

Abstract
Endoplasmic reticulum (ER) stress is a mechanism that allows to protect normal cellular functions in response to both internal perturbations, such as accumulation of unfolded proteins, and external perturbations, for example redox stress, UVB irradiation, and infection. A hallmark of ER stress is the accumulation of misfolded and unfolded proteins. Physiological levels of ER stress trigger the unfolded protein response (UPR) which is required to restore normal ER functions. However, the UPR can also initiate a cell death program/apoptosis pathway in response to excessive or persistent ER stress. Recently, it has become evident that chronic ER stress occurs in several diseases, including skin diseases like Darier’s disease, rosacea, vitiligo, and melanoma; furthermore, it is suggested that ER stress is directly involved in the pathogenesis of these disorders. Here, we review the role of ER stress in skin function, and discuss its significance in skin diseases.
Keywords: endoplasmic reticulum stress, skin function, skin disease, unfolded protein response
Graphical Abstract
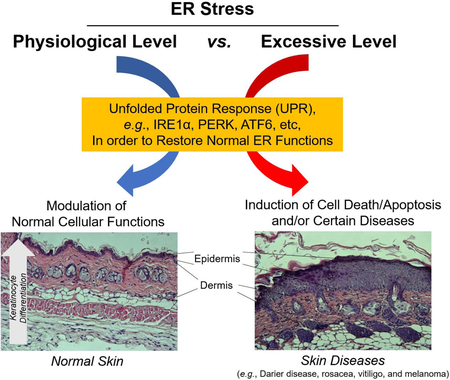
Physiological levels of endoplasmic reticulum (ER) stress are required for modulation of normal cellular functions in the skin, including keratinocyte differentiation, through the unfolded protein response (UPR). However, persistent or excessive levels of ER stress induce cell death and apoptosis signalling. Growing evidence describes chronic ER stress in several cutaneous diseases e.g., Darier’s disease, rosacea, vitiligo, and melanoma. In this review, we discuss the role of ER stress in normal skin function and disease.
Introduction
Endoplasmic reticulum (ER) stress is induced in cells following stress caused by both internal and external perturbations. In addition, chronic ER stress is evident in different human diseases, such as diabetes, immune disorders, cancers, neurodegeneration, pulmonary fibrosis, and rheumatoid arthritis [1]. In skin, subtoxic (physiological) levels of ER stress-induced unfolded protein response (UPR) is required for normal cellular function, including differentiation. Yet, a chronic, sustained ER stress-induced UPR has deleterious effects on cells; UPR becomes a cell death mechanism [2]. Furthermore, as emerging evidence reveals that a continuously active UPR is involved in the pathogenesis of certain skin diseases; i.e., Darier’s disease, rosacea, vitiligo, and melanoma, recent studies have highlighted ER stress and UPR as potential therapeutic targets for treatment of such diseases [3-8]. This review article discusses roles of ER stress in normal skin function and in skin disease.
Function and Structure of the Skin
The skin is the interface between the external and internal environment [9], and competent barriers deployed in the skin protect our bodies from insults, such as UV irradiation, chemicals, pathogenic microorganisms and dryness [9, 10]. The skin consists of epidermis, dermis and subcutaneous tissue (fat, sebaceous glands, sweat gland and hair). The epidermis, which is the outermost layer of the skin, is further divided into four histologically-distinct layers, dependent on different stages of keratinocyte (KC) differentiation. KCs are the dominant cell species (over 95%) in the epidermis [11, 12]. KCs proliferate at the innermost layer of epidermis, the stratum basale (SB), differentiate to the stratum spinosum (SS), then to the stratum granulosum (SG), and finally terminally differentiate to the stratum corneum (SC) [12]. During differentiation, KCs migrate towards the outer layer epidermis [12]. As KCs transition from the SG to the SC, they become enucleated corneocytes. Different from nucleated cells, the plasma membrane, which is formed by a lipid bilayer, is replaced by a protein cross-linked cornified envelope that resists mechanical and chemical stress [13, 14]. In addition, during the transition from SG to SC, intracellular organelles, called lamellar granules, which contain lipids, protein and hydrolytic enzymes, are secreted into the extracellular domain in the SC to form lamellar membrane structures that are responsible for permeability barrier function [12]. The permeability barrier prevents excess water and ion loss from the body and conversely prevents invasion of exogenous substances from the external environment [12]. Lipid species, cholesterol, free fatty acids and ceramides are the predominant constituents of lamellar membrane structures [14]. In addition, ceramide metabolites serve as lipid mediators to enhance innate immunity in the nucleated layers of epidermis (see below, “Physiological ER Stress Is Required for Normal Cellular Functions in Skin” section).
ER Stress and Unfolded Protein Response
The ER has the central machinery responsible for the synthesis, secretion, modification, and folding of proteins [1]. Various cellular stresses caused by external/internal circumstances or excessive protein production cause an inadequate folding of client proteins, leading to the accumulation of misfolding or unfolding proteins in the ER, which is referred to as “ER stress” [1, 15]. An unfolded protein response (UPR) initiates to restore normal ER functions by reducing ER stress through previously-demonstrated mechanisms [1, 15, 16]: i) shutting down cap-dependent translation; ii) increasing ubiquitin-proteasome-mediated degradation of misfolded/unfolded proteins via ER-associated degradation (ERAD); and iii) increasing expression of ER chaperones and folding enzymes that enhance the overall efficiency of protein folding. In fact, UPR is controlled by three ER transmembrane sensor proteins, including inositol-requiring enzyme 1 alpha (IRE1α), double-stranded RNA-dependent protein kinase (PER)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Fig.1). UPR activation is prevented when these three ER sensor proteins are bound by glucose-regulated protein 78/binding immunoglobulin protein (GRP78/BiP), the ER resident chaperone, in unstressed conditions; whereas, accumulation of unfolded/misfolded proteins within the ER lumen cause GRP78/BiP to dissociate from these three ER sensor proteins, leading to UPR activation [1, 15-17].
Fig. 1. The three branches of the unfolded protein response (UPR).

