

Abstract
Background
The symptoms of eczema can lead to sleeplessness and fatigue and may have a substantial impact on quality of life. Use of oral H1 antihistamines (H1 AH) as adjuvant therapy alongside topical agents is based on the idea that combining the anti‐inflammatory effects of topical treatments with the blocking action of histamine on its receptors in the skin by H1 AH (to reduce the principal symptom of itch) might magnify or intensify the effect of treatment. Also, it would be unethical to compare oral H1 AH alone versus no treatment, as topical treatment is the standard management for this condition.
Objectives
To assess the effects of oral H1 antihistamines as 'add‐on' therapy to topical treatment in adults and children with eczema.
Search methods
We searched the following databases up to May 2018: the Cochrane Skin Group Specialised Register, CENTRAL, MEDLINE, Embase, and the GREAT database (Global Resource of EczemA Trials; from inception). We searched five trials registers and checked the reference lists of included and excluded studies for further references to relevant randomised controlled trials (RCTs). We also searched the abstracts of four conference proceedings held between 2000 and 2018.
Selection criteria
We sought RCTs assessing oral H1 AH as 'add‐on' therapy to topical treatment for people with eczema compared with topical treatment plus placebo or no additional treatment as add‐on therapy.
Data collection and analysis
We used standard Cochrane methodological procedures. Primary outcome measures were 'Mean change in patient‐assessed symptoms of eczema' and 'Proportion of participants reporting adverse effects and serious adverse events'. Secondary outcomes were 'Mean change in physician‐assessed clinical signs', 'Mean change in quality of life', and 'Number of eczema flares'.
Main results
We included 25 studies (3285 randomised participants). Seventeen studies included 1344 adults, and eight studies included 1941 children. Most studies failed to report eczema severity at baseline, but they were conducted in secondary care settings, so it is likely that they recruited patients with more severe cases of eczema. Trial duration was between three days and 18 months. Researchers studied 13 different H1 AH treatments. We could not undertake pooling because of the high level of diversity across studies in terms of duration and dose of intervention, concomitant topical therapy, and outcome assessment. Risk of bias was generally unclear, but five studies had high risk of bias in one domain (attrition, selection, or reporting bias). Only one study measured quality of life, but these results were insufficient for statistical analysis.
Although this review assessed 17 comparisons, we summarise here the results of three key comparisons in this review.
Cetirizine versus placebo
One study compared cetirizine 0.5 mg/kg/d against placebo over 18 months in 795 children. Study authors did not report patient‐assessed symptoms of eczema separately for pruritus. Cetirizine is probably associated with fewer adverse events (mainly mild) (risk ratio (RR) 0.68, 95% confidence interval (CI) 0.46 to 1.01) and the need for slightly less additional H1 AH use as an indication of eczema flare rate (P = 0.035; no further numerical data given). Physician‐assessed clinical signs (SCORing Atopic Dermatitis index (SCORAD)) were reduced in both groups, but the difference between groups was reported as non‐significant (no P value given). Evidence for this comparison was of moderate quality.
One study assessed cetirizine 10 mg/d against placebo over four weeks in 84 adults. Results show no evidence of differences between groups in patient‐assessed symptoms of eczema (pruritus measured as part of SCORAD; no numerical data given), numbers of adverse events (RR 1.11, 95% CI 0.50 to 2.45; mainly sedation, other skin‐related problems, respiratory symptoms, or headache), or physician‐assessed changes in clinical signs, amount of local rescue therapy required, or number of applications as an indicator of eczema flares (no numerical data reported). Evidence for this comparison was of low quality.
Fexofenadine versus placebo
Compared with placebo, fexofenadine 120 mg/d taken in adults over one week (one study) probably leads to a small reduction in patient‐assessed symptoms of pruritus on a scale of 0 to 8 (mean difference (MD) ‐0.25, 95% CI ‐0.43 to ‐0.07; n = 400) and a greater reduction in the ratio of physician‐assessed pruritus area to whole body surface area (P = 0.007; no further numerical data given); however, these reductions may not be clinically meaningful. Results suggest probably little or no difference in adverse events (mostly somnolence and headache) (RR 1.05, 95% CI 0.74 to 1.50; n = 411) nor in the amount of 0.1% hydrocortisone butyrate used (co‐intervention in both groups) as an indicator of eczema flare, but no numerical data were given. Evidence for this comparison was of moderate quality.
Loratadine versus placebo
A study of 28 adults compared loratadine 10 mg/d taken over 4 weeks versus placebo. Researchers found no evidence of differences between groups in patient‐assessed pruritus, measured by a 100‐point visual analogue scale (MD ‐2.30, 95% CI ‐20.27 to 15.67); reduction in physician‐assessed clinical signs (SCORAD) (MD ‐4.10, 95% CI ‐13.22 to 5.02); or adverse events. Study authors reported only one side effect (folliculitis with placebo) (RR 0.25, 95% CI 0.01 to 5.76). Evidence for this comparison was of low quality. Number of eczema flares was not measured for this comparison.
Authors' conclusions
Based on the main comparisons, we did not find consistent evidence that H1 AH treatments are effective as 'add‐on' therapy for eczema when compared to placebo; evidence for this comparison was of low and moderate quality. However, fexofenadine probably leads to a small improvement in patient‐assessed pruritus, with probably no significant difference in the amount of treatment used to prevent eczema flares. Cetirizine was no better than placebo in terms of physician‐assessed clinical signs nor patient‐assessed symptoms, and we found no evidence that loratadine was more beneficial than placebo, although all interventions seem safe.
The quality of evidence was limited because of poor study design and imprecise results. Future researchers should clearly define the condition (course and severity) and clearly report their methods, especially participant selection and randomisation; baseline characteristics; and outcomes (based on the Harmonising Outcome Measures in Eczema initiative).
Plain language summary
Do H1 antihistamine tablets or liquids help improve eczema symptoms in people who are already using creams or ointments for their eczema?
Review question
Are H1 antihistamines (which inhibit the action of chemicals released as part of an allergic reaction; known as 'histamines'), taken as tablets or liquid, effective and safe in people of any age with diagnosed eczema, if given in addition to creams and ointments, compared to treatment with an inactive substance (placebo) or nothing added to creams and ointments?
Background
Eczema (also known as 'atopic eczema/dermatitis') is a skin disorder frequently affecting both children and adults. In developed countries, 10% to 20% of all people are affected by eczema during their lifetime. The main symptom is itch, which results in scratching and, together with skin inflammation, frequently produces reddening of the skin. The symptoms of eczema can lead to sleeplessness and fatigue, lowering quality of life. Antihistamines are frequently given for itch (specifically H1 antihistamines taken by mouth), and they may alleviate the symptoms of eczema when given in addition to conventional treatments directly applied to the skin (e.g. emollients, moisturisers, steroid creams), although they are not thought to cure it. Many antihistamines are available without prescription, for instance, cetirizine or loratadine. Although H1 antihistamines are frequently prescribed for treating eczema, we do not know whether they are effective and safe.
Study characteristics
We searched for relevant studies up to May 2018. We included 25 randomised controlled trials with 3285 participants of all ages with diagnosed eczema. Eight studies included children or adolescents, and 17 included adults. The gender of participants and the severity of symptoms often were not reported. All studies were conducted in secondary care settings, including hospital clinics, research clinics, dermatology centres, and surgery centres, meaning that participants were likely to have more severe eczema than if recruitment occurred from first point of contact settings (i.e. primary care). All but one study compared H1 antihistamine versus placebo. Researchers studied 13 different H1 antihistamines, most of which were less sedating H1 antihistamines (known as 'second‐generation antihistamines'). Studies lasted between three days and 18 months. Seven trials received funding from pharmaceutical companies; they are the largest trials included in this review.
Key results
We found no convincing evidence that H1 antihistamines help patients with eczema.
One study compared cetirizine 0.5 mg/kg/d versus placebo (in children over a period of 18 months). No data were provided on patient‐assessed itch symptoms of eczema. Cetirizine is probably associated with fewer (mainly mild) adverse events and the need for slightly less additional H1 antihistamine to prevent flares. Even though physician‐assessed clinical signs were reduced in both groups, results show no differences between groups (all moderate‐quality evidence).
When compared with placebo, we found no evidence that an increased dose of cetirizine 10 mg/d over four weeks makes a difference in terms of patient‐assessed itch, number of side effects, physician‐assessed signs, or number of eczema flares as measured by the amount of treatment used (all low‐quality evidence). Side effects reported in both groups included drowsiness, skin‐related problems, breathing issues, and headaches.
Compared with placebo, fexofenadine 120 mg/d given to adults for one week probably slightly improves patient‐assessed itch, as well as producing a greater reduction in the area of itch, as assessed by a physician, and it probably makes little or no difference in the number of participants experiencing side effects (mostly drowsiness and headaches) or in the extent of treatment required as an indicator of the number of eczema flares (all moderate‐quality evidence).
We found no evidence of a difference between placebo and loratadine 10 mg/d given to adults for four weeks in patient‐assessed itch, occurrence of side effects, or physician‐assessed signs of eczema (all low‐quality evidence). This study did not measure the number of eczema flares. Study authors reported only one side effect (folliculitis), which occurred with placebo.
Only one study measured quality of life, but results could not be analysed.
Quality of the evidence
For all outcomes across key comparisons, evidence was of low to moderate certainty. Reasons for lowering the quality of evidence include concerns over how studies were carried out and inclusion of too few participants, leading to less accurate results.
Summary of findings
Background
Description of the condition
For unfamiliar terms, see the Glossary of Terms provided in Table 6.
1. Glossary of terms.
| Term | Definition |
| Adverse event | The occurrence of an unwanted effect due to a medication or a medical procedure |
| Antihistamines | Antihistamines (or histamine antagonists/histamine blockers) are drugs that block the action of histamine (an endogenous messenger substance released, e.g. in allergic inflammatory reactions). H1 antihistamines are predominantly used in the treatment of allergies |
| Arrhythmia | Irregular heartbeat |
| Atopy | Describes a genetic predisposition for hypersensitivity to common antigens (e.g. pollen, house dust, animal dander). Atopy can manifest in atopic diseases such as hay fever, allergic asthma, or eczema. Atopy can be associated with increased immunoglobulin E (IgE) levels |
| Autonomic nervous system | A division of the peripheral nervous system that regulates smooth muscle and gland functioning. As such, it influences the function of internal organs. This is a control system that acts largely unconsciously and regulates bodily functions such as heart rate, digestion, respiratory rate, pupillary response, urination, and sexual arousal |
| Axon reflex flare | Vasodilation and increased redness and sensitivity of the skin caused by stimulation of peripheral nerves |
| Cardiotoxic | Pertaining to the occurrence of heart electrophysiology dysfunction or muscle damage. The heart becomes weaker and is not as efficient in pumping and therefore in circulating blood |
| Clinical signs | Physician‐assessed visible signs of eczema, such as erythema (redness) or dryness |
| Corticosteroids | A group of naturally occurring hormones or synthetic analogues of hormones. Corticosteroids are produced in the adrenal glands. According to their biological function, corticosteroids can be classified into: (1) mineralocorticoids (hormones that influence the salt and water balance within the body), (2) glucocorticosteroids (=> see below), and (3) androgen/oestrogen (male/female sex hormones) |
| Cross‐over study | A type of clinical trial in which participants under study receive a sequence of different treatments in a randomly selected order |
| Eosinophils | White blood cells ‐ one of the immune system components ‐ that control mechanisms associated with allergy and asthma as well as parasitosis |
| Emollients | Emollients (or moisturisers) are complex mixtures of chemical agents designed to soften the epidermis (external layers of the skin) |
| Epidermal | Referring to the epidermis. The epidermis is the protective outer layer of the skin |
| Epithelial | Pertaining to the thin tissue forming the outer layer of a body's surface and lining the alimentary canal and other hollow structures |
| Erythema | Redness of the skin (or mucous membranes) that is caused by increased blood flow into superficial vessels and capillaries. This can occur, for example, with skin injury, infection, or inflammation |
| Excoriations | Skin lesions caused by repetitive, compulsive scratching of the skin. Of different colours (e.g. yellow or red caused by tissue fluid and light bleeding), they are frequently found in patients suffering from itch |
| Filaggrin | A protein that binds to keratin fibres in the cells to form a functional barrier at the skin surface. There is defective permeability of the barrier function (skin‐protecting function) in eczema. Defects of the filaggrin gene contribute to the defective permeability of the outermost layer of the skin |
| Flexural | Related to the action of bending or curving (e.g. of the skin or a joint) |
| Gene polymorphism | A gene is said to be polymorphic if more than 1 allele (1 of 2 or more forms of a gene) occupies that gene’s locus (location of a gene on a chromosome) within a population |
| Glucocorticosteroids | Particular class of corticosteroids. They have various functions within the human body (e.g. in the immune system, in metabolism) |
| IgE | A class of antibodies (immunoglobulins, Ig). The primary function of IgE within the immune system is to fight endoparasites. However, it is also responsible for some allergic reactions (e.g. local inflammation in eczema, airway constriction in asthma). People with an atopic predisposition often have increased IgE levels |
| IL‐31 | Belongs to the group of cytokines. These are mediators that communicate between different cell types. IL stands for interleukin |
| Immunomodulator | Pharmacologically active substance that affects the immune system. Examples of topical immunomodulators used in the treatment of eczema are tacrolimus and pimecrolimus |
| Lichenification | Describes extensive thickening of the skin accompanied by noticeable skin markings. Lichenification is a symptom of a number of chronic skin diseases (e.g. eczema) |
| Lipid | An organic compound (e.g. fats, oils, hormones) grouped together because components are not soluble in water |
| Mediators | Intermediary substances or agents that are released from cells to regulate or cause physiological consequences |
| Metabolites | Small molecules that are intermediate or final products of metabolism |
| Neuropeptides | Small molecules that can be found in the nervous tissue. They serve as messenger substances used by nerve cells (neurons) to communicate with one another |
| Nodule | A palpable solid skin lesion usually 0.5 cm or larger in diameter. Can involve different depths of the skin and can vary in shape and surface |
| Oedema | Abnormal accumulation of fluid in or under the skin |
| Papule | A solid elevated palpable lesion usually less than 0.5 cm in diameter. Can have different shapes and surfaces (e.g. flat‐topped and rough) |
| Pimecrolimus | An immunomodulating agent used in the treatment of eczema. Available as a topical cream |
| Polymorphism | A variation in the DNA that is too common to be due merely to new mutation. A polymorphism must have a frequency of at least 1% in the population |
| Prurigo | Prurigo and prurigo nodularis may be used synonymously. Prurigo describes a condition of nodular cutaneous lesions that itch intensely (i.e. are pruritic). Prurigo nodules usually reflect a chronic state of itch and eczema |
| Pruritogen | Any substance that causes pruritus |
| Psychosomatic | Refers to the processes of psychological factors that influence physiological functioning |
| Long QT interval | Abnormal pattern seen on the records of the heart's electrical activity measured by electrocardiography (ECG). Long QT‐syndrome is a disorder of the heart's electrical activity that can cause fast and chaotic heartbeats, leading to a sudden fainting spell or, in some cases, death |
| RCT | Randomised controlled trial. A type of scientific study in which participants in the study population are allocated randomly to an experimental, placebo, or control group. RCTs are often conducted to test the effectiveness of a new medication or medical procedure |
| Somnolence | A state of strong desire for sleep, or sleeping for unusually long periods |
| Symptom | Departure from normal function or feeling that is noticed by a patient, indicating the presence of disease or abnormality (Knol 2011) (e.g. sleep loss, itching) |
| Tachycardia | A heart rate greater than 100 beats per minute (BPM) in adults. It means that the heart beats too fast |
| Tacrolimus | An immunosuppressive drug used mainly after organ transplantation to lower the risk of organ rejection. Tacrolimus is also used, among other indications, in a topical preparation for the treatment of eczema |
| Th2 helper cells | A functionally distinct subclass of T helper cells (lymphocytes, white blood cells) that play an important role in the immune system. They can stimulate IgE synthesis and are critical in favouring the differentiation of eosinophilic granulocytes. They activate the release of local mediators that cause sneezing, coughing, or eczema |
| Tryptase | A protein‐splitting enzyme (proteinase) that is produced by mast cells. These cells are located in the human dermis. Proteinases play a role in regulation of the immune response and cutaneous inflammation |
| Vasodilation | Refers to widening of blood vessels |
| Ventricular tachycardia | A type of tachycardia that arises from improper electrical activity in the ventricles of the heart |
| Vesicle | A cavity (smaller than 0.5 cm) filled with fluid. It may be non‐palpable in thicker skin (e.g. palms, soles), breaks easily, and releases its fluid onto the skin |
BPM: beats per minute; ECG: electrocardiogram; Ig: immunoglobulin; IL: interleukin; RCT: randomised controlled trial.
Definition
Eczema is a chronic, inflammatory, non‐infectious skin disease. It is among the most common skin diseases, which, although predominantly affecting children, may also persist into adulthood (Thomas 2008). The main sites of eczema migrate from infancy to childhood. The final typical location is in the folds of the elbows, in the hollows of the knees, and frequently also in the neck during adolescence and adulthood (Archer 2000; Thomas 2008).
Eczema often occurs in families with atopic diseases including asthma, allergic rhinitis/hay fever (and food allergy), and atopic eczema. These diseases share a common pathogenesis and are frequently present together in the same individual and family. The word 'atopy' refers to the genetic tendency to produce immunoglobulin E (IgE) antibodies in response to small quantities of common environmental proteins such as pollen, house dust mites, and food allergens (Stone 2002; Thomsen 2015). Around 30% of people with eczema develop asthma, and 35% develop allergic rhinitis (Luoma 1983). However, it is known that atopy does not concurrently occur in all patients with atopic eczema. In view of this, it has been proposed recently that the term 'eczema' should be used to define patients both with and without atopy. In keeping with the 'Revised nomenclature for allergy for global use' (Johansson 2004), and similar to other Cochrane Reviews evaluating eczema therapies (van Zuuren 2017), we will therefore use the term 'eczema' throughout this review.
Clinical features and symptoms
Eczema is characterised by an itchy, red rash, but it may also present with a wide spectrum of clinical signs such as erythema, papules, vesicles, prurigo nodules, crusts, scales, dry skin, and lichenification (Hanifin 1980). Burning, pain, and itch are the most frequent and distressing symptoms. It has been stated that "the diagnosis of eczema cannot be made if there is no history of itch" (Hanifin 1980). People with eczema often feel a strong urge to scratch as a consequence of itching (Darsow 2012). Scratching, together with skin inflammation, frequently produces reddening of the skin. The symptoms of eczema can lead to sleeplessness and fatigue and may have a substantial impact on the quality of life of those affected (Carroll 2005).
Various sets of diagnostic criteria for eczema have been developed and validated, but no consensus about their use has been reached (Brenninkmeijer 2008). The most extensively validated and widely used set of diagnostic criteria has been proposed by a United Kingdom (UK) Working Party (Williams 1994). According to these criteria, an individual must have an itchy skin condition in the last 12 months and must meet three or more of the following criteria: onset in the first two years of life, a history of flexural involvement, a history of generalised dry skin, a history of other atopic disease, or visible flexural dermatitis. An image of the typical lesions found in eczema is shown in Figure 1.
1.
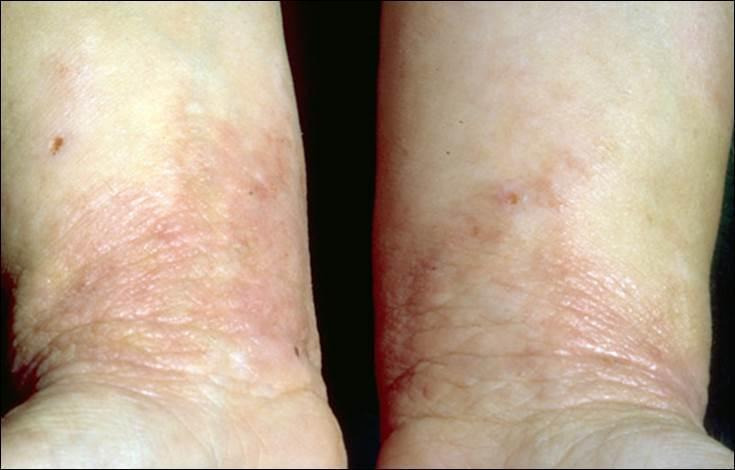
Typical eczema lesions on wrist insides and forearms.
Epidemiology and causes
In many countries, eczema is the most frequently encountered chronic condition during infancy, but there is wide variation in the prevalence of eczema at a global level (Mallol 2013; Odhiambo 2009). Recent data from the International Study of Asthma and Allergies in Childhood (ISAAC Phase 1) have shown that the prevalence of eczema (itchy flexural rash in the past 12 months) ranges from 0.9% to 22.5% in six‐ and seven‐year‐old children, and from 0.2% to 24.6% in those aged 13 to 14 years (Odhiambo 2009). The global total prevalence in children aged six to seven years was 7.9%, and in the 13‐ to 14‐year‐old age group, 7.3% (ISAAC Phase 3) (Mallol 2013). For adults, recent studies have estimated 12‐month prevalence (i.e. the percentage of people who had the condition within the last 12 months) at 14.3% in Vinding 2014, and at 10.2% in Silverberg 2013a, in Denmark and the United States (USA), respectively.
The onset of eczema usually occurs in early childhood, with most manifestations arising before the age of six (Bieber 2008; Nutten 2015; Weidinger 2016). Eczema can persist over long periods of time. However, its course can show a relapsing‐remitting pattern including repeated flares (Garmhausen 2013; Illi 2004). Eczema used to be considered mainly a childhood disease (Weidinger 2016), but recent evidence suggests that a considerable proportion of individuals do not encounter remittance (Weidinger 2016). A recent cohort study found persistent eczema in 50% of those given the diagnosis at school age (Mortz 2015), and another large‐scale study reported a lifetime prevalence of eczema of 40.7% among adults (Rönmark 2012).
Eczema is a complex disease that is most likely to be caused by gene and environmental interactions (Bieber 2008). In addition to allergic sensitisation and gene polymorphisms, a large number of environmental and lifestyle factors are potentially involved in the aetiology (Weidinger 2016). These include pet exposure, breastfeeding and tobacco smoke (Apfelbacher 2011), climate factors (Silverberg 2013b), and microbial exposure (Flohr 2014).
Biology
It has been thought that an immunological defect in Th2 helper cells may give rise to increased IgE production (Bieber 2008). However, although increased IgE levels are often found in patients with eczema, wide variation has been noted in the presence of allergies to aeroallergens (Flohr 2014).
Moreover, as a result of abnormal lipid metabolism or epidermal protein formation (e.g. caused by filaggrin mutation; Baurecht 2007), the skin barrier in eczema functions poorly, giving rise to dry skin, which is prone to epidermal barrier disruption. Skin inflammation and epidermal barrier disruptions are phenomena that reinforce each other (Weidinger 2016). Furthermore, damage to the epithelial barrier increases immune exposure to environmental allergens, stimulating the formation of IgE (Bieber 2008).
The rate of microbial colonisation of the skin is caused by a number of defects in innate cutaneous immunology, for example, with Staphylococcus aureus colonisation in eczema (Ring 2012a). This can lead to disease exacerbations and may require additional antimicrobial treatment (Ring 2012a). The cutaneous innate immune system is a key determinant of the physical, chemical, microbial, and immunological barrier functions of the epidermis (Kuo 2013). The innate cutaneous immunology (as well as the adaptive immunology of the skin) in patients with eczema is thought to be malfunctional (Biedermann 2015), leading to an inadequate host response to a pathogen or a persistent inflammatory state (Kuo 2013).
Psychological factors such as stress can influence the clinical course of eczema and the itch‐scratch cycle (Ring 2012b). Other co‐factors can have an effect, such as abnormal microbial colonisation leading to skin infection and psychosomatic factors influencing the autonomic nervous system and production of mediators like, for example, neuropeptides and eosinophils (Bieber 2009; Buddenkotte 2010; Ring 2012a; Ring 2012b).
Description of the intervention
The standard approach to treating eczema revolves around the restoration of epidermal barrier function with the use of emollients and moisturisers (Weidinger 2016). Topical corticosteroids are still the favoured therapy for acute flares, but they are also used proactively along with topical calcineurin inhibitors to maintain remission. Non‐specific immunosuppressive drugs, such as cyclosporine or azathioprine, are used in severe refractory cases (Weidinger 2016; Werfel 2016). Use of emollients, moisturisers, and topical corticosteroids is often supplemented by add‐on therapies such as ultraviolet (UV) therapy or antihistamines, but immunomodulatory systemic drugs such as cyclosporine may be prescribed in severe cases (Werfel 2016; Ring 2012b; Sidbury 2014). Add‐on therapy in general is an approach that is used to enhance a therapeutic effect through use of an additional drug.
Oral H1 antihistamines (H1 receptor antagonists) are widely used as a systemic 'add‐on' therapy to topical treatments, which include glucocorticosteroids, immunomodulators such as pimecrolimus or tacrolimus, tar preparations, anti‐infective topicals, and doxepin (Ring 2012a). Although four distinct types of histamine receptors are recognised (H1 involved in allergic responses, H2 in gastric acid secretion, H3 in neurotransmission, and H4 in immune responses), in human skin, only H1 and H2 receptors have been demonstrated (Greaves 2005). H1 antihistamines (H1 AH) are prescribed especially with the intention of alleviating the itch that accompanies eczema (Greaves 2005; He 2018).
Antihistamines in general are used to counteract the release of histamines. When histamines are released, they bind to specific receptors, and typical reactions (allergic reactions or allergy symptoms) occur (Mann 1989). Histamine induces pruritus, sneezing, increased vascular permeability, and smooth muscle contraction of the respiratory and gastrointestinal tracts (Takahashi 2004). H1 AH, which are not structurally related to histamine, do not antagonise the binding of histamine but bind to different sites on the receptor to produce an effect opposite to the effect that histamines would produce (Church 2013; Simons 2011). Thus, H1 AH are not receptor antagonists but are inverse agonists, in that they produce the opposite effect on the receptor to histamine (Leurs 2002; Simons 2008). As a consequence, the preferred term used to define these drugs is 'H1 antihistamines' rather than 'histamine antagonists' (Church 2013).
According to their chemical structure and properties, H1 AH are usually classified as first‐ or second‐generation H1 AH. First‐generation antihistamines may cause sedation and consequently can be useful for treating sleep disturbances due to itching. Second‐generation antihistamines are less sedating, as the molecule is less likely to cross the blood‐brain barrier; however, they are not given without the possibility of sedative effects, and some (particularly terfenadine and astemizole, which are no longer in use) may cause irregularities in heart rhythm (cardiac arrhythmia). A category of third‐generation antihistamines has been used to describe some of the later antihistamines. This term is not generally agreed upon, as it has been noted that such agents do not differ sufficiently from earlier compounds in terms of desirable and undesirable effects (Greaves 2005; Handley 1998; Holgate 2003; Simons 2004; Yanai 2012).
Agitation, confusion, somnolence, tachycardia, constipation, urinary retention, and adverse cardiac reactions have been reported as adverse effects of antihistamines, particularly of first‐generation antihistamines such as chlorpheniramine (Yanai 2012). Because of side effects such as sedation and drowsiness, use of these drugs in the daytime has been limited (Greaves 2005).
The aim of developing second‐generation antihistamines, such as astemizole or terfenadine, which are no longer in use, or loratadine, which does not penetrate the blood‐brain barrier, was to produce more tolerable agents without sedative side effects but with the same effectiveness as first‐generation antihistamines (Kay 2000). Motor and cognitive functions are substantially less impaired with the use of second‐generation antihistamines (Herman 2003). However, astemizole and terfenadine have been found to cause ventricular tachycardia, although this occurs rarely (Woosley 1996). Hence, they are no longer in use.
The goal of developing third‐generation antihistamines was to use therapeutically active metabolites of established parent drugs to produce agents that are not cardiotoxic (Handley 1998). Fexofenadine, for instance, is the active metabolite of terfenadine.
However, it has been argued that the sedative effects of H1 AH may be beneficial. The Sidbury 2014 review noted that short‐term, intermittent use of sedating antihistamines (not stated which class) may help those with disturbed sleep due to itch. Another review concluded that a non‐sedating H1 AH (cetirizine) was effective in reducing itch when given at a high sedating dose (Klein 1999). Again, itch reduction was linked to induction of drowsiness.
Current guidelines for the treatment of eczema do not provide recommendations in terms of a dose range for H1 AH nor a minimum duration of treatment (Werfel 2016). However, dosage and scheduling should be based on the drug profile of each individual medication (Sidbury 2014).
How the intervention might work
The pathophysiology of itch (pruritus) in eczema is complex and still is not fully understood (Ständer 2002; Tominaga 2016). Although histamines have been implicated as a mediator of pruritus, many other pruritogens have also been identified (Akiyama 2013). Of these, interleukin‐31 (IL‐31), thymic stromal lymphopoietin (TSLP), and lysophosphatidic acid (LPA) may underlie pruritus in patients with eczema (Tominaga 2016). Histamine‐independent mechanisms triggered by, for example, neuropeptides, proteinases, cytokines, and opioids, are thought to play a role in the pathophysiology of itch in atopic dermatitis (Ständer 2002; Tominaga 2016). Interleukin‐31, a cytokine produced by T cells, is currently suggested as the most important pruritogen identified in human and animal models (Tominaga 2016). Neuropeptides, proteinases, and opioids are also thought to play a role in the release of histamine (Tominaga 2016). In summary, pruritus in eczema is most likely a result of peripheral and central mechanisms including pruritogenic mediators and modulators, sensory nerve fibres, and nerve fibre density in skin and spine, and central itch transmission (Tominaga 2016).
Although it is not exactly clear how itch in eczema is mediated, clinical use of H1 AH remains frequent (Herman 2003). Antihistamines used in eczema are histamine 1 (H1) blockers. H1 AH are thought to suppress actions of histamine such as itch, increased vascular permeability of the skin, and local vasodilation (Greaves 2005). All of these responses are features of the acute allergic reaction of the skin (Takahashi 2004), which is characterised by visible wheals and flares. Wheals and flares may itch; thus, it is thought that H1 AH may be able to suppress the cardinal symptom itch, as well as other symptoms in eczema, by blocking H1 AH receptors.
Evidence from animal models suggests that H1 AH may be able to reduce scratch behaviour in mice (Murota 2010). Whether this can be applied to humans needs to be shown. However, evidence is available for psychomotor impairment after oral H1 AH intake (Takahashi 2004). Scratching exacerbates itch, as is commonly expressed by the itch‐scratch cycle. It may be hypothesised that H1 AH influence this cycle, but evidence for this remains to be put forward.
Due to sedating properties of at least first‐generation H1 AH, as well as the sedating quality of high doses of newer‐generation H1 AH (Klein 1999), it is also possible that patients with eczema who have sleep problems as a result of itch may benefit from the induced drowsiness and consequently better sleep quality.
Use of systemic antihistamines as adjuvant therapy together with topical agents such as, for example, topical corticosteroids or topical immune modulators in eczema (Kawashima 2003), is based on the idea that combining the anti‐inflammatory effects of topical treatments with blocking action of histamine on its receptors in the skin by H1 AH might magnify or intensify the effect of treatment (Greaves 2005; Kawashima 2003). In other words, it is expected that effects resulting from topical anti‐inflammatory treatments are augmented.
Why it is important to do this review
As part of a comprehensive health technology assessment (HTA) of treatments available for eczema, randomised controlled trials (RCTs) investigating the effects of antihistamines in eczema were summarised in a HTA report (Hoare 2000). Authors of this report identified 21 RCTs of any oral antihistamines for treatment of eczema. Of these, studies investigating sedating antihistamines and studies investigating H2 antihistamines (alone or combined with H1 AH) were unable to demonstrate clear benefit of the intervention. Studies investigating less‐sedating antihistamines show conflicting results. For instance, one RCT investigated the effects of various doses of cetirizine, demonstrating a possible effect of a very high dose (Hannuksela 1993). Generally, authors of the HTA report stated that study reporting was poor, rendering it difficult to draw firm conclusions. We believe it is important to update the evidence base.
We conducted a systematic search of the literature to inform the guideline of care for management of eczema put forth by the American Academy of Dermatology (AAD) (Sidbury 2014). This guideline concluded that short‐term, intermittent use of sedating antihistamines (not stated which class) may bring some benefit for sleeping problems that arise as a consequence of itch. No recommendation was made for non‐sedating antihistamines as routine treatment for eczema. Another review concluded that a non‐sedating H1 AH (cetirizine) was effective in reducing itch when given at a high sedating dose (Klein 1999). Again itch reduction was linked to induction of drowsiness.
A recent systematic review did not identify any RCT investigating single use of oral H1 AH versus placebo for eczema, so currently no evidence is available to support or refute the effectiveness of H1 AH used alone (Apfelbacher 2013). It appears as though physicians and researchers alike are reluctant to withhold anti‐inflammatory topical treatment from patients for ethical reasons on the grounds of established effectiveness. However, in clinical practice, antihistamines are often used as 'add‐on' therapy to topical treatment in the management of eczema. It is therefore important to systematically review studies investigating whether adding H1 AH to topical treatment improves eczema over and above the improvement due to topical treatment alone.
As outlined above in the How the intervention might work section, no unanimity has been reached regarding the possible pathways H1 AH may travel in the treatment of eczema. However, irrespective of whether it is known how they work, their frequent use in clinical practice is of paramount importance to at least enable judgement of whether or not patients with eczema benefit from their use (Herman 2003). This is why it is important to systematically review existing studies to ascertain whether or not use of oral H1 AH as add‐on therapy is justified.
The proposed outline for this review was published as a protocol 'Oral H1 antihistamines as ‘add‐on’ therapy to topical treatment for eczema' (Apfelbacher 2016).
Objectives
To assess the effects of oral H1 AH as 'add‐on' therapy to topical treatment in adults and children with eczema.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs), including cross‐over trials, that used oral H1 AH as add‐on therapy to topical treatments for eczema.
Types of participants
We included people of all ages with a clinical diagnosis of eczema, identified as 'atopic eczema' or 'eczema', made by a dermatologist or a physician. If the diagnosis was confirmed by a general medical practitioner, this must have involved the use of standardised diagnostic criteria such as the Hanifin and Rajka definitions ‐ Hanifin 1980 ‐ or the UK modification ‐ Williams 1994.
We excluded other forms of eczema, such as those caused by contact allergens or skin irritants, along with mixed forms of eczema.
We also excluded studies that included people with other diagnoses, if investigators did not report separately results for those with a diagnosis of eczema.
Types of interventions
Interventions included oral antihistamines (H1 antagonists) of all classes (sedating, non‐sedating) given as add‐on therapy to topical treatments for eczema (e.g. topical corticosteroids, topical immunomodulators, other topical eczema therapies, either alone or combined).
Comparators included placebo as add‐on therapy to topical treatment, or no additional treatment as add‐on therapy to topical treatment.
Types of outcome measures
We did not consider these prespecified outcomes as criteria for study inclusion eligibility (see Section 5.1.2, of the Cochrane Handbook for Systematic Reviews of Interventions, version 5.1 (Higgins 2011)). Outcomes included the core outcome set as suggested by the Harmonising Outcome Measures in Eczema (HOME) initiative (Schmitt 2012; Schmitt 2014): clinical signs, symptoms, quality of life, and long‐term control of eczema flares.
We considered separately outcomes measured up to one week (short term), at more than one week to six weeks (medium term), and at more than six weeks (long term).
Primary outcomes
Mean change in patient‐assessed symptoms of eczema, as measured by a standardised or validated eczema symptoms score (e.g. visual analogue scale for itching, patient‐oriented eczema measure (Charman 2004))
Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Secondary outcomes
Mean change in physician‐assessed clinical signs, as measured by a standardised or validated eczema signs score (e.g. Eczema Area and Severity Index (Hanifin 2001), SCORing Atopic Dermatitis index (SCORAD) (ETFAD 1993))
Mean change in quality of life, as measured by a standardised or validated quality of life measure (e.g. Dermatology Life Quality Index ‐ DLQI (Finlay 1994))
Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ (Thomas 2015)
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
The Cochrane Skin Information Specialist searched the following databases up to 9 May 2018.
Cochrane Skin Group Specialised Register via the search strategy presented in Appendix 1.
Cochrane Central Register of Controlled Trials (CENTRAL; 2018, Issue 4), in the Cochrane Library, via the strategy presented in Appendix 2.
MEDLINE via Ovid (from 1946) via the strategy presented in Appendix 3.
Embase via Ovid (from 1974) via the strategy provided in Appendix 4.
The Global Resource of EczemA Trials ‐ Centre of Evidence Based Dermatology (accessed at http://www.greatdatabase.org.uk, on 9 May 2018, browsed to results for 'antihistamines and mast cell stabilisers').
Trials registers
Two review authors (UM, CA) searched the following databases using the terms 'eczema, atopic eczema, atopic dermatitis' on 10 May 2018.
International Standard Randomized Controlled Trials Number (ISRCTN) registry (www.isrctn.com).
ClinicalTrials.gov (www.clinicaltrials.gov).
Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/AdvSearch.aspx).
EU Clinical Trials Register (www.clinicaltrialsregister.eu/).
Searching other resources
Reference lists
Two review authors (UM, CA) examined the bibliographies of included and excluded studies for further references to potentially eligible RCTs.
Adverse effects
We did not perform a separate search for adverse effects of the target intervention. However, we examined data on adverse effects from the included studies that we identified.
