

The reemergence of chikungunya virus (CHIKV), an alphavirus that is transmitted to humans by Aedes mosquitoes, is a serious global health threat. In the absence of effective antiviral drugs, CHIKV infection has a significant impact on human health, with chronic arthritis being one of the most serious complications. The molecular understanding of host-virus interactions is a prerequisite to the development of targeted therapeutics capable to interrupt viral replication and transmission. Here, we identify the host cell DHX9 DExH-Box helicase as an essential cofactor for early CHIKV genome translation. We demonstrate that CHIKV nsP3 protein acts as a key factor for DHX9 recruitment to replication complexes. Finally, we establish that DHX9 behaves as a switch that regulates the progression of the viral cycle from translation to genome replication. This study might therefore have a significant impact on the development of antiviral strategies.
KEYWORDS: chikungunya virus, DHX9, RNA helicase, nsP3, viral replication
ABSTRACT
Beyond their role in cellular RNA metabolism, DExD/H-box RNA helicases are hijacked by various RNA viruses in order to assist replication of the viral genome. Here, we identify the DExH-box RNA helicase 9 (DHX9) as a binding partner of chikungunya virus (CHIKV) nsP3 mainly interacting with the C-terminal hypervariable domain. We show that during early CHIKV infection, DHX9 is recruited to the plasma membrane, where it associates with replication complexes. At a later stage of infection, DHX9 is, however, degraded through a proteasome-dependent mechanism. Using silencing experiments, we demonstrate that while DHX9 negatively controls viral RNA synthesis, it is also required for optimal mature nonstructural protein translation. Altogether, this study identifies DHX9 as a novel cofactor for CHIKV replication in human cells that differently regulates the various steps of CHIKV life cycle and may therefore mediate a switch in RNA usage from translation to replication during the earliest steps of CHIKV replication.
IMPORTANCE The reemergence of chikungunya virus (CHIKV), an alphavirus that is transmitted to humans by Aedes mosquitoes, is a serious global health threat. In the absence of effective antiviral drugs, CHIKV infection has a significant impact on human health, with chronic arthritis being one of the most serious complications. The molecular understanding of host-virus interactions is a prerequisite to the development of targeted therapeutics capable to interrupt viral replication and transmission. Here, we identify the host cell DHX9 DExH-Box helicase as an essential cofactor for early CHIKV genome translation. We demonstrate that CHIKV nsP3 protein acts as a key factor for DHX9 recruitment to replication complexes. Finally, we establish that DHX9 behaves as a switch that regulates the progression of the viral cycle from translation to genome replication. This study might therefore have a significant impact on the development of antiviral strategies.
INTRODUCTION
The chikungunya virus (CHIKV), a mosquito-borne alphavirus transmitted by Aedes mosquitoes, represents an ongoing challenge to medicine and public health. The clinical manifestation of CHIKV infection is an acute syndrome (high fever, rash, myalgia, and intense arthralgia) that coincides with high viremia. In the absence of targeted therapeutics the infection evolves into a chronic incapacitating arthralgia in the distal joints in more than half of the cases, with patients requiring long-term administration of anti-inflammatory and immunosuppressive treatment (for a review, see reference 1). Because CHIKV recently caused major outbreaks worldwide with a disastrous socioeconomic impact and because antiviral molecules are still lacking, there is an urgent need to identify the mechanisms of infection that might be targeted therapeutically.
CHIKV genome is a 5′-m7GpppG capped and 3′-polyadenylated 11.8-kb positive-sense single-stranded RNA that contains two open reading frames encoding four nonstructural proteins (nsP1 to nsP4), three structural proteins (capsid and envelope glycoproteins E1 and E2), and three small cleavage products (E3, 6K, and TF). Once delivered in the host cell, the RNA genome is translated directly as the P1234 and P123 polyproteins which, after self-cleavage, will produce mature nonstructural proteins (nsPs): the RNA capping enzyme, nsP1; the RNA helicase/triphosphatase/NTPase/proteinase, nsP2; nsP3, which possesses phosphatase and RNA-binding activities; and the RNA-dependent RNA polymerase, nsP4 (2). The replication of the viral genome is initiated by the P123+nsP4 complex that synthesizes a negative-strand RNA [(–)RNA] copied from the incoming genome. During this step, nsPs/RNA complexes are targeted to host plasma membrane, where they anchor in the lipid bilayer thanks to membrane binding motifs in nsP1 (3–5). Further maturation of the P123 precursor then converts the replicase into a positive-strand RNA [(+)RNA] replicase to transcribe the (–)RNA into new full-length viral genomes and into subgenomic (+)RNAs used for capsid and envelope synthesis (5).
Several proteomic analysis have established nsP interaction with host proteins involved in RNA transport, splicing, and translation, thereby suggesting a close interplay of the virus replication machinery with the host RNA metabolism (6–9). One of these host proteins, DHX9, an essential nucleoside triphosphate (NTP)-dependent DExH box helicase that is also known as nuclear DNA helicase I and RNA helicase A, coimmunoprecipitates with Sindbis virus (SINV) nsPs (9) and copurifies with membranes of cells, supporting Semliki Forest virus (SFV) replication (8). This helicase has also been identified as a binding partner of CHIKV nsP3 when used as bait in yeast two-hybrid experiments (6). DHX9 is a ubiquitously expressed RNA helicase that is maintained at steady-state levels in the nucleus (10), while a fraction shuttles back and forth to the cytoplasm, where it associates with polyribosomes (11, 12). Its natural function is to unwind DNA and RNA structures thanks to its ability to bind nucleic acids with its N-terminal tandem double-stranded RNA (dsRNA)-binding domains and to hydrolyze NTPs with its two conserved RecA-like helicase domains (13, 14). DHX9 is, however, multifunctional and organizes most cellular processes implicating RNAs, including transcription, splicing, ribosome biogenesis, transport, miRNA processing, and translation of selected 5′ untranslated region (UTR)-structured mRNA (15–17). Because of its pleiotropic hallmarks, DHX9 has also been identified as a privileged partner during the replication of RNA viruses (Picornaviridae [18, 19], Orthomyxoviridae [20], Flaviviridae [21], and pestiviruses [22, 23]) even when they encode their own RNA helicases. The implication of cellular DHX9 has been particularly well documented in nearly every step of HIV-1 replication where its RNA unwinding activity contributes to the enhancement of viral gene transcription (24–27), and its expression assists the export of retroviral RNAs from the nucleus (26) and stimulates the translation of unspliced mRNA (15, 28). Nevertheless, since DHX9 senses dsRNA, cooperates with the antiviral signaling protein (IPS-1) to induce alpha interferon (IFN-α) secretion, and also binds the dsRNA binding kinase PKR, it may instead link foreign RNA recognition and the establishment of a type I IFN-dependent antiviral state (29, 30).
On account of the dual function that DHX9 may play in virus replication/sensing, the present study was designed to investigate DHX9-CHIKV interactions in infected cells and to establish the functional role played by this RNA helicase in CHIKV multiplication cycle. We demonstrate here that while DHX9 nuclear expression is significantly decreased in CHIKV-infected cells, a fraction of this protein is redistributed to plasma membrane-proximal cytoplasmic foci that stain positive for nsP3 and dsRNA replication forms. Using immunoprecipitation experiments, we established that DHX9 complexes with CHIKV genomic RNA and nsPs in infected cells and confirmed the capacity of DHX9 to directly bind nsP3, mainly in the hypervariable domain (HVD) in the absence of infection. Reducing the cellular pool of DHX9 with small interfering RNA or CRISPR/Cas9 genome editing significantly enhanced viral RNA synthesis in the long term, independently of type I IFN modulation. In contrast, early nsP expression is dramatically decreased by DHX9 knockdown, suggesting that the RNA helicase may play a proviral role during the earliest steps of the CHIKV life cycle. Altogether, this study identifies DHX9 as a new nsP3 binding partner that is recruited to replication complexes to regulate the various steps of CHIKV replication. Because of its capacity to promote nsP synthesis while interfering with RNA synthesis, DHX9 may coordinate nsP synthesis and RNA replication.
RESULTS
DHX9 is a binding partner of CHIKV nsPs in infected cells.
Despite having been identified as a CHIKV nsP3 binding partner in yeast two-hybrid experiments (6), interaction of the DHX9 helicase with nsP3 has not been investigated in infected cells. We therefore immunoprecipitated endogenous DHX9 from extracts of HeLa human epithelial cells infected with CHIKV for 8 h and separated DHX9-bound proteins using SDS-PAGE. Antisera directed to nsPs revealed the capacity of DHX9 to bind nsP2 and nsP3 components of the CHIKV replicase (Fig. 1A). Reciprocal immunoprecipitation reactions performed with antibodies raised against nsP3 allowed the detection of nsP1 and nsP2, as expected. It also revealed an enrichment in DHX9 (Fig. 1B), thereby confirming the capacity of DHX9 to be recruited by nsP3-containing complexes formed in infected cells. To determine which of the CHIKV nsPs physically interacts with DHX9, we individually overexpressed each one, fused to green fluorescent protein (GFP), together with a hemagglutinin (HA)-tagged DHX9 protein (HA-DHX9). Immunoprecipitation with anti-HA antibodies revealed that DHX9-HA efficiently bound GFP-nsP2 and GFP-nsP3 (Fig. 1C). No interaction and a faint background signal were observed for nsP4 and nsP1, respectively. Our data therefore show that DHX9 is a binding partner of the CHIKV replicase complex in infected cells that can preferentially bind nsP2 and nsP3 in the absence of other viral proteins or RNA.