In unstressed conditions, stress sensor proteins activating transcription factor (ATF) 6, inositol-requiring enzyme (IRE1) α, and RNA-dependent protein kinase-like ER-resident kinase (PERK), representing the three branches of the UPR, are associated with the folding chaperone glucose regulated protein/binding immunoglobulin protein (GRP78/BiP) in the ER. Accumulation of unfolded/misfolded proteins within the ER lumen causes GRP78/BiP disassociation from these three sensor proteins, leading to UPR activation. Each pathway uses a different mechanism of signal transduction. Activated IRE1α mediates unconventional splicing of X-box binding protein (XBP) 1 to produce spliced, active isoform of XBP1. IRE1α recruits TNF receptor associated factor (TRAF) 2 to activate the downstream signal mediators, NF-κB/JNK. IRE1α-mediated activations of XBP1 and TRAF2/NF-κB/JNK regulate UPR target genes associated with lipid metabolism, immune, inflammatory response, and differentiation, as well as structural/ functional expansion of ER and ER-associated protein degradation (ERAD). In addition, IRE1α can reduce the ER protein folding load by the IRE1-dependent decay of mRNA (RIDD) causing degradation of ER membrane-bound mRNAs. Activated PERK recruits and phosphorylates eukaryotic initiation factor (eIF2) α reduce global protein synthesis and thereby reduce protein folding load in ER-stressed cells. Paradoxically, however, PERK/eIF2α-translation of ATF4 increases certain UPR gene transcriptions, including CCAAT-enhancer-binding protein homologous protein (CHOP). Lastly, activated ATF6 is exported to the Golgi apparatus where it is cleaved by the Golgi-resident proteases SP1 and SP2 to produce the functional fragment of ATF6. The functional ATF6 is then translocated to the nucleus where it transactivates UPR genes associated with ER homeostasis.
IRE1 signaling
IRE1α is a highly conserved mediator of the UPR [16, 18]. Dissociated GRP78/BiP from IRE1α preferentially binds to unfolded/misfolded proteins, causing dimerization and auto-phosphorylation of IRE1α through its kinase activity [16, 19]. This leads to an increase in nuclease activity of IRE1α, leading to catalyzing the excision of an unconventional intron with 26 nucleotides in length from the X-box binding protein 1 (XBP1) mRNA to produce spliced isoform of XBP1 (XBP1s) [19]. While the unspliced isoform of XBP1 (XBP1u) is unable to activate gene expression due to a lack of transactivation domain, XBP1s can direct the transcription of a broad range of target genes involved in lipid metabolism, immune and inflammatory responses, and cellular differentiation as well as genes related to structural/ functional expansion of ER and ER-associated protein degradation (ERAD), in order to reduce ER stress and restore homeostasis [20-24]. In addition, IRE1α activation caused by phosphorylation induces the recruitment of tumor necrosis factor receptor-associated factor 2 (TRAF2), forming IRE1α-TRAF2 signaling complex [25]. Phosphorylated IRE1α-TRAF2 complex simultaneously activates both JNK and NF-κB, which signals to modulate IRE1α-mediated cell death [25, 26]. Prior studies using NF-κB and/or JNK1/2 knockout cells suggest that TRAF2-mediated activation of NF-κB and JNK1/2 protects cells from apoptosis by attenuating ROS production [27], whereas studies using cells overexpressed mutant IκB revealed that blockade of NF-κB activation made cells resistant to ER stress-mediated cell death[28]. Therefore, whether IRE1α-TRAF2-dependent NF-κB and JNK pathways affect cell survival or death remains unclear. Moreover, in addition to XBP1 mRNA, IRE1α also cleaves other mRNAs localized in the ER membrane and processes their degradation through a process known as regulated IRE1-dependent mRNA decay (RIDD) [29]. Emerging evidence suggests that RIDD has a critical role in the maintenance of ER homeostasis by alleviating ER client protein load through mRNA degradation and inhibition of protein synthesis by cleavage of 28S rRNA [16, 29]. See references [29] and [16] for more details on the role of IRE1α under ER stress.
PERK signaling
Similar to IRE1α activation, detachment of GRP78/BiP from PERK in the ER luminal domain leads to activation of PERK through its dimerization and auto-phosphorylation [16, 30]. Activated PERK recruits and phosphorylates a translation initiation factor, eukaryotic translation initiation factor 2α (eIF2α), through its kinase activity [30, 31]. eIF2α is a subunit of the heterotrimeric eIF2 complex which regulates protein synthesis initiation by promoting the binding of the initiator tRNA to 40S ribosomal subunits [30, 31]. However, phosphorylated eIF2α inhibits eukaryotic translation initiation factor 2B (eIF2B) activity, leading to attenuation of cap-dependent protein synthesis and thereby reducing protein folding load in ER-stressed cells. In addition, phosphorylation of eIF2α selectively induces translation of activating transcription factor 4 (ATF4), whose transcript contains regulatory sequences such as short upstream open reading frames [30, 31]. ATF4 controls expression of adaptive genes associated with protecting mechanisms which protect cells against ER stress; i.e., amino acid metabolism, anti-oxidant response, protein homeostasis and autophagy [30, 31]. However, overactivation of PERK due to sustained or unresolved ER stress shifts its adaptive response toward a pro-death response. This change is mediated by upregulation of CAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) that in turn enhances oxidative stress and ATP depletion, leading to cell death. See references [30] and [31] for more details on the role of PERK under ER stress.
ATF6 signaling
ATF6 expression is limited to the ER, however under ER stress it is exported to the Golgi apparatus where it is cleaved by the Golgi-resident proteases in order to produce the functional fragment of ATF6 [16, 19]. Fragmented AFT6 is then translocated into the nucleus to exert its function as a potent transcription factor, activating gene programs involved in restoring ER homeostasis [19]. See references [19] and [30] for more details on the role of PERK under ER stress.
Calcium signaling
ER is the largest calcium (Ca2+) store in the cell and it tightly regulates ER/cytosolic Ca2+ concentration through Ca2+ channels, Ca2+ transporters, Ca2+ pumps, or Ca2+-binding proteins [16, 32-34]. A balance between ER Ca2+-release and -uptake is crucial for the regulation of Ca2+ signaling-dependent normal cellular functions; e.g., proliferation, differentiation, apoptosis, and gene expression, in response to physiological stimuli [33, 34]. An increase in ER Ca2+-release leads to stimulation of cytosolic Ca2+ concentration, which induces ER stress, triggering UPR to either restore normal ER Ca2+ concentration and associated cellular functions or to eliminate the cells by apoptosis pathways [33, 34]. Depletion of Ca2+ concentration in the ER leads to a rapid accumulation of unfolded/misfolded proteins, which promote dissociation of GRP78/BiP from three ER transmembrane sensor proteins, IRE1 α, PERK and ATF6, resulting in activation of the UPR pathway [32, 34]. See references [34] and [32] for more details on the importance of maintaining Ca2+ homeostasis and appropriate adaptation to ER stress.
