

Abstract
Current nanoparticle (NP) drug carriers mostly depend on the the enhanced permeability and retention (EPR) effect for selective drug delivery to solid tumors. However, in the absence of persistent EPR effect, the peritumoral endothelium can function as an access barrier to tumors and negatively affect the effectiveness of NPs. In recognition of the peritumoral endothelium as a potential barrier in drug delivery to tumors, poly(lactic-co-glycolic acid) NPs are modified with a quinic acid (QA) derivative, synthetic mimic of selectin ligands. QA-decorated NPs (QA-NP) interact with human umbilical vein endothelial cells (HUVECs) expressing E-/P-selectins and induce transient increase in endothelial permeability to translocate across the layer. QA-NP reach selectin-upregulated tumors, achieving greater tumor accumulation and paclitaxel (PTX) delivery than polyethylene glycol-decorated NPs (PEG-NP). PTX-loaded QA-NP show greater anti-cancer efficacy than Taxol® or PTX-loaded PEG-NP at the equivalent PTX dose in different animal models and dosing regimens. Repeated dosing of PTX-loaded QA-NP for two weeks result in complete tumor remission in 40-60% of MDA-MB-231 tumor-bearing mice, while those receiving control treatments succumb to death. QA-NP can exploit the interaction with selectin-expressing peritumoral endothelium and deliver anti-cancer drugs to tumors to a greater extent than the level currently possible with the EPR effect.
Keywords: polymeric nanoparticles, drug delivery, quinic acid, selectin, tumor microenvironment
Graphical Abstract
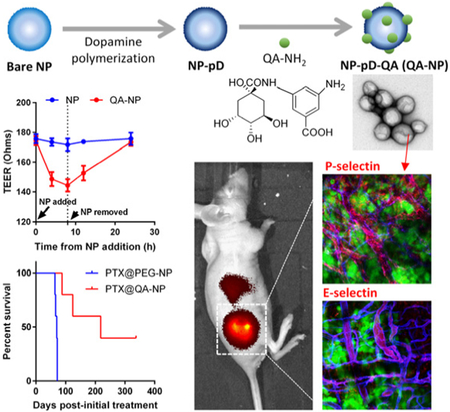
To overcome endothelial barrier in accessing tumors, polymeric nanoparticles are modified with a quinic acid (QA) derivative, synthetic mimic of selectin ligands, via polydopamine. The QA-decorated NPs interact with selectin-overexpressing endothelial cells and induce transient increase in endothelial permeability to translocate across the endothelial layer. QA-NP reach selectin-upregulated tumors, achieving greater tumor accumulation and paclitaxel delivery than polyethylene glycol-decorated NPs.
1. Introduction
Nanoparticles (NPs) have been considered a promising carrier of chemotherapeutic drugs.[1] The premise of NP-based chemotherapy is based on the notion that tumors tend to develop hypervasculature and poor lymphatic systems and, thus, provide selective access for NPs.[2] This phenomenon, called the enhanced permeability and retention (EPR) effect, has become the governing principle of most NP-based drug delivery. Nevertheless, the utility of NP-based chemotherapy has recently been challenged, due to the low tumor distribution of NPs[3] and the lack of evidence supporting the clinical benefits of NPs.[4] One of the potential reasons that limit NP delivery to tumors is the complex nature of the disease and high inter- and intra-subject differences, which result in variable efficiency of the EPR effect.[5] For the clinical success of NP-based chemotherapy, there should be additional means to leverage the EPR effect and enhance the delivery efficiency of NPs beyond the level currently possible.
We envision that the vascular endothelium – the first cell layer that circulating NPs encounter – serves as an access barrier to the underlying tumors; therefore, NPs that can actively interact with the endothelial barrier will have a greater chance to extravasate into the tumors. In this regard, we note E- and P-selectins, endothelial adhesion molecules expressed in response to local inflammatory stimuli. In the vasculature surrounding inflamed tissues, selectins mediate the interaction of leukocytes with the endothelium and their trafficking into the underlying tissues.[6] In addition to the well-known roles in inflammation, selectins are also implicated in cancer as a mediator of cell adhesion in metastasis and angiogenesis.[7] On the peritumoral endothelium, E-selectin promotes angiogenesis[8] and tumor proliferation[9] and serves as an adhesion point for circulating tumor cells and endothelial progenitor cells to support metastasis.[10] P-selectin is expressed constitutively in endothelial cells and platelets and stored in Weibel-Pallade bodies and α-granules, respectively.[11] Upon activation, P-selectin translocates from the intracellular granules to the cell membrane to mediate the adhesion and activation of leukocytes and platelets.[12] In tumor, P-selectin mediates endothelial interaction with circulating cancer cells as well as their aggregates with platelets.[13] Consistent with their roles in tumor progression, selectins are shown to be overexpressed in various human cancers[14] including breast,[15] kidney,[16] and lung cancers.[17] A recent study reports that among 420 clinical samples examined by immunohistochemistry, selective P-selectin expression is observed in multiple tumor types, including lung (19%), ovarian (68%), lymphoma (78%), and breast (49%) tumors.[14] In addition, selectin expression in tumor can be artificially induced by local ionizing radiation,[12b, 18] which is a clinically used cancer treatment protocol. Although the application for patients with other inflammatory diseases may be limited, the prevalent selectin expression in various types of tumor and the potential to induce local selectin expression in tumors suggest that NPs with affinity for selectins may preferentially bind to peritumoral endothelium and afford an increased chance to extravasate into tumors.
One of the natural ligands for selectins is sialyl Lewis-x (sLex), an oligosaccharide consisting of fucose, galactose, N-acetyllactosamine, and N-Acetylneuraminic acid.[19] sLex and its analogs have been widely explored as selectin antagonists for the therapy of inflammatory diseases[20] and the prevention of metastasis.[21] In particular, quinic acid (QA) and its derivatives have been noted as promising sLex mimics due to the ease of chemical modification.[22] These sLex mimics have been used as targeting ligands of macromolecular and NP drug carriers for the targeted delivery of chemotherapeutic drugs to the peritumoral endothelium.[14, 21b, 22b, 23] The affinity of QA derivatives for selectins is not high, comparable to sLex, with a millimolar range of the half maximal inhibitory concentration (IC50), in the inhibition of P-selectin binding to its natural ligand.[20h] We reason that the weak affinity ligand-receptor interactions may be beneficial for NP delivery to tumors as they will not prevent the NP transport beyond the endothelial binding. Here, the anticipated role of QA ligand on NP surface is to transiently populate NPs on the peritumoral endothelium, permitting the NPs to extravasate into the tumors.
Ligand-decorated NPs may be produced in various ways. First, NPs can be incubated with functional ligands to allow their physisorption on the NP surface.[24] However, the physisorbed ligands have limited stability in blood, where multiple molecular interactions can interfere with the NP-ligand interactions. Alternatively, ligands may be covalently conjugated to the surface via chemical reactions between functional groups of ligands and NP surface.[25] However, this method requires the presence of reactive functional groups on NPs, which are not always available in a desirable quantity. This leads to an increasingly complex adaptation of NP platforms, such as pre-functionalization of constituent polymers or chemical activation of the NP surface, imposing additional difficulties on the product development. To address this challenge, we employed a simple surface functionalization method based on dopamine polymerization, applicable to a variety of NP platforms irrespective of their chemical reactivity.[26] The dopamine polymerization method involves a simple incubation of NPs with dopamine in an oxidizing condition, which allows dopamine to polymerize and form a chemically reactive layer on the NP surface that accommodates ligand molecules.[27] This method allows for conjugation of various types of ligands, including a QA derivative, and flexible control of QA density on the NP surface. Such versatility and flexibility enable us to investigate the optimal conditions to conjugate QA on NPs for their transendothelial transport and compare the QA-modified NPs (QA-NP) with those containing typical stealth coating such as polyethylene glycol (PEG).
In this study, we demonstrate that QA mediates NP interaction with activated endothelial cells expressing selectins, promotes the NP transport across the activated endothelial layer in vitro, and brings more NPs to tumors than PEG-modified NPs (PEG-NP) in animal models. Consequently, paclitaxel (PTX) loaded in QA-NP (PTX@QA-NP) demonstrates greater anti-cancer effects than Taxol or PTX loaded in PEG-NP (PTX@PEG-NP) at the equivalent dose in multiple animal models and dosing regimens.
2. Results and Discussion
2.1. Synthesis of QA-NH2, synthetic mimic of sLex
QA-NH2, a synthetic mimic of sLex, was synthesized according to the previously reported scheme with slight modification (Figure 1a).[23a] The overall yield was 62%. 1H NMR confirmed the structure of the compound (Figure S1). Electron spray ionization mass spectrometry (ESI-MS) found the mass to charge (m/z) ratio of the compound to be 324.9 (−MS mode) as expected (Figure S2). QA-NH2 had no adverse effects on HUVEC proliferation (Figure S3).
Figure 1.

(a) Schematic of a quinic acid derivative, QA-NH2, a synthetic mimic of sLex. Overall yield: 62%. (b) Transmission electron micrographs of bare PLGA NPs (NP); pD-coated PLGA NPs (NP-pD); and QA-conjugated NP-pD (NP-pD-QA or QA-NP). Negative staining with 2% uranyl acetate. Scale bar: 100 nm.
2.2. Development and characterization of QA-NP
QA-NP was prepared with the dopamine-mediated surface modification method.[26] PLGA NPs (bare NP) were first prepared by the single emulsion solvent evaporation method. The bare NP was covered with a polymerized dopamine (pD) layer, which accommodated QA-NH2 via Michael addition and/or Schiff base reactions,[27-28] to form QA-NP. The ESI-MS analysis of QA-NP (Figure S4) provided qualitative evidence for successful conjugation of QA-NH2 on QA-NP. PLGA NPs simply incubated with QA-NH2 without pD functionalization (QA/NP) had no QA-NH2 signature peaks on ESI-MS, confirming the essential role of pD coating as the mediator of QA-NH2 conjugation. Transmission electron micrographs (TEM) (Figure 1b) identified a layer on the surface of the core NPs due to pD coating. However, there was no visible difference between NP-pD and NP-pD-QA (QA-NP) in the size and surface morphology, similar to previous examples using other ligands.[26] The number of QA-NH2 conjugated to the surface of QA-NP was indirectly determined by subtracting the amount of QA-NH2 remaining in the reaction medium from the original QA-NH2 feed. The QA-NH2 conjugated to unit surface area of QA-NP increased linearly with QA-NH2 feed (Figure S5) and did not saturate in the tested range. We attribute the efficient conjugation to hydrogen bonding between multiple hydroxyl groups of QA, which may allow for cooperative addition to the surface.
QA-NP had a z-average of 151 nm in diameter and a zeta potential of −11.8 mV according to dynamic light scattering (DLS) (Table 1, based on n = 3 independent batches). The particle size was larger than that estimated by TEM (100-120 nm) (Figure 1b). This difference is greater than the thickness of the typically attributed hydration layer, which is no more than a few nanometers, suggesting that the NPs underwent a mild degree of aggregation in the buffer in which the DLS measurement was performed. Prior to the biological evaluation of QA-NP, their size distribution in 50% FBS was tested at a NP concentration ranging from 0.05 to 0.2 mg/mL. Irrespective of the concentration, QA-NP showed a consistent peak at 120 nm (Figure S6a), indicating the resolution of NP aggregation. The two distinctive peaks at 10 nm and 80 nm were identified to be serum proteins and their aggregates (Figure S6b). Over 2 h in 50% FBS, no aggregation or agglomeration of QA-NP occurred. QA-NP could be stored as lyophilized solid in 4 °C for up to one month without substantial changes in the physical properties (data not shown).
