

Abstract
The inflammatory airway disease cystic fibrosis (CF) is characterized by airway obstruction due to mucus hypersecretion, airway plugging, and bronchoconstriction. The cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel is dysfunctional in CF, leading to defects in epithelial transport. Although CF pathogenesis is still disputed, activation of alternative Cl− channels is assumed to improve lung function in CF. Two suitable non-CFTR Cl− channels are present in the airway epithelium, the Ca2+ activated channel TMEM16A and SLC26A9. Activation of these channels is thought to be feasible to improve hydration of the airway mucus and to increase mucociliary clearance. Interestingly, both channels are upregulated during inflammatory lung disease. They are assumed to support fluid secretion, necessary to hydrate excess mucus and to maintain mucus clearance. During inflammation, however, TMEM16A is upregulated particularly in mucus producing cells, with only little expression in ciliated cells. Recently it was shown that knockout of TMEM16A in ciliated cells strongly compromises Cl− conductance and attenuated mucus secretion, but does not lead to a CF-like lung disease and airway plugging. Along this line, activation of TMEM16A by denufosol, a stable purinergic ligand, failed to demonstrate any benefit to CF patients in earlier studies. It rather induced adverse effects such as cough. A number of studies suggest that TMEM16A is essential for mucus secretion and possibly also for mucus production. Evidence is now provided for a crucial role of TMEM16A in fusion of mucus-filled granules with the apical plasma membrane and cellular exocytosis. This is probably due to local Ca2+ signals facilitated by TMEM16A. Taken together, TMEM16A supports fluid secretion by ciliated airway epithelial cells, but also maintains excessive mucus secretion during inflammatory airway disease. Because TMEM16A also supports airway smooth muscle contraction, inhibition rather than activation of TMEM16A might be the appropriate treatment for CF lung disease, asthma and COPD. As a number of FDA-approved and well-tolerated drugs have been shown to inhibit TMEM16A, evaluation in clinical trials appears timely.
Keywords: TMEM16A, anoctamin 1, mucus secretion, cystic fibrosis, asthma, COPD, Ca2+ signaling
Introduction
The inflammatory airway disease cystic fibrosis (CF) is characterized by airway obstruction due to mucus hypersecretion, mucus plugging, and bronchoconstriction. The cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel is dysfunctional in CF, leading to a loss of fluid secretion and probably impaired bicarbonate transport, along with Na+ hyperabsorption (Boucher, 2007; Stoltz et al., 2015). Dehydration of the airway surface (periciliary) fluid layer (ASL) covering the airway epithelium is thought to be the crucial factor leading to abnormal rheological properties of the mucus and occlusion of smaller airways (Boucher, 2007). Independent of the underlying precise molecular defect, tremendous effort was put into identification of small molecules, and natural compounds that would correct the basic defect by restoring CFTR function (Amaral and Kunzelmann, 2007; Verkman and Galietta, 2009). This, however, turned out to be a long and stony path that has finally provided effective drugs that correct and potentiate mutant CFTR. Moreover, the search is on to identify compounds that inhibit salt hyperabsorption in CF, and to find molecules that activate alternative Cl− secretory pathways. What has not been in the minds of CF researchers is to look for inhibitors of certain types of Cl− channels as a treatment for inflammatory airway disease (Li et al., 2017).
Still Unresolved: The Pathogenesis of CF
Lack of appropriate Cl− secretion due to defective CFTR was long regarded as the essential, if not only cause for CF lung disease. However, a pathogenic concept proposing Na+ hyperabsorption relative to attenuated Cl− secretion, leading to airway dehydration and mucus plugging, has also been put forward in a number of studies (Boucher et al., 1986; Grubb et al., 1994; Mall et al., 1998a, 2004). We detected enhanced Na+ conductances in nasal ex vivo tissue and freshly isolated intestinal cells from CF patients (Mall et al., 1998a, 2000). Along this line, reduced ASL would lead to thickened airway mucus, airway plugging and impaired mucociliary clearance with subsequent chronic bacterial infections. Yet, this concept has been questioned by Welsh and collaborators as well as other investigators, who did not find evidence for Na+ hyperabsorption. In contrast, reduced airway Na+ absorption in CF was claimed, leading to salt accumulation in the ASL, which under normal conditions might be even hypotonic when compared with the interstitial fluid. Thus, hypertonic ASL was blamed to inactivate ß-defensins, thereby causing a predisposition toward bacterial infections (Zabner et al., 1998; Chen et al., 2010; Itani et al., 2011). In contrast, the Boucher team and others found neither evidence for a hypotonic ASL under normal conditions, nor any salt concentration (hypertonic ASL) in CF airways (Matsui et al., 1998). Given the fact that the airway epithelium is relatively leaky and has a large hydraulic conductivity, it appears somewhat unlikely that it maintains a large transepithelial osmotic gradient.
A similar controversy arose around the pH value of the ASL. It had been shown that CFTR is permeable for bicarbonate (), or contributes to transport as a Cl− recycling pathway in a number of epithelial organs [reviewed in (Kunzelmann et al., 2017)]. To what extend is conducted by CFTR or rather operates indirectly as a Cl− recycling channel that drives secretion by Cl−/ exchangers, is still a matter of debate. At any rate, Smith and Welsh were among the first to show defective cAMP-induced bicarbonate secretion in airways of CF patients (Smith and Welsh, 1992), while others showed that CFTR is permeable for (Poulsen et al., 1994; Tang et al., 2009). It should be noted that patch clamp and other types of experiments with isotonic concentrations of are not trivial and may be compromised by pH fluctuations (Kunzelmann et al., 1991). Attenuated fluid/ secretion in CF airways was shown to have adverse effects on the biophysical properties of airway mucus (Trout et al., 1998). Quinton and others provided further evidence that bicarbonate transport is essential for proper mucus release and viscosity (Choi et al., 2001; Quinton, 2001). In fact, transport is impaired in a number of different epithelial tissues derived from CF patients (Kunzelmann et al., 2017). Importantly, human lung pathology was brilliantly reproduced in a CF pig model. Using this CF pig model, reduced airway surface liquid pH, impaired bacterial killing, and mucus abnormalities were demonstrated (Pedersen et al., 1999; Stoltz et al., 2010; Pezzulo et al., 2012; Hoegger et al., 2014). Interestingly, Hoegger et al. demonstrated abnormal mucociliary transport in CF in submerged epithelia, which somewhat questions the role of surface dehydration in CF (Hoegger et al., 2014). In sharp contrast to these results, Schultz and coworkers found no evidence for acidic airway surface liquid pH in lungs of CF children, using a novel optical pH probe and a specialized bronchoscope (Schultz et al., 2017).
Stick and Schulz claimed that only a small fraction of infants diagnosed early with CF through the Australian AREST CF early surveillance program, present lower airway infection (Stick and Schultz, 2018). This may support the “inflammation first” concept, proposing that inflammation in CF lungs is present early in life and clearly before airway infection (Khan et al., 1995; Doring and Worlitzsch, 2000). Thus, CF epithelia appear to be in an inflammatory, pro-proliferative, and constantly remodeling state, even without any bacterial infection (Hajj et al., 2007; Rottner et al., 2007, 2009; Martins et al., 2011). CF epithelial cells were also shown to have a compromised anti-oxidant defense by superoxide dismutase (Rottner et al., 2011). Notably, some CFTR knockout mouse models do show signs of lung/airway inflammation even in the absence of mucus obstruction (Tirkos et al., 2006; Wilke et al., 2011). Moreover, airways of newborn CF pigs demonstrate developmental defects, such as cartilage abnormalities, muscle bundles, and smaller airways, which may support progression into a CF lung disease (Chen et al., 2010; Stoltz et al., 2010; Klymiuk et al., 2012). Considerable insight into epithelial ion transport and its defect in CF, was also obtained in studies with wt and transgenic mice, although mouse airways show differences in structure, distribution of submucosal glands and contribution of CFTR to Cl− transport. While identical ion channels and transporters are expressed in human and mouse airway, intestinal epithelium, and other epithelial organs, the course of lung disease is mild in CF mice and airway plugging is not observed (Wilke et al., 2011). Nevertheless, because of the available broad range of tools in mouse genetics, valuable insights into organ physiology and pathological changes in CF have been gained in the different mouse CF models.
Is Abnormal Ion Transport Relevant in CF or Only an Epiphenomenon?
Others argue that airway inflammation is second to the transport defect and to bacterial colonization (Ribeiro et al., 2005). At any rate, the most impressive clinical symptom in CF lung disease is accumulation of large amounts of airway mucus. Yet, are low pH, Na+ hyperabsorption, or lack of Cl− secretion are truly the cause for mucus plugging and obstruction of airways? If we compare the changes in absorptive and secretory ion transport present in ßENaC-overexpressing mice (Mall et al., 2004), with the changes in ion transport in kcne3 knockout mice (Preston et al., 2010), or mice lacking TMEM16A in ciliated epithelial cells (Benedetto et al., 2017), the contribution of ion transport appears less clear. In the three mouse models we find a shift toward enhanced net absorptive transport: (i) pronounced increase in Na+ absorption with unchanged Cl− secretion (Mall et al., 2004), (ii) milder increase in Na+ absorption but strongly reduced Cl− secretion (Preston et al., 2010), (iii) partial reduction in Na+ absorption but pronounced inhibition of Cl− secretion (Benedetto et al., 2017) (Table 1). Only the ßENaC mice develop a CF like lung phenotype, which may suggest that indeed Na+ absorption by ENaC is rate limiting and determines the direction of net flux of ions and water through an otherwise relatively leaky airway epithelium (Cotton et al., 1983; Donaldson and Boucher, 2007). This may explains why loss of Cl− secretion in the airways of adult kcne3 and tmem16A knockout mice does not lead to a CF like lung phenotype.
Table 1.
Ion transport assessed by the measurement of short circuit currents in different adult mouse models [wild type (wt) littermates vs. transgenic (trans) mice].
| Mouse model | Amiloride-sensitive Isc (μA/cm2) | cAMP-activated Isc | Ca2+-activated Isc | Phenotype | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| wt | trans | %↓↑ | wt | trans | %↓↑ | wt | trans | %↓↑ | ||
| ßENaC overexpressing mouse (Mall et al., 2004) | 22 | 59 | ↑168 | 25 | 27 | ↑11 | 79 | 84 | ↑6 | CF-like lung disease |
| Kcne3-knockout mouse (Preston et al., 2010) | 48 | 84 | ↑ 75 | 83 | 17 | ↓80 | 294 | 50 | ↓83 | No lung phenotype |
| FOXJ1-Cre-TMEM16Aflox/flox mice (Benedetto et al., 2017) | 104 | 49 | ↓ 47 | 99 | 58 | ↓42 | 176 | 74 | ↓58 | No lung phenotype |
Shown are approximate Isc values. %↓↑, percent decrease or increase in unidirectional ion transport.
As discussed below, basal airway mucus secretion is attenuated in mice lacking TMEM16A in ciliated epithelial cells. This leads to mucus accumulation in secretory epithelial cells (Benedetto et al., 2019) (Figure 1). The mechanism of crosstalk between the two cell types is currently unclear. However, it is likely that ciliated cells secrete a factor that is required to release mucus from club cells. This factor could be ATP, which is found in the airway surface liquid, and which is assumed to control basal mucus secretion. Evidence has been presented that TMEM16A supports vesicular/granular exocytosis and subsequent insertion of transmembrane proteins into the plasma membrane. Moreover, it was also shown to control paracrine release of inflammatory mediators (Benedetto et al., 2016, 2017, 2019; Cabrita et al., 2017). We would favor a pathogenic mechanism that initiates the disease by intrinsic inflammation caused by dyslocalization/dysfunction of CFTR, and in the absence of bacterial infection. Intrinsic inflammation is followed by upregulation of TMEM16A, particularly in mucus producing cells, with consecutive mucus hyperproduction/hypersecretion. Accordingly, inflammation and mucus hyperproduction/hypersecretion should be pharmacologically targeted. Along this line it appears noteworthy that treatment with the anti-inflammatory drug ibuprofen was able to rescue trafficking mutant F508del-CFTR (Carlile et al., 2015).
Figure 1.

Accumulation of mucus in airways of TMEM16A−/− mice. Mucus staining (PAS) in bronchi of wild type littermates (TMEM16Aflox/flox; TMEM16A+/+) and mice with an airway epithelial-specific knockout of TMEM16A (FOXJ1-Cre-TMEM16Aflox/flox; TMEM16A−/−). Strong accumulation of mucus in airway epithelial cells is observed in TMEM16A−/− mice. Bars indicate 20 μm (Benedetto et al., 2019).