FIG 1.

DHX9 helicase interacts with CHIKV nsPs in infected cells. Total lysates prepared from uninfected HeLa cells (NI) or from cells infected with CHIKV for 8 h were immunoprecipitated with anti-DHX9 (A) or anti-nsP3 serum (B). Samples of lysates (Input) and immunoprecipitates (IP) were separated by SDS-PAGE and probed with antisera against DHX9, CHIKV nsP1, nsP2, nsP3, or GAPDH MAbs as indicated. (C) HEK293T cells were cotransfected to express HA-DHX9, together with GFP or GFP-fused nsPs, as indicated. Cell lysates were prepared 36 h after transfection, precipitated with anti-HA agarose affinity beads, and separated by SDS-PAGE. Lysates and precipitated complexes were probed with MAbs against GFP, HA, or GAPDH. For each panel, the molecular masses are indicated.
NsP2 and nsP3 regulate DHX9 expression and subcellular distribution.
Next, we examined the subcellular localization of endogenous DHX9 with respect to exogenously expressed GFP-nsPs. Confocal imaging established that GFP-nsP1 distributed to the plasma membrane where it generated filopodium-like protrusions (Fig. 2A). GFP-nsP2 and GFP-nsP4 showed a diffuse cytoplasmic localization with GFP-nsP2 also present in the nucleus. Finally, GFP-nsP3 was detected as discrete cytoplasmic foci. These localization patterns of nsPs fused to a GFP tag resemble those of nsPs in CHIKV-infected cells (31–33). Immunofluorescence labeling of endogenous DHX9 established that the RNA helicase was mainly nuclear in control cells expressing the GFP protein. This profile was unchanged upon expression of GFP-nsP1 or GFP-nsP4. In contrast, while DHX9 was also mainly nuclear in cells expressing GFP-nsP3, a small fraction of the RNA helicase was also detected as cytoplasmic aggregates. This signal colocalized with the GFP-nsP3 fluorescence. Finally, when analyzed in cells expressing GFP-nsP2, DHX9 was exclusively detected in the nucleus. However, the fluorescent signal was significantly lower than in all other conditions tested. Statistical analysis of DHX9 staining in nsP2-expressing cells and in cells expressing GFP alone (Fig. 2B and C) confirmed that fluorescence intensity was significantly decreased upon nsP2 expression (P < 0.0001). Immunoblotting with anti-DHX9 antibodies confirmed that endogenous DHX9 level was reduced by 62 to 78% in lysates prepared from cells expressing GFP-nsP2 compared to lysates from the GFP control condition (Fig. 2D and E). Taken together, these results therefore show that the isolated expression of nsP2 decreases DHX9 nuclear detection, perhaps reflecting a cytotoxic effect of nsP2 (34, 35), while that of nsP3 relocalized a pool of DHX9 to cytoplasmic aggregates.
FIG 2.

GFP-nsP2 and GFP-nsP3 modulate DHX9 nuclear expression and subcellular localization, respectively. (A) HEK293T cells transfected to express either GFP or each of the GFP-fused nsPs (green) were stained for endogenous DHX9 (red) and analyzed by confocal microscopy. Nuclei were stained with DAPI (blue). Colocalized signals are indicated by arrows, and enlargements are shown in the inset. Scale bars, 5 μm. (B) Wide-field fluorescence microscopy of cells expressing the GFP-nsP2 protein or GFP alone and stained for endogenous DHX9. (C) Intensity of DHX9 nuclear staining was determined from cultures shown in panel B using ImageJ software. The number of cells analyzed for each condition is indicated. Mean values were compared using a Student t test. ****, P < 0.0001. (D) The endogenous DHX9 level in HEK293T cells was monitored by immunoblot analysis at various times after transfection of GFP or GFP-nsP2 expression plasmids. (E) The DHX9 band intensity in panel D was determined using ImageJ software. The results were normalized to the GAPDH signal and are expressed as percentages of the GFP condition.
DHX9 is a binding partner of the nsP3 hypervariable domain.
Increasing evidence shows that nsP3 function in alphavirus replication mainly relates to its capacity to recruit host factors required for optimal replication, including members of the Ras-GAP SH3 domain-binding protein (G3BP) family (36, 37) and the fragile X syndrome (FXR) protein family (38, 39), especially through its C-terminal hypervariable domain (HVD). To investigate which regions in nsP3 display DHX9 binding affinity, plasmids encoding the nsP3 N-terminal macrodomain (MD), the central unique domain (AUD) or the C-terminal HVD (Fig. 3A) with a N-terminal GFP tag were generated and cotransfected with an HA-DHX9 expression plasmid. While full-length GFP-nsP3 mainly forms discrete cytoplasmic foci, the GFP-MD and GFP-HVD fusion proteins generated a diffuse fluorescence both in the cytoplasm and in the nucleus (Fig. 3B). In contrast, the GFP-AUD protein was detected as fluorescent filament-like structures surrounding the nucleus reminiscent of profiles reported by others for nsP3 protein with its N-terminal moiety deleted (36, 40). Staining with anti-HA antibodies showed that HA-DHX9 localized to the nucleus in cells expressing GFP-MD or GFP-AUD and therefore remained unchanged compared to GFP-expressing cells. In contrast, a diffuse cytoplasmic fluorescence that colocalized to some extent with the green fluorescent signal was observed in the cytoplasm of cells expressing GFP-HVD (Fig. 3B). Next, HA-DHX9 binding affinity for each nsP3 domain was analyzed by immunoprecipitation. Cell lysates were prepared from each culture condition, immunoprecipitated with anti-HA and immunoblotted with anti-GFP antibodies. The full-length GFP-fused nsP3 protein and HVD were the only proteins reproducibly immunoprecipitated with HA-DHX9. Under less-stringent conditions, the GFP-MD protein was, however, also detected as a DHX9 binding domain (Fig. 3C). These results therefore identify HVD as a major contributor in nsP3/DHX9 interaction and suggest the possible additional contribution of MD in this interaction.
FIG 3.

CHIKV nsP3 binds DHX9 through its C-terminal HVD. (A) Schematic representation of GFP-nsP3 and its truncated derivatives. (B) HEK293T cells transfected to coexpress HA-DHX9, together with proteins depicted in panel A or with GFP, were stained with anti-DHX9 serum. Nuclei were stained with DAPI, and the cells were imaged by confocal microscopy. Scale bar, 5 μm. (C) Total cell extracts were prepared for each condition and precipitated with anti-HA agarose affinity beads. Proteins and bound complexes were visualized using antibodies against GFP, HA, or GAPDH, as indicated. Molecular masses are indicated.
DHX9 is relocalized to active replication complexes in CHIKV-infected cells.
To determine the relevance of nsP3/DHX9 interactions in the context of CHIKV infection, we assessed the localization pattern of endogenous DHX9 in cells infected with the CHIKV-377-mCherry reporter virus expressing an nsP3-mCherry protein (41). After 8 h of infection, DHX9 analyzed by immunofluorescence labeling and confocal microscopy mainly localized in the nuclear compartment (Fig. 4A). However, fluorescence was significantly less intense (70% reduction, P = 0.0007) in the nucleus of cells that stained positive for nsP3-mCherry and dsRNA (CHIKV replicating cells) compared to cells from the noninfected control or with the cells that remained nsP3 negative in the CHIKV condition due to the multiplicity of infection (MOI) of 0.5 used in this experiment (Fig. 4B). Immunoblot analysis of the corresponding cell lysates further confirmed the dramatic decrease in the steady-state level of DHX9 as CHIKV infection progresses over time (Fig. 4D). This depletion was prevented by the addition of ribavirin, an inhibitor of alphavirus replication, to the culture medium (Fig. 4C). It was also prevented by the proteasome inhibitor N-acetyl-Leu-Leu-Norleu-al (ALLN), suggesting that DHX9 depletion occurring during CHIKV replication is proteasome dependent (Fig. 4D). Concomitantly with its nuclear depletion, DHX9 was also detected at low levels in the proximity of the plasma membrane in cells that were positive for CHIKV infection. This signal overlapped both nsP3-mCherry fluorescence and dsRNA foci detected by means of specific monoclonal antibodies (MAbs) (Fig. 4A) and could not be observed either in mock-infected cells or in nsP3-mCherry-negative cells in CHIKV-infected cultures. In Old World alphaviruses, including CHIKV, nsP3 binds the GTPase-activating protein (SH3 domain)-binding protein 1 (G3BP), an essential stress granule component (37, 38, 42, 43). We exploited this by overexpressing a G3BP-GFP protein to investigate DHX9 relocalization with respect to this known nsP3-binding partner. When the cells were infected with CHIKV, the G3BP-GFP protein was recruited to nsP3-, dsRNA-positive foci, as expected (Fig. 5A). When DHX9 was analyzed with respect to these markers in CHIKV-infected cells, we found that the endogenous RNA helicase redirected to the cytoplasm and colocalized with nsP3 foci also overlaid the G3BP-GFP fluorescence (Fig. 5B). Altogether, these observations attest that in CHIKV-infected cells, a fraction of DHX9 is relocated to bona fide nsP3+/dsRNA+ functional CHIKV replication complexes and to G3BP-containing cytoplasmic foci.
