

ABSTRACT
Recent metagenomic analysis has revealed that our gut microbiota plays an important role in not only the maintenance of our health but also various diseases such as obesity, diabetes, inflammatory bowel disease, and allergy. However, most intestinal bacteria are considered ‘unculturable’ bacteria, and their functions remain unknown. Although culture-independent genomic approaches have enabled us to gain insight into their potential roles, culture-based approaches are still required to understand their characteristic features and phenotypes. To date, various culturing methods have been attempted to obtain these ‘unculturable’ bacteria, but most such methods require advanced techniques. Here, we have tried to isolate possible unculturable bacteria from a healthy Japanese individual by using commercially available media. A 16S rRNA (ribosomal RNA) gene metagenomic analysis revealed that each culture medium showed bacterial growth depending on its selective features and a possibility of the presence of novel bacterial species. Whole genome sequencing of these candidate strains suggested the isolation of 8 novel bacterial species classified in the Actinobacteria and Firmicutes phyla. Our approach indicates that a number of intestinal bacteria hitherto considered unculturable are potentially culturable and can be cultured on commercially available media. We have obtained novel gut bacteria from a healthy Japanese individual using a combination of comprehensive genomics and conventional culturing methods. We would expect that the discovery of such novel bacteria could illuminate pivotal roles for the gut microbiota in association with human health.
KEYWORDS: Human gut microbiota, commercially available media, 16S rRNA gene metagenomic sequencing, whole genome sequencing, novel bacteria, unculturable bacteria
Introduction
A typical human intestine has around 1,000 bacterial species,1 and these ca. 38-trillion cells constitute the gut microbiota,2 showing that the number of bacteria is of the same order as the number of human cells (ca. 30-trillion cells). Recently, metagenomic analysis of bacterial 16S rRNA (ribosomal RNA) gene sequences targeted at the human gut microbiota using next generation sequencing (NGS) has resulted in detailed compositional information about the human gut microbiota, including uncultured bacterial species. Gordon et al. reported a correlation between obesity and the gut microbiota in 2006 and 2008;3,4 the gut microbiota has also been suggested to correlate not only with the maintenance of our health but also with bowel disease and systemic diseases including autoimmune disease and metabolic disease.5 Furthermore, recent studies have indicated that host genetics influence the composition of the human gut microbiome,6 while diet and environments appear to have a greater impact than host genetics,7 and a particular gut bacterial species has been notably correlated with other bacterial infections.8 Therefore, as well as human genetics, it becomes increasingly important to investigate the details of the gut microbiota. Because some intestinal bacteria are generally considered to be unculturable or minor population, such bacteria have not been well characterized yet, and are difficult to identify in the laboratory. To overcome this point, Nichols et al. designed an isolation chip (ichip) for high-throughput bacterial cultivation, consisting of several hundred miniature diffusion chambers, each inoculated with a single environmental cell.9 Bioinformatics researchers have succeeded in inferring the complete genome sequences of gut bacteria from metagenomic sequence data and thus gained insights, including metabolic profiles, into their ecological niches, nutrient requirements, and potential roles in maintaining our health.10 Thus, microbial culture techniques have been de-emphasized to date; however, there are limitations to the information that can be obtained from molecular approaches alone, and the pure culture of intestinal microbiota remains essential to define the roles of specific bacteria in the intestine. In addition, culture using selective media allows for the growth and detection of less abundant bacteria that may be missed by insufficient sequencing depth in culture-independent studies.11 Recently, several approaches have been established for the culture-based isolation of previously unculturable bacteria.
Tanaka et al. focused on interbacterial communications by using soluble substances that could diffuse among viable gut bacteria, designed a co-culture system utilizing soft agar layers separated by membrane filters (0.22 μm pore size) and successfully isolated three previously uncultured strains.12 Compared with 1.5% agar medium, soft agar contributes to maintaining ideal growth conditions, because molecules diffuse more quickly in the latter. This system is simple and can be easily and economically applied to conventional instrumentation and various culture conditions including aerobic and anaerobic cultures.
Goodman and coworkers developed new culture media to mimic the native gut habitat using easily obtained reagents such as peptone, yeast extract, meat extract, various carbohydrates, and growth factors including vitamins, minerals, and short chain fatty acids. Using these media, they demonstrated that it was possible to isolate and cultivate a large proportion of the fecal microbiota.13 This system can be easily and economically employed by using commercially available reagents. They suggested that other variations including stool extracts and rumen fluids undoubtedly will allow additional members of the human gut microbiota to be cultured in vitro.
Rettedal et al. reported 10 carefully designed and conditioned media using commercially available media including M9, GMM, Gifu anaerobic medium (GAM), and GAM (modified) with agar or gellan gum to enable the cultivation of a representative proportion of human gut bacteria and perform rapid multiplex phenotypic profiling. They used this approach to determine the phylogenetic distribution of antibiotic tolerance phenotypes for 16 antibiotics in the human gut microbiota. Based on the phenotypic mapping, they tailored antibiotic combinations to specifically select for previously uncultivated bacteria.14
Browne et al. designed a new workflow based on targeted phenotypic culturing (spore formation induced by ethanol treatment) linked to large-scale whole genome sequencing, phylogenetic analysis and computational modeling, and they demonstrated that a substantial proportion of intestinal bacteria are culturable.15
Lau et al. designed 66 culture conditions using 33 media, including commercially available media and supplements with aerobic/anaerobic culture conditions.16 They found that the majority of the bacteria in the human gut microbiota can be cultured, compared to culture-independent sequencing, by using this method. Applying this method to target Lachnospiraceae strains resulted in the recovery of 79 isolates, 12 of which are on the Human Microbiome Project’s “Most Wanted” list.16
Lagier et al. introduced microbial culturomics, a culturing approach that uses high-throughput culture conditions and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) and 16S rRNA gene sequencing for identification.11,17 MALDI-TOF is a recently available technique for microbial identification that provides results more rapidly by simpler sample preparation at a lower cost than those of other technologies. They also designed 70 culture conditions and obtained various samples from healthy individuals or patients with various diseases from different geographical origins to increase diversity. As Lagier et al. found, sampling from different geographical origins is one of the important factors for success in culturing many more unculturable bacteria due to the distinct dietary cultures. Recent studies have suggested that the human gut microbiome is affected by various factors such as diet and host genetic background.6,7 Essentially, human gut microbiomes can be classified into ‘enterotypes’ on the basis of differences in the abundance of a few major species largely linked with dietary habits.18,19 Nishijima et al. analyzed microbiome data from healthy individuals selected from 12 countries, including Japan, and they found that the gut microbiota of Japanese people was characterized by various unique functional features, including the highest abundance of phylum Actinobacteria because of the highest abundance of Bifidobacterium and high carbohydrate metabolism capacity.20 Furthermore, Hehemann et al. found that Japanese gut microbiomes contain more genes for aquatic-plant-derived polysaccharide-degrading enzymes than those of Americans do.21 Indeed, the Japanese have a unique dietary culture and habits that differ greatly from those of Western people, and the Japanese exhibit the highest average life span and very low body mass index (BMI) [World Health Organization 2015, Global Health Observatory (GHO) data: overweight and obesity. http://www.who.int/gho/ncd/risk_factors/overweight/en/and Life expectancy http://apps.who.int/gho/data/node. main.688?lang = en/]. Therefore, these findings raise a possibility that some of ethnically unculturable bacteria could be inherent in the Japanese gut microbiota.