Conference proceedings
One review author (UM) scanned abstracts from the International Research Workshops on Eczema (George Rajka International Symposia on Atopic Dermatitis (ISAD)) from 2000 to 2018, and conference proceedings of the European Academy of Dermatology and Venereology (EADV) from 2000 to 2017, the European Academy of Allergy and Clinical Immunology (EAACI) from 2000 to 2017, and the American Academy of Dermatology (AAD) from 2012 to 2017, to identify further potentially relevant RCTs. We conducted this search on 30 May 2018.
Correspondence
We contacted trial investigators and requested that they provide missing data or clarify study details.
Data collection and analysis
Some portions of the Methods section of this review use text that was originally published in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), as well as text that was originally published in other Cochrane protocols co‐authored by review authors BC and Esther J van Zuuren and Zbys Fedorowicz (predominantly van Zuuren 2013).
Selection of studies
Two review authors (UM, CA) determined trial eligibility and resolved disagreements by discussion.
Two review authors (UM, CA) independently assessed titles and abstracts of trials identified during literature searches. We obtained full‐text copies of studies appearing to meet the inclusion criteria, and of studies with insufficient detail to allow a clear decision based on title and abstract review. Three review authors (UM, CA, MB) independently reviewed the full‐text papers and resolved disagreements over eligibility through discussion, consensus, or consultation with a third review author (UM, CA, MB). Hsin‐Wen Wu, Toshiya Ebata, Masaki Futamura, Anneke Steens, Michal Gina, Ángela Merchán, and Stanislav Iakhno assessed studies that were published in a language that was not English or German.
We have presented details of studies excluded after assessment of full‐text copies along with reasons for their exclusion in the Characteristics of excluded studies table.
Data extraction and management
Three review authors (UM, CA, MB) extracted data using a previously developed and piloted data extraction form. We resolved disagreements on data extraction by consensus. Toshiya Ebata, Masaki Futamura, and Ángela Merchán extracted data from studies that were published in a language that was not English or German. Two review authors (UM, CA) checked and entered the data into RevMan 5.3 (Review Manager (RevMan), including into the Characteristics of included studies tables. We contacted trial investigators and asked them to provide missing data or to clarify study details when necessary.
Assessment of risk of bias in included studies
We assessed the risk of bias of included studies according to the guidelines provided in Chapter 8, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Two review authors (UM, CA) independently assessed included studies using a simple contingency form and in accordance with the domain‐based evaluation method (Higgins 2011). In a further step, we compared the results of these evaluations. When we detected inconsistencies, we discussed and resolved these within the whole review author group.
We assessed the following domains as having low, unclear, or high risk of bias.
(Random) sequence generation (selection bias).
Allocation concealment (selection bias).
Blinding of participants and personnel (performance bias).
Blinding of outcome assessment (detection bias).
Incomplete outcome data (attrition bias). We considered dropouts/withdrawals < 20% for short‐ and medium‐term studies, and < 30% for long‐term studies, as adequate. We also took into account reasons for dropouts and withdrawals.
Selective outcome reporting (reporting bias).
Other bias (including potential conflicts of interest and pharmaceutical funding).
We have reported assessments for each trial in the Characteristics of included studies tables and in associated 'Risk of bias' tables.
Two review authors (UM, CA) also categorised and reported the overall risk of bias of each of the included studies according to the following criteria.
Low risk of bias (plausible bias unlikely to seriously alter the results) if all criteria were met.
Unclear risk of bias (plausible bias that raises some doubts about the results) if one or more criteria were assessed as unclear.
High risk of bias (plausible bias that seriously weakens confidence in the results) if risk of bias was rated as high for any one of the six domains.
We reported these assessments in the risk of bias tables in the Characteristics of included studies section of this review.
Measures of treatment effect
We have presented dichotomous outcomes data as risk ratios (RRs) with associated 95% confidence intervals (CIs). We used any outcomes that were statistically significant (P < 0.05) to calculate the number needed to treat for an additional beneficial outcome (NNTB) or the number needed to treat for an additional harmful outcome (NNTH).
We presented continuous outcomes as mean differences (MDs) with 95% CIs. If different instruments were used to measure an outcome, we calculated standardised mean differences (SMDs).
Unit of analysis issues
Cross‐over trials
Unit of analysis issues can arise in studies that have randomised participants to multiple treatments in multiple periods, or in studies that reported an inadequate washout period between interventions. We planned to analyse these data based on advice provided in Section 16.4.4, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We also assessed carry‐over and period effects descriptively. However, all cross‐over trials included in this review did not adequately report data to allow such analyses; therefore, we reported these results narratively.
Studies with multiple treatment groups
For studies with multiple treatment groups, which may have provided participant data that contributed to multiple comparisons, we assessed which comparisons were clinically meaningful. If multiple pair‐wise comparisons were identified, we followed Section 16.5.4, of the Cochrane Handbook for Systematic Reviews of Interventions, and created a 'shared' group (Higgins 2011).
Dealing with missing data
For studies less than 10 years old, we contacted study investigators or sponsors when we encountered missing data. We inspected missing data for evidence against the assumption that data are missing completely at random (Higgins 2011). If we made this assumption, we planned to re‐analyse the data when possible using an intention‐to‐treat (ITT) principle (Section 16.1, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011)).
When analysing dichotomous outcomes, we examined the imbalance in dropout rates between study arms to identify attrition bias (Section 16.1, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011)), and we used per‐protocol data.
When analysing continuous outcomes, we conducted a per‐protocol analysis instead of using the ITT population.
Assessment of heterogeneity
We planned to consider the clinical diversity of studies, as well as their statistical heterogeneity, by examining the I² summary statistic. We would pool studies if the I² statistic was less than 60%, but we would not pool them if I² was greater than 80%. When I² was between 60% and 80% and could be explained by a clinical and coherent argument, we may have pooled studies into a meta‐analysis. However, due to clinical diversity in terms of duration of the intervention, H1 AH used, varying doses applied in the trial, variation in concomitant topical treatments allowed, and outcome assessment, studies were not comparable, and we could not pool any of the studies identified for inclusion in this review.
Assessment of reporting biases
We planned to assess reporting bias by following the recommendations for funnel plot asymmetry (Egger 1997), as described in Section 10.4.3.1, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We planned to create these plots for outcomes if we had pooled at least 10 studies. If we noted asymmetry, we planned to explore possible sources by performing sensitivity analyses. However, because we had undertaken no pooling, we could not generate plots.
Data synthesis
Three review authors (CA, MB, BC) analysed the data in RevMan and reported results in accordance with advice provided in Chapter 9, of the Cochrane Handbook for Systematic Reviews of Interventions (Review Manager (RevMan); Higgins 2011). If we found an adequate number of studies (n > 2) that were clinically homogeneous, we would enter them into a meta‐analysis (Treadwell 2006). For dichotomous data, we planned to apply a Mantel‐Haenszel analysis method using a random‐effects model to calculate the effect estimate. For continuous data, we used an inverse variance analysis method with a random‐effects model. However, due to the aforementioned clinical diversity, studies included in this review were not comparable, and we were not able to pool any of the studies identified.
When results for individual studies were estimated with low numbers of outcomes (< 10 in total), or when the total sample size was less than 30 participants and a risk ratio was used, we also reported the proportion of outcomes in each group together with a P value from Fisher’s exact test.
Subgroup analysis and investigation of heterogeneity
We planned to carry out the following subgroup analyses if we identified moderate to substantial heterogeneity (as defined above).
Atopic versus non‐atopic participants (i.e. sensitised showing elevated levels of IgE antibodies to inhalant allergens).
Concomitant hay fever versus no concomitant hay fever.
Children versus adults.
Sedating versus non‐sedating antihistamines.
However, as no pooling was undertaken, we could not carry out any subgroup analyses.
Sensitivity analysis
We planned to conduct sensitivity analyses to assess the robustness of review results by examining the following.
If we found enough studies with low risk of bias (n > 2), we would enter these into an additional meta‐analysis.
We would look separately at studies that were prospectively registered in clinical trials registers.
However, as no pooling occurred, we conducted no sensitivity analyses.
'Summary of findings' table
We included five 'Summary of findings' (SoF) tables in this review to summarise the most important comparisons.
Cetirizine 0.5 mg/kg/d versus placebo.
Cetirizine 10 mg/d versus placebo.
Fexofenadine 120 mg/d versus placebo.
Loratadine 10 mg/d versus placebo.
Loratadine 10 mg/d versus placebo.
We included the following outcomes in the SoF tables.
Mean change in eczema symptoms score (reported for "pruritus", as this was the only patient‐oriented measure assessed in these studies).
Proportion of participants reporting adverse events and proportion reporting serious adverse events.
Mean change in eczema clinical signs score.
Mean change in quality of life.
Number of eczema flares.
The rationale for selection of studies reported in the SoF tables (main comparisons) was based first on clinical relevance/opinion, then on the standard dose of treatment, then on the duration of follow‐up (favouring longer‐term follow‐up over short‐term follow‐up) and the number of participants included, and then finally, on overall risk of bias and quality of evidence. Altogether, the decision was based on a combination of the aforementioned items, and trade‐offs had to be made.
We assessed the quality of the body of evidence using the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias), according to Section 12.2.1, of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and we reported the quality of evidence for each outcome in the SoF tables and descriptively in the Effects of interventions section for non‐key comparisons. The quality of evidence could be rated as high, moderate, low, or very low.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
Results of the search
Electronic searches up to 10 May 2018 retrieved 658 records in total, and 656 after removal of duplicates. Screening of abstracts (up to 2018) from the International Research Workshops on Eczema (George Rajka International Symposia on Atopic Dermatitis (ISAD)) and of conference proceedings of the European Academy of Dermatology and Venereology (EADV) from 2000 to 2017, the European Academy of Allergy and Clinical Immunology (EAACI) from 2010 to 2017, and the American Academy of Dermatology (AAD) from 2012 to 2017 revealed no additional eligible studies.
Searches of trials registers revealed eight ongoing trials that matched our inclusion criteria. We have described ongoing trials identified through this search in the Characteristics of ongoing studies section. We will include these in future updates if study results become available. Among these eight, we found two completed studies from this search that met the inclusion criteria (Cambazard 2001; Jung 1989), but they had not been published. However, we were able to obtain the unpublished study reports, and we have included them. We found one study in a trials register ‐ NCT00160563 ‐ but the register stated that it had been terminated. We contacted study authors but have not heard from them yet. We placed this study in the studies awaiting classification section (Characteristics of studies awaiting classification).
Two review authors (UM, CA) examined the bibliographies of included and excluded (other than in protocol) studies for further references to potentially eligible RCTs. This search yielded two additional trials for further assessment (Schmoeckel 1992; Simons 2003).
After removal of duplicates, we found that we had obtained a total of 666 records via searches of all our sources.
We excluded 577 records based on review of titles and abstracts. We obtained full texts of the remaining 89 records. Two native speakers with a scientific background independently assessed references that had not been published in English or German. They documented study findings in English on the standardised eligibility form that we had used throughout the eligibility screening process. Those studies were reported as Chinese (e.g. Liu 2008), Dutch (e.g. DeBeule 1974a), Japanese (e.g. Ohtani 2004), Polish (e.g. Bogdaszewska 1968), Russian (e.g. Balabolkin 1983), and Spanish publications (e.g. Leon 1989). At least two members of the review author group (CA, UM) assessed all studies published in English or German for eligibility.
Based on assessment of full texts, we excluded 42 studies (see Characteristics of excluded studies), moved five to the ongoing trials section (see Characteristics of ongoing studies), and moved six to studies awaiting classification (see Characteristics of studies awaiting classification), leaving 36 references reporting on 25 included studies (see Characteristics of included studies).
For further details of our screening process, see the study flow diagram provided in Figure 2.
2.
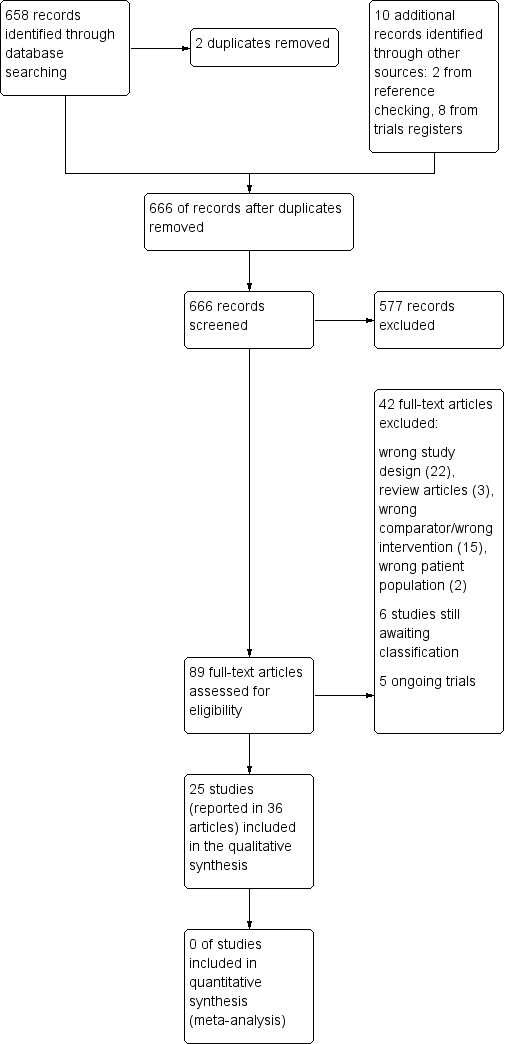
Study flow diagram.
Included studies
Thirty‐six references referred to a total of 25 studies, which met the criteria for inclusion of studies in this review.
We have presented in Table 7 a summary of the specific H1 AH investigated in the included studies.
2. H1 antihistamines under investigation in the included studies.
|
# of studies |
Study ID |
Type of study /grouping |
Duration of intervention |
Dose | Concomitant treatment |
n randomised |
|
| Acrivastine | 1 | Doherty 1989 | Parallel | 10 days | 24 mg/d | Twice‐daily application of 0.05% clobetasone butyrate ointment and aqueous cream as a soap substitute |
27 adults |
| Azelastine | 1 | Henz 1998 | Parallel | 2 weeks | 4 mg/d | Unspecified rescue hydrocortisone cream | 56 adults |
| Cetirizine | 7 | Hannuksela 1993 | Parallel | 4 weeks | 10, 20, 40 mg/d | "The use of emollients (Aqualan L or Novalan, Orion Pharmaceuticals) [was] allowed when it was felt necessary. Patients were given an emollient supply of 100g/week. In the event of failure hydrocortisone cream 1% ...could be used" |
178 adults |
| Cambazard 2001 | Parallel | 8 weeks | 0.25 mg/kg bw/d; 0.50 mg/kg bw/d; 0.75 mg/kg bw/d; bw = bodyweight |
"Patients were allowed to use 1% hydrocortisone (Cremicor) cream as rescue treatment for facial atopic dermatitis and clobetasone butyrate (Eumovatem) as rescue treatment for atopic dermatitis on other parts of the body" |
223 children | ||
| Jung 1989 | Parallel | 1 week | 5 and 10 mg/d | "Throughout the duration of the study the patients were asked to apply an antibiotic ointment (Fucicdin) to all their lesions; a topical corticosteroid (Eumovat) was to be used only when absolutely necessary" |
98 children | ||
| LaRosa 1994 | Parallel | 8 weeks | 5 to 10 mg/d | This study used disodium cromoglycate and procaterol, and 9 participants (82%) in the placebo group received other drugs, mainly disodium cromoglycate aerosol and nasal and cutaneous administration of topical steroids |
23 children | ||
| Henz 1998 | Parallel | 2 weeks | 10 mg | Unspecified rescue hydrocortisone cream | 29 adults | ||
| Tharp 1998 | Parallel | 12 weeks | 20 mg/d | This study allowed triamcinolone acetonide | 106 adults | ||
| Diepgen 2002 | Parallel | 18 months | 0.5 mg/kg/d | "All concomitant medications were allowed but had to be recorded by the parents/guardians on the diary card and by the investigator in the case report form" | 795 children | ||
| Chlorpheniramine or chlorpheniramine maleate |
3 | Frosch 1984 | Cross‐over | 4 weeks | 12 mg/d | "The patients were allowed to use at will a bland greasy ointment which did not contain an antiinflammatory active substance (e.g. vaseline or eucerin). They also received weighed containers of 0.1% betamethasone ointment...." |
18 adults |
| Munday 2002 | Parallel | 1 month | 2 to 4 mg/d (age dependent) or twice that amount |
This study allowed concomitant therapy with 1% hydrocortisone |
151 children | ||
| Nuovo 1992* | Cross‐over | 2 weeks | 16 and 24 mg/d | Concomitant therapy with topical corticosteroid | 1 adult | ||
| Fexofenadine | 1 | Kawashima 2003 | Parallel | 1 week | 120 mg/d | "In addition to 0.1% hydrocortisone butyrate twice daily, other topical agents necessary as emollients for the treatment of atopic dermatitis, including preparations containing heparinoids and zinc oxide ointment, were also permitted for concomitant use. All topical drugs for use on the face and head were allowed, along with agents that were considered to have no effect on the drug under investigation. Patients recorded in their diaries the use of 0.1% hydrocortisone butyrate and investigators recorded in clinical records the use of topical drugs or concomitant treatment" |
411 adults |
| Hydroxyzine | 1 | Monroe 1992 | Parallel | 1 week | 75 mg/d | This study permitted, but did not specify, concomitant treatment |
27 adults |
| Ketotifen | 3 | Leon 1989 | Parallel | 9 weeks | 2 mg/d | "Only the topical application of cold cream and baths with oat colloid were allowed in both groups" |
20 children |
| Iikura 1992 | Parallel | 52 weeks | 0.8 to 1.2 mg/d | "Beta2 agonists (by inhaled or oral route) and theophylline were permitted. Use of external preparations (nonsteroidal ointments and mild steroid preparations) was allowed" |
121 children | ||
| Falk 1993 | Parallel | 3 months | 2 mg/d | All participants received concomitant 1% hydrocortisone cream and 2 mg dexchlorpheniramine maleate (Polaramine) twice a day systemically |
60 adults | ||
| Levocetirizine | 2 | Kircik 2013 | Parallel | 4 weeks | 5 mg/d | Not reported | 40 adults |
| Simons 2007 | Parallel | 18 months | 0.25 mg/kg/d | Not reported | 510 children | ||
| Loratadine | 4 | Monroe 1992 | Parallel | 1 week | 10 mg/d | This study permitted, but did not specify, concomitant treatment |
27 adults |
| Langeland 1994 | Cross‐over | 2 weeks | 10 mg/d | "emollients and if needed, mild topical steroid ointment (hydrocortisone 17alpha‐butyras)" |
16 adults | ||
| Ruzicka 1998 | Parallel | 2 weeks | 5 to 20 mg/d | This study permitted, but did not specify, concomitant treatment |
159 adults | ||
| Kimura 2009 | Parallel | 4 weeks | 10 mg/d | This study permitted the use of moisturisers | 28 adults | ||
| Olopatadine | 1 | Kuniyuki 2009 | Parallel | 8 weeks | 10 mg/d | Moisturisers were permitted. This study also allowed topical corticosteroids |
99 adults |
| Tazifylline LN 2974 | 1 | Savin 1986 | Cross‐over | variable (≥ 3 days) | 30 mg/d | Unspecified routine topical treatment | 10 adults |
| Terfenadine | 4 | Hjorth 1988 | Cross‐over | 2 weeks | 120 mg/d | Not reported | 30 adults |
| Doherty 1989 | Parallel | 10 days | 180 mg/d | Twice‐daily application of 0.05% clobetasone butyrate ointment and aqueous cream as a soap substitute |
30 adults | ||
| Berth Jones 1989 | Cross‐over | 1 week | 240 mg/d | Unspecified steroids and emollients | 28 adults | ||
| Nuovo 1992a | Cross‐over | 2 weeks | 240 mg/d | Concomitant therapy with topical corticosteroid | 1 adult |
aNuovo 1992 is a single‐patient randomised study comparing chlorpheniramine and terfenadine versus placebo.
Design
All included studies were randomised controlled trials (RCTs). None used a quasi‐randomised design. Of the 25 included studies, 19 used a parallel design (Cambazard 2001; Diepgen 2002; Doherty 1989; Falk 1993; Hannuksela 1993; Henz 1998; Iikura 1992; Jung 1989; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; LaRosa 1994; Leon 1989; Monroe 1992; Munday 2002; Ruzicka 1998; Simons 2007; Tharp 1998). Five were cross‐over studies (Berth Jones 1989; Frosch 1984; Hjorth 1988; Langeland 1994; Savin 1986), and one was a single‐patient RCT using a cross‐over design (Nuovo 1992). All studies apart from three provided one comparison of an H1 AH versus placebo. Doherty 1989 compared terfenadine and acrivastine each with placebo. Henz 1998 compared cetirizine and azelastine each with placebo. Monroe 1992 compared loratadine with placebo and hydroxyzine with placebo.
Twelve of the included studies were conducted as a part of multi‐centre trials (Cambazard 2001; Diepgen 2002; Hannuksela 1993; Henz 1998; Iikura 1992; Jung 1989; Kawashima 2003; Kuniyuki 2009; Munday 2002; Ruzicka 1998; Simons 2007; Tharp 1998). The remainder (n = 13) were single‐centre trials (Berth Jones 1989; Doherty 1989; Falk 1993; Frosch 1984; Hjorth 1988; Kimura 2009: Kircik 2013; Langeland 1994; LaRosa 1994; Leon 1989; Monroe 1992; Nuovo 1992; Savin 1986).
Participants and sample sizes
We included 25 studies with a total of 3285 randomly assigned participants. We have provided a detailed summary of all included studies in the Characteristics of included studies tables. The number of participants ranged from one to 795. Studied samples were usually small. Only four studies investigated the effects of H1 AH in a sizable sample (n ≥ 200) (Cambazard 2001; Diepgen 2002; Kawashima 2003; Simons 2007). Ten trials investigated participants in samples ranging from 50 to 199 (Falk 1993; Hannuksela 1993; Henz 1998; Iikura 1992; Jung 1989; Kuniyuki 2009; Monroe 1992; Munday 2002; Ruzicka 1998; Tharp 1998), and 10 studies included fewer than 50 participants (Berth Jones 1989; Doherty 1989; Frosch 1984; Hjorth 1988; Kimura 2009; Kircik 2013; Langeland 1994; LaRosa 1994; Leon 1989; Savin 1986). As mentioned, only Nuovo 1992 was a single‐patient RCT.
Eight studies (participants = 1941) investigated samples of children (aged 0 to 12 years) or adolescents (aged 12 to 18 years), or both (Cambazard 2001; Diepgen 2002; Iikura 1992; Jung 1989; LaRosa 1994; Leon 1989; Munday 2002; Simons 2007). Seventeen studies (participants = 1325) were conducted with adults (Berth Jones 1989; Doherty 1989; Falk 1993; Frosch 1984; Hannuksela 1993; Henz 1998; Hjorth 1988; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; Langeland 1994; Monroe 1992; Nuovo 1992; Ruzicka 1998; Savin 1986; Tharp 1998).
Most studies failed to report on the severity of eczema (Berth Jones 1989; Cambazard 2001; Doherty 1989; Falk 1993; Frosch 1984; Henz 1998; Hjorth 1988; Jung 1989; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; LaRosa 1994; Leon 1989; Munday 2002; Nuovo 1992; Ruzicka 1998; Simons 2007; Tharp 1998). Two studies included individuals with at least moderate eczema (Monroe 1992; Savin 1986), two with moderate to severe eczema (Hannuksela 1993; Langeland 1994), one with moderate eczema (Iikura 1992), and one with mild to moderate eczema (Diepgen 2002).
Setting
All included studies were conducted in secondary care settings, including hospital clinics, research clinics, dermatology centres, and surgery centres. None were carried out in primary care settings. A vast majority of studies were carried out in Europe and North America (Berth Jones 1989; Cambazard 2001; Diepgen 2002; Doherty 1989; Falk 1993; Frosch 1984; Hannuksela 1993; Henz 1998; Hjorth 1988; Jung 1989; Kircik 2013; Langeland 1994; LaRosa 1994; Monroe 1992; Munday 2002; Nuovo 1992; Ruzicka 1998; Savin 1986; Tharp 1998); three were conducted in Japan (Kawashima 2003; Kimura 2009; Kuniyuki 2009), one in Japan and Brazil (Iikura 1992), and one in Mexico (Leon 1989). Simons 2007 was conducted in 10 European countries, Australia, and South Africa.
Seven studies were published up until 1990 (Berth Jones 1989; Doherty 1989; Frosch 1984; Hjorth 1988; Jung 1989; Leon 1989; Savin 1986), 10 between 1991 and 2000 (Falk 1993; Hannuksela 1993; Henz 1998; Iikura 1992; Langeland 1994; LaRosa 1994; Monroe 1992; Nuovo 1992; Ruzicka 1998; Tharp 1998), and eight between 2001 and 2017 (Cambazard 2001; Diepgen 2002; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; Munday 2002; Simons 2007).
Interventions
In our analyses (trials yielding outcome data for analysis or for a narrative description), we included the following H1 AH.
-
First‐generation H1 AH.
Chlorpheniramine (Frosch 1984; Nuovo 1992).
Chlorpheniramine maleate (Munday 2002).
Hydroxyzine (Monroe 1992).
Ketotifen (Falk 1993; Iikura 1992; Leon 1989).
-
Second‐generation or newer H1 AH, or both.
Acrivastine (Doherty 1989).
Azelastine (no longer in use) (Henz 1998).
Cetirizine (Cambazard 2001; Diepgen 2002; Hannuksela 1993; Henz 1998; Jung 1989; LaRosa 1994; Tharp 1998).
Levocetirizine (Kircik 2013; Simons 2007).
Fexofenadine (Kawashima 2003).
Loratadine (Kimura 2009; Langeland 1994; Monroe 1992; Ruzicka 1998).
Olapatadine (Kuniyuki 2009).
Tazifylline LN2974 (Savin 1986).
Terfenadine (no longer in use) (Berth Jones 1989; Doherty 1989; Hjorth 1988; Nuovo 1992).
The duration of the intervention and the respective dosages are shown in Table 7. An oral H1 AH had to be used as an add‐on therapy to topical treatment with, for example, topical corticosteroids, topical immunomodulators or other topical eczema therapy, or both. Use of emollients or moisturisers was also considered a topical treatment. Table 7 provides the topical treatments used in each included study.
Twenty‐four of the 25 included studies compared the respective H1 AH versus placebo, and in one study, no additional treatment was the comparator (Kuniyuki 2009).
Duration of the interventions
Table 7 shows the duration of treatment for each included study. The minimum duration was three days (Savin 1986); the maximum duration was 18 months (Diepgen 2002).
Trials were categorised according to how long participants received the intervention.
The duration of the oral application of H1 AH was short term (up to one week) in five studies (Berth Jones 1989; Jung 1989; Kawashima 2003; Monroe 1992; Savin 1986), medium term (from one to six weeks) in 11 studies (Doherty 1989; Frosch 1984; Hannuksela 1993; Henz 1998; Hjorth 1988; Kimura 2009; Kircik 2013; Langeland 1994; Munday 2002; Nuovo 1992; Ruzicka 1998), and long term (over more than six weeks) in nine studies (Cambazard 2001; Diepgen 2002; Falk 1993; Iikura 1992; Kuniyuki 2009; LaRosa 1994; Leon 1989; Simons 2007; Tharp 1998).
Outcomes
We had stated that we would consider separately outcomes measured up to one week (short term), from one to six weeks (medium term), and over more than six weeks (long term). As no pooling was conducted, all reported outcomes were described individually for each intervention. Table 8 summarises the outcomes from included studies considered in this review.
3. Outcomes.
| Study ID | Primary outcome 1 | Primary outcome 2 | Secondary outcome 1 |
Secondary outcome 2 |
Secondary outcome 3 |
| Berth Jones 1989 | VAS pruritus (0 to 10) | Adverse events | Severity of excoriations | Not assessed |
Not assessed |
| Cambazard 2001 | Pruritus as part of SCORAD but only total SCORAD reported |
Adverse events | SCORAD | Not assessed |
Amount of topical corticosteroid used |
| Diepgen 2002 | Pruritus as part of SCORAD but only total SCORAD reported |
Adverse events | SCORAD | Not assessed |
Use of a variety of topical and systemic medications including oral use of H1 antihistamines |
| Doherty 1989 | VAS pruritus (0 to 100) | Not assessed | Number of participants for whom treatment helped itching (physician‐ assessed) |
Not assessed |
Not assessed |
| Falk 1993 | Pruritus by Likert scale (scaling unclear) |
Not assessed | Combination of 5 signs and symptoms (severity of diseased skin, area affected, erythema, lichenification, and pruritus) to a total score |
Not assessed |
Not assessed |
| Frosch 1984 | Pruritus by VAS (0 to 100) | Adverse events | Number of participants with overall improvement, no overall improvement or overall worsening |
Not assessed |
Amount of betamethasone used in grams |
| Hannuksela 1993 | Pruritus as part of SCORAD (0 to 100) |
Adverse events | Total symptom score | Not assessed |
Amount of local rescue therapy (emollient or 1% hydrocortisone) |
| Henz 1998 | Pruritus on 4‐point Likert scale |
Adverse events (but not reported) |
Mean overall response rate (number of participants who responded to treatment) |
Not assessed |
Not assessed |
| Hjorth 1988 | Pruritus severity (no further details given) |
Not assessed | Not assessed | Not assessed |
Not assessed |
| Iikura 1992 | Not assessed | Adverse events | Complete physical examination | Not assessed |
Not assessed |
| Jung 1989 | Pruritus by 4‐point Likert scale |
Adverse events | Percentage of improvement rated as "good or excellent" |
Not assessed |
Not assessed |
| Kawashima 2003 | Pruritus 0 to 8 score | Adverse events | Investigator‐assessed change in the ratio of pruritus area to body surface area |
Not assessed |
Amount of 0.1% hydrocortisone butyrate cream used during the study |
| Kimura 2009 | Pruritus by VAS (0 to 100) | Adverse events | SCORAD | Not assessed |
Not assessed |
| Kircik 2013 | Pruritus by VAS (0 to 100) | Adverse events | Not assessed | Not assessed |
Not assessed |
| Kuniyuki 2009 | Pruritus by VAS (0 to 100) | Adverse events | SCORAD | Not assessed |
Topical steroid score |
| Langeland 1994 | Pruritus by VAS (0 to 10) | Adverse events | Not assessed | Not assessed |
Mean number of days with use of local steroids during the 6 treatment periods and severity of rash at the end of each treatment period |
| LaRosa 1994 | Pruritus by diary (no further details given) |
Adverse events | Assessment by clinical examination | Not assessed |
Amount of disodium cromoglycate, procaterol, steroids |
| Leon 1989 | Pruritus by 4‐point Likert scale |
Not assessed | Not assessed | Not assessed |
Not assessed |
| Monroe 1992 | Pruritus percentage score |
Adverse events (but not reported) |
Not assessed | Not assessed |
Not assessed |
| Munday 2002 | Pruritus by 4‐point Likert scale |
Adverse events | Composite score consisting of 5 symptoms (erythema, excoriation, dryness, lichenification, exudation and crusting) |
Not assessed |
Amount of 1% hydrocortisone used in grams |
| Nuovo 1992 | Pruritus on 7‐point Likert scale |
Adverse events | Not assessed | Not assessed |
Number of daily applications of the topical steroid triamcinolone |
| Ruzicka 1998 | Pruritus by VAS (0 to 10) | Adverse events | Number for whom treatment success was judged as good or very good |
Not assessed |
Not assessed |
| Savin 1986 | Pruritus by VAS (no further details given) |
Not assessed | Not assessed | Not assessed |
Not assessed |
| Simons 2007 | Not assessed | Adverse events | Not assessed | Not assessed |
Not assessed |
| Tharp 1998 | Pruritus (no further details given) |
Adverse events | Clinical signs and symptoms | Not assessed |
Use of triamcinolone acetonide 0.1% cream (corticosteroid) |
SCORAD: SCORing Atopic Dermatitis index; VAS: visual analogue scale.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema, as measured by a standardised or validated eczema symptoms score (e.g. visual analogue scale for itching, patient‐oriented eczema measure (Charman 2004))
With regard to our prespecified primary outcome, 23 studies reported a measure of pruritus (Berth Jones 1989; Cambazard 2001; Diepgen 2002; Doherty 1989; Falk 1993; Frosch 1984; Hannuksela 1993; Henz 1998; Hjorth 1988; Jung 1989; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; Langeland 1994; LaRosa 1994; Leon 1989; Monroe 1992; Munday 2002; Nuovo 1992; Ruzicka 1998; Savin 1986; Tharp 1998). Nine studies assessed pruritus by VAS (Berth Jones 1989; Doherty 1989; Frosch 1984; Kimura 2009; Kircik 2013; Kuniyuki 2009; Langeland 1994; Ruzicka 1998; Savin 1986). Three studies assessed pruritus as part of the SCORAD assessment (Cambazard 2001; Diepgen 2002; Hannuksela 1993). Four studies used a 4‐point Likert scale for assessment of pruritus (Jung 1989; Henz 1998; Leon 1989; Munday 2002). A 7‐point Likert scale was used by Nuovo 1992. Kawashima 2003 assessed pruritus on a scale from 0 to 8. Falk 1993 used a Likert scale but the scaling was unclear. A percentage score of pruritus was used by Monroe 1992. Three further studies assessed pruritus but failed to provide details on its assessment (Hjorth 1988; LaRosa 1994; Tharp 1998). Two of the included studies did not assess pruritus (Iikura 1992; Simons 2007).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Eighteen of the included studies assessed the occurrence of adverse events (Berth Jones 1989; Cambazard 2001; Diepgen 2002; Frosch 1984; Hannuksela 1993; Iikura 1992; Jung 1989; Kawashima 2003; Kimura 2009; Kircik 2013; Kuniyuki 2009; Langeland 1994; LaRosa 1994; Munday 2002; Nuovo 1992; Ruzicka 1998; Simons 2007; Tharp 1998). Five studies did not assess this outcome (Doherty 1989; Falk 1993; Hjorth 1988; Leon 1989; Savin 1986). Two studies assessed adverse events but failed to report them separately for patients with eczema (Henz 1998; Monroe 1992).
Secondary outcome 1. Mean change in physician‐assessed clinical signs, as measured by a standardised or validated eczema signs score (e.g. Eczema Area and Severity Index (Hanifin 2001), SCORing Atopic Dermatitis index (SCORAD) (ETFAD 1993))
Four studies assessed this outcome by SCORAD (Cambazard 2001; Diepgen 2002; Kimura 2009; Kuniyuki 2009). Thirteen studies assessed this outcome by various assessments (Berth Jones 1989; Doherty 1989; Falk 1993; Frosch 1984; Hannuksela 1993; Henz 1998; Iikura 1992; Jung 1989; Kawashima 2003; LaRosa 1994; Munday 2002; Ruzicka 1998; Tharp 1998) (see Table 8 for details).
Eight studies did not employ a measure of physician‐assessed clinical signs (Hjorth 1988; Kircik 2013; Langeland 1994; Leon 1989; Monroe 1992; Nuovo 1992; Savin 1986; Simons 2007).
Secondary outcome 2. Mean change in quality of life, as measured by a standardised or validated quality of life measure (e.g. Dermatology Life Quality Index ‐ DLQI (Finlay 1994))
The protocol for this review stated that we would assess this outcome; however, only one study provided quality of life outcomes of any kind (Kircik 2013), and these were insufficient for statistical analysis.
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ (Thomas 2015)
Eleven studies assessed this outcome (Cambazard 2001; Diepgen 2002; Frosch 1984; Hannuksela 1993; Kawashima 2003; Kuniyuki 2009; Langeland 1994; LaRosa 1994; Munday 2002; Nuovo 1992; Tharp 1998) (see Table 8 for details).
Declaration of interest and funding
Only one study stated that study authors were free from potential conflicts of interest (Kawashima 2003). None of the other trials provided any information on potential conflicts of interest. Seven studies declared that they had received funding from a pharmaceutical company (Cambazard 2001; Diepgen 2002; Hannuksela 1993; Jung 1989; Kawashima 2003; Kircik 2013; Simons 2007). One study did not declare its source of funding (Tharp 1998), but because the last author of the publication was an employee of Pfizer Inc., this pharmaceutical company is likely to be the funding body for this study. One study report declared receipt of funding from the National Center for Health Services Research ‐ Office of the Assistant Secretary for Health (OASH) (Nuovo 1992). As many of the reviewed studies were published before reporting of declarations of interests and/or funding became more common, it is difficult to infer whether reviewed studies that did not provide this information were truly free from potential conflicts of interest.
Excluded studies
Eighty‐nine studies were still potential candidates for inclusion after screening of titles or abstracts, or both. We excluded 42 studies after assessment of the full text and documented the reasons for their exclusion in the Characteristics of excluded studies table.
We excluded most of these studies on grounds of having the wrong study design (e.g. lack of randomisation). We excluded the remainder for having the wrong comparator (e.g. one H1 AH vs one or several other H1 AH but not vs placebo or no treatment as an 'add‐on' therapy) or the wrong patient population (e.g. participants not having received a diagnosis of eczema), or because they were review articles (e.g. articles not reporting a single trial but discussing several trials).