Physiological ER Stress Is Required for Normal Cellular Functions in Skin
ER stress occurs in both physiological and pathological conditions, which modulate multiple cellular responses, including pro-survival or pro-apoptotic mechanisms. In skin, epidermal barrier perturbation, as well as external stress, such as UV irradiation and other types of oxidative stress, induce ER stress, which triggers UPR to regulate normal cellular functions through modulation of multiple intracellular mediators; e.g., ER chaperones, protein kinases, signaling lipids, and transcriptional factors [2, 35, 36]. Importantly, physiological ER stress is required for the maintenance of normal biological functions in skin, including KC differentiation [2]. KC differentiation is a vital process for the proper formation of a competent skin barrier [12]. Both 1,25-dihydroxyvitamin D3, an active form of vitamin D3, and Ca2+ play important roles in the regulation of the KC differentiation process [37]. Maintaining the Ca2+ gradient within the epidermis, with lowest levels in the SB and the highest levels in the SG, is important for both epidermal permeability barrier homeostasis and epidermal differentiation. Moreover, prior studies revealed that ER stress-signaled UPR is activated during epidermal KC differentiation [2, 38, 39]. Expression levels of ER stress/UPR activation markers, such as spliced forms of XBP1, CHOP, and GRP78/BiP, in undifferentiated/proliferative stage of KC are increased during KC differentiation [2, 35]. In addition, specific pharmacological ER stressors, thapsigargin (a SERCA2 Ca2+ pump inhibitor that depletes ER Ca2+), tunicamycin (a specific inhibitor of N-linked glycosylation that blocks glycoprotein synthesis thereby inducing UPR), and Brefeldin A (an ER-Golgi transport inhibitor that causes accumulation of proteins in the ER, causing ER stress), stimulate expression of differentiation-related genes (ABCA12 and KLF4) through a XBP1-mediated mechanism. These gene inductions by ER stressors were significantly suppressed in KC treated with siRNA against UPR makers; e.g., ATF4 and XBP-1 [2, 39].
Finally, ceramide metabolites, sphingosine-1-phosphate and ceramide-1-phosphate, signal to stimulate the key antimicrobial peptide, cathelicidin antimicrobial peptide, and human beta-defensin 2 and 3, respectively, in keratinocytes to enhance antimicrobial defense in response to physiological levels of ER stress induced by external perturbations such as UV irradiation and other types of oxidative stress [35, 36, 40]. Moreover, sphingosine-1-phosphate-dependent increases in cathelicidin antimicrobial peptide production are likely linked to an increase in physiological ER stress during keratinocyte differentiation [41].
ER Stress and Associated Skin Diseases
Darier’s Disease
Darier’s disease is a disease associated with impaired ER calcium homeostasis that induces ER stress (Fig. 2). It is an autosomal dominant genodermatosis caused by mutations of the gene encoding sarco/endoplasmic reticulum Ca2+-ATPase 2 (SERCA2), an intracellular calcium pump that transports Ca2+ from the cytoplasm to the ER against a calcium gradient [8]. Darier’s disease is characterized by the symmetrical eruption of keratotic papules predominantly in seborrheic areas. Loss of cell-to-cell adhesion due to abnormal keratinocyte differentiation and dyskeratosis are the histological hallmarks of Darier’s disease [42], and ER stress response has been proposed to play a crucial role in the pathogenesis of this disease [43, 44]. Darier keratinocytes have been shown to have a chronic depletion of stored ER calcium, with a constitutive ER stress response [45]. Chronic decreases in ER calcium levels cause dysregulation of cargo protein processing and trafficking. Indeed, SERCA2-inhibited keratinocytes show i) impaired translocation of desmosomal cadherins such as desmoglein 3 and desmocollin 3, desmoplakin, and components of adherens junctions, to the cell membrane; and ii) ER retention of desmosomal cadherins and E-cadherin. These findings implicate that ER stress-induced abnormal trafficking of junctional components is a mechanism of acantholysis in Darier’s disease [43, 44]. In addition to the prolonged ER calcium depletion, SERCA2 mutant protein itself also contributes to the development of the ER stress response in this disease. SERCA2 mutant proteins have been shown to trigger and to enhance the UPR leading to apoptosis of keratinocytes [2]. These findings suggest that the accumulation of the mutant SERCA2-induced activation of the pro-apoptotic branches of the UPR, CHOP upregulation, is a mechanism of Darier’s disease dyskeratosis. The role of ER stress in the pathomechanism of Darier’s disease is further supported by the therapeutic effect of Miglustat, a drug with a chemical chaperone that reduces ER stress during the structural and functional restoration of desmosomes and adherens junctions in the Darier keratinocyte [43].
Fig. 2. Role of ER stress in Darier’s disease.

In Darier’s disease, mutations in the ATP2A2 gene, which encodes the SERCA2, cause impaired transport of calcium from cytosol to ER, thereby leading to chronic ER stress in keratinocytes. ER calcium is an important regulator of the reorganization of adherens junctions and desmosomes. Defective ER calcium homeostasis in keratinocytes of Darier’s disease may contribute to abnormal cell-to-cell adhesion via defective reorganization of junctional components, causing acantholysis. In addition, chronic ER stress triggers the disproportionate activation of the apoptotic component of the UPR. PERK-dependent apoptotic signaling can contribute to the non-physiologic and premature keratinocyte apoptosis which can be observed as dyskeratotic keratinocytes (“corp ronds”) in Darier’s disease. Taken together, ER stress is implicated in the pathogenesis of Darier’s disease characterized histologically by acantholytic dyskeratosis.
Keratinization Disorders
Prior studies have addressed the potential role of ER stress and UPR in keratinocyte differentiation and keratinization [2], and UPR has been demonstrated to be activated during normal epidermal keratinocyte differentiation [8]. These findings suggest that abnormal UPR may be associated with skin diseases characterized by abnormal keratinization, and differentiation. ER stress is known to play an important role in the pathomechanisms of several rare hereditary keratinization disorders.
Erythrokeratoderma variabilis
Erythrokeratoderma variabilis (EKV) is a rare hereditary disorder belonging to the heterogeneous group of skin diseases called the erythrokeratodermia which presents with migratory erythema and fixed hyperkeratotic plaques. EKV is caused by the mutations in GJB3 encoding the gap junction protein, Connexin 31 [4]. Connexin proteins are the main components of gap junctions which mediate epidermal keratinocyte communications. Several studies have demonstrated that microinjection of the skin disease-associated Connexin 31 mutants R42P, C86S, and G12D into keratinocytes showed a high incidence of cell death, but the precise mechanism is not known [46-48]. Recent study revealed that EKV-associated mutants of Connexin 31 have cytoplasmic localization with defective trafficking and leads to upregulation of UPR in keratinocytes. Despite lack of direct evidence, these findings suggest that mutant Connexin 31-induced pathological ER stress is associated with cell death in EKV [46]. The exact mechanism of abnormal keratinocyte differentiation and hyperproliferation in EKV is not yet defined, but it is postulated that the defective Connexin 31 may affect the functions of other connexins or other cellular components, thereby leading to the abnormal pathologies of EKV [49]. Further studies are needed to define the exact role and therapeutic potential of ER stress in EKV.