As shown in the various culturing studies, it has been deduced that only a minor part of the gut microbiota can be cultivated, but Browne et al have demonstrated that a substantial proportion of the intestinal bacteria are culturable with a novel workflow based on targeted phenotypic culturing linked to large-scale genomics and computational modelling.15
Instead of multiomics approaches, we speculated that a reasonably affordable standard culturing method with commercially available media could still have sufficient performance to grow ethnically novel gut microbes from a healthy Japanese donor. Intriguingly, our metagenomic analysis by 16S rRNA gene sequencing suggested that novel bacterial species could be isolated at very high probability on the commercially available media. We further performed a single colony isolation, followed by whole genome sequencing and phylogenetic analysis to characterize those isolates. Here, we report the isolation and characterization of a number of novel bacterial species from the Japanese gut microbiota.
Results
Selection of 26 media for the isolation of novel gut microbes
To explore novel members of the gut microbiota by utilizing the selectivity of commercially available media, 26 media were chosen for further analysis (Table S1; see abbreviations for each medium in Table 1). To be readily available in a reproducible fashion, most media consist of commercially available products, including selective and non-selective media. They have various features including culturing condition (pH, temperature, and aerobic/anaerobic), concentration of salt, food ingredients as energy source, and various reagents affecting their selectivity. Gut microbiota media (GMM) was also used, since Goodman et al.13 reported that GMM captures a remarkable proportion of the human fecal microbiota by using commercially available reagents.
Table 1.
The number of sequencing reads and OTUs in each culture media used in this study.
| Media | The number of joined fastq pair | The number of OTUs |
|---|---|---|
| Original fecal sample | 76,572 | 259 |
| Gifu Anaerobic Medium Agar, Modified (GAM) | 58,337 | 105 |
| Mueller Hinton Agar (MH) | 51,709 | 104 |
| Phenylethyl Alcohol Agar with 5% Sheep Blood (PEA) | 1,50,489 | 140 |
| Brain Heart Infusion (BHI) | 50,487 | 127 |
| BY-Chocolate Agar (Choco) | 62,625 | 128 |
| BY-Chocolate Agar (Choco-pasteurized) | 77,709 | 71 |
| Columbia CNA Agar with 5% Sheep Blood (CNA) | 65,893 | 121 |
| Gut Microbiota Medium (GMM) | 1,00,078 | 135 |
| Lactobacilli MRS Agar (MRS) | 1,62,287 | 51 |
| Tomato Juice Agar (Tomato) | 1,85,161 | 53 |
| Drigalski Lactose Agar (BTB) | 56,513 | 38 |
| Potato Dextrose Agar (Potato) | 85,607 | 29 |
| Mitis Salivarius Agar (Mitis) | 63,769 | 22 |
| DHL Agar (DHL) | 2,33,413 | 18 |
| Selenite Broth (Selenite) | 1,48,646 | 85 |
| FM Agar, Modified (FM) | 2,80,019 | 33 |
| CIN Agar (CIN) | 5,39,410 | 43 |
| Listeria Enrichment Broth (Listeria) | 86,810 | 43 |
| Bacteroides Bile Esculin Agar (BBE) | 37,048 | 26 |
| Thayer-Martin Selective Agar (TM) | 19,397 | 38 |
| CCFA Agar (CCFA) | 35,635 | 61 |
Sabouraud and Skirrow media, which are usually used for aerobic and microaerophilic culture, were also used for anaerobic culture in this study to investigate the possibility that they could isolate intestinal bacteria. For culturing heat-stable bacteria including spore-forming bacteria, the original fecal sample was pasteurized (65°C, 30 min) and cultured on BY chocolate agar anaerobically. Therefore, we attempted to culture the fecal sample–derived human gut bacteria by using 27 culture conditions in total.
Investigation of optimal site for isolation of viable bacterial cells
To be able to culture large amounts of live gut microbes from human feces, it is important to isolate more viable bacterial cells from fecal samples. The human gut microbiota has been widely reported using fecal samples to date, but there has been no investigation of which part of a fecal sample is a better site to collect viable bacteria.
We performed a metagenomic analysis of the surface and interior of the fecal sample and of the cultured bacteria derived from those parts on six media (Choco, CNA, DHL, GAM, GMM, and MRS) (Figure 1). The metagenomic analysis showed that there was no significant difference in the variety of bacterial species between surface and interior samples (coefficient of determination, R2: 0.98). Additionally, the surface sample culture plate showed more culturable colonies than the interior sample plate did (Fig. S1), indicating that the surface part contains more viable cells. Thus, we investigated the surface part of the fecal sample for further analysis.
Figure 1.
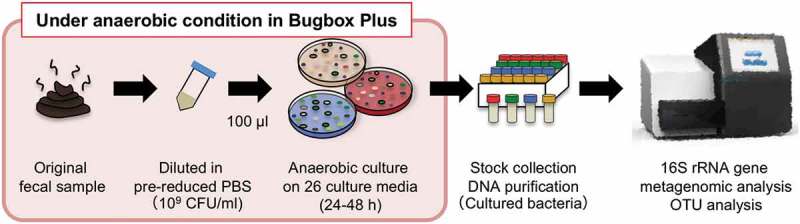
Schematic representation of the experimental procedures from sampling to metagenomics analysis. The original fecal sample was obtained immediately, and the subsequent procedures were performed under strict anaerobic conditions in a Bugbox Plus cabinet.
16S rrna gene metagenomic sequencing for analysis of human gut microbes from healthy human feces
To examine the population and the variety of cultured bacteria in 27 culture conditions (26 media and pasteurized sample), the fresh fecal sample was immediately stored in an anaerobic pack and cultured on each culture medium for 24 ~ 48 h. Since MSA, KF, Sabouraud agar (anaerobic culture), TT, TCBS and Skirrow agar (microaerophilic/anaerobic culture) did not support enough bacterial growth, showing few colonies even after 48 h incubation, we removed those media from further analysis. For the other media, colonies were harvested from each plate after 24 h culture (Figure 1). Frozen stocks were reserved for future bacterial isolation. A 16S rRNA gene metagenomic analysis was performed using purified DNA from original fecal sample and from the cultured samples. 16S rRNA gene deep sequencing resulted in 119,437 ± 115,333 reads as the average number of sequencing reads for each sample (Table 1). After removal of low-quality reads and chimeric sequences, the sequencing reads were grouped into OTUs by Deblur22 program against the Greengenes database (version 13_8), resulting in 445 OTUs (Figure 2).