Studies awaiting classification
Several studies are still awaiting classification because they provided limited information and review authors could not discern whether they should be included in or excluded from this review. Whether two studies were randomised remains unclear (Borelli 1969; Chunharas 2002). Three studies were conducted in samples of patients with various diagnoses (eczema was one of them) (DeBeule 1974; Laugier 1978; Simons 2003), and study authors presented results only for the whole sample ‐ not separately by diagnostic group. One trial was registered in 2005 and then was terminated (NCT00160563). However, we do not know whether it was terminated before the study commenced, or whether it was started and then terminated for reasons we would like to establish. We tried to contact the study authors in all cases, requesting additional information, but we have received no responses.
Ongoing studies
We classified five studies as ongoing (CRT EU 23679; CTR EU 23697; CTR EU 29342; CTR EU 39698; UMIN000010519); we will assess them again, once their results have been published.
Risk of bias in included studies
In this review, we assessed each of the included studies for risk of bias (RoB), and we report the judgements for each individual domain in the risk of bias table associated with each study (see Characteristics of included studies). Domains of interest are allocation, blinding, incomplete outcome data, selective outcome reporting, and other sources of bias. Please refer to Figure 3, which shows our judgements about each RoB item expressed as percentages of included studies in each category of risk, and Figure 4, which shows the judgement for each domain by study.
3.
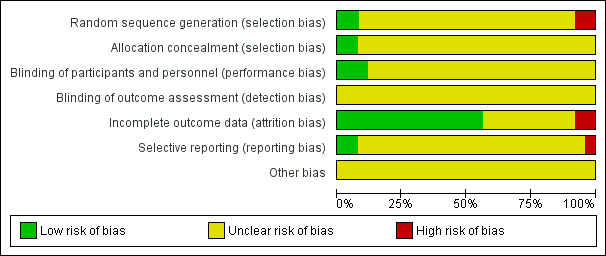
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.
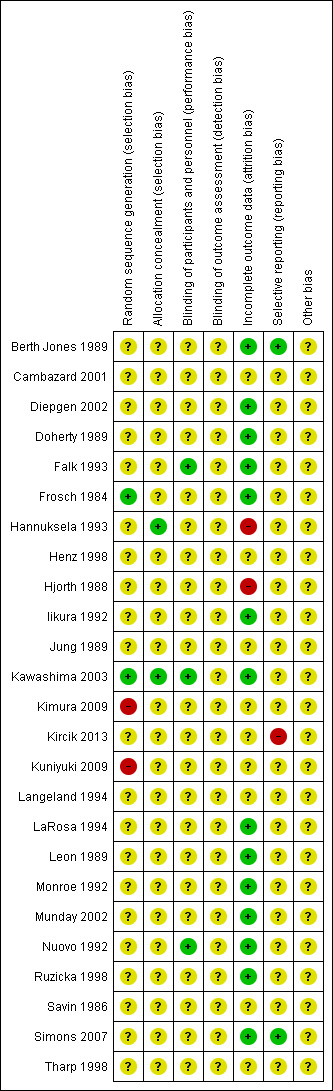
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Most studies lacked sufficient information to judge whether the methods used were capable of eliminating threats to their validity. No study provided complete clarity on every item in our ’RoB’ assessment, suggesting widespread inadequate reporting of methods or results. One trial at least provided sufficient information on four of the seven domains considered in this review (Kawashima 2003). However, we could not assess three domains due to insufficient reporting.
Of the 25 studies for which we assessed RoB, six were associated with unclear RoB in all domains (Cambazard 2001; Henz 1998; Jung 1989; Langeland 1994; Savin 1986; Tharp 1998). We judged eight as unclear in all domains but one, which we judged as low risk (Diepgen 2002; Doherty 1989; Iikura 1992; LaRosa 1994; Leon 1989; Monroe 1992; Munday 2002; Ruzicka 1998). Four studies were associated with unclear RoB in all domains but one, which we judged as high risk (Hjorth 1988; Kimura 2009; Kircik 2013; Kuniyuki 2009). We judged five studies as having unclear RoB in five domains but low risk in two domains (Berth Jones 1989; Falk 1993; Frosch 1984; Nuovo 1992; Simons 2007). One study was associated with unclear RoB in most domains, and one each with low and high risk (Hannuksela 1993). We judged only one study as having low RoB in four domains (Kawashima 2003).
Summarising RoB within the 25 studies for which RoB was assessed revealed that five (25%) had at least one domain that was rated as having high RoB. In two instances, this referred to incomplete outcome data (attrition bias), in another two, to selection bias, and once, to reporting bias. For all other studies, we judged at least one domain to be at unclear RoB. Across the 25 studies included in this review, we considered five as having an overall high risk of bias (n = 5; 20%) and 20 as having an overall unclear risk of bias (n = 20; 80%).
Allocation
Random sequence generation
In most of the 25 trials in this review, study authors did not describe the method of sequence generation at all or at best provided unclear information. Two studies described the method used to generate the allocation sequence in sufficient detail (Frosch 1984; Kawashima 2003); therefore, we judged this domain as having low risk of bias for these studies. For two studies, we judged the method to be at high RoB (Kimura 2009; Kuniyuki 2009).
Allocation concealment
Most studies did not provide sufficient information regarding how the allocation sequence was concealed; hence we judged them as having unclear risk in this domain. Two studies assured that allocation concealment was ensured. Both Kawashima 2003 and Hannuksela 1993 reported that sealed envelopes had been used to disguise group membership.
Blinding
Blinding of participants and personnel (performance bias)
Most of the included studies insufficiently described the procedures utilised to blind study participants and personnel from knowing which intervention a participant received, and consequently we judged them to be at unclear risk in this domain. Falk 1993, Kawashima 2003, and Nuovo 1992 achieved blinding by ensuring that the study medication had an identical appearance, and thus we judged them to have low risk of bias in this domain.
Blinding of outcome assessment (detection bias)
None of the included trials demonstrated adequate blinding of outcome assessment. Due to lack of information, we consequently rated risk of bias due to lack of blinding of outcome assessment as unclear in all trials.
Incomplete outcome data
We rated one parallel RCT as being at high risk with regard to attrition bias (Hannuksela 1993). The attrition rate was 28.7%. We also rated one cross‐over trial as high risk due to a large number of dropouts (14/30) (Hjorth 1988). None of these studies could sufficiently show that dropouts or losses to follow‐up did not systematically differ from participants remaining in the trial. We judged 14 studies to be at low risk (Berth Jones 1989; Diepgen 2002; Doherty 1989; Falk 1993; Frosch 1984; Iikura 1992; Kawashima 2003; LaRosa 1994; Leon 1989; Monroe 1992; Munday 2002; Nuovo 1992; Ruzicka 1998; Simons 2007). Nuovo 1992 was a single‐patient trial reporting all data points from this cross‐over trial; thus we judged that it had low RoB. Remaining studies (9/25) did not provide sufficient information to allow judgement of low or high risk (Cambazard 2001; Henz 1998; Jung 1989; Kimura 2009; Kircik 2013; Kuniyuki 2009; Langeland 1994; Savin 1986; Tharp 1998); consequently we rated them as having unclear risk.
Selective reporting
According to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), most studies are likely to fall into the category of unclear for this domain because the difficulty involved in establishing what outcomes may have been collected and not reported. Publication of study protocols before study commencement or at least before study completion has contributed significantly to our improved ability to detect selective outcome reporting; however, dissemination of study protocols was fairly uncommon until a few years ago.
For most studies, it is unlikely that a protocol would have been published. We attempted to obtain protocols for larger trials (n > 200) (Diepgen 2002; Kawashima 2003), but we did not succeed in doing so. As a consequence, we judged 22 studies as having unclear risk of reporting bias. We tried to resort to information from trial registrations when possible (see Table 9). We considered two studies to be at low risk of reporting bias (Berth Jones 1989; Simons 2007). We judged one study as having high risk of reporting bias because reporting of data was extremely rudimentary in the report and did not allow us to run necessary analyses (Kircik 2013). We contacted the study authors, but they failed to respond. Although some data were available (medians and interquartile ranges for primary outcome 1 (patient‐assessed symptoms) and secondary outcome 2 (quality of life)), researchers presented no group comparisons and provided insufficient data for us to run the statistical analyses ourselves.
4. Registered trials.
| Study ID | Trial registration number |
| Berth Jones 1989 | unable to obtain or not registered respectively |
| Cambazard 2001 | a protocol number was provided (MPCE97B2501/lV ‐11 Dec. 97 Amdt 1 (29.01.98) / Amdt 2 (06.03.98)), but we could not find it |
| Diepgen 2002 | unable to obtain or not registered respectively |
| Doherty 1989 | unable to obtain or not registered respectively |
| Falk 1993 | unable to obtain or not registered respectively |
| Frosch 1984 | unable to obtain or not registered respectively |
| Hannuksela 1993 | unable to obtain or not registered respectively |
| Henz 1998 | unable to obtain or not registered respectively |
| Hjorth 1988 | unable to obtain or not registered respectively |
| Iikura 1992 | unable to obtain or not registered respectively |
| Jung 1989 | a protocol number was provided (PCE87F291), but we could not find it |
| Kawashima 2003 | unable to obtain or not registered respectively |
| Kimura 2009 | unable to obtain or not registered respectively |
| Kircik 2013 | NCT00884325 Management of pruritus with Xyzal in atopic dermatitis |
| Kuniyuki 2009 | unable to obtain or not registered respectively |
| Langeland 1994 | unable to obtain or not registered respectively |
| LaRosa 1994 | unable to obtain or not registered respectively |
| Leon 1989 | unable to obtain or not registered respectively |
| Monroe 1992 | unable to obtain or not registered respectively |
| Munday 2002 | unable to obtain or not registered respectively |
| Nuovo 1992 | unable to obtain or not registered respectively |
| Ruzicka 1998 | unable to obtain or not registered respectively |
| Savin 1986 | unable to obtain or not registered respectively |
| Simons 2007 | NCT00152464 Prevention of Asthma With Levocetirizine 18 Month Treatment in Infants (12 ‐ 24 Months) Suffering From Eczema (Atopic Dermatitis) and Sensitized to Grass Pollen and/or House Dust Mite (HDM) |
| Tharp 1998 | unable to obtain or not registered respectively |
Other potential sources of bias
We assessed all included studies for additional potential sources of bias. We checked each study for inclusion of a conflict of interest statement, whether information about the source of funding could be obtained, and whether the study was initiated by a pharmacological company (Bero 2013). However, all trials failed to provide this information adequately, and consequently we judged them as having unclear risk of bias. Thus, further risk of bias may be present but information provided is insufficient for judgement of whether additional bias exists.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings for the main comparison. Cetirizine 0.5 mg/kg/d versus placebo (18‐month intervention).
| Cetirizine 0.5 mg/kg/d compared with placebo for eczema | ||||||
|
Patient or population: children with eczema Settings: secondary Intervention: cetirizine 0.5 mg/kg/d Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Cetirizine 0.5 mg/kg/d | |||||
|
Primary outcome 1. Mean change in patient‐assessed symptoms Pruritus was assessed once as part of SCORAD Follow‐up: 18 months |
See comment | See comment | ‐ | ‐ | ‐ | This outcome was assessed as part of the SCORAD assessment, but the data were not presented separately for pruritus. Thus, no data for analysis of this outcome were available from this study. |
|
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period Follow‐up: 18 months |
Study population |
RR 0.68 (0.46 to 1.01) |
795 (1 study) | ⊕⊕⊕⊝ moderatea | The difference was non‐significant and most reported symptoms and events were mild and were attributed to intercurrent respiratory or gastrointestinal infection or exacerbations of allergic disorders. | |
| 136 per 1000 |
93 per 1000 (63 to 138) |
|||||
|
Secondary outcome 1. Mean change in physician‐assessed clinical signs Measured by SCORAD Follow‐up: 18 months |
See comment | See comment | ‐ | 795 (1 study) | ⊕⊕⊕⊝ moderatea | No evidence for a treatment‐related reduction in SCORAD scores between baseline and the final visit was observed by study authors, as the difference between groups was reported to be non‐significant (from 24.9 to 15.2 in the cetirizine group and from 25.1 to 15.7 in the placebo group). Data could not be analysed because standard deviations were missing. |
| Secondary outcome 2. Mean change in quality of life | See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
|
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ Follow‐up: 18 months |
See comment | See comment | ‐ | 795 (1 study) | ⊕⊕⊕⊝ moderatea | This outcome assessed the use of a variety of topical and systemic medications including oral use of H1 antihistamines. No significant difference between groups was observed, with 1 exception: participants in the placebo group were more likely to have used H1 antihistamines (P = 0.035). No data for analysis were available from the study. In other words, the intervention probably makes little or no difference in the use of additional H1 antihistamines to prevent flares. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SCORAD: SCORing Atopic Dermatitis index. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded by one level for limitations in design due to unclear judgement for most domains.
Summary of findings 2. Cetirizine 10 mg/d versus placebo (4‐week intervention).
| Cetirizine 10 mg/d compared with placebo for eczema | ||||||
|
Patient or population: adults with eczema Settings: secondary Intervention: cetirizine 10 mg/d Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Cetirizine 10 mg/d | |||||
|
Primary outcome 1. Mean change in patient‐assessed symptoms Pruritus was assessed once as part of SCORAD and once by patient case report form (CRF) Follow‐up: 4 weeks |
See comment | See comment | ‐ | 84 (1 study) | ⊕⊕⊝⊝ lowa | Pruritus as part of SCORAD: no significant differences were observed between the intervention group and the placebo group Pruritus by patient CRF: according to study authors, no significant difference was observed between groups. It is of note that values reported in Table 2 of the publication actually increased from baseline to last visit, although the legend states that "lower values indicate less pruritus". We tried to contact the study authors to inquire about this but did not succeed in doing so. |
|
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period Follow‐up: 4 weeks |
Study population |
RR 1.11 (0.50 to 2.45) |
84 (1 study) | ⊕⊕⊝⊝ lowb | Non‐significant difference between groups in the occurrence of at least 1 adverse event. Most side effects pertained to sedation, others to skin‐related problems, respiratory symptoms or headache. | |
| 214 per 1000 |
238 per 1000 (107 to 524) |
|||||
|
Secondary outcome 1. Mean change in physician‐assessed clinical signs Total sum of 4 symptoms (signs) scores, each measured on 4‐point Likert scale (0 = absent; 3 = severe) Follow‐up: 4 weeks |
See comment | See comment | ‐ | 84 (1 study) | ⊕⊕⊝⊝ lowc | It was reported that the total symptom/signs score decreased in both groups but there was no significant difference between groups. |
| Secondary outcome 2. Mean change in quality of life | See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
|
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ Follow‐up: 4 weeks |
See comment | See comment | ‐ | 84 (1 study) | ⊕⊕⊝⊝ lowc | No difference in amount of local rescue therapy (emollient or 1% hydrocortisone) nor in the number of applications was observed among groups. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CRF: case report form; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded by two levels: one level due to limitations in study design (all domains judged as having unclear or high risk of bias) and irreproducible reporting of outcomes (legend states that lower values indicate less pruritus but pruritus increased in all groups from baseline to last visit; study author was contacted to clarify this issue but did not respond); and one level due to imprecision (small sample size).
bDowngraded by two levels: one level due to limitations in study design (all domains judged as having unclear or high risk of bias); and one level due to serious imprecision (wide CI due to small sample size).
cDowngraded by two levels: one for limitations in design (all domains judged as having unclear or high risk of bias), and one level due to serious imprecision (wide CI due to small sample size).
Summary of findings 3. Fexofenadine 120 mg/d versus placebo (1‐week intervention).
| Fexofenadine 120 mg/d compared with placebo for eczema | ||||||
|
Patient or population: adults with eczema Settings: secondary Intervention: fexofenadine 120 mg/d Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Fexofenadine 120 mg/d | |||||
|
Primary outcome 1. Mean change in patient‐assessed symptoms This outcome was measured as the mean change in patient‐assessed pruritus (range 0 to 8) between baseline and the night of day 6 Follow‐up: 1 week |
Mean change was ‐0.5 |
Mean change was 0.25 lower (0.43 lower to 0.07 lower) |
‐ | 400 (1 study) |
⊕⊕⊕⊝ moderatea | A significantly larger reduction was observed in the fexofenadine group compared to the placebo group. However, the effect observed appeared to be small. |
|
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period Follow‐up: 1 week |
Study population |
RR 1.05 (0.74 to 1.50) |
411 (1 study) | ⊕⊕⊕⊝ moderatea | No significant difference between groups. The most frequent adverse events referred to somnolence and headache. | |
| 221 per 1000 |
232 per 1000 (164 to 332) |
|||||
|
Secondary outcome 1. Mean change in physician‐assessed clinical signs This outcome was assessed as investigator‐assessed change in the ratio of pruritus area to body surface area Follow‐up: 1 week |
See comment | See comment | ‐ | Number of participants not reported (1 study) |
⊕⊕⊕⊝ moderatea | It was stated that in terms of investigator‐assessed change in the ratio of pruritus area to body surface area, significantly more patients in the fexofenadine group than in the placebo group experienced a reduction in pruritus (P = 0.007). No further numerical data were provided. |
| Secondary outcome 2. Mean change in quality of life | See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
|
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ Amount of 0.1% hydrocortisone butyrate cream used during the study Follow‐up: 1 week |
See comment | See comment | ‐ | Number of participants not reported (1 study) |
⊕⊕⊕⊝ moderatea | Study authors reported no significant difference between the 2 groups in the amount of 0.1% hydrocortisone butyrate cream used during the study. No data for analysis could be extracted from this study. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded by one level for limitations in design due to insufficient follow‐up period (1 week) (indirectness).
Summary of findings 4. Loratadine 10 mg/d versus placebo (4‐week intervention).
| Loratadine 10 mg/d compared with placebo for eczema | ||||||
|
Patient or population: male and female adults aged 15 years and older with eczema Settings: secondary Intervention: loratadine 10 mg/d Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No additional treatment | Loratadine 10 mg/d | |||||
|
Primary outcome 1. Mean change in patient‐assessed symptoms Reduction in pruritus on 100‐mm VAS from baseline Follow‐up: 4 weeks |
Mean reduction was 29.4 | Mean reduction was 2.30 lower (20.27 lower to 15.67 higher) |
‐ | 28 (1 study) | ⊕⊕⊝⊝ lowa | The difference was non‐significant. However, baseline differences were not adjusted for. |
|
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period Follow‐up: 4 weeks |
Study population | RR 0.25 (0.01 to 5.76) | 28 (1 study) | ⊕⊕⊝⊝ lowa | There was only 1 event (folliculitis) in the placebo group and 0 events in the intervention group. | |
| 83 per 1000 |
21 per 1000 (1 to 480) |
|||||
|
Secondary outcome 1. Mean change in physician‐assessed clinical signs SCORAD Follow‐up: 4 weeks |
Mean reduction was 16.9 | Mean reduction was 4.1 lower (13.22 lower to 5.02 higher) | ‐ | 28 (1 study) | ⊕⊕⊝⊝ lowa | The difference was non‐significant. |
| Secondary outcome 2. Mean change in quality of life | See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
|
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ Follow‐up: 4 weeks |
See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SCORAD: SCORing Atopic Dermatitis index; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded by two levels: one level for limitations of design due to unclear judgement of all but one domain, which was judged as low (selection bias), and one level due to imprecision (small sample size).
Summary of findings 5. Loratadine 10 mg/d versus placebo (2‐week intervention).
| Loratadine 10 mg/d compared with placebo for eczema | ||||||
|
Patient or population: male and female adults with eczema Settings: secondary Intervention: loratadine 10 mg/d Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Loratadine 10 mg/d | |||||
|
Primary outcome 1. Mean change in patient‐assessed symptoms VAS pruritus 10 cm Follow‐up: 2 weeks |
Mean reduction was 1.32 | Mean reduction was 0.96 higher (2.01 higher to 0.09 lower) |
‐ | 71 (1 study) | ⊕⊕⊝⊝ lowa | The difference in the pruritus sum between day 0 and day 13 did not significantly differ between the 10 mg/d group and the placebo group. However, baseline scores were not equally distributed between groups, and this analysis does not account for baseline differences. |
|
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period Follow‐up: 2 weeks |
See comment | See comment | ‐ | 73 (1 study) | ‐ | Study authors reported the occurrence of 16 adverse events in all 4 groups but failed to report them for each group separately. |
|
Secondary outcome 1. Physician‐assessed clinical signs Number for whom treatment success was judged as good or very good Follow‐up: 2 weeks |
211 per 1000 |
219 more per 1000 (from 2 fewer to 674 more) |
RR 2.04 (0.99 to 4.20) |
73 (1 study) | ⊕⊕⊝⊝ lowa | Participants in the 10 mg/d group were about twice as likely to be judged to have good or very good treatment success but the difference was not significant. |
| Secondary outcome 2. Mean change in quality of life | See comment | See comment | ‐ | ‐ | ‐ | No study addressed this outcome. |
| Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ | See comment | See comment | ‐ | ‐ | ‐ | Not assessed |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence. High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
aDowngraded by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
Due to clinical diversity among studies in terms of duration of the intervention, the H1 AH used, and doses provided, as well as variation in the concomitant topical treatment allowed (see Table 7) and in outcome assessment (see Table 8), we were unable to pool any of the studies that we identified for inclusion in this review. Consequently, we have reported the effects of interventions for each trial individually. For the main comparisons, see Table 1Table 2Table 3 and Table 4.
Comparison 1
Acrivastine 24 mg/d versus placebo
We identified one parallel study that compared this intervention in adults (Doherty 1989). Researchers reported an intermediate‐term intervention (10 days) but assessed the first primary outcome seven days after baseline (short‐term).
Primary outcome 1. Patient‐assessed symptoms: VAS pruritus seven days after baseline
Visual analogue scale (VAS) pruritus score (seven days after baseline and adjusted for baseline scores as a covariate; range 0 to 100) increased more in the placebo group; however, the 95% confidence interval (CI) associated with the mean difference (MD) was wide and included zero, meaning that the result was non‐significant (MD ‐11.40, 95% CI ‐29.44 to 6.64; P = 0.22; participants = 23; studies = 1; Analysis 1.1). We downgraded the outcome from high to low quality of evidence because of unclear risk of bias for most domains and because of imprecision (wide CI due to small sample size or high variability in outcome measurement).
1.1. Analysis.
Comparison 1 Acrivastine 24 mg/d versus placebo, Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus 7 days after baseline adjusted for baseline score as covariate.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
This study provided data not for mean change but for the number of participants for whom treatment helped to relieve itching (physician‐assessed). The comparison favoured acrivastine over placebo (risk ratio (RR) 2.69, 95% CI 1.12 to 6.49; P = 0.03; participants = 27; studies = 1; Analysis 1.2). Fisher's exact test confirmed this analysis (P = 0.021). The number needed to treat for an additional beneficial outcome (NNTB) was 2. The 95% CI for the NNTB ranged from 1 to 7. We downgraded the outcome from high to low quality of evidence because of unclear risk of bias for most domains and imprecision (wide CI due to small sample size).
1.2. Analysis.
Comparison 1 Acrivastine 24 mg/d versus placebo, Outcome 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment helped itching.
Comparison 2
Azelastine 4 mg/d versus placebo
We identified one parallel study that compared this intervention in adults (Henz 1998). Investigators reported an intermediate‐term intervention (two weeks). The study yielded retrievable/extractable data only in part, as study authors combined the data presented to include a variety of dermatological conditions including eczema. Attempts to contact the main study author were not successful.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
This study measured reduction in pruritus (assessed at baseline, week 1, week 2) marked at night on diary cards (itching recorded every night on a 4‐point scale: 0 = none, 1 = mild, 2 = moderate, 3 = severe). Study authors presented results in a graph separately for patients with eczema and showed no statistically significant differences between azelastine and placebo. They did not report exact P values nor mean changes. Hence, no study data were available for analysis. We downgraded the quality of evidence from high to low due to unclear risk of bias in all domains and serious imprecision (small sample size).
Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) two weeks after baseline
Although this study assessed a measure of 'physician‐assessed clinical signs', researchers presented not a mean change measure but the mean overall response rate (number of participants who responded to treatment). They presented data separately for participants with eczema. Response rates were higher in the azelastine group, but researchers observed no significant differences between intervention and placebo groups (RR 1.75, 95% CI 0.64 to 4.82; P = 0.28; participants = 55; studies = 1; Analysis 2.1). We downgraded the quality of evidence from high to low due to unclear risk of bias in all domains and serious imprecision (wide CI due to small sample size).
2.1. Analysis.
Comparison 2 Azelastine 4 mg/d versus placebo, Outcome 1 Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) 2 weeks after baseline.
Comparison 3
Cetirizine versus placebo
We identified seven parallel studies that compared this intervention (Cambazard 2001; Diepgen 2002; Hannuksela 1993; Henz 1998; Jung 1989; LaRosa 1994; Tharp 1998).
One study provided a short‐term intervention (up to one week) (Jung 1989), two an intermediate‐term intervention (one to six weeks) (Hannuksela 1993; Henz 1998), and four long‐term (more than six weeks) interventions (Cambazard 2001; Diepgen 2002; LaRosa 1994; Tharp 1998).
The dosage given ranged from 5 mg/d to 40 mg/d, or from 0.25 mg/kg bodyweight per day to 0.75 mg/kg bodyweight per day.
Short‐term interventions
Jung 1989 reported a short‐term intervention (one week; n = 98) conducted in children three to six years of age. One group received 5 mg/d (n = 33), another 10 mg/d (n = 36), and a third placebo (n = 29).
Primary outcome 1. Mean change in patient‐assessed symptoms
Study results show that pruritus was reduced by 39% in the 5 mg/d group, by 44% in the 10 mg/d group, and by 39% in the placebo group (no significant difference). No data from the study were available for analysis. We downgraded the quality of evidence from high to low due to serious risk of bias (all domains judged as having unclear risk of bias) and insufficient reporting of findings.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
The number of adverse events did not differ significantly between the 5 mg/d group and the placebo group (RR 0.59, 95% CI 0.11 to 3.27; P = 0.54; participants = 62; studies = 1; Analysis 3.1), or between the 10 mg/d group and the placebo group (RR 0.81, 95% CI 0.18 to 3.70; P = 0.78; participants = 65; studies = 1; Analysis 3.1). We downgraded the quality of evidence from high to low due to serious risk of bias (all domains judged as having unclear risk of bias) and serious imprecision (wide CI due to small sample size).
3.1. Analysis.
Comparison 3 Cetirizine 5 and 10 mg/d versus placebo (1‐week intervention), Outcome 1 Primary outcome 2. Adverse events.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
We assessed global evaluation of treatment efficacy as the percentage of improvement as "good or excellent". Study authors determined that 46% in the 5 mg/d group had improved, along with 40% in the 10 mg/d group and 20% in the placebo group, and that no significant differences had emerged. No data were available from this study for analysis. We downgraded the quality of evidence from high to moderate due to serious risk of bias (all domains judged as having unclear risk of bias).
Intermediate‐term interventions
Henz 1998 reported an intermediate‐term intervention (two weeks) comparing 10 mg/d of cetirizine (n = 16) versus placebo (n = 11) in adults. This study yielded retrievable/extractable data only in part, as researchers combined data presented to include a variety of dermatological conditions, including eczema. Attempts to contact the main author were not successful.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
This study measured reduction in pruritus (assessed at baseline, week 1, week 2) marked at night on diary cards (itching recorded every night on a 4‐point scale: 0 = none, 1 = mild, 2 = moderate, 3 = severe). Investigators presented results in a graph separately for patients with eczema and showed no statistically significant differences between cetirizine and placebo. No data from the study were available for analysis. We downgraded the quality of evidence from high to low due to serious risk of bias (all domains judged as having unclear risk of bias) and serious imprecision (small sample size).
Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) two weeks after baseline
Although this study employed a measure of 'physician‐assessed clinical signs', researchers presented not a mean change measure but the mean overall response rate (number of participants who responded to treatment). They presented data separately for patients with eczema and showed no significant differences between intervention and placebo groups (RR 0.69, 95% CI 0.17 to 2.80; P = 0.60; participants = 27; studies = 1; Analysis 4.1). Fisher's exact test yielded a P value of 0.662. We downgraded the quality of evidence from high to low because of serious risk of bias (all domains judged as having unclear risk of bias) and serious imprecision (small sample size).
4.1. Analysis.
Comparison 4 Cetirizine 10 mg versus placebo (2‐week intervention), Outcome 1 Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) 2 weeks after baseline.
Hannuksela 1993 reported an intermediate‐term intervention (four weeks; n = 178) and compared 10 mg/d (n = 42), 20 mg/d (n = 45), or 40 mg/d of cetirizine (n = 47) versus placebo (n = 42) in adults. We judged this study as having high risk regarding attrition bias.
Primary outcome 1. Mean change in patient‐assessed symptoms
Investigators assessed pruritus once as part of SCORAD and once by patient case report form (CRF). No data from the study were available for analysis.
Pruritus as part of SCORAD: all groups showed significant improvement between baseline and the last visit. Researchers observed no significant differences between any of the active treatments and placebo.
Pruritus by patient CRF (case report form): again, all groups were significantly improved at the last visit compared to baseline. Study authors reported that the 20 mg/d (P = 0.04) and 40 mg/d (P = 0.05) groups were significantly better than the placebo group.
The values reported in Table 2 of the study publication show an increase from baseline to the last visit, although the legend states that "lower values indicate less pruritus". We contacted the study authors but were not able to resolve this inconsistency. No data from the study were available for analysis. Caution is needed in interpreting these study findings.
We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (high for the domain incomplete outcome data and for several domains judged as having unclear risk of bias) and due to reporting of outcomes that could not be reproduced (i.e. the legend states lower values indicate less pruritus, but pruritus increased in all groups from baseline to last visit). We contacted study authors to clarify this issue but received no response (Table 2).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
In all, 10 of 42 participants in the 10 mg/d group and 9 of 42 in the placebo group reported the occurrence of at least one adverse event (RR 1.11, 95% CI 0.50 to 2.45; P = 0.79; participants = 84; studies = 1; Analysis 5.1; Table 2). A total of 14 of 45 participants in the 20 mg/d group reported at least one adverse event (RR 1.45, 95% CI 0.70 to 3.00; P = 0.31; participants = 87; studies = 2; Analysis 5.1). We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (high for the domain incomplete outcome data and for several domains judged as having unclear risk of bias) and serious imprecision (wide CI due to low sample size).
5.1. Analysis.
Comparison 5 Cetirizine 10, 20, 40 mg/d versus placebo (4‐week intervention), Outcome 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period.
However, results show a significant difference in occurrence of adverse events between 40 mg/d and placebo, in favour of the placebo group. In total, 24 of 47 in the 40 mg group and 9 of 42 in the placebo group reported at least one adverse event (RR 2.38, 95% CI 1.25 to 4.53; P = 0.008; participants = 89; studies = 1; Analysis 5.1). The number needed to treat for an additional harmful outcome (NNTH) was 3 (95% CI 2 to 9). We downgraded the quality of evidence from high to low due to serious risk of bias (several domains judged as having unclear risk of bias) and serious imprecision (wide CI due to small sample size). Most side effects pertained to sedation, others to skin‐related problems, respiratory symptoms, or headache.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
The total symptom score significantly decreased in all groups between baseline and the last visit according to study authors, who also reported that the decrease was significantly more marked for the 40 mg/d group than for the placebo group, thus favouring the 40 mg intervention group. No data from the study were available for analysis. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (high for the domain incomplete outcome data and for several domains judged as having unclear risk of bias) and serious imprecision (due to small sample size).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
Study authors observed no differences among groups in the amount of local rescue therapy (emollient or 1% hydrocortisone) nor in the number of applications. No data from the study were available for analysis. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (high for the domain incomplete outcome data and for several domains judged as having unclear risk of bias) and serious imprecision (due to small sample size).
Long‐term interventions
Cambazard 2001 reported the results of a long‐term intervention (eight weeks; n = 223) of 0.5 mg/d per kg bodyweight versus placebo conducted in children between 11 and 71 months of age.
Primary outcome 1. Mean change in patient‐assessed symptoms
Investigators assessed pruritus as part of the SCORAD. However, they did not report this outcome separately but provided only the total SCORAD score. No data from the study were available for analysis.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors observed very few adverse events. Comparison of the four treatment groups on the basis of frequency of adverse events did not reveal a statistically significant difference. No data from this study were available for analysis. We downgraded the quality of the evidence by two levels to low: one level for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias), and one level for reporting bias due to non‐publication of results.
Secondary outcome 1. Physician‐assessed clinical signs
Investigators assessed this outcome by using a modified SCORAD. Scores decreased by 7.9 units in the placebo group and by 9.4, 9.1, and 9.1 units in the 0.25, 0.50, and 0.75 mg/kg body weight each day (bw/d) cetirizine dosage groups, respectively. The difference observed between placebo and the reference dosage of 0.50 mg/kg bw/d cetirizine was not significant. No significant dose‐effect relationship between change in modified SCORAD index and dosage of study medication was evident. No data from the study were available for analysis. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias), and one level for reporting bias due to non‐publication of results.
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
On average, participants in the placebo group used 7.2 g 1% hydrocortisone cream and those in the 0.50 mg/kg bw/d cetirizine dosage group used 6.9 g. On average, those in the placebo group consumed 39.0 g clobetasone butyrate cream and those in the 0.50 mg/kg bw/d cetirizine dosage group consumed 52.0 g of clobetasone butyrate cream. No significant differences emerged when groups were compared with regard to topical anti‐inflammatory medication use. No data from the study were available for analysis. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias), and one level for reporting bias due to non‐publication of results.
Diepgen 2002 reported the results of a long‐term intervention (18 months) in children 12 to 24 months of age. Interventions comprised 0.50 mg/kg bw/d cetirizine and placebo. Participants were allowed to use other H1 AH during the study, and study authors reported that a higher percentage in the placebo group used them (Simons 1999).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Investigators assessed this outcome as part of the SCORAD assessment. They did not present data separately for pruritus but provided only the total SCORAD score. Thus, no data from the study were available for analysis of this outcome.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Data from an earlier publication of the same study show that 37 of 399 in the cetirizine group and 54 of 396 in the placebo group reported the occurrence of adverse events (RR 0.68, 95% CI 0.46 to 1.01; P = 0.06; participants = 795; studies = 1; moderate‐quality evidence; Analysis 6.1; Table 1) (Simons 1999). The difference was non‐significant, and most reported symptoms and events were mild and were attributed to intercurrent (occurring during and modifying the course of another disease) respiratory or gastrointestinal infection or to exacerbations of allergic disorders.
6.1. Analysis.
Comparison 6 Cetirizine 0.5 mg/d per kg versus placebo, Outcome 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Researchers assessed this outcome by using SCORAD. They found no evidence of a treatment‐related reduction in SCORAD scores between baseline and the final visit and reported that the difference between groups was non‐significant (from 24.9 to 15.2 in the cetirizine group and from 25.1 to 15.7 in the placebo group; moderate‐quality evidence). No data from the study were available for analysis (Table 1).
We had stated in the section Subgroup analysis and investigation of heterogeneity that we would compare participants with atopic versus non‐atopic dermatitis (i.e. sensitised showing elevated levels of IgE antibodies to inhalant allergens) if data became available. Study authors stated that participants with elevated IgE aeroallergens at baseline showed a decrease in the average SCORAD index during the study period from 30.3 to 22.3 in the placebo group and from 32.6 to 18.8 in the cetirizine group (from baseline to last visit). However, they reported the comparison between groups, and we could extract no data for analysis.
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
For this outcome, researchers assessed the use of a variety of topical and systemic medications including oral use of H1 AH. Results show no significant differences between groups, with one exception: participants in the placebo group were more likely to have used H1 AH (P = 0.035; moderate‐quality evidence; Table 1). No data from this study were available for analysis.
LaRosa 1994 reported the results of a long‐term intervention (eight weeks; n = 23) conducted in children six to 12 years of age. Investigators compared 5 mg cetirizine for children ≤ 30 kg and 10 mg for children > 30 kg versus placebo.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Study authors stated that cetirizine showed a significant advantage over placebo at week 8 (Chi² 4.55; P < 0.05) with regard to pruritus assessed by a diary, which favours the intervention group. However, results as presented are not reproducible, as they are shown in figures or are given in a rudimentary way in the text. We could extract no data for analysis from this study. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to unclear judgement for all other domains apart from the domain incomplete outcome data (low risk), and one level due to imprecision (small sample size).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Investigators observed no adverse events and provided no study data for analysis. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to unclear judgement for all other domains apart from the domain incomplete outcome data (low risk), and one level due to imprecision (small sample size).
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Researchers assessed this outcome by clinical examination at baseline, at four weeks of treatment, and at eight weeks of treatment. Although they observed a significant reduction in scores within groups, they noted no significant differences between groups. No data from this study were available for analysis. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to unclear judgement for all other domains apart from the domain incomplete outcome data (low risk), and one level due to imprecision (small sample size).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
For this outcome, investigators measured the use of concomitant therapy. In all, 18% in the active treatment group and 82% in the placebo group reported use of concomitant therapy (disodium cromoglycate, procaterol, steroids). A Chi² test attested to the significance of this difference (P < 0.01). Our analysis confirmed the significance of the reported result (RR 0.22, 95% CI 0.06 to 0.80; P = 0.02; participants = 22; studies = 1; Analysis 7.1), thus favouring the intervention group. We downgraded the quality of evidence by two levels to low: one level for limitations in design due to unclear judgement for all other domains apart from the domain incomplete outcome data (low risk), and one level due to imprecision (small sample size).