Correcting and Potentiating CFTR
Pharmacological restoration of defective CFTR chloride transport has been the primary goal over the past decade and has led to considerable success in correcting the gating mutant G551D-CFTR by the potentiator compound VX-770 (ivacaftor, kalydeco) (Van Goor et al., 2009; Accurso et al., 2010). G551D accounts for only a small fraction (about 5%) of all CF cases (De Boeck et al., 2014). Thus, US-based Vertex developed the small molecule CFTR-corrector VX-809 (lumacaftor), a compound that is able to rescue a fraction of the class two mutant F508del-CFTR in vitro (Van Goor et al., 2011; Pranke et al., 2017). Large phase 3 studies demonstrated a moderate improvement in the percentage of predicted FEV1 between 2.6 and 4.0% (Wainwright et al., 2015). Studies with next generation compounds, e.g., VX-661 (Tezacaftor), reported improvement in FEV1 in the range of 7% (Rowe et al., 2017; Taylor-Cousar et al., 2017; Donaldson et al., 2018). Recent clinical trials reported great therapeutic success with triple combinations of the corrector VX661 with VX-445 or VX-659, together with the potentiator VX-770. The combinations increased the percentage of predicted FEV1 by more than 10% (Davies et al., 2018; Keating et al., 2018). Despite such great success in correcting mutant CFTR, critical voices were raised regarding a combinatorial drug treatment. For example, adverse effects of VX-770 were reported on VX-809-corrected F508del-CFTR in the combinatorial preparation Orkambi. Moreover, long-term effects of these drugs on CFTR expression could be negative (Cholon et al., 2014; Veit et al., 2014; Chin et al., 2018). Finally, the costs for treatment by these drugs are often prohibitive (Ferkol and Quinton, 2015). Thus, there is a need for alternative drug treatments.
A number of other strategies to correct biosynthesis of misfolded CFTR are currently under investigation Recent screening efforts identified the translation initiation factor 3a (eIF3a) as a potentially druggable central hub for the biogenesis of CFTR (Hutt et al., 2018). FDA-approved histone deacetylase (HDAC) inhibitors such as panobinostat (LBH-589) and romidepsin (FK-228) can help to correct misfolded CFTR, particularly in combination with other correctors such as VX809 (Angles et al., 2018). Promising results were also obtained with combinations of pharmacological chaperones with different sites of action, such as VX-809, RDR1, and MCG1516A (Carlile et al., 2018). All these recent results are rather encouraging, however, as not every CFTR mutation is accessible to such a CFTR-based therapy, activation of other airway epithelial Cl− channels was proposed to compensate for defective CFTR (De Boeck and Amaral, 2016).
Airway Chloride Channels: SLC26A9
Apart from CFTR, there are two other major Cl− channels present in human and mouse airways, namely SLC26A9 and TMEM16A. How much both Cl− channels quantitatively contribute to production of the ASL and support mucociliary clearance is currently not known (Figure 2). SLC26 proteins typically operate as anion exchangers, but for SLC26A9 it was shown to function as a Cl− channel (Mount and Romero, 2004; Ousingsawat et al., 2011b; Bertrand et al., 2017). In contrast to CFTR, SLC26A9 is spontaneously active, once inserted into the apical membrane of airway epithelial cells. However, it is also regulated by Cl− feedback/WNK kinases and surprisingly is controlled by the Cl− channel CFTR (Dorwart et al., 2007; Bertrand et al., 2009). SLC26A9 is likely to provide the basal Cl− conductance that is found in airways in the absence of any secretagogue (Bertrand et al., 2017). Absence of basal Cl− secretion in airways of CF patients carrying the type II mutation F508del-CFTR (De Boeck and Amaral, 2016), is probably due to the lack of expression of SLC26A9 in the apical membrane (Figure 2B) (El Khouri and Toure, 2014; Bertrand et al., 2017). SLC26A9 and CFTR form a complex with the help of PDZ scaffold proteins such as NHERF1 (Figure 3). When coexpressed with wtCFTR, SLC26A9 is co-trafficked together with CFTR from the ER to the plasma membrane. However, if coexpressed with F508del-CFTR, SLC26A9 will remain intracellularly and will be degraded (Bertrand et al., 2017). As shown recently, also CFTR and TMEM16A do interact via PDZ-domain proteins, and TMEM16A has an impact on plasma membrane expression of CFTR (Benedetto et al., 2017).
Figure 2.
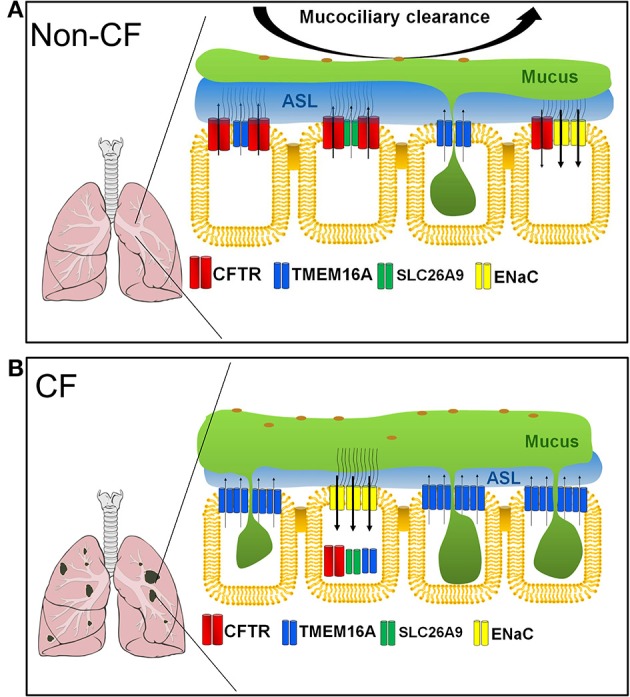
Ion channels contributing to fluid/mucus balance in non-CF and CF airways. (A) In non-CF airways, CFTR is expressed in ciliated epithelial cells and ionocytes (not shown). Ca2+ activated Cl− channels are sparsely expressed in both ciliated and secretory club and goblet cells. SLC26A9, TMEM16A and CFTR located in ciliated epithelial cells and probably in ionocytes are in charge of fluid secretion, while TMEM16A expressed in secretory club/goblet cells support mucus secretion. Epithelial Na+ channels reabsorb Na+ thereby causing fluid absorption. (B) In inflamed CF airways, CFTR is dysfunctional and often dislocated intracellularly together with SLC26A9 and TMEM16A in ciliated epithelial cells. TMEM16A is upregulated in secretory cells strongly contributing to mucus secretion. Na+ absorption by ENaC is augmented. Inflammation and transport abnormalities lead to excessive mucus production/secretion, airway plugging, and reduced water secretion, strongly reducing mucociliary clearance.
Figure 3.

Molecular and functional relationship of CFTR, SLC26A9 and TMEM16A. Both SLC26A9 and TMEM16A interact with CFTR via their C-terminal PDZ (PSD-95/Discs-large/ZO1) -binding domains with the help of the PDZ-protein NHERF1 (Na+/H+ exchanger regulatory factor 1).
Furthermore, SLC26A9 may be regulated by the R domain of CFTR through STAS domain interaction (Chang et al., 2009). Indeed, SLC26A9 has been demonstrated to be a genetic modifier in CF (Sun et al., 2012; Miller et al., 2015), and the CFTR corrector VX-809 partially rescued SLC26A9, probably by facilitating trafficking of F508del-CFTR to the plasma membrane (Strug et al., 2016). Similar to TMEM16A, SLC26A9 is upregulated during airway inflammation and exposure of the airway cells to IL-13 (Anagnostopoulou et al., 2012). It will be interesting to learn more about the regulation of SLC26A9 expression, and whether SLC26A9 is upregulated in CF airways in ciliated or in mucus secreting cells. Taken together, SLC26A9 could potentially serve as an alternative Cl− channel in CF, but compromised biosynthesis of CFTR carrying type two mutations need to be corrected. Identifying small molecules that would interfere with the formation of the F508del-CFTR/SLC26A9 complex could be an interesting therapeutic option in CF.
Airway Chloride Channels: TMEM16A
TMEM16A is a Ca2+ activated Cl− channel (CaCC) that belongs to a larger family of 10 paralogous proteins (TMEM16A-K), also called anoctamins (ANO1-ANO10) (Kunzelmann et al., 2011; Pedemonte and Galietta, 2014). The majority of these double-barreled channels are operating as phospholipid scramblases, i.e., they transport phospholipids from one site of the bilayer membrane to the other, once activated by a strong increase in intracellular Ca2+ (Suzuki et al., 2010). TMEM16A and B are solely CaCCs, whose structure and gating has been largely uncovered in cryo-EM studies (Paulino et al., 2017a,b). TMEM16A is typically localized in the apical plasma membrane of epithelial cells. However, it is also found to be expressed basolateral and in intracellular compartments (Schreiber et al., 2010, 2014). TMEM16F is a phospholipid scramblase that also conducts Cl− and other ions (Yang et al., 2012; Grubb et al., 2013; Shimizu et al., 2013; Kunzelmann et al., 2014; Ousingsawat et al., 2015; Scudieri et al., 2015; Drumm et al., 2017; Schreiber et al., 2018). Although endogenous TMEM16 proteins are mostly localized intracellularly, overexpression of these proteins together with purinergic receptors allows partial trafficking to the plasma membrane (Tian et al., 2012a). TMEM16A is clearly the epithelial airway CaCC (Benedetto et al., 2017), but a number of other TMEM16 paralogues are also coexpressed in mouse airways and in human large and small bronchi, bronchiole and alveoli, such as TMEM16C, F, J (Kunzelmann et al., 2012). Particularly TMEM16F may participate as well in epithelial Cl− transport.
Upregulation of TMEM16A During Inflammatory Lung Disease: Good or Bad?
TMEM16A is strongly upregulated during inflammation, a fact that was utilized to identify the molecular nature of CaCC (Galietta et al., 2002; Caputo et al., 2008). TMEM16A is strongly upregulated in CF and asthma, which parallels goblet cell metaplasia and mucus hypersecretion (Huang et al., 2012; Kondo et al., 2017), and is also upregulated by bacterial components (Caci et al., 2015). Upregulation of TMEM16A is predominant in mucus producing cells and to a much lesser degree in ciliated epithelial cells (Huang et al., 2012; Scudieri et al., 2012). Expression of TMEM16A is almost undetectable by immunocytochemistry in normal adult human and mouse airways; although CaCC is clearly present (Huang et al., 2009, 2012; Benedetto et al., 2017). While this may be explained by the limited sensitivity of the available antibodies, it also raises questions as to what degree other members of the TMEM16 family might participate in CaCC. As mentioned above, TMEM16C, F, and J are also expressed in mouse airway epithelium (Kunzelmann et al., 2012). On the other hand, knockout of TMEM16A completely abolished CaCC activated by purinergic stimulation (Benedetto et al., 2017). We will discuss below that TMEM16A has a strong impact on intracellular Ca2+ signals triggered by stimulation of G-protein coupled receptors (GPCRs), which then activate phospholipase C, increase inositol trisphosphate and intracellular Ca2+ (Kunzelmann et al., 2016).
As mentioned above, expression of TMEM16A in normal adult airways is hardly detectable by immunohistochemistry (Ousingsawat et al., 2009; Benedetto et al., 2017) (Figure 4). However, induction of airway inflammation in an ovalbumin asthma model induced a pronounced upregulation of TMEM16A in mucus producing club/goblet cells, but induced little expression in ciliated epithelial cells (Benedetto et al., 2019) (Figure 4). Ciliated epithelial cells, and particularly the recently identified ionocytes express CFTR and are in charge of fluid secretion (Montoro et al., 2018; Plasschaert et al., 2018). Expression of TMEM16A is low in ciliated cells when compared to mucus producing club and goblet cells. The contribution of TMEM16A to overall fluid secretion by the airway epithelium might therefore be limited, while it plays a central role for basal mucus secretion (Benedetto et al., 2019). This will be further outlined below.
Figure 4.