Figure 2.
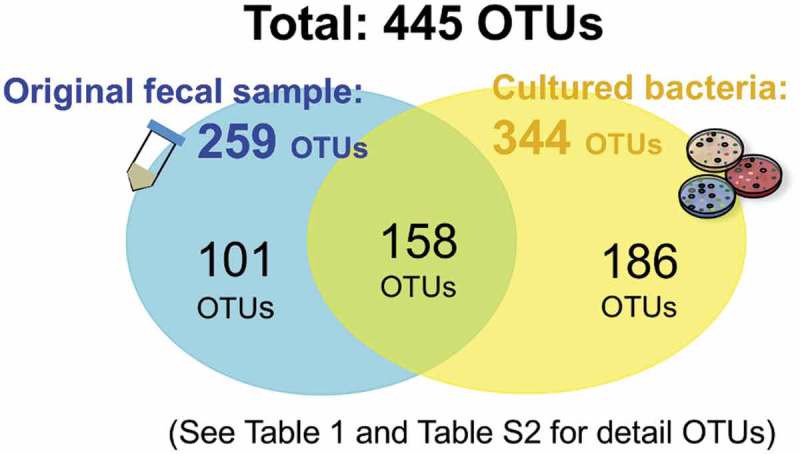
Operational taxonomic unit (OTU) analysis. All detected 16S rRNA V3-V4 regions from the original fecal sample and the cultured bacteria were summarized, yielding 445 OTUs in total. See the detected OTU number for each medium and sample in Table 2 and the detailed OTU information in Table S2.
The numbers of OTUs found in each culture medium are listed in Table 1. Of the total 445 OTUs, only 259 OTUs were observed in the original fecal sample (Figure 2). Among these 259 OTUs, 158 OTUs could be detected after media cultivation, suggesting that the culture methods used in this study could cover 61.0% of the OTUs in the fecal sample, while 186 OTUs might be minor populations highlighted by cultivation (Figure 2). The remaining 186 OTUs were not populous enough to be detectable in the original fecal sample but were also found in the above cultured samples, although their proportions were less than 1% (Table S2). These included as follows: the genera Acidaminococcus and Ruminococcus faecis (GAM, MH, PEA, BHI, and Choco); Catenibacterium mitsuokai (GMM), [Clostridium] sordellii (Choco-pasteurized); genera Enterococcus and Megasphaera (Potato); Anaerostipes hadrus (MRS); genera Mitsuokella and Megasphaera (MRS, Tomato, BTB, Potato); genera Bifidobacterium and Streptococcus salivarius (Mitis); Fusobacterium (CIN, Selenite, FM, DHL); [Eubacterium] hallii (Listeria); genera Coprobacillus (CCFA) (Table S2). Moreover, some OTUs were cultured only on distinct culture media other than CNA, MH, GAM, TM, BBE, and Potato.
In this study, we observed 344 OTUs across all summarized cultured samples, whereas 259 OTUs were found from the original fecal sample. This result indicates that cultivation enabled rather minor populations of bacteria in the original fecal sample to grow on the media. Thus, this culture method enabled us to highlight greater bacterial diversity through medium-specific selective features. Importantly, 101 of the 259 OTUs from the original fecal sample could not be cultured, suggesting that those 101 OTUs require other ingredients for their growth or might be unculturable bacteria with the methods used in this study. These 101 OTUs include representatives of the genera Collinsella, Coprobacter, Butyricicoccus, Ihubacter, Desulfotomaculum, Anaeromassilibacillus, Anaerotruncus, Flintibacter and Faecalicoccus (Table S2). In addition, some of the OTUs with < 98.7% nucleotide identity suggested that novel bacterial species23 were mainly found in non-selective media (Table S2). Therefore, we performed bacterial isolation for each medium that suggested the presence of novel bacteria in ≥ 1% proportion.
Differential gut bacterial growth on the available media
We examined to what extent which OTUs were cultured in each culture condition, and a list of the OTUs and their proportions in each culture condition are shown in Table S2. For clustering analysis, we conducted heatmap analysis of bacterial microbiota composition profiles represented by 16S rRNA OTU at each medium (Figure 3).
Figure 3.
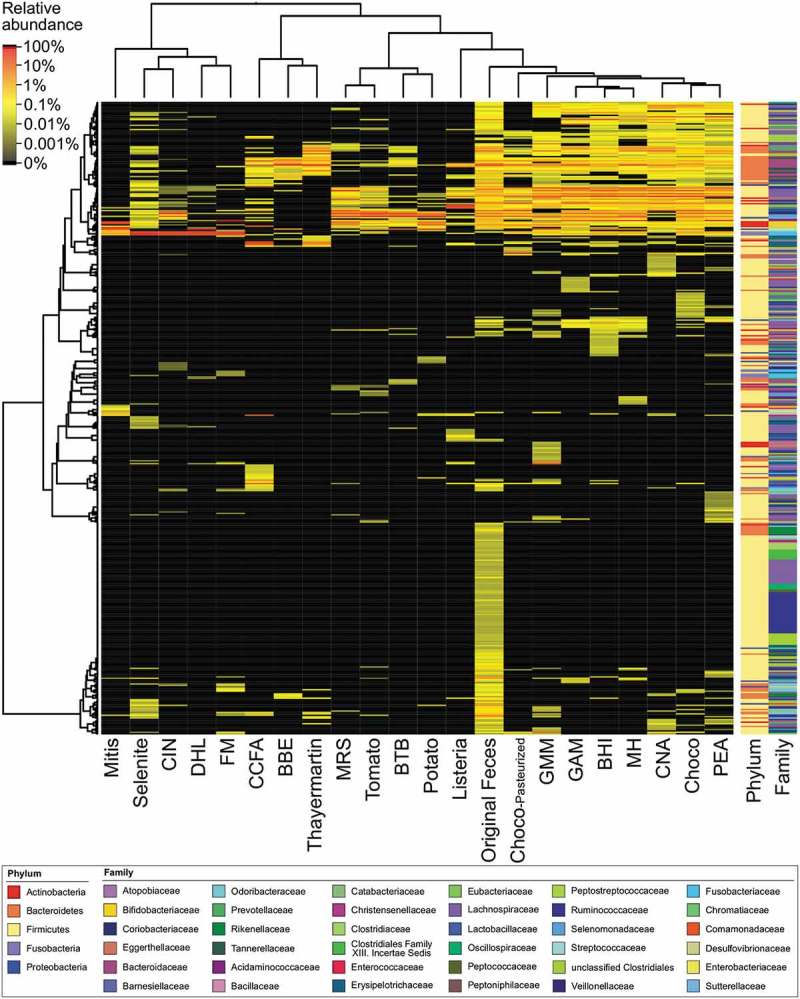
Heatmap of hierarchical clustering of bacterial microbiota composition profiles represented by 16S rRNA OTU at each medium. The tested media in the columns and OTUs in the rows were clustered using R package dist program with Pearson distribution method and hclust program with Ward’s method, followed by displaying heatmap dendrogram clustering with heatmap.2. Relative abundance of each OTU in the tested media is shown as the scale at upper left. Media abbreviations below the heatmap are shown in Table 1. Taxonomic classification based on phylum or family are shown to the right, and the color represents the bacterial taxonomies at the bottom panel.
