,1027 In complexes (B) and (C) an axial pyridine is appended to the porphyrin at the mesityl position. In the case of (C), this axial base can coordinate to the heme iron; however, for (B), the axial base is not bound, most likely due to geometric constraints arising from the tether properties. The O2-reduction capabilities of these species were probed electrocatalytically on graphite electrodes in aqueous solution, and while all the models reduce dioxygen via 2e− and 4e− pathways, only for (A) does the selectivity improve on inclusion of a copper ion. The presence of an axial base for (B) and (C) leads to low catalyst stability under these conditions. To expand upon this work, complex (D) was synthesized and contains no axial base but, rather, a phenolic moiety at the mesityl position. This system binds both O2 and CO and reduces oxygen via 2e− and 4e− mechanisms, although further characterization has been hampered by low stability on graphite electrodes. In another approach to ligand design, the Coutsolelos group utilized 5,10,15,20-tetrakis(2-amino- phenylporphyrin) (TAPP) as a precursor to incorporate a TREN derivative type copper chelate, (N1-benzyl-N2,N2-bis- [2-(benzylamino)-ethyl]ethane-1,2-diamine) and a tyrosine mimic above the parent porphyrin (E.i).1028 Rotating ring disk electrochemistry (RRDE) on an edge plane graphite electrode showed that both the 2e− and 4e− reduction mechanisms for O2 reduction to H2O2 and H2O were active. In this case, the presence of the copper ion, did not appear to increase the electrocatalysts selectivity for the 4e− process. Additionally, protection of the phenol OH by methylation in (E.ii) resulted in the same catalytic activity as the unprotected version (E.i). The lack of a notable effect by the phenol was attributed to improper orientation of the phenolic OH group away from the binuclear site of the catalyst.1028
Over time, the Collman group continued to modify their model systems to what they have referred to as more “accurate” biomimics (vide infra). Inspired by the tris-histidyl ligation of the copper ion in the active site of CcO, they incorporated three imidazolyl ligands for copper and pyridyl or imidazolyl axial bases onto the opposite side the porphyrin (Figure 124, A and B).1029 These compounds are selective for the 4e− reduction of O2 to H2O with no detectable leakage of H2O2. The onset of the electrocatalytic reduction by (B) occurs at ~50 mV higher than that of (A), and thus it can be inferred that (B) better replicates the active site properties of cytochrome c oxidase, which reduces dioxygen at positive potentials. In 2002, constructs containing N-methyl imidazolyl ligands were incorporated into the tetrakis(aminophenyl)- porphyrinate core as ligands for copper, and in line with previous results, an imidazolyl axial base was inserted on the opposite side (Figure 124, C).1030 When paired with an electrode possessing fast electron flux, these model systems are highly efficient dioxygen reduction catalysts, competent for at least 10000 turnovers at physiologically relevant pH and potential (>50 mV vs NHE).1031 Under these conditions, the presence of the copper ion did not appear to affect the dioxygen reduction mechanism, turnover frequency, or turnover number, although the selectivity for the 4e− reduction pathway is higher in the Fe/Cu systems.
Figure 124.

“Picket fence” porphyrin-derivatized heme-copper model systems from the Collman group featuring “bioinspired” tris-histidyl ligation for copper. See text for discussion.
Collman and co-workers, when referring to more “accurate” models, we believe are referring to the fact that their complexes possess features closer to those in the enzyme, for example, imidazolyl versus pyridyl donors for copper, or possession of a nitrogenous proximal ligand or distal phenolic moiety. But we remind the readers that having “accuracy” in this way does not mean it is a better model. A model can never allow one to absolutely deduce an enzyme mechanism; only, by definition, can a mechanism or absolute conclusion of any kind be made upon experiments with the enzyme itself. Models are meant to sharpen questions and/or isolate fundamental questions which can more likely be answered through the study of the crude bioimimic.747 There are numerous cases135,1032–1037 where less “accurate” models (i) provide greater activity (if activity/reactivity is judged to be the main point of interest), (ii) provide an answer to a specific question,747 or (iii) provide answers of critical importance from direct comparisons of systematically varied ligand environments (e.g., giving different metal ion geometries, different ligand–metal E° values, or different M⋯Mˊ distances). For example these types of models allow for elucidation of those factors which give rise to homolytic versus heterolytic O–O reductive cleavage, etc.47,50,457,589,594,595,1038 And, while many or all of Collman and co-workers’ metal–ligand complex constructs are leading-edge research examples and beautifully designed models which possess great sophistication, they (and those models of others) do not, for example, possess such qualities as the known enzyme Cu-ligand juxtaposition, high Cu E°, known biological natural porphyrinates, nor the known natural histamine containing imidazole moiety as proximal heme- or copper-ligand connected through the imidazole 4,5-position and not the imidazolyl non metal-binding N-atom.
In order to evaluate the O2-reduction reaction at slow rates of electron transfer, which is rate-limiting in the enzyme, the authors incorporated the catalysts at low molar fraction (>3%) into a 1,2-dimyristolyl-L-α-phosphatidylcholine (PC) lipid matrix deposited on the electrode surface. In doing so, the catalyst molecules are mobile and isolated close to the electrode surface, in a matrix composed of biologically relevant lipids. This enabled detection of catalytic current that was independent of the O2 concentration and rotating disc electrode (RDE) disc rotation frequency (that in the rotating ring disc electrochemistry (RRDE) technique), and thus allowing for the evaluation of variable electron transfer rates.1039 In these cases, in the absence of copper, the catalysts (C) (Figure 124) displayed low activity and undergo rapid degradation. With coordinated CuI, the catalyst is still active, and the Cu ion is proposed to serve as an electron storage site, which avoids the formation of a ferryl, FeIV=O+•, species which would degrade the organic ligand. Under conditions of slow electron transport, this highly oxidizing cationic intermediate species persists long enough to damage the ligand, causing nucleophilic reactions with the solvent, and hence the copper ion is necessary for catalyst integrity. This work indicates the importance of copper (as a mimic for the CuB site in HCOs) within the regime of slow electron transport; however, the details of the mechanism are difficult to confirm without spectroscopic evidence.56
Stoichiometric addition of dioxygen to (C), Figure 124 yields a ferric superoxide species, FeIII– (O2•−)⋯CuI, described in section 2.2.2 (Figure 40).524 In a single turnover experiment (Figure 125), this was reported to react intermolecularly with a sterically hindered Tyr244 mimic (2,6-di-tert-butyl-4-methox- yphenol) to give a stable phenoxyl radical, and an Fe=O species, as has long been discussed as what occurs during CcO enzyme turnover (see section 4). The mechanism proposed involves hydrogen atom abstraction by the FeIII-superoxo species yielding an FeIII-OOH product, which is subsequently cleaved homolytically to yield the Cmpd II type high-valent FeIV=O complex. This intermediate in turn oxidizes a second equivalent of phenol, yielding the CuII-OH, FeIII-OH, and two equivalents of the phenoxyl radical products (Figure 125).525 A small deuterium kinetic isotope effect (KIE) (~2) was observed for the addition of ArOH(D), consistent with formation of a hydroperoxo product species. Further evidence for formation of the oxyferryl intermediate, making this system a model for the CcO A to Pm, is derived from the observation that O=PPh3 formed following addition of PPh3, and mass spectrometry indicated the presence of a peak corresponding to a FeIV=O/Cu form of the complex.
Figure 125.

A single-turnover experiment wherein a superoxide-bound heme-copper binuclear complex reacts with sterically hindered phenols to yield an FeIV=O species. This represents a possible model for the A to PM conversion in Cytochrome c Oxidase. See text for discussion.
A next logical step in catalyst design aimed at O2-reduction chemistry is to append a Tyr244 mimic into the tris-imidazolyl picket fence porphyrin periphery in order to promote an intramolecular O–O bond cleavage reaction via HAT or PCET from the phenol to a metal-bound O2 moiety which led Collman and co-workers to design (A) (Figure 126).1025 The same chemistry as described for the construct lacking the internal phenol (see Figure 125) was reported to occur, as shown in Figure 126. This model system was the first to contain all three redox centers, the heme, the Cu, and the internal phenol, plus the proximal imidazole ligand.
Figure 126.

Intramolecular hydrogen atom abstraction by heme-bound superoxide (oxy-A) to yield a PM model (C).1025.
It should be noted that the electrocatalysis studies thus far described utilized edge plane (EPG) electrodes, or pyrolytic graphite, where the rate of electron transfer to the catalyst (>104 s−1) is much faster than the rate of electron delivery to the active site of CcO (~2 s−1).923 The surface of edge plane graphite electrodes is not uniform, so there is no site isolation for each catalyst on the surface. However, the development and application of “click chemistry” enabled the immobilization of heme–copper models such as (A) (Figure 126) onto electrode surfaces using self-assembled monolayers (SAMs) as outlined in Figure 127, right.1040 This approach is advantageous for electrocatalytic models because the “click” reaction works under mild conditions, it is selective, it works in aqueous media, and is itself a catalytic process. Additionally, the electron transfer properties of the SAM can be altered by adjusting its length and degree of conjugation.1041–1043
Figure 127.

(Left) Covalent attachment of heme-copper model systems to gold electrode surfaces via “click” chemistry. (Right) Several different linker molecules, which allow for tunable electron transfer rates. Adapted from ref 1040. Copyright 2001 Wiley-VCH.
This newer methodology enables probing of the role of the copper and phenol moieties on dioxygen reduction activity and selectivity, now at slow electron transfer rates. Toward this goal, complex (A) from Figure 126 was again modified to possess a methoxyphenyl group in its secondary coordination sphere, yielding (A) in Figure 128 (top left), which was then attached to a SAM-coated gold electrode.1044 Two different linker/SAM combinations were employed; one assembled from a 1-azidohexadecanethiol/hexadecanethiol SAM combination (“Slow SAM S1”, Figure 128, left), and the other assembled from an azidophenylethynylenebenzyl thiol/octane thiolacetylene SAM combination (“Fast SAM S2”, Figure 128, right), in order to vary the electron transfer rate for heme–Cu electrocatalytic O2-reduction experiments. Under conditions of rate-limiting electron transfer with SAM S1, the presence of copper and the phenol moiety reduced the amount of PROS produced by over 30% relative to SAM S2, where a second electron can be delivered to the Fe center at an abiological rate, much faster than that of SAM S1 (Figure 128). These results were concluded by Collman and co-workers to firmly indicate that under biologically relevant conditions, the copper ion and phenol moiety are essential in alleviating the production of PROS. Thus, complex (A) (Figure 126) studied via electrocatalytic O2-reduction, reasonably mimics the HCO binuclear center, in its metal ion redox potentials at the Fe/Cu core, its selectivity for full 4e− reduction of O2 to water under biologically relevant electron delivery rates, and additionally is active in both aqueous and hydrophobic media.
Figure 128.

Plot in the bottom, left shows the percentage of partially reduced oxygen species produced during electrocatalytic O2 reduction at slow and fast electron transfer rates. The effect of the Cu ion, tyrosine mimic, and linker between the Fe-Cu system and the electrode were evaluated with respect to catalyst selectivity, stability, and rates of electron transfer. Adapted with permission from ref 1044. Copyright 2007 American Association for the Advancement of Science.
Collman and co-workers evaluated the effect of hydrogen sulfide on the model complex and O2-reduction catalyst shown in Figures 125 and 127 by studying the chemistry carried out in a NaSH buffer solution as a source of H2S.1045 At higher H2S concentrations, the catalytic current gradually decreased by up to 60%. However, when the NaSH buffer was replaced with air-saturated buffer, the catalytic reduction of O2 was subsequently restored. This gas, now known to be a signaling agent (i.e., a gasotransmitter), can induce hibernation in mice at similar concentrations to those used in this work.1045 Also, addition of a nongaseous ligand, tetrazole, as a possible metal binding ligand also inhibited O2-reduction in this model system. This was determined to be particularly interesting, as tetrazole is known to have an effect on respiratory inhibition in platelet mitochondria leading to platelet deactivation, which is an important part of blood coagulation processes.1046
These researchers also examined the binding and chemistry of small molecules which are known physiologically important signaling (i.e., NO, H2S) or cytotoxic agents (i.e., CO, CN−) to their heme-copper constructs. As CcO is important in biological respiration, it was desirable to see the effects of these potential ligands on their CcO model for O2-reduction activity. Such complementary studies could provide information useful in inhibitor design, considering a therapeutic perspective (Figure 129, A–E depict the adducts which were studied). Here, nitric oxide acts as a CcO inhibitor via formation of a stable iron nitrosyl species, which can only be replaced in the presence of superoxide (which could be generated via O2- binding to the CuI ion. Therefore, inhibition of this model by nitric oxide is mitigated by the presence of CuI and a constant supply of electrons and dioxygen (Figure 129).1047–1049 Interestingly, nitric oxide reverses the inhibition of the model by CO and CN− by displacing them from the ferrous heme, and the subsequent reaction with superoxide yields nitrate which diffuses away to generate the uninhibited oxidized state of the catalyst. Hydrogen sulfide was observed to weakly bind to the FeII center (D, Figure 129), and it could be displaced by dioxygen to yield the oxy bound form of the catalyst (A, Figure 129).
Figure 129.

Small molecule, binucleating ligands evaluated for their effect on O2 reduction chemistry by “refined” heme-copper models developed by the Collman group. See text for further details.
Karlin and co-workers have also employed several of their model system constructs for application to electrocatalytic O2- reduction studies. Although the ligand systems utilized have been mostly employed in homogeneous solution (and cryogenic conditions) for the formation and detection of catalytic intermediates and stoichiometric reactivity, they were worthwhile candidates for electrocatalytic studies, given their ease of synthesis and robust nature. The models consisted of an Fe/Cu binucleating ligand with a heme and a covalently tethered tris(2-pyridyl)methylamine copper chelate, [(6L)- FeIICuI]+ (A), its Fe-only analogue [(6L)FeII] (B), and the “parent” F8 porphyrinate [(F8)FeII] (C) (Figure 130). Previous work (see sections 2.2 and 5.1) has shown that these models form stable FeIII–(O22−)– CuII and FeIII– (O2•−) species, and given that chemical and theoretical research efforts have proposed peroxo and superoxo species to be key intermediates in reductive O–O bond scission, it is reasonable to expect that these models can be competent O2-reduction electrocatalysts. Indeed, the tethered heme–copper model reduces O2 to water with good efficiency (74%), possibly via a peroxo intermediate species. The parent F8-heme (C) exhibits only 20% efficiency for O2 reduction, which is even lower than the Fe-only analogue of 6L (B), which reduced O2 at 59% efficiency. It was proposed that this is due to FeIII–(O2•−) stabilization by protonated pyridyl groups (of the Cu-ligand) in the acidic media employed (triflic acid). The lower selectivity of the parent heme (F8; Figure 130, C) for the four-electron pathway may have been a result of the formation of heme-peroxo-heme adducts, which on protonation yield hydrogen peroxide as a 2e− reduced byproduct, given the highly acidic proton source employed here.1050
Figure 130.

Heme copper and heme-only systems developed by the Karlin group to be first evaluated as O2 reduction catalysts. See text for details.
More recently, Sengupta and Dey1051 investigated the same heme–Cu or heme-only complexes but now with advanced techniques, allowing for the coupling of surface-enhanced resonance raman spectroscopy (SERRS) and RRDE to probe intermediates formed in situ at the electrode surface during catalyst turnover.1052 Expanding on the previous electrocatalytic (vide supra)1050 and stoichiometric (vide infra)1053 work by Karlin and co-workers, the authors employed [(6L)FeIII(OH)] (A, Figure 131) and [(6L)FeIII–(OH) – CuII]+ (B) as the initial form of the electrocatalyst and studied its efficiency and selectivity for dioxygen reduction under varying electron delivery rates.
Figure 131.

Surface enhanced resonance raman spectroscopy (SERRS) coupled to rotating ring-disc-electrochemistry (RRDE) developed by Sengupta and Dey, applied to synthetic heme-copper model systems developed by the Karlin group (A, B, and C). The resonance Raman data (bottom right) show the presence of a peroxidic intermediate during electrocatalytic O2 reduction. Adapted from ref 1051. Copyright 2015 American Chemical Society.
With rapid electron delivery, with the catalyst adsorbed on an edge plane graphite electrode, a very high kcat [(4.49 ± 0.9) × 106 M−1 cm−1] for O2-reduction was reported for [(6L)FeIII– (OH)–CuII]+ (B, Figure 131), in fact about 1 order of magnitude greater than some of Collman's complexes, previously studied for electrocatalysis. High selectivity for O2 reduction to water (only 6% PROS) was also reported with [(6L)FeIII–(OH)– CuII]+, see Figure 131, top. Bridging peroxide intermediates (see rRaman spectra, Figure 131) were detected during turnover for both the parent, [(6L)FeIII– (OH)–CuII]+ (B, Figure 131), and its imidazole ligated analogue, [(Im)(6L)FeIII-(OH)-CuII]+ (C, Figure 131), although the latter displayed a significantly higher percentage of PROS production (~25%). The detection of these peroxidic species on an EPG electrode in pH 7 buffer was enabled by utilizing mass-transfer limiting conditions for catalysis, where only species whose decay does not involve electron transfer can have steady-state accumulation. The rRaman features identified during turnover for the parent [(6L)FeIII–(OH) – CuII]+ (B, Figure 131) complex support a side-on, end-on heme-peroxo-copper intermediate, [(6L)FeIII– (η1: η 2-O22–) – CuII]+, [v(O–O): 819 cm−1 (∆18O2 = −60 cm−1), as depicted in Figure 132 (Scheme 1, species B]. Here, it is proposed that an iron-bound water molecule and hydrogen-bonding interactions favor end-on (η1) binding to low-spin FeIII and side-on (η2) coordination to the CuII.
Figure 132.

(Top) Sengupta and Dey evaluated the role of the Cu ion and heme axial base ligation in three electrode-immobilized heme(-Cu) systems on electrocatalytic selectivity (%PROS released) for the reduction of O2 at both fast (EPG electrode) and slow (C8SH and C16SH SAM coated gold electrodes) electron transfer rates. Distinct pathways for PROS production were proposed for hydroxo-bridged heme-copper complexes without (Scheme 1) and with (Scheme 2) an axial imidazole ligand. Adapted from ref 1051. Copyright 2015 American Chemical Society.
When an exogenous axial imidazole ligand is incorporated into the distal side of the 6L ligand scaffold, as in [(Im)(6L)FeIII(OH)-CuII]+ (C, Figure 131), a peroxide binding mode where the O2 fragment is end-on with respect to both metal ions was proposed based on the rRaman spectra in Figure 131 [v(O–O) = 847 cm−1 (∆18O2 = −61 cm−1)], this intermediate is depicted in Figure 132, Scheme 2, C. As also discussed below, the high-spin heme-peroxo-Cu complexes not possessing an axial proximal ligand switches to low- spin analogs when adding an imidazole ligand, as here.
Distinct pathways for PROS production were proposed to account for the difference in selectivity of the parent catalyst [(6L)FeIII-(OH)-CuII]+ (Figure 131, B), and its imidazole bound analogue, [(Im)(6L)FeIII-(OH)-CuII]+ (Figure 131, C) at fast (EPG) versus slow (SAM) electron transfer rates (Figure 132).1051 For B (Figure 131), reduction of the low-spin iron(III) resting state yields a 5-coordinate, high spin ferrous heme (Figure 132, Scheme 1, complex A), which benefits from binuclear site accessibility and resulting fast dioxygen binding kinetics. A detectable peroxidic intermediate B forms (Figure 132, Scheme 1), which was discussed above. Subsequent protonation cleaves the peroxide O–O bond, yielding the high valent intermediate D (Scheme 1, Figure 132, D) which is not detected under steady state turnover due to its rapid reduction/protonation to give back A. Dioxygen binding to the proximal site of FeII in this case most likely results in PROS production upon electron transfer and protonation (C → A, Scheme 1, Figure 132). In the case of complex [(Im)(6L)FeIII-(OH)-CuII]+, SERRS-RDE indicates an accumulation of a six coordinate LS FeII species (Figure 132, Scheme 2, A) as the resting state. The much lower selectivity of starting electrocatalyst C (Figure 131) for O2 reduction at fast electron transfer rates can be attributed to the slower dioxygen binding kinetics of low spin ferrous hemes (such as A, Figure 132, Scheme 2). When electron transfer is fast, the copper ion can reduce dioxygen to yield PROS (B to A, Scheme 2). When the rate of electron transfer is comparable to, or slower than O2 binding to the LS FeII, then the dioxygen reduction reaction preferentially proceeds via a bridging heme- peroxo-copper intermediate, for a selective 4e−/4H+ oxygen reduction reaction. These two proposed mechanistic scenarios help to explain the opposite selectivity behavior of [(6L)FeIII– (OH)–CuII]+ and [(Im)(6L)FeIII(OH)-CuII]+ (Figure 131, B and C, respectively) at fast and slow electron transfer rates.
This work is significant because it shows that FeIII–(O22−) – CuII species can form in aqueous media, under relatively harsh turnover conditions (relative to self-assembly studies in organic media at low temperature), in a physiologically relevant pH range. The dependence of the selectivity for 4e−/4H+ dioxygen reduction pathways on the electron transfer rate and coordination properties (and spin state) of the ferrous heme is also enlightening. This work provides insight into the factors that favor selective dioxygen reduction with earth abundant transition metals such as iron and copper, in man-made systems. Bridging peroxide intermediate species have been proposed in theoretical studies and can be crystallized in the enzyme, so the involvement of such species during catalytic O2-reduction by HCOs remains to be a subject of debate. The effect of hydrophobic residues, or phospholipids, on catalytic activity, and selectivity with this model could be an interesting future direction.
The development of electrocatalytically competent small molecule biomimics for heme–copper oxidases has provided insight into several factors that affect the feasibility and efficiency of the dioxygen reduction reaction with earth- abundant transition metals such as iron and copper. The copper/iron primary and secondary coordination environments and redox potentials, solvent polarity, and electron delivery rate are critical determinants of catalyst activity. Classic work in this subfield has been enhanced by new spectroscopic techniques (RDE-SERRS) that allow for the characterization of intermediates formed during catalytic turnover at the electrode surface. This new approach will undoubtedly help to bridge the two design approaches of electrocatalytic model systems and stoichiometric dioxygen reduction chemistry.
5.2.5. Catalytic Dioxygen Reduction by a CcO Model System in the Presence of Proton and Electron Sources.
A homogeneous catalytic study of O2 reduction to water provided further insight into the role of the copper ion during O2 reduction and assessed the rate-limiting step during turnover as a function of temperature.1053 The catalyst [(6L)FeII–CuI]+ and its Fe-only analogue [(6L)FeII] in acetone were exposed to dioxygen in the presence of trifluoroacetic/decamethylferrocene (H+/e−) at –60 °C or at room temperature to achieve O–O reductive cleavage. Detailed mechanistic studies reveal that the rate-limiting step during turnover changes as a function of temperature, as evidenced by a change in the nature of the detected steady-state intermediate (Figure 133). Namely, at room temperature the rate-limiting step is dioxygen binding, as indicated by the presence of [(6L)FeII⋯CuI]+, and [(6L)FeII] during steady state turnover, although it is important to note that the rate of dioxygen binding is twice as fast for [(6L)FeII⋯CuI]+. However, at –60 °C in acetone, a new FeIII–OOH species is observed for both [(6L)FeII⋯CuI]+ and [(6L)FeII] during steady state turnover. Under these low-temperature conditions, the rate of O–O bond cleavage for [(6L)FeIII(OOH)] and [(6L)FeIII–(OOH)–CuII]+ were nearly the same. This led the authors to suggest that the copper ion does not necessarily need to be involved in O–O bond cleavage via a bridging peroxo species. It may instead be involved in facilitating faster O2 binding at noncryogenic temperatures. This result is consistent with O2 binding to copper complexes at diffusion controlled rate940,1002 and CcO enzyme studies which implicate CuB as the initial binding site of O2 during catalysis.975,976
Figure 133.

(A) Proposed mechanism for the catalytic reduction of O2 to water by [(6L)FeII–CuI]+. (B) Proposed mechanism for the catalytic reduction of O2 to water by [(6L)FeII] (iron-only). In both cases, the steady state changes from O2 binding to room temperature (RT) to reductive O–O bond cleavage at cryogenic temperatures (LT). Adapted with permission from ref 1053. Copyright 2011 National Academy of Sciences.
5.2.6. PCET Reactivity of an HCO Model Highlighting Critical Factors for O2-Reduction.
In CcO, although the precise PCET mechanism involved in the O–O cleavage step (i.e., relative timing and method of delivery of protons and electrons) is not entirely known, controlled addition of known substrates to achieve activation and reduction of O2 with synthetic model systems can be used to probe such questions. Important substrate considerations for inducing protonation/reduction of a peroxo moiety in heme–(O22−)–Cu entities undoubtedly include local steric factors, peroxo O-atom pKb (for both the Fe-bound and Cu-bound O atoms), O–O bond dissociation energy (BDE), and heme and/or copper ion E0; however, analogous properties of the model complex are equally influential for directing their reactivity. Perhaps not surprisingly, the LS-3DCHIm and LS-4DCHIm show identical reactivity toward strong proton or electron sources along (i.e., they are not reduced by reductants as strong as decamethyl- ferrocene) (Fc*; E0 = +20 mV vs SCE in MeTHF; E0 = −110 mV vs SCE in MeCN)1054,1055 alone, and addition of sufficiently strong acids such as [H(DMF)+]OTf− (pKa = 6.1 in CH3CN)1056,1057 results in protonation (metal–O cleavage) and release of a full equivalent of H2O2 (see Figure 134). The oxidized, uncoupled metal complexes and H2O2 products were confirmed and quantified by EPR spectroscopy and a horseradish peroxidase enzyme assay (HRP test), respectively.919
Figure 134.

(Top) Scheme showing the H+ and/or e− reactivity of LS-4DCHIm with proton sources of varying strengths and Fc* as the electron- donor. (Middle) UV-vis monitoring of the reaction of LS-4DCHIm (black) following addition of p-NO2-phenol (blue) and Fc* (green); rR spectra showing the changes in Fe-O and O–O stretching frequencies upon formation of the H-bonded adduct, LS-4DCHIm(ArOH); and EPR spectra of the LS-4DCHIm, LS-4DCHIm(ArOH) (both silent), and final LS-FeIII/CuII products (green) which match that of a mixture of the authentically generated products shown in the reaction scheme. (Bottom) Kinetic plots and analysis showing saturation behavior of kobs with respect to increasing [phenol] and linear dependence of kobs on [Fc*]. See text for further details. Adapted from ref 919. Copyright 2017 American Chemical Society.
Proton-coupled reductive cleavage of a metal-bound peroxide moiety has been shown in at least one other chemical system to be favored over H2O2 evolution when weak, rather than strong acids, are employed.1058 Indeed, the cross-linked Tyr244 (bovine) at the binuclear active site of CcO (which is widely accepted to provide both a proton and the fourth required electron during enzymatic turnover, see section 4) is relatively more acidic (pKa estimated to be approximately 8.5) than Tyr without an imidazolyl cross-link (pKa ≈ 10) as the ortho-His cross-link substitution displays weak σ-electron withdrawing character.850,853 Interestingly, while phenols having low O–H BDEs [typically H-atom (H+ and e−) sources] show no reaction with the model complexes, LS-3DCHIm and LS-4DCHIm, a weakly acidic phenol does exhibit distinct reactivity toward each of them. No spectroscopic changes occur when even excess acidic phenol [4-NO2-phenol; pKa(CH3CN) = 20.7]1059 in the presence, or not, of Fc* as a reductant, are added to a solution of LS-3DCHIm. However, this pair of phenol and reductant have been shown to react with LS-4DCHIm via a biomimetic mechanism (Figure 134, top).919 First, addition of 4-NO2- phenol in 10-fold excess results in spectroscopic changes consistent with formation of an adduct in which the phenolic O–H hydrogen bonds to the copper-bound O atom of the peroxo moiety, effectively “activating” the O–O bond. The optical spectrum of the new adduct species displays features at 415 nm (Soret), 540 nm (Q-band), and the low-energy features diagnostic of the LS-4DCHIm peroxo complex (~ 845 nm) change and somewhat decrease in intensity (Figure 134, middle left). Coupled to these observations, the vibrational frequencies observed by rR spectroscopy are shifted upon phenol association: v(Fe–O) = 598 cm−1, v(O–O) = 827 cm−1, and v4 = 1367 cm−1 (shifts of +10, –42, and +3 cm−1, respectively, for the phenol adduct relative to the “parent” LS-4DCHIm complex) (Figure 134, middle). The vibrational data are consistent with an H-bonding interaction with the distal O atom of the peroxide (relative to Fe),574,1060,1061 and partial oxidation of the Fe(III) center,1062 which has been corroborated by extensive DFT calculations (and see Figure 134 for a DFT structure of this phenol adduct).919 Furthermore, it is important to note that the entire Fe- (O22−)-Cu complex remains intact during this association (DFT, HRP test, UV–vis evidence) likely owing to the “mobile/adjustable” quality of the monodentate DCHIm copper ligands. Reductive cleavage of this “activated” adduct, (DCHIm)FeIII-O-(O∙∙HOAr)-CuII(DCHIm)4, is then possible with addition of Fc* as the reductant. At that time, the protons and the two stoichiometrically required electrons are transferred, as determined by UV–vis observation of the 4-NO2- ArO− (λmax = 393 nm) and quantification of the Fc*+ (λmax = 785 nm, ε = 580 M−1 cm−1)1063 products and lack of H2O2 evolved (HRP test) (Figure 134). Uncoupling of the metal centers occurs following reductive cleavage resulting in an EPR spectrum of overlapping Cu(II) and mostly (>95%) low-spin Fe(III) signals (Figure 134, middle right) which match an authentic spectrum of the product mixture shown in Figure 134, top.
Thorough kinetic investigation of this mechanism coupled with control experiments using an authentic FeIV=O complex,1064 it has been revealed that (i) an equilibrium phenol-association step with KArOH = 604 ± 21 M−1 precedes (ii) the rate determining single electron transfer with ket1 = 1.6 ± 0.1 M−1 s−1 (see Figure 134, bottom), which is followed by (iii) a rapid 1 e− reduction of the resultant Cmpd II-like ferryl species with ket2 = 5.0 M−1 s−1 (see Figure 134). Kinetic isotope studies with 4-NO2-ArOD confirm the involvement of the O–H/D bond in both the H-bond association and rate-determining ET steps of the reaction, with a KH/D = 1.61 ± 0.06 and ket1(H/D) = 1.84 ± 0.09, in the range for phenol O–H bond activation by metal–oxygen species.1065 Through comparison to a structurally similar complex, LS-3DCHIm (DFT structures), the stepwise PCET reactivity observed for LS-4DCHIm with weakly acidic phenol and reductant highlights the fact that while the substrate H+/e− properties of course play a role in directing the chemistry of these complexes, perhaps more notably, the ability for the heme-peroxo-Cu complex to accommodate/accept an H-bond donor to prime the peroxo for reductive cleavage is a critical factor (in the case of LS-3DCHIm, the compressed metal–metal distance is proposed to prevent such H-bond adduct formation).919 Importantly, these results illustrate the value of cryogenic, synthetic, active-site modeling chemistry, in that mechanistic insights could be gained from kinetic studies and relevant intermediates along an O–O reductive cleavage pathway could be characterized.
5.2.7. Reaction Coordinate for a Phenol Induced O– O Reductive Cleavage in a Heme-Peroxo Synthetic Construct (from DFT).
None of the high spin heme-peroxo-copper adducts shown in Figure 135 react with strong phenolic hydrogen atom donors, such as 2,4-(tBu)2-phenol or 2,4,6- (tBu)3-phenol, in contrast to reported work from Collman's group.525,1025 This difference is possibly due to the low spin ferric heme in Collman's work and the high spin ferric heme in these complexes. This is consistent with some observed chemistry of nonheme FeIII–OOR complexes, wherein the low-spin, as opposed to high-spin configuration is amenable to reductive O–O cleavage chemistry, as a result of the presence of a stronger Fe–O bond and a weaker O–O bond.1066,1067
Figure 135.

(Top) Evaluated mechanistic pathways for the reaction of a heme-peroxo-copper complex, LS-AN, with 4-OMe-phenol which proceed through one of two transition states, denoted TSpi for the proton-initiated mechanism and TShb for the hydrogen bond assisted pathway (see text for discussion). (Bottom) The potential energy surfaces for the PI and HB pathways. The red and blue lines in each plot correspond to the progress of the reaction leading to the products shown in the top, right, with the highest point on each surface being the transition state for each reaction. Dashed lines denote where the pathway is behind the surface. Adapted from ref 747. Copyright 2017 American Chemical Society.
Computational methods have proved to be valuable for understanding the CcO enzymatic mechanism (vide supra, section 4); however, they can also be directed toward understanding the fundamental chemistry of model complexes (and evaluating how they relate to the enzyme's activity). Recently, the Solomon and Karlin research groups combined extensive density functional theory calculations and experimental/kinetic analysis of the reaction of a heme-peroxo copper system with an electron-rich phenol: it was found that excess 4-OMe-phenol reacts with the low-spin, LS-AN complex (Figure 135, left) at low temperatures via an H-atom transfer reaction (UV–vis, radical trap/ESI-MS evidence).747 The importance of H-bonding and/or protonation events in inducing PCET O–O reductive cleavage in HCOs has been documented in studies of the enzyme as well as model systems (see just above),919 therefore, determining the nuances of acid strength and timing of PT and ET are critical for furthering our understanding of the HCO mechanism.
In the case of {LS-AN + 4-OMe-phenol}, two different mechanistic pathways were considered: (i) a proton-initiated pathway, PI, where the phenolic proton transfers early in the reaction coordinate and mostly before the reaction barrier, and (ii) an H-bond-initiated pathway, HB, where the proton transfers largely after the barrier (pathways and transition state structures, TSPI and TSHB (Figure 135). Potential energy surfaces were generated for each reaction coordinate as a function of intraperoxo O–O distance and Cu-bound O atom to phenolic proton distance (Figure 135, bottom). The overall energetic barrier for these reactions is very similar; however, since the reactions differ by the amount of proton transfer in the transition state (i.e., PI involves predominantly proton transfer; HB involves minimal proton transfer), and therefore how much the phenolic O–H bond is participating in the ratedetermining step, they can be distinguished by experimental kinetic isotope effect measurements. Regardless of the functional used, DFT calculations predict a large, primary KIE for the PI mechanism [10.2 (B3LYP); 7.7 (BP86)], whereas a small secondary KIE is expected for the HB mechanism [1.2 (B3LYP); 1.6 (BP86)]. Using protonated and deuterated 4-OMe-phenol, a KIE of 1.7 was found experimentally, aligning closely with the predicted value for the HB mechanism, therefore suggesting the mechanism of action proceeds via an H-bonded intermediate/transition state which precedes PCET. “The overall electronic structure of TSHB is best described as an FeIV=O/CuII–O•/PhOH”.747
To understand the relevance of such a system with respect to HCOs, the same methods were applied to a theoretical model of the CcO active site, where it was determined that the active site Tyr residue likely provides a proton followed by donation of an electron from the phenolate to cleave the O–O bond, and this PCET process is aided by an H-bonding network of water molecules in the active site which function to lower the barrier in a similar manner to the H-bonding phenol in the {LS-AN + 4-OMe-phenol} study. Finally, the authors compare the viability of several different DFT functionals in analyzing such a reaction by comparing the calculated results to the experimental results and find that B3LYP offers the closest estimation of the reaction's energetic barrier (∆G‡HB = 16.2 kcal/mol) versus the barrier calculated following variable temperature kinetic experiments under substrate-saturated (Michaelis–Menten-type) conditions (∆G‡expt = 14.9 kcal/mol).747 These results together embody a thorough investigation of phenol-induced O–O cleavage in a biomimetic system in parallel to the biological inspiration itself and highlight the potential capabilities of cooperative experimental model studies and theoretical analysis of complex chemical reactions.
5.3. Heme-μ-oxo/μ-Hydroxo-Copper Model Systems
Prior to the classification of the heme-peroxo-copper complexes discussed above, the Karlin group synthesized and characterized a μ-oxo heme FeIII– (O2–)– CuII(L) compound obtained from the reaction of an equimolar mixture of the (F8)FeII and [(TMPA)CuI]+ complexes with dioxygen at room temperature (Figure 136).502,1068 The formation and kinetic stability of this FeIII– (O2–)– CuII complex is intriguing; such species might be expected to convert to the known highly stable μ-oxo iron(III) binuclear (P)FeIII–(O2–)– FeIII(P) complexes, those typically formed as a result of oxygenation of ferrous hemes at RT (see Scheme 9 and related text).536,1069 The heterobinuclear μ-oxo compound, [(F8)FeIIIO-CuII(TMPA)]+, can also be formed via acid–base chemistry, mixing (porphyrinate)-FeIII-OH and CuII(L) species in the presence of a base (Figure 136).502,1068
Figure 136.

Heme-copper dioxygen or acid-base chemistry leading to μ-peroxo, μ-oxo, or μ-hydroxo-bridged high-spin [(F8)FeIII-X- CuII(TMPA)]n+ (X = O or OH; n = 1 or 2) complexes. In these compounds, the iron(III) ion lies well above the porphryinate plane. Similar chemistry occurs when utilizing other tetra- or tridentate nitrogen-containing ligands for the copper ion. See the text for discussion.
Thus, over the years a large variety of related μ-oxo and protonated μ-hydroxo [(porphryinate)FeIII-O(H)-CuII(ligand)]2+ complexes have been generated. In some sense, such reactions are biomimetic in that we are fully reducing dioxygen by four electrons with cleavage of the O–O bond, utilizing a heme–Cu assembly, as in HCOs. However, in the many cases where we have carefully determined products and yields, for tetradentate or tridentate (vide infra) chelates for the copper ion, we find that the reaction occurs with the following stoichiometry (eq 7):
| (7) |
Since, at low temperatures, heme-peroxo-Cu complexes form and are then stable, the overall RT reaction takes place by a disproportionation, by an as yet unknown mechanism, releasing O2(g) (eq 8).513,927
| (8) |
The μ-oxo and μ-hydroxo complexes have mostly been characterized spectroscopically (for example, they possess well red-shifted strong Soret absorption bands, λmax ~ 435–455 nm)943 and by X-ray crystallography or X-ray absorption spectroscopic (XAS) methodologies. Holm and co-workers1070,1071 also used acid–base approaches to synthesize and characterize some μ-oxo and μ-hydroxo complexes utilizing octaethylporphyrinate (OEP). All of these are depicted in Figure 137, where M–Ooxo and Fe⋯Cu distances, along with ∠FeIIIO-CuII values, are provided.
Figure 137.

Depiction of heme-FeIII-X–CuII(ligand) complexes (X = μ-oxo or μ-hydroxo) which have been characterized spectroscopically and by X-ray crystallography or XAS studies. Note that for tetradentate CuII chelates (A, B, C, and G), the μ-oxo complexes possess a near-linear ∠FeIII–O–CuII, while for (ligand)CuII species with tridentate chelates (D, E, and F), the same structural moiety is very bent. Φ is the distance (above + , below – ) of the iron from plane of the porphyrin. Created using data from refs 502, 505, 513, 928, 1023, and 1071 – 1073.
Notably, with tripodal tetradentate chelates bound to the copper(II) ion, the ∠FeIII–O–CuII is near-linear. Changing to tridentate ligands such as LMe2N or MePY2 (Figure 137) results in significant bending, ∠FeIII–O–CuII is 142–150°.513,1073 With protonation, which is kinetically very slow,1023 bending and FeIII–OH and CuII–OH bond lengthening occurs; for the μ-hydroxo complexes that could be studied, [(F8)FeIII-OH-CuII(TMPA)]+, bending occurs where ∠FeIII–O–CuII ~ 178° changes to ~157° upon protonation of [(F8)FeIII-O- CuII(TMPA)] + (Figure 137). Bending upon protonation can be explained by rehybridization at the bridging O atom, changing from sp in the near-linear μ-oxo complexes to sp2 in the μ-hydroxo compounds; for other systems undergoing μ-oxo protonation, Norton and co-workers1074,1075 have also ascribed this kinetically slow process to this rehybridization.
An interesting structural observation comes when comparing [(F8)FeIIIO-CuII(TMPA)]+ and [(TMPP)FeIII-O- CuII(TMPA)]+, the latter possessing p-methoxy substituents on the porphyrinate aryl groups. They both possess similar ∠FeIII–O–CuII core angles, yet the copper environments differ. Notably, in [(TMPP)FeIIIO-CuII(TMPA)]+, the copper geometry is nearly perfectly trigonal bipyramidal; however, in [(F8)FeIIIO-CuII(TMPA)]+, the copper(II) ion geometry is distorted square pyramidal. This difference can be attributed to the steric influence of the heme fluorine atoms in the F8 containing complex, which favors the orientation of the three pyridyl donors of TMPA to lie over three of the four pyrrole groups and between the fluorinated aryl meso substituents of the heme.943
As mentioned, the μ-oxo ligand on all of the complexes we have made are highly basic and thus protonate to give corresponding μ-hydroxide species, many of them reversibly (Figure 136).496,523,943,1023 We posit that the basicity of intermediates containing reduced O2-derived ligands (e.g., oxo and hydroxo) coordinated to metal ions may be critical to the proton transfer/movement in HCOs, either during formation of water from dioxygen or in a role related to HCO protontranslocation. In fact, the idea of Fea3-X–CuB (X = O22−, O2−, or −OH) resting state or even possibly turnover intermediates in HCOs is not new.1023,1076–1079 At the time of our study of the μ-oxo and μ-hydroxo complexes, [(F8)FeIII-O(H)- CuII(TMPA)]+,2+, we also spent some discussion on such aspects,1023 as a spectroscopic investigation of the quinol-oxidizing cytochrome aa3 from B. subtilis suggested that a μ-hydroxo Fea3-(−OH)-CuB resting binuclear center was detectable.1076
Much more recently, computational/theoretical investigations have further led to discussions and results which have invoked the possibility or presence of such μ-hydroxo or even μ-oxo Fea3-O(H)-CuB species occurring as intermediates in HCO enzyme turnover, possibly in the “reductive phase” relating to the formation of intermediate OH and/or transformation to EH (see Scheme 23, above). Two examples are shown in Figure 138.901,903 Note the strong similarity in structure to those μ-hydroxo complexes which have been fully characterized as synthetically derived heme-Cu constructs (Figure 137). A protein oxidase X-ray structure with μ-hydroxo (or μ-aqua) Fea3-(OH(H))-CuB center has even been obtained;989 however, this will be further discussed in section 6, along with descriptions of how coordination chemistry aspects of heme-O(H)-Cu(ligand) complexes could relate to HCO turnover.
Figure 138.

Theoretical/computational structures of plausible intermediates in the HCO reaction cycle. Structure (A) is Blomberg and co-workers’ μ-oxo OB state (with Y288 as a neutral phenol) which is believed to be a high-potential species (a form of intermediate OH), poised to facilitate uptake of a proton and electron and give intermediate EH (see Scheme 23). This conversion is accompanied by membrane proton translocation. Previously, Sharma, Karlin, and Wikström proposed the “strained” μ-hydroxo species, with Y244 as a deprotonated tyrosinate (B), as one possible structure for OH.901 See Scheme 23, the text, and further discussion in section 6.3. Φ is the distance (above +, below –) of the iron from plane of the porphyrin. Created using data from refs 901 and 903.
As per the discussion above and related to bridging oxo or hydroxo heme-Cu complexes (see above, Figure 137), we also had the opportunity to investigate proton-transfer chemistry. Thus, the pKa of the bridging hydroxide ligand in some of the FeIII-OH-CuII synthetic systems has been determined through acid-base titration experiments.496,523,1023 In general, these values range between 14 and 18.5 in acetonitrile (between 6.5 and 11 in H2O, using the estimate1080,1081 that aqueous pKa values are 7.5 ± 1 pKa units lower than those found in acetonitrile). This data shows that synthetic (heme)FeIIIO-CuII species can be highly basic, especially when compared to other bridging oxides containing Mn, Fe, and Ru, which have aqueous pKa values < 7, many far less than 7.496,1080,1082,1083
Interestingly, the observed ∠Fe-O-Cu correlates with the basicity of the bridging oxo ligand. This striking fact was demonstrated in the complexes [(F8)FeIIIO-CuII(TMPA)]+, [(6L)FeIIIO-CuII]+, and [(5L)FeIIIO-CuII]+ (Figure 139).523 Due to the ligand constraints imposed by the linker in the 5L construct, this μ-oxo complex is bent, with a Fe-O-Cu angle of ~141°. It is possible that this bent geometry allows for the bridging oxo ligand to contain partial sp2 orbital hybridization, facilitating faster and easier protonation to the μ-hydroxo complex upon addition of an acid. In fact, the basicity of the μ-oxo ligand increases from [(F8)FeIIIO-CuII(TMPA)]+ to [(5L)FeIIIO-CuII]+ (Figure 139).523 [(5L)FeIIIO-CuII]+ is so basic, or so strained, that even addition of a weak acid (N-methylmorpholinium triflate) results in cleavage of the bridge giving [(5L)FeIIIOH (triflate)-CuII]+.
Figure 139.

Synthetic heme-copper μ-oxo complexes. The pKa values given are for H2O, estimated from those determined in MeCN by subtracting 7.5 pKa units. Except for complex C, all other μ-oxo complexes can be protonated with weak acids to give μ-hydroxo species. However, in all cases, the bridge breaks if stronger acids are added, even one equiv, to yield species such as [(5L)FeIIIOH (S)-CuII]+, where S = solvent or counteranion which is present. Thus, ligand environment can measurably influence the acid-base properties of μ-hydroxo/oxo heme-copper assemblies. See text and also section 6.3 for discussion of possible implications for HCO active site chemistry. See text for details.
The denticity of the copper chelate employed also has a clear effect on the structure and basicity of heme-copper μ-oxo complexes. The μ-oxo complexes using tridentate copper chelates have a bent Fe-O-Cu core, similar to that observed for [(5L)FeIIIO-CuII]+ (Figure 137).496 However, the linearity of the Fe-O-Cu core is not the only factor in determining the basicity. For instance, [(F8)FeIII-O-CuII(AN)] is more basic than both [(6L)FeIII-O-CuII]+ and [(F8)FeIIIO-CuII(MePy2)]+, although its Fe-O–Cu angle is larger (Figure 137). These μ-oxo complexes containing tridentate copper chelates show how such a species might be important in the catalytic cycle of CcOs (as further discussed in section 6.3).
5.4. Re-Engineered Protein Mimics: Insights into HCO Structure–Function Relationships
The bulk of enzyme structure, to a synthetic inorganic chemist, is essentially a complex “bio-ligand” for metal cofactors, which are considered to be the site(s) where most of the metalloenzyme chemistry of interest occurs. Accordingly, biosynthetic models, wherein protein constructs are engineered into existing proteins, are undeniably attractive targets for extensive investigations. Wild-type transmembrane proteins are especially difficult to isolate/crystallize and study; however, through miniaturization, modification, or redesign of existing protein scaffolds (or indeed, de novo design), it is possible to engineer non-natural metal binding sites and/or probe specific function(s) of residues, which may be redox-active or involved in substrate and/or product mobility.24,25,1084 Yi Lu and co-workers in particular have championed this method in a highly successful manner, producing a large amount of literature with investigations using various modified myoglobin (Mb) models; these efforts have resulted in elegant engineered protein constructs where numerous insights into HCO activity and mechanism have been obtained. Myoglobin in particular is useful due to its relative ease of crystallization, modification via site directed mutagenesis, and replacement/exchange of hemes or other cofactors.1085–1088 One of Lu's recent review articles very nicely compares and contrasts the significant insights obtained and limitations of direct studies of HCOs versus the generation and examination of synthetic models or versus biosynthetic models.1085
5.4.1. Engineering the CuB Site and Incorporation/Effects of Copper.
Myoglobin was chosen, in part because it already contains a high-spin heme b cofactor with an axial His ligand and a high O2-binding affinity but none of the other metal cofactors found in HCOs (making it relatively more accessible for spectroscopic studies). The most widely utilized Mb template in these investigations is an engineered sperm whale Mb enzyme, which the Lu research group has termed “CuBMb” to indicate that a CuB binding site like that of CcO has been built into the existing distal heme pocket. This is achieved by two X-to-His mutations, Leu29His and Phe43His, which, in combination with the naturally existing, nearby His64 make up a binding site which can aptly bind a single Cu ion at a distance of ~5 Å, from the heme iron center.1089,1090 The crystal structure of this model overlays closely with that of WT bovine CcO (Figure 140A), suggesting that the aforementioned mutations only effect little structural change in the model (which may or may not physically contain a metal ion in the CuB site and or other amino acid mutations depending on the intent of the specific study). Systematic studies on this type of biosynthetic model can, and have, shed light on how nature has tuned the properties of the CcO active site (i.e., redox properties of metal cofactors and active site residues and H-bonding interactions) to effect efficient O2-reduction chemistry.1084
Figure 140.

(A) Overlaid structures of the Lu group's model, CuBMb (gray), with the wild-type bovine CcO crystal structure (green), and (B, C, and D) engineered myoglobins to mimic the active site of heme/nonheme containing nitric oxide reductases (NORs) used as probes for metal- identity-dependent O2-reduction chemistry. This figure has been created using data from references (A) 832, 1091, 1092 and (B, C, and D) 1093. A note from our analysis of PDB 3M39 (C) and 3M3A (D): regarding the bonds drawn from the lower O atom of Glu68, we have chosen to draw a bond to the heme-Fe based on its shorter distance to that metal ion.
In the case of CcO, determining how Fe and Cu function together/cooperate is complicated; both heme-only and Cu-only enzymes (or coordination complexes) (see sections 2 and 3) are known to be capable of reducing O2 to water. Although the O2-bound (A/oxy) intermediate in the CcO cycle is often compared to the oxy-Mb species [rR spectroscopy for both give v(Fe-O) ~ 570 cm−1],483,1094 myoglobin is simply a dioxygen-transport protein (section 2.1.1), whereas CcO binds and reduces O2. Thus, one of the many important questions, which can be probed by CuBMb-type modeling, is what is the role of copper in effecting O2 reduction? Interestingly, UV–vis spectroscopy reveals that when the copper-free, deoxy-CuBMb model (434 nm) is exposed to air, incomplete formation of the six-coordinate oxy-form (418 nm) occurs, in contrast to WT-Mb where exposure to the same conditions yields exclusively oxy-WT-Mb. This result suggests that the active site mutations in CuBMb decrease the heme O2-binding affinity. Insertion of a AgI ion as a redox inactive mimic for CuI in the CuB site resulted in complete oxy-(AgI)CuBMb formation, an indication of regained/enhanced dioxygen binding affinity.1090 Additional evidence for cooperative heme-Fe and Cu interactions in CcO is that it is known to bind CN− in a bridging fashion,854,870,871,1095 resulting in spin-coupling of the metal centers. Copper(II)-bound CuBMb also binds CN− with high affinity (Kb = 143.9 mM−1; 20-fold greater than the Cu-free derivative), concomitant with a heme spin state change to low-spin (UV-vis Soret band shift from 407 to 425 nm) and evidence of antiferromagnetic coupling of the FeIII and CuII centers via quantitative EPR spectroscopic titration.1089
It is also of interest to uncover why exactly nature has chosen copper as the heme-iron partner in HCOs, when, for example, an analogous heme/nonheme iron motif is involved in biological NO reduction [by NORs, which likely share a common ancestor with CcO (see section 6.5.5)],1096 and its synthetic models1097 have been shown to effect O2-cleavage. Myoglobin scaffolds modified to contain a distal metal binding site with His29-His43-His64-Glu68 ligation (similar to the nonheme site in NORs, and therefore named FeBMb) were shown to uptake a single copper, iron, or zinc ion at that site with preserved structural arrangement, lending themselves as apt models for such a study (see Figure 140, B, C, and D). The oxygen reduction rates (k) and selectivity (S = % of O2 reduced to water rather than ROS such as hydrogen peroxide) for the CuI (k = 2.72 ± 0.1 μMs1-; S = 94%), FeII (k = 1.15 ± 0.07 μMs1-; S = 96%), and ZnII (k = 0.22 ± 0.02 μMs1-; S = 57%) variants highlight the importance of a redox-active metal in the nonheme or CuB site for controlling the rate and selectivity of the O2-reduction chemistry, with copper performing the best.1093 The enhanced reactivity observed is postulated to be due to the higher Eo of copper as compared to iron, resulting in the heme-copper derivative having the highest driving force for fast electron transfer in a reaction where ET is rate-limiting.1093 The presence of a copper ion was also shown to facilitate faster O2-binding in a chemical model system in homogeneous solution where catalytic O2-reduction to water was studied using a heme versus heme-copper construct and ferrocenyl complexes as one-electron outer-sphere reductants.1053 Together, these results indicate that the CuB site plays a critical role in enhancing the binuclear center's affinity for O2 and donates an electron to activate the (possibly metal-bridging) oxy species.
Recently, the insertion of other first row transition metals into the NOR model FeBMb template uncovered further important details pertaining to the role of the nonheme metal in the O2-reduction reaction.1098 Both cobalt and manganese are known to be active in metalloenzyme redox chemistry and importantly can bind in the nonheme site of FeBMb (UV-vis and crystallographic evidence), thereby making MnII-FeBMb and CoII-FeBMb good candidates for a systematic study with the previously described FeII-FeBMb (see just above). The binding affinity of these metals in the engineered nonheme site follows the Irving-Williams series trend (MnII < FeII < CoII) and interestingly also correlates well with total turnover numbers and selectivity (Figure 141). However, XANES spectroscopy confirmed that unlike the FeII-FeBMb and CuI-FeBMb models, in which the nonheme metals donate an electron to the heme-oxy intermediate, the MnII and CoII variants are not oxidized following subjection to single turnover conditions, similar to the nonredox-active ZnII variant.1093 It was also ruled out using spectroelectrochemical methods that the identity of the nonheme metal may be regulating the O2 chemistry by impacting the heme-iron reduction potential, given the small shifts in E°'heme regardless of the nonheme metal and the lack of correlation to the O2-reduction activity of the models.1093,1098,1099 To further understand the reason for the differences in selectivity across nonheme metal ion identities, the authors looked beyond the impact of tight metal binding which is undoubtedly important for stable, continuous turnover. The two factors which stand out with respect to the most selective model, CoII-FeBMb, are that (i) this variant forms the heme-oxy intermediate the slowest, and (ii) the CoII ion is the smallest and most Lewis acidic of the three ions employed in this study.1100,1101 The fact that the initial oxy intermediate is formed slowest in this case indicates the lack of electron transfer from the Co to generate PROS, the release of which can be detrimental to the enzyme. This study shows, however, that rather than using ET to activate the bound O2 moiety, the Lewis acidic nature of the CoII can impart a similar effect, with smaller risk of ROS release. Indeed, resonance Raman and X-ray crystallographic data, with support from DFT calculations, show a strengthen- ing/shortening of the Fe-O bond and weakening/lengthening of the O–O bond as well as an enhanced interaction with the distal O-atom as compared with the Fe or Mn variants.1098 Since many of the previous studies within the modified Mb paradigm, as well as other models and enzyme studies, have focused on the redox/electronic influences of the nonheme metal ion on HCO or NOR activity, the observations garnered from this study with Mn and Co have demonstrated that indeed other factors are important in determining O2 activation and selective four-electron reduction (i.e., tight metal binding in the nonheme pocket, Lewis acidic nature of the metal ion).
Figure 141.

(A) Plot showing O2-consumption over time for the MnII-, FeII-, and CoII-bound FeBMb models as well as for the case in which the nonheme site is empty (black trace); initial rates are included in (C). (B) O2-consumption rates for the same four variants as a function of the final products, H2O versus reactive oxygen species such as superoxide or peroxide. (C) Tabulated parameters including metal dissociation constants, initial rates of O2-consumption, total turnover numbers, and percentage of ROS formation for the four variants showing the trends described in the text. Adapted from ref 1098. Copyright 2017 American Chemical Society.
However, the question remains as to exactly how the Cu ion native to HCOs is involved in active turnover situations with dioxygen and how it may contribute to high selectivity for 4e− reduction to produce water. When CuSO4 is added to the oxy-CuBMb in the presence of a reductant (ascorbate) and redox mediator TMPD (TMPD = N,N,N',N'-tetramethyl-p-phenyl-enediamine) (plus catalase which inhibits the documented O2-reduction by Mb under these reducing conditions), the observed decay of the oxy-CuBMb Soret band at 418 nm follows biphasic kinetics (k1 = 0.028 s−1; k2 = 0.0083 s−1). The products, characterized by UV-vis spectroscopy (623 nm), and ESI-MS (619 m/z), could be assigned to predominantly CO-bound verdoheme, which is a product of oxidative heme degradation similar to those observed in heme oxygenase (HO) biochemistry (see Figure 142 and section 2.1.2.2).832,1090 Addition of the nonredox-active place-holders, AgI or ZnII, did not cause any spectral changes, which suggests that copper indeed plays a role in O2-reduction, here acting as a one-electron reductant to take the Fe-superoxide to a hemedegrading peroxide. Molecular HCO models have supported the importance of Cu in O2 binding and reduction selectivity by heme/copper systems as well.1031 However, when exploiting the “peroxide shunt reaction”, via addition of H2O2 to met-CuBMb (408, 508, 624 nm), the ferryl-heme was observed (419, 546, and 581 nm), regardless of whether the CuB-site was occupied with CuII, AgI, ZnII, or metal-free, as each of these situations have similar rates of ferryl heme formation (see Figure 142).1090 This result, where important redox steps are effectively skipped, highlights the fact that, while CuB plays a role in the redox chemistry, perhaps the more challenging and pertinent questions pertain to how and when protons are shuttled to the active site to direct the mechanism away from ROS release (vide infra). This chemistry also interestingly demonstrates how the local active site environment of a single model system can be modified to direct the mechanism/activity [i.e., O2-reversible binding (Mb chemistry), heme oxidation/degradation (HO chemistry), or O2-reductive cleavage (HCO or P450 chemistry)].
Figure 142.

Copper- and proton-dependent reaction pathways of CuBMb beginning with met-CuBMb (bottom, left). In the presence of CuII, oxy-CuBMb decays and self-oxidizes to yield verdoheme (HO-type heme degradation chemistry, top pathway and spectra). If there are protons available in the active site (provided by H2O2), the product is ferryl-heme (“peroxide shunt” mechanism known for P450s, HCOs), suggesting that proton shuttling to the active site and H-bonding residues are important for directing the mechanism toward O–O cleavage rather than ROS release. Adapted with permission from ref 832. Copyright 2008 Verlag Helvetica Chimica Acta AG.
5.4.2. Heme Environment Perturbations and Their Effects.
Another relevant distinction between CcO and the CuBMb derivatives is that they contain different heme cofactors [heme a in bovine CcO (and other hemes, b or o, in other organisms) and heme b in the original CuBMb], which have different reduction potentials (see Figure 143, top).1089,1102 An additional redox consideration stems from the fact that second coordination sphere residues have also been shown to influence metal ion reduction potentials or activity toward dioxygen.1103–1106 Interestingly, a single mutation of Ser29 in F33Y-CuBMb (which H-bonds to the iron axial His ligand and heme propionate) to an alanine residue results in an ~40 mV increase of heme Eo from +95 to +123 mV versus SHE with little change to the active site structure.1107 Note that the (S29A)F33Y-CuBMb model used in these studies contains a mutated active site Tyr and was crystallized for these studies without a Cu ion. Notably, heme a in bovine CcO has a 14-carbon farnesyl chain and an electron-withdrawing formyl group on a pyrrole ring, resulting in a large, positive heme Eo = +365 mV versus SHE. Attempts to directly exchange heme b for heme a in the biosynthetic model, S29A-F33Y-CuBMb, resulted in misfolding, so a series of smaller heme a analogues were used, including diacetyl, monoformyl, and diformyl derivatives (see Figure 143, top). Spectroelectrochemical measurements revealed that all three cases showed significantly more positive heme Eo relative to the Mb-native heme b, achieving the most positive reduction potential with the diformyl heme having Eo = +320 mV vs SHE. The O2-reducing capabilities of these mutants were evaluated in the presence of ascorbate as a reductant and TMPD as a redox mediator and reported in terms of selectivity toward 4e− reduction (as opposed to 1 or 2e− reduction and release of ROS).1108 With increasing heme Eo of the variants, O2-reduction activity and selectivity both improved, with the diformyl version showing a more than 5-fold increase over the initial F33Y-CuBMb (Figure 140, bottom) and remaining active for over 1000 turnovers.1107 Electron flux to the active site in CcO has been proposed to be a rate-limiting and product-directing factor during enzymatic turnover,679,876,1109 and the results from these experiments with engineered Mb-derived mimics of CcO suggest that the heme Eo is so positive in CcO to render it a better electron acceptor and thus able to drive O2-reduction. Interestingly, analogous transformations in WT-Mb also showed increased rates of O2-reduction; however, they also resulted in higher ratios of ROS formation, implicating the roles of H-bonding, CuB, and proximal Tyr in selectivity (vide infra).
Figure 143.

(Top) Myoglobin crystal structure and the heme-types which can be incorporated into modified Mb models (including hemes b and a, native to Mb and CcOs, respectively, plus synthetic variations) and their reduction potentials (vs SHE). (Bottom) Plot of oxidase activity (measured using an O2 electrode) as a function of heme reduction potential, in reference to wild-type Mb and bovine CcO. Adapted from ref 1107. Copyright 2014 American Chemical Society. Adapted with permission from ref 1102. Copyright 2017 Wiley-VCH.
Recently, heme reduction potential has further been probed in these modified Mb systems to determine exactly why and how such tuning of the heme cofactor affects oxidase activity. Several mutants were evaluated, and it was concluded that modification of heme reduction potential serves to control four key properties: (1) electron transfer rates, (2) O2-binding, and (3) dissociation rates, which all increase with increasing heme E°, and (4) O2-affinities, which decrease with increasing heme E° (dioxygen binding and dissociation rates were determined by flow-flash methods, wherein reduced heme-CO species, in the presence of solution O2, were flashed, so as to observe fast O2-binding kinetics).1102 These conclusions are additionally supported by observations from kinetic studies on HCOs from different species (i.e., containing different hemes), implicating evolutionary/adaptive advancements, for example, organisms that use specific hemes to adjust for low O2-concentrations in their environments.1102,1110,1111
It has been observed that HCOs with heme a or o have slightly different structural and/or functional properties from those containing heme b (i.e., O2 binding affinity);840,1112,1113 however, the most striking difference between a/o-type hemes and b-type hemes is the presence of a pyrrole hydroxyethyl-farnesyl group (see Figure 89C), alluding to the importance of this functional group in O2-reduction chemistry. The long hydrophobic farnesyl chain is believed to play a purely structural role, anchoring the heme in the proper place at the active site. Therefore, an investigation into the importance of the hydroxyl moiety was carried out by replacing the native heme b of CuBMb with mimics of heme o which contained a small monohydroxyethyl group on the pyrrole in place of the hydroxyfarnesyl.1114 While there was no observed correlation between the different heme reduction potentials (Ehemeb = 80 mV vs NHE; Ehemeomimic = 62 mV vs NHE) and their reactivities with dioxygen, another important conclusion was reached based on the combination of kinetic studies and computational modeling.1114 In the presence of reductant, and with CuI in the engineered CuB site, the CuBMb variant containing the o-type heme was reported to have significantly reduced (by ~20-fold) heme oxygenase activity (i.e., formation of verdoheme). In contrast, it was established that the heme b model reacts with H2O2 to instead give the ferryl oxo (and HCO biomimetic intermediate) instead of heme degradation, suggesting the presence of extra protons and/or proton delivery to the active site is crucial for directing the reaction outcome. Additionally, a computational model of the CuBMb active site containing the heme o mimic indicated bond distances consistent with an extended H-bonding network involving the heme peripheral hydroxyl group.1114 In native HCOs, the importance of such a hydrogen-bonding network (between the heme hydroxyfarnesyl group, cross-linked Tyr residue, and one or more water molecules) has been well-documented (see sections 4 and 5.4.4) for its role in gating of the proton pump and timely proton delivery to the bound O2 moiety during catalytic turnover. This heme replacement study with modified Mb proteins isolated this cause-effect relationship and cemented the participation of heme periphery H- bonding as an equally important factor as heme reduction potential in directing the O2-reduction reactivity of heme- copper systems (for further discussion, see section 5.4.4).
A thorough understanding of the bonding relationship between a heme-iron center and an O2 moiety is central to both heme-only and heme-copper systems due to the fact that the varying natures of this Fe-O bond give rise to the array of functions carried out by such proteins (i.e., O2 transport vs reduction). As discussed at length in this review, especially throughout this section, spectroscopic characterization of (heme)Fe-O2 intermediates in enzymes such as Hb, Mb, and HCOs are quite comprehensive, yet debate remains as to what distinguishes their electronic configurations and causes the different reaction outcomes. A very recent study involving the modified Mb models investigated the role of the heme iron center (especially the Fe redox potential) in directing heme/nonheme diiron systems toward either HCO or NOR activity. It is known that many HCO enzymes are capable of nitric oxide (NO) reduction and many NOR enzymes can reduce O2 (vide supra), but the fact that they are structurally very similar further complicates the task of deciphering the origin of their different functions. In the modified Mb models, by changing the identity of the incorporated heme or the H-bonding to the proximal His ligand (which effectively lowers the heme redox potential),309 they were able to tune the heme iron E°' and, in doing so, observed a significant enhancenment in NOR activity at lower heme potentials.1115 The key to NO reduction in NORs is balancing the energetic favorability of (i) NO binding, (ii) Fe-NO decay, and (iii) electron transfer to the (heme)Fe-NO intermediate, analogous to the necessity for HCOs to control O2 binding, ROS release, and PCET to the bound oxy ligand. All of these processes are affected by heme redox potentials. In this study, the results of various vibrational, spectroscopic, electrochemical, and computational methods performed on the different heme-containing model systems showed that these rate determining factors for NO reduction can be controlled by tuning heme E°ˊ to achieve ~35 turnovers (based on quantification of N2O production using GC/MS), which is more than any previous synthetic (small molecule) or engineered protein system.1115 This result was obtained with the FeBMb(MF-heme) protein which contains a monoformylated (Figure 143) heme with E°' = +53 ± 7 mV versus SHE. The significance of these results is further relevant for understanding the even wider variation in functions observed with other heme proteins as well (O2-transporting globins, vs O2-activating HCOs and P450s, vs NORs), which all have evolutionarily tuned heme environments and redox potentials.
5.4.3. Influence of His-Tyr Cross-Link Mimics.
Since the first reported CcO crystal structure,982 the active site tyrosine residue has been implicated as being integral to the mechanism of the O2-reduction reaction, as the His-Tyr covalent cross-link is unique to, and conserved in, HCOs and is essential for function (see section 4). Site-directed mutagenesis in enzyme redesign experiments from the Lu group have represented a valuable method for studying how and through what mechanism the Tyr affects this chemistry. Of the four electrons required to fully reduce dioxygen to water, two come from the iron (FeII → FeIV), one comes from the copper (CuI → CuII), and it is now widely accepted that the fourth derives from the tyrosine, a known redox-active amino acid, which importantly has also been considered as a proton source (via PCET, possibly utilizing an H-bonding network of active-site water molecules; see section 4, and vide infra).747,878 Investigations of its role using biosynthetic models have matured systematically, beginning with addition of Tyr in spatially logical positions of CuBMb. Interestingly, a Tyr placed 4 residues away from the cross-linked His as in bovine CcO (F33Y-CuBMb) pointed toward the CuB binding site, but the overall structure did not overlay well with the CcO active site; however, a Gly65Tyr mutation resulted in a better structural mimic, based on its computational model.1091 In the presence of excess reductant, quantification of the O2-reduction rates of these two modified Mbs was carried out following the consumption of dioxygen with time, using an O2 electrode and monitoring the products using 17O NMR spectroscopy (H217O) or ROS scavengers (O2•−, O22−), to determine the effect of active-site Tyr presence and positioning on functionality (with and without different metal ions present in the CuB site). Independent of CuB-site occupation, the G65Y-CuBMb model (with the larger degree of HCO structural overlap) showed the fastest overall rate (28 min−1), most turnovers (TON = 1056), and greatest selectivity for production of water versus ROS, as compared with WT-swMb, CuBMb, or F33Y-CuBMb.1091 These results demonstrated that the active site Tyr has the potential to influence the O2-reduction chemistry at the BNC in a positive manner, and further, highlighted the importance of its specific orientation.
In addition to defining the position/orientation of the Tyr residue, further studies were conducted to determine the significance of the covalent (Tyr)C-N(His) cross-link. Genetic incorporation of an unnatural amino acid, imiTyr (tyrosine with o-imidazolyl substitution), at position 33 (plus the Leu29His mutation used previously), afforded a new model, imiTyrCuBMb, which exhibited improved structural similarity with CcO, and comparable Tyr placement to the previously characterized F33Y-CuBMb (based on modeling), and offers the opportunity to compare {His and Tyr} versus {His-Tyr} (Figure 144, top). Dioxygen-reactivity studies showed that the presence of the physical covalent cross-link (as opposed to the presence but separate His, Tyr residues) significantly enhanced the selectivity of O2 reduction to water (rather than ROS) (see Figure 144, bottom), and that the imiTyrCuBMb model was catalytic to a TON > 1000 with repeated additions of O2.1108 This study paves the way for investigation into the evolutionary incorporation of this unique post-translational modification in HCOs and begs the question: how does the C-N bond modulate the chemical properties of the two residues (pKa/Eo values, and relatedly, H-bonding interactions, Tyr radical formation/stability, and changes to the CuB properties) to effect such enhancements in O2 reactivity?
Figure 144.