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome (KLICK syndrome [MIM 601952]) is a rare autosomal-recessive skin disorder characterized by palmoplantar keratoderma, linear hyperkeratotic papules, and ichthyosiform scaling and is causes by POMP (proteasome maturation protein) gene mutations [50]. Altered distribution of POMP and proteasome subunits during formation of the horny layer have been shown to induce persistent ER stress in keratinocytes, suggesting that proteasome insufficiency-induced abnormal UPR contributes to the abnormal terminal differentiation in KLIKC syndrome [2, 50].
Ichthyosis follicularis, alopecia, and photophobia syndrome
Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome is a rare X-linked disease caused by mutations in membrane-bound transcription factor protease, site 2 (MBTPS2), a membrane-embedded zinc metalloprotease. MBTPS2 is involved in the cholesterol homeostasis and ER stress response [51]. It has been suggested that mutated MBTPS2-induced impairment in the cleavage of ATF6 and sterol regulatory element-binding proteins (SREBP), and consequent impairment of cholesterol metabolism and UPR is the mechanism of the abnormal keratinization in IFAP syndrome [51, 52].
Psoriasis
Abnormal ER stress response in epidermal keratinocytes is also reported in a common inflammatory skin disease, psoriasis [2]. The abnormal differentiation and hyperproliferation of epidermal keratinocytes are important parts of the psoriasis pathogenesis. Recent studies demonstrated that UPR induced proteins BiP/GRP78 and HRD1, which are normally induced during keratinocyte differentiation, were poorly expressed in lesional epidermis, suggesting that impaired activation of the UPR in psoriasis KCs might contribute to abnormal epidermal keratinocyte differentiation [45]. However, how ER stress contributes to the pathophysiology of psoriasis remains unknown.
Rosacea
Rosacea is a chronic inflammatory skin disease characterized by transient or persistent erythema, papules and pustules, and telangiectasia on the facial skin. Facial flushing, burning, or tingling sensations are frequent in affected individuals, especially with exposure to various substances including cosmetics [3]. The pathophysiology of rosacea is diverse, while aberrant innate immune responses and neurovascular dysregulation are evident [53]. Increased expression of both cathelicidin antimicrobial peptide and the pattern recognition receptor, toll-like receptor 2 (TLR2) occurs in rosacea keratinocytes [54]. TLR2 activation by various triggering factors such as ultraviolet (UV), demodex and other microbes enhances the production of a serine protease, kallikrein 5 (KLK5), which cleaves cathelicidin to LL-37 and smaller peptides, thereby triggering pro-inflammatory events and angiogenesis [54]. ER stress by several inducers such as thapsigargin, tunicamycin, and dithiothreitol has been shown to increase the expression of TLR2 via transcription factor ATF4 signaling pathway and Ligand-responsiveness of TLR2 in epithelial cells [55]. Although further studies are needed to confirm that ER stress-induced UPR signaling is responsible for the upregulation of TLR2 in the lesional skin of rosacea, it can be postulated that ER stress plays a role in rosacea via upregulating TLR2 and triggering a TLR2-KLK5-LL37 inflammatory cascade (Fig. 3) [55-57]. Enhanced ER stress also can promote TLR2 signaling in neurons, which could trigger neurogenic inflammation [57]. Moreover, ER stress directly upregulates cathelicidin, the precursor of LL-37, via sphingosine-1-phosphate (S1P)-NF-κB-C/EBPα-dependent pathway [36]. Vitamin D is an important regulator of cathelicidin expression, but a recent study demonstrated that serum vitamin D is lower in patients with rosacea, although serum cathelicidin is higher than that of the controls, suggesting that in rosacea, ER stress is essential for production of cathelicidin [58].
Fig. 3. Role of ER stress in Rosacea.

Rosacea is a chronic inflammatory condition, in which both innate and adaptive immune responses are activated by multiple environmental factors. Many triggering factors of rosacea can induce ER stress and UPR signaling pathways in keratinocytes. ATF4-mediated signaling induces TLR2 expression and TLR2-mediated innate immune responses. Subsequently, TLR2 increases KLK5 expression in keratinocytes. ER stress can also induce cathelicidin production by S1P signaling pathway in keratinocytes. Excess cathelicidin and their proteolytic processing by KLK5 play a central role in the innate immune activation of rosacea.
In addition, various trigger factors for rosacea such as UV exposure, skin irritants that perturbate skin barrier, heat, and some foods, induce ER stress in keratinocytes. ER stress likely contributes to the aberrant innate immune responses and neurovascular dysregulation in rosacea pathogenesis, and inhibition of ER stress responses may provide a potential therapeutic strategy in rosacea.
Vitiligo
Vitiligo is a melanocyte-specific autoimmune disease of the skin affecting melanocytes that leads to depigmentation in the affected area of skin, mucosa, and hair. Recent study shows that IFNγ-induced chemokines and cytotoxic CD8 T cells play a key role in the autoimmune responses in vitiligo [5]. Melanocytes are constantly exposed to environmental factors, such as UV exposure and certain chemicals; e.g., phenolic and catecholic chemicals, that induce oxidative stress [5]. In addition, it has been suggested that melanocytes from vitiligo patients have intrinsic defects that reduce the capacity to manage cellular stress, resulting in increases in ROS production and UPR induction, which in turn activate innate inflammation [5, 59]. Genetic association studies of XBP1 SNPs in patients suggest a role of ER stress in vitiligo [60]. These findings illuminate a possible role of ER stress-UPR signaling in melanocytes in the pathogenesis of vitiligo.
Pemphigus
Pemphigus is an autoimmune mucocutaneous blistering disease caused by autoantibodies against desmosomal cadherins, desmoglein (Dsg) 1 and Dsg3, which induce loss of cell-to-cell adhesion (acantholysis) and intraepidermal blisters [61]. The activation of cellular signaling pathways including p38 mitogen activated protein kinase (p38 MAPK) have been suggested as pathomechanisms of autoantibodies-induced acantholysis [62]. Recently, emerging evidence suggests the possible role of ER stress in the pathophysiology of pemphigus [63-65], i.e., 1) PERK is activated in keratinocytes exposed to pemphigus vulgaris serum by non-IgG serum factors, thereby eliciting the reduction of metabolic activity and cell viability in keratinocytes, and these changes were almost absent in PERK-deficient cells [63], and 2). anti-Dsg1 autoantibodies specifically induce ER stress marker CHOP expression [64]. Interestingly, MAPK signaling can also drive ER stress, and ER stress is known to induce stress kinases such as p38 MAPK, indicating that activation of PERK-CHOP pathway can be a novel signaling mechanism of pemphigus acantholysis via its acting as a positive regulator of p38 MAPK pathway and inducing apoptosis [65].