The non-selective media (MH, BHI, CNA, PEA, GMM, Choco, Choco-pasteurized, and GAM) were closely clustered next to each other (Figure 3), suggesting that the cultured bacterial species in those media were highly correlated to each other. Actually, the major OTUs in each non-selective medium showed predominantly almost the same bacterial species, including Acidaminococcus intestini, Anaerostipes hadrus, genus Bacteroides (B. coprocola, B. faecis, and B. plebeius), Catenibacterium mitsuokai, genus Clostridium (C. sordellii, C. spiroforme, C. lactatifermentans), genus Dorea (D. formicigenerans, D. longicatena), Megasphaera elsdenii, Mitsuokella jalaludinii, Prevotella copri, and Ruminococcus faecis. On the other hand, selective media (BBE, BTB, CCFA, CIN, DHL, FM, Listeria, Mitis, MRS, Potato, Selenite, TM, Tomato) showed potentially unique selections of cultured bacteria species (Figure 3); such a differential clustering appears to be basically associated with basal nutrients and selective agents for growth purposes.
MRS/Tomato/BTB/Potato were clustered closely and mainly cultured Mitsuokella jalaludinii (Figure 3 and Table S2); previous studies have reported that acidic pH is appropriate for the growth of Mitsuokella jalaludinii.24 While MRS was close to the Tomato/BTB/Potato cluster, MRS mainly supported the culture of Anaerostipes hadrus and Megasphaera elsdenii instead of Mitsuokella jalaludinii (Table S2). Mitis supported the cultivation of Escherichia fergusonii and g Bifidobacterium pseudocatenulatum as well as Streptococcus salivarius (Table S2), although it includes crystal violet and potassium tellurite for inhibition of Gram-negative rod-shaped bacterial growth (Table S1).
DHL/Selenite/FM/CIN were clustered closely and mainly cultured Fusobacterium mortiferum (Figure 3 and Table S2). FM, which is a medium for the isolation of genus Fusobacterium, mainly supported the cultivation of Fusobacterium mortiferum. Acidaminococcus intestini was cultured in FM in the second position in this study (Table S2), but there was no or very low detection in DHL/Selenite/CIN; these results suggested that FM could be a potentially good medium to select Acidaminococcus intestini because of the rather low selective pressure. Selenite media for the enrichment of Salmonella species from various materials inhibits coliforms and fecal streptococci by its toxicity. Selenite supported the majority growth of Fusobacterium mortiferum (97.3% proportion, Table S2), indicating that its growth reduced the selenite, leading to reduced toxicity. DHL, a medium generally used for the isolation of pathogenic enteric bacteria from fecal samples, inhibits the growth of Gram-positive cocci with sodium deoxycholate. DHL supported the growth of Fusobacterium mortiferum (57.6%, Table S2) and Escherichia fergusonii (42.9%, Table S2); no other bacterial species were found. CIN, a medium for the isolation of Yersinia and Aeromonas, inhibits the growth of major enteric bacteria using crystal violet, sodium deoxycholate, irgasan, cefsulodin, and novobiocin. Basically, CIN inhibits genus Staphylococcus (S. aureus, S. epidermidis), Pseudomonas aeruginosa, and Escherichia coli. In this study, CIN supported the growth of Fusobacterium mortiferum (50.5%, Table S2) and Megasphaera elsdenii (37.1%, Table S2); intriguingly, Tomato also supported the growth of Megasphaera elsdenii (33.9%, Table S2), suggesting that the tomato juice in Tomato medium might contain good supporting agents in common with CIN medium.
Listeria, a medium including acriflavine for inhibition of Gram-positive bacteria, generally cultures Listeria but in this study mainly cultured [Eubacterium] hallii (80.3%, Table S2). We expected the enrichment of [Eubacterium] hallii and its related species with Listeria medium, but regrowth from the frozen stock did not result in a colony, possibly because of freeze-thaw damage when preparing the stocks.
BBE/TM were clustered closely (Figure 3) and mainly cultured genus Bacteroides (B. coprocola, B. uniformis, B. faecis, B. xylanisolvens, B. dorei, and B. caccae) (Table S2). BBE mainly cultured genus Bacteroides by its selectivity.
CCFA, a medium that facilitates the isolation and differentiation of Clostridium difficile from fecal specimens using cefoxitin and cycloserine, cultured mainly genera Coprobacillus and Clostridium.
Exploration of novel gut microbes from human feces
To explore novel bacteria, each frozen stock was cultured on each medium for 72 h, and ~ 198 colonies were picked up and their 16S rRNA gene sequences (V3-V4 regions: 341F-926R) were subjected to Sanger sequencing to enable taxonomic characterization. The isolates showing < 98.7% identity to the NCBI BLAST 16S rRNA gene sequence database were considered candidate novel bacterial species,23 and whole genome sequencing was performed. Based on the selection of those genome-derived 16S rRNA gene sequences, we successfully isolated 8 novel bacterial species classified as Actinobacteria or Firmicutes (Figure 4 and Table 2). These strains were isolated from GAM (4 strains), Choco (2 strains), CNA (1 strain), and PEA (1 strain). Those nutrient requirements were investigated further, revealing that GAM sufficiently supported the growth of all those 8 strains in this study. The 8 strains show significantly low nucleotide identity at ≤ 96% of 16S-rRNA, hsp60, dnaJ and rpoB genes to the most closely related type strains (Table 2), indicating that these strains can illuminate the phylogeny of Actinobacteria or Firmicutes as novel species (Figure 4). A phylogenetic tree of those 16S rRNA gene sequences compared with the type strains also suggested that the eight strains are located in significantly distinct lineages from most type strains. For instance, the GAM79 (OTU117) strain showed the highest nucleotide identity of 16S rRNA gene at 91.9% to Butyrivibrio crossotus ATCC 29175T (Table 2), and the other tested genes also showed lower homology, below 80%, indicating that this strain could be potentially assigned as a novel genus candidate rather than a species.
Figure 4.