Table 1.
Particle size, zeta potential, and polydispersity index (PDI) of NPs.
| Name | Z-average (d, nm) |
Zeta Potential (mV) |
PDI |
|---|---|---|---|
| Bare NP | 134 ± 12 | −10.4 ± 9.3 | 0.1 ± 0.02 |
| NP-pD | 142 ± 17 | −11.2 ± 8.1 | 0.2 ± 0.06 |
| PEG-NP | 163 ± 11 | −10.7 ± 6.2 | 0.2 ± 0.04 |
| PEG-NP (PLGA-DyLight 594) | 167 ± 14 | −12.1 ± 4.9 | 0.2 ± 0.07 |
| QA-NP | 151 ± 14 | −11.8 ± 4.9 | 0.1 ± 0.03 |
| QA-NP (PLGA-FITC) | 149 ± 7 | −9.3 ± 5.5 | 0.1 ± 0.03 |
| QA-NP (PLGA-Alexa 555) | 147 ± 11 | −10.4 ± 6.1 | 0.1 ± 0.05 |
| QA-NP (PLGA-ICG) | 154 ± 20 | −10.8 ± 5.7 | 0.1 ± 0.06 |
| QA-NP (PLGA-DyLight 594) | 158 ± 14 | −11.6 ± 6.4 | 0.2 ± 0.03 |
| Bare NP (PLGA-TPGS) | 171 ± 33 | −12.3 ± 6.6 | 0.2 ± 0.09 |
| PTX@PEG-NP (PLGA-TPGS) | 184 ± 41 | −11.5 ± 5.7 | 0.2 ± 0.07 |
| PTX@QA-NP (PLGA-TPGS) | 179 ± 26 | −10.8 ± 6.4 | 0.1 ± 0.04 |
Data: mean ± s.d. (n = 3 independently and identically prepared batches)
2.3. QA-NP interact with activated HUVECs
QA-NP interactions with endothelial cell layer was investigated with human umbilical vein endothelial cells (HUVECs). The HUVEC model was chosen on the basis of reported use as a cell model in the investigation of tumor-endothelial cell interactions.[29] QA-NP-HUVEC interaction was examined with confocal microscopy and flow cytometry (Figure 2a-c). Specifically, HUVECs were incubated with FITC-labeled QA-NP or bare NP with or without tumor necrosis factor-alpha (TNF-α) treatment. A common condition for endothelial expression of selectins (10 ng/mL of TNF-α for 4 h) was followed.[30] Bare NPs were also tested in the same manner. Figure 2a shows that QA-NP were associated with the HUVECs with the majority on the cell membrane whereas bare NPs showed minimal non-specific binding. QA-NP was also observed in empty space between cells. Given that QA-NP did not bind to the plate without cells (Figure S7), we speculate that soluble E-selectin secreted by the activated HUVECs[31] deposited on the plate surface and interacted with QA-NP. To quantify the NPs retained with the cells, fluorescence intensity of the cell suspension was measured by flow cytometry. Activated HUVECs incubated with QA-NP showed the greatest fluorescence intensity compared to control groups (non-activated HUVECs with QA-NP (or bare NP) or activated HUVECs with bare NP), in agreement with confocal imaging. These results suggest that QA-NP have the affinity for the activated HUVECs and thus the potential to interact with the peritumoral endothelium.
Figure 2.

(a) Confocal imaging of FITC-labeled NP and QA-NP incubated with resting or TNF-α-activated HUVECs. Green: NPs; blue: cell nuclei; red: plasma membrane. The box in QA-NP/activated HUVEC image was magnified at the bottom in two channels. (b) Representative flow cytometry histogram. (c) Quantitative measurement of HUVECs interacting with NP or QA-NP (n = 3 tests of a representative batch, mean ± s.d.). ****: p<0.0001 vs. QA-NP with TNF-α activated HUVEC by Dunnett’s multiple comparisons test. Competitive inhibition of QA-NP binding to HUVECs by (d) free QA-NH2; (e) anti-E-selectin antibody; and (f) anti-P-selectin antibody. QA-NP to HUVEC binding was quantified by flow cytometry. (n = 3 tests of a representative batch, mean ± s.d.). ****: p<0.0001 by Dunnett’s multiple comparisons test. The experiment was repeated with an independently and identically prepared batch of QA-NP, and the results are presented in Figure S8.
To confirm the mediators of QA-NP and HUVEC interactions, the incubation was performed after pre-treatment with free QA-NH2, anti-E-selectin or anti-P-selectin antibodies. QA-NP binding to the activated HUVECs was reduced by free QA-NH2 (Figure 2d, Figure S8a), anti-E-selectin (Figure 2e, Figure S8b), and anti-P-selectin antibodies (Figure 2f, Figure S8c) in a dose-dependent manner, which indicates that free QA-NH2 competes with QA-NP and that the blockade of E/P-selectin interferes with QA-NP binding to the activated HUVECs, respectively. These results confirm that the binding of QA-NP to the activated HUVECs was mediated by the QA interaction with E/P-selectin.
2.4. QA-NP translocate across the activated HUVEC layer inducing transient increase in endothelial permeaiblity
We hypothesized that the interactions between QA-NP and the activated HUVEC would increase the chance for NPs to travel through the endothelial layer and reach the underlying tissues. To estimate this potential in vitro, we assessed the QA-NP transport across the endothelial cell layer grown on a Transwell insert (Figure 3a). We placed QA-NP and control NPs on the apical side of the Transwell containing a confluent HUVEC layer indicated by the transendothelial electrical resistance (TEER) values (Figure 3b), with and without TNF-α pre-treatment, and quantified the NPs remaining in the apical side and those recovered from the basolateral side. As expected, the transport across the endothelial layer was observed only with QA-NP and TNF-α-activated (i.e., E/P-selectin expressing) HUVECs (Figure 3c).
Figure 3.

(a) Schematic diagram of transendothelial transport of NPs. (b) Representative TEER plot of HUVEC monolayer. The arrow indicates the time of NP addition. (c) The percentage of NP transport across the confluent HUVEC monolayer with and without TNF-α activation (n = 3 test of a representative batch, mean ± s.d.). ****: p<0.0001 vs. NP with non-activated HUVECs in each compartment by Holm-Sidak’s multiple comparisons test. (d) Representative TEER plot of HUVEC monolayer with values measured at shorter intervals from the time NP was added. The plot in a dashed box was magnified on the right. (e) Relationship between the ligand density and the transendothelial transport of QA-NP (n = 3 tests of a representative batch, mean ± s.d.).
Literature suggests that the binding of antibodies or cancer cells to E-selectin enhances the transendothelial permeability and cell migration[32] by activating extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase, which initiate the disassembly of vascular endothelial-cadherin complexes.[33] We suspect that the binding of QA-NP to activated HUVECs may have enhanced transendothelial transport of QA-NP in a similar manner. To test if QA-NP treatment increased transendothelial permeability, we repeated the same experiment measuring TEER values after QA-NP or bare NP treatment with shorter intervals. With the addition of QA-NP (but not with bare NP), the TEER value immediately started to decrease over 8 h (Figure 3d). Once QA-NP was removed, the TEER value was gradually restored over the next 16 h. This result suggests that the transendothelial transport of QA-NP may be mediated by the transient increase in paracellular permeability of the endothelium due to the QA-NP binding to E/P-selectins.
To examine the relationship between ligand density and the transendothelial transport, we measured the transport of QA-NP across the activated HUVECs varying the ligand densities (Figure 3e). QA-NP transport increased as the ligand density of QA-NP increased from 8 to 36 QA-NH2/nm2 but rather decreased with further increase of the ligand density to 74 QA-NH2/nm2. We speculate that excess QA-NH2 may have formed intermolecular clustering due to hydrogen bonding instead of interacting with selectins. Overall, the Transwell experiment supports that QA-NH2 ligands with an optimal surface density help transport QA-NP across the activated HUVECs.
2.5. QA-NP accumulate in tumors with forced selectin expression by focal irradiation
To test the distribution of QA-NP in vivo, we first used an animal model with forced E/P-selectin expression via focal ionizing radiation.[12b, 18, 34] For comparison with traditional PEG-modified NPs that depend mostly on the EPR effect, PEG-NP were prepared in the same manner as QA-NP except for the replacement of QA-NH2 with PEG-NH2 (Figure S9). Balb/c mice were inoculated with syngeneic CT26 colon cancer cells on both hind limbs. One side was treated with 6 Gy of X-irradiation, and the other was left untreated. One day after intravenous (IV) injection of QA-NP or PEG-NP, tumors were collected and optically cleared for confocal imaging with the immunofluorescence staining of endothelial cells (anti-CD31), P-selectin, E-selectin, and macrophages (anti-F4/80).[35] QA-NP displayed greater accumulation in tumor than PEG-NP, even in the absence of irradiation (Figure 4, Figure S10). The difference apparently increased in the irradiated tumors, which manifested stronger E- and P-selectin stains, indicating that QA-NP accumulation in tumor increased with selectin expression. The irradiated tumors also showed an increased population of macrophages (a response to irradiation-induced inflammation[36]); however, we exclude the possibility of the tumor-associated macrophages mediating NP uptake[37] for the following reasons: (i) PEG-NP accumulation remained negligible in the irradiated tumor despite the increased macrophage population (Figure 4, Figure S10a), (ii) there is little overlap between QA-NP and F4/80+ cells (Figure S10b), and (iii) our in vitro study suggested that the surface QA suppress the macrophage uptake of QA-NP (Figure S11), likely due to the multiple hydroxyl groups increasing the hydrophilicity of the surface. It is worth noting that, at the time of observation, both E- and P-selectin signals were not colocalized with CD31 but rather seen with macrophages. This may reflect the transient nature of selectin expression on endothelial surface[38, 39] and the macrophage uptake of the secreted selectins. To observe QA-NP transport in cancer models with more stable expression of endothelial selectins, we next screened cancer cells that may induce selectin expression in the endothelial cells.
Figure 4.

Confocal imaging of optically cleared, antibody-stained CT26 tumor sections from animals receiving QA-NP (Top) and PEG-NP (Bottom), with or without 6 Gy X-irradiation (± ionizing radiation, ± IR). Tumors were sampled one day after injection. Different views of each section are shown in Figure S10. n = 2 mice per NP, 2 sections per mouse.
2.6. Cancer cells induce endothelial selectin expression
Endothelial expression of selectins is induced by cytokines from the neighboring cells.[15a, 40] To examine whether cancer cell lines have such paracrine effects on endothelial cells, we incubated endothelial cells in the media conditioned with various cancer cells of matching species and measured the E/P-selectin expression. All the tested human cell lines induced the expression of selectins in HUVECs (Figure S12a, b). MDA-MB-231-conditioned medium induced E-selectin expression to the greatest extent, followed by MCF-7-conditioned medium. P-selectin expression was the greatest with MCF-7-conditioned medium, then with MDA-MB-231 and A2780-conditioned media. Similarly, murine B16F10 and 4T1 cells induced significant expression of E/P-selectin in mouse hemangioendothelioma (EOMA) cells (Figure S12c, d). These results support broad applicability of E/P-selectin targeting. As the MDA-MB-231-conditioned medium induced both E- and P-selectin expression, we chose a mouse model of MDA-MB-231 xenograft for subsequent in vivo evaluation of QA-NP. Among murine cell lines, B16F10 cells were used to make a syngeneic model of melanoma for additional evaluation of PTX@QA-NP. E/P-selectin expression in MDA-MB-231 tumor was confirmed by immunohistochemistry (Figure S13) as well as intravital imaging in live animals (Figure S14). In the intravital imaging, E/P-selectin expression was mainly observed in the blood vessel unlike immunohistochemistry of MDA-MB-231 model or clinical samples,[14] which showed broader selectin distribution. This difference may be explained by the way the selectins were stained. While immunohistochemistry stains the sectioned tissues letting the tumor tissues contact antibodies, intravital imaging provides antibodies through circulation, where the antibodies are likely to bind first to the endothelium without proceeding further.