FIG 4.

Consequences of CHIKV infection on DHX9 levels and its subcellular localization. (A) HeLa cells infected for 8 h with the CHIKV-377-mCherry virus (MOI = 0.5) were fixed and stained with antibodies against DHX9 and dsRNA. Uninfected cells (NI) are shown as a control. Scale bars, 10 μm. (B) The intensity of DHX9 nuclear staining in nsP3+ or nsP3– cells from CHIKV infection condition or in uninfected (NI) culture was determined using ImageJ software. Mean values were compared using a Student t test. ***, P < 0.001. (C) Total lysate of uninfected cells or that from cells infected with CHIKV for 16 h in the absence or presence of ribavirin were probed with anti-DHX9, anti-nsP3, and anti-capsid antibodies; anti-tubulin MAb was used as a loading control. (D) Infected cells maintained in medium alone (CHIKV) or culture in the presence of 10 μM ALLN (CHIKV+ALLN) were collected at different times postinfection and analyzed by immunoblotting with antibodies specific to DHX9, nsP3, and GAPDH.
FIG 5.

DHX9 is recruited to nsP3/G3BP complexes formed in the cytoplasm of CHIKV-infected cells. HeLa cells transfected to express the G3BP-GFP protein were left uninfected (NI) or were infected with the CHIKV-377-mCherry virus for 8 h. Cells were stained with anti-dsRNA antibodies (A) or anti-DHX9 serum (B) and processed for confocal imaging. Arrows indicate cytoplasmic foci with colocalized signals. For each panel, the lower lane shows an enlargement of the boxed zone. (C) Protein extracts prepared from uninfected cells or cells infected with the CHIKV-377-mCherry reporter virus for the indicated time were processed for immunoblot analysis of DHX9, nsP3, and G3PB expression (Input). Proteins complexed with nsP3 were immunoprecipitated (IP) with anti-mCherry MAbs and revealed by specific antibodies, as indicated.
DHX9 is recruited to viral RNA/nsPs complexes in CHIKV-infected cells.
Based on the evidence for colocalization reported above, we next tested the capacity of DHX9 to physically interact with CHIKV replication complexes. Lysates derived from cultures infected for 4 to 8 h (the DHX9 steady state is only poorly affected at this time [Fig. 4D]) with a CHIKV encoding a nsP3-mCherry fusion protein were immunoprecipitated using anti-mCherry MAbs, and electrophoretically separated and bound complexes were analyzed by immunoblotting. Under these conditions, endogenous DHX9 was detected as an nsP3-mCherry binding protein (Fig. 5C). Moreover, despite endogenous G3BP protein being barely detectable in total cell lysates (Fig. 5C), this CHIKV cofactor was also enriched in nsP3/DHX9 complexes. The presence of viral RNA in these complexes was addressed by RNA immunoprecipitation assays. To optimize the detection of CHIKV RNA, the cytosolic compartment was fractionated from cells infected for 6 h and subjected to immunoprecipitation with anti-DHX9 antiserum (Fig. 6). Reverse transcription-PCR (RT-PCR) analysis detected the presence of the viral genome in DHX9-containing immunocomplexes (Fig. 6D). RT-PCR signals recorded from noninfected cells immunoprecipitated with anti-DHX9 antiserum or from lysates of CHIKV-infected cells incubated with irrelevant antibodies unable to precipitate DHX9 (Fig. 6B) were below the detection limits of the assay, demonstrating the specificity of our results. These data therefore provide consistent evidence that in CHIKV-infected cells DHX9 is recruited to viral complexes that are positive for nsP3 and viral (+)RNA.
FIG 6.
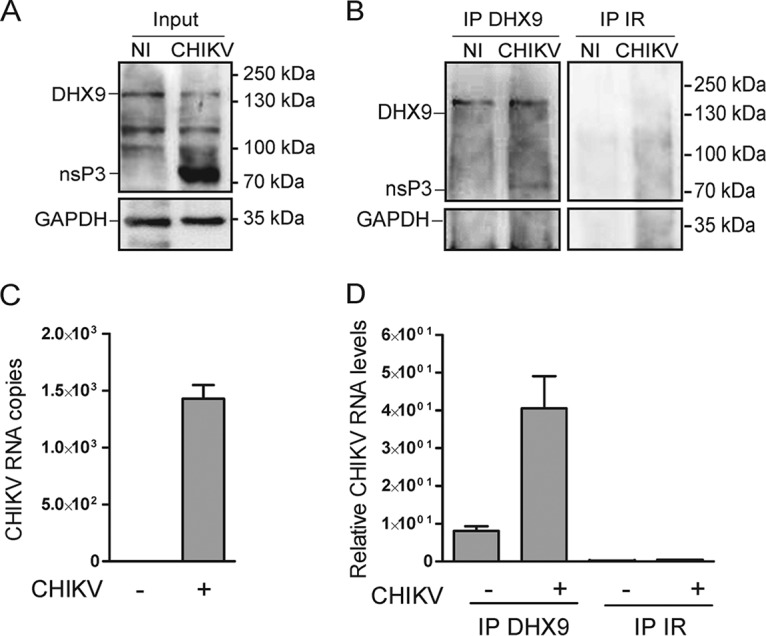
DHX9/nsP3 complexes formed in CHIKV-infected cells contain viral RNA. (A) Cytosolic extracts prepared from uninfected cells (NI) or from cells infected for 6 h with CHIKV-LR-5'GFP (CHIKV) were analyzed for DHX9, nsP3, and GAPDH expression. (B) Complexes immunoprecipitated with anti-DHX9 rabbit polyclonal serum or nonspecific IgG (IR) were analyzed for the presence of DHX9, nsP3, and GAPDH using specific antibodies. (C and D) RNA isolated from cytosolic fractions (C) or from immunoprecipitates prepared using the same concentration of cytosolic proteins for each condition (D) was subjected to qRT-PCR with primers specific for CHIKV RNA. For input samples, values were normalized according to GAPDH mRNA copies. Values are means of duplicates.
DHX9 negatively impacts CHIKV (–)RNA and (+)RNA synthesis.
A growing amount of evidence indicates that, among the DExD/H box helicase protein family, DHX9 can modulate the replication of a variety of RNA viruses, acting either as a beneficial factor and/or instead by creating an antiviral state, depending on the viral family considered (19, 20, 22, 28, 44, 45). To evaluate the contribution of DHX9 to the replication of CHIKV, we used two strategies. First, the genome of HEK293T cells was edited using CRISPR/Cas9 technology. All cell clones isolated expressed DHX9 at reduced levels (Fig. 7A, left panel). Such incomplete DHX9 extinction may reflect a heterozygous edit at the dhx9 locus, a situation that may corroborate previous evidence that complete inhibition of DHX9 expression has deleterious effects on cell cycle and viability (46, 47). These cells, referred to as DHX9+/–, were infected with CHIKV-LR-5′GFP in parallel with the parental cell line. CHIKV replication, determined by quantification of a virus-encoded GFP reporter in the cell lysate (Fig. 7B), was significantly higher in DHX9+/– cells than in the wild-type cells (P = 0.001) (Fig. 7B). The consequences of DHX9 inactivation on CHIKV infection were further validated by a one-step growth curve (Fig. 7C). A comparison of this curve with the parental HEK293T cell line revealed that DHX9 overexpression reduced the final CHIKV titers in culture supernatant by 3-fold at 24 h postinfection (P = 0.002). These data are therefore consistent with the prominent stimulating effect that silencing this protein has on CHIKV multiplication. Since cell growth and metabolism can be significantly modified by partial DHX9 knockout and/or during selection and amplification of new cell lines (47), these experiments were repeated using transient transfection of DHX9-specific shRNAs. After 48 h, DHX9 expression was reduced by 64 to 81% in the transfected culture (Fig. 7A, right panel). The cells were then infected with CHIKV-LR-5’GFP. Coherent with data obtained using DHX9+/– cells, CHIKV replication monitored by quantification of the virus-encoded GFP reporter was significantly higher in cells transfected with DHX9 shRNA than in control cells (Fig. 7D). To definitively validate the functional importance of DHX9 during CHIKV infection, we next questioned the consequences of DHX9 overexpression on CHIKV multiplication. HEK293T cells that were transfected either with an empty vector or with a plasmid encoding an HA-DHX9 protein (Fig. 7E) were infected with CHIKV-LR. Infectious progeny titers of CHIKV in culture supernatants determined overtime were 2- to 3-fold higher in the parental cell line than in cells expressing the HA-DHX9 protein (P = 0.0025) (Fig. 7F). The consequences of DHX9 inactivation were next studied at the level of CHIKV (–)RNA and (+)RNA synthesis. Coherent with monitoring of GFP expression in DHX9+/– cells, quantitative RT-PCR (qRT-PCR) analysis revealed that (+)RNA levels were higher in these cells than in the parental cells (Fig. 8B). Quantification of (–)RNA replication forms from the same samples mirrored that of (+)RNA (Fig. 8A). Finally, analyzing IFN-β mRNA levels from the same samples revealed no correlation with CHIKV RNA levels, thereby rejecting that DHX9 inhibitory effect may result from its previously reported function in the establishment of a type I IFN-dependent antiviral state (Fig. 8C) (29, 30). Taken together, the data obtained from DHX9 silencing and overexpression experiments indicated that this protein negatively regulated CHIKV growth, likely inhibiting the earliest steps of RNA genome replication.