Graft versus Host Disease
GvHD is a fatal complication following allogeneic hematopoietic stem cell transplantation in which immune cells from donor recognize the host as foreign, leading to adaptive immune responses and tissue damage. There are two clinical forms of GvHD, acute and chronic GvHD, which differ in their pathophysiology [66, 67]. Donor T cells are the primary immunocompetent cells that induce both acute and chronic form of GvHD, but in chronic GvHD, B cell signaling pathways are persistently activated and play an important role in pathophysiology by the production of antibodies to HY and nuclear antigens that can cause tissue damage [68]. Inhibition of B cell signaling was reported to reverse tissue injury in murine models of chronic GvHD [68]. ER stress is important in B cell function and autoimmunity [69]. Recently, conditioned knockdown of XBP-1 in B cells was shown to prevent chronic GvHD and to preserve the graft-versus-leukemia in chronic GvHD mice model [70]. These findings suggest a possibility that IRE-1α/XBP-1 pathway can be a new therapeutic target of chronic GvHD.
Hypopigmentation of tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is a genetic disease caused by mutations in the TSC1 and TSC2 tumor suppressor genes resulting in hyperactivation of the mammalian target of rapamycin (mTOR) signaling pathway [71]. The mTOR is a central regulator of cellular proliferation and metabolic homoeostasis; therefore, hyperactivation of mTORC1 signaling is the key mechanism of hamartomas occurring in multiple organ systems [71]. Cutaneous manifestations including facial angiofibromas or forehead plaque, nontraumatic ungual or periungual fibroma, hypomelanotic macules, and shagreen patches are the major diagnosis criteria of TSC. Among them, angiofibromas, ungulal or periungual fibroma, and shagreen patches are connective tissue hamartomas related to mtoc1 overactivation, however, the exact contribution of mTORC1 signaling to cutaneous hypopigmentation is not fully understood. A recent study demonstrated that constitutive hyperactivation of mTORC1 by conditional TSC2-knockout in melanocytes induces ER damage and enhances ER stress markers in melanocytes, which in turn, reduce skin pigmentation in mice by showing that alleviation of ER stress partially reversed the reduced pigmentation in these mice [72]. These findings suggest that ER stress-induced UPR is involved in the mTORC1 signaling mediated regulation of cutaneous pigmentation and hypopigmentation in TSC.
Melanoma
Recent studies show an involvement of ER stress in tumorigenesis. Various physiologic and pathological stimuli causing ER stress, such as hypoxia, hypoglycemia, genome instability, and cytotoxic compound administration, occur in the uncontrolled proliferation of cancer cells [7]. Cellular adaptation to ER stress is regarded as cancer cell survival strategy, by which the cells escape from apoptosis and host anti-tumor immune systems [73]. ER stress is often evident in melanoma [74]. The high expression of GRP78/BiP, which increases in ER-stressed cells, correlates with melanoma malignancy [75]. ER stress-induced autophagy is a potential pro-survival mechanism that contributes to melanoma progression and a protective mechanism of melanoma cells to overcome BRAF inhibitor resistance [6, 76]. This evidence suggests that targeting adaptation to ER stress can be a potential therapeutic strategy for melanoma. In addition, recently, forkhead family transcription factor (FOXO), which is an important transcriptional regulator of tumor growth and progression, has been shown to interact with ER stress and UPR signals, including PERK and IRE-1 pathways [77, 78]. It has been demonstrated that FOXO activity is controlled in melanoma cells through PI3K/Akt activation, TRIB2 (tribbles homolog 2) and microRNA and is involved in the proliferation and invasion of melanoma, and suppression of FOXO3 promotes survival and metastasis of melanoma cells [79-81]. Taken together, these findings suggest that the inhibition of ER-stress/UPR signaling and its FOXO link can have therapeutic potentials in melanoma treatment.
Conclusion
The ER is a multifunctional signaling organelle that regulates a variety of biological processes; e.g., protein folding and Ca2+ signaling, through evolutionary-conserved signaling pathways, termed the UPR. Previous studies provide important evidence of how the UPR pathway can have a “Yin-Yang” role in cells in response to various ER stress levels triggered by diverse conditions. As we discussed in this review, physiological ER stress is required for the maintenance of normal biological functions in skin, including KC differentiation, a vital process in competent skin barrier formation. In contrast, excessive ER stress is involved in the pathogenesis of certain skin diseases; i.e., Darier’s disease, rosacea, vitiligo, and melanoma (Table 1). Because disease phenotype and symptoms caused by dissimilar conditions are expressed differently, regulation of ER stress could be a potent therapeutic strategy for the treatment of a number of skin diseases in which various pathomechanisms are involved. Moreover, management of ER stress can reduce the risk of developing certain health conditions, including aging. To apply strategies that target ER stress and the UPR pathway to the treatment of these diseases, a comprehensive understanding of what the UPR pathway is associated with in the etiology of each disease, and how it contributes to each disease pathomechanism at the molecular level is needed. Moreover, 1) involvement of the intensity, type, and duration of ER stress in epidermal barrier homeostasis; and 2) the underlying pathomechanism of skin disease associated with ER stress are still unknown.
Table 1.
ER stress and skin disorders
| Disease | Possible mechanism of ER stress | Possible implication of ER stress in the disease pathomechanism |
|---|---|---|
| Darier disease | Chronic depletion of ER calcium store due to mutations in the gene encoding SERCA2 causes constitutive ER stress in keratinocytes [31] | • Abnormal trafficking of junctional components implicates in the acantholysis [30, 31] • SERCA2-induced activation of the pro-apoptotic branches of the UPR implicates in the dyskeratosis [46] |
| Erythrokeratoderma variabilis | Mutant Connexin 31 with defective trafficking causes UPR in keratinocytes [4, 32] | Cell death, abnormal keratinocyte differentiation and hyperproliferation [32] |
| Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome | POMP (proteasome maturation protein) gene mutations proteasome insufficiency induces persistent elevated ER stress in keratinocytes [11] | Abnormal terminal differentiation [11] |
| Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome | Mutated MBTPS2 (a membrane-embedded zinc metalloprotease)-induced impairment in the cleavage of ATF6 induces UPR in keratinocytes [34, 35] | Abnormal keratinization [34, 35] |
| Psoriasis | Undefined | Abnormal epidermal keratinocyte differentiation [2] |
| Rosacea | Various triggering factors of rosacea such as UV exposure, skin irritants, heat, and some foods induce ER stress in keratinocytes [38, 39] | Upregulation of TLR2 which triggers TLR2-KLK5-LL37 inflammatory cascade [37-39] |
| Vitiligo | Environmental factors which induce oxidative stress such as UV exposure and certain chemicals can induce UPR in melanocytes [5, 40, 41] | Activation of innate inflammation which triggers autoimmunity targeting melanocytes [5, 40, 41] |
| Melanoma | Hypoxia, hypoglycemia, genome instability,and cytotoxic compounds [7, 42] | • Cellular adaptation to ER stress can be the survival strategies of melanoma cells [6] • ER stress-induced autophagy can be a pro-survival mechanism of melanoma cells to overcome BRAF inhibitor resistance [45] |
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07050504), the Korea Institute for Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA)(117063031SB101)(to K.P.), the Ministry of Trade, Industry and Energy (MOTIE) and Korea Institute for Advancement of Technology (KIAT) through the Encouragement Program for the Industries of Economic Cooperation Region (R0004132)(to S.E.L), and the National Rosacea Society, the San Francisco Foundation, and National Institutes of Health Grants AR051077 and AR062025 (the National Institute of Arthritis and Musculoskeletal and Skin Diseases) (to Y.U.). We acknowledge the support of the Medical Research Services of the Veterans Affairs Medical Center, San Francisco.