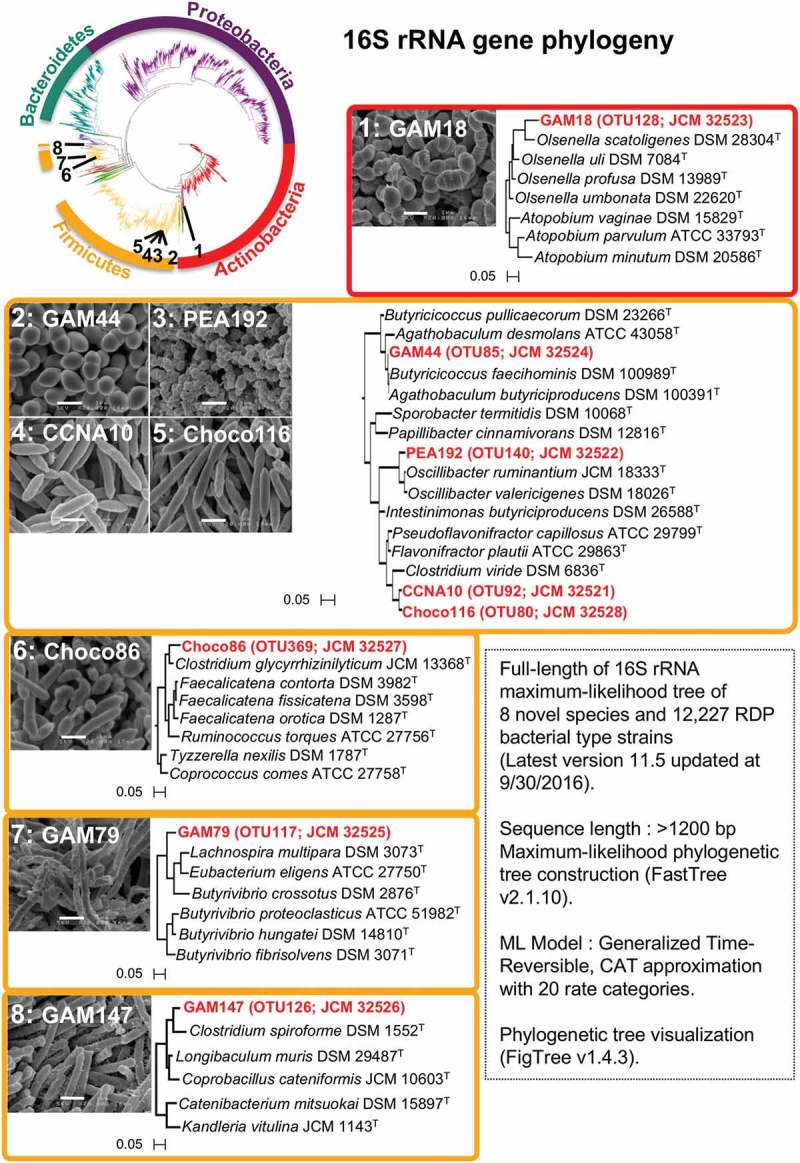
Characterization of 8 novel bacterial species based on the phylogenetic analysis. The comprehensive bacterial phylogeny was investigated using post-aligned 16S rRNA sequences from 12,227 RDP bacterial type strains (latest version 11.5, updated Sept. 30th, 2016) and 8 novel species in this study. The 8 successfully isolated novel bacterial species were classified as Actinobacteria or Firmicutes (see also Table 2). The tree scale indicates sequence differences. The white scale bar on the scanning electron microscopy (SEM) image indicates 1 µm.
Table 2.
Classification of isolates cultured in this study.
| |
|
|
|
|
|
|
|
nucleotide sequence Identity (%) |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | OTU IDa | JCM IDb | GenBank ID | Genome size (Mb) | G + C (%) | SCFAc | Closest relative type straind | 16S rRNA | hsp60 (groEL) | dnaJ | rpoB |
| GAM18 | 128 | JCM 32523 | SAMD00113754 | 2.07 | 65.6 | a, b, p | Olsenella uli ATCC 49627T | 94.1 | 87.1 | 74.0 | 86.9 |
| GAM44 | 85 | JCM 32524 | SAMD00113755 | 3.48 | 57.9 | a, p | Butyricicoccus pullicaecorum DSM23266T | 95.5 | 81.0 | 78.3 | 82.9 |
| PEA192 | 140 | JCM 32522 | SAMD00113757 | 4.41 | 53.3 | a, p | Oscillibacter valericigenes DSM 18026T | 95.8 | 84.5 | 78.2 | 84.5 |
| CCNA10 | 92 | JCM 32521 | SAMD00113750 | 3.48 | 57.9 | a, b, p | Flavonifractor plautii ATCC 29863T | 95.5 | 87.7 | 76.6 | 85.8 |
| Choco116 | 80 | JCM 32528 | SAMD00113751 | 3.68 | 56.6 | a, b, p | Flavonifractor plautii ATCC 29863T | 95.0 | 86.4 | 76.8 | 85.1 |
| Choco86 | 369 | JCM 32527 | SAMD00113752 | 2.75 | 44.0 | a, p | Faecalicatena contorta ATCC 25540T | 95.2 | 81.5 | 75.9 | 81.9 |
| GAM79 | 117 | JCM 32525 | SAMD00113756 | 4.06 | 46.2 | a, b, p | Butyrivibrio crossotus ATCC 29175T | 91.9 | 76.4 | 69.0 | 73.4 |
| GAM147 | 126 | JCM 32526 | SAMD00113753 | 2.70 | 29.5 | a, p | Clostridium spiroform DSM 1552T | 91.5 | 79.5 | 78.0 | 83.5 |
a See Table S2.
b JCM: Japan Collection of Microorganisms in RIKEN BioResource Research Center.
c Short chain fatty acid (SCFA) production predicted by whole genome sequence using KEGG; a:acetic acid, b:butyric acid, p:propionic acid
d Type stain of which four gene sets (16S rRNA, hsp60, dnaJ, and rpoB) are available in public database was selelcted for the nucleotide homology search.
In addition, the results of the 16S rRNA gene metagenomic analysis suggested that we were unable to isolate further novel bacteria showing low homology to known 16S rRNA gene sequences. For instance, OTU96 (94.8% similar to [Clostridium] lactatifermentans, which is also designated Anaerotignum lactatifermentans DSM 14214T25) was detected in PEA and CNA with high proportions of 10.5% and 8.1%, respectively, but the corresponding bacteria could not be reproducibly isolated from PEA or CNA media, implying that freezing or other technical procedures might have affected the viability and stability of this bacterium. In addition, coexistence with other bacteria might be critical for the survival and proliferation of some bacteria, which may lose their ability to survive with other bacteria by single isolation.
Source distribution and country specificity of novel bacterial isolates
The 16S rRNA sequence for each eight novel isolate was searched with megablast homology search against retrieved 20,508 BioSample entries metagenomic short read archives (SRA) for 16S rRNA gene sequencing. Source distribution of those eight novel isolates (Figure 5A) suggested that four Firmicutes isolates (GAM44, PEA192, GAM79 and GAM147) were detected in half of Human sources, while other three Firmicutes isolates (CCNA10, Choco116 and Choco86) show rather less detection in Human sources. Intriguingly, Choco116 shows significant detection to animal and environmental sources, indicating that Choco116 might be one of general bacteria in mammalian sources. In contrast, Actinobacteria GAM18 shows low prevalence to all sources.