(Top) (A) Heme a3/CuB site of bovine CcO and (B) overlaid structural models of imiTyrCuBMb (green) containing the imidazole-phenol cross-linked, imiTyr, and F33YCuBMb (gray) which contains non-cross-linked His and Tyr residues proximal to the CuB site. (Bottom) (C) Rate of O2 consumption to yield water or ROS (superoxide, peroxide) in the reactions of WT-Mb and the two different mutants shown in (B), each with and without CuII in the Cub site. Conditions: [Mb variant] = 6 μM; [TMPD] = 0.6 mM; [ascorbic acid] = 6 mM. Adapted with permission from ref 1108. Copyright 2012 Wiley-VCH.
As the efficient delivery of H+/e− to the BNC oxy form in CcO is crucial to preventing ROS release, and the cross-linked Tyr244 is proposed to be a source of a proton and an electron, the effect of the cross-link on the fundamental properties of the Tyr-OH moiety have garnered significant interest. Independent analytical and computational studies on isolated phenolimidazole compounds have shown that an ortho-imidazolyl-phenol has a lower pKa (Δexpt = −1.4, Δcalc = −1.8), slightly more positive Eo (Δexpt = +69, Δcalc = +3 mV vs NHE), and a weaker O-H BDE (Δexpt = −0.3, Δcalc = −2.4 kcal/mol), with the values shown here representing the variation from the properties of the parent, unsubstituted phenol.853 These effects are supported by other phenol-imidazole model studies,851,852,1116,1117 and computational active site studies, which additionally estimated that such a cross-link stabilizes CuBII over CuBI by ~100 mV.1118 Genetic incorporation of unnatural tyrosine analogs (i.e., Tyr with various phenolic substituents) into the 33rd position of the established CuBMb1089 scaffold allows systematic investigation into the effects of pKa and Eo of the phenol-OH moiety (within the protein environment) on O2-reduction chemistry, including the apparent increase in selectivity (vide supra). As the independent model studies have shown, the most drastic effect of an o-imidazole is the observed decrease in pKa, so three Tyr analogs with electron-withdrawing substituents (2,3,6-trifluoro, 2,6-difluoro, and 2-chloro) and unsubstituted-Tyr (Figure 145, left) were the suitable candidates employed at the 33rd position (in addition to the two His mutations which help make up the CuB site) to establish a relationship between Tyr acidity and the O2-reduction activity/selectivity. These mimics encompassed a pKa range of 6.4 to 10.0 and a Ep range of 850 to 672 mV (for F3Tyr to Tyr), where Ep is the anodic peak potential at pH = 13. Interestingly, both the oxidase activity and selectivity for H2O production by the four variants showed an inverse relationship with pKa and (Figure 145, right) supporting the role of the Tyr as a proton donor and product regulator during turnover.1119 At physiological pH, the Ep values reported are uninformative, since reduction potential and pKa are interrelated, and at pH 7, where Tyr oxidation would be coupled to proton transfer, no trend was apparent. Therefore, to interrogate the Tyr's potential as an electron donor, CuBMb was modified to include a 3-methoxy tyrosine (OMeY) in the 33rd position, which has a similar pKa but a lower reduction potential (by 179 mV at pH 7) as compared to Tyr. The rate of O2-reduction and selectivity of F33OMeY- CuBMb was far superior to F33Y-CuBMb (15.0 μM min−1/82% H2O vs. 6.5 μM min−1/51%) and could perform more than twice as many turnovers (TON > 1100 vs < 500).1120 The results of the halogenated and methoxy Tyr mimics taken together strongly support the importance of the Tyr in O2 reduction by CcO, as a good proton and electron donor (with logical extension to activity under physiological conditions) and, in doing so, illustrate nature’s ingenuity in evolving the His-Tyr cross-link to accomplish efficient and selective cellular respiration.
Figure 145.

(Left) Crystal structure of the Phe33Tyr-CuBMb variant (PDB ID: 4FWX) and the pKa values of Tyr and its non-natural mimics which can be substituted in the 33rd position. (Right) Plots showing the linear correlations between the Mb variants with various Tyr33 analogs and their O2-reduction activity (top) or water production (middle) as a function of phenol moiety pKa, and O2-reduction activity as a function of phenol peak potential, Ep at pH = 13 (bottom). Adapted from ref 1119. Copyright 2015 American Chemical Society.
Direct observation of a tyrosyl radical (by EPR spectroscopy), which is postulated to exist in the PM state of enzyme turnover, is complicated by the presence of numerous redox-active amino acid residues in the large transmembrane protein (i.e., tyrosine, tryptophan, and cysteine) as well as the other paramagnetic metal sites (i.e., CuA, low-spin heme a). In fact, EPR signals ascribed to neighboring Tyr (other than the His-Tyr active site Tyr)887 or Trp889–891 residues can be generated and observed in native CcOs. However, the CuBMb biosynthetic models are easier to handle and modify, to specifically interrogate/probe the chemistry at the active site Tyr during reactions with O2. The tyrosine-containing F33Y-CuBMb scaffold, expressed without a Cu ion, was employed in reactions toward O2 (oxidase activity) and H2O2 (“peroxide-shunt” reaction) (see Figure 146A), and the reactions were monitored by X- and Q-band EPR spectroscopy. Following addition of O2-saturated buffer (and freeze quenching after <100 ms) or 1 equiv H2O2, a protein radical peak was observed (g ≈ 2) concomitant with a significant disappearance of the high-spin ferric resting state signal.1121 The hyperfine splitting of the g ≈ 2 signal resembled that reported in HCO,860 matched simulation of a Tyr• (Figure 146B), and was not present in reactions of O2/H2O2 species with WT-swMb. Mutagenic control experiments ruled out this radical character lying on any other redox-active residues in the Mb protein backbone, once again supporting the redox role of the active-site Tyr residue in O2-reduction by HCOs.1121
Figure 146.

(A) Reactions of the ferric or ferrous F33Y-CuBMb model with either H2O2 (peroxide shunt) or O2, respectively, showing a new X-band EPR signal at g ≈ 2, having the hyperfine (due to H-atoms) structure shown. (B) The experimental data can be simulated and matches well with the expected EPR signal for a Tyr radical species. Adapted from ref 1121. Copyright 2014 American Chemical Society.
5.4.4. Proton Delivery to the BNC and Importance of H-Bonding Networks.
Both the proton and electron flow into the binuclear active site are important considerations which effect O2-reduction. A wealth of experimental and computational data (reviewed herein) support the involvement of the Tyr in providing both a proton and an electron in order to effect O–O cleavage and therefore contributing to the pH balance and redox flux within the enzyme. Simulations of simultaneous H+/e− transfer from the Tyr indicate that this mechanism involves a reaction barrier over 10 kcal/mol higher than that calculated based on transition state theory and the experimentally observed lifetime of intermediate A/Oxy (Scheme 23).855,881 Importantly, introduction of an additional proton in the active site led to an agreement between experiment and theory on this point, and accordingly, an H-bonding network was implicated in achieving the proton transfer.747 In support of these theoretical and mechanistic studies, Lu’s biosynthetic Mb-models were able to contribute to this discussion with spectroscopic and crystallographic support. The model enzyme containing a Tyr near to the heme and engineered (empty) CuB binding site, F33Y-CuBMb, was previously shown to be capable of reducing O2,1091 (vide supra) using a chemical reductant (ascorbate). Tandem use of cryoreduction and subsequent stepwise annealing with EPR spectroscopy allowed for the observation and characterization of EPR-active intermediates trapped along the O2-reduction pathway, including a ferric peroxo (g = 2.24, 2.13, and 1.95), ferric hydroperoxo (g = 2.34, 2.19, and 1.94), inferred EPR-silent ferryl-oxo, and finally ferric-hydroxo (g = 2.52, 2.15, and 1.90) species (see Figure 147, right). X-ray crystallographic data was obtained for the oxy-F33Y-CuBMb at 1.27 Å resolution showing an H-bond network of two water molecules linking the engineered Tyr33 and His29 to the terminal O atom of the oxy-ligand (Figure 147, left).1122 These interactions are believed to facilitate the necessary proton delivery to the oxy-heme to initiate oxidase activity, as they are not observed in the WT-swMb crystal structure which is incapable of O2-reduction.
Figure 147.

(Left) Crystal structure of the oxy-F33Y-CuBMb (1.27 Å resolution; PDB ID: 5HAV) with two water molecules in the active site pocket. (Center) Cryoreduction method for formation and trapping of (hydro)peroxo intermediates (X = active site His or Tyr). (Right) The EPR spectra of oxy-F33Y-CuBMb cryoreduced at 77 K showing a ferric-hydroperoxo (H-bonded to an active site water, green) which decays (and loses H-bond stabilization) upon stepwise annealing. Adapted from ref 1122. Copyright 2016 American Chemical Society.
5.4.5. Electron Flux and Electrochemical Results.
Timely and controlled electron flow to the BNC is important to achieve efficient and complete O2 reduction to water and avoid the release of detrimental partially reduced reactive oxygen species, and this phenomenon has been successfully studied with small molecule model systems appended onto electrodes.1044 Incredibly, entire hemoproteins131,1123 from the Lu group (the modified myoglobin biosynthetic models) can also be studied in this manner, following apoprotein reconstitution around a heme group tethered to a self-assembled monolayer (SAM) on a gold electrode (Figure 148).1124 Fast and efficient electrocatalytic O2-reduction by the appended G65Y-CuBMb biosynthetic model was followed by the recently established surface-enhanced resonance raman spectroscopy-rotating disk electrode (SERRS-RDE) method by the research group of A. Dey.1052 In this reaction, the final catalytic step (slow ferric-hydroxide reduction/dissociation), is hurdled in an electron transfer “shunt” mechanism with the overall effect being faster, more efficient reduction,1125 all supporting the notion of the importance of electron flow to the active site in maintaining the correct chemistry.
Figure 148.

Scheme describing the procedure for tethering a biosynthetic Mb model to a mixed self-assembled thiolate monolayer on a gold electrode for electrochemical experiments. A modified heme (“Hemin-yne”) cofactor is covalently attached to the monolayer via a “click” reaction, and the apoprotein is reconstituted in situ around the heme. Adapted from ref 1125. This material is licensed under a Creative Commons license, Attribution 4.0 International (https://creativecommons.org/licenses/by/4.0/).
5.5. Conclusions from the Chemistry of HCO Model Systems
Overwhelming evidence from small-molecule, and biosynthetic model studies (with computational support) emphasize nature's purposeful construction of certain details and seemingly minor interactions within a complex enzyme, CcO, which have critical importance for accomplishing one of the most important reactions in biology. Certain insights into the four-electron reduction of dioxygen, including the difficult O–O bond cleavage step, are attainable only via systematic and rigorous investigation of structure–function relationships in model systems, be they small molecular weight synthetic systems or engineered protein constructs. From defining the roles of metal centers and other redox cofactors to identifying steric and H-bonding interactions unique and necessary to the chemistry of the BNC, scientific modeling allows for a considerably enhanced chemical and spectroscopic understanding of catalytic mechanism. In addition to building upon fundamental biochemical knowledge of cellular respiration, the oxygen reduction reaction (ORR) and its related reverse, water splitting or the oxygen evolution reaction (OER), are extremely important in energy research as the anodic fuel half-cell reaction and the basis for (artificial) photosynthesis, respectively. It is no surprise then that we look to nature for inspiration when designing man-made systems.6,1126,1127 To meet an exponentially increasing demand for devices to be faster, smaller, more efficient, etc., the molecular basis for catalysis in systems such as O2-reduction must be acutely understood, so the recent advances in biomimetic modeling reviewed here, represent a broadening potential to reach or surpass nature’s efficiency.
6. COORDINATION CHEMISTRY PERSPECTIVES ON HCO HEME-CU ACTIVE-SITE STRUCTURES AND O2-REDUCTION
6.1. General Perspectives
To fundamentally understand the factors which govern the O–O bond reductive cleavage chemistry carried out by heme-copper oxidases during cellular respiration, it is certainly beneficial to consider the chemistry of dioxygen in related biological systems. For this reason, we have reviewed here important recent advances in synthetic inorganic chemistry pertaining to heme-O2 and Cu-O2 complex properties and reactivities, in addition to those of heterobinuclear heme-Cu systems. The key findings of these studies will aid in a pursuit of understanding why Nature precisely constructed the unique heme-copper active site in HCOs to achieve this seemingly simple yet undoubtedly complicated and important biological reaction of dioxygen reduction to water. As discussed, the manner by which heme and copper centers cooperate to facilitate the O–O cleavage event is surely complex; however, the incorporation of insights gained from copper-O2 and heme-O2 systems into heme-copper models may hold the key in determining the fundamental properties which promote O2 reductive cleavage and thus inspire the design of artificial systems to achieve this chemistry for practical applications.1128 Again, we are compelled to remind the reader that the goal of model chemistry is not to recreate an exact active site under physiological conditions, but rather, it is to mimic one or more aspects in a system which can be manipulated in order to ascertain certain structure-function relationships which may govern enzymatic reactivity when applied in those systems.
Heme enzymes are known to bind and reduce dioxygen, forming a variety of interesting reactive intermediates. In both enzyme and model systems, certain factors have been found to determine (i.e., hinder or enhance) the strength of binding and mode of O–O cleavage (i.e., homolytic vs heterolytic), and these details are of great interest, not only since they shed light on mechanistic details of biological systems but also due to the crucial roles of practical catalytic dioxygen reduction in alternative energy applications such as fuel cells. Not surprisingly, the electronic structure at the iron center plays a large role in the stability of certain intermediates as well as reaction outcomes, and it can be modulated (as seen in model systems, section 2) by substitution(s) on the porphyrin itself or by changing the axial sixth ligand donor type or strength.1129 Additionally, second coordination sphere effects have proved to be important for stabilizing certain iron-oxy derivatives, such as Fe-OO(H) species (see section 2), which is related to the putative IP intermediate in the proposed HCO catalytic cycle and discussed further in this section as well as in section 4.2.3. Importantly, computational and spectroscopic methods employed in systematic studies of model systems have identified trends (i.e., in rR spectroscopic data, vide infra) which can aid in identification of enzymatic intermediates and characterization of the active site environment during catalytic turnover. Understanding all of these electronic and structural/secondary factors can help build upon our understanding of HCOs, which (i) may contain a variety of hemes based on the organism/subfamily, (ii) have an axial histidine residue rather than Cys as in cyt P-450 monooxygenase and many heme peroxidases, and (iii) are believed to employ a hydrogenbonding network to facilitate delivery of H+/e− to the O2-derived moiety.
The exact role of the critical CuB site in the unique heterobinuclear active site of HCOs has long been debated, and therefore, it is of utmost importance to understand how the copper center in heme-Cu model systems influences O2-reactivity and the accompanying electron- and proton-transfer processes required for the O–O bond breaking step. Evidence from structural studies and various spectroscopic inquiries lend support to a scenario in which O2 binds to the fully reduced state (R) CuB before it transfers to heme a3. The CuB ion is what an O2 molecule “sees” first, and its affinity, in terms of both kinetics and thermodynamics, is tuned depending precisely on its ligation; a trigonal planar His3 coordination makes the metal ion less accessible, thus less reactive, as opposed to a pyramidal His3 ligation which would have open access to a fourth ligand (i.e., O2) and be very reactive. As described, synthetic model studies have shown that O2-binding to Cu in a (tetradentate-ligand)CuI complex can occur in a near diffusion controlled manner, consistent with the known extremely rapid binding of dioxygen to CcO at the CuB and/or Fea3 metal ions at the BNC.940,975,976,978,1053 Also recall discussions in section 5.2.3.2, which summarize critically important recent investigations implicating CuB as a “sensor”, closely linking geometric changes and enhanced molecular oxygen binding to CuB (in order to initiate the O–O reductive cleavage process) only when sufficient water/protons have moved nearby.879,981 As was reviewed further in section 5, there are two functional catalytic O2-reduction (to water) heme-Cu model systems, one homogeneous1053 and the other electrochemical,1044 that implicate the copper ion as being critical in O2-binding. Following transfer of O2 from CuB to the reduced heme, the remaining copper(I) may transfer its electron(s) and/or facilitate stabilization of O–O cleaved products, such as oxide, −OH, or H2O. So, is copper simply the chosen −OH or OH2 acceptor, or rather is its purpose related to the proton-pumping function of HCOs?843,846 Taking perspective from Cu-only systems (biological and inorganic models),625 the unique coordination environment and positive reduction potential has recently suggested that CuB’s function is primarily as a regulatory control center for efficient electron flow and eventual rereduction of the active site in order to repeat/turn over the catalytic cycle.
These and other possibilities have been the focus of studies on heme- and copper-containing model systems described herein (section 5), as well as the motivation for future investigations of this type (see section 6.4). While, as described here, mononuclear or bridged O2-derived fragments bind to copper in a variety of geometries and types, in fact, the requirements for Cu-ligation (donor atom types or numbers, coordination geometry, or reduction potential) or the nature/properties of ideal proton and/or electron-donating substrates have not been elucidated in detail with respect to O–O cleavage preferences. However, even thus far, ligand design in copper-containing model systems has aided in discovering and optimizing structure-function relationships, and some of these findings (e.g., as described in sections 5.2.6 and 5.2.7) may be directly applied to synthetic heme-Cu systems as well as understanding of various biological metalloenzyme systems.
Undoubtedly, the active site Tyr residue presents an important aspect to be incorporated into model systems, as the proposed H-bonding and eventual PCET to the O atom proximal to the Cu ion leads to successful homolytic O–O reductive cleavage and formation of the PM intermediate, FeIV=O/CuII-OH/TyrO•. The His-Tyr cross-link itself is expected to alter both the electronic structure of the Cu ion as well as the pKa and O–H BDE of the TyrOH residue, so these aspects of HCO modeling and reactivity studies are important when it comes to analyzing the reactivity in the context of the H+ and/or e− donating substrates. Finally, in the context of HCO chemistry, it will be useful to understand the origin and/or purpose of the very positive CuB reduction potential (vide supra, section 4).
As it has been reviewed above, both heme-only and copper-only systems are capable of reducing O2 to water, so why has Nature partnered them in heme–copper oxidases, how do these two metal ions cooperate, and why the Fe/Cu combination, since it is known that the related NORs (heme/nonheme di-iron) can also perform O2-reduction, although not with great efficiency.834,1096 Figure 149 shows a generalized view of conceivable heme–O2–Cu derived intermediates (i.e., superoxo or peroxo), based on precedence from dicopper and/or dicobalt coordination chemistry and partly from known heme chemistry. Low-spin μ-1,2-peroxo heme–Cu synthetic constructs, which contain a sixth axial heme ligand, are now well-known (see section 5). A transient heme(FeIV)-(bis-μ-oxo)-CuIII species (Figure 149, bottom left), intuitively seems unlikely to form, in part because it would require an unreasonably short Fe⋯Cu distance, the likes of which has never been observed in CcO X-ray or computational BNC structures, and therefore, it has never been considered as a possible initial O–O cleavage product during the catalytic cycle in HCOs. Compounds with μ-η2:η2 side-on and μ-η2:η1 side-on/end-on peroxo bridged structures are also known, but only for high-spin systems where no strong proximal axial ligand (e.g., an imidazolyl group) is present (see section 5). As concerns HCO BNC-mediated O2 reductive-cleavage, we speculate that many new scenarios become part of the discussion if the proximal histidine imidazole ligand can or does dissociate during turnover.
Figure 149.

On the basis of established dicopper, dicobalt, and some heme-containing systems, depicted here are generalized possibilities for Fe– Cu heterobinuclear structures possessing dioxygen-derived bridged groups.
6.2. What is the Real Identity of the Proclaimed Peroxo-Bridged Heme-Cu "As-Isolated" Cytochrome c Oxidase Protein Structures?
Detailed biochemical and biophysical experiments, in concert with advanced computational methods have contributed to detailed structural and electronic descriptions of the various intermediates during the oxidative (Figure 150) and reductive phases in the CcO catalytic cycle. Dioxygen binding to the reduced BNC, R, leads to generation of A, which most likely goes through a hydroperoxide (IP) on the way to cleaving the O–O bond.873,909,918,920,1130 However, for the sake of discussions here and later in this section, we have added another plausible intermediate, Peroxy, which precedes IP,747,855,909,917,919 and by adding this, we imply that reduction by CuBI and protonation (the H+ ion derived from the cross-linked Tyrosine) may occur stepwise. The development of more sophisticated computational methods, coupled with higher quality crystal structures, has enabled meticulous analysis of intermediates both on and off the enzymatic pathway. Reanalysis of the structural assignments of these intermediates have led to both clarification and more questions as to the chemical nature and identities of some of these species and their physiological relevance.
Figure 150.

First steps in the oxidative phase of the CcO catalytic mechanism. Dioxygen binding to the reduced BNC, R, leads to generation of oxy intermediate, A. An electron transfer from CuB leads to the formation of the putative bridging Peroxy intermediate. Protonation of the Peroxy intermediate would lead to the generation of a (possibly bridging) hydroperoxide, IP. See the text for further discussion.
As described in detail, synthetic systems with a peroxide moiety bridging a heme-copper system have been spectroscopically characterized and are useful tools for gaining insight into the coordination chemistry (i.e., structures) and O–O cleavage reactions facilitated by such systems (see section 5). There are likewise many “resting state” (also called “as-isolated” or “oxidized” form) CcO protein structures described in detail and represented as peroxide-bridged heme-copper BNC species. In this section, a coordination chemistry perspective is taken, via discussions of specific structural parameters (O–O, M–O, and M··· M distances), of these supposed heme-peroxo-copper X-ray structures. As discussed below, it is concluded that in most, if not all, cases, these published CcO structures do not contain a dianionic peroxide (O22−) moiety bridging the iron and copper metal ions.
6.2.1. Known Small Molecule Copper, Cobalt, Heme, and Heme-Copper Peroxo Structures.
As has already been discussed in previous sections of this review, there have been multiple crystallographically characterized metal-“oxy” complexes for both enzymatic and model systems. For a direct comparison of structural features of these numerous species, selected distances and angles are presented in Table 5. It is often appropriate to compare the metal-bound reduced dioxygen species to that of its ionic or protonated form. The O–O bond length is a good indicator as to the redox state (i.e., dioxygen, superoxide, or peroxide) of the metal-bound O2- fragment. As is evident from Table 5, the O–O bond lengths in iron(III)- and copper(II)-bound superoxide complexes (1.23–1.29 Å, entries 1–5) are in line with that known for potassium superoxide (1.28 Å). The O–O bond lengths in heme superoxide complexes are somewhat shorter than that of potassium superoxide due to the fact that the iron in these complexes is proposed to be highly covalent (and therefore the O2-fragment is not fully reduced to superoxide) as discussed in section 2.2.2. X-ray structure determination of hydroperoxo (−OOH) moieties bound to metal ions have also been reported for heme and (di)copper systems. The O–O bond lengths in these complexes (1.46–1.52 Å, entries 6 and 9–11) are similar to that of free hydrogen peroxide (1.47 Å). Structures of a small molecule acylperoxo dicopper(II) compound, and hydrogen peroxide (as H2O2) bound to a zinc(II) ion complex have also been reported and have similar O–O bond lengths (Table 5, entries 12 and 13). Multiple bimetallic bridging peroxide species have been reported in synthetically derived systems containing copper (see section 3) and cobalt (Figure 151; Table 5, entries 14–18, 23–26, and 29–34), and these complexes also possess O–O bond lengths close to that observed for hydrogen peroxide. A survey of dicobalt complexes has been included in our discussion/analyses because many dicobalt(III) peroxo and superoxo complexes are known and structurally characterized, and cobalt(III) stands as a good surrogate for when iron(III) would be present in heme a3 structures, having the same ionic charge and very similar size.
Figure 151.

Depiction of crystal structures of synthetically derived trans- and cis-μ-1,2-peroxodicobalt(lIl) species with listings of relevant structural parameters. Created using data from refs 1135–1140.
While the O–O bond length confirms the presence of a bridging peroxide ligand, it is also interesting to examine the metal–oxygen and metal–metal distances. As is expected, Com–Operoxo bond lengths (1.88 Å) are, on average, slightly shorter than those for copper(II)–Operoxo (1.92 Å) due to the additional positive charge on the cobalt(III) ions. The range of metal-metal distances is similar for both dicobalt(III)- and dicopper(II)-peroxides but is dependent on the orientation of the two metal ions with regard to the peroxide ligand (cis or trans). Bridging peroxides with the cis conformation tend to have smaller metal–metal distances (3.03–3.80 Å) than those with the trans conformation (3.98–4.69 Å) (Table 5).
Two X-ray structures have been reported for peroxide bridging between four (instead of two) copper(II) ions (Figure 152). These complexes possess both cis- and trans-peroxide moieties (Table 5, entries 19–22, 27, and 28), which have comparable structural parameters as their dicopper counter-parts. However, [CuII4(L−)2(O22−)(OH)2]2+ has the longest copper–copper distances of all reported copper bridging peroxides (4.85 Å, entry 22, Table 5, longer by more than 0.1 Å for both cis and trans conformers), presumably due to the geometry enforced by ligand constraints (Figure 152 B).
Figure 152.

Depiction of crystal structures of synthetically derived trans- and cis-μ-1,2-peroxotetracopper(ll) species with listings of relevant structural parameters. Created using data from refs 1133 and 1134.
As described in section 5, there is a plethora of spectroscopic data for both high- and low-spin heme-peroxo-copper complexes but only one single-crystal X-ray structure, that from Naruta's high-spin complex lacking a proximal (axial) base ligand (Figure 95C). However, there are detailed and reliable computational lowest-energy structures available for many of the model compounds, including all of the low-spin examples (Table 5, entries 35 –41),497,747,919 and for one (entry 35),932 XAS investigations have complemented and were found to be consistent with the DFT calculations. These calculated heterobimetallic peroxides have O–O, M–O, and M···M distances which are all similar to the homobimetallic trans-peroxides mentioned above, with Fe–O bond lengths shorter than the Cu–O bonds (Figure 153), as expected, since higher-oxidation state iron(lIl) versus copper(ll) ions, respectively, are involved. Notably, for iron and copper, the M–Operoxo bond lengths are less than 2.0 Å and often considerably less. Remarkably, the longest metal–metal distance in these structures is 4.48 Å (Figure 153C, entry 37 in Table 5), which is shorter than the longest reported metal– metal distances for dicobalt(III) (4.62 Å) and dicopper(II) trans-peroxides (4.69 Å with one other unusually long example, vide supra).
Figure 153.

Calculated structures of model system heme-peroxo-copper complexes (A–F) and calculated bridging peroxide in CcO (H), starting with the X-ray structure of reduced CcO (G), with relevant structural parameters listed. Created using data from refs 497, 747, and 919.
6.2.2. Structures of CcO Reported to be Peroxo-Dianion Bridged Heme-Copper Species.
X-ray structural determinations for the resting state of CcO described as containing a bridging heme-peroxo-copper species have been reported by multiple research groups (Figure 154, entries 42– 50 in Table 6).842,911,985,988,1141,1142 These complexes are not believed to be relevant to catalytic turnover, but their structures could still provide fundamental insight into the binding of O2-derived species which are possible in the BNC of CcO. The exact nature of the O2-reduced moiety and its formation in these “as isolated” structures has been disputed.20,535,1142 Furthermore, the comparisons of structural, along with rR spectroscopic data of one “as isolated” CcO containing bridging heme-copper peroxides presented below are not consistent with numerous protein and model complex examples of heme, copper, heme-copper, and cobalt (hydro)-peroxides (Figures 155, 156, and Table 6). If these protein X- ray structures do contain a peroxide ligand between the two metal centers, how exactly could a peroxide fragment be generated within the BNC? This matter has merited some discussion: for example, the oxidized metal ions present are capable of being reduced by the X-ray beam,1143 giving the reduced state (FeII and CuI) which might react with dioxygen to form a bridging peroxide complex. Similarly, intense X-ray irradiation may otherwise result in the formation of peroxide from direct reduction of O2 or (as has been suggested) result from the coupling of hydroxyl radicals which may have derived from the irradiation of water.20,988,989
Figure 154.

CcO enzyme X-ray structures proposed to contain peroxo (O22– )-bridged heme-copper centers, with relevant structural parameters listed. Numbers under each structure correspond to entry numbers in Table 6. See the text for further discussion. Created using data from refs 842, 911, 985, 988, 1141, and 1142.
Figure 155.

Comparisons of (A) O−O, (B) M−O, and (C) M···M distances found in “oxidized” (also referred to as “as-isolated” or “resting-state”) CcO crystals reported to contain a bridging peroxide moiety in the BNC. The red dots represent the particular protein Xray structures shown in Figure 154 and listed in Table 6. Bonding parameters reported for putative peroxo bridged heme-Cu BNC protein X-ray structures, some reported at quite high resolution, are outside the range established for peroxo-bridged small molecules. An exception appears to be protein structure entry 50, PDB: 3HB3. See the text for further discussion.
Figure 156.

Selected rR v(O−O) values for various protein and synthetically derived heme systems, taken from Tables 3 and 7. See the text for further discussion and explanation.
In order to add insight from a fundamental, structural inorganic point of view, average values of bond distances and angles for metal-“oxy” species are presented in Table 6 along with the corresponding data for reported CcO “heme-peroxo-copper” centers. For model system bridging peroxide complexes, average O–O bond distances range from 1.40 to 1.43 Å, slightly shorter than that found in hydrogen peroxide (1.47 Å). However, the O–O bond distances in the published CcO peroxide structures represented in Figure 154 and Table 6 (with the exception of entries 49 and 50; Table 6) have an average value of 1.61 Å and are all greater than 1.5 Å; therefore, these O–O distances are inconsistent with having a peroxide character. The short O–O bond ascribed to the structure for entry 49 (1.11 Å) is even less than that of dioxygen (1.21 Å) and therefore cannot be a peroxide. The metal–oxygen bonds in the CcO X-ray structures (again, except for entry 50 in Table 6) are also elongated compared to model complexes; all are greater than 2.1 Å and have an average value of 2.28 and 2.23 Å, for Fe–O and Cu–O, respectively. These are considerably longer than the average metal–oxygen distances found for synthetic peroxide-bridged systems, including the [(porphyrinate)FeIII-(O22−)- CuII(ligand)]+ and (one) protein BNC structures characterized by DFT calculations (see Figure 153 and Table 6). These inconsistencies in metal ligand bond lengths lead us to suggest that the oxygen atoms in these “as isolated” protein structures, entries 42–49, may not even be coordinated to either metal center. It should be realized that in “true” iron(lIl), cobalt(lIl), and copper(ll) peroxo-bridged synthetic complexes, where low molecular weight compounds allow for highly accurate structural characterization, the Fe–Operoxo, Co–Operoxo, O– O(peroxo), and Cu–Operoxo bond lengths lie in a narrow range. Additionally, these are strong (and short) bonds because of the +3 or +2 metal ion oxidation state and the “hard” anionic character of the Operoxo atoms. FeIII–Operoxo bonds are under 1.85 Å (down to even 1.80 Å; Table 5) and CuII–Operoxo bonds average 1.93 Å from a total of 26 structures (Tables 5 and 6; determined by small molecule X-ray crystallography or computational studies). Further, high-resolution X-ray crystallographic and DFT computational structures reveal bond distances consistent with our summaries here (i.e., for a chloroperoxidase protein hydroperoxo structure and myoglobin-peroxo protein structures, O–O bond lengths are less than 1.50 Å and Fe–O bond distances average 1.83 Å) (Table 5).
It is important to recognize that in protein X-ray crystallography, especially for very large proteins such as HCOs, determining the precise identity of small molecules (e.g., O2 or reduced derivatives such as water, hydroxide, or other) which lie between heme a3 and CuB is not an exact science, and the determination of highly accurate bond- distances (metal–O and especially O–O) is very difficult. However, one can consider metal–metal distances as a more precise structural indicator, since these heavy (or heavier) atoms possess very large electron densities (compared to O atoms), and thus the exact position of the Fe and Cu atoms in the protein matrix (and therefore the Fea3···CuB distance) are accurately determined in the enzyme structures. All of the CcO peroxide X-ray crystal structures represented in Figure 154 (except entry 50 in Table 6) have Fea3···CuB separations of greater than 4.8 Å (4.87 Å avg). These distances are measurably longer than all of the small molecule bimetallic bridging peroxide complexes presented here which (again) lie in a very narrow range with M1···M2 = 4.42 Å; this is an average of 28 examples of trans peroxides, putting the two metal ions as far away as possible, constrained by normal bonds associated with the electronic structure and bonding for a peroxide moiety containing a O–O single bond (Tables 5 and 6). There is one case with a large CuII-(O22–)-CuII distance of 4.691 Å, where multiple hydrogen bond donors to the peroxide moiety, and ligand steric effects in the secondary coordination sphere, may contribute to the observed Cu···Cu elongation (entry 18 in Table 5). There is only one exception to our statement that peroxo-bridged dimetal complexes have significantly shorter M···M separations compared to those observed for the proposed peroxo-bridged [hemea3-(O22−)-CuBII structures; Figure 154]: this is one of the tetracopper(ll)-peroxide complexes, [CuII4(L−)2(O22−)- (OH)2]2+ (Figure 152B, entries 21 and 22 in Table 5), whose long Cu··· Cu distance (>4.8 Å) is likely due to ligand constraints. Taken from the viewpoint of structural inorganic/coordination chemistry, we believe the assignment of these protein structures as “peroxo-bridged” (Figure 154) is a misrepresentation.
The possible exception for the “as isolated” crystal structures of CcO as containing a peroxo-bridged BNC is entry 50, which may indeed comprise a heme-peroxo-copper species (Table 6). This X-ray structure reported by Michel and co-workers (PDB: 3HB3)985 has structural parameters more closely matching those of the bimetallic, small molecule, synthetically derived model systems. The O–O, M–O, and M···M distances all reside in the ranges of bimetallic bridging peroxides (Figure 155) and this is the only structure that fits with all three parameters. This complex is the only one reported that contains an O–O bond that is close to that of hydrogen peroxide (1.49 vs 1.47 Å, respectively). The Fe–O and Cu–O bond distances are in line with metal–oxygen bonds found in dicobalt(III) and dicopper(II) trans-peroxides, as well as the Cu–O bonds in the calculated heme-peroxo-copper complexes (Figure 155B). While the metal–metal distance is somewhat long (4.62 Å), it is shorter than the longest distance in dicopper(ll) trans-peroxide species (4.69 Å) and only slightly longer than the longest distance in dicobalt(lIl) trans-peroxide complexes (4.53 Å), see Figure 155 C. From an examination of the experimental procedures for generating the X-ray quality crystals of the putative peroxo complexes (42–50, Table 6), we wish to note that it is only for entry 50, published by Michel and co-workers,985 that a “redox-cycling” procedure was used, wherein dithionite was added to the CcO preparation (to fully reduce the enzyme) and this was followed by air-oxidation, all of which was repeated several times prior to a final chromatographic purification step.
To summarize the above discussion, we can say that with the detailed analysis given above, and from our inorganic/coordination chemistry perspective, we suggest that the assignments for the above X-ray structures as bridging peroxides (with the possible exception of entry 50 in Table 6) may have been misinterpretations. The structural parameters (O–O, M–O, and M···M distances), when compared to those of well-characterized coordination complexes, do not allow for the designation of a peroxide ligand bridging the heme and copper ions (see below for discussion of what these species may be). However, the crystal structure described by Michel and co-workers (entry 50 in Table 6)985 fits more closely with the parameters from model systems, and its formulation as a heme–peroxo–copper species may be correct.
Noodleman and co-workers have also computationally questioned the nature of at least one example: the proposed peroxide-bridged CcO “as-isolated” protein X-ray structure determined at 1.95 Å resolution, entry 45 (Table 6).915 Their results showed that an assignment of a peroxide dianion bridging oxidized iron and copper centers is unlikely since the geometrically optimized structures of Fea3III-(O22−)-CuBII exhibited large deviations in the Fe···Cu or O–O distances relative to the experimentally observed crystal structure.988 Instead, the Noodleman group's best calculated fit, consistent with empirical data, was a reduced Fea3II-(HO–O−)-CuBI species, where the Fea3 site consists of a mixture of low-spin, intermediate-spin, and high-spin states, and where the peroxidic oxygen coordinating Fea3 is protonated. Entry 45 may indeed contain a (hydro)peroxide moiety, as the O–O bond length falls within the range of O–O distances for globin and copper(II) hydroperoxides and near those found in heme protein hydroperoxides (Table 5). Our opinion is that the computational fitting leads to a reduced metal ions state because for the lower oxidation state and thus lesser positive charge, M–Operoxo bonds would be considerably longer, which would increase the computationally derived Fe···Cu distance to be closer to that found for the protein X-ray structure. However, as a relevant aside, we note that dianionic or monoanionic (hydro)peroxo groups rarely, if ever, exist with metal ions in a reduced state. These authors commented that the origins of this putative bridging peroxide structure was that the (hydro)peroxo ligand was most likely formed due to X-ray radiation, perhaps through the generation and recombination of hydroxyl radicals in the active site to form H2O2 and therefore is not likely to be of physiological importance.988
6.2.3. Resonance Raman Spectroscopy of an "As Isolated" CcO Peroxo Complex.
Resonance Raman spectroscopy has long been used to elucidate the extent of reductive O2-activation in CcO during the oxidative phase of the enzymatic cycle.322,894,895,1144 Typically when coupled with isotope labeling, v(O–O) obtained from rR spectroscopy can give an accurate description as to the degree of O2 reduction in the solution phase and provide a method for interpreting metal-dioxygen intermediates. Unfortunately, when studying enzymatic systems, v(O–O) values are not always easily obtained, and instead, the corresponding v(Fe–O) values are at times used to identify an observed intermediate (Table 7).Iron–oxygen stretches are not as diagnostic of metal-dioxygen intermediates as O–O stretches, however, and may lead to potentially dubious assignments (see below).
Table 7.
Tabulated v(O–O) and v(Fe–O) Values for Ferric Heme Superoxo, Peroxo, and Hydroperoxo Entities Characterized for Enzymes and Model Systemsa
| entry no. | complex | v(O–O) (cm−1)c | v(Fe–O) (cm−1)c | ref |
|---|---|---|---|---|
| ferric heme superoxo | ||||
| (1) | Mb WT | 1103 | 578 (−29) | 1145 and 1146 |
| (2) | Mb H64L | 580 (−25) | 1147 | |
| (3) | Cyt P450 WT | 1139 (−66) | 546 (−31) | 573 |
| (4) | Cyt P450 D251N | 1136 (−66) | 537 (−30) | 574 |
| (5) | LPO pH 8.2b | 534 (−22) | 461 and 1148 | |
| (6) | LPO pH 5.6b | 558 (−24) | 1148 | |
| (7) | [(THF)(F8)FeIII-O2• −] | 1178 (−64) | 568 (−24) | 513 |
| (8) | [(PIm)FeIII-O2• −] | 1180 (−56) | 575 (–23) | 518 |
| (9) | [(6L)FeIII-O2• − FeII-Cl] | 1176 (−63) | 574 (–25) | 1149 |
| (10) | [(6L)FeIII-O2• −] | 1176 (−64) | 572 (−24) | 1149 |
| (11) | [(NMePr)FeIII–O2• −] | 575 (−21) | 524 | |
| (12) | [(NMePr)FeIII–O• −···Cu1] | 570 (−26) | 524 | |
| (13) | [(TMPImXO2EtFeIIIO• −] | 582 (−26) | 122 | |
| (14) | [(MeTHF)(F8)FeIII-O2• −] | 1175 (−63) | 574 (−24) | 497 |
| (15) | [(α4-Fe(CO2Me)4)–O2• −] | 1004 (−53) | 581 (−22) | 1237 |
| 967 (−56) | ||||
| (16) | [(α4-FeEs4)–O2• −] | 581 (−22) | 1238 | |
| (17) | [(α4-FeEs4)–O2• −] | 585 (−24) | 1238 | |
| (18) | [(FeIII(OPhP))–O2• −] | 1147 (–59) | 570 (−22) | 1239 |
| (19) | [(FeIII(HOPhP))–O2• −] | 584 (−27) | 1239 | |
| (20) | [Fe(PIM)–O2• −] | 578 (−26) | 1239 | |
| (21) | [(TMP)FeIII(O2• −)···CuI(LN3-OH)] | 576 (−26) | 935 | |
| ferric heme end-on peroxo | ||||
| (22) | Cyt P450 D251N | 792 (−38) | 553 (−27) | 574 |
| (23) | LPO pH 8.2b | 570 (−22) | 1148 | |
| (24) | [(TMPImXO2Et)FeIII-O22−] | 808 (−37) | 585 (–25) | 122 |
| ferric heme hydroperoxo | ||||
| (25) | Mb WT | 617 (−15) | 1146 | |
| (26) | Cyt P450 WT | 799 (−40) | 559 (−27) | 573 |
| (27) | Cyt P450 D251N | 774 (−37) | 564 (−28) | 574 |
| (28) | LPO pH 5.6b | 604 (−24) | 1148 | |
| (29) | [(TMPIm)FeIII-OOH] | 810 (–47) | 570 (−26) | 490 |
| (30) | [(TMPImXO2Et)FeIII-OOH] | 807 (−31) | 575 (–25) | 122 |
| (31) | [(TMPImAOH)FeIII-OOH] | 807 (−40) | 579 (−28) | 568 |
| (32) | [(TMPImAOEt)FeIII-OOH] | 811 | 576 | 568 |
| (33) | [(α4-FeEs4)-OOH] | 830 (−47) | 631 (–37) | 1052 |
Additional entries for biological and synthetic oxy-heme FeIII-superoxo complexes are available in the review by Momenteau and Reed.79
Lactoperoxidase.
Values in parentheses are Δ18O2 shifts.
Both model systems and isolated protein intermediates containing oxy-heme, heme–(hydro)peroxide, and heme– copper peroxide complexes have been studied using rR spectroscopy. Synthetic FeIII–O2•− complexes, representing the oxy-heme state found in enzymes, show a v(O–O) spanning a narrow range from 1147 to 1180 cm−1 (Table 7, entries 7–10, 14 and 18), reflecting significant covalency in the Fe–O2 bond, and reduction of dioxygen to a superoxide moiety (i.e., a species with an O–O bond-order (BO) of 1.5 (BO = 2.0 for dioxygen and 1.0 for a peroxide moiety). A notable exception is reported by Dey and co-workers1237 who characterized a synthetic heme FeIII–O2•– supported by a hydrogen bond scaffold (Table 7, entry 15), and found it to exhibit a very low v(O–O) relative to the aforementioned synthetic examples. In actual oxy-heme proteins, the observed v(O–O) frequency, where available, is still assignable as a superoxo moiety but is lower relative to model compounds, spanning values from 1103 to 1139 cm−1 (Table 7, entries 1– 3), suggesting that a greater degree of charge transfer from FeII to O2 is occurring, weakening the O–O bond. Further reduction of the oxy-heme complex yields a (hydro)peroxo species. There are relatively few examples of mononuclear end-on ferric heme peroxo complexes, most likely due to the strong basicity of the terminal, anionic peroxide O atom; it may be stabilized through H-bonding (which could be the case for protein entries 22 and 23 in Table 7) or it should readily undergo protonation and formation of a hydroperoxo entity. Of the few examples where rR spectroscopic data exists for peroxy-anion species, both model and protein structures appear to give similar v(O–O) frequencies of 792 and 808 cm−1, respectively (Table 7, entries 22 and 24). Protonation of peroxides to form hydroperoxides generally results in a decrease in the v(O–O) in both model and protein systems, compare entries 22 to 27 and 24 to 30 (Table 7). These shifts in the v(O–O) are much less dramatic (i.e., as small as 1 cm−1) as compared to conversion from the oxy form (FeIII– O2•–) to (hydro)peroxide;122 this is expected since, for the former case, there is no change in BO. Reported v(O–O) values for heme peroxo and hydroperoxo species show considerable overlap (Figure 156), and as such, it is almost impossible to distinguish between the two solely from the O– O stretching frequency, unless both compounds can be generated and otherwise unambiguously identified for the same system.
Resonance Raman spectroscopy has also been extensively utilized for synthetic heme–copper peroxide compounds to evaluate the structure of the O22− core (see section 5.1). Reported v(O–O) values for these low-spin peroxides range from 758 to 876 cm−1 (Figure 156 and Table 7). At least one “as-isolated” structure putatively containing peroxide in the BNC (based on X-ray crystallography) has been studied using rR spectroscopy and found to have v(O–O) of 755 cm−1; this was assigned to the peroxo O–O stretching frequency of a bridging Fea3III-(O22−)-CuBII moiety (Figures 156 and 157).1150 In lieu of an 18O2 isotope labeling experiment, this v(O–O) parameter was tentatively assigned by adding cyanide to displace the putative peroxide ligand in this as-isolated protein, in order to generate a difference spectrum. Unfortunately, no v(Fe–O) stretch could be identified using this experimental approach, and without an 18O2 isotope labeling experiment the reported 755 cm−1 assigned as an O– O peroxo stretch is not conclusive. It should be noted that a v(O–O) of 755 cm−1 for this putative peroxo-protein BNC species is lower (thus possessing a weaker O–O bond) than all of the reported protein and synthetic heme superoxides and end-on (hydro)peroxides, as well as synthetic low-spin heme- copper peroxides (see section 5.1.1). The 755 cm−1 species could possibly be formulated as a high-spin ferric heme– copper peroxide, which is reported to exhibit v(O–O) frequencies ranging from 747 to 812 cm−1 for heme-peroxo- Cu constructs possessing tridentate copper ligation (Table 3, Figure 157). To achieve such a structure would require the protein axial histidine donor to dissociate from the iron (the axial histidine is 2.13 Å away in that particular protein crystal structure: Figure 154, A; PDB: 5B1A; entry 42 in Table 6), thus converting from a low-spin to high-spin state, and for Fea3III and CuBII to accommodate μ-η2:η2-O22–(side-on) coordination (see section 5.1.1). This has not been observed in the protein BNC and is unlikely to occur, we believe, given the steric constraints of the active site.
Figure 157.

Selected rR v(O–O) and v(Fe-O) values for low-spin synthetic systems plus bovine heart CcO (see Table 3 for complete overview). The 755* cm−1 O–O stretching frequency has been observed for one example of an “as-isolated” protein structure putatively possessing a bridging peroxo group. A **571 cm−1 Fe–O stretching frequency, or values very close to this, are found for the oxy-heme, intermediate A, occurring in CcO turnover. See text for further discussion and interpretations.
6.2.4. What Could These Protein Structures Really Be?
We do not know! All the CcO protein X-ray structures are beautiful, and they provide a great deal of information beyond the hemea3···CuB BNC, such as insights into water channels, juxtaposition of helices or protein subunits, etc. However, as detailed above, and based primarily on geometric-structural criteria such as Fea3···CuB separations, and Fe–Operoxo, CuB– Operoxo, and peroxo dianion O–O bond distances reported from structural refinement, we do not believe that they are O22− bridged structures where the peroxo group ligates both to Fea3 and CuB. Mainly, for all of these protein structures (Figure 154), the Fea3···CuB distance is too long and proper chemical bonds to the metals could not form. We cannot determine exactly what ligand(s) are present instead of the peroxo dianion; however, in Chart 3, we illustrate some structures that perhaps may better describe these enzyme X-ray structural determinations.
Chart 3.

Plausible Formulations for Structures Which Were Otherwise Formulated As Peroxo-Bridged Fea3···CuB Protein Structures
If the putative peroxo-bridged Fea3···CuB protein X-ray structures are not peroxo dianions, what if they are just hydroperoxo groups, bound to one or the other metal ion (Chart 3, A and B). The freedom of such a ligand not being bridged to both metal ions simultaneously should allow for the observed Fea3···CuB separations and avoid the structure solutions which have unreasonably long M–Operoxo bonds. As discussed (vide supra, sections 6.2.2–6.2.3), any peroxo formulation for the protein structures in Figure 154 is not unambiguously supported by spectroscopic measurements, but this “negative” observation cannot rule out a peroxo species. Further, we could speculate that the group(s) between heme and Cu are just interstitial (i.e., molecules that were “trapped” there upon crystal formation), so the neutral H2O2 molecule containing structure (Chart 3, C) is one possibility.
Another possibility is that the intervening ligands consist of two hydroxide groups, one bound to each metal ion (Chart 3, structure D); of course, this should lead to structure refinement solutions giving short (i.e., < 2.0 Å) M–Ohydroxide bonds, which was not the case. Or perhaps, a ligand or group to be found at the BNC residing between heme a3 and CuB is H3O2−, which is, in fact, a well-known entity in structural inorganic chemistry.1151–1156 Of course, H3O2− is equivalent to H2O/−OH, but the arrangement of H3O2− is distinctive in bridged dimetal complexes; we illustrate this and provide a known crystal structure picture in Chart 4. As the data summarized in this figure shows, there exists a near-linear O·· H··O moiety (where the H atom is halfway between the O atoms). This O···O separation lies between 2.4 and 2.5 Å, and it is known that exceptionally short distances 2.3–2.4 Å can occur for bent O–H··O hydrogen bonds resulting from environmental and/or solid state effects.1157,1158 Distinguishing such a moiety from that of a peroxidic O–O bond at ~1.45 Å should be clear. As regards, M···M separation in these H3O2−, bridged dimetal complexes, they vary but are always over 5.1 Å and usually longer (Chart 4). Recall that in the protein structures claimed to contain a peroxide bridge, the Fea3···CuB separation is between 4.8 and 4.9 Å, with one exception (Figure 154; Table 6). Thus, perhaps an H3O2− formulation for the protein structures is not valid.
Chart 4.

H3O2− Bridging Group in Inorganic Coordination Chemistrya
aSee the text for discussion and relevant implications. (B) was created using data from ref 1152.
Two other points concerning the possible presence of H2O/−OH molecules between Fea3 and CuB are as follows: (i) Wikström and co-workers suggest such a species as the formulation for the “relaxed” (inactive for enzyme turnover), oxidized species O (see section 6.3). Noodleman and coworkers have derived a computational structure with H2O/−OH, which is also discussed in section 6.3. In fact, Noodleman's structure does appear to have an H3O2− bridging, with an O···O separation of 2.6 Å and a Fea3···CuB distance of 5.53 Å (a much longer distance than in the putative peroxo bridged BNC protein X-ray structures shown in Figure 154).
We would also like to point out something about the structure of water molecules in protein X-ray structure determinations, which perhaps bears on the difficulty of unambiguously assigning or identifying similarly small groups such as peroxo (O22−), hydroperoxo (−OOH), hydroxo (−OH), or water. In looking over a sampling of protein X- ray crystal structure refinements, to our admittedly untrained perspective (as we are not X-ray crystallographers), we see publications which report the “finding” of water molecules with very short O···O separations, much smaller than expected in the normal structure of water. In fact, oxygen atoms from two H2O molecules with one H atom in between are almost always found in a near-linear arrangement, (H)O-H···O(H)2 with O···O = 2.6–2.8 Å.1159 Values down to ~2.4 Å are known; however, even “bent” or “bifurcated” H bonds do not get the O atoms much closer.1160–1162 These arrangements and atom separations also apply to ions [e.g., aquated hydronium ions1163 or H3O2− anions (vide supra)]. Thus, we suggest that water–water O···O separations in protein structures that were found to be far less than 2.6 Å (Figure 158, highlighted in green) are likely not correct. The van der Waals radius of an oxygen atom is 1.52 Å, and so, O atoms from two nearby H2O molecules cannot approach each other to closer than ~3 Å unless an H atom lies in between. Of course, X-ray structures may show disorder of water molecules (i.e., partial occupation of more than one x, y, and z position), and this might explain those apparently short O···O distances. Authors of papers reporting the protein structures shown here in Figure 158 did not note disordered waters among those shown. In structure 3S8F, HOH605 (a) is said to be H-bonded to the peroxo O atom bound to CuB as shown; however, 3.57 Å is not an H- bonding distance, and as we have discussed above, we do not in the first place think that any of the protein structures in Figure 154 are peroxo complexes (see discussion in section 6.2.2). HOH664 (b) is found 2.26 Å away from HOH605 (a). This is an untenable O···O separation; it is too short. We suggest that these findings for water molecule definition in protein structures highlight the difficulty in assigning O atom containing fragments in proteins. This of course includes noting that it is very difficult to unambiguously assign and locate molecules such as O2, HO2, H2O2, HO2−, O22–, H2O/−OH, or −OH/−OH moieties expected to bind between heme a3 and CuB in cytochrome c oxidase BNCs. This is all to say that the factors discussed in this section most certainly have contributed to what we feel has been the misrepresentation of the protein structures in Figure 154, as peroxo-bridged species.
Figure 158.

Representation of heme centers in several protein X-ray structures, highlighting clusters of water molecules defined by the structure solutions. Highlighted in green are distances between water O atoms which we think are chemically unreasonable. See text for further explanation and discussion.
6.3. Coordination Chemistry Suggestions Concerning Other Intermediates in the CcO Catalytic Cycle
6.3.1. Identity of the Initial Oxy-Compound (Intermediate A).
The original term “compound A” (subsequently also referred to as intermediate A) was first coined by Chance et al. when reporting the initial intermediate observed following flash photolysis of frozen (T = −100 °C) CO coordinated CcO in the presence of dioxygen.1164,1165 This initial oxygenated species, intermediate A, is short-lived (t1/2 = 12 s, −100 °C) possessing absorption features at 425 and 591 nm.1164 The electronic structure of intermediate A is best described in the continuum between a ferric superoxide and a ferrous dioxygen adduct, the same description as that found in oxy-globin proteins1164 (see section 2.2 for discussion on oxy- heme protein electronic structure). This interpretation is widely accepted by the broad community of scientists who study HCOs. However, we think there is merit in re-examining the experimental and theoretical lines of evidence used to characterize intermediate A and wish to posit that this assignment as a ferric superoxide is not as solid as it appears to be.
6.3.1.1. Optical Spectroscopic Evidence for Intermediate A.
Initial assignment of intermediate A was partially made based on similarities in the optical spectra relative to other well-studied oxy-heme proteins. Chance et al. noted the observed Q-band (591 nm) of intermediate A was similar to oxy-Mb (577 nm) and oxy-Hb (582 nm) (Table 8). Additional support for this assignment include (1) an EPR spectrum showing no significant oxidation of the heme or copper center, and (2) treatment of enzyme with ferricynanide, where all metal centers except heme a3 are oxidized, resulting in identical optical and EPR spectra, implying redox chemistry by CuB does not occur during formation of intermediate A.1164 Taken together these results favor an FeIII-(O2•–)···CuI description for intermediate A. Further examination of these results open the possibility for other interpretations. The 591 nm band for intermediate A is indeed similar to oxy-heme proteins (Table 8, entries 1–4) and several synthetic heme copper models corroborate this feature (Table 8, entries 11–12). However, the absence of an EPR signal does not solely support an FeIII- (O2•–)···CuI description but also magnetically coupled FeIII/CuII (hydro)peroxide species. Additionally, one would expect a copper signal in the EPR spectrum for the ferricyanide treated intermediate A; the resulting Fea3III–(O2•–)–CuBII formulation would have at least one unpaired spin. The absence of both iron- and copper-based signals leaves open the possibility that intermediate A might be a magnetically coupled Fea3III/CuII peroxy-like intermediate. Another explanation could be ferricyanide is not a strong-enough oxidant to oxidize the high potential CuB center, and one is simply observing an identical binuclear center as compared to the untreated samples.
Table 8.
Tabulated Soret and Q-Band Values for Heme and Heme-Copper Complexes in Both Protein and Synthetic Model Systems
| entry no. | complex | Soret band (nm) | Q-band (nm) | ref |
|---|---|---|---|---|
| ferric superoxidef | ||||
| (1) | Oxy-Mb | 415 | 541, 577 | 1167 and 1168 |
| (2) | Oxy-Hb | 418 | 544, 582 | 1169 |
| (3) | Oxy-Cyt P450 | 417 | 556, 580(sh) | 1170 |
| (4) | Oxy-CPOa,b | 428 | 553, 587 | 1171 |
| (5) | [(THF)(F8)FeIII-(O2• −)] | 416 | 536 | 498 |
| (6) | [(PIm)FeIII-(O2• −)] | 423 | 534 | 420 |
| (7) | [(PPy)FeIII-(O2• −)]b | 419 | 555 | 420 |
| (8) | [(6L)FeIII-O2• −] | 422 | 542 | 504 |
| (9) | [(6L)FeIII-O2• − FeII-Cl] | 418 | 536 | 1149 |
| (10) | [(TMPIm)FeIII-(O2• −)-CuI(LN4-OH)]+b | 425 | 550, 590 | 485 |
| (11) | [(TMPImXO2Et)FeIII(O2• −)]b | 428 | 550, 592 | 122 |
| (12) | [(NMePr)FeIII-(O2• −)-CuI)+b | 422 | 542 | 524 |
| (13) | [(MeTHF)(F8)FeIII-O2·−] | 416 | 535 | 497 |
| (14) | [(TMP)FeIII(O2• −)···CuI(LN3-OH)] | 414 | 935 | |
| (15) | [α4-Fe(CO2Me)4)-O2• −] | 522, 545 | 1237 | |
| (16) | [(S4-FeFc4)-O2• −] | 425 | 522, 545, 584 | 1238 |
| (17) | [(α4-FeEs4)-O2• −] | 426 | 543 | 1238 |
| ferric peroxides | ||||
| (18) | Mb-(O22−) | 433 | 540, 567 | 571 |
| (19) | Cyt. P450-(O22−) | 439 | 530, 561 | 574 |
| (20) | [(TMPImXO2Et)FeIII(O22−)]− | 430 | 568, 610 | 122 |
| (21) | [(TMPIm)FeIII(O22−)]− | 440 | 574, 615 | 490 |
| (22) | [(TMP)FeIII(O22−)]− | 439 | 569, 614 | 490 |
| (23) | [(TPP)FeIII(O22−)]− | 437 | 565, 610 | 539 |
| (24) | [(OEP)FeIII(O22−)]− | 423 | 543, 573 | 539 |
| (25) | [(F8)FeIII(O22−)]− | 435 | 540, 560 | 495 |
| ferric hydroperoxide | ||||
| (26) | Mb-(OOH) | 413 | ~540, 573 | 1172 |
| (27) | HRP-(OOH)c | 419 | 526, 556 | 1173 |
| (28) | HO-(OOH)d | 421 | 530, 557 | 180 |
| (29) | CPO-(OOH) | 449 | 546, 576 | 1171 |
| (30) | Cyt P450-(OOH) | 437 | ~530, 561 | 574 |
| (31) | [(TMPIm)FeIII-OOH] | 428 | 532, 562, 608 | 490 |
| (32) | [(TMPImXO2Et)FeIII-OOH] | 429 | 566, 608 | 122 |
| (33) | [(TMPImAOH)FeIII-OOH] | 429 | 538, 568 | 568 |
| (34) | [(TMPImAOEt)FeIII-OOH] | 430 | 570, 610 | 568 |
| high-spin heme-copper(L)3 peroxos | 938 | |||
| (35) | [(2L)FeIII-(O22−)-CuII] + | 419 | 488, 544, 575 | 938 |
| (36) | [(TMP)FeIII-(O22−)-CuII(LN3-H)] + | 419 | 543 | 935 |
| (37) | [(TMP)FeIII-(O22−)-CuII(LN3-ONa)] + | N.R. | 546, 570 | 935 |
| (38) | [(F8)FeIII-(O22−)-CuII(AN)] + | 418 | 538, 561 | 516 |
| (39) | [(F8)FeIII-(O22−)-CuII(LMe2N)]+ | 420 | 540, 567 | 513 |
| (40) | [(F8)FeIII-(O22−)-CuII(MeTHF)3]+ | 418 | 480, 538, 565 | 497 |
| low-spin heme-copper(L)3 peroxos | ||||
| (41) | [(DCHIm)(F8)FeIII-(O22−)-CuII(ANE)] + | 421 | 535 | 928 |
| (42) | [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)3] + | 421 | 531 | 497 |
| high-spin heme-copper(L)4 peroxos | ||||
| (43) | [(TPP)FeIII-(O22−)-CuII(tpa)] + | 420 | 557, 612 | 934 |
| (44) | [(TPP)FeIII-(O22−)-CuII(5-Metpa)]+ | 930 | ||
| (45) | [(TMP)FeIII-(O22−)-CuII(5-Metpa)] + | 420 | 557, 612 | 931 |
| (46) | [(TMP)FeIII-(O22−)-CuII(LOH)] + | 429 | 558, 612 | 486 and 935 |
| (47) | [(TMP)FeIII-(O22−)-CuII(LOMOM)] + | 421 | 557, 615 | 935 |
| (48) | (6L)FeIII-(O22−)-CuII] + | 418 | 561, 632 | 936 |
| (49) | [(sL)FeIII-(O22−-)-CuII] + | 936 | ||
| (50) | [(F8)FeIII-(O22−)-CuII(TMPA)] + | 418 | 560 | 498 |
| (51) | [(F8)FeIII-(O22−)-CuII(LN4OH)] + | 417 | 560 | 937 |
| (52) | [(F8)FeIII-(O22−)-CuII(LN4OMe)] + | 937 | ||
| low-spin hem-copper(L) 4 peroxos | ||||
| (53) | [(DCHIm)(TACNAcr)FeIII-(O22−)-CuII] + | 428 | 933 | |
| (54) | [(TMPIm)FeIII-(O22−)-CuII(LN4-OH)]+ | 429 | 538 | 485 |
| (55) | [(TMPIm)FeIII-(O22−)-CuII(LN4-OMOM)] + | 429 | 537 | 485 |
| (56) | [(DCHIm)(F8)FeIII-(O22−)-CuII(TMPA)] + | 426 | 535 | 497 |
| (57) | [(DCHIm)(F8)FeIII-(O22−)-CuII(DCHIm)4] + | 423 | 537 | 497 |
| carbonyl | ||||
| (58) | Hb-CO | 419 | 539, 569 | 1174 |
| cytochrome c oxidase | ||||
| (59) | Oxy-CcO | 425 | 591 | 1164 and 1166 |
| (60) | CO-CcO | 430 | 589 | 1164 and 1166 |
Chloroperoxidase.
v(O-O) not reported.
Horseradish peroxidase.
Heme oxygenase.
Not reported.
See also the “Note Added in Proof’, for reference to additional examples.
Wikström and co-workers1166 have reported kinetic studies on the first 200 μs following oxygenation of CcO using similar flash photolysis techniques utilized by Chance and observed a Soret feature for intermediate A at 425–430 nm.873,1166 In the same study, the O2 binding kinetics were measured, and a large dissociation constant (koff) of 40000 s-1 was calculated. Comparison of the reported Soret band with other oxy-heme proteins reveal it is red-shifted relative to oxy-globins containing axial histidine ligation but is in the range of oxidase heme proteins containing axial cysteine ligation and synthetic heme superoxides (Figure 159). However, several heme-only and heme-copper peroxides contain Soret bands which overlap with intermediate A and thereby obfuscates assignment via optical spectroscopy. The large koff rate for O2 binding is comparable to the upper bounds found for myoglobin (ca. 27000 s-1)79 and much greater than that found in hemocyanin (ca. 2750 s-1),19 the dicopper protein which binds O2 as a η2:η2 peroxide (See section 3.2).
Figure 159.

Reported Soret band values for heme and heme-copper dioxygen adducts found in protein and synthetic model systems. See text for further discussion.
6.3.1.2. Resonance Raman Spectroscopic Evidence for Intermediate A.
Resonance Raman spectroscopic studies of intermediate A reveal an 18O sensitive stretch at 571 cm−1, which is assigned as the v(Fe–O) for the now O2-coordinated compound, A/oxy (Figure 160, A/oxy). Mixed 18O16O labeling experiments pointed to O2 being coordinated to Fea3 in a bent, end-on fashion similar to O2 coordination in globin proteins.895 The observed v(Fe–O) stretch of 571 cm−1 (Figures 157 and 160) is very similar to the v(Fe–O) detected in oxy-myoglobin and oxy-picket-fence-porphyrin (569 cm−1 1145,1175 and 572 cm−1 1010 respectively), and based on these similarities, A was assigned as an FeIII–(O2•–) species (also see section 4).20
Figure 160.

Selected rR v(Fe–O) values for various protein and model heme systems, taken from Tables 3 and 7. See the text for further discussion and explanation.
Unambiguous assignment of intermediate A based solely on the Fe–O stretching frequency may not be possible as the observed value overlaps with end-on (hydro)peroxides found in proteins and our well-characterized synthetic low-spin heme–copper peroxo complexes (Figure 160). Further, even the 18O2 derived shift for superoxo v(Fe–O) and peroxo v(Fe–O) parameters are the same, all in the 20–30 cm−1 range, see Table 7. Without the accompanying v(O–O) value, and given the current data, one can only unambiguously conclude that an O2 derived fragment (oxidation state unknown) is coordinated in an end-on fashion to iron.
However, we note that the v(O–O) is notoriously observed to not be resonance-enhanced in oxy-heme proteins,20,524,1176 and such is also the case for CcO, lending further support to the assignment of A as an oxy-heme (ferric-superoxide) species. As such, Collman and co-workers found that when replacing Fe for Co in their CcO model complex, both v(O– O) and v(Co–O) values were observed for the oxy intermediate, while only the v(Fe–O) was observed for the iron analog.524 On the other hand, the degree of resonance enhancement is difficult to assess in intermediate A due to the short lifetime of the compound. All vibrational data is evaluated based on time-resolved difference spectra where the extent of accumulation of intermediate A is unknown. The lack of empirical O–O vibrational data does not allow for assessment of the degree of O2 reduction for intermediate A, and while the most probable structure is a ferric superoxide, the so-called ferrous oxy-heme, as found in other oxy-heme proteins, the body of work in our opinion does not fully rule out other peroxide level intermediates. As stated by Yoshikawa and Shimada,20 a definitive structure of intermediate A is needed.
6.3.1.3. Computational Investigations into Intermediate A.
Computational studies have been thoroughly utilized in studying the distinct catalytic intermediates and transition state structures in the enzymatic cycle of CcO.873,1130,1177 Calculations regarding the BNC structure of the oxy form have yielded Fe–O distances in line with other oxy-globin intermediates, consistent with an assignment of a FeIII– (O2•–)---CuI species.903,909 Yet, the same (two) calculated oxy- heme structures yield O–O distances (1.31 A) which are elongated compared to enzyme oxy-heme structures [e.g., myo- or hemoglobins, Tables 5 and 6, 1.24 Å (avg)] and lie closer to bond distances observed in heme-(hydro)peroxides (average O–O distance of 1.36 A; Table 6, also see Figure 155). If one were to suppose that A was in fact a bridging peroxide intermediate [i.e., FeIII– (O22-)– CuII] (e.g., Figures 150 and 161, C, peroxy), proton transfer from the nearby cross-linked tyrosine would give a bridging hydroperoxide intermediate that we have referred to as putative CcO turnover intermediate Ip, (Figure 161 D). As already mentioned above, solely from the observed bond stretching frequency v(Fe–O) = 571 cm−1, we see that it is not only in the range for oxy-heme FeIII-superoxides but also is within the range of a number of known peroxo FeIII– (O22–)– CuII complexes (Table 3). The ambiguity in the assignment for intermediate A is thus again highlighted by these observations.
Figure 161.

Top left protein structure, (A), depicts a fully reduced form of the BNC, referred to as intermediate R in CcO reaction sequences (see Scheme 23). The top right structure, (B), is a computational model of CcO reaction intermediate A (from Blomberg and co-workers) which is formulated in the cytochrome c oxidase literature as an oxy-heme {Fea3-(O2•–)} complex. The other four structures shown (C–F) are optimized computational models of peroxo or hydroperoxo-bridged heme-Cu assemblies, all previously discussed in the present review article. Species (D) is what we have referred to as CcO intermediate, IP (also see Scheme 23). A comparison of structural parameters (as shown) might suggest that intermediate A could well be formulated as an Fea3III–(O22−)-CubII; complex. Created using data from refs 747, 903, 917, 919, and 1178. See the text for further discussion.
However, some DFT calculations on the CcO catalytic cycle also supply an argument against this assignment of A as a bridging peroxide based on calculated relative energies of a ferric superoxide versus a (hydro)peroxo-bridged BNC. The O–O cleavage step has been extensively studied, with some debate as to possible species involved going from intermediate A to intermediate p,855,909,1179 with all calculations being examined in light of the experimentally determined O–O cleavage total activation energy (12.4 kcal/mol)881 and also the enthalpic barrier (6.3 kcal/mol), which is important to note since calculated activation energies correspond to the enthalpy of activation.1177 Multiple groups have calculated the energy of the IP intermediate (formally Fea3III-OOH-CuBII ) and determined that it is significantly higher in energy than intermediate A (Fea3III–O2•– CuBI) by 6–20 kcal/mol.920,1177,1179 Thus, it is not reasonable to assume that such a peroxidic intermediate would be more highly populated than the ferric superoxide species. It is interesting to note that in one recent theoretical treatment from Noodleman and coworkers, formation of peroxidic intermediates is overall downhill from a ferric superoxide (Figure 162; 3 → 4).909
Figure 162.