7.1. Analysis.
Comparison 7 Cetirizine 5 mg/d for children < 30 kg and 10 mg/d for children > 30 kg versus placebo, Outcome 1 Secondary outcome 3. Use of concomitant therapy (disodium cromoglycate, procaterol, steroids).
Tharp 1998 compared 20 mg/d given to adults in the active treatment group as a long‐term intervention versus placebo (12 weeks; n = 106) and published these findings in abstract format.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Study authors stated that patient‐reported pruritus (no information about scaling provided) improved in the active treatment group significantly more than in the placebo group (P ≤ 0.05) at weeks 3, 5, and 6. The global evaluation of symptoms by patients (no precise information provided on how this was assessed) indicated significantly more improvement for patients treated with cetirizine over placebo (P < 0.05). Both analyses thus favoured the intervention. We could extract no data for analysis from this study. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported no significant differences between groups in the occurrence of this outcome. We could extract no data for analysis from this study. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias).
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Researchers reported that the mean improvement in investigator‐assessed clinical signs and symptoms was significantly greater in the cetirizine group than in the placebo group (P = 0.04), thus favouring the intervention. Again, no data from this study were available for analysis. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
Study authors observed no significant differences between groups in the use of triamcinolone acetonide 0.1% cream (corticosteroid). We could extract no data from this study for analysis. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (all domains judged as having unclear risk of bias).
Comparison 4
Chlorpheniramine 12 mg/d versus placebo
We identified one cross‐over study that compared this intervention in adults (Frosch 1984). Researchers reported an intermediate‐term intervention (four weeks; n = 18).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
For this outcome, study authors measured pruritus by VAS (100 mm) during the day and during the night, observing no significant differences between groups. No data from this study were available for analysis, and no P values were given. We downgraded the quality of evidence by two levels from high to low due to unclear judgement for most domains and two that were judged low (selection and attrition bias) with imprecision (small sample size).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Three participants reported adverse events during treatment with chlorpheniramine; none reported adverse events during the placebo period. No data from this study were available for analysis. We downgraded the quality of evidence by two levels from high to low due to unclear judgement of most domains and two that were judged low (selection and attrition bias) with imprecision (small sample size).
Secondary outcome 1. Mean change in physician‐assessed clinical signs
This global assessment reported the numbers of participants with overall improvement, no overall improvement, and overall worsening. Study authors reported no significant differences between groups based on the Kruskal‐Wallis test. No data from this study were available for analysis, and no P values were given. We downgraded the quality of evidence by two levels from high to low due to unclear judgement of most domains and two that were judged low (selection and attrition bias) with imprecision (small sample size).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
For this outcome, researchers assessed the amount of betamethasone used in grams. Study authors observed no significant differences between groups. We could extract no data for analysis from this study, and no P values were given. We downgraded the quality of evidence by two levels from high to low due to unclear judgement of most domains and two that were judged low (selection and attrition bias) with imprecision (small sample size).
Comparison 5
Chlorpheniramine maleate BP (2 to 4 mg/d (age dependent) or twice that amount) versus placebo
We included in this comparison one parallel‐group study conducted in children (Munday 2002). Researchers reported the results of an intermediate‐term (one month) intervention. One group received 2 mg/5 mL chlorpheniramine maleate for those one to five years of age, or 10 mL (4 mg) for six‐ to 12‐year‐olds. After two weeks, investigators administered double the bedtime dose (5.0 mL for those aged one to five years, and 10 mL for participants six to 12 years old) to participants who continued to experience sleeplessness. The total dose was 2 or 4 mg (age dependent) or twice that amount after two weeks. The other group received placebo. Study authors concluded that chlorpheniramine maleate is no more effective than placebo for relieving pruritus and controlling symptoms of eczema.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Study authors did not assess this outcome as 'Mean change in patient‐assessed symptoms', but participants rated the severity of pruritus (ranked) as none, minimal, mild, or moderate between days 1 and 29. Results show no significant differences (P = 0.745 based on the Cochran‐Mantel‐Haenzsel test) between intervention and placebo groups (stratified for age groups and controlling for baseline differences) in severity of night‐time pruritus. Note that modal response is defined as the most common response over a specified period. This combined with the fact that the statistical test used is not very sensitive makes this analysis not very powerful. No data for this outcome were available for analysis. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (most domains judged as having unclear risk of bias).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Researchers noted no significant differences between groups (RR 0.95, 95% CI 0.49 to 1.82; P = 0.87; participants = 151; studies = 1; Analysis 8.1). We downgraded this outcome by one level from high to moderate because of serious risk of bias (most domains judged as having unclear risk of bias).
8.1. Analysis.
Comparison 8 Chlorpheniramine maleate BP (2 to 4 mg/d (age dependent) or twice that amount) versus placebo, Outcome 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Investigators presented this outcome as a composite score consisting of five symptoms (erythema, excoriation, dryness, lichenification, exudation and crusting). Data show no significant differences between groups at day 1 (P = 0.479), day 15 (P = 0.33), or day 29 (P = 0.53). No data were available for analysis. We downgraded the quality of evidence by one level from high to moderate for limitations in design due to serious risk of bias (most domains judged as having unclear risk of bias).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
Study authors assessed this outcome as the amount of 1% hydrocortisone in grams used and analysed data separately for age groups one to five years and six to 12 years. Results show no significant differences between intervention and placebo groups, neither in the age group one to five years (MD ‐1.30, 95% CI ‐5.96 to 3.36; P = 0.58; participants = 61; studies = 1; Analysis 8.2) nor in the age group six to 12 years (MD 1.60, 95% CI ‐2.53 to 5.73; P = 0.45; participants = 90; studies = 1; Analysis 8.2). We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (most domains judged as having unclear risk of bias) with serious imprecision (wide CI due to small sample size or high variability in outcome measurements).
8.2. Analysis.
Comparison 8 Chlorpheniramine maleate BP (2 to 4 mg/d (age dependent) or twice that amount) versus placebo, Outcome 2 Secondary outcome 3. Amount of 1% hydrocortisone in grams used.
Comparison 6
Fexofenadine 120 mg/d versus placebo
We identified one parallel‐group study that compared this intervention in adults (Kawashima 2003). Investigators reported the results of a short‐term intervention (one week) and concluded that fexofenadine led to rapid, significant improvement in pruritus associated with eczema, and that the safety profile was similar to that of placebo.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema: pruritus difference between baseline and night of day 6
Researchers measured this outcome as the mean change in patient‐assessed pruritus (range 0 to 8) between baseline and the night of day 6. The mean change was significantly larger in the fexofenadine group than in the placebo group; therefore, the comparison favours the intervention group (MD ‐0.25, 95% CI ‐0.43 to ‐0.07; P = 0.006; participants = 400; studies = 1; moderate‐quality evidence; Analysis 9.1; Table 3).
9.1. Analysis.
Comparison 9 Fexofenadine 120 mg/d versus placebo, Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: mean pruritus difference (0 to 8) between baseline and night of day 6.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
In all, 48 of 207 participants in the fexofenadine group and 45 of 204 in the placebo group experienced an adverse event during the trial (RR 1.05, 95% CI 0.74 to 1.50; P = 0.78; participants = 411; studies = 1; moderate‐quality evidence; Analysis 9.2; Table 3). This difference was non‐significant, and the most frequently reported adverse events were somnolence and headache.
9.2. Analysis.
Comparison 9 Fexofenadine 120 mg/d versus placebo, Outcome 2 Primary outcome 2. Adverse events.
Secondary outcome 1. Physician‐assessed clinical signs
Study authors stated that in terms of investigator‐assessed change in the ratio of pruritus area to body surface area, significantly more participants in the fexofenadine group than in the placebo group experienced a reduction in this ratio (P = 0.007). This comparison favours the intervention group (moderate‐quality evidence). No data from this study were available for analysis.
Secondary outcome 3. Number of eczema flares/amount of 0.1% hydrocortisone butyrate in grams
Study authors reported no significant differences between groups in the amount of 0.1% hydrocortisone butyrate cream used during the study (moderate‐quality evidence). We could extract from this study no data for analysis.
Comparison 7
Hydroxyzine 75 mg/d versus placebo
We identified one parallel‐group trial conducted in adults that performed this comparison (Monroe 1992). Researchers reported the results of a short‐term (one week) intervention.
Primary outcome 1. Patient‐assessed symptoms: global evaluation of the antipruritic effect of treatment: number of participants who reported a marked or complete response
Investigators assessed this outcome not as mean change but as percentage change in pruritus scores (0 to 3) from baseline to endpoint. The daily pruritus score decreased by 38% in the hydroxyzine group and by 33% in the placebo group. Study authors reported that the difference was not significant but gave no P value. According to the global evaluation of the antipruritic effect of treatment, six participants in the hydroxyzine group and three in the placebo group reported a marked or complete response (RR 1.86, 95% CI 0.58 to 5.94; P = 0.30; participants = 27; studies = 1; Analysis 10.1). Fisher's exact test analysis yielded a P value of 0.42. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (most domains judged as having unclear risk of bias) and due to serious imprecision (wide CI due to small sample size).
10.1. Analysis.
Comparison 10 Hydroxyzine 75 mg/d versus placebo, Outcome 1 Primary outcome 1. Patient‐assessed symptoms: global evaluation of the antipruritic effect of treatment: number of participants who reported a marked or complete response.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors did not report occurrence of adverse events separately for participants with eczema but provided data only for the whole sample, including those with urticaria. No data for analysis were available from this study.
Comparison 8
Ketotifen versus placebo
We identified three parallel‐group studies for this comparison (Falk 1993; Iikura 1992; Leon 1989).
Ketotifen 2 mg/d versus placebo
We identified two studies that performed this comparison (Falk 1993; Leon 1989).
Falk 1993 reported the results of a long‐term intervention in adults (three months; n = 56).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Study authors reported a significant reduction in pruritus in both intervention (2 mg/d) and placebo groups but conducted no test to detect differences between groups. It is not possible to extract data from this study for analysis. We downgraded the quality of evidence from high to low due to unclear judgement of most domains and two that were judged low (performance and attrition bias) with imprecision (small sample size).
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Researchers assessed this outcome by combining five signs and symptoms (severity of diseased skin, area affected, erythema, lichenification, and pruritus) for a total score ranging from 0 to 4. After treatment, the ketotifen group had a mean score of 2.8 and the placebo group had a mean score of 1.2. Study authors reported the difference to be significant at P < 0.01 and to favour the intervention group. No data from this study were available for analysis. We downgraded the quality of evidence from high to low due to unclear judgement of most domains and two that were judged low (performance and attrition bias) with imprecision (low sample size).
Leon 1989 investigated a long‐term intervention (nine weeks) of ketotifen (2 mg/d) in a small sample of children (n = 20).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Investigators assessed the intensity of day and night pruritus on a scale from 0 to 3 (absent = 0, mild = 1, moderate = 2, intense = 3). They provided data for the ketotifen group (day pruritus: visit 1: Mean (M) = 2.1 (standard deviation (SD) = 0.74), week 9: M = 0.7 (SD 0.67); night pruritus: visit 1: M = 1.7 (SD = 1.25), week 9: M = 0.2 (SD = 0.42)) but not for the placebo group. Study authors stated that differences in both daytime and night‐time pruritus between visit 1 and week 9 were not significant for the placebo group but showed significant improvement for the ketotifen group (P = 0.01 for night‐time and P = 0.005 for daytime pruritus comparisons). However, investigators carried out no comparison between groups, and as we could extract no data from the study, no inference could be made about whether ketotifen has an effect on pruritus over placebo. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (most domains judged as having unclear risk of bias) and imprecision (small sample size).
Ketotifen (0.8 mg/d (< 14 kg) or 1.2 mg/d (≥ 14 kg)) versus placebo
Iikura 1992 investigated the effect of ketotifen (0.8 mg/d (< 14 kg) or 1.2 mg/d (≥ 14 kg)) in a long‐term intervention (52 weeks) against the onset of asthma in children.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported the occurrence of adverse events in 5 of 61 in the ketotifen group and in 0 of 60 in the placebo group (RR 10.82, 95% CI 0.61 to 191.53; P = 0.10; participants = 121; studies = 1; Analysis 11.1); due to the small sample size, Fisher's exact test revealed a significant difference between the two groups (P = 0.04) in favour of placebo. We downgraded the quality of evidence by two levels from high to low because of serious risk of bias (most domains judged as having unclear risk of bias) and serious imprecision (wide CI due to small sample size).
11.1. Analysis.
Comparison 11 Ketotifen 0.8 to 1.2 mg/d versus placebo, Outcome 1 Primary outcome 2. Adverse events.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
Researchers assessed this outcome by performing a complete physical examination one year after randomisation. They scored data as marked improvement, moderate improvement, slight improvement, unchanged, and aggravated. Fisher's exact test compared the distribution of marked/moderate improvement between groups, and another compared the distribution of marked/moderate/slight improvement between groups. Both analyses favoured ketotifen over placebo (P < 0.05 and P < 0.001). No data from this study were available for analysis. We downgraded the quality of evidence by one level from high to moderate because of serious risk of bias (most domains judged as having unclear risk of bias).
Comparison 9
Levocetirizine versus placebo
Levocetirizine 5 mg/d versus placebo
We identified one parallel‐group study that compared this intervention in adults (Kircik 2013). Researchers reported the results of an intermediate‐term intervention (four weeks) in 20 participants in the intervention group and 20 participants in the placebo group.
Primary outcome 1. Patient‐assessed symptoms of eczema: VAS pruritus
Researchers reported medians and interquartile ranges for baseline, two weeks, and four weeks after randomisation. Baseline values were 7.8 (6.5 to 8.5) for the intervention group and 7.5 (7.00 to 8.00) for the placebo group. At two weeks, the values were 6.10 (1.6 to 7.5) for the intervention group and 5.9 (3.65 to 7.75) for the placebo group. At four weeks, the values were 4.15 (2.4 to 6.9) and 6.4 (2.5 to 7.3). We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (all domains judged as having unclear or high risk of bias) and serious imprecision (due to small sample size).
The same study published an interim report with fewer participants, which yielded data for the analysis of primary outcome 1, showing no significant differences in pruritus between groups two weeks after randomisation (MD ‐0.79, 95% CI ‐3.54 to 1.96; P = 0.57; participants = 21; studies = 1; Analysis 12.1) or 4 weeks after randomisation (MD ‐0.59, 95% CI ‐3.46 to 2.28; P = 0.69; participants = 21; studies = 1; Analysis 12.1). We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (all domains judged as having unclear or high risk of bias) and serious imprecision (wide CI due to small sample size).
12.1. Analysis.
Comparison 12 Levocetirizine 5 mg/d versus placebo, Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: mean difference in VAS pruritus between baseline and 2 and 4 weeks after randomisation.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported that they observed no treatment‐related adverse events in either group. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (all domains judged as having unclear or high risk of bias) and serious imprecision (due to small sample size).
Secondary outcome 2. Mean change in quality of life, as measured by a standardised or validated quality of life measure (e.g. Dermatology Life Quality Index ‐ DLQI)
Study authors reported median DLQI scores and interquartile ranges at baseline, two weeks, and four weeks after randomisation. Baseline values were 7.5 (4.0 to 14.0) for the intervention group and 8.0 (7.0 to 13.0) for the placebo group. Two weeks after randomisation, the values were 3.5 (2.0 to 6.5) for the intervention group and 5.5 (2.0 to 9.0) for the placebo group. Four weeks after randomisation, the values were 2.0 (1.0 to 5.0) for the intervention group and 4.0 (1.0 to 7.5) for the placebo group. Trial authors provided no analysis nor could we extract any data from the study for analysis. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (all domains judged as having unclear or high risk of bias) and serious imprecision (due to small sample size).
Levocetirizine 0.25 mg/kg/d versus placebo
We identified one parallel‐group study that compared this intervention in children (Simons 2007). Researchers reported the results of a long‐term intervention (18 months).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors provided a detailed analysis of the occurrence of adverse events during the trial, noting no significant differences between groups in occurrence of one or more adverse events (RR 1.01, 95% CI 0.98 to 1.05; P = 0.48; participants = 510; studies = 1; Analysis 13.1) nor of treatment‐attributed adverse events (RR 0.81, 95% CI 0.40 to 1.65; participants = 510; studies = 1; P = 0.57; Analysis 13.1) nor of serious adverse events (RR 0.84, 95% CI 0.54 to 1.31; participants = 510; studies = 1; P = 0.44; Analysis 13.1) nor of treatment‐attributed serious adverse events (RR 0.33, 95% CI 0.01 to 8.14; participants = 510; studies = 1; P = 0.50; Analysis 13.1) nor of adverse events that led to discontinuation of the study medication (RR 1.67, 95% CI 0.40 to 6.90; P = 0.48; participants = 510; studies = 1; Analysis 13.1). We downgraded the quality of evidence by one level from high to moderate due to serious risk of bias (all but one domain judged as having unclear risk of bias).
13.1. Analysis.
Comparison 13 Levocetirizine 0.25 mg/kg/d, Outcome 1 Primary outcome 2. Adverse events.
Comparison 10
Loratadine versus placebo
We identified three parallel‐group studies ‐ Ruzicka 1998,Monroe 1992, and Kimura 2009 ‐ and one cross‐over trial ‐ Langeland 1994 ‐ that compared this intervention.
Ruzicka 1998 reported the results of an intermediate‐term intervention (two weeks) in adults. Participants consisted of three groups receiving 5 mg/d, 10 mg/d, or 20 mg/d of loratadine and one placebo group. Baseline values of pruritus were unequally distributed among the groups, with higher baseline values in the 20 mg/d group than in the other groups.
Loratadine 5 mg/d
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema: VAS pruritus (difference in pruritus sum between day 0 and day 13)
The difference in the pruritus sum between day 0 and day 13 did not differ significantly between the 5 mg/d group and the placebo group (MD ‐1.10, 95% CI ‐2.30 to 0.10; P = 0.07; participants = 72; studies = 1; Analysis 14.1). However, baseline scores were not equally distributed between groups, and this analysis does not account for baseline differences. We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
14.1. Analysis.
Comparison 14 Loratadine 5 mg/d versus placebo (2‐week intervention), Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported the occurrence of 16 adverse events in all four groups but failed to report them for each group separately. No data from this study were available for analysis.
Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation
Researchers reported physician‐assessed clinical signs not as mean change but as the number of participants for whom treatment success was judged as good or very good. Again, they noted no significant differences in this outcome between groups (RR 1.58, 95% CI 0.73 to 3.42; P = 0.24; participants = 74; studies = 1; Analysis 14.2). We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
14.2. Analysis.
Comparison 14 Loratadine 5 mg/d versus placebo (2‐week intervention), Outcome 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation.
Loratadine 10 mg/d
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema: VAS pruritus (difference in pruritus sum between day 0 and day 13)
The difference in the pruritus sum between day 0 and day 13 did not differ significantly between the 10 mg/d group and the placebo group (MD ‐0.96, 95% CI ‐2.01 to 0.09; P = 0.07; participants = 71; studies = 1; Analysis 15.1; Table 5). We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size) .
15.1. Analysis.
Comparison 15 Loratadine 10 mg/d versus placebo (2‐week intervention), Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported the occurrence of 16 adverse events in all four groups but failed to report them for each group separately. No data from this study were available for analysis (Table 5).
Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation
Researchers reported physician‐assessed clinical signs not as mean change but as the number of participants for whom treatment success was judged as good or very good. They noted a marginally significant difference in this outcome between groups (RR 2.04, 95% CI 0.99 to 4.20; P = 0.05; participants = 73; studies = 1; Analysis 15.2; Table 5). Participants in the 10 mg/d group were about twice as likely to be judged as having good or very good treatment success. The NNTB was 5 (95% CI 2 to 111). We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
15.2. Analysis.
Comparison 15 Loratadine 10 mg/d versus placebo (2‐week intervention), Outcome 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation.
Loratadine 20 mg/d
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema: VAS pruritus (difference in pruritus sum between day 0 and day 13)
The difference in the pruritus sum between day 0 and day 13 was significantly larger in the 20 mg/d group than in the placebo group (MD ‐1.41, 95% CI ‐2.49 to ‐0.33; P = 0.01; participants = 72; studies = 1; Analysis 16.1). The comparison favours the intervention group. We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
16.1. Analysis.
Comparison 16 Loratadine 20 mg/d versus placebo (2‐week intervention), Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported the occurrence of 16 adverse events in all four groups but failed to report them for each group separately. No data from this study were available for analysis.
Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation
Researchers reported physician‐assessed clinical signs not as mean change but as the number of participants for whom treatment success was judged as good or very good. They noted a marginally significant difference in this outcome between groups (RR 2.04, 95% CI 0.99 to 4.20; P = 0.05; participants = 73; studies = 1; Analysis 16.2). Participants in the 20 mg/d group were about twice as likely to be judged as having good or very good treatment success. The NNTB was 5 (95% CI 2 to 111). We downgraded the quality of evidence by two levels from high to low due to unclear judgement in most domains and serious imprecision (wide CI due to small sample size).
16.2. Analysis.
Comparison 16 Loratadine 20 mg/d versus placebo (2‐week intervention), Outcome 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good.
Monroe 1992 reported the results of a short‐term intervention (one week) in adults consisting of 10 mg/d of loratadine (n = 14) versus placebo (n = 13).
Primary outcome 1. Patient‐assessed symptoms of eczema
Investigators did not report this outcome as mean change but measured pruritus as the percentage change in scores (0 to 3) from baseline to endpoint. The daily pruritus score decreased by 57% in the loratadine group and by 33% in the placebo group. Study authors reported that this difference was not significant but gave no P value. According to the global evaluation of the antipruritic effect of treatment, nine participants in the loratadine group and three in the placebo group reported a marked or complete response (RR 2.79, 95% CI 0.96 to 8.09; participants = 27; studies = 1; P = 0.06; Analysis 17.1). The P value from Fisher's exact test was 0.06. This underpowered analysis was not statistically significant but suggested that loratadine may improve pruritus compared to placebo. We downgraded the quality of evidence by two levels from high to low due to limitations in design (most domains judged as having unclear risk of bias) and due to serious imprecision (wide CI due to small sample size).
17.1. Analysis.
Comparison 17 Loratadine 10 mg/d versus placebo (1‐week intervention), Outcome 1 Primary outcome 1. Global evaluation of the antipruritic effect of treatment: number of participants who reported a marked or complete response.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Investigators did not report the occurrence of adverse events separately for participants with eczema but included results for all participants, including those with urticaria. No data from this study were available for analysis.
Kimura 2009 reported the results of an intermediate‐term intervention (four weeks) in adults. One group received 10 mg/d loratadine and the other no additional treatment.
Primary outcome 1. Patient‐assessed symptoms of eczema: VAS pruritus four weeks after randomisation
For this outcome, study authors assessed pruritus by VAS four weeks after randomisation, noting no significant differences between groups (MD ‐2.30, 95% CI ‐20.27 to 15.67; P = 0.80; participants = 28; studies = 1; Analysis 18.1; Table 4). We downgraded the quality of evidence by two levels to low: one level for limitations of design due to unclear judgement of all but one domain, which we judged as low (selection bias), and one level due to imprecision (small sample size).
18.1. Analysis.
Comparison 18 Loratadine 10 mg/d versus no additional treatment (4‐week intervention), Outcome 1 Primary outcome 1. Patient‐assessed symptoms: VAS pruritus 4 weeks after randomisation.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
The occurrence of adverse events did not differ significantly between groups (RR 0.25, 95% CI 0.01 to 5.76; P = 0.39; participants = 28; studies = 1; Analysis 18.2; Table 4). Fisher's exact test confirmed this analysis (P = 0.429). One participant in the placebo arm reported folliculitis. We downgraded the quality of evidence by two levels to low: one level for limitations of design due to unclear judgement of all but one domain, which we judged as low (selection bias), and one level due to imprecision (small sample size).
18.2. Analysis.
Comparison 18 Loratadine 10 mg/d versus no additional treatment (4‐week intervention), Outcome 2 Primary outcome 2. Adverse events.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
For this outcome, researchers assessed the SCORAD after four weeks and found that the difference between groups was not significant (MD ‐4.10, 95% CI ‐13.22 to 5.02; P = 0.38; participants = 28; studies = 1; Analysis 18.3; Table 4). We downgraded the quality of evidence by two levels to low: one level for limitations of design due to unclear judgement of all but one domain, which we judged as low (selection bias), and one level due to imprecision (small sample size)
18.3. Analysis.
Comparison 18 Loratadine 10 mg/d versus no additional treatment (4‐week intervention), Outcome 3 Secondary outcome 1. Physician‐assessed clinical signs: SCORAD after 4 weeks.
Langeland 1994 reported the results of an intermediate‐term intervention conducted in adults in a complex cross‐over trial comparing the effects of 10 mg/d loratadine versus placebo in six consecutive periods, each lasting two weeks (six periods, each lasting two weeks = 12 weeks total duration of study; n = 16). Statistical methods used for analysis were inappropriate, as study authors used methods for independent data and not for related data. Therefore these findings must be interpreted with caution. For instance, Hotelling's T² test was specified to compare the effects of loratadine versus placebo. Hence, no control was provided for carry‐over and period effects.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
For this outcome, study authors assessed pruritus during the day and during the night on a 10‐cm VAS recorded daily by the participant. Study authors stated that they detected significant effects of loratadine as compared with placebo with regard to this outcome, favouring the intervention. However, the statistical test used was inappropriate, as it did not account for the within‐subject design. Reported changes in mean VAS scores varied little across treatment periods. No data from this study were available for analysis. We downgraded the quality of evidence by two levels: one level for limitations of design due to serious risk of bias (all domains judged as having unclear risk of bias), and one level due to imprecision (small sample size).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Study authors reported that they noted no adverse effects irrespective of treatment. We downgraded the quality of evidence by two levels from high to low: one level for limitations in design (all domains judged as having unclear risk of bias), and one level due to imprecision (small sample size).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
Study authors reported that the mean number of days with use of local steroids during the six treatment periods and the severity of rash at the end of each treatment period showed a similar pattern to those reported for pruritus during the day and at night. Again, caution is needed in interpreting these findings. We could extract from this study no data for analysis. We downgraded the quality of evidence by two levels from high to low: one level for limitations in design (all domains judged as having unclear risk of bias), and one level due to imprecision (small sample size).
Comparison 11
Olopatadine 10 mg/d versus no additional treatment
We identified one parallel‐group study (n = 99) that compared this intervention in adults (Kuniyuki 2009), which reported the results of a long‐term intervention (eight weeks).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
For this outcome, researchers assessed pruritus once by VAS (0 to 100) scale, and once by the 5‐point Likert scale (Shiratori scale) (0 = none to 4 = severe). Both instruments were found to be significantly different between groups at weeks 2, 4, and 8. Study authors reported significantly lower values for the olopatadine group than for those given no additional treatment. The comparison thus favours the intervention group. We could extract no data from this study for analysis. We downgraded the quality of evidence from high to moderate due to limitations in design (all domains judged as having unclear or high risk of bias).
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
Investigators reported that 8% (4/49) of those in the olopatadine group and 0% (0/50) in the no additional treatment group reported mild sleepiness (RR 9.18, 95% CI 0.51 to 166.10; P = 0.13; participants = 99; studies = 1; Analysis 19.1). We downgraded the quality of evidence from high to low due to limitations in design (all domains judged as having unclear or high risk of bias) and serious imprecision (wide CI due to low sample size).
19.1. Analysis.
Comparison 19 Olopatadine 10 mg/d versus no additional treatment, Outcome 1 Primary outcome 2. Adverse events 8 weeks after randomisation.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
For this outcome, researchers measured results by SCORAD. At week 8, they observed significantly lower values in the olopatadine group than in the no additional treatment group. The comparison thus favours the intervention group. No data from this study were available for analysis. We downgraded the quality of evidence from high to moderate due to limitations in design (all domains judged as having unclear or high risk of bias).
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
Investigators assessed this outcome by using a topical steroid score. Its respective potency (0.5 to 3) and the amount (g) of topical steroids applied were multiplied to obtain the score. Study authors reported that the decrease in score was significantly lower in the olopatadine group than in the no additional treatment group (P < 0.05) eight weeks after randomisation. The comparison thus favours the intervention group. No data from this study were available for analysis. We downgraded the quality of evidence from high to moderate due to limitations in design (all domains judged as having unclear or high risk of bias).
Comparison 12
Terfenadine versus placebo
We identified three studies that performed this comparison. One study used a parallel‐group design (Doherty 1989), and two used a cross‐over design (Berth Jones 1989; Hjorth 1988).
Doherty 1989 reported an intermediate‐term intervention (10 days) of 180 mg/d in adults but assessed primary outcome 1 seven days after baseline (short‐term).
Primary outcome 1. Patient‐assessed symptoms of eczema: VAS pruritus seven days after baseline
VAS pruritus scores (seven days after baseline and adjusted for baseline scores as a covariate; range 0 to 100) were higher in the placebo group than in the terfenadine group, but the 95% CI was wide and included one showing uncertainty (MD ‐15.20, 95% CI ‐33.03 to 2.63; P = 0.09; participants = 27; studies = 1; Analysis 20.1). We downgraded the quality of evidence by two levels from high to low because of unclear risk of bias for most domains and because of imprecision (wide CI due to small sample size or high variability in outcome measurement).
20.1. Analysis.
Comparison 20 Terfenadine 180 mg/d versus placebo, Outcome 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus 7 days after baseline adjusted for baseline score as covariate.
Secondary outcome 1. Mean change in physician‐assessed clinical signs
This study provided data not in the form of mean change but as the number of participants for whom treatment helped itching (physician‐assessed). Study authors reported more events in the intervention group but showed no significant difference between terfenadine and placebo groups due to the wide 95% CI, including 1 (RR 2.19, 95% CI 0.88 to 5.44; P = 0.09; participants = 30; studies = 1; Analysis 20.2). We downgraded the quality of evidence by two levels from high to low because of unclear judgement for most domains and serious imprecision (wide CI due to small sample size).
20.2. Analysis.
Comparison 20 Terfenadine 180 mg/d versus placebo, Outcome 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment helped itching (1 week after randomisation).
Berth Jones 1989 reported the results of a short‐term intervention (one week) of 240 mg/d of terfenadine versus placebo in adults using a cross‐over design. Researchers used appropriate statistical tests to attempt to account for possible carry‐over and period effects (Wilcoxon rank test = paired text accounting for within‐subject design) and as such was an exception among the cross‐over trials considered for inclusion in this review.
Primary outcome 1. Patient‐assessed symptoms of eczema: VAS pruritus after one week
Study authors reported no evidence of differences between intervention and placebo. Mean scores were 23.95 (standard error (SE) = 4.9) in the terfenadine group and 23.13 (SE = 5.1) in the placebo group. Study authors reported no significant differences between groups based on a Wilcoxon signed rank matched‐pair test. We downgraded the quality of evidence by two levels from high to low due to limitations in design (unclear judgement for most domains) and imprecision (small sample size). No data from this study were available for analysis.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
One participant given placebo and one taking terfenadine reported drowsiness. We downgraded the quality of evidence by two levels from high to low due to limitations in design (unclear judgement for most domains) and imprecision (small sample size).
Secondary outcome 1. Mean change in physician‐assessed clinical signs
This study assessed the severity of excoriations and found no significant differences between intervention and placebo groups. No data from this study were available for analysis. We downgraded the quality of evidence by two levels from high to low due to limitations in design (unclear judgement for most domains) and imprecision (small sample size).
Another cross‐over trial conducted in adults reported the results of an intermediate‐term intervention (two weeks) of 120 mg/d terfenadine versus placebo (Hjorth 1988).
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
Researchers assessed this outcome as severity of itch (no further details given). They stated that terfenadine reduced the severity of itch in approximately 52% of participants; 34% reported no change, and 14% reported increased severity of itch. However, study authors provided no test for significance and no data for placebo. Hence, we could extract no data from this study for analysis. We downgraded the quality of evidence by two levels from high to low due to serious risk of bias (all domains judged as having unclear or high risk of bias) and imprecision (small sample size).
Comparison 13
LN2974 (tazifylline) 15 mg/d versus placebo
We identified one cross‐over study that compared this intervention in adults (Savin 1986). Study authors reported a short‐term intervention upon randomising participants to receive placebo for seven days, placebo or tazifylline for three days, placebo for seven days, and placebo or tazifylline for three days.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
For this outcome, researchers assessed pruritus by VAS, noting no significant differences between treatment and placebo. No data from this study were available for analysis. We judged the quality of evidence to be low, as we downgraded the outcome by one level due to serious risk of bias (all domains judged as having unclear risk of bias) and one level due to imprecision (small sample size).
Comparison 14
One single‐patient trial compared in a cross‐over fashion the effects of chlorpheniramine and terfenadine versus placebo (Nuovo 1992). The trial consisted of four periods, each lasting two weeks, during which placebo, chlorpheniramine 16 mg/d, chlorpheniramine 24 mg/d, or terfenadine 240 mg/d was administered. We downgraded the quality of evidence from high to very low due to very serious imprecision (single‐patient trial = small sample size) for all comparisons.
Primary outcome 1. Mean change in patient‐assessed symptoms of eczema
For this outcome, researchers assessed pruritus on a 7‐point Likert scale. Mean scores during placebo were 1.7, during chlorpheniramine 16 mg/d 0.7, during chlorpheniramine 24 mg/d 0.7, and during terfenadine 240 mg/d 1.4. Results show a marginally significant difference (P = 0.06). No data from this study were available for analysis.
For this outcome, researchers also assessed the severity of symptoms on 7‐point Likert scale. Mean scores during placebo were 2.0, during chlorpheniramine 16 mg/d 2.6, during chlorpheniramine 24 mg/d 3.0, and during terfenadine 240 mg/d 1.5. Study authors observed no significant differences (P = 0.10). No data from this study were available for analysis.
Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period
For this outcome, investigators assessed the severity of drowsiness on a 7‐point Likert scale. Mean scores during placebo were 2.6, during chlorpheniramine 16 mg/d 1.9, during chlorpheniramine 24 mg/d 2.7, and during terfenadine 240 mg/d 3.1. Study authors observed no significant differences (P = 0.30). No data from this study were available for analysis.
Secondary outcome 3. Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
For this outcome, investigators assessed the number of daily applications of the topical steroid triamcinolone. Mean scores during placebo were 0.8, during chlorpheniramine 16 mg/d 0.9, during chlorpheniramine 24 mg/d 0.3, and during terfenadine 240 mg/d 0.3. Study authors observed a marginally significant difference (P = 0.06). No data from this study were available for analysis.
Discussion
It is assumed that about 40 different H1 antihistamines (AH) have been developed (Simons 2004; Simons 2011). How many of these have been applied orally to relieve the symptoms and signs of eczema is not known, and little is known about their efficacy and safety despite the fact that oral H1 AH are widely prescribed for patients with eczema.
Summary of main results
We identified 25 randomised controlled trials (RCTs) that tested oral H1 AH as an 'add‐on' treatment for eczema against placebo or no add‐on treatment; these trials randomised 3285 participants with eczema. Here, we summarise results for the four main comparisons.
For the comparison 'Cetirizine 0.5 mg/kg/d versus placebo' (18‐month intervention) in 795 children (Table 1), researchers assessed the outcome 'Patient‐assessed symptom reduction' as part of the SCORing Atopic Dermatitis index (SCORAD) assessment but did not present data separately for pruritus. Adverse events (mainly mild) were probably less frequent in the intervention group than in the placebo group, but this finding is based on moderate‐quality evidence. With regard to the outcome 'Physician‐assessed clinical signs', moderate‐quality evidence indicates there is probably no difference in treatment‐related reduction in SCORAD scores between baseline and the final visit (only change scores were provided, without standard deviations). For the outcome 'Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’, study authors assessed the use of a variety of topical and systemic medications, yielding moderate‐quality evidence to show that the intervention probably leads to slightly less additional H1 AH use to prevent flares.
For the comparison 'Cetirizine 10 mg/d versus placebo' (four‐week intervention) in 84 adults (Table 2), investigators provided low‐quality evidence for 'Patient‐assessed symptom reduction'; thus, we are uncertain whether cetirizine 10 mg/d improves pruritus. It is noteworthy that the values reported in Table 2 of the publication actually increased from baseline to the last visit, although the legend stated that "lower values indicate less pruritus". We tried to contact the study authors to inquire about this but were not successful in doing so. Low‐quality evidence suggests that adverse events may be slightly higher in the intervention group than in the placebo group, but this result was highly imprecise (due to the small number of participants; n = 84). Adverse events reported included sedation, other skin‐related problems, respiratory symptoms, and headache. Low‐quality evidence suggests no difference in 'physician‐assessed signs' between the cetirizine and placebo groups (but no numerical data were provided). With regard to the outcome 'Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’', low‐quality evidence suggests no difference in the amount of local rescue therapy (emollient or 1% hydrocortisone) nor in the number of applications observed among groups.
For the comparison 'Fexofenadine 120 mg/d versus placebo' (1‐week intervention) in 411 adults (Table 3), moderate‐quality evidence shows there is probably a larger 'patient‐assessed symptom reduction' (pruritus) in the fexofenadine group than in the placebo group. Moderate‐quality evidence shows there is probably little or no difference between groups in occurrence of adverse events (mostly somnolence and headache) nor in the number of eczema flares, measured by the amount of 0.1% hydrocortisone butyrate cream used during the study. Moderate‐quality evidence also shows probably a greater reduction in 'physician‐assessed clinical signs' with fexofenadine, in terms of investigator‐assessed change in the ratio of pruritus area to body surface area, where significantly more participants in the fexofenadine group than in the placebo group experienced a reduction in pruritus. We could not ascertain the magnitude of this effect because study authors reported no coefficient. The clinical meaningfulness of the effect was small, however.