2.7. In vivo fluorescence imaging shows QA-NP distribution
On the basis of the above in vitro results, we hypothesized that QA-NP might develop active interactions with peritumoral endothelium via E/P-selectin, translocate across the activated endothelium, as it did with the activated HUVEC, and accumulate in MDA-MB-231 tumors to a greater extent than PEG-NP. To trace the NP distribution over time by whole body fluorescence imaging, we labeled QA-NP with indocyanine green (ICG), a near infrared fluorescence dye, by conjugating the dye to PLGA via carbodiimide chemistry. NPs made with the PLGA-ICG conjugate (ICG-NP) remained stable in 50% FBS in contrast to those physically encapsulating ICG (ICG/NP) (Figure S15), indicating that the fluorescence of the ICG-NPs would represent NPs in the whole body imaging.
The ICG-labeled (indicated as *) QA-NP and PEG-NP or free ICG with equivalent fluorescence intensity were injected via tail vein and imaged over 24 h (Figure 5a, Figure S16). Free ICG spread throughout the body immediately following the administration and gradually disappeared by hepatobiliary elimination.[41] On the other hand, QA-NP* and PEG-NP* showed up in the liver immediately after the injection. Significant tumor accumulation of QA-NP* was observed starting at 2 h post injection, whereas nearly no fluorescence was detected in animals treated with PEG-NP*, let alone free ICG. The QA-NP* signal in the tumor gradually decreased over time but persisted throughout the 24-h experiment period (Figure S17). Animals receiving PEG-NP* and free ICG did not show fluorescence in tumors at all time points. It is curious that PEG-NP* was not observed at all in the tumor. Although not as vascularized as highly angiogenic LS174T xenografts[42] (Figure S18), MDA-MB-231 xenograft is still more vascularized than muscle tissues, according to the extent of Evans blue accumulation (LS174T/muscle = 1.34, MDA-MB-231/muscle = 1.04), in accordance with the literature.[43] We speculate that the fluorescence detection threshold was set too high to capture weak fluorescence intensity of tumors with PEG-NP*. Major organs were excised and imaged ex vivo 24 h post injection. Consistent with live whole body imaging, animals receiving PEG-NP* showed the greatest fluorescence signal in the liver, followed by the lung and spleen (Figure 5b). QA-NP* showed less accumulation in the reticuloendothelial system (RES) and significantly higher tumor signals than PEG-NP*. This result was confirmed in another set of animals (Figure S19). The ex vivo imaging obtained at 6 h post injection showed a similar trend as 24 h post injection, except for the relative distribution of PEG-NP* in the RES organs. Although the contribution of the EPR effect may not be excluded, the superior tumor accumulation of QA-NP* relative to PEG-NP*, consistently shown in Figures 4 and 5, indicates that the selectin-mediated transport plays a dominant role in tumor distribution of QA-NP.
Figure 5.

(a) Representative whole body imaging of animals over 24 h from the injection of each treatment. Animals bearing subcutaneous MDA-MB-231 tumor xenografts were treated with QA-NP* (top), PEG-NP* (middle), and free ICG (bottom) by tail vein injection. See Figure S16 for all animals receiving the treatments. (b) Fluorescence intensity of ex vivo images of major organs retrieved at the end of whole body imaging (24-h post-injection). n=5 mice per group. ****: p<0.0001 by Bonferroni's multiple comparisons test following two-way ANOVA.
To investigate whether the interaction between QA-NP and E/P-selectin helped QA-NP to extravasate and enter tumors, we used intravital microscopy to visualize QA-NP in mice with GFP-expressing MDA-MB-231 tumors. QA-NP was observed from 2 h post injection near the blood vessel, and the signal increased over time indicating gradual accumulation of QA-NP in the tumor (Figure 6). This observation agrees with in vitro Transwell result and provides an in vivo proof-of-concept that QA-NP interaction with E/P-selectin improved extravasation and enhanced tumor accumulation. It is worth noting that most QA-NP were nevertheless observed near the vasculature, which indicates limited intratumoral transport of the NPs. Given that efficient tumor penetration was reported with NPs of 50 nm or smaller,[44] further reduction of NP size may be necessary to improve intratumoral transport of QA-NP. Alternatively, conditionally-disintegrated assemblies of ultrasmall NPs[45] are also conceivable to afford both long-term circulation (favoring relatively large size[44c]) and deep intratumoral penetration (favoring small size[44c]).
Figure 6.

Intravital microscopy images of GFP-expressing MDA-MB-231 tumor in a dorsal skinfold chamber after 2, 6, and 24 h after IV injection of Alexa Fluor® 555-labeled QA-NP. Top: Overlays of blood vessels (blue), QA-NP* (red), and MDA-MB-231-GFP tumor cells (green). Bottom: Overlays of blood vessels and QA-NP only.
2.8. PTX-loaded QA-NP demonstrates greater in vivo anti-cancer efficacy than Taxol or PEGylated NPs
2.8.1. Production of PTX-loaded QA-NP
We next examined whether QA-NP would deliver paclitaxel (PTX), a model anti-cancer drug, better than PEG-NP and achieve superior anti-cancer efficacy. Typical PTX loading efficacy in PLGA NPs is no more than 5 wt%.[46] The low drug loading increases the NP dose requirement and thus the concentration and/or volume of injection, posing technical challenges in administration. To increase the drug loading capacity, we used a conjugate of PLGA and tocopheryl polyethylene glycol 1000 succinate (PLGA-TPGS) instead of PLGA following the reported method.[47] PLGA-TPGS was synthesized by ring opening polymerization and confirmed by 1H NMR (Figure S20) and Matrix-Assisted Laser Desorption/Ionization-MS (MALDI-MS). The molecular weight (MW) ranged 4-8 kDa with an average of 5 kDa according to MALDI-MS (Figure S21). NP made of PLGA-TPGS showed greater PTX loading (13.0 ± 0.2%) than those made of PLGA150k (loading efficiency: 4.0 ± 0.01%) or PLGA30k and physically stabilized by TPGS (PLGA30k/TPGS, loading efficiency: 9.6 ± 0.5%) (Figure S22a). Release kinetics performed in PBS containing 0.2% Tween 80 showed that in 48 h PTX@NP (PLGA-TPGS) released 59.8 ± 3.5% of the loaded PTX; PTX@NP (PLGA30k/TPGS) 79.1 ± 9.8%; and PTX@NP (PLGA150k) 46.8 ± 0.5% (Figure S22b, c). TEM images showed no significant morphological difference between NPs made of PLGA and those of PLGA-TPGS (Figure 1 vs. Figure S23). To confirm the functionality of QA-conjugated PLGA-TPGS NPs, QA-NPs made of regular PLGA or PLGA-TPGS (and bare NPs made of respective polymers as negative controls) were used as competitive inhibitors for the binding of FITC-labeled QA-NP to the activated HUVECs. The binding was not affected by the added bare NPs but decreased with the addition of QA-NPs in a dose-dependent manner, irrespective of the polymer type used for the core NPs (Figure S24). This result indicates that core NPs did not affect the surface functionality and PLGA-TPGS NPs might serve as a valid substitute for PLGA NPs as core particles. It also demonstrates the robustness and broad applicability of QA conjugation process and suggests the potential for modular design of QA-modified NPs. On the basis of high drug loading and consistent surface functionality, we used PLGA-TPGS in producing PTX-loaded QA-NP (PTX@QA-NP) for the subsequent in vivo studies. Free PTX, a physical mixture of PTX and blank QA-NP, and PTX@QA-NP showed similar half maximal inhibitory concentration (IC50) values in MDA-MB-231 cells, confirming that QA-NP itself had no additional toxicity (Figure S25).
2.8.2. Design of anti-cancer efficacy studies
The anti-cancer efficacy of PTX@QA-NP was tested in comparison with Taxol, a commercial surfactant-solubilized PTX formulation, and PTX@PEG-NP, a control NP without QA modification. The maximum tolerated dose of PTX@QA-NP was first determined. Healthy female nude mice survived 10 injections of PTX@QA-NP at PTX 30 mg/kg/dose over 2 weeks without a significant weight loss (Figure S26). Since the reported maximum tolerated dose of Taxol was relatively low (20 mg/kg PTX equivalent, qd × 5),[48] the evaluation of QA-NP in vivo anti-cancer efficacy was performed in two different settings: one compared with Taxol at its known tolerated doses (20 mg/kg PTX equivalent, single administration or q3d × 5 at 20 mg PTX/kg/dose over 2 weeks) and the other compared with PTX@PEG-NP at the NP’s maximum tolerated dose (30 mg/kg/dose × 10 times over 2 weeks).
2.8.3. Anti-cancer efficacy: PTX@QA-NP vs. Taxol
The anti-cancer efficacy of PTX@QA-NP was first tested with a single dose (20 mg/kg PTX equivalent) in a female nude mouse model of subcutaneous MDA-MB-231 tumor. Taxol was used at the same dose as a reference. As shown in Figure 7a, PTX@QA-NP attenuated the growth of MDA-MB-231 tumors more efficiently than Taxol. The median survival time of animals receiving PTX@QA-NP was 62 days, significantly longer than that of Taxol (44 days, p < 0.01, Log-rank test). The same regimen was administered to male C57BL/6 mice with syngeneic B16F10 tumors (Figure S27). Reflecting the known aggressive nature,[49] the B16F10 tumors grew much faster than MDA-MB-231 tumors, but PTX@QA-NP still induced superior tumor attenuation to Taxol at the same dose.
Figure 7.

Anti-cancer efficacy of PTX@QA-NP and Taxol in female nude mice with MDA-MB-231 xenografts. (a) Treatments given as a single dose equivalent to PTX 20 mg/kg. Mice were sacrificed once the tumor volume (V) reached 2000 mm3 or showed any signs of morbidity defined in the animal protocol. n=5 mice per group. Tumor specific growth rate is defined as ΔlogV/Δt (t: time in days). *: p<0.05 by unpaired t-test. (b) Treatments given as five doses of 20 mg PTX equivalent/kg/dose over 2 weeks. n=5 mice per group. *: p<0.05 by Gehan-Breslow-Wilcoxon test. Inset graphs show the tumor growth and survival over 150 days.
Next, PTX@QA-NP was given every three days over 2 weeks (total five times: q3d × 5) at 20 mg/kg/dose (Figure 7b). PTX@QA-NP offered a significant survival benefit with complete remission in 60% (3 out of 5) animals by 357 days (as of the submission of this report), while only one surviving in the Taxol group during the same period. Two animals treated with Taxol reached the end point based on the tumor size, one with deteriorating health condition with an ulcerated tumor, and the other found dead with no apparent reason. These results collectively demonstrate that PTX@QA-NP brings greater anti-cancer efficacy than Taxol at the equivalent dose.