Calculated structures and relative energies of proposed intermediates in the oxidative phase of the catalytic cycle of CcO, from Noodleman and co-workers.909 The pathway from 2H (reduced binuclear center intermediate R) to 6 contains low-energy barriers and is overall energetically downhill. The three so-called “true peroxo” states (4, 5, and 6) are all calculated to be lower in energy that state 3 (intermediate A). Further descriptors of the various calculated intermediates are 2H: {H376H+, FeII_CuI, H282H, Y'OH}; 3: {H376H+, FeII–O2_CuI–H2O, H282H, Y'OH}; 4: {H376, H2O FeIII–O–O–CuII, H282H, Y'OH}; 5: {H376H+, FeIII–O–O–CuII, H282H, Y'O‒}; 6: {H376H+, FeIII–O–OH–CuII, H282H, Y'O‒}; 72: {H376, H2O FeIV=O, H2O–CuII, H282‒, Y'O·•}. H376 is a nearby histidine residue with H-bonding interactions with copper ligand H283 through an aspartate residue and water molecule. H282 is one of the histidine residues bound to Cub. Y'O(H) is the Tyrosine residue cross-linked to His233; it may be deprotonated (i.e., Y'O‒) or have lost an H atom and be in its oxidized radical form (i.e., Y'O•).
As previously described (vide supra), Ip, sometimes called the “true peroxide” has been predicted to form transiently via reduction of intermediate a 909,1130 As mentioned (section 4), experimental evidence now exists showing that a reduction reaction occurs prior to O–O bond scission to form P (FeIV= O); the electron transfer is proposed (with support from DFT calculations) to occur after formation of IP (Fea3III-OOH- CuBII), resulting in a new intermediate (dubbed IP−) that is formally described as Fea3III-OOH···CuIB.918 So, even though an electron transfer step has recently been confirmed to occur after formation of intermediate A and prior to O–O cleavage to form intermediate P, this does not preclude the possibility of the intermediacy of a peroxide species.
To conclude, our present supposition is that intermediate A could be a peroxo or hydroperoxo species. An oxy-heme species must form during the course of O2 reductive activation (i.e., binding to the reduced enzyme followed by reduction- protonation), but the first experimentally observed oxygenated intermediate need not be a ferric superoxide. It is possible intermediate A may be a mixture of superoxo and (hydro)- peroxo species owing to the incredible efficiency of the BNC to reduce O2; experimentally, the incredible speed of the reaction with dioxygen precludes observation of a single species. On the basis of the experimental evidence discussed in the previous subsections (6.3.1.1 and 6.3.1.2), intermediate A is very likely a ferric superoxide (oxy-heme) species, although this assignment is not absolutely verified.20
6.3.2. Could the Conversion of F to OH Occur Through a Bridging Heme-oxo-Copper (FeIII–O–CuII) Intermediate?
Mechanistic insight into the conversion of F to OH (Scheme 23, Scheme 24) in the catalytic cycle of CcO is of great interest since little information regarding the intricacies of this transformation has been gained using spectroscopic methods. However, there have been multiple computational studies,901,903,1180 and these suggest that in both the F and OH states, there is significant CuI/Tyr-O· character, which may be responsible for the high redox potential of the F state.859,875,901 Nevertheless, we will mainly consider the CuII/Tyr–O− resonance form in our discussions throughout this section. In the transition from F to OH, the ferryl intermediate is reduced and protonated to give a ferric hydroxide complex, although the exact structure of OH is still debated and may be (section 6.3.3) a bridging FeIII-(OH)-CuII species; these are both depicted in Scheme 24. Further, the exact timing of electron and proton transfer and whether they are coupled is unknown.
Scheme 24.

Conversion of Intermediate F to OH in the Catalytic Cycle of HCOs, Which Occurs with the Translocation of One Proton Across the Membrane
Given that enzymatic (heme)FeIV=O species containing axial His ligation are known to not be exceedingly basic [pKa(FeIV–OH) ≤ 4 in all cases reported thus far],127,1181,1182 it is proposed that electron transfer to the F state occurs prior to or concurrent with proton transfer. If ET occurs before proton transfer, the resulting heme complex would be a ferric oxide [FeIII–(O2−)]. Such entities are highly basic and will be readily protonated to give a ferric hydroxide (FeIII–OH). However, with the copper(II) ion close by in the catalytic site, we propose, based on our coordination chemistry perspective derived from heme–copper–dioxygen chemistry studies (section 5), that it is plausible for a bridging μ-oxo FeIII– O–CuII complex to be formed following initial electron transfer (Scheme 25). Studies of model systems of heme– copper μ-oxo species (see section 5.3) have been shown to be highly basic, especially when copper is bound by three nitrogen-donor ligands (aqueous pKa ~ 9.7 to 10.6).496 Thus, an FeIII–O–CuII intermediate, if formed, would be easily protonated, completing the reduction-protonation of F to give OH, which may still contain the bridging hydroxide.
Scheme 25.

Coordination Chemistry Derived Proposal for the Transformation of Intermediate F to OH in HCOs Which May Occur through a μ-Oxo, FeIII-O–CuII, Intermediate if Electron Transfer Occurs Prior to Proton Transfer
Supporting the possibility of μ-oxo species formation, we note that in a recent work by Noodleman and co-workers, there is a focus upon the movement of water molecules in the BNC and how they may affect conversions between the F and OH states.1180 The calculated lowest energy structure of F was [FeIV=O---CuII/Tyr–O−] but with what appears to be a weak interaction between the copper(II) ion and the oxide ligand on iron; the distance between CuII and the oxide ligand was calculated to be ~2.4 Å (Figure 163A). This short distance, certainly at least a weak interaction as considered for inorganic chemistry structures, would facilitate formation of a bridging hydroxide complex, [FeIII-(OH)-CuII/Tyr-O‒], which is a likely candidate for the structure of OH, which would be formed upon addition of the electron and proton to intermediate F (Figure 163B). However, if electron transfer were to occur prior to proton transfer, this interaction would also allow for the formation of a bridging oxide complex, [FeIII-O–CuII], as described above.
Figure 163.

Optimized structures of the (A) F and (B) OH states in the catalytic cycle of HCOs.1180 See the text for details.
6.3.3. Could a Bridging Heme-Hydroxo-Copper (FeIII- (OH)-CuII) Species be Important in OH to EH Conversion?
An important final part in the enzymatic cycle of CcO is to complete the reduction of the BNC Fea3···CuB metal ions, so as to be available again for dioxygen binding (see Scheme 23 in section 4). The first of such steps is the conversion of the OH species to the EH intermediate. This is an important transition (the final electron transfer to the CuB site in the catalytic cycle), and it occurs concomitantly with the pumping of one proton. While the OH complex has been proposed to be a ferric hydroxide, there is still some debate as to the actual formulation of the BNC at this state. For instance, since the cross-linked tyrosine residue is deprotonated in OH, there are two resonance forms that are possible, with either the cross- linked Tyr or CuB being reduced (i.e., CuII/Tyr–O− or CuI/Tyr–O•). The oxidation state of the copper ion in OH may determine whether the single hydroxide ligand is bridging between the two metal sites [i.e., FeIII– (OH)– CuII] or bound to only a single metal center.
Wikström and co-workers have recently investigated the conversion of the OH to the EH state in aa3- and ba3-type CcOs using time-resolved absorption spectroscopy.1183,1184 In aa3- type CcO, an electron is transferred from the CuA site to heme a in an initial fast step, followed by ET from heme a to heme a3 and, finally, to CuB (Scheme 26A). These two subsequent steps occur with proton pumping across the membrane. In contrast, in the ba3-type CcO, the initial fast ET step occurs from CuA to both the heme b and heme a3 sites (with both heme sites being partially reduced, Scheme 26B). Wikstrom and co-workers propose that this indicates that heme a3 has a high reduction potential, implying that the hydroxide ligand in OH for ba3- type CcO is bound to the copper(ll) ion instead of iron; Fe(ll) does not readily bind an anionic hydroxide ligand. Electron transfer from heme b to heme a3 in turn results in reduction of CuB via ET from heme a3, giving the EH state (Scheme 26B).1184
Scheme 26.

Electron Transfer through the Metal Centers of CcO for the Conversion of OH to EH for (A) aa3-type and (B) ba3-type CcOa
aSee the text and ref 1184 for details. Colors indicate oxidation state of the metals (orange for Cu1, green for CuII, red for FeII, brown for FeIII, and brown/red for partially reduced Fe sites).
An accurate and detailed picture of the structure of the fully oxidized OH state of CcO is important for understanding key steps in electron transfer and proton pumping in the final steps of the catalytic cycle. There have been multiple computational studies that suggest that the structure of OH contains a μ- hydroxo core (Figure 164, panels A–C).901,903,916 These optimized structures have similar physical characteristics to heme–Cu model systems containing a hydroxide bridge (see section 5.3). However, no crystal structures of CcO unambiguously containing a bridging hydroxide or water ligand have been published to date (and see further discussion below).
Figure 164.

Computationally derived structures for the (A-C) OH state and (D) OB μ-oxo state in CcO, all containing a bridging hydroxide or oxide ligand between heme a3 and Cub. Φ is the distance of the iron above the plane of the porphyrin. Created using data from refs 901, 903, and 916.
While many of the as-isolated crystal structures of CcO have been proposed, most likely incorrectly (vide supra), to have peroxide bridged between the heme and copper ions (see section 6.2 above), some have been postulated to have hydroxide or water bound (Figure 165, panels A and b).987,989 However, when compared with known heme–copper model systems containing bridging hydroxide ions (Figure 165, C and D) , one sees that the metal–OH(H) separations in the CcO structures are by comparison drastically elongated (by greater than 2 Å). Such distances are not consistent with normal bonds between Lewis acidic metal ions like Fe(III) or Cu(II) and a negatively charged ligand like hydroxide. One might consider taking into account that the given metal–oxygen bonds in these protein structures may not be entirely accurate because of their resolutions. Once again, comparing the CcO X-ray structures to the structural parameters known for model complexes and calculated for the enzyme, the metal-metal distances for synthetic systems (3.66–3.81 Å) and computational structures (3.58–3.97 Å) are much shorter than those found in the enzyme (4.39–4.83 Å) (Figure 165). Thus, even if the formulation of a single-oxygen species such as hydroxide or water between the two metal centers in the BNC of CcO structures A and B (in Figure 165) is correct, it is highly unlikely that it is bridging the heme and copper moieties in these protein structures. We suggest that the hydroxide or water ligand is interstitial and either not binding any metal ion or, at best, weakly coordinated to just one of the metal ions. Certainly, based on the structure parameters obtained for PDB: 5NDC, no bonding occurs to Fea3, while there may be a weak interaction of the H2O/−OH group with CuB.
Figure 165.

X-ray structures of proposed μ-hydroxo (or μ-aquo) heme-copper centers proposed in CcO X-ray structures (A and B) or found in a small molecule model complex X-ray structure (C). For a different synthetic complex, [(F8)FeIII-(OH)-CuII(TMPA)]2+ with a tetradentate Cu center (D), structural parameters were determined from extended X-ray absorption fine structure (EXAFS) spectroscopy. Φ is the distance (above + , below −3) of the iron from plane of the porphyrin. Notably, the protein structure 5NDC (A) is said to be free of any possible damage derived from X-ray radiation, as it was determined utilizing an X-ray free electron laser (XFEL). Created using data from refs 987, 989, 1023, and 1071.
Blomberg has also proposed a μ-oxo structure, labeled OB, calculated to be similar in energy to the proposed μ-hydroxo structure (μ-oxo is 3.5 kcal/mol higher) (Figure 164D).902,903 The proton coupled reduction potential for the conversion of OB to Eh (i.e., [FeIII-O-CuII/Tyr-OH] to [FeIII-OH-CuI/Tyr-OH]) was calculated to be 0.93 V,902 which represents a very large driving force (i.e., enough to move protons across the membrane) against the proton electrochemical gradient.901 The nature of the origin of the high reduction potential OH state will be further discussed in section 6.3.4.
6.3.4. Could a Classical Electron-Transfer by an "Inner-Sphere" Mechanism be Involved for the OH to EH Transformation?
On the basis of known redox mechanisms in inorganic (coordination) chemistry, we posit that a hydroxide ligand could well be involved in mediating the electron-transfer (ET) step occurring between heme a3 and CuB in the OH to EH transformation, and that this possibility should be considered by the biochemists/bioenergeticists/computational-chemists. “Inner-sphere” electron transfer is known to occur in complexes that can form a bridging ligand between two metal ions. Compared to the very well-developed “outer-sphere” Marcus theory of electron-transfer, the scientific principles and details of such “inner-sphere” ET reactions are not fully developed. However, Henry Taube's well-known classical research with coordination complexes does provide certain mechanistic insight possibly relevant to the present discussion.1185,1186 In his seminal work, 1187,1188 Taube and co-workers demonstrated that a chromium(II) coordination complex could reduce a cobalt(III) compound under acidic conditions via the following reaction steps:
| (9) |
| (10) |
| (11) |
| (12) |
Unlike the more common outer-sphere electron transfer, the two reactants first form an activated complex, [(NH3)5CoIII···Cl···CrII(H2O)5]4+, here with chloride as a ligand bridging the two metal centers (eq 9). Electron transfer from CrII to CoIII occurs “through” the chloride ion (eq 10) and then the activated complex breaks apart (eqs 11 and 12). Such reactions seem to mainly or only occur with ligands that are able and known to bridge two metal ions; the hydroxide anion is certainly one such ligand, and bridging hydroxide complexes can occur with most, if not all, transition metal ions. Formally, this overall ET-reaction involves a transfer of a chlorine atom, Cl•.
As discussed above, Wikström and co-workers propose that, in the conversion of the OH to the EH state, a CuII-OH species in the heterobinuclear active site possibly forms in the OH state for ba3-type CcO.1184 However, upon reduction of CuII to CuI, the hydroxide ligand is transferred to the iron center, resulting in FeIII-OH···CuI species. Because of this, as well as accounting for the considerable amount of computational evidence for the formation of an FeIII-(OH)-CuII species, we suggest that reduction of OH to EH in CcO via electron transfer from heme a3 to CuB occurs through a bridging hydroxide ligand (Scheme 27) (i.e., the OH to EH conversion involves an “inner-sphere” electron-transfer mechanism).
Scheme 27.

Conversion of OH to EH in CcO May Occur via Initial Electron Transfer from Heme b to Heme a3a
aWe suggest that the subsequent electron transfer from heme a3 to CuB may occur through an “inner-sphere” mechanism utilizing a bridging hydroxide ligand, as shown. Formally, the CuBII reduction occurs by transfer of a hydroxyl radical (•OH) moiety.
In fact, Karlin and co-workers have reported an inner-sphere electron transfer reaction occurring in a heme-Cu model system.1189 Using a variety of tetradentate copper chelates (TMPA, PMEA, PMAP, and TEPA, see Figure 75 in section 3.3.2), the reaction of [(F8)FeIII-Cl] with cuprous complexes was examined. For all [(L)CuI]+ complexes, except when L = TEPA, the reactions resulted in reduction of [(F8)FeIII(Cl)] to [(F8)FeII] and formation of the [(L)CuII(Cl)]+ species. Investigations into the kinetics of these reactions determined that the trend in relative rates does not follow the trend in redox potentials of the [(L)CuI]+ complexes; the copper complex which is the strongest reductant [i.e., with the most negative reduction potential (easiest to oxidize)] does not react the fastest. Because of this, an inner-sphere mechanism of Cl• transfer from [(F8)FeIII(Cl)] to [(L)CuI]+ was pro- posed.1189
Similar experiments have been carried out with the binucleating ligand 6L. Addition of CuI to [(6L)FeIII(Cl)] resulted in the establishment of an equilibrium between the two species [(6L)FeIII-Cl···CuI]+ and [(6L)FeII···Cl–CuII]+ (Scheme 28). By varying experimental conditions (i.e., solvent, temperature, or addition of an axial base, dioxygen, or carbon monoxide), the equilibrium could be shifted, alternatively favoring either the FeIII-Cl or the CuII–Cl form.1189 At room temperature, the equilibrium was approximately 1:1, [(6L)- FeIII–Cl···CuI]+:[(6L)FeII···Cl–CuII]+; however, [(6L)Fen···Cl-CuII]+ was favored upon decreasing the reaction temperature or by addition of carbon monoxide or an axial base, forming stable FeII-X(2) complexes (i.e., FeII–CO or FeII– (base)2).
Scheme 28.

On the Basis of Redox Chemistry Observations Made for (i) F8, the Well-Studied Heme Built into the Binucleating Ligand 6L and (ii) Copper-TMPA Species, Where TMPA is the Tetradentate Chelate for Copper Appended to the Heme Base in 6L, the Observed Conversion of [(6L)FeIII–Cl···CuI]+ (left) to [(6L)FeII···Cl-CuII]+ (right) is Proposed to Occur by an “Inner-Sphere” Electron-Transfer Mechanisms, Via Formation of a Bridging Chloride Intermediate (Middle).1189
The structural and redox implications of chloride (or hydroxide) binding at a heme-Cu center have also been investigated using the modified myoglobin model, CuBMb (see section 5.4), since it is known that chloride can bind to the oxidized BNC in HCOs, concomitant with uptake of one proton.1190,1191 It was found that within this protein template, Cl− does not bind to the heme center but only to the CuB, which induces important structural changes within the BNC which are importantly related to electron transfer events.1192 A redox titration of CuBMb in the FeIII–CuII state in the presence of Cl− ions (the concentration of which at physiological pH can be up to 25 mM) resulted in decoordination of one of the CuB-bound His ligands upon the first reduction (due to binding of Cl− at CuB), which subsequently coordinated to the heme, forming a low-spin, six- coordinate FeIII center (UV–vis, EPR evidence).1192 These structural changes, which are not observed when Cl− is rigorously excluded, are consistent with proposals that redox changes and/or binding of anionic ligands in the BNC can cause significant structural changes around the active site such as Cu-His dissociation.854,1193,1194 An additional reduction to a FeII-CuI state results in decoordination of the His from the iron, returning the heme center to a high-spin, five-coordinate environment. This study clearly demonstrated that Cl− can bind to the CuB center in close mimics of the HCO binuclear center, and that binding event can be linked to redox-induced structural changes and likely also to the proton deliveries which are coupled to the electron transfers.
6.3.5. What are the Identities of the Relaxed O and High Potential OH States?
The reduction potential of isolated fully oxidized intermediates has been reported to be 0.28–0.35 V.876,877 However, the bioenergeticists have concluded that this does not allow for enough of a thermodynamic driving force that is needed to translocate a proton in the conversion of OH to EH (Scheme 23), and the enzyme may return to the reduced state with only two protons being translocated instead of four (if going through O and E, instead; these are off-pathway enzyme forms, which may be referred to as “relaxed”; also see discussions below).878,907 Thus, there has been considerable deliberation as to the possible electronic and geometric structure of the isolable relaxed complex (O) and the actual high potential metastable turnover intermediate oH.861,901,902
6.3.5.1. Possible Structures of Fully Oxidized, Isolated Relaxed O State.
One way in which the O state may be formed, proposed by Wikstrom and co-workers, is that the water molecule formed from reduction and protonation of P may not dissociate away from the copper(II) ion and instead remain bound.901 The ferric hydroxide species produced after further H+/e‒ transfer then deprotonates the cupric-aquo complex, forming a [FeIII-OH2···HO-CuII] intermediate (Scheme 29A). In fact, one X-ray crystal structure of CcO derivative has been reported and described as having this same formulation (Figure 166a). For the purpose of matching a possible BNC structure to earlier Mössbauer spectroscopic studies, Noodleman and co-workers916,1195 computationally optimized such an Fea3–OH2···HO–CuB CcO structure; it is shown as Figure 166B. However, the match between protein X-ray structures and computational models (A vs B, Figure 166) is not good; structure A displays an unreasonably short O···O distance between the hydroxide and water ligand O atoms as discussed in section 6.2.4). The O···O separation in such fragments (e.g., O···H···OH2) should be greater than 2.4 Å, and in Noodleman's structure (Figure 166B), this distance is ~2.6 Å. The result is that the computed Fea3III–OH2···HO–CuBII structure (B) has a much longer and reasonable Fea3··· CuB separation (5.53 Å). Also, as previously mentioned, the H3O2− moiety is a well-known bridging ligand group in inorganic chemistry, necessitating M···M(M') separations of well over 5 Å. Thus, the true identity of the groups or ligand binding between the hemea3 and CuB in structure PDB: 2GSM is unclear.
Scheme 29.

Proposed Formation of Possible “Relaxed” Oxidized O Intermediate States by (A) Wikström and Co-workers901 and (B) Blomberg et al.902
Figure 166.

(A) X-ray1196 and (B) calculated916 structures showing the catalytic core of CcO with the formulation [(heme a3)FeIII- OH2···HO–CuBII/Tyr–O−]+, which has been proposed by Wikstrom and co-workers to be the relaxed oxidized species o.901 However, due to the short and unreasonable O···O distance between the hydroxide and water ligand O atoms in the protein structure (A), its formulation comes into question. See text for discussion.
Blomberg et al. proposed a separate structure for the O state (called OP+ by the authors).902 They suggest that proton transfer to OH would result in protonation of the nearby cross- linked tyrosine molecule, making the complex to be formulated as a hydroxide-bridged species [FeIII-(OH)-CuII/Tyr-OH] (Scheme 29B). The proton coupled reduction potential of OP+ was calculated to be 0.49 V, much lower than that for the complex without the extra proton (OB in Figure 164D)902 and less than 0.2 V higher than the experimentally determined reduction potentials for the O intermediate.
6.3.5.2. Role of CuB Ligation Contributing to the Required High Potential Active OH Intermediates.
As discussed above, the “as isolated” form of oxidized CcO, O, does not possess the requisite reduction potential to couple electron transfer to the BNC and proton translocation across the membrane, and it is speculated to be an off-pathway intermediate.878 Photoejection studies have shown that a high potential “active” oxidized state, called OH (vide supra), is the probable catalytically competent intermediate, where reduction of EH is coupled to the pumping of a proton across the lipid membrane.907,1183 Theoretical- computational studies have led to the description of OH as discussed above. Still, experimental evidence giving an exact chemical and structural description of OH remains elusive. Here, we provide information and suggestions for how the coordination environment around CuB very well could play a critical role in establishing a high potential state; such proposals may possibly be investigated using synthetic models.
In copper coordination chemistry, it has been long established that the ligand environment (donor number and type) and overall geometry (including chelate size) around a copper ion has a profound impact on the observed reduction potential (see Table 9).1218 High potential copper centers can be attained through reducing the number of ligand donors in order to stabilize low coordinate CuI complexes; CuII complexes possessing low coordination numbers are typically very oxidizing and exhibit more positive reduction potentials relative to their higher coordinate analogs. When comparing copper complexes bearing the polypyridyl tetradentate (TMPA) versus tridentate (MePY2) ligands (section 3.3), a 0.3 V increase in the CuII/I reduction potential is observed [dimethylformamide (DMF) as solvent], when the coordination number is reduced from four to three. In some rare cases two-coordinate, linear copper compounds can be stabilized (Figure 167) and are reported to possess Cu centers with very positive reduction potentials (Table 9). In fact the linear, two- coordinate bis-histidine model compound [LδCuI]+ (Figure 167, B) is reported to have a reduction potential which is 0.64 V more positive compared to MePY2.633 Furthermore, addition of exogenous N-methylimidazole to [LδCuI]+ leads to the generation of a three-coordinate compound, and a huge decrease in the measured reduction potential (ΔΕ1/2 =–0.61 V) relative to the two-coordinate parent is observed. EXAFS spectroscopy confirms the formation of a three-coordinate compound with an average Cu-N distance increasing to 1.933 Å (two short N scatterers at 1.896 Å and one long N scatterer at 2.008 Å) from 1.876 Å. These synthetic chemistry examples and others in the literature1202,1218 show that copper is able to accommodate a reduction potential range on the order of ~1V by simple changes in the primary coordination sphere. Further, it is notable that Yi Lu and co-workers1103 have shown that a huge range of CuII/I reduction potentials (E°) (700 mV or more) can be achieved simply by modification (i.e., mutagenesis) of the second-sphere around the copper ion center (nonligand amino acid changes), which effects H bonding, active-site hydrophobicity and small Cu ion geometries. Through the lens of these fundamental coordination chemistry principles, we wish to discuss the role of CuB ligation in the high potential OH state.
Table 9.
Selected Reduction Potentials of Various Copper(ligand) Complexes and Enzyme Active Sites and Their Reported Geometries
| copper complex | Ln | Cu(l) geometry | E1/2 (CuII/CuI) vs Fc+/0 (V)e | E1/2 (CuII/CuI) vs NHE (V)e |
|---|---|---|---|---|
| Cu(aq) | – | – | 0.15 (H2O)1197 | |
| four-coordinate complexes | ||||
| (MeCN)4Cu | 4N | tetrahedral1198 | 1.18 (MeCN)1199 | |
| (Phen)2Cu | 4N | distorted square planar1200 | 0.17 (H2O)1197 | |
| (2,9-Me2phen)2Cu | 4N | distorted tetrahedral1201 | 0.19 (DMF)1202 | 0.82 (DMF)g |
| 0.60 (H2O)1097 | ||||
| (TMPA)(MeCN)Cu | 4N | distorted tetrahedral1203 | −0.41 (CH3CN)1002 | 0.22 (CH3CN)g |
| 0.02 (DMF)g | ||||
| −0.61 (DMF)771 | −0.15 (H2O)1204 | |||
| (TEPA)Cu | 4N | distorted tetrahedral1254 | −0.11 (DMF)1205 | 0.68 (DMF)g |
| (PMAS)Cu | 2N, 2S | trigonal pyramidal1205 | −0.16 (DMF)1205 | 0.47 (DMF)g |
| three-coordinate complexes | ||||
| (MePY2)Cu | 3N | distorted Tc | −0.31c (DMF)509 | 0.32 (DMF)g |
| (AN)Cu | 3N | distorted T1206 | −0.07 (MeCN)f | 0.56 (MeCN)g |
| (MeAN)Cu | 3N | distorted T1206 | −0.20 (DMF)1207 | 0.43 (DMF)g |
| (L5)(MeIm)Cu | 3N | b | −0.28 (DMF)633 | 0.35 (DMF)g |
| two-coordinate Cu(I) complexes | ||||
| (Lδ)Cu | 2N | linear,633d | 0.33 (DMF)633 | 0.96 (DMF)g |
| (Im)2Cu | 2N | probably linearb | −0.10 (H2O)1208 | 0.53 (H2O)b |
| (Py)2Cu | 2N | probably linearb | −0.18 (H2O)1208 | 0.45 (H2O)g |
| copper enzymes | ||||
| azurin | 2N, 1S | trigonal planar1209 | 0.31 (H2O)1210 | |
| rusticyanin | 2N, 1S | trigonal planar1211 | 0.68 (H2O)1212 | |
| T1 site, fungal laccase | 2N, 1S | trigonal planar1213 | 0.76–0.79 (H2O)1214 | |
| CuH, (PHMcc) | 3N | distorted T1215 | 0.27 (H2O)1216 | |
| CuB, CcO, bovine | 3N | distorted T842 | 0.28–0.35 (H2O)876,877 | |
| CuB calculated potentialsa | ||||
| CuB “relaxed” (calc) | 3N | distorted Y902d | 0.49 (calc)902 | |
| CuB “active” (calc) | 3N | distorted Y902d | 0.93 (calc)902 |
A “proton coupled” reduction potential is reported.
LCuI X-ray structure not determined.
Data not available for MePY2, geometry based on X- ray crystal structure for the (BnPY2)Cu derivative.
Computational structure.
DMF = dimethylformamide.
Measured for this review.
An estimated value for E1/2 vs NHE by adding 0.63 V to the observed E1/2 potentials reported vs Fc/Fc.+1217
Figure 167.

(A) The crystal structure of bis-1,2-dimethylimidazole copper(I) shows a near-perfect linear N–Cu–N geometry and short Cu-imidazole bonds. (B) The calculated (DFT) structure of (truncated) [LδCuI]+ also displays linear N–Cu–N coordination and short Cu–N bonds through the Nδ atom of the histidines.633
Experimentally, the BNC in the OH state is observed to contain a high spin ferric heme, an EPR silent CuB, and a deprotonated Y244.877 Computational analysis of this state has generally yielded two descriptions: a high-spin, antiferromag- netically coupled, bridging FeIII-(−OH)-CuII compound along with a tyrosinate or a high-spin ferric-hydroxide, antiferromagnetically coupled to a copper(I)-tyrosyl radical moiety (see discussion of OH in section 6.3, Scheme 24, and here in Figure 168). As discussed in section 6.3, the CuBII/Tyr–O− description should play a pivotal role in the OH to EH transition, as it is directly reduced by the incoming electron.
Figure 168.

Conversion of metastable “active” oxidized intermediate OH (far left) to EH (far right) through the reduction of CuB or the cross-linked tyrosine.916 The numbering scheme for CcO isolated from T. thermophilus is used. Structures for proposed high potential CuB sites through changes in coordination number are proposed. (A) Dissociation of the coordinated hydroxide moiety leads to formation of a planar 3-coordinate CuI complex following protonation and reduction. (B) Deligation of H283 and H233 and addition of an electron leads to formation of a linear H282- CuI-OH moiety. (C) Dissociation of hydroxide and H283 gives a linear Nε coordinated H282-Cu-H233 Cub state following protonation and reduction. (D) A tautomeric change in H283 and H282 coordination and dissociation of H233 and hydroxide leads to formation of a N<5 ligated H283-Cu-H282 complex after proton and electron transfer. Recall (section 3.1) that for adjacent bis-histidine dipeptides and single-copper sites, formation of a linear 2-coordinate copper(I) structure is highly favored when both imidazole groups ligate via their Nδ nitrogen-atoms. The inspiration for proposals B and c derive from the computational studies of Noodleman and co-workers.916 See text for details.
A computational study has examined OH states where removal of the oxygenous ligand from CuB enforced a planar three-coordinate geometry around the copper ion,901 which possesses a high CuII/I reduction potential (Table 9) and could possibly be the source of the high potential found for OH (Figure 168, pathway A). Noodleman and co-workers have conducted DFT studies on the change in the CuB coordination state for the BNC in the reduced form and found that CuBI can support linear, two-coordinate geometries, either through coordination of H282 and a hydroxide ion or through H282 and H233 (Figure 169).916 It is interesting to consider if similar low-coordinate copper intermediates may form along the pathway from OH to EH. Noodleman's calculated linear two-coordinate states strongly favor copper(I) and would account for the high reduction potential found for OH (Figure 168, pathways B and C). Low-coordinate compounds featuring bis-His chelation through the Nε nitrogens of the histidines, as shown in pathway C, have been observed in a model compound where EXAFS and DFT calculations confirmed linear two-coordinate copper geometry.633 A fourth possibility leading to a high potential CuB site is through a tautomeric switch in the ligation of His 282 and 283, forming a linear H282-Cu-H283 complex via coordination through the NÅ nitrogens of histidine (Figure 168, pathway D). Again, such model compounds have been observed both in synthetic systems and enzyme active sites (see section 3.1), where chelation of adjacent bis-His residues imparts a strong preference for linear Cu geometry and formation of strong and short Cu–N bonds (~1.88 Å).633
Figure 169.

Noodleman and co-workers' DFT calculated structures suggest that Cub ligands His233 and His 283 are “mobile”, allowing for the formation of linear two-coordinate CuB(I) intermediates, (A) H283 and a water molecule dissociate from CuB yielding a linear two-coordinate bis-histidyl cuprous structure with short Cu-NHis distances. (B) H283 and H233 dissociate from Cub to give a linear H282-CuI-OH moiety. Both Cu- NHis and Cu-OH bonds are shortened relative to the “relaxed” oxidized state (with CuBII-His3(OH(H)) coordination). Created using data from ref 916.
Blomberg and co-workers have proposed that CuB inherently possesses a high reduction potential, requiring no change in ligation, and favors an electronic description of OH where CuB and active-site tyrosine Y233 exists as CuI and tyrosyl radical.875,902 Their DFT calculations of the BNC model reveal a “proton coupled” reduction potential of 0.93 V. Also see Table 9 and section 6.3.
6.4. Bridging the Gaps: Observations and Conclusions From Heme-Copper Synthetic Analog Investigations and Implications for HCO Biochemistry
Structural (X-ray crystallographic and often also computational) and spectroscopic (UV-vis, EPR, and rR) data are available for metal (heme, copper, and cobalt) superoxo, peroxo, or hydroperoxo coordination complexes (or in some cases proteins), wherein O2-derived ligands may be bridging two metal centers, which delineate important structural aspects of various plausible HCO intermediates. Similar types of data are also available for nonheme iron synthetic complexes and a few proteins, but we have chosen to leave these out of the discussions. All together, these findings have led us to boldly question some structural presumptions made based on HCO enzyme crystal structures and their forthcoming analysis. Our motivation as synthetic inorganic chemists, in bringing up such doubts and suggestions, is to inspire interdisciplinary lines of thinking which may lead to new experiments and perspectives for our biological research colleagues. These suggestions are outlined just below, followed by our future projections and hopes for the ways in which model chemistry may contribute to the discussions and scientific insights at the forefront of HCO O2-reduction chemistry.
-
The Putative Peroxo-Bridged BNC Protein X-ray Structures: we have questioned the assignment, or, if you will, the description, applied to the well more than a dozen published CcO protein X-ray structures as possessing a peroxo- bridging ligand present as a dianionic −O–O− fragment wherein one O atom is binding to Fea3 and the other to CuB. Just in the past few years, we have been able to structurally characterize synthetically derived, low-spin, peroxo-bridged heme-copper assemblies (see sections 5 and 6.2). With knowledge concerning these structures and with reference to quite an extensive literature of small molecule inorganic complexes where a peroxide dianion bridges two +2 or +3 first- row transition metal ions, we can state that the protein structures have Fea3···CuB separations which are too large relative to “authentic” peroxo-bridged dimetal complexes. Further, the putative M-Operoxo bond lengths which were determined are quite “off”, and overall, the peroxo-bridging descriptions of these protein structures are not chemically reasonable.
We are not X-ray crystallographers, but we do understand that even when protein crystals are of excellent quality and/or the methodology utilized leads to high resolution (for protein crystallography), structure model refinement most often does not lead to unambiguous identification of small mobile molecules (i.e., their exact atom location or arrangement is not necessarily defined based on precedents). For example, unlike defining the location of a rigid five-membered imidazole ring of a histidine amino acid which is attached in a known way to the backbone of a protein α helix, identification and spatial definition of hydroxides, waters, or (hydro)peroxides, which may also be juxtaposed with other similar small molecules, often cannot be done unambiguously. This is to say that structure refinement and determination of accurate bond lengths between light atoms of such molecules is a very difficult undertaking.
So, what are these structures, if not peroxo bridged Fea3-CuB moieties? We do not know, but have discussed some possibilities in section 6.2.
The Identity of Enzyme Turnover Intermediate ‘A': what will be a controversial, and perhaps a not well-received suggestion from us, the present authors, mainly the senior author, is that we question a basic dogma in the field of heme- copper oxidase research. Rather, we would suggest further consideration regarding the issue of the assignment for intermediate A of HCO catalytic cycles. As laid out in Scheme 23 and Figure 150 and accompanying discussions, intermediate A is the first O2-derived species that can be characterized following O2-addition via fast time-resolved resonance Raman spectroscopy. This has always been described as an oxy-heme adduct, a ferric-superoxo (FeIII-O2•–) species, which is essentially the same as that which is observed in hemoglobins or myoglobins. The conclusion is based on the observations by highly prominent time-resolved resonance Raman spectroscopy research groups of an Fe-O stretch observed at 571 cm−1 (plus or minus a few wavenumbers) and the shift of that spectroscopically observed band upon isotopic substitution with 18O2 (see section 6.2). An unambiguous assignment in fact could be made if there was ever a protein turnover intermediate v(O–O) stretch detected, as this parameter is very different for heme superoxidic versus peroxidic O2-derived species. We believe that at the present time, one cannot unambiguously conclude that HCO turnover intermediate A is an oxy heme, and rather, it might be a peroxy type complex (Figure 150).
Bridging μ-hydroxo and μ-oxo heme-Cu synthetic complexes and proposed relevance to HCOs. The syntheses, interrogation of spectroscopic properties, and interconversion of a variety of such complexes has been explored, as described in section 5.3. Through the measurement of pKa values for the FeIII-(OH)-CuII species, it has been determined that the heme-(μ-oxo)-copper compounds are quite basic, and factors such as copper coordination (3- vs 4-coordinate) and the linearity of the Fe-O-Cu vector can affect the basicity. We propose that a heme-(μ-oxo)-Cu species may be prevalent in the transformation of the F state to OH (section 6.3.2 and Scheme 25), and the high basicity of this intermediate would readily result in the proposed heme-(μ- hydroxo)-Cu formulation of OH (Figure 170A). The μ-hydroxide complex may be deprotonated by the nearby histidine cross-linked tyrosinate (e.g., Tyr-O−) residue, resulting in a heme-copper bridging oxide moiety and tyrosine, TyrOH (Figure 170B). These intermediates have also been the target of computational investigations aimed at determining the exact structure of the high potential OH state in the catalytic cycle of CcO. Wikström901 and Noodleman,916,1180 with their respective co-workers, have both proposed that the μ-hydroxo structure (Figure 170A) is catalytically active, while Blomberg and co-workers902,903 have proposed that the μ-oxo complex (Figure 170B) allows for the thermodynamic driving force required for proton translocation during the conversion of OH to Eh (see Scheme 23 and section 6.3) for the catalytic cycle of CcO.
A μ-H(OH) Fea3-CuB Enzyme X-ray Structure: as already discussed extensively with respect to the many CcO protein X-ray structures described as peroxo-bridged, we feel compelled as inorganic/coordination chemists to comment on the description of the enzyme metal-containing active site which we believe to be represented in an incorrect manner. Here, we repeat our assertion that the putative hydroxide or water-bridged heme–Cu structure PDB: 5NDC (Figure 165) is not well-described. The Fea3···CuB separation here is very long, ~4.8 Å, whereas for known small molecule hydroxide bridged heme-FeIII–(OH)-CuII(ligand) complexes with “normal” FeIII–hydroxide and CuII–hydroxide bonds, metal- metal separations are well under 4.0 Å. Computationally derived structures of hydroxo-bridged CcO analogs Fea3– (OH)–CuB species also possess Fe···Cu distances which are under 4.0 Å. Thus, if a hydroxide or water molecule lies between heme a3 and CuB in the protein structure, it is either an interstitial group not binding either metal, or it is ligated to just one of the metal ions. Further details are given in section 6.3.
Heme a3···CuB separations and large BNC motions occurring in HCO Turnover? A survey of HCO X-ray crystal structures in a variety of forms [reduced, oxidized, with or without ligands such as CO, NO, azide (N3−), O2 (or HO2 or HO2− or O22−), hydroxide, or water] reveals that for the vast majority, the distance between the Fea3 and CuB BNC metal ions is 4.7 Å or greater, and many exhibit a separation of over 5 Å (see information throughout this Review and the following refs 19 and 20). These large M···M distances are in stark contrast with many synthetic and computational model systems as outlined here. (i) For O2-derived, peroxo-bridged heme–copper synthetic constructs, Fe···Cu separations vary between 4.2 and 4.5 Å. (ii) For theoretical-computational modeling of intermediate A (a superoxo or peroxo species, vide supra) or a hydroperoxo-bridged protein model with a coordination essentially the same as that of a peroxo-dianion, the Fea3···CuB distances are 4.46 and 4.38 Å, respectively (Figure 161). ( iii) For a variety of hydroxo- or oxo-bridged synthetic heme–copper complexes which have been characterized by X-ray crystallography, the (heme)Fe---Cu spacings are between 3.4 and 3.8 Å, and three computational groups have derived structures proposed to be a key intermediate, OH (see sections 6.3.1 and 6.3.2), wherein the protein BNC Fea3··· CuB separations are 3.8 to 4.0 Å (Figure 164).
Figure 170.

Heme-Cu μ-hydroxo (A) and μ-oxo (B) species, which have been modeled in synthetic systems (see section 5.3) and explored computationally (see section 6.3), have been proposed to be intermediates in the catalytic turnover of HCOs (see Scheme 23). Specifically, these species are postulated to represent the high potential OH state, a property required from a bioenergetics perspective in order to drive proton translocation concomitant with the OH to Eh conversion (see section 6.3). These two complexes are tautomers, with the proton residing on either the bridging oxide ligand (A) or the nearby histidine cross-linked tyrosine residue (B).
What does this all suggest? We see this contrast as perhaps indicating huge structural changes in and around the heme a3- CuB BNC, occurring during enzyme turnover where molecular oxygen is reduced to water and protons are pumped across the mitochondrial inner-membrane. The fully reduced BNC seems to have a large metal spacing of 5.0 Å or greater, yet later small molecule or atom-bridged intermediates may “squeeze down to Fea3···CuB separations of (well) under 4 Å. Such movements, as accompanied by resulting motions of connected protein helices, surely constitute significant aspects of the protein mechanism of action. This is not a new insight and the protein and proton movements, electron transfer processes, and associated bioenergetics are far more intricate then we can possibly express.20,817,873 However, we would like to emphasize how these supramolecular adaptations relate to the important aspects of coordination chemistry concomitantly playing out in the binuclear heme–copper center during turnover. Perhaps, at least we can say that more details of the exact movements of the heme, CuB, and their ligated and mobile histidine imidazole groups can and should be recognized, appreciated, and further analyzed.
6.5. Future Directions for Synthetic Modeling of HCO Chemistry
Studies of small molecule mimics of the BNC in HCOs have already contributed significantly to the current understanding of biological O2 reduction. Merging advanced computational techniques and state of the art spectroscopic methods, recent advancements in the field of synthetic inorganic chemistry related to HCO activity have uncovered important fundamental structure–function relationships related to selective O2 reduction chemistry and have even accomplished biomimetic O–O cleavage chemistry within model constructs (see section 5). Continuing research in this area will likely draw inspiration from and build upon many of the studies reviewed herein, and therefore, we close this section 6 and this review article with a brief outlook on future directions concerning O2-reduction reactivity of heme–copper systems, which can and should now be addressed using model chemistry.
6.5.1. Expanding the Scope of Heme-Peroxo-Cu Model Substrate Reactivities: Fundamental (Kinetic and Thermodynamic) Studies.
The most basic/fundamen- tal processes which ultimately give rise to the biochemically elaborate O2-reduction in HCOs are single (though likely often coupled) proton and electron transfer events. Detailing the timing, source, and destination of these protons and electrons as well as the parameters associated with their successful transfer (i.e., pKa’s, BDEs, E°’s) are crucial in developing understanding of how O–O bond cleavage occurs, how the enzymatic cycle turns over, and how O2 can be efficiently and selectively reduced to water, preventing ROS release. Using small molecule model systems, these types of questions can be directly probed using rational, systematic substrate selection in mechanistic reactivity studies. As was shown by several studies of heme-peroxo-Cu complexes reacting with various phenols (see section 5), applying appropriate experimental techniques (i.e., low temperature, spectral and kinetic monitoring, and radical trapping experiments)747,919 allows for detection of reaction intermediates and mechanistic understanding (with support from DFT) of O–O cleavage reactivity. The phenolic substrates in these studies were inspired by the active site Tyr residue in the BNC; however, in these cases, the chosen phenol is either (i) a poor electron donor (i.e., 4-NO2-phenol), requiring a separate electron source for PCET reactivity, or (ii) an electron-rich phenol with a weak O–H BDE (i.e., 4-OMe-phenol) but which requires large excesses of substrate to carry out the multiproton–electron chemistry. Importantly, both of these studies highlighted the activating influence of H-bonding interactions with a bound (bridging) peroxide moiety (as proposed/seen in HCOs) and the structural/steric demands to allow for such interactions to occur. Additionally, the modified Mb systems developed by the Lu group, which included systematic modifications of incorporated Tyr analogs, showed correlations between both phenolic pKa and E° with respect to O2-reduction reactivity.1119
With these advancements in mind, as well as the stoichiometry of peroxide reduction (O22– + 4H+ + 2e−→ 2H2O), one can imagine other biorelevant substrates (i.e., catechols/polyphenols, aminophenols) which, with proper substitution, can be tuned to provide a balance of H-bond donation as well as H+/e− (possibly as H•) donation to a bridging peroxide (Figure 171). On the basis of the properties of the substrate (pKa, BDE, and possible other factors such as intramolecular H-bonding between ortho-OH/NH groups) and results of reactivity studies, insights into the thermodynamics of successful O2-reduction chemistry stand to be revealed. Again, strategies such as low temperatures and fast time scale kinetic monitoring will be important to characterize intermediates (i.e., H-bonded adducts) within such model studies which may not be detectable in analogous studies on HCOs.
Figure 171.

A proposal for a reactivity study involving multielectron-donating substrates with heme-peroxo-copper model complexes alluding to proposed mechanistic investigations.
Given the specific desired chemical outcome of O–O reductive cleavage, the question which is equally as important as “How are protons and electrons transferred?”, is the question of “Where do they end up?” [i.e., to which O atom of a (possibly bridging) O2 moiety are they transferred]. In many of the enzymatic, small molecule, and computational studies reported herein, there is evidence of hydrogen bonding and/or proton transfer to the distal O atom (that which is not bound to Fe) of a (heme–O–O···Cu) structure. Small molecule models are best-suited to resolve thermodynamic parameters of such metal-oxy species such as the basicities (pKb's) of each O atom, the relationship between proton donors [which, as described just above, can be easily chosen or systematically studied based on their known properties (pKa's)], and resulting (bridging) hydroperoxo structures can be acutely followed. The ability to accurately assign bond distances (by high-resolution X-ray crystallography) or vibrational frequencies (rR data) within a M–O–O–M model construct before and after proton transfer(s) is extremely important for building an understanding of structural and electronic changes which occur during HCO turnover, and how proton and electron transfers are directed by the changing active site. We have pointed out this difficulty in dealing with large enzyme structures and addressed potential consequences of ambiguous structural assignments earlier in this section (section 6.2). Further, model systems lend themselves to fine-tuning and small modifications which can extend thermodynamic understandings to include the ways by which heme and copper ligand environments may influence such parameters as pKb's (vide infra).
6.5.2. Probing Ligand Effects in Small Molecule Model Chemistry of Heme-Cu Systems.
The heterobimetallic active site in heme–copper oxidases is quite unique, and while this poses challenges in biomimicry in the literal sense (i.e., constructing identical structural analogs), it also presents opportunities for creative design of model complexes which not only can provide insights into the biochemical activity of HCOs but also make strides toward understanding structure–function relationships as they relate to coordination chemistry in general. From a synthetic perspective, the chemist can define and modify the ligands for each metal ion to tune coordination geometry and electronics, and in some cases, can even incorporate functionalities that pose secondary coordination sphere interactions. For example, in terms of the copper ion environment, the use of N-donating ligands with different denticities, varying imposed coordination geometry or donor strengths (due to donor type or ligand substituents), can govern reduction potentials and O2-reactivities drastically, as presented in section 3. Modifications of the heme environment can include derivatization of the heme itself and/or the identity of the axial base ligand; both such alterations, as outlined in section 2, impact the extent of possible electron donation to a bound O2 moiety and, therefore, the basicities of the O atoms and the reduction capability of the model as a whole. Additionally, sterically encumbered hemes may (and have) also contributed to the enhanced stability of transient iron– oxygen intermediates. These approaches will continue to be important in moving the field forward. Here we outline our outlook and proposals for (1) how the synthetic inorganic chemistry community can gain insights via various purposeful modifications of the copper ligand, (2) incorporation of secondary coordination sphere elements related to the active site Tyr, and (3) alterations at the heme iron site.
Ligand environment and redox properties of the copper center. The profound impact of ligand denticity and geometry in copper chemistry leads us to propose several modes of expansion in the design of copper ligands for heme–Cu models. Interestingly, in a case of CN-bound CcO, it was observed that one of the copper His ligands could dissociate [d(Cu–N) = 2.8 Å] in order to optimize the active site conditions to accommodate the diatomic molecule.854 The authors then suggested a similar lability occurring during the enzymatic cycle, possibly allowing for the formation of a transient peroxide bridged intermediate such as IP (vide supra, Scheme 23). To probe this type of ligand self-optimization, and with new synthetic methods reviewed in section 5,497 it is possible to study heme–copper assemblies with varying numbers and types of monodentate (flexible) donor ligands. Further investigations into such complexes and utilization of advanced structural characterization techniques could provide insights into how the BNC adapts to different conditions (i.e., the presence of partially reduced O2 moieties (O2•‒, O22–, and HO2−), and/or hydrogen-bonding interactions as probed with solvents or substrate modifications). Along those lines, we believe that the intermediates following O–O bond cleavage deserve more attention from a synthetic modeling perspective, and an inorganic chemist can also use systematic ligand design to study aspects of the CuB site utilizing systems which support bridging oxo or hydroxo moieties, possibly building upon Cu2–O2 chemistry; relevant bis-μ-(hydr)oxo species are known to exist in Cu2–O2 chemistry (see section 3).18,612 As the structural properties and identities of the later activated intermediates (such as OH and EH; see Scheme 23 and section 6.3) are still debated and have therefore been the focus of mostly computational investigations, synthetic models can certainly contribute experimental evidence in support of or to argue against some of the current theoretical deductions. Undoubtedly, a very important part of this later phase of the HCO cycle is proton pumping, and while it is extremely difficult, if not impossible, to use small molecule models to understand such phenomena, it has been suggested and supported by calculations that a high (positive) reduction potential of CuB is necessary in this phase for the proton pumping function.902 With the use of what is known about ligand characteristics which control/tune Cu redox potentials (much of which has been reviewed here in section 3 or moreso in section 6.3), these design strategies can be incorporated into heme-Cu complexes in order to better understand this proton pumping motive force.
-
Modeling the formation and involvement of the His-Tyr cofactor. The tyrosine in the heme-copper active site represents several concepts worth noting to a synthetic model chemist, and these are essentially born out of the fact that the protonation state, and therefore also the H-bonding capability, of this phenolic residue is known to change several times throughout the HCO catalytic cycle (see Scheme 23 in section 4.2). As the phenol is covalently cross-linked through the ortho position of one of the Cu-ligated histidine residues, its presence and protonation state may significantly impact the electronic structure around CuB (although direct studies to probe these effects are clearly extremely difficult in enzyme studies). It is known from simple organic models of imidazolephenol compounds, that in an analog of the His-Tyr moiety, the imidazole “substituent” reduces the pKa of the phenol O–H by >1 pKa unit while also increasing the E° by 69 mV.852,853 These studies did not, however, touch on the changes in the properties of the imidazole group which would alter its donating ability to CuB and thus modify at least to a small extent the CuBII/I standard reduction potential.
Several heme-copper model complexes reviewed in section 5 employed copper ligands with imidazole-phenol cross-link mimics, but we propose that more in-depth investigations are required to deduce the effects of such substitutions on the redox properties of the CuB center, and further, how those influences are altered when the phenol moiety is either deprotonated, giving tyrosinate (TyrO−), or possibly existing in a radical form (i.e., tyrosyl radical, TyrO•). These studies will require clever experimental designs as the deprotonated or radical species are likely unstable. That being said, uncovering the links between proton/H atom movements and concomitant changes in metal-based redox properties would not only offer significant contributions to biomimetics but also to inorganic coordination chemistry in general. Another perspective to take when considering modeling of the BNCs tyrosine moiety is the H-bonding aspect that it provides, which computational and experimental studies have both supported. There is significant precedence in both Fe-O2 and Cu-O2 chemistries (see sections 2 and 3, respectively) for the importance of H-bonding effects in stabilizing different metal-oxygen species and/or in taking part in the actual O–O scission process itself (i.e., facilitating or “triggering” reductive cleavage by PCET). Such ligand designs that have already been used in those Fe-only or Cu-only systems can be incorporated into heme–copper models to study the details of secondary coordination sphere H-bonding interactions in a CcO dioxygen reduction mechanism. Again, intermolecular H- bonding effects have been observed for recently described heme-Cu model systems;747,919 however, investigations of intramolecular H-bonding interactions could add a level of cooperative effects in that insights can be gained related to the interplay of H-bonding extent and other aspects such as metal ion redox potentials. This type of information would be exceedingly difficult to extract from studies on native HCOs; however, model studies provide a facile platform to examine these complicated and interrelated phenomena which are essential pieces of efficient O2 reduction chemistry.
A final point related to this interesting His-Tyr moiety in HCOs is the actual biochemical formation of the cross-link CTyr-NHis bond. It has been shown in enzyme and model studies (Yi Lu's modified Mbs, see section 5.4.3) that the covalent bond linking these two residues (beyond simply the presence of the two residues at the BNC) is necessary for enzymatic function, and further, it has been proposed that this unique post-translational modification may form during the first turnover of the enzyme.612 Studying this single bond formation in native HCOs is therefore extremely difficult; however, model systems which do not include macromolecular protein structure (which may have issues folding and/or functioning during experiments involving this cross-link formation) may provide an opportunity to understand this important process and investigate the possibility that it could form, and how, during O2-reduction by heme and Cu. For example, a bioinspired heme-Cu system (especially those known already to reduce dioxygen, see section 5) with Cu- ligated imidazole containing an “unprotected” Νε-H bond can be studied in the presence of various phenolic substrates as tyrosine mimics. Using techniques in the synthetic inorganic “toolbox”, perhaps the study of synthetic constructs, one may be able to determine how and when (if during O2-reduction, i.e., in the presence of which metal-oxygen species/intermediates) the cross-link may form in the natural system.
Heme center and axial ligation. One of the structural factors distinguishing the heme portion of cytochrome P450 and HCO active sites (which heterolytically and homolytically cleave the O–O bond, respectively) is the proximal axial ligand bound to the heme iron center. In cyt P450, it is a Cys (thiolate), whereas in HCOs it is a histidine (imidazole). The nature of the axial ligand in heme model systems as well as enzymes has been shown to affect the iron spin state and redox properties, spatial equilibrium of the iron atom (i.e., in reference to the plane of the porphyrin ring), and small molecule binding affinities and O–O cleavage patterns (see section 2); all of these factors may influence the O2-reduction chemistry of such systems. Small molecule models can systematically probe these relationships which fall under the umbrella of what is often referred to as the “push-pull” concept (see section 2), with the copper ion potentially also exerting Lewis acid-type interactions, underscoring the importance of systematic approaches in investigating these complicated chemical phenomena. As has been outlined in this review, very simple or very complex heme scaffolds which employ tethered or free axial ligands can be utilized for such studies. We hope that such investigations can shed light on how subtle aspects (i.e., orientation/flexibility, donating ability, etc.) or large aspects (i.e., overall charged, neutral, charge delocalized, and/or prominent steric effects) of the heme axial ligand can shape the details of O2-reduction chemistry. As stated in section 4, changes in the protein matrix which effect the local structure at the proximal side of the heme may alter the aforementioned subtle aspects of the axial His in HCOs, especially during catalytic transformations involving proton pumping. Many heme-focused model studies have altered the substituents on the heme itself (at the meso or pyrrole positions, see section 2), in order to analyze electronic effects that originate from the iron center. On the other hand, some other heme substituents offer secondary sphere noncovalent interactions, as in the case of “picket-fence” and “Hangman” platforms (see section 2). The expansion of such studies/ideas carries great potential as well, and is left to the design creativity of the chemist, and reaches another level of complexity and potential understanding within heme–Cu constructs. Finally, related to both points made here regarding axial ligand influences and variations about the heme center of model complexes, synthetic studies can/should be aimed at understanding the timing of electron transfer from the low-spin heme which is tethered to heme a3 via the axial histidine (see section 4.1). These studies perhaps may best be approached from an electrochemistry perspective (via electrocatalysis studies, see section 5.2.4) but must be carefully thought out when multiple redox-active moieties are contained within the same model (as in heme–Cu systems possibly with Tyr mimics). Overall, with the plethora of information about heme and heme–O2 chemistry, there is much more to be explored and interpreted within heterobimetallic Fe–Cu–dioxygen reactivities.
6.5.3. Electrocatalysis Studies Utilizing Heme-Cu or Dicopper Synthetic Complexes.
The evaluation of small molecule synthetic models for cytochrome c oxidase as mounted on electrode surfaces to carry out electrocatalytic investigations on the four-electron, four-proton reduction of dioxygen to water, the ORR reaction, has very much enhanced our understanding of the factors that increase the efficiency and selectivity for the four- versus two-electron pathways. Such studies have been carried out by a number of research groups, but primarily led by Collman (and Chidsey) and co-workers (see section 5.2.4).56,921,1011,1044 Many of these studies have benefited from advanced electrochemical techniques, such as use of rotating ring disc electrode (RRDE) instrumentation to detect H2O2 evolved (as a partially reduced oxygen species, PROS), novel methods of attachment to electrode surfaces, and the ability to measurably vary electron fluxes reaching the heme–Cu catalyst.
However, as seen in a study we carried out in collaboration with A. Dey and co-workers (section 5.2.4),1051 it is now possible to also carry out detailed interrogation of electrochemical (catalytic) processes while also employing either resonance Raman or infrared spectroscopies [i.e., surface enhanced resonance raman spectroscopy (SERRS),1219 or surface-enhanced infrared absorption (SEIRA) spectrosco- py].1220 In our own work, we used simpler heme–Cu constructs, efficiently reducing dioxygen to water, while also detecting vibrations of peroxo intermediates (employing SERRS) (section 5.2.4). Thus, electrocatalysis studies in the future should be very powerful in terms of elucidation of key intermediates and mechanism of action of synthetically derived inorganic complex catalysts for the ORR, contributing in a complementary manner to our basic understanding of the O2- binding and reduction chemistry occurring within heme–Cu binuclear complexes.
6.5.4. Studies on Homolytic vs Heterolytic O–O Reductive Cleavage in Heme–Cu Peroxo Complexes.
Utilizing heme–copper synthetic systems, it has been shown that O–O cleavage of a bridging peroxo heme–Cu synthetic assembly can be achieved by addition of an H atom (H•) equivalent, either through hydrogen-atom abstraction (HAA) from p-methoxyphenol or protonation/reduction from separate H+ and e− entities (i.e., 4-nitrophenol and decamethyl- ferrocene, respectively) (see section 5.2). This reaction results in two equivalents of water being formed, proceeding through an intermediate containing Cmpd II and a copper(II)– hydroxide complex, although the evidence for this FeIV=O··· HO–CuII(ligand) intermediate has yet to be directly observed in synthetic systems. From a combined kinetic and theoretical- computational analysis, the HAA mechanism that was favored for O–O reductive cleavage was proposed to proceed through an H-bonded transition state (TSHB′ in Scheme 30). The homolytic O–O bond cleavage occurs prior to HAA, giving a Cmpd II entity and what was best described as a copper(II)- oxyl species (cupryl, CuII–O•; Scheme 30); the latter moiety performs HAA, giving a state that is equivalent to PM in the catalytic cycle of CcO (Scheme 30; also see Scheme 23 in section 4.2). Thus, investigations into the spectroscopic characterization and substrate reactivity of such copper(II)- oxyl species is of great interest.
Scheme 30.

Proposed Transition States (TSHB′), in the O–O Cleavage Step of the Catalytic Cycle of CcO (Peroxy to PM), May Possess Copper(II)-Oxyl (Highlighted in Green) Charactera
aSuch a species would be highly reactive towards hydrogen atom abstraction from a nearby phenol (possibly the histidine cross-linked tyrosine residue at the BNC).
Because of their highly reactive nature, a copper(II)-oxyl coordination complex has not yet been characterized or isolated in solution or the solid state (also see discussions in section 3.3.1.3). However, O–O cleavage of copper alkylperoxos (CuII–OOR) compounds has been investigated as a synthetic approach to gain insights into the fundamental understanding of the formation, properties, and reactivity of CuII–O•· species. The use of specific alkylperoxos allows for the determination of how the O–O bond is being broken (i.e., heterolytic vs homolytic cleavage). Homolytic cleavage would result in a CuII–O•· intermediate, while heterolytic cleavage could presumably give an even more reactive CuIII–O· compound; however, such is an unknown species.
Itoh and co-workers have reported on the thermal decay of CuII–OOR complexes in the absence and presence of organic substrates.610,745,1221,1222 Identification of the organic products after reactions indicated that homolytic O–O cleavage had occurred, and, in the presence of substrate, hydrogen atom abstraction took place. Kinetic studies showed that O–O bond cleavage was faster in the presence of substrates, suggesting that HAA and homolytic scission of the O–O bond occurred in the same reaction step. Therefore, no discrete CuII–O•· intermediate appears to form in these reactions, but some insight into the O–O cleavage mechanism was achieved.
We suggest that further future research in this area is important. Studies might be carried out with copper-only synthetic compounds but also with newly designed heme–Cu complexes.
-
Copper-only systems. Modification of the ligand environments around copper may allow for future insight into the possibility of direct characterization of a CuII–O•· species in reductive O–O bond cleavage for CuII–OOR complexes. As has already been addressed, factors such as denticity, donor atom (N vs O vs S), chelate ring size, and ligand charge all affect the reduction potential of copper compounds (section 3.3). A complex having a very negative reduction potential (i.e., favoring a higher oxidation state) may be able to stabilize a high-valent cupryl species. In addition, synthetic input of secondary coordination sphere groups may also facilitate the stabilization of a copper(II)-oxyl complexes. Hydrogen-bonding moieties have been shown to stabilize copper(II)-superoxo and −3hydroperoxo compounds (see sections 3.3.1 and 3.3.2) and may also have a similar effect for a CuII–O•· species.
However, the investigations into O–O cleavage by copper- only systems need not only concern CuII–OOR complexes. Multiple mono- and binuclear copper(II)-hydroperoxo (CuIIn–OOH, where n = 1 or 2, respectively) have been reported (section 3.3). In the same way as heme-only systems (section 2.1), hydroperoxo species can cleave via heterolytic or homolytic pathways, resulting in different high-valent species. One can imagine that systems of Cu2II–OOH compounds may be the most competent in research efforts aimed to isolate and characterize high-valent copper species, as electrons can be taken from each copper ion, resulting in either a Cu2III-oxo (i.e., oxide dianion) intermediate product after heterolytic O– O cleavage or, if an electron derives from only one copper ion, giving a mixed-valent CuIICuIII-oxo moiety (with •O(H) byproduct) following homolytic cleavage.
Heme-Cu systems. Likewise to the copper-only systems mentioned above, the ligand environment around copper can be modulated in heme–Cu constructs as well. Further, aspects of the heme center physical properties may also be synthetically modified. As has been discussed in section 2.1.4, factors such as the identity of the coordinated axial base, changing porphyrinate substituents so as to modify the inherent heme redox potential, as well as synthetic input of secondary coordination interactions in the distal pocket (e.g., by placing H-bonding donor groups on the periphery of the ligand groups binding to the copper ion) may lead to the favoring of either heterolytic or homolytic cleavage. Synthetic model complexes have the advantage of being highly tunable, allowing for drastic changes in both the primary and secondary coordination sphere. This approach would likely be important for stabilizing a high-valent iron-oxo species and may allow for directing the chemistry toward preferential formation of Cmpd I vs Cmpd II.
While it is assumed that reductive O–O cleavage in the catalytic cycle of CcO is homolytic, giving [Fea3IV=O···HO– CuBII/Tyr–O·] (PM), there is the possibility that heterolytic O–O cleavage occurs, resulting in Cmpd I instead (i.e., [•+Fea3IV=O···HO–CuBII/Tyr–O−]). In fact, Rousseau, Gerfen, and co-workers have demonstrated that Cmpd I may be formed prior to the oxidation of the nearby histidine cross- linked tyrosine residue, when high-valent species are generated by addition of hydrogen peroxide to an oxidized form of bovine CcO. 884,887 Such chemistry may also be investigated for heme–copper complexes of synthetic design followed by reactivity studies, all contributing to our knowledge of the coordination chemistry of HCO BNC coordination chemistry.
6.5.5. Modeling Nitric Oxide Reductase Activity of Heme–Cu Oxidases.
Nitric oxide reductase (NOR) is an evolutionary relative of CcO, which possesses a binuclear heme/nonheme di-iron active site,1096 with close similarities to that of CcO (Figure 172). The proximal ligand on heme at the NOR active site is also a His residue, with the nonheme iron center (referred to as FeB) being coordinated to the three conserved His residues and a distal glutamate residue. NOR is an anaerobic respiratory enzyme found in bacteria, which catalyzes the reductive coupling of nitric oxide, NO(g), in the presence of protons to produce nitrous oxide, N2O(g), and water (Figure 172).1223,1224 Although nitric oxide reductases are involved in crucial physiological processes (particularly as related to pathological situations), mechanistic details of their NO(g) coupling reaction are not fully understood; coupling of nitric oxide molecules requires N–N bond formation, and then an N–O cleavage reaction must occur. The timing of proton and/or electron transfer events is not clear; however, the intermediacy of a hyponitrito (N2O22−) species (Figure 172) has been postulated.
Figure 172.