Figure 5.
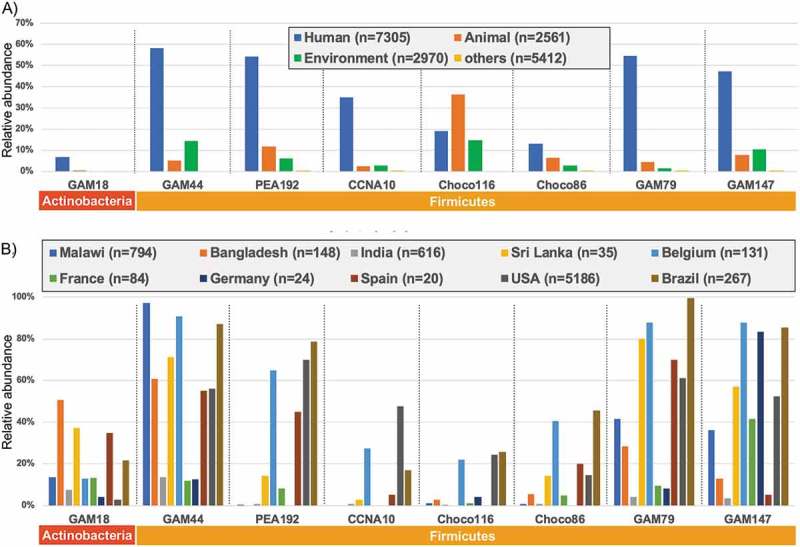
Source distribution and country specificity of novel bacterial isolates. Source distribution A) and country specificity B) for each isolate are shown as relative abundance (%) for total number of respective keywords.
Country-specific detection (Figure 5B) suggested that most isolates, except GAM18, are significantly positive in Belgium, USA and Brazil. Three Firmicutes isolates (CCNA10, Choco116 and Choco86) display a low prevalence in the human source of the tested countries, while Actinobacteria GAM18 shows significant positivity in Malawi in Africa, indicating that such a country-specificity might be associated with ethnically-related gut microbiota.
Discussion
Unculturable bacteria have made it difficult to characterize the important role of the gut microbiota in our health and diseases.3–6,8 To solve this problem, many approaches have been attempted for the isolation of unculturable bacteria from fecal samples and have successfully isolated novel bacteria. For instance, simple and affordable methods can be effective to uncover a diverse array of previously uncultured gut microbiota.13,15 Further elaborate approaches might be required to complete whole picture of gut microbiota.17
In this study, we have investigated the gut microbiota derived from a healthy Japanese individual by using the available media. A 16S rRNA gene metagenomic analysis revealed that each of the 26 culture media showed differential bacterial growth depending on their selective features. Of the total 445 OTUs, 259 OTUs were derived from the original fecal sample, including 158 OTUs that could be detected after culture in the media. In addition, 186 OTUs from 26 media could not be detected in the original fecal sample by the culture-independent method, indicating that cultivation enabled rather minor populations of bacteria in the original fecal sample to grow on those media and be detected by 16S rRNA gene metagenomic sequencing.
In conjunction with 158 OTUs, 344 OTUs (77.3% of all 445 OTUs) could be detected among all the media. The culture method enabled us to highlight a greater bacterial diversity via the specific selective features of the media. On the other hand, 101 OTUs could not be detected in any of the culture media, whereas they were found in the original fecal sample (Table S2). This result raises the possibility that the proportion of those bacterial cells in the original fecal sample may be very low (all were less than 0.1%), that those bacteria could not germinate from spores or that they were slow-growing because of insufficient nutrients or loss of coexistent bacteria to support their growth. Although 101 OTUs were not observed in our culture method, other cutting-edge method such as a membrane filter-based co-culture system and culturomics12,17 might enable their culture.
Surprisingly, OTU analysis suggested that possible novel species (< 98.7% nucleotide identity against the NCBI 16S rRNA gene database) were observed in non-selective media, with very unexpected high proportions of over 6% (Table S2). This finding encouraged us to isolate novel species with conventional growth methods. In this study, we attempted to isolate ‘uncharacterized’ bacteria from a fecal sample derived from a healthy Japanese individual by using the available media. 16S rRNA gene metagenomic analysis and whole genome sequencing resulted in the isolation of 8 novel bacterial species. Generally, uncharacterized bacteria might be unculturable because of nutrient limitations, but we successfully isolated 8 novel bacterial species using commercially available media, suggesting that the most culturable novel bacteria could remain to be identified from our intestinal bacterial microbiota.
In fact, we expected to be able to isolate more novel bacteria, but only 8 strains were isolated (Figure 4 and Table 2). Indeed, as described above, OTU96, which was closely related to A. lactatifermentans DSM 14214T (94.8% nucleotide identity to 16S rRNA gene), was cultured at first propagation on PEA at 10.5% detection, but we were unable to further propagate the isolate, suggesting that such novel bacteria could require strict conditions including other bacterial communities for their support.
Conclusions
We have obtained novel gut bacteria from a healthy Japanese individual in combination with comprehensive genomics and conventional culture methods. We expect that the discovery of such novel bacteria could illuminate pivotal roles of the gut microbiota in association with human health. Regarding the Japanese-specific gut microbiota, a previous study has suggested that the Japanese gut microbiome is remarkably richer in Bacteroides plebeius, which carries aquatic plant-derived polysaccharide-degrading enzymes, than that of Americans,21 indicating that polysaccharide/fiber-rich Japanese diets might support a specific composition of the microbiota. Indeed, B. plebeius (OTU103, see Table S2) was found at 4.2% proportion of the total 31.2% of genus Bacteroides, indicating that B. plebeius was the second most common Bacteroides species in the fecal sample of the volunteer. Indeed, country-specific differences were found in each isolate in this study, such findings might suggest a pivotal contribution of those isolates in gut microbiota. The source of the sample could be a critical factor to explore the novel bacterial species, and samples from additional volunteers would be required to characterize the whole picture of the Japanese gut microbiota.
Materials and methods
Preparation of human fecal sample
A fecal sample was collected from a healthy Japanese adult who had not taken any antimicrobial agents for five years. Written informed consent was obtained from the healthy volunteer. The study protocol was approved by the ethics committee of the National Institute of Infectious Diseases in Japan (Approval No. 642, 11/Dec/2015). It was conducted according to the principles of the Declaration of Helsinki.
The fresh fecal sample was transferred to a sterile dish immediately (within 1 min following defecation) and stored in an airtight plastic bag with Anaero Pack (Mitsubishi Gas Chemical Co., Japan) until being transferred to an anaerobic glove box, UM-017 Bugbox Plus (Ruskinn Technology, UK), filled with 5.5% H2, 20% CO2, and 74.5% N2.