Predominant expression of TMEM16A in mucus producing cells. Distal airways of a control mouse and an ovalbumin-sensitized (asthmatic) mouse. Expression of TMEM16A (green fluorescence) is hardly detectable in control airways, but is clearly detectable in mucus producing club/goblet cells (G) of asthmatic mice with inflamed airways. Little expression of TMEM16A is found in ciliated epithelial cells (C). Blue, DAPI staining of nuclei. Bars indicate 20 μm (Benedetto et al., 2019).
F508del-CFTR Attenuates Expression of TMEM16A in the Apical Membrane
A functional coupling between TMEM16A and CFTR has been described in a number of previous publications (Kunzelmann et al., 1997; Wei et al., 1999; Ousingsawat et al., 2011a). Subsequent studies reported attenuated expression of TMEM16A in the apical membrane of airway epithelial cells by coexpressed F508del-CFTR (Ruffin et al., 2013; Benedetto et al., 2017). We demonstrated that TMEM16A and CFTR directly interact through PSD-95/Dlg/ZO-1 (PDZ) domain proteins, similar to SLC26A9 (Figure 3). The functional interaction between TMEM16A and CFTR is also demonstrated by a crosstalk of intracellular Ca2+ and cAMP-dependent signaling. This compartmentalized crosstalk is facilitated by exchange protein directly activated by cAMP (EPAC1) and Ca2+ -sensitive adenylate cyclase type 1 (ADCY1). Assembly of such a local signalosome was shown to depend on the number of phospholipase C coupled GPCRs (Lerias et al., 2018).
These studies suggest a significant overlap of cAMP- and Ca2+ activated Cl− currents. Along this line, our early studies also showed that epithelial cAMP-dependent and Ca2+-activated Cl− currents cannot be easily separated based on apparently specific ion channel inhibitors (Kunzelmann et al., 1992; Benedetto et al., 2017; Lerias et al., 2018). Moreover, in both airway and intestinal epithelium, most of the Ca2+ activated Cl− secretion is in fact through CFTR and not through TMEM16A Cl− channels (Mall et al., 1998b; Namkung et al., 2010a; Billet and Hanrahan, 2013; Benedetto et al., 2017). The actual purinergic (ATP-activated) Cl− secretion via TMEM16A is very transient, as TMEM16A deactivates quickly within 1–5 min after activation by ATP when examined in Xenopus oocytes (Faria et al., 2009), HEK293 cells (Tian et al., 2012b), mouse trachea (Kunzelmann et al., 2002), and human airways (Mall et al., 2003). Analysis of freshly isolated human nasal epithelial cells demonstrates ATP-induced steady-state secretion only in non-CF cells, but not in CF nasal cells. Thus, the direct contribution of TMEM16A to the epithelial secretory Cl− transport is small. However, the non-transient steady component of purinergic Cl− secretion that is produced by CFTR is essential for fluid secretion (Mall et al., 2003; Benedetto et al., 2017). The traffic mutant F508del is by far the most common mutation in CF that also compromises biosynthesis of TMEM16A. Therefore, the pro-secretory function of TMEM16A in CF is probably limited, and inhibition of TMEM16A may not much reduce Cl− secretion in CF (Ruffin et al., 2013; Benedetto et al., 2017).
Upregulation of TMEM16A in Airway Smooth Muscles
Upon induction of asthma, we also observed an upregulation of TMEM16A in mouse airway smooth muscle (ASM) cells (Benedetto et al., 2017; Miner et al., 2019) (Figure 4). This has already been described in a number of previous studies (Huang et al., 2009, 2012; Gallos et al., 2013; Danielsson et al., 2015). Inhibitors of TMEM16A were shown to induce hyperpolarization of ASM and airway relaxation (Yim et al., 2013; Danielsson et al., 2017; Miner et al., 2019). Airway inflammation is well-known to induce hyperresponsiveness of ASM (Brightling et al., 2002; Galli et al., 2008). Inflammatory mediators binding to GPCRs activate TMEM16A channels; depolarize the membrane voltage and cause airway contraction, a process that is upregulated in asthma (Wang et al., 2018). Expression of TMEM16A is not only upregulated in allergic asthma, but also in airway epithelial cells and probably ASM of CF patients (Caci et al., 2015). Moreover, the signaling cascade comprising GPCR - TMEM16A - intracellular Ca2+ is further augmented by inflammatory mediators and cholinergic stimuli.
TMEM16A Controls Ca2+ Signals, Membrane Exocytosis and Mucus Secretion
As pointed out above, membrane expression and activity of CFTR strongly relies on TMEM16A (Benedetto et al., 2017; Lerias et al., 2018). We showed that augmentation of apical Ca2+ signals in the presence of TMEM16A activates adenylate cyclase type 1, enhances local cAMP levels and boosts CFTR activity (Figure 5A). The enhanced plasma membrane expression of CFTR in the presence of TMEM16A may be caused by enhanced Ca2+ levels in the apical submembranous compartment, which triggers the exocytic machinery and membrane insertion of CFTR in ciliated epithelial cells and possibly ionocytes (Benedetto et al., 2017) (Figure 5B). A similar exocytic mechanism may apply to the process of mucus secretion by club and goblet cells. We found that ATP-induced mucus secretion by secretory cells is strongly compromised in the absence of TMEM16A. Without TMEM16A, intracellular Ca2+ concentrations in the apical pole of club and goblet cells are attenuated. These Ca2+ ions are required for basal and acute ATP-activated mucus release (Benedetto et al., 2019) (Figure 5C). ATP-dependent mucus secretion is characterized by Ca2+ dependent single granule docking to the apical membrane which requires Munc13 proteins and the SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) machinery (Fahy and Dickey, 2010). Taken together, in healthy non-CF airways TMEM16A may support CFTR-driven fluid secretion in ciliated cells and possibly ionocytes, and supports basal mucus release by club and goblet cells. In inflammatory CF airway disease, the function of TMEM16A may be marginal in ciliated cells and ionocytes, but may be pronounced in secretory cells due to strong upregulation of expression.
Figure 5.
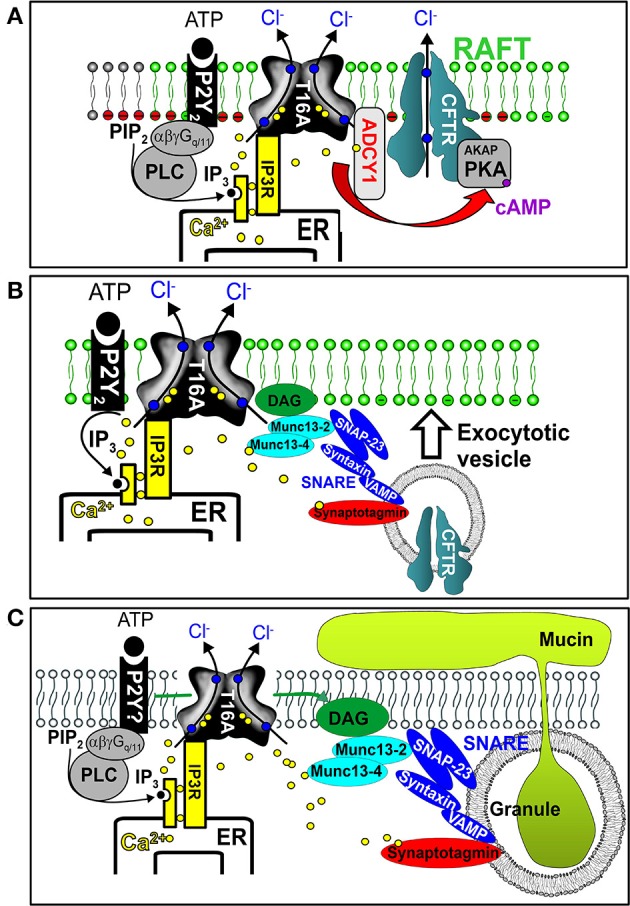
Impact of TMEM16A on intracellular Ca2+, exocytosis, and mucus secretion. (A) TMEM16A is colocalized with purinergic P2Y receptors and CFTR within the apical membrane. Stimulation of purinergic receptors leads to endoplasmic reticulum (ER) Ca2+ store release through IP3 receptors (IP3R). Ca2+ not only activates TMEM16A (T16A) but also stimulates adenylate cyclase type 1 (ADCY1) to produce cAMP and to activate CFTR via protein kinase A (PKA). TMEM16A binds to IP3 receptors and tethers the ER to the apical membrane, thereby facilitating effective compartmentalized Ca2+ signaling. (B) Effective apical Ca2+ signaling by TMEM16A leads to activation of the exocytic SNARE machinery insertion and improved expression of CFTR in the apical plasma membrane. (C) Effective apical Ca2+ signaling by TMEM16A leads to fusion of mucus containing granules, exocytosis, and release of mucus.
According to this, pharmacological activation of TMEM16A in CF and asthma patients could have adverse effects on lung function due to its prosecretory effect on mucus release. Correspondingly, we found in OVA-sensitized asthmatic mice that activation of TMEM16A by the compound Eact (Namkung et al., 2011b) induced massive mucus release and a considerable expiratory stridor, suggesting airway contraction. Airway narrowing was confirmed by analysis of the airway cross section (Benedetto et al., 2019) (Figure 6). It may be argued that Eact raises intracellular Ca2+ and therefore induces adverse effects independent of TMEM16A. However, increase in intracellular Ca2+ by Eact is expected. As outlined below, activation of TMEM16A is known to increase intracellular Ca2+ (Cabrita et al., 2017). Activation of TMEM16A by Eact depolarizes the membrane voltage, which leads to an increase in intracellular Ca2+. Notably, increase in intracellular Ca2+ by Eact was inhibited by 1 μM of the TMEM16A blocker niclosamide (Benedetto et al., 2019).
Figure 6.

Activation of TMEM16A leads to mucus release and airway contraction. Control mouse airways show very little mucus (alcian blue). In contrast, airways from ovalbumin-sensitized mice show pronounced mucus accumulation. Acute exposure of an ovalbumin-sensitized mouse to the known activator of TMEM16A, Eact (4.8 μg, tracheal instillation), induced a rapid mucus release and airway contraction (Benedetto et al., 2019). Bars indicate 20 μm.
Inhibitors and Activators of TMEM16A
Meanwhile a larger number of inhibitors of TMEM16A has been identified, but there is only one published group of N-aroylaminothiazole “activators” (Eact) (Namkung et al., 2011b), apart from some herbal compounds like Ginsenoside Rb1 and Resveratrol, which apparently activate TMEM16A in a Ca2+ independent manner (Chai et al., 2017; Guo et al., 2017) (Table 2). Enterprise therapeutics (Sussex, UK; http://www.enterprisetherapeutics.com/) is currently working on potentiators of TMEM16A, but details on potentiating molecules are not yet available. Silurian pharmaceuticals (Oakland, US; http://www.silurianpharma.com/index.php) reported brevenal to activate Ca2+ activated Cl− channels, possibly in a Ca2+ independent fashion. Denufusol was developed by Inspire pharmaceuticals (later taken over by Merck). Denufosol is a deoxycytidine-uridine dinucleotide with enhanced metabolic stability, to activate purinergic P2Y2 receptors which stimulate TMEM16A (Yerxa et al., 2002) and inhibit ENaC (Kunzelmann et al., 2002) (c.f. below). An interesting group of lipids was identified originally to uncouple GPCR-mediated Ca2+ increase from inactivation/desensitization of Ca2+ activated Cl− channels. These D-myo-inositol 3,4,5,6-tetrakisphosphate [Ins(3,4,5,6)P4] (Vajanaphanich et al., 1994) were synthetically modified to result in the membrane permeable analog INO4995, which was shown to inhibit ENaC (Moody et al., 2005) and to activate TMEM16A (Tian et al., 2012b). INO4995 did not increase intracellular Ca2+. It activated overexpressed TMEM16A directly, but potentiated ATP-dependent activation of TMEM16A expressed endogenously. Preliminary data suggested enhanced membrane localization of TMEM16A induced by INO4995 (Tian et al., 2012b) (Table 3).
Table 2.