For the comparison 'Loratadine 10 mg/d versus placebo' (four‐week intervention) in 28 adults (Table 4), the quality of evidence was low for all outcomes. The outcome 'Patient‐assessed symptom reduction' may be slightly higher in the intervention group, but the result was very imprecise. Study authors reported only one adverse event (folliculitis), and it occurred in the placebo group. The mean change in physician‐assessed clinical signs (SCORAD) was greater in the intervention group, but again, this was based on imprecise data. Investigators did no assess the number of eczema flares.
Additional summary of results
Results show no significant differences in patient‐assessed symptoms for acrivastine, azelastine, chlorpheniramine and chlorpheniramine maleate BP, hydroxyzine, ketotifen, levocetirizine, LN2974 (tazifylline), and terfenadine. For most comparisons of cetirizine, low‐ to moderate‐quality evidence shows no significant differences in patient‐assessed symptoms. For an eight‐week intervention in children (low‐quality evidence) and a two‐week intervention in adults (moderate‐quality evidence), cetirizine significantly improved patient‐assessed symptoms compared to placebo. In four comparisons of loratadine, researchers observed non‐significant differences, and for the other two comparisons ‐ loratadine (low‐quality evidence) and olopatadine (moderate‐quality evidence) ‐ patient‐assessed symptoms were significantly better in the intervention group than in the placebo group.
Results show significantly more adverse events in the intervention group than in the placebo group for one ketotifen comparison (low‐quality evidence), but two others did not measure adverse events. For one cetirizine comparison (40 mg/d; low‐quality evidence), results show significantly more adverse events in the intervention group; for all other cetirizine comparisons, researchers observed no significant differences, or we could extract no data. For comparisons of acrivastine, azelastine, chlorpheniramine or chlorpheniramine maleate BP, hydroxyzine, levocetirizine, olopatadine, LN2974 (tazifylline), olopatadine, and terfenadine, study authors observed no significant differences, or we could extract no data.
Investigators reported significantly greater improvement in physician‐assessed signs in the intervention groups for the comparison acrivastine (low‐quality evidence) and olopatadine (moderate‐quality evidence), and two ketotifen comparisons (low‐ or moderate‐quality evidence) but not for the others, and for two cetirizine comparisons (both‐low quality evidence) but not for the others. Results show no significant differences in physician‐assessed signs, or investigators did not assess this difference for azelastine, chlorpheniramine or chlorpheniramine maleate BP, hydroxyzine, levocetirizine, loratadine, LN2974 (tazifylline), or terfenadine.
Results show significantly better flare control with the intervention for one cetirizine (low‐quality evidence) and one olopatadine (moderate‐quality evidence) comparison. For all other comparisons, differences were non‐significant, or we could extract no data.
Overall completeness and applicability of evidence
Applicability of the evidence described in this review may be restricted by a variety of factors.
Most studies considered in this review were conducted in high‐income countries; thus, the generalisability of study findings may need to be considered in light of socioeconomic issues related to education or access to health care.
Even within the scope of socioeconomically restricted populations, it is not clear whether study findings can be generalised to all groups within the population from which the specific sample was drawn. Little information on sociodemographic features was available from most included studies, and very rarely did study authors attempt to establish that no differences in these features were apparent between groups before commencement of the intervention. However, compliance with therapeutic regimens or adherence to medication is, for instance, associated with sociodemographic variables such as old age, male gender, low education level, physical and mental status, and health literacy (Jin 2016). Health behaviour in general is also affected by sociodemographic and socioeconomic variables (Wang 2011).
Populations from which samples were drawn may have differed across studies with regard to diagnostic characteristics, as a standardised approach to obtaining the diagnosis of eczema based on established criteria such as the Hanifin and Rajka definitions or the UK modification was not evident in all studies (Hanifin 1980; Williams 1994). All trials were conducted in secondary settings. However, a large proportion of trials recruited their sample from within the setting of university hospitals. These are not distributed evenly across countries, and it is likely that patients with more severe cases of eczema may attend clinics there. Thus, these trials may have investigated samples with different clinical features compared to samples recruited in dermatology or general practitioner practices. Further, most trials appear to have used convenience samples rather than random samples.
Although trials have studied many interventions (such as acrivastine, azelastine, cetirizine, chlorpheniramine, chlorpheniramine maleate, fexofenadine, hydroxyzine, ketotifen, levocetirizine, loratadine, olopatadine, tazifylline LN2974, and terfenadine) as oral add‐on treatments for eczema, other H1 AH have not been investigated as add‐on eczema treatments. For instance, ebastine, rupatadine, desloratadine, diphenhydramine, dimetindene, clemastine, cyproheptadine, mizolastine, and promethazine are H1 AH that are used in clinical practice for treatment of eczema, although their efficacy and safety have not yet been examined.
Duration of treatment in the included studies ranged between three days and 18 months, which allowed us to assess the effects of H1 AH over a variety of time frames. However, for many specific H1 AH, only one study considered the effect of the specific H1 AH medication and dose, but for others, such as loratadine and cetirizine, several studies provided data over a variety of time frames. Overall, the duration of treatment may have been insufficient in some studies to establish treatment effects, especially for long‐term eczema control. The quality of most evidence was low, so caution should be applied when attempting to generalise observed effects.
Conclusions drawn from these studies should apply only to short‐term relief and consideration of potential adverse events, respectively, in this short time frame or observational period.
Many studies were conducted in small samples, thus reducing the statistical power associated with the likelihood of increased type II error, and some used a cross‐over design with sometimes undefined washout periods. Although we would have liked to conduct meta‐analyses to arrive at effect sizes over many studies, it was not possible to do so, as researchers used 13 different H1 AH and applied widely varying dosages and intervention durations.
Most included studies assessed our primary outcomes of interest well but did not evaluate our secondary outcomes as well: less than half of the included studies measured the number of eczema flares, and only one study measured ‘mean change in quality of life, as measured by a standardised or validated quality of life measure'.
Although we had planned to analyse cross‐over data adequately by accounting for the correlation of data points measured within participants, lack of respective coefficients in the publication and our inability to contact study authors meant that we could provide only narrative accounts of these results.
Additionally, we had intended to pursue the study of H1 AH interventions in patients with eczema with allergic comorbidity as assessed by allergic sensitisations (known as "atopic" eczema). However, no data were available, and we were not able to do so.
Quality of the evidence
Our overall judgement of the quality of the body of evidence based on the GRADE approach was low or very low (Higgins 2011). In a few instances, we believed a moderate judgement was merited. We felt that downgrading was necessary most often because of incomplete outcome data, selection bias, unclear judgement of several or all other risk of bias domains, or serious imprecision. These reasons in addition to the details provided below may impact the quality of the evidence and must be considered when conclusions are drawn on the basis of study findings considered in this review.
Limitations in study design and implementation
Assessment of risk of bias according to Cochrane's 'Risk of bias' tool showed that no single study was at low risk of bias across all domains (Figure 3; Figure 4). Most studies failed to provide sufficient information to allow a comprehensive assessment of the seven domains of risk of bias. Hence, we judged that many studies were at unclear risk of bias for most or all domains. We judged only a few studies to be at low risk of bias for one or more domains. We rated five (25%) included studies as having high risk of bias for at least one domain. In two instances, this referred to incomplete outcome data (attrition bias), in two others, to selection bias, and once, to selective outcome reporting (reporting bias). For all other studies, we judged at least one domain to be at unclear risk of bias. Across studies, we obtained the information collated in this review from studies with overall high risk of bias (25%) or overall unclear risk of bias (75%).
With regard to general quality of the experimental design, further problems arose. No single study reported that a manipulation check had been carried out (e.g. blood samples to check whether the experimental group took H1 AH or used other means to observe compliance).
Another issue deserving discussion revolves around the question of whether intervention and placebo groups were balanced with regard to important clinical and sociodemographic characteristics. Although several study investigators analysed their study data to demonstrate that groups did not differ at baseline, this could not be taken as evidence that allocation bias was absent. First of all, the variables used to analyse any differences were often based on very basic sociodemographic data ‐ not on relevant clinical variables ‐ or study authors reported no differences but did not provide data, or it is not clear on what variable(s) the comparison was based.
Inconsistency of results
Most of the comparisons included in this review were performed in single studies, which did not allow us to assess consistency of results across studies for different H1 AH treatments. When several single studies investigated the same H1 AH, the duration, the dosage, or the population differed. Differences also occurred with regard to concomitant topical treatment. This heterogeneity meant that we were unable to conduct any meta‐analyses.
Indirectness of evidence
In addition, the quality of outcome measures cannot be said to be of high quality across trials. Some measures such as the SCORAD have been used widely and have undergone more rigorous attempts to establish their measurement quality than others (Gerbens 2017; Rehal 2011; Schmitt 2013).
Nonetheless, the quality of several of the other measures summarised under secondary outcome 1 ('Mean change in physician‐assessed clinical signs') remains questionable, as study authors failed to provide information on their measurement properties.
Researchers often use the visual analogue scale (VAS) to assess pruritus (Pereira 2017). However, little is known about its measurement properties, particularly responsiveness, when used in trials of patients with eczema because validation studies have not been conducted or measurement properties remain unclear (Gerbens 2017). In the most comprehensive systematic review evaluating the measurement properties of symptom measurement instruments for eczema, review authors put the VAS into category D (A highest, D lowest). Category D meant that the VAS has (almost) not been validated at all (Gerbens 2017). Kido‐Nakahara 2015 was referred to as providing limited evidence for indeterminate construct validity of VAS pruritus.
Nine of the 23 included studies providing a measure of pruritus utilised a VAS. The remainder used a variety of measures, most of which failed to report on their measurement properties.
Neither was a standardised assessment of eczema control (Secondary outcome 3. 'Number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'') used in any of the studies considered in this review. International consensus suggests that long‐term control of eczema flares should be included as a core outcome domain in future eczema trials (Schmitt 2012). However, it is unclear how this should be defined and measured (Langan 2006; Langan 2014); included studies varied in terms of individual assessment.
In summary, the validity of outcome measures continues to be an important consideration for the present evidence base, given the variety of tools utilised and their unclear quality. Although many studies reported the validity of their measurement tools, many others did not.
Imprecision of results
Many studies were conducted with small samples, thus reducing the statistical power associated with increased likelihood of type II error; some used a cross‐over design with sometimes undefined washout periods.
Publication bias
Publication bias was difficult to assess because few published studies have assessed the same H1 AH, the same dosage, similar treatment duration, and comparable concomitant treatment. However, because we were also able to include the results of unpublished studies, we were able to minimise any existent publication bias. We remain uncertain about whether this review was able to include all data that have been accumulated so far.
Potential biases in the review process
Studies that met our criteria for inclusion in this review were carried out all over the world. We believe we have searched exhaustively and comprehensively and have identified studies conducted in geographically diverse populations, including those from the USA, Australia, South Africa, several European countries, Central America, and Japan. We also searched abstracts from the International Research Workshops on eczema (George Rajka International Symposia on Atopic Dermatitis (ISAD)) and conference proceedings of the European Academy of Dermatology and Venereology (EADV) from 2000 to 2017, the European Academy of Allergy and Clinical Immunology (EAACI) from 2010 to 2015, and the American Academy of Dermatology (AAD) from 2012 to 2015, to identify further potentially relevant RCTs. We searched for reports on clinical trials in progress and for data from completed but unpublished clinical trials, and we checked (handsearched) the reference sections of included and excluded studies for further relevant trials. Our ongoing trials search yielded unpublished studies ‐ Jung 1989 and Cambazard 2001 ‐ that had not been included in previous systematic reviews on the topic ‐ Hoare 2000 and Klein 1999 ‐ nor were these detected in a comprehensive literature review that occurred before these reviews (Sidbury 2014). We did all of this to minimise the risk of potential publication bias in this review.
We included only randomised trials; thus addressing all possible outcomes that may have been investigated in uncontrolled trials was not possible within the constraints of this review. Choice of outcomes was based on both clinical meaningfulness and the core outcome set consented to by the Harmonising Outcome Measures in Eczema (HOME) initiative (clinician‐reported signs, patient‐reported symptoms, quality of life, and long‐term control) (Schmitt 2012; Schmitt 2014). Selection of RCTs for inclusion, however, was not based on prespecified outcomes, but rather on types of studies, participants, and interventions.
Although we assessed risk of bias for all studies, it was often impossible to determine whether studies were at high or low risk of bias due to insufficient reporting of information necessary for the respective risk of bias judgement. Contacting study authors for additional information proved difficult, as all contacted authors failed to respond to our queries. Hence, we judged a large number of studies in this review as having unclear risk of bias for many, sometimes even all, domains.
Agreements and disagreements with other studies or reviews
We identified several literature reviews and systematic reviews (Hoare 2000; Klein 1999; Sidbury 2014), and our findings are in concordance with them. However, although one review stated that the efficacy of antihistamines remains to be adequately investigated (Klein 1999), our recommendations are slightly different (see Implications for practice). We found that there is now a significant body of clinical trial evidence on this topic, and we concluded that trials of antihistamines were unable to demonstrate a clear benefit of the intervention. A recent study analysing secondary data concluded that H1 AH, although widely prescribed, especially by non‐dermatologists, are not effective in reducing pruritus in eczema (He 2018). These findings are consistent with the findings of this review.
Authors' conclusions
Implications for practice.
We found little conclusive evidence to establish the effectiveness of H1 AH as add‐on therapy to topical treatment for eczema. We derived this conclusion from careful consideration of the quality of included trials and their reporting, as well as the wide variation in comparisons and the fact that we found no opportunities to combine results in meta‐analyses.
Details of the specific dose and regimen of the interventions described in this section are found in Effects of interventions. All interventions in this section were compared against placebo.
With regard to the primary outcome ‘Patient‐assessed symptoms’, which we narrow here specifically to participant‐assessed pruritus, we found no evidence that cetirizine 10 mg/d or loratadine makes a difference in this outcome (both low‐quality evidence). Fexofenadine probably slightly improves this outcome (moderate‐quality evidence), but the clinical meaning of this slight improvement is uncertain. Trials assessing cetirizine 0.5 mg/kg/d did not report this outcome.
Fexofenadine probably makes little or no difference in the occurrence of adverse effects, and cetirizine 0.5 mg/kg/d is probably associated with fewer adverse events (both moderate‐quality evidence). We found no evidence that cetirizine 10 mg or loratadine makes a difference in this outcome (both low‐quality evidence).
In terms of physician‐assessed clinical signs, we found no evidence that loratadine or cetirizine 10 mg/d or 0.5 mg/kg/d makes a difference (low‐quality evidence). Fexofenadine probably improves this outcome (moderate‐quality evidence), but the clinical meaning of this improvement is uncertain.
Fexofenadine probably makes little or no difference in terms of the number of eczema flares (measured by ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’) (moderate‐quality evidence). We found no evidence that cetirizine 10 mg makes a difference in the amount of treatment required (low‐quality evidence), but less cetirizine 0.5 mg/kg/d is probably needed, hence reducing the number of eczema flares (moderate‐quality evidence). Trials assessing loratadine did not report this outcome.
We could draw no conclusions about effects on quality of life as only one study assessed this outcome. The data provided by this single study were insufficient for statistical analysis.
Implications for research.
One general implication of this review is that some uncertainty remains with regard to the question of whether H1 AH are effective in the treatment of eczema. Further, review findings are not robust. In light of these limitations, little evidence from the 25 included trials suggests that H1 AH have a clinically significant impact on eczema.
As the quality of evidence was often low and our risk of bias assessment arrived frequently at a judgement of unclear, we would like to recommend several methodological requirements for future studies, which should be randomised controlled trials. First, research would benefit from a clear definition of the condition examined, including its course and severity. In intervention studies, it is important to know who exactly was studied, which is why a standardised assessment should be carried out and appropriately reported. Many of the reviewed studies failed to provide this important information. Other relevant baseline data should be obtained and reported. For instance, a clear sociodemographic description should be provided. In our review, most studies failed to do so.
Second, we often noted insufficient detail on how randomisation was implemented, how allocation concealment was achieved, and how incomplete outcome data (attrition) were dealt with. Adherence to guidelines, such as the CONSORT (Consolidated Standards of Reporting Trials) statement (Moher 1998), would help in ensuring complete reporting. For example, to safeguard against incomplete outcome data, investigators need to routinely anticipate attrition and incorporate various measures to minimise dropouts during trial design (Hui 2013). Study participants should be well informed, and expectations with respect to study procedures should be realistic (Matsui 2012). Should substantial attrition occur, it is of paramount importance to assess whether attrition may have affected any effect estimates (Hui 2013). To ensure that intervention and placebo groups are balanced with regard to important clinical and demographic variables, investigators should employ and report adequate randomisation procedures and should check the distribution of these variables between groups.
Third, future research would benefit from a standardised assessment of outcomes. Guidance for choice of reliable and valid outcome measures may be obtained from the HOME initiative (Schmitt 2014). Future availability of more trials using the same standardised outcome measurement, similar intervention periods, and comparable dosages may allow pooling of data ‐ something we could not do. We also recommend that future studies should be conducted in a multi‐arm fashion with regard to dosages, such that several different dosages of the same H1 AH are compared against placebo. Further, concomitant treatment should be applied in a standardised way. For instance, studies should mimic clinical practice in accordance with existing guidelines, consisting of moisturisers and/or emollients in combination with either corticosteroids or other immunomodulatory agents (such as tacrolimus or pimecrolimus) (Werfel 2016). Additionally, we suggest that H1 AH interventions should be studied in patients with eczema with allergic comorbidity as assessed by allergic sensitisations (known as 'atopic' eczema).
Several of the included trials studied small or very small samples. As a consequence, we downgraded the quality of the evidence from such trials. Future studies should state what effect is considered clinically useful and should base the sample size calculation on this effect to achieve adequate power. This would greatly enhance confidence in effect estimates.
An important issue pertains to the generalisability of reported findings. Most studies appear to have based their selection rationale on easy availability of potential participants in clinical settings. In other words, the samples can be said to be convenience samples ‐ a fact that greatly limits the extent to which findings can be generalised to the population of patients with eczema on the whole. Authors of high‐quality intervention studies should design their trials to maximise the generalisability of study findings.
Moreover, although the duration of the intervention and of follow‐up was as long as 18 months for cetirizine and levocetirizine, up to 52 weeks for ketotifen, or eight weeks for olopatadine, only short‐ or medium‐term interventions and follow‐ups were available for acrivastine, azelastine, chlorpheniramine, fexofenadine, hydroxyzine, loratadine, terfenadine, or tazifylline. Future research should ensure a sufficient duration of the intervention to assess impact on eczema severity.
However, as current guidelines recommend the use of H1 AH as add‐on to conventional topical treatment during acute flares only (Werfel 2016), future studies could greatly benefit from comparing H1 add‐on treatment in groups of patients with acute flares that are comparable.
Only one study assessed quality of life, but the data it provided were insufficient for statistical analysis. It must be borne in mind though that many studies were conducted at a time when health‐related patient‐reported outcomes were given minor credence compared to today. It is recommended that future trials should include one such measure, in line with the consented core outcome set of the HOME initiative (Heinl 2016; Heinl 2017; Schmitt 2012).
There is scope for replication studies. For instance, olopatadine and fexofenadine were found to have positive effects, albeit of little clinical significance. Further confirmatory trials may be informative. There is also scope for investigating other H1 AH as oral add‐on interventions for eczema. For instance, other H1 AH, such as ebastine, rupatadine, desloratadine, diphenhydramine, dimetindene, clemastine, cyproheptadine, mizolastine, and promethazine, are used in clinical practice for the treatment of eczema. However, the efficacy and safety of these agents have not yet been studied in clinical trials.
Should future studies establish more robust evidence for specific H1 AH treatment effects on eczema and adequate safety, subsequent trials could try to compare H1 AH among each other.
In light of recent findings that not only histamine‐related pathways lead to mediation of pruritus (Ständer 2002; Tominaga 2016; Yosipovitch 2008), it may be worthwhile to study combination treatments in the future.
Acknowledgements
The review authors wish to thank the Cochrane Skin editorial base and the peer referees for their support and advice during preparation of this review.
We greatly appreciate the help of Hsin‐Wen Wu, Toshiya Ebata, Masaki Futamura, Anneke Steens, Michal Gina, Ángela Merchán, and Stanislav Iakhno, in assessing study eligibility and extracting data from articles published in languages other than English or German.
We acknowledge the support of the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and the Maudsley NHS Foundation Trust and King’s College London.
The Cochrane Skin editorial base wishes to thank Bob Boyle, who was the Cochrane Dermatology Editor for this review; Matthew Grainge and Ching‐Chi Chi, who were the Statistical and Methods Editors, respectively; the clinical referees, Gil Yosipovitch and Hywel Williams; and the consumer referee, Amanda Roberts. We would also like to thank Dolores P Matthews for copy‐editing the review.
Disclaimer
The research underlying this report was funded by the German Federal Ministry of Education and Research (file number FKG 01KG1401). The review authors are responsible for the content of this review.
This project was supported by the National Institute for Health Research (NIHR) via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, the National Health Service (NHS), or the UK Department of Health.
Appendices
Appendix 1. Cochrane Skin Group Specialised Register (CRS) search strategy
((atopic and dermatitis) or eczema or (atopic and ekzema) or neurodermatitis) AND ((histamine and antagonist*) or (H1 and histamine and antagonist*) or (H‐1 and histamine and antagonist*) or ethanolamine or diphenhydramine or clemastine or dimenhydrinate or doxylamine or piperazine or hydroxyzine or hydroxyzine or meclizine or cyclizine or chlorcyclizine or phenothiazine or chlorpromazine or promethazine or trimeprazine or ethylenediamine or mepyramine or tripelenamine or alkylamine or brompheniramine or chlorpheniramine or carbinoxamine or teldane or terfenadine or astemizole or hismanal or loratadine or mequitazine or mebhydroline or semprex or fexofenadine or telfast or mizolastine or mizollen or azatadine or cyproheptadine or atosil or norastemizole or dimetapp or tavist or dramamine or benadryl or atarax or vistaril or actifed or thorazine or dimetane or zyrtec or cetirizine or periactin or claritine or seldane or allegra or desloratadine or aerius or urtimed or rupatadine or acrivastine or promethazine or chlorpromazine or cyproheptadine)
Appendix 2. CENTRAL (the Cochrane Library) search strategy
#1 atopic dermatitis:ti,ab,kw #2 eczema:ti,ab,kw #3 neurodermatitis:ti,ab,kw or atopic ekzema:ti,ab,kw #4 MeSH descriptor: [Dermatitis, Atopic] this term only #5 MeSH descriptor: [Neurodermatitis] this term only #6 MeSH descriptor: [Eczema] this term only #7 {or #1‐#6} #8 histamine H1 antagonist*:ti,ab,kw or histamine antagonist*:ti,ab,kw #9 MeSH descriptor: [Histamine H1 Antagonists] explode all trees #10 (ethanoliamine or diphenhydramine or clemastine or dimenhydrinate or doxylamine or piperazine or hydroxizine or hydroxyzine or meclizine or cyclizine or chlorcyclizine or phenothiazine or chlorpromazine or promethazine or trimeprazine or ethylenediamine or mepyramine or tripelenamine or alkylamine or brompheniramine or chlorpheniramine or carbinoxamine or teldane or terfenadine or astemizole or hismanal or loratidine or mequitazine or mebhydroline or semprex or fexofenadine or telfast or mizolastine or mizollen or azatadine or cyproheptadine or atosil or norastemizole or dimetapp or tavist or dramamine or benadryl or atarax or vistaril or actifed or thorazine or dimetane or zyrtec or cetirizine or periactin or claritine or seldane or allegra or desloratadine or aerius or urtimed or rupatadine or acrivastine or promethazine or chlorpromazine or cyproheptadine):ti,ab,kw #11 [mh Ethanolamine] or [mh Diphenhydramine] or [mh Clemastine] or [mh Dimenhydrinate] or [mh Doxylamine] or [mh Piperazines] or [mh Meclizine] or [mh Cyclizine] or [mh Phenothiazines] or [mh Chlorpromazine] or [mh Promethazine] or [mh Trimeprazine] or [mh Ethylenediamines] or [mh Pyrilamine] or [mh Brompheniramine] or [mh Chlorpheniramine] or [mh Terfenadine] or [mh Astemizole] or [mh Loratadine] or [mh Cyproheptadine] or [mh Cetirizine] #12 {or #8‐#11} #13 #7 and #12
Appendix 3. MEDLINE (Ovid) search strategy
1. atopic dermatitis.mp. 2. exp Dermatitis, Atopic/ 3. neurodermatitis.mp. 4. exp Neurodermatitis/ 5. exp Eczema/ 6. atopic ekzema.mp. 7. eczema.mp. 8. or/1‐7 9. histamine H1 antagonist$.mp. 10. exp Histamine H1 Antagonists/ 11. histamine antagonists/ 12. exp Ethanolamine/ or ethanolamine.mp. 13. diphenhydramine.mp. or exp Diphenhydramine/ 14. clemastine.mp. or exp Clemastine/ 15. dimenhydrinate.mp. or exp Dimenhydrinate/ 16. doxylamine.mp. or exp Doxylamine/ 17. piperazine$.mp. or exp Piperazines/ 18. (hydroxizine or hydroxyzine).mp. 19. meclizine.mp. or exp Meclizine/ 20. cyclizine.mp. or exp Cyclizine/ 21. chlorcyclizine.mp. 22. phenothiazine$.mp. or exp Phenothiazines/ 23. chlorpromazine.mp. or exp Chlorpromazine/ 24. promethazine.mp. or exp Promethazine/ 25. trimeprazine.mp. or exp Trimeprazine/ 26. ethylenediamine$.mp. or exp Ethylenediamines/ 27. mepyramine.mp. or exp Pyrilamine/ 28. tripelenamine.mp. 29. alkylamine.mp. 30. brompheniramine.mp. or exp Brompheniramine/ 31. chlorpheniramine.mp. or exp Chlorpheniramine/ 32. carbinoxamine.mp. 33. (teldane or terfenadine).mp. 34. exp Terfenadine/ 35. astemizole.mp. 36. exp Astemizole/ 37. hismanal.mp. 38. loratadine.mp. 39. exp Loratadine/ 40. mequitazine.mp. 41. mebhydroline.mp. 42. semprex.mp. 43. fexofenadine.mp. 44. telfast.mp. 45. mizolastine.mp. 46. mizollen.mp. 47. azatadine.mp. 48. cyproheptadine.mp. 49. exp Cyproheptadine/ 50. atosil.mp. 51. norastemizole.mp. 52. dimetapp.mp. 53. tavist.mp. 54. dramamine.mp. 55. benadryl.mp. 56. atarax.mp. 57. vistaril.mp. 58. actifed.mp. 59. thorazine.mp. 60. dimetane.mp. 61. (zyrtec or cetirizine).mp. 62. exp Cetirizine/ 63. periactin.mp. 64. claritine.mp. 65. seldane.mp. 66. allegra.mp. 67. desloratadine.mp. 68. aerius.mp. 69. urtimed.mp. 70. rupatadine.mp. 71. or/9‐70 72. randomized controlled trial.pt. 73. controlled clinical trial.pt. 74. randomized.ab. 75. placebo.ab. 76. clinical trials as topic.sh. 77. randomly.ab. 78. trial.ti. 79. 72 or 73 or 74 or 75 or 76 or 77 or 78 80. exp animals/ not humans.sh. 81. 79 not 80 82. 8 and 71 and 81
[Lines 72‐81: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision)]
Appendix 4. Embase (Ovid) search strategy
1. exp ECZEMA/ 2. eczema.ti,ab. 3. exp atopic dermatitis/ 4. exp NEURODERMATITIS/ 5. neurodermatitis.ti,ab. 6. atopic dermatitis.mp. 7. atopic ekzema.mp. 8. or/1‐7 9. exp histamine H1 receptor antagonist/ 10. exp antihistaminic agent/ 11. exp diphenhydramine/ or diphenhydramine.mp. 12. ethanolamine.mp. or exp ethanolamine/ 13. histamine H1 antagonist$.mp. 14. clemastine.mp. or exp clemastine/ 15. dimenhydrinate.mp. or exp dimenhydrinate/ 16. doxylamine.mp. or exp doxylamine/ 17. exp piperazine/ 18. piperazine$.mp. 19. (hydroxizine or hydroxyzine).mp. 20. meclizine.mp. or exp meclozine/ 21. exp cyclizine/ or cyclizine.mp. 22. exp chlorcyclizine/ or chlorcyclizine.mp. 23. exp phenothiazine/ 24. Phenothiazine$.mp. 25. exp chlorpromazine/ or chlorpromazine.mp. 26. exp promethazine/ or promethazine.mp. 27. trimeprazine.mp. or exp alimemazine/ 28. exp ethylenediamine/ 29. Ethylenediamine$.mp. 30. exp mepyramine/ or mepyramine.mp. 31. Pyrilamine.mp. 32. tripelenamine.mp. or exp tripelennamine/ 33. alkylamine.mp. or exp aliphatic amine/ 34. exp brompheniramine/ or brompheniramine.mp. 35. exp chlorpheniramine/ or chlorpheniramine.mp. 36. carbinoxamine.mp. or exp carbinoxamine/ 37. (teldane or terfenadine).mp. 38. exp terfenadine/ 39. astemizole.mp. or exp astemizole/ 40. hismanal.mp. 41. loratadine.mp. or exp loratadine/ 42. mequitazine.mp. or exp mequitazine/ 43. mebhydroline.mp. or exp mebhydrolin/ 44. semprex.mp. or exp acrivastine/ 45. exp fexofenadine/ or fexofenadine.mp. 46. telfast.mp. 47. mizolastine.mp. or exp mizolastine/ 48. mizollen.mp. 49. exp azatadine/ or azatadine.mp. 50. exp cyproheptadine/ or cyproheptadine.mp. 51. atosil.mp. or exp promethazine/ 52. norastemizole.mp. or exp norastemizole/ 53. dimetapp.mp. or exp dimetapp/ 54. tavist.mp. or exp clemastine fumarate/ 55. dramamine.mp. 56. benadryl.mp. 57. atarax.mp. 58. exp hydroxyzine/ 59. vistaril.mp. or exp hydroxyzine embonate/ 60. actifed.mp. or exp pseudoephedrine plus triprolidine/ 61. thorazine.mp. or exp chlorpromazine/ 62. dimetane.mp. 63. (zyrtec or cetirizine).mp. 64. exp cetirizine/ 65. periactin.mp. or exp cyproheptadine/ 66. claritine.mp. 67. seldane.mp. 68. allegra.mp. 69. exp desloratadine/ or desloratadine.mp. 70. aerius.mp. 71. urtimed.mp. 72. rupatadine.mp. or exp rupatadine/ 73. or/9‐72 74. crossover procedure.sh. 75. double‐blind procedure.sh. 76. single‐blind procedure.sh. 77. (crossover$ or cross over$).tw. 78. placebo$.tw. 79. (doubl$ adj blind$).tw. 80. allocat$.tw. 81. trial.ti. 82. randomized controlled trial.sh. 83. random$.tw. 84. or/74‐83 85. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 86. human/ or normal human/ 87. 85 and 86 88. 85 not 87 89. 84 not 88 90. 8 and 73 and 89
Data and analyses
Comparison 1. Acrivastine 24 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus 7 days after baseline adjusted for baseline score as covariate | 1 | 23 | Mean Difference (IV, Random, 95% CI) | ‐11.40 [‐29.44, 6.64] |
| 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment helped itching | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [1.12, 6.49] |
Comparison 2. Azelastine 4 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) 2 weeks after baseline | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.64, 4.82] |
Comparison 3. Cetirizine 5 and 10 mg/d versus placebo (1‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Cetirizine 5 mg/d | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.11, 3.27] |
| 1.2 Cetirizine 10 mg/d | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.18, 3.70] |
Comparison 4. Cetirizine 10 mg versus placebo (2‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Secondary outcome 1. Physician‐assessed clinical signs: mean overall response rate (number of participants who responded to treatment) 2 weeks after baseline | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.17, 2.80] |
Comparison 5. Cetirizine 10, 20, 40 mg/d versus placebo (4‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 10 mg/d vs placebo | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.50, 2.45] |
| 1.2 20 mg/d vs placebo | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.70, 3.00] |
| 1.3 40 mg/d vs placebo | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 2.38 [1.25, 4.53] |
Comparison 6. Cetirizine 0.5 mg/d per kg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period | 1 | 795 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.46, 1.01] |
Comparison 7. Cetirizine 5 mg/d for children < 30 kg and 10 mg/d for children > 30 kg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Secondary outcome 3. Use of concomitant therapy (disodium cromoglycate, procaterol, steroids) | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.06, 0.80] |
Comparison 8. Chlorpheniramine maleate BP (2 to 4 mg/d (age dependent) or twice that amount) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Proportion of participants reporting adverse effects and serious adverse events throughout the study period | 1 | 151 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.49, 1.82] |
| 2 Secondary outcome 3. Amount of 1% hydrocortisone in grams used | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 days 1 to 29 ages 1 to 5 years | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐5.96, 3.36] |
| 2.2 days 1 to 29 ages 6 to 12 years | 1 | 90 | Mean Difference (IV, Random, 95% CI) | 1.60 [‐2.53, 5.73] |
Comparison 9. Fexofenadine 120 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: mean pruritus difference (0 to 8) between baseline and night of day 6 | 1 | 400 | Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.43, ‐0.07] |
| 2 Primary outcome 2. Adverse events | 1 | 411 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.74, 1.50] |
Comparison 10. Hydroxyzine 75 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Patient‐assessed symptoms: global evaluation of the antipruritic effect of treatment: number of participants who reported a marked or complete response | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.58, 5.94] |
Comparison 11. Ketotifen 0.8 to 1.2 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Adverse events | 1 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 10.82 [0.61, 191.53] |
Comparison 12. Levocetirizine 5 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: mean difference in VAS pruritus between baseline and 2 and 4 weeks after randomisation | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 2 weeks after randomisation | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐0.79 [‐3.54, 1.96] |
| 1.2 4 weeks after randomisation | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐0.59 [‐3.46, 2.28] |
Comparison 13. Levocetirizine 0.25 mg/kg/d.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 One or more adverse events | 1 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.98, 1.05] |
| 1.2 Treatment‐attributed adverse events | 1 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.40, 1.65] |
| 1.3 Serious adverse events | 1 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.54, 1.31] |
| 1.4 Treatment‐attributed serious adverse events | 1 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
| 1.5 Adverse events that led to discontinuation | 1 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.40, 6.90] |
Comparison 14. Loratadine 5 mg/d versus placebo (2‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13) | 1 | 72 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐2.30, 0.10] |
| 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation | 1 | 74 | Risk Ratio (M‐H, Random, 95% CI) | 1.58 [0.73, 3.42] |
Comparison 15. Loratadine 10 mg/d versus placebo (2‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13) | 1 | 71 | Mean Difference (IV, Random, 95% CI) | ‐0.96 [‐2.01, 0.09] |
| 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good 13 days after randomisation | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.99, 4.20] |
Comparison 16. Loratadine 20 mg/d versus placebo (2‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus (difference in pruritus sum between day 0 and day 13) | 1 | 72 | Mean Difference (IV, Random, 95% CI) | ‐1.41 [‐2.49, ‐0.33] |
| 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment success was judged as good or very good | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.99, 4.20] |
Comparison 17. Loratadine 10 mg/d versus placebo (1‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Global evaluation of the antipruritic effect of treatment: number of participants who reported a marked or complete response | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.79 [0.96, 8.09] |
Comparison 18. Loratadine 10 mg/d versus no additional treatment (4‐week intervention).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Patient‐assessed symptoms: VAS pruritus 4 weeks after randomisation | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐2.30 [‐20.27, 15.67] |
| 2 Primary outcome 2. Adverse events | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.01, 5.76] |
| 3 Secondary outcome 1. Physician‐assessed clinical signs: SCORAD after 4 weeks | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐4.10 [‐13.22, 5.02] |
Comparison 19. Olopatadine 10 mg/d versus no additional treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 2. Adverse events 8 weeks after randomisation | 1 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 9.18 [0.51, 166.10] |
Comparison 20. Terfenadine 180 mg/d versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome 1. Mean change in patient‐assessed symptoms: VAS pruritus 7 days after baseline adjusted for baseline score as covariate | 1 | 27 | Mean Difference (IV, Random, 95% CI) | ‐15.2 [‐33.03, 2.63] |
| 2 Secondary outcome 1. Physician‐assessed clinical signs: number of participants for whom treatment helped itching (1 week after randomisation) | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 2.19 [0.88, 5.44] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Berth Jones 1989.