Abbreviations:
- ATF
activating transcription factor
- Ca2+
calcium
- CHOP
C/EBP homologous protein
- Dsg
desmoglein
- eIF2
eukaryotic translation initiation factor 2α
- EKV
erythrokeratoderma variabilis
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- FOXO
forkhead family transcription factor
- GRP78/BiP
glucose-regulated protein 78/binding immunoglobulin protein
- IFAP
ichthyosis follicularis, alopecia, and photophobia
- IRE1 α
inositol-requiring enzyme 1 alpha
- KC
keratinocytes
- KLK
kallikrein
- MBTPS2
membrane-bound transcription factor protease, site 2
- mTOR
mammalian target of rapamycin
- PERK
protein kinase R-like endoplasmic reticulum kinase
- POMP
proteasome maturation protein
- S1P
sphingosine-1-phosphate
- SB
stratum basale
- SC
stratum corneum
- SERCA2
sarco/endoplasmic reticulum Ca2+-ATPase 2
- SG
stratum granulosum
- SREBP
sterol regulatory element-binding protein
- SS
stratum spinosum
- TLR2
toll-like receptor 2
- TRIB
tribbles homolog 2
- TSC
Tuberous sclerosis complex
- UPR
unfolded protein response
- UV
ultraviolet
- XPB1
X-box-binding protein 1
Footnotes
Disclosure statement
The authors state no conflict of interest.
References:
- 1.Yoshida H (2007) ER stress and diseases, Febs j. 274, 630–58. [DOI] [PubMed] [Google Scholar]
- 2.Sugiura K, Muro Y, Futamura K, Matsumoto K, Hashimoto N, Nishizawa Y, Nagasaka T, Saito H, Tomita Y & Usukura J (2009) The unfolded protein response is activated in differentiating epidermal keratinocytes, J Invest Dermatol. 129, 2126–35. [DOI] [PubMed] [Google Scholar]
- 3.Woo YR, Lim JH, Cho DH & Park HJ (2016) Rosacea: Molecular Mechanisms and Management of a Chronic Cutaneous Inflammatory Condition, Int J Mol Sci. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Common JE, O’Toole EA, Leigh IM, Thomas A, Griffiths WA, Venning V, Grabczynska S, Peris Z, Kansky A & Kelsell DP (2005) Clinical and genetic heterogeneity of erythrokeratoderma variabilis, J Invest Dermatol. 125, 920–7. [DOI] [PubMed] [Google Scholar]
- 5.Frisoli ML & Harris JE (2017) Vitiligo: Mechanistic insights lead to novel treatments, J Allergy Clin Immunol. 140, 654–662. [DOI] [PubMed] [Google Scholar]
- 6.Meng XX, Yao M, Zhang XD, Xu HX & Dong Q (2015) ER stress-induced autophagy in melanoma, Clin Exp Pharmacol Physiol. 42, 811–6. [DOI] [PubMed] [Google Scholar]
- 7.Corazzari M, Gagliardi M, Fimia GM & Piacentini M (2017) Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate, Front Oncol. 7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O’Donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T & Hovnanian A (1999) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease, Nat Genet. 21, 271–7. [DOI] [PubMed] [Google Scholar]
- 9.Elias PM (2005) Stratum corneum defensive functions: an integrated view, J Invest Dermatol. 125, 183–200. [DOI] [PubMed] [Google Scholar]
- 10.Elias PM (2007) The skin barrier as an innate immune element, Semin Immunopathol. 29, 3–14. [DOI] [PubMed] [Google Scholar]
- 11.Elias PM, Ahn SK, Denda M, Brown BE, Crumrine D, Kimutai LK, Komuves L, Lee SH & Feingold KR (2002) Modulations in epidermal calcium regulate the expression of differentiation-specific markers, J Invest Dermatol. 119, 1128–36. [DOI] [PubMed] [Google Scholar]
- 12.Elias PM & Feingold KR (2001) Coordinate regulation of epidermal differentiation and barrier homeostasis, Skin Pharmacol Appl Skin Physiol. 14 Suppl 1, 28–34. [DOI] [PubMed] [Google Scholar]
- 13.Uchida Y, Behne M, Quiec D, Elias PM & Holleran WM (2001) Vitamin C stimulates sphingolipid production and markers of barrier formation in submerged human keratinocyte cultures, J Invest Dermatol. 117, 1307–13. [DOI] [PubMed] [Google Scholar]
- 14.Elias PM & Feingold KR (1988) Lipid-related barriers and gradients in the epidermis, Annals of the New York Academy of Sciences. 548, 4–13. [DOI] [PubMed] [Google Scholar]
- 15.Hampton RY (2000) ER stress response: getting the UPR hand on misfolded proteins, Curr Biol. 10, R518–21. [DOI] [PubMed] [Google Scholar]
- 16.Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, Luis A, McCarthy N, Montibeller L, More S, Papaioannou A, Puschel F, Sassano ML, Skoko J, Agostinis P, de Belleroche J, Eriksson LA, Fulda S, Gorman AM, Healy S, Kozlov A, Munoz-Pinedo C, Rehm M, Chevet E & Samali A (2018) Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications, The FEBS journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paschen W (2003) Shutdown of translation: lethal or protective? Unfolded protein response versus apoptosis, J Cereb Blood Flow Metab. 23, 773–9. [DOI] [PubMed] [Google Scholar]
- 18.Schroder M (2008) Endoplasmic reticulum stress responses, Cellular and molecular life sciences : CMLS. 65, 862–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida H, Matsui T, Yamamoto A, Okada T & Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor, Cell. 107, 881–91. [DOI] [PubMed] [Google Scholar]
- 20.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS & Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation, Cell. 101, 249–58. [DOI] [PubMed] [Google Scholar]
- 21.Iwakoshi NN, Lee AH & Glimcher LH (2003) The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response, Immunol Rev. 194, 29–38. [DOI] [PubMed] [Google Scholar]
- 22.Covino R, Hummer G & Ernst R (2018) Integrated Functions of Membrane Property Sensors and a Hidden Side of the Unfolded Protein Response, Molecular cell. 71, 458–467. [DOI] [PubMed] [Google Scholar]
- 23.Sriburi R, Jackowski S, Mori K & Brewer JW (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum, The Journal of cell biology. 167, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.So JS (2018) Roles of Endoplasmic Reticulum Stress in Immune Responses, Molecules and cells. 41, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP & Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1, Science. 287, 664–6. [DOI] [PubMed] [Google Scholar]
- 26.Kaneko M, Niinuma Y & Nomura Y (2003) Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2, Biol Pharm Bull. 26, 931–5. [DOI] [PubMed] [Google Scholar]
- 27.Mauro C, Crescenzi E, De Mattia R, Pacifico F, Mellone S, Salzano S, de Luca C, D’Adamio L, Palumbo G, Formisano S, Vito P & Leonardi A (2006) Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis, J Biol Chem. 281, 2631–8. [DOI] [PubMed] [Google Scholar]