Inhibitors of TMEM16A.
| Inhibitor | IC50 (μM) | Structural formula | References |
|---|---|---|---|
| 10bm | 0.03 | 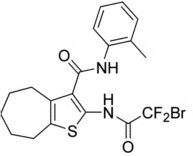 |
Truong et al., 2017 |
| Monna | 0.08 | 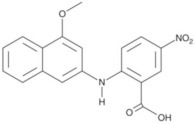 |
Oh et al., 2013 |
| Niclosamide | 0.7 | 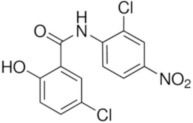 |
Miner et al., 2019 |
| Ani9 | 0.1 | 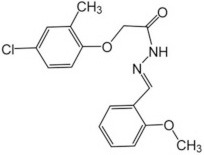 |
Seo et al., 2016 |
| Tannic acid | 0.323 | 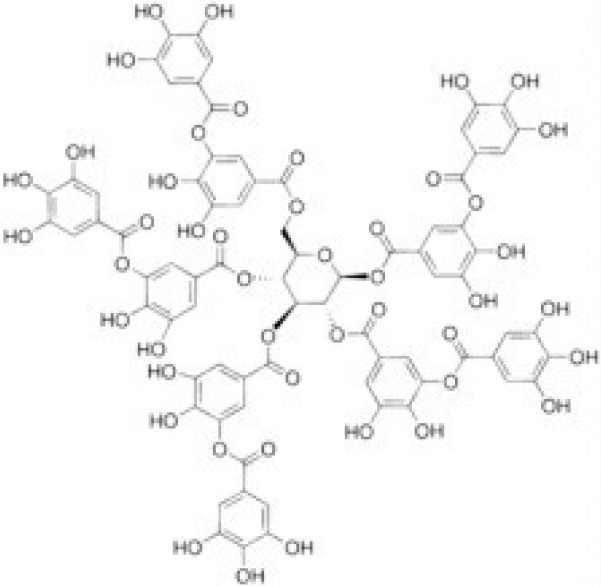 |
Namkung et al., 2010b |
| T16A-A01 | 1.1 | 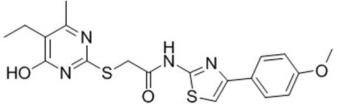 |
Namkung et al., 2011a; Fedigan et al., 2017 |
| Dichlorophen | 5.49 | 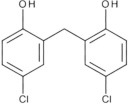 |
Huang et al., 2012 |
| Idebenone | 5.52 | 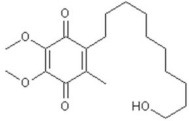 |
Seo et al., 2015 |
| Shikonin | 6.5 | 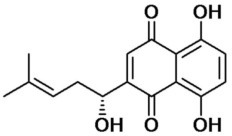 |
Jiang et al., 2016 |
| Benzbromarone | 9.97 | 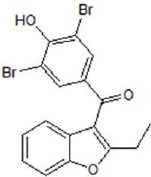 |
Huang et al., 2012 |
| CaCC-A01 | 10 | 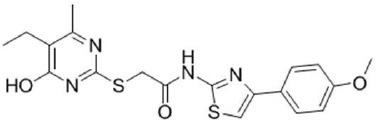 |
de La Fuente et al., 2007; Gianotti et al., 2016; Fedigan et al., 2017 |
| 9-Phenanthrol | 12 | 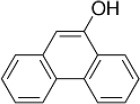 |
Burris et al., 2015) |
| Niflumic Acid | 12 | 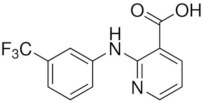 |
Hogg et al., 1994 |
| Flufenamic acid | 28 | 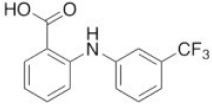 |
White and Aylwin, 1990 |
| Talniflumate | – | 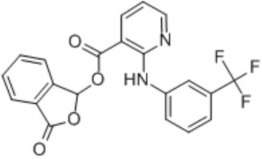 |
Walker et al., 2006 |
| A9C | 58 | 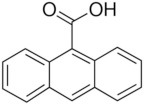 |
Baron et al., 1991 |
| Dehydroandrographolide | 20–30 | 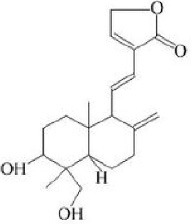 |
Sui et al., 2015 |
| DIDS | 10–100 | 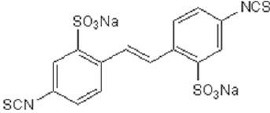 |
Kubitz et al., 1992 |
| NPPB | 15–150 | 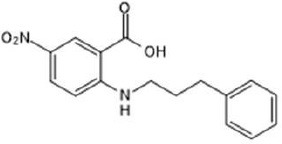 |
Kubitz et al., 1992; Yang et al., 2008 |
| Rice bran extract | Sharm et al., 2017 | ||
| Matrine | 28 μM | 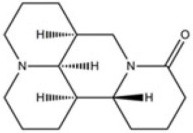 |
Guo et al., 2018 |
| (Ani9 derivative) 5f | 22 nM | 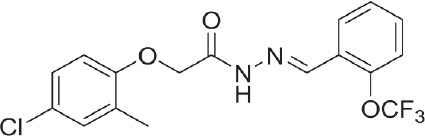 |
Seo et al., 2018 |
Table 3.
Activators of TMEM16A.
| Activator | IC50 (μM) | Structural formula | References |
|---|---|---|---|
| Brevenal | – |  |
http://www.silurianpharma.com/index.php |
| Eact | 3 | 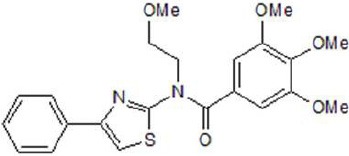 |
Namkung et al., 2011b |
| INO-4995 | 5 | 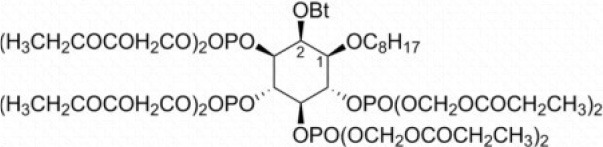 |
Tian et al., 2012b |
| Denufosol | 10 | 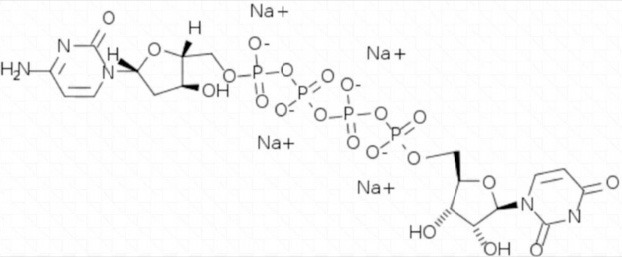 |
Yerxa et al., 2002 |
| Cinnamaldehyde | 10 | 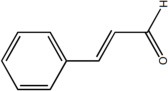 |
Huang et al., 2018 |
| Ginsenoside Rb1 | 38.4 | 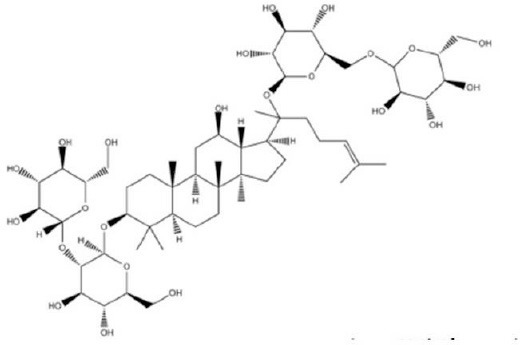 |
Guo et al., 2017 |
| Resveratrol | 47.9 | 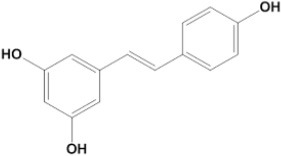 |
Chai et al., 2017 |
| A9C | 100–1000 | 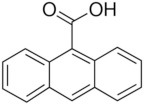 |
Ta et al., 2016 |
Biological Effects of Inhibitors and Activators of TMEM16A
TMEM16A is broadly expressed in many epithelial and non-epithelial tissues. It is therefore expected that inhibition or activation of TMEM16A through systemic drug application might have a number of side effects. Apart from inhibiting mucus production and mucus secretion, TMEM16A-inhibitors will induce bronchorelaxation by blocking TMEM16A in airway smooth muscle (Huang et al., 2012; Gallos et al., 2013; Zhang et al., 2013; Danielsson et al., 2015; Wang et al., 2018; Miner et al., 2019). Another desired lung-specific effect of TMEM16A-inhibitors is the inhibition of release of inflammatory mediators (Knight, 2004; Danielsson et al., 2017; Benedetto et al., 2019).
Some inhibitors of TMEM16A have additional beneficial effects, such as protection from reactive oxygen species (idebenone, Villalba et al., 2010), and general inhibition of inflammation (Knight, 2004; Schreiber et al., 2018). Many studies demonstrate inhibition of proliferation and anti-cancer effects by blocking TMEM16A (Wanitchakool et al., 2014; Wang et al., 2017). General anti-hypertensive effects are likely (Namkung et al., 2010b; Heinze et al., 2014), as well as inhibition of nociception, itching, and heat perception (Cho et al., 2012; Lee et al., 2014; Pusch and Zifarelli, 2014; Deba and Bessac, 2015). Inhibition of saliva production and dry mouth may occur mediation with TMEM16A-inhibitors (Ousingsawat et al., 2009; Catalan et al., 2015). Inhibition of TMEM16A may also attenuate intestinal contraction and abdominal peristalsis (Sanders et al., 2012; Singh et al., 2014), and therefore could have antidiarrheal effects (Tradtrantip et al., 2010; Namkung et al., 2011b; Jiang et al., 2016). Finally, inhibition of renal TMEM16A could potentially lead to proteinuria and acidosis (Faria et al., 2014; Schenk et al., 2018). For activators of TMEM16A, opposite effects are possible, which is why local application via aerosol may be recommended for the treatment of CF lung disease. Nevertheless, oral application of a TMEM16A-blocker was shown to increase survival of CFTR-knockout mice (Walker et al., 2006).
Activating or Inhibiting TMEM16A?
Under physiological conditions, airway mucus represents an innate defense mechanism against pathogens. It traps inhaled pathogens and particles and is part of the mucociliary clearance (Knowles and Boucher, 2002). However, mucus becomes a serious problem when hypersecreted in inflammatory lung diseases, such as asthma, COPD, and CF (Dunican et al., 2018). Despite many pathological findings and pathogenic mechanisms proposed for CF lung disease (c.f. above), the single most prominent finding is the excessive overproduction of highly viscous mucus with adhesive and cohesive properties in CF patients (Button et al., 2018). It causes airway obstruction and a reduced mucociliary clearance, and it thereby drives chronic inflammatory lung disease (Fahy and Dickey, 2010). Thus, inhibition of mucus production/secretion is likely to be the most effective treatment of CF lung disease, normalizing the imbalance between excessive mucus secretion and reduced ASL (Figure 7). Our recent data show that TMEM16A and other TMEM16 proteins are essential for mucus production and basal secretion of mucus in airways and intestine (Benedetto et al., 2019). These findings open up a new avenue for the therapy of inflammatory airway diseases, and particularly for the treatment of airway mucus plugging and CF lung disease.
Figure 7.

Activation of inhibition of TMEM16A in airways of CF patients. (A) Activators of TMEM16A (green arrow, +) will find only few TMEM16A channels in the apical membrane of ciliated epithelial cells, which could enhance fluid secretion by ciliated epithelial cells. (B) In contrast, activators of TMEM16A will prominently activate TMEM16A strongly upregulated in mucus producing cells and airway smooth muscle cells during airway inflammation. This leads to hypersecretion of mucus along with airway contraction. (C) Inhibitors of TMEM16A will have an only limited (adverse) effect on TMEM16A channels expressed (fluid secreting) ciliated epithelial cells. In contrast, inhibition of TMEM16 channels in mucus producing cells and airway smooth muscle cells will inhibit mucus production/secretion and induce bronchodilation. (D) Inhibition of TMEM16 channels are likely to restore the balance between mucus and fluid in CF airways.
In contrast, activation of TMEM16A in CF to facilitate fluid secretion may be rather ineffective due to the reasons outlined above (Figure 7A). An earlier clinical trial was performed using stabilized dinucleotides (Denufosol) to induce purinergic Ca2+ dependent Cl− secretion and to restore ASL with the goal of improving lung function in CF. However, the clinical trial failed to demonstrate any benefit of denufosol (Ratjen et al., 2012; Moss, 2013). As an adverse effect, the aerosol induced cough in 52% of all patients. It may be speculated that denufosol may have induced additional mucus secretion and even additional airway obstruction. Being strongly upregulated in inflammatory airway disease in secretory cells and in ASM, activation of TMEM16A could induce adverse effects by augmenting mucus secretion and bronchoconstriction (Figure 7B).