2.8.4. Anti-cancer efficacy: PTX@QA-NP vs. PTX@PEG-NP
PTX@QA-NP was compared with PTX@PEG-NP (both made with PTX@PLGA-TPGS core particles) at the maximum tolerated dose. Female nude mice with subcutaneous MDA-MB-231 animals were treated with 10 injections of one of the following treatments over the period of 2 weeks: (i) saline; (ii) blank QA-NP; (iii) blank PEG-NP; (iv) PTX@PEG-NP at 30 mg PTX/kg/dose; and (v) PTX@QA-NP at 30 mg PTX/kg/dose. Only the PTX@QA-NP group showed a survival benefit compared to the saline group, with 40% complete remission as of 367 days (as of the submission of this report) (Figure 8a). All other groups succumbed to death with a median survival time of 68 days (saline), 86 days (blank QA-NP), 80 days (blank PEG-NP), and 70 days (PTX@PEG-NP). It was surprising that the PTX@PEG-NP group was no better than the negative control groups and had no surviving animals. We reasoned that PTX@PEG-NP might have induced the production of anti-PEG antibodies. The accelerated blood clearance (ABC) phenomemon due to anti-PEG IgM is well documented with PEGylated liposomes and shown to be responsible for rapid elimination of repeatedly administered liposomes.[50] The extent of ABC may vary with the extent of modification, and the length and density of PEG, but since it has been reported with different types of NP systems,[51] we infer that anti-PEG antibodies may have accelerated the clearance of subsequently-dosed PTX@PEG-NPs. To investigate this possibility, we treated healthy nude mice with saline, blank PEG-NP or blank QA-NP, collected blood 5 days later, and added the serum to a plate coated with mPEG or QA to determine the presence of antibodies. Animals receiving PEG-NP produced antibodies that bound to PEG-decorated surface, while other treatments (saline, QA-NPs) did not (Figure 8b). QA-NP injection did not induce the production of anti-QA antibodies. These results demonstrate that QA-NP delivered PTX to tumors more efficiently than PEG-NP by repeated administration.
Figure 8.

(a) Anti-cancer efficacy of PTX@QA-NP, compared with those of saline, blank PEG-NP, blank QA-NP, and PTX@PEG-NP. Treatments were given as 10 doses of 30 mg PTX equivalent/kg/dose over 2 weeks. n=5 mice per treatment. *: p<0.05 by Log-rank (Mantel-Cox) test. Inset graphs show the tumor growth and survival over 150 days. (b) Formation of antibodies to each ligand (PEG, QA), detected at 5 days after a single injection of PEG-NP, QA-NP, or saline (as a negative control). n = 3 mice per group. (c) Plasma PTX concentration profile after a single injection of PTX@QA-NP or PTX@PEG-NP at a dose equivalent to 20 mg/kg PTX in MDA-MB-231 xenograft-bearing mice. n=3 mice per time per group. (d) Biodistribution of PTX in major organs 24 h after a single injection of PTX@QA-NP or PTX@PEG-NP at a dose equivalent to 20 mg/kg PTX in MDA-MB-231 xenograft-bearing mice. n=3 mice per group. ****: p<0.0001 by Sidak’s multiple comparisons test.
2.8.5. Pharmacokinetics and biodistribution of PTX delivered by QA-NP and PEG-NP
Pharmacokinetics and biodistribution of PTX were compared following a single IV injection of PTX@QA-NP and PTX@PEG-NP at a dose equivalent to PTX 20 mg/kg. The two NPs showed similar PK profiles and comparable plasma PK parameters upon non-compartmental analyses (Table 2 and Figure 8c). However, PTX@QA-NP resulted in a significantly higher PTX concentration in tumor than PTX@PEG-NP (7119 ng/g for PTX@QA-NP and 145.5 ng/g for PTX@PEG-NP), when measured at 24 h after IV injection, whereas PTX concentration in other major organs remained comparable (Figure 8d). This result is consistent with the superior anti-cancer efficacy of PTX@QA-NP relative to PTX@PEG-NP (Figure 8a). The PTX distribution did not exactly mirror the ex vivo imaging, which showed the semiquantitative NP distribution at 24 h post-injection (Figure 5b). This discrepancy may be explained by the preferential removal of PTX from the RES organs, such as hepatic metabolism[52] and/or PTX redistribution following phagocytic degradation.
Table 2. Plasma pharmacokinetic parameters of PTX@PEG-NP, and PTX@QA-NP.
| PK Parameter | Formulation | |
|---|---|---|
| PTX@PEG-NP | PTX@QA-NP | |
| t1/2 (h) | 7.7 | 5.4 |
| Cmax (μg/mL) | 3.4 (0.5) | 2.4 (0.05) |
| AUC0-last (h•μg/mL) | 10.6 (0.4) | 11.0 (0.1) |
| AUC0-∞ (h•μg/mL) | 11.3 | 11.6 |
| MRT (h) | 5.8 | 6.3 |
| CL (L/h/kg) | 1.8 | 1.7 |
| Vss (L/kg) | 10.4 | 11.0 |
PK parameters were obtained from non-compartmental analyses. The numbers in parentheses denote the standard errors (SE) associated with estimated parameters.
3. Conclusion
QA was conjugated on the surface of PLGA NPs to promote the extravasation of the NPs via the interaction with selectin-expressing peritumoral endothelium. The QA-decorated NPs (QA-NP) interacted with selectin-expressing HUVECs and translocated across the confluent HUVEC layer. QA-NP showed greater accumulation and PTX delivery in selectin-expressing tumors as compared to PEG-NP. PTX-loaded QA-NP showed greater anti-cancer efficacy than Taxol or PTX-loaded PEG-NP at equivalent PTX doses, leading to complete tumor remission in 40-60% of MDA-MB-231 tumor-bearing mice by repeated dosing over 2 weeks. The superior anti-cancer efficacy of QA-NP is also partly due to the lack of antibody formation. Our results support that QA-NP can exploit the interaction with selectin-expressing peritumoral endothelium and deliver anti-cancer drugs to tumors to a greater extent than conventional PEG-NP.
4. Experimental Section
Materials
PLGA (LA:GA = 85:15, acid endcap, MW: 150 kDa), PLGA (LA:GA = 50:50, acid endcap, MW: 30kDa), fluorescein (FITC)-conjugated PLGA (LA:GA = 48:52, MW: 5 kDa), and PLGA-ethylenediamine (PLGA-NH2, LA:GA = 57:43, MW: 5 kDa) were purchased from Akina, Inc. (West Lafayette, IN, USA). Indocyanine green-N-succinimidyl ester (ICG-NHS) was purchased from Intrace medical (Lausanne, Switzerland). Hoechst 33342 and recombinant human tumor necrosis factor-alpha (TNF-α) were purchased from Invitrogen (Eugene, OR, USA). Methoxy-polyethylene glycol-amine (mPEG-NH2, MW: 5 kDa) was purchased from JenKem Technology USA (Allen, TX, USA). Collagen I rat-tail, Vacutainer® PST™ Tubes with spray-coated lithium heparin and a gel for plasma separation, Vacutainer® Plus Plastic Serum Tubes with spray-coated silica for serum separation, and Calcein AM were purchased BD Biosciences (San Jose, CA, USA). Dopamine hydrochloride was purchased from Alfa Aesar (Ward Hill, MA, USA). CellMask™ deep red membrane staining dye, Alexa Fluor® 555 NHS ester (succinimidyl ester), DyLight® 594-NHS ester, goat anti-mouse IgM secondary antibody, HRP-conjugated, Gibco® Dulbecco’s modified eagle’s medium (DMEM), Gibco® Eagle’s minimum essential complete medium (EMEM), Gibco® RPMI 1640 medium (RPMI) Gibco® Iscove’s modified Dulbecco’s medium (IMDM) and were purchased from ThermoFisher Scientific (Waltham, MA, USA). Endothelial cell growth medium EGM™−2 BulletKit™ was purchased from Lonza, Inc. (Williamsport, PA, USA). E-selectin antibody (P2H3) FITC, E-selectin antibody (UZ5) FITC, P-selectin antibody (CTB201), and P-selectin antibody (CTB201) FITC were purchased from Santa Cruz (Dallas, TX, USA). Transwell polycarbonate membrane cell culture inserts (6.5 mm) with 3.0 μm pore were purchased from Corning (Corning, NY, USA). Paclitaxel (PTX) was a generous gift from Samyang Biopharm (Seongnam, South Korea). All other materials were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Synthesis of QA-NH2 (Supplementary Scheme 1)
(1S,3R,4S,5R)-1,3,4,5-tetraacetoxycyclohexane-1-carboxylic acid (Compound 2):
Quinic acid (compound 1, 961 mg, 5 mmol) was dissolved in 12 mL of acetic anhydride - pyridine 1:2 mixture. The solution was mixed with 4-dimethylaminopyridine (20 mg, 0.16 mmol) and stirred for 12 h at 5 °C. The reaction mixture was then added to ice water, acidified to pH 3 and extracted with dichloromethane (DCM). The extract was dried with sodium sulfate and concentrated to give a white solid (1.75 g, 97% Yield). ESI: (M+H)+: 361.
(1R,2S,3R,5S)-5-((3-amino-5-(methoxycarbonyl)phenyl)carbamoyl) cyclohexane-1,2,3,5-tetrayl tetraacetate (Compound 3):
Compound 2 (958 mg, 2.7 mmol) was dissolved in 15 mL of dimethylformamide. The solution was mixed with N-hydroxybenzotrizole (432 mg, 3.2 mmol) and stirred for 5 min at 0 °C. (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (612 mg, 3.2 mmol) and triethyl amine (0.5 mL, 4.3 mmol) were then added to the mixture and stirred for 1 h at 0 °C. Finally, a solution of methyl 3,5-diaminobenzoate (1.7 g, 7.7 mmol) in dimethylformamide (5 mL) was added to the mixture, which was warmed up to room temperature and stirred for 72 h. The reaction mixture was then added to ice water and extracted with DCM. The extract was dehydrated with sodium sulfate, filtered and further evaporated. The residue was purified by column chromatography on silica gel (hexane-ethyl acetate 3:7) to yield a brown solid compound (1.3 g, 92% Yield). ESI: (M+H)+: 509.
3-amino-5-((1S,3R,4S,5R)-1,3,4,5-tetrahydroxycyclohexane-1-carboxamido)benzoic acid (Compound 4):
A solution of compound 3 (102 mg, 0.2 mmol) in tetrahydrofuran (5 mL) was stirred with lithium hydroxide monohydrate (63 mg, 1.5 mmol) for 48 h at room temperature. The reaction mixture was then acidified to pH 5 using Amberlite acidic resin, filtered, and purified by reverse column chromatography on C18-reversed phase silica gel (water-acetonitrile 1:2) to yield a white solid compound (45 mg, 69% Yield). ESI: (M+H)+: 327.
Synthesis of ICG-conjugated PLGA, Alexa Fluor® 555-conjugated PLGA, and DyLight® 594-conjugated PLGA
Two hundred milligrams of PLGA-ethylene diamine were dissolved in 2 mL DMSO at room temperature. 1.5 mg of ICG-NHS, Alexa Fluor® 555-NHS, or DyLight® 594-NHS was dissolved in 1 mL DMSO at room temperature with rigorous vortex mixing. One milliliter of N,N-diisopropylethylamine (DIPEA) was added to PLGA solution, followed by dropwise addition of ICG-NHS, Alexa Fluor® 555-NHS or DyLight® 594-NHS solution. The mixture was constantly stirred for 2 h, put in a regenerated cellulose dialysis bag with a MW cut off 3,500 Da (Spectrum Labs, Rancho Dominguez, CA, USA) and dialyzed against excess DMSO 3 times and DCM 3 times. The purified sample was collected via rotary evaporation and stored at −20 °C.