A side-by-side comparison of (A) NOR1096 and (B) CcO1236 active site structures (top), and the nitric oxide coupling reaction catalyzed by these enzymes (bottom) depicting the intermediacy of a hyponitrito dianionic species following N–N bond formation. Upon protonation, the hyponitrite intermediate produces N2O(g) and water. The electrons for the first step are believed to originate from the redox-active metal centers within the active site.
On the other hand, HCOs are also capable of reductively coupling NO(g), albeit with much lower efficiency than NORs. Therefore, interrogating the essential details of NO(g) coupling chemistry of HCO model systems is a topic of current interest, especially given the fact that NO(g) is a common inhibitor for CcO. A handful of small molecule NOR model systems that can couple NO(g) and produce N2O(g) have been reported by several research groups including our own.230,1048,1049,1225–1228 Importantly, Lu and co-workers have developed re-engineered myoglobin derived models of NOR,1099,1229–1232 which can catalytically couple NO(g) to N2O(g) with high efficiency.1229 These protein-based models have also revealed significant insights into mechanistic details,1230,1232 which not only benefit in comprehending the actual NOR turnover events but also serve as useful reference points in the rational design of small-molecule model systems. As with NORs, critical details pertaining to the nature of the intermediates, their protonation state(s) and coordination modes, and the influence of metal redox properties on reactivity remain unclear. To this end, heme-Cu model systems can and will serve as useful platforms to investigate such chemical problems, and the findings from these studies may serve in therapeutic applications,1233,1234 or in aiding the development of environmentally benign catalytic convertors for detoxifying NO(g) in industrial and automobile exhausts.1235
6.6. Final Remarks
It is evident from the results reviewed herein, which span studies in metalloenzyme biochemistry and synthetic inorganic coordination chemistry, that in order to develop a detailed understanding of biochemical processes (such as O2-reduction during cellular respiration), it is best to enlist perspectives and techniques from many subdisciplines. We have highlighted the important and diverse recent advancements contributed by investigations of bioinspired small molecule model systems and discussed them in the context of heme-copper oxidases. While the aim of designing, synthesizing, and investigating the small molecule activation chemistry of synthetic constructs is not to replicate the exact enzyme active site, they comprise various analogous characteristics which allow for directed interrogations of structure–function relationships and/or the nature of proton/electron transfer events. As we point out, especially in section 6, a well-rounded inorganic chemistry perspective based in experiment and theory is invaluable for critical evaluations of biochemical studies as well as elucidations of certain details which are inaccessible in direct enzyme studies (just as biochemical results inspire the design of new, more effective model systems). Of course, some aspects, such as the redox-coupled proton-pumping function of HCOs cannot (yet) be handled with synthetic models. However, we have also presented a brief, yet exciting, outlook for the many ways in which the application of inorganic coordination chemistry can be directed to provide even further important insights and inspire the design of systems to lead to a better fundamental understanding of the O2-reduction reaction necessary for sustaining aerobic life or even lead to the development of stand-alone, potentially catalytic, systems to carry out this fuel cell reaction.
ACKNOWLEDGMENTS
We gratefully acknowledge the USA National Institutes of Health (Research Grants R01 GM60353 and RO1 GM28962 to K.D.K.) for the support of the works from our laboratories described herein and the writing of this article.
ABBREVIATIONS
- 5C
Five coordinate
- 6C
six coordinate
- AO
amine oxidase
- APX
ascorbate peroxidase
- ATR-MIR
attenuated total reflection in mid-infrared
- BDE
bond dissociation energy
- BDFE
bond dissociation free energy
- BNC
binuclear center
- BO
bond order
- CcO
Cytochrome c Oxidase
- CCP
Cytochrome c Peroxidase
- Cmpd 0
Compound 0; (P)FeIII-OOH
- Cmpd I
Compound I; (P•+)FeIV=O
- Cmpd II
Compound II; (P)FeIV=O
- COx
catechol oxidase
- CPO
chloroperoxidase
- CV
cyclic voltammetry
- Cyt. P450
Cytochrome P450
- DFT
density functional theory
- DMAP
4-dimethylaminopyridine
- DβM
dopamine β-monooxygenase
- ENDOR
electron-nuclear double resonance
- EPG
edge plane pyrolytic graphite
- EPR
electron paramagnetic resonance
- ESI-MS
electrospray ionization mass spectrometry
- ET
electron transfer
- EXAFS
extended X-ray absorption fine structure
- FGE
Formylglycine generating enzyme
- FTIR
Fourier-transform infrared
- GO
galactose oxidase
- H-bonding
hydrogen bonding
- H.S.
high-spin
- HAA
hydrogen atom abstraction
- HAT
hydrogen atom transfer
- Hb
hemoglobin
- Hc
hemocyanin
- HCO
heme-copper oxidase
- HO
heme oxygenase
- HRP
horseradish peroxidase
- IDO
indoleamine 2,3-dioxygenase
- KIE
kinetic isotope effect
- L.S.
low-spin
- LMCT
ligand to metal charge transfer
- LPMO
lytic polysaccharide monooxygenase
- LPO
lactoperoxidase
- MALDI-TOF-MS
matrix-assisted laser desorption ionization time of flight mass spectrometry
- Mb
myoglobin
- MCD
magnetic circular dichroism
- MOF
metal organic framework
- MPO
myeloperoxidase
- NFK
N-formylkynureine
- NMR
nuclear magnetic resonance
- NOR
nitric oxide reductase
- NOS
nitric oxide synthase
- OER
oxygen-evolving reaction
- ORR
oxygen reduction reaction
- P
porphyrinate
- PCET
proton coupled electron transfer
- PHM
peptidylglycine α-hydroxylating monooxy-genase
- pMMO
particulate methane monooxygenase
- PROS
partially reduced oxygen species
- PT
proton transfer
- ROS
reactive oxygen species
- rR/rRaman
resonance Raman
- RRDE
rotating ring disc electrochemistry
- SAM
self-assembled monolayers
- SEIRA
surface-enhanced infrared absorption
- SERRS
surface-enhanced resonance Raman spectroscopy
- SF-ROX
serial femtosecond rotational crystallography
- sMMO
soluble methane monooyxgenase
- TDO
tryptophan 2,3-dioxygenase
- TS
transition state
- Ty
tyrosinase
- TβM
tyramine β-monooxygenase
- XANES
X-ray absorption near edge spectroscopy
- XAS
X-ray absorption spectroscopy
- XFEL
X-ray free electron laser
- XRD
X-ray diffraction
Biography
Suzanne M. Adam received her B.S. degree in chemistry from Lehigh University in 2012, during which time she also began her research career in the lab of Dr. Gregory S. Ferguson. She completed her Ph.D. work at Johns Hopkins University in 2017 under the guidance of Dr. Kenneth D. Karlin, wherein she studied the mechanism of O2- reduction and structure–function relationships involved in PCET reactions of bioinspired synthetic heme–Cu systems. She is currently completing postdoctoral research with Carole Duboc at the Université Grenoble Alpes and Vincent Artero at CEA-Grenoble, involving CO2 chemical valorization using bioinspired Ni/Fe catalysts inspired by carbon monoxide dehydrogenase and acetyl-CoA synthase.
Gayan B. Wijeratne was born in Kandy, Sri Lanka, and received his B.S. degree in chemistry from the University of Colombo, where his research on transition metal complexes of Sri Lankan natural products was recognized by the Professor R. S. Ramakrishna Memorial Gold Medal in inorganic chemistry in 2009. He moved to the United States in 2010 and carried out his graduate research in the laboratory of Professor Timothy A. Jackson at the University of Kansas, working on bioinspired manganese complexes that bind dioxygen and its reduced derivatives, as well as on thermodynamic and kinetic studies of proton-coupled electron transfer reactivity of manganese-based oxidants. There he won the Higuchi Doctoral Progress Award as the superior advanced graduate student of the University of Kansas in 2015. He was a postdoctoral research associate in the laboratory of Professor Kenneth D. Karlin at Johns Hopkins University from 2015 to 2018, where his research interests were focused on understanding heme and/or copper interactions with nitrogen oxides, primarily the mechanistic aspects involved with the reductive coupling of nitric oxide to nitrous oxide. He was also involved in dioxygen reduction and substrate oxidation reactivity studies of heme/copper assemblies. Dr. Wijeratne is currently an Assistant Professor of Chemistry at The University of Alabama at Birmingham, where his independent research program is geared toward interrogating key mechanistic details pertaining to various crucial bioinorganic processes, and comprehending their implications in human health and therapeutics, sustainable catalysis, and alternative energy applications.
Patrick J. Rogler received his B.S. degree in chemistry from Rutgers University, The State University of New Jersey, in 2012. He is currently working toward his Ph.D. in chemistry at The Johns Hopkins University in Baltimore, MD, in the research group of Professor Kenneth D. Karlin. His doctoral work is concerned with small molecule synthetic models for the active site of Cytochrome c Oxidase.
Daniel E. Diaz was born in Santiago, Chile, and received his B.S. degree in chemistry from Universidad de Santiago de Chile. He came to the USA with a Fulbright fellowship and joined Johns Hopkins University, where he is currently working toward his Ph.D. in Chemistry under the direction of Professor Kenneth D. Karlin. His research focuses on the biomimetic study of primary copper (di) oxygen adducts present in the proposed catalytic mechanisms of copper-containing monooxygenases, such as PHM and LPMOs.
David A. Quist received his B.S. degree in chemistry from the University of Michigan in 2013. He is currently a Ph.D. candidate at Johns Hopkins University, working in the lab of Dr. Karlin. His research is aimed toward the understanding of binuclear copper model complex O2-adducts, their redox interconversions, and their O–O bond cleavage processes.
Jeffrey J. Liu received his B.S. degree in chemistry from Boston University in 2013 and went on to Johns Hopkins University to pursue his Ph.D. under the supervision of Professor Kenneth D. Karlin. His current research in the Karlin Lab involves understanding the reactivity of copper-dioxygen intermediates with nitric oxide, including investigations of copper-peroxynitrite species which may form from Cu-O2 and NO.
Kenneth D. Karlin received his B.S. degree in chemistry from Stanford University, and went on to obtain a Ph.D. from Columbia University in 1975 under the direction of Stephen J. Lippard. He then completed a NATO postdoctoral fellowship at the University of Cambridge, UK (1975–1977), before beginning his independent career at the State University of New York (SUNY) at Albany in 1977. In 1990, he moved to Baltimore, MD and began building his research program at Johns Hopkins University, where he currently holds the position of Ira Remsen Chair in Chemistry. His research interests include the coordination chemistry of bioinspired copper and heme/Cu systems, with an emphasis on mechanistic studies of their small molecule (O2, NOx) activation reactivities.
Footnotes
Since completion of this Review, a number of excellent articles have appeared, which are of relevance to sub-topics described here. An important paper on 2nd sphere hydrogen-bonding for O2-reduction by hemes appeared, see ref 1240. A further study on pMMO affirms the hypothesis that the active site consists of a mono-copper center including a “histidine brace” chelate (ref 1241). An excellent perspective article on LPMOs was recently published (ref 1242). The importance of methane and its oxidation to methanol has been highlighted for chemistry occurring in zeolites (refs 1243, 1244, 1245, 1246, 1247, 1248) or in Metal-Organic Frameworks (MOFs) (refs 1249, 1250). Two new CcO X-ray structures (refs 1251, 1252) have been published and new data on a CcO reaction intermediate appears in a recent report (ref 1253).
The authors declare no competing financial interest.
REFERENCES
- (1).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Armstrong DA; Huie RE; Koppenol WH; Lymar SV; Merenyi G; Neta P; Ruscic B; Stanbury DM; Steenken S; Wardman P Standard Electrode Potentials Involving Radicals in Aqueous Solution: Inorganic Radicals (IUPAC Technical Report). Pure Appl. Chem 2015, 87, 1139–1150. [Google Scholar]
- (3).Zhang W; Lai W; Cao R Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin- and Corrole-Based Systems. Chem. Rev 2017, 117, 3717–3797. [DOI] [PubMed] [Google Scholar]
- (4).Su DS; Sun G Nonprecious-Metal Catalysts for Low-Cost Fuel Cells. Angew. Chem., Int. Ed 2011, 50, 11570–11572. [DOI] [PubMed] [Google Scholar]
- (5).Dy ES; Roman TA; Kubota Y; Miyamoto K; Kasai H Exploring Haem-Based Alternatives for Oxygen Reduction Catalysis in Fuel Cells–A Status Report of Our First Principles Calculations. J. Phys.: Condens. Matter 2007, 19, 445010. [Google Scholar]
- (6).Shao M; Chang Q; Dodelet J-P; Chenitz R Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev 2016, 116, 3594–3657. [DOI] [PubMed] [Google Scholar]
- (7).Cracknell JA; Vincent KA; Armstrong FA Enzymes as Working or Inspirational Electrocatalysts for Fuel Cells and Electrolysis. Chem. Rev 2008, 108, 2439–2461. [DOI] [PubMed] [Google Scholar]
- (8).Chung HT; Cullen DA; Higgins D; Sneed BT; Holby EF; More KL; Zelenay P Direct Atomic-Level Insight into the Active Sites of a High-Performance PGM-Free ORR Catalyst. Science 2017, 357, 479–484. [DOI] [PubMed] [Google Scholar]
- (9).Mano N; de Poulpiquet A O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev 2018, 118, 2392. [DOI] [PubMed] [Google Scholar]
- (10).Hammes-Schiffer S; Stuchebrukhov AA Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev 2010, 110, 6939–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Siegbahn PEM; Blomberg MRA Quantum Chemical Studies of Proton-Coupled Electron Transfer in Metalloenzymes. Chem. Rev 2010, 110, 7040–7061. [DOI] [PubMed] [Google Scholar]
- (12).Migliore A; Polizzi NF; Therien MJ; Beratan DN Biochemistry and Theory of Proton-Coupled Electron Transfer. Chem. Rev 2014, 114, 3381–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rosenthal J; Nocera DG Role of Proton-Coupled Electron Transfer in O–O Bond Activation. Acc. Chem. Res 2007, 40, 543–553. [DOI] [PubMed] [Google Scholar]
- (14).Usharani D; Janardanan D; Li C; Shaik S A Theory for Bioinorganic Chemical Reactivity of Oxometal Complexes and Analogous Oxidants: The Exchange and Orbital-Selection Rules. Acc. Chem. Res 2013, 46, 471–482. [DOI] [PubMed] [Google Scholar]
- (15).Yee GM; Tolman WB Transition Metal Complexes and the Activation of Dioxygen In Metal Ions in Life Sciences; Kroneck PMH, Sosa Torres ME, Eds.; Springer International Publishing: Cham, 2015; Vol. 15, pp 131–204. [DOI] [PubMed] [Google Scholar]
- (16).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron-Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper-Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yoshikawa S; Shimada A Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- (21).Warren JJ; Mayer JM Moving Protons and Electrons in Biomimetic Systems. Biochemistry 2015, 54, 1863–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Boulatov R Understanding the Reaction That Powers This World: Biomimetic Studies of Respiratory O2 Reduction by Cytochrome Oxidase. Pure Appl. Chem 2004, 76, 303–319. [Google Scholar]
- (23).Yu F; Cangelosi VM; Zastrow ML; Tegoni M; Plegaria JS; Tebo AG; Mocny CS; Ruckthong L; Qayyum H; Pecoraro VL Protein Design: Toward Functional Metalloenzymes. Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nastri F; Chino M; Maglio O; Bhagi-Damodaran A; Lu Y; Lombardi A Design and Engineering of Artificial Oxygen-Activating Metalloenzymes. Chem. Soc. Rev 2016, 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bhagi-Damodaran A; Hosseinzadeh P; Mirts E; Reed J; Petrik ID; Lu Y Design of Heteronuclear Metalloenzymes In Methods in Enzymology; Pecoraro VL, Ed.; Elsevier Inc., 2016; Vol. 580, pp 501–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fukuzumi S; Lee Y-M; Nam W Mechanisms of Two- Electron versus Four-Electron Reduction of Dioxygen Catalyzed by Earth-Abundant Metal Complexes. ChemCatChem 2018, 10, 9–28. [Google Scholar]
- (27).Pegis ML; Roberts JAS; Wasylenko DJ; Mader EA; Appel AM; Mayer JM Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylfor-mamide. Inorg. Chem 2015, 54, 11883–11888. [DOI] [PubMed] [Google Scholar]
- (28).Watanabe Y; Nakajima H; Ueno T Reactivities of Oxo and Peroxo Intermediates Studied by Hemoprotein Mutants. Acc. Chem. Res 2007, 40, 554–562. [DOI] [PubMed] [Google Scholar]
- (29).Smith LJ; Kahraman A; Thornton JM Heme Proteins–Diversity in Structural Characteristics, Function, and Folding. Proteins: Struct. Fund., Genet 2010, 78, 2349–2368. [DOI] [PubMed] [Google Scholar]
- (30).Mukai M; Savard P-Y; Ouellet H; Guertin M; Yeh S-R Unique Ligand-Protein Interactions in a New Truncated Hemoglobin from Mycobacterium Tuberculosis. Biochemistry 2002, 41 , 3897–3905. [DOI] [PubMed] [Google Scholar]
- (31).Wittenberg JB; Bolognesi M; Wittenberg BA; Guertin M Truncated Hemoglobins: A New Family of Hemoglobins Widely Distributed in Bacteria, Unicellular Eukaryotes, and Plants. J. Biol. Chem 2002, 277, 871–874. [DOI] [PubMed] [Google Scholar]
- (32).Lama A; Pawaria S; Dikshit KL Oxygen Binding and NO Scavenging Properties of Truncated Hemoglobin, HbN, of Mycobacterium Smegmatis. FEBS Lett. 2006, 580, 4031–4041. [DOI] [PubMed] [Google Scholar]
- (33).Ouellet H; Ouellet Y; Richard C; Labarre M; Wittenberg B; Wittenberg J; Guertin M Truncated Hemoglobin HbN Protects Mycobacterium Bovis from Nitric Oxide. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 5902–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hill DR; Belbin TJ; Thorsteinsson MV; Bassam D; Brass S; Ernst A; Boger P; Paerl H; Mulligan ME; Potts M GlbN (Cyanoglobin) Is a Peripheral Membrane Protein That Is Restricted to Certain Nostoc Spp. J. Bacteriol 1996, 178, 6587–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Royer WE; Sharma H; Strand K; Knapp JE; Bhyravbhatla B Lumbricus Erythrocruorin at 3.5 A Resolution: Architecture of a Megadalton Respiratory Complex. Structure 2006, 14, 1167–1177. [DOI] [PubMed] [Google Scholar]
- (36).Shimizu T; Huang D; Yan F; Stranava M; Bartosova M; Fojtíkova V; Martínkova M Gaseous O2, NO, and CO in Signal Transduction: Structure and Function Relationships of Heme-Based Gas Sensors and Heme-Redox Sensors. Chem. Rev 2015, 115, 6491–6533. [DOI] [PubMed] [Google Scholar]
- (37).Aono S Novel Bacterial Gas Sensor Proteins with Transition Metal-Containing Prosthetic Groups as Active Sites. Antioxid. Redox Signaling 2012, 16, 678–686. [DOI] [PubMed] [Google Scholar]
- (38).Aono S Metal-Containing Sensor Proteins Sensing Diatomic Gas Molecules. Dalton Trans 2008, 0, 3137–3146. [DOI] [PubMed] [Google Scholar]
- (39).Lombardi A; Nastri F; Pavone V Peptide-Based HemeProtein Models. Chem. Rev 2001, 101, 3165–3190. [DOI] [PubMed] [Google Scholar]
- (40).Sono M; Roach MP; Coulter ED; Dawson JH Heme-Containing Oxygenases. Chem. Rev 1996, 96, 2841–2888. [DOI] [PubMed] [Google Scholar]
- (41).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- (42).Shaik S; Kumar D; de Visser SP; Altun A; Thiel W Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev 2005, 105, 2279–2328. [DOI] [PubMed] [Google Scholar]
- (43).Meunier B; de Visser SP; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (44).Karlin KD Model Offers Intermediate Insight. Nature 2010, 463, 168–169. [DOI] [PubMed] [Google Scholar]
- (45).Poulos TL Peroxidase and Catalases In Biological Inorganic Chemistry: Structure and Reactivity; Bertini I, Gray HB, Stiefel EI, Valentine JS, Eds.; University Science Books, 2007; pp 343–353. [Google Scholar]
- (46).Shaik S; Cohen S; Wang Y; Chen H; Kumar D; Thiel W P450 Enzymes: Their Structure, Reactivity, and Selectivity–Modeled by QM/MM Calculations. Chem. Rev 2010, 110, 949–1017. [DOI] [PubMed] [Google Scholar]
- (47).Bruice TC Reactions of Hydroperoxides with Metal-lotetraphenylporphyrins in Aqueous Solutions. Acc. Chem. Res 1991, 24, 243–249. [Google Scholar]
- (48).Traylor TG; Tsuchiya S; Byun YS; Kim C High-Yield Epoxidations with Hydrogen Peroxide and Tert-Butyl Hydroperoxide Catalyzed by Iron(III) Porphyrins: Heterolytic Cleavage of Hydroperoxides. J. Am. Chem. Soc 1993, 115, 2775–2781. [Google Scholar]
- (49).Oszajca M; Franke A; Brindell M; Stochel G; van Eldik R Redox Cycling in the Activation of Peroxides by Iron Porphyrin and Manganese Complexes. “Catching” Catalytic Active Intermediates. Coord. Chem. Rev 2016, 306, 483–509. [Google Scholar]
- (50).Groves JT; Watanabe Y Reactive Iron Porphyrin Derivatives Related to the Catalytic Cycles of Cytochrome P-450 and Peroxidase. Studies of the Mechanism of Oxygen Activation. J. Am. Chem. Soc 1988, 110, 8443–8452. [Google Scholar]
- (51).Nam W; Lim MH; Oh S-YY Effect of Anionic Axial Ligands on the Formation of Oxoiron(IV) Porphyrin Intermediates. Inorg. Chem 2000, 39, 5572–5575. [DOI] [PubMed] [Google Scholar]
- (52).Machii K; Watanabe Y; Morishima I Acylperoxo-Iron(III) Porphyrin Complexes: A New Entry of Potent Oxidants for the Alkene Epoxidation. J. Am. Chem. Soc 1995, 117, 6691–6697. [Google Scholar]
- (53).Newcomb M; Toy PH Hypersensitive Radical Probes and the Mechanisms of Cytochrome P450-Catalyzed Hydroxylation Reactions. Acc. Chem. Res 2000, 33, 449–455. [DOI] [PubMed] [Google Scholar]
- (54).Perutz MF; Rossmann MG; Cullis AF; Muirhead H; Will G; North ACT Structure of Haemoglobin: A ThreeDimensional Fourier Synthesis at 5.5-Å Resolution, Obtained by X-Ray Analysis. Nature 1960, 185, 416–422. [DOI] [PubMed] [Google Scholar]
- (55).Kendrew JC; Dickerson RE; Strandberg BE; Hart RG; Davies DR; Phillips DC; Shore VC Structure of Myoglobin: A Three-Dimensional Fourier Synthesis at 2 A Resolution. Nature 1960, 185, 422–427. [DOI] [PubMed] [Google Scholar]
- (56).Collman JP; Boulatov R; Sunderland CJ; Fu L Functional Analogues of Cytochrome c Oxidase, Myoglobin, and Hemoglobin. Chem. Rev 2004, 104, 561–588. [DOI] [PubMed] [Google Scholar]
- (57).Hoard JL; Hamor MJ; Hamor TA; Caughey WS The Crystal Structure and Molecular Stereochemistry of Methoxyiron(III) Mesoporphyrin-IX Dimethyl Ester. J. Am. Chem. Soc 1965, 87, 2312–2319. [Google Scholar]
- (58).Hoard JL; Cohen GH; Glick MD The Stereochemistry of the Coordination Group in an Iron(III) Derivative of Tetraphenylporphine. J. Am. Chem. Soc 1967, 89, 1992–1996. [Google Scholar]
- (59).Countryman RP; Collins DM; Hoard JL Stereochemistry of the Low-Spin Iron Porphyrin, bis-(imidazole)-α,β,γ,δ-tetraphenylporphinatoiron(III) Chloride. J. Am. Chem. Soc 1969, 91, 5166–5167. [DOI] [PubMed] [Google Scholar]
- (60).Hu C; An J; Noll BC; Schulz CE; Scheidt WR Electronic Configuration of High-Spin Imidazole-Ligated Iron(II) Octaethylporphyrinates. Inorg. Chem 2006, 45, 4177–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ellison MK; Schulz CE; Scheidt WR Structure of the Deoxymyoglobin Model [Fe(TPP)(2-MeHIm)] Reveals Unusual Porphyrin Core Distortions. Inorg. Chem 2002, 41, 2173–2181. [DOI] [PubMed] [Google Scholar]
- (62).Perutz MF Regulation of Oxygen Affinity of Hemoglobin: Influence of Structure of the Globin on the Heme Iron. Annu. Rev. Biochem 1979, 48, 327–386. [DOI] [PubMed] [Google Scholar]
- (63).Jameson GB; Ibers JA Biological and Synthetic Dioxygen Carriers In Bioinorganic Chemistry; Bertini I, Gray HB, Lippard SJ, Valentine JS, Eds.; University Science Books, 1994; pp 167–252. [Google Scholar]
- (64).Braun W; Wagner G; Worgotter E; Vasak M; Kagi JHR; Wuthrich K Polypeptide Fold in the Two Metal Clusters of Metallothionein-2 by Nuclear Magnetic Resonance in Solution. J. Mol. Biol 1986, 187, 125–129. [DOI] [PubMed] [Google Scholar]
- (65).Kepp KP; Dasmeh P Effect of Distal Interactions on O2 Binding to Heme. J. Phys. Chem. B 2013, 117, 3755–3770. [DOI] [PubMed] [Google Scholar]
- (66).Das TK; Couture M; Ouellet Y; Guertin M; Rousseau DL Simultaneous Observation of the O–O and Fe–O2 Stretching Modes in Oxyhemoglobins. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Paoli M; Liddington R; Tame J; Wilkinson A; Dodson G Crystal Structure of T State Haemoglobin with Oxygen Bound At All Four Haems. J. Mol. Biol 1996, 256, 775–792. [DOI] [PubMed] [Google Scholar]
- (68).Phillips SEV Structure of Oxymyoglobin. Nature 1978, 273, 247–248. [DOI] [PubMed] [Google Scholar]
- (69).Shaanan B The Iron-Oxygen Bond in Human Oxy- haemoglobin. Nature 1982, 296, 683–684. [DOI] [PubMed] [Google Scholar]
- (70).Chen H; Ikeda-Saito M; Shaik S Nature of the Fe-O2 Bonding in Oxy-Myoglobin: Effect of the Protein. J. Am. Chem. Soc 2008, 130, 14778–14790. [DOI] [PubMed] [Google Scholar]
- (71).Basolo F; Hoffman BM; Ibers JA Synthetic Oxygen Carriers of Biological Interest. Acc. Chem. Res 1975, 8, 384–392. [Google Scholar]
- (72).Jameson GB; Molinaro FS; Ibers JA; Collman JP; Brauman JI; Rose E; Suslick KS Models for the Active Site of Oxygen-Binding Hemoproteins. Dioxygen Binding Properties and the Structures of (2-Methylimidazole)-Meso-tetra(α,α,α,α–o–pivalamidophenyl)porphyrinatoiron(II)-Ethanol and Its Dioxygen Adduct. J. Am. Chem. Soc 1980, 102, 3224–3237. [Google Scholar]
- (73).Perdew JP; Tao J; Staroverov VN; Scuseria GE MetaGeneralized Gradient Approximation: Explanation of a Realistic Nonempirical Density Functional. J. Chem. Phys 2004, 120, 6898–6911. [DOI] [PubMed] [Google Scholar]
- (74).Reiher M Theoretical Study of the Fe(phen)2(NCS)2 SpinCrossover Complex with Reparametrized Density Functionals. Inorg. Chem 2002, 41, 6928–6935. [DOI] [PubMed] [Google Scholar]
- (75).Lawson Daku LM; Vargas A; Hauser A; Fouqueau A; Casida ME Assessment of Density Functionals for the High-Spin/Low-Spin Energy Difference in the Low-Spin Iron(II) Tris(2,2′-Bipyridine) Complex. ChemPhysChem 2005, 6, 1393–1410. [DOI] [PubMed] [Google Scholar]
- (76).Jameson GB; Ibers JA Dioxygen Carriers In Biological Inorganic Chemistry: Structure and Reactivity; Bertini I, Gray HB, Stiefel EI, Valentine JS, Eds.; University Science Books, 2007; pp 354–388. [Google Scholar]
- (77).Samuni U; Juszczak L; Dantsker D; Khan I; Friedman AJ; Perez-Gonzalez-de-Apodaca J; Bruno S; Hui HL; Colby JE; Karasik E; et al. Functional and Spectroscopic Characterization of Half-Liganded Iron-Zinc Hybrid Hemoglobin: Evidence for Conformational Plasticity within the T State. Biochemistry 2003, 42, 8272–8288. [DOI] [PubMed] [Google Scholar]
- (78).Venkatesh B; Manoharan PT; Rifkind JM Metal Ion Reconstituted Hybrid Hemoglobins In Progress in Inorganic Chemistry; John Wiley & Sons, Inc., 2007; pp 563–684. [Google Scholar]
- (79).Momenteau M; Reed CA Synthetic Heme-Dioxygen Complexes. Chem. Rev 1994, 94, 659–698. [Google Scholar]
- (80).Heidner EJ; Ladner RC; Perutz MF Structure of Horse Carbonmonoxyhaemoglobin. J. Mol. Biol 1976, 104, 707–722. [DOI] [PubMed] [Google Scholar]
- (81).Bucci E; Gryczynski Z; Razynska A; Kwansa H Entropy-Driven Intermediate Steps of Oxygenation May Regulate the Allosteric Behavior of Hemoglobin. Biophys. J 1998, 74, 2638–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Umbreit J Methemoglobin–It’s Not Just Blue: A Concise Review. Am. J. Hematol 2007, 82, 134–144. [DOI] [PubMed] [Google Scholar]
- (83).Wallace WJ; Houtchens RA; Maxwell JC; Caughey WS Mechanism of Autooxidation for Hemoglobins and Myoglobins. Promotion of Superoxide Production by Protons and Anions. J. Biol. Chem 1982, 257, 4966–4977. [PubMed] [Google Scholar]
- (84).Tsuruga M; Matsuoka A; Hachimori A; Sugawara Y; Shikama K The Molecular Mechanism of Autoxidation for Human Oxyhemoglobin: Tilting of the Distal Histidine Causes Nonequivalent Oxidation in the β Chain. J. Biol. Chem 1998, 273, 8607–8615. [DOI] [PubMed] [Google Scholar]
- (85).Bren KL; Eisenberg R; Gray HB Discovery of the Magnetic Behavior of Hemoglobin: A Beginning of Bioinorganic Chemistry. Proc. Natl. Acad. Sci U. S. A 2015, 112, 13123–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Pauling L; Coryell CD The Magnetic Properties and Structure of Hemoglobin, Oxyhemoglobin and Carbonmonoxyhemoglobin. Proc. Natl. Acad. Sci U. S. A 1936, 22, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Weiss JJ Nature of the Iron-Oxygen Bond in Oxy- haemoglobin. Nature 1964, 202, 83–84. [DOI] [PubMed] [Google Scholar]
- (88).McClure DS Electronic Structure of Transition-Metal Complex Ions. Radiat. Res., Suppl 1960, 2, 218–242. [Google Scholar]
- (89).Harcourt RD Increased-Valence Formulae and the Bonding of Oxygen to Haemoglobin. Int. J. Quantum Chem 1971, 5, 479–495. [Google Scholar]
- (90).Harcourt RD Comment on a CASSCF Study of the Fe-O2 Bond in a Dioxygen Heme Complex. Chem. Phys. Lett 1990, 167, 374–377. [Google Scholar]
- (91).Goddard WA; Olafson BD Ozone Model for Bonding of an O2 to Heme in Oxyhemoglobin. Proc. Natl. Acad. Sci. U. S. A 1975, 72, 2335–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Schuth N; Mebs S; Huwald D; Wrzolek P; Schwalbe M; Hemschemeier A; Haumann M; et al. Effective Intermediate-Spin Iron in O2-Transporting Heme Proteins. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 8556–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Hersleth H-P; Andersson KK How Different Oxidation States of Crystalline Myoglobin Are Influenced by X-Rays. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 785–796. [DOI] [PubMed] [Google Scholar]
- (94).Wilson SA; Kroll T; Decreau RA; Hocking RK; Lundberg M; Hedman B; Hodgson KO; Solomon EI Iron L-Edge X-Ray Absorption Spectroscopy of Oxy-Picket Fence Porphyrin: Experimental Insight into Fe-O2 Bonding. J. Am. Chem. Soc 2013, 135, 1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Wilson SA; Green E; Mathews II; Benfatto M; Hodgson KO; Hedman B; Sarangi R X-Ray Absorption Spectroscopic Investigation of the Electronic Structure Differences in Solution and Crystalline Oxyhemoglobin. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 16333–16338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Grinstaff MW; Hill MG; Labinger JA; Gray HB Mechanism of Catalytic Oxygenation of Alkanes by Halogenated Iron Porphyrins. Science 1994, 264, 1311–1314. [DOI] [PubMed] [Google Scholar]
- (97).Potter WT; Tucker MP; Houtchens RA; Caughey WS Oxygen Infrared Spectra of Oxyhemoglobins and Oxymyoglobins. Evidence of Two Major Liganded Oxygen Structures. Biochemistry 1987, 26, 4699–4707. [DOI] [PubMed] [Google Scholar]
- (98).Spiro TG; Strekas TC Resonance Raman Spectra of Heme Proteins. Effects of Oxidation and Spin State. J. Am. Chem. Soc 1974, 96, 338–345. [DOI] [PubMed] [Google Scholar]
- (99).Hayaishi O; Katagiri M; Rothberg S Mechanism of the Pyrocatechase Reaction. J. Am. Chem. Soc 1955, 77, 5450–5451. [Google Scholar]
- (100).Mason HS; Fowlks WL; Peterson E Oxygen Trasfer and Electron Transport by the Phenolase Complex. J. Am. Chem. Soc 1955, 77, 2914–2915. [Google Scholar]
- (101).Hamilton GA Chemical Models and Mechanisms for Oxygenases In Molecular Mechanisms of Oxygen Activation; Hayaishi O, Ed.; Academic Press: New York, NY, 1974; pp 405–451. [Google Scholar]
- (102).Mak PJ; Gregory MC; Denisov IG; Sligar SG; Kincaid JR Unveiling the Crucial Intermediates in Androgen Production. Proc. Natl. Acad. Sci U. S. A 2015, 112, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Mak PJ; Duggal R; Denisov IG; Gregory MC; Sligar SG; Kincaid JR Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J. Am. Chem. Soc 2018, 140, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Cole PA; Robinson CH Peroxide Model Reaction for Placental Aromatase. J. Am. Chem. Soc 1988, 110, 1284–1285. [Google Scholar]
- (105).Cole PA; Bean JM; Robinson CH Conversion of a 3-Desoxysteroid to 3-Desoxyestrogen by Human Placental Aromatase. Proc. Natl. Acad. Sci U. S. A 1990, 87, 2999–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Ghosh D; Griswold J; Erman M; Pangborn W Structural Basis for Androgen Specificity and Oestrogen Synthesis in Human Aromatase. Nature 2009, 457, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Waterman MR Anticancer Drug Target Pictured. Nature 2009, 457, 159–160. [DOI] [PubMed] [Google Scholar]
- (108).Yoshimoto FK; Guengerich FP Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase. J. Am. Chem. Soc 2014, 136, 15016–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Poulos TL Thirty Years of Heme Peroxidase Structural Biology. Arch. Biochem. Biophys. 2010, 500, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Hannemann F; Bichet A; Ewen KM; Bernhardt R Cytochrome P450 Systems–Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta, Gen. Subj 2007, 1770, 330–344. [DOI] [PubMed] [Google Scholar]
- (111).Ueyama N; Yamada Y; Okamura T; Kimura S; Nakamura A Structure and Properties of [Fe4S4{2,6-Bis(acylamino)-benzenethiolato-S}4]2- and [Fe2S2{2,6-Bis(acylamino)-benzenethiolato-S}4]2-: Protection of the Fe-S Bond by Double. Inorg. Chem 1996, 35, 6473–6484. [DOI] [PubMed] [Google Scholar]
- (112).Langen R; Jensen GM; Jacob U; Stephens PJ; Warshel A Protein Control of Iron-Sulfur Cluster Redox Potentials. J. Biol. Chem 1992, 267, 25625–25627. [PubMed] [Google Scholar]
- (113).Adman E; Watenpaugh KD; Jensen LH NH–S Hydrogen Bonds in Peptococcus Aerogenes Ferredoxin, Clostridium Pasteurianum Rubredoxin, and Chromatium High Potential Iron Protein. Proc. Natl. Acad. Sci. U. S. A 1975, 72, 4854–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Raag R; Poulos TL The Structural Basis for Substrate-Induced Changes in Redox Potential and Spin Equilibrium in Cytochrome P-450CAM. Biochemistry 1989, 28, 917–922. [DOI] [PubMed] [Google Scholar]
- (115).Haines DC; Tomchick DR; Machius M; Peterson JA Pivotal Role of Water in the Mechanism of P450BM-3. Biochemistry 2001, 40, 13456–13465. [DOI] [PubMed] [Google Scholar]
- (116).Shiro Y; Makino R; Sato F; Oyanagi H; Matsushita T; Ishimura Y; Iizuka T Structural and Electronic Characterization of Heme Moiety in Oxygenated Hemoproteins by Using XANES Spectroscopy. Biochim. Biophys. Acta, Gen. Subj 1991, 1115, 101–107. [DOI] [PubMed] [Google Scholar]
- (117).Bonfils C; Debey P; Maurel P Highly Purified Microsomal P-450: The Oxyferro Intermediate Stabilized at Low Temperature. Biochem. Biophys. Res. Commun 1979, 88, 1301–1307. [DOI] [PubMed] [Google Scholar]
- (118).Bangcharoenpaurpong O; Rizos AK; Champion PM; Jollie D; Sligar SG Resonance Raman Detection of Bound Dioxygen in Cytochrome P-450cam. J. Biol. Chem 1986, 261, 8089–8092. [PubMed] [Google Scholar]
- (119).Sharrock M; Debrunner PG; Schulz C; Lipscomb JD; Marshall V; Gunsalus IC Cytochrome P450cam and Its Complexes, Mossbauer Parameters of the Heme Iron. Biochim. Biophys. Acta, Protein Struct 1976, 420, 8–26. [DOI] [PubMed] [Google Scholar]
- (120).Schlichting I; Berendzen J; Chu K; Stock AM; Maves SA; Benson DE; Sweet RM; Ringe D; Petsko GA; Sligar SG The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science 2000, 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- (121).Davydov R; Makris TM; Kofman V; Werst DE; Sligar SG; Hoffman BM Hydroxylation of Camphor by Reduced Oxy- Cytochrome P450cam: Mechanistic Implications of EPR and ENDOR Studies of Catalytic Intermediates in Native and Mutant Enzymes. J. Am. Chem. Soc 2001, 123, 1403–1415. [DOI] [PubMed] [Google Scholar]
- (122).Liu J-G; Shimizu Y; Ohta T; Naruta Y Formation of an End-on Ferric Peroxo Intermediate upon One-Electron Reduction of a Ferric Superoxo Heme. J. Am. Chem. Soc 2010, 132, 3672–3673. [DOI] [PubMed] [Google Scholar]
- (123).Ohta T; Liu J-G; Naruta Y Resonance Raman Characterization of Mononuclear Heme-Peroxo Intermediate Models. Coord. Chem. Rev 2013, 257, 407–413. [Google Scholar]
- (124).Nagano S; Poulos TL Crystallographic Study on the Dioxygen Complex of Wild-Type and Mutant Cytochrome P450cam. J. Biol. Chem 2005, 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- (125).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (126).Rittle J; Younker JM; Green MT Cytochrome P450: The Active Oxidant and Its Spectrum. Inorg. Chem 2010, 49, 3610–3617. [DOI] [PubMed] [Google Scholar]
- (127).Yosca TH; Ledray AP; Ngo J; Green T A New Look at the Role of Thiolate Ligation in Cytochrome P450. J. Biol. Inorg. Chem 2017, 22, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Korzekwa KR; Trager WF; Mancewicz J; Osawa Y Studies on the Mechanism of Aromatase and Other Cytochrome P450 Mediated Deformylation Reactions. J. Steroid Biochem. Mol. Biol 1993, 44, 367–373. [DOI] [PubMed] [Google Scholar]
- (129).Ahmed S; Davis PJ The Mechanism of Aromatase – a Molecular Modelling Perspective. Bioorg. Med. Chem. Lett 1995, 5, 2789–2794. [Google Scholar]
- (130).Akhtar M; Corina D; Pratt J; Smith T Studies on the Removal of C-19 in Oesterogen Biosynthesis Using 18O2. J. Chem. Soc., Chem. Commun 1976, 854–856. [Google Scholar]
- (131).Davydov R; Hoffman BM Active Intermediates in Heme Monooxygenase Reactions as Revealed by Cryoreduction/Annealing, EPR/ENDOR Studies. Arch. Biochem. Biophys 2011, 507, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)Hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Fujii H Model Complexes of Heme Peroxidases In Heme Peroxidases; Raven E, Dunford B, Eds.; Royal Society of Chemistry: Cambridge, UK, 2016; pp 181–217. [Google Scholar]
- (134).Fujii H Electronic Structure and Reactivity of High-Valent Oxo Iron Porphyrins. Coord. Chem. Rev 2002, 226, 51–60. [Google Scholar]
- (135).Huang X; Groves JT Beyond Ferryl-Mediated Hydrox- ylation: 40 Years of the Rebound Mechanism and C-H Activation. JBIC, J. Biol. Inorg. Chem 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Groves JT; McClusky GA; White RE; Coon MJ Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P-450. Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun 1978, 81, 154–160. [DOI] [PubMed] [Google Scholar]
- (137).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Cryle MJ; Ortiz de Montellano PR; De Voss JJ Cyclopropyl Containing Fatty Acids as Mechanistic Probes for Cytochromes P450. J. Org. Chem 2005, 70, 2455–2469. [DOI] [PubMed] [Google Scholar]
- (141).Shaik S; de Visser SP; Ogliaro F; Schwarz H; Schröder D Two-State Reactivity Mechanisms of Hydroxylation and Epoxidation by Cytochrome P-450 Revealed by Theory. Curr. Opin. Chem. Biol 2002, 6, 556–567. [DOI] [PubMed] [Google Scholar]
- (142).Shaik S; Lai W; Chen H; Wang Y The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs. Acc. Chem. Res 2010, 43, 1154–1165. [DOI] [PubMed] [Google Scholar]
- (143).Newcomb M; Shen R; Lu Y; Coon MJ; Hollenberg PF; Kopp DA; Lippard SJ Evaluation of Norcarane as a Probe for Radicals in Cytochome P450- and Soluble Methane Monooxygenase-Catalyzed Hydroxylation Reactions. J. Am. Chem. Soc 2002, 124, 6879–6886. [DOI] [PubMed] [Google Scholar]
- (144).Porter TD; Coon MJ Cytochrome P-450. Multiplicity of Isoforms, Substrates, and Catalytic and Regulatory Mechanisms. J. Biol. Chem 1991, 266, 13469–13472. [PubMed] [Google Scholar]
- (145).Ortiz de Montellano PR Cytochrome P450: Structure, Mechanism, and Biochemistry, 2nd ed.; Ortiz de Montellano PR, Ed.; Plenum: New York, 1995. [Google Scholar]
- (146).White RE; Coon MJ Oxygen Activation by Cytochrome P-450. Annu. Rev. Biochem 1980, 49, 315–356. [DOI] [PubMed] [Google Scholar]
- (147).Wang X; Ullrich R; Hofrichter M; Groves JT Heme-Thiolate Ferryl of Aromatic Peroxygenase Is Basic and Reactive. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 3686–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Wang Y; Lan D; Durrani R; Hollmann F Peroxygenases En Route to Becoming Dream Catalysts. What Are the Opportunities and Challenges? Curr. Opin. Chem. Biol 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- (149).Wang X; Peter S; Kinne M; Hofrichter M; Groves JT Detection and Kinetic Characterization of a Highly Reactive Heme–Thiolate Peroxygenase Compound I. J. Am. Chem. Soc 2012, 134, 12897–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Bernhardt R Cytochrome P450: Structure, Function, and Generation of Reactive Oxygen Species. Rev. Physiol. Biochem. Pharmacol 1996, 127, 137–221. [DOI] [PubMed] [Google Scholar]
- (151).Loida PJ; Sligar SG Molecular Recognition in Cytochrome P-450: Mechanism for the Control of Uncoupling Reactions. Biochemistry 1993, 32, 11530–11538. [DOI] [PubMed] [Google Scholar]
- (152).Kadkhodayan S; Coulter ED; Maryniak DM; Bryson TA; Dawson JH Uncoupling Oxygen Transfer and Electron Transfer in the Oxygenation of Camphor Analogues by Cytochrome P450-CAM. J. Biol. Chem 1995, 270, 28042–28048. [DOI] [PubMed] [Google Scholar]
- (153).Mueller EJ; Loida PJ; Sligar SG Twenty-Five Years of P450cam Research: Mechanistic Insights into Oxygenase Catalysis In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano PR, Ed.; Plenum Press: New York, 1995; pp 83–124. [Google Scholar]
- (154).Tenhunen R; Marver HS; Schmid R The Enzymatic Conversion of Heme to Bilirubin by Microsomal Heme Oxygenase. Proc. Natl. Acad. Sci. U. S. A 1968, 61, 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Williams SEJ; Wootton P; Mason HS; Bould J; Iles DE; Riccardi D; Peers C; Kemp PJ Hemoxygenase-2 Is an Oxygen Sensor for a Calcium-Sensitive Potassium Channel. Science 2004, 306, 2093–2097. [DOI] [PubMed] [Google Scholar]
- (156).Suematsu M; Ishimura Y The Heme Oxygenase-Carbon Monoxide System: U Regulator of Hepatobiliary Function. Hepatology 2000, 31, 3–6. [DOI] [PubMed] [Google Scholar]
- (157).Poss KD; Tonegawa S Reduced Stress Defense in Heme Oxygenase 1-Deficient Cells. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 10925–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Maines MD The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol 1997, 37, 517–554. [DOI] [PubMed] [Google Scholar]
- (159).Yoshida T; Kikuchi G Features of the Reaction of Heme Degradation Catalyzed by the Reconstituted Microsomal Heme Oxygenase System. J. Biol. Chem 1978, 253, 4230–4236. [PubMed] [Google Scholar]
- (160).Caza M; Kronstad JW Shared and Distinct Mechanisms of Iron Acquisition by Bacterial and Fungal Pathogens of Humans. Front. Cell. Infect. Microbiol 2013, 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Matsui T; Unno M; Ikeda-Saito M Heme Oxygenase Reveals Its Strategy for Catalyzing Three Successive Oxygenation Reactions. Acc. Chem. Res 2010, 43, 240–247. [DOI] [PubMed] [Google Scholar]
- (162).Ortiz de Montellano PR Heme Oxygenase Mechanism: Evidence for an Electrophilic, Ferric Peroxide Species. Acc. Chem. Res 1998, 31, 543–549. [Google Scholar]
- (163).Schacter BA; Nelson EB; Marver HS; Masters BSS Immunochemical Evidence for an Association of Heme Oxygenase with the Microsomal Electron Transport System. J. Biol. Chem 1972, 247, 3601–3607. [PubMed] [Google Scholar]
- (164).Cornejo J; Willows RD; Beale SI Phytobilin Biosynthesis: Cloning and Expression of a Gene Encoding Soluble Ferredoxin-Dependent Heme Oxygenase from Synechocystis Sp. PCC 6803. PlantJ 1998, 15, 99–107. [DOI] [PubMed] [Google Scholar]
- (165).Bianchetti CM; Yi L; Ragsdale SW; Phillips GN Comparison of Apo- and Heme-Bound Crystal Structures of a Truncated Human Heme Oxygenase-2. J. Biol. Chem 2007, 282, 37624–37631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Schuller DJ; Wilks A; Ortiz de Montellano PR; Poulos TL Crystal Structure of Human Heme Oxygenase-1. Nat. Struct. Biol 1999, 6, 860–867. [DOI] [PubMed] [Google Scholar]
- (167).Unno M; Matsui T; Chu GC; Couture M; Yoshida T; Rousseau DL; Olson JS; Ikeda-Saito M Crystal Structure of the Dioxygen-Bound Heme Oxygenase from Corynebacterium Diphtheriae: Implications for Heme Oxygenase Function. J. Biol. Chem 2004, 279, 21055–21061. [DOI] [PubMed] [Google Scholar]
- (168).Takahashi S; Rousseau DL; Ishikawa K; Yoshida T; Takeuchi N; Ikeda-Saito M Oxygen-Bound Heme-Heme Oxygenase Complex: Evidence for a Highly Bent Structure of the Coordinated Oxygen. J. Am. Chem. Soc 1995, 117, 6002–6006. [Google Scholar]
- (169).Matsui T; Furukawa M; Unno M; Tomita T; Ikeda-Saito M Roles of Distal Asp in Heme Oxygenase from Corynebacterium Diphtheriae, HmuO. A Water-Driven Oxygen Activation Mechanism. J. Biol. Chem 2005, 280, 2981–2989. [DOI] [PubMed] [Google Scholar]
- (170).Sugishima M; Omata Y; Kakuta Y; Sakamoto H; Noguchi M; Fukuyama K Crystal Structure of Rat Heme Oxygenase-1 in Complex with Heme. FEBS Lett. 2000, 471, 61–66. [DOI] [PubMed] [Google Scholar]
- (171).Hirotsu S; Chu GC; Unno M; Lee D-S; Yoshida T; Park S-Y; Shiro Y; Ikeda-Saito M The Crystal Structures of the Ferric and Ferrous Forms of the Heme Complex of HmuO, a Heme Oxygenase of Corynebacterium Diphtheriae. J. Biol. Chem 2004, 279, 11937–11947. [DOI] [PubMed] [Google Scholar]
- (172).Colas C; Ortiz de Montellano PR Autocatalytic Radical Reactions in Physiological Prosthetic Heme Modification. Chem. Rev 2003, 103, 2305–2332. [DOI] [PubMed] [Google Scholar]
- (173).Unno M; Matsui T; Ikeda-Saito M Structure and Catalytic Mechanism of Heme Oxygenase. Nat. Prod. Rep 2007, 24, 553–570. [DOI] [PubMed] [Google Scholar]
- (174).Matsui T; Nakajima A; Fujii H; Matera KM; Migita CT; Yoshida T; Ikeda-Saito M O2- and H2O2-Dependent Verdoheme Degradation by Heme Oxygenase: Reaction Mechamisms and Potential Physiological Roles of the Dual Pathway Degradation. J. Biol. Chem 2005, 280, 36833–36840. [DOI] [PubMed] [Google Scholar]
- (175).Matsui T; Omori K; Jin H; Ikeda-Saito M Alkyl Peroxides Reveal the Ring Opening Mechanism of Verdoheme Catalyzed by Heme Oxygenase. J. Am. Chem. Soc 2008, 130, 4220–4221. [DOI] [PubMed] [Google Scholar]
- (176).Davydov R; Chemerisov S; Werst DE; Rajh T; Matsui T; Ikeda-Saito M; Hoffman BM Proton Transfer at Helium Temperatures during Dioxygen Activation by Heme Monooxygenases. J. Am. Chem. Soc 2004, 126, 15960–15961. [DOI] [PubMed] [Google Scholar]
- (177).Matsui T; Kim SH; Jin H; Hoffman BM; Ikeda-Saito M Compound I of Heme Oxygenase Cannot Hydroxylate Its Heme Meso-Carbon. J. Am. Chem. Soc 2006, 128, 1090–1091. [DOI] [PubMed] [Google Scholar]
- (178).Wilks A; Ortiz de Montellano PR Rat Liver Heme Oxygenase. High Level Expression of a Truncated Soluble Form and Nature of the Meso-Hydroxylating Species. J. Biol. Chem 1993, 268, 22357–22362. [PubMed] [Google Scholar]
- (179).Davydov RM; Yoshida T; Ikeda-Saito M; Hoffman BM Hydroperoxy-Heme Oxygenase Generated by Cryoreduction Catalyzes the Formation of α-Meso-Hydroxyheme as Detected by EPR and ENDOR J. Am. Chem. Soc 1999, 121, 10656–10657. [Google Scholar]
- (180).Denisov IG; Ikeda-Saito M; Yoshida T; Sligar SG Cryogenic Absorption Spectra of Hydroperoxo-Ferric Heme Oxygenase, the Active Intermediate of Enzymatic Heme Oxygenation. FEBS Lett. 2002, 532, 203–206. [DOI] [PubMed] [Google Scholar]
- (181).Garcia-Serres R; Davydov RM; Matsui T; Ikeda-Saito M; Hoffman BM; Huynh BH Distinct Reaction Pathways Followed upon Reduction of Oxy-Heme Oxygenase and Oxy-Myoglobin as Characterized by Mossbauer Spectroscopy. J. Am. Chem. Soc 2007, 129, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Davydov R; Matsui T; Fujii H; Ikeda-Saito M; Hoffman, Kinetic AM Isotope Effects on the Rate-Limiting Step of Heme Oxygenase Catalysis Indicate Concerted Proton Transfer/Heme Hydroxylation. J. Am. Chem. Soc 2003, 125, 16208–16209. [DOI] [PubMed] [Google Scholar]
- (183).Kumar D; de Visser SP; Shaik S Theory Favors a Stepwise Mechanism of Porphyrin Degradation by a Ferric Hydroperoxide Model of the Active Species of Heme Oxygenase. J. Am. Chem. Soc 2005, 127, 8204–8213. [DOI] [PubMed] [Google Scholar]
- (184).Chen H; Moreau Y; Derat E; Shaik S Quantum Mechanical/Molecular Mechanical Study of Mechanisms of Heme Degradation by the Enzyme Heme Oxygenase: The Strategic Function of the Water Cluster. J. Am. Chem. Soc 2008, 130, 1953–1965. [DOI] [PubMed] [Google Scholar]
- (185).Sharma PK; Kevorkiants R; de Visser SP; Kumar D; Shaik S Porphyrin Traps Its Terminator! Concerted and Stepwise Porphyrin Degradation Mechanisms Induced by Heme-Oxygenase and Cytochrome P450. Angew. Chem., Int. Ed 2004, 43, 1129–1132. [DOI] [PubMed] [Google Scholar]
- (186).Morishima I; Fujii H; Shiro Y; Sano S Studies on the Iron(II) Meso-Oxyporphyrin π-Neutral Radical as a Reaction Intermediate in Heme Catabolism. Inorg. Chem 1995, 34, 1528–1535. [Google Scholar]
- (187).Matera KM; Takahashi S; Fujii H; Zhou H; Ishikawa K; Yoshimura T; Rousseau DL; Yoshida T; Ikeda-Saito M Oxygen and One Reducing Equivalent Are Both Required for the Conversion of α-Hydroxyhemin to Verdoheme in Heme Oxygenase. J. Biol. Chem 1996, 271 , 6618–6624. [DOI] [PubMed] [Google Scholar]
- (188).Sakamoto H; Omata Y; Hayashi S; Harada S; Palmer G; Noguchi M The Reactivity of α-Hydroxyhaem and Verdohaem Bound to Haem Oxygenase-1 to Dioxygen and Sodium Dithionite. Eur. J. Biochem 2002, 269, 5231–5239. [DOI] [PubMed] [Google Scholar]
- (189).Liu Y; Ortiz de Montellano P R Reaction Intermediates and Single Turnover Rate Constants for the Oxidation of Heme by Human Heme Oxygenase-1. J. Biol. Chem 2000, 275, 5297–5307. [DOI] [PubMed] [Google Scholar]
- (190).Fuhrhop J-H; Kruger P 1- or 19-Methoxy-, 1- or 19-Amino-and 1- or 19-Thio- Deoxybiliverdins. Justus Liebigs Ann. Chem 1977, 1977, 360–370. [Google Scholar]
- (191).Saito S; Itano HA Verdohemochrome IXa: Preparation and Oxidoreductive Cleavage to Biliverdin IXα. Proc. Natl. Acad. Sci. U. S. A 1982, 79, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Sano S; Sano T; Morishima I; Shiro Y; Maeda Y On the Mechanism of the Chemical and Enzymic Oxygenations of α-Oxyprotohemin IX to Fe-biliverdin IXα. Proc. Natl. Acad. Sci. U. S. A 1986, 83, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Takahashi S; Matera KM; Fujii H; Zhou H; Ishikawa K; Yoshida T; Ikeda-Saito M; Rousseau DL Resonance Raman Spectroscopic Characterization of α-Hydroxyheme and Verdoheme Complexes of Heme Oxygenase. Biochemistry 1997, 36, 1402–1410. [DOI] [PubMed] [Google Scholar]
- (194).Damaso CO; Bunce RA; Barybin MV; Wilks A; Rivera M The Ferrous Verdoheme-Heme Oxygenase Complex Is Six-Coordinate and Low-Spin. J. Am. Chem. Soc 2005, 127, 17582–17583. [DOI] [PubMed] [Google Scholar]
- (195).Lai W; Chen H; Matsui T; Omori K; Unno M; Ikeda-Saito M; Shaik S Enzymatic Ring-Opening Mechanism of Verdoheme by the Heme Oxygenase: A Combined X-Ray Crystallography and QM/MM Study. J. Am. Chem. Soc 2010, 132, 12960–12970. [DOI] [PubMed] [Google Scholar]
- (196).Baranano DE; Rao M; Ferris CD; Snyder SH Biliverdin Reductase: A Major Physiologic Cytoprotectant. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16093–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Skaar EP; Gaspar AH; Schneewind O IsdG and IsdI, Heme-Degrading Enzymes in the Cytoplasm of Staphylococcus Aureus. J. Biol. Chem 2004, 279, 436–443. [DOI] [PubMed] [Google Scholar]
- (198).Skaar EP; Gaspar AH; Schneewind O Bacillus Anthracis IsdG, a Heme-Degrading Monooxygenase. J. Bacteriol 2006, 188, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Chim N; Iniguez A; Nguyen TQ; Goulding CW Unusual Diheme Conformation of the Heme-Degrading Protein from. J. Mol. Biol 2010, 395, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Matsui T; Nambu S; Ono Y; Goulding CW; Tsumoto K; Ikeda-Saito M Heme Degradation by Staphylococcus Aureus IsdG and IsdI Liberates Formaldehyde Rather Than Carbon Monoxide. Biochemistry 2013, 52, 3025–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Nambu S; Matsui T; Goulding CW; Takahashi S; Ikeda-Saito M A New Way to Degrade Heme. J. Biol. Chem 2013, 288, 10101–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Avila L; Huang H; Damaso CO; Lu S; Moënne-Loccoz P; Rivera M Coupled Oxidation vs Heme Oxygenation: Insights from Axial Ligand Mutants of Mitochondrial Cytochrome b5. J. Am. Chem. Soc 2003, 125, 4103–4110. [DOI] [PubMed] [Google Scholar]
- (203).Sigman JA; Wang X; Lu Y Coupled Oxidation of Heme by Myoglobin Is Mediated by Exogenous Peroxide. J. Am. Chem. Soc 2001, 123, 6945–6946. [DOI] [PubMed] [Google Scholar]
- (204).Suits MDL; Pal GP; Nakatsu K; Matte A; Cygler M; Jia Z Identification of an Escherichia Col O157:H7 Heme Oxygenase with Tandem Functional Repeats. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 16955–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Lee WC; Reniere ML; Skaar EP; Murphy MEP Ruffling of Metalloporphyrins Bound to IsdG and IsdI, Two Heme-Degrading Enzymes in Staphylococcus Aureus. J. Biol. Chem 2008, 283, 30957–30963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Ukpabi G; Takayama SJ; Mauk AG; Murphy MEP Inactivation of the Heme Degrading Enzyme IsdI by an Active Site Substitution That Diminishes Heme Ruffling. J. Biol. Chem 2012, 287, 34179–34188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Matsui T; Nambu S; Goulding CW; Takahashi S; Fujii H; Ikeda-Saito M Unique Coupling of Mono- and Dioxygenase Chemistries in a Single Active Site Promotes Heme Degradation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 3779–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Wilks A; Ikeda-Saito M Heme Utilization by Pathogenic Bacteria: Not All Pathways Lead to Biliverdin. Acc. Chem. Res 2014, 47, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Hayaishi O; Rothberg S; Mehler AH; Saito Y Studies on Oxygenases; Enzymatic Formation of Kynurenine from Tryptophan. J. Biol. Chem 1957, 229, 889–896. [PubMed] [Google Scholar]
- (210).Hirata F; Hayaishi O; Tokuyama T; Senoh S In Vitro and in Vivo Formation of Two New Metabolites of Melatonin. J. Biol. Chem 1974, 249, 1311–1313. [PubMed] [Google Scholar]
- (211).Efimov I; Basran J; Thackray SJ; Handa S; Mowat CG; Raven EL Structure and Reaction Mechanism in the Heme Dioxygenases. Biochemistry 2011, 50, 2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Raven ELA Short History of Heme Dioxygenases: Rise, Fall and Rise Again. JBIC, J. Biol. Inorg. Chem 2017, 22, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Uyttenhove C; Pilotte L; Theate I; Stroobant V; Colau D; Parmentier N; Boon T; Van den Eynde BJ Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-Dioxygenase. Nat. Med 2003, 9, 1269–1274. [DOI] [PubMed] [Google Scholar]
- (214).Löb S; Königsrainer A; Rammensee H-G; Opelz G; Terness P Inhibitors of Indoleamine-2,3-Dioxygenase for Cancer Therapy: Can We See the Wood for the Trees? Nat. Rev. Cancer 2009, 9, 445–452. [DOI] [PubMed] [Google Scholar]
- (215).Chen W IDO: More than an Enzyme. Nat. Immunol 2011, 12, 809–811. [DOI] [PubMed] [Google Scholar]
- (216).Capece L; Lewis-Ballester A; Yeh S-R; Estrin DA; Marti MA Complete Reaction Mechanism of Indoleamine 2,3-Dioxygenase as Revealed by QM/MM Simulations. J. Phys. Chem. B 2012, 116, 1401–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Sugimoto H; Oda S-I; Otsuki T; Hino T; Yoshida T; Shiro Y Crystal Structure of Human Indoleamine 2,3-Dioxygenase: Catalytic Mechanism of O2 Incorporation by a Heme-Containing Dioxygenase. Proc. Natl. Acad. Sci U. S. A 2006, 103, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Forouhar F; Anderson JLR; Mowat CG; Vorobiev SM; Hussain A; Abashidze M; Bruckmann C; Thackray SJ; Seetharaman J; Tucker T; et al. Molecular Insights into Substrate Recognition and Catalysis by Tryptophan 2,3-Dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Lewis-Ballester A; Forouhar F; Kim S-M; Lew S; Wang Y; Karkashon S; Seetharaman J; Batabyal D; Chiang B-Y; Hussain M; et al. Molecular Basis for Catalysis and Substrate-Mediated Cellular Stabilization of Human Tryptophan 2,3-Dioxygenase. Sci. Rep 2016, 6, 35169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Thackray SJ; Bruckmann C; Anderson JLR; Campbell LP; Xiao R; Zhao L; Mowat CG; Forouhar F; Tong L; Chapman SK Histidine 55 of Tryptophan 2,3-Dioxygenase Is Not an Active Site Base but Regulates Catalysis by Controlling Substrate Binding. Biochemistry 2008, 47, 10677–10684. [DOI] [PubMed] [Google Scholar]
- (221).Hamilton GA Mechanisms of Two- and Four-Electron Oxidations Catalyzed by Some Metalloenzymes. Adv. Enzymol. Relat. Areas Mol. Biol 2006, 32, 55–96. [DOI] [PubMed] [Google Scholar]
- (222).Cady SG; Sono M 1-Methyl-DL-Tryptophan, β-(3-Benzofuranyl)-DL-Alanine (the Oxygen Analog of Tryptophan), and β-[3-Benzo(b)thienyl]-DL-Alanine (the Sulfur Analog of Tryptophan) Are Competitive Inhibitors for Indoleamine 2,3-Dioxygenase. Arch. Biochem. Biophys 1991, 291, 326–333. [DOI] [PubMed] [Google Scholar]
- (223).Terentis AC; Thomas SR; Takikawa O; Littlejohn TK; Truscott RJW; Armstrong RS; Yeh S-R; Stocker R The Heme Environment of Recombinant Human Indoleamine 2,3-Dioxygenase. J. Biol. Chem 2002, 277, 15788–15794. [DOI] [PubMed] [Google Scholar]
- (224).Chung LW; Li X; Sugimoto H; Shiro Y; Morokuma K Density Functional Theory Study on a Missing Piece in Understanding of Heme Chemistry: The Reaction Mechanism for Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase. J. Am. Chem. Soc 2008, 130, 12299–12309. [DOI] [PubMed] [Google Scholar]
- (225).Basran J; Booth ES; Lee M; Handa S; Raven EL Analysis of Reaction Intermediates in Tryptophan 2,3-Dioxygenase: A Comparison with Indoleamine 2,3-Dioxygenase. Biochemistry 2016, 55, 6743–6750. [DOI] [PubMed] [Google Scholar]
- (226).Lewis-Ballester A; Batabyal D; Egawa T; Lu C; Lin Y; Marti MA; Capece L; Estrin DA; Yeh S-R Evidence for a Ferryl Intermediate in a Heme-Based Dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 17371–17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Chung LW; Li X; Sugimoto H; Shiro Y; Morokuma K ONIOM Study on a Missing Piece in Our Understanding of Heme Chemistry: Bacterial Tryptophan 2,3-Dioxygenase with Dual Oxidants. J. Am. Chem. Soc 2010, 132, 11993–12005. [DOI] [PubMed] [Google Scholar]
- (228).Forstermann U; Sessa WC; Forstermann U; Sessa WC Nitric Oxide Synthases: Regulation and Function. Eur. Heart J 2012, 33, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Alderton WK; Cooper CE; Knowles RG Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J 2001, 357, 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Wang J; Schopfer MP; Puiu SC; Sarjeant AAN; Karlin KD Reductive Coupling of Nitrogen Monoxide (•NO) Facilitated by Heme/Copper Complexes. Inorg. Chem 2010, 49, 1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Bredt DS; Snyder SH Nitric Oxide: A Physiologic Messenger Molecule. Annu. Rev. Biochem 1994, 63, 175–195. [DOI] [PubMed] [Google Scholar]
- (232).Griffith OW; Stuehr DJ Nitric Oxide Synthases: Properties and Catalytic Mechanism. Annu. Rev. Physiol 1995, 57, 707–734. [DOI] [PubMed] [Google Scholar]
- (233).Lehnert N; Berto TC; Galinato MGI; Goodrich LE The Role of Heme-Nitrosyls in the Biosynthesis, Transport, Sensing, and Detoxification of Nitric Oxide (NO) in Biological Systems: Enzymes and Model Complexes In The Handbook of Porphyrin Science; Kadish KM, Smith K, Guilard R, Eds.; World Scientific: Singapore, 2011; Vol. 14, pp 1–247. [Google Scholar]