Inhibition of TMEM16A in CF may appear counterintuitive in the first place (assuming a potential inhibition of fluid secretion), however, the available data suggest otherwise. As outlined above, in CF TMEM16A membrane expression is compromised (Ruffin et al., 2013; Benedetto et al., 2017), Cl− secretion through CFTR is missing (Namkung et al., 2010a; Billet and Hanrahan, 2013; Benedetto et al., 2017; Lerias et al., 2018), and most TMEM16A is expressed in mucus secreting cells (Benedetto et al., 2019). Therefore, inhibition of TMEM16A may not substantially lower fluid secretion. In contrast, interfering with mucus and cytokine secretion by blocking upregulated TMEM16A is likely to improve mucociliary clearance and to improve lung function (Figure 7C). Along this line, talniflumate, the anti-inflammatory pro-drug of the common TMEM16A-inhibitor NFA that was originally developed by Argentinian Laboratorios Bago, increased survival of CF mice remarkably (Walker et al., 2006). Talniflumate had been further developed by the former company Genaera, as a mucoregulator for cystic fibrosis, chronic obstructive pulmonary disease, and asthma. Sadly, phase II trials have never been finished due to the shutdown of the company (Knight, 2004). In a large number of studies, mucus production, and secretion, as well as airway constriction were inhibited by niflumic acid and other inhibitors of TMEM16A (Kondo et al., 2012; Yim et al., 2013; Lin et al., 2015; Danielsson et al., 2017; Miner et al., 2019). The inhibitors niflumic acid, CaCCinhAO1, T16Ainh-A01, 17 benzbromarone, or niclosamide have been examined in vivo as well as in vitro in the low micromolecular range. Despite voltage dependence of TMEM16A-inhibition by NFA and other inhibitors, in vivo application to airway epithelial cells maintaining their intrinsic hyperpolarized membrane voltage demonstrated remarkable biological effects. Similar has been observed when TMEM16A inhibitors were applied to tracheal ring preparations ex vivo. Presumably TMEM16A is partially active in the airways, as low levels of ATP in the airway surface liquid maintain a basal activity of TMEM16A.
Because TMEM16A currents are blocked by niflumic acid, the published reports suggest that TMEM16A is in charge of both production and secretion of mucus. Niflumic acid, however, is a rather non-specific drug that inhibits a number of ion channels and blocks intracellular Ca2+ signals (Cabrita et al., 2017). Suppression of Ca2+ signals by niflumic acid is probably due to inhibition of TMEM16A. Other TMEM16A inhibitors such as CaCCinhAO1, T16Ainh-A01, benzbromarone, or niclosamide also inhibit intracellular Ca2+ and mucus release (de La Fuente et al., 2007; Kondo et al., 2017; Miner et al., 2019). It is important to note that other TMEM16 paralogues are also blocked by inhibitors of TMEM16A (Sirianant et al., 2015; Wanitchakool et al., 2016). Because several TMEM16 paralogues are expressed in airway epithelial cells, the possible contribution of other TMEM16 proteins to Ca2+ signaling and mucus production/secretion is currently unknown. Using different TMEM16 knockout mice and TMEM16 knockout cell lines, we found that most TMEM16 paralogues affect intracellular Ca2+ signals (Kunzelmann et al., 2016; Cabrita et al., 2017). It is currently assumed that at least two TMEM16 members, TMEM16A and TMEM16F control mucus production and mucus secretion (Benedetto et al., 2019).
Conclusion
Despite significant progress in the development of CFTR–specific treatments for CF lung disease, it appears reasonable to search for alternative drug targets in CF. Potentiators and correctors of mutant CFTR show benefit in patients carrying the common F508del mutation. Improvement of lung function by the recent combinatorial drugs can be as high as 13%. Insight into mode of action of these compounds is still limited, and the costs for treatment may exclude some patients from therapy (Ferkol and Quinton, 2015). Moreover, a fraction of patients with particular CFTR mutations [type 1,5,6,7 mutations (De Boeck and Amaral, 2016)] will not respond to such a treatment.
Restoration of the mucus/liquid balance has been the driving force behind the search for novel openers of secretory Cl− channels (SLC26A9 and TMEM16A) and basolateral pro-secretory K+ channels, as well as for inhibitors of reabsorptive Na+ channels. The counterintuitive idea of using inhibitors of TMEM16 channels is based on their role for mucus production and mucus secretion, which were uncovered only recently. Overwhelming mucus production and mucus plugging is the central problem in CF lung disease. Therefore, potent and well-tolerated TMEM16-inhibitors, which have FDA-approval for other diseases, should be further examined in preclinical and clinical studies to be use in CF lung disease and other inflammatory airway diseases.
Ethics Statement
All animals studies were approved by the local ethical board.
Author Contributions
KK wrote manuscript, analyzed data, and designed experiments. RS, JO, and IC analyzed data, designed experiments, and performed experiments. TD, AB, and MJ wrote manuscript. RB wrote manuscript, analyzed data, designed experiments, and performed experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by Cystic Fibrosis Trust, SRC 013, INOVCF, DFG KU756/14-1, DFG KU756/14-1, and Gilead Stiftung 2018: Inhibition of TMEM16A to reduce mucus plugging in CF.
References
- Accurso F. J., Rowe S. M., Clancy J. P., Boyle M. P., Dunitz J. M., Durie P. R., et al. (2010). Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 363, 1991–2003. 10.1056/NEJMoa0909825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M. D., Kunzelmann K. (2007). Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol. Sci. 28, 334–341. 10.1016/j.tips.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Anagnostopoulou P., Riederer B., Duerr J., Michel S., Binia A., Agrawal R., et al. (2012). SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J. Clin. Invest. 122, 3629–3634. 10.1172/JCI60429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angles F., Hutt D., Balch W. E. (2018). HDAC inhibitors rescue multiple disease-causing CFTR variants. bioRxiv. 10.1101/399451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron A., Pacaud P., Loirand G., Mironneau C., Mironneau J. (1991). Pharmacological block of Ca2+-activated Cl− current in rat vascular smooth muscle cells in short-term primary culture. Pflugers Arch. 419, 553–558. 10.1007/BF00370294 [DOI] [PubMed] [Google Scholar]
- Benedetto R., Cabrita I., Schreiber R., Kunzelmann K. (2019). TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. [Epub ahead of print]. 10.1096/fj.201801333RRR [DOI] [PubMed] [Google Scholar]
- Benedetto R., Ousingsawat J., Wanitchakool P., Zhang Y., Holtzman M. J., Amaral M., et al. (2017). Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep. 7:12397. 10.1038/s41598-017-10910-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetto R., Sirianant L., Pankonien I., Wanitchakool P., Ousingsawat J., Cabrita I., et al. (2016). Relationship between TMEM16A/anoctamin 1 and LRRC8A. Pflugers Arch. 468, 1751–1763. 10.1007/s00424-016-1862-1 [DOI] [PubMed] [Google Scholar]
- Bertrand C. A., Mitra S., Mishra S. K., Wang X., Zhao Y., Pilewski J. M., et al. (2017). The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L912–L925. 10.1152/ajplung.00178.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand C. A., Zhang R., Pilewski J. M., Frizzell R. A. (2009). SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J. Gen. Physiol. 133, 421–438. 10.1085/jgp.200810097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billet A., Hanrahan J. W. (2013). The secret life of CFTR as a calcium-activated chloride channel. J. Physiol. 591, 5273–5278. 10.1113/jphysiol.2013.261909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher R. C. (2007). Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 58, 157–170. 10.1146/annurev.med.58.071905.105316 [DOI] [PubMed] [Google Scholar]
- Boucher R. C., Stutts M. J., Knowles M. R., Cantley L., Gatzy J. T. (1986). Na+ transport in cystic fibrosis respiratory epithelia: abnormal basal rate and response to adenylate cyclase. J. Clin. Invest. 78, 1245–1252. 10.1172/JCI112708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling C. E., Bradding P., Symon F. A., Holgate S. T., Wardlaw A. J., Pavord I. D. (2002). Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 346, 1699–1705. 10.1056/NEJMoa012705 [DOI] [PubMed] [Google Scholar]
- Burris S. K., Wang Q., Bulley S., Neeb Z. P., Jaggar J. H. (2015). 9-phenanthrol inhibits recombinant and arterial myocyte TMEM16A channels. Br. J. Pharmacol. 172, 2459–2468. 10.1111/bph.13077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Button B., Goodell H. P., Atieh E., Chen Y. C., Williams R., Shenoy S., et al. (2018). Roles of mucus adhesion and cohesion in cough clearance. Proc. Natl. Acad. Sci. U.S.A. 115, 12501–12506. 10.1073/pnas.1811787115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita I., Benedetto R., Fonseca A., Wanitchakool P., Sirianant L., Skryabin B. V., et al. (2017). Differential effects of anoctamins on intracellular calcium signals. FASEB J. 31, 2123–2134. 10.1096/fj.201600797RR [DOI] [PubMed] [Google Scholar]
- Caci E., Scudieri P., Di Carlo E., Morelli P., Bruno S., De F., et al. (2015). Upregulation of TMEM16A protein in bronchial epithelial cells by bacterial pyocyanin. PLoS ONE 10:e0131775. 10.1371/journal.pone.0131775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., et al. (2008). TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594. 10.1126/science.1163518 [DOI] [PubMed] [Google Scholar]
- Carlile G. W., Robert R., Goepp J., Matthes E., Liao J., Kus B., et al. (2015). Ibuprofen rescues mutant cystic fibrosis transmembrane conductance regulator trafficking. J. Cyst. Fibros. 14, 16–25. 10.1016/j.jcf.2014.06.001 [DOI] [PubMed] [Google Scholar]
- Carlile G. W., Yang Q., Matthes E., Liao J., Radinovic S., Miyamoto C., et al. (2018). A novel triple combination of pharmacological chaperones improves F508del-CFTR correction. Sci. Rep. 8:11404. 10.1038/s41598-018-29276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalan M. A., Kondo Y., Pena-Munzenmayer G., Jaramillo Y., Liu F., Choi S., et al. (2015). A fluid secretion pathway unmasked by acinar-specific Tmem16A gene ablation in the adult mouse salivary gland. Proc. Natl. Acad. Sci U.S.A. 112, 2263–2268. 10.1073/pnas.1415739112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai R., Chen Y., Yuan H., Wang X., Guo S., Qi J., et al. (2017). Identification of resveratrol, an herbal compound, as an activator of the calcium-activated chloride channel, TMEM16A. J. Membr. Biol. 250, 483–492. 10.1007/s00232-017-9975-9 [DOI] [PubMed] [Google Scholar]
- Chang M. H., Plata C., Sindic A., Ranatunga W. K., Chen A. P., Zandi-Nejad K., et al. (2009). Slc26a9 is inhibited by the R-region of CFTR via the STAS domain. J. Biol. Chem. 284, 28306–28318. 10.1074/jbc.M109.001669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. H., Stoltz D. A., Karp P. H., Ernst S. E., Pezzulo A. A., Moninger T. O., et al. (2010). Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143, 911–923. 10.1016/j.cell.2010.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin S., Hung M., Won A., Wu Y. S., Ahmadi S., Yang D., et al. (2018). Lipophilicity of the cystic fibrosis drug, ivacaftor (VX-770), and its destabilizing effect on the major CF-causing mutation: F508del. Mol. Pharmacol. 94, 917–925. 10.1124/mol.118.112177 [DOI] [PubMed] [Google Scholar]
- Cho H., Yang Y. D., Lee J., Lee B., Kim T., Jang Y., et al. (2012). The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat. Neurosci. 15, 1015–1021. 10.1038/nn.3111 [DOI] [PubMed] [Google Scholar]
- Choi J. Y., Muallem D., Kiselyov K., Lee M. G., Thomas P. J., Muallem S. (2001). Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 410, 94–97. 10.1038/35065099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholon D. M., Quinney N. L., Fulcher M. L., Esther C. R., Jr., Das J., Dokholyan N. V., et al. (2014). Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 6:246ra296. 10.1126/scitranslmed.3008680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton C. U., Lawson E. E., Boucher R. C., Gatzy J. T. (1983). Bioelectric properties and ion transport of airways excised from adult and fetal sheep. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 55, 1542–1549. 10.1152/jappl.1983.55.5.1542 [DOI] [PubMed] [Google Scholar]