- 28.Hu P, Han Z, Couvillon AD, Kaufman RJ & Exton JH (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression, Molecular and cellular biology. 26, 3071–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maurel M, Chevet E, Tavernier J & Gerlo S (2014) Getting RIDD of RNA: IRE1 in cell fate regulation, Trends in biochemical sciences. 39, 245–54. [DOI] [PubMed] [Google Scholar]
- 30.Walter P & Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation, Science (New York, NY). 334, 1081–6. [DOI] [PubMed] [Google Scholar]
- 31.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA & Majsterek I (2016) The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress, Current molecular medicine. 16, 533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carreras-Sureda A, Pihan P & Hetz C (2018) Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses, Cell Calcium. 70, 24–31. [DOI] [PubMed] [Google Scholar]
- 33.Bahar E, Kim H & Yoon H (2016) ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players, Int J Mol Sci. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krebs J, Agellon LB & Michalak M (2015) Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling, Biochem Biophys Res Commun. 460, 114–21. [DOI] [PubMed] [Google Scholar]
- 35.Park K, Elias PM, Oda Y, Mackenzie D, Mauro T, Holleran WM & Uchida Y (2011) Regulation of Cathelicidin Antimicrobial Peptide Expression by an Endoplasmic Reticulum (ER) Stress Signaling, Vitamin D Receptor-independent Pathway, J Biol Chem. 286, 34121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park K, Ikushiro H, Seo HS, Shin KO, Kim YI, Kim JY, Lee YM, Yano T, Holleran WM, Elias P & Uchida Y (2016) ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex, Proc Natl Acad Sci U S A. 113, E1334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bikle DD, Xie Z & Tu CL (2012) Calcium regulation of keratinocyte differentiation, Expert Rev Endocrinol Metab. 7, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Celli A, Sanchez S, Behne M, Hazlett T, Gratton E & Mauro T (2010) The epidermal Ca(2+) gradient: Measurement using the phasor representation of fluorescent lifetime imaging, Biophys J. 98, 911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Celli A, Mackenzie DS, Crumrine DS, Tu CL, Hupe M, Bikle DD, Elias PM & Mauro TM (2011) Endoplasmic reticulum Ca2+ depletion activates XBP1 and controls terminal differentiation in keratinocytes and epidermis, Br J Dermatol. 164, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim YI, Park K, Kim JY, Seo HS, Shin KO, Lee YM, Holleran WM, Elias PM & Uchida Y (2014) An endoplasmic reticulum stress-initiated sphingolipid metabolite, ceramide-1-phosphate, regulates epithelial innate immunity by stimulating beta-defensin production, Mol Cell Biol. 34, 4368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin KO, Kim KP, Cho Y, Kang MK, Kang YH, Lee YM, Ikushiro H, Yokota M, Yano T, Choe SJ, Choi EH, Lim CJ, Park K, Holleran WM, Park K & Uchida Y (2018) Both sphingosine kinase 1 and 2 coordinately regulate cathelicidin antimicrobial peptide production during keratinocyte differentiation, J Invest Dermatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hovnanian A (2004) Darier’s disease: from dyskeratosis to endoplasmic reticulum calcium ATPase deficiency, Biochem Biophys Res Commun. 322, 1237–44. [DOI] [PubMed] [Google Scholar]
- 43.Savignac M, Simon M, Edir A, Guibbal L & Hovnanian A (2014) SERCA2 dysfunction in Darier disease causes endoplasmic reticulum stress and impaired cell-to-cell adhesion strength: rescue by Miglustat, J Invest Dermatol. 134, 1961–1970. [DOI] [PubMed] [Google Scholar]
- 44.Li N, Park M, Xiao S, Liu Z & Diaz LA (2017) ER-to-Golgi blockade of nascent desmosomal cadherins in SERCA2-inhibited keratinocytes: Implications for Darier’s disease, Traffic. 18, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onozuka T, Sawamura D, Goto M, Yokota K & Shimizu H (2006) Possible role of endoplasmic reticulum stress in the pathogenesis of Darier’s disease, J Dermatol Sci. 41, 217–20. [DOI] [PubMed] [Google Scholar]
- 46.Tattersall D, Scott CA, Gray C, Zicha D & Kelsell DP (2009) EKV mutant connexin 31 associated cell death is mediated by ER stress, Hum Mol Genet. 18, 4734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di WL, Monypenny J, Common JE, Kennedy CT, Holland KA, Leigh IM, Rugg EL, Zicha D & Kelsell DP (2002) Defective trafficking and cell death is characteristic of skin disease-associated connexin 31 mutations, Human molecular genetics. 11, 2005–14. [DOI] [PubMed] [Google Scholar]
- 48.Diestel S, Richard G, Doring B & Traub O (2002) Expression of a connexin31 mutation causing erythrokeratodermia variabilis is lethal for HeLa cells, Biochemical and biophysical research communications. 296, 721–8. [DOI] [PubMed] [Google Scholar]
- 49.Mese G, Richard G & White TW (2007) Gap junctions: basic structure and function, The Journal of investigative dermatology. 127, 2516–24. [DOI] [PubMed] [Google Scholar]
- 50.Dahlqvist J, Klar J, Tiwari N, Schuster J, Torma H, Badhai J, Pujol R, van Steensel MA, Brinkhuizen T, Gijezen L, Chaves A, Tadini G, Vahlquist A & Dahl N (2010) A single-nucleotide deletion in the POMP 5’ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis, Am J Hum Genet. 86, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS & Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs, Mol Cell. 6, 1355–64. [DOI] [PubMed] [Google Scholar]
- 52.Oeffner F, Fischer G, Happle R, Konig A, Betz RC, Bornholdt D, Neidel U, Boente Mdel C, Redler S, Romero-Gomez J, Salhi A, Vera-Casano A, Weirich C & Grzeschik KH (2009) IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response, Am J Hum Genet. 84, 459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, Morhenn VB & Gallo RL (2007) Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea, Nat Med. 13, 975–80. [DOI] [PubMed] [Google Scholar]