FIG 7.

Consequences of DHX9 expression on CHIKV infection. (A) Parental HEK293T cells, cells edited by CRISPR/Cas9 (DHX9+/–), or cells transfected with nontargeting shRNA (shCtrl) or shRNA against DHX9 (#1 and #2) were analyzed for DHX9 and GAPDH expression by Western blotting. The DHX9/GAPDH ratio determined by band intensity analysis is indicated for each condition. (B) Replication of the CHIKV-LR-5′GFP reporter virus in DHX9+/– cells and in the parental cell line was monitored by quantification of GFP expression in total cell extracts. Values are a mean of triplicate experiments. (C) Each cell line was infected with CHIKV-LR. Viral production in culture supernatants was determined by a standard plaque assay at the indicated times postinfection. Values are means of triplicates. (D) Replication of the CHIKV-LR-5′GFP reporter virus in cells transfected with shCtrl or shRNA against DHX9 was monitored by quantification of GFP expression in total cell extracts. Values are a mean of triplicate experiments. (E) Cells transfected with an empty plasmid (Mock) or with a plasmid encoding a HA-DHX9 protein were analyzed by immunoblotting with the indicated antibodies. (F) Cells were infected with CHIKV-LR, and virus production was monitored overtime in culture supernatant by a plaque assay. Mock-transfected cells are shown as a control. Values are means of triplicates; values were compared using a Student t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
FIG 8.

DHX9 overexpression negatively regulates viral RNA synthesis independently of type I IFN expression. DHX9+/– cells and the parental HEK293T cells infected with the CHIKV-LR-5′GFP reporter virus for the indicated times were processed for qRT-PCR amplification of CHIKV (–)RNA (A) or (+)RNA (B). Noninfected cells (NI) are shown as a control. RNA levels were normalized according to GAPDH mRNA levels in the samples. Values were then normalized against an internal control, and results are expressed as arbitrary units. Analyses were performed in triplicate, and the error bars represent the standard deviations. (C) IFN-β mRNA was quantified from the same samples, as previously reported (74). GAPDH was used as an mRNA housekeeping control. Values are means of duplicates, and the error bars represent the standard deviations.
DHX9 expression is required for optimal nsP translation.
Next, we addressed DHX9 contribution in incoming genome translation. Uncoupling alphavirus genome translation from replication remains technically challenging. For this, we took advantage of a conditionally lethal mutation at position W258 in nsP1. The W258 residue of CHIKV corresponds to the W259 residue of the related SFV, where it acts as a key determinant for nsP1 interaction with membranes (48, 49). Alanine substitution at this site has a severe effect on the MT activity of nsP1 (4) and renders SFV defective in RNA replication (49). For CHIKV, it results in a severe temperature sensitivity phenotype: while having little effect at 28°C, the mutation hampers activity of CHIKV replicase at 37 to 39°C (50). At this temperature, viruses harboring the mutation are unable to replicate their genome but support the translation of nsPs. We therefore used a CHIKV-nsP1W258A-NanoLuc virus encoding an nsP3-fused nanoluciferase reporter gene (50) (Fig. 9A, upper panel) for parallel infection of cells transfected with either nontargeting or with DHX9-specific shRNAs. After 4 h in culture at a restrictive temperature (37°C), nsP translation (as measured by nanoluciferase activity in the cell lysates) was significantly less efficient in cells with reduced DHX9 expression (Fig. 9B). When the same experimental conditions were used to infect cells overexpressing the HA-DHX9 protein, the CHIKV-nsP1W258A-NanoLuc reporter virus generated an enhanced nanoluciferase expression with regard of cells transfected with an empty plasmid (Fig. 9C), attesting for an enhanced CHIKV RNA translation. DHX9 functional importance in CHIKV translation in the absence of replication was finally validated in the CRISPR/Cas9 edited cells. These cells were transfected with an in vitro-transcribed CHIKV RNA genome deleted of structural genes and most of the nsP4 sequence (amino acids [aa] 374 to 551 in nsP4) (Fig. 9D). Because the polymerase region has been deleted in this construct, this RNA cannot be replicated but serves as a template for nsP translation. At 36 h posttransfection, monitoring nsPs expression by immunoblotting showed a 80% reduction in DHX9+/– cells compared to the parental cell line. Our data therefore clearly illustrate that cellular DHX9 expression is required to ensure optimal expression of nsPs from incoming CHIKV RNA. Taken together, these results highlight a complex role for DHX9 during CHIKV multiplication cycle: while inhibiting CHIKV RNA replication, it enhances translation of the incoming virus genome. This dual role confers on DHX9 a potential key function in regulating the transition between different stages of the infectious cycle of this poorly known pathogen.
FIG 9.

DHX9 expression regulates nonstructural protein translation. (A) Mock-transfected HEK293T cells (WT) and cells transfected with the indicated shRNAs were analyzed for DHX9 and GAPDH expression. The cells were infected with CHIKV-nsP1W258A-NanoLuc (depicted in the upper panel). (B) After 4 h at a nonpermissive temperature (37°C), the nanoluciferase activity was determined in cell lysates and normalized to the protein concentration in the samples. ****, P < 0.0001; **, P < 0.01. (C) HEK293T cells transfected to overexpress HA-DHX9 were infected CHIKV-nsP1W258A-NanoLuc and maintained at 37°C for 4 h. NsP translation was monitored by quantification of the nanoluciferase activity in the cell lysate, and values were normalized according to the protein content in the sample. The expression of the HA-DHX9 transgene was controlled by immunoblotting the total cell extracts with the indicated antibodies. (D) Parental (WT) or DHX9 CRISPR/Cas9-edited HEK293T cells were transfected with the ΔnsP4 CHIKV replicon (depicted in the upper panel). At 8 h after transfection, cell lysates were analyzed for DHX9, GAPDH, and nsP expression. The band intensity was determined, and the nsP2/GAPDH ratios are indicated.
DISCUSSION
While our understanding of alphavirus-host protein interactions is still in its infancy, most of the nsP binding factors identified thus far, including members of the heterogeneous nuclear ribonucleoprotein family (hnRNP) (6, 8, 51), the HuR protein (52), ribosomal components such as RpS6 (53), and the stress granule components G3BPs (7, 54, 55), are proteins with function in RNA metabolism. In the present study, we focused on DHX9, a cellular RNA helicase belonging to the list of CHIKV nsP-binding factors (6) and investigated its function during the CHIKV infection cycle. Our first aim was to validate the nsPs/DHX9 interaction revealed by yeast two-hybrid analyses in infected human cells. We found that both nsP2 and nsP3 independently bound DHX9 in the absence of any other viral component. These proteins had a significant and different impact on the outcome of DHX9 even in the absence of infection. Indeed, nsP2 reduced nucleus DHX9 levels, indicating that nsP2 may be the factor responsible for the proteasome-dependent degradation of DHX9 observed in CHIKV-infected cells. This observation is reminiscent of the nsP2-triggered, Rpb1 RNA polymerase II (Pol II) subunit proteasome-dependent degradation (56) proposed as a basis for the inhibition of host gene expression and for Old World alphavirus-induced cytopathic effects (34, 57, 58). Since DHX9 is a component of the Pol II complex that can be excluded from the nucleus in the absence of Pol II complex association (12, 59), the decreased nuclear DHX9 level observed in nsP2-expressing cells may either reflect the direct nsP2/DHX9 interaction and/or the consequences of nsP2-induced loss of Rpb1. The respective contributions of these mechanisms therefore need to be evaluated.
In contrast to nsP2, the ectopic expression of nsP3 did not affect DHX9 detection in the nucleus. However, a fraction of DHX9 was relocalized to nsP3-positive cytoplasmic foci, where it colocalized and interacted with nsP3. This phenomenon was mainly triggered by the nsP3 HVD identified as a DHX9 binding domain. This result further extends the role played by alphavirus nsP3 in the recruitment, to replication complexes, of RNA-binding proteins required for optimal genome replication, especially through their intrinsically disordered HVD (38, 39, 54). This DHX9/nsP3 interaction is conserved in infected cells, in which it redirects a pool of DHX9 to discrete cytoplasmic foci, where it colocalizes and/or binds with at least two nsPs (we could not detect nsP4 due to its very low level in infected cells and the poor quality of anti-nsP4 antibodies), viral RNA, and the well-described CHIKV cofactor G3BP. These data therefore extend to CHIKV the capacity of phylogenetically distinct viruses, including picornaviruses (18, 19), arteriviruses (45) and pestiviruses (23), to relocalize DHX9 to cytoplasmic region where the viral genome is replicated.