Synthesis of TPGS-conjugated PLGA (PLGA-TPGS)
Glycolide 1 g, lactide 1 g, D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) 400 mg were dissolved in toluene containing 0.5 wt% stannous octoate. The reaction was carried out at 145 °C under oxygen- and moisture-free environment for 16 h. The resulting product was dissolved in DCM and then precipitated in excess cold methanol to separate unreacted reagents. The final product, PLGA-TPGS, was collected by filtration, washed twice with distilled water and dried under vacuum at 60 °C for 72 h.
Preparation of Bare NP
Bare NP was produced by the single emulsion solvent evaporation method. In brief, 100 mg of PLGA was dissolved in 4 mL of DCM. The polymer solution was added to 12 mL of a 5% polyvinyl alcohol (PVA) solution and emulsified using a Sonics Vibracell probe sonicator (Sonics, Newtown, CT, USA) for 2 min. The emulsion was added to 40 mL of deionized (DI) water and stirred for 4 h to evaporate DCM. The NP was then collected by an Optima MAX-XP ultracentrifuge (Beckman Coulter, Brea, CA, USA) at 34000 ×g and washed with DI water three times. For confocal microscopy and flow cytometry, NP was prepared with FITC-conjugated PLGA. For imaging of NPs in focally irradiated tumors, NP was prepared with DyLight®
594-conjugated PLGA. For optical in vivo imaging, NP was prepared with ICG-conjugated PLGA. For intravital microscopy, NP was made with Alexa Fluor® 555-conjugated PLGA. PTX-encapsulated NPs (PTX@NP) were prepared with PLGA150k, PLGA30k, and PLGA-TPGS5k. For PTX@NP with PLGA150k (PTX@NP (PLGA150k)), 20 mg of PLGA150k and 2 mg of PTX were dissolved in 1 mL of DCM, homogenized in 4 mL of 5% PVA by probe sonication (2 min, with 4s on, 2s off at 40% amplitude), and stirred in 20 mL of DI water overnight. For PTX@NP with PLGA30k, 20 mg of PLGA30k and 2 mg of PTX dissolved in 1 mL of DCM was homogenized in 4 mL aqueous solution containing 0.5% PVA and 0.1% TPGS, and stirred in 20 mL of 0.5% PVA overnight. To indicate the use of TPGS as an emulsifier, we called the NPs PTX@NP (PLGA30k/TPGS). PTX@NP with PLGA-TPGS5k (PTX@NP (PLGA-TPGS5k)) was prepared by homogenizing 1 mL DCM solution of 20 mg of PLGA-TPGS5k and 2 mg of PTX in 4 mL of 5% PVA and stirring in 20 mL of DI water. The NPs were collected via ultracentrifugation, and washed three times prior to freeze drying.
NP Surface Modification
The core NPs were coated with pD by 3 h incubation in 2 mL of dopamine hydrochloride solution in Tris buffer (10 mM, pH 8.5) at room temperature with rotation. The pD-coated PLGA NPs (NP-pD) were collected by ultracentrifugation and washed twice with DI water. For further surface functionalization, NP-pD were resuspended in Tris buffer containing QA-NH2 or mPEG-NH2, maintaining the weight ratio of NP-pD to the ligands at 1:2 unless specified otherwise. After 30 min incubation, the NPs were collected via ultracentrifugation and washed with DI water twice. The surface-functionalized NPs were designated as QA-NP (or NP-pD-QA) and PEG-NP (or NP-pD-PEG), respectively.
NP Characterization
Physical characterization:
NPs were dispersed in phosphate buffer (1 mM, pH 7.4), and their sizes and zeta potentials were measured by a Malvern Zetasizer Nano ZS90 (Worcestershire, UK). NP morphology was observed by Tecnai™ transmission electron microscopy (FEI, Hillsboro, OR, USA) after negative staining with 2% uranyl acetate. QA-NH2 surface decoration was confirmed by ESI-MS analysis of QA-NP. PEG-NH2 surface decoration was confirmed by MALDI-TOF MS.
QA-NH2 Ligand Density:
A known amount of NP-pD was resuspended in Tris buffer (10 mM, pH 8.5) containing a known amount of QA-NH2 (QA-NH2 feed). After 30 min incubation, the NP was collected via ultracentrifugation, and the supernatant was analyzed by an Agilent 1100 HPLC system (Palo Alto, CA, USA) equipped with Ascentis C18 column (25 cm × 4.6 mm, particle size: 5μm) to quantify unconjugated free QA-NH2. The mobile phase was a 90:10 volume mixture of water and ACN and run at 0.5 mL/min. QA-NH2 was detected by a UV detector at the wavelength of 227 nm. The surface-conjugated QA-NH2 was calculated by subtracting the unconjugated QA-NH2 from the QA-NH2 feed. The QA-NH2 ligand density (QA-NH2/nm2) was calculated as the number of surface-conjugated QA-NH2 divided by the total surface area of NP-pD.
Serum stability:
For evaluation of size stability, 5 mg of QA-NP were suspended in 5 mL of 50% FBS and incubated for 2 h at room temperature. Particle size distribution was measured by a Malvern Zetasizer Nano ZS90. For evaluation of fluorescence stability, ICG-labeled QA-NP was suspended in 50% FBS. NP pellet and supernatant were separated by ultracentrifugation at pre-specified time points and imaged with IVIS Lumina to detect fluorescence intensity.
PTX loading efficiency:
NPs with a premeasured mass were dissolved in 50% ACN at a concentration of 50 μg/mL and filtered with 0.45 μm PVDF syringe filter. HPLC analysis was performed with an Agilent 1100 HPLC system equipped with Ascentis C18 column (25 cm × 4.6 mm, particle size: 5 μm). The mobile phase was a 50:50 volume mixture of water and ACN and run at 1 mL/min. PTX was detected by a UV detector at a wavelength of 227 nm. Drug loading efficiency was defined as the PTX amount divided by the NP mass.
PTX release kinetics:
NPs equivalent to 10 μg of PTX were suspended in 1 mL of PBS containing 0.2% Tween 20 and incubated at 37 °C under constant agitation. At timed intervals, NP suspension was ultracentrifuged at 34000 μg for 15 min. 0.8 mL of supernatant was sampled for HPLC analysis and replaced with 0.8 mL of fresh release medium. NPs were resuspended in the medium by brief ultrasonication and returned to incubation.
Cell Culture
Human umbilical vein endothelial cells (HUVECs, ATCC, Manassas, VA, USA) at passage 4 were grown in EGM-2 complete medium. Culture plates were pre-coated with 5 μg/cm2 of rat tail collagen type I. Human breast adenocarcinoma cells (MDA-MB-231, ATCC; MCF-7, ATCC) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Human colon adenocarcinoma cells (LS174T, ATCC) were cultured in EMEM supplemented with 10% FBS. Human ovarian cancer cells (A2780, ATCC) were cultured in RPMI supplemented with 10% FBS. Human ovarian cancer cells (NCI/ADR-RES) were cultured in DMEM supplemented with 10% FBS. Mouse hemangioendothelioma cells (EOMA, ATCC) were cultured in DMEM supplemented with 10% FBS. Mouse mammary carcinoma cells (4T1, ATCC) were cultured in RPMI supplemented with 10% FBS. Mouse melanoma cells (B16F10, ATCC) were cultured in RPMI supplemented with 10% FBS. Mouse monocyte macrophages (J774A.1, ATCC) were cultured in DMEM supplemented with 10% FBS. All media were supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin. All cells were subcultured at a ratio of 1:7.5 when they became 70-80% confluent. HUVECs was used between 4-7 passages, and all other cells were used in less than 20 passages.
Cytotoxicity
Cytotoxicity of QA-NH2 on HUVECs:
HUVECs were seeded in a 96 well plate at a density of 10,000 cells per well coated with rat tail collagen type I. A group of cells were incubated with 10 ng/mL of TNF-α for 4 h and then treated with QA-NH2 (1 nM to 1 mM) for 24 h. The cell viability was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Cells were treated with 75 μg of MTT and incubated for 4 h. The formazan crystals were dissolved in stop/solubilization solution (50% DMF, 20% SDS, pH 5) and quantified by a SpectraMax M3 microplate reader (Molecular Devices, CA, USA) at the wavelength of 562 nm. The measured absorbance was normalized to the absorbance of control cells that did not receive any treatment.
Cytotoxicity of PTX@QA-NP on MDA-MB-231 cells:
MDA-MB-231 cells were seeded in a 96 well plate at a density of 10,000 cells per well. After overnight incubation, the culture medium was replaced with fresh DMEM, to which PTX, PTX + QA-NP, and PTX@QA-NP were added to provide the final PTX concentration ranging from 0.1 nM to 100 μM. After 72 h incubation, the cell viability was determined by the MTT assay.
Confocal Microscopy of In Vitro NP-cell Interactions
HUVECs were seeded in a 35-mm dish with a glass window (MatTek Corporation, Ashland, MA, USA) coated with rat tail collagen type I and grown in EGM-2 complete medium. When HUVECs were 80% confluent, cells were treated with 10 ng/mL of TNF-α for 4 h. The medium was replaced with EGM-2 containing 0.2 mg/mL of FITC-labeled NP suspension and incubated for 2 h at 37 °C. The suspension was replaced with fresh EGM-2 medium containing 0.5 μM CellMask™ deep red membrane staining dye. After 10 min, cells were gently rinsed with PBS twice. Hoechst nuclear stain (10 μL at the concentration of 0.2 mg/mL) was added 10 min prior to the imaging. Imaging was performed with a Nikon A1R confocal microscope (Melville, NT, USA) equipped with a Spectra Physics 163C argon ion laser and a Coherent CUBE diode laser. The NPs, cell nuclei, and cell membrane were excited using 495, 361, and 649 nm laser, respectively, and detected at 519, 497, and 666 nm.
Flow Cytometry for Quantitative analysis of NP-cell Interactions
For quantitative analysis of NP-cell interaction, HUVECs were treated with FITC-labeled NPs in the same way as confocal microscopy and gently washed with PBS twice to remove free or loosely bound NPs. The cells were then trypsinized, collected by centrifugation at 930 μg for 5 min, resuspended in 0.2 mL of PBS at 4 °C, and analyzed by an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA) equipped with an FL-1 detector (λex/λem = 488/525 nm).
For competition assay, free QA-NH2, E-selectin antibody, or P-selectin antibody were pre-incubated with HUVECs for 30 min prior to the NP treatment and flow cytometry analysis. In another competition assay, known concentrations of NP, NP (PLGA-TPGS5k), QA-NP, and QA-NP (PLGA-TPGS5k) were pre-incubated with the activated HUVECs for 30 min. The pre-treated HUVECs were incubated with FITC-labeled QA-NP for 2 h and analyzed by flow cytometry.
Macrophage interactions of bare NP and QA-NP were compared. J774A.1 macrophages were seeded at a density of 100,000 cells/cm2 in a 6-well plate. When 70-80% confluent, the cells were incubated with 0.1 mg/mL of FITC-labeled QA-NP, NP, or NP-pD at an equivalent fluorescence level in DMEM and incubated for 2 h at 37 °C. The cells were washed with PBS twice and analyzed by flow cytometry.