| Methods |
Study design: randomised controlled trial Study grouping: cross‐over |
|
| Participants |
Baseline characteristics Terfenadine
Placebo
Inclusion criteria: (1) diagnostic: eczema as defined by Hanifin and Rajka (1980); (2) severity of condition: not specified; (3) duration of condition: not specified; (4) site evaluated (e.g. face/back): physician‐assessed over entire body Exclusion criteria: not reported Group differences: cross‐over study |
|
| Interventions |
Intervention characteristics Terfenadine
Placebo
|
|
| Outcomes |
Primary outcome: patient‐assessed symptoms: pruritus VAS (0 to 10)
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
|
|
| Identification |
Sponsorship source: not stated Country: UK Setting: secondary Comments: not specified, presumably outpatient hospital (i.e. secondary care), probably Leicester Royal Infirmary as affiliation of main study author Author's name: Berth‐Jones, John, Dr. Institution: Department of Dermatology, University Hospitals Coventry and Warwickshire NHS Trust Email: johnberthjones@aol.com Address: Department of Dermatology, University Hospitals Coventry and Warwickshire NHS Trust, Coventry CV2 2DX, UK |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: study authors merely state 'in randomised order'. Insufficient information about the sequence generation process to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Although study authors state that they report "the results of a double‐blind, placebo‐controlled" study, whether the placebo was identically matched to the terfenadine tablets remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: apart from stating "double‐blind", information is insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: loss to follow‐up: 4/28; no ITT analysis. Attrition less than 20%, so not substantial |
| Selective reporting (reporting bias) | Low risk | Comment: no comparison possible with protocol, as study protocol is not available, but it is clear that the published report includes all expected outcomes, including those that were prespecified |
| Other bias | Unclear risk | Comment: there may be further risk of bias but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Cambazard 2001.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Cetirizine 0.25; 0.50; 0.75 mg/kg bw/d
Placebo
Total
Inclusion criteria: (1) diagnostic: children, male or female, 1 to 5 years old, who suffer from atopic dermatitis, with written consent to participate in the trial obtained from parents/legal representatives, who satisfy all inclusion/exclusion criteria; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: not reported Group differences: not reported |
|
| Interventions |
Intervention characteristics Cetirizine 3 groups
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: not reported Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs: modified SCORAD
Secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications'
|
|
| Identification |
Sponsorship source: UCB Pharma, Belgium Country: 26 centres: France (10), Russia (8), Germany (4), Belgium (2), The Netherlands (1), United Kingdom (1) Setting: secondary Comments: Author's name: F. Cambazard Institution: not reported Email: not reported Address: Professor F. Cambazard, 42055 Saint‐Etienne, France |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk. Number lost to follow‐up not reported |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided); also, the study was funded in part by UCB, SA (Brussels, Belgium), and study medication was provided by that company |
Diepgen 2002.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Cetirizine
Placebo
Inclusion criteria: (1) diagnostic: AD, infants (1 to 2 years of age), at least 1 parent or sibling with history of AD, allergic rhinitis or asthma; (2) severity of condition: active symptoms; (3) duration of condition: at least 1 month; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: infants with asthma, or with a history (beyond the age of 6 months) of 1 or more episodes of wheezing or nocturnal cough, as well as any conditions that might obscure the diagnosis of asthma (14), were excluded from the study. Further exclusion criteria were the following: weight below the third percentile, chronic pulmonary disease, severe neurological or psychological disorder, any third disease likely to interfere with the study drug, clinically relevant cardiac disease, any anomaly of the Q–T interval on electrocardiogram (ECG) tracing, history of sleep apnoea in the participant or in siblings, neonatal distress, prior desensitisation or immunotherapy, prior treatment with medicines interfering with the immune system, hypersensitivity to cetirizine or other piperazines or parabens, participation in a clinical study within 3 months before randomisation Group differences: groups were comparable at baseline according to study authors |
|
| Interventions |
Intervention characteristics Cetirizine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus as part of SCORAD
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
|
|
| Identification |
Sponsorship source: UCB, SA, Pharma sector (Brussels, Belgium) Country: 12 European countries and Canada Setting: dermatological or paediatric centres in 12 countries (i.e. secondary) Comments: Author's name: Thomas L. Diepgen Institution: University Hospital Heidelberg Email: thomas.diepgen@med.uni‐heidelberg.de Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Informatio about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: incomplete outcome data, as 817 were randomised but ITT population consists of only 795 participants. 12.45% further unaccounted attrition. However, attrition was less than 30% (long‐term trial) so was considered to be low risk |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | There may be further risk of bias, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided); also, the study was funded in part by UCB, SA (Brussels, Belgium), and study medication was provided by that company |
Doherty 1989.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: clinical diagnosis of AD; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: not reported Group differences: not reported |
|
| Interventions |
Intervention characteristics Acrivastine
Terfenadine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus VAS
Primary outcome 2: adverse events
Primary outcome 2: adverse events: number of flares
Secondary outcome 1: physician‐assessed clinical signs: no. for whom treatment helped itching
|
|
| Identification |
Sponsorship source: not reported Country: UK Setting: secondary Comments: UK outpatient hospital Author's name: Valerie Doherty Institution: Royal Infirmary, Edinburgh Email: val.doherty@nhslothian.scot.nhs.uk. Address: Valerie Doherty, Consultant Dermatologist, Royal Infirmary, Edinburgh EH3 9YW |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Apart from stating "double‐blind", information is insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 'as‐treated’ analysis done with departure of the intervention received from that assigned at randomisation. Attrition (10.2%) and no statement of analysis based on ITT principles. Cases with missing data were deleted from analyses. However, attrition was below 20% and medium‐term duration intervention, so considered as low risk |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Falk 1993.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Ketotifen
Placebo
Inclusion criteria: (1) diagnostic: eczema according to Hanifin and Rajka (1980); (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: pregnant women and patients concurrently using drugs or vaccines (hyposensitisation) that could influence the disease Group differences: "...and the two parallel groups were comparable as to age, sex, severity and duration of the disease" |
|
| Interventions |
Intervention characteristics Ketotifen
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
|
|
| Identification |
Sponsorship source: not reported Country: Norway Setting: secondary Comments: likely secondary setting, probably outpatient university hospital Author's name: Falk, Edvard S Institution: Department of Dermatology, University Hospital, Tromso, Norway Email: not reported Address: Professor Dr. med Edvard S. Falk, Hudlegekontoret Akutten,Skippergata 7A, 9008 Tromsø; Tel.: 77 68 52 87 |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In a double blind study...., patients received placebo capsules identical to ketotifen capsules...." Comment: no further information provided about personnel, but it is unlikely that their blinding could have been broken |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis, but as per protocol, dropouts = 4 (2 in each group) due to increased severity during the study. Dropout rate 6.6% in each group, which is fairly low |
| Selective reporting (reporting bias) | Unclear risk | Comment: no comparison between protocol and article possible |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company?) |
Frosch 1984.
| Methods |
Study design: randomised controlled trial Study grouping: cross‐over |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: history of AD of at least 3 years (however, neither according to Hanifin and Rajka nor UK criteria) but study conducted in Department of Dermatology; (2) severity of condition: not specified; (3) duration of condition: at least 3 years; (4) site evaluated (e.g. face/back): not specified Exclusion criteria: treatment with systemic corticosteroids or ACTH within the last 2 months; change in topical medication in the last month; history of very distinct seasonal variations in atopic dermatitis; presence of any non‐atopic dermatitis‐related pathological finding, detected by routine laboratory screening; pregnancy or lactation Group differences: not reported but cross‐over |
|
| Interventions |
Intervention characteristics Chlorpheniramine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: VAS pruritus
Primary outcome 2: adverse events
Secondary outcome 1: global assessment: number of patients with overall improvement, no overall improvement, or overall worsening
Secondary outcome 3: amount of betamethasone used in grams
|
|
| Identification |
Sponsorship source: not reported Country: Germany Setting: secondary Comments: not stated, but likely outpatients treated at hospital Author's name: P.J. Frosch Institution: Department of Dermatology, University of Münster Email: not reported Address: Department of Dermatology, University of Münster, D‐4400 Münster, Federal Republic of Germany |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation was conducted according to a Latin square design" Comment: randomisation procedure was adequate and unlikely to introduce selection bias |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo tablets were identical with intervention 1, and placebo tablets were identical with intervention 2 (i.e. 2 types of placebo), whether study personnel were blinded remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: as‐treated analysis done with departure of the intervention received from that assigned at randomisation (11.11% attrition) |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, Did study authors receive funding from any source?) Statement was made regarding receipt of technical assistance and support from Smith, Kline, and Dauelsberg (Göttingen, FRG) |
Hannuksela 1993.
| Methods |
Study design: randomised controlled trial Study grouping: parallel |
|
| Participants |
Baseline characteristics Overall (not given for groups)
Inclusion criteria: (1) diagnostic: adults with AD (18 years and older); (2) severity of condition: moderate to severe AD needing oral therapy and defined by the presence of pruritus and typical atopic dermatitis morphology and typical cutaneous lesion distribution and chronicity; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: pregnancy, pregnancy potential and lactation, renal or hepatic sufficiency, known allergy to piperazines, atopic dermatitis requiring systemic treatment other than antihistamines, severe infection complicating atopic dermatitis, systemic infection requiring antibacterial treatment, any diseases that may interfere with treatment such as dermal mycoses, use of antidepressants, needing a specific treatment for asthma, starting hyposensitisation, UVA or UVB therapy, use of depot corticosteroids during the last month, antimicrobial drugs during the last 7 days, topical or systemic antihistamines during the last 7 days (exceptions: astemizole, 6 weeks, and ketotifen, 2 weeks) Group differences: all groups were comparable in terms of sex, age, weight, and prior use of emollients and hydrocortisone. They were also comparable at baseline for symptom scores |
|
| Interventions |
Intervention characteristics Cetirizine 3 groups
Placebo
|
|
| Outcomes |
Primary outcome 1a: mean change in patient‐assessed symptoms: pruritus as part of SCORAD
Primary outcome 1b: mean change in patient‐assessed symptoms: pruritus assessed by patient CRF
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
|
|
| Identification |
Sponsorship source: UCB Pharmaceutical Sector, Belgium Country: Finland Setting: secondary Comments: likely outpatients treated at University Hospital Turku and Tampere, Finland Author's name: Matti Hannuksela Institution: The Allergy and Asthma Federation, Helsinki, Finland Email: not reported Address: not reported |
|
| Notes | The direction of change appears reversed; study authors were contacted to clarify the nature of this, but we received no reply | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: study authors state that they had randomised by blocks of 4, but it is unclear what this means. Information about the sequence generation process was insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Low risk | Comment: sealed envelopes were used, so unlikely to introduce selection bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: in a double‐blind study...., "indistinguishable tablets of either cetirizine ...or placebo were administered"...no further information about personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: ‘as‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation'; attrition was 28.7%, and no ITT was stated |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided), although statement was made that study was funded by UCB Pharma. However, whether this may have affected authors' decisions remains unclear |
Henz 1998.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Azelastine
Cetirizine
Placebo
Inclusion criteria: (1) diagnostic: AD accompanied by moderate to severe pruritus (not according to Hanifin and Rajka or UK criteria) but study conducted in department of dermatology; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: not reported Group differences: not reported |
|
| Interventions |
Intervention characteristics Azelastine
Cetirizine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus reduction assessed at night on diary card
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs: mean overall response rate
Secondary outcome 2: secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
|
|
| Identification |
Sponsorship source: not reported Country: Austria and Germany Setting: secondary Comments: 30 centres in Austria and Germany (i.e. outpatient hospitals) Author's name: B.M. Henz Institution: Charite‐Virchow Klinikum Email: henz@prolink.de Address: Prof. Dr. med. Beate M. Henz, Charite‐Virchow Klinikum, Medizinische Fakultiit der Humboldt‐Universität zu Berlin, Dermatologische Universitiitsklinik und Poliklinik Augustenburger Platz 1, D‐13353 Berlin, Germany |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: study authors state that randomisation was done centrally, but it is not clear what this means. Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: study authors state "double‐blind", but whether placebo was identical to azelastine or cetirizine remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. information insufficient to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: as it is not clear how many participants were randomised, we cannot judge whether any attrition bias occurred |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Hjorth 1988.
| Methods |
Study design: randomised controlled trial Study grouping: cross‐over |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: no information given on how atopic dermatitis was diagnosed; (2) severity of condition: not really defined, just “patients with atopic dermatitis whose history indicated contact urticaria”; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: not reported Group differences: not applicable |
|
| Interventions |
Intervention characteristics Terfenadine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
|
|
| Identification |
Sponsorship source: not reported Country: Denmark Setting: secondary Comments: dermatologist's practice Author's name: Niels Hjorth Institution: Department of Dermatology, KAS Gentofte, Copenhagen, Denmark Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: apart from "a double‐blind..." study, only insufficient information to permit judgement of low risk or high risk. Study authors did not declare whether medication was identical in appearance and dosage to placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. insufficient information to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It is stated that 30 participants were randomised, assuming 16/30 reported reduced itching. Assumed an ITT was not carried out |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did authors receive funding from any source?) |
Iikura 1992.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Ketotifen
Placebo
Inclusion criteria: (1) diagnostic: those having more than moderate dermal symptoms of AD in both groups; (2) duration: for at least 1 month; (3) severity: more than moderate dermal symptoms; (4) site evaluated: at least 2 sites on the body Exclusion criteria: history of or displaying recurrent cough and/or wheezing or with condition diagnosed as asthma; cardiac, hepatic, or renal disorders; patients considered unsuitable by the examining physician for some other reason Group differences: study authors report that based on their statistical analyses, there were no significant differences in age, sex. weight, allergic family history, onset of AD, duration of AD, or laboratory tests between groups |
|
| Interventions |
Intervention characteristics Ketotifen
Placebo
|
|
| Outcomes |
Secondary outcome 1: physician‐assessed clinical signs obtained from complete physical examination
Primary outcome 2: adverse events
|
|
| Identification |
Sponsorship source: not reported Country: Brazil and Japan Setting: secondary Comments: Author's name: Yoji Iikura Institution: Department of Allergology, National Pediatric Hospital, Tokyo, Japan Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although study authors stated that "a placebo syrup indistinguishable from the active syrup was administered", how personnel were blinded remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: study authors report to have studied 121 children but report data for only 91 in their final evaluation. No information is given as to how these missing data came about or how they were treated, respectively. Although attrition was 25%, this was a long‐term study so was considered to be at low risk |
| Selective reporting (reporting bias) | Unclear risk | Information was insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Jung 1989.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Cetirizine 5 mg/d
Cetirizine 10 mg/d
Placebo
Total
Inclusion criteria: (1) diagnostic: atopic dermatitis with the following symptoms: pruritus or a burning sensation; morphology and distribution of skin lesions typical for children over 2 years of age (i.e. occurring at the flexure of the arms and legs, nape and sides of the neck, retro‐auricular groove, hands, and perioral area). A minimum score of 4 for the 2 main symptoms ‐ pruritus and erythema ‐ and of 6 for the symptoms considered as a whole, both main and accessory (i.e. vesicles, lichenification, and crusting required for inclusion in the study). This score was obtained based on a 5‐point scale where 0 = “none” to 4 = “very severe”, for the main symptoms, and on a 2‐point scale, where 0 = “none” and 1 = “present”, for the accessory symptoms; (2) severity of condition: chronic or chronically relapsing condition; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): see above Exclusion criteria: children with a non‐atopic dermatitis or dermatitis so severe that recourse to systemic therapy with a drug other than an antihistamine was necessary; all generalised or local (e.g. impetigo, herpes) infection requiring antibiotics; known allergy to piperazines; chronic renal, hepatic cardiovascular, or haematological problems Group differences: comparison of groups at the time of the start of the study did not reveal any statistically significant differences regarding age (…), weight (…), height or sex distribution |
|
| Interventions |
Intervention characteristics Cetirizine 5 mg/d
Cetirizine 10 mg/d
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus measured on 5‐point Likert scale (0 = none, 4 = severe)
Primary outcome 2: adverse events (number of patient‐reported adverse events)
Secondary outcome 1: physician‐assessed clinical signs: global evaluation measured on 5‐point Likert scale (0 = none, 4 = severe)
|
|
| Identification |
Sponsorship source: UCB Pharmaceutical Sector Country: Germany, Belgium, and France Setting: secondary Comments: multi‐centre study Author's name: Prof. E. G. Jung Institution: not reported Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were allocated, in each centre, according to a randomisation list to one of the 3 following treatment groups..." Comment: no other information available |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: although study authors report no missing outcome data, whether all randomised participants contributed to data collection remains unclear |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol could not be obtained |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided); also, the study was funded in part by UCB, SA (Brussels, Belgium), and study medication was provided by that company |
Kawashima 2003.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Fexofenadine
Placebo
Total
Inclusion criteria: (1) diagnostic: male and female outpatients aged 16 years or older who had a diagnosis of atopic dermatitis based on the criteria of the Japanese Dermatological Association (similar to those of Hanifin and Rajka); (2) severity of condition: self‐assessed mean pruritus score ≥ 4 points and < 8 points for the last 3 days of the placebo lead‐in period (baseline); (3) duration of condition: not specified; (4) site evaluated (e.g. face/back): not specified Exclusion criteria: using a topical steroid preparation other than 0.1% hydrocortisone butyrate twice daily within the 1‐week placebo lead‐in period; had taken antiallergic agents, antihistamines, non‐steroidal anti‐inflammatory agents, gamma‐globulin preparations, anticholinergics, tranquillisers, hypnotics, antipsychotic drugs, cold remedies containing antihistamine agents, and any other antiallergic or antipruritic drugs in the 6 days preceding the day of enrolment; use of steroids and immunosuppressive agents not allowed for 2 weeks before and during the 3‐day participant selection period; sustained‐release steroid depot preparations and astemizole not allowed for 4 weeks before and during the 3‐day participant selection period; if pruritus was associated only with the face and head; history of contact dermatitis induced by a topical steroid; history of complications including severe hepatic disorder, renal disorder, cardiopathy, or blood disease; prolonged QTc interval; non‐compliance during the 3‐day participant selection period; a change in the method of use or in the class of topical preparation during the 1‐week placebo run‐in period; skin infection induced by bacteria, fungi, or viruses; history of epileptic attacks or organic brain lesions; history of drug allergy to antihistamines and antiallergic agents; receiving specific hyposensitisation, immunomodulation, or light therapy; had participated in other clinical studies in the past 3 to 6 months; history of fexofenadine use; pregnant or nursing women Group differences: no statistically significant differences between groups in terms of baseline characteristics (Table 2) or symptom assessments. However, analyses were based on n = 400 after exclusion of 11 participants |
|
| Interventions |
Intervention characteristics Fexofenadine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
Primary outcome 2: adverse events (number of patient‐reported adverse events)
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: number of eczema flares/amount of 0.1% hydrocortisone butyrate in grams
|
|
| Identification |
Sponsorship source: Aventis Pharma Ltd. (Japan) Country: Japan Setting: secondary Comments: 52 centres in Japan (i.e. outpatient hospitals) Author's name: M. Kawashima Institution: Department of Dermatology, Tokyo Women’s Medical University Email: m‐kawash@derm.twmu.ac.jp Address: Department of Dermatology, Tokyo Women’s Medical University, 8‐1, Kawada‐cho, Shinjuku‐ku, Tokyo, 162‐8666, Japan |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: randomisation was performed with a uniform block size of 8 |
| Allocation concealment (selection bias) | Low risk | Comment: treatment was allocated using an "ordinal pre‐printed schedule", and code listing was secured in a sealed envelope |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In order to maintain a double‐blind protocol, different individuals in the same Clinical Research Organization (CRO) department acted as the generator and executor of assignment. The similarity of treatment characteristics was confirmed by the CRO. Successful blinding was guaranteed by the CRO’s standard operating practice (code listing was secured in a sealed envelope stored in the CRO). The emergency code was also stored in the CRO" Comment: blinding was done |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. insufficient information to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: in the fexofenadine group, 6 (2.9%) of 207, and in the placebo group, 5 (2.5%) of 204 dropped out |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Although study authors disclose receipt of payments from the study sponsor (Aventis Pharma Ltd. ‐ Japan) for conducting and publicising this study and state that they have no commercial associations that might pose a conflict of interest, we do not know whether this may have impacted the process of conducting the study |
Kimura 2009.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Loratadine
No additional treatment
Inclusion criteria: (1) diagnostic: atopic dermatitis; (2) severity of condition: mild to moderate, aged 15 or older; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): whole body Exclusion criteria: pregnant or in lactation; treated with systemic corticosteroids; epilepsy or other brain disorders Group differences: study authors report no differences, but the data are not shown |
|
| Interventions |
Intervention characteristics Loratadine
No additional treatment
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: VAS (0 to 100)
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs: SCORAD after 4 weeks
|
|
| Identification |
Sponsorship source: not reported Country: Japan Setting: secondary Comments: dermatological clinic at the university hospital in Japan (a single centre) Author's name: Utako Kimura Institution: Department of Dermatology and Allergology, Juntendo University, Tokyo, Japan Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Participants were allocated to each group whether the clinical ID number was odd (Group A) or even (Group B)" Comment: randomisation method likely to introduce selection bias |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Informaiton insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: although study authors report no missing outcome data, whether all randomised participants contributed to data collection remains unclear |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Other bias | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
Kircik 2013.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: no information given on how atopic dermatitis was defined; (2) severity of condition: "subjects with uncomplicated atopic dermatitis"; (3) duration of condition: no information given; (4) site evaluated (e.g. face/back); no information given Exclusion criteria: not reported Group differences: information from interim report: "the treatment groups were balanced with respect to all demographic and baseline characteristics" |
|
| Interventions |
Intervention characteristics Levocetirizine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
Primary outcome 2: adverse events
Secondary outcome 2: mean change in quality of life, as measured by a standardised or validated quality of life measure (e.g. Dermatology Life Quality Index ‐ DLQI (Finlay 1994))
|
|
| Identification |
Sponsorship source: UCB Pharmaceuticals Country: USA Setting: secondary Comments: single‐centre Author's name: Leon Kircik Institution: Mount Sinai Health System Address: 1169 Eastern Pkwy # 2310, Louisville, KY 40217, United States |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: it is not clear whether medication was identical in appearance to placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. information insufficient to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient reporting of attrition/exclusions to permit judgement of low risk or high risk. No information given about attrition given. Although study authors report no missing outcome data, whether all randomised participants contributed to data collection remains unclear |
| Selective reporting (reporting bias) | High risk | Comment: reporting of data was rudimentary in the report and did not allow us to run necessary analyses. Contacting the study author failed as the study author did not respond |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, but study was funded by UCB Pharmaceuticals) |
Kuniyuki 2009.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Olapatadine
No additional treatment
Inclusion criteria: (1) diagnostic: chronic eczema/dermatitis; (2) severity of condition: not mentioned; (3) duration of condition: longer than 1 month; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: severe systemic disorders; treated with systemic corticosteroids, immune modulators, or antihistamine within 2 weeks; hepatic or nephrotic disorder; pregnant or in lactation Group differences: groups appear comparable from the figures |
|
| Interventions |
Intervention characteristics Olapatadine
No additional treatment
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: VAS and 5‐grade scale
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
|
|
| Identification |
Sponsorship source: not reported Country: Japan Setting: secondary Comments: A dermatology clinic at the general hospital and 5 private dermatology clinics in Japan (6 centres) Author's name: Shuichi Kuniyuki Institution: Division of Dermatology, Osaka City General Hospital Email: ga28547@wa2.so‐net.ne.jp; kuniyuki@ocgh.hospital.city.osaka.jp Address: Miyakojima‐Hondori 2‐13‐22, Miyakojima‐ku, 534‐0021, Osaka, Japan |
|
| Notes | Data fully extracted by 2 native speakers | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Comment: participants were allocated to each group according to their age: odd to olopatadine and even to placebo |
| Allocation concealment (selection bias) | Unclear risk | Comment: although age was used to allocate individuals to groups, we do not know whether this action was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: final numbers of evaluable participants not stated |
| Selective reporting (reporting bias) | Unclear risk | No comparison possible with protocol, as a study protocol is not available, but it is clear that the published report includes all expected outcomes, including those that were prespecified |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Langeland 1994.
| Methods |
Study design: randomised controlled trial Study grouping: cross‐over |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: pruritus caused by moderate to severe AD according to Rajka and Langeland (1989); (2) severity of condition: moderate to severe; (3) duration of condition: mean = 24 years; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: pregnant or lactating women; patients receiving concomitant medication expected to interfere with skin disorders or with known mechanisms of pruritus Group differences: n/a since cross‐over trial |
|
| Interventions |
Intervention characteristics Loratadine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus during the night on 10 cm VAS daily recorded by patient
Primary outcome 2: proportion of participants reporting adverse effects and serious adverse events throughout the study period
Secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
|
|
| Identification |
Sponsorship source: not reported Country: Norway Setting: probably secondary (i.e. outpatient university hospital (Oslo, Norway)) Comments: Author's name: Dr. med. Tor Langeland Institution: Department of Dermatology, National Hospital, Rikshospitalet, Oslo Email: torl@online.no Address: St. Olavs plass 3, 0165 Oslo, Norway |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information about the sequence generation process insufficient to permit judgement of low risk or high risk, although a 'block‐randomised' design is mentioned |
| Allocation concealment (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “a block‐randomised, double blind…” Comment: participants received identical placebo tablets...no further information about personnel, but it is unlikely that their blinding could have been broken |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: apart from: "double blind ...", no information given as to how outcome assessor was blinded; however it is unlikely that blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: although study authors report no missing outcome data, whether all randomised participants contributed to data collection remains unclear |
| Selective reporting (reporting bias) | Unclear risk | Comment: no comparison possible with protocol, as a study protocol is not available, but it is clear that the published report includes all expected outcomes, including those that were prespecified |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
LaRosa 1994.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Cetirizine
Placebo
Inclusion criteria: (1) diagnostic: diagnosis of AD based on presence of at least 3 major and minor criteria according to Hanifin and Rajka; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: chronic kidney disease; liver or cardiovascular disease; treatment with parenteral steroids, ketotifen, oxatomide, other histamines; cutaneous or other infection Group differences: "the randomisation procedure resulted in a balancing of the two groups with respect to" ...age, weight, height, and sex. However, there were differences in clinical parameters between groups (e.g. family history of allergy) |
|
| Interventions |
Intervention characteristics Cetirizine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’
|
|
| Identification |
Sponsorship source: not reported Country: Italy Setting: secondary Comments: Day Hospital of the Pediatric Clinic of Catania, University (i.e. secondary setting) (outpatients at university hospital) Author's name: M. LaRosa Institution: Department of Pediatrics, University of Catania, Italy Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind study"; "the therapy consisted of identical 20‐ml ampules containing placebo or cetirizine" Comment: no further information provided about personnel, but it is unlikely that their blinding could have been broken |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind study" Comment: no information given as to how outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: only 1 participant randomised to cetirizine group withdrew "voluntarily"; no other reason for dropout given ‐ but risk of attrition bias considered low |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided; although provision of information regarding origin of drugs is reported, we do not know whether study was initiated by pharmacological company? Did study authors receive funding from any source?) |
Leon 1989.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Ketotifen
Placebo
Inclusion criteria: (1) diagnostic: clinical diagnosis of atopic eczema; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): whole body Exclusion criteria: chronic disease of the gastrointestinal system, liver, or kidneys that could influence the absorption, metabolism, and excretion of medication; diabetes mellitus, heart disease, or pre‐existing or intermittent pneumopathy Group differences: not reported |
|
| Interventions |
Intervention characteristics Ketotifen
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus during the day and the night
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
|
|
| Identification |
Sponsorship source: not reported Country: Mexico Setting: secondary Comments: Author's name: Gladys Leon Institution: Oficina Farmacologia Clinica, Servicio de Investigacion Clinica Hospitalaria. Adscrita al Servicio de Dermatologigia del Hospital General de México, S.S. y Dermatologia de la UNAM y Universidad La Salle Email: not reported Address: Oficina Farmacologia Clinica, Servicio de Investigacion Clinica Hospitalaria. Adscrita al Servicio de Dermatologigia del Hospital General de México, S.S. y Dermatologia de la UNAM y Universidad La Salle; Petén 316 Col. Navarte. México, DF 03020 |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: although study authors stated that placebo medication was identical in appearance and taste to the ketotifen medication, they failed to elucidate the blinding procedure for personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. information insufficient to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: the number of participants randomised equals the number of participants evaluated |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Monroe 1992.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: atopic dermatitis diagnosed by a physician (department of dermatology); (2) severity of condition: atopic dermatitis with at least moderate itching and skin lesions; (3) duration of condition: skin condition had to be “in an active state at least 3 weeks prior to the study”; (4) site evaluated (e.g. face/back): whole body Exclusion criteria: "patients were excluded from the study if they were totally unresponsive to previous treatment with AH or they had taken AH in the previous 24 h, oral corticosteroids in the previous 10 days, or depot corticosteroids in the previous 28 days, or were taking any medication that could have a clinical effect on the course of the skin disorder" Group differences: not reported |
|
| Interventions |
Intervention characteristics Loratadine
Hydroxyzine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus (percentage change in scores (0 to 3) from baseline to endpoint
Primary outcome 2: adverse events: somnolence or sedation during treatment
|
|
| Identification |
Sponsorship source: not reported Country: USA Setting: secondary Comments: outpatients at Department of Dermatology, Milwaukee Medical Clinic, and Medical College of Wisconsin, Milwaukee, Wisonsin Author's name: Eugene W. Monroe Institution: Department of Dermatology, Milwaukee Medical Clinic, and Medical College of Wisconsin, Milwaukee, Wisonsin Email: not reported Address: not reported |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although "all drugs were supplied as identical capsules" the frequency of usage varied across the 3 groups. Thus whether participants could have shared this knowledge among one other remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. information insufficient to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: number randomised was the number analysed |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Munday 2002.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: established AD with nocturnal itching and scratching (not stated whether according to Hanifin & Rajka or UK criteria) but study group consisted of dermatologists; (2) severity of condition: not reported; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: had received therapy with any systemic antihistamines within the 2‐week period before the date of screening (day 1); history of epilepsy, glaucoma, or hepatic disease; hypersensitivity or contraindications to the study medication (including hydrocortisone and Unguentum Merck emollient); any other clinical abnormalities that the investigator felt were likely to affect participation in the trial Group differences: no statistically significant difference in relation to duration of condition, condition status, or prior use of eczema therapies |
|
| Interventions |
Intervention characteristics Chlorpheniramine maleate BP
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: severity of pruritus (none, minimal, mild, moderate) between days 1 and 29
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: number of eczema flares/amount of 1% hydrocortisone in grams
|
|
| Identification |
Sponsorship source: not reported Country: UK and Poland Setting: secondary Comments: multi‐centre trial; 3 outpatient centres in UK and Poland (i.e. secondary setting) Author's name: James Munday Institution: Pharmax Ltd., Bexley Email: imunday@intercern.com Address: James Munday, 18 Brockley Park, Forest Hill, London SE23 1PS (UK) |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk. Study authors failed to state whether placebo elixir was identical in appearance and taste to chlorpheniramine maleate BP |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all analyses were performed using the intention‐to‐treat (ITT) population" |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as no study protocol appears to have been registered |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) Main study author worked for Pharmax Ltd. |
Nuovo 1992.
| Methods |
Study design: single‐patient randomised trial Study grouping: cross‐over single‐patient |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: (2) severity of condition: not reported; (3) duration of condition: lifelong; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: n.a. Group differences: n.a. |
|
| Interventions |
Intervention characteristics Chlorpheniramine 16 mg/d
Chlorpheniramine 24 mg/d
Terfenadine 240 mg/d
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus assessed on 7‐point Likert scale
Primary outcome 1: mean change in patient‐assessed symptoms: patient‐assessed severity of symptoms assessed on 7‐point Likert scale
Primary outcome 2: adverse events (severity of drowsiness) on 7‐point Likert scale
Secondary outcome 3: number of eczema flares/frequency of triamcinolone applications Outcome type: continuous outcome
|
|
| Identification |
Sponsorship source: the paper was supported by a grant (HS06006) from the National Center for Health Services Research‐OASH Country: USA Setting: secondary Comments: Author's name: James Nuovo Institution: Departments of Family Medicine, Pharmacy Practice, and Medicine, Schools of Medicine and Phannacy, University of Washington, Seattle Email: not reported Address: Department of Family Medicine, RF‐30, University of Washington, Seattle, WA 98195 |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the order of treatment was determined by random allocation" Comment: no further information given (i.e. information about the sequence generation process insufficient to permit judgement of low risk or high risk) |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the patient and the investigator were blinded to the treatment sequence"; "A pharmacist prepared all study medications to appear identical as pink capsules and maintained the drug code" Comment: participants and personnel were blinded to study groups |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all data from the cross‐over trial were reported |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company?) However, a source of funding was provided |
Ruzicka 1998.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic AD according to Hanifin and Rajka, age 18 to 65 years; (2) severity of condition: pruritus score 4 to 7 (VAS), moderate severity of condition according to EASI score (Eczema Area and Severity Index) subtotal score A ≥ 18 and ≤ 40, and B ≥ 2 and ≤ 10; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: cardiovascular disease; neurological and other systemic diseases; administration of depot corticosteroids within previous 3 months, systemic corticosteroids within previous 28 days, or topical corticosteroids within previous 7 days Group differences: no baseline comparisons were provided; however, study authors report that baseline pruritus was not equally distributed among groups |
|
| Interventions |
Intervention characteristics Loratadine 5 mg
Loratadine 10 mg
Loratadine 20 mg
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: VAS pruritus (0 to 10 cm) (difference in pruritus sum between day 0 and day 13)
Patient‐assessed symptoms: VAS pruritus: sum of days 7 to 13
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs: no. for whom treatment success was judged as good or very good
|
|
| Identification |
Sponsorship source: not reported Country: Germany Setting: secondary Comments: 8 centres Author's name: T. Ruzicka Institution: Klinik und Poliklinik für Dermatologie und Allergologie, Frauenlobstraße 9–11, 80337 München Email: Marion.Michl@med.uni‐muenchen.de Address: Klinik und Poliklinik für Dermatologie und Allergologie, Frauenlobstraße 9–11, 80337 MünchenKlinikum München Thalkirchner Straße, Thalkirchner Straße 48, 80337 München |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: whether medication was identical in appearance to placebo remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: of 160 participants randomised, only 138 were assessed at the end of the study (i.e. attrition rate was 13.7%). Study authors did not provide reasons for these dropouts and did not describe the missing data ‐ but the risk of attrition bias was considered low based on the attrition rate |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: no comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Savin 1986.