- 54.Yamasaki K, Kanada K, Macleod DT, Borkowski AW, Morizane S, Nakatsuji T, Cogen AL & Gallo RL (2011) TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes, J Invest Dermatol. 131, 688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimasaki S, Koga T, Shuto T, Suico MA, Sato T, Watanabe K, Morino-Koga S, Taura M, Okada S, Mori K & Kai H (2010) Endoplasmic reticulum stress increases the expression and function of toll-like receptor-2 in epithelial cells, Biochemical and biophysical research communications. 402, 235–40. [DOI] [PubMed] [Google Scholar]
- 56.Melnik BC (2014) Endoplasmic reticulum stress: key promoter of rosacea pathogenesis, Exp Dermatol. 23, 868–73. [DOI] [PubMed] [Google Scholar]
- 57.Melnik BC (2016) Rosacea: The Blessing of the Celts - An Approach to Pathogenesis Through Translational Research, Acta Derm Venereol. 96, 147–56. [DOI] [PubMed] [Google Scholar]
- 58.Park BW, Ha JM, Cho EB, Jin JK, Park EJ, Park HR, Kang HJ, Ko SH, Kim KH & Kim KJ (2018) A Study on Vitamin D and Cathelicidin Status in Patients with Rosacea: Serum Level and Tissue Expression, Annals of dermatology. 30, 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rashighi M & Harris JE (2017) Vitiligo Pathogenesis and Emerging Treatments, Dermatol Clin. 35, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Birlea SA, Jin Y, Bennett DC, Herbstman DM, Wallace MR, McCormack WT, Kemp EH, Gawkrodger DJ, Weetman AP, Picardo M, Leone G, Taieb A, Jouary T, Ezzedine K, van Geel N, Lambert J, Overbeck A, Fain PR & Spritz RA (2011) Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP, J Invest Dermatol. 131, 371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bystryn JC & Rudolph JL (2005) Pemphigus, Lancet (London, England). 366, 61–73. [DOI] [PubMed] [Google Scholar]
- 62.Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA & Rubenstein DS (2006) p38MAPK inhibition prevents disease in pemphigus vulgaris mice, Proceedings of the National Academy of Sciences of the United States of America. 103, 12855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanza A, Lanza M, Santoro R, Soro V, Prime SS & Cirillo N (2011) Deregulation of PERK in the autoimmune disease pemphigus vulgaris occurs via IgG-independent mechanisms, The British journal of dermatology. 164, 336–43. [DOI] [PubMed] [Google Scholar]
- 64.Mihailidou C, Katsoulas N, Panagiotou E, Farmaki E, Sklavounou A, Kiaris H & Chatzistamou I (2016) Endoplasmic reticulum stress is associated with the pathogenesis of pemphigus vulgaris, Experimental dermatology. 25, 731–3. [DOI] [PubMed] [Google Scholar]
- 65.Cipolla GA, Park JK, Lavker RM & Petzl-Erler ML (2017) Crosstalk between Signaling Pathways in Pemphigus: A Role for Endoplasmic Reticulum Stress in p38 Mitogen-Activated Protein Kinase Activation?, Frontiers in immunology. 8, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Billingham RE (1966) The biology of graft-versus-host reactions, Harvey lectures. 62, 21–78. [PubMed] [Google Scholar]
- 67.Blazar BR, Murphy WJ & Abedi M (2012) Advances in graft-versus-host disease biology and therapy, Nature reviews Immunology. 12, 443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarantopoulos S, Blazar BR, Cutler C & Ritz J (2015) B cells in chronic graft-versus-host disease, Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 21, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang K, Wong HN, Song B, Miller CN, Scheuner D & Kaufman RJ (2005) The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis, The Journal of clinical investigation. 115, 268–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schutt SD, Wu Y, Tang CH, Bastian D, Nguyen H, Sofi MH, Zhang M, Liu C, Helke K, Wilson C, Schnapp LM, Del Valle JR, Hu CC & Yu XZ (2018) Inhibition of the IRE-1alpha/XBP-1 pathway prevents chronic GVHD and preserves the GVL effect in mice, Blood advances. 2, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwartz RA, Fernandez G, Kotulska K & Jozwiak S (2007) Tuberous sclerosis complex: advances in diagnosis, genetics, and management, Journal of the American Academy of Dermatology. 57, 189–202. [DOI] [PubMed] [Google Scholar]
- 72.Yang F, Yang L, Wataya-Kaneda M, Yoshimura T, Tanemura A & Katayama I (2018) Uncoupling of ER/Mitochondrial Oxidative Stress in mTORC1 Hyperactivation-Associated Skin Hypopigmentation, The Journal of investigative dermatology. 138, 669–678. [DOI] [PubMed] [Google Scholar]
- 73.Riha R, Gupta-Saraf P, Bhanja P, Badkul S & Saha S (2017) Stressed Out - Therapeutic Implications of ER Stress Related Cancer Research, Oncomedicine. 2, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hersey P & Zhang XD (2008) Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma, Pigment Cell Melanoma Res. 21, 358–67. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu A, Kaira K, Yasuda M, Asao T & Ishikawa O (2017) Clinical and Pathological Significance of ER Stress Marker (BiP/GRP78 and PERK) Expression in Malignant Melanoma, Pathol Oncol Res. 23, 111–116. [DOI] [PubMed] [Google Scholar]
- 76.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, Lazova R, Klump V, Pawelek JM, Xu X, Xu W, Schuchter LM, Davies MA, Herlyn M, Winkler J, Koumenis C & Amaravadi RK (2014) Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma, J Clin Invest. 124, 1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hornsveld M, Dansen TB, Derksen PW & Burgering BMT (2018) Re-evaluating the role of FOXOs in cancer, Seminars in cancer biology. 50, 90–100. [DOI] [PubMed] [Google Scholar]
- 78.Alasiri G, Fan LY, Zona S, Goldsbrough IG, Ke HL, Auner HW & Lam EW (2018) ER stress and cancer: The FOXO forkhead transcription factor link, Molecular and cellular endocrinology. 462, 67–81. [DOI] [PubMed] [Google Scholar]
- 79.Zanella F, Renner O, Garcia B, Callejas S, Dopazo A, Peregrina S, Carnero A & Link W (2010) Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells, Oncogene. 29, 2973–82. [DOI] [PubMed] [Google Scholar]
- 80.Ren JW, Li ZJ & Tu C (2015) MiR-135 post-transcriptionally regulates FOXO1 expression and promotes cell proliferation in human malignant melanoma cells, International journal of clinical and experimental pathology. 8, 6356–66. [PMC free article] [PubMed] [Google Scholar]
- 81.Tsitsipatis D, Klotz LO & Steinbrenner H (2017) Multifaceted functions of the forkhead box transcription factors FoxO1 and FoxO3 in skin, Biochimica et biophysica acta. 1861, 1057–1064. [DOI] [PubMed] [Google Scholar]