- (234).Toledo JC; Augusto O Connecting the Chemical and Biological Properties of Nitric Oxide. Chem. Res. Toxicol 2012, 25, 975–989. [DOI] [PubMed] [Google Scholar]
- (235).Wasser IM; de Vries S; Moënne-Loccoz P; Schröder I; Karlin KD Nitric Oxide in Biological Denitrification: Fe/Cu Metalloenzyme and Metal Complex NOx Redox Chemistry. Chem. Rev 2002, 102, 1201–1234. [DOI] [PubMed] [Google Scholar]
- (236).Taylor EL; Megson IL; Haslett C; Rossi AG Nitric Oxide: A Key Regulator ofMyeloid Inflammatory Cell Apoptosis. Cell Death Differ. 2003, 10, 418–430. [DOI] [PubMed] [Google Scholar]
- (237).Fukumura D; Kashiwagi S; Jain RK The Role of Nitric Oxide in Tumour Progression. Nat. Rev. Cancer 2006, 6, 521–534. [DOI] [PubMed] [Google Scholar]
- (238).Gorren ACF; Mayer B Nitric-Oxide Synthase: A Cytochrome P450 Family Foster Child. Biochim. Biophys. Acta, Gen. Subj 2007, 1770, 432–445. [DOI] [PubMed] [Google Scholar]
- (239).Abu-Soud HM; Stuehr DJ Nitric Oxide Synthases Reveal a Role for Calmodulin in Controlling Electron Transfer. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 10769–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Brunner K; Tortschanoff A; Hemmens B; Andrew PJ; Mayer B; Kungl AJ Sensitivity of Flavin Fluorescence Dynamics in Neuronal Nitric Oxide Synthase to Cofactor-Induced Conformational Changes and Dimerization. Biochemistry 1998, 37, 17545–17553. [DOI] [PubMed] [Google Scholar]
- (241).Werner ER; Gorren ACF; Heller R; Werner-Felmayer G; Mayer B Tetrahydrobiopterin and Nitric Oxide: Mechanistic and Pharmacological Aspects. Exp. Biol. Med 2003, 228, 1291–1302. [DOI] [PubMed] [Google Scholar]
- (242).Wei C-C; Wang Z-Q; Wang Q; Meade AL; Hemann C; Hille R; Stuehr DJ Rapid Kinetic Studies Link Tetrahydrobiopterin Radical Formation to Heme-Dioxy Reduction and Arginine Hydroxylation in Inducible Nitric-Oxide Synthase. J. Biol. Chem 2001, 276, 315–319. [DOI] [PubMed] [Google Scholar]
- (243).Hurshman AR; Krebs C; Edmondson DE; Huynh BH; Marietta MA Formation of a Pterin Radical in the Reaction of the Heme Domain of Inducible Nitric Oxide Synthase with Oxygen. Biochemistry 1999, 38, 15689–15696. [DOI] [PubMed] [Google Scholar]
- (244).Huang H; Hah J-M; Silverman R B. Mechanism of Nitric Oxide Synthase. Evidence That Direct Hydrogen Atom Abstraction from the O–H Bond of NG-Hydroxyarginine Is Not Relevant to the Mechanism. J. Am. Chem. Soc. 2001, 123, 2674–2676. [DOI] [PubMed] [Google Scholar]
- (245).Wei C-C; Wang Z-Q; Hemann C; Hille R; Stuehr DJ A Tetrahydrobiopterin Radical Forms and Then Becomes Reduced during N^-Hydroxyarginine Oxidation by Nitric-Oxide Synthase. J. Biol. Chem. 2003, 278, 46668–46673. [DOI] [PubMed] [Google Scholar]
- (246).Sies H Role of Metabolic H2O2 Generation. J. Biol. Chem 2014, 289, 8735–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Rhee SG H2O2, a Necessary Evil for Cell Signaling. Science 2006, 312, 1882–1883. [DOI] [PubMed] [Google Scholar]
- (248).Murthy MRN; Reid TJ; Sicignano A; Tanaka N; Rossmann MG Structure of Beef Liver Catalase. J. Mol. Biol 1981, 152, 465–499. [DOI] [PubMed] [Google Scholar]
- (249).Valentine JS; Sheridan RP; Allen LC; Kahn PC Coupling between Oxidation State and Hydrogen Bond Conformation in Heme Proteins. Proc. Natl. Acad. Sci U. S. A 1979, 76, 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Finzel BC; Poulos TL; Kraut J Crystal Structure of Yeast Cytochrome c Peroxidase Refined at 1.7-A Resolution. J. Biol. Chem 1984, 259, 13027–13036. [PubMed] [Google Scholar]
- (251).Chang CK; Traylor TG Proximal Base Influence on the Binding of Oxygen and Carbon Monoxide to Heme. J. Am. Chem. Soc 1973, 95, 8477–8479. [DOI] [PubMed] [Google Scholar]
- (252).Schonbaum GR; Lo S Interaction of Peroxidases with Aromatic Peracids and Alkyl Peroxides: Product Analysis. J. Biol. Chem 1972, 247, 3353–3360. [PubMed] [Google Scholar]
- (253).Rutter R; Hager LP The Detection of Two Electron Paramagnetic Resonance Radical Signals Associated with Chloroperoxidase Compound I. J. Biol. Chem 1982, 257, 7958–7961. [PubMed] [Google Scholar]
- (254).Chance B; Powers L; Ching Y; Poulos T; Schonbaum GR; Yamazaki I; Paul KG X-Ray Absorption Studies of Intermediates in Peroxidase Activity. Arch. Biochem. Biophys 1984, 235, 596–611. [DOI] [PubMed] [Google Scholar]
- (255).Edwards SL; Nguyen Huu X; Hamlin RC; Kraut J Crystal Structure of Cytochrome c Peroxidase Compound I. Biochemistry 1987, 26, 1503–1511. [DOI] [PubMed] [Google Scholar]
- (256).Bonagura CA; Bhaskar B; Shimizu H; Li H; Sundaramoorthy M; McRee DE; Goodin DB; Poulos TL High-Resolution Crystal Structures and Spectroscopy of Native and Compound I Cytochrome c Peroxidase. Biochemistry 2003, 42, 5600–5608. [DOI] [PubMed] [Google Scholar]
- (257).Fulop V; Phizackerley RP; Soltis SM; Clifton IJ; Wakatsuki S; Erman J; Hajdu J; Edwards SL Laue Diffraction Study on the Structure of Cytochrome c Peroxidase Compound I. Structure 1994, 2, 201–208. [DOI] [PubMed] [Google Scholar]
- (258).Coulson AFWW; Yonetani T Oxidation of Cytochrome c Peroxidase with Hydrogen Peroxide: Identification of the “Endogenous Donor. Biochem. Biophys. Res. Commun 1972, 49, 391–398. [DOI] [PubMed] [Google Scholar]
- (259).Sivaraja M; Goodin DB; Smith M; Hoffman BM Identification by ENDOR of Trp191 as the Free-Radical Site in Cytochrome c Peroxidase Compound ES. Science 1989, 245, 738–740. [DOI] [PubMed] [Google Scholar]
- (260).Dolphin D; Forman A; Borg DC; Fajer J; Felton RH Compounds I of Catalase and Horse Radish Peroxidase:π −3Cation Radicals. Proc. Natl. Acad. Sci U. S. A 1971, 68, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Musah RA; Goodin DB Introduction of Novel Substrate Oxidation into Cytochrome c Peroxidase by Cavity Complementation: Oxidation of 2-Aminothiazole and Covalent Modification of the Enzyme. Biochemistry 1997, 36, 11665–11674. [DOI] [PubMed] [Google Scholar]
- (262).Sprangler BD; Erman JE Cytochrome c Peroxidase Compound I: Formation of Covalent Protein Crosslinks during the Endogenous Reduction of the Active Site. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1986, 872, 155–157. [DOI] [PubMed] [Google Scholar]
- (263).Erman JE; Vitello LB; Miller MA; Shaw A; Brown KA; Kraut J Histidine 52 Is a Critical Residue for Rapid Formation of Cytochrome c Peroxidase Compound I. Biochemistry 1993, 32, 9798–9806. [DOI] [PubMed] [Google Scholar]
- (264).Howes BD; Rodriguez-Lopez JN; Smith AT; Smulevich G Mutation of Distal Residues of Horseradish Peroxidase: Influence on Substrate Binding and Cavity Properties. Biochemistry 1997, 36, 1532–1543. [DOI] [PubMed] [Google Scholar]
- (265).Poulos TL; Kraut J The Stereochemistry of Peroxidase Catalysis. J. Biol. Chem 1980, 255, 8199–8205. [PubMed] [Google Scholar]
- (266).Vidossich P; Fiorin G; Alfonso-Prieto M; Derat E; Shaik S; Rovira C On the Role of Water in Peroxidase Catalysis: A Theoretical Investigation of HRP Compound I Formation. J. Phys. Chem. B 2010, 114, 5161–5169. [DOI] [PubMed] [Google Scholar]
- (267).Berglund GI; Carlsson GH; Smith AT; Szöke H; Henriksen A; Hajdu J The Catalytic Pathway of Horseradish Peroxidase at High Resolution. Nature 2002, 417, 463–468. [DOI] [PubMed] [Google Scholar]
- (268).Meharenna YT; Doukov T; Li H; Soltis SM; Poulos TL Crystallographic and Single-Crystal Spectral Analysis of the Peroxidase Ferryl Intermediate. Biochemistry 2010, 49, 2984–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Casadei CM; Gumiero A; Metcalfe CL; Murphy EJ; Basran J; Concilio MG; Teixeira SCM; Schrader TE; Fielding AJ; Ostermann A; et al. Neutron Cryo-Crystallography Captures the Protonation State of Ferryl Heme in a Peroxidase. Science 2014, 345, 193–197. [DOI] [PubMed] [Google Scholar]
- (270).Groves JT; Boaz NC Fishing for Peroxidase Protons. Science 2014, 345, 142–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Reczek CM; Sitter AJ; Terner J Resonance Raman Characterization of Heme Fe(IV)=O Groups of Intermediates of Yeast Cytochrome c Peroxidase and Lactoperoxidase. J. Mol. Struct 1989, 214, 27–41. [Google Scholar]
- (272).Sitter AJ; Reczek CM; Terner J Observation of the Fe(IV)=O Stretching Vibration of Ferryl Myoglobin by Resonance Raman Spectroscopy. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1985, 828, 229–235. [DOI] [PubMed] [Google Scholar]
- (273).Hashimoto S; Tatsuno Y; Kitagawa T Resonance Raman Evidence for the Presence of the FeIV=O Bond in Horseradish Peroxidase Compound II. Proc. Jpn. Acad., Ser. B 1984, 60, 345–348. [Google Scholar]
- (274).Chance M; Powers L; Poulos T; Chance B Cytochrome c Peroxidase Compound ES Is Identical with Horseradish Peroxidase Compound I in Iron-Ligand Distances. Biochemistry 1986, 25, 1266–1270. [DOI] [PubMed] [Google Scholar]
- (275).Penner-Hahn JE; Smith Eble K; McMurry TJ; Renner M; Balch AL; Groves JT; Dawson JH; Hodgson KO Structural Characterization of Horseradish Peroxidase Using EXAFS Spectroscopy. Evidence for Fe = O Ligation in Compounds I and II. J. Am. Chem. Soc 1986, 108, 7819–7825. [DOI] [PubMed] [Google Scholar]
- (276).Hersleth H-P; Dalhus B; Görbitz C; Andersson K An Iron Hydroxide Moiety in the 1.35 A Resolution Structure of Hydrogen Peroxide Derived Myoglobin Compound II at pH 5.2. JBIC, J. Biol. Inorg. Chem 2002, 7, 299–304. [DOI] [PubMed] [Google Scholar]
- (277).Murshudov GN; Grebenko AI; Brannigan JA; Antson AA; Barynin VV; Dodson GG; Dauter Z; Wilson KS; Melik-Adamyan WR The Structures of Micrococcus Lysodeikticus Catalase, Its Ferryl Intermediate (Compound II) and NADPH Complex. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1972–1982. [DOI] [PubMed] [Google Scholar]
- (278).Green MT Application of Badger’s Rule to Heme and Non-Heme Iron–Oxygen Bonds: An Examination of Ferryl Protonation States. J. Am. Chem. Soc 2006, 128, 1902–1906. [DOI] [PubMed] [Google Scholar]
- (279).Hersleth H-P; Hsiao Y-W; Ryde U; Gorbitz CH; Andersson KK The Influence of X-Rays on the Structural Studies of Peroxide-Derived Myoglobin Intermediates. Chem. Biodiversity 2008, 5, 2067–2089. [DOI] [PubMed] [Google Scholar]
- (280).Gumiero A; Metcalfe CL; Pearson AR; Raven EL; Moody PCE Nature of the Ferryl Heme in Compounds I and II. J. Biol. Chem 2011, 286, 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Kwon H; Basran J; Casadei CM; Fielding AJ; Schrader TE; Ostermann A; Devos JM; Aller P; Blakeley MP; Moody PCE; et al. Direct Visualization of a Fe(IV)-OH Intermediate in a Heme Enzyme. Nat. Commun 2016, 7, 13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Vitello LB; Erman JE; Miller MA; Wang J; Kraut J Effect of Arginine-48 Replacement on the Reaction Between Cytochrome c Peroxidase and Hydrogen Peroxide. Biochemistry 1993, 32, 9807–9818. [DOI] [PubMed] [Google Scholar]
- (283).Rodriguez-Lopez JN; Smith AT; Thorneley RNF Role of Arginine 38 in Horseradish Peroxidase. J. Biol. Chem 1996, 271, 4023–4030. [DOI] [PubMed] [Google Scholar]
- (284).Houseman ALP; Doan PE; Goodwin DB; Hoffman BM Comprehensive Explanation of the Anomalous EPR Spectra of Wild-Type and Mutant Cytochrome c Peroxidase Compound ES. Biochemistry 1993, 32, 4430–4443. [DOI] [PubMed] [Google Scholar]
- (285).Morris DR; Hager LP Chloroperoxidase I. Isolation and Properties of the Crystalline Glycoprotein. J. Biol. Chem 1966, 241, 1763–1768. [PubMed] [Google Scholar]
- (286).Morris DR; Hager LP Mechanism of the Inhibition of Enzymatic Halogenation by Antithyroid Agents. J. Biol. Chem 1966, 241, 3582–3589. [PubMed] [Google Scholar]
- (287).Hofrichter M; Ullrich R Heme-Thiolate Haloperoxidases: Versatile Biocatalysts with Biotechnological and Environmental Significance. Appl. Microbiol. Biotechnol 2006, 71, 276–288. [DOI] [PubMed] [Google Scholar]
- (288).Hager LP; Morris DR; Brown FS; Eberwein H Chloroperoxidase: II. Utilization of Halogen Anions. J. Biol. Chem 1966, 241, 1769–1777. [PubMed] [Google Scholar]
- (289).Zhang R; He Q; Chatfield D; Wang X Paramagnetic Nuclear Magnetic Resonance Relaxation and Molecular Mechanics Studies of the Chloroperoxidase-Indole Complex: Insights into the Mechanism of Chloroperoxidase-Catalyzed Regioselective Oxidation of Indole. Biochemistry 2013, 52, 3688–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Sundaramoorthy M; Terner J; Poulos TL The Crystal Structure of Chloroperoxidase: A Heme Peroxidase-Cytochrome P450 Functional Hybrid. Structure 1995, 3, 1367–1378. [DOI] [PubMed] [Google Scholar]
- (291).Osborne RL; Raner GM; Hager LP; Dawson JHC Fumago Chloroperoxidase Is Also a Dehaloperoxidase: Oxidative Dehalogenation of Halophenols. J. Am. Chem. Soc 2006, 128, 1036–1037. [DOI] [PubMed] [Google Scholar]
- (292).Kedderis GL; Koop DR; Hollenberg PF N-Demethylation Reactions Catalyzed by Chloroperoxidase. J. Biol. Chem 1980, 255, 10174–10182. [PubMed] [Google Scholar]
- (293).Allain EJ; Hager LP; Deng L; Jacobsen EN Highly Enantioselective Epoxidation of Disubstituted Alkenes with Hydrogen Peroxide Catalyzed by Chloroperoxidase. J.Am. Chem. Soc 1993, 115, 4415–4416. [Google Scholar]
- (294).Carmichael R; Fedorak PM; Pickard MA Oxidation of Phenols by Chloroperoxidase. Biotechnol. Lett 1985, 7, 289–294. [Google Scholar]
- (295).Colonna S; Gaggero N; Manfredi A; Casella L; Gullotti M; Carrea G; Pasta P Enantioselective Oxidations of Sulfides Catalyzed by Chloroperoxidase. Biochemistry 1990, 29, 10465–10468. [DOI] [PubMed] [Google Scholar]
- (296).Corbett MD; Baden DG; Chipko BR Arylamine Oxidations by Chloroperoxidase. Bioorg. Chem 1979, 8, 91–95. [Google Scholar]
- (297).Geigert J; Dalietos DJ; Neidleman SL; Lee TD; Wadsworth J Peroxide Oxidation of Primary Alcohols to Aldehydes by Chloroperoxidase Catalysis. Biochem. Biophys. Res. Commun 1983, 114, 1104–1108. [DOI] [PubMed] [Google Scholar]
- (298).Kiljunen E; Kanerva LT Chloroperoxidase-Catalysed Oxidation of Alcohols to Aldehydes. J. Mol. Catal B: Enzym 2000, 9, 163–172. [Google Scholar]
- (299).Sundaramoorthy M; Terner J; Poulos TL Stereochemistry of the Chloroperoxidase Active Site: Crystallographic and Molecular-Modeling Studies. Chem. Biol 1998, 5, 461–473. [DOI] [PubMed] [Google Scholar]
- (300).Wagenknecht H-A; Woggon W-D Identification of Intermediates in the Catalytic Cycle of Chloroperoxidase. Chem. Biol 1997, 4, 367–372. [DOI] [PubMed] [Google Scholar]
- (301).Pardillo AD; Morozov AN; Chatfield DC Proximal Pocket Hydrogen Bonds Significantly Influence the Mechanism of Chloroperoxidase Compound I Formation. J. Phys. Chem. B 2015, 119, 12590–12602. [DOI] [PubMed] [Google Scholar]
- (302).Kühnel K; Derat E; Terner J; Shaik S; Schlichting I Structure and Quantum Chemical Characterization of Chloroperoxidase Compound 0, a Common Reaction Intermediate of Diverse Heme Enzymes. Proc. Natl. Acad. Sci U. S. A 2007, 104, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Manoj KM; Hager LP Chloroperoxidase, a Janus Enzyme. Biochemistry 2008, 47, 2997–3003. [DOI] [PubMed] [Google Scholar]
- (304).Sundaramoorthy M Chloroperoxidase In Handbook of Metalloproteins; John Wiley & Sons, Ltd: Chichester, 2006. [Google Scholar]
- (305).Griffin BW Chloroperoxidase: A Review. In Peroxidases in Chemistry and Biology; Everse J, Everse KE, Grisham MB, Eds.; 1990; Vol. 2, pp 85–138. [Google Scholar]
- (306).Artero V Bioinspired Catalytic Materials for Energy-Relevant Conversions. Nat. Energy 2017, 2, 17131. [Google Scholar]
- (307).Xia Z Bioinspired Catalysts for Energy Conversion in Fuel Cells, Metal-Air Batteries and Water Splitting. Sci Lett. J 2015, 4, 190. [Google Scholar]
- (308).Naik RR; Singamaneni S Introduction: Bioinspired and Biomimetic Materials. Chem. Rev 2017, 117, 12581–12583. [DOI] [PubMed] [Google Scholar]
- (309).Moody PCE; Raven EL The Nature and Reactivity of Ferryl Heme in Compounds I and II. Acc. Chem. Res 2018, 51, 427–435. [DOI] [PubMed] [Google Scholar]
- (310).Newmyer SL; de Montellano P R O. Horseradish Peroxidase His-42 →, His-42 →, and Phe-41 → Mutants. J. Biol. Chem 1995, 270, 19430–19438. [DOI] [PubMed] [Google Scholar]
- (311).Rodríguez-López JN; Lowe DJ; Hernández-Ruiz J; Hiner AN; García-Cánovas F; Thorneley RN Mechanism of Reaction of Hydrogen Peroxide with Horseradish Peroxidase: Identification of Intermediates in the Catalytic Cycle. J. Am. Chem. Soc 2001, 123, 11838–11847. [DOI] [PubMed] [Google Scholar]
- (312).Ramanan R; Dubey KD; Wang B; Mandal D; Shaik S Emergence of Function in P450-Proteins: A Combined Quantum Mechanical/Molecular Mechanical and Molecular Dynamics Study of the Reactive Species in the H2O2-Dependent Cytochrome P450Spα and Its Regio- and Enantioselective H. J. Am. Chem. Soc 2016, 138, 6786–6797. [DOI] [PubMed] [Google Scholar]
- (313).Roth JP; Cramer CJ Direct Examination of H2O2 Activation by a Heme Peroxidase. J. Am. Chem. Soc 2008, 130, 7802–7803. [DOI] [PubMed] [Google Scholar]
- (314).Dawson JH; Holm RH; Trudell JR; Barth G; Linder RE; Bunnenberg E; Djerassi C; Tang SC Oxidized Cytochrome P-450. Magnetic Circular Dichroism Evidence for Thiolate Ligation in the Substrate-Bound Form. Implications for the Catalytic Mechanism. J. Am. Chem. Soc 1976, 98, 3707–3709. [DOI] [PubMed] [Google Scholar]
- (315).Sono M; Dawson JH Formation of Low Spin Complexes of Ferric Cytochrome P-450-CAM with Anionic Ligands. Spin State and Ligand Affinity Comparison to Myoglobin. J. Biol. Chem 1982, 257, 5496–5502. [PubMed] [Google Scholar]
- (316).Liu HI; Sono M; Kadkhodayan S; Hager LP; Hedman B; Hodgson KO; Dawson JH X-Ray Absorption Near Edge Studies of Cytochrome P-450-CAM, Chloroperoxidase, and Myoglobin. J. Biol. Chem 1995, 270, 10544–10550. [DOI] [PubMed] [Google Scholar]
- (317).Dawson JH Probing Structure-Function Relations in Heme-Containing Oxygenases and Peroxidases. Science 1988, 240, 433–439. [DOI] [PubMed] [Google Scholar]
- (318).Poulos TL Heme Enzyme Crystal Structures. Adv. Inorg. Biochem 1988, 7, 1–36. [PubMed] [Google Scholar]
- (319).Groves JT Using Push to Get Pull. Nat. Chem 2014, 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Green MT; Dawson JH; Gray HB Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (321).Adachi S; Nagano S; Ishimori K; Watanabe Y; Morishima I; Egawa T; Kitagawa T; Makino R Roles of Proximal Ligand in Heme Proteins: Replacement of Proximal Histidine of Human Myoglobin with Cysteine and Tyrosine by Site-Directed Mutagenesis as Models for P-450, Chloroperoxidase, and Catalase. Biochemistry 1993, 32, 241–252. [DOI] [PubMed] [Google Scholar]
- (322).Proshlyakov DA; Ogura T; Shinzawa-Itoh K; Yoshikawa S; Appelman EH; Kitagawa T Selective Resonance Raman Observation of the “607nm” Form Generated in the Reaction of Oxidized Cytochrome c Oxidase with Hydrogen Peroxide. J. Biol. Chem 1994, 269, 29385–29388. [PubMed] [Google Scholar]
- (323).Paulus A; Rossius SGH; Dijk M; de Vries S Oxoferryl-Porphyrin Radical Catalytic Intermediate in Cytochrome Bd Oxidases Protects Cells from Formation of Reactive Oxygen Species. J. Biol. Chem 2012, 287, 8830–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Ozaki S; Roach MP; Matsui T; Watanabe Y Investigations of the Roles of the Distal Heme Environment and the Proximal Heme Iron Ligand in Peroxide Activation by Heme Enzymes via Molecular Engineering of Myoglobin. Acc. Chem. Res 2001, 34, 818–825. [DOI] [PubMed] [Google Scholar]
- (325).Matsui T; Ozaki S; Watanabe Y Formation and Catalytic Roles of Compound I in the Hydrogen Peroxide-Dependent Oxidations by His64 Myoglobin Mutants. J. Am. Chem. Soc 1999, 121, 9952–9957. [Google Scholar]
- (326).Matsui T; Ozaki S; Watanabe Y On the Formation and Reactivity of Compound I of the His-64 Myoglobin Mutants. J. Biol. Chem 1997, 272, 32735–32738. [DOI] [PubMed] [Google Scholar]
- (327).Boso B; Lang G; McMurry TJ; Groves JT Mössbauer Effect Study of Tight Spin Coupling in Oxidized Chloro-5,10,15,20- tetra(mesityl)porphyrinatoiron(IH). J. Chem. Phys 1983, 79, 1122–1126. [Google Scholar]
- (328).Groves JT; Haushalter RC; Nakamura M; Nemo TE; Evans BJ High-Valent Iron-Porphyrin Complexes Related to Peroxidase and Cytochrome P-450. J. Am. Chem. Soc 1981, 103, 2884–2886. [Google Scholar]
- (329).Mandon D; Weiss R; Jayaraj K; Gold A; Terner J; Bill E; Trautwein AX Models for Peroxidase Compound I: Generation and Spectroscopic Characterization of New Oxoferryl Porphyrin π Cation Radical Species. Inorg. Chem 1992, 31, 4404–4409. [Google Scholar]
- (330).Rutter R; Hager LP; Dhonau H; Hendrich M; Valentine M; Debrunner P Chloroperoxidase Compound I: Electron Paramagnetic Resonance and Moessbauer Studies. Biochemistry 1984, 23, 6809–6816. [DOI] [PubMed] [Google Scholar]
- (331).Balch AL; Latos-Grazynski L; Renner MW Oxidation of Red Ferryl [(FeIVO)2+] Porphyrin Complexes to Green Ferryl [(FeIVO)2+] Porphyrin Radical Complexes. J. Am. Chem. Soc 1985, 107, 2983–2985. [Google Scholar]
- (332).Woiowiec S; Latos-Grażyński L Oxidation of Iron(III) Tetramesitylporphyrin with Dimethyldioxirane. Inorg. Chem 1998, 37, 2984–2988. [Google Scholar]
- (333).Sugimoto H; Tung HC; Sawyer DT The Formation, Characterization, and Reactivity of the Oxene Adduct of [tetrakis(2,6-dichlorophenyl)porphinato]iron(III) Perchlorate in Acetonitrile. Model for the Reactive Intermediate of Cytochrome P-450. J. Am. Chem. Soc 1988, 110, 2465–2470. [Google Scholar]
- (334).Fujii H; Ichikawa K Preparation and Characterization of an A1u (Oxo) Iron(IV) Porphyrin π-Cation-Radical Complex. Inorg. Chem 1992, 31, 1110–1112. [Google Scholar]
- (335).Penner-Hahn JE; McMurry TJ; Renner M; Latos-Grazynsky L; Eble KS; Davis 1M; Balch AL; Groves JT; Dawson JH; Hodgson KO X-Ray Absorption Spectroscopic Studies of High Valent 1ron Porphyrins. Horseradish Peroxidase Compounds I and II and Synthetic Models. J. Biol. Chem 1983, 258, 12761–12764. [PubMed] [Google Scholar]
- (336).Hashimoto S; Tatsuno Y; Kitagawa T Observation of the FeIV = O Stretching Raman Band for a Ferryl Porphyrin π Cation Radical. J. Am. Chem. Soc 1987, 109, 8096–8097. [Google Scholar]
- (337).Kincaid JR; Schneider AJ; Paeng KJ The Resonance Raman Spectrum of a Ferrylporphyrin Cation Radical and Its Photodegradation in the Presence of Methanol. J. Am. Chem. Soc 1989, 111 , 735–737. [Google Scholar]
- (338).Hashimoto S; Mizutani Y; Tatsuno Y; Kitagawa T Resonance Raman Characterization of Ferric and Ferryl Porphyrin π Cation Radicals and the FeIV=O Stretching Frequency. J. Am. Chem. Soc 1991, 113, 6542–6549. [Google Scholar]
- (339).Dolphin D; Felton RH Biochemical Significance of Porphyrin π Cation Radicals. Acc. Chem. Res 1974, 7, 26–32. [Google Scholar]
- (340).Fujii H Effects of the Electron-Withdrawing Power of Substituents on the Electronic Structure and Reactivity in Oxoiron-(IV) Porphyrin π-Cation Radical Complexes. J. Am. Chem. Soc 1993, 115, 4641–4648. [Google Scholar]
- (341).Fujii H; Yoshimura T; Kamada H ESR Studies of A1u and A2u Oxoiron(IV) Porphyrin π-Cation Radical Complexes. Spin Coupling between Ferryl Iron and A1u/A2u Orbitals. Inorg. Chem 1996, 35, 2373–2377. [DOI] [PubMed] [Google Scholar]
- (342).Thanabal V; La Mar GN; De Ropp JS A Nuclear Overhauser Effect Study of the Heme Crevice in the Resting State and Compound I of Horseradish Peroxidase: Evidence for Cation Radical Delocalization to the Proximal Histidine. Biochemistry 1988, 27, 5400–5407. [DOI] [PubMed] [Google Scholar]
- (343).Benecky MJ; Frew JE; Scowen N; Jones P; Hoffman BM EPR and ENDOR Detection of Compound 1 from Micrococcus Lysodeikticus Catalase. Biochemistry 1993, 32, 11929–11933. [DOI] [PubMed] [Google Scholar]
- (344).Watanabe Y; Fujii H Characterization of High-Valent Oxo-Metalloporphyrins In Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; Springer: Berlin, 2000; Vol. 97, pp 61–89. [Google Scholar]
- (345).Goto Y; Watanabe Y; Fukuzumi S; Jones JP; Dinnocenzo JP Mechanisms of N-Demethylations Catalyzed by High-Valent Species of Heme Enzymes: Novel Use of 1sotope Effects and Direct Observation of Intermediates. J.Am. Chem. Soc 1998, 120, 10762–10763. [Google Scholar]
- (346).Goto Y; Matsui T; Ozaki S; Watanabe Y; Fukuzumi S Mechanisms of Sulfoxidation Catalyzed by High-Valent Intermediates of Heme Enzymes: Electron-Transfer vs Oxygen-Transfer Mechanism. J. Am. Chem. Soc 1999, 121, 9497–9502. [Google Scholar]
- (347).Groves JT; Watanabe Y Preparation and Characterization of an (acylperoxo)iron(III) Porphyrin. Inorg. Chem 1987, 26, 785–786. [Google Scholar]
- (348).Kudrik EV; Afanasiev P; Alvarez LX; Dubourdeaux P; Clemancey M; Latour J-M; Blondin G; Bouchu D; Albrieux F; Nefedov SE; et al. An N-Bridged High-Valent diiron-Oxo Species on a Porphyrin Platform That Can Oxidize Methane. Nat. Chem 2012, 4, 1024–1029. [DOI] [PubMed] [Google Scholar]
- (349).Song WJ; Ryu YO; Song R; Nam W Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions. JBIC, J. Biol. Inorg. Chem 2005, 10, 294–304. [DOI] [PubMed] [Google Scholar]
- (350).Takahashi A; Kurahashi T; Fujii H Activation Parameters for Cyclohexene Oxygenation by an Oxoiron(IV) Porphyrin π-Cation Radical Complex: Entropy Control of an Allylic Hydroxylation Reaction. Inorg. Chem 2007, 46, 6227–6229. [DOI] [PubMed] [Google Scholar]
- (351).Bell SR; Groves JT A Highly Reactive P450 Model Compound 1. J. Am. Chem. Soc 2009, 131 , 9640–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Cong Z; Kinemuchi H; Kurahashi T; Fujii H Factors Affecting Hydrogen-Tunneling Contribution in Hydroxylation Reactions Promoted by Oxoiron(IV) Porphyrin π-Cation Radical Complexes. Inorg. Chem 2014, 53, 10632–10641. [DOI] [PubMed] [Google Scholar]
- (353).Boaz NC; Bell SR; Groves JT Ferryl Protonation in Oxoiron(1V) Porphyrins and 1ts Role in Oxygen Transfer. J. Am. Chem. Soc 2015, 137, 2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Asaka M; Fujii H Participation of Electron Transfer Process in Rate-Limiting Step of Aromatic Hydroxylation Reactions by Compound I Models of Heme Enzymes. J. Am. Chem. Soc 2016, 138, 8048–8051. [DOI] [PubMed] [Google Scholar]
- (355).De Visser SP; Shaik S A Proton-Shuttle Mechanism Mediated by the Porphyrin in Benzene Hydroxylation by Cytochrome P450 Enzymes. J. Am. Chem. Soc 2003, 125, 7413–7424. [DOI] [PubMed] [Google Scholar]
- (356).Hanzlik RP; Ling KHJ Active Site Dynamics of Xylene Hydroxylation by Cytochrome P-450 As Revealed by Kinetic Deuterium Isotope Effects. J. Am. Chem. Soc 1993, 115, 9363–9370. [Google Scholar]
- (357).Shiota Y; Suzuki K; Yoshizawa K Mechanism for the Direct Oxidation of Benzene to Phenol by FeO+. Organometallics 2005, 24, 3532–3538. [Google Scholar]
- (358).Vannelli T; Hooper AB N1H Shift in the Hydroxylation of Aromatic Compounds by the Ammonia-Oxidizing Bacterium Nitrosomonas Europaea. Evidence against an Arene Oxide Intermediate. Biochemistry 1995, 34, 11743–11749. [DOI] [PubMed] [Google Scholar]
- (359).Korzekwa KR; Swinney DC; Trager WF Isotopically Labeled Chlorobenzenes as Probes for the Mechanism of Cytochrome P-450 Catalyzed Aromatic Hydroxylation. Biochemistry 1989, 28, 9019–9027. [DOI] [PubMed] [Google Scholar]
- (360).Burka LT; Plucinski TM; Macdonald TL Mechanisms of Hydroxylation by Cytochrome P-450: Metabolism of Monohalobenzenes by Phenobarbital-Induced Microsomes. Proc. Natl. Acad. Sci. U. S. A 1983, 80, 6680–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Sato H; Guengerich FP Oxidation of 1,2,4,5-Tetramethoxybenzene to a Cation Radical by Cytochrome P450. J. Am. Chem. Soc 2000, 122, 8099–8100. [Google Scholar]
- (362).Kang M-J; Song WJ; Han A-R; Choi YS; Jang HG; Nam W Mechanistic Insight into the Aromatic Hydroxylation by High-Valent Iron(IV)-Oxo Porphyrin π-Cation Radical Complexes. J. Org. Chem 2007, 72, 6301–6304. [DOI] [PubMed] [Google Scholar]
- (363).Gross Z The Effect of Axial Ligands on the Reactivity and Stability of the Oxoferryl Moiety in Model Complexes of Compound I of Heme-Dependent Enzymes. JBIQ J. Biol. Inorg. Chem 1996, 1, 368–371. [Google Scholar]
- (364).Takahashi A; Yamaki D; Ikemura K; Kurahashi T; Ogura T; Hada M; Fujii H Effect of the Axial Ligand on the Reactivity of the Oxoiron(IV) Porphyrin π-Cation Radical Complex: Higher Stabilization of the Product State Relative to the Reactant State. Inorg. Chem 2012, 51, 7296–7305. [DOI] [PubMed] [Google Scholar]
- (365).Chin D-H; Balch AL; La Mar GN Formation of Porphyrin Ferryl (FeO2+) Complexes through the Addition of Nitrogen Bases to Peroxo-Bridged Iron(III) Porphyrins. J. Am. Chem. Soc 1980, 102, 1446–1448. [Google Scholar]
- (366).La Mar GN; De Ropp JS; Latos-Grazynski L; Balch AL; Johnson RB; Smith KM; Parish DW; Cheng RJ Proton NMR Characterization of the Ferryl Group in Model Heme Complexes and Hemoproteins: Evidence for the FeIVO Group in Ferryl Myoglobin and Compound II of Horseradish Peroxidase. J. Am. Chem. Soc 1983, 105, 782–787. [Google Scholar]
- (367).Balch AL; Chan YW; Cheng RJ; La Mar GN; Latos- Grazynski L; Renner MW Oxygenation Patterns for Iron(II) Porphyrins. Peroxo and Ferryl (FeIVO) Intermediates Detected by Proton Nuclear Magnetic Resonance Spectroscopy during the Oxygenation of (tetramesitylporphyrin)iron(II). J. Am. Chem. Soc 1984, 106, 7779–7785. [Google Scholar]
- (368).Balch AL; La Mar GN; Latos-Grazynski L; Renner MW; Thanabal V Nuclear Magnetic Resonance Studies of Axial Amine Coordination in Synthetic Ferryl (FeIVO)2+ Porphyrin Complexes and in Ferryl Myoglobin. J. Am. Chem. Soc 1985, 107, 3003–3007. [Google Scholar]
- (369).Groves JT; Quinn R; McMurry TJ; Nakamura M; Lang G; Boso B Preparation and Characterization of a Dialkoxyiron(IV) Porphyrin. J. Am. Chem. Soc 1985, 107, 354–360. [Google Scholar]
- (370).Jeong YJ; Kang Y; Han A-R; Lee Y-M; Kotani H; Fukuzumi S; Nam W Hydrogen Atom Abstraction and Hydride Transfer Reactions by Iron(IV)-Oxo Porphyrins. Angew. Chem., Int. Ed 2008, 47, 7321–7324. [DOI] [PubMed] [Google Scholar]
- (371).Pan Z; Newcomb M Kinetics and Mechanism of Oxidation Reactions of Porphyrin-Iron(IV)-Oxo Intermediates. Inorg. Chem 2007, 46, 6767–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Groves JT; Gross Z; Stern MK Preparation and Reactivity of Oxoiron(IV) Porphyrins. Inorg. Chem 1994, 33, 5065–5072. [Google Scholar]
- (373).Takahashi A; Kurahashi T; Fujii H Redox Potentials of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Participation of Electron Transfer Process in Oxygenation Reactions. Inorg. Chem 2011, 50, 6922–6928. [DOI] [PubMed] [Google Scholar]
- (374).Lee WA; Calderwood TS; Bruice TC Stabilization of Higher-Valent States of Iron Porphyrin by Hydroxide and Methoxide Ligands: Electrochemical Generation of Iron(IV)-Oxo Porphyrins. Proc. Natl. Acad. Sci U. S. A 1985, 82, 4301–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Calderwood TS; Bruice TC Spectral and Electrochemical Identification of Iron(IV)-Oxo Porphyrin and Iron(IV)-Oxo Porphyrin π-Cation Species. J.Am. Chem. Soc 1985, 107, 8272–8273. [Google Scholar]
- (376).Swistak C; Mu XH; Kadish KM Electrochemistry of Hydroxo- and Methoxo[tetrakis(2,4,6-Trimethylphenyl)-porphyrinato]iron in Dichloromethane. Electrogeneration of Iron(IV) and Iron(II) Porphyrins. Inorg. Chem 1987, 26, 4360–4366. [Google Scholar]
- (377).Groves JT; Gilbert JA Electrochemical Generation of an Iron(IV) Porphyrin. Inorg. Chem 1986, 25, 123–125. [Google Scholar]
- (378).Proniewicz LM; Bajdor K; Nakamoto K Resonance Raman Spectra of Ferrylporphyrins and Related Compounds in Dioxygen Matrixes. J. Phys. Chem 1986, 90, 1760–1766. [Google Scholar]
- (379).Proniewicz LM; Paeng IR; Nakamoto K Resonance Raman Spectra of Two Isomeric Dioxygen Adducts of Iron(II) Porphyrins and π-Cation Radical and Nonradical Oxoferryl Porphyrins Produced in Dioxygen Matrixes: Simultaneous Observation of More than Seven Oxygen Isotope Sensitive Bands. J. Am. Chem. Soc 1991, 113, 3294–3303. [Google Scholar]
- (380).Fujii H; Yoshimura T; Kamada H ESR Studies of Oxochromium(V) Porphyrin Complexes: Electronic Structure of the CrVO Moiety. Inorg. Chem 1997, 36, 1122–1127. [DOI] [PubMed] [Google Scholar]
- (381).Fujii H; Kurahashi T; Tosha T; Yoshimura T; Kitagawa T 17O NMR Study of Oxo Metalloporphyrin Complexes: Correlation with Electronic Structure of MO Moiety. J. Inorg. Biochem 2006, 100, 533–541. [DOI] [PubMed] [Google Scholar]
- (382).Czarnecki K; Proniewicz LM; Fujii H; Kincaid JR Resonance Raman Spectrum of a 2A1u Ferryl Porphyrin π-Cation Radical. J. Am. Chem. Soc 1996, 118, 4680–4685. [Google Scholar]
- (383).Paeng IR; Shiwaku H; Nakamoto K Detection of the Fe-O–O-Fe Intermediate in the Oxidation Reaction of Ferrous Porphyrins by Resonance Raman Spectroscopy. J. Am. Chem. Soc 1988, 110, 1995–1996. [Google Scholar]
- (384).Mizutani Y; Hashimoto S; Tatsuno Y; Kitagawa T Resonance Raman Pursuit of the Change from FeII-O2 to FeIII-OH via FeIV=O in the Autoxidation of Ferrous Iron-Porphyrin. J. Am. Chem. Soc 1990, 112, 6809–6814. [Google Scholar]
- (385).Czarnecki K; Kincaid JR; Fujii H Resonance Raman Spectra of Legitimate Models for the Ubiquitous Compound I Intermediates of Oxidative Heme Enzymes. J. Am. Chem. Soc 1999, 121, 7953–7954. [Google Scholar]
- (386).Kean RT; Oertling WA; Babcock GT Characterization of Six-Coordinate Ferryl Protoheme by Resonance Raman and Optical Absorption Spectroscopy. J.Am. Chem. Soc 1987, 109, 2185–2187. [Google Scholar]
- (387).Gold A; Jayaraj K; Doppelt P; Weiss R; Chottard G; Bill E; Ding X; Trautwein AX Oxoferryl Complexes of the Halogenated (Porphinato)iron Catalyst [tetrakis(2,6-Dichlorophenyl)porphinato]iron. J. Am. Chem. Soc 1988, 110, 5756–5761. [Google Scholar]
- (388).Chin D-H; La Mar GN; Balch AL Role of Ferryl (FeO2+) Complexes in Oxygen Atom Transfer Reactions. Mechanism of Iron(II) Porphyrin Catalyzed Oxygenation of Triphenylphosphine. J. Am. Chem. Soc 1980, 102, 5945–5947. [Google Scholar]
- (389).Dunford HB Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology and Physiology, 2nd ed.; Wiley & Sons, Ltd.: New York, 2010. [Google Scholar]
- (390).Dunford HB Heme Peroxidases; John Wiley & Sons, Inc.: New York, 1999. [Google Scholar]
- (391).Poulos TL Peroxidases and Catalases In Biological Inorganic Chemistry-Structure and Reactivity; Bertini I, Gray HB, Stiefel EI, Valentine JS, Eds.; University Science Books: Sausaito, CA, 2007; pp 343–353. [Google Scholar]
- (392).Cong Z; Kurahashi T; Fujii H Oxidation of Chloride and Subsequent Chlorination of Organic Compounds by Oxoiron(IV) Porphyrin π-Cation Radicals. Angew. Chem. Int. Ed 2011, 50, 9935–9939. [DOI] [PubMed] [Google Scholar]
- (393).Brown FS; Hager LP Chloroperoxidase. IV. Evidence for an Ionic Electrophilic Substitution Mechanism. J. Am. Chem. Soc 1967, 89, 719–720. [DOI] [PubMed] [Google Scholar]
- (394).Cong Z; Kurahashi T; Fujii H Formation of Iron(lIl) Meso-Chloro-Isoporphyrin as a Reactive Chlorinating Agent from Oxoiron(IV) Porphyrin π-Cation Radical. J. Am. Chem. Soc 2012, 134, 4469–4472. [DOI] [PubMed] [Google Scholar]
- (395).Dolphin D; Felton RH; Borg DC; Fajer J Isoporphyrins. J. Am. Chem. Soc 1970, 92, 743–745. [DOI] [PubMed] [Google Scholar]
- (396).Cong Z; Yanagisawa S; Kurahashi T; Ogura T; Nakashima S; Fujii H Synthesis, Characterization, and Reactivity of Hypochloritoiron(III) Porphyrin Complexes. J. Am. Chem. Soc 2012, 134, 20617–20620. [DOI] [PubMed] [Google Scholar]
- (397).Collman JP; Gagne RR; Reed CA; Robinson WT; Rodley GA Structure of an Iron(II) Dioxygen Complex; A Model for Oxygen Carrying Hemeproteins. Proc. Natl. Acad. Sci. U. S. A 1974, 71, 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Eaton WA; Henry ER; Hofrichter J; Mozzarelli A Is Cooperative Oxygen Binding by Hemoglobin Really Understood? Nat. Struct. Biol 1999, 6, 351–358. [DOI] [PubMed] [Google Scholar]
- (399).Cohen IA; Caughey WS Substituted Deuteroporphyrins. IV. Kinetics and Mechanism of Reactions of Iron(II) Porphyrins with Oxygen. Biochemistry 1968, 7, 636–641. [DOI] [PubMed] [Google Scholar]
- (400).Sadasivan N; Eberspaecher HI; Fuchsman WH; Caughey WS Substituted Deuterophorphyrins. VI. Ligand-Exchange and Dimerization Reactions of Deuterohemins. Biochemistry 1969, 8, 534–541. [DOI] [PubMed] [Google Scholar]
- (401).Weschler CJ; Anderson DL; Basolo F Kinetics and Thermodynamics of Oxygen and Carbon Monoxide Binding to Simple Ferrous Porphyrins at Low Temperatures. J. Am. Chem. Soc 1975, 97, 6707–6713. [DOI] [PubMed] [Google Scholar]
- (402).Mincey T; Traylor TG Anion Complexes of Ferrous Porphyrins. J. Am. Chem. Soc 1979, 101, 765–766. [Google Scholar]
- (403).Latos-Grazynski L; Cheng RJ; La Mar GN; Balch AL Oxygenation Patterns for Substituted Meso-Tetraphenylporphyrin Complexes of Iron(II). Spectroscopic Detection of Dioxygen Complexes in the Absence of Amines. J. Am. Chem. Soc 1982, 104, 5992–6000. [Google Scholar]
- (404).Chin D-H; Del Gaudio J; La Mar GN; Balch AL Detection and Characterization of the Long-Postulated Iron-Dioxygen-Iron Intermediate in the Autoxidation of Ferrous Porphyrins. J. Am. Chem. Soc 1977, 99, 5486–5488. [DOI] [PubMed] [Google Scholar]
- (405).Shikama K The Molecular Mechanism of Autoxidation for Myoglobin and Hemoglobin: A Venerable Puzzle. Chem. Rev 1998, 98, 1357–1374. [DOI] [PubMed] [Google Scholar]
- (406).Brantley RE; Smerdon SJ; Wilkinson AJ; Singleton EW; Olson JS The Mechanism of Autooxidation of Myoglobin. J. Biol. Chem 1993, 268, 6995–7010. [PubMed] [Google Scholar]
- (407).Springer BA; Sligar SG; Olson JS; Phillips GN Jr. Mechanisms of Ligand Recognition in Myoglobin. Chem. Rev 1994, 94, 699–714. [Google Scholar]
- (408).Dickerson VRE; Geis I Hemoglobin: Structure, Function, Evolution, and Pathology; Benjamin/Cunnings: Menlo Park, CA, 1983. [Google Scholar]
- (409).Lavalette D; Tetreau C; Mispelter J; Momenteau M; Lhoste J-M Linear Free-Energy Relationships in Binding of Oxygen and Carbon Monoxide with Heme Model Compounds and Heme Proteins. Eur. J. Biochem 1984, 145, 555–565. [DOI] [PubMed] [Google Scholar]
- (410).Lexa D; Momenteau M; Saveant JM; Xu F Redox Properties and Stability of Hydroxy Complexes of Protected Iron(III) and Iron(II) Porphyrins. Inorg. Chem 1985, 24, 122–127. [Google Scholar]
- (411).Gerothanassis IP; Momenteau M; Loock B Hydrogen-Bond Stabilization of Dioxygen: Conformation Excitation and Autoxidation Mechanism in Hemoprotein Models as Revealed by 17O NMR Spectroscopy. J. Am. Chem. Soc 1989, 111, 7006–7012. [Google Scholar]
- (412).Brault D; Rougee M Ferrous Porphyrins in Organic Solvents. I. Preparation and Coordinating Properties. Biochemistry 1974, 13, 4591–4597. [DOI] [PubMed] [Google Scholar]
- (413).Radonovich LJ; Bloom A; Hoard JL Stereochemistry of Low-Spin Iron Porphyrins. II. Bis(piperidine)-α,β, γ, δ-tetraphenylporphinatoiron(II). J. Am. Chem. Soc 1972, 94, 2073–2078. [DOI] [PubMed] [Google Scholar]
- (414).Spartalian K; Lang G; Collman JP; Gagne RR; Reed CA Mossbauer Spectroscopy of Hemoglobin Model Compounds: Evidence for Conformational Excitation. J. Chem. Phys 1975, 63, 5375–5382. [Google Scholar]
- (415).Collman JP; Reed CA Syntheses of Ferrous-Porphyrin Complexes. Hypothetical Model for Deoxymyoglobin. J. Am. Chem. Soc 1973, 95, 2048–2049. [DOI] [PubMed] [Google Scholar]
- (416).Hoard JL Stereochemistry of Hemes and Other Metalloporphyrins. Science 1971, 174, 1295–1302. [DOI] [PubMed] [Google Scholar]
- (417).Hu C; Noll BC; Schulz CE; Scheidt WR Electronic Configuration of Five-Coordinate High-Spin Pyrazole-Ligated Iron-(II) Porphyrinates. Inorg. Chem 2010, 49, 10984–10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Momenteau M; Scheidt WR; Eigenbrot CW; Reed CA A Deoxymyoglobin Model with a Sterically Unhindered Axial Imidazole. J. Am. Chem. Soc 1988, 110, 1207–1215. [Google Scholar]
- (419).Momenteau M; Loock B; Bisagni E; Rougee M Five-Coordinate Iron(II) Porphyrins Derived from Meso-α,β,γ,δ Tetraphenylporphin: Synthesis, Characterization, and Coordinating Properties. Can. J. Chem 1979, 57, 1804–1813. [Google Scholar]
- (420).Li Y; Sharma SK; Karlin KD New Heme-Dioxygen and Carbon Monoxide Adducts Using Pyridyl or Imidazolyl Tailed Porphyrins. Polyhedron 2013, 58, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Sharma SK; Rogler PJ; Karlin KD Reactions of a Heme-Superoxo Complex toward a Cuprous Chelate and •NO(g):CcO and NOD Chemistry. J. Porphyrins Phthalocyanines 2015, 19, 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Geibel J; Cannon J; Campbell D; Traylor TG Model Compounds for R-State and T-State Hemoglobins. J. Am. Chem. Soc 1978, 100, 3575–3585. [Google Scholar]
- (423).Sharma SK; Kim H; Rogler PJ; Siegler MA; Karlin KD Isocyanide or Nitrosyl Complexation to Hemes with Varying Tethered Axial Base Ligand Donors: Synthesis and Characterization. JBIC, J. Biol. Inorg. Chem 2016, 21, 729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Momenteau M; Rougee M; Loock B Five-Coordinate Iron-Porphyrin as a Model for the Active Site of Hemoproteins. Characterization and Coordinating Properties. Eur. J. Biochem 1976, 71, 63–76. [DOI] [PubMed] [Google Scholar]
- (425).Collman JP; Brauman JI; Doxsee KM; Halbert TR; Suslick KS Model Compounds for the T State of Hemoglobin. Proc. Natl. Acad. Sci. U. S. A 1978, 75, 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Collman JP; Brauman JI; Doxsee KM; Halbert TR; Bunnenberg E; Linder RE; LaMar GN; Del Gaudio J; Lang G; Spartalian K Synthesis and Characterization of “Tailed Picket Fence” Porphyrins. J. Am. Chem. Soc 1980, 102, 4182–4192. [Google Scholar]
- (427).Perutz MF; Hasnain SS; Duke PJ; Sessler JL; Hahn JE Stereochemistry of Iron in Deoxyhaemoglobin. Nature 1982, 295, 535–538. [DOI] [PubMed] [Google Scholar]
- (428).Collman JP; Gagne RR; Halbert TR; Marchon JC; Reed CA Reversible Oxygen Adduct Formation in Ferrous Complexes Derived from a Picket Fence Porphyrin. Model for Oxymyoglobin. J. Am. Chem. Soc 1973, 95, 7868–7870. [DOI] [PubMed] [Google Scholar]
- (429).Jameson GB; Molinaro FS; Ibers JA; Collman JP; Brauman JI; Rose E; Suslick KS Structural Changes upon Oxygenation of an Iron(II)(porphyrinato)(imidazole) Complex. J. Am. Chem. Soc 1978, 100, 6769–6770. [Google Scholar]
- (430).Jameson GB; Rodley GA; Robinson WT; Gagne RR; Reed C; Collman JP Structure of a Dioxygen Adduct of (1-Methylimidazole)-Meso-Tetrakis(α,α,α,α-O-pivalamidophenyl)-porphinatoiron(II). An Iron Dioxygen Model for the Heme Component of Oxymyoglobin. Inorg. Chem 1978, 17, 850–857. [Google Scholar]
- (431).Pauling L Nature of the Iron-Oxygen Bond in Oxyhemoglobin. Nature 1964, 203, 182–183. [PubMed] [Google Scholar]
- (432).Cramer CJ; Tolman WB; Theopold KH; Rheingold AL Variable Character of O–O and M–O Bonding in Side-on (η2) 1:1 Metal Complexes of O2. Proc. Natl. Acad. Sci U. S. A 2003, 100, 3635–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Li J; Noll BC; Oliver AG; Schulz CE; Scheidt WR Correlated Ligand Dynamics in Oxyiron Picket Fence Porphyrins: Structural and Mossbauer Investigations. J. Am. Chem. Soc 2013, 135, 15627–15641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Godbout N; Sanders LK; Salzmann R; Havlin RH; Wojdelski M; Oldfield E Solid-State NMR, Mossbauer, Crystallographic, and Density Functional Theory Investigation of Fe-O2 and Fe-O2 Analogue Metalloporphyrins and Metalloproteins. J. Am. Chem. Soc 1999, 121, 3829–3844. [Google Scholar]
- (435).Collman JP; Zhang X; Wong K; Brauman JI Dioxygen Binding in Iron and Cobalt Picnic Basket Porphyrins. J. Am. Chem. Soc 1994, 116, 6245–6251. [Google Scholar]
- (436).Mispelter J; Momenteau M; Lavalette D; Lhoste JM Hydrogen-Bond Stabilization of Oxygen in Hemoprotein Models. J. Am. Chem. Soc 1983, 105, 5165–5166. [Google Scholar]
- (437).Collman JP; Herrmann PC; Fu L; Eberspacher TA; Eubanks M; Boitrel B; Hayoz P; Zhang X; Brauman JI; Day VW Aza-Crown-Capped Porphyrin Models of Myoglobin: Studies of the Steric Interactions of Gas Binding. J. Am. Chem. Soc 1997, 119, 3481–3489. [Google Scholar]
- (438).Quillin ML; Arduini RM; Olson JS; Phillips GN High-Resolution Crystal Structures of Distal Histidine Mutants of Sperm Whale Myoglobin. J. Mol. Biol 1993, 234, 140–155. [DOI] [PubMed] [Google Scholar]
- (439).Lamb DC; Ostermann A; Prusakov VE; Parak FG From Metmyoglobin to Deoxy Myoglobin: Relaxations of an Intermediate State. Eur. Biophys. J 1998, 27, 113–125. [DOI] [PubMed] [Google Scholar]
- (440).Collman JP; Decreau RA; Dey A; Yang Y Water May Inhibit Oxygen Binding in Hemoprotein Models. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4101–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Collman JP Synthetic Models for the Oxygen-Binding Hemoproteins. Acc. Chem. Res 1977, 10, 265–272. [Google Scholar]
- (442).Collman JP; Fu L Synthetic Models for Hemoglobin and Myoglobin. Acc. Chem. Res 1999, 32, 455–463. [Google Scholar]
- (443).Li J; Peng Q; Barabanschikov A; Pavlik JW; Alp EE; Sturhahn W; Zhao J; Schulz CE; Sage JT; Scheidt WR New Perspectives on Iron-Ligand Vibrations of Oxyheme Complexes. Chem. −3 Eur. J 2011, 17, 11178–11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Rovira C; Parrinello M Factors Influencing Ligand-Binding Properties of Heme Models: A First Principles Study of Picket-Fence and Protoheme Complexes. Chem. −3 Eur. J 1999, 5, 250–262. [Google Scholar]
- (445).Nasri H; Wang Y; Huynh Boi Hanh; Scheidt WR Nitrite-Bound Five-Coordinate Low-Spin Iron(II) Model Complex for the Prosthetic Group of Nitrite Reductase with an Unusually Large Quadrupole Splitting. Synthesis, Moessbauer Properties, and Molecular Structure of the Complex (Nitro)(α,α,α,α-Tetrakis(o-P. J. Am. Chem. Soc 1991, 113, 717–719. [Google Scholar]
- (446).Nasri H; Wang Y; Boi Hanh H; Walker FA; Scheidt WR Reactions of Bis(nitro)(α,α,α,α-Tetrakis(o-pivalamidophenyl)-porphinato)ferrate(III) with Pyridine and Imidazole. EPR and Moessbauer Spectra and Molecular Structures of the Mixed-Ligand Species. Inorg. Chem 1991, 30, 1483–1489. [Google Scholar]
- (447).Nasri H; Ellison MK; Chen S; Huynh BH; Scheidt WR Sharing the π-Bonding. An Iron Porphyrin Derivative with Trans, π-Accepting Axial Ligands. Synthesis, EPR and Mossbauer Spectra, and Molecular Structure of Two Forms of the Complex Nitronitrosyl-(α,α,α,α-Tetrakis(o-Pivalamidophenyl)-porphinato)ferrate(II). J. Am. Chem. Soc 1997, 119, 6274–6283. [Google Scholar]
- (448).Nasri H; Ellison MK; Shang M; Schulz CE; Scheidt WR Variable π-Bonding in Iron(II) Porphyrinates with Nitrite, CO, and Tert-Butyl Isocyanide: Characterization of[Fe(TpivPP)(NO2)(CO)]. Inorg. Chem 2004, 43, 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Li J; Noll BC; Oliver AG; Scheidt WR Structural Insights into Ligand Dynamics: Correlated Oxygen and Picket Motion in Oxycobalt Picket Fence Porphyrins. J. Am. Chem. Soc 2012, 134, 10595–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Rovira C; Parrinello M Harmonic and Anharmonic Dynamics of Fe–CO and Fe–O2 in Heme Models. Biophys. J 2000, 78, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Price DC Vibronic Effects in Mossbauer Spectra: The 57Fe Quadrupole Splitting in FeCO3. Aust. J. Phys 1978, 31, 397–420. [Google Scholar]
- (452).Scheidt WR; Osvath SR; Lee YJ Crystal and Molecular Structure of Bis(imidazole)(meso-tetraphenylporphinato)iron(III) Chloride. A Classic Molecule Revisited. J. Am. Chem. Soc 1987, 109, 1958–1963. [Google Scholar]
- (453).Kossanyi A; Tani F; Nakamura N; Naruta Y Properties of a Binaphthyl-Bridged Porphyrin-Iron Complex Bearing Hydroxy Groups Inside Its Cavity. Chem. −3 Eur. J 2001, 7, 2862–2872. [DOI] [PubMed] [Google Scholar]
- (454).Matsu-ura M; Tani F; Naruta Y Formation and Characterization of Carbon Monoxide Adducts of Iron “Twin Coronet” Porphyrins. Extremely Low CO Affinity and a Strong Negative Polar Effect on Bound CO. J. Am. Chem. Soc 2002, 124, 1941–1950. [DOI] [PubMed] [Google Scholar]
- (455).Tani F; Matsu-ura M; Ariyama K; Setoyama T; Shimada T; Kobayashi S; Hayashi T; Matsuo T; Hisaeda Y; Naruta Y Iron Twin-Coronet Porphyrins as Models of Myoglobin and Hemoglobin: Amphibious Electrostatic Effects of Overhanging Hydroxyl Groups for Successful CO/O2 Discrimination. Chem. −3 Eur. J 2003, 9, 862–870. [DOI] [PubMed] [Google Scholar]
- (456).Traylor TG; Koga N; Deardurff LA Structural Differentiation of Carbon Monoxide and Oxygen Binding to Iron Porphyrins: Polar Pocket Effects. J. Am. Chem. Soc 1985, 107, 6504–6510. [Google Scholar]
- (457).Traylor TG; Traylor PS Reactions of Dioxygen and Its Reduced Forms with Heme Proteins and Model Porphyrin Complexes In Active Oxygen in Biochemistry; Valentine JS, Foote CS, Greenberg A, Liebman JF, Eds.; Springer US: Boston, MA, 1995; pp 84–187. [Google Scholar]
- (458).Tani F; Matsu-ura M; Nakayama S; Ichimura M; Nakamura N; Naruta Y Synthesis and Characterization of Alkanethiolate-Coordinated Iron Porphyrins and Their Dioxygen Adducts as Models for the Active Center of Cytochrome P450: Direct Evidence for Hydrogen Bonding to Bound Dioxygen. J. Am. Chem. Soc 2001, 123, 1133–1142. [DOI] [PubMed] [Google Scholar]
- (459).El-Kasmi D; Tetreau C; Lavalette D; Momenteau M Chemical Models of Hemoglobins and Cytochromes P-450: Influence of the Basicity of the Proximal Ligand on O2 and CO Binding Kinetics. J. Am. Chem. Soc 1995, 117, 6041–6047. [Google Scholar]
- (460).Schappacher M; Ricard L; Fischer J; Weiss R; Bill E; Montiel-Montoya R; Winkler H; Trautwein AX Synthesis, Structure and Spectroscopic Properties of Two Models for the Active Site of the Oxygenated State of Cytochrome P540. Eur. J. Biochem 1987, 168, 419–429. [DOI] [PubMed] [Google Scholar]
- (461).Hu S; Schneider AJ; Kincaid JR Resonance Raman Studies of Oxycytochrome P450cam: Effect of Substrate Structure on v(O–O) and v(Fe-O2). J. Am. Chem. Soc 1991, 113, 4815–4822. [Google Scholar]
- (462).Macdonald IDG; Sligar SG; Christian JF; Unno M; Champion PM Identification of the Fe-O–O Bending Mode in Oxycytochrome P450cam by Resonance Raman Spectroscopy. J. Am. Chem. Soc 1999, 121, 376–380. [Google Scholar]
- (463).Egawa T; Ogura T; Makino R; Ishimura Y; Kitagawa T Observation of the O–O Stretching Raman Band for Cytochrome P450cam under Catalytic Conditions. J. Biol. Chem 1991, 266, 10246–10248. [PubMed] [Google Scholar]
- (464).Kitagawa T; Ondrias MR; Rousseau DL; Ikeda-Saito M; Yonetani T Evidence for Hydrogen Bonding of Bound Dioxygen to the Distal Histidine of Oxycobalt Myoglobin and Haemoglobin. Nature 1982, 298, 869–871. [DOI] [PubMed] [Google Scholar]
- (465).Bruha A; Kincaid JR Resonance Raman Studies of Dioxygen Adducts of Cobalt-Substituted Heme Proteins and Model Compounds. Vibrationally Coupled Dioxygen and the Issues of Multiple Structures and Distal Side Hydrogen Bonding. J. Am. Chem. Soc 1988, 110, 6006–6014. [DOI] [PubMed] [Google Scholar]
- (466).Proniewicz LM; Kincaid JR Vibrational Coupling Effects in the Resonance Raman Spectra of O2 Adducts of Heme Proteins and Model Compounds. Coord. Chem. Rev 1997, 161, 81–127. [Google Scholar]
- (467).Phillips SEV; Schoenborn BP Neutron Diffraction Reveals Oxygen-Histidine Hydrogen Bond in Oxymyoglobin. Nature 1981, 292, 81–82. [DOI] [PubMed] [Google Scholar]
- (468).Olson JS; Phillips GN Jr. Myoglobin Discriminates between O2, NO, and CO by Electrostatic Interactions with the Bound Ligand. JBIQ J. Biol. Inorg. Chem 1997, 2, 544–552. [Google Scholar]
- (469).Spiro TG; Kozlowski PM Will the Real FeCO Please Stand Up? JBIC, J. Biol. Inorg. Chem 1997, 2, 516–520. [Google Scholar]
- (470).Kachalova GS A Steric Mechanism for Inhibition of CO Binding to Heme Proteins. Science 1999, 284, 473–476. [DOI] [PubMed] [Google Scholar]
- (471).Spiro TG; Kozlowski PM Is the CO Adduct of Myoglobin Bent, and Does It Matter? Acc. Chem. Res 2001, 34, 137–144. [DOI] [PubMed] [Google Scholar]
- (472).Chu K; Vojtchovsky J; McMahon BH; Sweet R M.; Berendzen, J.; Schlichting, I. Structure of a Ligand-Binding Intermediate in Wild-Type Carbonmonoxy Myoglobin. Nature 2000, 403, 921–923. [DOI] [PubMed] [Google Scholar]
- (473).Wittenberg JB; Appleby CA; Wittenberg BA The Kinetics of the Reactions of Leghemoglobin with Oxygen and Carbon Monoxide. J. Biol. Chem 1972, 247, 527–531. [PubMed] [Google Scholar]
- (474).Kloek AP; Yang J; Mathews FS; Goldberg DE Expression, Characterization, and Crystallization of Oxygen-Avid Ascaris Hemoglobin Domains. J. Biol. Chem 1993, 268, 17669–17671. [PubMed] [Google Scholar]
- (475).Yang J; Kloek AP; Goldberg DE; Mathews FS The Structure of Ascaris Hemoglobin Domain I at 2.2 Å Resolution: Molecular Features of Oxygen Avidity. Proc. Natl. Acad. Sci. U. S. A 1995, 92, 4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Huang S; Huang J; Kloek AP; Goldberg DE; Friedman JM Hydrogen Bonding of Tyrosine B10 to Heme-Bound Oxygen in Ascaris Hemoglobin. J. Biol. Chem 1996, 271, 958–962. [DOI] [PubMed] [Google Scholar]
- (477).Momenteau M; Loock B; Lavalette D; Tétreau C; Mispelter J Iron(II) “Hanging Imidazole” Porphyrin: Synthesis and Proximal Ligand Effect on CO and O2 Binding. J. Chem. Soc. Chem. Commun 1983, 962–964. [Google Scholar]
- (478).Boitrel B; Hijazi I; Roisnel T; Oohora K; Hayashi T Iron-Strapped Porphyrins with Carboxylic Acid Groups Hanging over the Coordination Site: Synthesis, X-Ray Characterization, and Dioxygen Binding. Inorg. Chem 2017, 56, 7373–7383. [DOI] [PubMed] [Google Scholar]
- (479).Collman JP; Brauman JI; Doxsee KM; Sessler JL; Morris RM; Gibson QH Effect of Axial Base on Dioxygen and Carbon Monoxide Affinities of iron(II) Porphyrins. Imidazole vs. Pyridine. Inorg. Chem 1983, 22, 1427–1432. [Google Scholar]
- (480).Desbois A; Momenteau M; Lutz M Resonance Raman Spectroscopy of iron(II) Superstructured Porphyrins: Influence of Porphyrin Distortions on Carbonyl and Dioxygen Ligand Dissociation. Inorg. Chem 1989, 28, 825–834. [Google Scholar]
- (481).Walters MA; Spiro TG; Suslick KS; Collman JP Resonance Raman Spectra of (Dioxygen)(porphyrinato)(hindered imidazole)iron(II) Complexes: Implications for Hemoglobin Coop- erativity. J. Am. Chem. Soc 1980, 102, 6857–6858. [Google Scholar]
- (482).Oertling WA; Kean RT; Wever R; Babcock GT Factors Affecting the Iron-Oxygen Vibrations of Ferrous Oxy and Ferryl Oxo Heme Proteins and Model Compounds. Inorg. Chem 1990, 29, 2633–2645. [Google Scholar]
- (483).Hirota S; Ogura T; Appelman EH; Shinzawa-Itoh K; Yoshikawa S; Kitagawa T Observation of a New Oxygen-Isotope-Sensitive Raman Band for Oxyhemoproteins and Its Implications in Heme Pocket Structures. J. Am. Chem. Soc 1994, 116, 10564–10570. [Google Scholar]
- (484).Walters MA; Spiro TG Resonance Raman Spectroscopic Studies of Axial Ligation in Oxyhemoglobin and Oxymyoglobin, and Nitrosylmyoglobin. Biochemistry 1982, 21, 6989–6995. [DOI] [PubMed] [Google Scholar]
- (485).Liu J-G; Naruta Y; Tani F A Functional Model of the Cytochrome c Oxidase Active Site: Unique Conversion of a Heme-μ-Peroxo-CuII Intermediate into Heme-Superoxo/CuI. Angew. Chem., Int. Ed 2005, 44, 1836–1840. [DOI] [PubMed] [Google Scholar]
- (486).Liu J-G; Naruta Y; Tani F; Chishiro T; Tachi Y Formation and Spectroscopic Characterization of the Dioxygen Adduct of a Heme-Cu Complex Possessing a Cross-Linked Tyrosine-Histidine Mimic: Modeling the Active Site of Cytochrome c Oxidase. Chem. Commun 2004, 120–121. [DOI] [PubMed] [Google Scholar]
- (487).Ohta T; Liu J-G; Nagaraju P; Ogura T; Naruta Y A Cryo-Generated Ferrous-Superoxo Porphyrin: EPR, Resonance Raman and DFT Studies. Chem. Commun 2015, 51, 12407–12410. [DOI] [PubMed] [Google Scholar]
- (488).Bartlett N; Symons MCR Electron Addition to the (FeO2) Unit of Oxyhaemoglobin Glycera. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1983, 744, 110–114. [DOI] [PubMed] [Google Scholar]
- (489).Davydov R; Satterlee JD; Fujii H; Sauer-Masarwa A; Busch DH; Hoffman BM A Superoxo-Ferrous State in a Reduced Oxy-Ferrous Hemoprotein and Model Compounds. J. Am. Chem. Soc 2003, 125, 16340–16346. [DOI] [PubMed] [Google Scholar]
- (490).Liu J-G; Ohta T; Yamaguchi S; Ogura T; Sakamoto S; Maeda Y; Naruta Y Spectroscopic Characterization of a Hydro- peroxo Heme Intermediate: Conversion of a Side-On Peroxo to an End-On Hydroperoxo Complex. Angew. Chem. Int. Ed 2009, 48, 9262–9267. [DOI] [PubMed] [Google Scholar]
- (491).Kieber-Emmons MT; Annaraj J; Seo MS; Van Heuvelen KM; Tosha T; Kitagawa T; Brunold TC; Nam W; Riordan CG Identification of an “End-On” Nickel-Superoxo Adduct, [Ni-(tmc)(O2)]+. J. Am. Chem. Soc 2006, 128, 14230–14231. [DOI] [PubMed] [Google Scholar]
- (492).Duerr K; Olah J; Davydov R; Kleimann M; Li J; Lang N; Puchta R; Hübner E; Drewello T; Harvey JN; et al. Studies on an Iron(III)-Peroxo Porphyrin. Iron(III)-Peroxo or iron(II)-Superoxo? Dalton Trans 2010, 39, 2049. [DOI] [PubMed] [Google Scholar]
- (493).Dürr K; Macpherson BP; Warratz R; Hampel F; Tuczek F; Helmreich M; Jux N; Ivanović-Burmazović I Iron(III) Complex of a Crown Ether-Porphyrin Conjugate and Reversible Binding of Superoxide to Its Iron(II) Form. J. Am. Chem. Soc 2007, 129, 4217–4228. [DOI] [PubMed] [Google Scholar]
- (494).Duerr K; Troeppner O; Olah J; Li J; Zahl A; Drewello T; Jux N; Harvey JN; Ivanovic-Burmazovic I Solution Behavior of Iron(III) and Iron(II) Porphyrins in DMSO and Reaction with Superoxide. Effect of Neighboring Positive Charge on Thermodynamics, Kinetics and Nature of Iron-(Su)peroxo Product. Dalton Trans 2012, 41, 546–557. [DOI] [PubMed] [Google Scholar]
- (495).Chufán EE; Karlin KD An Iron–Peroxo Porphyrin Complex: New Synthesis and Reactivity Toward a Cu(II) Complex Giving a Heme–Peroxo–Copper Adduct. J. Am. Chem. Soc 2003, 125, 16160–16161. [DOI] [PubMed] [Google Scholar]
- (496).Kopf M-A; Neuhold Y-M; Zuberbuhler AD; Karlin KD Oxo- and Hydroxo-Bridged Heme-Copper Assemblies Formed from Acid-Base or Metal-Dioxygen Chemistry. Inorg. Chem 1999, 38, 3093–3102. [Google Scholar]
- (497).Garcia-Bosch I; Adam SM; Schaefer AW; Sharma SK; Peterson RL; Solomon EI; Karlin KDA “Naked” FeIII-(O22-)- CuII Species Allows for Structural and Spectroscopic Tuning of Low-Spin Heme-Peroxo-Cu Complexes. J. Am. Chem. Soc 2015, 137, 1032–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Ghiladi RA; Kretzer RM; Guzei I; Rheingold AL; Neuhold Y-M; Hatwell KR; Zuberbuhler AD; Karlin KD (F8TPP)FeII/O2) Reactivity Studies [F8TPP = tetrakis(2,6-difluorophenyl)porphyrinate(2-)]: Spectroscopic (UV-Visible and NMR) and Kinetic Study of Solvent-Dependent (Fe/O2 = 1:1 or 2:1) Reversible O2-Reduction and Ferryl Formation. Inorg. Chem 2001, 40, 5754–5767. [DOI] [PubMed] [Google Scholar]
- (499).Collman JP; Brauman JI; Halbert TR; Suslick KS Nature of O2 and CO Binding to Metalloporphyrins and Heme Proteins. Proc. Natl. Acad. Sci. U. S. A 1976, 73, 3333–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Chin D-H; La Mar GN; Balch AL Mechanism of Autoxidation of iron(II) Porphyrins. Detection of a Peroxo-Bridged iron(III) Porphyrin Dimer and the Mechanism of Its Thermal Decomposition to the Oxo-Bridged iron(III) Porphyrin Dimer. J. Am. Chem. Soc 1980, 102, 4344–4350. [Google Scholar]
- (501).Shirazi A; Goff HM Characterization of Superoxide-Metalloporphyrin Reaction Products: Effective Use of Deuterium NMR Spectroscopy. J. Am. Chem. Soc 1982, 104, 6318–6322. [Google Scholar]
- (502).Karlin KD; Nanthakumar A; Fox S; Murthy NN; Ravi N; Huynh BH; Orosz RD; Day EP X-Ray Structure and Physical Properties of the Oxo-Bridged Complex [(F8-TPP)Fe-O-Cu(TMPA)]+, Fg-TPP = Tetrakis(2,6-difluorophenyl)-porphyrinate(2-), TMPA = Tris(2-Pyridylmethyl)amine: Modeling the Cytochrome c Oxidase Fe. J. Am. Chem. Soc 1994, 116, 4753–4763. [Google Scholar]