Transendothelial NP Transport Across Confluent HUVECs
HUVECs were seeded in a Corning Transwell insert coated with rat tail collagen type I and grown in EGM-2 complete medium. The transendothelial electrical resistance (TEER) across the HUVEC layer in the Transwell insert was monitored daily by EVOM2™ epithelial voltohmmeter (World Precision Instruments, Sarasota, FL, USA). When the TEER value reached a plateau (indicating confluency), the HUVEC layer was treated with 10 ng/mL of TNF-α for 4 h, followed by incubation with 0.1 mg/mL of FITC-labeled NP or QA-NP for 8 h. The fluorescence intensity of total NPs prior to incubation and those of NPs in the apical and basolateral sides of a Transwell insert after incubation period were measured by FluoroMax®-3 spectrofluorometer (Horiba Scientific, Edison, NJ, USA). In a separate study, the confluent, TNF-α activated HUVECs were treated with 0.1 mg/mL of NP or QA-NP for 8 h, followed by 16 h incubation in NP-free medium. The TEER values were measured at 0, 4, 8, 12, and 24 h from the NP treatment.
Endothelial Expression of E-selectin and P-selectin by Tumor-Conditioned Media
Human cell lines:
HUVECs were seeded in a 6-well plate coated with rat tail collagen type I at a density of 10,000 cells/cm2. When HUVECs became 70-80% confluent, cells were exposed to the culture media conditioned with MCF-7, MDA-MB-231, LS174T, A2780, or NCI-ADR cells for 4 h. Respective cell-free media were used as medium controls, and TNF-α (10 ng/mL) was used as a positive control. The HUVECs were trypsinized and collected via centrifugation. The cell pellets were resuspended in PBS and stained with FITC-labeled antihuman E-selectin or anti-human P-selectin antibody according to the manufacturer’s protocol. The extent of E-selectin or P-selectin expression was quantified by flow cytometry.
Mouse cell lines:
EOMAs at passage 2 were seeded in a 6-well plate at a density of 10,000 cells/cm2. When EOMAs became 70-80% confluent, cells were exposed to the culture media conditioned with 4T1 or B16F10 cells for 4 h. Respective cell-free media were used as medium controls, and TNF-α (10 ng/mL) and lipopolysaccharide (LPS O111:B4 from E. coli, 10 ng/mL) were used as positive controls. EOMAs were collected in the same way as above, stained with FITC-labeled anti-mouse E-selectin or anti-mouse P-selectin antibody, and analyzed by flow cytometry.
Immunohistochemistry of E-/P-selectin expression in MDA-MB-231 xenograft model
MDA-MB-231 xenograft tumors were fixed in formalin and embedded in paraffin blocks. The tissue blocks were cut in 3 μm thick sections, de-paraffinized by xylene, and treated with 0.3% hydrogen peroxide and the antigen retrieval buffer (10 mM Tris, 1 mM EDTA, 0.03% Tween 20). The sections were blocked with PBS containing 4% BSA and 0.03% Tween 20. The sections were incubated with anti-E-selectin antibody (1:800 dilution, AF575, R&D system) and anti-P-selectin antibody (1:200 dilution, AF737, R&D system) for 1 h, followed by corresponding secondary antibodies conjugated with horse peroxidase (One-step or Two-step Polymer Detection Systems, Nichirei, Japan). The selectin signals were visualized by incubation with 3, 3'-diaminobenzidine (DAB) for 7 min. The sections were counterstained with hematoxylin and imaged with an Eclipse 80i microscope (Nikon, Japan).
Confocal imaging of QA-NP accumulation in X-irradiated Tumor
Balb/c female mice (6-8 week old) were purchased from Envigo (Indianapolis, IN, USA). All mice were maintained under specific pathogen-free conditions. The study has been approved by the Institutional Animal Care and Use Committee of the University of Chicago, and all experiments conformed to the relevant regulatory standards. CT26 cells (ATCC® CRL-2638™) were cultured in 5% CO2 and maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, and 100 units/ml penicillin. To form subcutaneous tumors, 5 × 105 CT26 cells were injected into both hind limbs of a mouse. Ten days after tumor implantation, CT26 tumor on the right hind limb was X-irradiated with 6 Gy using an X-RAD 225 Cx image-guided radiotherapy system (Precision X-Ray) at 225 kV, 13 mV, 1.1 mm Cu half-value layer and a dose rate of 2.5 Gy/min. At 2 days after radiation, DyLight® 594-labeled QA-NP or PEG-NP (1 mg in 0.2 mL PBS) solution was IV injected to the mouse. At 1 day-post injection of NPs, CT26 tumors were harvested, washed with cold PBS, fixed with 2% paraformaldehyde in PBS for 10 min at room temperature, and washed with PBS. The washed tumors were cast in 2% agarose gel (dissolved in distilled water, LE Quick Dissolve Agarose, GeneMate) in 24 well plates. The gel plugs containing tumors were mounted on a vibrating microtome (VT1200S, Leica) equipped with a buffer tray. Sections (in 0.4 mm thickness) were collected in order in cold PBS. The macrosections were stained with DyLight® 488-anti-F4/80 (BioXcell, clone CI:A3-1), DyLight® 550-anti-E-selectin (BioLegend, clone RME-1/CD62E), DyLight® 633-anti-CD31 (BioLegend, clone MEC13.3), and DyLight® 680-anti-P-selectin (BioLegend, clone RMP-1) antibodies in staining buffer (SB, RPMI 1640 media with 10 mg/mL BSA and 0.1% Triton X-100) at 4°C overnight. The macrosections were incubated in 10 mL of 80% D-fructose solution for 1 h at 25°C with gentle agitation. The stained sections were imaged by a Leica TCS SP8 confocal laser scanning microscope, equipped with a white light laser, a HC PL APO 40X/1.25 NA oil objective (0.24 mm working distance), 488 nm excitation and 495-528 nm emission filter for DyLight® 488, 550 nm excitation and 563-579 nm emission filter for DyLight® 550, 594 nm excitation and 600-620 nm filters for DyLight® 594, 633 nm excitation and 637-655 nm filter for DyLight® 633, and 670 nm excitation and 680-700 nm filter for DyLight® 680.
Whole Body Imaging
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks were purchased from Envigo (Indianapolis, IN, USA) and acclimatized for 3 days prior to the procedure. All animal procedures were approved by the Purdue Animal Care and Use Committee, in compliance with the NIH guidelines for the care and use of laboratory animals. Each mouse received 5 × 106 MDA-MB-231 cells in the flank of hind leg by subcutaneous injection. When the averaged tumor volume reached 200 mm3, each mouse received 6 mg of ICG-labeled QA-NP or PEG-NP in 0.9% saline via tail vein injection. The animals were imaged with IVIS Lumina II system (Caliper Life Sciences, Hopkinton, MA, USA) to detect near infrared fluorescence (NIR) signal of ICG over 24 h. After 24 h, mice were sacrificed, and tumors and major organs were retrieved and imaged with IVIS Lumina system. The experiment was repeated with another set of tumor-bearing animals receiving 6 mg of ICG-labeled QA-NP or PEG-NP in 0.9% saline via tail vein injection. The image of ex vivo organs was taken at 6 h post injection using IVIS Lumina system.
Intravital Microscopy
Female Balb/c nude mice at the age of 8 weeks were purchased from OrientBio (Suwon, Korea). All animal experiments were performed in accordance with the standard guidelines for the care and use of laboratory animals and were approved by Institutional Animal Care and Use Committee of KAIST (protocol no. KA2013-30). All surgeries were performed under anesthesia. A dorsal skinfold chamber was implanted into a mouse.[53] Immediately after the implantation, a mixture of 3 × 106 MDA-MB-231-GFP cells and Corning Matrigel matrix was injected to the center of dorsal skinfold window chamber. After 1 week from the implantation, intravital imaging was performed. Two milligrams of Alexa Fluor® 555-labeled QA-NP was IV injected via tail vein through a custom-built catheter composed of 30 gauge needle tip and a polyethylene tube. At 2, 6, and 24 h after the NP injection, tumor microenvironment was repeatedly visualized to locate the NPs. At 3 h before intravital imaging, 25 μg of anti-CD31 monoclonal antibody (553370, BD) conjugated to Alexa Fluor® 647 (A20106, Invitrogen) was IV injected to label the blood vessels. For P-selectin (CD62P) or E-selectin (CD62E) labeling, 25 μg of anti-P-selectin monoclonal antibody (553742, BD) or anti-E-selectin monoclonal antibody (553749, BD) conjugated to Alexa Fluor® 647 (A20106, Invitrogen) was IV injected at 3 h before the intravital imaging. During intravital imaging, the body temperature of the mouse was continuously monitored and maintained at 37 °C by the homeothermic system (RightTemp, Kent Scientific).
A custom-built video-rate laser-scanning confocal microscopy system[54] was used for intravital imaging. Three laser modules with output wavelengths at 488 nm (MLD488, Cobolt), 561 nm (Jive, Cobolt), and 640 nm (MLD640, Cobolt) were used as excitation light sources. Three photomultiplier tubes (PMT; R9110, Hamamatsu) with bandpass filters (FF02-525/50, FF01-600/37, FF01-685/40, Semrock) were used for multi-color confocal fluorescence imaging. To achieve two-dimensional laser beam scanning at video-rate of 30Hz, a rapidly rotating polygonal mirror with 36 facets (MC-5, aluminum coated, Lincoln Laser) for x-axis scanning and a galvanometer scanning mirror (6230H, Cambridge Technology) for y-axis scanning were used. A commercial objective lens (CFI Plan Apo lambda, 20X, NA 0.75, Nikon) was used to capture confocal fluorescence images.
Determination of Maximum Tolerated Dose of PTX@QA-NP
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks were administered with PTX@QA-NP varying the frequency of the dosing. Each time, animals received the NPs at a PTX dose of 30 mg/kg through tail vein injection and monitored over 15 days. The body weight was measured every other day. Weight loss in excess of 20% was considered a humane endpoint.
In vivo Anti-cancer Efficacy
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks received 5 × 106 MDA-MB-231 cells in the flank of hind leg by subcutaneous injection. When the averaged tumor volume reached 100 mm3, each mouse received saline, bare QA-NP, bare PEG-NP, PTX@PEG-NP, or PTX@QA-NP at a dose equivalent to 30 mg/kg PTX 10 times over a 2-week period via tail vein injection. The tumor volume was measured every other day. The length (L) and width (W) of each tumor were measured by a digital caliper, and the volume (V) was calculated by the modified ellipsoid formula: V= (L × W2) / 2.[55] Specific growth rate of a tumor was calculated as ΔlogV/ΔΔt (t: time in days).[56] Mice with ulcerated tumors or tumors greater than 2000 mm3 were euthanized in a humane manner. Two other sets of experiments were carried out to compare PTX@QA-NP with Taxol: One group received a single dose at 20 mg/kg PTX equivalent, and the other received 5 doses of 20 mg/kg PTX equivalent every 3 days over 2 weeks.
Male C57BL/6 mice at the age of 5-6 weeks were purchased from Envigo (Indianapolis, IN, USA) and acclimatized for 3 days prior to the procedure. Each mouse received 5 × 106 B16F10 cells in the flank of hind leg by subcutaneous injection. When the averaged tumor volume reached 100 mm3, each mouse received a single dose of PTX@QA-NP or Taxol at 20 mg/kg PTX equivalent and was monitored in the same way as above.
Comparison of Tumor Vascularization
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks received 5 × 106 MDA-MB-231 cells or LS174T cells in the flank of hind leg by subcutaneous injection. When the averaged tumor volume reached 200 mm3, 200 μL of 0.25% Evans blue (Abcam, Cambridge, UK) in PBS was injected via tail vein. After 1 h, the mice were sacrificed, and the tumors and muscle tissues were imaged.