Evaluating the functional importance of DHX9 recruitment to replication complexes, we found that it negatively regulates CHIKV (–)RNA and (+)RNA synthesis. Such a negative effect is unlikely to be due to the previously reported function of DHX9 in IFN signaling (29, 60), since the type I IFN-β mRNA levels did not differ in CHIKV-infected HEK293T cells with wild-type or reduced DHX9 expression. Since we suspected that DHX9 could interfere with the earliest steps of the CHIKV life cycle, we next considered its contribution in genomic RNA translation. This could be achieved using the CHIKV-nsP1W258A-NanoLuc mutant recently reported to be temperature dependent for replication (50). Unexpectedly, we found that DHX9 expression was clearly required for optimal nsPs expression from viral RNAs. Such a stimulatory effect may be assigned either to the stabilization of the incoming viral genomic RNA or the translated nsPs or, instead, to an improved translation of nsPs. Indeed, in addition to its known function in gene transcription (24, 59), RNA splicing (25), and export (26), the DHX9 unwinding activity also stimulates canonical cap-dependent translation of selected cellular and viral RNAs (15, 17, 61) by remodeling the highly structured posttranscriptional control element (PCE) in noncoding regions of JunD (15), p53 and type I collagen (17) mRNAs. Consistent with this capacity to overcome structural barriers to efficient ribosome scanning, DHX9 is also recruited to retroviral RNA PCE and to the structured 5′ and 3′ UTRs in viral genomes (19, 22, 62, 63), where it enhances translation (28, 63). The 5′ UTR in the alphavirus genome is predicted to be highly structured (64), a characteristic that is generally incompatible with efficient ribosome scanning during the cap-dependent translation mechanism used by these viruses (65). While the direct interaction of DHX9 with CHIKV RNA in the absence of nsP still remains to be investigated, DHX9 hijacking by reorganizing the structured RNA domains could be part of a unique strategy to ensure the optimal translation of its genome (Fig. 10). Instead, DHX9 could contribute to remodeling of the translation ribonucleoprotein complex to favors 43S preinitiation ribosome complex recruitment and subsequent scanning of mRNA (15, 17, 66). The potential contribution of these mechanisms in DHX9-dependent enhancement of CHIKV RNA translation therefore deserves further investigation.
FIG 10.

Model of DHX9-dependent regulation of the alphavirus translation-to-replication switch. DHX9 is recruited to the plasma membrane-bound replication complexes through interactions with the HVD domain in nsP3, where it enhances nsP translation. As translation and nsP precursor cleavage proceeds, the concentration of mature nsP2 increases, accounting for DHX9 unloading from the replication complex and redirection to proteasomal degradation. In the absence of DHX9, translation is shut down, and replication starting with (–)RNA synthesis is favored. G3BP recruitment to replication complexes is depicted in this model.
Unexpectedly, we observed that DHX9 has opposing effects on nsP translation and viral RNA replication. Interestingly, mutations disrupting RNA hairpins in alphavirus 5′ UTRs have been reported to decrease (–)RNA synthesis while increasing translation efficiency (64), mirroring the opposing roles played by DHX9 in the various steps of the CHIKV life cycle. Both RNA structures and DHX9 unwinding activities may therefore regulate the transition between the different replication steps, enhancing translation of the incoming genome while slowing down the progression to RNA synthesis. With regard to DHX9 function, transition from RNA translation to replication would therefore require DHX9 unloading from the viral RNA. Evidence provided for the requirement of a poliovirus 3CD protease-dependent digestion of viral RNA-bound PCBP2 to enable the switch from genome translation to replication argues in favor of this hypothesis (67). This may parallel the degradation of DHX9 observed as the CHIKV life cycle progresses. However, CHIKV may also use its protease (nsP2) for this purpose; though, unlike picornaviral 3CD, nsP2 does not cleave DHX9 but instead induces its degradation in proteasomes. Although the exact mechanisms involved still need to be elucidated, such regulation would confer on DHX9 a key function in orchestrating the dynamics of the earliest steps of the CHIKV multiplication cycle (Fig. 10).
In conclusion, our study provides new information on host cell factors recruited to the site of CHIKV replication and on the role played by host cell RNA helicases in Old World alphavirus replication. Our results parallel the role played by related DDX1 and DDX3 DExD/H box RNA helicases, two nsP3-binding partners, in the translation of the New World alphavirus VEEV (68). Recruitment by distantly related alphaviruses of various DExD/H helicases sharing similar RNA-binding/unwinding functions and therefore competent for reshaping structured RNA domains may therefore be part of an evolution process that ensures viral RNA translation. Because of the established involvement of DExD/H helicases in various human cancers, human senescence, and autoimmune diseases, strategies to produce inhibitors of their activities are currently undergoing development (16). Such inhibitors might also be considered for the future elaboration of antiviral strategies against alphaviruses.
MATERIALS AND METHODS
Cells.
HEK293T cells (ATCC ACS-4500), HeLa cells (ATCC CRM-CCL2), and BHK21 cells (ATCC CCL-10) used for propagation and Vero cells (ATCC CCL-81) used for titration of the CHIKV strains were cultured in Dulbecco modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (FCS; Lonza) and grown at 37°C, unless specified otherwise, in a 5% CO2 atmosphere.
Viruses and replicons.
The pCHIKV-LR-5′GFP (69), CHIKV-377-mCherry (41), CHIKV-nsP1W258A-NanoLuc (50), CHIKV-LRic (69) full-length CHIKV molecular clones and the ΔnsP4CHIKV replicon were transcribed in vitro from the SP6 promoter or the T7 promoter using an mMESSAGE mMACHINE kit (Ambion-Life Technologies). RNA (0.5 μg) was then electroporated into 5 × 106 BHK-21 cells, and the cells were incubated at 37°C for 24 h or at 28°C for 3 days in the case of CHIKV-nsP1W258A-NanoLuc. At indicated time, culture medium was collected, filtered through 0.45-μm-pore size membrane, divided into aliquots, and stored at –80°C. Titers of viral stocks were determined using a plaque assay, as previously reported (70).
Infection with CHIKV reporter viruses.
The cells (70 to 80% confluence) were rinsed once with phosphate-buffered saline (PBS) before viruses diluted to the desired MOI were added to the cells. For quantitative analysis of GFP reporter expression, the cells were lysed with radioimmunoprecipitation assay buffer. Fluorescence was measured directly from the cell lysate using an Infinite F200PRO fluorometer (Tecan). Luciferase expression was determined using a Dual-Glo luciferase assay system (Promega). Values were normalized according to protein content in the sample determined using a BCA assay (Pierce).
Virus titration assay.
Supernatants were titrated as previously described (70). Briefly, Vero cells, grown to 70 to 80% confluence, were incubated with four separate, 10-fold dilutions of viral supernatant in DMEM. After 2 h, the culture was overlaid with a mix of nutriment solution with agar (Lonza) and maintained at 37°C for 6 days. For plaque counting, the cells were incubated with 3.7% formaldehyde and 0.5% crystal violet in 20% ethanol. Results are expressed as PFU/ml.
Reagents and antibodies.
dsRNA was detected using mouse MAb J2 from Scicons. Anti-DHX9 antibodies (A300-855A) were from Bethyl Laboratories, anti-HA MAbs (H9658) from were Sigma-Aldrich, and anti-GAPDH serum (sc-25778) and anti-GFP (C-2) MAbs were from Santa Cruz Biotechnologies. Anti-CHIKV nsPs sera were rabbit polyclonal sera prepared in house. Secondary Alexa 647- or Alexa 488-conjugated or horseradish peroxidase (HRP)-coupled antibodies were purchased from Jackson/Life Technologies. Nuclei were stained with Hoechst 33342 (Sigma-Aldrich). Ribavirin, N-acetyl-Leu-Leu-Norleu-al (ALLN), and cycloheximide (Sigma-Aldrich) were used at final concentrations of 200 μM, 10 μM, and 50 μg/ml, respectively.
Plasmids.
Expression vector for the full-length HA-tagged DHX9 helicase (29) was kindly provided by Yong-Jun Liu (Center for Cancer Immunology Research, University of Texas). The G3BP1-GFP construct was a generous gift from Jamal Tazi (IGMM, CNRS, France). The sequences coding for CHIKV nsPs were subcloned into peGFP-C1 vectors using PCR amplification with the primers shown in Table 1 and pCHIKV-LR-5′GFP as a template. The fragments of nsP3, corresponding to three functional subdomains, were generated and cloned using the same approach; the corresponding primers are listed in Table 1.
TABLE 1.