- (503).Schappacher M; Weiss R; Montiel-Montoya R; Trautwein A; Tabard A Formation of an Iron(IV)-Oxo “Picket-Fence” Porphyrin Derivative via Reduction of the Ferrous Dioxygen Adduct and Reaction with Carbon Dioxide. J. Am. Chem. Soc 1985, 107, 3736–3738. [Google Scholar]
- (504).Ghiladi RA; Karlin KD Low-Temperature UV-Visible and NMR Spectroscopic Investigations of O2 Binding to (6L)FeII, a Ferrous Heme Bearing Covalently Tethered Axial Pyridine Ligands. Inorg. Chem 2002, 41, 2400–2407. [DOI] [PubMed] [Google Scholar]
- (505).Ju TD; Ghiladi RA; Lee D-H; van Strijdonck GPF; Woods AS; Cotter RJ; Young VG; Karlin KD Dioxygen Reactivity of Fully Reduced [LFeII···CuI]+ Complexes Utilizing Tethered Tetraarylporphyrinates: Active Site Models for Heme-Copper Oxidases. Inorg. Chem 1999, 38, 2244–2245. [Google Scholar]
- (506).Kim E; Helton ME; Lu S; Moënne-Loccoz P; Incarvito CD; Rheingold AL; Kaderli S; Zuberbuhler AD; Karlin KD Tridentate Copper Ligand Influences on Heme-Peroxo-Copper Formation and Properties: Reduced, Superoxo, and μ-Peroxo Iron/Copper Complexes. Inorg. Chem 2005, 44, 7014–7029. [DOI] [PubMed] [Google Scholar]
- (507).Chufan EE; Puiu SC; Karlin KD Heme-Copper/Dioxygen Adduct Formation, Properties, and Reactivity. Acc. Chem. Res 2007, 40, 563–572. [DOI] [PubMed] [Google Scholar]
- (508).Liang H-C; Karlin KD; Dyson R; Kaderli S; Jung B; Zuberbuhler AD Dioxygen-Binding Kinetics and Thermodynamics of a Series of Dicopper(I) Complexes with Bis[2-(2-Pyridyl)ethyl]-amine Tridendate Chelators Forming Side-On Peroxo-Bridged Dicopper(II) Adducts. Inorg. Chem 2000, 39, 5884–5894. [DOI] [PubMed] [Google Scholar]
- (509).Zhang CX; Liang H-C; Kim E; Shearer J; Helton ME; Kim E; Kaderli S; Incarvito CD; Zuberbuhler AD; Rheingold AL; et al. Tuning Copper-Dioxygen Reactivity and Exogenous Substrate Oxidations via Alterations in Ligand Electronics. J. Am. Chem. Soc 2003, 125, 634–635. [DOI] [PubMed] [Google Scholar]
- (510).Pidcock E; DeBeer S; Obias HV; Hedman B; Hodgson KO; Karlin KD; Solomon EI A Study of Solid [{Cu-(MePY2)}2O2]2+ Using Resonance Raman and X-Ray Absorption Spectroscopies: An Intermediate Cu2O2 Core Structure or a Solid Solution? J. Am. Chem. Soc 1999, 121, 1870–1878. [Google Scholar]
- (511).Obias HV; Lin Y; Murthy NN; Pidcock E; Solomon EI; Ralle M; Blackburn NJ; Neuhold Y-M; Zuberbuhler AD; Karlin KD Peroxo−, Oxo−, and Hydroxo-Bridged Dicopper Complexes: Observation of Exogenous Hydrocarbon Substrate Oxidation. J. Am. Chem. Soc 1998, 120, 12960–12961. [Google Scholar]
- (512).Pidcock E; Obias HV; Abe M; Liang H-C; Karlin KD; Solomon EI Spectroscopic and Theoretical Studies of Oxygenated Dicopper(I) Complexes Containing Hydrocarbon-Linked Bis[2-(2-Pyridyl)ethyl]amine Units: Investigation of a Butterfly [Cu(μ-η2:η2)(O2)]2+ Core. J. Am. Chem. Soc 1999, 121, 1299–1308. [Google Scholar]
- (513).Kim E; Helton ME; Wasser IM; Karlin KD; Lu S; Huang H; Moenne-Loccoz P; Incarvito CD; Rheingold AL; Honecker M; et al. Superoxo, μ-Peroxo, and μ-Oxo Complexes from heme/O2 and Heme-Cu/O2 Reactivity: Copper Ligand Influences in Cytochrome c Oxidase Models. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3623–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Karlin KD; Qingfen G; Farooq A; Shuncheng L; Zubieta J Synthesis and X-Ray Crystal Structure of a Trinuclear copper(I) Cluster. Inorg. Chim. Acta 1989, 165, 37–39. [Google Scholar]
- (515).Karlin KD; Tyeklar Z; Farooq A; Haka MS; Ghosh P; Cruse RW; Gultneh Y; Hayes JC; Toscano PJ; Zubieta J Dioxygen-Copper Reactivity and Functional Modeling of Hemocya- nins. Reversible Binding of O2 and Carbon Monoxide to Dicopper(I) Complexes [CuI2(L)]2+ (L = Dinucleating Ligand) and the Structure of a Bis(carbonyl). Inorg. Chem 1992, 31, 1436–1451. [Google Scholar]
- (516).Chufán EE; Mondal B; Gandhi T; Kim E; Rubie ND; Moënne-Loccoz P; Karlin KD Reactivity Studies on FeIII–(O22-)–CuII Compounds: Influence of the Ligand Architecture and Copper Ligand Denticity. Inorg. Chem 2007, 46, 6382–6394. [DOI] [PubMed] [Google Scholar]
- (517).Berto TC; Praneeth VKK; Goodrich LE; Lehnert N Iron-Porphyrin NO Complexes with Covalently Attached N-Donor Ligands: Formation of a Stable Six-Coordinate Species in Solution. J. Am. Chem. Soc 2009, 131, 17116–17126. [DOI] [PubMed] [Google Scholar]
- (518).Sharma SK; Schaefer AW; Lim H; Matsumura H; Moënne-Loccoz P; Hedman B; Hodgson KO; Solomon EI; Karlin KD A Six-Coordinate Peroxynitrite Low-Spin Iron(III) Porphyrinate Complex–The Product of the Reaction of Nitrogen Monoxide (·NO(g)) with a Ferric-Superoxide Species. J. Am. Chem. Soc 2017, 139, 17421–17430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Thompson DW; Kretzer RM; Lebeau EL; Scaltrito DV; Ghiladi RA; Lam K-C; Rheingold AL; Karlin KD; Meyer GJ Synthesis, Characterization, and Laser Flash Photolysis Reactivity of a Carbonmonoxy Heme Complex. Inorg. Chem 2003, 42, 5211–5218. [DOI] [PubMed] [Google Scholar]
- (520).Silvernail NJ; Noll BC; Schulz CE; Scheidt WR Coordination of Diatomic Ligands to Heme: Simply CO. Inorg. Chem 2006, 45, 7050–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Silvernail NJ; Roth A; Schulz CE; Noll BC; Scheidt WR Heme Carbonyls: Environmental Effects on vc-o and Fe-C/C-O Bond Length Correlations. J. Am. Chem. Soc 2005, 127, 14422–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Kopf M-A; Karlin KD Dioxygen Reactivity of Reduced Heme and Heme-Copper Complexes Utilizing Tetraarylporphyrinates Tethered with Both a Pyridyl Axial Ligand and N,N-Bis[2-(2-Pyridyl)ethyl]amine Chelate. Inorg. Chem 1999, 38, 4922–4923. [DOI] [PubMed] [Google Scholar]
- (523).Obias HV; van Strijdonck GPF; Lee D-H; Ralle M; Blackburn NJ; Karlin KD Heterobinucleating Ligand-Induced Structural and Chemical Variations in [(L)FeIII–O–CuII]+ μ-Oxo Complexes. J. Am. Chem. Soc 1998, 120, 9696–9697. [Google Scholar]
- (524).Collman JP; Sunderland CJ; Berg KE; Vance MA; Solomon EI Spectroscopic Evidence for a Heme-Superoxide/Cu(I) Intermediate in a Functional Model of Cytochrome c Oxidase. J. Am. Chem. Soc 2003, 125, 6648–6649. [DOI] [PubMed] [Google Scholar]
- (525).Collman JP; Decréau RA; Sunderland CJ Single-Turnover Intermolecular Reaction Between a FeIII-Superoxide-CuI Cytochrome c Oxidase Model and Exogeneous Tyr244 Mimics. Chem. Commun 2006, 3894–3896. [DOI] [PubMed] [Google Scholar]
- (526).Collman JP; Berg KE; Sunderland CJ; Aukauloo A; Vance MA; Solomon EI Distal Metal Effects in Cobalt Porphyrins Related to CcO. Inorg. Chem 2002, 41, 6583–6596. [DOI] [PubMed] [Google Scholar]
- (527).Oliveira R; Zouari W; Herrero C; Banse F; Schollhorn B; Fave C; Anxolabehere-Mallart E Characterization and Subsequent Reactivity of an Fe-Peroxo Porphyrin Generated by Electrochemical Reductive Activation of O2. Inorg. Chem 2016, 55, 12204–12210. [DOI] [PubMed] [Google Scholar]
- (528).Dürr K; Jux N; Zahl A; van Eldik R; Ivanović-Burmazović I Volume Profile Analysis for the Reversible Binding of Superoxide to Form Iron(II)-Superoxo/Iron(III)-Peroxo Porphyrin Complexes. Inorg. Chem 2010, 49, 11254–11260. [DOI] [PubMed] [Google Scholar]
- (529).Selke M; Valentine JS Switching on the Nucleophilic Reactivity of a Ferric Porphyrin Peroxo Complex. J. Am. Chem. Soc 1998, 120, 2652–2653. [Google Scholar]
- (530).Nakamoto K; Watanabe T; Ama T; Urban MW Matrix Isolation Infrared Spectra of Oxy-tetraphenylporphyrinatoiron(II). J. Am. Chem. Soc 1982, 104, 3744–3745. [Google Scholar]
- (531).Watanabe T; Ama T; Nakamoto K Matrix-Isolation Infrared Spectra of Dioxygen Adducts of iron(II) Porphyrins and Related Compounds. J. Phys. Chem 1984, 88, 440–445. [Google Scholar]
- (532).Kincaid JR; Urban MW; Watanabe T; Nakamoto K Infrared Spectra of Matrix-Isolated Metal Complexes of Octaethyl-porphine. J. Phys. Chem. 1983, 87, 3096–3101. [Google Scholar]
- (533).Chang S; Blyholder G; Fernandez J Iron-Oxygen Interactions in an Argon Matrix. Inorg. Chem 1981, 20, 2813–2817. [Google Scholar]
- (534).Anderson JS; Gallagher AT; Mason JA; Harris TD A Five-Coordinate Heme Dioxygen Adduct Isolated within a Metal-Organic Framework. J. Am. Chem. Soc 2014, 136, 16489–16492. [DOI] [PubMed] [Google Scholar]
- (535).Kim E; Chufan EE; Kamaraj K; Karlin KD Synthetic Models for Heme-Copper Oxidases. Chem. Rev 2004, 104, 1077–1134. [DOI] [PubMed] [Google Scholar]
- (536).Balch AL The Reactivity of Spectroscopically Detected Peroxy Complexes of Iron Porphyrins. Inorg. Chim. Acta 1992, 198–200, 297–307. [Google Scholar]
- (537).Miksztal AR; Valentine JS Reactivity of the Peroxo Ligand in Metalloporphyrin Complexes. Reaction of Sulfur Dioxide with Iron and Titanium Porphyrin Peroxo Complexes to Give Sulfato Complexes of Sulfate. Inorg. Chem 1984, 23, 3548–3552. [Google Scholar]
- (538).Ibrahim M; Kincaid J R Spectroscopic Studies of Peroxo/Hydroperoxo Derivatives of Heme Proteins and Model Compounds. J. Porphyrins Phthalocyanines 2004, 08, 215–225. [Google Scholar]
- (539).McCandlish E; Miksztal AR; Nappa M; Sprenger AQ; Valentine JS; Stong JD; Spiro TG Reactions of Superoxide with Iron Porphyrins in Aprotic Solvents. A High Spin Ferric Porphyrin Peroxo Complex. J. Am. Chem. Soc 1980, 102, 4268–4271. [Google Scholar]
- (540).Burstyn JN; Roe JA; Miksztal AR; Shaevitz BA; Lang G; Valentine JS Magnetic and Spectroscopic Characterization of an Iron Porphyrin Peroxide Complex. Peroxoferrioctaethylporphyrin(1-). J.Am. Chem. Soc 1988, 110, 1382–1388. [Google Scholar]
- (541).Welborn CH; Dolphin D; James BR One-Electron Electrochemical Reduction of a Ferrous Porphyrin Dioxygen Complex. J. Am. Chem. Soc 1981, 103, 2869–2871. [Google Scholar]
- (542).Friant P; Goulon J; Fischer J; Ricard L; Schappacher M; Weiss R; Momenteau M Structural Investigation by EXAFS of Peroxo-Titanium(IV) and Iron(III) Porphyrinates. Nouv. J. Chim 1985, 9, 33–40. [Google Scholar]
- (543).Regen SL; Whitesides GM Reactions of Transition Metal Peroxides with N-Butyllithium. J. Organomet. Chem 1973, 59, 293–297. [Google Scholar]
- (544).Watanabe Y; Ishimura Y A Model Study on Aromatase Cytochrome P-450 Reaction: Transformation of Androstene-3,17,19-Trione to 10β-Hydroxyester-4-Ene-3,17-Dione. J. Am. Chem. Soc 1989, 111, 8047–8049. [Google Scholar]
- (545).Watanabe Y; Ishimura Y Aromatization of Tetralone Derivatives by FeIIIPFP(Cl)/PhIO and Cytochrome P-450cam: A Model Study on Aromatase Cytochrome P-450 Reaction. J. Am. Chem. Soc 1989, 111, 410–411. [Google Scholar]
- (546).Sheldon RA; Van Doorn JA Cycloaddition of Group VIII Metal-Dioxygen Complexes to Electrophilic Olefins. J. Organomet. Chem 1975, 94, 115–129. [Google Scholar]
- (547).Sisemore MF; Burstyn JN; Valentine JS Epoxidation of Electron-Deficient Olefins by a Nucleophilic Iron(III) Peroxo Porphyrinato Complex, Peroxo(tetramesitylporphyrinato)-ferrate(1–). Angew. Chem. Int. Ed. Engl 1996, 35, 206–208. [Google Scholar]
- (548).Selke M; Sisemore MF; Valentine JS The Diverse Reactivity of Peroxy Ferric Porphyrin Complexes of Electron-Rich and Electron-Poor Porphyrins. J. Am. Chem. Soc 1996, 118, 2008–2012. [Google Scholar]
- (549).Sisemore MF; Selke M; Burstyn JN; Valentine JS Metalloporphyrin Peroxo Complexes of Iron(III), Manganese(III), and Titanium(IV). Comparative Studies Demonstrating That the Iron(III) Complex Is Extremely Nucleophilic. Inorg. Chem 1997, 36, 979–984. [DOI] [PubMed] [Google Scholar]
- (550).Wertz DL; Sisemore MF; Selke M; Driscoll J; Valentine JS Mimicking Cytochrome P-450 2B4 and Aromatase: Aromatization of a Substrate Analogue by a Peroxo Fe(III) Porphyrin Complex. J. Am. Chem. Soc 1998, 120, 5331–5332. [Google Scholar]
- (551).Goto Y; Wada S; Morishima I; Watanabe Y Reactivity of Peroxoiron(III) Porphyrin Complexes: Models for Deformylation Reactions Catalyzed by Cytochrome P-450. J. Inorg. Biochem 1998, 69, 241–247. [Google Scholar]
- (552).Khenkin AM; Shteinman AA The Mechanism of Oxidation of Alkanes by Peroxo Complexes of Iron Porphyrins in the Presence of Acylating Agents: A Model for Activation of O2 by Cytochrome P-450. J. Chem. Soc. Chem. Commun 1984, 1219–1220. [Google Scholar]
- (553).Mandon D; Weiss R; Franke M; Bill E; Trautwein AX Oxoiron Porphyrin Species with High-Valent Iron: Formation by Solvent-Dependent Protonation of a Peroxoiron(III) Porphyrinate Derivative. Angew. Chem. Int. Ed. Engl 1989, 28, 1709–1711. [Google Scholar]
- (554).Groves JT; Krishnan S; Avaria GE; Nemo TE Studies of the Mechanism of Oxygen Activation and Transfer Catalyzed by Cytochrome P 450. In Biomimetic Chemistry; American Chemical Society, 1980; Vol. 191, pp 277–289. [Google Scholar]
- (555).Groves JT; Watanabe Y Oxygen Activation by Metal-loporphyrins Related to Peroxidase and Cytochrome P-450. Direct Observation of the Oxygen-Oxygen Bond Cleavage Step. J. Am. Chem. Soc 1986, 108, 7834–7836. [DOI] [PubMed] [Google Scholar]
- (556).Tajima K; Ishizu K; Sakurai H; Ohya-Nishiguchi H A Possible Model of Hemoprotein-Peroxide Complexes Formed in an Iron-Tetraphenylporphyrin System. Biochem. Biophys. Res. Commun 1986, 135, 972–978. [DOI] [PubMed] [Google Scholar]
- (557).Tajima K; Jinno J; Ishizu K; Sakurai H; Ohya-Nishiguchi H Direct Evidence of Heme-Tert-Butyl Peroxide Adduct Formation Demonstrated by Simultaneous ESR and Optical Measurements. Inorg. Chem 1989, 28, 709–715. [Google Scholar]
- (558).Zipplies MF; Lee WA; Bruice TC Influence of Hydrogen Ion Activity and General Acid-Base Catalysis on the Rate of Decomposition of Hydrogen Peroxide by a Novel Nonaggregating Water-Soluble iron(III) Tetraphenylporphyrin Derivative. J. Am. Chem. Soc 1986, 108, 4433–4445. [Google Scholar]
- (559).Tajima K A Possible Model of a Hemoprotein-Hydrogen Peroxide Complex. Inorg. Chim. Acta 1989, 163, 115–122. [Google Scholar]
- (560).Gaffney BJ High Resolution EPR; Berliner L, Hanson G, Eds.; Biological Magnetic Resonance; Springer: New York, NY, 2009; Vol. 28. [Google Scholar]
- (561).Rotilio G; Federici G; Calabrese L; Costa M; Cavallini D An Electron Paramagnetic Resonance Study of the Nonheme Iron of Cysteamine Oxygenase. J. Biol. Chem 1970, 245, 6235–6236. [PubMed] [Google Scholar]
- (562).Chasteen ND; Grady JK; Skorey KI; Neden KJ; Riendeau D; Percival MD Characterization of the Non-Heme Iron Center of Human 5-Lipoxygenase by Electron Paramagnetic Resonance, Fluorescence, and Ultraviolet-Visible Spectroscopy: Redox Cycling between Ferrous and Ferric States. Biochemistry 1993, 32, 9763–9771. [DOI] [PubMed] [Google Scholar]
- (563).Tajima K; Shigematsu M; Jinno J; Ishizu K; Ohya-Nishiguchi H Generation of FeIIIOEP-Hydrogen Peroxide Complex (OEP = Octaethylporphyrinato) by Reduction of FeIIOEP-O2 with Ascorbic Acid Sodium Salt. J. Chem. Soc., Chem. Commun 1990, 144–145. [Google Scholar]
- (564).Tajima K; Oka S; Edo T; Miyake S; Mano H; Mukai K; Sakurai H; Ishizu K Optical Absorption and EPR Studies on a Six- Coordinate Iron(III)-Tetramesitylporphyrin-Hydrogen Peroxide Complex Having a Nitrogenous Axial Ligand. J. Chem. Soc., Chem. Commun 1995, 0, 1507–1508. [Google Scholar]
- (565).Chang CJ; Chng LL; Nocera DG Proton-Coupled O–O Activation on a Redox Platform Bearing a Hydrogen-Bonding Scaffold. J. Am. Chem. Soc 2003, 125, 1866–1876. [DOI] [PubMed] [Google Scholar]
- (566).Rosenthal J; Chng LL; Fried SD; Nocera DG Stereochemical Control of H2O2 Dismutation by Hangman Porphyrins. Chem. Commun 2007, 2642–2644. [DOI] [PubMed] [Google Scholar]
- (567).Soper JD; Kryatov SV; Rybak-Akimova EV; Nocera DG Proton-Directed Redox Control of O–O Bond Activation by Heme Hydroperoxidase Models. J. Am. Chem. Soc 2007, 129, 5069–5075. [DOI] [PubMed] [Google Scholar]
- (568).Nagaraju P; Ohta T; Liu J-G; Ogura T; Naruta Y The Secondary Coordination Sphere Controlled Reactivity of a Ferric-Superoxo Heme: Unexpected Conversion to a Ferric Hydroperoxo Intermediate by Reaction with a High-Spin Ferrous Heme. Chem. Commun 2016, 52, 7213–7216. [DOI] [PubMed] [Google Scholar]
- (569).de Visser SP; Valentine JS; Nam W A Biomimetic Ferric Hydroperoxo Porphyrin Intermediate. Angew. Chem., Int. Ed 2010, 49, 2099–2101. [DOI] [PubMed] [Google Scholar]
- (570).Unno M; Chen H; Kusama S; Shaik S; Ikeda-Saito M Structural Characterization of the Fleeting Ferric Peroxo Species in Myoglobin: Experiment and Theory. J. Am. Chem. Soc 2007, 129, 13394–13395. [DOI] [PubMed] [Google Scholar]
- (571).Hersleth H-P; Hsiao Y-W; Ryde U; Görbitz CH; Andersson KK The Crystal Structure of Peroxymyoglobin Generated through Cryoradiolytic Reduction of Myoglobin Compound III during Data Collection. Biochem. J 2008, 412, 257–264. [DOI] [PubMed] [Google Scholar]
- (572).Leibl W; Nitschke W; Huttermann J Spin-Density Distribution in the [FeO2]− Complex. Electron Spin Resonance of Myoglobin Single Crystals. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1986, 870, 20–30. [DOI] [PubMed] [Google Scholar]
- (573).Mak PJ; Denisov IG; Victoria D; Makris TM; Deng T; Sligar SG; Kincaid JR Resonance Raman Detection of the Hydroperoxo Intermediate in the Cytochrome P450 Enzymatic Cycle. J. Am. Chem. Soc 2007, 129, 6382–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR Resonance Raman Characterization of the Peroxo and Hydroperoxo Intermediates in Cytochrome P450. J. Phys. Chem. A 2008, 112, 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (575).Ogliaro F; de Visser SP; Cohen S; Sharma PK; Shaik S Searching for the Second Oxidant in the Catalytic Cycle of Cytochrome P450: A Theoretical Investigation of the Iron(III)-Hydroperoxo Species and Its Epoxidation Pathways. J. Am. Chem. Soc 2002, 124, 2806–2817. [DOI] [PubMed] [Google Scholar]
- (576).Sharma PK; de Visser SP; Shaik S Can a Single Oxidant with Two Spin States Masquerade as Two Different Oxidants? A Study of the Sulfoxidation Mechanism by Cytochrome P450. J. Am. Chem. Soc 2003, 125, 8698–8699. [DOI] [PubMed] [Google Scholar]
- (577).Kamachi T; Shiota Y; Ohta T; Yoshizawa K Does the Hydroperoxo Species of Cytochrome P450 Participate in Olefin Epoxidation with the Main Oxidant, Compound I? Criticism from Density Functional Theory Calculations. Bull. Chem. Soc. Jpn 2003, 76, 721–732. [Google Scholar]
- (578).Park MJ; Lee J; Suh Y; Kim J; Nam W Reactivities of Mononuclear Non-Heme Iron Intermediates Including Evidence That Iron(III)-Hydroperoxo Species Is a Sluggish Oxidant. J. Am. Chem. Soc 2006, 128, 2630–2634. [DOI] [PubMed] [Google Scholar]
- (579).Seo MS; Kamachi T; Kouno T; Murata K; Park MJ; Yoshizawa K; Nam W Experimental and Theoretical Evidence for Nonheme Iron(III) Alkylperoxo Species as Sluggish Oxidants in Oxygenation Reactions. Angew. Chem., Int. Ed 2007, 46, 2291–2294. [DOI] [PubMed] [Google Scholar]
- (580).Han A-R; Jin Jeong Y; Kang Y; Yoon Lee J; Sook Seo M; Nam W Direct Evidence for an iron(IV)-Oxo Porphyrin π-Cation Radical as an Active Oxidant in Catalytic Oxygenation Reactions. Chem. Commun 2008, 1076. [DOI] [PubMed] [Google Scholar]
- (581).Wang B; Li C; Cho K-B; Nam W; Shaik S The FeIII(H2O2) Complex as a Highly Efficient Oxidant in Sulfoxidation Reactions: Revival of an Underrated Oxidant in Cytochrome P450. J. Chem. Theory Comput 2013, 9, 2519–2525. [DOI] [PubMed] [Google Scholar]
- (582).Gross Z; Nimri S A Pronounced Axial Ligand Effect on the Reactivity of Oxoiron(IV) Porphyrin Cation Radicals. Inorg. Chem 1994, 33, 1731–1732. [Google Scholar]
- (583).Nam W; Lim MH; Oh S-Y; Lee JH; Lee HJ; Woo SK; Kim C; Shin W Remarkable Anionic Axial Ligand Effects of Iron(III) Porphyrin Complexes on the Catalytic Oxygenations of Hydrocarbons by H2O2 and the Formation of Oxoiron(IV) Porphyrin Intermediates by M-Chloroperoxybenzoic Acid. Angew. Chem 2000, 112, 3792–3795. [DOI] [PubMed] [Google Scholar]
- (584).Nam W; Jin SW; Lim MH; Ryu JY; Kim C Anionic Ligand Effect on the Nature of Epoxidizing Intermediates in Iron Porphyrin Complex-Catalyzed Epoxidation Reactions. Inorg. Chem 2002, 41, 3647–3652. [DOI] [PubMed] [Google Scholar]
- (585).Franke A; Fertinger C; van Eldik R Axial Ligand and Spin- State Influence on the Formation and Reactivity of Hydroperoxo-Iron(III) Porphyrin Complexes. Chem. −3 Eur. J 2012, 18, 6935–6949. [DOI] [PubMed] [Google Scholar]
- (586).Bill E; Ding X-Q; Bominaar EL; Trautwein AX; Winkler H; Mandon D; Weiss R; Gold A; Jayaraj K; Hatfield WE; et al. Evidence for Variable Metal-Radical Spin Coupling in Oxoferrylporphyrin Cation Radical Complexes. Eur. J. Biochem 1990, 188, 665–672. [DOI] [PubMed] [Google Scholar]
- (587).Newcomb M; Shen R; Choi S-Y; Toy PH; Hollenberg PF; Vaz ADN; Coon MJ Cytochrome P450-Catalyzed Hydroxylation of Mechanistic Probes That Distinguish between Radicals and Cations. Evidence for Cationic but Not for Radical Intermediates. J. Am. Chem. Soc 2000, 122, 2677–2686. [Google Scholar]
- (588).Newcomb M; Aebisher D; Shen R; Chandrasena REP; Hollenberg PF; Coon MJ Kinetic Isotope Effects Implicate Two Electrophilic Oxidants in Cytochrome P450-Catalyzed Hydroxyla- tions. J. Am. Chem. Soc 2003, 125, 6064–6065. [DOI] [PubMed] [Google Scholar]
- (589).Bruice TC Chemical Studies Pertaining to the Chemistry of Cytochrome P-450 and the Peroxidases. Ann. N. Y. Acad. Sci 1986, 471, 83–98. [DOI] [PubMed] [Google Scholar]
- (590).Murata K; Panicucci R; Gopinath E; Bruice TC The Reaction of 5,10,15,20-tetrakis(2,6-Dichloro-3-sulfonatophenyl)-porphinatoiron(III) Hydrate with Alkyl and Acyl Hydroperoxides. The Dynamics of Reaction of Water-Soluble and Non μ-Oxo Dimer Forming Iron(III) Porphyrins in Aqueous Solution. J. Am. Chem. Soc 1990, 112, 6072–6083. [Google Scholar]
- (591).Gopinath E; Bruice TC Dynamics of Reaction of [Meso-tetrakis(2,6-Dimethyl-3-Sulfonatophenyl)porphinato]-Iron(III) Hydrate with Various Alkyl Hydroperoxides in Aqueous Solution. Comparison of Kinetic Parameters and D2O Solvent Isotope Effects. J. Am. Chem. Soc 1991, 113, 6090–6094. [Google Scholar]
- (592).Almarsson O; Bruice TC A Homolytic Mechanism of O–O Bond Scission Prevails in the Reactions of Alkyl Hydroperoxides with an Octacationic Tetraphenylporphinato-Iron(III) Complex in Aqueous Solution. J. Am. Chem. Soc 1995, 117, 4533–4544. [Google Scholar]
- (593).Traylor TG; Kim C; Fann W-P; Perrin CL Reactions of Hydroperoxides with iron(III) Porphyrins: Heterolytic Cleavage Followed by Hydroperoxide Oxidation. Tetrahedron 1998, 54, 7977–7986. [Google Scholar]
- (594).Traylor TG; Lee WA; Stynes DV Model Compound Studies Related to Peroxidases. Mechanisms of Reactions of Hemins with Peracids. J. Am. Chem. Soc 1984, 106, 755–764. [Google Scholar]
- (595).Balasubramanian PN; Lee RW; Bruice TC Reaction of [Meso-tetrakis(2,6-Dimethyl-3-sulfonatophenyl)porphinato]iron(III) Hydrate with Various Acyl and Alkyl Hydroperoxides in Aqueous Solution. J. Am. Chem. Soc 1989, 111, 8714–8721. [Google Scholar]
- (596).Bruice TC; Balasubramanian PN; Lee RW; Smith JRL The Mechanism of Hydroperoxide Oxygen-Oxygen Bond Scission on Reaction of Hydroperoxides with iron(III) Porphyrins. J. Am. Chem. Soc 1988, 110, 7890–7892. [Google Scholar]
- (597).Nam W; Choi HJ; Han HJ; Cho SH; Lee HJ; Han S-Y Use of 2-Methyl-1-Phenylpropan-2-Yl Hydroperoxide (MPPH) as a Mechanistic Probe for the Heterolytic versus Homolytic O–O Bond Cleavage of Tert-Alkyl Hydroperoxide by Iron(III) Porphyrin Complex. Chem. Commun 1999, 387–388. [Google Scholar]
- (598).Yang SJ; Nam W Water-Soluble Iron Porphyrin Complex-Catalyzed Epoxidation of Olefins with Hydrogen Peroxide and Tert- Butyl Hydroperoxide in Aqueous Solution. Inorg. Chem 1998, 37, 606–607. [Google Scholar]
- (599).Traylor TG; Kim C; Richards JL; Xu F; Perrin CL Reactions of Iron(III) Porphyrins with Oxidants. Structure-Reactivity Studies. J. Am. Chem. Soc 1995, 117, 3468–3474. [Google Scholar]
- (600).Lee YJ; Goh YM; Han S-Y; Kim C; Nam W Epoxidation of Olefins with H2O2 Catalyzed by an Electronegatively-Substituted Iron Porphyrin Complex in Aprotic Solvent. Chem. Lett 1998, 27, 837–838. [Google Scholar]
- (601).Bartoli JF; Battioni P; De Foor WR; Mansuy D Synthesis and Remarkable Properties of Iron β-Polynitroporphyrins as Catalysts for Monooxygenation Reactions. J. Chem. Soc., Chem. Commun 1994, 23–24. [Google Scholar]
- (602).Wolak M; van Eldik R Mechanistic Studies on Peroxide Activation by a Water-Soluble Iron(III)-Porphyrin: Implications for O–O Bond Activation in Aqueous and Nonaqueous Solvents. Chem. −3 Eur. J 2007, 13, 4873–4883. [DOI] [PubMed] [Google Scholar]
- (603).Liu M; Su YO Selective Electrocatalysis of Alkene Oxidations in Aqueous Media. Electrochemical and Spectral Characterization of Oxo-Ferryl Porphyrin, Oxo-Ferryl Porphyrin Radical Cation and Their Reaction Products with Alkenes at Room Temperature. J. Electroanal. Chem 1998, 452, 113–125. [Google Scholar]
- (604).Franke A; Wolak M; van Eldik R Factors That Affect the Nature of the Final Oxidation Products in “Peroxo-Shunt” Reactions of Iron-Porphyrin Complexes. Chem. −3 Eur. J 2009, 15, 10182–10198. [DOI] [PubMed] [Google Scholar]
- (605).Oszajca M; Drzewiecka-Matuszek A; Franke A; Rutkowska-Zbik D; Brindell M; Witko M; Stochel G; van Eldik R Mechanistic Insight into Peroxo-Shunt Formation of Biomimetic Models for Compound II, Their Reactivity toward Organic Substrates, and the Influence of N −3Methylimidazole Axial Ligation. Chem. −3 Eur. J 2014, 20, 2328–2343. [DOI] [PubMed] [Google Scholar]
- (606).Stochel G; van Eldik R Elucidation of Inorganic Reaction Mechanisms through Volume Profile Analysis. Coord. Chem. Rev 1999, 187, 329–374. [Google Scholar]
- (607).Oszajca M; Franke A; Drzewiecka-Matuszek A; Brindell M; Stochel G; van Eldik R Temperature and Pressure Effects on C-H Abstraction Reactions Involving Compound I and II Mimics in Aqueous Solution. Inorg. Chem 2014, 53, 2848–2857. [DOI] [PubMed] [Google Scholar]
- (608).Keown W; Gary JB; Stack TD P. High-Valent Copper in Biomimetic and Biological Oxidations. JBIQ J. Biol. Inorg. Chem 2017, 22, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (609).Gagnon N; Tolman WB [CuO]+ and [CuOH]2+ Complexes: Intermediates in Oxidation Catalysis? Acc. Chem. Res 2015, 48, 2126–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (610).Itoh S Developing Mononuclear Copper-Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reactions. Acc. Chem. Res 2015, 48, 2066–2074. [DOI] [PubMed] [Google Scholar]
- (611).Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of Dioxygen by Copper Metalloproteins and Insights from Model Complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (613).Choi M; Davidson VL Cupredoxins – A Study of How Proteins May Evolve to Use Metals for Bioenergetic Processes. Metallomics 2011, 3, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Solomon EI; Szilagyi RK; DeBeer George S; Basumallick L Electronic Structures of Metal Sites in Proteins and Models : Contributions to Function in Blue Copper Proteins. Chem. Rev 2004, 104, 419–458. [DOI] [PubMed] [Google Scholar]
- (615).Wilmot CM Visualization of Dioxygen Bound to Copper During Enzyme Catalysis. Science 1999, 286, 1724–1728. [DOI] [PubMed] [Google Scholar]
- (616).Ito N; Phillips SEV; Stevens C; Ogel ZB; McPherson MJ; Keen JN; Yadav KDS; Knowles PF Novel Thioether Bond Revealed by a 1.7 A Crystal Structure of Galactose Oxidase. Nature 1991, 350, 87–90. [DOI] [PubMed] [Google Scholar]
- (617).Rokhsana D; Shepard EM; Brown DE; Dooley DM Amine Oxidase and Galactose Oxidase In Copper-Oxygen Chemistry; Itoh KDKS, Ed.; John Wiley & Sons, Inc, 2011; pp 53–106. [Google Scholar]
- (618).Pintus F; Sabatucci A; Maccarrone M; Dainese E; Medda R Amine Oxidase from Euphorbia Charadas: Kinetic and Structural Characterization. Biotechnol. Appl Biochem 2018, 65, 81. [DOI] [PubMed] [Google Scholar]
- (619).Johnson BJ; Yukl ET; Klema VJ; Klinman JP; Wilmot CM Structural Snapshots from the Oxidative Half-Reaction of a Copper Amine Oxidase. J. Biol. Chem 2013, 288, 28409–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (620).Liu Y; Mukherjee A; Nahumi N; Ozbil M; Brown D; Angeles-Boza AM; Dooley DM; Prabhakar R; Roth JP Experimental and Computational Evidence of Metal-O2 Activation and Rate-Limiting Proton-Coupled Electron Transfer in a Copper Amine Oxidase. J. Phys. Chem. B 2013, 117, 218–229. [DOI] [PubMed] [Google Scholar]
- (621).Shepard EM; Dooley DM Inhibition and Oxygen Activation in Copper Amine Oxidases. Acc. Chem. Res 2015, 48, 1218–1226. [DOI] [PubMed] [Google Scholar]
- (622).Rao G; Bansal S; Law WX; O’Dowd B; Dikanov SA; Oldfield E Pulsed Electron Paramagnetic Resonance Insights into the Ligand Environment of Copper in Drosophila Lysyl Oxidase. Biochemistry 2017, 56, 3770–3779. [DOI] [PubMed] [Google Scholar]
- (623).Klinman JP The Copper-Enzyme Family of Dopamine Beta-Monooxygenase and Peptidylglycine Alpha-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J. Biol. Chem 2006, 281, 3013–3016. [DOI] [PubMed] [Google Scholar]
- (624).Prigge ST; Eipper B; Mains R; Amzel LM Dioxygen Binds End-On to Mononuclear Copper in a Precatalytic Enzyme Complex. Science 2004, 304, 864–867. [DOI] [PubMed] [Google Scholar]
- (625).Cowley RE; Tian L; Solomon EI Mechanism of O2 Activation and Substrate Hydroxylation in Noncoupled Binuclear Copper Monooxygenases. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 12035–12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (626).Rudzka K; Moreno DM; Eipper B; Mains R; Estrin DA; Amzel LM Coordination of Peroxide to the CuM Center of Peptidylglycine Alpha-Hydroxylating Monooxygenase (PHM): Structural and Computational Study. JBIQ J. Biol. Inorg. Chem 2013, 18, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (627).Holm RH; Solomon EI Introduction: Bioinorganic Enzymology II. Chem. Rev. 2014, 114, 4039–4040. [DOI] [PubMed] [Google Scholar]
- (628).Chufan EE; Prigge ST; Siebert X; Eipper BA; Mains RE; Amzel LM Differential Reactivity between Two Copper Sites in Peptidylglycine Alpha-Hydroxylating Monooxygenase. J. Am. Chem. Soc 2010, 132, 15565–15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (629).Himes RA; Park GY; Sutha Siluvai G; Blackburn NJ; Karlin KD Structural Studies of Copper(I) Complexes of Amyloid-β Peptide Fragments: Formation of Two-Coordinate Bis(histidine) Complexes. Angew. Chem., Int. Ed 2008, 47, 9084–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (630).Shearer J; Szalai VA The Amyloid-β Peptide of Alzheimer’s Disease Binds CuI in a Linear Bis-His Coordination Environment: Insight into a Possible Neuroprotective Mechanism for the Amyloid-β Peptide. J. Am. Chem. Soc 2008, 130, 17826–17835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Conklin SE; Bridgman EC; Su Q; Riggs-Gelasco P; Haas KL; Franz KJ Specific Histidine Residues Confer Histatin Peptides with Copper-Dependent Activity against Candida Albicans. Biochemistry 2017, 56, 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (632).Haas KL; Putterman AB; White DR; Thiele DJ; Franz KJ Model Peptides Provide New Insights into the Role of Histidine Residues as Potential Ligands in Human Cellular Copper Acquisition via Ctr1. J. Am. Chem. Soc 2011, 133, 4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (633).Himes RA; Park GY; Barry AN; Blackburn NJ; Karlin KD Synthesis and X-Ray Absorption Spectroscopy Structural Studies of Cu(I) Complexes of HistidylHistidine Peptides: The Predominance of Linear 2-Coordinate Geometry. J. Am. Chem. Soc 2007, 129, 5352–5353. [DOI] [PubMed] [Google Scholar]
- (634).Gaggelli E; Kozlowski H; Valensin D; Valensin G Copper Homeostasis and Neurodegenerative Disorders (Alzheimer’s, Prion, and Parkinson’s Diseases and Amyotrophic Lateral Sclerosis). Chem. Rev 2006, 106, 1995–2044. [DOI] [PubMed] [Google Scholar]
- (635).Kline CD; Mayfield M; Blackburn NJ HHM Motif at the CuH-Site of Peptidylglycine Monooxygenase Is a pH-Dependent Conformational Switch. Biochemistry 2013, 52, 2586–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (636).Jaron S; Mains RE; Eipper BA; Blackburn NJ The Catalytic Role of the Copper Ligand H172 of Peptidylglycine α-Hydroxylating Monooxygenase (PHM): A Spectroscopic Study of the H172A Mutant. Biochemistry 2002, 41, 13274–13282. [DOI] [PubMed] [Google Scholar]
- (637).Blackburn NJ; Rhames FC; Ralle M; Jaron S Major Changes in Copper Coordination Accompany Reduction of Peptidylglycine Monooxygenase: Implications for Electron Transfer and the Catalytic Mechanism. JBIQ J. Biol. Inorg. Chem 2000, 5, 341–353. [DOI] [PubMed] [Google Scholar]
- (638).Hemsworth GR; Johnston EM; Davies GJ; Walton PH Lytic Polysaccharide Monooxygenases in Biomass Conversion. Trends Biotechnol. 2015, 33, 747–761. [DOI] [PubMed] [Google Scholar]
- (639).Li X; Beeson WT; Phillips CM; Marletta MA; Cate JHD Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure 2012, 20, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Chaplin AK; Wilson MT; Hough MA; Svistunenko DA; Hemsworth GR; Walton PH; Vijgenboom E; Worrall JAR Heterogeneity in the Histidine-Brace Copper Coordination Sphere in Auxiliary Activity Family 10 (AA10) Lytic Polysaccharide Monooxygenases. J. Biol. Chem 2016, 291, 12838–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (641).Bacik J-P; Mekasha S; Forsberg Z; Kovalevsky AY; Vaaje-Kolstad G; Eijsink VGH; Nix JC; Coates L; Cuneo MJ; Unkefer CJ; et al. Neutron and Atomic Resolution X-Ray Structures of a Lytic Polysaccharide Monooxygenase Reveal CopperMediated Dioxygen Binding and Evidence for N-Terminal Deprotonation. Biochemistry 2017, 56, 2529–2532. [DOI] [PubMed] [Google Scholar]
- (642).Meier KK; Jones SM; Kaper T; Hansson H; Koetsier MJ; Karkehabadi S; Solomon EI; Sandgren M; Kelemen B Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chem. Rev 2018, 118, 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (643).Frandsen KEH; Simmons TJ; Dupree P; Poulsen J-CN; Hemsworth GR; Ciano L; Johnston EM; Tovborg M; Johansen KS; von Freiesleben P; et al. The Molecular Basis of Polysaccharide Cleavage by Lytic Polysaccharide Monooxygenases. Nat. Chem. Biol 2016, 12, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (644).Span EA; Suess DLM; Deller MC; Britt RD; Marletta MA The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem. Biol 2017, 12, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (645).O’Dell WB; Agarwal PK; Meilleur F Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angew. Chem., Int. Ed 2017, 56, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (646).Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)-Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (647).Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (648).Kim S; StÅhlberg J; Sandgren M; Paton RS; Beckham GT Quantum Mechanical Calculations Suggest That Lytic Polysaccharide Monooxygenases Use a Copper-Oxyl, Oxygen-Rebound Mechanism. Proc. Natl. Acad. Sci U. S. A 2014, 111, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (649).Kjaergaard CH; Qayyum MF; Wong SD; Xu F; Hemsworth GR; Walton DJ; Young NA; Davies GJ; Walton PH; Johansen KS; et al. Spectroscopic and Computational Insight into the Activation of O2 by the Mononuclear Cu Center in Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 8797–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (650).Bissaro B; Røhr ÅK; Müller G; Chylenski P; Skaugen M; Forsberg Z; Horn SJ; Vaaje-Kolstad G; Eijsink VGH Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol 2017, 13, 1123–1128. [DOI] [PubMed] [Google Scholar]
- (651).Wang B; Johnston EM; Li P; Shaik S; Davies GJ; Walton PH; Rovira C QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal. 2018, 8, 1346–1351. [Google Scholar]
- (652).Kuusk S; Bissaro B; Kuusk P; Forsberg Z; Eijsink VGH; Sørlie M; Väljamäe P Kinetics of H2O2-Driven Degradation of Chitin by a Bacterial Lytic Polysaccharide Monooxygenase. J. Biol. Chem 2018, 293, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (653).Hangasky JA; Iavarone AT; Marletta MA Reactivity of O2 versus H2O2 with Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. U.SA 2018, 115 (19), 4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Culpepper MA; Rosenzweig AC Architecture and Active Site of Particulate Methane Monooxygenase. Crit. Rev. Biochem. Mol. Biol 2012, 47, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Ross MO; Rosenzweig AC A Tale of Two Methane Monooxygenases. JBIC, J. Biol. Inorg. Chem 2017, 22, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (656).Cao L; Caldararu O; Rosenzweig AC; Ryde U Quantum Refinement Does Not Support Dinuclear Copper Sites in Crystal Structures of Particulate Methane Monooxygenase. Angew. Chem. Int. Ed 2018, 57, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Wang VC-C; Maji S; Chen PP-Y; Lee HK; Yu SS-F; Chan SI Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev 2017, 117, 8574–8621. [DOI] [PubMed] [Google Scholar]
- (658).Tinberg CE; Lippard SJ Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res 2011, 44, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (659).Peng J; Alam S; Radhakrishnan K; Mariappan M; Rudolph MG; May C; Dierks T; von Figura K; Schmidt B Eukaryotic Formylglycine-Generating Enzyme Catalyses a Monooxygenase Type of Reaction. FEBS J. 2015, 282, 3262–3274. [DOI] [PubMed] [Google Scholar]
- (660).Knop M; Dang TQ; Jeschke G; Seebeck FP Copper Is a Cofactor of the Formylglycine-Generating Enzyme. ChemBioChem 2017, 18, 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (661).Meury M; Knop M; Seebeck FP Structural Basis for Copper-Oxygen Mediated C-H Bond Activation by the Formylglycine-Generating Enzyme. Angew. Chem. Int. Ed 2017, 56, 8115–8119. [DOI] [PubMed] [Google Scholar]
- (662).Pretzler M; Rompel A What Causes the Different Functionality in Type-III-Copper Enzymes? A State of the Art Perspective. Inorg. Chim. Acta 2017, DOI: 10.1016/j.ica.2017.04.041. [DOI] [Google Scholar]
- (663).Gerdemann C; Eicken C; Krebs B The Crystal Structure of Catechol Oxidase: New Insight into the Function of Type-3 Copper Proteins. Acc. Chem. Res 2002, 35, 183–191. [DOI] [PubMed] [Google Scholar]
- (664).Yoon J; Fujii S; Solomon EI Geometric and Electronic Structure Differences Between the Type 3 Copper Sites of the Multicopper Oxidases and Hemocyanin/Tyrosinase. Proc. Natl. Acad. Sci U. S. A 2009, 106, 6585–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (665).Loehr JS; Freedman TB; Loehr TM Oxygen Binding to Hemocyanin: A Resonance Raman Spectroscopic Study. Biochem. Biophys. Res. Commun 1974, 56, 510–515. [DOI] [PubMed] [Google Scholar]
- (666).Fry HC; Lucas HR; Narducci Sarjeant AA; Karlin KD; Meyer GJ Carbon Monoxide Coordination and Reversible Photodissociation in copper(I) Pyridylalkylamine Compounds. Inorg. Chem 2008, 47, 241–256. [DOI] [PubMed] [Google Scholar]
- (667).Bonaventura C; Sullivan B; Bonaventura J; Bourne S Carbon Monoxide Binding by Hemocyanins of Limulus Polyphemus, Busycon Carica, and Callinectes Sapidus. Biochemistry 1974, 13, 4784–4789. [DOI] [PubMed] [Google Scholar]
- (668).Suzuki K; Shimokawa C; Morioka C; Itoh S Monooxygenase Activity of Octopus Vulgaris Hemocyanin. Biochemistry 2008, 47, 7108–7115. [DOI] [PubMed] [Google Scholar]
- (669).Decker H; Schweikardt T; Tuczek F The First Crystal Structure of Tyrosinase: All Questions Answered? Angew. Chem. Int. Ed 2006, 45, 4546–4550. [DOI] [PubMed] [Google Scholar]
- (670).Herres-Pawlis S; Verma P; Haase R; Kang P; Lyons CT; Wasinger EC; Florke U; Henkel G; Stack TDP Phenolate Hydroxylation in a Bis(μ-oxo)dicopper(III) Complex: Lessons from the Guanidine/Amine Series. J. Am. Chem. Soc 2009, 131, 1154–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (671).Gasparetti C Biochemical and Structural Characterisation of the Copper Containing Oxidoreductases Catechol Oxidase, Tyrosinase, and Laccase from Ascomycete Fungi. Ph.D. Thesis, Aalto University; 2012. [Google Scholar]
- (672).Buitrago E; Hardre R; Haudecoeur R; Jamet H; Belle C; Boumendjel A; Bubacco L; Reglier M Are Human Tyrosinase and Related Proteins Suitable Targets for Melanoma Therapy? Curr. Top. Med. Chem 2016, 16, 3033–3047. [DOI] [PubMed] [Google Scholar]
- (673).Lai X; Wichers HJ; Soler-Lopez M; Dijkstra BW Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chem. −3 Eur. J 2018, 24, 47–55. [DOI] [PubMed] [Google Scholar]
- (674).Ginsbach JW; Kieber-Emmons MT; Nomoto R; Noguchi A; Ohnishi Y; Solomon EI Structure/Function Correlations Among Coupled Binuclear Copper Proteins Through Spectroscopic and Reactivity Studies of NspF. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 10793–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (675).Noguchi A; Kitamura T; Onaka H; Horinouchi S; Ohnishi Y A Copper-Containing Oxidase Catalyzes C-Nitrosation in Nitrosobenzamide Biosynthesis. Nat. Chem. Biol 2010, 6, 641–643. [DOI] [PubMed] [Google Scholar]
- (676).Kersten RD; Dorrestein PC Metalloenzymes: Natural Product Nitrosation. Nat. Chem. Biol 2010, 6, 636–637. [DOI] [PubMed] [Google Scholar]
- (677).Suzuki H; Furusho Y; Higashi T; Ohnishi Y; Horinouchi S A Novel o-Aminophenol Oxidase Responsible for Formation of the Phenoxazinone Chromophore of Grixazone. J. Biol. Chem 2006, 281, 824–833. [DOI] [PubMed] [Google Scholar]
- (678).Sheng Y; Abreu IA; Cabelli DE; Maroney MJ; Miller A-F; Teixeira M; Valentine JS Superoxide Dismutases and Superoxide Reductases. Chem. Rev 2014, 114, 3854–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (679).Garcia-Bosch I; Karlin KD Copper Peroxide Bioinorganic Chemistry: From Metalloenzymes to Bioinspired Synthetic Systems In The Chemistry of Peroxides; Greer A, Liebman JF, Eds.; John Wiley & Sons, Ltd, 2014; pp 805–856. [Google Scholar]
- (680).Hatcher LQ; Karlin KD Oxidant Types in Copper- Dioxygen Chemistry: The Ligand Coordination Defines the Cun-O2 Structure and Subsequent Reactivity. JBIC, J. Biol. Inorg. Chem 2004, 9, 669–683. [DOI] [PubMed] [Google Scholar]
- (681).Hatcher QL; Karlin DK Ligand Influences in Copper-Dioxygen Complex-Formation and Substrate Oxidations In Advances in Inorganic Chemistry; van Eldik R, Reedijk JBT-A, Eds.; Academic Press, 2006; Vol. 58, pp 131–184. [Google Scholar]
- (682).Citek C; Herres-Pawlis S; Stack TDP Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc. Chem. Res 2015, 48, 2424–2433. [DOI] [PubMed] [Google Scholar]
- (683).Lanci MP; Smirnov VV; Cramer CJ; Gauchenova EV; Sundermeyer J; Roth JP Isotopic Probing of Molecular Oxygen Activation at Copper(I) Sites. J. Am. Chem. Soc 2007, 129, 14697–14709. [DOI] [PubMed] [Google Scholar]
- (684).Woertink JS; Tian L; Maiti D; Lucas HR; Himes RA; Karlin KD; Neese F; Wurtele C; Holthausen MC; Bill E; et al. Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(II) Complex: Geometric and Electronic Factors That Determine the Ground State. Inorg. Chem 2010, 49, 9450–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (685).Ginsbach JW; Peterson RL; Cowley RE; Karlin KD; Solomon EI Correlation of the Electronic and Geometric Structures in Mononuclear Copper(II) Superoxide Complexes. Inorg. Chem 2013, 52, 12872–12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (686).Kunishita A; Kubo M; Sugimoto H; Ogura T; Sato K; Takui T; Itoh S Mononuclear Copper(II)-Superoxo Complexes That Mimic the Structure and Reactivity of the Active Centers of PHM and D^M. J. Am. Chem. Soc 2009, 131, 2788–2789. [DOI] [PubMed] [Google Scholar]
- (687).Tano T; Okubo Y; Kunishita A; Kubo M; Sugimoto H; Fujieda N; Ogura T; Itoh S Redox Properties of a Mononuclear Copper(II)-Superoxide Complex. Inorg. Chem 2013, 52, 10431–10437. [DOI] [PubMed] [Google Scholar]
- (688).Maiti D; Fry HC; Woertink JS; Vance MA; Solomon EI; Karlin KDA 1:1 Copper-Dioxygen Adduct Is an End-on Bound Superoxo Copper(II) Complex Which Undergoes Oxygenation Reactions with Phenols. J. Am. Chem. Soc 2007, 129, 264–265. [DOI] [PubMed] [Google Scholar]
- (689).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric-Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (690).Kim S; Lee JY; Cowley RE; Ginsbach JW; Siegler MA; Solomon EI; Karlin KD A N3S(thioether)-Ligated CuII-Superoxo with Enhanced Reactivity. J. Am. Chem. Soc 2015, 137, 2796–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (691).Zhang CX; Kaderli S; Costas M; Kim E; Neuhold Y; Karlin KD; Zuberbuhler AD Copper(I)-Dioxygen Reactivity of [(L)CuI]+ (L = Tris(2-Pyridylmethyl)amine): Kinetic/Thermody- namic and Spectroscopic Studies Concerning the Formation of Cu-O2 and Cu2-O2 Adducts as a Function of Solvent Medium and 4-Pyridyl Ligand Substituent Variations. Inorg. Chem 2003, 42, 1807–1824. [DOI] [PubMed] [Google Scholar]
- (692).Bhadra M; Lee JYC; Cowley RE; Kim S; Siegler MA; Solomon EI; Karlin KD Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc 2018, 140, 9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (693).Karlin KD; Wei N; Jung B; Kaderli S; Zuberbuhler AD Kinetic, Thermodynamic and Spectral Characterization of the Primary Cu-O2 Adduct in a Reversibly Formed and Structurally Characterized Peroxo-Dicopper(II) Complex. J. Am. Chem. Soc 1991, 113, 5868–5870. [Google Scholar]
- (694).Kobayashi Y; Ohkubo K; Nomura T; Kubo M; Fujieda N; Sugimoto H; Fukuzumi S; Goto K; Ogura T; Itoh S Copper(I)-Dioxygen Reactivity in a Sterically Demanding Tripodal Tetradentate Tren Ligand: Formation and Reactivity of a Mononuclear Copper(II) End-On Superoxo Complex. Eur. J. Inorg. Chem 2012, 2012, 4574–4578. [Google Scholar]
- (695).Wurtele C; Gaoutchenova E; Harms K; Holthausen MC; Sundermeyer J; Schindler S Crystallographic Characterization of a Synthetic 1:1 End-On Copper Dioxygen Adduct Complex. Angew. Chem., Int. Ed 2006, 45, 3867–3869. [DOI] [PubMed] [Google Scholar]
- (696).Paria S; Morimoto Y; Ohta T; Okabe S; Sugimoto H; Ogura T; Itoh S Copper(I)-Dioxygen Reactivity in the Isolated Cavity of a Nanoscale Molecular Architecture. Eur. J. Inorg. Chem 2018, 2018, 1976–1983. [Google Scholar]
- (697).Sánchez-Eguía BN; Flores-Alamo M; Orio M; Castillo I Side-on Cupric-superoxo Triplet Complexes as Competent Agents for H-Abstraction Relevant to the Active Site of PHM. Chem. Commun 2015, 51, 11134–11137. [DOI] [PubMed] [Google Scholar]
- (698).Maiti D; Lee D-H; Gaoutchenova K; Würtele C; Holthausen MC; Narducci Sarjeant AA; Sundermeyer J; Schindler S; Karlin KD Reactions of a Copper (II) Superoxo Complex Lead to C-H and O-H Substrate Oxygenation : Modeling Copper-Monooxygenase C-H Hydroxylation. Angew. Chem. Int. Ed 2008, 47, 82–85. [DOI] [PubMed] [Google Scholar]
- (699).Donoghue PJ; Gupta AK; Boyce DW; Cramer CJ; Tolman WB An Anionic, Tetragonal Copper(II) Superoxide Complex. J. Am. Chem. Soc 2010, 132, 15869–15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (700).Yamaguchi S; Nagatomo S; Kitagawa T; Funahashi Y; Ozawa T; Jitsukawa K; Masuda H Copper Hydroperoxo Species Activated by Hydrogen-Bonding Interaction with Its Distal Oxygen. Inorg. Chem 2003, 42, 6968–6970. [DOI] [PubMed] [Google Scholar]
- (701).Wada A; Harata M; Hasegawa K; Jitsukawa K; Masuda H; Mukai M; Kitagawa T; Einaga H Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo-Copper(II) Complex with Tripodal Pyridylamine Ligands. Angew. Chem. Int. Ed 1998, 37, 798–799. [DOI] [PubMed] [Google Scholar]
- (702).Peterson RL; Himes RA; Kotani H; Suenobu T; Tian L; Siegler MA; Solomon EI; Fukuzumi S; Karlin KD Cupric Superoxo-Mediated Intermolecular C-H Activation Chemistry. J. Am. Chem. Soc 2011, 133, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (703).Mareque-Rivas JC; Hinchley SL; Metteau L; Parsons S The Strength of Hydrogen Bonding to Metal-Bound Ligands Can Contribute to Changes in the Redox Behaviour of Metal Centres. Dalton Trans 2006, 2316–2322. [DOI] [PubMed] [Google Scholar]
- (704).Fujisawa K; Tanaka M; Moro-oka Y; Kitajima N A Monomeric Side-On Superoxocopper(II) Complex: Cu(O2)(HB(3-tBu-5-iPrpz)3). J.Am. Chem. Soc 1994, 116, 12079–12080. [Google Scholar]
- (705).Chen P; Root DE; Campochiaro C; Fujisawa K; Solomon EI Spectroscopic and Electronic Structure Studies of the Diamagnetic Side-On CuII-Superoxo Complex Cu(O2)[HB(3-R-5-iPrpz)3]: Antiferromagnetic Coupling versus Covalent Delocalization. J. Am. Chem. Soc 2003, 125, 466–474. [DOI] [PubMed] [Google Scholar]
- (706).Iovan DA; Wrobel AT; McClelland AA; Scharf AB; Edouard GA; Betley TA Reactivity of a Stable Copper-Dioxygen Complex. Chem. Commun 2017, 53, 10306–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (707).Sarangi R; Aboelella NW; Fujisawa K; Tolman WB; Hedman B; Hodgson KO; Solomon EI X-Ray Absorption Edge Spectroscopy and Computational Studies on LCuO2 Species: Superoxide-CuII versus Peroxide-CuIII Bonding. J. Am. Chem. Soc 2006, 128, 8286–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (708).Aboelella NW; Kryatov SV; Gherman BF; Brennessel WW; Young VG; Sarangi R; Rybak-Akimova EV; Hodgson KO; Hedman B; Solomon EI; et al. Dioxygen Activation at a Single Copper Site: Structure, Bonding, and Mechanism of Formation of 1:1 Cu–O2 Adducts. J. Am. Chem. Soc 2004, 126, 16896–16911. [DOI] [PubMed] [Google Scholar]
- (709).Reynolds AM; Gherman BF; Cramer CJ; Tolman WB Characterization of a 1:1 Cu-O2 Adduct Supported by an Anilido Imine Ligand. Inorg. Chem 2005, 44, 6989–6997. [DOI] [PubMed] [Google Scholar]
- (710).Aboelella NW; Lewis EA; Reynolds AM; Brennessel WW; Cramer CJ; Tolman WB Snapshots of Dioxygen Activation by Copper: The Structure of a 1:1 Cu/O2 Adduct and Its Use in Syntheses of Asymmetric Bis(μ-Oxo) Complexes. J. Am. Chem. Soc 2002, 124, 10660–10661. [DOI] [PubMed] [Google Scholar]
- (711).Pirovano P; Magherusan AM; McGlynn C; Ure A; Lynes A; McDonald A R Nucleophilic Reactivity of a Copper(II)-Superoxide Complex. Angew. Chem., Int. Ed 2014, 53, 5946–5950. [DOI] [PubMed] [Google Scholar]
- (712).Yamaguchi S; Masuda H Basic Approach to Development of Environment-Friendly Oxidation Catalyst Materials. Mononuclear Hydroperoxo copper(II) Complexes. Sci. Technol. Adv. Mater 2005, 6, 34–47. [Google Scholar]
- (713).Neisen BD; Gagnon NL; Dhar D; Spaeth AD; Tolman WB Formally Copper(III)-Alkylperoxo Complexes as Models of Possible Intermediates in Monooxygenase Enzymes. J. Am. Chem. Soc 2017, 139, 10220–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (714).Shook RL; Borovik AS Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (715).Yamaguchi S; Wada A; Funahashi Y; Nagatomo S; Kitagawa T; Jitsukawa K; Masuda H Thermal Stability and Absorption Spectroscopic Behavior of (μ-Peroxo)dicopper Complexes Regulated with Intramolecular Hydrogen Bonding Interactions. Eur. J. Inorg. Chem 2003, 2003, 4378–4386. [Google Scholar]
- (716).Wada A; Honda Y; Yamaguchi S; Nagatomo S; Kitagawa T; Jitsukawa K; Masuda H Steric and Hydrogen-Bonding Effects on the Stability of Copper Complexes with Small Molecules. Inorg. Chem 2004, 43, 5725–5735. [DOI] [PubMed] [Google Scholar]
- (717).Yamaguchi S; Wada A; Nagatomo S; Kitagawa T; Jitsukawa K; Masuda H Thermal Stability of Mononuclear Hydroperoxocopper(II) Species. Effects of Hydrogen Bonding and Hydrophobic Field. Chem. Lett 2004, 33, 1556–1557. [Google Scholar]
- (718).Fujii T; Yamaguchi S; Funahashi Y; Ozawa T; Tosha T; Kitagawa T; Masuda H Mononuclear Copper(II)–hydroperoxo Complex Derived from Reaction of Copper(I) Complex with Dioxygen as a Model of DβM and PHM. Chem. Commun 2006, 4428–4430. [DOI] [PubMed] [Google Scholar]
- (719).Fujii T; Yamaguchi S; Hirota S; Masuda H H-Atom Abstraction Reaction for Organic Substrates via Mononuclear Copper(II)-Superoxo Species as a Model for DβM and PHM. Dalton Trans 2008, 164–170. [DOI] [PubMed] [Google Scholar]
- (720).Kim S; Saracini C; Siegler MA; Drichko N; Karlin KD Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent. Inorg. Chem 2012, 51, 12603–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (721).Mann SI; Heinisch T; Ward TR; Borovik AS Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins. J. Am. Chem. Soc 2017, 139, 17289–17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (722).Harata M; Jitsukawa K; Masuda H; Einaga H Preparations, Structures, and Properties of Cu(II) Complexes with Tripodal Tetradentate Ligand, tris(6-Pivaloylamino-2-Pyridylmethyl)-amine (Htppa), and Reaction of Its Cu(I) Complex with Dioxygen. Bull. Chem. Soc. Jpn 1998, 71, 637–645. [Google Scholar]
- (723).Harata M; Hasegawa K; Jitsukawa K; Masuda H; Einaga H Preparations, Structures, and Properties of copper(II) Complexes with a New Tripodal Tetradentate Ligand, N-(2-pyridylmethyl)bis(6- Pivalamido-2-Pyridylmethyl)amine, and Reactivities of the Cu(I) Complex with Dioxygen. Bull. Chem. Soc. Jpn 1998, 71, 1031–1038. [Google Scholar]
- (724).Osako T; Nagatomo S; Tachi Y; Kitagawa T; Itoh S Low-Temperature Stopped-Flow Studies on the Reactions of Copper(II) Complexes and H2O2: The First Detection of a Mononuclear Copper(II)-Peroxo Intermediate. Angew. Chem. Int. Ed 2002, 41, 4325–4328. [DOI] [PubMed] [Google Scholar]
- (725).Itoh S Mononuclear Copper Active-Oxygen Complexes. Curr. Opin. Chem. Biol 2006, 10, 115–122. [DOI] [PubMed] [Google Scholar]
- (726).Osako T; Nagatomo S; Kitagawa T; Cramer CJ; Itoh S Kinetics and DFT Studies on the Reaction of Copper(II) Complexes and H2O2. JBIC, J. Biol. Inorg. Chem 2005, 10, 581–590. [DOI] [PubMed] [Google Scholar]
- (727).Maiti D; Narducci Sarjeant AA; Karlin KD Copper(II)- Hydroperoxo Complex Induced Oxidative N-Dealkylation Chemistry. J. Am. Chem. Soc 2007, 129, 6720–6721. [DOI] [PubMed] [Google Scholar]
- (728).Maiti D; Lucas HR; Sarjeant AAN; Karlin KD Aryl Hydroxylation from a Mononuclear Copper-Hydroperoxo Species. J. Am. Chem. Soc 2007, 129, 6998–6999. [DOI] [PubMed] [Google Scholar]
- (729).Maiti D; Narducci Sarjeant AA; Karlin KD Copper–Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases. Inorg. Chem 2008, 47, 8736–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (730).Kim S; Ginsbach JW; Lee JY; Peterson RL; Liu JJ; Siegler MA; Sarjeant AA; Solomon EI; Karlin KD Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. J. Am. Chem. Soc 2015, 137, 2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (731).Hirao H; Li F; Que L; Morokuma K Theoretical Study of the Mechanism of Oxoiron(IV) Formation from H2O2 and a Nonheme Iron(II) Complex: O–O Cleavage Involving Proton-Coupled Electron Transfer. Inorg. Chem 2011, 50, 6637–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (732).Fujii T; Naito A; Yamaguchi S; Wada A; Funahashi Y; Jitsukawa K; Nagatomo S; Kitagawa T; Masuda H Construction of a Square-Planar Hydroperoxo-copper(II) Complex Inducing a Higher Catalytic Reactivity. Chem. Commun 2003, 2700–2701. [DOI] [PubMed] [Google Scholar]
- (733).Choi YJ; Cho K-B; Kubo M; Ogura T; Karlin KD; Cho J; Nam W Spectroscopic and Computational Characterization of CuII-OOR (R = H or Cumyl) Complexes Bearing a Me6-Tren Ligand. Dalton Trans 2011, 40, 2234–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (734).Chen P; Fujisawa K; Solomon EI Spectroscopic and Theoretical Studies of Mononuclear Copper(II) Alkyl- and Hydroperoxo Complexes: Electronic Structure Contributions to Reactivity. J.Am. Chem. Soc 2000, 122, 10177–10193. [Google Scholar]
- (735).Kodera M; Kita T; Miura I; Nakayama N; Kawata T; Kano K; Hirota S Hydroperoxo-Copper(II) Complex Stabilized by N3S-Type Ligand Having a Phenyl Thioether. J. Am. Chem. Soc 2001, 123, 7715–7716. [DOI] [PubMed] [Google Scholar]
- (736).Kamachi T; Yoshizawa K Water-Assisted Oxo Mechanism for Heme Metabolism. J. Am. Chem. Soc 2005, 127, 10686–10692. [DOI] [PubMed] [Google Scholar]
- (737).Yoshizawa K; Kihara N; Kamachi T; Shiota Y Catalytic Mechanism of Dopamine β-Monooxygenase Mediated by Cu(III)-Oxo. Inorg. Chem 2006, 45, 3034–3041. [DOI] [PubMed] [Google Scholar]
- (738).Crespo A; Martí MA; Roitberg AE; Amzel LM; Estrin DA The Catalytic Mechanism of Peptidylglycine α-Hydroxylating Monooxygenase Investigated by Computer Simulation. J. Am. Chem. Soc 2006, 128, 12817–12828. [DOI] [PubMed] [Google Scholar]
- (739).Yoshizawa K Quantum Chemical Studies on Dioxygen Activation and Methane Hydroxylation by Diiron and Dicopper Species as Well as Related Metal-Oxo Species. Bull. Chem. Soc. Jpn 2013, 86, 1083–1116. [Google Scholar]
- (740).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C-H Bond Activation by a Monocopper(III)-Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (741).Spaeth AD; Gagnon NL; Dhar D; Yee GM; Tolman WB Determination of the Cu(III)-OH Bond Distance by Resonance Raman Spectroscopy Using a Normalized Version of Badger’s Rule. J. Am. Chem. Soc 2017, 139, 4477–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (742).Dhar D; Yee GM; Markle TF; Mayer JM; Tolman WB Reactivity of the copper(III)-Hydroxide Unit with Phenols. Chem. Sci 2017, 8, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (743).Dietl N; Schlangen M; Schwarz H Thermal HydrogenAtom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry. Angew. Chem. Int. Ed 2012, 51, 5544–5555. [DOI] [PubMed] [Google Scholar]
- (744).Rijs NJ; Gonzalez-Navarrete P; Schlangen M; Schwarz H Penetrating the Elusive Mechanism of Copper-Mediated Fluoromethylation in the Presence of Oxygen through the Gas-Phase Reactivity of Well-Defined [LCuO]+ Complexes with Fluoromethanes (CH(4-n)Fn, N = 1–3). J. Am. Chem. Soc 2016, 138, 3125–3135. [DOI] [PubMed] [Google Scholar]
- (745).Kunishita A; Ishimaru H; Nakashima S; Ogura T; Itoh S Reactivity of Mononuclear Alkylperoxo Copper(II) Complex. O–O Bond Cleavage and C–H Bond Activation. J. Am. Chem. Soc 2008, 130, 4244–4245. [DOI] [PubMed] [Google Scholar]
- (746).Kunishita A; Ishimaru H; Nakashima S; Ogura T; Itoh S Reactivity of Mononuclear Alkylperoxo Copper(II) Complex. O–O Bond Cleavage and C-H Bond Activation. J. Am. Chem. Soc 2008, 130, 4244–4245. [DOI] [PubMed] [Google Scholar]
- (747).Schaefer AW; Kieber-Emmons MT; Adam SM; Karlin KD; Solomon EI Phenol-Induced O–O Bond Cleavage in a Low-Spin Heme-Peroxo-Copper Complex: Implications for O2 Reduction in Heme-Copper Oxidases. J. Am. Chem. Soc 2017, 139, 7958–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (748).Kieber-Emmons MT; Ginsbach JW; Wick PK; Lucas HR; Helton ME; Lucchese B; Suzuki M; Zuberbuhler AD; Karlin KD; Solomon EI Observation of a CuIII2(μ-1,2-peroxo)/CuIII2(μ-oxo)2 Equilibrium and Its Implications for Copper-Dioxygen Reactivity. Angew. Chem. Int. Ed 2014, 53, 4935–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (749).Stack TDP Complexity with Simplicity: A Steric Continuum of Chelating Diamines with copper(I) and Dioxygen. Dalton Trans 2003, 0, 1881–1889. [Google Scholar]
- (750).Lewis EA; Tolman WB Reactivity of Dioxygen-Copper Systems. Chem. Rev 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]
- (751).Steiner RA; Kalk KH; Dijkstra BW Anaerobic Enzyme.substrate Structures Provide Insight into the Reaction Mechanism of the Copper-Dependent Quercetin 2,3-Dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16625–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (752).Matsumoto T; Ohkubo K; Honda K; Yazawa A; Furutachi H; Fujinami S; Fukuzumi S; Suzuki M Aliphatic C–H Bond Activation Initiated by a (μ-η2:η2-Peroxo)dicopper(II) Complex in Comparison with Cumylperoxyl Radical. J. Am. Chem. Soc 2009, 131, 9258–9267. [DOI] [PubMed] [Google Scholar]