| Methods |
Study design: randomised controlled trial Study grouping: cross‐over |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: not reported which diagnostic criteria were used for the definition of atopic dermatitis; (2) severity of condition: not reported; (3) duration of condition: long‐standing (not further defined); (4) site evaluated (e.g. face/back): not reported Exclusion criteria: not reported Group differences: no baseline group differences (cross‐over design) |
|
| Interventions |
Intervention characteristics LN2974 (tazifylline)
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
|
|
| Identification |
Sponsorship source: not reported Country: UK Setting: secondary Comments: Author's name: John Andrew Savin Institution: Department of Dermatology, The Royal Infirmary, Edinburgh, Scotland |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Informationn insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "in a double blind ....trial" Comment: whether medication was identical remains unclear. No information given regarding blinding of clinician |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given as to how assessor was blinded (i.e. information insufficient to permit judgement of low risk or high risk) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reporting of attrition/exclusions insufficient to permit judgement of low risk or high risk |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
Simons 2007.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Levocetirizine
Placebo
Inclusion criteria: (1) diagnostic: atopic dermatitis (no further details given); (2) severity of condition: not specified; (3) duration of condition: not specified; (4) site evaluated (e.g. face/back): not reported Exclusion criteria: children with asthma or any other systemic disease; height or body mass below the 5th percentile; any severe neurological or psychological disorder requiring medical treatment; known to be intolerant of levocetirizine or any other piperazine antihistamine, or to the parabens used as preservatives in H1 antihistamine liquid formulations; personal history or sibling history of sleep apnoea; renal insufficiency or any metabolic condition that might affect elimination of levocetirizine Group differences: ‐ |
|
| Interventions |
Intervention characteristics Levocetirizine
Placebo
|
|
| Outcomes |
Primary outcome 2: adverse events
|
|
| Identification |
Sponsorship source: UCB Pharma (information obtained from trial registration site) Country: Multi‐centre: 10 European countries, Australia, and South Africa Setting: secondary Comments: Author's name: Simons F.E.R. Institution: Children’s Hospital Research Institute of Manitoba Email: lmcniven@hsc.mb.ca Address: Children’s Hospital Research Institute of Manitoba, 513 – 715 McDermot Avenue, Winnipeg MB R3E 3P4 |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: although assignment to treatment was made according to preselected randomisation factors at baseline, including status of sensitisation to grass pollen or house dust mite, sensitisation to egg, maternal history of asthma, and country of residence, whether this eliminated selection bias remains unclear |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: ITT analyses were conducted for presented outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the trial registration were reported |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) Acknowledgement to UCB is made, but no further details are given |
Tharp 1998.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: (1) diagnostic: no information given on how atopic dermatitis was defined; (2) severity of condition: patients with atopic dermatitis involving 20% to 50% of total body surface area and located at 2 or more non‐contiguous areas; (3) duration of condition: not reported; (4) site evaluated (e.g. face/back): whole body Exclusion criteria: not reported Group differences: not reported |
|
| Interventions |
Intervention characteristics Cetirizine
Placebo
|
|
| Outcomes |
Primary outcome 1: mean change in patient‐assessed symptoms: pruritus
Primary outcome 2: adverse events
Secondary outcome 1: physician‐assessed clinical signs
Secondary outcome 3: secondary outcome 3: number of eczema flares, measured by, for example, ‘escalation of treatment’ or ‘use of topical anti‐inflammatory medications’ Amount of triamcinolone used
|
|
| Identification |
Sponsorship source: not reported, but because the last author of the abstract was an employee of Pfizer Inc., this pharmaceutical company might be the funding body for this study Country: USA Setting: secondary Comments: multi‐centre study Author's name: Michael D. Tharp Institution: Rush University Medical Center Email: not reported Address: 1725 W. Harrison St., Suite 264, Chicago, IL 60612 USA. Phone: (312) 942‐2195; fax: (312) 563‐2263 |
|
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Information about the sequence generation process insufficient to permit judgement of low risk or high risk |
| Allocation concealment (selection bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk Comment: whether medication was identical in appearance to placebo remains unclear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Information insufficient to permit judgement of low risk or high risk |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient reporting of attrition/exclusions to permit judgement of low risk or high risk. No information regarding dropouts was given |
| Selective reporting (reporting bias) | Unclear risk | Information insufficient to permit judgement of low risk or high risk. No comparison with protocol possible, as a study protocol is not available |
| Other bias | Unclear risk | Further risk of bias is possible, but information is insufficient to judge whether additional bias exists (e.g. no conflict of interest statement provided, no provision of information regarding origin of drugs, Was study initiated by pharmacological company? Did study authors receive funding from any source?) |
ACTH: adrenocorticotropic hormone; AD: atopic dermatitis; AE: adverse event; bw/d: body weight each day; CRO: clinical research organisation; EASI: Eczema Area and Severity Index; ECG: electrocardiogram; ITT: intention‐to‐treat; M: mean; n.a.: not applicable; SCORAD: SCORing Atopic Dermatitis index; SD: standard deviation; SE: standard error; UVA: ultraviolet A; UVB: ultraviolet B; VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Anton'ev 1988 | Wrong study design: study not described as randomised |
| Balabolkin 1983 | Wrong study design: study not described as randomised |
| Bogdaszewska 1968 | Wrong study design: study not described as randomised |
| CastroTorres 1987 | Wrong indication/wrong patient population: study included participants with different pruritic skin diseases, varying from insect bites to psoriasis. However, study authors did not give clear indications that the study included participants with eczema |
| Eberhartinger 1969 | Wrong study design: study not described as randomised |
| Foulds 1981 | Wrong comparator/wrong intervention: Cimetidene vs promethazine hydrochloride but not vs placebo |
| Furue 2009 | Wrong study design: study not described as randomised |
| Grabner 1970 | Wrong study design: study not described as randomised |
| Imaizumi 2003 | Wrong study design: study not described as randomised |
| Kawakami 2006 | Wrong comparator/wrong intervention: comparison of fexofenadine combined with emollient vs fexofenadine combined with steroid treatment |
| Kleine Natrop 1970 | Wrong study design: study not described as randomised |
| Kori Lindner 1997 | Review article |
| Korossy 1972 | Wrong study design: study not described as randomised |
| Liu 2008 | Wrong study design: study not described as randomised |
| Lutsky 1993 | Wrong comparator/wrong intervention: comparison of loratadine vs terfenadine but not vs placebo |
| May 1966 | Wrong study design: study not described as randomised |
| Mensing 1991 | Wrong comparator/wrong intervention: comparison of loratadine vs dimethindene but not vs placebo |
| Mesquita 1994 | Wrong comparator/wrong intervention: comparison of dimethindene vs astemizole but not vs placebo |
| Miller 1963 | Wrong study design: study not described as randomised |
| Nitzschner 1970 | Wrong study design: study not described as randomised |
| Ohsawa 2014 | Review article |
| Ohtani 2004 | Wrong comparator/wrong intervention: comparison of cetirizine vs epinastine hydrochloride vs vitamin B2 but not vs placebo |
| Pacor 1992 | Wrong comparator/wrong intervention: comparison of oxatomide vs cromoglycate but not vs placebo |
| Patel 1997 | Wrong comparator/wrong intervention: comparison of loratadine vs cetirizine but not vs placebo |
| Pfab 2012 | Wrong study design: comparison of effects of several interventions on magnitude of experimentally induced itch |
| Rajka 1965 | Wrong study design: study not described as randomised |
| Rajka 1968a | Wrong study design: study not described as randomised |
| Rajka 1968b | Wrong study design: study not described as randomised |
| Schmoeckel 1992 | Wrong study design: observational drug safety study (i.e. study not described as randomised) |
| Sheng 2009 | Wrong comparator/wrong intervention: comparison of mizolastine in combination with dyclonine cream vs cetirizine and dyclonine cream vs vitamin C and dyclonine cream |
| Simons 1984 | Wrong comparator/wrong intervention: comparison of hydroxyzine 0.7 mg/kg 3 times daily vs hydroxyzine 1.4 mg/kg 3 times daily but not vs placebo |
| Sugai 2003 | Wrong comparator/wrong intervention: comparison of tacrolimus ointment alone for 2 weeks followed by complementary use of cetirizine for another 2 weeks vs tacrolimus ointment and cetirizine for 2 weeks followed by tacrolimus alone for 2 weeks |
| Tarchalska 1988 | Review article: a literature review |
| Veien 1995 | Wrong patient population: sample consisted of patients with current hand eczema who had or previously had eczema |
| Voigtlander 1989 | Wrong study design: study not described as randomised |
| Wahlgren 1990 | Wrong study design: study not described as randomised |
| Wahlgren 1991 | Wrong study design: study not described as randomised |
| Weitgasser 1967 | Wrong comparator/wrong intervention: comparison of tavegyl vs another unspecified antihistamine |
| Wolfram 1967 | Wrong study design: study not described as randomised |
| Zhang 1998 | Wrong comparator/wrong intervention: comparison of oral terfenadine, injection of interferon gamma (up to 7 to 11 weeks), and rinse with calamine lotion vs cyproheptadine tablets and rinse with calamine lotion |
| Zhang 2010 | Wrong comparator/wrong intervention: comparison of Qushi decoction oral and bathed vs oral chlorpheniramine 0.35 mg/kg 3×/d combined with Kangfu ointment |
| Zuluaga de Cadena 1989 | Wrong study design: study not described as randomised |
Wrong study design (e.g. lack of randomisation); wrong comparator (e.g. one H1 AH vs one or several other H1 AH but not vs placebo); wrong patient population (e.g. participants with no diagnosis of eczema); review article (e.g. not a trial but an article discussing several trials).
Characteristics of studies awaiting assessment [ordered by study ID]
Borelli 1969.
| Methods | Controlled clinical comparison of BP 400 vs placebo |
| Participants | 200 patients with eczema and other pruritogenic skin conditions |
| Interventions | BP 400 (synonyms: 1‐methyl‐4‐thioxanthen‐9‐ylidene‐piperidine; 9‐(1‐methyl‐4‐piperidinylidene)thioxanthene; 9‐(N‐methyl‐4’‐piperidylene)thioxanthene; 9‐(N‐methyl‐4’‐piperidylidene)‐thioxanthene; BP 400S; CGA 123427; Calmixen; Mepithiathene; Pimetixene) vs placebo |
| Outcomes | Antiprurigogenic and sedative effects of treatment |
| Notes | Not clear whether this was a randomised study. We contacted study authors but received no response to our queries |
Chunharas 2002.
| Methods | Double‐blind placebo controlled trial; however, whether a random allocation took place remains unclear |
| Participants | 48 patients with eczema (children) with a mean age of 73.67 months |
| Interventions | Oral loratadine (5 mL/30 kg bodyweight/d) vs placebo as add‐on to once‐a‐day topical mometasone fuorate 0.1% ointment |
| Outcomes | Severity of eczema as measured by SCORAD; physician global assessment of improvement; pruritus |
| Notes | Not clear whether this was a randomised study. We contacted study authors but received no response to our queries |
DeBeule 1974.
| Methods | This is a randomised controlled trial |
| Participants | 8 patients with eczema; 40 with other dermatological conditions, mainly hay fever |
| Interventions | Oral primalan (mequitazine) (10 mg/d) vs placebo |
| Outcomes | Clinical signs of eczema |
| Notes | Although the study was also conducted in patients with eczema, results were not reported separately for these. We tried to contact study authors to request additional information but received no response |
Laugier 1978.
| Methods | This is a randomised controlled trial |
| Participants | 10 patients with eczema, 20 with urticaria, and 10 with other skin conditions |
| Interventions | Oral mequitazine 10 mg/d vs placebo |
| Outcomes | Overall clinical assessment of response to treatment |
| Notes | Although the study was also conducted in patients with eczema, results were not reported separately for these. We tried to contact study authors to request additional information but received no response |
NCT00160563.
| Methods | This is a randomised controlled trial |
| Participants | Inclusion criteria that must be verified at the end of the first 18 months of treatment (visit 9) Having completed the previous 18‐month treatment period of the EPAAC trial ‐ NCT00152464 |
| Interventions | Drug: levocetirizine 5 mg/mL oral drops, 0.125 mg/kg body weight, bid for 18 months; other name: Xyzal® Other: placebo oral drops, bid for 18 months Other: placebo oral drops, bid for 18 months |
| Outcomes | Primary outcome measures: time to onset of asthma [time frame: 36 months (from randomisation visit to preceding A00309 ‐ NCT00152464 trial onwards)] [designated as safety issue: no] Secondary outcome measures: time to onset of asthma in the subset of subjects still asthma free after first 18 months [time frame: 18 months (from end of the preceding A00309 ‐ NCT00152464 trial onwards)] [designated as safety issue: no] |
| Notes | Study has been terminated, but we do not know whether this took place before commencement of the study or afterwards |
Simons 2003.
| Methods | This is a randomised controlled trial |
| Participants | Infants who had a history of H1‐antihistamine treatment for allergic rhinitis, urticaria, atopic dermatitis (eczema), or other disorders |
| Interventions | Drug: oral cetirizine 0.5 mg/d Other: matched placebo |
| Outcomes | Adverse events and electrocardiographic measurements |
| Notes | Although the study was also conducted in patients with eczema, results were not reported separately for these. We tried to contact study authors to request additional information but received no response |
SCORAD: SCORing Atopic Dermatitis index.
Characteristics of ongoing studies [ordered by study ID]
CRT EU 23679.
| Trial name or title | Prednicarbat and cetirizine dihydrochloride in the treatment of acute stage of atopic eczema in children |
| Methods | Not reported |
| Participants | Children with eczema (age 2 to 11 years) |
| Interventions | Prednicarbat and cetirizine dihydrochloride |
| Outcomes | Not reported |
| Starting date | Not reported |
| Contact information | Not reported |
| Notes | ‐ |
CTR EU 23697.
| Trial name or title | Effect of long‐term treatment with the H1 ‐receptor antagonist cetirizine in the prevention of urticaria in children with atopic dermatitis |
| Methods | Not reported |
| Participants | Children with eczema (age 2 to 11 years) |
| Interventions | H1‐receptor antagonist cetirizine |
| Outcomes | Not reported |
| Starting date | Not reported |
| Contact information | Not reported |
| Notes | May be related to Simons 2007 |
CTR EU 29342.
| Trial name or title | Hydroxyzine: pharmacokinetics and antipruritic effect in children with atopic dermatitis |
| Methods | Not reported |
| Participants | Children with eczema (age 2 to 11 years) |
| Interventions | Hydroxyzine |
| Outcomes | Not reported |
| Starting date | Not reported |
| Contact information | Not reported |
| Notes | ‐ |
CTR EU 39698.
| Trial name or title | Evaluation of the efficacy and the safety of DCL syrup in childhood atopic dermatitis |
| Methods | Not reported |
| Participants | Children with eczema (age range unclear) |
| Interventions | Desloratadine |
| Outcomes | Not reported |
| Starting date | Not reported |
| Contact information | Not reported |
| Notes | ‐ |
UMIN000010519.
| Trial name or title | Impact of olopatadine hydrochloride (Allelock(R)) against scratching behavior of atopic dermatitis patients at night |
| Methods | This is a randomised controlled trial |
| Participants | Adults suffering from atopic dermatitis Inclusion criteria:
|
| Interventions | Olapatadine versus placebo |
| Outcomes | Measurement of the scratching ratio and the depth of sleep Measurement will be performed on days ‐2, ‐1, and 7 for patients with atopic dermatitis Measurement on day 7 day can be extended up to 3 additional days, in case the participant is not available on day 7 VAS value of itch and dermatitis score (EASI, ELQI) are measured for patients with atopic dermatitis Medication and measurement on day 7 will not be performed for healthy individuals |
| Starting date | 18/02/2013 |
| Contact information | Keiichi Yamanaka, MD, PhD; Mie University, Graduate School of Medicine; Department of Dermatology; 2‐174 Edobashi, Tsu, Mie 514‐8507, Japan |
| Notes | ‐ |
DCL: denotes desloratadine; EASI: Eczema Area and Severity Index; ELQI: authors did not provide what ELQI stands for, the abbreviation may contain a spelling mistake and actually refer to DLQI: Dermatology Quality of Life Index.
Differences between protocol and review
It was stated in the protocol that "Two review authors (MB and EW) will search for ongoing trials in the following databases using the terms: eczema, atopic eczema, atopic dermatitis", but for this review, UM and CA conducted this search.
It was also stated in the protocol that "Two review authors (EW and MB) will examine the bibliographies of the included and excluded studies for further references to potentially‐eligible RCTs", but for this review, UM and CA carried out the examination.
The protocol stated that "Two authors (CA and MB) will independently assess the abstracts of trials resulting from the searches." However, UM carried out the conference proceedings search for this review.
The protocol stated that "Two authors (CA and MB) will independently assess the abstracts of trials resulting from the searches", but UM and CA performed these assessments for this review.
It was stated in the protocol that "Two authors (CA and MB) will independently review the full‐text papers and disagreements over eligibility will be resolved by discussion, consensus, or with a third review author (BC)". However, UM, CA, and MB reviewed the full‐text papers and resolved disagreements among themselves.
The protocol stated "Two authors (CA and MB) will enter details of the included trials into the ’Characteristics of included studies’ tables in RevMan 5.3 (Review Manager (RevMan)", but for this review, UM and CA performed this task.
"Four review authors (CA, MB, AJ, EW) will extract data using a previously developed and piloted data extraction form and will resolve disagreements on data extraction by consensus". UM, CA, and MB actually performed this task for this review.
The protocol stated that "Two authors (CA and MB) will check and enter the data into RevMan 5.3 (Review Manager (RevMan))", but UM and CA performed this task for this review.
We had stated in the protocol that we would search the bibliographies of included studies for further references to potentially eligible randomised controlled trials. We extended this search also to the bibliographies of excluded studies.
The protocol stated that risk of bias would be assessed by CA and MB, but in this review, UM and CA performed this task.
The protocol stated that "any outcomes that are statistically significant (P < 0.05) were used to calculate a number needed to treat for an additional beneficial outcome (NNTB)", but we also reported the number needed to treat for an additional harmful outcome (NNTH), for instance, for adverse events.
In the protocol, we had stated that "the 'Mean change in physician‐assessed clinical signs' would be based on standardised or validated measures." However, we also considered other measures provided by the respective study for this outcome and described the outcome assessment in detail.
We had stated in the protocol that "If we feel there are several major comparisons or that our findings need to be summarised for different populations, we would include further 'Summary of findings' tables (Higgins 2011)." However, we did not summarise findings for different populations.
The protocol stated that 'Summary of findings' tables would report mean change in eczema symptoms scores. We narrowed this outcome to look only at pruritus in the 'Summary of findings' table because no other eczema symptom score was assessed in the studies summed up in the 'Summary of findings' table.
Because we were unable to pool data in a meta‐analysis due to clinical diversity, we could not undertake subgroup and sensitivity analyses to assess heterogeneity, as planned in our protocol. Neither were we able to study H1 AH interventions in patients with eczema with allergic comorbidity as assessed by allergic sensitisations (known as "atopic" eczema), as no data were available.
We had originally planned to analyse cross‐over data based on advice provided in Section 16.4.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We had particularly planned to assess carry‐over and period effects descriptively. The protocol stated that if there was a limited suggestion of these effects and data were adequate, we would carry out a paired analysis. However, for all cross‐over trials, data were not adequately reported to allow such analyses. Instead we described narratively the data as reported by study authors.
Contributions of authors
CA secured funding to carry out this review. CA, AJ, BC, EW, and MB drafted the protocol. UM was the contact person with the editorial base. UM co‐ordinated contributions from coauthors. UM and CA wrote the final draft of the review with contributions from all review authors. UM, CA, and MB screened papers against eligibility criteria. UM obtained data on ongoing and unpublished studies. UM and CA appraised the quality of papers. UM, CA, MB, and AJ extracted data for the review and sought additional information about papers. UM, CA, MB, and AJ assessed the risk of bias. UM entered data into RevMan. UM, CA, and BC analysed and interpreted data. UM, CA, and BC worked on the methods sections. CA, EW, UM, and AJ drafted the clinical sections of the background and responded to the clinical comments of referees. UM, CA, and BC responded to the methods and statistics comments of referees. CA is the guarantor of the update.
Sources of support
Internal sources
No sources of support supplied
External sources
-
The National Institute for Health Research (NIHR), UK.
The NIHR, UK, is the largest single funder of the Cochrane Skin Group. This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Skin Group.
-
Federal Ministry of Education and Research, Germany.
The project was funded by the 'Bundesministerium für Bildung und Forschung' (Federal Ministry of Education and Research) (project name: ANTEplus, project number: 01KG1401).
Declarations of interest
Uwe Matterne: none known. Merle Margarete Böhmer: none known. Elke Weisshaar: my institution received money from Menlo Therapeutics and Trevi Therapeutics for my participation in advisory board meetings and publication steering committee meetings for itch trials. My institution also received money from Menlo Therapeutics and Trevi Therapeutics for recruiting participants into clinical studies with new antipruritic substances for prurigo nodularis, none of which are relevant to this review. However, antihistamines are a complementary (add‐on) treatment, so eczema treatment is not a direct competitor (i.e. relevant) to this review’s intervention of interest; thus, this declaration of interest is compliant with the Cochrane Commercial sponsorship policy. The last date funding was received was 18 December 2018. Aldrin Jupiter: I am a shareholder of Bayer and Novartis. Ben Carter: none known. Christian J Apfelbacher: my institution received a grant from Dr. August Wolff GmbH (which produces products that are used for atopic eczema and other forms of dermatitis, such as emollients) for research related to work on patient‐reported outcome validation for hyperhidrosis. However, antihistamines are a complementary (add‐on) treatment, so eczema treatment is not a direct competitor (i.e. relevant) to this review’s intervention of interest; thus, this declaration of interest is compliant with the Cochrane Commercial sponsorship policy. I also received money for consultancy from Dr. August Wolff GmbH, and travel and meeting expenses from LKC Medicine, NTU Singapore, the European Academy of Allergy and Clinical Immunology (EAACI), and the European League Against Rheumatism (EULAR). The last date funding was received was 28 November 2018.
New
References
References to studies included in this review
Berth Jones 1989 {published data only}
- Berth‐Jones J, Graham‐Brown RA. Failure of terfenadine in relieving the pruritus of atopic dermatitis. British Journal of Dermatology 1989;121(5):635‐7. [CENTRAL: CN‐00064362; PUBMED: 2574594] [DOI] [PubMed] [Google Scholar]
Cambazard 2001 {unpublished data only}
- Cambazard F, UCB Pharma. Efficacy and safety of cetirizine drops at 3 doses (0.25, 0.50, 0.75 mg/kg bw/day) in an 8‐week treatment of atopic dermatitis in 1 to 5 year old children: a multicentric, multi‐country, double‐blind, placebo‐controlled, randomised study, 2001. art45‐paediatric‐studies‐docs.ema.europa.eu/GROUP%20C/UCB_Cetirizine/Clinical%20Study%20Reports%20‐%20Cetirizine%20(part%203)/9708%20‐%20Cetirizine%20‐%20MRCE00H0202.pdf (accessed 26 April 2017).
Diepgen 2002 {published data only}
- Businco L. Early treatment of the atopic child: first results of the clinical trial. Pediatric Pulmonology. Supplement 1997;23(S16):73. [CENTRAL: CN‐00147078; PUBMED: 9443210] [DOI] [PubMed] [Google Scholar]
- Diepgen T. Impact of cetirizine on atopic dermatitis. Journal of the European Academy of Dermatology and Venereology 1999;12(Suppl 2):S133. [CENTRAL: CN‐00478509] [DOI] [PubMed] [Google Scholar]
- Diepgen TL, Early Treatment of the Atopic Child Study Group. Long‐term treatment with cetirizine of infants with atopic dermatitis: a multi‐country, double‐blind, randomized, placebo‐controlled trial (the ETAC trial) over 18 months. Pediatric Allergy and Immunology 2002;13(4):278‐86. [CENTRAL: CN‐00434564; PUBMED: 12390444] [DOI] [PubMed] [Google Scholar]
- Estelle F, Simons R. Prospective, long‐term safety evaluation of the H1‐receptor antagonist cetirizine in very young children with atopic dermatitis. Allergologie 2000;23(5):244‐55. [CENTRAL: CN‐00424063] [DOI] [PubMed] [Google Scholar]
- Simons FER, ETAC Study Group on Early Treatment of the Atopic Child. Prospective, long‐term safety evaluation of the H1‐receptor antagonist cetirizine in very young children with atopic dermatitis. Journal of Allergy and Clinical Immunology 1999;104(2 Pt 1):433‐40. [CENTRAL: CN‐00414410; PUBMED: 10452767] [DOI] [PubMed] [Google Scholar]
- Stevenson J, Cornah D, Evrard P, Vanderheyden V, Billard C, Bax M, et al. Long‐term evaluation of the impact of the H1‐receptor antagonist cetirizine on the behavioral, cognitive, and psychomotor development of very young children with atopic dermatitis. Pediatric Research 2002;52(2):251‐7. [CENTRAL: CN‐00409300; PUBMED: 12149503] [DOI] [PubMed] [Google Scholar]
Doherty 1989 {published data only}
- Doherty V, Sylvester DG, Kennedy CT, Harvey SG, Calthrop JG, Gibson JR. Treatment of itching in atopic eczema with antihistamines with a low sedative profile. BMJ 1989;298(6666):96. [CENTRAL: CN‐00058292; PUBMED: 2563952] [DOI] [PMC free article] [PubMed] [Google Scholar]
Falk 1993 {published data only}
- Falk ES. Ketotifen in the treatment of atopic dermatitis. Results of a double blind study. European Review for Medical and Pharmacological Sciences 1993;15(2):63‐6. [CENTRAL: CN‐00100944; PUBMED: 8171216] [PubMed] [Google Scholar]
Frosch 1984 {published data only}
- Frosch PJ, Schwanitz HJ, Macher E. A double blind trial of H1 and H2 receptor antagonists in the treatment of atopic dermatitis. Archives of Dermatological Research 1984;276(1):36‐40. [CENTRAL: CN‐00033953; PUBMED: 6142700] [DOI] [PubMed] [Google Scholar]
- Schwanitz HJ, Frosch PJ, Macher E. Results of a double blind study with H1‐ and H2‐histaminereceptor antagonists in atopic dermatitis. Archives of Dermatological Research 1992;273(1‐2):164. [CENTRAL: CN‐00350456] [DOI] [PubMed] [Google Scholar]
Hannuksela 1993 {published data only}
- Hannuksela M, Kalimo K, Lammintausta K, Mattila T, Turjanmaa K, Varjonen E, et al. Dose ranging study: cetirizine in the treatment of atopic dermatitis in adults. Annals of Allergy 1993;70(2):127‐33. [CENTRAL: CN‐00090744; PUBMED: 8430920] [PubMed] [Google Scholar]
Henz 1998 {published data only}
- Henz BM, Metzenauer P, O'Keefe E, Zuberbier T. Differential effects of new‐generation H1‐receptor antagonists in pruritic dermatoses. Allergy 1998;53(2):180‐3. [CENTRAL: CN‐00684205; PUBMED: 9534917] [DOI] [PubMed] [Google Scholar]
Hjorth 1988 {published data only}
- Hjorth N. Terfenadine in the treatment of chronic idiopathic urticaria and atopic dermatitis. Cutis; Cutaneous Medicine for the Practitioner 1988;42(4A):29‐30. [CENTRAL: CN‐00268216; PUBMED: 2903817] [PubMed] [Google Scholar]
Iikura 1992 {published data only}
- Iikura Y, Baba M, Mikawa H, Nishima S, Maeda K, Akasaka T, et al. A double blind study of the effectiveness of ketotifen in preventing the development of asthma in atopic dermatitis patients. Arerugi [Allergy] 1991;40(2):132‐40. [CENTRAL: CN‐00487852; PUBMED: 2069512] [PubMed] [Google Scholar]
- Iikura Y, Naspitz CK, Mikawa H, Talaricoficho S, Baba M, Sole D, et al. Prevention of asthma by ketotifen in infants with atopic dermatitis. Annals of Allergy 1992;68(3):233‐6. [CENTRAL: CN‐00082427; PUBMED: 1546818] [PubMed] [Google Scholar]
Jung 1989 {unpublished data only}
- Jung EG, Kaymer FG, Ring J, Casimir G, Bersaques J, Brassine M, et al. Multicentre double‐blind study in parallel groups, comparing to placebo 5 and 10 mg cetirizine administered in two daily doses for a week, to children aged 3 – 6 years suffering from atopic dermatitis – determination of the antipruritic effect. UCB Pharmaceutical Sector, 1989. http://art45‐paediatric‐studies‐docs.ema.europa.eu/GROUP%20C/UCB_Cetirizine/Clinical%20Study%20Reports%20‐%20Cetirizine%20(part%203)/A103%20‐%20Cetirizine%20‐%20CE89G241.pdf (accessed 26 April 2017).
Kawashima 2003 {published data only}
- Kawashima M, Nakagawa H, Tango T, Harada S. Fexofenadine is effective at reducing itching associated with atopic dermatitis. Journal of Allergy, Asthma and Immunolgy 2002;109(Suppl 1):S160‐1. [Google Scholar]
- Kawashima M, Nakagawa H, Tango T, Harada S, Noguchi T, Inagi M. Fexofenadine is effective at reducing itching associated with atopic dermatitis. Annales de Dermatologie et de Venereologie 2002;129(Suppl 1 Pt 2):1S413. [CENTRAL: CN‐00454385] [Google Scholar]
- Kawashima M, Tango T, Noguchi T, Inagi M, Nakagawa H, Harada S. Addition of fexofenadine to a topical corticosteroid reduces the pruritus associated with atopic dermatitis in a 1‐week randomized, multicentre, double‐blind, placebo‐controlled, parallel‐group study. British Journal of Dermatology 2003;148(6):1212‐21. [CENTRAL: CN‐00438764; PUBMED: 12828751] [DOI] [PubMed] [Google Scholar]
Kimura 2009 {published data only}
- Kimura U, Matsuba S, Chikenji T, Haruna K, Kamano M, Suga Y, et al. Usefulness of TARC in serum as treatment marker and combination treatment with loratadine and steroid ointment for atopic dermatitis. Skin Research 2009;8(2):125‐31. [CENTRAL: CN‐00802955] [Google Scholar]
Kircik 2013 {published and unpublished data}
- Kircik L. Management of pruritus with Xyzal in atopic dermatitis, 2013. https://clinicaltrials.gov/ct2/show/results/NCT00884325?term=levocetirizine&recrs=e&rslt=With&type=Intr&cond=Atopic+Dermatitis&rank=1§=X70156.