Primer sequences
| Primer | Sequence (5′–3′) | Positions (nt) |
|---|---|---|
| nsP1-Fw | AGATCTCGAGCTCAAGCTTCGATGGATCCTGTGTACGTG | 77–94 |
| nsP1-Rev | TTAACCGTCGACTGCAGATCCTGCGCCCGCTCTGTC | 1667–1686 |
| nsP2-Fw | TCCGGACTCAGATCTCGAGCTATAATAGAGACTCCGAGAGGA | 1685–1705 |
| nsp2-Rev | GGATCCCGGGCCCCGCGGTACCACATCCTGCTCGGGTGAC | 4058–4076 |
| nsP3-Fw | TCCGGACTCAGATCTCGAGCTGCACCGTCGTACCGGGTA | 4075–4093 |
| nsP3-Rev | GGATCCCGGGCCCGCGGTACCCACCTGCCCTGTCTAG | 5648–5665 |
| nsP4-Fw | TCCGGACTCAGATCTCGAGCTTATATATTCTCGTCGGAC | 5666–5683 |
| nsp4-Rev | GGATCCCGGGCCCGCGTACCCTATTAGGACCGCCGTA | 7484–7507 |
| Macro-Fw | CAAGCTTCGAATTGTGCACCGTCGTACCGGGTAAAA | 4083–4096 |
| Macro-Rev | AGATCCGGTGGATCCTCACAGCTCTACTTGGGTCCGCAT | 4547–4570 |
| AUD-Fw | CAAGCTTCGAATTCTCTGGATAGCACATCTCCTAG | 4564–4585 |
| AUD-Rev | AGATCCGGAGGATCCTCAATATTCCCTTGGACTTACG | 5029–5047 |
| HVD-Fw | CAAGCTTCGAATTCTAGTCTTCCCAGGAGTCTGC | 5051–5067 |
| HVD-Rev | AGATCCGGTGGATCCTCAACCTGCCCTGTCTAGTCTTAA | 5660–5680 |
| CHIKV-F | GGCAGTGGTCCCAGATAATTCAAG | 3170–3193 |
| CHIKV-R | GCTGTCTAGATCCACCCCATACATG | 3229–3253 |
| CHIKV(–) | GGCAGTATCGTGAATTCGATGCCGCTGTACCGTCCCCATTCC | 6220–6239 |
| CHIKV(–)F | GGCAGTATCGTGAATTCGATGC | 6354–6364 |
| CHIKV(–)R | ACTGCTGAGTCCAAAGTGGG | 6311–6330 |
Immunoprecipitation analysis.
Cells transfected with JetPei transfection reagent (PolyPlus transfection) were lysed in 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM MgCl2, 1% Triton X-100, and protease inhibitors (Complete; Roche). A portion (50 μg) of total protein was kept as an input, and the remaining cell lysate was incubated with antibodies for 1 h at 4°C and with protein G-Sepharose beads (GE Healthcare) overnight at 4°C. Immunoprecipitation of HA-tagged proteins was performed by incubating 150 μg of total cellular proteins with EZview Red anti-HA affinity gel (Sigma-Aldrich) for 16 h at 4°C. The bead-immune complexes were washed six times with 1 ml of lysis buffer and released from the beads by boiling in 1× Laemmli buffer.
Western blot analyses.
Material separated using SDS-PAGE was transferred to polyvinylidene difluoride membrane (Immobilon P; Millipore). The membrane was incubated for 2 h with appropriate primary antibodies, washed with PBS–0.1% Tween 20 (pH 7.5), and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After extensive washings, the protein bands were detected using Luminata Forte detection reagent (Merck Millipore) and a GeneGnome XRQ device (Syngene).
Immunofluorescence labeling and confocal microscopy.
Cells grown on a glass coverslip were fixed with 4% formaldehyde/PBS (Sigma-Aldrich), permeabilized with 0.1% Triton X-100 in PBS, and blocked in PBS–2% FCS. Incubation with primary antibody was performed at 37°C for 1 h at room temperature, and secondary reagents were added for 30 min at 37°C. DAPI (4′,6′-diamidino-2-phenylindole; Sigma-Aldrich) was used to stain the nucleus. After the final washes, coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Images were acquired using a Leica SP5-SMD scanning confocal microscope equipped with a 63×, 1.4 numerical aperture Leica Apochromat oil lens at the Montpellier Resources Imaging platform. Image processing and colocalization analysis were performed using ImageJ software.
RNA immunoprecipitation.
RNA immunoprecipitation was performed using the Magna RIP RNA-binding protein immunoprecipitation kit protocol (Merck Millipore). A total of 2 × 107 HEK293T cells were infected with CHIKV at an MOI of 5 for 8 h and then harvested. The cells were subjected to subcellular fractionation using a ProteoJET cytoplasmic and nuclear protein extraction kit (Fermentas). One-tenth of the obtained lysate was kept as an input, and the remaining lysate was brought to a volume of 1 ml with immunoprecipitation buffer supplemented with 0.5 M EDTA and 5 μl of RNase inhibitor. Samples were incubated overnight at 4°C in the presence of 5 μg of anti-DHX9 rabbit polyclonal serum (A300-855A; Bethyl Laboratories) or irrelevant antibodies and 50 μl of magnetic beads. Bound complexes were captured with a magnetic separator. After washing steps, one-tenth of the immunocomplexes was separated using SDS-PAGE and visualized using anti-DHX9 serum (A300-855A; Bethyl Laboratories) or anti-nsP3 in-house rabbit polyclonal serum or anti-GAPDH MAbs (sc-25778; Santa Cruz Biotechnologies). The remaining samples were subjected to RNA extraction and qRT-PCR quantification of CHIKV genome.
Quantitative RT-PCR detection.
Viral and cellular RNAs were extracted using a TRIzol-based protocol and tested for mRNA purity and concentration using light absorbance. RNA (0.1 μg) was converted to cDNA with an oligo(dT12–18) primer (Invitrogen) and Superscript III reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. PCR amplification of (+)RNA was carried out on one-tenth of the cDNA in a reaction mix containing 0.4 μM CHIKV-F and CHIKV-R primers matching the nsP2 encoding region of the virus genome, as well as 5 μl of SYBR Green master amplification mix (Faststart DNA Master plus SYBR Green amplification kit; Roche Diagnostics). Reactions were subjected to a first cycle of 10 min at 95°C, followed by 40 amplification cycles of 15 s at 95°C and 60 s at 60°C on a Rotor Gene System (Labgene Scientific). The fluorescence signal was recorded at the end of each cycle. A standard curve was generated from 101 to 106 copies of pCHIKV-LR-5′GFP plasmid. To detect CHIKV (–)RNA, RNA samples were reverse transcribed using CHIKV(–)R primer. cDNAs were treated with exonucleases for 20 min and then subjected to PCR amplification with CHIKV(–)F and CHIKV(–)R primers, as previously described (71). The primers used are listed in Table 1.
RNA interference.
shRNA targeting DHX9 mRNA was achieved by transfection of HEK293T cells with the following pLKO plasmids: sh#1 (5′-CCGGACGACAATGGAAGCGGATATACTCGAGTATATCCGCTTCCATTGTCGTTTTTTG-3′) and sh#2 (5′-CCGGGGGCTATATCCATCGAAATTTCTCGAGAAATTTCGATGGATATAGCCCTTTTTG-3′) (Sigma-Aldrich). Briefly, subconfluent cultures of HEK293T cells were transfected with plasmids (3 μg) using Lipofectamine 2000 reagent (Thermo Fisher). At 48 h posttransfection, an aliquot of the cells was harvested to determine the silencing efficiency by Western blotting. The remaining cells were infected with CHIKV at an MOI of 0.5.
DHX9 knockout by CRISPR/Cas9 genome edition.
The plasmids for CRISPR-Cas9 were obtained from the Montpellier Genomic Collection Platform (Biocampus, Montpellier, France). Guide RNA targeting DHX9 DNA was designed using three online gRNA-optimizing softwares: CRISPR design (http://crispr.mit.edu), CRISPR RNA Configurator (http://dharmacon.gelifesciences.com/gene-editing/edit-r/custom-crrna), and CRISPR gRNA Design tool (https://www.dna20.com/eCommerce/cas9). gFH9 (5′-CACCGCATCTTCCTTTTGCCACACC-3′) and gRH9 (5′-AAACGGTGTGGCAAAAGGAAGATGC-3′) oligonucleotides (nucleotides [nt] 207 to 225 of human DHX9 mRNA [NM_001357]) were hybridized and cloned into pUC57 attB U6 gRNA vectors (72). The generated plasmid pUC57 attB U6 DHX9gRNA was transfected into HEK293T cells with Lipofectamine 2000, along with the pSpCas9(BB)-2A-GFP (PX458) plasmid (73). At 6 h after transfection, the cells were trypsin treated and resuspended in complete DMEM at 2 × 104 cells per ml. Portions (200 μl) of the cell suspension (4 × 103 cells) were used to inoculate 96-well plates and to isolate single cell-derived clones by serial dilution. Isolated green fluorescent protein (GFP)-positive clones were amplified to cell lines and analyzed by Western blotting for DHX9 expression.