- (753).Mirica LM; Ottenwaelder X; Stack TDP Structure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1046. [DOI] [PubMed] [Google Scholar]
- (754).Rolff M; Schottenheim J; Tuczek F Monooxygenation of External Phenolic Substrates in Small-Molecule Dicopper Complexes: Implications on the Reaction Mechanism of Tyrosinase. J. Coord. Chem 2010, 63, 2382–2399. [Google Scholar]
- (755).Rolff M; Schottenheim J; Decker H; Tuczek F Copper–O2 Reactivity of Tyrosinase Models towards External Monophenolic Substrates: Molecular Mechanism and Comparison with the Enzyme. Chem. Soc. Rev 2011, 40, 4077–4098. [DOI] [PubMed] [Google Scholar]
- (756).Rolff M; Tuczek F How Do Copper Enzymes Hydroxylate Aliphatic Substrates? Recent Insights from the Chemistry of Model Systems. Angew. Chem. Int. Ed 2008, 47, 2344–2347. [DOI] [PubMed] [Google Scholar]
- (757).Serrano-Plana J; Garcia-Bosch I; Company A; Costas M Structural and Reactivity Models for Copper Oxygenases: Cooperative Effects and Novel Reactivities. Acc. Chem. Res 2015, 48, 2397–2406. [DOI] [PubMed] [Google Scholar]
- (758).Würtele C; Sander O; Lutz V; Waitz T; Tuczek F; Schindler S Aliphatic C-H Bond Oxidation of Toluene Using Copper Peroxo Complexes That Are Stable at Room Temperature. J. Am. Chem. Soc 2009, 131, 7544–7545. [DOI] [PubMed] [Google Scholar]
- (759).Tyeklar Z; Jacobson RR; Wei N; Murthy NN; Zubieta J; Karlin KD Reversible Reaction of Dioxygen (and Carbon Monoxide) with a copper(I) Complex. X-Ray Structures of Relevant Mononuclear Cu(I) Precursor Adducts and the Trans-(μ-1,2- peroxo)dicopper(II) Product. J. Am. Chem. Soc 1993, 115, 2677–2689. [Google Scholar]
- (760).Kitajima N; Fujisawa K; Morooka Y; Toriumi K μ-η2:η2- Peroxo Binuclear Copper Complex, [Cu(HB(3,5-(Me2CH)2pz)3)]2(O2). J.Am. Chem. Soc 1989, 111, 8975–8976. [Google Scholar]
- (761).Ross PK; Solomon EI An Electronic Structural Comparison of Copper-Peroxide Complexes of Relevance to Hemocyanin and Tyrosinase Active Sites. J. Am. Chem. Soc 1991, 113, 3246–3259. [Google Scholar]
- (762).Ross PK; Solomon EI Electronic Structure of Peroxide Bridged Copper Dimers of Relevance to Oxyhemocyanin. J. Am. Chem. Soc 1990, 112, 5871–5872. [Google Scholar]
- (763).Solomon EI; Ginsbach JW; Heppner DE; Kieber-Emmons MT; Kjaergaard CH; Smeets PJ; Tian L; Woertink JS Copper Dioxygen (Bio)inorganic Chemistry. Faraday Discuss. 2011, 148, 11–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (764).Solomon EI Dioxygen Binding, Activation, and Reduction to H2O by Cu Enzymes. Inorg. Chem 2016, 55, 6364–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).Halfen JA; Mahapatra S; Wilkinson EC; Kaderli S; Young VG; Que L; Zuberbuhler AD; Tolman WB Reversible Cleavage and Formation of the Dioxygen O–O Bond Within a Dicopper Complex. Science 1996, 271, 1397–1400. [DOI] [PubMed] [Google Scholar]
- (766).Jacobson RR; Tyeklar Z; Farooq A; Karlin KD; Liu S; Zubieta J A Cu2-O2 Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System. J. Am. Chem. Soc 1988, 110, 3690–3692. [Google Scholar]
- (767).Komiyama K; Furutachi H; Nagatomo S; Hashimoto A; Hayashi H; Fuj inami S; Suzuki M; Kitagawa T Dioxygen Reactivity of Copper(I) Complexes with Tetradentate Tripodal Ligands Having Aliphatic Nitrogen Donors: Synthesis, Structures, and Properties of Peroxo and Superoxo Complexes. Bull. Chem. Soc. Jpn 2004, 77, 59–72. [Google Scholar]
- (768).Hoppe T; Schaub S; Becker J; Wurtele C; Schindler S Characterization of a Macrocyclic End-on Peroxido Copper Complex. Angew. Chem. Int. Ed 2013, 52, 870–873. [DOI] [PubMed] [Google Scholar]
- (769).Dahl EW; Dong HT; Szymczak NK Phenylamino Derivatives of tris(2-Pyridylmethyl)amine: Hydrogen-Bonded Peroxodicopper Complexes. Chem. Commun 2018, 54, 892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (770).Hatcher LQ; Lee D-H; Vance MA; Milligan AE; Sarangi R; Hodgson KO; Hedman B; Solomon EI; Karlin KD Dioxygen Reactivity of a Copper(I) Complex with a N3S Thioether Chelate; Peroxo-Dicopper(II) Formation Including Sulfur-Ligation. Inorg. Chem 2006, 45, 10055–10057. [DOI] [PubMed] [Google Scholar]
- (771).Lee Y; Lee D-H; Park GY; Lucas HR; Narducci Sarjeant AA; Kieber-Emmons MT; Vance MA; Milligan AE; Solomon EI; Karlin KD Sulfur Donor Atom Effects on Copper(I)/O2 Chemistry with Thioanisole Containing Tetradentate N3S Ligand Leading to μ-1,2-Peroxo-Dicopper(Π) Species. Inorg. Chem 2010, 49, 8873–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (772).Maiti D; Woertink JS; Narducci Sarjeant AA; Solomon EI; Karlin KD Copper Dioxygen Adducts: Formation of Bis(μ-oxo)dicopper(III) versus (μ-1,2)Peroxodicopper(II) Complexes with Small Changes in One Pyridyl-Ligand Substituent. Inorg. Chem 2008, 47, 3787–3800. [DOI] [PubMed] [Google Scholar]
- (773).Schatz M; Becker M; Thaler F; Hampel F; Schindler S; Jacobson RR; Tyeklar Z; Murthy NN; Ghosh P; Chen Q; et al. Copper(I) Complexes, Copper(I)/O2 Reactivity, and Copper(II) Complex Adducts, with a Series of Tetradentate Tripyridylalkylamine Tripodal Ligands. Inorg. Chem 2001, 40, 2312–2322. [DOI] [PubMed] [Google Scholar]
- (774).Zubieta J; Karlin KD; Hayes JC Structural Systematics of Cu(I) and Cu(II) Derivatives of Tripodal Ligands. In Copper Coordination Chemistry: Biochemical and Inorganic Perspectives; Karlin KD, Zubieta J, Eds.; Adenine Press: Albany, N.Y, 1983; pp 97–108. [Google Scholar]
- (775).Ambundo EA; Deydier M-V; Grall AJ; Aguera-Vega N; Dressel LT; Cooper TH; Heeg MJ; Ochrymowycz LA; Rorabacher DB Influence of Coordination Geometry upon Copper(II/I) Redox Potentials. Physical Parameters for Twelve Copper Tripodal Ligand Complexes. Inorg. Chem 1999, 38, 4233–4242. [Google Scholar]
- (776).Augustin MA; Yandell JK; Addison AW; Karlin KD Rates of Electron Transfer and Redox Potentials of Some copper(II) Thioether Complexes. Inorg. Chim. Acta 1981, 55, L35–L37. [Google Scholar]
- (777).Lee Y; Park GY; Lucas HR; Vajda PL; Kamaraj K; Vance MA; Milligan AE; Woertink JS; Siegler MA; Narducci Sarjeant AA; et al. Copper(I)/O2 Chemistry with Imidazole Containing Tripodal Tetradentate Ligands Leading to μ-1,2-Peroxo–Dicopper(II) Species. Inorg. Chem 2009, 48, 11297–11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (778).Lee D-HD-H; Wei N; Murthy NN; Tyeklar Z; Karlin KD; Kaderli S; Jung B; Zuberbuhler AD; Tyeklar Z; Karlin KD; et al. Reversible O2 Binding to a Dinuclear Copper(I) Complex with Linked Tris(2-Pyridylmethyl)amine Units: Kinetic-Thermodynamic Comparisons with Mononuclear Analogs. J. Am. Chem. Soc 1995, 117, 12498–12513. [Google Scholar]
- (779).Karlin KD; Lee D-H; Kaderli S; Zuberbühler AD Copper Dioxygen Complexes Stable at Ambient Temperature: Optimization of Ligand Design and Solvent. Chem. Commun 1997, 475–476. [Google Scholar]
- (780).Karlin KD; Cruse RW; Gultneh Y; Hayes JC; Zubieta J Peroxide Coordination to a dicopper(II) Center. Dioxygen Binding to a Structurally Characterized Phenoxide-Bridged Binuclear copper(I) Complex. J. Am. Chem. Soc 1984, 106, 3372–3374. [Google Scholar]
- (781).Blackburn NJ; Strange RW; Cruse RW; Karlin KD Dioxygen-Copper Reactivity: EXAFS Studies of a Peroxo-dicopper(II) Complex. J.Am. Chem. Soc 1987, 109, 1235–1237. [Google Scholar]
- (782).Pate JE; Cruse RW; Karlin KD; Solomon EI Vibrational, Electronic, and Resonance Raman Spectral Studies of [Cu2(XYL-O-)O2]+, a copper(II) Peroxide Model Complex of Oxyhemocyanin. J. Am. Chem. Soc 1987, 109, 2624–2630. [Google Scholar]
- (783).Cao R; Saracini C; Ginsbach JW; Kieber-Emmons MT; Siegler MA; Solomon EI; Fukuzumi S; Karlin KD Peroxo and Superoxo Moieties Bound to Copper Ion: Electron-Transfer Equilibrium with a Small Reorganization Energy. J. Am. Chem. Soc 2016, 138, 7055–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (784).Karlin KD; Cruse RW; Gultneh Y Dioxygen-Copper Reactivity: A Hydroperoxo-Dicopper(II) Complex. J. Chem. Soc., Chem. Commun 1987, 599–600. [Google Scholar]
- (785).Karlin KD; Ghosh P; Cruse RW; Farooq A; Gultneh Y; Jacobson RR; Blackburn NJ; Strange RW; Zubieta J Dioxygen-Copper Reactivity: Generation, Characterization, and Reactivity of a Hydroperoxodicopper(II) Complex. J. Am. Chem. Soc 1988, 110, 6769–6780. [Google Scholar]
- (786).Ghosh P; Tyeklar Z; Karlin KD; Jacobson RR; Zubieta J Dioxygen-Copper Reactivity: X-Ray Structure and Characterization of an (Acylperoxo)dicopper Complex. J. Am. Chem. Soc 1987, 109, 6889–6891. [Google Scholar]
- (787).Mahroof-Tahir M; Murthy NN; Karlin KD; Blackburn NJ; Shaikh SN; Zubieta J New Thermally Stable Hydroperoxo-and Peroxo-Copper Complexes. Inorg. Chem 1992, 31, 3001–3003. [Google Scholar]
- (788).Murthy NN; Mahroof-Tahir M; Karlin KD Dicopper(I) Complexes of Unsymmetrical Binucleating Ligands and Their Dioxygen Reactivities. Inorg. Chem 2001, 40, 628–635. [DOI] [PubMed] [Google Scholar]
- (789).Kindermann N; Dechert S; Demeshko S; Meyer F Proton-Induced, Reversible Interconversion of a μ-1,2-Peroxo and a μ-1,1-Hydroperoxo Dicopper(II) Complex. J. Am. Chem. Soc 2015, 137, 8002–8005. [DOI] [PubMed] [Google Scholar]
- (790).Dalle KE; Gruene T; Dechert S; Demeshko S; Meyer F Weakly Coupled Biologically Relevant CuII2(μ-η1:η1-O2) Cis-Peroxo Adduct That Binds Side-On to Additional Metal Ions. J. Am. Chem. Soc 2014, 136, 7428–7434. [DOI] [PubMed] [Google Scholar]
- (791).Kindermann N; Bill E; Dechert S; Demeshko S; Reijerse EJ; Meyer F A Ferromagnetically Coupled (S = 1) Peroxodicopper(II) Complex. Angew. Chem., Int. Ed 2015, 54, 1738–1743. [DOI] [PubMed] [Google Scholar]
- (792).Kindermann N; Gunes C-J; Dechert S; Meyer F Hydrogen Atom Abstraction Thermodynamics of a μ-1,2-Superoxo Dicopper(II) Complex. J. Am. Chem. Soc 2017, 139, 9831–9834. [DOI] [PubMed] [Google Scholar]
- (793).Uozumi K; Hayashi Y; Suzuki M; Uehara A Reactivity of Copper(I) Complexes Containing Various Tetradentate Tripodal Ligands with Molecular Oxygen. Chem. Lett 1993, 22, 963–966. [Google Scholar]
- (794).Karlin KD; Haka MS; Cruse RW; Gultneh Y Dioxygen-Copper Reactivity. Reversible Oxygen and Carbon Monoxide Binding by a New Series of Binuclear copper(I) Complexes. J. Am. Chem. Soc 1985, 107, 5828–5829. [Google Scholar]
- (795).Kitajima N; Fujisawa K; Fujimoto C; Morooka Y; Hashimoto S; Kitagawa T; Toriumi K; Tatsumi K; Nakamura A A New Model for Dioxygen Binding in Hemocyanin. Synthesis, Characterization, and Molecular Structure of the μ-η2:η2 Peroxo Dinuclear Copper(II) Complexes, [Cu(HB(3,5-R2pz)3)]2(O2) (R = Isop. J.Am. Chem. Soc 1992, 114, 1277–1291. [Google Scholar]
- (796).Yachandra V; DeRose V; Latimer M; Mukerji I; Sauer K; Klein M Where Plants Make Oxygen: A Structural Model for the Photosynthetic Oxygen-Evolving Manganese Cluster. Science 1993, 260, 675–679. [DOI] [PubMed] [Google Scholar]
- (797).Zang Y; Dong Y; Que L; Kaufmann K; Muenck E The First Bis(μ-oxo)diiron(III) Complex. Structure and Magnetic Properties of [Fe2(μ-O)2(6TLA)2](ClO4)2. J. Am. Chem. Soc 1995, 117, 1169–1170. [Google Scholar]
- (798).Mahadevan V; Hou Z; Cole AP; Root DE; Lal TK; Solomon EI; Stack TDP Irreversible Reduction of Dioxygen by Simple Peralkylated Diamine-Copper(I) Complexes: Characterization and Thermal Stability of a [Cu2(μ-O)2]2+ Core. J. Am. Chem. Soc 1997, 119, 11996–11997. [Google Scholar]
- (799).Henson MJ; Vance MA; Zhang CX; Liang H-C; Karlin KD; Solomon EI Resonance Raman Investigation of Equatorial Ligand Donor Effects on the Cu2O22+ Core in End-On and Side-On μ-Peroxo-Dicopper(II) and Bis-μ-Oxo-Dicopper(III) Complexes. J. Am. Chem. Soc 2003, 125, 5186–5192. [DOI] [PubMed] [Google Scholar]
- (800).Karahalis GJ; Thangavel A; Chica B; Bacsa J; Dyer RB; Scarborough CC Synthesis and Catalytic Reactivity of a Dicopper(II) μ-η2:η2 −3Peroxo Species Supported by 1,4,7-Tri-Tert-Butyl-1,4,7-Triazacyclononane. Inorg. Chem 2016, 55, 1102–1107. [DOI] [PubMed] [Google Scholar]
- (801).Saracini C; Ohkubo K; Suenobu T; Meyer GJ; Karlin KD; Fukuzumi S Laser-Induced Dynamics of Peroxodicopper(II) Complexes Vary with the Ligand Architecture. One-Photon Two- Electron O2 Ejection and Formation of Mixed-Valent CuICuII- Superoxide Intermediates. J. Am. Chem. Soc 2015, 137, 15865–15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (802).Lieberman RL; Rosenzweig AC Crystal Structure of a Membrane-Bound Metalloenzyme That Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. [DOI] [PubMed] [Google Scholar]
- (803).Smith SM; Rawat S; Telser J; Hoffman BM; Stemmler TL; Rosenzweig AC Crystal Structure and Characterization of Particulate Methane Monooxygenase from Methylocystis Species Strain M. Biochemistry 2011, 50, 10231–10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (804).Sirajuddin S; Barupala D; Helling S; Marcus K; Stemmler TL; Rosenzweig AC Effects of Zinc on Particulate Methane Monooxygenase Activity and Structure. J. Biol. Chem 2014, 289, 21782–21794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (805).Ipek B; Wulfers MJ; Kim H; Göltl F; Hermans I; Smith JP; Booksh KS; Brown CM; Lobo RF Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal 2017, 7, 4291–4303. [Google Scholar]
- (806).Woertink JS; Smeets PJ; Groothaert MH; Vance MA; Sels BF; Schoonheydt RA; Solomon EIA [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci U. S. A 2009, 106, 18908–18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (807).Haack P; Limberg C Molecular CuII-O-CuII Complexes: Still Waters Run Deep. Angew. Chem., Int. Ed 2014, 53, 4282–4293. [DOI] [PubMed] [Google Scholar]
- (808).Snyder BER; Bols ML; Schoonheydt RA; Sels BF; Solomon EI Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev 2018, 118, 2718–2768. [DOI] [PubMed] [Google Scholar]
- (809).Karlin KD; Gultneh Y; Hayes JC; Zubieta J Copper(I) Dioxygen Reactivity, 2. Reaction of a Three-Coordinate Cu(I) Complex with O2 with Evidence for a Binuclear Oxo-Cu(II) Species: Structural Characterization of a Parallel-Planar Di-Hydroxo Bridged Dimer. Inorg. Chem 1984, 23, 519–521. [Google Scholar]
- (810).Sanyal I; Mahroof-Tahir M; Nasir S; Ghosh P; Cohen BI; Gultneh Y; Cruse R; Farooq A; Karlin KD; Liu S; et al. Reaction of Dioxygen (O2) with Mononuclear Copper(I) Complexes: Temperature-Dependent Formation of Peroxo- or Oxo (and Dihydroxo) Bridged Dicopper(II) Complexes. Inorg. Chem 1992, 31, 4322–4332. [Google Scholar]
- (811).Ali G; VanNatta PE; Ramirez DA; Light KM; Kieber- Emmons MT Thermodynamics of a μ-Oxo Dicopper(II) Complex for Hydrogen Atom Abstraction. J. Am. Chem. Soc 2017, 139, 18448–18451. [DOI] [PubMed] [Google Scholar]
- (812).Smeets PJ; Hadt RG; Woertink JS; Vanelderen P; Schoonheydt RA; Sels BF; Solomon EI Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5. J. Am. Chem. Soc 2010, 132, 14736–14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (813).Lyons TW; Reinhard CT; Planavsky NJ The Rise of Oxygen in Earth’s Early Ocean and Atmosphere. Nature 2014, 506, 307–315. [DOI] [PubMed] [Google Scholar]
- (814).Schirrmeister BE; de Vos JM; Antonelli A; Bagheri HC Evolution of Multicellularity Coincided with Increased Diversification of Cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (815).Sessions AL; Doughty DM; Welander PV; Summons RE; Newman DK The Continuing Puzzle of the Great Oxidation Event. Curr. Biol 2009, 19, R567–R574. [DOI] [PubMed] [Google Scholar]
- (816).Hoganson CW; Pressler MA; Proshlyakov DA; Babcock GT From Water to Oxygen and Back Again: Mechanistic Similarities in the Enzymatic Redox Conversion between Water and Dioxygen. Biochim. Biophys. Acta, Bioenerg 1998, 1365, 170–174. [DOI] [PubMed] [Google Scholar]
- (817).Rich PR Mitochondrial Cytochrome c Oxidase: Catalysis, Coupling and Controversies. Biochem. Soc. Trans 2017, 45, 813–829. [DOI] [PubMed] [Google Scholar]
- (818).Robinson BH Human Cytochrome Oxidase Deficiency. Pediatr. Res. 2000, 48, 581–585. [DOI] [PubMed] [Google Scholar]
- (819).DiMauro S; Tanji K; Schon EA The Many Clinical Faces of Cytochrome c Oxidase Deficiency In Mitochondrial Oxidative Phosphorylation: Advances in Experimental Medicine and Biology; Kadenbach B, Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, 2012; Vol. 748, pp 341–357. [DOI] [PubMed] [Google Scholar]
- (820).Chaturvedi RK; Flint Beal M Mitochondrial Diseases of the Brain. Free Radical Biol. Med 2013, 63, 1–29. [DOI] [PubMed] [Google Scholar]
- (821).Diaz F Cytochrome c Oxidase Deficiency: Patients and Animal Models. Biochim. Biophys. Acta, Mol. Basis Dis 2010, 1802, 100–110. [DOI] [PubMed] [Google Scholar]
- (822).Maurer IA Selective Defect of Cytochrome c Oxidase Is Present in Brain of Alzheimer Disease Patients. Neurobiol Aging 2000, 21, 455–462. [DOI] [PubMed] [Google Scholar]
- (823).Davis RE; Miller S; Herrnstadt C; Ghosh SS; Fahy E; Shinobu LA; Galasko D; Thal LJ; Beal MF; Howell N; et al. Mutations in Mitochondrial Cytochrome c Oxidase Genes Segregate with Late-Onset Alzheimer Disease. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 4526–4531. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (824).Swerdlow R H.; Burns, J. M.; Khan, S. M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta, Mol. Basis Dis 2014, 1842, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (825).Zhu Z; Yao J; Johns T; Fu K; De Bie I; Macmillan C; Cuthbert AP; Newbold RF; Wang J; Chevrette M; et al. SURF1, Encoding a Factor Involved in the Biogenesis of Cytochrome c Oxidase, Is Mutated in Leigh Syndrome. Nat. Genet 1998, 20, 337–343. [DOI] [PubMed] [Google Scholar]
- (826).Srinivasan S; Avadhani NG Cytochrome c Oxidase Dysfunction in Oxidative Stress. Free Radical Biol. Med 2012, 53, 1252–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (827).Leadsham JE; Sanders G; Giannaki S; Bastow EL; Hutton R; Naeimi WR; Breitenbach M; Gourlay CW Loss of Cytochrome c Oxidase Promotes RAS-Dependent ROS Production from the ER Resident NADPH Oxidase, Yno1p, in Yeast. Cell Metab. 2013, 18, 279–286. [DOI] [PubMed] [Google Scholar]
- (828).Ren J-C; Rebrin I; Klichko V; Orr WC; Sohal R S. Cytochrome c Oxidase Loses Catalytic Activity and Structural Integrity during the Aging Process in Drosophila Melanogaster. Biochem. Biophys. Res. Commun 2010, 401, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Paradies G; Ruggiero FM; Petrosillo G; Quagliariello E Age-Dependent Decline in the Cytochrome c Oxidase Activity in Rat Heart Mitochondria: Role of Cardiolipin. FEBS Lett. 1997, 406, 136–138. [DOI] [PubMed] [Google Scholar]
- (830).Borisov VB; Verkhovsky MI Oxygen as Acceptor. EcoSal Plus 2009, 3, 1–32. [DOI] [PubMed] [Google Scholar]
- (831).Hemp J; Gennis RB Diversity of the Heme–Copper Superfamily in Archaea: Insights from Genomics and Structural Modeling In Bioenergetics; Springer: Berlin, 2008; Vol. 45, pp 1–31. [DOI] [PubMed] [Google Scholar]
- (832).Yeung N; Lu Y One Heme, Diverse Functions: Using Biosynthetic Myoglobin Models to Gain Insights into Heme–Copper Oxidases and Nitric Oxide Reductases. Chem. Biodiversity 2008, 5, 1437–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (833).Garcia-Horsman JA; Barquera B; Rumbley J; Ma J; Gennis RB The Superfamily of Heme-Copper Respiratory Oxidases. J. Bacteriol. 1994, 176, 5587–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (834).Shiro Y Structure and Function of Bacterial Nitric Oxide Reductases. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 1907–1913. [DOI] [PubMed] [Google Scholar]
- (835).Sousa FL; Alves RJ; Ribeiro MA; Pereira-Leal JB; Teixeira M; Pereira MM The Superfamily of Heme-copper Oxygen Reductases: Types and Evolutionary Considerations. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 629–637. [DOI] [PubMed] [Google Scholar]
- (836).Belevich I; Verkhovsky MI Molecular Mechanism of Proton Translocation by Cytochrome c Oxidase. Antioxid. Redox Signaling 2008, 10, 1–30. [DOI] [PubMed] [Google Scholar]
- (837).Agmon N The Grotthuss Mechanism. Chem. Phys. Lett 1995, 244, 456–462. [Google Scholar]
- (838).Jones AJY; Blaza JN; Varghese F; Hirst J Respiratory Complex I in Bos Taurus and Paracoccus Denitrificans Pumps Four Protons across the Membrane for Every NADH Oxidized. J. Biol. Chem 2017, 292, 4987–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (839).Choi SK; Schurig-Briccio L; Ding Z; Hong S; Sun C; Gennis RB Location of the Substrate Binding Site of the Cytochrome bo3 Ubiquinol Oxidase from Escherichia Coli. J. Am. Chem. Soc 2017, 139, 8346–8354. [DOI] [PubMed] [Google Scholar]
- (840).Abramson J; Riistama S; Larsson G; Jasaitis A; Svensson- Ek M; Laakkonen L; Puustinen A; Iwata S; Wikström M; Wikstrom M; et al. The Structure of the Ubiquinol Oxidase from Escherichia Coli and Its Ubiquinone Binding Site. Nat. Struct. Biol 2000, 7, 910–917. [DOI] [PubMed] [Google Scholar]
- (841).Fedor JG; Jones AJY; Di Luca A; Kaila VRI; Hirst J Correlating Kinetic and Structural Data on Ubiquinone Binding and Reduction by Respiratory Complex I. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 12737–12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (842).Yano N; Muramoto K; Shimada A; Takemura S; Baba J; Fujisawa H; Mochizuki M; Shinzawa-Itoh K; Yamashita E; Tsukihara T; et al. The Mg2+-Containing Water Cluster of Mammalian Cytochrome c Oxidase Collects Four Pumping Proton Equivalents in Each Catalytic Cycle. J. Biol. Chem 2016, 291, 23882–23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (843).Belevich I; Gorbikova E; Belevich NP; Rauhamaki V; Wikstrom M; Verkhovsky MI Initiation of the Proton Pump of Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18469–18474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Hemp J; Robinson DE; Ganesan KB; Martinez TJ; Kelleher NL; Gennis RB Evolutionary Migration of a Post- Translationally Modified Active-Site Residue in the Proton-Pumping Heme-Copper Oxygen Reductases. Biochemistry 2006, 45, 15405–15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (845).Das TK; Pecoraro C; Tomson FL; Gennis R B.; Rousseau, D. L. The Post-Translational Modification in Cytochrome c Oxidase Is Required To Establish a Functional Environment of the Catalytic Site. Biochemistry 1998, 37, 14471–14476. [DOI] [PubMed] [Google Scholar]
- (846).Kaila VRI; Johansson MP; Sundholm D; Laakkonen L; Wikstrom M The Chemistry of the CuB Site in Cytochrome c Oxidase and the Importance of Its Unique His-Tyr Bond. Biochim. Biophys. Acta, Bioenerg 2009, 1787, 221–233. [DOI] [PubMed] [Google Scholar]
- (847).Pinakoulaki E; Pfitzner U; Ludwig B; Varotsis C The Role of the Cross-Link His-Tyr in the Functional Properties of the Binuclear Center in Cytochrome c Oxidase. J. Biol. Chem 2002, 277, 13563–13568. [DOI] [PubMed] [Google Scholar]
- (848).Uchida T; Mogi T; Nakamura H; Kitagawa T Role of Tyr-288 at the Dioxygen Reduction Site of Cytochrome Bo Studied by Stable Isotope Labeling and Resonance Raman Spectroscopy. J. Biol. Chem 2004, 279, 53613–53620. [DOI] [PubMed] [Google Scholar]
- (849).Proshlyakov DA; Pressler MA; DeMaso C; Leykam JF; DeWitt DL; Babcock GT Oxygen Activation and Reduction in Respiration: Involvement of Redox-Active Tyrosine 244. Science 2000, 290, 1588–1591. [DOI] [PubMed] [Google Scholar]
- (850).Gorbikova EA; Belevich I; Wikstrom M; Verkhovsky MI The Proton Donor for O–O Bond Scission by Cytochrome c Oxidase. Proc. Natl. Acad. Sci U. S. A 2008, 105, 10733–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (851).Aki M; Ogura T; Naruta Y; Le TH; Sato T; Kitagawa T UV Resonance Raman Characterization of Model Compounds of Tyr 244 of Bovine Cytochrome c Oxidase in Its Neutral, Deprotonated Anionic, and Deprotonated Neutral Radical Forms: Effects of Covalent Binding Between Tyrosine and Histidine. J. Phys. Chem. A 2002, 106, 3436–3444. [Google Scholar]
- (852).McCauley KM; Vrtis JM; Dupont J; van der Donk WA Insights into the Functional Role of the Tyrosine–Histidine Linkage in Cytochrome c Oxidase. J. Am. Chem. Soc 2000, 122, 2403–2404. [Google Scholar]
- (853).Pratt DA; Pesavento RP; van der Donk WA Model Studies of the Histidine-Tyrosine Cross-Link in Cytochrome c Oxidase Reveal the Flexible Substituent Effect of the Imidazole Moiety. Org. Lett 2005, 7, 2735–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (854).Yoshikawa S; Muramoto K; Shinzawa-Itoh K The O2 Reduction and Proton Pumping Gate Mechanism of Bovine Heart Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2011, 1807, 1279–1286. [DOI] [PubMed] [Google Scholar]
- (855).Blomberg MRA; Siegbahn PEM; Wikstrom M Metal-Bridging Mechanism for O–O Bond Cleavage in Cytochrome c Oxidase. Inorg. Chem 2003, 42, 5231–5243. [DOI] [PubMed] [Google Scholar]
- (856).Dominelli-Whiteley N; Brown JJ; Muchowska KB; Mati IK; Adam C; Hubbard TA; Elmi A; Brown AJ; Bell IAW; Cockroft SL Strong Short-Range Cooperativity in Hydrogen-Bond Chains. Angew. Chem., Int. Ed 2017, 56, 7658–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (857).Ravikiran B; Mahalakshmi R Unusual Post-Translational Protein Modifications: The Benefits of Sophistication. RSC Adv. 2014, 4, 33958–33974. [Google Scholar]
- (858).Yoshikawa S; Muramoto K; Shinzawa-Itoh K; Mochizuki M Structural Studies on Bovine Heart Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 579–589. [DOI] [PubMed] [Google Scholar]
- (859).Sharma V; Wikstrom M The Role of the K-Channel and the Active-Site Tyrosine in the Catalytic Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2016, 1857, 1111–1115. [DOI] [PubMed] [Google Scholar]
- (860).MacMillan F; Kannt A; Behr J; Prisner T; Michel H Direct Evidence for a Tyrosine Radical in the Reaction of Cytochrome c Oxidase with Hydrogen Peroxide. Biochemistry 1999, 38, 9179–9184. [DOI] [PubMed] [Google Scholar]
- (861).Wikström M; Sharma V; Kaila VRI; Hosler JP; Hummer G New Perspectives on Proton Pumping in Cellular Respiration. Chem. Rev 2015, 115, 2196–2221. [DOI] [PubMed] [Google Scholar]
- (862).Brzezinski P; Adelroth P Pathways of Proton Transfer in Cytochrome c Oxidase. J. Bioenerg. Biomembr 1998, 30, 99–107. [DOI] [PubMed] [Google Scholar]
- (863).Ädelroth P; Svensson Ek M; Mitchell DM; Gennis RB; Brzezinski P Glutamate 286 in Cytochrome aa3 from Rhodobacter Sphaeroides Is Involved in Proton Uptake during the Reaction of the Fully-Reduced Enzyme with Dioxygen. Biochemistry 1997, 36, 13824–13829. [DOI] [PubMed] [Google Scholar]
- (864).Konstantinov AA; Siletsky S; Mitchell D; Kaulen A; Gennis RB The Roles of the Two Proton Input Channels in Cytochrome c Oxidase from Rhodobacter Sphaeroides Probed by the Effects of Site-Directed Mutations on Time-Resolved Electrogenic Intraprotein Proton Transfer. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (865).Liang R; Swanson JMJ; Wikstrom M; Voth GA Understanding the Essential Proton-Pumping Kinetic Gates and Decoupling Mutations in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 5924–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (866).Sharma V; Enkavi G; Vattulainen I; Róg T; Wikström M Proton-Coupled Electron Transfer and the Role of Water Molecules in Proton Pumping by Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (867).Supekar S; Gamiz-Hernandez AP; Kaila VRI A Protonated Water Cluster as a Transient Proton-Loading Site in Cytochrome c Oxidase. Angew. Chem., Int. Ed 2016, 55, 11940–11944. [DOI] [PubMed] [Google Scholar]
- (868).Siegbahn PEM; Blomberg MRA A Combined Picture from Theory and Experiments on Water Oxidation, Oxygen Reduction and Proton Pumping. Dalton Trans 2009, 0, 5832. [DOI] [PubMed] [Google Scholar]
- (869).Kubo M; Nakashima S; Yamaguchi S; Ogura T; Mochizuki M; Kang J; Tateno M; Shinzawa-Itoh K; Kato K; Yoshikawa S Effective Pumping Proton Collection Facilitated by a Copper Site (CuB) of Bovine Heart Cytochrome c Oxidase, Revealed by a Newly Developed Time-Resolved Infrared System. J. Biol. Chem 2013, 288, 30259–30269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (870).Muramoto K; Ohta K; Shinzawa-Itoh K; Kanda K; Taniguchi M; Nabekura H; Yamashita E; Tsukihara T; Yoshikawa S Bovine Cytochrome c Oxidase Structures Enable O2 Reduction with Minimization of Reactive Oxygens and Provide a Proton-Pumping Gate. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 7740–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (871).Qin L; Liu J; Mills DA; Proshlyakov DA; Hiser C; Ferguson-Miller S Redox-Dependent Conformational Changes in Cytochrome c Oxidase Suggest a Gating Mechanism for Proton Uptake. Biochemistry 2009, 48, 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (872).Ishigami I; Hikita M; Egawa T; Yeh S-R; Rousseau DL Proton Translocation in Cytochrome c Oxidase: Insights from Proton Exchange Kinetics and Vibrational Spectroscopy. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (873).Wikstrom M; Krab K; Sharma V Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev 2018, 118, 2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (874).Kaila VRI; Sharma V; Wikstrom M The Identity of the Transient Proton Loading Site of the Proton-Pumping Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2011, 1807, 80–84. [DOI] [PubMed] [Google Scholar]
- (875).Blomberg MRA; Siegbahn PEMM How Cytochrome c Oxidase Can Pump Four Protons per Oxygen Molecule at High Electrochemical Gradient. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 364–376. [DOI] [PubMed] [Google Scholar]
- (876).Babcock GT; Wikstrom M Oxygen Activation and the Conservation of Energy in Cell Respiration. Nature 1992, 356, 301–309. [DOI] [PubMed] [Google Scholar]
- (877).Jancura D; Berka V; Antalik M; Bagelova J; Gennis RB; Palmer G; Fabian M Spectral and Kinetic Equivalence of Oxidized Cytochrome c Oxidase as Isolated and “Activated” by Reoxidation. J. Biol. Chem. 2006, 281, 30319–30325. [DOI] [PubMed] [Google Scholar]
- (878).Kaila VRI; Verkhovsky MI; Wikstrom M Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev 2010, 110, 7062–7081. [DOI] [PubMed] [Google Scholar]
- (879).Nakashima S; Ogura T; Kitagawa T Infrared and Raman Spectroscopic Investigation of the Reaction Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 86–97. [DOI] [PubMed] [Google Scholar]
- (880).Ogura T Resonance Raman Applications in Investigations of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 575–578. [DOI] [PubMed] [Google Scholar]
- (881).Karpefors M; Adelroth P; Namslauer A; Zhen Y; Brzezinski P Formation of the “Peroxy” Intermediate in Cytochrome c Oxidase Is Associated with Internal Proton/Hydrogen Transfer. Biochemistry 2000, 39, 14664–14669. [DOI] [PubMed] [Google Scholar]
- (882).Karpefors M; Adelroth P; Aagaard A; Smirnova IA; Brzezinski P The Deuterium Isotope Effect as a Tool to Investigate Enzyme Catalysis: Proton-Transfer Control Mechanisms in Cytochrome c Oxidase. Isr. J. Chem 1999, 39, 427–437. [Google Scholar]
- (883).Ogura T; Kitagawa T Resonance Raman Characterization of the P Intermediate in the Reaction of Bovine Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 290–297. [DOI] [PubMed] [Google Scholar]
- (884).Yu MA; Egawa T; Shinzawa-Itoh K; Yoshikawa S; Yeh S-R; Rousseau DL; Gerfen GJ Radical Formation in Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2011, 1807, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (885).Morgan JE; Verkhovsky MI; Palmer G; Wikström M Role of the PR Intermediate in the Reaction of Cytochrome c Oxidase with O2. Biochemistry 2001, 40, 6882–6892. [DOI] [PubMed] [Google Scholar]
- (886).Wikström M Active Site Intermediates in the Reduction of O2 by Cytochrome Oxidase, and Their Derivatives. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 468–475. [DOI] [PubMed] [Google Scholar]
- (887).Yu MA; Egawa T; Shinzawa-Itoh K; Yoshikawa S; Guallar V; Yeh S -R R; Rousseau, D. L.; Gerfen, G. J. Two Tyrosyl Radicals Stabilize High Oxidation States in Cytochrome c Oxidase for Efficient Energy Conservation and Proton Translocation. J. Am. Chem. Soc 2012, 134, 4753–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (888).MacMillan F; Budiman K; Angerer H; Michel H The Role of Tryptophan 272 in the Paracoccus Denitrificans Cytochrome c Oxidase. FEBS Lett. 2006, 580, 1345–1349. [DOI] [PubMed] [Google Scholar]
- (889).Wiertz FGM; Richter OMH; Ludwig B; De Vries S Kinetic Resolution of a Tryptophan-Radical Intermediate in the Reaction Cycle of Paracoccus Denitrificans Cytochrome c Oxidase. J. Biol. Chem 2007, 282, 31580–31591. [DOI] [PubMed] [Google Scholar]
- (890).de Vries S The Role of the Conserved Tryptophan272 of the Paracoccus Denitrificans Cytochrome c Oxidase in Proton Pumping. Biochim. Biophys. Acta, Bioenerg 2008, 1777, 925–928. [DOI] [PubMed] [Google Scholar]
- (891).Al-Attar S; de Vries S Energy Transduction by Respiratory Metallo-Enzymes: From Molecular Mechanism to Cell Physiology. Coord. Chem. Rev 2013, 257, 64–80. [Google Scholar]
- (892).Pinakoulaki E; Daskalakis V; Ohta T; Richter O-MH; Budiman K; Kitagawa T; Ludwig B; Varotsis C The Protein Effect in the Structure of Two Ferryl-Oxo Intermediates at the Same Oxidation Level in the Heme Copper Binuclear Center of Cytochrome c Oxidase. J. Biol. Chem 2013, 288, 20261–20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (893).Rigby SEJ; Junemann S; Rich PR; Heathcote P Reaction of Bovine Cytochrome c Oxidase with Hydrogen Peroxide Produces a Tryptophan Cation Radical and a Porphyrin Cation Radical? Biochemistry 2000, 39, 5921–5928. [DOI] [PubMed] [Google Scholar]
- (894).Ogura T; Hirota S; Proshlyakov DA; Shinzawa-Itoh K; Yoshikawa S; Kitagawa T Time-Resolved Resonance Raman Evidence for Tight Coupling between Electron Transfer and Proton Pumping of Cytochrome c Oxidase upon the Change from the FeV Oxidation Level to the FeIV Oxidation Level. J. Am. Chem. Soc 1996, 118, 5443–5449. [Google Scholar]
- (895).Ogura T; Takahashi S; Hirota S; Shinzawa-Itoh K; Yoshikawa S; Appelman EH; Kitagawa T Time-Resolved Resonance Raman Elucidation of the Pathway for Dioxygen Reduction by Cytochrome c Oxidase. J. Am. Chem. Soc 1993, 115, 8527–8536. [Google Scholar]
- (896).Karpefors M; Adelroth P; Zhen Y; Ferguson-Miller S; Brzezinski P Proton Uptake Controls Electron Transfer in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 13606–13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (897).Proshlyakov DA; Ogura T; Shinzawa-itoh K; Yoshikawa S; Kitagawa T Resonance Raman/Absorption Characterization of the Oxo Intermediates of Cytochrome c Oxidase Generated in Its Reaction with Hydrogen Peroxide: pH and H2O2 Concentration Dependence. Biochemistry 1996, 35, 8580–8586. [DOI] [PubMed] [Google Scholar]
- (898).Belevich I; Verkhovsky MI; Wikstrom M Proton-Coupled Electron Transfer Drives the Proton Pump of Cytochrome c Oxidase. Nature 2006, 440, 829–832. [DOI] [PubMed] [Google Scholar]
- (899).Gorbikova EA; Wikstrom M; Verkhovsky MI The Protonation State of the Cross-Linked Tyrosine during the Catalytic Cycle of Cytochrome c Oxidase. J. Biol. Chem 2008, 283, 34907–34912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (900).Han S; Takahashi S; Rousseau DL Time Dependence of the Catalytic Intermediates in Cytochrome c Oxidase. J. Biol. Chem 2000, 275, 1910–1919. [DOI] [PubMed] [Google Scholar]
- (901).Sharma V; Karlin KD; Wikstrom M Computational Study of the Activated OH State in the Catalytic Mechanism of Cytochrome c Oxidase. Proc. Natl. Acad. Sci U. S. A 2013, 110, 16844–16849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (902).Blomberg MRAA; Siegbahn PEM Protonation of the Binuclear Active Site in Cytochrome c Oxidase Decreases the Reduction Potential of CuB. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 1173–1180. [DOI] [PubMed] [Google Scholar]
- (903).Blomberg MRA Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 2016, 55, 489–500. [DOI] [PubMed] [Google Scholar]
- (904).Jancura D; Antalik M; Berka V; Palmer G; Fabian M Filling the Catalytic Site of Cytochrome c Oxidase with Electrons: Reduced CuB Facilitates Internal Electron Transfer to Heme a3. J. Biol. Chem 2006, 281, 20003–20010. [DOI] [PubMed] [Google Scholar]
- (905).Verkhovsky MI; Tuukkanen A; Backgren C; Puustinen A; Wikström M Charge Translocation Coupled to Electron Injection into Oxidized Cytochrome c Oxidase from Paracoccus Denitrificans. Biochemistry 2001, 40, 7077–7083. [DOI] [PubMed] [Google Scholar]
- (906).Brändén G; Gennis R B.; Brzezinski, P. Transmembrane Proton Translocation by Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2006, 1757, 1052–1063. [DOI] [PubMed] [Google Scholar]
- (907).Bloch D; Belevich I; Jasaitis A; Ribacka C; Puustinen A; Verkhovsky MI; Wikstrom M The Catalytic Cycle of Cytochrome c Oxidase Is Not the Sum of Its Two Halves. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (908).Wikström M; Verkhovsky MI Proton Translocation by Cytochrome c Oxidase in Different Phases of the Catalytic Cycle. Biochim. Biophys. Acta, Bioenerg 2002, 1555, 128–132. [DOI] [PubMed] [Google Scholar]
- (909).Noodleman L; Han Du W-G; Fee JA; Götz AW; Walker RC Linking Chemical Electron-Proton Transfer to Proton Pumping in Cytochrome c Oxidase: Broken-Symmetry DFT Exploration of Intermediates along the Catalytic Reaction Pathway of the Iron–Copper Dinuclear Complex. Inorg. Chem 2014, 53, 6458–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (910).Kieber-Emmons MT; Li Y; Halime Z; Karlin KD; Solomon EI Electronic Structure of a Low-Spin heme/Cu Peroxide Complex: Spin-State and Spin-Topology Contributions to Reactivity. Inorg. Chem 2011, 50, 11777–11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (911).Aoyama H; Muramoto K; Shinzawa-Itoh K; Hirata K; Yamashita E; Tsukihara T; Ogura T; Yoshikawa S A Peroxide Bridge between Fe and Cu Ions in the O2 Reduction Site of Fully Oxidized Cytochrome c Oxidase Could Suppress the Proton Pump. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2165–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (912).Szundi I; Funatogawa C; Fee JA; Soulimane T; Einarsdóttir Ó CO Impedes Superfast O2 Binding in ba3 Cytochrome Oxidase from Thermus Thermophilus. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (913).Proshlyakov DA; Pressler MA; Babcock GT Dioxygen Activation and Bond Cleavage by Mixed-Valence Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 8020–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (914).Blomberg MRA; Siegbahn PEM; Babcock GT; Wikstrom MM Modeling Cytochrome Oxidase: A Quantum Chemical Study of the O–O Bond Cleavage Mechanism. J. Am. Chem. Soc 2000, 122, 12848–12858. [Google Scholar]
- (915).Han Du W-G; Noodleman L; Du W-GH; Noodleman L Density Functional Study for the Bridged Dinuclear Center Based on a High-Resolution X-Ray Crystal Structure of ba3 Cytochrome c Oxidase from Thermus Thermophilus. Inorg. Chem 2013, 52, 14072–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (916).Han Du W-G; Noodleman L Broken Symmetry DFT Calculations/Analysis for Oxidized and Reduced Dinuclear Center in Cytochrome c Oxidase: Relating Structures, Protonation States, Energies, and Mössbauer Properties in ba3 Thermus Thermophilus. Inorg. Chem 2015, 54, 7272–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (917).Han Du W-G; Götz AW; Yang L; Walker R C.; Noodleman, L. A Broken-Symmetry Density Functional Study of Structures, Energies, and Protonation States along the Catalytic O–O Bond Cleavage Pathway in ba3 Cytochrome c Oxidase from Thermus Thermophilus. Phys. Chem. Chem. Phys 2016, 18, 21162–21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (918).Poiana F; von Ballmoos C; Gonska N; Blomberg MRA; Ädelroth P; Brzezinski P Splitting of the O–O Bond at the Heme-Copper Catalytic Site of Respiratory Oxidases. Sci. Adv 2017, 3, No. e1700279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (919).Adam SM; Garcia-Bosch I; Schaefer AW; Sharma SK; Siegler MA; Solomon EI; Karlin KD Critical Aspects of Heme-Peroxo-Cu Complex Structure and Nature of Proton Source Dictate Metal-Operoxo Breakage versus Reductive O–O Cleavage Chemistry. J. Am. Chem. Soc 2017, 139, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (920).Sharma V; Wikstrom M; Kaila VRI Stabilization of the Peroxy Intermediate in the Oxygen Splitting Reaction of Cytochrome cbb3 Biochim. Biophys. Acta, Bioenerg 2011, 1807, 813–818. [DOI] [PubMed] [Google Scholar]
- (921).Coliman JP; Decréau RA Functional Biomimetic Models for the Active Site in the Respiratory Enzyme Cytochrome c Oxidase. Chem. Commun 2008, 5065–5076. [DOI] [PubMed] [Google Scholar]
- (922).Karlin K Metalloenzymes, Structural Motifs, and Inorganic Models. Science 1993, 261, 701–708. [DOI] [PubMed] [Google Scholar]
- (923).Decréau RA; Collman JP; Hosseini A Electrochemical Applications. How Click Chemistry Brought Biomimetic Models to the Next Level: Electrocatalysis Under Controlled Rate of Electron Transfer. Chem. Soc. Rev 2010, 39, 1291–1301. [DOI] [PubMed] [Google Scholar]
- (924).Ricard D; Didier A; L’Her M; Boitrel B Application of 3-Quinolinoyl Picket Porphyrins to the Electroreduction of Dioxygen to Water: Mimicking the Active Site of Cytochrome c Oxidase. ChemBioChem 2001, 2, 144–148. [DOI] [PubMed] [Google Scholar]
- (925).Collman JP; Fu L; Herrmann PC; Wang Z; Rapta M; Bröring M; Schwenninger R; Boitrel B A Functional Model of Cytochrome c Oxidase: Thermodynamic Implications. Angew. Chem. Int. Ed 1998, 37, 3397–3400. [DOI] [PubMed] [Google Scholar]
- (926).Ghiladi RA; Hatwell KR; Karlin KD; Huang HW; Moënne-Loccoz P; Krebs C; Huynh BH; Marzilli LA; Cotter RJ; Kaderli S; et al. Dioxygen Reactivity of Mononuclear Heme and Copper Components Yielding a High-Spin Heme-Peroxo-Cu Complex. J. Am. Chem. Soc 2001, 123, 6183–6184. [DOI] [PubMed] [Google Scholar]
- (927).Ghiladi RA; Chufán EE; del Río D; Solomon EI; Krebs C; Huynh BH; Huang H; Moenne-Loccoz P; Kaderli S; Honecker M; et al. Further Insights into the Spectroscopic Properties, Electronic Structure, and Kinetics of Formation of the Heme-Peroxo- Copper Complex [(F8TPP)FeI I I-(O22–)–CuII(TMPA)]+. Inorg. Chem 2007, 46, 3889–3902. [DOI] [PubMed] [Google Scholar]
- (928).Halime Z; Kieber-Emmons MT; Qayyum MF; Mondal B; Gandhi T; Puiu SC; Chufán EE; Sarjeant AAN; Hodgson KO; Hedman B; et al. Heme-Copper-Dioxygen Complexes: Toward Understanding Ligand-Environmental Effects on the Coordination Geometry, Electronic Structure, and Reactivity. Inorg. Chem 2010, 49, 3629–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (929).del Río D; Sarangi R; Chufán EE; Karlin KD; Hedman B; Hodgson KO; Solomon EI Geometric and Electronic Structure of the Heme-Peroxo-Copper Complex [(F8TPP)FeIII-(O22-)–CuII(TMPA)](ClO4). J. Am. Chem. Soc 2005, 127, 11969–11978. [DOI] [PubMed] [Google Scholar]
- (930).Naruta Y; Sasaki T; Tani F; Tachi Y; Kawato N; Nakamura N Heme-Cu Complexes as Oxygen-Activating Functional Models for the Active Site of Cytochrome c Oxidase. J. Inorg. Biochem 2001, 83, 239–246. [DOI] [PubMed] [Google Scholar]
- (931).Chishiro T; Shimazaki Y; Tani F; Tachi Y; Naruta Y; Karasawa S; Hayami S; Maeda Y 1solation and Crystal Structure of a Peroxo-Bridged Heme–Copper Complex. Angew. Chem. Int. Ed 2003, 42, 2788–2791. [DOI] [PubMed] [Google Scholar]
- (932).Kieber-Emmons MT; Qayyum MF; Li Y; Halime Z; Hodgson KO; Hedman B; Karlin KD; Solomon E 1. Spectroscopic Elucidation of a New Heme/Copper Dioxygen Structure Type: 1mplications for O···O Bond Rupture in Cytochrome c Oxidase. Angew. Chem 2012, 124, 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (933).Collman JP; Herrmann PC; Boitrel B; Zhang X; Eberspacher TA; Fu L; Wang J; Rousseau DL; Williams ER A Synthetic Analogue for the Oxygen Binding Site in Cytochrome c Oxidase. J. Am. Chem. Soc 1994, 116, 9783–9784. [Google Scholar]
- (934).Sasaki T; Nakamura N; Naruta Y Formation and Spectroscopic Characterization of the Peroxo FeIII-CuII Complex. A Modeling Reaction of the Heme-Cu Site in Cytochrome c Oxidase. Chem. Left 1998, 27, 351–352. [Google Scholar]
- (935).Liu J-G; Naruta Y; Tani F Synthetic Models of the Active Site of Cytochrome c Oxidase: Influence of Tridentate or Tetradentate Copper Chelates Bearing a His-Tyr Linkage Mimic on Dioxygen Adduct Formation by heme/Cu Complexes. Chem. −3 Eur. J 2007, 13, 6365–6378. [DOI] [PubMed] [Google Scholar]
- (936).Ghiladi RA; Ju TD; Lee D-H; Moënne-Loccoz P; Kaderli S; Neuhold Y-M; Zuberbuhler AD; Woods AS; Cotter RJ; Karlin KD Formation and Characterization of a High-Spin Heme-Copper Dioxygen (Peroxo) Complex. J. Am. Chem. Soc 1999, 121, 9885–9886. [Google Scholar]
- (937).Kim E; Kamaraj K; Galliker B; Rubie ND; Moënne-Loccoz P; Kaderli S; Zuberbuhler AD; Karlin KD Dioxygen Reactivity of Copper and Heme-Copper Complexes Possessing an 1midazole–Phenol Cross-Link. Inorg. Chem 2005, 44, 1238–1247. [DOI] [PubMed] [Google Scholar]
- (938).Kim E; Shearer J; Lu S; Moënne-Loccoz P; Helton ME; Kaderli S; Zuberbühler AD; Karlin KD Heme/Cu/O2 Reactivity: Change in FeIII-(O22-)–CuII Unit Peroxo Binding Geometry Effected by Tridentate Copper Chelation. J. Am. Chem. Soc 2004, 126, 12716–12717. [DOI] [PubMed] [Google Scholar]
- (939).Ghiladi RA; Huang H; Moenne-Loccoz P; Stasser J; Blackburn NJ; Woods AS; Cotter RJ; 1ncarvito, C. D.; Rheingold, A. L.; Karlin, K. D. Heme-Copper/Dioxygen Adduct Formation Relevant to Cytochrome c Oxidase: Spectroscopic Characterization of [(6L)FeIII-(O22-)-CuII]+. JBIC, J. Biol. Inorg. Chem 2005, 10, 63–77. [DOI] [PubMed] [Google Scholar]
- (940).Fry HC; Scaltrito DV; Karlin KD; Meyer GJ The Rate of O2 and CO Binding to a Copper Complex, Determined by a “Flash-and-Trap” technique, Exceeds That for Hemes. J. Am. Chem. Soc 2003, 125, 11866–11871. [DOI] [PubMed] [Google Scholar]
- (941).Kretzer RM; Ghiladi RA; Lebeau EL; Liang H-C; Karlin KD Synthesis and Characterization of Reduced Heme and Heme/Copper Carbonmonoxy Species. Inorg. Chem 2003, 42, 3016–3025. [DOI] [PubMed] [Google Scholar]
- (942).Ferguson-Miller S; Babcock GT Heme/Copper Terminal Oxidases. Chem. Rev 1996, 96, 2889–2907. [DOI] [PubMed] [Google Scholar]
- (943).Hematian S; Garcia-Bosch, 1.; Karlin, K. D. Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase 1nteractions with Dioxygen and Nitrogen Oxides. Acc. Chem. Res 2015, 48, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (944).Citek C; Lyons CT; Wasinger EC; Stack TDP Self-Assembly of the Oxy-Tyrosinase Core and the Fundamental Components of Phenolic Hydroxylation. Nat. Chem 2012, 4, 317–322. [DOI] [PubMed] [Google Scholar]
- (945).Marlin DS; Olmstead MM; Mascharak PK Structure-Spectroscopy Correlation in Distorted Five-Coordinate Cu(11) Complexes: A Case Study with a Set of Closely Related Copper Complexes of Pyridine-2,6-Dicarboxamide Ligands. Inorg. Chem 2001, 40, 7003–7008. [DOI] [PubMed] [Google Scholar]
- (946).Yoshikawa S; Shinzawa-1toh K; Nakashima R; Yaono R; Yamashita E; 1noue N; Yao M; Fei MJ; Libeu CP; Mizushima T; et al. Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science 1998, 280, 1723–1729. [DOI] [PubMed] [Google Scholar]
- (947).Koutsoupakis C; Pinakoulaki E; Stavrakis S; Daskalakis V; Varotsis C Time-Resolved Step-Scan Fourier Transform 1nfrared 1nvestigation of Heme-Copper Oxidases: 1mplications for O2 1nput and H2O/H+ Output Channels. Biochim. Biophys. Ada, Bioenerg 2004, 1655, 347–352. [DOI] [PubMed] [Google Scholar]
- (948).McMahon BH; Fabian M; Tomson F; Causgrove TP; Bailey JA; Rein FN; Dyer RB; Palmer G; Gennis RB; Woodruff WH FT1R Studies of 1nternal Proton Transfer Reactions Linked to 1nter-Heme Electron Transfer in Bovine Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 321–331. [DOI] [PubMed] [Google Scholar]
- (949).Kim Y; Shinzawa-1toh, K.; Yoshikawa, S.; Kitagawa, T. Presence of the Heme-Oxo 1ntermediate in Oxygenation of Carbon Monoxide by Cytochrome c Oxidase Revealed by Resonance Raman Spectroscopy. J. Am. Chem. Soc 2001, 123, 757–758. [DOI] [PubMed] [Google Scholar]
- (950).Simonneaux G Phosphines as Structural and Functional Probes of Hemoproteins. Coord. Chem. Rev 1997, 165, 447–474. [Google Scholar]
- (951).Dallacosta C; Alves WA; da Costa Ferreira AM; Monzani E; Casella L A New Dinuclear Heme-Copper Complex Derived from Functionalized Protoporphyrin 1X. Dalton Trans 2007, 0, 2197. [DOI] [PubMed] [Google Scholar]
- (952).Pirota V; Gennarini F; Dondi D; Monzani E; Casella L; Dell’Acqua S Dinuclear Heme and Non-Heme Metal Complexes as Bioinspired Catalysts for Oxidation Reactions. New J. Chem 2014, 38, 518–528. [Google Scholar]
- (953).Birukou I; Soman J; Olson JS Blocking the Gate to Ligand Entry in Human Hemoglobin. J. Biol. Chem 2011, 286, 10515–10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (954).Abbruzzetti S; Spyrakis F; Bidon-Chanal A; Luque FJ; Viappiani C Ligand Migration through Hemeprotein Cavities: Insights from Laser Flash Photolysis and Molecular Dynamics Simulations. Phys. Chem. Chem. Phys 2013, 15, 10686–10701. [DOI] [PubMed] [Google Scholar]
- (955).Hargrove MS A Flash Photolysis Method to Characterize Hexacoordinate Hemoglobin Kinetics. Biophys. J 2000, 79, 2733–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (956).Zhang L; Andersen EME; Khajo A; Magliozzo RS; Koder RL Dynamic Factors Affecting Gaseous Ligand Binding in an Artificial Oxygen Transport Protein. Biochemistry 2013, 52, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (957).Babcock GT How Oxygen Is Activated and Reduced in Respiration. Proc. Natl. Acad. Sci U. S. A 1999, 96, 12971–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (958).Kitagawa T; Ogura T Oxygen Activation Mechanism at the Binuclear Site of Heme–Copper Oxidase Superfamily as Revealed by Time-Resolved Resonance Raman Spectroscopy. Prog. Inorg. Chem 2007, 45, 431–479. [Google Scholar]
- (959).Szundi I; Liao G-L; Einarsdóttir Ó Near-Infrared Time- Resolved Optical Absorption Studies of the Reaction of Fully Reduced Cytochrome c Oxidase with Dioxygen. Biochemistry 2001, 40, 2332–2339. [DOI] [PubMed] [Google Scholar]
- (960).Giuffrè A; Forte E; Antonini G; D’Itri E; Brunori M; Soulimane T; Buse G Kinetic Properties of Ba 3 Oxidase from Thermus Thermophilus : Effect of Temperature. Biochemistry 1999, 38, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (961).Woodruff WH Coordination Dynamics of Heme-Copper Oxidases. The Ligand Shuttle and the Control and Coupling of Electron Transfer and Proton Translocation. J. Bioenerg. Biomembr 1993, 25, 177–188. [DOI] [PubMed] [Google Scholar]
- (962).Einarsdottir O; Dyer RB; Lemon DD; Killough PM; Hubig SM; Atherton SJ; Lopez-Garriga JJ; Palmer G; Woodruff WH; et al. Photodissociation and Recombination of Carbonmonoxy Cytochrome Oxidase: Dynamics from Picoseconds to Kiloseconds. Biochemistry 1993, 32, 12013–12024. [DOI] [PubMed] [Google Scholar]
- (963).Pinakoulaki E; Soulimane T; Varotsis C Fourier Transform Infrared (FTIR) and Step-Scan Time-Resolved FTIR Spectroscopies Reveal a Unique Active Site in Cytochrome caa3 Oxidase from Thermus Thermophilus. J. Biol. Chem 2002, 277, 32867–32874. [DOI] [PubMed] [Google Scholar]
- (964).Tsuchida E; Komatsu T; Arai K; Nishide H Synthesis and Dioxygen-Binding Properties of Double-Sided Porphyrinatoiron(II) Complexes Bearing Covalently Bound Axial Imidazole. J. Chem. Soc., Dalton Trans 1993, 0, 2465–2469. [Google Scholar]
- (965).Linard JE; Ellis PE; Budge JR; Jones RD; Basolo F Oxygenation of iron(II) and cobalt(II) “capped” porphyrins. J. Am. Chem. Soc 1980, 102, 1896–1904. [Google Scholar]
- (966).Rondelez Y; Sénèque O; Rager M-N; Duprat AF; Reinaud O Biomimetic Copper(I)-CO Complexes: A Structural and Dynamic Study of a Calix[6]arene-Based Supramolecular System. Chem. −3 Eur. J 2000, 6, 4218–4226. [DOI] [PubMed] [Google Scholar]
- (967).Hirota S; Iwamoto T; Tanizawa K; Adachi O; Yamauchi O Spectroscopic Characterization of Carbon Monoxide Complexes Generated for Copper/Topa Quinone-Containing Amine Oxidases. Biochemistry 1999, 38, 14256–14263. [DOI] [PubMed] [Google Scholar]
- (968).Fry HC; Cohen AD; Toscano JP; Meyer GJ; Karlin KD Photoinduced Carbon Monoxide Migration in a Synthetic Heme-Copper Complex. J. Am. Chem. Soc 2005, 127, 6225–6230. [DOI] [PubMed] [Google Scholar]
- (969).Lucas HR; Meyer GJ; Karlin KD Carbon Monoxide and Nitrogen Monoxide Ligand Dynamics in Synthetic Heme and Heme–Copper Complex Systems. J. Am. Chem. Soc 2009, 131 , 13924–13925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (970).Gibson QH; Greenwood C Reactions of Cytochrome Oxidase with Oxygen and Carbon Monoxide. Biochem. J 1963, 86, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (971).Fiamingo FG; Altschuld RA; Moh PP; Alben JO Dynamic Interactions of CO with a3Fe and CuB in Cytochrome c Oxidase in Beef Heart Mitochondria Studied by Fourier Transform Infrared Spectroscopy at Low Temperatures. J. Biol. Chem 1982, 257, 1639–1650. [PubMed] [Google Scholar]
- (972).Treuffet J; Kubarych KJ; Lambry J-C; Pilet E; Masson J-B; Martin J-L; Vos MH; Joffre M; Alexandrou A Direct Observation of Ligand Transfer and Bond Formation in Cytochrome c Oxidase by Using Mid-Infrared Chirped-Pulse Upconversion. Proc. Natl. Acad. Sci U. S. A 2007, 104, 15705–15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (973).Ohta K; Muramoto K; Shinzawa-Itoh K; Yamashita E; Yoshikawa S; Tsukihara T X-Ray Structure of the NO-Bound CuB in Bovine Cytochrome c Oxidase. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun 2010, 66, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (974).Liu B; Zhang Y; Sage JT; Soltis SM; Doukov T; Chen Y; Stout CD; Fee JA Structural Changes That Occur upon Photolysis of the Fe(II)a3–CO Complex in the Cytochrome ba3-Oxidase of Thermus Thermophilus: A Combined X-Ray Crystallographic and Infrared Spectral Study Demonstrates CO Bindin. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (975).Oliveberg M; Malmstrom BG Reaction of Dioxygen with Cytochrome c Oxidase Reduced to Different Degrees: Indications of a Transient Dioxygen Complex with Copper-B. Biochemistry 1992, 31 , 3560–3563. [DOI] [PubMed] [Google Scholar]
- (976).Blackmore RS; Greenwood C; Gibson QH Studies of the Primary Oxygen Intermediate in the Reaction of Fully Reduced Cytochrome Oxidase. J. Biol. Chem 1991, 266, 19245–19249. [PubMed] [Google Scholar]
- (977).Karlin KD; Wei N; Jung B; Kaderli S; Niklaus P; Zuberbuhler AD Kinetics and Thermodynamics of Formation of Copper-Dioxygen Adducts: Oxygenation of Mononuclear Cu(I) Complexes Containing Tripodal Tetradentate Ligands. J. Am. Chem. Soc 1993, 115, 9506–9514. [Google Scholar]
- (978).Babcock GT; Floris R; Nilsson T; Pressler M; Varotsis C; Vollenbroek E Dioxygen Activation in Enzymatic Systems and in Inorganic Models. Inorg. Chim. Acta 1996, 243, 345–353. [Google Scholar]
- (979).Varotsis C; Zhang Y; Appelman EH; Babcock GT Resolution of the Reaction Sequence during the Reduction of O2 by Cytochrome Oxidase. Proc. Natl. Acad. Sci U. S. A 1993, 90, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (980).Egawa T; Chen Y; Fee JA; Yeh S-R; Rousseau DL The Rate-Limiting Step in O2 Reduction by Cytochrome ba3 from Thermus Thermophilus. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (981).Shimada A; Kubo M; Baba S; Yamashita K; Hirata K; Ueno G; Nomura T; Kimura T; Shinzawa-Itoh K; Baba J; et al. A Nanosecond Time-Resolved XFEL Analysis of Structural Changes Associated with CO Release from Cytochrome c Oxidase. Sci. Adv 2017, 3, e1603042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (982).Tsukihara T; Aoyama H; Yamashita E; Tomizaki T; Yamaguchi H; Shinzawa-Itoh K; Nakashima R; Yaono R; Yoshikawa S Structures of Metal Sites of Oxidized Bovine Heart Cytochrome c Oxidase at 2.8 A. Science 1995, 269, 1069–1074. [DOI] [PubMed] [Google Scholar]
- (983).Tsukihara T; Aoyama H; Yamashita E; Tomizaki T; Yamaguchi H; Shinzawa-Itoh K; Nakashima R; Yaono R; Yoshikawa S The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 Á. Science 1996, 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- (984).Iwata S; Ostermeier C; Ludwig B; Michel H Structure at 2.8 Å Resolution of Cytochrome c Oxidase from Paracoccus Denitrificans. Nature 1995, 376, 660–669. [DOI] [PubMed] [Google Scholar]
- (985).Koepke J; Olkhova E; Angerer H; Müller H; Peng G; Michel H High Resolution Crystal Structure of Paracoccus Denitrificans Cytochrome c Oxidase: New Insights into the Active Site and the Proton Transfer Pathways. Biochim. Biophys. Acta, Bioenerg 2009, 1787, 635–645. [DOI] [PubMed] [Google Scholar]
- (986).Svensson-Ek M; Abramson J; Larsson G; Tornroth S; Brzezinski P; Iwata S The X-Ray Crystal Structures of Wild-Type and EQ(I-286) Mutant Cytochrome c Oxidases from Rhodobacter Sphaeroides. J. Mol. Biol 2002, 321, 329–339. [DOI] [PubMed] [Google Scholar]
- (987).Soulimane T Structure and Mechanism of the Aberrant ba3-Cytochrome c Oxidase from Thermus Thermophilus. EMBO J. 2000, 19, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (988).Tiefenbrunn T; Liu W; Chen Y; Katritch V; Stout CD; Fee JA; Cherezov V High Resolution Structure of the ba3 Cytochrome c Oxidase from Thermus Thermophilus in a Lipidic Environment. PLoS One 2011, 6, e22348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (989).Andersson R; Safari C; Dods R; Nango E; Tanaka R; Yamashita A; Nakane T; Tono K; Joti Y; Båth P; et al. Serial Femtosecond Crystallography Structure of Cytochrome c Oxidase at Room Temperature. Sci Rep. 2017, 7, 4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (990).Einarsdóttir Ó; Funatogawa C; Soulimane T; Szundi I Kinetic Studies of the Reactions of O2 and NO with Reduced Thermus Thermophilus ba3 and Bovine aa3 Using Photolabile Carriers. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (991).Einarsdóttir Ó; McDonald W; Funatogawa C; Szundi I; Woodruff WH; Dyer RB The Pathway of O2 to the Active Site in Heme-copper Oxidases. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (992).Stavrakis S; Koutsoupakis K; Pinakoulaki E; Urbani A; Saraste M; Varotsis C Decay of the Transient CuB-CO Complex Is Accompanied by Formation of the Heme Fe-CO Complex of Cytochrome cbb3-CO at Ambient Temperature: Evidence from Time-Resolved Fourier Transform Infrared Spectroscopy. J. Am. Chem. Soc 2002, 124, 3814–3815. [DOI] [PubMed] [Google Scholar]
- (993).Iwase T; Varotsis C; Shinzawa-Itoh K; Yoshikawa S; Kitagawa T Infrared Evidence for CuB Ligation of Photodissociated CO of Cytochrome c Oxidase at Ambient Temperatures and Accompanied Deprotonation of a Carboxyl Side Chain of Protein. J.Am. Chem. Soc 1999, 121, 1415–1416. [Google Scholar]
- (994).Pinakoulaki E; Koutsoupakis C; Stavrakis S; Aggelaki M; Papadopoulos G; Daskalakis V; Varotsis C Structural Dynamics of Heme-Copper Oxidases and Nitric Oxide Reductases: Time-Resolved Step-Scan Fourier Transform Infrared and Time-Resolved Resonance Raman Studies. J. Raman Spectrosc 2005, 36, 337–349. [Google Scholar]
- (995).Kitagawa T; Ogura T Oxygen Activation Mechanism at the Binuclear Site of Heme-Copper Oxidase Superfamily as Revealed by Time-Resolved Resonance Raman Spectroscopy Progress in Inorganic Chemistry; John Wiley & Sons, Inc., 1997; Vol. 45, pp 431–479. [Google Scholar]
- (996).Larsen RW; Miksovska J Time Resolved Thermodynamics of Ligand Binding to Heme Proteins. Coord. Chem. Rev 2007, 251, 1101–1127. [Google Scholar]
- (997).Vos MH Ultrafast Dynamics of Ligands within Heme Proteins. Biochim. Biophys. Acta, Bioenerg 2008, 1777, 15–31. [DOI] [PubMed] [Google Scholar]