Detection of Anti-PEG Antibody in NP-treated Animals
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks received tail-vein injection of saline or PEG-NP or QA-NP (30 mg/kg). Five days later, blood was obtained by cardiac puncture and collected in a BD Vacutainer® Plus Plastic Serum Tube. Serum was separated by centrifugation at 930 ×g for 15 min. In the meantime, 200 μL of 2 mg/mL dopamine Tris solution was added to each well of a 96-well plate for surface coating. After 4 h incubation at room temperature, the plate was triple rinsed with DI water and incubated with 200 μL of 2 mg/mL mPEG-NH2 or QA-NH2 for each ligand decoration. After 1 h, the plate was triple rinsed with DI water and treated with 100 μL of 1% HSA blocking buffer (Tris buffered saline + 1% HSA + 0.05% Tween 20) for 1 h, followed by quintuple rinse. The blocked well was incubated with 100 μL of diluted sera from the NP-treated animals (1:20 dilution) for 1 h and rinsed 5 times. Goat anti-mouse IgM IgG-HRP conjugate (0.8 μg/mL or 4 μg/mL) was added to each well for 1 h incubation followed by quintuple rinse. Another 100 μL of 1% HSA blocking buffer was added to each well for 1 h incubation and rinsed 5 times. Finally, each well was treated with 50 μL of 3,3’,5,5’-tetramethylbenzidine (TMB) for 5 min to develop a blue color corresponding to the amount of the bound IgG-HRP, followed by quenching with 2 M sulfonic acid. The substrate solution was transferred to another plate for absorbance measurement at 450 nm.
Pharmacokinetics and Biodistribution of PTX
Female athymic nude mice (Foxn1nu) at the age of 6-7 weeks were inoculated with 5 × 106 MDA-MB-231 cells in the flank of hind leg by subcutaneous injection. When the average tumor size reached 100 mm3, animals were administered with PTX@PEG-NP, PTX@QA-NP, and Taxol at a PTX dose of 20 mg/kg by tail vein injection. At pre-determined time points, 3 mice were randomly sacrificed for the collection of blood and major organs. Blood was obtained by cardiac puncture and collected in a BD Vacutainer® PST™ tube. Plasma was separated from whole blood via centrifugation at 3000 ×g for 15 min.
Plasma and tissue concentrations of PTX was quantified using docetaxel (DTX) as the internal standard. Samples were prepared by liquid-liquid extraction. Specifically, plasma samples were diluted with water and extracted with methyl tertiary butyl ether. The samples were mixed and spun at high speed. The organic layer was transferred to a tube, evaporated to dryness, the tube was reconstituted with methanol and a small volume was injected into the HPLC-MS/MS. For tissues samples, frozen tissue was weighed, phosphate buffer (100 mM, pH 7.4) was added, and the tissue was homogenized using a Qiagen TissueRuptor with disposable probes. A volume of homogenate was removed, internal standard added, and the tissue sample was extracted using the same procedure that was used for plasma. Standards containing known amounts of PTX were prepared to estimate the concentrations in the samples. The standards ranged from 0.1-1000 ng/mL (n=10 standards including a 0) for mouse plasma samples. The standards ranged from 0.8 to 80 ng/sample (n=6 including a 0) for tumor, spleen, lung, kidney, and heart tissues, and from 1.8 to 180 ng/sample (n=6 including a 0) for liver tissue. The tissues from mice receiving no treatment were used as the matrix in the standard preparation for the respective tissues. The samples were analyzed by HPLC-MS/MS (ABSciex 4000). The mass spectrometer utilized an electrospray ionization probe and was run in positive mode. The multiple reaction monitoring (MRM) Q1/Q3 (m/z) transitions were 854.3/286.0 and 808.2/527.1, for PTX and DTX respectively.
The PK parameters were obtained via non-compartmental analyses using the sparse data option of WinNonlin (version 7.0, Certara, NJ, USA), allowing for the computation of the standard errors (SE) associated with estimated parameters.
Statistical Analysis
All statistical analysis was performed with GraphPad Prism 7 (La Jolla, CA, USA). All data were analyzed with one-way or two-way ANOVA test to determine the statistical difference of means among various groups, followed by the recommended multiple comparisons tests. A p-value less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health NIBIB R01EB017791, the Lilly Endowment Gift Graduate Research Award (J.X.), and NIBIB K99 EB022636 (S.S.L.). This work was also supported by the Purdue Research Foundation and the Indiana Clinical and Translational Sciences Institute, funded in part by grant #UL1 TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. HPLC-MS/MS analysis was performed by the Clinical Pharmacology Analytical Core laboratory, a core laboratory of the Indiana University Melvin and Bren Simon Cancer Center supported by the National Cancer Institute grant P30 CA082709. We thank Samyang Biopharm (Seoul, Korea) for the kind donation of paclitaxel and Prof. You-Yeon Won and Hyun Chang Kim for the assistance with PLGA-TPGS synthesis.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Jun Xu, Department of Industrial and Physical Pharmacy, Purdue University, 575 Stadium Mall Drive, West Lafayette, IN 47907, USA, yyeo@purdue.edu
Dr. Steve Seung-Young Lee, Ludwig Center for Metastasis Research, The University of Chicago, 5758 South Maryland Avenue, MC 9006, and Department of Molecular Genetics and Cellular Biology, The University of Chicago, 929 East 57th Street, GCIS W519, Chicago, IL 60637, USA
Howon Seo, Graduate School of Nanoscience and Technology and KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea.
Liang Pang, Department of Pharmaceutics, School of Pharmacy, Fudan University, Shanghai, 201203, People’s Republic of China.
Yearin Jun, College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, 08826, Republic of Korea.
Dr. Ruo-Yu Zhang, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 720 Clinic Drive, West Lafayette, IN 47907, USA
Prof. Zhong-Yin Zhang, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 720 Clinic Drive, West Lafayette, IN 47907, USA
Prof. Pilhan Kim, Graduate School of Nanoscience and Technology and KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea; Graduate School of Medical Science and Engineering, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea
Prof. Wooin Lee, College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, 08826, Republic of Korea
Prof. Stephen J. Kron, Ludwig Center for Metastasis Research, The University of Chicago, 5758 South Maryland Avenue, MC 9006, and Department of Molecular Genetics and Cellular Biology, The University of Chicago, 929 East 57th Street, GCIS W519, Chicago, IL 60637, USA
Prof. Yoon Yeo, Department of Industrial and Physical Pharmacy, Purdue University, 575 Stadium Mall Drive, West Lafayette, IN 47907, USA, yyeo@purdue.edu; Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA
References
- [1].a) Peer D, Karp J, Hong S, Farokhzad OC, Margalit R, Langer R, Nature Nanotechnology 2007, 2, 751–760; [DOI] [PubMed] [Google Scholar]; b) Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Matsumura Y, Maeda H, Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- [3].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nat. Rev. Mater 2016, 1, 1–12 [Google Scholar]
- [4].Ledford H, Nature 2016, 533, 304–305. [DOI] [PubMed] [Google Scholar]
- [5].a) Karathanasis E, Chan L, Karumbaiah L, McNeeley K, D'Orsi CJ, Annapragada AV, Sechopoulos I, Bellamkonda RV, PLoS ONE 2009, 4, e5843; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maeda H. Adv. Drug Delivery Rev 2015. [Google Scholar]
- [6].a) Kansas GS, Blood 1996, 88, 3259–3287; [PubMed] [Google Scholar]; b) Lawrence MB, Springer TA, Cell 1991, 65, 859–873. [DOI] [PubMed] [Google Scholar]
- [7].a) Laubli H, Borsig L, Semin. Cancer Biol 2010, 20, 169–177; [DOI] [PubMed] [Google Scholar]; b) Makrilia N, Kollias A, Manolopoulos L, Syrigos K, Cancer Invest. 2009, 27, 1023–1037. [DOI] [PubMed] [Google Scholar]
- [8].a) Nguyen M, Strubel NA, Bischoff J, Nature 1993, 365, 267; [DOI] [PubMed] [Google Scholar]; b) Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ, Nature 1995, 376, 517. [DOI] [PubMed] [Google Scholar]
- [9].Barthel SR, Gavino JD, Descheny L, Dimitroff CJ, Expert Opin. Ther. Targets 2007, 11, 1473–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Gassmann P, Enns A, Haier J, Onkologie 2004, 27, 577–582; [DOI] [PubMed] [Google Scholar]; b) Kobayashi H, Boelte KC, Lin PC, Curr. Med. Chem 2007, 14, 377–386; [DOI] [PubMed] [Google Scholar]; c) Rice GE, Bevilacqua MP, Science 1989, 246, 1303; [DOI] [PubMed] [Google Scholar]; d) Liu ZJ, Tian R, Li Y, An W, Zhuge Y, Livingstone AS, Velazquez OC, Ann. Surg 2011, 254, 450–456; discussion 456-457. [DOI] [PubMed] [Google Scholar]
- [11].Bevilacqua MP, Nelson RM, J. Clin. Invest 1993, 91, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Bonfanti BFR, Furie B and Wagner DD, Blood 1989, 73, 1109–1112; [PubMed] [Google Scholar]; b) Hallahan DE, Virudachalam S, Radiat. Res 1999, 152, 6–13. [PubMed] [Google Scholar]
- [13].Gay LJ, Felding-Habermann B, Nat. Rev. Cancer 2011, 11, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shamay Y, Elkabets M, Li H, Shah J, Brook S, Wang F, Adler K, Baut E, Scaltriti M, Jena PV, Gardner EE, Poirier JT, Rudin CM, Baselga J, Haimovitz-Friedman A, Heller DA, Sci. Transl. Med 2016, 8, 345ra387–345ra387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Nguyen M, Corless CL, Kraling BM, Tran C, Atha T, Bischoff J, Barsky SH, Am. J. Pathol 1997,150, 1307–1314; [PMC free article] [PubMed] [Google Scholar]; b) Shaker OG, El-Deen MAA, El-Rahim MTA, Talaat RM, Tumori. 2006, 92, 524–530; [DOI] [PubMed] [Google Scholar]; c) Fox SB, Turner GD, Gatter KC, Harris AL, J. Pathol 1995, 177, 369–376. [DOI] [PubMed] [Google Scholar]
- [16].Hemmerlein B, Scherbening J, Kugler A, Radzun HJ, Histopathology 2000, 37, 78–83. [DOI] [PubMed] [Google Scholar]