Statistical analysis.
All of the analyses (unpaired Student t test) were performed using GraphPad Prism version 6 (GraphPad Software). A P value of <0.05 was considered statistically significant (**, P < 0.05; ***, P < 0.001; ****, P < 0.0001).
ACKNOWLEDGMENTS
We thank the Montpellier Rio Imaging facility for imaging assistance. We also thank Ian Robbins (IGMM, Montpellier, France) for critical reading of the manuscript and William Bakhache for technical assistance with confocal imaging.
This study was supported by the Agence Nationale de la Recherche (ANR) (L.B.), Government of New Caledonia (R.M.), and the Fondation pour la Recherche Médicale (FRM; ING20130526799) (S.F.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
The authors declare that no competing interests exist.
REFERENCES
- 1.Thiberville SD, Moyen N, Dupuis-Maguiraga L, Nougairede A, Gould EA, Roques P, de Lamballerie X. 2013. Chikungunya fever: epidemiology, clinical syndrome, pathogenesis, and therapy. Antiviral Res 99:345–370. doi: 10.1016/j.antiviral.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupp JC, Sokoloski KJ, Gebhart NN, Hardy RW. 2015. Alphavirus RNA synthesis and nonstructural protein functions. J Gen Virol 96:2483–2500. doi: 10.1099/jgv.0.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spuul P, Balistreri G, Kaariainen L, Ahola T. 2010. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest virus replication complexes from the plasma membrane to modified lysosomes. J Virol 84:7543–7557. doi: 10.1128/JVI.00477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahola T, Lampio A, Auvinen P, Kaariainen L. 1999. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J 18:3164–3172. doi: 10.1093/emboj/18.11.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemm JA, Rumenapf T, Strauss EG, Strauss JH, Rice CM. 1994. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: a model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J 13:2925–2934. doi: 10.1002/j.1460-2075.1994.tb06587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourai M, Lucas-Hourani M, Gad HH, Drosten C, Jacob Y, Tafforeau L, Cassonnet P, Jones LM, Judith D, Couderc T, Lecuit M, Andre P, Kummerer BM, Lotteau V, Despres P, Tangy F, Vidalain PO. 2012. Mapping of chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J Virol 86:3121–3134. doi: 10.1128/JVI.06390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, MacDonald MR. 2006. Tracking and elucidating alphavirus-host protein interactions. J Biol Chem 281:30269–30278. doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- 8.Varjak M, Saul S, Arike L, Lulla A, Peil L, Merits A. 2013. Magnetic fractionation and proteomic dissection of cellular organelles occupied by the late replication complexes of Semliki Forest virus. J Virol 87:10295–10312. doi: 10.1128/JVI.01105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. 2010. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J Virol 84:6720–6732. doi: 10.1128/JVI.01983-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchsova B, Novak P, Kafkova J, Hozak P. 2002. Nuclear DNA helicase II is recruited to IFN-alpha-activated transcription sites at PML nuclear bodies. J Cell Biol 158:463–473. doi: 10.1083/jcb.200202035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aratani S, Oishi T, Fujita H, Nakazawa M, Fujii R, Imamoto N, Yoneda Y, Fukamizu A, Nakajima T. 2006. The nuclear import of RNA helicase A is mediated by importin-α3. Biochem Biophys Res Commun 340:125–133. doi: 10.1016/j.bbrc.2005.11.161. [DOI] [PubMed] [Google Scholar]
- 12.Fujita H, Ohshima T, Oishi T, Aratani S, Fujii R, Fukamizu A, Nakajima T. 2005. Relevance of nuclear localization and functions of RNA helicase A. Int J Mol Med 15:555–560. [PubMed] [Google Scholar]
- 13.Zhang S, Grosse F. 2004. Multiple functions of nuclear DNA helicase II (RNA helicase A) in nucleic acid metabolism. Acta Biochim Biophys Sin (Shanghai) 36:177–183. doi: 10.1093/abbs/36.3.177. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Grosse F. 1997. Domain structure of human nuclear DNA helicase II (RNA helicase A). J Biol Chem 272:11487–11494. doi: 10.1074/jbc.272.17.11487. [DOI] [PubMed] [Google Scholar]
- 15.Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris-Lawrie K. 2006. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol 13:509–516. doi: 10.1038/nsmb1092. [DOI] [PubMed] [Google Scholar]
- 16.Lee T, Pelletier J. 2016. The biology of DHX9 and its potential as a therapeutic target. Oncotarget 7:42716–42739. doi: 10.18632/oncotarget.8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manojlovic Z, Stefanovic B. 2012. A novel role of RNA helicase A in regulation of translation of type I collagen mRNAs. RNA 18:321–334. doi: 10.1261/rna.030288.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence P, Conderino JS, Rieder E. 2014. Redistribution of demethylated RNA helicase A during foot-and-mouth disease virus infection: role of Jumonji C-domain containing protein 6 in RHA demethylation. Virology 452-453:1–11. doi: 10.1016/j.virol.2013.12.040. [DOI] [PubMed] [Google Scholar]
- 19.Lawrence P, Rieder E. 2009. Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J Virol 83:11356–11366. doi: 10.1128/JVI.02677-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin L, Li Y, Pyo HM, Lu X, Raman SN, Liu Q, Brown EG, Zhou Y. 2012. Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J Virol 86:1942–1954. doi: 10.1128/JVI.06362-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Masaki T, Shimakami T, Lemon SM. 2014. hnRNP L and NF90 interact with hepatitis C virus 5′-terminal untranslated RNA and promote efficient replication. J Virol 88:7199–7209. doi: 10.1128/JVI.00225-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng C, Yao Y, Chen B, Wang Y, Chen J, Xiao M. 2013. RNA helicase is involved in the expression and replication of classical swine fever virus and interacts with untranslated region. Virus Res 171:257–261. doi: 10.1016/j.virusres.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Jefferson M, Donaszi-Ivanov A, Pollen S, Dalmay T, Saalbach G, Powell PP. 2014. Host factors that interact with the pestivirus N-terminal protease, Npro, are components of the ribonucleoprotein complex. J Virol 88:10340–10353. doi: 10.1128/JVI.00984-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujii R, Okamoto M, Aratani S, Oishi T, Ohshima T, Taira K, Baba M, Fukamizu A, Nakajima T. 2001. A role of RNA helicase A in cis-acting transactivation response element-mediated transcriptional regulation of human immunodeficiency virus type 1. J Biol Chem 276:5445–5451. doi: 10.1074/jbc.M006892200. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Tang H, Mullen TM, Westberg C, Reddy TR, Rose DW, Wong-Staal F. 1999. A role for RNA helicase A in posttranscriptional regulation of HIV type 1. Proc Natl Acad Sci U S A 96:709–714. doi: 10.1073/pnas.96.2.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy TR, Tang H, Xu W, Wong-Staal F. 2000. Sam68, RNA helicase A and Tap cooperate in the posttranscriptional regulation of human immunodeficiency virus and type D retroviral mRNA. Oncogene 19:3570–3575. doi: 10.1038/sj.onc.1203676. [DOI] [PubMed] [Google Scholar]
- 27.Xing L, Niu M, Kleiman L. 2014. Role of the OB-fold of RNA helicase A in the synthesis of HIV-1 RNA. Biochim Biophys Acta 1839:1069–1078. doi: 10.1016/j.bbagrm.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Bolinger C, Sharma A, Singh D, Yu L, Boris-Lawrie K. 2010. RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res 38:1686–1696. doi: 10.1093/nar/gkp1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. 2011. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol 187:4501–4508. doi: 10.4049/jimmunol.1101307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Wang L, Berman M, Kong YY, Dorf ME. 2011. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peranen J, Rikkonen M, Liljestrom P, Kaariainen L. 1990. Nuclear localization of Semliki Forest virus-specific nonstructural protein nsP2. J Virol 64:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorchakov R, Garmashova N, Frolova E, Frolov I. 2008. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol 82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Utt A, Quirin T, Saul S, Hellstrom K, Ahola T, Merits A. 2016. Versatile trans-replication systems for chikungunya virus allow functional analysis and tagging of every replicase protein. PLoS One 11:e0151616. doi: 10.1371/journal.pone.0151616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garmashova N, Gorchakov R, Frolova E, Frolov I. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol 80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fros JJ, van der Maten E, Vlak JM, Pijlman GP. 2013. The C-terminal domain of chikungunya virus nsP2 independently governs viral RNA replication, cytopathicity, and inhibition of interferon signaling. J Virol 87:10394–10400. doi: 10.1128/JVI.00884-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panas MD, Ahola T, McInerney GM. 2014. The C-terminal repeat domains of nsP3 from the Old World alphaviruses bind directly to G3BP. J Virol 88:5888–5893. doi: 10.1128/JVI.00439-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim DY, Reynaud JM, Rasalouskaya A, Akhrymuk I, Mobley JA, Frolov I, Frolova EI. 2016. New World and Old World alphaviruses have evolved to exploit different components of stress granules, FXR and G3BP proteins, for assembly of viral replication complexes. PLoS Pathog 12:e1005810. doi: 10.1371/journal.ppat.1005810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frolov I, Kim DY, Akhrymuk M, Mobley JA, Frolova EI. 2017. Hypervariable domain of eastern equine encephalitis virus nsP3 redundantly utilizes multiple cellular proteins for replication complex assembly. J Virol 91:e00371-17. doi: 10.1128/JVI.00371-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fros JJ, Geertsema C, Zouache K, Baggen J, Domeradzka N, van Leeuwen DM, Flipse J, Vlak JM, Failloux AB, Pijlman GP. 2015. Mosquito Rasputin interacts with chikungunya virus nsP3 and determines the infection rate in Aedes albopictus. Parasit Vectors 8:464. doi: 10.1186/s13071-015-1070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kummerer BM, Grywna K, Glasker S, Wieseler J, Drosten C. 2012. Construction of an infectious chikungunya virus cDNA clone and stable insertion of mCherry reporter genes at two different sites. J Gen Virol 93:1991–1995. doi: 10.1099/vir.0.043752-0. [DOI] [PubMed] [Google Scholar]