- Danielsson J., Mikami M., Emala C. W. (2017). Antagonism of the Tmem16a calcium-activated chloride channel attenuates allergic lung inflammation. Am. J. Respir. Crit. Care Med. 195:A5290. [Google Scholar]
- Danielsson J., Perez-Zoghbi J., Bernstein K., Barajas M. B., Zhang Y., Kumar S., et al. (2015). Antagonists of the TMEM16A calcium-activated chloride channel modulate airway smooth muscle tone and intracellular calcium. Anesthesiology 123, 569–581. 10.1097/ALN.0000000000000769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C., Moskowitz S. M., Brown C., Horsley A., Mall M. A., Mckone E. F., et al. (2018). VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 379, 1599–1611. 10.1056/NEJMoa1807119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck K., Amaral M. D. (2016). Progress in therapies for cystic fibrosis. Lancet Respir. Med. 4, 662–674. 10.1016/S2213-2600(16)00023-0 [DOI] [PubMed] [Google Scholar]
- De Boeck K., Zolin A., Cuppens H., Olesen H. V., Viviani L. (2014). The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 13, 403–409. 10.1016/j.jcf.2013.12.003 [DOI] [PubMed] [Google Scholar]
- de La Fuente R., Namkung W., Mills A., Verkman A. S. (2007). Small molecule screen identifies inhibitors of a human intestinal calcium activated chloride channel. Mol. Pharmacol. 73, 758–768. 10.1124/mol.107.043208 [DOI] [PubMed] [Google Scholar]
- Deba F., Bessac B. F. (2015). Anoctamin-1 Cl channels in nociception: activation by an N-aroylaminothiazole and capsaicin and inhibition by T16A[inh]-A01. Mol. Pain 11:55. 10.1186/s12990-015-0061-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson S. H., Boucher R. C. (2007). Sodium channels and cystic fibrosis. Chest 132, 1631–1636. 10.1378/chest.07-0288 [DOI] [PubMed] [Google Scholar]
- Donaldson S. H., Pilewski J. M., Griese M., Cooke J., Viswanathan L., Tullis E., et al. (2018). Tezacaftor/Ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 197, 214–224. 10.1164/rccm.201704-0717OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring G., Worlitzsch D. (2000). Inflammation in cystic fibrosis and its management. Paediatr. Respir. Rev. 1, 101–106. 10.1053/prrv.2000.0030 [DOI] [PubMed] [Google Scholar]
- Dorwart M. R., Shcheynikov N., Wang Y., Stippec S., Muallem S. (2007). SLC26A9 is a Cl(-) channel regulated by the WNK kinases. J. Physiol. 584, 333–345. 10.1113/jphysiol.2007.135855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drumm B. T., Hennig G. W., Battersby M. J., Cunningham E. K., Sung T. S., Ward S. M., et al. (2017). Clustering of Ca2+) transients in interstitial cells of cajal defines slow wave duration. J. Gen. Physiol. 149, 703–725. 10.1085/jgp.201711771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunican E. M., Elicker B. M., Gierada D. S., Nagle S. K., Schiebler M. L., Newell J. D., et al. (2018). Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J. Clin. Invest. 128, 997–1009. 10.1172/JCI95693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khouri E., Toure A. (2014). Functional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): physiological and pathophysiological relevance. Int. J. Biochem. Cell Biol. 52, 58–67. 10.1016/j.biocel.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Fahy J. V., Dickey B. F. (2010). Airway mucus function and dysfunction. N. Engl. J. Med. 363, 2233–2247. 10.1056/NEJMra0910061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria D., Schlatter E., Witzgall R., Grahammer F., Bandulik S., Schweda F., et al. (2014). The calcium activated chloride channel Anoctamin 1 contributes to the regulation of renal function. Kindey Int. 85, 1369–1381. 10.1038/ki.2013.535 [DOI] [PubMed] [Google Scholar]
- Faria D., Schreiber R., Kunzelmann K. (2009). CFTR is activated through stimulation of purinergic P2Y2 receptors. Pflügers Arch. 457, 1373–1380. 10.1007/s00424-008-0606-2 [DOI] [PubMed] [Google Scholar]
- Fedigan S., Bradley E., Webb T., Large R. J., Hollywood M. A., Thornbury K. D., et al. (2017). Effects of new-generation TMEM16A inhibitors on calcium-activated chloride currents in rabbit urethral interstitial cells of cajal. Pflugers Arch. 469, 1443–1455. 10.1007/s00424-017-2028-5 [DOI] [PubMed] [Google Scholar]
- Ferkol T., Quinton P. (2015). Precision medicine: at what price? Am. J. Respir. Crit. Care Med. 192, 658–659. 10.1164/rccm.201507-1428ED [DOI] [PubMed] [Google Scholar]
- Galietta L. J., Pagesy P., Folli C., Caci E., Romio L., Costes B., et al. (2002). IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J. Immunol. 168, 839–845. 10.4049/jimmunol.168.2.839 [DOI] [PubMed] [Google Scholar]
- Galli S. J., Tsai M., Piliponsky A. M. (2008). The development of allergic inflammation. Nature 454, 445–454. 10.1038/nature07204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallos G., Remy K. E., Danielsson J., Funayama H., Fu X. W., Chang H. Y., et al. (2013). Functional expression of the TMEM16 family of calcium activated chloride channels in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 305, L625–L634. 10.1152/ajplung.00068.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianotti A., Ferrera L., Philp A. R., Caci E., Zegarra-Moran O., Galietta L. J., et al. (2016). Pharmacological analysis of epithelial chloride secretion mechanisms in adult murine airways. Eur. J. Pharmacol. 787, 100–108. 10.1016/j.ejphar.2016.04.007 [DOI] [PubMed] [Google Scholar]
- Grubb B. R., Vick R. N., Boucher R. C. (1994). Hyperabsorption of Na+ and raised Ca2+ mediated Cl− secretion in nasal epithelia of CF mice. Am. J. Physiol. 266, C1478–C1483. 10.1152/ajpcell.1994.266.5.C1478 [DOI] [PubMed] [Google Scholar]
- Grubb S., Poulsen K. A., Juul C. A., Kyed T., Klausen T. K., Larsen E. H., et al. (2013). TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 141, 585–600. 10.1085/jgp.201210861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Chen Y., Pang C., Wang X., Qi J., Mo L., et al. (2017). Ginsenoside Rb1, a novel activator of the TMEM16A chloride channel, augments the contraction of guinea pig ileum. Pflugers Arch. 469, 681–692. 10.1007/s00424-017-1934-x [DOI] [PubMed] [Google Scholar]
- Guo S., Chen Y., Pang C., Wang X., Shi S., Zhang H., et al. (2018). Matrine is a novel inhibitor of the TMEM16A chloride channel with antilung adenocarcinoma effects. J. Cell Physiol. [Epub ahead of print]. 10.1002/jcp.27529 [DOI] [PubMed] [Google Scholar]
- Hajj R., Lesimple P., Nawrocki-Raby B., Birembaut P., Puchelle E., Coraux C. (2007). Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 211, 340–350. 10.1002/path.2118 [DOI] [PubMed] [Google Scholar]
- Heinze C., Seniuk A., Sokolov M. V., Huebner A. K., Klementowicz A. E., Szijarto I. A., et al. (2014). Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J. Clin. Invest. 124, 675–686. 10.1172/JCI70025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegger M. J., Fischer A. J., Mcmenimen J. D., Ostedgaard L. S., Tucker A. J., Awadalla M. A., et al. (2014). Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345, 818–822. 10.1126/science.1255825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg R. C., Wang Q., Large W. A. (1994). Action of niflumic acid on evoked and spontaneous calcium-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br. J. Pharmacol. 112, 977–984. 10.1111/j.1476-5381.1994.tb13177.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Rock J. R., Harfe B. D., Cheng T., Huang X., Jan Y. N., et al. (2009). Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl. Acad. Sci. U.S.A. 106, 21413–21418. 10.1073/pnas.0911935106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Zhang H., Wu M., Yang H., Kudo M., Peters C. J., et al. (2012). Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 109, 16354–16359. 10.1073/pnas.1214596109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Guo S., Ren S., Chen Y., Zhan Y., An H. (2018). The natural compound cinnamaldehyde is a novel activator of calcium-activated chloride channel. J. Membr. Biol. 251, 747–756. 10.1007/s00232-018-0052-9 [DOI] [PubMed] [Google Scholar]
- Hutt D. M., Loguercio S., Roth D. M., Su A. I., Balch W. E. (2018). Correcting the F508del-CFTR variant by modulating eukaryotic translation initiation factor 3-mediated translation initiation. J. Biol. Chem. 293, 13477–13495. 10.1074/jbc.RA118.003192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani O. A., Chen J. H., Karp P. H., Ernst S., Keshavjee S., Parekh K., et al. (2011). Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. U.S.A. 108, 10260–10265. 10.1073/pnas.1106695108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Yu B., Yang H., Ma T. (2016). Shikonin inhibits intestinal calcium-activated chloride channels and prevents rotaviral diarrhea. Front. Pharmacol. 7:270. 10.3389/fphar.2016.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D., Marigowda G., Burr L., Daines C., Mall M. A., Mckone E. F., et al. (2018). VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 379, 1612–1620. 10.1056/NEJMoa1807120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T. Z., Wagener J. S., Bost T., Martinez J., Accurso F. J., Riches D. W. (1995). Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 151, 1075–1082. [DOI] [PubMed] [Google Scholar]
- Klymiuk N., Mundhenk L., Kraehe K., Wuensch A., Plog S., Emrich D., et al. (2012). Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. J. Mol. Med. 90, 597–608. 10.1007/s00109-011-0839-y [DOI] [PubMed] [Google Scholar]
- Knight D. (2004). Talniflumate (Genaera). Curr. Opin. Investig. Drugs 5, 557–562. [PubMed] [Google Scholar]
- Knowles M. R., Boucher R. C. (2002). Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109, 571–577. 10.1172/JCI0215217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M., Nakata J., Arai N., Izumo T., Tagaya E., Takeyama K., et al. (2012). Niflumic acid inhibits goblet cell degranulation in a guinea pig asthma model. Allergol. Int. 61, 133–142. 10.2332/allergolint.11-OA-0307 [DOI] [PubMed] [Google Scholar]
- Kondo M., Tsuji M., Hara K., Arimura K., Yagi O., Tagaya E., et al. (2017). Chloride ion transport and overexpression of TMEM16A in a guinea pig asthma model. Clin. Exp. Allergy 47, 795–804. 10.1111/cea.12887 [DOI] [PubMed] [Google Scholar]
- Kubitz R., Warth R., Kunzelmann K., Grolik M., Greger R. (1992). Small conductance Cl− channels induced by cAMP, Ca2+, and hypotonicity in HT29 cells: ion selectivity, additivity and stilbene sensitivity. Pflügers Arch. 421, 447–454. 10.1007/BF00370255 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Cabrita I., Wanitchakool P., Ousingsawat J., Sirianant L., Benedetto R., et al. (2016). Modulating Ca2+signals: a common theme for TMEM16, Ist2, and TMC. Pflügers Arch. 468, 475–490. 10.1007/s00424-015-1767-4 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Gerlach L., Fröbe U., Greger R. (1991). Bicarbonate permeability of epithelial chloride channels. Pflügers Arch. 417, 616–621. 10.1007/BF00372960 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Kubitz R., Grolik M., Warth R., Greger R. (1992). Small conductance Cl− channels in HT29 cells: activation by Ca2+, hypotonic cell swelling and 8-Br-cGMP. Pflügers Arch. 421, 238–246. 10.1007/BF00374833 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Mall M., Briel M., Hipper A., Nitschke R., Ricken S., et al. (1997). The cystic fibrosis transmembrane conductance regulator attenuates the endogenous Ca2+ activated Cl− conductance in Xenopus ooyctes. Pflügers Arch. 434, 178–181. 10.1007/s004240050498 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Nilius B., Owsianik G., Schreiber R., Ousingsawat J., Sirianant L., et al. (2014). Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel or phospholipid scramblase? Pflügers Arch. 466, 407–414. 10.1007/s00424-013-1305-1 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Schreiber R., Cook D. I. (2002). Mechanisms for inhibition of amiloride-sensitive Na+ absorption by extracellular nuceotides in mouse trachea. Pflügers Arch. 444, 220–226. 10.1007/s00424-002-0796-y [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Schreiber R., Hadorn H. B. (2017). Bicarbonate in cystic fibrosis. J. Cyst. Fibros. 16, 653–662. 10.1016/j.jcf.2017.06.005 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Tian Y., Martins J. R., Faria D., Kongsuphol P., Ousingsawat J., et al. (2011). Anoctamins. Pflugers Arch. 462, 195–208. 10.1007/s00424-011-0975-9 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Tian Y., Martins J. R., Faria D., Kongsuphol P., Ousingsawat J., et al. (2012). Cells in focus: airway epithelial cells-Functional links between CFTR and anoctamin dependent Cl(-) secretion. Int. J. Biochem. Cell Biol. 44, 1897–1900. 10.1016/j.biocel.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Lee B., Cho H., Jung J., Yang Y. D., Yang D. J., Oh U. (2014). Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. Pain 10:5. 10.1186/1744-8069-10-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerias J., Pinto M., Benedetto R., Schreiber R., Amaral M., Aureli M., et al. (2018). Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell. Signal. 44, 10–19. 10.1016/j.cellsig.2018.01.008 [DOI] [PubMed] [Google Scholar]