- (998).Cassano JA; Choi SK; McDonald W; Szundi I; Villa Gawboy TR; Gennis RB; Einarsdottir O The CO Photodissociation and Recombination Dynamics of the W172Y/F282T Ligand Channel Mutant of Rhodobacter Sphaeroides aa3 Cytochrome c Oxidase. Photochem. Photobiol 2016, 92, 410–419. [DOI] [PubMed] [Google Scholar]
- (999).von Ballmoos C; Smirnova I; Poiana F; Gonska N; Chang H-Y; Gennis RB; Brzezinski P; Adelroth P Dynamics of the KB Proton Pathway in Cytochrome ba3 from Thermus Thermophilus. Isr. J. Chem 2017, 57, 424–436. [Google Scholar]
- (1000).Mahinthichaichan P; Gennis RB; Tajkhorshid E All the O2 Consumed by Thermus Thermophilus Cytochrome ba3 Is Delivered to the Active Site through a Long, Open Hydrophobic Tunnel with Entrances within the Lipid Bilayer. Biochemistry 2016, 55, 1265–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1001).McDonald W; Funatogawa C; Li Y; Szundi I; Chen Y; Fee JA; Stout CD; Einarsdottir O Ligand Access to the Active Site in Thermus Thermophilus ba3 and Bovine Heart aa3 Cytochrome Oxidases. Biochemistry 2013, 52, 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1002).Lucas HR; Meyer GJ; Karlin KD CO and O2 Binding to Pseudo-Tetradentate Ligand-Copper(I) Complexes with a Variable N-Donor Moiety: Kinetic/Thermodynamic Investigation Reveals Ligand-Induced Changes in Reaction Mechanism. J. Am. Chem. Soc 2010, 132, 12927–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1003).Karlin KD; Kaderli S; Zuberbühler AD Kinetics and Thermodynamics of Copper(I)/Dioxygen Interaction. Acc. Chem. Res 1997, 30, 139–147. [Google Scholar]
- (1004).Kunishita A; Ertem MZ; Okubo Y; Tano T; Sugimoto H; Ohkubo K; Fujieda N; Fukuzumi S; Cramer CJ; Itoh S Active Site Models for the CuA Site of Peptidylglycine α-Hydroxylating Monooxygenase and Dopamine β-Monooxygenase. Inorg. Chem 2012, 51, 9465–9480. [DOI] [PubMed] [Google Scholar]
- (1005).Szundi I; Kittredge C; Choi SK; McDonald W; Ray J; Gennis RB; Einarsdóttir Ó Kinetics and Intermediates of the Reaction of Fully Reduced Escherichia Coli bo3 Ubiquinol Oxidase with O2. Biochemistry 2014, 53, 5393–5404. [DOI] [PubMed] [Google Scholar]
- (1006).Niederhoffer EC; Timmons JH; Martell AE Thermodynamics of Oxygen Binding in Natural and Synthetic Dioxygen Complexes. Chem. Rev 1984, 84, 137. [Google Scholar]
- (1007).Jones RD; Summerville DA; Basolo F Review on Cobalt Oxygen Complexes. Chem. Rev 1979, 79, 139–179. [Google Scholar]
- (1008).Slebodnick C; Ibers JA Myoglobin Models and Steric Origins of the Discrimination between O2 and CO. JBIC, J. Biol. Inorg. Chem 1997, 2, 521–525. [Google Scholar]
- (1009).Collman JP; Gagne RR; Reed C; Halbert TR; Lang G; Robinson WT Picket Fence Porphyrins. Synthetic Models for Oxygen Binding Hemoproteins. J. Am. Chem. Soc 1975, 97, 1427–1439. [DOI] [PubMed] [Google Scholar]
- (1010).Burke JM; Kincaid JR; Peters S; Gagne RR; Collman JP; Spiro TG Structure-Sensitive Resonance Raman Bands of Tetraphenyl and “Picket Fence” porphyrin-Iron Complexes, Including an Oxyhemoglobin Analog. J. Am. Chem. Soc 1978, 100, 6083–6088. [Google Scholar]
- (1011).Halime Z; Karlin KD; Rokia SE Cytochrome c Oxidase and Models In Copper-Oxygen Chemistry; Itoh S, Karlin KD, Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2011; Vol. 4, pp 283–319. [Google Scholar]
- (1012).Collman JP; Fu L; Herrmann PC; Zhang X Functional Model Related to Cytochrome c Oxidase and Its Electrocatalytic Four-Electron Reduction of O2. Science 1997, 275, 949–951. [DOI] [PubMed] [Google Scholar]
- (1013).Collman JP; Schwenninger R; Rapta M; Bröring M; Fu L New 1,4,7-Triazacyclononane-Based Functional Analogues of the Fe/Cu Active Site of Cytochrome c Oxidase: Structure, Spectroscopy and Electrocatalytic Reduction of Oxygen. Chem. Commun 1999, 137–138. [Google Scholar]
- (1014).Collman JP Functional Analogs of Heme Protein Active Sites. Inorg. Chem 1997, 36, 5145–5155. [Google Scholar]
- (1015).Ruzié C; Even-Hernandez P; Boitrel B Efficient and Versatile Synthesis of New Porphyrins Bearing an N3O Moiety: Ligands for Mimicking Cytochrome c Oxidase. Org. Lett 2008, 10, 2673–2676. [DOI] [PubMed] [Google Scholar]
- (1016).Lo M; Mahajan D; Wytko JA; Boudon C; Weiss J Toward Generic Models of Hemoproteins. Org. Lett 2009, 11 , 2487–2490. [DOI] [PubMed] [Google Scholar]
- (1017).Melin F; Boudon C; Lo M; Schenk KJ; Bonin M; Ochsenbein P; Gross M; Weiss J Electrocatalytic Reduction of Oxygen with Phenanthroline-Strapped Porphyrins. J. Porphyrins Phthalocyanines 2007, 11, 212–221. [Google Scholar]
- (1018).Melin F; Trivella A; Lo M; Ruzie C; Hijazi I; Oueslati N; Wytko JA; Boitrel B; Boudon C; Hellwig P; et al. Comparative Studies in Series of Cytochrome c Oxidase Models. J. Inorg. Biochem 2012, 108, 196–202. [DOI] [PubMed] [Google Scholar]
- (1019).Ricard D; Andrioletti B; Boitrel B; L’Her M Electrocatalytic Reduction of Dioxygen to Water by Tren-Capped Porphyrins, Functional Models of Cytochrome c Oxidase. Chem. Commun 1999, 1523–1524. [Google Scholar]
- (1020).Vorburger P; Lo M; Choua S; Bernard M; Melin F; Oueslati N; Boudon C; Elhabiri M; Wytko JA; Hellwig P; et al. A Question of Flexibility in Cytochrome c Oxidase Models. Inorg. Chim. Acta 2017, 468, 232–238. [Google Scholar]
- (1021).Kitagishi H; Shimoji D; Ohta T; Kamiya R; Kudo Y; Onoda A; Hayashi T; Weiss J; Wytko JA; Kano K A Water- Soluble Supramolecular Complex That Mimics the Heme/copper Hetero-Binuclear Site of Cytochrome c Oxidase. Chem. Sci 2018, 9, 1989–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1022).Kano K; Kitagishi H; Tamura S; Yamada A Anion Binding to a Ferric Porphyrin Complexed with Per-O-Methylated-Cyclodextrin in Aqueous Solution. J. Am. Chem. Soc 2004, 126, 15202–15210. [DOI] [PubMed] [Google Scholar]
- (1023).Fox S; Nanthakumar A; Wikström M; Karlin KD; Blackburn NJ XAS Structural Comparisons of Reversibly Interconvertible Oxo- and Hydroxo-Bridged Heme-Copper Oxidase Model Compounds. J. Am. Chem. Soc 1996, 118, 24–34. [Google Scholar]
- (1024).Varotsis C; Woodruff WH; Babcock GT Direct Detection of a Dioxygen Adduct of Cytochrome a3 in the Mixed Valence Cytochrome Oxidase/Dioxygen Reaction. J. Biol. Chem 1990, 265, 11131–11136. [PubMed] [Google Scholar]
- (1025).Collman JP; Decmau R A.; Yan, Y.; Yoon, J.; Solomon, E. I. Intramolecular Single-Turnover Reaction in a Cytochrome c Oxidase Model Bearing a Tyr244 Mimic. J. Am. Chem. Soc 2007, 129, 5794–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1026).Ladomenou K; Charalambidis G; Coutsolelos AG Spectroscopic and Electrochemical Studies of Novel Model Compounds for Cytochrome c Oxidase. Inorg. Chim. Acta 2010, 363, 2201–2208. [Google Scholar]
- (1027).Ladomenou K; Charalambidis G; Coutsolelos AG CO and O2 Binding Studies of New Model Complexes for CcO. Polyhedron 2013, 54, 47–53. [Google Scholar]
- (1028).Charalambidis G; Ladomenou K; Boitrel B; Coutsolelos AG Synthesis and Studies of a Super-Structured Porphyrin Derivative −3 A Potential Building Block for CcO Mimic Models. Eur. J. Org. Chem 2009, 2009, 1263–1268. [Google Scholar]
- (1029).Collman JP; Rapta M; Bröring M; Raptova L; Schwenninger R; Boitrel B; Fu L; L’Her M Close Structural Analogues of the Cytochrome c Oxidase Fe a3/CuB Center Show Clean 4e− Electroreduction of O2 to H2O at Physiological pH. J. Am. Chem. Soc 1999, 121, 1387–1388. [Google Scholar]
- (1030).Collman JP; Sunderland CJ; Boulatov R Biomimetic Studies of Terminal Oxidases: Trisimidazole Picket Metalloporphyr- ins. Inorg. Chem 2002, 41, 2282–2291. [DOI] [PubMed] [Google Scholar]
- (1031).Boulatov R; Collman JP; Shiryaeva IM; Sunderland CJ Functional Analogues of the Dioxygen Reduction Site in Cytochrome Oxidase: Mechanistic Aspects and Possible Effects of Cub. J. Am. Chem. Soc 2002, 124, 11923–11935. [DOI] [PubMed] [Google Scholar]
- (1032).Ray K; Pfaff FF; Wang B; Nam W Status of Reactive Non-Heme Metal-Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc 2014, 136, 13942–13958. [DOI] [PubMed] [Google Scholar]
- (1033).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1034).de Visser SP; Nam W High-Valent Iron-Oxo Porphyrins in Oxygenation Reactions In Handbook of Porphyrin Science; Handbook of Porphyrin Science; World Scientific Publishing Company, 2010; Vol. 10, pp 85–139. [Google Scholar]
- (1035).McLain JL; Lee J; Groves JT Biomimetic Oxygenations Related to Cytochrome P450: Metal-Oxo and Metal-Peroxo Intermediates In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier B, Ed.; Imperial College Press: London, UK, 2000; pp 91–170. [Google Scholar]
- (1036).Kal S; Que L Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. JBIQ J. Biol. Inorg. Chem 2017, 22, 339–365. [DOI] [PubMed] [Google Scholar]
- (1037).Fiedler AT; Fischer AA Oxygen Activation by Mononuclear Mn, Co, and Ni Centers in Biology and Synthetic Complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 407–424. [DOI] [PubMed] [Google Scholar]
- (1038).Groves JT Models and Mechanisms of Cytochrome P450 Action In Cytochrome P450; Ortiz de Montellano PR, Ed.; Springer US: Boston, MA, 2005; pp 1–43. [Google Scholar]
- (1039).Collman JP; Boulatov R Electrocatalytic O2 Reduction by Synthetic Analogues of the Heme/Cu Site of Cytochrome Oxidase Incorporated in a Lipid Film. Angew. Chem., Int. Ed 2002, 41 , 3487–3489. [DOI] [PubMed] [Google Scholar]
- (1040).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (1041).Finklea HO; Hanshew DD Electron-Transfer Kinetics in Organized Thiol Monolayers with Attached Pentaammine(pyridine) ruthenium Redox Centers. J. Am. Chem. Soc 1992, 114, 3173–3181. [Google Scholar]
- (1042).Chidsey CE Free Energy and Temperature Dependence of Electron Transfer at the Metal-Electrolyte Interface. Science 1991, 251, 919–922. [DOI] [PubMed] [Google Scholar]
- (1043).Creager S; Yu CJ; Bamdad C; O’Connor S; MacLean T; Lam E; Chong Y; Olsen GT; Luo J; Gozin M; et al. Electron Transfer at Electrodes through Conjugated “Molecular Wire” Bridges. J. Am. Chem. Soc 1999, 121, 1059–1064. [Google Scholar]
- (1044).Collman JP; Devaraj NK; Decmau RA; Yang Y; Yan Y-L; Ebina W; Eberspacher TA; Chidsey CED A Cytochrome c Oxidase Model Catalyzes Oxygen to Water Reduction Under Rate-Limiting Electron Flux. Science 2007, 315, 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1045).Collman JP; Ghosh S; Dey A; Decreau RA Using a Functional Enzyme Model to Understand the Chemistry behind Hydrogen Sulfide Induced Hibernation. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 22090–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1046).Collman JP; Herrmann PC; Tyvoll DA; Decreau RA; Bull BS; Barile CJ Reducing Platelet Activation, Aggregation and Platelet-Stimulated Thrombosis or Blood Coagulation by Reducing Mitochondrial Respiration. U.S. Patent, USPTO Application 20110301180, 2011.
- (1047).Collman JP; Dey A; Decreau RA; Yang Y; Hosseini A; Solomon EI; Eberspacher TA Interaction of Nitric Oxide with a Functional Model of Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 9892–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1048).Collman JP; Yang Y; Dey A; Decreau RA; Ghosh S; Ohta T; Solomon EI A Functional Nitric Oxide Reductase Model. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15660–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1049).Collman JP; Dey A; Yang Y; Decréau RA; Ohta T; Solomon EI Intermediates Involved in the Two Electron Reduction of NO to N2O by a Functional Synthetic Model of Heme Containing Bacterial NO Reductase. J. Am. Chem. Soc 2008, 130, 16498–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1050).Shin H; Lee D-H; Kang C; Karlin KD Electrocatalytic Four-Electron Reductions of O2 to H2O with Cytochrome c Oxidase Model Compounds. Electrochim. Acta 2003, 48, 4077–4082. [Google Scholar]
- (1051).Chatterjee S; Sengupta K; Hematian S; Karlin KD; Dey A Electrocatalytic O2-Reduction by Synthetic Cytochrome c Oxidase Mimics: Identification of a “Bridging Peroxo” Intermediate Involved in Facile 4e–/4H+ O2-Reduction. J. Am. Chem. Soc 2015, 137, 12897–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1052).Sengupta K; Chatterjee S; Samanta S; Dey A Direct Observation of Intermediates Formed during Steady-State Electrocatalytic O2 Reduction by Iron Porphyrins. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 8431–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1053).Halime Z; Kotani H; Li Y; Fukuzumi S; Karlin KD Homogeneous Catalytic O2 Reduction to Water by a Cytochrome c Oxidase Model with Trapping of Intermediates and Mechanistic Insights. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 13990–13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1054).Peterson RL; Ginsbach JW; Cowley RE; Qayyum MF; Himes RA; Siegler MA; Moore CD; Hedman B; Hodgson KO; Fukuzumi S; et al. Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper-Dioxygen Adduct. J. Am. Chem. Soc 2013, 135, 16454–16467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1055).Lee EJ; Wrighton MS Competitive Energy and Electron-Transfer Quenching of Excited Ruthenium Polypyridyls by Ferrocene Derivatives. J. Am. Chem. Soc 1991, 113, 8562–8564. [Google Scholar]
- (1056).Carver CT; Matson BD; Mayer JM Electrocatalytic Oxygen Reduction by Iron Tetra-Arylporphyrins Bearing Pendant Proton Relays. J. Am. Chem. Soc 2012, 134, 5444–5447.22394189 [Google Scholar]
- (1057).Favier I; Duñach E New Protic Salts of Aprotic Polar Solvents. Tetrahedron Lett. 2004, 45, 3393–3395. [Google Scholar]
- (1058).Ching HYV; Anxolabéhère-Mallart E; Colmer HE; Costentin C; Dorlet P; Jackson TA; Policar C; Robert M Electrochemical Formation and Reactivity of a Manganese Peroxo Complex: Acid Driven H2O2 Generation vs. O–O Bond Cleavage. Chem. Sci 2014, 5, 2304–2310. [Google Scholar]
- (1059).Eckert F; Leito I; Kaljurand I; Kutt A; Diedenhofen M; Klamt A Prediction of Acidity in Acetonitrile Solution with COSMO-RS. J. Comput. Chem 2009, 30, 799–810. [DOI] [PubMed] [Google Scholar]
- (1060).Gregory M; Mak PJ; Sligar SG; Kincaid J R Differential Hydrogen Bonding in Human CYP17 Dictates Hydrox- ylation versus Lyase Chemistry. Angew. Chem., Int. Ed 2013, 52, 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1061).Spiro TG; Soldatova AV; Balakrishnan G CO NO and O2 as Vibrational Probes of Heme Protein Interactions. Coord. Chem. Rev 2013, 257, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1062).Kitagawa T; Abe M; Kyogoku Y; Ogoshi H; Sugimoto H; Yoshida Z Resonance Raman Spectra of 15-N Enriched Metallooctaethylorphyrins. Characterization of the Oxidation State Marker Bands of Hemoproteins. Chem. Phys. Lett 1977, 48, 55–58. [Google Scholar]
- (1063).Kakuda S; Peterson RL; Ohkubo K; Karlin KD; Fukuzumi S Enhanced Catalytic Four-Electron Dioxygen (O2) and Two-Electron Hydrogen Peroxide (H2O2) Reduction with a Copper(II) Complex Possessing a Pendant Ligand Pivalamido Group. J. Am. Chem. Soc 2013, 135, 6513–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1064).Garcia-Bosch I; Sharma SK; Karlin KD A Selective Stepwise Heme Oxygenase Model System: An Iron(IV)-Oxo Porphyrin π-Cation Radical Leads to a Verdoheme-Type Compound via an Isoporphyrin Intermediate. J. Am. Chem. Soc 2013, 135, 16248–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1065).Kundu S; Miceli E; Farquhar ER; Ray K Mechanism of Phenol Oxidation by Heterodinuclear Ni Cu Bis(^-Oxo) Complexes Involving Nucleophilic Oxo Groups. Dalton Trans 2014, 43, 4264–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1066).Lehnert N; Ho RYN; Que L; Solomon EI Electronic Structure of High-Spin Iron(III)-Alkylperoxo Complexes and Its Relation to Low-Spin Analogues: Reaction Coordinate of O–O Bond Homolysis. J. Am. Chem. Soc 2001, 123, 12802–12816. [DOI] [PubMed] [Google Scholar]
- (1067).Ho RYN; Roelfes G; Feringa BL; Que L Raman Evidence for a Weakened O–O Bond in Mononuclear Low-Spin Iron(III)-Hydroperoxides. J. Am. Chem. Soc 1999, 121, 264–265. [Google Scholar]
- (1068).Nanthakumar A; Fox S; Murthy NN; Karlin KD; Ravi N; Huynh H; Orosz RD; Day EP; Hagen KS; Blackburn NJ; et al. Oxo- and Hydroxo-Bridged (Porphyrin)iron(III)-Copper(II) Species as Cytochrome c Oxidase Models: Acid-Base Interconversions and X-Ray Structure of the Fe(III)-(O2-)-Cu(II) Complex. J. Am. Chem. Soc 1993, 115, 8513–8514. [Google Scholar]
- (1069).Kurtz DM Jr. Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem. Rev 1990, 90, 585–606. [Google Scholar]
- (1070).Lee SC; Holm R H. Synthesis and Characterization of an Asymmetric Bridged Assembly Containing the Unsupported [FeIII-O-CuII] Bridge: An Analogue of the Binuclear Site in Oxidized Cytochrome c Oxidase. J. Am. Chem. Soc 1993, 115, 11789–11798. [Google Scholar]
- (1071).Scott MJ; Zhang HH; Lee SC; Hedman B; Hodgson KO; Holm RH Oxygen-Bridged Iron-Copper Assemblies Pertinent to Heme-Copper Oxidases: Synthesis and Structure of an [FeIII-(OH)-CuII] Bridge and EXAFS Multiple-Scattering Effects of Linear Oxo and Nonlinear Hydroxo Bridges. J. Am. Chem. Soc 1995, 117, 568–569. [Google Scholar]
- (1072).Lee SC; Holm RH A Synthetic Analog of the Iron/copper Bridged Assembly in Cytochrome c Oxidase. J. Am. Chem. Soc 1993, 115, 5833–5834. [Google Scholar]
- (1073).Hematian S; Kenkel I; Shubina TE; Dürr M; Liu JJ; Siegler MA; Ivanovic-Burmazovic I; Karlin KD Nitrogen Oxide Atom-Transfer Redox Chemistry; Mechanism of NO(g) to Nitrite Conversion Utilizing μ-Oxo Heme-FeIII–O–CuII(L) Constructs. J. Am. Chem. Soc 2015, 137, 6602–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1074).Carroll JM; Norton J R Protonation of a Bridging Oxo Ligand Is Slow. J. Am. Chem. Soc 1992, 114, 8744–8745. [Google Scholar]
- (1075).Kramarz KW; Norton JR Slow Proton-Transfer Reactions in Organometallic and Bioinorganic Chemistry. Prog. Inorg. Chem 2007, 42, 1–65. [Google Scholar]
- (1076).Fann YC; Ahmed I; Blackburn NJ; Boswell JS; Verkhovskaya ML; Hoffman BM; Wikstrom M Structure of Cub in the Binuclear Heme-Copper Center of the Cytochrome aa3-Type Quinol Oxidase from Bacillus Subtilis: An ENDOR and EXAFS Study. Biochemistry 1995, 34, 10245–10255. [DOI] [PubMed] [Google Scholar]
- (1077).Morgan JE; Verkhovsky MI; Wikstrom M The Histidine Cycle: A New Model for Proton Translocation in the Respiratory Heme-Copper Oxidases. J. Bioenerg. Biomembr 1994, 26, 599–608. [DOI] [PubMed] [Google Scholar]
- (1078).Wikström M; Morgan JE; Verkhovsky MI On the Mechanism of Proton Translocation by Respiratory Enzyme. J. Bioenerg. Biomembr 1998, 30, 139–145. [DOI] [PubMed] [Google Scholar]
- (1079).Wikström M Cytochrome Oxidase. In Encyclopedia of Inorganic Chemistry, 2nd ed.; King RB, Ed.; John Wiley & Sons Ltd.: Chichester, 2005; Vol. 2, pp 1242–1253. [Google Scholar]
- (1080).Baldwin MJ; Gelasco A; Pecoraro VL The Effect of Protonation on [MnIV(μ2-O)]2 Complexes. Photosynth. Res 1993, 38, 303–308. [DOI] [PubMed] [Google Scholar]
- (1081).Moore EJ; Sullivan JM; Norton JR Kinetic and Thermodynamic Acidity of Hydrido Transition-Metal Complexes. 3. Thermodynamic Acidity of Common Mononuclear Carbonyl Hydrides. J. Am. Chem. Soc 1986, 108, 2257–2263. [DOI] [PubMed] [Google Scholar]
- (1082).Turowski PN; Armstrong WH; Liu S; Brown SN; Lippard SJ Synthesis and Characterization of Hydroxo-Bridged diiron(III) Complexes Containing Carboxylate or Phosphate Ester Bridges: Comparisons to diiron(III) Proteins. Inorg. Chem 1994, 33, 636–645. [Google Scholar]
- (1083).Hage R; Krijnen B; Warnaar JB; Hartl F; Stufkens DJ; Snoeck TL Proton-Coupled Electron-Transfer Reactions in [MnIV2(μ-O)3L’2]2+ (L’ = 1,4,7-Trimethyl-1,4,7-Triazacyclononane). Inorg. Chem 1995, 34, 4973–4978. [Google Scholar]
- (1084).Petrik ID; Liu J; Lu Y Metalloenzyme Design and Engineering through Strategic Modifications of Native Protein Scaffolds. Curr. Opin. Chem. Biol 2014, 19, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1085).Bhagi-Damodaran A; Petrik I; Lu Y Using Biosynthetic Models of Heme-Copper Oxidase and Nitric Oxide Reductase in Myoglobin to Elucidate Structural Features Responsible for Enzymatic Activities. Isr. J. Chem 2016, 56, 773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1086).Lim AR; Sishta BP; Grant Mauk A Contribution of the Heme Propionate Groups to the Electron Transfer and Electrostatic Properties of Myoglobin. J. Inorg. Biochem 2006, 100, 2017–2023. [DOI] [PubMed] [Google Scholar]
- (1087).Watanabe Y Construction of Heme Enzymes: Four Approaches. Curr. Opin. Chem. Biol 2002, 6, 208–216. [DOI] [PubMed] [Google Scholar]
- (1088).Lu Y Biosynthetic Inorganic Chemistry. Angew. Chem., Int. Ed 2006, 45, 5588–5601. [DOI] [PubMed] [Google Scholar]
- (1089).Sigman JA; Kwok BC; Lu Y From Myoglobin to Heme-Copper Oxidase: Design and Engineering of a CuB Center into Sperm Whale Myoglobin. J. Am. Chem. Soc 2000, 122, 8192–8196. [Google Scholar]
- (1090).Sigman JA; Kim HK; Zhao X; Carey JR; Lu Y The Role of Copper and Protons in Heme-Copper Oxidases: Kinetic Study of an Engineered Heme-Copper Center in Myoglobin. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1091).Miner KD; Mukherjee A; Gao Y-G; Null EL; Petrik ID; Zhao X; Yeung N; Robinson H; Lu Y A Designed Functional Metalloenzyme That Reduces O2 to H2O with Over One Thousand Turnovers. Angew. Chem., Int. Ed 2012, 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1092).Vojtěchovský J; Chu K; Berendzen J; Sweet R M.; Schlichting, I. Crystal Structures of Myoglobin-Ligand Complexes at Near-Atomic Resolution. Biophys. J 1999, 77, 2153–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1093).Bhagi-Damodaran A; Michael MA; Zhu Q; Reed J; Sandoval BA; Mirts EN; Chakraborty S; Moënne-Loccoz P; Zhang Y; Lu Y Why Copper Is Preferred over Iron for Oxygen Activation and Reduction in Haem-Copper Oxidases. Nat. Chem 2017, 9, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1094).Kerr EA; Yu NT; Bartnicki DE; Mizukami H Resonance Raman Studies of CO and O2 Binding to Elephant Myoglobin (Distal His(E7)––Gln). J. Biol. Chem 1985, 260,8360–8365. [PubMed] [Google Scholar]
- (1095).Decréau RA; Collman JP Three Toxic Gases Meet in the Mitochondria. Front. Physiol 2015, 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1096).Hino T; Matsumoto Y; Nagano S; Sugimoto H; Fukumori Y; Murata T; Iwata S; Shiro Y Structural Basis of Biological N2O Generation by Bacterial Nitric Oxide Reductase. Science 2010, 330, 1666–1670. [DOI] [PubMed] [Google Scholar]
- (1097).Collman JP; Dey A; Yang Y; Ghosh S; Decreau RA O2 Reduction by a Functional Heme/Nonheme Bis-Iron NOR Model Complex. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 10528–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1098).Reed JH; Shi Y; Zhu Q; Chakraborty S; Mirts EN; Petrik ID; Bhagi-Damodaran A; Ross M; Moënne-Loccoz P; Zhang Y; et al. Manganese and Cobalt in the Nonheme-MetalBinding Site of a Biosynthetic Model of Heme-Copper Oxidase Superfamily Confer Oxidase Activity through Redox-Inactive Mechanism. J. Am. Chem. Soc 2017, 139, 12209–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1099).Yeung N; Lin Y-W; Gao Y-G; Zhao X; Russell BS; Lei L; Miner KD; Robinson H; Lu Y Rational Design of a Structural and Functional Nitric Oxide Reductase. Nature 2009, 462, 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1100).Fukuzumi S; Ohkubo K Quantitative Evaluation of Lewis Acidity of Metal Ions Derived from Theg Values of ESR Spectra of Superoxide: Metal Ion Complexes in Relation to the Promoting Effects in Electron Transfer Reactions. Chem. −3 Eur. J 2000, 6, 4532–4535. [DOI] [PubMed] [Google Scholar]
- (1101).Shannon RD Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr 1976, 32, 751–767. [Google Scholar]
- (1102).Bhagi-Damodaran A; Kahle M; Shi Y; Zhang Y; Adelroth P; Lu Y Insights Into How Heme Reduction Potentials Modulate Enzymatic Activities of a Myoglobin-Based Functional Oxidase. Angew. Chem., Int. Ed 2017, 56, 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1103).Marshall NM; Garner DK; Wilson TD; Gao Y-G; Robinson H; Nilges MJ; Lu Y Rationally Tuning the Reduction Potential of a Single Cupredoxin beyond the Natural Range. Nature 2009, 462, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1104).Rauhamaki V; Bloch DA; Verkhovsky MI; Wikström M Active Site of Cytochrome cbb3. J. Biol. Chem 2009, 284, 11301–11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1105).Uchida T; Dojun N; Sekine Y; Ishimori K Heme Proximal Hydrogen Bonding between His170 and Asp132 Plays an Essential Role in the Heme Degradation Reaction of HutZ from Vibrio Cholerae. Biochemistry 2017, 56, 2723–2734. [DOI] [PubMed] [Google Scholar]
- (1106).Jasaitis A; Backgren C; Morgan JE; Puustinen A; Verkhovsky MI; Wikström M Electron and Proton Transfer in the Arginine-54-Methionine Mutant of Cytochrome c Oxidase from Paracoccus Denitrificans. Biochemistry 2001, 40, 5269–5274. [DOI] [PubMed] [Google Scholar]
- (1107).Bhagi-Damodaran A; Petrik ID; Marshall NM; Robinson H; Lu Y Systematic Tuning of Heme Redox Potentials and Its Effects on O2 Reduction Rates in a Designed Oxidase in Myoglobin. J. Am. Chem. Soc 2014, 136, 11882–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1108).Liu X; Yu Y; Hu C; Zhang W; Lu Y; Wang J Significant Increase of Oxidase Activity through the Genetic Incorporation of a Tyrosine-Histidine Cross-Link in a Myoglobin Model of Heme-Copper Oxidase. Angew. Chem. Int. Ed 2012, 51, 4312–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1109).Brunori M; Giuffre A; Sarti P Cytochrome Oxidase, Ligands and Electrons. J. Inorg. Biochem 2005, 99, 324–336. [DOI] [PubMed] [Google Scholar]
- (1110).Khalimonchuk O; Rödel G Biogenesis of Cytochrome c Oxidase. Mitochondrion 2005, 5, 363–388. [DOI] [PubMed] [Google Scholar]
- (1111).Ekici S; Pawlik G; Lohmeyer E; Koch H-G; Daldal F Biogenesis of cbb3-Type Cytochrome c Oxidase in Rhodobacter Capsulatus. Biochim. Biophys. Acta, Bioenerg 2012, 1817, 898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1112).Pitcher RS; Watmough NJ The Bacterial Cytochrome cbb3 Oxidases. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 388–399. [DOI] [PubMed] [Google Scholar]
- (1113).Gier J-WL; Schepper M; Reijnders WNM; Dyck SJ; Slotboom DJ; Warne A; Saraste M; Krab K; Finel M; Stouthamer AH; et al. Structural and Functional Analysis of aa3-Type and cbb3-Type Cytochrome c Oxidases of Paracoccus Denitrificans Reveals Significant Differences in Proton-Pump Design. Mol. Microbiol 1996, 20, 1247–1260. [DOI] [PubMed] [Google Scholar]
- (1114).Wang N; Zhao X; Lu Y Role of Heme Types in Heme-Copper Oxidases: Effects of Replacing a Heme B with a Heme O Mimic in an Engineered Heme-Copper Center in Myoglobin §. J. Am. Chem. Soc 2005, 127, 16541–16547. [DOI] [PubMed] [Google Scholar]
- (1115).Bhagi-Damodaran A; Reed JH; Zhu Q; Shi Y; Hosseinzadeh P; Sandoval BA; Harnden KA; Wang S; Sponholtz MR; Mirts EN; et al. Heme Redox Potentials Hold the Key to Reactivity Differences between Nitric Oxide Reductase and Heme-Copper Oxidase. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 6195–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1116).Cappuccio JA; Ayala I; Elliott GI; Szundi I; Lewis J; Konopelski JP; Barry BA; Einarsdottir O Modeling the Active Site of Cytochrome Oxidase: Synthesis and Characterization of a Cross-Linked Histidine-Phenol. J. Am. Chem. Soc 2002, 124, 1750–1760. [DOI] [PubMed] [Google Scholar]
- (1117).Himo F; Noodleman L; Blomberg MRA; Siegbahn PEM Relative Acidities of Ortho-Substituted Phenols, as Models for Modified Tyrosines in Proteins. J. Phys. Chem. A 2002, 106, 8757–8761. [Google Scholar]
- (1118).Colbran SB; Paddon-Row MN Could the Tyrosine-Histidine Ligand to CuB in Cytochrome c Oxidase Be Coordinatively Labile? Implications from a Quantum Chemical Model Study of Histidine Substitutional Lability and the Effects of the Covalent Tyrosine-Histidine Cross-L. JBIC, J. Biol. Inorg. Chem 2003, 8, 855–865. [DOI] [PubMed] [Google Scholar]
- (1119).Yu Y; Lv X; Li J; Zhou Q; Cui C; Hosseinzadeh P; Mukherjee A; Nilges MJ; Wang J; Lu Y Defining the Role of Tyrosine and Rational Tuning of Oxidase Activity by Genetic Incorporation of Unnatural Tyrosine Analogs. J. Am. Chem. Soc 2015, 137, 4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1120).Yu Y; Zhou Q; Wang L; Liu X; Zhang W; Hu M; Dong J; Li J; Lv X; Ouyang H; et al. Significant Improvement of Oxidase Activity through the Genetic Incorporation of a Redox-Active Unnatural Amino Acid. Chem. Sci 2015, 6, 3881–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1121).Yu Y; Mukherjee A; Nilges MJ; Hosseinzadeh P; Miner KD; Lu Y Direct EPR Observation of a Tyrosyl Radical in a Functional Oxidase Model in Myoglobin during Both H2O2 and O2 Reactions. J. Am. Chem. Soc 2014, 136, 1174–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1122).Petrik ID; Davydov R; Ross M; Zhao X; Hoffman B; Lu Y Spectroscopic and Crystallographic Evidence for the Role of a Water-Containing H-Bond Network in Oxidase Activity of an Engineered Myoglobin. J. Am. Chem. Soc 2016, 138, 1134–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1123).Davydov R; Laryukhin M; Ledbetter-Rogers A; Sono M; Dawson JH; Hoffman BM Electron Paramagnetic Resonance and Electron-Nuclear Double Resonance Studies of the Reactions of Cryogenerated Hydroperoxoferric–Hemoprotein Intermediates. Biochemistry 2014, 53, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1124).Mukherjee S; Sengupta K; Das MR; Jana SS; Dey A Site-Specific Covalent Attachment of Heme Proteins on Self- Assembled Monolayers. JBIC, J. Biol. Inorg. Chem 2012, 17, 1009–1023. [DOI] [PubMed] [Google Scholar]
- (1125).Mukherjee S; Mukherjee A; Bhagi-Damodaran A; Mukherjee M; Lu Y; Dey A A Biosynthetic Model of Cytochrome c Oxidase as an Electrocatalyst for Oxygen Reduction. Nat. Commun 2015, 6, 8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1126).Quah T; Milton RD; Abdellaoui S; Minteer SD Bioelectrocatalytic NAD+/NADH Inter-Conversion: Transformation of an Enzymatic Fuel Cell into an Enzymatic Redox Flow Battery. Chem. Commun 2017, 53, 8411–8414. [DOI] [PubMed] [Google Scholar]
- (1127).Que L; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- (1128).Decker A; Solomon EI Dioxygen Activation by Copper, Heme and Non-Heme Iron Enzymes: Comparison of Electronic Structures and Reactivities. Curr. Opin. Chem. Biol 2005, 9, 152–163. [DOI] [PubMed] [Google Scholar]
- (1129).Walker FA; Simonis U Iron Porphyrin Chemistry–A Ten-Year Update In Encyclopedia of Inorganic Chemistry, 2nd ed.; King RB, Ed.; Wiley & Sons, Ltd., 2005; pp 2390–2521. [Google Scholar]
- (1130).Blomberg MRA; Borowski T; Himo F; Liao R-Z; Siegbahn PEM Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev 2014, 114, 3601–3658. [DOI] [PubMed] [Google Scholar]
- (1131).Phillips SEV Structure and Refinement of Oxymyoglobin at 1.6 Å Resolution. J. Mol. Biol 1980, 142, 531–554. [DOI] [PubMed] [Google Scholar]
- (1132).Wallen CM; Bacsa J; Scarborough CC Hydrogen Peroxide Complex of Zinc. J. Am. Chem. Soc 2015, 137, 14606–14609. [DOI] [PubMed] [Google Scholar]
- (1133).Reim J; Krebs B A Thermally Stable Peroxocopper(ll) Complex with Unusual μ4-Coordination of the Peroxo Ligand. Angew. Chem., Int. Ed. Engl 1994, 33, 1969–1971. [Google Scholar]
- (1134).Meyer F; Pritzkow H μ4-Peroxo versus Bis(μ2-Hydroxo) Cores in Structurally Analogous Tetracopper(ll) Complexes. Angew. Chem., Int. Ed 2000, 39, 2112–2115. [PubMed] [Google Scholar]
- (1135).Leznoff DB; Katz MJ; Cheng LKL; Draper ND; Batchelor RJ The Perils and Opportunities of Reactive Building Blocks: Attempted Synthesis of New Hg(CN)2-Based Coordination Polymers and the Structures of the Resulting Products. J. Mol. Struct 2006, 796, 223–229. [Google Scholar]
- (1136).Corona T; Padamati SK; Duboc C; Browne WR; Company A; Acuña-Parés F Trapping of Superoxido Cobalt and Peroxido Dicobalt Species Formed Reversibly from CoII and O2. Chem. Commun 2017, 53, 11782–11785. [DOI] [PubMed] [Google Scholar]
- (1137).Givaja G; Volpe M; Edwards MA; Blake AJ; Wilson C; Schroder M; Love JB Dioxygen Reduction at Dicobalt Complexes of a Schiff Base Calixpyrrole Ligand. Angew. Chem. Int. Ed 2007, 46, 584–586. [DOI] [PubMed] [Google Scholar]
- (1138).Wang H-Y; Mijangos E; Ott S; Thapper A Water Oxidation Catalyzed by a Dinuclear Cobalt-Polypyridine Complex. Angew. Chem., Int. Ed 2014, 53, 14499–14502. [DOI] [PubMed] [Google Scholar]
- (1139).Gavrilova AL; Qin CJ; Sommer RD; Rheingold AL; Bosnich B Bimetallic Reactivity. One-Site Addition Two-Metal Oxidation Reaction of Dioxygen with a Bimetallic dicobalt(II) Complex Bearing Five- and Six-Coordinate Sites. J. Am. Chem. Soc 2002, 124, 1714–1722. [DOI] [PubMed] [Google Scholar]
- (1140).Seo JS; Hynes R; Williams D; Chin J; Sung ND Structure and Reactivity of Dinuclear cobalt(III) Complexes with Peroxide and Phosphate Diester Analogues Bridging the Metal Ions. J. Am. Chem. Soc 1998, 120, 9943–9944. [Google Scholar]
- (1141).Hirata K; Shinzawa-Itoh K; Yano N; Takemura S; Kato K; Hatanaka M; Muramoto K; Kawahara T; Tsukihara T; Yamashita E; et al. Determination of Damage-Free Crystal Structure of an X-Ray-sensitive Protein Using an XFEL. Nat. Methods 2014, 11, 734–736. [DOI] [PubMed] [Google Scholar]
- (1142).Kaila VRI; Oksanen E; Goldman A; Bloch DA; Verkhovsky MI; Sundholm D; Wikstrom M A Combined Quantum Chemical and Crystallographic Study on the Oxidized Binuclear Center of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2011, 1807, 769–778. [DOI] [PubMed] [Google Scholar]
- (1143).Liu B; Chen Y; Doukov T; Soltis SM; Stout CD; Fee JA Combined Microspectrophotometric and Crystallographic Examination of Chemically Reduced and X-Ray Radiation-Reduced Forms of Cytochrome ba3 Oxidase from Thermus Thermophilus: Structure of the Reduced Form of the Enzyme. Biochemistry 2009, 48, 820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1144).Ogura T; Takahashi S; Shinzawa-Itoh K; Yoshikawa S; Kitagawa T Observation of the iron(II)-Oxygen Stretching Raman Band for Cytochrome Oxidase Compound A at Ambient Temperature. J. Am. Chem. Soc 1990, 112, 5630–5631. [PubMed] [Google Scholar]
- (1145).Tsubaki M; Nagai K; Kitagawa T Resonance Raman-Spectra of Myoglobins Reconstituted with Spirographis and Isospirographis Hemes and Iron 2,4-Diformylprotoporphyrin-IX-Effect of Formyl Substitution at the Heme Periphery. Biochemistry 1980, 19, 379–385. [DOI] [PubMed] [Google Scholar]
- (1146).Ibrahim M; Denisov IG; Makris TM; Kincaid JR; Sligar SG Resonance Raman Spectroscopic Studies of Hydro-peroxo-Myoglobin at Cryogenic Temperatures. J. Am. Chem. Soc 2003, 125, 13714–13718. [DOI] [PubMed] [Google Scholar]
- (1147).Wang Y; Mak PJ; Zhu Q; Kincaid JR Application of Resonance Raman Spectroscopy for Interrogation of Cryoradiolytically Reduced Oxygenated Heme Proteins. J. Raman Spectrosc 2017, 48, 180–190. [Google Scholar]
- (1148).Mak PJ; Thammawichai W; Wiedenhoeft D; Kincaid JR Resonance Raman Spectroscopy Reveals pH-Dependent Active Site Structural Changes of Lactoperoxidase Compound 0 and Its Ferryl Heme O–O Bond Cleavage Products. J. Am. Chem. Soc 2015, 137, 349–361. [DOI] [PubMed] [Google Scholar]
- (1149).Wasser IM; Huang HW; Moenne-Loccoz P; Karlin KD Heme/Non-Heme Diiron(II) Complexes and O2, CO, and NO Adducts as Reduced and Substrate-Bound Models for the Active Site of Bacterial Nitric Oxide Reductase. J. Am. Chem. Soc 2005, 127, 3310–3320. [DOI] [PubMed] [Google Scholar]
- (1150).Sakaguchi M; Shinzawa-Itoh K; Yoshikawa S; Ogura T A Resonance Raman Band Assignable to the O–O Stretching Mode in the Resting Oxidized State of Bovine Heart Cytochrome c Oxidase. J. Bioenerg. Biomembr 2010, 42, 241–243. [DOI] [PubMed] [Google Scholar]
- (1151).Ardon M; Bino A A New Aspect of Hydrolysis of Metal Ions: The Hydrogen-Oxide Bridging Ligand (H3O2–) In Solid State Chemistry; Springer-Verlag: Berlin/Heidelberg, 1987; Vol. 65, pp 1–28. [Google Scholar]
- (1152).Puerta DT; Cohen SM [(TpMe,Ph)2Zn2(H3O2)]ClO4: A New H3O2 Species Relevant to Zinc Proteinases. Inorg. Chim. Acta 2002, 337, 459–462. [Google Scholar]
- (1153).Ardon M; Bino A; Michelsen K; Pedersen E Long-Distance Magnetic Exchange Between Chromium(III) Atoms Bridged by H3O2-Ligands. J. Am. Chem. Soc 1987, 109, 5855–5856. [Google Scholar]
- (1154).Ardon M; Bino A; Jackson WG The Structure of Aqua-Hydroxo Double Salts. Polyhedron 1987, 6, 181–187. [Google Scholar]
- (1155).Justel T; Bendix J; Metzler-Nolte N; Weyhermüller T; Nuber B; Wieghardt K Ruthenium Complexes Containing “Non-innocent” o-Benzoquinone Diimine/o-Phenylenediamide(2–) Ligands. Synthesis and Crystal Structure of the Nitrido-Bridged Complex [{LRu(o-C6H4(NH)2)}2(μ-N)](PF6)2·3CH3CN·C6H5CH3. Inorg. Chem 1998, 37, 35–43. [DOI] [PubMed] [Google Scholar]
- (1156).Bino A; Gibson D A New Bridging Ligand, the Hydrogen Oxide Ion (H3O2–). J. Am. Chem. Soc 1981, 103, 6741–6742. [Google Scholar]
- (1157).Beran A; Giester G; Libowitzky E The Hydrogen Bond System in Natrochalcite-Type Compounds? An FTIR Spectroscopic Study of the H3O2 Unit. Mineral. Petrol 1997, 61, 223–235. [Google Scholar]
- (1158).Bond AD; Derossi S; Jensen F; Larsen FB; McKenzie CJ; Nelson J Squeezing the [Cu-OH···H2O-Cu]3+ Bridge by Cryptate Encapsulation. Inorg. Chem 2005, 44, 5987–5989. [DOI] [PubMed] [Google Scholar]
- (1159).Keutsch FN; Saytally RJ Water Clusters: Untangling the Mysteries of the Liquid, One Molecule at a Time. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 10533–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1160).Giguère PA The Bifurcated Hydrogen-bond Model of Water and Amorphous Ice. J. Chem. Phys 1987, 87, 4835–4839. [Google Scholar]
- (1161).Chidambaram R; Sikka SK; Bent O -H–O. Hydrogen Bonds in Crystals. Chem. Phys. Lett 1968, 2, 162–165. [Google Scholar]
- (1162).Scheiner S Bent Hydrogen Bonds and Proton Transfers. Acc. Chem. Res 1994, 27, 402–408. [Google Scholar]
- (1163).Stoyanov ES; Stoyanova IV; Reed CA The Structure of the Hydrogen Ion (Haq+) in Water. J. Am. Chem. Soc 2010, 132, 1484–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1164).Chance B; Saronio C; Leigh JS Jr. Functional Intermediates in the Reaction of Membrane-Bound Cytochrome Oxidase with Oxygen. J. Biol. Chem 1975, 250, 9226–9238. [PubMed] [Google Scholar]
- (1165).Chance B; Saronio C; Leigh JS Jr. Functional Intermediates in Reaction of Cytochrome Oxidase with Oxygen. Proc. Natl. Acad. Sci. U. S. A 1975, 72, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1166).Verkhovsky MI; Morgan JE; Wikström M Oxygen Binding and Activation : Early Steps in the Reaction of Oxygen with Cytochrome c Oxidase. Biochemistry 1994, 33, 3079–3086. [DOI] [PubMed] [Google Scholar]
- (1167).Yamazaki I; Yokota KN; Shikama K Preparation of Crystalline Oxymyoglobin from Horse Heart. J. Biol. Chem 1964, 239, 4151–4153. [PubMed] [Google Scholar]
- (1168).Hardman KD; Eylar EH; Banaszak LJ; Gurd FR Isolation of Sperm Whale Myoglobin By Low Temperature Factionation With Ethanol and Metallic Ions. J. Biol. Chem 1966, 241, 432–442. [PubMed] [Google Scholar]
- (1169).Philo JS; Adams ML; Schuster TM Association-Dependent Absorption Spectra of Oxyhemoglobin A and Its Subunits. J. Biol. Chem 1981, 256, 7917–7924. [PubMed] [Google Scholar]
- (1170).Larroque C; Van Lier JE The Subzero Temperature Stabilized Oxyferro Complex of Purified Cytochrome P450 SCC. FEBS Lett. 1980, 115, 175–177. [DOI] [PubMed] [Google Scholar]
- (1171).Denisov IG; Dawson JH; Hager LP; Sligar SG The Ferric-Hydroperoxo Complex of Chloroperoxidase. Biochem. Biophys. Res. Commun 2007, 363, 954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1172).Cooper CE; Jurd M; Nicholls P; Wankasi MM; Svistunenko DA; Reeder BJ; Wilson MT On the Formation, Nature, Stability and Biological Relevance of the Primary Reaction Intermediates of Myoglobins with Hydrogen Peroxide. Dalton Trans 2005, 21, 3483. [DOI] [PubMed] [Google Scholar]
- (1173).Denisov IG; Makris TM; Sligar SG Formation and Decay of Hydroperoxo-Ferric Heme Complex in Horseradish Peroxidase Studied by Cryoradiolysis. J. Biol. Chem 2002, 277, 42706–42710. [DOI] [PubMed] [Google Scholar]
- (1174).Wang S; Yuan F; Chen G; Tu K; Wang H; Wang L-Q Dextran-Based Thermo-Responsive Hemoglobin-polymer Conjugates with Oxygen-Carrying Capacity. RSC Adv. 2014, 4, 52940–52948. [Google Scholar]
- (1175).Van Wart HE; Zimmer J Resonance Raman Evidence for the Activation of Dioxygen in Horseradish Oxyperoxidase. J. Biol. Chem 1985, 260, 8372–8377. [PubMed] [Google Scholar]
- (1176).Nagai K; Kitagawa T; Morimoto H Quaternary Structures and Low Frequency Molecular Vibrations of Haems of Deoxy and Oxyhaemoglobin Studied by Resonance Raman Scattering. J. Mol. Biol 1980, 136, 271–289. [DOI] [PubMed] [Google Scholar]
- (1177).Blomberg MRA; Siegbahn PEM Quantum Chemistry as a Tool in Bioenergetics. Biochim. Biophys. Acta, Bioenerg 2010, 1797, 129–142. [DOI] [PubMed] [Google Scholar]
- (1178).Tsukihara T; Shimokata K; Katayama Y; Shimada H; Muramoto K; Aoyama H; Mochizuki M; Shinzawa-Itoh K; Yamashita E; Yao M; et al. The Low-Spin Heme of Cytochrome c Oxidase as the Driving Element of the Proton-Pumping Process. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 15304–15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1179).Blomberg MRA; Siegbahn PEM Quantum Chemistry Applied to the Mechanisms of Transition Metal Containing enzymes– Cytochrome c Oxidase, a Particularly Challenging Case. J. Comput. Chem. 2006, 27, 1373–1384. [DOI] [PubMed] [Google Scholar]
- (1180).Han Du W-G; Gotz AW; Noodleman L A Water Dimer Shift Activates a Proton Pumping Pathway in the PR → F Transition of Ba 3 Cytochrome c Oxidase. Inorg. Chem 2018, 57, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1181).Behan RK; Green MT On the Status of Ferryl Protonation. J. Inorg. Biochem 2006, 100, 448–459. [DOI] [PubMed] [Google Scholar]
- (1182).Yosca TH; Behan RK; Krest CM; Onderko EL; Langston MC; Green MT Setting an Upper Limit on the Myoglobin Iron(IV)Hydroxide pKa: Insight into Axial Ligand Tuning in Heme Protein Catalysis. J. Am. Chem. Soc 2014, 136, 9124–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1183).Belevich I; Bloch DA; Belevich N; Wikström M; Verkhovsky MI Exploring the Proton Pump Mechanism of Cytochrome c Oxidase in Real Time. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2685–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1184).Siletsky SA; Belevich I; Belevich NP; Soulimane T; Wikstrom M Time-Resolved Generation of Membrane Potential by ba3 Cytochrome c Oxidase from Thermus Thermophilus Coupled to Single Electron Injection into the O and OH States. Biochim. Biophys. Acta, Bioenerg 2017, 1858, 915–926. [DOI] [PubMed] [Google Scholar]
- (1185).Taube H Electron Transfer between Metal Complex- A Retrospective View. Angew. Chem., Int. Ed. Engl 1984, 23, 329–339. [Google Scholar]
- (1186).Creutz C; Ford PC; Meyer TJ Henry Taube: Inorganic Chemist Extraordinaire. Inorg. Chem 2006, 45, 7059–7068. [DOI] [PubMed] [Google Scholar]
- (1187).Taube H; Myers H; Rich RL Observations on the Mechanism of Electron Transfer in Solution. J. Am. Chem. Soc 1953, 75, 4118–4119. [Google Scholar]
- (1188).Taube H; Myers H Evidence for a Bridged Activated Complex for Electron Transfer Reactions. J. Am. Chem. Soc 1954, 76, 2103–2111. [Google Scholar]
- (1189).Fry HC; Lucas HR; Zakharov LN; Rheingold AL; Meyer GJ; Karlin KD Intermolecular versus Intramolecular Electron-/Atom- (Cl) Transfer in Heme-Iron and Copper Pyridy-lalkylamine Complexes. Inorg. Chim. Acta 2008, 361, 1100–1115. [Google Scholar]
- (1190).Fabian M; Skultety L; Jancura D; Palmer G Implications of Ligand Binding Studies for the Catalytic Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg 2004, 1655, 298–305. [DOI] [PubMed] [Google Scholar]
- (1191).Fabian M; Skultety L; Brunel C; Palmer G Cyanide Stimulated Dissociation of Chloride from the Catalytic Center of Oxidized Cytochrome c Oxidase. Biochemistry 2001, 40, 6061–6069. [DOI] [PubMed] [Google Scholar]
- (1192).Zhao X; Nilges MJ; Lu Y Redox-Dependent Structural Changes in an Engineered Heme–Copper Center in Myoglobin: Insights into Chloride Binding to CuB in Heme Copper Oxidases. Biochemistry 2005, 44, 6559–6564. [DOI] [PubMed] [Google Scholar]
- (1193).Ralle M; Verkhovskaya ML; Morgan JE; Verkhovsky MI; Wikstrom M; Blackburn NJ Coordination of CuB in Reduced and CO-Liganded States of Cytochrome bo3 from Escherichia Coli. Is Chloride Ion a Cofactor? Biochemistry 1999, 38, 7185–7194. [DOI] [PubMed] [Google Scholar]
- (1194).Forte E; Barone MC; Brunori M; Sarti P; Giuffrè A Redox-Linked Protonation of Cytochrome c Oxidase: The Effect of Chloride Bound to CuB. Biochemistry 2002, 41, 13046–13052. [DOI] [PubMed] [Google Scholar]
- (1195).Zimmermann BH; Nitsche CI; Fee JA; Rusnak F; Munck E Properties of a Copper-Containing Cytochrome ba3: A Second Terminal Oxidase from the Extreme Thermophile Thermus Thermophilus. Proc. Natl. Acad. Sci U. S. A 1988, 85, 5779–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1196).Qin L; Hiser C; Mulichak A; Garavito RM; Ferguson-Miller S Identification of Conserved Lipid/Detergent-Binding Sites in a High-Resolution Structure of the Membrane Protein Cytochrome c Oxidase. Proc. Natl. Acad. Sci U. S. A 2006, 103, 16117–16122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1197).Yandell JK Copper(II)-Copper(I) Outer-Sphere Electron Transfer In Copper Coordination Chemistry: Biochemical and Inorganic Perspectives; Karlin KD, Zubieta J, Eds.; Adenine Press: New York, 1983; pp 157–166. [Google Scholar]
- (1198).Black JR; Levason W; Webster M Tetrakis(acetonitrile-N)copper(I) Hexafluorophosphate(V) Acetonitrile Solvate. Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1995, 51, 623–625. [Google Scholar]
- (1199).Cox BG; Wojciech J; Palou J Oxidation of Ascorbic Acid by Copper(II) and the Ferrocenium Ion in Acetonitrile-Water Mixtures. J. Chem. Soc., Dalton Trans 1988, 733–740. [Google Scholar]
- (1200).Healy PC; Engelhardt LM; Patrick VA; White AH Lewis-Base Adducts of Group 1B metal(I) Compounds. Part 19. Crystal Structures of bis(1,10-phenanthroline)copper(I) Perchlorate and dibromocuprate(I). J. Chem. Soc., Dalton Trans 1985, 12, 2541. [Google Scholar]
- (1201).Kovalevsky AY; Gembicky M; Novozhilova IV; Coppens P Solid-State Structure Dependence of the Molecular Distortion and Spectroscopic Properties of the Cu(I) Bis(2,9- Dimethyl-1,10-Phenanthroline) Ion. Inorg. Chem. 2003, 42, 8794–8802. [DOI] [PubMed] [Google Scholar]
- (1202).Patterson GS; Holm RH Structural and Electronic Effects on the Polarographic Half-Wave Potentials of Copper(II) Chelate Complexes. Bioinorg. Chem 1975, 4, 257–275. [DOI] [PubMed] [Google Scholar]
- (1203).Lim BS; Holm RH Molecular Heme–Cyanide–Copper Bridged Assemblies: Linkage Isomerism, Trends in Vcn Values, and Relation to the Heme-a3/CuB Site in Cyanide-Inhibited Heme−Copper Oxidases. Inorg. Chem 1998, 37, 4898–4908. [DOI] [PubMed] [Google Scholar]
- (1204).Baek HK; Holwerda RA An Sulfur-Bonded Adduct of Cysteine with [tris(2-pyridylmethyl)amine]copper(II) Ion. Inorg. Chem 1983, 22, 3452–3456. [Google Scholar]
- (1205).Karlin KD; Sherman SE Redox Comparisons of Pseudotetrahedral Copper(I) Complexes Containing Tripod Ligands. Inorg. Chim. Acta 1982, 65, L39–L40. [Google Scholar]
- (1206).Liang H-CH-C; Zhang CX; Henson MJ; Sommer RD; Hatwell KR; Kaderli S; Zuberbuhler AD; Rheingold AL; Solomon EI; Karlin KD Contrasting Copper Dioxygen Chemistry Arising from Alike Tridentatte Alkyltriamine Copper(I) Complexes. J. Am. Chem. Soc 2002, 124, 4170–4171. [DOI] [PubMed] [Google Scholar]
- (1207).Park GY; Lee Y; Lee D-H; Woertink JS; Narducci Sarjeant AA; Solomon EI; Karlin KD Thioether S-Ligation in a Side-on μ-η2:η2-peroxodicopper(II) Complex. Chem. Commun 2010, 46, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1208).Hawkins CJ; Perrin DD Oxidation–reduction Potentials of Metal Complexes in Water: Some Copper Complexes. J. Chem. Soc 1962, 0, 1351–1357. [Google Scholar]
- (1209).Shepard WEB; Anderson BF; Lewandoski DA; Norris GE; Baker EN Copper Coordination Geometry in Azurin Undergoes Minimal Change on Reduction of Copper(II) to Copper(I). J.Am. Chem. Soc 1990, 112, 7817–7819. [Google Scholar]
- (1210).Battistuzzi G; Borsari M; Loschi L; Righi F; Sola M Redox Thermodynamics of Blue Copper Proteins. J. Am. Chem. Soc 1999, 121, 501–506. [Google Scholar]
- (1211).Botuyan MV; Toy-Palmer A; Chung J; Blake RC II; Beroza P; Case DA; Dyson HJ NMR Solution Structure of Cu(I) Rusticyanin from Thiobacillus Ferrooxidans: Structural Basis for the Extreme Acid Stability and Redox Potential. J. Mol. Biol 1996, 263, 752–767. [DOI] [PubMed] [Google Scholar]
- (1212).Ingledew WJ; Cobley JG A Potentiometric and Kinetic Study on the Respiratory Chain of Ferrous-Iron-Grown Thiobacillus Ferrooxidans. Biochim. Biophys. Acta, Bioenerg 1980, 590, 141 – 158. [DOI] [PubMed] [Google Scholar]
- (1213).Bento I; Martins LO; Lopes GG; Carrondo MA; Lindley PF Dioxygen Reduction by Multi-Copper Oxidases; A Structural Perspective. Dalton Trans 2005, 3507–3513. [DOI] [PubMed] [Google Scholar]
- (1214).Xu F; Shin W; Brown SH; Wahleithner JA; Sundaram UM; Solomon EI A Study of a Series of Recombinant Fungal Laccases and Bilirubin Oxidase That Exhibit Significant Differences in Redox Potential, Substrate Specificity, and Stability. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1996, 1292, 303–311. [DOI] [PubMed] [Google Scholar]
- (1215).Prigge ST; Kolhekar AS; Eipper BA; Mains RE; Amzel LM Amidation of Bioactive Peptides: The Structure of Peptidylglycine-Hydroxylating Monooxygenase. Science 1997, 278, 1300–1305. [DOI] [PubMed] [Google Scholar]
- (1216).Chauhan S; Hosseinzadeh P; Lu Y; Blackburn NJ Stopped-Flow Studies of the Reduction of the Copper Centers Suggest a Bifurcated Electron Transfer Pathway in Peptidylglycine Monooxygenase. Biochemistry 2016, 55, 2008–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1217).Pavlishchuk VVV; Addison AWW Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25 Degrees C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar]
- (1218).Rorabacher DB Electron Transfer by Copper Centers. Chem. Rev 2004, 104, 651–697. [DOI] [PubMed] [Google Scholar]
- (1219).Sengupta K; Chatterjee S; Dey A In Situ Mechanistic Investigation of O2 Reduction by Iron Porphyrin Electrocatalysts Using Surface-Enhanced Resonance Raman Spectroscopy Coupled to Rotating Disk Electrode (SERRS-RDE) Setup. ACS Catal 2016, 6, 6838–6852. [Google Scholar]
- (1220).Ly HK; Wrzolek P; Heidary N; Gotz R; Horch M; Kozuch J; Schwalbe M; Weidinger IM; et al. 2nd Coordination Sphere Controlled Electron Transfer of Iron Hangman Complexes on Electrodes Probed by Surface Enhanced Vibrational Spectroscopy. Chem. Sci 2015, 6, 6999–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1221).Tano T; Ertem MZ; Yamaguchi S; Kunishita A; Sugimoto H; Fujieda N; Ogura T; Cramer CJ; Itoh S Reactivity of copper(II)-Alkylperoxo Complexes. Dalton Trans 2011, 40, 10326–10336. [DOI] [PubMed] [Google Scholar]
- (1222).Abe T; Morimoto Y; Mieda K; Sugimoto H; Fujieda N; Ogura T; Itoh S Geometric Effects on O–O Bond Scission of Copper(II)-Alkylperoxide Complexes. J. Inorg. Biochem 2017, 177, 375–383. [DOI] [PubMed] [Google Scholar]
- (1223).Moënne-Loccoz P; Fee JA Catalyzing NO to N2O in the Nitrogen Cycle. Science 2010, 330, 1632–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1224).Vázquez-Torres A; Bäumler AJ Nitrate, Nitrite and Nitric Oxide Reductases: From the Last Universal Common Ancestor to Modern Bacterial Pathogens. Curr. Opin. Microbiol 2016, 29, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1225).Wang J; Schopfer MP; Sarjeant AAN; Karlin KD Heme-Copper Assembly Mediated Reductive Coupling of Nitrogen Monoxide (-NO). J. Am. Chem. Soc 2009, 131, 450–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1226).Schopfer MP; Wang J; Karlin KD Bioinspired Heme, Heme/Nonheme Diiron, Heme/Copper, and Inorganic NOx Chemistry: •NO(g) Oxidation, Peroxynitrite–Metal Chemistry, and •NO(g) Reductive Coupling. Inorg. Chem 2010, 49, 6267–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1227).Wijeratne GB; Hematian S; Siegler MA; Karlin KD Copper(I)/NO(g) Reductive Coupling Producing a Trans-Hyponitrite Bridged Dicopper(II) Complex: Redox Reversal Giving Copper(I)/NO(g) Disproportionation. J. Am. Chem. Soc 2017, 139, 13276–13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1228).Dey A; Ghosh S Model Compounds for Nitric Oxide Reductase In Metalloenzymes in Denitrification: Applications and Environmental Impacts; The Royal Society of Chemistry, 2017; pp 185–224. [Google Scholar]
- (1229).Zhao X; Yeung N; Russell BS; Garner DK; Lu Y Catalytic Reduction of NO to N2O by a Designed Heme Copper Center in Myoglobin: Implications for the Role of Metal Ions. J. Am. Chem. Soc 2006, 128, 6766–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1230).Matsumura H; Hayashi T; Chakraborty S; Lu Y; Moënne-Loccoz P The Production of Nitrous Oxide by the Heme/Nonheme Diiron Center of Engineered Myoglobins (FeBMbs) Proceeds through a trans-Iron-Nitrosyl Dimer. J. Am. Chem. Soc 2014, 136, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1231).Chakraborty S; Reed J; Ross M; Nilges MJ; Petrik ID; Ghosh S; Hammes-Schiffer S; Sage JT; Zhang Y; Schulz CE; et al. Spectroscopic and Computational Study of a Nonheme Iron Nitrosyl Center in a Biosynthetic Model of Nitric Oxide Reductase. Angew. Chem., Int. Ed 2014, 53, 2417–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1232).Chakraborty S; Reed J; Sage JT; Branagan NC; Petrik ID; Miner KD; Hu MY; Zhao J; Alp EE; Lu Y Recent Advances in Biosynthetic Modeling of Nitric Oxide Reductases and Insights Gained from Nuclear Resonance Vibrational and Other Spectroscopic Studies. Inorg. Chem 2015, 54, 9317–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1233).Kakishima K; Shiratsuchi A; Taoka A; Nakanishi Y; Fukumori Y Participation of Nitric Oxide Reductase in Survival of Pseudomonas Aeruginosa in LPS-Activated Macrophages. Biochem. Biophys. Res. Commun 2007, 355, 587–591. [DOI] [PubMed] [Google Scholar]
- (1234).Laver JR; Stevanin TM; Messenger SL; Lunn AD; Lee ME; Moir JWB; Poole RK; Read RC Bacterial Nitric Oxide Detoxification Prevents Host Cell S-Nitrosothiol Formation: A Novel Mechanism of Bacterial Pathogenesis. FASEB J. 2010, 24, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1235).Wright AM; Hayton TW Understanding the Role of Hyponitrite in Nitric Oxide Reduction. Inorg. Chem 2015, 54, 9330–9341. [DOI] [PubMed] [Google Scholar]
- (1236).Hunsicker-Wang LM; Pacoma RL; Chen Y; Fee JA; Stout CD A Novel Cryoprotection Scheme for Enhancing the Diffraction of Crystals of Recombinant Cytochrome ba3 Oxidase from Thermus Thermophilus. Acta Crystallogr., Sect. D: Biol. Crystallogr 2005, 61, 340–343. [DOI] [PubMed] [Google Scholar]
- (1237).Mittra K; Chatterjee S; Samanta S; Sengupta K; Bhattacharjee H; Dey A A Hydrogen Bond Scaffold Supported Synthetic Heme FeIII– O2– Adduct. Chem. Commun 2012, 48, 10535–10537. [DOI] [PubMed] [Google Scholar]
- (1238).Samanta S; Mittra K; Sengupta K; Chatterjee S; Dey A Second Sphere Control of Redox Catalysis: Selective Reduction of O2 to O2– or H2O by an Iron Porphyrin Catalyst. Inorg. Chem 2013, 52, 1443–1453. [DOI] [PubMed] [Google Scholar]
- (1239).Das PK; Mittra K; Dey A Spectroscopic Characterization of a Phenolate Bound FeII–O2 Adduct: Gauging the Relative “Push” Effect of a Phenolate Axial Ligand. Chem. Commun 2014, 50, 5218–5220. [DOI] [PubMed] [Google Scholar]
- (1240).Bhunia S; Rana A; Roy P; Martin DJ; Pegis ML; Roy B; Dey A Rational Design of Mononuclear Iron Porphyrins for Facile and Selective 4e−/4H+ O2 Reduction: Activation of O–O Bond by 2nd Sphere Hydrogen Bonding. J. Am. Chem. Soc 2018, 140, 9444–9457. [DOI] [PubMed] [Google Scholar]
- (1241).Ro SY; Ross MO; Deng YW; Batelu S; Lawton TJ; Hurley JD; Stemmler TL; Hoffman BM; Rosenzweig AC From Micelles to Bicelles: Effect of the Membrane on Particulate Methane Monooxygenase Activity. J. Biol. Chem 2018, 293, 10457–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1242).Ciano L; Davies GJ; Tolman WB; Walton PH Bracing Copper for the Catalytic Oxidation of C–H Bonds. Nat. Catal 2018, 1, 571–577. [Google Scholar]
- (1243).Tomkins P; Ranocchiari M; van Bokhoven JA Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res 2017, 50, 418–425. [DOI] [PubMed] [Google Scholar]
- (1244).Mahyuddin MH; Staykov A; Shiota Y; Miyanishi M; Yoshizawa K Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged AEI, CHA, AFX, and MFI Zeolites. ACS Catal. 2017, 7, 3741–3751. [Google Scholar]
- (1245).Mahyuddin MH; Tanaka T; Shiota Y; Staykov A; Yoshizawa K Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu2(μ-O3)]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar]
- (1246).Snyder BER; Vanelderen P; Schoonheydt R A.; Sels, B. F.; Solomon, E. I. Second-Sphere Effects on Methane Hydroxylation in Cu-Zeolites. J. Am. Chem. Soc 2018, 140, 9236–9243. [DOI] [PubMed] [Google Scholar]
- (1247).Sushkevich Vitaly L; Palagin D; van Bokhoven Jeroen A The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem., Int. Ed 2018, 57, 8906–8910. [DOI] [PubMed] [Google Scholar]
- (1248).Newton MA; Knorpp AJ; Pinar AB; Sushkevich VL; Palagin D; van Bokhoven JA On the Mechanism Underlying the Direct Conversion of Methane to Methanol by Copper Hosted in Zeolites; Braiding Cu K-Edge XANES and Reactivity Studies. J. Am. Chem. Soc 2018, 140, 10090–10093. [DOI] [PubMed] [Google Scholar]
- (1249).Ikuno T; Zheng J; Vjunov A; Sanchez-Sanchez M; Ortuno MA; Pahls DR; Fulton JL; Camaioni DM; Li ZY; Ray D; Mehdi BL; Browning ND; Farha OK; Hupp JT; Cramer CJ; Gagliardi L; Lercher JA Methane Oxidation to Methanol Catalyzed by Cu-Oxo Clusters Stabilized in NU-1000 Metal-Organic Framework. J. Am. Chem. Soc 2017, 139, 10294–10301. [DOI] [PubMed] [Google Scholar]
- (1250).Doan HA; Li Z; Farha OK; Hupp JT; Snurr R Q. Theoretical Insights into Direct Methane to Methanol Conversion over Supported Dicopper Oxo Nanoclusters. Catal. Today 2018, 312, 2–9. [Google Scholar]
- (1251).Luo F; Shinzawa-Itoh K; Hagimoto K; Shimada A; Shimada S; Yamashita E; Yoshikawa S; Tsukihara T Structure of Bovine Cytochrome c Oxidase Crystallized at a Neutral pH Using a Fluorinated Detergent. Acta Crystallogr., Sect. F: Struct. Biol. Commun 2017, 73, 416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1252).Luo F; Shinzawa-Itoh K; Hagimoto K; Shimada A; Shimada S; Yamashita E; Yoshikawa S; Tsukihara T Structure of Bovine Cytochrome c Oxidase in the Ligand-Free Reduced State at Neutral pH. Acta Crystallogr., Sect. F: Struct. Biol. Commun 2018, 74, 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1253).Vilhjálmsdóttir J; Gennis RB; Brzezinski P The Electron Distribution in the “Activated” State of Cytochrome C. Sci. Rep 2018, 8, 7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1254).Karlin KD; Hayes JC; Hutchinson JP; Hyde JR; Zubieta J Synthesis and X-ray Structural Characterization of Cu(I) and Cu(II) Derivatives of a New Symmetric Tripodal Ligand N(CH2CH2-py)3, (py = 2-pyridyl). Inorg. Chim. Acta 1982, 64, L219–L220. [Google Scholar]
- (1255).Antonini E; Brunori M Hemoglobin and Myoglobin in Their Reactions with Ligands; North-Holland Publishing Co.: Amsterdam, 1971. [Google Scholar]