- [17].a) Young J. Kim LB, Varki Nissi M., and Varki Ajit, Proc. Natl. Acad. Sci. U. S. A 1998, 95, 9325–9330; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gong L, Mi HJ, Zhu H, Zhou X, Yang H, Mol. Med. Rep 2012, 5, 935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mollà M, Gironella M, Salas A, Miquel R, Pérez-del-Pulgar S, Conill C, Engel P, Biete A, Piqué JM, Panés J, Int. J. Cancer 2001, 96, 99–109. [DOI] [PubMed] [Google Scholar]
- [19].Phillips M, Nudelman E, Gaeta F, Perez M, Singhal A, Hakomori S, Paulson J, Science 1990, 250, 1130–1132. [DOI] [PubMed] [Google Scholar]
- [20].a) Pedatella S, Nisco M. De, Ernst B, Guaragna A, Wagner B, Woods RJ, Palumbo G, Carbohydr. Res 2008, 343, 31–38; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robina I, Moreno-Vargas A, Carmona A, Ferrali A, Molina L, Abstracts of Papers, 233rd ACS National Meeting, Chicago, IL, United States, March 25-29, 2007 2007, CARB-067; [Google Scholar]; c) Huang H, Wong C-H, J. Org. Chem 1995, 60, 3100–3106; [Google Scholar]; d) Rao BN, Anderson MB, Musser JH, Gilbert JH, Schaefer ME, Foxall C, Brandley BK, J. Biol. Chem 1994, 269, 19663–19666; [PubMed] [Google Scholar]; e) Cappi MW, Moree WJ, Qiao L, Marron TG, Weitz-Schmidt G, Wong C-H, Bioorg. Med. Chem 1997, 5, 283–296; [DOI] [PubMed] [Google Scholar]; f) DeFrees SA, Phillips L, Guo L, Zalipsky S, J. Am. Chem. Soc 1996, 118, 6101–6104; [Google Scholar]; g) Kaila N, IV BET, Med. Res. Rev 2002, 22, 566–601; [DOI] [PubMed] [Google Scholar]; h) Kaila N, J. Med. Chem 2005, 48, 4346–4357. [DOI] [PubMed] [Google Scholar]
- [21].a) Zeisig R, Stahn R, Wenzel K, Behrens D, Fichtner I, Biochim. Biophys. Acta, Biomembr 2004,1660, 31; [DOI] [PubMed] [Google Scholar]; b) Shamay Y, Raviv L, Golan M, Voronov E, Apte RN, David A, J. Controlled Release 2015, 217, 102–112. [DOI] [PubMed] [Google Scholar]
- [22].a) Girard C, Dourlat J, Savarin A, Surcin C, Leue S, Escriou V, Largeau C, Herscovici J, Scherman D, Bioorg. Med. Chem. Lett 2005,15, 3224–3228; [DOI] [PubMed] [Google Scholar]; b) Shamay Y, Paulin D, Ashkenasy G, David A, J. Med. Chem 2009, 52, 5906–5915. [DOI] [PubMed] [Google Scholar]
- [23].a) Amoozgar Z, Park J, Lin Q, Weidle JH 3rd, Yeo Y, Biomacromolecules 2013, 14, 2389–2395; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shamay Y, Paulin D, Ashkenasy G, David A, Biomaterials 2009, 30, 6460–6468. [DOI] [PubMed] [Google Scholar]
- [24].Liu Y, Zhong R, Zhang P, Ma Y, Yun X, Gong P, Wei J, Zhao X, Zhang F, ACS Appl. Mater. Interfaces 2016, 8, 2478–2485. [DOI] [PubMed] [Google Scholar]
- [25].Rao W, Wang H, Han J, Zhao S, Dumbleton J, Agarwal P, Zhang W, Zhao G, Yu J, Zynger DL, Lu X, He X, ACS Nano 2015, 6, 5725–5740. [DOI] [PubMed] [Google Scholar]
- [26].Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y, ACS Nano 2014, 8, 3347–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee H, Miller WM, Messersmith PB, Science 2007, 318, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee H, Rho J, Messersmith PB, Adv. Mater 2009, 21, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Peyri N, Berard M, Fauvel-Lafeve F, Trochon V, Arbeille B, Lu H, Legrand C, Crepin M, Anticancer Res. 2009, 29, 2347–2355; [PubMed] [Google Scholar]; b) Shoval H, Karsch-Bluman A, Brill-Karniely Y, Stem T, Zamir G, Hubert A, Benny O, Sci. Rep 2017, 7, 10428; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee E, Pandey NB, Popel AS, Sci. Rep 2014, 4, 5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Stocker CJ, Sugars KL, Harari OA, Landis RC, Morley BJ, Haskard DO, J. Immunol 2000,164, 3309–3315; [DOI] [PubMed] [Google Scholar]; b) Bhaskar V, Law DA, Ibsen E, Breinberg D, Cass KM, DuBridge RB, Evangelista F, Henshall SM, Hevezi P, Miller JC, Pong M, Powers R, Senter P, Stockett D, Sutherland RL, von Freeden-Jeffry U, Willhite D, Murray R, Afar DE, Ramakrishnan V, Cancer Res. 2003, 63, 6387–6394; [PubMed] [Google Scholar]; c) Lu Y, Yu T, Liang H, Wang J, Xie J, Shao J, Gao Y, Yu S, Chen S, Wang L, Jia L, Sci Rep 2014, 4, 4344; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Magder S, Neculcea J, Neculcea V, Sladek R, J. Vase. Res 2006, 43, 447–461. [DOI] [PubMed] [Google Scholar]
- [31].a) Harrington EO, Stefanec T, Newton J, Rounds S, Lung 2006,184, 259–266; [DOI] [PubMed] [Google Scholar]; b) Leeuwenberg JF, Smeets EF, Neefjes JJ, Shaffer MA, Cinek T, Jeunhomme TM, Ahern TJ, Buurman WA, Immunology 1992, 77, 543–549. [PMC free article] [PubMed] [Google Scholar]
- [32].a) Zhong L, Simoneau B, Huot J, Simard MJ, Oncotcirget 2017, 8, 1678–1687; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Laferriere J, Houle F, Hout J, Ann. N. Y. Acad. Sci 2002, 973, 562–572. [DOI] [PubMed] [Google Scholar]
- [33].a) Tremblay PL, Auger FA, Huot J, Oncogene 2006, 25, 6563–6573; [DOI] [PubMed] [Google Scholar]; b) Khanna P, Yunkunis T, Muddana HS, Peng HH, August A, Dong C. Am. J. Physiol.: Cell Physiol 2010, 298, C1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nubel T, Dippold W, Kaina B, Fritz G, Carcinogenesis 2004, 25, 1335–1344. [DOI] [PubMed] [Google Scholar]
- [35].Lee SS, Bindokas VP, Kron SJ, Sci Rep 2017, 7, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wu Q, Allouch A, Martins I, Modjtahedi N, Deutsch E, Perfettini JL, Biomed. J 2017, 40, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Miller MA, Chandra R, Cuccarese MF, Pfirschke C, Engblom C, Stapleton S, Adhikary U, Kohler RH, Mohan JF, Pittet MJ, Weissleder R, Sci. Transl. Med 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hariri G, Zhang Y, Fu A, Han Z, Brechbiel M, Tantawy MN, Peterson TE, Mernaugh R, Hallahan D, Ann. Biomed. Eng 2008, 36, 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smeets EF, De Vries T, Leeuwenberg JFM, van den Eijnden DH, Buurman WA, Neefjes JJ, Eur. J. Immunol 1993, 23, 147–151. [DOI] [PubMed] [Google Scholar]
- [40].Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, Lin CW, Usami S, Chien S, Blood 2007, 110, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Desmettre T, Devoisselle JM, Mordon S, Surv. Ophthalmol 2000, 45, 15–27; [DOI] [PubMed] [Google Scholar]; b) Yaseen MA, Yu J, Jung B, Wong MS, Anvari B, Mol. Pharmaceutics 2009, 6, 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jarzyna PA, Deddens LH, Kann BH, Ramachandran S, Calcagno C, Chen W, Gianella A, Dijkhuizen RM, Griffioen AW, Fayad ZA, Mulder WJM, Neoplasia (N. Y., NY, U. S.) 2012, 14, 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bumpers H, Huang MB, Katkoori V, Manne U, Bond V, J. Cancer Ther 2013, 4, 898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a) Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Nat. Ncinotechnol 2011, 6, 815–823; [DOI] [PubMed] [Google Scholar]; b) Lee H, Fonge H, Hoang B, Reilly RM, Allen C, Mol. Pharmaceutics 2010, 7, 1195–1208; [DOI] [PubMed] [Google Scholar]; c) Tang L, Yang X, Yin Q, Cai K, Wang H, Chaudhury I, Yao C, Zhou Q, Kwon M, Hartman JA, Dobrucki IT, Dobrucki LW, Borst LB, Lezmi S, Helferich WG, Ferguson AL, Fan TM, Cheng J, Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15344–15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Su Y-L, Yu T-W, Chiang W-H, Chiu H-C, Chang C-H, Chiang C-S, Hu S-H, Adv. Funct. Mater 2017, 27, 1700056. [Google Scholar]
- [46].Abouelmagd SA, Ku YJ, Yeo Y, J. Drug Targeting 2015, 23, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ma Y, Zheng Y, Liu K, Tian G, Tian Y, Xu L, Yan F, Huang L, Mei L, Nanoscale Res. Lett 2010, 5, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang X, Burt HM, Mangold G, Dexter D, Von Hoff D, Mayer L, Hunter WL, Anticancer Drugs 1997, 8, 696–701. [DOI] [PubMed] [Google Scholar]
- [49].Willem W, Overwijk NPR, Curr. Protoc. Immunol 2001, 20.1, 1–20.18432774 [Google Scholar]
- [50].a) Mima Y, Hashimoto Y, Shimizu T, Kiwada H, Ishida T, Mol. Pharmaceutics 2015, 12, 2429–2435; [DOI] [PubMed] [Google Scholar]; b) Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H, J. Controlled Release 2003, 88, 35–42; [DOI] [PubMed] [Google Scholar]; c) Ishida T, Masuda K, Ichikawa T, Ichihara M, Irimura K, Kiwada H, Int. J. Pharm (Amsterdam, Neth.) 2003, 255, 167–174; [DOI] [PubMed] [Google Scholar]; d) Ishida T, Ichihara M, Wang X, Kiwada H, J. Controlled Release 2006, 115, 243–250; [DOI] [PubMed] [Google Scholar]; e) Ishida T, Ichihara M, Wang X, Yamamoto K, Kimura J, Majima E, Kiwada H, J. Controlled Release 2006, 112, 15–25; [DOI] [PubMed] [Google Scholar]; f) Ishida T, Atobe K, Wang X, Kiwada H, J. Controlled Release 2006, 115, 251–258. [DOI] [PubMed] [Google Scholar]
- [51].Abu Lila AS, Kiwada H, Ishida T, J. Controlled Release 2013,172, 38–47. [DOI] [PubMed] [Google Scholar]
- [52].a) Spratlin J, Sawyer MB, Crit. Rev. Oncol. Hematol 2007, 61, 222–229; [DOI] [PubMed] [Google Scholar]; b) Monsarrat B, Mariel E, Cros S, Garès M, Guénard D, Guéritte-Voegelein F, Wright M, Drug Metab. Dispos 1990, 18, 895–901; [PubMed] [Google Scholar]; c) Panday VR, Huizing MT, Willemse PH, Graeff A. De, Bokkel Huinink W. W. ten, Vermorken JB, Beijnen JH, Semin. Oncol 1997, 24, S11–S34; [PubMed] [Google Scholar]; d) Monsarrat B, Alvinerie P, Wright M, Dubois J, Gueritte-Voegelein F, Guenard D, Donehower RC, Rowinsky EK, J. Natl. Cancer Inst Monogr. 1993, 39–46. [PubMed] [Google Scholar]
- [53].Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R, Nat. Commun 2013, 4, 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].a) Veilleux I, Spencer JA, Biss DP, Cote D, Lin CP, IEEE J. Sel. Top. Quantum Electron 2008, 14, 10–18; [Google Scholar]; b) Choe K, Hwang Y, Seo H, Kim P, J. Biomed. Opt 2013, 18; [DOI] [PubMed] [Google Scholar]; c) Ahn J, Choe K, Wang T, Hwang Y, Song E, Kim KH, Kim P, Biomed. Opt. Express 2015, 6, 3963–3972; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Seo H, Hwang Y, Choe K, Kim P, Biomed. Opt. Express 2015, 6, 2158–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tomayko MM, Reynolds CP, Cancer Chemother. Pharmacol 1989, 24, 148–154. [DOI] [PubMed] [Google Scholar]
- [56].Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P, Cancer Res. 2007, 67, 3970–3975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.