- Li H., Salomon J. J., Sheppard D. N., Mall M. A., Galietta L. J. (2017). Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr. Opin. Pharmacol. 34, 91–97. 10.1016/j.coph.2017.10.002 [DOI] [PubMed] [Google Scholar]
- Lin J., Jiang Y., Li L., Liu Y., Tang H., Jiang D. (2015). TMEM16A mediates the hypersecretion of mucus induced by Interleukin-13. Exp. Cell Res. 334, 260–269. 10.1016/j.yexcr.2015.02.026 [DOI] [PubMed] [Google Scholar]
- Mall M., Bleich M., Greger R., Schreiber R., Kunzelmann K. (1998a). The amiloride inhibitable Na+ conductance is reduced by CFTR in normal but not in CF airways. J. Clin. Invest. 102, 15–21. 10.1172/JCI2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M., Bleich M., Greger R., Schürlein M., Kühr J., Seydewitz H. H., et al. (1998b). Cholinergic ion secretion in human colon requires co-activation by cAMP. Am. J. Physiol. 275, G1274–G1281. [DOI] [PubMed] [Google Scholar]
- Mall M., Gonska T., Thomas J., Schreiber R., Seydewitz H. H., Kuehr J., et al. (2003). Modulation of Ca2+ activated Cl− secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatr. Res. 53, 608–618. 10.1203/01.PDR.0000057204.51420.DC [DOI] [PubMed] [Google Scholar]
- Mall M., Grubb B. R., Harkema J. R., O'neal W. K., Boucher R. C. (2004). Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10, 487–493. 10.1038/nm1028 [DOI] [PubMed] [Google Scholar]
- Mall M., Wissner A., Kühr J., Gonska T., Brandis M., Kunzelmann K. (2000). Inhibition of amiloride sensitive epithelial Na+ absorption by extracellular nucleotides in human normal and CF airways. Am. J. Respir. Cell Mol. Biol. 23, 755–761. 10.1165/ajrcmb.23.6.4207 [DOI] [PubMed] [Google Scholar]
- Martins J. R., Kongsuphol P., Sammels E., Daimène S., Aldehni F., Clarke L., et al. (2011). F508del-CFTR increases intracellular Ca2+ signaling that causes enhanced calcium-dependent Cl− conductance in cystic fibrosis. Biochim. Biophys. Acta 1812, 1385–1392. 10.1016/j.bbadis.2011.08.008 [DOI] [PubMed] [Google Scholar]
- Matsui H., Grubb B. R., Tarran R., Randell S. H., Gatzy J. T., Davis C. W., et al. (1998). Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95, 1005–1015. 10.1016/S0092-8674(00)81724-9 [DOI] [PubMed] [Google Scholar]
- Miller M. R., Soave D., Li W., Gong J., Pace R. G., Boelle P. Y., et al. (2015). Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J. Pediatr. 166, 1152–1157.e1156. 10.1016/j.jpeds.2015.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner K., Mohn D., Elliot R., Powers D., Chen J., Liu B., et al. (2019). The anthelminthic niclosamide and related compounds represent potent Tmem16a antagonists that fully relax mouse and human airway rings. Front. Pharmacol. (in press). [Google Scholar]
- Montoro D. T., Haber A. L., Biton M., Vinarsky V., Lin B., Birket S. E., et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. 10.1038/s41586-018-0393-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody M., Pennington C., Schultz C., Caldwell R., Dinkel C., Rossi M. W., et al. (2005). Inositol polyphosphate derivative inhibits Na+ transport and improves fluid dynamics in cystic fibrosis airway epithelia. Am. J. Physiol. Cell Physiol. 289, C512–C520. 10.1152/ajpcell.00591.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R. B. (2013). Pitfalls of drug development: lessons learned from trials of denufosol in cystic fibrosis. J. Pediatr. 162, 676–680. 10.1016/j.jpeds.2012.11.034 [DOI] [PubMed] [Google Scholar]
- Mount D. B., Romero M. F. (2004). The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 447, 710–721. 10.1007/s00424-003-1090-3 [DOI] [PubMed] [Google Scholar]
- Namkung W., Finkbeiner W. E., Verkman A. S. (2010a). CFTR-Adenylyl cyclase I association is responsible for UTP activation of CFTR in well-differentiated primary human bronchial cell cultures. Mol. Biol. Cell 21, 2639–2648. 10.1091/mbc.e09-12-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W., Phuan P. W., Verkman A. S. (2011a). TMEM16A inhibitors reveal TMEM16A as a minor component of CaCC conductance in airway and intestinal epithelial cells. J. Biol. Chem. 286, 2365–2374. 10.1074/jbc.M110.175109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W., Thiagarajah J. R., Phuan P. W., Verkman A. S. (2010b). Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 24, 4178–4186. 10.1096/fj.10-160648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W., Yao Z., Finkbeiner W. E., Verkman A. S. (2011b). Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 25, 4048–4062. 10.1096/fj.11-191627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. J., Hwang S. J., Jung J., Yu K., Kim J., Choi J. Y., et al. (2013). MONNA, a potent and selective blocker for TMEM16A/Anoctamin-1. Mol. Pharmacol. 84, 726–735. 10.1124/mol.113.087502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousingsawat J., Kongsuphol P., Schreiber R., Kunzelmann K. (2011a). CFTR and TMEM16A are separate but functionally related Cl channels. Cell. Physiol. Biochem. 28, 715–724. 10.1159/000335765 [DOI] [PubMed] [Google Scholar]
- Ousingsawat J., Martins J. R., Schreiber R., Rock J. R., Harfe B. D., Kunzelmann K. (2009). Loss of TMEM16A causes a defect in epithelial Ca2+ dependent chloride transport. J. Biol. Chem. 284, 28698–28703. 10.1074/jbc.M109.012120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousingsawat J., Schreiber R., Kunzelmann K. (2011b). Differential contribution of SLC26A9 to Cl(-) conductance in polarized and non-polarized epithelial cells. J. Cell. Physiol. 227, 2323–2329. 10.1002/jcp.22967 [DOI] [PubMed] [Google Scholar]
- Ousingsawat J., Wanitchakool P., Kmit A., Romao A. M., Jantarajit W., Schreiber S., et al. (2015). Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7-receptors in macrophages. Nat. Commun. 6:6245. 10.1038/ncomms7245 [DOI] [PubMed] [Google Scholar]
- Paulino C., Kalienkova V., Lam A. K. M., Neldner Y., Dutzler R. (2017a). Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 552, 421–425. 10.1038/nature24652 [DOI] [PubMed] [Google Scholar]
- Paulino C., Neldner Y., Lam A. K., Kalienkova V., Brunner J. D., Schenck S., et al. (2017b). Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. Elife 6:e26232. 10.7554/eLife.26232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N., Galietta L. J. (2014). Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94, 419–459. 10.1152/physrev.00039.2011 [DOI] [PubMed] [Google Scholar]
- Pedersen K. A., Schroder R. L., Skaaning-Jensen B., Strobaek D., Olesen S. P., Christophersen P. (1999). Activation of the human intermediate-conductance Ca2+)-activated K+ channel by 1-ethyl-2-benzimidazolinone is strongly Ca2+-dependent. Biochim. Biophys. Acta 1420, 231–240. 10.1016/S0005-2736(99)00110-8 [DOI] [PubMed] [Google Scholar]
- Pezzulo A. A., Tang X. X., Hoegger M. J., Alaiwa M. H., Ramachandran S., Moninger T. O., et al. (2012). Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487, 109–113. 10.1038/nature11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert L. W., Zilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., et al. (2018). A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381. 10.1038/s41586-018-0394-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen J. H., Fischer H., Illek B., Machen T. E. (1994). Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. U.S.A 91, 5340–5344. 10.1073/pnas.91.12.5340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranke I. M., Hatton A., Simonin J., Jais J. P., Le Pimpec-Barthes F., Carsin A., et al. (2017). Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 7:7375. 10.1038/s41598-017-07504-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston P., Wartosch L., Gunzel D., Fromm M., Kongsuphol P., Ousingsawat J., et al. (2010). Disruption of the K+ channel beta-subunit KCNE3 reveals an important role in intestinal and tracheal Cl− transport. J. Biol. Chem. 285, 7165–7175. 10.1074/jbc.M109.047829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M., Zifarelli G. (2014). Thermal sensitivity of CLC and TMEM16 chloride channels and transporters. Curr. Top. Membr. 74, 213–231. 10.1016/B978-0-12-800181-3.00008-7 [DOI] [PubMed] [Google Scholar]
- Quinton P. M. (2001). The neglected ion: . Nat. Med. 7, 292–293. 10.1038/85429 [DOI] [PubMed] [Google Scholar]
- Ratjen F., Durham T., Navratil T., Schaberg A., Accurso F. J., Wainwright C., et al. (2012). Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 11, 539–549. 10.1016/j.jcf.2012.05.003 [DOI] [PubMed] [Google Scholar]
- Ribeiro C. M., Paradiso A. M., Schwab U., Perez-Vilar J., Jones L., O'neal W., et al. (2005). Chronic airway infection/Inflammation Induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 280, 17798–17806. 10.1074/jbc.M410618200 [DOI] [PubMed] [Google Scholar]
- Rottner M., Freyssinet J. M., Martinez M. C. (2009). Mechanisms of the noxious inflammatory cycle in cystic fibrosis. Respir. Res. 10:23. 10.1186/1465-9921-10-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner M., Kunzelmann C., Mergey M., Freyssinet J. M., Martinez M. C. (2007). Exaggerated apoptosis and NF-kappaB activation in pancreatic and tracheal cystic fibrosis cells. FASEB J. 21, 2939–2948. 10.1096/fj.06-7614com [DOI] [PubMed] [Google Scholar]
- Rottner M., Tual-Chalot S., Mostefai H. A., Andriantsitohaina R., Freyssinet J. M., Martinez M. C. (2011). Increased oxidative stress induces apoptosis in human cystic fibrosis cells. PLoS ONE 6:e24880. 10.1371/journal.pone.0024880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M., Daines C., Ringshausen F. C., Kerem E., Wilson J., Tullis E., et al. (2017). Tezacaftor-Ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med. 377, 2024–2035. 10.1056/NEJMoa1709847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffin M., Voland M., Marie S., Bonora M., Blanchard E., Blouquit-Laye S., et al. (2013). Anoctamin 1 dysregulation alters bronchial epithelial repair in cystic fibrosis. Biochim. Biophys. Acta 1832, 2340–2351. 10.1016/j.bbadis.2013.09.012 [DOI] [PubMed] [Google Scholar]
- Sanders K. M., Zhu M. H., Britton F., Koh S. D., Ward S. M. (2012). Anoctamins and gastrointestinal smooth muscle excitability. Exp. Physiol. 97, 200–206. 10.1113/expphysiol.2011.058248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk L. K., Buchholz B., Henke S. F., Michgehl U., Daniel C., Amann K., et al. (2018). Nephron-specific knockout of TMEM16A leads to reduced number of glomeruli and albuminuria. Am. J. Physiol. Renal Physiol. [Epub ahead of print]. 10.1152/ajprenal.00638.2017 [DOI] [PubMed] [Google Scholar]
- Schreiber R., Faria D., Skryabin B. V., Rock J. R., Kunzelmann K. (2014). Anoctamins support calcium-dependent chloride secretion by facilitating calcium signaling in adult mouse intestine. Pflügers Arch. 467, 1203–1213. 10.1007/s00424-014-1559-2 [DOI] [PubMed] [Google Scholar]