Culturing of fecal sample
Inside the Bugbox Plus anaerobic chamber, fecal samples were collected from the surface or inside of the feces by rolling a cotton swab 2 or 3 times and diluted with pre-reduced phosphate-buffered saline (PBS) to approximately 109 CFU/ml. The debris was precipitated briefly with 5 min standing at room temperature. A 100 µl of the supernatant was spread on agar plates of each medium, and the plates were incubated in the anaerobic glove box UM-017 Bugbox Plus at 37°C for 24 h. The starting fecal supernatant was stored as a frozen glycerol stock, and the genomic DNA was purified as a control sample. We selected 26 types of media (Table S1) for bacterial culturing under 3 different conditions. For some liquid culture media, 1.5% of bacto agar (Difco) was added to prepare agar plates.
For the Skirrow plate and Sabouraud plate, the spread plates were incubated at 37°C with an Anaero Pack for microaerophilic culture and aerobic culture, respectively.
To isolate heat-stable bacteria, including spore-forming bacteria, the supernatant was pasteurized at 65°C for 30 min and then spread on a BY chocolate agar plate (Beckton Dickinson).
After 24 h cultivation, colonies were harvested with 1 ml PBS, the bacterial suspension was frozen at final concentration of 20% glycerol and 5% skim milk at −80°C, and the genomic DNA was prepared for further experiments.
DNA purification from fecal sample
Genomic DNA purification from fecal sample and cultured colonies was performed by using the ZR Fecal DNA MiniPrep Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. The DNA concentration was determined by a Qubit 2.0 Fluorometer (Invitrogen, USA) with the HS dsDNA fluorescence assay.
Metagenome analysis
The DNA-Seq library was prepared by using Nextera XT DNA Sample Preparation Kit (Illumina; San Diego, CA, USA) following the manufacturer’s instructions. Metagenomic sequencing was performed with the MiSeq platform (Illumina) using Reagent Kit v3 chemistry with 150-mer single-end reads on a MiSeq instrument (Illumina).
16S rrna gene metagenome sequencing and analysis
Library preparation followed the 16S Metagenomic Sequencing Library Preparation protocol (27 November 2013 release) (Illumina). Metagenomic sequencing of the 16S rRNA gene V3-V4 region was performed by using the Illumina MiSeq platform with v3 chemistry and 350-mer and 250-mer (for read 1 and read 2, respectively) paired-end reads on the MiSeq instrument (Illumina) according to the manufacturer’s instructions.
Sequencing reads were analyzed by Sickle to remove the adapters and low-quality reads (≤ Q20). Trimmed paired-end reads (≥ 150 bp) were assembled by fastq-join, and low-quality reads (≤ Q20) were removed. Chimeric sequences were removed by USEARCH (version 6.1)26 and the Greengenes reference database (version 13_8).27 Operational taxonomic units (OTUs) were binned using Deblur at default parameter (-t 425 -O 8 – min-reads 2 – min-size 2 – indel-prob 0.01 – mean-error 0.005, instead of – min-reads “10”)22 and taxonomy assigned against the NCBI 16S rRNA sequence database (16SMicrobial updated at 20/May 20th, 2018/) using blastn homology search. OTUs showing non-16S rRNA gene sequences (blast search “No hits”), those with low coverage (≤ 95%) and singletons were discarded. OTUs showing < 97% nucleotide identity were considered novel bacterial species for further identification.
For heatmap clustering of bacterial composition profiles represented by 16S rRNA OTU at each medium, the tested media and OTUs were clustered using R’s gplots package dist program with Pearson distribution method and hclust prograz2m with Ward’s method, followed by displaying heatmap dendrogram clustering with heatmap.2.
Isolation and identification of bacterial strains
The frozen glycerol stocks from each culture medium were resuspended in GAM broth, and 100 µl of 10–5 to 10–8 dilutions were plated on the original selected medium to obtain a single colony (suspensions of glycerol stock obtained from GAM-medium growth were spread on GAM agar plates). Based on the 16S rRNA gene metagenomic analysis data, we isolated up to 200 colonies from each medium, and single colonies were streaked on fresh agar plates for replication. Genomic DNA was purified using the ZR-96 Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA, USA). The V3-V4 regions of the 16S rRNA gene were amplified using the 341F (5ʹ-CCTACGGGAGGCAGCAG-3ʹ) and 926R (5ʹ-CCGTCAATTCCTTTRAGTTT-3ʹ) primers. Purified PCR products were subjected to Sanger sequencing by a 3730xl DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA). Taxonomic assignments of isolates were made with NCBI BLAST using the 16S rRNA gene sequence database; we considered candidate strains with < 97% nucleotide identity as novel bacterial species. For those candidates, single colonies from the replica plates were streaked onto fresh plates, and colonies were frozen at final concentration of 20% glycerol and 5% skim milk at −80°C.
Whole genome sequencing and genome annotation
Novel bacterial strains were cultured with GAM broth, and the cell pellet was mechanically homogenized with ZR BashingBead Lysis tubes (Zymo Research, Irvine, CA, USA) in 500 µl Tris-HCl (pH 8.0), 50 µl 1% sodium dodecyl sulfate (SDS), and 500 µl phenol-chloroform-isoamyl alcohol (25:24:1). The upper phase was purified by using the Qiagen MinElute PCR Purification Kit. Whole genome sequencing (WGS) was performed as follows. For short-read sequencing, a DNA-Seq library was prepared by using the Nextera XT DNA Sample Preparation Kit (Illumina), followed by sequencing using the Illumina MiSeq and NextSeq 500 systems according to the manufacturer’s instructions. For long-read sequencing by the PacBio RSII system (Pacific Biosciences; Menlo Park, CA, USA), SMRTbell library preparation was performed as previously described.28 The DNA sequencing was carried out with P6-C4 chemistry, and the movie length was 360 min. De novo assembly was conducted by using HGAP3 (SMRT Analysis version 2.2.0), further error correction with the Illumina short reads was performed by Pilon. 29 Genome annotation was performed by PROKKA version 1.11 30
Phylogenetic analysis of novel bacterial isolates
To examine the properties of the novel bacterial isolates, genome-based phylogenetic and morphological analysis was performed. Phylogenetic analysis was done by using the genome-derived full-length 16S rRNA, hsp60 (groEL), dnaJ, and rpoB gene sequences. For the 16S rRNA gene, the sequences of its 29 closest relatives were retrieved from the NCBI 16S rRNA gene sequence database (last updated Feb 15th, 2018), and multiply aligned using ClustalW 31 in MEGA 7 software (version 7.0.16). 32 Based on those 30 sequences, a phylogenetic tree was constructed with the maximum-likelihood method,33 and evolutionary distances were computed by the Tamura-Nei method34 using MEGA 7. The statistical reliability of the tree was evaluated by a bootstrap analysis of 1,000 replicates. For the sequences of the hsp60, dnaJ, and rpoB genes, genome-derived full-length nucleotide sequences were aligned against the NCBI non-redundant nt sequence database, and a phylogenetic tree was constructed as in the 16S rRNA gene multiple alignment analysis.