- 42.Foy NJ, Akhrymuk M, Akhrymuk I, Atasheva S, Bopda-Waffo A, Frolov I, Frolova EI. 2013. Hypervariable domains of nsP3 proteins of New World and Old World alphaviruses mediate formation of distinct, virus-specific protein complexes. J Virol 87:1997–2010. doi: 10.1128/JVI.02853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Remenyi R, Gao Y, Hughes RE, Curd A, Zothner C, Peckham M, Merits A, Harris M. 2018. Persistent replication of a chikungunya virus replicon in human cells is associated with presence of stable cytoplasmic granules containing nonstructural protein 3. J Virol 92:e00477-18. doi: 10.1128/JVI.00477-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolinger C, Yilmaz A, Hartman TR, Kovacic MB, Fernandez S, Ye J, Forget M, Green PL, Boris-Lawrie K. 2007. RNA helicase A interacts with divergent lymphotropic retroviruses and promotes translation of human T-cell leukemia virus type 1. Nucleic Acids Res 35:2629–2642. doi: 10.1093/nar/gkm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Tian J, Nan H, Tian M, Li Y, Xu X, Huang B, Zhou E, Hiscox JA, Chen H. 2016. Porcine reproductive and respiratory syndrome virus nucleocapsid protein interacts with Nsp9 and cellular DHX9 to regulate viral RNA synthesis. J Virol 90:5384–5398. doi: 10.1128/JVI.03216-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee T, Di Paola D, Malina A, Mills JR, Kreps A, Grosse F, Tang H, Zannis-Hadjopoulos M, Larsson O, Pelletier J. 2014. Suppression of the DHX9 helicase induces premature senescence in human diploid fibroblasts in a p53-dependent manner. J Biol Chem 289:22798–22814. doi: 10.1074/jbc.M114.568535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee T, Paquet M, Larsson O, Pelletier J. 2016. Tumor cell survival dependence on the DHX9 DExH-box helicase. Oncogene 35:5093–5105. doi: 10.1038/onc.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lampio A, Kilpelainen I, Pesonen S, Karhi K, Auvinen P, Somerharju P, Kaariainen L. 2000. Membrane binding mechanism of an RNA virus-capping enzyme. J Biol Chem 275:37853–37859. doi: 10.1074/jbc.M004865200. [DOI] [PubMed] [Google Scholar]
- 49.Spuul P, Salonen A, Merits A, Jokitalo E, Kaariainen L, Ahola T. 2007. Role of the amphipathic peptide of Semliki Forest virus replicase protein nsP1 in membrane association and virus replication. J Virol 81:872–883. doi: 10.1128/JVI.01785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bartholomeeusen K, Utt A, Coppens S, Rausalu K, Vereecken K, Arien KK, Merits A. 2018. A chikungunya virus trans-replicase system reveals the importance of delayed nonstructural polyprotein processing for efficient replication complex formation in mosquito cells. J Virol 92:e00152-18. doi: 10.1128/JVI.00152-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. 2006. Formation of nsP3-specific protein complexes during Sindbis virus replication. J Virol 80:4122–4134. doi: 10.1128/JVI.80.8.4122-4134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sokoloski KJ, Dickson AM, Chaskey EL, Garneau NL, Wilusz CJ, Wilusz J. 2010. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 8:196–207. doi: 10.1016/j.chom.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montgomery SA, Berglund P, Beard CW, Johnston RE. 2006. Ribosomal protein S6 associates with alphavirus nonstructural protein 2 and mediates expression from alphavirus messages. J Virol 80:7729–7739. doi: 10.1128/JVI.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scholte FE, Tas A, Albulescu IC, Zusinaite E, Merits A, Snijder EJ, van Hemert MJ. 2015. Stress granule components G3BP1 and G3BP2 play a proviral role early in chikungunya virus replication. J Virol 89:4457–4469. doi: 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akhrymuk I, Kulemzin SV, Frolova EI. 2012. Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol 86:7180–7191. doi: 10.1128/JVI.00541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamm K, Merits A, Sarand I. 2008. Mutations in the nuclear localization signal of nsP2 influencing RNA synthesis, protein expression and cytotoxicity of Semliki Forest virus. J Gen Virol 89:676–686. doi: 10.1099/vir.0.83320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fros JJ, Major LD, Scholte FE, Gardner J, van Hemert MJ, Suhrbier A, Pijlman GP. 2015. Chikungunya virus nonstructural protein 2-mediated host shut-off disables the unfolded protein response. J Gen Virol 96:580–589. doi: 10.1099/vir.0.071845-0. [DOI] [PubMed] [Google Scholar]
- 59.Aratani S, Fujii R, Oishi T, Fujita H, Amano T, Ohshima T, Hagiwara M, Fukamizu A, Nakajima T. 2001. Dual roles of RNA helicase A in CREB-dependent transcription. Mol Cell Biol 21:4460–4469. doi: 10.1128/MCB.21.14.4460-4469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rahman MM, Liu J, Chan WM, Rothenburg S, McFadden G. 2013. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog 9:e1003465. doi: 10.1371/journal.ppat.1003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halaby MJ, Harris BR, Miskimins WK, Cleary MP, Yang DQ. 2015. Deregulation of internal ribosome entry site-mediated p53 translation in cancer cells with defective p53 response to DNA damage. Mol Cell Biol 35:4006–4017. doi: 10.1128/MCB.00365-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paingankar MS, Arankalle VA. 2015. Identification and characterization of cellular proteins interacting with hepatitis E virus untranslated regions. Virus Res 208:98–109. doi: 10.1016/j.virusres.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692. doi: 10.1261/rna.594207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nickens DG, Hardy RW. 2008. Structural and functional analyses of stem-loop 1 of the Sindbis virus genome. Virology 370:158–172. doi: 10.1016/j.virol.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Joubert PE, Stapleford K, Guivel-Benhassine F, Vignuzzi M, Schwartz O, Albert ML. 2015. Inhibition of mTORC1 enhances the translation of chikungunya proteins via the activation of the MnK/eIF4E pathway. PLoS Pathog 11:e1005091. doi: 10.1371/journal.ppat.1005091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin J, Jing W, Lei XX, Feng C, Peng S, Boris-Lawrie K, Huang Y. 2011. Evidence that Lin28 stimulates translation by recruiting RNA helicase A to polysomes. Nucleic Acids Res 39:3724–3734. doi: 10.1093/nar/gkq1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chase AJ, Daijogo S, Semler BL. 2014. Inhibition of poliovirus-induced cleavage of cellular protein PCBP2 reduces the levels of viral RNA replication. J Virol 88:3192–3201. doi: 10.1128/JVI.02503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amaya M, Brooks-Faulconer T, Lark T, Keck F, Bailey C, Raman V, Narayanan A. 2016. Venezuelan equine encephalitis virus nonstructural protein 3 (nsP3) interacts with RNA helicases DDX1 and DDX3 in infected cells. Antiviral Res 131:49–60. doi: 10.1016/j.antiviral.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. 2006. Infectious clones of chikungunya virus (La Reunion isolate) for vector competence studies. Vector Borne Zoonotic Dis 6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- 70.Bernard E, Solignat M, Gay B, Chazal N, Higgs S, Devaux C, Briant L. 2010. Endocytosis of chikungunya virus into mammalian cells: role of clathrin and early endosomal compartments. PLoS One 5:e11479. doi: 10.1371/journal.pone.0011479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Plaskon NE, Adelman ZN, Myles KM. 2009. Accurate strand-specific quantification of viral RNA. PLoS One 4:e7468. doi: 10.1371/journal.pone.0007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC. 2014. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 73.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernard E, Hamel R, Neyret A, Ekchariyawat P, Moles JP, Simmons G, Chazal N, Despres P, Misse D, Briant L. 2014. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 476C:1–10. doi: 10.1016/j.virol.2014.11.013. [DOI] [PubMed] [Google Scholar]