- Schreiber R., Ousingsawat J., Wanitchakool P., Sirianant L., Benedetto R., Reiss K., et al. (2018). Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J. Physiol. 596, 217–229. 10.1113/JP275175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R., Uliyakina I., Kongsuphol P., Warth R., Mirza M., Martins J. R., et al. (2010). Expression and function of epithelial anoctamins. J. Biol. Chem. 285, 7838–7845. 10.1074/jbc.M109.065367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz A., Puvvadi R., Borisov S. M., Shaw N. C., Klimant I., Berry L. J., et al. (2017). Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat. Commun. 8:1409 10.1038/s41467-017-00532-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudieri P., Caci E., Bruno S., Ferrera L., Schiavon M., Sondo E., et al. (2012). Association of TMEM16A chloride channel overexpression with airway goblet cells metaplasia. J. Physiol. 590, 6141–6155. 10.1113/jphysiol.2012.240838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudieri P., Caci E., Venturini A., Sondo E., Pianigiani G., Marchetti C., et al. (2015). Ion channel and lipid scramblase activity associated with expression of tmem16F/ANO6 isoforms. J. Physiol. 593, 3829–3848. 10.1113/JP270691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y., Kim J., Chang J., Kim S. S., Namkung W., Kim I. (2018). Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur. J. Med. Chem. 160, 245–255. 10.1016/j.ejmech.2018.10.002 [DOI] [PubMed] [Google Scholar]
- Seo Y., Lee H. K., Park J., Jeon D. K., Jo S., Jo M., et al. (2016). Ani9, a novel potent small-molecule ANO1 inhibitor with negligible effect on ANO2. PLoS ONE 11:e0155771. 10.1371/journal.pone.0155771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y., Park J., Kim M., Lee H. K., Kim J. H., Jeong J. H., et al. (2015). Inhibition of ANO1/TMEM16A chloride channel by idebenone and its cytotoxicity to cancer cell lines. PLoS ONE 10:e0133656. 10.1371/journal.pone.0133656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharm K., Sung J., Kim H. J., Oak M. H., Yi E. (2017). Rice bran extract inhibits TMEM16A-Involved activity in the neonatal rat cochlea. J. Nanosci. Nanotechnol. 17, 2390–2393. 10.1166/jnn.2017.13333 [DOI] [PubMed] [Google Scholar]
- Shimizu T., Iehara T., Sato K., Fujii T., Sakai H., Okada Y. (2013). TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am. J. Physiol. Cell Physiol. 304, C748–C759. 10.1152/ajpcell.00228.2012 [DOI] [PubMed] [Google Scholar]
- Singh R. D., Gibbons S. J., Saravanaperumal S. A., Du P., Hennig G. W., Eisenman S. T., et al. (2014). Ano1, a Ca2+-activated Cl− channel coordinates contractility in mouse intestine by ca2+ transient coordination between interstitial cells of cajal. J. Physiol. 592, 4051–4068. 10.1113/jphysiol.2014.277152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianant L., Ousingsawat J., Wanitchakool P., Schreiber R., Kunzelmann K. (2015). Cellular volume regulation by anoctamin 6:Ca2+, phospholipase A2, osmosensing. Pflügers Arch. 468, 335–349. 10.1007/s00424-015-1739-8 [DOI] [PubMed] [Google Scholar]
- Smith J. J., Welsh M. J. (1992). cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J. Clin. Invest. 89, 1148–1153. 10.1172/JCI115696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stick S. M., Schultz A. (2018). CrossTalk opposing view: mucosal acidification does not drive early progressive lung disease in cystic fibrosis. J. Physiol. 596, 3439–3441. 10.1113/JP275426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltz D. A., Meyerholz D. K., Pezzulo A. A., Ramachandran S., Rogan M. P., Davis G. J., et al. (2010). Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2:29ra31. 10.1126/scitranslmed.3000928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltz D. A., Meyerholz D. K., Welsh M. J. (2015). Origins of cystic fibrosis lung disease. N. Engl. J. Med. 372, 351–362. 10.1056/NEJMra1300109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strug L. J., Gonska T., He G., Keenan K., Ip W., Boelle P. Y., et al. (2016). Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 25, 4590–4600. 10.1093/hmg/ddw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y., Wu F., Lv J., Li H., Li X., Du Z., et al. (2015). Identification of the novel TMEM16A Inhibitor dehydroandrographolide and its anticancer activity on SW620 cells. PLoS ONE 10:e0144715. 10.1371/journal.pone.0144715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Rommens J. M., Corvol H., Li W., Li X., Chiang T. A., et al. (2012). Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat. Genet. 44, 562–569. 10.1038/ng.2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J., Umeda M., Sims P. J., Nagata S. (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- Ta C. M., Adomaviciene A., Rorsman N. J., Garnett H., Tammaro P. (2016). Mechanism of allosteric activation of TMEM16A/ANO1 channels by a commonly used chloride channel blocker. Br. J. Pharmacol. 173, 511–528. 10.1111/bph.13381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Fatehi M., Linsdell P. (2009). Mechanism of direct bicarbonate transport by the CFTR anion channel. J. Cyst. Fibros. 8, 115–121. 10.1016/j.jcf.2008.10.004 [DOI] [PubMed] [Google Scholar]
- Taylor-Cousar J. L., Munck A., Mckone E. F., Van Der Ent C. K., Moeller A., Simard C., et al. (2017). Tezacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med. 377, 2013–2023. 10.1056/NEJMoa1709846 [DOI] [PubMed] [Google Scholar]
- Tian Y., Schreiber R., Kunzelmann K. (2012a). Anoctamins are a family of Ca2+ activated Cl− channels. J. Cell Sci. 125, 4991–4998. 10.1242/jcs.109553 [DOI] [PubMed] [Google Scholar]
- Tian Y., Schreiber R., Wanitchakool P., Kongsuphol P., Sousa M., Uliyakina I., et al. (2012b). Control of TMEM16A by INO-4995 and other inositolphosphates. Br. J. Pharmacol. 168, 253–265. 10.1111/j.1476-5381.2012.02193.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirkos S., Newbigging S., Nguyen V., Keet M., Ackerley C., Kent G., et al. (2006). Expression of S100A8 correlates with inflammatory lung disease in congenic mice deficient of the cystic fibrosis transmembrane conductance regulator. Respir. Res. 7:51. 10.1186/1465-9921-7-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L., Namkung W., Verkman A. S. (2010). Crofelemer, an antisecretory antidiarrheal proanthocyanidin oligomer extracted from Croton lechleri, targets two distinct intestinal chloride channels. Mol. Pharmacol. 77, 69–78. 10.1124/mol.109.061051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trout L., King M., Feng W., Inglis S. K., Ballard S. T. (1998). Inhibition of airway liquid secretion and its effect on the physical properties of airway mucus. Am. J. Physiol. 274, L258–L263. 10.1152/ajplung.1998.274.2.L258 [DOI] [PubMed] [Google Scholar]
- Truong E. C., Phuan P. W., Reggi A. L., Ferrera L., Galietta L. J. V., Levy S. E., et al. (2017). Substituted 2-acylaminocycloalkylthiophene-3-carboxylic acid arylamides as inhibitors of the calcium-activated chloride channel transmembrane protein 16A (TMEM16A). J. Med. Chem. 60, 4626–4635. 10.1021/acs.jmedchem.7b00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajanaphanich M., Schultz C., Rudolf M. T., Wasserman M., Enyedi P., Craxton A., et al. (1994). Long-term uncoupling of chloride secretion from intracellular calcium levels by Ins(3,4,5,6)P4. Nature 371, 711–714. 10.1038/371711a0 [DOI] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., et al. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 106, 18825–18830. 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Stack J. H., Straley K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848. 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G., Avramescu R. G., Perdomo D., Phuan P. W., Bagdany M., Apaja P. M., et al. (2014). Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 6:246ra297. 10.1126/scitranslmed.3008889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman A. S., Galietta L. J. (2009). Chloride channels as drug targets. Nat. Rev. Drug Discov. 8, 153–171. 10.1038/nrd2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba J. M., Parrado C., Santos-Gonzalez M., Alcain F. J. (2010). Therapeutic use of coenzyme Q10 and coenzyme Q10-related compounds and formulations. Expert Opin. Investig. Drugs 19, 535–554. 10.1517/13543781003727495 [DOI] [PubMed] [Google Scholar]
- Wainwright C. E., Elborn J. S., Ramsey B. W., Marigowda G., Huang X., Cipolli M., et al. (2015). Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 373, 220–231. 10.1056/NEJMoa1409547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker N. M., Simpson J. E., Levitt R. C., Boyle K. T., Clarke L. L. (2006). Talniflumate increases survival in a cystic fibrosis mouse model of distal intestinal obstructive syndrome. J. Pharmacol. Exp. Ther. 317, 275–283. 10.1124/jpet.105.094847 [DOI] [PubMed] [Google Scholar]
- Wang H., Zou L., Ma K., Yu J., Wu H., Wei M., et al. (2017). Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol. Cancer 16:152. 10.1186/s12943-017-0720-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Zhao W., Sun J., Tao T., Chen X., Zheng Y. Y., et al. (2018). Inflammatory mediators mediate airway smooth muscle contraction through a GPCR-TMEM16A-VDCC axis and contribute to bronchial hyperresponsiveness in asthma. J. Allergy Clin. Immunol. 141, 1259–1268. 10.1016/j.jaci.2017.05.053 [DOI] [PubMed] [Google Scholar]
- Wanitchakool P., Ousingsawat J., Sirianant L., Macaulay N., Schreiber R., Kunzelmann K. (2016). Cl− channels in apoptosis. Eur. Biophys. J. 45, 599–610. 10.1007/s00249-016-1140-3 [DOI] [PubMed] [Google Scholar]
- Wanitchakool P., Wolf L., Koehl G., Sirianant L., Gaumann A., Schreiber R., et al. (2014). Role of anoctamins in cancer and apoptosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130096. 10.1098/rstb.2013.0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Vankeerberghen A., Cuppens H., Eggermont J., Cassiman J. J., Droogmans G., et al. (1999). Interaction between calcium-activated chloride channels and the cystic fibrosis transmembrane conductance regulator. Pflugers Arch. 438, 635–641. 10.1007/s004249900108 [DOI] [PubMed] [Google Scholar]
- White M. M., Aylwin M. (1990). Niflumic and flufenamic acids are potent reversible blockers of Ca2+-activated Cl− channels in Xenopus oocytes. Mol. Pharmacol. 37, 720–724. [PubMed] [Google Scholar]
- Wilke M., Buijs-Offerman R. M., Aarbiou J., Colledge W. H., Sheppard D. N., Touqui L., et al. (2011). Mouse models of cystic fibrosis: phenotypic analysis and research applications. J. Cyst. Fibros. 10 (Suppl. 2), S152–171. 10.1016/S1569-1993(11)60020-9 [DOI] [PubMed] [Google Scholar]
- Yang H., Kim A., David T., Palmer D., Jin T., Tien J., et al. (2012). TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122. 10.1016/j.cell.2012.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. D., Cho H., Koo J. Y., Tak M. H., Cho Y., Shim W. S., et al. (2008). TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215. 10.1038/nature07313 [DOI] [PubMed] [Google Scholar]
- Yerxa B. R., Sabater J. R., Davis C. W., Stutts M. J., Lang-Furr M., Picher M., et al. (2002). Pharmacology of INS37217 [P(1)-(uridine 5')-P(4)- (2'-deoxycytidine 5')tetraphosphate, tetrasodium salt], a next-generation P2Y(2) receptor agonist for the treatment of cystic fibrosis. J Pharmacol. Exp. Ther. 302, 871–880. 10.1124/jpet.102.035485 [DOI] [PubMed] [Google Scholar]
- Yim P. D., Gallos G., Perez-Zoghbi J. F., Trice J., Zhang Y., Siviski M., et al. (2013). Chloride channel blockers promote relaxation of TEA-induced contraction in airway smooth muscle. J. Smooth Muscle Res. 49, 112–124. 10.1540/jsmr.49.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabner J., Smith J. J., Karp P. H., Widdicombe J. H., Welsh M. J. (1998). Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol. Cell 2, 397–403. 10.1016/S1097-2765(00)80284-1 [DOI] [PubMed] [Google Scholar]
- Zhang C. H., Li Y., Zhao W., Lifshitz L. M., Li H., Harfe B. D., et al. (2013). The transmembrane protein 16A Ca2+-activated Cl− channel in airway smooth muscle contributes to airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 187, 374–381. 10.1164/rccm.201207-1303OC [DOI] [PMC free article] [PubMed] [Google Scholar]