The comprehensive bacterial phylogeny was investigated using the post-alignment 16S rRNA gene sequences from 12,227 RDP bacterial type strains (latest version 11.5, updated Sept. 30th, 2016) and 8 novel species in this study. A maximum-likelihood phylogenetic tree was constructed by FastTree v2.1.10 35 (ML Model: generalized time-reversible, CAT approximation with 20 rate categories), followed by phylogenetic tree visualization with FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
A Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using BlastKOALA 36 was performed to verify the production of short chain fatty acids (SCFA). Genome sizes and G + C contents of the isolates were estimated from the results of WGS.
Morphological analysis of novel bacterial isolates
Morphological characterization was performed using scanning electron microscopy (SEM) by JSM-6320F (Japan Electron Optics Laboratory Co., Tokyo, Japan) at Filgen (Filgen Inc., Nagoya, Japan). Gram-staining was assessed by using cells grown on culture media at 37°C for 48–96 h in anaerobic conditions with Gram-staining reagents (MUTO PURE CHEMICALS; Tokyo, Japan).
Source distribution and country specificity of novel bacterial isolates
Metagenomic short read archives (SRA) for 16S rRNA gene sequencing were retrieved at NCBI SRA BioSample database using keywords ‘16S’ and ‘Illumina’, resulting that all 20,508 BioSample entries have been obtained (23 May 2018). All entries were investigated by the OTU analysis with Deblur described above. The 16S rRNA sequence for each eight novel isolate was searched with megablast homology search against the above 20,508 entries at 99.0% nucleotide identity and 99.0% region coverage as threshold.
Funding Statement
Japan Agency for Medical Research and Development [JP16fk0108119]; Japan Agency for Medical Research and Development [JP17fk0108121]; Japan Agency for Medical Research and Development [JP17fk0108219]; Japan Agency for Medical Research and Development [JP16fk0108305].
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Authors’ contributions
MK designed the study concept. TI and NK performed genomic sequencing and bacterial isolation. TI, TS and AY analyzed the genomic data. TI wrote the manuscript, and MK revised the manuscript regarding intellectual content.
Acknowledgments
The authors would like to thank Nature Research Editing Service (http://authorservices.springernature.com/language-editing/) for the English language review, Filgen for the preparing samples and performing the scanning electron microscopy. The authors also thank to all members of the Pathogen Genomics Center lab for helpful discussions.
Declarations
Ethics approval and consent to participate
Written informed consent was obtained from the healthy volunteer. The study protocol was approved by the ethics committee of the National Institute of Infectious Diseases in Japan (Approval No. 642, 12/11/2015). It was conducted according to the principles of the Declaration of Helsinki.
Consent for publication
All authors approved the submission to BMC Microbiome.
Availability of data and material
The metagenome analysis short-read sequences have been deposited in the DNA Data Bank of Japan (DDBJ; accession numbers: DRA006636). Complete or draft genome sequences for novel 8 strains have been deposited in DDBJ (see Table 2). Bacterial isolates have been deposited at the Japan Collection of Microorganisms (JCM; http://jcm.brc.riken.jp/en/) (see Table 2).
Competing interests
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sender R, Fuchs S, Milo R.. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. [DOI] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonnenburg JL, Backhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. [DOI] [PubMed] [Google Scholar]
- 8.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols D, Cahoon N, Trakhtenberg EM, Pham L, Mehta A, Belanger A, Kanigan T, Lewis K, Epstein SS. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl Environ Microbiol. 2010;76:2445–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeraldo P, Hernandez A, Nielsen HB, Chen X, White BA, Goldenfeld N, Nelson H, Alhquist D, Boardman L, Chia N. Capturing one of the human gut microbiome’s most wanted: reconstructing the genome of a novel butyrate-producing, clostridial scavenger from metagenomic sequence data. Front Microbiol. 2016;7:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C, Bittar F, Fournous G, Gimenez G, Maraninchi M, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–1193. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka Y, Benno Y. Application of a single-colony coculture technique to the isolation of hitherto unculturable gut bacteria. Microbiol Immunol. 2015;59:63–70. [DOI] [PubMed] [Google Scholar]
- 13.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108:6252–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rettedal EA, Gumpert H, Sommer MO. Cultivation-based multiplex phenotyping of human gut microbiota allows targeted recovery of previously uncultured bacteria. Nat Commun. 2014;5:4714. [DOI] [PubMed] [Google Scholar]
- 15.Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533:543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau JT, Whelan FJ, Herath I, Lee CH, Collins SM, Bercik P, Surette MG. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 2016;8:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagier JC, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol. 2016;1:16203. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishijima S, Suda W, Oshima K, Kim SW, Hirose Y, Morita H, Hattori M. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 2016;23:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. [DOI] [PubMed] [Google Scholar]
- 22.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2 DOI: 10.1128/mSystems.00191-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–645. [DOI] [PubMed] [Google Scholar]
- 24.Lan GQ, Ho YW, Abdullah N. Mitsuokella jalaludinii sp. nov., from the rumens of cattle in Malaysia. Int J Syst Evol Microbiol. 2002;52:713–718. [DOI] [PubMed] [Google Scholar]
- 25.Ueki A, Goto K, Ohtaki Y, Kaku N, Ueki K. Description of Anaerotignum aminivorans gen. nov., sp. nov., a strictly anaerobic, amino-acid-decomposing bacterium isolated from a methanogenic reactor, and reclassification of Clostridium propionicum, Clostridium neopropionicum and Clostridium lactatifermentans as species of the genus Anaerotignum. Int J Syst Evol Microbiol. 2017;67:4146–4153. [DOI] [PubMed] [Google Scholar]
- 26.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 27.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato K, Hashino M, Ito T, Matsui M, Suzuki S, Kai K, Kitazume M, Sekizuka T, Kuroda M. Rapid and affordable size-selected PacBio single-molecule real-time sequencing template library construction using the bead-beating DNA extraction method. J Biol Methods. 2017;4:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 31.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2:Unit 2 3. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavalli-Sforza LL, Edwards AW. Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet. 1967;19:233–257. [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. [DOI] [PubMed] [Google Scholar]
- 35.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metagenome analysis short-read sequences have been deposited in the DNA Data Bank of Japan (DDBJ; accession numbers: DRA006636). Complete or draft genome sequences for novel 8 strains have been deposited in DDBJ (see Table 2). Bacterial isolates have been deposited at the Japan Collection of Microorganisms (JCM; http://jcm.brc.riken.jp/en/) (see Table 2).