- Kircik L. Management of pruritus with levocetirizine dihydrochloride in atopic dermatitis in a randomized, double‐blind, placebo controlled study ‐ Interim analysis of 21 subjects. Allergy and Asthma Proceedings 2010;31(2):166. [CENTRAL: CN‐00980752] [Google Scholar]
Kuniyuki 2009 {published data only}
- Kuniyuki S, Yoshida Y, Maekawa N, Yamanaka K. Examination of olopatadine hydrochloride for anti‐itching and topical‐steroid sparing effects in patients with chronic eczema or dermatitis. Nishinihon Journal of Dermatology 2009;71(5):526‐30. [CENTRAL: CN‐00754437; DOI: 10.2336/nishinihonhifu.71.526] [DOI] [Google Scholar]
Langeland 1994 {published data only}
- Langeland T, Fagertun HE, Larsen S. Therapeutic effect of loratadine on pruritus in patients with atopic dermatitis. A multi‐crossover‐designed study. Allergy 1994;49(1):22‐6. [CENTRAL: CN‐00101711; PUBMED: 8198236] [DOI] [PubMed] [Google Scholar]
LaRosa 1994 {published data only}
- Rosa M, Ranno C, Musarra I, Guglielmo F, Corrias A, Bellanti JA. Double‐blind study of cetirizine in atopic eczema in children. Annals of Allergy 1994;73(2):117‐22. [CENTRAL: CN‐00103861; PUBMED: 8067594] [PubMed] [Google Scholar]
Leon 1989 {published data only}
- Leon G, Arellano I. Tolerance and efficacy of ketotifen in children with atopic dermatitis [Tolerancia y eficacia del ketotifeno en ninos con dermatitis atopica]. Dermatologia Revista Mexicana 1989;33(3):153‐60. [CENTRAL: CN‐00510646] [Google Scholar]
Monroe 1992 {published data only}
- Monroe EW. Relative efficacy and safety of loratadine, hydroxyzine, and placebo in chronic idiopathic urticaria and atopic dermatitis. Clinical Therapeutics 1992;14(1):17‐21. [CENTRAL: CN‐00083752; PUBMED: 1349509] [PubMed] [Google Scholar]
Munday 2002 {published data only}
- Munday J, Bloomfield R, Goldman M, Robey H, Kitowska GJ, Gwiezdziski Z, et al. Chlorpheniramine is no more effective than placebo in relieving the symptoms of childhood atopic dermatitis with a nocturnal itching and scratching component. Dermatology (Basel, Switzerland) 2002;205(1):40‐5. [CENTRAL: CN‐00397742; PUBMED: 12145433] [DOI] [PubMed] [Google Scholar]
Nuovo 1992 {published data only}
- Nuovo J, Ellsworth AJ, Larson EB. Treatment of atopic dermatitis with antihistamines: lessons from a single‐patient, randomized clinical trial. Journal of the American Board of Family Practice 1992;5(2):137‐41. [CENTRAL: CN‐00083686; PUBMED: 1575065] [PubMed] [Google Scholar]
Ruzicka 1998 {published data only}
- Ruzicka T, Karmann B, Kowok K, Altmeyer P. Atopisches ekzem: behandlung mit loratadin. Der Deutsche Dermatologe 1998;46(4):1‐8. [Google Scholar]
Savin 1986 {published data only}
- Savin JA, Dow R, Harlow BJ, Massey H, Yee KF. The effect of a new non‐sedative Hsub 1‐receptor antagonist (LN2974) on the itching and scratching of patients with atopic eczema. Clinical and Experimental Dermatology 1986;11(6):600‐2. [CENTRAL: CN‐00240981; PUBMED: 2889550] [DOI] [PubMed] [Google Scholar]
Simons 2007 {published data only}
- Simons FE, Early Prevention of Asthma in Atopic Children (EPAAC) Study Group. Safety of levocetirizine treatment in young atopic children: an 18‐month study. Pediatric Allergy and Immunology 2007;18(6):535‐42. [CENTRAL: CN‐00610098; PUBMED: 17561929] [DOI] [PubMed] [Google Scholar]
- Simons FE, Early Prevention of Asthma in Atopic Children Study Group. H1‐antihistamine treatment in young atopic children: effect on urticaria. Annals of Allergy, Asthma & Immunology 2007;99(3):261‐6. [CENTRAL: CN‐00612359; PUBMED: 17910330] [DOI] [PubMed] [Google Scholar]
Tharp 1998 {published data only}
- Tharp MD, Lowe NJ, Soter N, Kramer B. A double blind parallel study of cetirizine C 20 mg and placebo P in the management of patients with atopic dermatitis AD. Annals of Allergy, Asthma and Immunology 1998;80:110. [CENTRAL: CN‐00285014] [Google Scholar]
References to studies excluded from this review
Anton'ev 1988 {published data only}
- Anton'ev AA, Kaminka ME, Absava GI, Poroshina IuA, Ushakov IV. Bikarfen ‐ a new Soviet antihistaminic and antiserotonin preparation in the therapy of allergic dermatoses. Vestnik Dermatologii i Venerologii 1988;10:68‐72. [PUBMED: 2906194] [PubMed] [Google Scholar]
Balabolkin 1983 {published data only}
- Balabolkin II, Matus NI. Effectiveness of the use of zaditen in allergic dermatoses and the dermorespiratory syndrome in children. Pediatriia 1983;7:37‐9. [PUBMED: 6138748] [PubMed] [Google Scholar]
Bogdaszewska 1968 {published data only}
- Bogdaszewska‐Czabanowska J, Wiehler E. Trial of clinical evaluation of Doxergan, a new antihistaminic drug. Przeglad Dermatologiczny 1968;55(3):349‐55. [PUBMED: 4385459] [PubMed] [Google Scholar]
CastroTorres 1987 {published data only}
- Castro Torres A, Martín Moreno L, Jiménez Reyes J, Riesgo Bucher Y. Terfenadine compared to clemastine in the treatment of pruriginous cutaneous diseases. Double‐blind comparative study. Revista Clínica Española 1987;181(8):410‐2. [CENTRAL: CN‐00052433; PUBMED: 2894043] [PubMed] [Google Scholar]
Eberhartinger 1969 {published data only}
- Eberhartinger C, Ebner H. Experience with a new antihistaminic in dermatology. Results of a clinical double blind trial [German]. Wiener Klinische Wochenschrift 1969;119(38):630‐2. [CENTRAL: CN‐00003768; PUBMED: 4390510] [PubMed] [Google Scholar]
Foulds 1981 {published data only}
- Foulds IS, MacKie RM. A double‐blind trial of the H2 receptor antagonist cimetidine, and the H1 receptor antagonist promethazine hydrochloride in the treatment of atopic dermatitis. Clinical Allergy 1981;11(4):319‐23. [CENTRAL: CN‐00026318; PUBMED: 7028312] [DOI] [PubMed] [Google Scholar]
Furue 2009 {published data only}
- Furue M. Efficacy of ebastine for treatment of pruritic skin disease: patient assessment with a self‐administered questionnaire. Nishinihon Journal of Dermatology 2009;71(3):321‐7. [DOI: 10.2336/nishinihonhifu.71.321; EMBASE: 2009395751] [DOI] [Google Scholar]
Grabner 1970 {published data only}
- Grabner K. Clinical and experimental studies with the antihistaminic Tavegyl in dermatologic patients. Wiener Klinische Wochenschrift 1970;82(36):625‐7. [PUBMED: 4396798] [PubMed] [Google Scholar]
Imaizumi 2003 {published data only}
- Imaizumi A, Kawakami T, Murakami F, Soma Y, Mizoguchi M. Effective treatment of pruritus in atopic dermatitis using H1 antihistamines (second‐generation antihistamines): changes in blood histamine and tryptase levels. Journal of Dermatological Science 2003;33(1):23‐9. [CENTRAL: CN‐00470067; PUBMED: 14527736] [DOI] [PubMed] [Google Scholar]
Kawakami 2006 {published data only}
- Kawakami T, Kaminishi K, Soma Y, Kushimoto T, Mizoguchi M. Oral antihistamine therapy influences plasma tryptase levels in adult atopic dermatitis. Journal of Dermatological Science 2006;43(2):127‐34. [DOI: 10.1016/j.jdermsci.2006.04.002; PUBMED: 16843643] [DOI] [PubMed] [Google Scholar]
Kleine Natrop 1970 {published data only}
- Kleine‐Natrop HE, Richter G. Testing and use of 9,9‐dioxopromethazine (Prothanon) as an antihistaminic and antipruritic agent in dermatology. Die Pharmazie 1970;25(2):108‐11. [PUBMED: 4393058] [PubMed] [Google Scholar]
Kori Lindner 1997 {published data only}
- Kori‐Lindner C, Beckers C, Phleps W. Dimethindene maleate (DMM) in a therapeutic comparison with 2nd generation antihistamines. Allergo Journal 1997;6(4):204‐12. [EMBASE: 1997194298] [Google Scholar]
Korossy 1972 {published data only}
- Korossy S, Doroszlay J, Munkácsi A, Dömötör A. Evaluation of peritol in the management of allergic diseases of the skin. Therapia Hungarica (English Edition) 1972;20(3):95‐103. [CENTRAL: CN‐00008197; PUBMED: 4405678] [PubMed] [Google Scholar]
Liu 2008 {published data only}
- Liu SP, Huang XL, Fan CY. Effect observation of eczema treated with combined traditional Chinese medicine for external use with cetirizine and hydrocortisone butyrate. Southern China Journal of Dermato‐venereology [ Lingnan Pi Fu Xing Bing Ke za Zhi ] 2008;15(3):161‐2. [CENTRAL: CN‐00792887] [Google Scholar]
Lutsky 1993 {published data only}
- Lutsky BN, Schuller JL, Cerio R, DeLourdes Chieira M, Giannetti A, Goncalves HM, et al. Comparative study of the efficacy and safety of loratadine syrup and terfenadine suspension in the treatment of chronic allergic skin diseases in a pediatric population. Arzneimittel‐Forschung 1993;43(11):1196‐9. [CENTRAL: CN‐00098523; PUBMED: 8292064] [PubMed] [Google Scholar]
May 1966 {published data only}
- May KL, Nelemans FA. The antipruritic effect of Fenistil (dimethypyrindene) in allergic conditions. Double blind clinical study. Acta Allergologica 1966;21(4):337‐42. [CENTRAL: CN‐00000897; PUBMED: 6012601] [PubMed] [Google Scholar]
Mensing 1991 {published data only}
- Mensing H, Neumann Y. Antihistamine therapy in allergic skin disease. Comparison between loratadine and dimetindene. Zeitschrift fur Allgemeinmedizin 1991;67(8):498‐503. [CENTRAL: CN‐00398579] [Google Scholar]
Mesquita 1994 {published data only}
- Mesquita GJ, Ramos S, Barros MA, De AL. Dimetindene versus astemizole in the treatment of itching eczematous disorders: a randomized, double‐blind, comparative trial. European Journal of Dermatology 1994;4(3):202‐6. [CENTRAL: CN‐00172070] [Google Scholar]
Miller 1963 {published data only}
- Miller J. Clinical trial of a multiphasic antihistaminic preparation (clemizole, quercetin, amphetamine). Annals of Allergy 1963;21:692‐7. [PUBMED: 14085799] [PubMed] [Google Scholar]
Nitzschner 1970 {published data only}
- Nitzschner H. The introduction of 9,9‐dioxopromethazine (Prothanon) into the treatment of skin diseases in hospitalized patients. Die Pharmazie 1970;25(2):111‐3. [CENTRAL: CN‐00004543; PUBMED: 4393059] [PubMed] [Google Scholar]
Ohsawa 2014 {published data only}
- Ohsawa Y, Hirasawa N. The role of histamine H1 and H4 receptors in atopic dermatitis: from basic research to clinical study. Allergology International 2014;63(4):533‐42. [CENTRAL: 2015832218; DOI: 10.2332/allergolint.13-RA-0675] [DOI] [PubMed] [Google Scholar]
Ohtani 2004 {published data only}
- Ohtani T, Furukawa F, Kanauchi H, Ishii T, Tsujioka K. Usefulness of anti‐histamine drugs in treatment of facial atopic dermatitis: their effect on the dose‐reduction maintenance of tacrolimus ointments used for facial lesions. Skin Research 2004;3(3):316‐22. [CENTRAL: CN‐00516357] [Google Scholar]
Pacor 1992 {published data only}
- Pacor ML, Peroli P, Favari F, Lunardi C. Controlled study of oxatomide vs disodium chromoglycate for treating adverse reactions to food. Drugs Under Experimental and Clinical Research 1992;18(3):119‐23. [CENTRAL: CN‐00088282; PUBMED: 1358563] [PubMed] [Google Scholar]
Patel 1997 {published data only}
- Patel P, Gratton D, Eckstein G, Aberer W, Pryzbilla B, Chelly M, et al. A double‐blind study of loratadine and cetirizine in atopic dermatitis. Journal of Dermatological Treatment 1997;8(4):249‐53. [CENTRAL: CN‐00195014; DOI: 10.3109/09546639709160530] [DOI] [Google Scholar]
Pfab 2012 {published data only}
- Pfab F, Kirchner MT, Huss‐Marp J, Schuster T, Schalock PC, Fuqin J, et al. Acupuncture compared with oral antihistamine for type I hypersensitivity itch and skin response in adults with atopic dermatitis ‐ a patient‐ and examiner‐blinded, randomized, placebo‐controlled, crossover trial. Revista Portuguesa de Imunoalergologia 2012;20(3):228. [EMBASE: 2013056602] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfab F, Kirchner MT, Huss‐Marp J, Schuster T, Schalock PC, Fuqin J, et al. Acupuncture compared with oral antihistamine for type I hypersensitivity itch and skin response in adults with atopic dermatitis: a patient‐ and examiner‐blinded, randomized, placebo‐controlled, crossover trial. Allergy: European Journal of Allergy and Clinical Immunology 2012;67(4):566‐73. [CENTRAL: CN‐00832880; DOI: 10.1111/j.1398-9995.2012.02789.x; PUBMED: 22313287] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rajka 1965 {published data only}
- Rajka E, Korossy S, Gozony M. Therapeutic trial with antipruritic drugs. Dermatologica 1965;130:113‐29. [PUBMED: 14300883] [PubMed] [Google Scholar]
Rajka 1968a {published data only}
- Rajka G. Evaluation of drug influence on the itch duration in the skin of patients with atopic dermatitis, various eczemas and psoriasis. I. Experiments in involved skin. Comparison with itch threshold technique. Acta Dermato‐Venereologica 1968;48(2):93‐7. [CENTRAL: CN‐00001850; PUBMED: 4171289 ] [PubMed] [Google Scholar]
Rajka 1968b {published data only}
- Rajka G. Evaluation of drug influence on the itch duration in the skin of patients with atopic dermatitis, various eczemas and psoriasis. II. Experiments in unaffected skin. Comparison with itch threshold technique and clinical evaluation. Acta Dermato‐Venereologica 1968;48(2):98‐102. [CENTRAL: CN‐00001851; PUBMED: 4171290] [PubMed] [Google Scholar]
Schmoeckel 1992 {published data only}
- Schmoeckel C. Antihistamine therapy of itching skin disorders‐results of a multi‐centre outpatient study [Antihistaminika‐Therapie juckender Hauterkrankungen‐Ergebnisse einer multizentrischen Praxisstudie]. Dt Dermatologe 1992;40:1442‐52. [Google Scholar]
Sheng 2009 {published data only}
- Sheng N, Zong WK, Yu MW, Lin L, Cui PG. The clinical and immunological study of the mizolastine on the eczema and atopic dermatitis. Journal of Clinical Dermatology 2009;38(7):440‐2. [CENTRAL: CN‐00803052] [Google Scholar]
Simons 1984 {published data only}
- Simons FE, Simons KJ, Becker AB, Haydey RP. Pharmacokinetics and antipruritic effects of hydroxyzine in children with atopic dermatitis. Journal of Pediatrics 1984;104(1):123‐7. [CENTRAL: CN‐00186501; PUBMED: 6361228] [DOI] [PubMed] [Google Scholar]
Sugai 2003 {published data only}
- Sugai J, Kakurai M, Otsuki M, Nakagawa H. Combination therapy with 0.1% tacrolimus ointment and cetirizine for facial atopic dermatitis. Journal of the European Academy of Dermatology and Venereology 2003;17(Suppl 3):174. [CENTRAL: CN‐00478771] [Google Scholar]
Tarchalska 1988 {published data only}
- Tarchalska B, Zawisza E. A new generation of antihistaminics (H1). Polski Tygodnik Lekarski 1988;43(51‐52):1672‐5. [CENTRAL: CN‐00063527; PUBMED: 2908342] [PubMed] [Google Scholar]
Veien 1995 {published data only}
- Veien NK, Kaaber K, Larsen PO, Nielsen AO, Thestrup‐Pedersen K. Ranitidine treatment of hand eczema in patients with atopic dermatitis: a double‐blind, placebo‐controlled trial. Journal of the American Academy of Dermatology 1995;32(6):1056‐7. [CENTRAL: CN‐00114123; PUBMED: 7751455] [DOI] [PubMed] [Google Scholar]
Voigtlander 1989 {published data only}
- Voigtlander V. The antihistamine loratadine in the treatment of urticaria and itching eczema. Aktuelle Dermatologie 1989;15(8):254‐7. [EMBASE: 1989191983] [Google Scholar]
Wahlgren 1990 {published data only}
- Wahlgren CF, Hägermark O, Bergström R. The antipruritic effect of a sedative and a non‐sedative antihistamine in atopic dermatitis. British Journal of Dermatology 1990;122(4):545‐51. [CENTRAL: CN‐00067491; PUBMED: 2110817] [DOI] [PubMed] [Google Scholar]
Wahlgren 1991 {published data only}
- Wahlgren CF. Itch and atopic dermatitis: clinical and experimental studies [A]. Acta Dermato‐venereologica. Supplementum 1991;165(Suppl):1‐53. [CENTRAL: CN‐00081621; PUBMED: 1686130] [PubMed] [Google Scholar]
Weitgasser 1967 {published data only}
- Weitgasser H. Ambulatory testing of a new antihistaminic. Experience with Tavegil. Munchener Medizinische Wochenschrift (1950) 1967;109(20):1134‐6. [PUBMED: 4384702] [PubMed] [Google Scholar]
Wolfram 1967 {published data only}
- Wolfram S. Clinical experience with a new antihistaminic. Wiener Medizinische Wochenschrift (1946) 1967;117(38):868‐70. [PUBMED: 4385553] [PubMed] [Google Scholar]
Zhang 1998 {published data only}
- Zhang SF, Zhang TD, Qiu XW, Zhan QS. 36 cases of terfenadine combining with IFN‐gamma for atopic dermatitis. Chinese Journal of Dermatovenereology 1998;12(2):85‐6. [CENTRAL: CN‐00454961] [Google Scholar]
Zhang 2010 {published data only}
- Zhang YF, Lin ZS. Observation of clinical efficacy of Chinese medicine for 30 cases with infantile eczema. Journal of Pediatics of Traditional Chinese Medicine 2010;6(5):18‐20. [CENTRAL: CN‐01067884] [Google Scholar]
Zuluaga de Cadena 1989 {published data only}
- Zuluaga de Cadena A, Ochoa de VA, Donado JH, Mejia JI, Chamah HM, Montoya de Restrepo F. Comparative study of the effect of the hidroxicina the terfenadina and the astemizol in children with atopic dermatitis: Hospital General de Medellin‐Centro de Especialistas cE.S. 1986‐1988. Revista CES Medicina 1989;3(1):7‐13. [CENTRAL: CN‐00499475] [Google Scholar]
References to studies awaiting assessment
Borelli 1969 {published data only}
- Borelli S, Ene‐Popescu C, Michailov P. Drug‐induced changes in allergic mediatory reactivity in constitutional neurodermatitis patients [Medikamentös* bedingte Veränderungen der allergisch‐mediatorischen Reaktivität bei Neurodermitits‐constitutionalis Kranken]. Zeitschrift für Haut‐ und Geschlechtskrankheiten 1969;44(3):93‐8. [CENTRAL: CN‐00004010; PUBMED: 4391262] [PubMed] [Google Scholar]
Chunharas 2002 {published data only}
- Chunharas A, Wisuthsarewong W, Wananukul S, Viravan S. Therapeutic efficacy and safety of loratadine syrup in childhood atopic dermatitis treated with mometasone furoate 0.1 per cent cream. Journal of the Medical Association of Thailand 2002;85(4):482‐7. [PUBMED: 12118496] [PubMed] [Google Scholar]
DeBeule 1974 {published data only}
- Beule R. A double blind experimental study with a new synthetic antihistamine: mequitazine. Ars Medici Internationaal Tijdschrift voor Praktische Therapie 1974;3(12):2463‐72. [EMBASE: 1975170367] [Google Scholar]
- Beule R. Double blind test with a new antihistamine: Primalan (Mequitazine). Ars Medici Internationaal Tijdschrift voor Praktische Therapie 1974;3(11):1965‐73. [EMBASE: 1975160502] [Google Scholar]
Laugier 1978 {published data only}
- Laugier P, Orusco M. Comparative trial of antihistamine, mequitazine, and placebo. Current Medical Research and Opinion 1978;5(5):371‐5. [DOI: 10.1185/03007997809111900] [DOI] [PubMed] [Google Scholar]
NCT00160563 {unpublished data only}
- NCT00160563. Prevention of asthma with levocetirizine (36 month treatment) in young children suffering from eczema (atopic dermatitis) and sensitized to grass pollen and house dust mite and having completed the previous EPAAC trial (NCT00152464). clinicaltrials.gov/ct2/show/NCT00160563 (first received 26 April 2017).
Simons 2003 {published data only}
- Simons FER, Silas P, Portnoy JM, Catuogno J, Chapman D, Olufade AO. Safety of cetirizine in infants 6 to 11 months of age: a randomized, double‐blind, placebo‐controlled study. Journal of Allergy and Clinical Immunology 2003;111(6):1244‐8. [PUBMED: 12789224] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
CRT EU 23679 {unpublished data only}
- CRT EU 23679. Prednicarbat and cetirizin dihydrochloride in the treatment of acute stage of atopic eczema in children. www.clinicaltrialsregister.eu/ctr‐search/viewArt45/23679 (accessed 26 April 2017).
CTR EU 23697 {unpublished data only}
- CTR EU 23697. Effect of long‐term treatment with the h 1 ‐receptor antagonist cetirizine in the prevention of urticaria in children with atopic dermatitis. www.clinicaltrialsregister.eu/ctr‐search/viewArt45/23697 (accessed 26 April 2017).
CTR EU 29342 {unpublished data only}
- CTR EU 29342. Hydroxyzine: pharmacokinetics and antipruritic effect in children with atopic dermatitis. www.clinicaltrialsregister.eu/ctr‐search/viewArt45/29342 (accessed 26 April 2017).
CTR EU 39698 {unpublished data only}
- CTR EU 39698. Evaluation of the efficacy and the safety of DCL syrup in childhood atopic dermatitis. www.clinicaltrialsregister.eu/ctr‐search/viewArt45/39698 (accessed 26 April 2017).
UMIN000010519 {unpublished data only}
- UMIN000010519. JPRN‐UMIN000010519 Impact of olopatadine hydrochloride (Allelock(R)) against scratching behavior of atopic dermatitis patients at night (Kuniyuki_2009 study as principal investigator Yamanaka but this registrations is from 2013 with anticipated start of trial 2013). upload.umin.ac.jp/cgi‐open‐bin/ctr_e/ctr_view.cgi?recptno=R000012289 (first received 17 April 2013).
Additional references
Akiyama 2013
- Akiyama T, Carstens E. Neural processing of itch. Neuroscience 2013;250:697‐714. [DOI: 10.1016/j.neuroscience.2013.07.035] [DOI] [PMC free article] [PubMed] [Google Scholar]
Apfelbacher 2011
- Apfelbacher CJ, Diepgen TL, Schmitt J. Determinants of eczema: population‐based cross‐sectional study in Germany. Allergy 2011;66(2):206‐13. [PUBMED: 20804468] [DOI] [PubMed] [Google Scholar]
Apfelbacher 2013
- Apfelbacher CJ, Zuuren EJ, Fedorowicz Z, Jupiter A, Matterne U, Weisshaar E. Oral H1 antihistamines as monotherapy for eczema. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD007770.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Archer 2000
- Archer CB. The pathophysiology and clinical features of atopic dermatitis. Atopic Dermatitis. Cambridge: Cambridge University Press, 2000:25‐40. [Google Scholar]
Baurecht 2007
- Baurecht H, Irvine AD, Novak N, Illig T, Bühler B, Ring J, et al. Toward a major risk factor for atopic eczema: meta‐analysis of filaggrin polymorphism data. Journal of Allergy and Clinical Immunology 2007;120(6):1406‐12. [PUBMED: 17980411] [DOI] [PubMed] [Google Scholar]
Bero 2013
- Bero LA. Why the Cochrane risk of bias tool should include funding source as a standard item [editorial]. Cochrane Database of Systematic Reviews 2013, issue 12. [DOI: 10.1002/14651858.ED000075] [DOI] [PMC free article] [PubMed]
Bieber 2008
- Bieber T. Atopic dermatitis. New England Journal of Medicine 2008;358(14):1483‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bieber 2009
- Bieber T, Leung D. Atopic Dermatitis. 2nd Edition. New York, London: Informa Healthcare, 2009. [Google Scholar]
Biedermann 2015
- Biedermann T, Skabytska Y, Kaesler S, Volz T. Regulation of T cell immunity in atopic dermatitis by microbes: the Yin and Yang of cutaneous inflammation. Frontiers in Immunology 2015;6:353. [DOI: 10.3389/fimmu.2015.00353; PUBMED: 26217343] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brenninkmeijer 2008
- Brenninkmeijer EE, Schram ME, Leeflang MM, Bos JD, Spuls PI. Diagnostic criteria for atopic dermatitis: a systematic review. British Journal of Dermatology 2008;158(4):754‐65. [PUBMED: 18241277] [DOI] [PubMed] [Google Scholar]
Buddenkotte 2010
- Buddenkotte J, Steinhoff M. Pathophysiology and therapy of pruritus in allergic and atopic diseases. Allergy 2010;65(7):805‐21. [PUBMED: 20384615] [DOI] [PubMed] [Google Scholar]
Carroll 2005
- Carroll CL, Balkrishnan R, Feldman SR, Fleischer AB Jr, Manuel JC. The burden of atopic dermatitis: impact on the patient, family, and society. Pediatric Dermatology 2005;22(3):192‐9. [DOI: 10.1111/j.1525-1470.2005.22303.x] [DOI] [PubMed] [Google Scholar]
Charman 2004
- Charman CR, Venn AJ, Williams HC. The patient‐oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Archives of Dermatology 2004;140(12):1513‐9. [PUBMED: 15611432] [DOI] [PubMed] [Google Scholar]
Church 2013
- Church MK, Church DS. Pharmacology of antihistamines. Indian Journal of Dermatology 2013;58(3):219‐24. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Darsow 2012
- Darsow U, Pfab F, Valet M, Tölle TR, Ring J. Itch and eczema. Chemical Immunology & Allergy 2012;96:81‐8. [PUBMED: 22433375] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [PUBMED: 9310563] [DOI] [PMC free article] [PubMed] [Google Scholar]
ETFAD 1993
- No authors listed. Severity scoring of atopic dermatitis: the SCORAD index. Consensus Report of the European Task Force on Atopic Dermatitis. Dermatology 1993;186(1):23‐31. [PUBMED: 8435513] [DOI] [PubMed] [Google Scholar]
Finlay 1994
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI) ‐ a simple practical measure for routine clinical use. Clinical and Experimental Dermatology 1994;19(3):210‐6. [PUBMED: 8033378] [DOI] [PubMed] [Google Scholar]
Flohr 2014
- Flohr C, Mann J. New insights into the epidemiology of childhood atopic dermatitis. Allergy 2014;69(1):3‐16. [PUBMED: 24417229] [DOI] [PubMed] [Google Scholar]
Garmhausen 2013
- Garmhausen D, Hagemann T, Bieber T, Dimitriou I, Fimmers R, Diepgen T, et al. Characterization of different courses of atopic dermatitis in adolescent and adult patients. Allergy 2013;68(4):498‐506. [DOI: 10.1111/all.12112; PUBMED: 23452057] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gerbens 2017
- Gerbens LA, Prinsen CA, Chalmers JR, Drucker AM, Kobyletzki LB, Limpens J, et al. Evaluation of the measurement properties of symptom measurement instruments for atopic eczema: a systematic review. Allergy 2017;72(1):146‐63. [DOI: 10.1111/all.12959; PUBMED: 27322918] [DOI] [PubMed] [Google Scholar]
Greaves 2005
- Greaves MW. Antihistamines in dermatology. Skin Pharmacology & Physiology 2005;18(5):220‐9. [PUBMED: 16015020] [DOI] [PubMed] [Google Scholar]
Handley 1998
- Handley DA, Magnetti A, Higgins AJ. Therapeutic advantages of third generation antihistamines. Expert Opinion on Investigational Drugs 1998;7(7):1945‐54. [PUBMED: 15992014] [DOI] [PubMed] [Google Scholar]
Hanifin 1980
- Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Dermato‐Venereologica 1980;92 Suppl:44‐7. [Google Scholar]
Hanifin 2001
- Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Experimental Dermatology 2001;10(1):11‐8. [PUBMED: 11168575] [DOI] [PubMed] [Google Scholar]
He 2018
- He A, Feldman SR, Fleischer AB Jr. An assessment of the use of antihistamines in the management of atopic dermatitis. Journal of the American Academy of Dermatology 2018;79(1):92‐6. [DOI: 10.1016/j.jaad.2017.12.077; PUBMED: 29317281] [DOI] [PubMed] [Google Scholar]
Heinl 2016
- Heinl D, Prinsen CA, Deckert S, Chalmers JR, Drucker AM, Ofenloch R, et al. Measurement properties of adult quality‐of‐life measurement instruments for eczema: a systematic review. Allergy 2016;71(3):358‐70. [DOI: 10.1111/all.12806; PUBMED: 26564008] [DOI] [PubMed] [Google Scholar]
Heinl 2017
- Heinl D, Prinsen CAC, Sach T, Drucker AM, Ofenloch R, Flohr C, et al. Measurement properties of quality‐of‐life measurement instruments for infants, children and adolescents with eczema: a systematic review. British Journal of Dermatology 2017;176(4):878‐89. [DOI: 10.1111/bjd.14966; PUBMED: 27543747] [DOI] [PubMed] [Google Scholar]
Herman 2003
- Herman SM, Vender RB. Antihistamines in the treatment of dermatitis. Journal of Cutaneous Medicine & Surgery 2003;7(6):467‐73. [PUBMED: 15926215] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoare 2000
- Hoare C, Li Wan Po A, Williams H. Systematic review of treatments of atopic eczema. Health Technology Assessment 2000;4(37):1‐191. [PUBMED: 11134919] [PMC free article] [PubMed] [Google Scholar]
Holgate 2003
- Holgate ST, Canonica GW, Simons E, Taglialatela M, Tharp M, Timmerman H, et al. Consensus Group on New‐Generation Antihistamines (CONGA): present status and recommendations. Clinical and Experimental Allergy 2003;33(9):1305‐24. [PUBMED: 12956754] [DOI] [PubMed] [Google Scholar]
Hui 2013
- Hui D, Glitza I, Chisholm G, Yennu S, Bruera E. Attrition rates, reasons, and predictive factors in supportive care and palliative oncology clinical trials. Cancer 2013;119(5):1098‐105. [PUBMED: 23132290] [DOI] [PMC free article] [PubMed] [Google Scholar]
Illi 2004
- Illi S, Mutius E, Lau S, Nickel R, Grüber C, Niggemann B, et al. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. Journal of Allergy and Clinical Immunology 2004;113(5):925‐31. [DOI: 10.1016/j.jaci.2004.01.778; PUBMED: 15131576] [DOI] [PubMed] [Google Scholar]
Jin 2016
- Jin H, Kim Y, Rhie SJ. Factors affecting medication adherence in elderly people. Patient Preference and Adherence 2016;10:2117‐25. [PUBMED: PMC5077271] [DOI] [PMC free article] [PubMed] [Google Scholar]
Johansson 2004
- Johansson SG, Bieber T, Dahl R, Friedmann PS, Lanier BQ, Lockey RF, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. Journal of Allergy & Clinical Immunology 2004;113(5):832‐6. [PUBMED: 15131563 ] [DOI] [PubMed] [Google Scholar]
Kay 2000
- Kay GG. The effects of antihistamines on cognition and performance. Journal of Allergy & Clinical Immunology 2000;105(6 Pt 2):S622‐7. [PUBMED: 10856168] [DOI] [PubMed] [Google Scholar]
Kido‐Nakahara 2015
- Kido‐Nakahara M, Katoh N, Saeki H, Mizutani H, Hagihara A, Takeuchi S, et al. Comparative cut‐off value setting of pruritus intensity in visual analogue scale and verbal rating scale. Acta Dermato‐Venereologica 2015;95(3):345‐6. [DOI: 10.2340/00015555-1972] [DOI] [PubMed] [Google Scholar]
Klein 1999
- Klein PA, Clark RA. An evidence‐based review of the efficacy of antihistamines in relieving pruritus in atopic dermatitis. Archives of Dermatology 1999;135(12):1522‐5. [PUBMED: 10606058] [DOI] [PubMed] [Google Scholar]
Knol 2011
- Knol DL, Terwee CB, Mokkink LB, Vet HCW. Measurement in Medicine: A Practical Guide. London, UK: Cambridge University Press, 2011. [Google Scholar]
Kuo 2013
- Kuo IH, Yoshida T, Benedetto A, Beck LA. The cutaneous innate immune response in patients with atopic dermatitis. Journal of Allergy and Clinical Immunology 2013;131(2):266‐78. [DOI: 10.1016/j.jaci.2012.12.1563; PUBMED: 23374259] [DOI] [PubMed] [Google Scholar]
Langan 2006
- Langan SM, Thomas KS, Williams HC. What is meant by a "flare" in atopic dermatitis? A systematic review and proposal. Archives of Dermatology 2006;142(9):1190‐6. [DOI: 10.1001/archderm.142.9.1190; PUBMED: 16983006] [DOI] [PubMed] [Google Scholar]
Langan 2014
- Langan SM, Schmitt J, Williams HC, Smith S, Thomas KS. How are eczema 'flares' defined? A systematic review and recommendation for future studies. British Journal of Dermatology 2014;170(3):548‐56. [DOI: 10.1111/bjd.12747; PUBMED: 24266741] [DOI] [PubMed] [Google Scholar]
Leurs 2002
- Leurs R, Church MK, Taglialatela M. H1‐antihistamines: inverse agonism, anti‐inflammatory actions and cardiac effects. Clinical & Experimental Allergy 2002;32:489‐98. [PUBMED: 11972592] [DOI] [PubMed] [Google Scholar]
Luoma 1983
- Luoma R, Koivikko A, Viander M. Development of asthma, allergic rhinitis and atopic dermatitis by the age of five years. A prospective study of 543 newborns. Allergy 1983;38(5):339–46. [PUBMED: 6614406] [DOI] [PubMed] [Google Scholar]
Mallol 2013
- Mallol J, Crane J, Mutius E, Odhiambo J, Keil U, Stewart A, et al. The international study of asthma and allergies in childhood (ISAAC) phase three: a global synthesis. Allergologia et Immunopathologia 2013;41(2):73‐85. [DOI: 10.1016/j.aller.2012.03.001; PUBMED: 22771150] [DOI] [PubMed] [Google Scholar]
Mann 1989
- Mann KV, Crowe JP, Tietze KJ. Nonsedating histamine H1‐receptor antagonists. Clinical Pharmacy 1989;8(5):331‐44. [PUBMED: 2568212] [PubMed] [Google Scholar]
Matsui 2012
- Matsui D. Strategies to measure and improve patient adherence in clinical trials. Pharmaceutical Medicine 2012;23(5‐6):289‐97. [DOI: 10.1007/BF03256784] [DOI] [Google Scholar]
Moher 1998
- Moher, D. CONSORT: an evolving tool to help improve the quality of reports of randomized controlled trials. Consolidated Standards of Reporting Trials. JAMA 1998;279(18):1489‐91. [PUBMED: 9600488] [DOI] [PubMed] [Google Scholar]
Mortz 2015
- Mortz CG, Andersen KE, Dellgren C, Barington T, Bindslev‐Jensen C. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence and comorbidities. Allergy 2015;70(7):836‐45. [DOI: 10.1111/all.12619] [DOI] [PubMed] [Google Scholar]
Murota 2010
- Murota H, El‐latif MA, Tamura T, Amano T, Katayama I. Olopatadine hydrochloride improves dermatitis score and inhibits scratch behavior in NC/Nga mice. International Archives of Allergy and Immunology 2010;153(2):121‐32. [DOI: 10.1159/000312629; PUBMED: 20407268] [DOI] [PubMed] [Google Scholar]
Nutten 2015
- Nutten, S. Atopic dermatitis: global epidemiology and risk factors. Annals of Nutrition & Metabolism 2015;66(Suppl 1):8‐16. [DOI: 10.1159/000370220; PUBMED: 25925336] [DOI] [PubMed] [Google Scholar]
Odhiambo 2009
- Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI, ISAAC Phase Three Study Group. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. Journal of Allergy & Clinical Immunology 2009;124(6):1251‐8. [PUBMED: 20004783] [DOI] [PubMed] [Google Scholar]
Pereira 2017
- Pereira MP, Ständer S. Assessment of severity and burden of pruritus. Allergology International 2017;66(1):3‐7. [DOI: 10.1016/j.alit.2016.08.009; PUBMED: 27634668] [DOI] [PubMed] [Google Scholar]
Rehal 2011
- Rehal B, Armstrong A. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality‐of‐life instruments 1985–2010. PloS One 2011;6(4):e17520. [DOI: 10.1371/journal.pone.0017520; PUBMED: 21533286] [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager (RevMan) [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ring 2012a
- Ring J, Alomar A, Bieber T, Deleuran M, Fink‐Wagner A, Gelmetti C, et al. Guidelines for treatment of atopic eczema (atopic dermatitis) part I. Journal of the European Academy for Dermatology & Venereology 2012;26(8):1045‐60. [PUBMED: 22805051] [DOI] [PubMed] [Google Scholar]
Ring 2012b
- Ring J, Alomar A, Bieber T, Deleuran M, Fink‐Wagner A, Gelmetti C, et al. Guidelines for treatment of atopic eczema (atopic dermatitis) part II. Journal of the European Academy for Dermatology & Venereology 2012;26(9):1176‐93. [PUBMED: 22813359] [DOI] [PubMed] [Google Scholar]
Rönmark 2012
- Rönmark EP, Ekerljung L, Lötvall J, Wennergren G, Rönmark E, Torén K, et al. Eczema among adults: prevalence, risk factors and relation to airway diseases. Results from a large‐scale population survey in Sweden. British Journal of Dermatology 2012;166(6):1301‐08. [DOI: 10.1111/j.1365-2133.2012.10904.x; PUBMED: 22372948 ] [DOI] [PubMed] [Google Scholar]
Schmitt 2012
- Schmitt J, Spuls P, Boers M, Thomas K, Chalmers J, Roekevisch E, et al. Towards global consensus on outcome measures for atopic eczema research: results of the HOME II meeting. Allergy 2012;67(9):1111‐7. [DOI: 10.1111/j.1398-9995.2012.02874.x; PUBMED: 22844983] [DOI] [PubMed] [Google Scholar]
Schmitt 2013
- Schmitt J, Langan S, Deckert S, Svensson A, Kobyletzki L, Thomas K, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. Journal of Allergy and Clinical Immunology 2013;132(6):1337‐47. [DOI: 10.1016/j.jaci.2013.07.008; PUBMED: 24035157] [DOI] [PubMed] [Google Scholar]
Schmitt 2014
- Schmitt J, Spuls PI, Thomas KS, Simpson E, Furue M, Deckert S, et al. The Harmonising Outcome Measures for Eczema (HOME) statement to assess clinical signs of atopic eczema in trials. Journal of Allergy and Clinical Immunology 2014;134(4):800‐7. [PUBMED: 25282560] [DOI] [PubMed] [Google Scholar]
Sidbury 2014
- Sidbury R, Davis DM, Cohen DE, Cordoro KM, Berger TG, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis. Section 3. Management and treatment with phototherapy and systemic agents. Journal of the American Academy of Dermatology 2014;71(2):327‐49. [PUBMED: 24813298] [DOI] [PMC free article] [PubMed] [Google Scholar]
Silverberg 2013a
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population‐based study. Journal of Allergy & Clinical Immunology 2013;132(5):1132‐8. [PUBMED: 24094544] [DOI] [PubMed] [Google Scholar]
Silverberg 2013b
- Silverberg JI, Hanifin J, Simpson EL. Climatic factors are associated with childhood eczema prevalence in the United States. Journal of Investigative Dermatology 2013;133(7):1752‐9. [PUBMED: 23334343] [DOI] [PMC free article] [PubMed] [Google Scholar]
Simons 1999
- Simons FE. Prospective, long‐term safety evaluation of the H1‐receptor antagonist cetirizine in very young children with atopic dermatitis. ETAC Study Group. Early Treatment of the Atopic Child. Journal of Allergy and Clinical Immunology 1999;104(2):433‐40. [PUBMED: 10452767] [DOI] [PubMed] [Google Scholar]
Simons 2004
- Simons FE. Advances in H1‐antihistamines. New England Journal of Medicine 2004;351(21):2203‐17. [PUBMED: 15548781] [DOI] [PubMed] [Google Scholar]
Simons 2008
- Simons FER, Simons KJ. Antihistamines. In: Kay AB, Kaplan AP, Bousquet J, Holt PG editor(s). Allergy and Allergic Diseases. Second Edition. Oxford: Wiley‐Blackwell, 2008:550‐65. [Google Scholar]
Simons 2011
- Simons FER, Simons KJ. Histamine and H1‐antihistamines: celebrating a century of progress. Journal of Allergy and Clinical Immunology 2011;128(6):1139‐50. [DOI: 10.1016/j.jaci.2011.09.005; PUBMED: 22035879] [DOI] [PubMed] [Google Scholar]
Stone 2002
- Stone KD. Atopic diseases of childhood. Current Opinion in Pediatrics 2002;14(5):634‐46. [PUBMED: 12352260] [DOI] [PubMed] [Google Scholar]
Ständer 2002
- Ständer S, Steinhoff M. Pathophysiology of pruritus in atopic dermatitis: an overview. Experimental Dermatology 2002;11(1):12‐24. [PUBMED: 11952824] [DOI] [PubMed] [Google Scholar]
Takahashi 2004
- Takahashi H, Ishida‐Yamamoto A, Iizuka H. Effects of bepotastine, cetirizine, fexofenadine and olopatidine on histamine induced wheal and flare‐response, sedation and psychomotor performance. Experimental Dermatology 2004;29(5):526‐32. [DOI: 10.1111/j.1365-2230.2004.01618.x; PUBMED: 15347340] [DOI] [PubMed] [Google Scholar]
Thomas 2008
- Thomas K, Bath‐Hextall F, Ravenscroft J, Charman C, Williams H. Atopic eczema. In: Williams H, Bigby M, Diepgen T, Herxheimer A, Naldi L, Rzany B editor(s). Evidence‐based Dermatology. 2nd Edition. Oxford: Blackwell Publishing, 2008:128‐63. [Google Scholar]
Thomas 2015
- Thomas KS, Stuart B, O'Leary CJ, Schmitt J, Paul C, Williams HC, et al. Validation of treatment escalation as a definition of atopic eczema flares. PLoS One 2015;10(4):e0124770. [PUBMED: 25897763] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thomsen 2015
- Thomsen SF. Epidemiology and natural history of atopic diseases. European Clinical Respiratory Journal 2015;2(1):24642. [PUBMED: 26557262] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tominaga 2016
- Tominaga M, Takamori K. Atopic dermatitis. In: Misery L, Ständer L editor(s). Pruritus. Second Edition. New York, NY: Springer, 2016:131‐44. [Google Scholar]
Treadwell 2006
- Treadwell JR, Tregear SJ, Reston JT, Turkelson CM. A system for rating the stability and strength of medical evidence. BMC Medical Research Methodology 2006;19(6):52. [PUBMED: 17052350] [DOI] [PMC free article] [PubMed] [Google Scholar]
van Zuuren 2013
- Zuuren EJ, Fedorowicz Z, Carter B. Interventions for hirsutism excluding laser and photoepilation therapy. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD010334] [DOI] [PMC free article] [PubMed] [Google Scholar]
van Zuuren 2017
- Zuuren EJ, Fedorowicz Z, Christensen R, Lavrijsen A, Arents BWM. Emollients and moisturisers for eczema. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.CD012119.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vinding 2014
- Vinding GR, Zarchi K, Ibler KS, Miller IM, Ellervik C, Jemec GB. Is adult atopic eczema more common than we think? A population‐based study in Danish adults. Acta Dermato‐Venereologica 2014;94(4):480‐2. [PUBMED: 24217962] [DOI] [PubMed] [Google Scholar]
Wang 2011
- Wang Y, Chen X. How much of racial/ethnic disparities in dietary intakes, exercise, and weight status can be explained by nutrition‐ and health‐related psychosocial factors and socioeconomic status among US adults?. Journal of the American Dietetic Association 2011;111(12):1904‐11. [PUBMED: 22117667] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weidinger 2016
- Weidinger S, Novak N. Atopic dermatitis. Lancet 2016;387(10023):1109‐22. [DOI: 10.1016/S0140-6736(15)00149-X] [DOI] [PubMed] [Google Scholar]
Werfel 2016
- Werfel T, Heratizadeh A, Aberer W, Ahrens F, Augustin M, Biedermann T, et al. S2k guideline on diagnosis and treatment of atopic dermatitis – short version. Journal der Deutschen Dermatologischen Gesellschaft [Journal of the German Society of Dermatology] 2016;14(1):92‐106. [DOI: 10.1111/ddg.12871; PUBMED: 26713654] [DOI] [PubMed] [Google Scholar]
Williams 1994
- Williams HC, Burney PG, Hay RJ, Archer CB, Shipley MJ, Hunter JJ, et al. The UK Working Party's diagnostic criteria for atopic dermatitis. British Journal of Dermatology 1994;131(3):383‐96. [PUBMED: 7918015] [DOI] [PubMed] [Google Scholar]
Woosley 1996
- Woosley RL. Cardiac actions of antihistamines. Annual Review of Pharmacology & Toxicology 1996;36:233‐52. [PUBMED: 8725389] [DOI] [PubMed] [Google Scholar]
Yanai 2012
- Yanai K, Rogala B, Chugh K, Paraskakis E, Pampura AN, Boev R. Safety considerations in the management of allergic diseases: focus on antihistamines. Current Medical Research & Opinion 2012;28(4):623‐42. [PUBMED: 22455874] [DOI] [PubMed] [Google Scholar]
Yosipovitch 2008
- Yosipovitch G, Papoiu ADP. What causes itch in atopic dermatitis?. Current Allergy and Asthma Reports 2008;8(4):306‐11. [DOI: 10.1007/s11882-008-0049-z; PUBMED: 18606082] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Apfelbacher 2016
- Apfelbacher CJ, Jupiter A, Carter B, Weisshaar E, Böhmer MM. Oral H1 antihistamines as ‘add‐on’ therapy to topical treatment for eczema. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD012167] [DOI] [PMC free article] [PubMed] [Google Scholar]